Submitted:
26 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
Keywords: Antisense Oligonucleotides, Cell Penetrating Peptides, Delivery, DG9 peptide, Phosphorodiamidate morpholino oligomers (PMO), Pip, R6G.
Keywords:
Antisense Oligonucleotides
; Cell Penetrating Peptides
; Delivery
; DG9 peptide
; Phosphorodi-amidate morpholino oligomers (PMO)
; Pip
; R6G
1. Introduction
The advancement of antisense oligonucleotides (ASOs) has brought about a profound change in the field of genetic therapeutics, offering a promising avenue for addressing a diverse array of diseases on a molecular level. ASOs are short synthetic nucleic acid analogs that offer a revolutionary means to modulate gene expression by precisely interacting with RNA transcripts. The history of ASO can be traced back to the pioneering work of Zamecnik and Stephenson in early 1970, who first proposed the concept of using synthetic oligonucleotides to regulate eukaryotic gene expression in cultured cells through sequence-specific hybridization with RNA [1,2]. Later, the pharmacokinetic properties of ASOs, such as stability, reduced susceptibility to nuclease degradation, specificity, and cellular absorption have been greatly improved by developments in oligonucleotide chemistry, including the introduction of chemical modifications and different backbone structures which transformed them from theoretical concepts into potentially effective therapeutic agents [3].
ASOs have been successfully employed in treating a wide range of diseases, including Duchenne muscular dystrophy (DMD), spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS), and many more which led to the regulatory approval of 10 ASO-based drugs till now [4] and many antisense drug candidates to clinical trials to treat cardiovascular, metabolic, endocrine, neurological, neuromuscular, inflammatory, and infectious diseases [5]. This demonstrates the dynamic nature of ASO-mediated therapy. Despite being a promising approach, it is widely accepted that delivery of ASO treatments to specific tissues is limited by factors such as intracellular trafficking, degradation in biological fluids, and transportation across cellular barriers [6]. Although chemical modifications have improved their metabolic stabilities significantly as well as their affinities for RNA targets and have to some extent reduced off-target effects, no chemical modification has significantly improved cellular uptake or tissue targeting.
Cell-penetrating peptides (CPPs) or peptide transduction domains (PTDs) are one of the many approaches that have been developed to improve the delivery of oligonucleotides. CPPs are small peptides with the ability to transport cargos, including ASOs across cellular barriers and hereby offer the potential to improve ASOs’ cellular uptake and intracellular distribution, enhancing the therapeutic outcomes and reducing the required dosage [7]. The first CPP was introduced in 1946 and since then there has been a continuous effort of developing a more efficient cell-penetrating peptide that can ensure increased delivery of oligonucleotides and better pharmacological properties [8].
Particularly in the context of phosphorodiamidate morpholino oligomers (PMOs), R6G, PiP (PNA/PMO Internalizing Peptides) and DG9 have captured interest among the CPPs for their potential to improve ASO-mediated therapy. PMOs have shown effectiveness in treating genetic diseases, but their poor cellular absorption continues to be a major drawback. Due to the high efficacy and low toxicity, DG9 has become a promising CPP for improving the intracellular transport of PMOs since it holds the prospect of improved therapeutic advantages [9,10]. This review offers a thorough analysis of ASO therapies and the difficulties they encounter, highlighting the potential contribution of CPPs, particularly DG9, to overcoming these difficulties and improving ASO efficacy. Through an exploration of CPP-mediated ASO delivery intricacies and focusing on the remarkable properties of DG9, this review seeks to highlight the potential of this approach to transform ASO-mediated therapy more effectively.
2. Why Antisense Technology?
With their ability to precisely target disease-causing genes at the RNA level, antisense oligonucleotides (ASOs) have become an important tool in the development of therapeutics. ASOs show promise in the treatment of a wide range of illnesses, such as cancer, viral infections, genetic disorders, and neurological problems. Currently, 15 ASOs have received approval from the Food and Drug Administration (FDA, USA), the European Medicines Agency (EMA), and/or the Japanese Ministry of Health, Labour, and Welfare and most of them have received approval in the past 4 years [11] (Table 1). Compared to the chemical compounds, antisense technology’s unprecedented specificity, ability to modulate gene expression, variety of target types, potential for personalized therapy, disease modification abilities, and documented clinical effectiveness make antisense technology an appealing strategy for therapeutic research.
3. The mode of action of Antisense Oligonucleotide
Antisense Oligonucleotides (ASOs) are synthetic, single-stranded nucleic acid molecules, generally comprised of ~18-30 nucleotides with a variety of chemical structures [12]. ASOs form a DNA-RNA hybrid by binding specific RNA sequences through Watson-Crick base pairing to modulate gene expression [13]. The functional mechanism of ASO can be broadly categorized into two main modes of action- RNase H-mediated degradation and Steric hindrance [13].
RNase H-mediated degradation: When DNA-based oligonucleotides, also known as gapmers bind to their respective mRNA sequence they can recruit endogenous RNase H enzyme. RNase H recognizes RNA-DNA duplex and catalyzes the degradation of RNA leading to the reduction of the target RNA and gene silencing (Figure 1a) [14]. This strategy has been employed widely to suppress disease-causing or disease-modifying genes. Fomivirsen, mipomersen, and inotersen are the three RNase H-competent ASOs that have so far acquired regulatory approval [14].
Steric hindrance: Apart from RNase H-mediated breakdown, ASOs can interfere with RNA-RNA or RNA-Protein interaction by blocking certain regions within the target transcript. This results in the prevention of translation rather than inducing the lowering of transcript level [15]. The best-known application of this mode of action is splicing modulation which can cause either exclusion (exon skipping) or retention (exon inclusion) of specific exon/exons by targeting splice sites or exonic/intronic inclusion signals respectively [16,17]. Typically, this approach can be used both for restoration of the translational reading frame to have functional protein synthesis or for disruption of translation of target gene [18,19] (Figure 1b). Eteplirsen, golodirsen, nusinersen, viltolarsen, casimersen, milasen and atipeksen are the splice-switching ASOs that have received FDA approval to date [12,20,21,22].
4. Molecular mechanism of cellular uptake and intracellular distribution of Antisense Oligonucleotides
The effectiveness of antisense oligonucleotides (ASOs) as therapeutic agents depends significantly on cellular uptake and intracellular distribution. To have the desired effects, ASOs must efficiently penetrate cells and locate their target locations. After intravenous, subcutaneous, or direct administration, ASOs reach the bloodstream where they can be broken down by nucleases [23]. Once they reach the target organ, the cellular uptake process can be achieved in several ways, such as phagocytosis, macropinocytosis, micropinocytosis via clathrin and caveolin-independent pathways, caveolar internalization and classical clathrin-mediated endocytosis. Following cellular uptake ASOs are internalized into early endosomes and then late endosomes regulated by Rab, SNARE, and tethering proteins. A percentage of ASO drugs, possibly a very tiny portion are released from late endosomes into the cytoplasm where they target mRNAs or pre-mRNAs in the cytoplasm or the nucleus to carry out their therapeutic effects. Nuclear entry can be actively mediated by nuclear pore mechanism or passively via simple diffusion [24]. Many small cellular proteins, such as COPII, can facilitate nuclear trafficking. However, the process is not entirely known [23]. The target of different ASOs is located at different subcellular sites. For RNase H-mediated mRNA degradation, the ASO drugs need to reach either the cytoplasm (ribosomes) or nucleus [25] whereas for exon skipping/inclusion ASOs must be present in the spliceosomes of the nucleus [26]. Another percentage of ASO medications enter lysosomes or are subsequently expelled from the cell by one of three hypothesized mechanisms: membrane leakage, back-fusion-mediated release, or vesicle-mediated release [23]. It is essential for ASOs to avoid or circumvent lysosomal degradation to maintain their integrity and efficacy. Apart from that cellular uptake and intracellular localization of ASOs can significantly vary based on the ASO chemistry, cell type, and specific cellular condition.
5. Challenges associated with ASOs delivery
Although antisense oligonucleotides (ASOs) have great potential as therapeutic agents, their efficient delivery faces several difficulties. These difficulties are associated with the physiochemical characteristics of ASO molecules, such as their large size, molecular weight (single-stranded ASOs are ~4–10 kDa, double-stranded siRNAs are ~14 kDa), and negative charge which hinders passive diffusion across the cell membrane. ASOs predominantly rely on endocytosis for cellular uptake which might be ineffective and lead to entrapment in endosomes or lysosomes, leading to lysosomal degradation. So, once inside the cell, ASO must escape endosomal entrapment to gain access to the target region in the cytoplasm or nucleus [27]. Apart from that, for the systemically administered ASOs to be effective, it needs to avoid renal clearance [28,29], resist nuclease degradation both in extracellular fluid and intracellular compartment [30] and avoid removal by the reticuloendothelial system, which includes mononuclear phagocytes, liver sinusoidal endothelial cells, and Kupffer cells [31]. A study reported that intravenous administration of an AON resulted in 40% and 18% accumulation in the liver and kidneys respectively [32]. Recently ASOs are also being developed for the treatment of Central Nervous System (CNS) related diseases. The additional barrier, in this case, ASOs have to cross the blood-brain barrier (BBB) or brain–cerebrospinal barrier, before they can distribute within the CNS. The vascular barriers of the nervous system are comprised of a monolayer of endothelial cells forming tight junctions through interactions of cell adhesion molecules which prevents most of the ASOs to reach the CNS after systemic injection [33] (Figure 2).
Due to these challenges, to date, most of the approved oligonucleotide treatments are delivered either locally (for example- to the eye or spinal cord) or to the liver. The eye is chosen as a target for ASO delivery (for example- Pegaptanib and Fomivirsen) due to its accessibility, anatomical considerations, and immune-privileged status [12]. Although ocular delivery of ASOs has benefits, there are still obstacles to be overcome, including getting through anatomical obstacles (such as the blood-retinal barrier), maximizing ASO stability, and pharmacokinetics for long-lasting therapeutic effects. For ASOs targeting the CNS, direct delivery into the cerebrospinal fluid via lumber puncture is most commonly used (for example-Nusinersen) [34]. It should be noted, however, that this method requires expertise, and specialized equipment, and carries a small risk of complications associated with invasive procedures.
6. Strategies to enhance the stability and delivery of Antisense Oligonucleotides
6.1. Chemical modification
Antisense oligonucleotides were initially employed as synthesized, unaltered DNA which turned out to be extremely vulnerable to exonuclease and endonuclease degradation [35] (Figure 3). Chemical modifications Antisense Oligonucleotides can enhance stability, improve target binding affinity and biodistribution, and provide protection against nuclease-mediated degradation. Modification of the nucleic acid backbone, the ribose sugar moiety, and the nucleobase itself have been extensively employed to improve the drug-like properties of Antisense Oligonucleotides [29,36].
6.1.1. Backbone modification
Backbone modifications involve changing the repeating sugar-phosphate units that make up the phosphodiester backbone of ASOs. Typical changes to the backbone include the incorporation of phosphorothioate (PS) linkages, in which one of the non-bridging oxygen atoms of the inter-nucleotide phosphate group is replaced with sulfur [37] (Figure 3c). Phosphorothioate (PS) belongs to the first generation of ASOs that work by an RNase H-mediated mRNA cleavage-based mechanism and do not disrupt RNase H activity [38]. Unmodified ASOs are reported to be degraded within 30 mins in serum [1] whereas oligonucleotides with these modifications are more stable with reported half-lives of 9 h in human serum [39]. Scavenger receptors (such as the stabilins STAB1 and STAB2) also take up sulfated molecules, such as oligonucleotides with PS linkages, and facilitate their internalization into organs like the liver [32,40,41]. Apart from that incorporation of PS linkages increases the binding of ASO to proteins in both plasma and within cells which significantly improves drug pharmacokinetics by reducing renal clearance and increasing circulation time [42]. However, high PS concentrations have been shown to cause cytotoxic effects, which are assumed to be due to protein binding [43,44,45]. In addition to that, PS backbone modifications have been found to reduce the binding affinity of the oligonucleotide for its target [12].
6.1.2. Ribose sugar modification
Oligonucleotides are frequently modified at the 2ʹ position of the ribose sugar to provide resistance to enzymatic degradation and improve stability in plasma, and to increase tissue half-lives. 2ʹ-O-methyl (2ʹ-OMe), 2ʹ-O-methoxyethyl (2ʹ-MOE) and 2ʹ-Fluoro (2ʹ-F) are among the most used 2ʹ substituents (Figure 3b) [46] 2ʹ-ribose modified ASO are used or steric block oligonucleotides, or for the flanking sequences in gapmer ASOs as these modifications are not compatible with RNase H activity [12].
In comparison to unmodified phosphorothioates, 2′-O-methyl (2OMe) and 2′-O-methoxy-ethyl (MOE) have been shown to increase hybridization affinity to their target RNA and decrease sequence-independent toxicity arising from the PS backbone [47,48,49,50]. Due to the extensive success and effectiveness, among 15 FDA-approved drugs, 5 use 2OMe or 2’MOE chemistry (for example- Pegaptanib, Mipomersen, Nusinersen, Patisiran, Inotersen) [12].
Locked nucleic acids (LNA) are a type of 2’-modification in which the 4’-carbon is linked to the 2’-hydroxyl group and have also been utilized in steric block ASOs, such as miRNA inhibitors (Figure 3b). LNAs offer increased resistance to nucleases [51] and exhibit significantly improved hybridization compared to other 2’-modifications [52,53]. However, they are associated with more severe toxicological issues in systemic treatment [54]. Furthermore, a loss in target specificity can also be due to the strong affinity of LNAs [53,55].
Other sugar modifications that are less frequently used are tricyclo-DNA (tc-DNA) and S-constrained-ethyl (cEt) (Figure 3b). For tc-DNA modification, this adds an ethylene bridge fused with a cyclopropane unit which results in a more stable duplex formation [56]. When tested in cells, Tc-DNA was found to be more effective at correcting splicing than a 2OMe-PS oligonucleotide and to be stable in serum [57]. However, tc-DNA has only been used in a very small number of studies up to this point. Finally, the cEt-modified antisense oligonucleotides show a similar binding affinity to LNA but a better toxicity profile [58] and have recently shown good promise in a humanized mouse model for HD (Huntington disease) [59].
6.1.3. Nucleobase modification
ASOs’ characteristics can also be enhanced by the introduction of nucleobase modifications is introduced in ASO to achieve optimized Watson–Crick base-pairing and thereby control the melting temperature of the ASOs [36] (Figure 3d). Alteration of the nucleobase chemistry results in a thermally more stable ASO–target duplex by increasing the affinity towards the target. The thermal stability of splice-switching ASOs plays a vital role, as a stronger hybridization between ASOs and its target can hinder the formation as it directly impacts their ability to effectively block the splice site or hinder the assembly of the ribosomal complex, ultimately preventing translation and executing the therapeutic effects [60]. Among nucleobase modifications, the incorporation of cytosine analogs has been used extensively. 5-methylcytidine and 5-methyluridine/ribothymidine) has the effect of increasing the oligonucleotide melting temperature by ~0.5 °C per substitution [36] (Figure 3d). Apart from that, 5’-methyl cytosine-based analogs were used to reduce the immunological stimulation caused by CpG dinucleotide-mediated toll-like receptor activation [61,62].
6.1.4. Other Oligonucleotide Modifications
Apart from the modifications mentioned above, other chemistries have also been explored for the improvement of ASOs’ drug-like properties, such as- PNA (Peptide Nucleic Acid) and PMO (Phosphorodiamidate Morpholino Oligomer) (Figure 3e). PNA is a synthetic nucleic acid analog in which the sugar-phosphate backbone is replaced with a peptide-like backbone which makes them uncharged [63]. As a result, PNAs have a high binding affinity and are resistant to enzymatic degradation [64,65]. These are mostly implemented in splicing modulation approaches or translation inhibition as they are unable to activate the RNase H enzyme. A clear shortcoming of this type of modification is its poor cellular uptake and water insolubility [66,67]. PNAs are found to be rapidly cleared when administered peripherally [68] and these poor pharmacokinetic properties are the main reason for their limited in vivo use thus far.
Another strategy that has been explored is the use of PMO synthetic backbone modification in which the five-membered ribose heterocycle is replaced by a six-membered morpholine ring structure with phosphorodiamidate linkages [69]. Similar to PNA, PMOs are neutrally charged and work by steric hindrance or splice modulation to provide an antisense effect [70]. The absence of a carbonyl group provides PMO resistance against protease and nuclease degradation [70] and neutral charge makes them less susceptible to activating immune response [71]. Apart from that, studies have shown that the administration of multiple high doses can be achieved with minimal toxicity [72].
To date, four PMO drugs have been approved by the FDA, eteplirsen, golodirsen viltolarsen, and casimersen (Eteplirsen targets exons 51, Golodirsen and Viltolarsen exon 53, and Casimersen targets exon 45 of the dystrophin mRNA) for Duchenne Muscular Dystrophy [73,74] (Table 1). The first in vivo exon skipping of Dmd using 2’-OMePS ASOs chemistry was documented in a work by Lu et al. [75]. In another work, exon 23 skipping was successfully induced, and dystrophin production was restored in mdx mice by the intramuscular administration of leashed PMOs (PMOs annealed to complementary anionic oligonucleotides to increase delivery). Even two weeks after the injection, the skipped transcript was observed, which was not the case with 2’-OMePS ASOs [76]. An additional investigation on CXMDJ dogs discovered that intramuscular or intravenous delivery of a 3-PMO cocktail promotes in-frame 6 to 8 exon skipping with 61-83% exon skipping efficiency two weeks after intramuscular injection, which results in approximately 25-50% dystrophin protein restoration [77].
In addition to the DMD, the neutrally charged PMO chemistry was also being studied by several groups for the treatment of Central Nervous System (CNS) disease. For Spinal Muscular Atrophy, a neurodegenerative disease, peripheral administration of Nusinersen, an MOE-based drug has already been proven to rescue the SMA phenotype [78,79] (Table 1). However, with peripheral delivery or intrathecal injection being more invasive, the focus is now to modify the ASO in such a way that systemic injection can reach the CNS by crossing the Blood Brain Barrier (BBB). There is a length-dependent effect of PMO ASO. Two separate studies showed that a single intravenous injection of longer PMO (25 mer) in a severe SMA mouse model can increase the survival rate. Based on the experimental result, they emphasized the superiority of the morpholino ASOs because of lower toxicity, increased SMN levels, and prolonged survival [80,81]. In addition to that another study proved PMO can more readily cross the immature BBB into the CNS [72].
However, the main pharmacokinetic shortcomings associated with PMO are low efficacy, which is related to its rapid clearance from the bloodstream, poor uptake in tissues like the skeletal muscle, and endosomal entrapment [82,83]. As PMOs are uncharged nucleic acid molecules, this provides the opportunity to covalently conjugate them to charged delivery-promoting moieties such as cell-penetrating peptides (CPPs) for enhanced delivery as mentioned below [69,84].
6.2. Bioconjugates
While chemical modifications are required to protect ASOs from exonucleases and prolong their stability, the next challenge becomes ASO passage across biological barriers, such as the vascular endothelial barrier, cell membranes, and intracellular compartments, specific cell/tissue targeting, and reduction of clearance from circulation [85]. Improving ASO delivery potential can be achieved through the conjugation of different moieties that can direct the drug to specific tissues and enhance internalization. Bioconjugates are distinct molecular entities with precise stoichiometry which ensures well-defined pharmacokinetic properties and simplifies large-scale synthesis. Additionally, bioconjugates tend to have a small size, which often results in favorable biodistribution profiles [12]. Bioconjugates usually promote interaction with cell-type associated receptors, consequently enhancing delivery to the target tissue and internalization by receptor-mediated endocytosis [86]. There are different types of conjugates available including- lipid-based bioconjugates (e.g.- cholesterols and its derivatives) [87,88,89], peptide-based bioconjugates (e.g.- cell penetrating peptides) [90,91,92,93,94,95], aptamers [96], antibodies [97,98], sugars (for example, N-acetylgalactosamine (GalNAc) [99,100], and polymer (e.g.- PEG) (Table 2). The selection of the appropriate bioconjugate depends on several factors, including the application goals, specific requirements of the ASO delivery system, the intended therapeutic application, and safety considerations. Due to the effectiveness of bioconjugates in increasing the efficacy of ASO delivery, bioconjugated compounds are present in four of the five FDA-approved siRNA medications [101].
6.2.1. Cell Penetrating Peptides
Cell-penetrating peptides (CPPs, also known as protein transduction domains)) are short (fewer than 30 amino acids) cationic, amphipathic, or hydrophobic peptides that translocate small drugs/cargo across cell membranes and biological barriers [101,102]. CPPs are not just recently discovered; they were first identified in 1988 when two research groups (Frankel and Pabo, 1988; Green and Loewenstein, 1988) identified that the transactivator of transcription (TAT) protein of HIV can cross the cell membranes and can be efficiently internalized by cells due to the strong electrostatic interactions with heparin sulfate during endocytosis [103]. Later, another group of scientists (Vives et al. 1997) found thirteen amino acid sequences that align with residues 48–60 of TAT play a key role in cellular uptake. Subsequently, peptides exhibiting cell membrane translocation like TAT were commonly categorized as cell-penetrating peptides (CPPs) [104].
CPPs can be classified into- cationic CPPs, amphipathic CPPs, and hydrophobic CPPs based on their physicochemical characteristics [105]. Cationic CPPs are mainly composed of basic amino acids such as arginine and lysine [104]. Poly-arginine stretches exhibit the utmost capacity for cellular uptake and hold considerable therapeutic potential [105]. However, higher values of arginine are related to irreversible side effects [106]. Several studies have shown that positively charged CPPs interact with negatively charged carboxylic, phosphate, and sulfate groups of the cell membrane and eventually mediate internalization by endocytic pathways [104,107]. TAT, penetratin, and polyarginine are some examples of cationic CPPs [104]. Amphipathic CPPs contain polar and non-polar amino acid regions. Generally, the nonpolar region is made up of valine, leucine, and alanine, whereas the polar region is made up of lysine and arginine. Amphipathic peptides have been found to play a role both in cellular internalization and endosomal escape [107]. This group of CPPs makes up more than 40% of all the CPPs that have so far been identified [105]. Some of the examples of amphipathic CPPs are MAP, transportan, and Pep-1 [103]. Compared to other types CPPs, there are relatively few numbers of hydrophobic CPPs that are typically composed of a large number of non-polar residues or only a few charged amino acids (less than 20% of the sequence) [105]. Hydrophobic CPPs usually interact with the hydrophobic region of the cellular membrane and probably translocate by an energy-independent mechanism as represented by K-FGF, C105Y, and gH625; some of the natural hydrophobic CPPs [104]. However, the peptide sequence of hydrophobic CPPs has not been found to significantly affect cell uptake [108]. CPPs can function either via receptor-mediated endocytosis where CPPs trigger endocytosis by directly binding to the specific cell surface receptor or via direct translocation where CPPs cross the cell by creating transient pores on the cell membrane and deliver the cargo inside the cytoplasm or nucleus [101].
7. Overcoming the limitations of PMO by conjugating it with Cell Penetrating Peptides
As mentioned earlier, PMOs shows low efficacy as therapeutic agent due to their poor cellular uptake, less permeability of membrane barriers, rapid clearance from the systemic circulation, inability to cross blood-brain barriers, and the requirement of repetitive administration and/or high dosage of the drug for executing its function. Apart from that, due to the hydrophobicity of the plasma membrane and the neutral charge of PMO, only small portions of internalized PMOs can escape endosomes and reach their intended target [83]. A promising utilization of CPP is their ability to directly conjugate with neutrally charged PMO and PNA and increase the delivery efficacy [109,110,111].
A promising utilization of CPP is their ability to directly conjugate with neutrally charged PMO and PNA by using several methods, including maleimide linkage, disulfide linkage, click chemistry or amide linkage, and enhances the pharmacokinetic properties of PMO and PNA. Among various therapeutic purposes, this approach has been extensively explored most for Duchenne muscular dystrophy (DMD), which affects approximately 1 in 3500 newborn boys and is caused by out-of-frame deletions in the Dmd gene resulting in the loss of dystrophin, the structural muscle protein [112]. Lack of dystrophin results in progressive muscular degeneration, which impairs ambulation and causes mortality from cardiac and respiratory failure [113]. The mRNA reading frame around the deletion can be restored by the "exon-skipping" approach where pre-mRNA splicing is modulated to produce smaller but functional proteins. This approach has been used successfully with naked PMO that resulted in the conditional approval of four PMO-based drugs for DMD (e.g.- eteplirsen, golodirsen viltolarsen, and casamirsen) [73,74]. Eteplirsen has been found to restore an average of 0.9% dystrophin of normal levels after 180 weeks of treatment which indicates low treatment efficacy despite the safe profile of this drug [102]. For Golodirsen, according to the trial results dystrophin expression increases by ~0.9% after the demonstration of the drug [73], and the casimersen-treated group saw a 0.81% increase in dystrophin production [114]. Whether such a tiny increase in dystrophin expression is enough to slow down disease progression and provide clinical benefits, is still a big question. Apart from that Vitolarsen has limited efficacy in cardiac tissue due to poor uptake. As the primary cause of mortality in the DMD patient population is cardiorespiratory complications, the low efficacy of the drug in the heart is a serious concern in exon-skipping therapy [115].
Therefore, there is still a need for a more potent substance to raise dystrophin levels and thereby maximize the functional advantages of this strategy.
Conjugation of CPPs to PMO is one such approach to improve PMO delivery. This strategy was first demonstrated with an arginine-rich peptide, (RXR)4 which was administered to the mdx mouse model of DMD in a variety of doses, time intervals, and delivery methods and it was observed that a single intravenous administration can high dystrophin exon skipping in skeletal muscle, the diaphragm, and for the first time in the heart [116]. Another arginine-rich peptide, (RXRRBR)2 peptide (B-peptide) identified from a screen using the EGFP-654 splicing reporter mouse model to ensure PPMO entry to cells and notable exon-skipping in the heart after retro-orbital injection resulting in improved cardiac function specifically end-systolic volume and end-diastolic volume and resistance to dobutamine [117]. In another study, intravenous injection of a single 25 mg/kg dose of B-peptide conjugated PMO to mdx mouse confirmed approximately 50% wild-type dystrophin levels along with restoration in cardiac function [116,117] and improved muscle function whereas weekly administration of naked PMO at 200 mg/kg for 12 weeks could only achieve 10% wild-type dystrophin levels [118]. Fusion of muscle-specific peptide (MSP) with B-peptide through a phage display has been found to improve activity 2- to 4-fold after multiple 6 mg/kg dosing [119]. Interestingly, another study revealed that a specific orientation (B-MSP-PMO) can lead to a 2–5-fold improvement in skeletal muscle restoration compared to B- PMO [120]. Additionally, B-PMO has also been used for research in canine models of DMD that better mimic the pathophysiology of human illness and serve as a more rigorous evaluation of the efficacy of CPP-PMOs in restoring dystrophin expression. Repeat low dose (4 mg/kg per ASO) B-PMO intravenous injection has been found to restore 5% dystrophin of wild-type levels throughout the body, including in the heart where improvement of cardiac conduction defects was seen after therapy [121]. Another arginine-rich peptide- R6G is also currently being explored for the treatment of DMD [122]. R6G peptide is a modification of the conventional R6 peptide with the glycine residue. that has been extensively studied for various neuromuscular disorders [123,124]. When conjugated with PMO, it has shown promise in exon skipping efficacy specifically in cardiac muscle [122].
Recent research has led to the development of several peptide series known as "Pip’s" (PMO/PNA internalization peptides), which are generated from the parent peptide penetratin [125,126] and consist of the amino acids arginine (R), 6-aminohexanoic acid (X), and ß-alanine spacer (B), with an internal core containing hydrophobic residues [12]. The most recent Pip-PMO conjugates are significantly more effective than naked PMO and, more critically, reach cardiac muscle following systemic administration in dystrophic animal models. A single intravenous injection of the Pip5e peptide conjugated PMO induced the highest amounts of exon skipping and dystrophin restoration throughout the body, including in the heart of mdx mice [127]. To increase homogenous dystrophin repair and to target the heart muscle more effectively, the Pip6 series of peptides were generated by further iterations of core design [128]. In a study, it was observed that inversion of the Pip5e-PMO hydrophobic core (Pip6a) resulted in a cardiac dystrophin recovery score of up to 37% in the mdx animal model [92]. In another study by the same group, it has been demonstrated that administering Pip6f-PMO (scrambled peptide core) can increase the levels of the protein dystrophin by up to 28% in the heart of mdx mice who had previously undergone a forced exercise regimen to cause changes resembling the DMD cardiac phenotype [129]. Additionally, injection of Pip2a or Pip2b conjugated PPMOs in the tibialis anterior of the mdx mouse has also been found to induce an effective exon 23 skipping and a noticeable increase in dystrophin rescue [130]. Another CPP created to target muscle is M12 which was discovered through phage display conducted on C2C12 myoblasts upon conjugation to PMO, M12 achieved approximately 10–25% of wild-type dystrophin levels following a single systemic administration although at dosing levels 5- to 6-fold higher than those required for comparable efficacy [102].
CPP-PMO has also been used as a therapeutic approach for myotonic dystrophy type I, where a CTG expansion in the DMPK gene’s 3′ untranslated region causes a pathogenic transcript that interacts with RNA-binding proteins like muscleblind-like 1 (MBNL1) to cause widespread aberrant splicing abnormalities. Systemic administration of B-PMO targeting this repeat element causes blocking of Mbnl1 sequestration, resulting in normal nuclear distribution and subsequent correction of abnormal RNA splicing, including for chloride channel 1 gene, which is a primary contributor to myotonia [131].
One of the biggest challenges of nucleic acid therapeutic is to cross the blood-brain barrier to reach the central nervous system (CNS) after systemic delivery. CPPs have been identified as promising medicines in the treatment of central nervous system (CNS) diseases due to their demonstrated transmembrane transporting ability. It is assumed that small-size cationic or amphipathic CPPs may exhibit greater affinity for negatively charged endothelial cells on the blood-brain barrier [132,133]. CPP-PMOs have recently been investigated in preclinical models of spinal muscular atrophy (SMA), an autosomal recessive neuromuscular disorder that results in premature death [134]. This disease is caused by mutations in the survival of the motor neuron 1 (SMN1) gene. A paralogous gene, SMN2, encodes a vital SMN protein but generates only minimal levels due to a sequence variant leading to the exclusion of exon 7 from approximately 90% of mature transcripts. Consequently, a truncated, non-functional protein is produced [135,136]. To address the functional deficiency caused by the loss of SMN1 protein in patients, ASOs have been employed to facilitate the inclusion of exon 7 in SMN2 transcripts, thereby enhancing the production of SMN2 protein [137]. However, the limited delivery of the currently used ASO in the rostral spinal and brain has reduced the therapeutic efficacy [104]. Nusinersen, a modified 2’-MOE PS ASO has been recently approved by FDA for the treatment of SMA. Intrathecal injection of Nusinersen can significantly improve motor function and increase the lifetime of SMA patients [138,139]. However, this procedure is invasive and is linked to unpleasant post-lumbar puncture adverse effects for the patients [137]. Therefore, to address this, PPMO trials have been conducted. Intravenous administration of Pip6A-PMO in the Taiwanese severe SMA mouse model increased mean survival and SMN2 expression in the brain and spinal cord, and improved neuromuscular junction morphology [140]. Due to the mouse model’s severity, the drug has to be administered before postnatal day 2 to demonstrate functional benefit. It is likely that the BBB may not be fully formed at that time, as a result, does not accurately represent the clinical condition for therapeutic intervention [102]. To prove the blood-brain-barrier crossing capacity of PPMO, a study has been conducted where symptomatic SMA mice were administered RXR-MO and r6-MO (morpholino oligomer) conjugates intraperitoneally at PD-5 with a completely closed BBB. The treated mice showed improved median survivals of 41.4 and 23 days, respectively which is significantly higher compared to the naked MO (~17 days). Additionally, RXR-MO and r6-MO conjugates were found in the central nervous system in a symptomatic phase. Pathological studies demonstrated that CPP-MOs mitigate the degradation of neuromuscular connections more efficiently than scrambled or naked MOs [124]. Another study demonstrated that a derivative of an ApoE could induce a 0.25-fold increase in exon 7 inclusion in the pre-mRNA of the spinal cord and to a lesser extent in the brain of a spinal muscular atrophy mouse model, improving the diseased mice’s phenotype [141].
CPP-PMO strategies have also been developed for the treatment of other neurological diseases like Huntington’s disease (HD) and Amyotrophic Lateral Sclerosis (ALS) [83] as well as for use as antibacterial agents because ASOs by themselves are not very effective at penetrating bacterial cell walls [102]. It is evident that CPPs have a great deal of therapeutic potential in delivering and increasing the efficacy of ASOs specifically the PMO-based strategy.
8. DG9: A CPP for enhancing the delivery and cellular uptake of ASO and proteins
Although CPPs hold promise in facilitating the transport of biologically active cargo across cell membranes, including the notorious blood-brain barrier and other challenging barriers within the body, CPPs also pose a number of difficulties and issues that require careful study. The primary obstacle to completing clinical trials for PPMO-based medications right now is their toxicity and immunogenicity. Toxicity can be variable depending on several factors, including species, treatment duration, frequency of systemic administration, dosage, exons skipped, and the cationic nature of the peptide [83]. Additionally, first-generation arginine-rich peptides were found to be more immunogenic than PMOs [142], suggesting that the toxicity may result from immunogenic processes such as complement activation [121,143]. Due to severe side effects, a pre-clinical experiment using an arginine-rich PPMO by Sarepta had to be stopped. It is assumed that the side effects were partially attributable to the high dosage employed [144]. In a separate study, rats given high doses of B-peptide-PMO experienced a loss of body weight and an increase in serum blood urea nitrogen and creatinine in a dose-dependent manner indicating decreased renal output [145]. Therefore, the quest for cell-penetrating peptides is still ongoing in order to overcome the challenges. A peptide found recently in this search is- DG9.
DG9 is a cell-penetrating peptide derived from the protein transduction domain (PTD) of the human Hph-1 transcription factor, which facilitates the cell membrane penetration of its protein cargos in the lungs (Figure 4). Two of these Hph-1 domains constitute DG9 [9]. In a study by Choi et al., it has been shown that intraperitoneal injection of fusion proteins conjugated with the Hph-1 domain has enhanced the delivery in a wide range of organs, including the heart and brain which are apparently challenging to be delivered. Additionally, according to the study, cell viability was not affected, and behavioral abnormalities, cytotoxic effects, and immunogenicity were not observed after 1.6 mg/kg of intravenous administration of Hph-1 fused protein into mice for 14 days or 100 ug of intraperitoneal injection two times a week for 2 weeks [146]. The same author later reported another study where they used the same protein transduction domain (PTD) of the human Hph-1 transcription factor, but this time with two tandem sequences (HHph-1-PTD) and fused it with Foxp3 (target protein of the study) protein to increase the cell permeability of Foxp3. In in vitro study, HHph-1-Foxp3 was detected in the nucleus as well as in the cytoplasm within 30 minutes of transduction, suggesting that Foxp3 protein is efficiently delivered to cells and is localized in the nucleus. The delivery efficacy of HHph-1 was also proved in vivo as HHph-1-Foxp3 treated mice lived longer and the phenotype got improved compared to the control groups. They also found that two repeats of Hph-1-PTD (HHph-1) resulted in optimal intracellular transduction and rapid delivery compared to one Hph1 domain [147].
A separate study reported that a conjugate of PMO and a unique peptide, derived from a human T cell and a near dimer of the PTD is at least 10- to 100-fold more efficient than the prior peptides at delivering the PMO into bacteria and ultimately causing the bacterial death. The only difference between the DG9 and the peptide used in the study is only L forms of amino acid residues were used in the peptide [148]. Kim et al. previously demonstrated that DG9 can deliver a PMO to the zebrafish heart and can cause strong exon skipping in the heart while also inducing exon skipping at significantly greater levels in the skeletal muscle [149].
The FDA has approved exon skipping as a promising therapy for DMD which utilizes phosphorodiamidate morpholino oligomer (PMO) to target and modulate gene expression. Yokota’s research group has identified DG9 peptide conjugation as a powerful way of enhancing the exon-skipping efficacy of PMO in vivo [9]. The positive aspect of the DG9 peptide used in this study is that it has a potentially better toxicity profile compared to other peptides. As mentioned earlier, Peptide-conjugated PMOs have been found to induce dose-dependent toxic effects in pre-clinical studies which is thought to be linked with their amino acid compositions [83,144,150]. It has also been reported that substituting D-amino acid for L-amino acid in polymer-peptide conjugates attenuated anti-polymer antibody generation and toxicity and exhibit good tolerance in vivo even after repeated administration [151]. Therefore, certain L-arginine residues in DG9 were converted to D- arginine (DG9 (sequence N-YArVRRrGPRGYArVRRrGPRr-C; uppercase: L-amino acids, lowercase: D-amino acids)) [9]. This conversion has been shown to improve the viability of peptide-conjugated PMO- treated cells in vitro along with increasing serum stability [152]. Additionally, this DG9 does not contain any 6-aminohexanoic acid residues (often represented by “X” in peptide sequences), which have also been linked to higher toxicity [152]. In the study of DG9-PMO mediated efficient exon skipping by Lim et al., it has been demonstrated that retro-orbital injection of DG9-conjugated PMO into hDMDdel52; mdx mice can increase skipping efficiency 2.2 to 12.3-fold and 14.4-fold compared to the unconjugated PMO with a dystrophin restoration amount of 3% and 2.5% of wild-type levels in skeletal muscles and heart, respectively. Skeletal muscles produced 2.8 to 3.9% more dystrophin and had an exon 51 skipping level of 55 to 71% after receiving repeated injections of DG9-PMO once each week for three weeks. Most notably, hDMDdel52;mdx mice treated repeatedly with DG9-PMO showed a considerable improvement in forelimb and total limb grip strength indicating the improvement of the muscle function of the treated mice. Additionally, the tibialis anterior DG9-PMO intramuscular injection was successful and demonstrated dystrophin restoration, suggesting the possibility of DG9-PMO for DMD therapy. There was no significant toxicity observed after the injection of DG9-PMO [9]
Another study by Yokota’s research group tested the effectiveness of DG9 peptide in an SMA mouse model (Figure 4). In the study, Tejal et al. showed that after a single subcutaneous administration of DG9-PMO into SMA mice (Taiwanese model) FL-SMN2 (Full-length SMN2) expression was increased ~5-fold compared to unconjugated PMO in the majority of the tissues including brain and spinal cord. The results indicated improved motor and breathing function and muscle strength with an increased mean survival of 58 days for DG9-PMO treated mice which were significantly higher compared to untreated (8 days) and unconjugated PMO treated mice (12 days). The fact that DG9 greatly improved the uptake of PMO in the CNS and peripheral tissues at PD7, despite subcutaneous treatment at PD5, indicates that DG9-PMO can assure extensive distribution of the PMO to both the peripheral and CNS tissues. The toxicological studies show that DG9-PMO does not appear to be adverse or to impair mice’s immune systems [10].
9. Conclusion
Despite their huge potential, the poor biodistribution of nucleic acids-based medications has prevented them from being used clinically, despite their enormous potential. As a result, cell-penetrating peptides have been thought of as a potential tactic to enhance passage across biological barriers and intracellular delivery, as covered in great detail in this chapter. However, there are a number of concerns that need to be resolved before CPPs are used in clinics, such as in vivo stability, immunogenicity, cellular toxicity, lack of selective intracellular uptake, and inability to escape from endosomes. Despite extensive research, underlying the mechanisms regulating the extravasation and cellular transport of these CPP-drug conjugates or complexes remain poorly understood. Undoubtedly, a deeper comprehension of these PPMOs’ limitations, pharmacodynamics, and mode of action will help create new CPP generations that are better able to target various tissues (and clinical diseases) and deliver their payload within the appropriate cellular compartment, giving patients hope for an improvement in their quality of life. The integration of DG9 into ASO-mediated therapy holds the potential to enhance cellular uptake and biodistribution of PMO, opening the door to more effective and precise treatments for a wide range of disorders. However, further research and development are necessary to fully realize the potential and long-term safety considerations. Continued research can provide the answers whilst making CPP-conjugated ASO-mediated therapy a potential tool in the arsenal of gene therapy.
Author Contributions
US.H. and T.Y. prepared the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
No specific grant support was received for this study.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Acknowledgments
We thank the University of Alberta Faculty of Medicine and Dentistry, the Canadian Institutes of Health Research, the Friends of Garrett Cumming Research Funds, HM Toupin Neurological Science Research Funds, Muscular Dystrophy Canada, and the Women and Children’s Health Research Institute for their support.
Conflicts of Interest
T.Y. is a co-founder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A 1978, 75, 285–288. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Sasso, J.M.; Ambrose, B.J.B.; Tenchov, R.; Datta, R.S.; Basel, M.T.; DeLong, R.K.; Zhou, Q.A. The Progress and Promise of RNA Medicine horizontal line An Arsenal of Targeted Treatments. J Med Chem 2022, 65, 6975–7015. [Google Scholar] [CrossRef] [PubMed]
- Kuijper, E.C.; Bergsma, A.J.; Pijnappel, W.; Aartsma-Rus, A. Opportunities and challenges for antisense oligonucleotide therapies. J Inherit Metab Dis 2021, 44, 72–87. [Google Scholar] [CrossRef]
- Sharma, V.K.; Sharma, R.K.; Singh, S.K. Antisense oligonucleotides: modifications and clinical trials. MedChemComm 2014, 5, 1454–1471. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ming, X.; Carver, K.; Laing, B. Cellular uptake and intracellular trafficking of oligonucleotides: implications for oligonucleotide pharmacology. Nucleic Acid Ther 2014, 24, 101–113. [Google Scholar] [CrossRef]
- Jarver, P.; Coursindel, T.; Andaloussi, S.E.; Godfrey, C.; Wood, M.J.; Gait, M.J. Peptide-mediated Cell and In Vivo Delivery of Antisense Oligonucleotides and siRNA. Mol Ther Nucleic Acids 2012, 1, e27. [Google Scholar] [CrossRef]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem 1996, 271, 18188–18193. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Woo, S.; Melo, D.; Huang, Y.; Dzierlega, K.; Shah, M.N.A.; Aslesh, T.; Roshmi, R.R.; Echigoya, Y.; Maruyama, R.; et al. Development of DG9 peptide-conjugated single- and multi-exon skipping therapies for the treatment of Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 2022, 119. [Google Scholar] [CrossRef]
- Aslesh, T.; Erkut, E.; Ren, J.; Lim, K.R.Q.; Woo, S.; Hatlevig, S.; Moulton, H.M.; Gosgnach, S.; Greer, J.; Maruyama, R.; et al. DG9-conjugated morpholino rescues phenotype in SMA mice by reaching the CNS via a subcutaneous administration. JCI Insight 2023, 8. [Google Scholar] [CrossRef]
- van Roon-Mom, W.; Ferguson, C.; Aartsma-Rus, A. From Failure to Meet the Clinical Endpoint to U.S. Food and Drug Administration Approval: 15th Antisense Oligonucleotide Therapy Approved Qalsody (Tofersen) for Treatment of SOD1 Mutated Amyotrophic Lateral Sclerosis. Nucleic Acid Ther 2023, 33, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv Drug Deliv Rev 2015, 87, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem 2004, 279, 17181–17189. [Google Scholar] [CrossRef]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 2012, 11, 125–140. [Google Scholar] [CrossRef]
- Dominski, Z.; Kole, R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc Natl Acad Sci U S A 1993, 90, 8673–8677. [Google Scholar] [CrossRef]
- Singh, R.N.; Singh, N.N. Mechanism of Splicing Regulation of Spinal Muscular Atrophy Genes. Adv Neurobiol 2018, 20, 31–61. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther 2017, 27, 251–259. [Google Scholar] [CrossRef]
- Wan, L.; Dreyfuss, G. Splicing-Correcting Therapy for SMA. Cell 2017, 170, 5. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Dolgin, E. News Feature: Gene therapy successes point to better therapies. Proc Natl Acad Sci U S A 2019, 116, 23866–23870. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Sinhari, A.; Jain, P.; Jadhav, H.R. A perspective on oligonucleotide therapy: Approaches to patient customization. Front Pharmacol 2022, 13, 1006304. [Google Scholar] [CrossRef] [PubMed]
- Migliorati, J.M.; Liu, S.; Liu, A.; Gogate, A.; Nair, S.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X.B. Absorption, Distribution, Metabolism, and Excretion of US Food and Drug Administration-Approved Antisense Oligonucleotide Drugs. Drug Metab Dispos 2022, 50, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ming, X.; Nakagawa, O. Cellular uptake and intracellular trafficking of antisense and siRNA oligonucleotides. Bioconjug Chem 2012, 23, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol Ther 2017, 25, 2075–2092. [Google Scholar] [CrossRef]
- Echevarria, L.; Aupy, P.; Goyenvalle, A. Exon-skipping advances for Duchenne muscular dystrophy. Hum Mol Genet 2018, 27, R163–R172. [Google Scholar] [CrossRef]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol 2013, 31, 653–658. [Google Scholar] [CrossRef]
- Iversen, F.; Yang, C.; Dagnaes-Hansen, F.; Schaffert, D.H.; Kjems, J.; Gao, S. Optimized siRNA-PEG conjugates for extended blood circulation and reduced urine excretion in mice. Theranostics 2013, 3, 201–209. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev 2015, 87, 46–51. [Google Scholar] [CrossRef]
- Tsui, N.B.Y.; Ng, E.K.O.; Lo, Y.M.D. Stability of Endogenous and Added RNA in Blood Specimens, Serum, and Plasma. Clinical Chemistry 2002, 48, 1647–1653. [Google Scholar] [CrossRef]
- Allen, T.M. The use of glycolipids and hydrophilic polymers in avoiding rapid uptake of liposomes by the mononuclear phagocyte system. Advanced Drug Delivery Reviews 1994, 13, 285–309. [Google Scholar] [CrossRef]
- Bijsterbosch, M.K.; Manoharan, M.; Rump, E.T.; De Vrueh, R.L.A.; van Veghel, R.; Tivel, K.L.; Biessen, E.A.L.; Bennett, C.F.; Cook, P.D.; van Berkel, T.J.C. In vivo fate of phosphorothioate antisense oligodeoxynucleotides: predominant uptake by scavenger receptors on endothelial liver cells. Nucleic Acids Research 1997, 25, 3290–3296. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M. The blood-brain barrier.
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef] [PubMed]
- Di Fusco, D.; Dinallo, V.; Marafini, I.; Figliuzzi, M.M.; Romano, B.; Monteleone, G. Antisense Oligonucleotide: Basic Concepts and Therapeutic Application in Inflammatory Bowel Disease. Front Pharmacol 2019, 10, 305. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.B.; Seth, P.P. The Medicinal Chemistry of Therapeutic Oligonucleotides. J Med Chem 2016, 59, 9645–9667. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther 2014, 24, 374–387. [Google Scholar] [CrossRef]
- Furdon, P.J.; Dominski, Z.; Kole, R. RNase H cleavage of RNA hybridized to oligonucleotides containing methylphosphonate, phosphorothioate and phosphodiester bonds. Nucleic Acids Res 1989, 17, 9193–9204. [Google Scholar] [CrossRef]
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol 2018, 14, 9–21. [Google Scholar] [CrossRef]
- Ezzat, K.; Aoki, Y.; Koo, T.; McClorey, G.; Benner, L.; Coenen-Stass, A.; O’Donovan, L.; Lehto, T.; Garcia-Guerra, A.; Nordin, J.; et al. Self-Assembly into Nanoparticles Is Essential for Receptor Mediated Uptake of Therapeutic Antisense Oligonucleotides. Nano Lett 2015, 15, 4364–4373. [Google Scholar] [CrossRef]
- Miller, C.M.; Donner, A.J.; Blank, E.E.; Egger, A.W.; Kellar, B.M.; Ostergaard, M.E.; Seth, P.P.; Harris, E.N. Stabilin-1 and Stabilin-2 are specific receptors for the cellular internalization of phosphorothioate-modified antisense oligonucleotides (ASOs) in the liver. Nucleic Acids Res 2016, 44, 2782–2794. [Google Scholar] [CrossRef]
- Gaus, H.J.; Gupta, R.; Chappell, A.E.; Ostergaard, M.E.; Swayze, E.E.; Seth, P.P. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res 2019, 47, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.A. A review of the issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim Biophys Acta 1999, 1489, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Kang, S.H.; Gryaznov, S.M.; DeDionisio, L.; Heidenreich, O.; Sullivan, S.; Xu, X.; Nerenberg, M.I. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. Journal of Biological Chemistry 1994, 269, 26801–26805. [Google Scholar] [CrossRef] [PubMed]
- Guvakova, M.A.; Yakubov, L.A.; Vlodavsky, I.; Tonkinson, J.L.; Stein, C.A. Phosphorothioate oligodeoxynucleotides bind to basic fibroblast growth factor, inhibit its binding to cell surface receptors, and remove it from low affinity binding sites on extracellular matrix. J Biol Chem 1995, 270, 2620–2627. [Google Scholar] [CrossRef]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res 2016, 44, 6518–6548. [Google Scholar] [CrossRef]
- Freier, S.M.; Altmann, K.H. The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res 1997, 25, 4429–4443. [Google Scholar] [CrossRef]
- Lubini, P.; Zurcher, W.; Egli, M. Stabilizing effects of the RNA 2’-substituent: crystal structure of an oligodeoxynucleotide duplex containing 2’-O-methylated adenosines. Chem Biol 1994, 1, 39–45. [Google Scholar] [CrossRef]
- McKay, R.A.; Miraglia, L.J.; Cummins, L.L.; Owens, S.R.; Sasmor, H.; Dean, N.M. Characterization of a potent and specific class of antisense oligonucleotide inhibitor of human protein kinase C-alpha expression. J Biol Chem 1999, 274, 1715–1722. [Google Scholar] [CrossRef]
- Prakash, T.P. An overview of sugar-modified oligonucleotides for antisense therapeutics. Chem Biodivers 2011, 8, 1616–1641. [Google Scholar] [CrossRef]
- Frieden, M.; Hansen, H.F.; Koch, T. Nuclease Stability of LNA Oligonucleotides and LNA-DNA Chimeras. Nucleosides, Nucleotides & Nucleic Acids 2003, 22, 1041–1043. [Google Scholar] [CrossRef]
- Braasch, D.A.; Liu, Y.; Corey, D.R. Antisense inhibition of gene expression in cells by oligonucleotides incorporating locked nucleic acids: effect of mRNA target sequence and chimera design. Nucleic Acids Research 2002, 30, 5160–5167. [Google Scholar] [CrossRef] [PubMed]
- Elayadi, A.N.; Braasch, D.A.; Corey, D.R. Implications of high-affinity hybridization by locked nucleic acid oligomers for inhibition of human telomerase. Biochemistry 2002, 41, 9973–9981. [Google Scholar] [CrossRef]
- Swayze, E.E.; Siwkowski, A.M.; Wancewicz, E.V.; Migawa, M.T.; Wyrzykiewicz, T.K.; Hung, G.; Monia, B.P.; Bennett, C.F. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res 2007, 35, 687–700. [Google Scholar] [CrossRef]
- Gruegelsiepe, H.; Brandt, O.; Hartmann, R.K. Antisense inhibition of RNase P: mechanistic aspects and application to live bacteria. J Biol Chem 2006, 281, 30613–30620. [Google Scholar] [CrossRef] [PubMed]
- Renneberg, D.; Leumann, C.J. Watson-Crick base-pairing properties of tricyclo-DNA. J Am Chem Soc 2002, 124, 5993–6002. [Google Scholar] [CrossRef] [PubMed]
- Renneberg, D.; Bouliong, E.; Reber, U.; Schumperli, D.; Leumann, C.J. Antisense properties of tricyclo-DNA. Nucleic Acids Res 2002, 30, 2751–2757. [Google Scholar] [CrossRef]
- Seth, P.P.; Siwkowski, A.; Allerson, C.R.; Vasquez, G.; Lee, S.; Prakash, T.P.; Kinberger, G.; Migawa, M.T.; Gaus, H.; Bhat, B.; et al. Design, synthesis and evaluation of constrained methoxyethyl (cMOE) and constrained ethyl (cEt) nucleoside analogs. Nucleic Acids Symp Ser (Oxf) 2008, 553–554. [Google Scholar] [CrossRef]
- Southwell, A.L.; Skotte, N.H.; Kordasiewicz, H.B.; Ostergaard, M.E.; Watt, A.T.; Carroll, J.B.; Doty, C.N.; Villanueva, E.B.; Petoukhov, E.; Vaid, K.; et al. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther 2014, 22, 2093–2106. [Google Scholar] [CrossRef]
- Deleavey, G.F.; Damha, M.J. Designing chemically modified oligonucleotides for targeted gene silencing. Chem Biol 2012, 19, 937–954. [Google Scholar] [CrossRef]
- Chenna, V.; Rapireddy, S.; Sahu, B.; Ausin, C.; Pedroso, E.; Ly, D.H. A simple cytosine to G-clamp nucleobase substitution enables chiral gamma-PNAs to invade mixed-sequence double-helical B-form DNA. Chembiochem 2008, 9, 2388–2391. [Google Scholar] [CrossRef]
- Rapireddy, S.; Bahal, R.; Ly, D.H. Strand invasion of mixed-sequence, double-helical B-DNA by gamma-peptide nucleic acids containing G-clamp nucleobases under physiological conditions. Biochemistry 2011, 50, 3913–3918. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Demidov, V.V.; Potaman, V.N.; Frank-Kamenetskii, M.D.; Egholm, M.; Buchard, O.; Sonnichsen, S.H.; Nielsen, P.E. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem Pharmacol 1994, 48, 1310–1313. [Google Scholar] [CrossRef]
- Schwarz, F.P.; Robinson, S.; Butler, J.M. Thermodynamic comparison of PNA/DNA and DNA/DNA hybridization reactions at ambient temperature. Nucleic Acids Res 1999, 27, 4792–4800. [Google Scholar] [CrossRef]
- Wittung, P.; Kajanus, J.; Edwards, K.; Nielsen, P.; Norden, B.; Malmstrom, B.G. Phospholipid membrane permeability of peptide nucleic acid. FEBS Lett 1995, 365, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Hyrup, B.; Nielsen, P.E. Peptide nucleic acids (PNA): synthesis, properties and potential applications. Bioorg Med Chem 1996, 4, 5–23. [Google Scholar] [CrossRef]
- McMahon, B.M.; Mays, D.; Lipsky, J.; Stewart, J.A.; Fauq, A.; Richelson, E. Pharmacokinetics and tissue distribution of a peptide nucleic acid after intravenous administration. Antisense Nucleic Acid Drug Dev 2002, 12, 65–70. [Google Scholar] [CrossRef]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev 1997, 7, 187–195. [Google Scholar] [CrossRef]
- Hudziak, R.M.; Barofsky, E.; Barofsky, D.F.; Weller, D.L.; Huang, S.B.; Weller, D.D. Resistance of morpholino phosphorodiamidate oligomers to enzymatic degradation. Antisense Nucleic Acid Drug Dev 1996, 6, 267–272. [Google Scholar] [CrossRef]
- Lee, J.J.; Yokota, T. Antisense therapy in neurology. J Pers Med 2013, 3, 144–176. [Google Scholar] [CrossRef]
- Sheng, L.; Rigo, F.; Bennett, C.F.; Krainer, A.R.; Hua, Y. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acids Res 2020, 48, 2853–2865. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Yokota, T. Golodirsen for Duchenne muscular dystrophy. Drugs Today (Barc) 2020, 56, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today (Barc) 2019, 55, 627–639. [Google Scholar] [CrossRef]
- Lu, Q.L.; Mann, C.J.; Lou, F.; Bou-Gharios, G.; Morris, G.E.; Xue, S.A.; Fletcher, S.; Partridge, T.A.; Wilton, S.D. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med 2003, 9, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Gebski, B.L.; Mann, C.J.; Fletcher, S.; Wilton, S.D. Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum Mol Genet 2003, 12, 1801–1811. [Google Scholar] [CrossRef]
- Yokota, T.; Lu, Q.L.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol 2009, 65, 667–676. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef]
- Hua, Y.; Liu, Y.H.; Sahashi, K.; Rigo, F.; Bennett, C.F.; Krainer, A.R. Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev 2015, 29, 288–297. [Google Scholar] [CrossRef]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet 2012, 21, 1625–1638. [Google Scholar] [CrossRef]
- Zhou, H.; Janghra, N.; Mitrpant, C.; Dickinson, R.L.; Anthony, K.; Price, L.; Eperon, I.C.; Wilton, S.D.; Morgan, J.; Muntoni, F. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum Gene Ther 2013, 24, 331–342. [Google Scholar] [CrossRef]
- Lehto, T.; Castillo Alvarez, A.; Gauck, S.; Gait, M.J.; Coursindel, T.; Wood, M.J.; Lebleu, B.; Boisguerin, P. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res 2014, 42, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.A.; Aoki, Y. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine 2019, 45, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Summerton, J.E. Invention and Early History of Morpholinos: From Pipe Dream to Practical Products. Methods Mol Biol 2017, 1565, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Thomas, O.S.; Weber, W. Overcoming Physiological Barriers to Nanoparticle Delivery-Are We There Yet? Front Bioeng Biotechnol 2019, 7, 415. [Google Scholar] [CrossRef] [PubMed]
- Nishina, K.; Unno, T.; Uno, Y.; Kubodera, T.; Kanouchi, T.; Mizusawa, H.; Yokota, T. Efficient In Vivo Delivery of siRNA to the Liver by Conjugation of alpha-Tocopherol. Mol Ther 2008, 16, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol 2007, 25, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef]
- Lorenz, C.; Hadwiger, P.; John, M.; Vornlocher, H.P.; Unverzagt, C. Steroid and lipid conjugates of siRNAs to enhance cellular uptake and gene silencing in liver cells. Bioorg Med Chem Lett 2004, 14, 4975–4977. [Google Scholar] [CrossRef]
- Klein, A.F.; Varela, M.A.; Arandel, L.; Holland, A.; Naouar, N.; Arzumanov, A.; Seoane, D.; Revillod, L.; Bassez, G.; Ferry, A.; et al. Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. J Clin Invest 2019, 129, 4739–4744. [Google Scholar] [CrossRef]
- Eguchi, A.; Meade, B.R.; Chang, Y.C.; Fredrickson, C.T.; Willert, K.; Puri, N.; Dowdy, S.F. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat Biotechnol 2009, 27, 567–571. [Google Scholar] [CrossRef]
- Betts, C.; Saleh, A.F.; Arzumanov, A.A.; Hammond, S.M.; Godfrey, C.; Coursindel, T.; Gait, M.J.; Wood, M.J. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates With Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol Ther Nucleic Acids 2012, 1, e38. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Ming, X.; Fisher, M.; Lackey, J.G.; Rajeev, K.G.; Manoharan, M.; Juliano, R.L. Multivalent cyclic RGD conjugates for targeted delivery of small interfering RNA. Bioconjug Chem 2011, 22, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Ammala, C.; Drury, W.J., 3rd; Knerr, L.; Ahlstedt, I.; Stillemark-Billton, P.; Wennberg-Huldt, C.; Andersson, E.M.; Valeur, E.; Jansson-Lofmark, R.; Janzen, D.; et al. Targeted delivery of antisense oligonucleotides to pancreatic beta-cells. Sci Adv 2018, 4, eaat3386. [Google Scholar] [CrossRef]
- Liu, X.; Wang, W.; Samarsky, D.; Liu, L.; Xu, Q.; Zhang, W.; Zhu, G.; Wu, P.; Zuo, X.; Deng, H.; et al. Tumor-targeted in vivo gene silencing via systemic delivery of cRGD-conjugated siRNA. Nucleic Acids Res 2014, 42, 11805–11817. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.O., 2nd; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol 2006, 24, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhu, P.; Lee, S.K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol 2005, 23, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Tushir-Singh, J. Antibody-siRNA conjugates: drugging the undruggable for anti-leukemic therapy. Expert Opin Biol Ther 2017, 17, 325–338. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc 2014, 136, 16958–16961. [Google Scholar] [CrossRef]
- Matsuda, S.; Keiser, K.; Nair, J.K.; Charisse, K.; Manoharan, R.M.; Kretschmer, P.; Peng, C.G.; A, V.K.i.; Kandasamy, P.; Willoughby, J.L.; et al. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem Biol 2015, 10, 1181–1187. [Google Scholar] [CrossRef]
- Anwar, S.; Mir, F.; Yokota, T. Enhancing the Effectiveness of Oligonucleotide Therapeutics Using Cell-Penetrating Peptide Conjugation, Chemical Modification, and Carrier-Based Delivery Strategies. Pharmaceutics 2023, 15. [Google Scholar] [CrossRef]
- McClorey, G.; Banerjee, S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med 2012, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.H.; Park, J.; Koo, H. Recent advances in selective and targeted drug/gene delivery systems using cell-penetrating peptides. Arch Pharm Res 2023, 46, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front Pharmacol 2020, 11, 697. [Google Scholar] [CrossRef]
- Verdurmen, W.P.; Brock, R. Biological responses towards cationic peptides and drug carriers. Trends Pharmacol Sci 2011, 32, 116–124. [Google Scholar] [CrossRef]
- Godfrey, C.; Desviat, L.R.; Smedsrod, B.; Pietri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: lessons learnt from developing splice-switching antisense therapies. EMBO Mol Med 2017, 9, 545–557. [Google Scholar] [CrossRef]
- Gomez, J.A.; Chen, J.; Ngo, J.; Hajkova, D.; Yeh, I.J.; Gama, V.; Miyagi, M.; Matsuyama, S. Cell-Penetrating Penta-Peptides (CPP5s): Measurement of Cell Entry and Protein-Transduction Activity. Pharmaceuticals (Basel) 2010, 3, 3594–3613. [Google Scholar] [CrossRef]
- Klabenkova, K.; Fokina, A.; Stetsenko, D. Chemistry of Peptide-Oligonucleotide Conjugates: A Review. Molecules 2021, 26. [Google Scholar] [CrossRef]
- Lehto, T.; Ezzat, K.; Wood, M.J.A.; El Andaloussi, S. Peptides for nucleic acid delivery. Adv Drug Deliv Rev 2016, 106, 172–182. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed Pharmacother 2018, 108, 1090–1096. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.M.; Amin, R.S.; Bach, J.R.; Benditt, J.O.; Eagle, M.; Finder, J.D.; Kalra, M.S.; Kissel, J.T.; Koumbourlis, A.C.; et al. The respiratory management of patients with duchenne muscular dystrophy: a DMD care considerations working group specialty article. Pediatr Pulmonol 2010, 45, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Manzur, A.Y.; Kinali, M.; Muntoni, F. Update on the management of Duchenne muscular dystrophy. Arch Dis Child 2008, 93, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Wilton-Clark, H.; Yokota, T. Casimersen for Duchenne muscular dystrophy. Drugs Today (Barc) 2021, 57, 707–717. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Viltolarsen: From Preclinical Studies to FDA Approval. Methods Mol Biol 2023, 2587, 31–41. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum Mol Genet 2008, 17, 3909–3918. [Google Scholar] [CrossRef]
- Wu, B.; Moulton, H.M.; Iversen, P.L.; Jiang, J.; Li, J.; Li, J.; Spurney, C.F.; Sali, A.; Guerron, A.D.; Nagaraju, K.; et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci U S A 2008, 105, 14814–14819. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med 2015, 21, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Samoylova, T.I.; Smith, B.F. Elucidation of muscle-binding peptides by phage display screening.
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum Mol Genet 2009, 18, 4405–4414. [Google Scholar] [CrossRef]
- Echigoya, Y.; Nakamura, A.; Nagata, T.; Urasawa, N.; Lim, K.R.Q.; Trieu, N.; Panesar, D.; Kuraoka, M.; Moulton, H.M.; Saito, T.; et al. Effects of systemic multiexon skipping with peptide-conjugated morpholinos in the heart of a dog model of Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 2017, 114, 4213–4218. [Google Scholar] [CrossRef]
- Therapeutics, P.E.O. Available online: https://investors.pepgen.com/static-files/7244e66d-1227-42b1-841fbf95306d84b1.
- Abes, S.; Turner, J.J.; Ivanova, G.D.; Owen, D.; Williams, D.; Arzumanov, A.; Clair, P.; Gait, M.J.; Lebleu, B. Efficient splicing correction by PNA conjugation to an R6-Penetratin delivery peptide. Nucleic Acids Res 2007, 35, 4495–4502. [Google Scholar] [CrossRef]
- Bersani, M.; Rizzuti, M.; Pagliari, E.; Garbellini, M.; Saccomanno, D.; Moulton, H.M.; Bresolin, N.; Comi, G.P.; Corti, S.; Nizzardo, M. Cell-penetrating peptide-conjugated Morpholino rescues SMA in a symptomatic preclinical model. Mol Ther 2022, 30, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. Journal of Biological Chemistry 1994, 269, 10444–10450. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; Joliot, A.; Bloch-Gallego, E.; Zahraoui, A.; Triller, A.; Prochiantz, A. Antennapedia homeobox as a signal for the cellular internalization and nuclear addressing of a small exogenous peptide. J Cell Sci 1992, 102 Pt 4, 717–722. [Google Scholar] [CrossRef]
- Yin, H.; Saleh, A.F.; Betts, C.; Camelliti, P.; Seow, Y.; Ashraf, S.; Arzumanov, A.; Hammond, S.; Merritt, T.; Gait, M.J.; et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol Ther 2011, 19, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Muses, S.; McClorey, G.; Wells, K.E.; Coursindel, T.; Terry, R.L.; Betts, C.; Hammond, S.; O’Donovan, L.; Hildyard, J.; et al. How much dystrophin is enough: the physiological consequences of different levels of dystrophin in the mdx mouse. Hum Mol Genet 2015, 24, 4225–4237. [Google Scholar] [CrossRef] [PubMed]
- Betts, C.A.; Saleh, A.F.; Carr, C.A.; Hammond, S.M.; Coenen-Stass, A.M.; Godfrey, C.; McClorey, G.; Varela, M.A.; Roberts, T.C.; Clarke, K.; et al. Prevention of exercised induced cardiomyopathy following Pip-PMO treatment in dystrophic mdx mice. Sci Rep 2015, 5, 8986. [Google Scholar] [CrossRef]
- Ivanova, G.D.; Arzumanov, A.; Abes, R.; Yin, H.; Wood, M.J.; Lebleu, B.; Gait, M.J. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res 2008, 36, 6418–6428. [Google Scholar] [CrossRef]
- Leger, A.J.; Mosquea, L.M.; Clayton, N.P.; Wu, I.H.; Weeden, T.; Nelson, C.A.; Phillips, L.; Roberts, E.; Piepenhagen, P.A.; Cheng, S.H.; et al. Systemic delivery of a Peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther 2013, 23, 109–117. [Google Scholar] [CrossRef]
- Gao, H. Progress and perspectives on targeting nanoparticles for brain drug delivery. Acta Pharm Sin B 2016, 6, 268–286. [Google Scholar] [CrossRef]
- Mae, M.; Langel, U. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr Opin Pharmacol 2006, 6, 509–514. [Google Scholar] [CrossRef]
- Faravelli, I.; Nizzardo, M.; Comi, G.P.; Corti, S. Spinal muscular atrophy--recent therapeutic advances for an old challenge. Nat Rev Neurol 2015, 11, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Feldkotter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002, 70, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Strasswimmer, J.; Yao, J.M.; Baleja, J.D.; Hahnen, E.; Wirth, B.; Le, T.; Burghes, A.H.; Androphy, E.J. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet 1998, 19, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Nakevska, Z.; Yokota, T. Challenges and future perspective of antisense therapy for spinal muscular atrophy: A review. Eur J Cell Biol 2023, 102, 151326. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci U S A 2016, 113, 10962–10967. [Google Scholar] [CrossRef]
- Shabanpoor, F.; Hammond, S.M.; Abendroth, F.; Hazell, G.; Wood, M.J.A.; Gait, M.J. Identification of a Peptide for Systemic Brain Delivery of a Morpholino Oligonucleotide in Mouse Models of Spinal Muscular Atrophy. Nucleic Acid Ther 2017, 27, 130–143. [Google Scholar] [CrossRef]
- Muntoni, F.; Wood, M.J. Targeting RNA to treat neuromuscular disease. Nat Rev Drug Discov 2011, 10, 621–637. [Google Scholar] [CrossRef]
- Henry, S.P.; Beattie, G.; Yeh, G.; Chappel, A.; Giclas, P.; Mortari, A.; Jagels, M.A.; Kornbrust, D.J.; Levin, A.A. Complement activation is responsible for acute toxicities in rhesus monkeys treated with a phosphorothioate oligodeoxynucleotide. Int Immunopharmacol 2002, 2, 1657–1666. [Google Scholar] [CrossRef]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim Biophys Acta 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Amantana, A.; Moulton, H.M.; Cate, M.L.; Reddy, M.T.; Whitehead, T.; Hassinger, J.N.; Youngblood, D.S.; Iversen, P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug Chem 2007, 18, 1325–1331. [Google Scholar] [CrossRef]
- Choi, J.M.; Ahn, M.H.; Chae, W.J.; Jung, Y.G.; Park, J.C.; Song, H.M.; Kim, Y.E.; Shin, J.A.; Park, C.S.; Park, J.W.; et al. Intranasal delivery of the cytoplasmic domain of CTLA-4 using a novel protein transduction domain prevents allergic inflammation. Nat Med 2006, 12, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Shin, J.H.; Sohn, M.H.; Harding, M.J.; Park, J.H.; Tobiasova, Z.; Kim, D.Y.; Maher, S.E.; Chae, W.J.; Park, S.H.; et al. Cell-permeable Foxp3 protein alleviates autoimmune disease associated with inflammatory bowel disease and allergic airway inflammation. Proc Natl Acad Sci U S A 2010, 107, 18575–18580. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, D.; Tae, H.S.; Gandotra, N.; Llopis, P.; Shen, N.; Altman, S. Basic peptide-morpholino oligomer conjugate that is very effective in killing bacteria by gene-specific and nonspecific modes. Proc Natl Acad Sci U S A 2011, 108, 16582–16587. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Clark, K.; Barton, C.; Tanguay, R.; Moulton, H. A Novel Zebrafish Model for Assessing In Vivo Delivery of Morpholino Oligomers. Methods Mol Biol 2018, 1828, 293–306. [Google Scholar] [CrossRef]
- Saar, K.; Lindgren, M.; Hansen, M.; Eiriksdottir, E.; Jiang, Y.; Rosenthal-Aizman, K.; Sassian, M.; Langel, U. Cell-penetrating peptides: a comparative membrane toxicity study. Anal Biochem 2005, 345, 55–65. [Google Scholar] [CrossRef]
- Sylvestre, M.; Lv, S.; Yang, L.F.; Luera, N.; Peeler, D.J.; Chen, B.M.; Roffler, S.R.; Pun, S.H. Replacement of L-amino acid peptides with D-amino acid peptides mitigates anti-PEG antibody generation against polymer-peptide conjugates in mice. J Control Release 2021, 331, 142–153. [Google Scholar] [CrossRef]
- Wu, R.P.; Youngblood, D.S.; Hassinger, J.N.; Lovejoy, C.E.; Nelson, M.H.; Iversen, P.L.; Moulton, H.M. Cell-penetrating peptides as transporters for morpholino oligomers: effects of amino acid composition on intracellular delivery and cytotoxicity. Nucleic Acids Res 2007, 35, 5182–5191. [Google Scholar] [CrossRef]
Figure 1.
Functional mechanism of Antisense Oligonucleotide mediated modulation of gene ex-pression. a) RNase H mediated degradation of RNA by Antisense Oligonucleotides. b) Suppress-ing the translation or splicing modulation by Antisense Oligonucleotide through steric hindrance mechanism. Created with BioRender.com.
Figure 1.
Functional mechanism of Antisense Oligonucleotide mediated modulation of gene ex-pression. a) RNase H mediated degradation of RNA by Antisense Oligonucleotides. b) Suppress-ing the translation or splicing modulation by Antisense Oligonucleotide through steric hindrance mechanism. Created with BioRender.com.
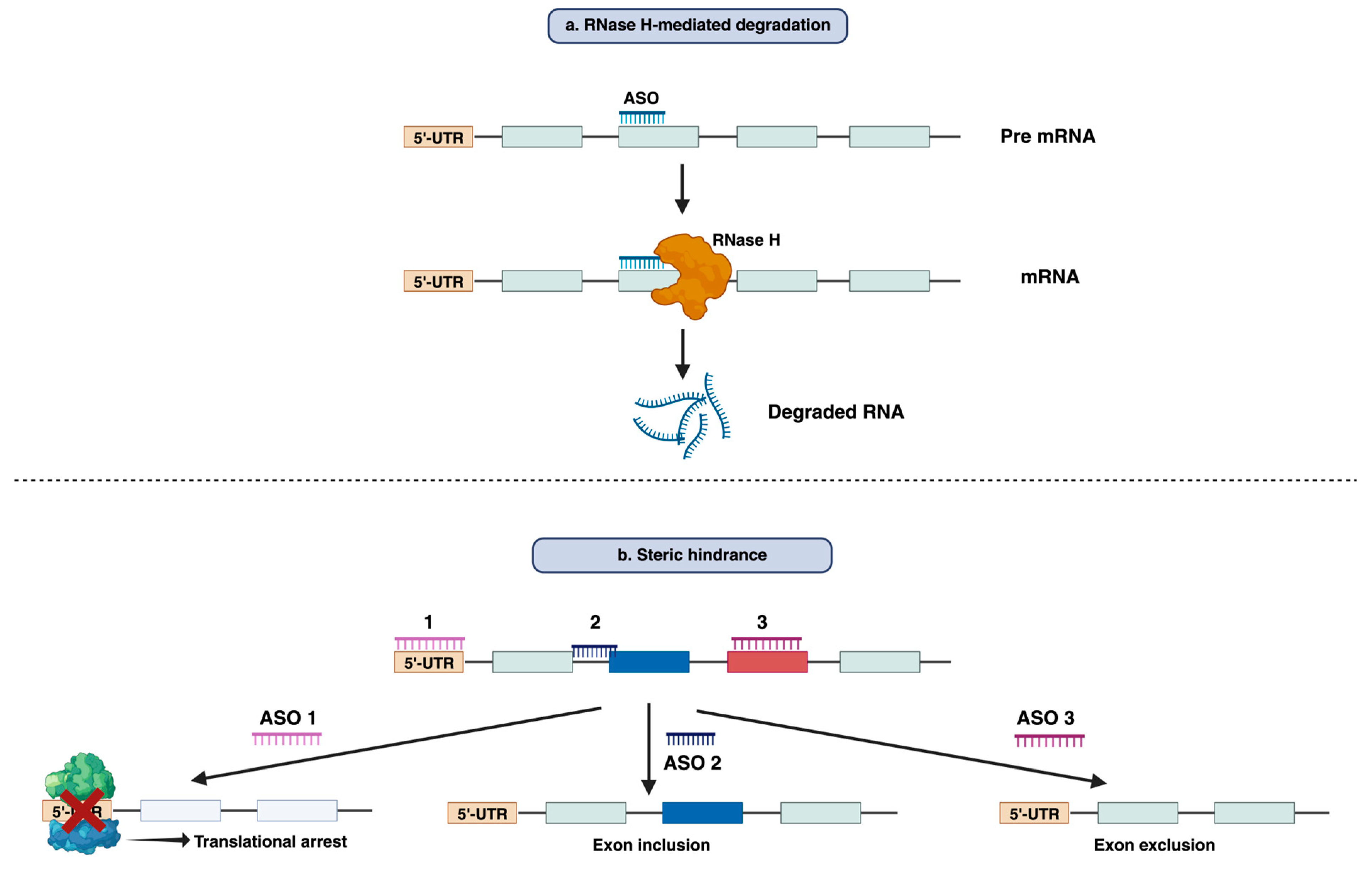
Figure 2.
Challenges in systemic delivery of ASO drugs. After systemic administration, Antisense Oligonucleotide drugs face difficulties in terms of reaching the central nervous system, heart, and other targeted tissue due to their accumulation in the liver and spleen and renal excretion. Most systemically administered naked ASO gets degraded by nuclease in the bloodstream. Upon reaching the target tissue, it gets internalized by the endosome. Endosomal escape is required to exhibit activity. Apart from that ASO gets degraded in the lysosome or gets out of the cell/tissue by the multi-vesicular system.
Figure 2.
Challenges in systemic delivery of ASO drugs. After systemic administration, Antisense Oligonucleotide drugs face difficulties in terms of reaching the central nervous system, heart, and other targeted tissue due to their accumulation in the liver and spleen and renal excretion. Most systemically administered naked ASO gets degraded by nuclease in the bloodstream. Upon reaching the target tissue, it gets internalized by the endosome. Endosomal escape is required to exhibit activity. Apart from that ASO gets degraded in the lysosome or gets out of the cell/tissue by the multi-vesicular system.
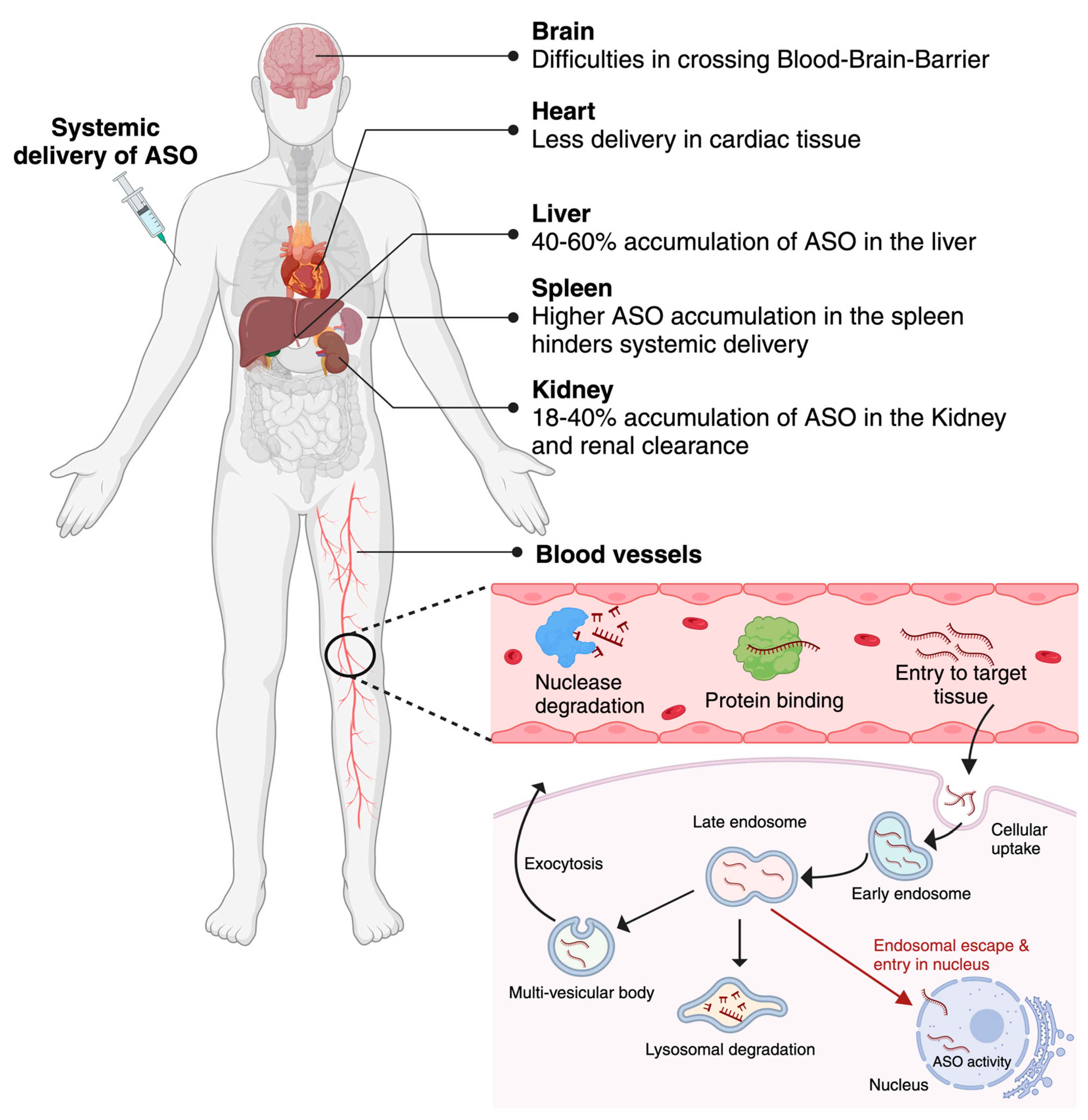
Figure 3.
Some common chemical modification used in Antisense Oligonucleotide chemistry. a) Schematic of an RNA nucleotide with common modification site. b) Ribose sugar modification: 2ʹ-OMe, 2ʹ-O-methyl; 2ʹ-MOE, 2ʹ-O-methoxyethyl; 2’-Fluoro; tcDNA, tricyclo DNA; LNA, locked nucleic acid; cEt, constrained ethyl bridged nucleic acid; ENA, ethylene-bridged nucleic acid. c) Backbone modification: PS, phosphorothioate. d) Nucleobase modification: 5-methylcytidine, 5-methyluridine. e) Alternative chemistries: PMO, phosphorodiamidate morpholino oligonucleotide; PNA, peptide nucleic acid. Created with BioRender.com.
Figure 3.
Some common chemical modification used in Antisense Oligonucleotide chemistry. a) Schematic of an RNA nucleotide with common modification site. b) Ribose sugar modification: 2ʹ-OMe, 2ʹ-O-methyl; 2ʹ-MOE, 2ʹ-O-methoxyethyl; 2’-Fluoro; tcDNA, tricyclo DNA; LNA, locked nucleic acid; cEt, constrained ethyl bridged nucleic acid; ENA, ethylene-bridged nucleic acid. c) Backbone modification: PS, phosphorothioate. d) Nucleobase modification: 5-methylcytidine, 5-methyluridine. e) Alternative chemistries: PMO, phosphorodiamidate morpholino oligonucleotide; PNA, peptide nucleic acid. Created with BioRender.com.
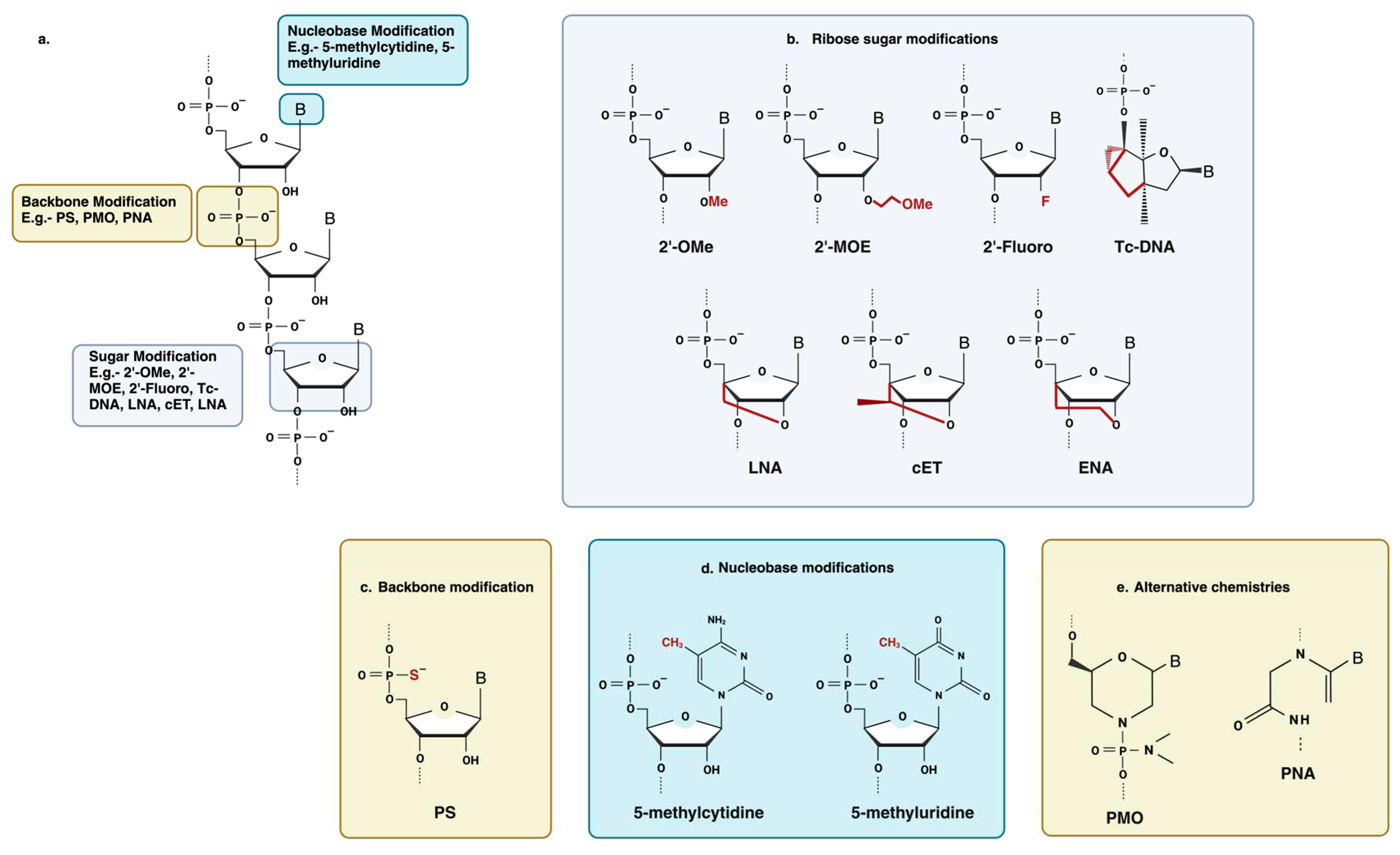
Figure 4.
Structural representation of DG9-PMO. C-terminal end of DG9 is conjugated to the 3’end of the PMO. Created with BioRender.com.
Figure 4.
Structural representation of DG9-PMO. C-terminal end of DG9 is conjugated to the 3’end of the PMO. Created with BioRender.com.
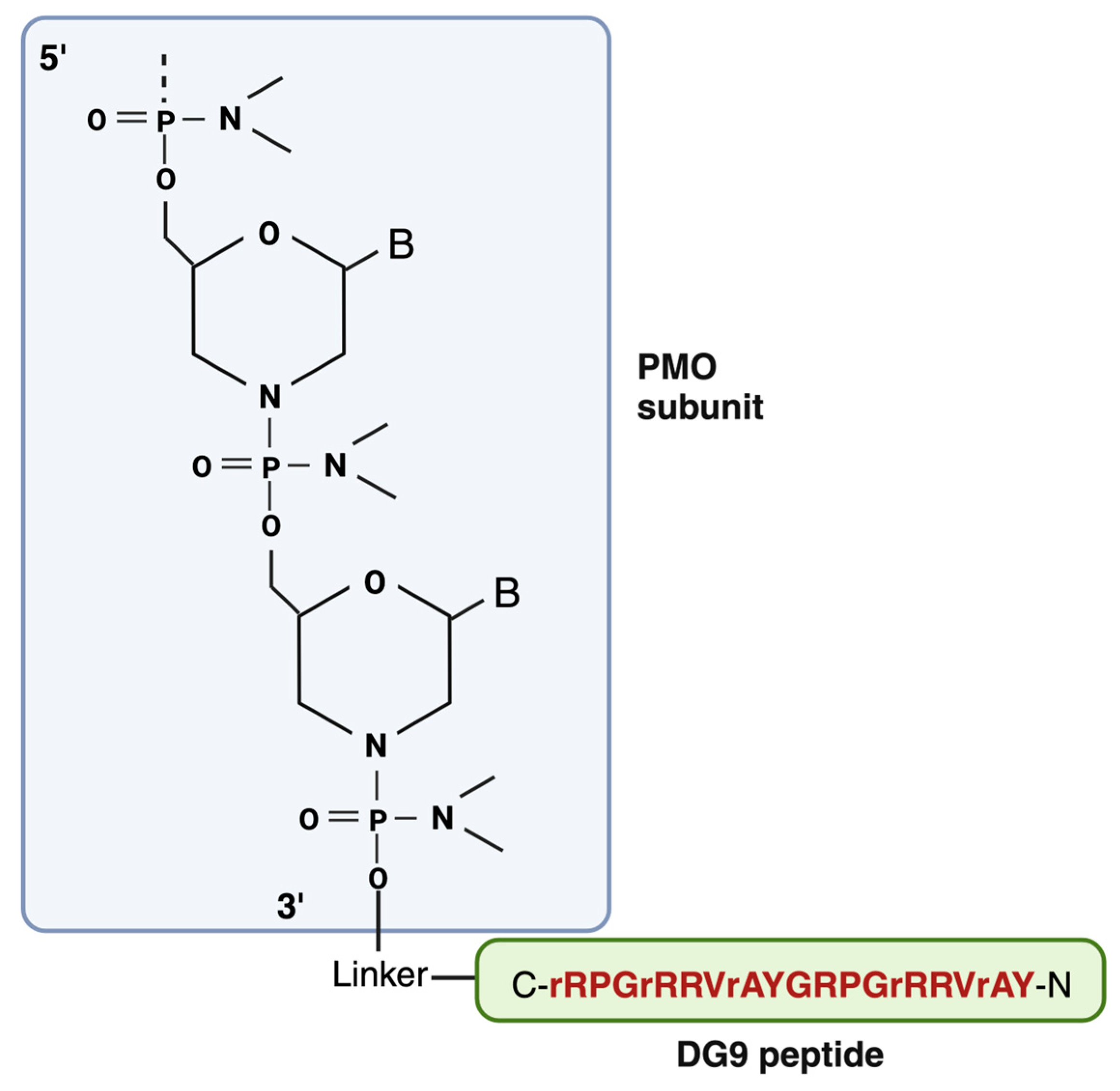

Table 1.
FDA-approved Oligonucleotide therapeutics. ASO, antisense oligonucleotide; dsDNA, double-stranded DNA; 2ʹ-F, 2ʹ-fluoro; GalNac, N-acetylgalactosamine; LNP, lipid nanoparticle; 2ʹ-MOE, 2ʹ-O-methoxyethyl; 2ʹ-OMe, 2ʹ-O-methyl; PMO, phosphorodiamidate morpholino oligonucleotide; PO, phosphodiester; PS, phosphorothioate; ROA, route of administration; siRNA, small interfering RNA; ssDNA, single-stranded DNA.
Table 1.
FDA-approved Oligonucleotide therapeutics. ASO, antisense oligonucleotide; dsDNA, double-stranded DNA; 2ʹ-F, 2ʹ-fluoro; GalNac, N-acetylgalactosamine; LNP, lipid nanoparticle; 2ʹ-MOE, 2ʹ-O-methoxyethyl; 2ʹ-OMe, 2ʹ-O-methyl; PMO, phosphorodiamidate morpholino oligonucleotide; PO, phosphodiester; PS, phosphorothioate; ROA, route of administration; siRNA, small interfering RNA; ssDNA, single-stranded DNA.
| Drug Name (Market name) | ROA | Target gene | Indication | Modality | Chemistry | Mechanism of action | Approval | Company |
|---|---|---|---|---|---|---|---|---|
| Fomivirsen (Vitravene) | Intraocular | IE-2 mRNA | Cytomegalovirus (CMV) retinitis | ASO | 21mer PS DNA | RNase H1 | FDA/EMA (1998) | Ionis Pharmaceuticals, Novartis |
| Pegaptanib (Macugen) | Intraocular | Heparin-binding domain of VEGF-165 | Neovascular age-related macular degeneration | Aptamer | 27mer 2ʹ-F/2ʹ-OMe pegylated | Binding and blocking | FDA (2004) | OSI Pharmaceuticals |
| Mipomersen (Kynamro) | Subcutaneous | Apolipoprotein B100 | Homozygous familial hypercholesterolemia | ASO (gapmer) | 20mer PS 2ʹ-MOE | RNase H1 | FDA (2013) | Kastle Therapeutics, Ionis Pharmaceuticals, Genzyme |
| Eteplirsen (Exondys 51) | Intravenous | Exon 51 of DMD | Duchenne muscular dystrophy | ASO | 30mer PMO | Splicing modulation | FDA (2016) | Sarepta Therapeutics |
| Nusinersen (Spinraza) | Intrathecal | Exon 7 of SMN2 | Spinal muscular atrophy | ASO | 18mer PS 2ʹ-MOE | Splicing modulation | FDA/EMA (2016) | Ionis Pharmaceuticals, Biogen |
| Defibrotide (Defitelio) | Intravenous | Adenosine A1/A2 receptor |
Veno-occlusive disease in liver | Aptamer | Mixture of PO ssDNA and dsDNA | Binding and activating | FDA (2016) | Jazz Pharmaceuticals |
| Inotersen (Tegsedi) | Subcutaneous | Transthyretin | Polyneuropathy caused by hereditary transthyretin-mediated (hATTR) amyloidosis | ASO (gapmer) | 20mer PS 2ʹ-MOE | RNase H1 | FDA (2018) | Akcea Therapeutics |
| Milasen* | Intrathecal | CLN7 | Mila Makovec’s CLN7 gene associated with Batten disease | ASO | 22 mer 2′-O-MOE, PS, 5-methyl cytosine | Splicing modulation | FDA (2018) | Boston Children’s Hospital* |
| Patisiran (Onpattro) | Intravenous | Transthyretin | Polyneuropathy caused by hATTR amyloidosis | siRNA (LNP formulation) | 19 + 2mer 2ʹ-OMe modified | RNAi | FDA/EMA (2018) | Alnylam Pharma |
| Golodirsen (Vyondys 53) | Intravenous | Exon 53 of DMD | Duchenne muscular dystrophy | ASO | 25mer PMO | Splicing modulation | FDA (2019) | Sarepta Therapeutics |
| Givosiran (Givlaari) | Subcutaneous | 5-aminolevulinic acid synthase |
Acute hepatic porphyria (AHP) | siRNA (GalNAc conjugate) | 21/23mer Dicer substrate siRNA | RNAi | FDA/EMA (2019) | Alnylam Pharma |
| Volanesorsen (Waylivra) | Subcutaneous | Apolipoprotein C3 | Familial chylomicronemia syndrome (FCS) | ASO | 20 mer PS, 2ʹ-MOE | RNase H1 | EMA (2019) | Akcea Therapeutics |
| Viltolarsen (Viltepso) | Intravenous | Exon 53 of DMD | Duchenne muscular dystrophy | ASO | 21 mer PMO | Splicing modulation | FDA (2020) | NS Pharma |
| Casimersen (Amondys 45) | Intravenous | Exon 45 of DMD | Duchenne muscular dystrophy | ASO | 22 mer PMO | Splicing modulation | FDA (2021) | Sarepta Therapeutics |
| Tofersen (Qalsody) | Intrathecal | SOD1 | Amyotrophic lateral sclerosis | ASO | 20 mer 2ʹ-MOE, gapmer | RNase H1 | FDA (2023) | Ionis Pharmaceuticals, Biogen |
| Valeriasen | Intrathecal | KCNT1 | Epilepsy | ASO | 2ʹ-MOE, gapmer | RNase H1 | FDA (2020) | Boston Children’s Hospital* |
| Atipeksen | Intrathecal | ATM | Ataxia telangiectasia | ASO | Splicing modulation | Boston Children’s Hospital* |
*Milasen, Valeriasen and Atipeksen are individualized medicine and are approved under investigational new drug by FDA.
Table 2.
Brief description of most used Bioconjugates in delivery of Antisense Oligonucleotides. The table is adapted from Roberts et al. and Anwar et al. [11,97].
| Bioconjugates | Brief introduction | Benefits |
|---|---|---|
| Lipid-based conjugates | Lipid-based moieties are usually cholesterol and its derivatives which are covalently conjugated to siRNA and antagomir ASOs to enhance the delivery. This group of bioconjugates enhances in vivo delivery by adhering to lipoprotein particles (such as HDL and LDL) in the circulation and therefore taking over the body’s natural system for lipid uptake and transport. The overall hydrophobicity of siRNAs governs their in vivo association with the various classes of lipoprotein, with the more hydrophobic conjugates preferentially attaching to LDL and primarily taken up by the liver. The less lipophilic conjugates preferentially bind to HDL and are consumed by the liver, adrenal glands, ovary, kidney, and small intestine. Another lipid derivatives α-tocopherol (vitamin E) was also found to increase the delivery of siRNA. |
|
| GalNac conjugates | Trimeric GalNac is the most clinically successful tissue-targeting ligand used in ASO delivery to date. GalNAc is a carbohydrate moiety that has a high affinity for the highly expressed asialoglycoprotein receptor 1 (ASGR1, ASPGR). This interaction promotes the endocytosis of PO ASOs and siRNAs into hepatocytes. Givosiran, a GalNAc-conjugated siRNA was granted FDA approval for the treatment of acute hepatic porphyria in November 2019 as a result its remarkable success. | |
| Antibody and Aptamer conjugates | Antibody–RNA bioconjugates offer a promising strategy for nucleic acid therapeutics, however, their utility for oligonucleotide delivery is still in the early stages of development. Antibodies are useful for the targeted delivery of oligonucleotides to cells or tissues that other methods cannot reach since they are very selective in recognizing target antigens. Similar to antibodies, aptamers bind to their respective target proteins with high affinity. Aptamers bind to their specific target proteins with high affinity, just like antibodies do. Aptamers are regarded as chemical antibodies and have demonstrated many advantages over antibodies, including being easier and less expensive to produce (i.e., through chemical synthesis), smaller size, and lower immunogenicity. | |
| Polymer conjugates | PEG is a non-ionic, hydrophilic polymer with a wide range of applications. It is widely used to prolong blood circulation time and improve drug efficacy. PEGylation, which involves covalently adding PEG to a drug, improves the stability of ASOs and reduces renal excretion by forming a protective hydration layer around them. PEG-conjugated drugs have been found to have better pharmacokinetic and pharmacodynamic properties in terms of the drug’s chemical aspects of absorption, distribution, metabolism, excretion, and toxicity (ADMET). Other polymers besides PEG have also received attention, including poly(glycerol), poly(2-oxazoline), poly (amino acid), and poly[N-(2-hydroxypropyl)methacrylamide] because they are more ADMET-enhancing and less immunogenic. | |
| Peptide-based conjugates | Peptides are short chains of amino acids that can serve as carriers for oligonucleotide delivery for its cell-specific targeting, cell-penetrating, or endosomolytic properties. More information about peptide conjugates is mentioned in section- 6.2.1. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



