
Submitted:
29 August 2023
Posted:
31 August 2023
You are already at the latest version
Abstract
The direct methane to methanol (MTM) oxidation is a grand challenge in catalysis, with profound economical implications for the modern chemical industry. Bioinspired metal-organic frameworks (MOFs) with active iron and copper sites have been emerging very recently as innovative catalytic platforms to accomplish the MTM conversion under mild conditions. This review discusses the current state of the art regarding the application of MOFs with iron and copper catalytic centers to perform the MTM reaction, with a focus on the diverse spectroscopic techniques used to unveil the electronic and structural properties of the MOF catalysts at a microscopic level. We explore the synthetic strategies employed to incorporate iron and copper sites into different MOF topologies, the efficiency and selectivity of the iron- and copper-bearing MOF catalysts, and the ensuing MTM reaction mechanisms proposed on the basis of spectroscopic characterization supported by theory. In particular, we evidence how the combination of complementary spectroscopic tools probing different regions of the electromagnetic spectrum is particularly useful to reach a satisfactory understanding of the key reaction pathways and intermediates. Finally, we provide a critical perspective on future directions to advance the use of MOFs to accomplish the MTM reaction.
Keywords:
Methane
; Methanol
; MOF
; Spectroscopy
; XAS
; IR
; Raman
; Mössbauer
; EPR
; XPS
1. Introduction
Methane (CH) is a highly abundant hydrocarbon, being the main constituent of shale and natural gas (where it amounts up to 80-90 % of all available reservoirs), as well as of biogas [1], a mixture of gases (mostly CH and CO) produced from biodegradable materials in an effort to implement alternative approaches for natural gas decarbonization [2]. Methane is also found in crystalline hydrates at the continental slopes of many oceans as well as in permafrost areas, and may be catalytically converted to numerous industrially relevant compounds, such as to methanol, synthesis gas, hydrogen cyanide, ethylene, formaldehyde, methyl chloride, methyl bromide and aromatics [3]. For these reasons methane plays a key role in the modern chemical industry as a widespread and renewable energy and chemical source. However, methane is also the second strongest anthropogenic greenhouse gas after CO, it possesses a global warming potential (GWP) of about 28-36 over 100 years (the GWP of CO is defined as equal to unity) [4], and its atmospheric burden has increased more than twice since 1850 [1]. Methane emission sources include natural ones (321 Tg CH4 yr), such as water, geological, insect and animal sources, as well as anthropogenic sources (696 Tg CH4 yr) which are mainly related to agriculture, petroleum, ruminants, biomass and biofuel burning [5,6]. Moreover, methane emissions caused by leakages or incomplete flaring during fossil fuel production and usage are especially troublesome, while approximately 144 billion cubic meters of methane gas are annually burned causing severe consequences to the global carbon footprint [7]. As a consequence, innovative methodologies and materials are urgently needed to more efficiently convert methane into useful energy or chemical sources and to mitigate global warming effects by decreasing methane emissions. Unfortunately, most of the current methane conversion processes suffer from the intrinsic inertness of the CH molecule [8,9] and require relatively elevated temperatures and pressures [6,10].
In particular, the oxidation of methane to methanol (MTM) is a central reaction for the modern chemical industry, with methanol and its derivatives being obtained almost entirely from natural gas (∼65%) and coal (∼35%) [11]. Global demand for methanol has nearly doubled in the last decade, reaching 106 million tonnes in 2021, and is expected to continue to grow [4,12]. Current demand is based on the use of methanol as a chemical feedstock (), predominantly to manufacture olefins (32%), formaldehyde (23%) and acetic acid (8%) [4]. In addition, transport fuels also employ methanol such as through methyl tert-butyl ether and biodiesel [12]. Common MTM conversion routes at an industrial scale involve the initial transformation of methane from natural gas into syngas (a mixture of CO and H). Subsequently, syngas is converted to methanol by a heterogeneous catalytic reaction, typically conducted over a bimetallic (Cu/Zn or Cu/Cr) catalyst at 220-250 C and high pressure (70-100 bar) [13,14]. The high temperature (> 800 C) and energy demands required by methane steam reforming are associated to elevated costs, and significant efforts have been devoted to develop alternative catalytic platforms to efficiently and sustainably accomplish the MTM oxidation reaction. Further, it is not yet possible to directly convert methane to methanol through an industrial route [9,15]. The lack of catalysts for such a process is due to the relatively large energy barriers associated with the activation of the nonpolar and highly symmetric CH molecule and to the higher relative reactivity of the methanol product. In fact, methane possesses a negligible electron affinity, a high ionization energy, a very large HOMO-LUMO gap and an elevated pKa value, it does not display a dipole moment or a significant polarizability [9], and large energies are required for both homo- and heterolytic methane C-H bond cleavages. In addition, the C-H bonds of methanol have a 0.4 eV lower bond dissociation energy (BDE) than those in methane and it is challenging to prevent product overoxidation [15]. To date, a wide variety of solid-state systems have been employed to investigate conversion reactions of methane to methanol and other oxygenated products. These include molybdenum oxide-based catalysts such as silica-supported MoO[16] and MoO materials [17,18], MoO/La-Co-O [19], VO/SiO[20], and microporous materials such as the Fe/ZSM-5 [21,22,23,24] and Cu-MOR [25,26,27,28,29] zeolites, as well as Au-based photocalysts supported on TiO [30,31]. However, one of the main issues of such heterogeneous catalysts is their reduced level of tunability, which severely limits how one may vary the chemical nature of the platform components while exploring the "chemical space" of effective MTM catalysts. Another issue affecting systems such as Fe- and Cu-exchanged zeolites is the heterogeneous metal speciation within the zeolitic framework, which is influenced by factors such as the original synthetic method, the employed activation procedure and the total metal content [32,33]. For instance, exhisting studies on the Fe-ZSM–5 zeolite report a wide range of first–shell Fe–Fe, Fe–Fe and Fe–Si/Al bond distances, due to the high heterogeneity in metal site distribution that severely undermines efforts to decypher the properties of the true active sites [34].
While electro- [35] and photoelectrocatalytic [36,37] systems have also been proposed for methane oxidation, an emerging approach to tackle the MTM reaction, a "holy grail" in the field, makes use of metal-organic frameworks (MOFs). As detailed in the following Sections, MOFs are unique porous materials with very high potential for methane chemistry [6].
2. Metal-organic frameworks as catalytic platforms for the MTM reaction
Figure 1.
Top: common organic MOF linkers. From left to right: benzene-1,4-dicarboxylate (BDC), 2,5-2,5-dioxido-1,4-benzenedicarboxylate (DOBDC), benzene-1,3,5-tricarboxylate (BTC), 3,3’,5,5’-azobenzene-tetracarboxylate (ABTC), 1,3,6,8-tetrakis(p-benzoate)-pyrene (TBAPy). Color code: oxygen, red; carbon, grey; nitrogen, blue; hydrogen, white. Middle: common MOF secondary building units (SBUs). Bottom: common MOF structures. MOF-5 (Reprinted with permission from Kaye et al., J. Am. Chem. Soc. 2007, 129, 46, 14176-14177 [38]. Copyright 2007 American Chemical Society.), MOF-74 (Reprinted with permission from Bloch et al., J. Am. Chem. Soc. 2011, 133, 37, 14814-14822 [39]. Copyright 2011 American Chemical Society.), MIL-53 (Reproduced from Bara et al., Mater. Horiz. 2021, 8, 12 3377-3386 [40]. Copyright 2021 Royal Society of Chemistry.), HKUST-1 (Reproduced from Hendon and Walsh, Chem. Sci. 2015, 6, 7 3674-3683 [41]. Copyright 2015 Royal Society of Chemistry.), MIL-100(Fe) (Reproduced with permission from Quijia et al., J. Drug. Deliv. Sci. Technol. 2021, 61, 102217 [42]. Copyright 2021 Elsevier.), UiO-66 (Reprinted with permission from Cavka et al., J. Am. Chem. Soc. 2008, 130, 42, 13850-13851 [43]. Copyright 2008 American Chemical Society.), PCN-250 (Reproduced with permission from Yuan et al., Joule 2017, 1, 4, 806-815 [44]. Copyright 2017 Elsevier.), MOF-505 (Adapted with permission from Chen 2005, Figure 1a, p. 4745 [45]. Copyright 2005 Oxford University Press.)
Figure 1.
Top: common organic MOF linkers. From left to right: benzene-1,4-dicarboxylate (BDC), 2,5-2,5-dioxido-1,4-benzenedicarboxylate (DOBDC), benzene-1,3,5-tricarboxylate (BTC), 3,3’,5,5’-azobenzene-tetracarboxylate (ABTC), 1,3,6,8-tetrakis(p-benzoate)-pyrene (TBAPy). Color code: oxygen, red; carbon, grey; nitrogen, blue; hydrogen, white. Middle: common MOF secondary building units (SBUs). Bottom: common MOF structures. MOF-5 (Reprinted with permission from Kaye et al., J. Am. Chem. Soc. 2007, 129, 46, 14176-14177 [38]. Copyright 2007 American Chemical Society.), MOF-74 (Reprinted with permission from Bloch et al., J. Am. Chem. Soc. 2011, 133, 37, 14814-14822 [39]. Copyright 2011 American Chemical Society.), MIL-53 (Reproduced from Bara et al., Mater. Horiz. 2021, 8, 12 3377-3386 [40]. Copyright 2021 Royal Society of Chemistry.), HKUST-1 (Reproduced from Hendon and Walsh, Chem. Sci. 2015, 6, 7 3674-3683 [41]. Copyright 2015 Royal Society of Chemistry.), MIL-100(Fe) (Reproduced with permission from Quijia et al., J. Drug. Deliv. Sci. Technol. 2021, 61, 102217 [42]. Copyright 2021 Elsevier.), UiO-66 (Reprinted with permission from Cavka et al., J. Am. Chem. Soc. 2008, 130, 42, 13850-13851 [43]. Copyright 2008 American Chemical Society.), PCN-250 (Reproduced with permission from Yuan et al., Joule 2017, 1, 4, 806-815 [44]. Copyright 2017 Elsevier.), MOF-505 (Adapted with permission from Chen 2005, Figure 1a, p. 4745 [45]. Copyright 2005 Oxford University Press.)
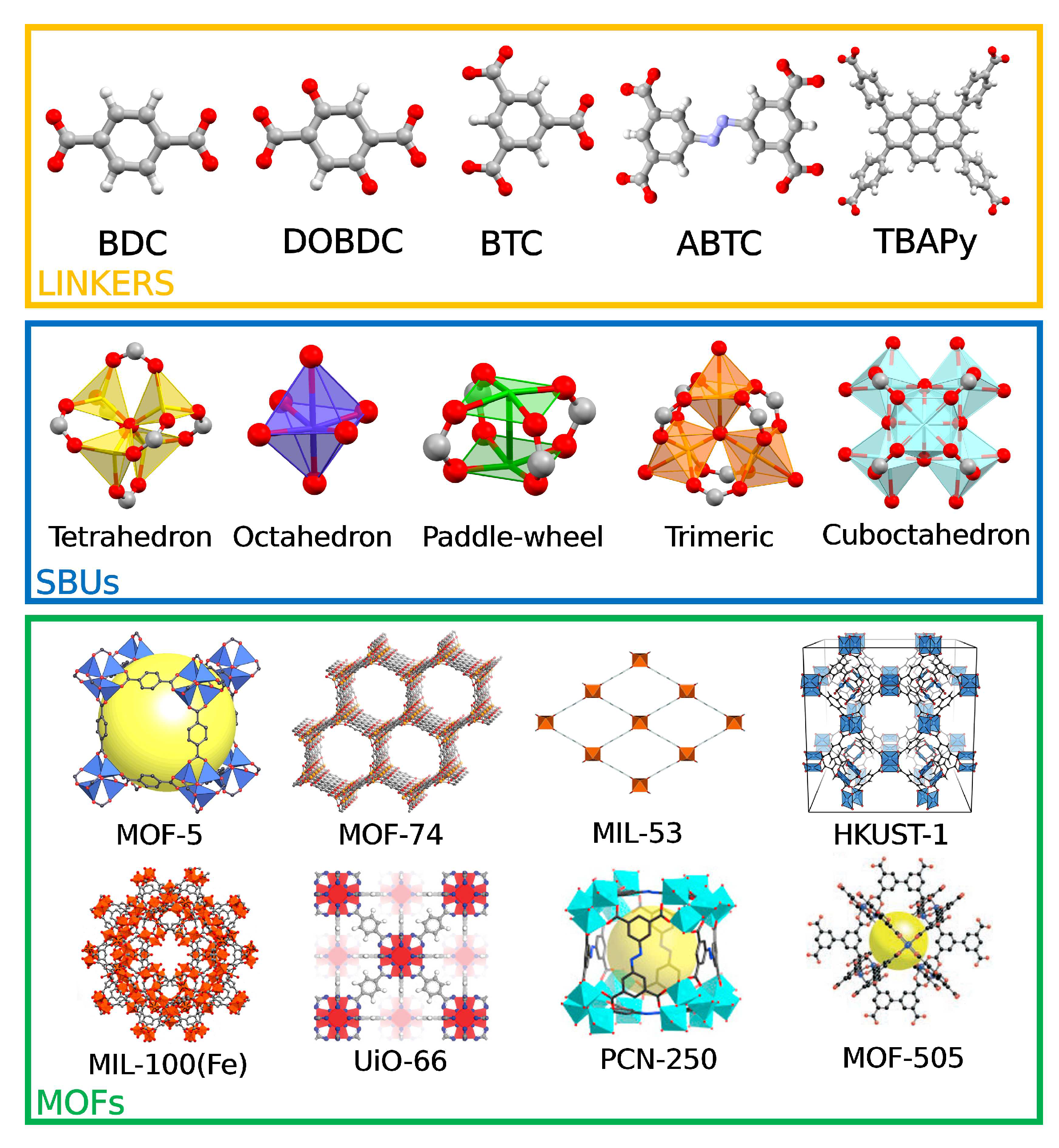
MOFs are a class of crystalline and permanently porous materials which have been attracting significant attention in the field of heterogeneous catalysis [46]. MOFs are composed of inorganic metal clusters (referred to as secondary building units or SBUs) bound to polydentate organic ligands (referred to as linkers) resulting in highly ordered structures of well-defined topologies [47]. Figure 1 presents a number of the most common linkers, SBUs and MOF structures, including some explored to accomplish the MTM reaction and discussed in this work. Owing to their hybrid nature, MOFs have sparked great interest for applications in catalysis. MOFs in fact possess an exceptional degree of structural and chemical tunability, significantly higher than that of other porous materials such as zeolites. This property allows one to design MOFs that are specially tailored for a given application by finely tuning the MOF composition [6,46,47,48,49,50,51,52,53,54]. For instance, it was shown that the pore size of the UiO-66 MOF could be regulated by increasing the linker length, yielding the UiO-67 and UiO-68 structures [43]. Further, a density functional theory (DFT)-based study demonstrated that the photocatalytic properties of MIL-125, a MOF containing Ti nodes and 1,4-benzene-dicarboxylate (BDC) moieties, were influenced by functional groups introduced on the terephtalate ligand, evidencing how the Ti band gap was reduced upon insertion of an amino group on the linker [55]. In addition, multivariate mixed-metal MOFs can be prepared to either exploit the interaction of adjacent metal sites improving catalytic activity [56], or to dilute the active metal site with the aim of increasing product selectivity [57]. MOF reactivity may also be tuned through a large number of post-synthetic modifications, which range from ligand or metal exchange to ligand functionalization strategies [58,59,60,61]. As an example, a bifunctionalized MIL-101(Cr)-SOH-NH catalyst was prepared by a series of post-synthetic modification steps, and the MOF showed excellent yields for the one-pot deacetalization-nitroaldol reaction of benzaldehyde dimethyl acetal into 2-nitrovinyl benzene with no loss of reactivity after three consecutive cycles [62]. MOFs can also be employed as catalytic platforms to support active single-atom metal sites or clusters [63,64,65,66,67,68]. For instance, sintering-resistant single Ni sites have been embedded into the Zr-based NU-1000 framework, acting as efficient ethylene hydrogenation catalysts [69]. Besides exhibiting a good thermal and chemical stability, MOF metal sites possess well-defined positions, local coordination geometries, oxidation and spin states. This allows one not only to exert a greater synthetic control over the MOF metal centers, but also to more easily rationalize the catalytic activity of the given MOF material.
The first application of MOF catalysts for alkane C-H bond activation was reported almost a decade ago by the group of Prof. Jeffrey Long [57]. In this pioneering work, it was shown that MOF-74(Fe) catalyzes the NO-based direct conversion of ethane to ethanol. The reaction mechanism was later demonstrated to involve a Fe(IV)=O intermediate generated via the oxidation of Fe(II) sites by NO, producing ethanol through a C-H bond cleavage-radical rebound process [70]. Such novel findings motivated further studies to investigate the MOF-based oxidation of light alkanes, with the aim of achieving the grand challenge of converting methane to methanol over MOFs [71,72]. Indeed, the MTM reaction has been successfully accomplished both over iron and copper centers present in a number of different MOFs, as a result of attempts to mimick the catalytic activity of methane monoxigenase enzymes which effectively perform the MTM reaction over iron and copper active sites. As far as Fe-based MOFs are concerned, MIL-100(Fe) and PCN-250 were proven to hydroxylate methane to methanol using NO as the oxidant [34,73], while the mixed-metal MIL-53(Al, Fe) material was shown to carry out the HO-based MTM reaction [74]. In addition, the UiO-67 MOF has been employed as a catalytic platform to build a complex photocatalytic system based on a mono-iron hydroxyl site, able to perform the MTM conversion with 100% selectivity by activating O in a liquid-phase process [75]. Active copper sites obtained through the insertion of copper dimers or clusters in highly stable Zr-MOFs also exhibit a promising degree of methane hydroxylation activity. For instance, MOF-808 has been employed as a platform containing bionspired active copper dimeric sites that react with CH and NO to yield methanol [76]. Cu-oxo dimers and clusters have also been embedded in the Nu-1000 framework through water-mediated cation exchange and atomic layer deposition techniques [77,78], allowing the activation of O for the MTM reaction in mild conditions.
In the following Sections, we discuss the studies that have investigated the use of iron and copper centers anchored in MOFs for the MTM conversion, as well as the main spectroscopic techniques employed to unravel the properties of the key reaction active sites and intermediates.
3. MOFs exhibiting iron active sites for the MTM conversion
Iron is a low cost metal with reduced toxicity that can shuttle between different oxidation states. Further, iron may form both mononuclear and polynuclear biomimetic clusters that when integrated in MOFs have been demonstrated to show high promise for the oxidation of methane. The application of Fe-based MOFs as catalysts for methane oxidation has been inspired by the presence of iron in catalytic enzyme pockets, such as, notably, in the soluble methane monooxygenase (sMMO) active site. Methanotrophic bacteria employ methane as their sole energy source and utilize sMMO to oxidize methane to methanol at the initial phase of methane metabolism [4]. In particular, the sMMO enzyme is constituted by (i) a hydroxylase (MMOH) which is responsible for MTM conversion, (ii) a reductase (MMOR) that guides the activated O towards the hydroxylase core, and (iii) a regulatory protein (MMOB) controling CH admission to the MMOH active pocket. Notably, the sMMO active site possesses a hydrophobic cavity running through the protein center which favors the release of methanol, being hydrophilic, and prevents its overoxidation. The active site for the oxidation of methane, solved recently by Banerjee and coworkers [79], is known as compound Q and is built from a diiron cluster, as shown in Figure 2. At the beginning of the sMMO MTM catalytic cycle, two electrons are transferred in the presence of a nicotinamide adenine dinucleotide cofactor (NADH) to the H intermediate, leading to the reduction of the Fe(III)-Fe(III) antiferromagnetically coupled, high spin, dimeric site to a Fe(II)-Fe(II) species (H intermediate) [80], as shown in Figure 3.
Subsequently, the MMOH-MMOB complex guides O to the diiron active site, leading to the formation of intermediate P* [82,83]. Molecular oxgen is then activated and the O–O bond is cleaved to yield compound Q, which is responsible for the hydroxylation of methane. Specifically, it has been proposed that the H-atom abstraction from CH proceeds via the formation of a hydroxyl radical, bound to a partially reduced Fe(III)-Fe(IV) cluster, and of a methyl radical, with the two radicals recombining in the methanol formation step [84]. Notably, sMMO possesses unrivaled selectivity by enclosing the hydrophilic CH molecule in a nonpolar environment near the active site that promotes the radical rebound mechanism and facilitates product removal. Further, the magnetic properties of the sMMO Fe(III)-Fe(III) dimer are believed to be crucial for the enzymatic reactivity [85]. It is important to note that while performing the MTM conversion all sMMO compartments display high surface area and porosity, and at the same time leverage a Fe-based active site with interchanging oxidation states in a concerted and hierarchical fashion. These are all properties that can be mimicked using MOFs, and a number of studies have been carried out to explore the MTM conversion over this highly tunable class of artificial materials.
In a work published in 2018, Gascon and coworkers [86] utilized isolated Fe sites incorporated into the MIL-53(Al,Fe) MOF framework (Figure 4a) to carry out the MTM conversion in the presence of HO. The MIL-53(Al) structure is built by chains of AlO octahedra connected by bridging BDC linkers and trans-located HO ions [87]. This MOF was chosen as the support matrix due to the inertness of the AlO units towards redox reactions and since its internal pore cavities are built from a hydrophobic organic linker that could favor rapid methanol desorption. Two distinct synthetic strategies were pursued to incorporate Fe sites into the Al-based MOF framework, namely, a post-synthetic cation exchange by placing into contact MIL-53(Al) and different FeCl solutions (HTS samples), as well as an electrochemical synthesis of MIL-53(Al) from an Al electrode and a terephtalic acid solution containing FeCl aliquots (ECS samples) [86]. It was found that between 0.15 and 2 wt % (wt %) Fe could be successfully incorporated into MIL-53(Al) using cation exchange at a relatively low temperature (80 C) while electrochemical synthesis allowed between 0.3 and 5.5 wt % Fe incorporation. In these samples with low Fe loading (LL samples) small MOF nanoparticles form by agglomeration and deplete the well-known breathing effect of the MIL-53 material [87]. Catalytic methane oxidation tests were performed in water using HO as the oxidant over both the HTS and ECS samples at temperatures below 60 C, leading in both cases to selective methanol formation with no C–C coupling products [86]. Conversely, performing the post-synthetic Fe incorporation at higher temperature (120 C) allowed the introduction of larger Fe amounts (up to 16.6 wt %) into high Fe loading samples (HL samples), but also yielded extraframework FeO and led to the formation of a higher amount of side products during the methane oxidation catalytic tests. Altogether, the ECS samples were the most catalytically active, with selectivities towards oxygenates of about 80% (see Figure 4b), while suppressing the formation of undesired extraframework iron species, FeO. H-NMR and gas chromatography analyses evidenced the formation of methanol, methyl peroxide, formic acid and CO as sole reaction products [86]. As detailed in Section 5, a series of different spectroscopic techniques were employed to better understand the nature and stability of the iron catalytic sites.
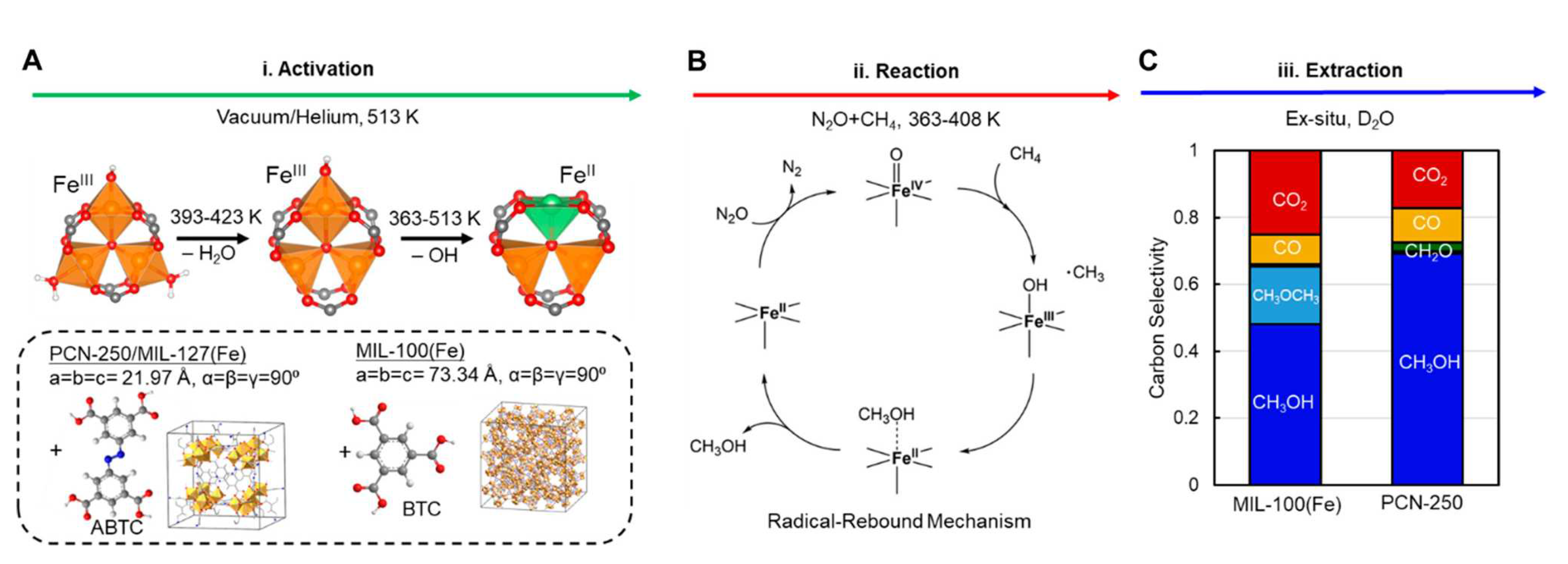
In 2020, Bollini and coworkers [34] reported the successful low-temperature MTM oxidation over MIL-100(Fe), a MOF comprised of tri-iron SBUs first discovered by the group of Gérard Férey [88,89]. In particular, MIL-100(Fe) is made up of trimeric iron nodes interconnected by trimesate linkers to produce a framework of overall MTN topology, as shown in Figure 5a. In the MOF SBU there are three Fe sites bridged by a -oxo center and coordinated in an octahedral environment, with water molecules located in two thirds of the apical coordination positions, while a monovalent anion (such as HO or F) occupies the third available site. Notably, thermally treating the hydroxyl-containing version of MIL-100(Fe) in vacuum or under an inert gas flow generates either Fe(III) or Fe(II) sites via the elimination of water molecules or of HO groups, respectively [34,89]. By co-feeding CH and the NO oxidant at 200 C over a Fe(II)-containing activated MOF sample, and by then exposing the post-reaction sample to a He stream containing water, the production of 0.34 mol (mol Fe) of methanol was observed. Interestingly, this value was quite similar to the cumulative moles of reacted methane (0.32 mol (mol Fe)) [34]. The produced quantity of methanol was also close to a value of 1 mole of CHOH per mole of Fe(II), whose abundance in the MOF was found to approach the theoretical maximum density. Further, by employing NO as a selective probe of the Fe(II) sites, it was demonstrated that methanol formation occurs exclusively over Fe(II) and not Fe(III) centers. Finally, the formation of methoxy intermediates within MIL-100(Fe) which react with water to yield gas phase methanol was strongly supported by exposing the post-reaction MOF to increasing fractions of DO and by measuring increasing fractions of produced CHOD in a 1:1 correspondence [34].
In 2021, Simons and coworkers [73] disclosed that PCN-250, a MOF also built from Fe--oxo nodes incorporating a maximum of one high-spin (S=2) Fe(II) site per node (see Figure 5a), catalyzes the NO-based MTM reaction at temperatures inferior to 125 C. Altogether, the Fe(II) sites arising from a thermal activation of the MOF initially form Fe(IV)=O centers by reacting with NO, while the methane homolytic activation was proposed to occur through a radical rebound mechanism often invoked when monuclear Fe(II) sites are exposed to mixtures of NO + CH (Figure 5b). However, the findings that (i) the NO + CH reaction on PCN-250 yields various overoxidation products such as CHO, CO, CO, as well as CHOCH (see Figure 5c), and that (ii) the methanol product is observed solely when washing the material ex situ with water led the authors to assume underlying pathways of methanol product protection, i.e. invoking reaction steps beyond the radical rebound mechanism. On the basis of in situ IR measurements and DFT calculations, the authors proposed that methanol is stabilized within the MOF during the MTM reaction as methoxy groups located on the Fe-based SBUs. Further, a composite constituted by the MOF and the MFI zeolite was exploited to improve methanol selectivity, yielding an amount of methanol equal to ca. 210 mol g at 1.1 bar and at temperatures between 105 and 135 C using a recirculating batch reactor [73].
Figure 5.
(a) Molecular structure of the Fe--oxo secondary building unit and of its ligand modifications upon thermal activation. The unit cells of PCN-250 and MIL-100(Fe) are displayed in the inset, along with the corresponding ABTC and BTC linkers. (b) Schematic representation of the commonly proposed radical-rebound mechanism followed by methane activation on mononuclear Fe(II) centers exposed to mixtures of NO + CH. (c) Cumulative carbon selectivity of products observed upon exposure of MIL-100(Fe)/PCN-250 to 80 kPa of NO, 10 kPa of CH, and 10 kPa of Ar at 120 C for 4 h. The product selectivity is reported in ascending order: CHOH (dark blue), CHOCH (light blue), CHO (green), CO (yellow), and CO (red). [Reprinted with permission from Ref.[73]. Copyright 2021, American Chemical Society].
Figure 5.
(a) Molecular structure of the Fe--oxo secondary building unit and of its ligand modifications upon thermal activation. The unit cells of PCN-250 and MIL-100(Fe) are displayed in the inset, along with the corresponding ABTC and BTC linkers. (b) Schematic representation of the commonly proposed radical-rebound mechanism followed by methane activation on mononuclear Fe(II) centers exposed to mixtures of NO + CH. (c) Cumulative carbon selectivity of products observed upon exposure of MIL-100(Fe)/PCN-250 to 80 kPa of NO, 10 kPa of CH, and 10 kPa of Ar at 120 C for 4 h. The product selectivity is reported in ascending order: CHOH (dark blue), CHOCH (light blue), CHO (green), CO (yellow), and CO (red). [Reprinted with permission from Ref.[73]. Copyright 2021, American Chemical Society].
More recently, in 2022 An and coworkers [75] reported that mono-iron hydroxyl sites immobilized in a Ru- and Fe-based MOF, termed PMOF-RuFe(OH), photocatalitically convert methane to methanol in the presence of HO and O with 100% selectivity. Here, we observe that the use of O is particularly relevant, as molecular oxygen is more environmentally benign and less expensive than other common oxidants such as HO or NO. The employed photocatalytic system integrated four different components: (i) a MOF based on the UiO-67 architecture to serve as a support platform; (ii) the photosensitizer [Ru(II)(bpy)(bpydc)] (bpy=2,2’-bipyridine; Hbpydc=2,2’-bipyridine-5,5’-dicarboxylic acid) to absorb light; (iii) a polyvanadotungstate [PWVO] to activate O; (iv) a mono-iron hydroxyl site to activate methane [75]. The Ru photosensitizer and polyvanatotungstate moiety were introduced into the UiO-67 pores by a one-pot synthesis, while the Fe active sites were incorporated through a post-synthetic treatment using FeCl·6HO to yield PMOF-RuFe(Cl), which in turn afforded the active catalyst PMOF-RuFe(OH) upon pre-treatment in water with light irradiation for 2h (the wavelength of light used was 400-780 nm). The presence of Fe–OH species was demonstrated by combining a series of different experimental techniques, including inelastic neutron scattering (INS), X-ray photoelectron spectroscopy (XPS), electron paramagnetic resonance (EPR) and X-ray absorption spectroscopy (XAS), supported by DFT calculations (refer to Section 5 for a more detailed discussion). In order to perform the MTM reaction, a continuous-flow photo-oxidation setup was implemented, where a packed layer of PMOF-RuFe(OH) catalyst was exposed to a flow of CH/O-saturated water under irradiation. The dynamic gas/solid/liquid interface maximizes contact between CH, O, HO and the MOF and, notably, leads to a methanol time yield of 8.81±0.34 mmol gh under ambient conditions, even outperforming the Methylococcus capsulatus (Bath) MMO (5.05 · mmol· gh) [90].
Finally, a very recent study by Rungtaweevoranit and coworkers [91] reported a MOF with isolated Fe sites supported on the Zr-based UiO-66 structure, which may perform the MTM reaction under continuous gas-phase flow conditions in the presence of O and water vapor. In the synthesized UiO-66 material there is approximately 1 acetate molecule and one HO/HO pair per Zr oxide cluster replacing the linker in missing-linker defects, which were leveraged to anchor Fe atoms onto the MOF. The activation of the Fe/UiO-66 catalyst was conducted by heating up to 250 C under Ar and by exposing the MOF to (i) 10% O/Ar at 250 C for 1 h, (ii) 5% water vapor in Ar for 1 h at 250 C, 1 bar and (iii) Ar while decreasing the temperature to a final value of 180 C. The Fe/UiO-66 MOF was then exposed to the reaction gas mixture (10% CH, 5% O + 0.2% HO, balance Ar) at 5 bar. During the first 250 min of the reaction the rate of methanol formation increased up to 16.7 × 10mol gFe s (Figure 6a). Subsequently, the MOF catalyst entered a deactivation phase over 850 min, while increasing the reaction temperature to 190 C resulted in a short increase of catalytic activity up to 10.3 mol g s (see Figure 6a). Conversely, CO formation reached a rate of 9.5 mol g s over 650 min and was not affected by the temperature increase to 190 C. Under the steady-state conditions at 180 C (Figure 6b), the Fe/UiO-66 catalytic platform exhibited a selectivity towards the formation of methanol of 62%.
The methane hydroxylation performance of the Fe-contanining MOFs illustrated in this Section is summarized in Table 1.
4. MOF-supported Cu clusters for the MTM conversion
Particulate methane monoxygenase (pMMO) is a copper-based membrane enzyme expressed by nearly all methanotrophic organisms and constitutes the insoluble counterpart of the sMMO enzyme [92]. Organisms than may express both sMMO and pMMO exhibit a preferential pMMO expression in high copper availability conditions [93,94], despite the better performance of sMMO as for methanol turnover frequency and catalytic efficiency [95].
However, unlike sMMO, many properties of pMMO are not yet fully understood due to the low resolution of the available enzyme crystal structures (< 2.68 Å [97]). For instance, the exact structure of the copper active site(s) is yet to be disclosed [98], and evidence supporting the presence of both monocopper and dicopper sites has been presented often proposing their first-shell coordination with histidine residues [97,99]. As an example, Figure 7 shows putative monocopper and dicopper pMMO active site structures optimized though hybrid quantum mechanics/molecular mechanics (QM/MM) methods [96]. Such degree of uncertainty regarding the pMMO active site, together with the absence of dicopper sites in similar enzymes with well-determined structures, has hindered the full comprehension of the pMMO-mediated MTM reaction mechanism.
Nonetheless, numerous reaction schemes have been proposed to explain the reactivity of pMMO during the MTM process. A DFT-based study by Yoshizawa and coworkers modeled the reaction pathway over a dicopper site (see Figure 8), assuming a radical rebound mechanism [96]. Following O activation, a superoxo-bridged doublet dicopper active species (Oxo) is formed. This species interacts with methane leading to intermediate R, which subsequently undergoes a H-bond cleavage process through transition state TS (18.9 kcal/mol) to form the methyl radical (Int). Then, a radical rebound step (TS2, 25.9 kcal/mol) finally yields CHOH and a monoxygenated dicopper site P while releasing 45.1 kcal/mol.
Many attempts at replicating the reactivity of pMMO have been carried out, starting from model dicopper complexes that can oxidize weak C-H bonds [100]. Similarly, Cu-exchanged zeolites that exhibit mononuclear, dinuclear and even trinuclear copper active sites have been studied for the MTM conversion showing good yields [101,102,103].
At variance with the inorganic skeleton of zeolites, the hybrid MOF structures are believed to more closely resemble the environments offered by polypeptidic chains in enzymes [76], enabling one to pursue biomimetic reactivity while employing MOF catalysts. Following this principle, in 2018 Baek and coworkers installed three different imidazole-based ligands (5-benzimidazolecarboxylate, L-histidine and 4-imidazole acrylate) on the Zr-based MOF-808 framework [76], subsequently loading Cu(I) centers on the ligands to form dicopper sites almost identical to those proposed in pMMO (see Figure 9). Although the structure of these slightly disordered dimeric sites is elusive to crystallographic methods, their presence was confirmed via energy-dispersive X-ray spectroscopy (EDS), N K-edge and Cu K-edge XAS, UV-Vis diffuse reflectance spectroscopy (DRS), as well as by resonance Raman spectroscopy measurements. Catalytic tests showed that these copper sites effectively oxidize methanol at 150 °C using NO as the oxidant, with a methanol productivity of up to 71.8 ± 23.4 mol g when 5-benzimidazolecarboxylate acts as the support (MOF-808-Bzz-Cu). The other two imidazole ligands showed reduced activity, with L-histidine (MOF-808-His-Cu) displaying the lowest methanol productivity of 31.7 ± 13.0 mol g. Notably, only methanol and water were observed as reaction products during methanol steam desorption experiments, although a certain amount of CO was formed as a reaction byproduct. The recyclability of MOF-808-Bzz-Cu was also explored, and subsequent reaction cycles over the catalyst showed a large degree of deactivation with methanol productivity being reduced from 71.8 to 7.5 mol g between the first and the second reaction cycle. This effect was attributed to strongly adsorbed water molecules that hamper the catalytic process [76]. Powder X-ray diffraction (PXRD) analyses evidenced the structural stability of all three MOF catalyst under reactive conditions.
Different strategies have also been explored to realize Cu-loaded Zr-based MOFs. In 2017, Ikuno and coworkers inserted oligomeric copper-oxo clusters within the NU-1000 MOF (see Figure 10) using the atomic layer deposition (ALD) technique [78]. In this way a 10 % wt copper-loaded Cu-NU-1000 MOF was obtained, possessing an average of 4 copper atoms per node and a crystal structure identical to that of its pristine counterpart, as confirmed by PXRD and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analyses. The structure and oxidation state of the copper-oxo clusters, whose proposed DFT-derived geometry is depicted in Figure 11a, were investigated using XAS, XPS and pair distribution function (PDF) measurements, and closely resemble those of copper sites present in Cu-NU-1000 and Cu(OH) [104]. Most of the copper species (∼85%) were determined to be in a octahedrally coordinated Cu(II) state, while ∼15% of Cu(I) sites in two- or three-coordinated geometries was detected. A number of MTM reaction conditions were explored for the O-based MTM conversion at 150 °C over this MOF, following pretreatment in O flow at 200 °C for 3 h to remove physisorbed water. When exposing the pretreated Cu-NU-1000 to a CH flow at 150 °C for 3 h and then to a 10% steam He flux at 135 °C, methanol was produced with a yield of 17.7 mol g. Dimethyl ether (DME), a valuable oxygenate of high industrial relevance, was also detected in small quantities (2.0 mol g), for a total carbon selectivity (methanol + DME) of ∼45%. Two subsequent reaction cycles were performed, showing reduced reactivity with yields of 15.8 and 13.2 mol g, respectively. When increasing the water content of the steam He flow to 50%, methanol yield and carbon selectivity were both drastically reduced (6.9 mol g and ∼14%). This was speculated to be due to the decarboxylation of the Cu-NU-1000 linkers, as 60% MOF pore volume was lost while a reduction of only 1-2% was observed while exposing the system to a 10% water He flux.
In 2019, Zheng and coworkers also exploited NU-1000 as a platform for copper ion loading through various degrees of cation exchange over the terminal -OH groups present in the zirconium oxide SBUs [77]. Three different copper loadings were explored, namely 2.9 wt % (Cu-2.9-NU-1000), 1.9 wt % (Cu-1.9-NU-1000) and 0.6 wt % (Cu-0.6-NU-1000), and also in this case the obtained MOFs were found to be isostructural to the pristine Zr-based one. Scanning transmission electron microscope-energy dispersive X-ray spectroscopy (STEM-EDS) measurements evidenced a uniform distribution of copper sites. Extended X-ray absorption fine structure (EXAFS) measurements were leveraged to probe their local structure, which was again found to be nearly identical to that of copper within Cu(OH), with almost all copper species displaying the +2 oxidation state in monomeric (prevalent in Cu-0.6-NU-1000) or dimeric units (prevalent in Cu-2.9-NU-1000), as shown in Figure 11b. Further, the methanol yield was demonstrated to correlate with the copper loading. In particular, the highest yield (4.4 mol g) was obtained while using Cu-2.9-NU-1000, and after activating the catalyst with 1 bar O at 200 °C and exposing it to 1 bar CH at 150 °C. Further, Cu-2.9-NU-1000 produced approximately 0.01 methanol molecules per single copper site, a degree of activity comparable to that of the ALD-synthetized Cu-NU-1000 MOF (vide supra) and higher than that obtained with copper-exchanged zeolites such as Cu-MOR or Cu-ZSM [106,107]. Moreover, although the previously reported ALD-prepared Cu-NU-1000 outperforms Cu-2.9-NU-1000 in terms of methanol yield, the latter can be more readily prepared and displays a very high methanol selectivity of 70%. Remarkably, recyclability tests over five consecutive cycles also indicated only slight catalyst deactivation after the first cycle with stable methanol selectivity, and DME was not observed as a byproduct. Methanol selectivity was confirmed through isotope labeling experiments that made use of C-methane as the reactant under the same conditions: C-labeled methanol and C-labeled CO were found to be the main products with only minor amounts of CO being released. Increasing the methane pressure to 40 bar during the reaction step resulted in increased methanol productivity by a factor of ∼3.5, together with the formation of DME in small quantities. Conversely, the increase in methanol overoxidation to CO was negligible and the total carbon selectivity at 150 °C and 40 bar CH was found to be as high as ∼90% (with 0.04 methanol molecules produced per copper site). Since the methanol yield in dicopper-dominated Cu-2.9-NU-1000 was more than six times higher than that of monocopper-dominated Cu-0.6-NU-1000, it was concluded that dimeric copper oxyl species were responsible for most of the MTM reactivity over Cu-exchanged NU-1000. The MTM reaction mechanism was investigated with DFT calculations, which indicated that the reaction initially proceeds through a homolitic C-H bond dissociation over Cu(II)(OH) clusters, producing a CH radical and a HO ligand (21.2 kcal/mol). In turn, the methyl radical then abstracts an HO radical from the HO ligand to form methanol (14.7 kcal/mol). DFT modeling also predicted the formation of Cu(II)(O)(OH) copper-oxyl species in relatively small quantities. These species were also predicted to hydroxylate methane following a mechanism based on H-atom abstraction over the oxyl moiety, while requiring a significantly smaller activation energy (18.9 kcal/mol) if compared to that required by the reaction occurring over Cu(II)(OH) sites (35.3 kcal/mol). This result suggested that the very low MTM reactivity of Cu-2.9-NU-1000 is due to the fact that in this MOF copper oxyl clusters, the most active species, are only present in very limited quantities.
In 2021, Ren and coworkers exploited a ligand-coordination strategy to insert square-planar CuCl clusters coordinated by the 2,2’-bipyridine-5,5’-dicarboxylate (bpydc) ligand within the UiO-bpy MOF [108]. Such Cu-based clusters have been shown to produce linearly coordinated CuO species upon activation in O flow at 200 °C [109], and XPS measurements indicated that their structure and oxidation state were very close to those observed in CuO. The MTM reaction was then carried out on the activated system and involved isothermal CH loading for 3h and methanol desorption by exposing the system to a steam/He flux. The reported methanol yield was equal to 24.33 mol g, with a selectivity of 88.1% during the first reaction cycle. Ethanol was formed as the main reaction byproduct while no amount of CO was detected in these conditions. During subsequent reaction cycles, the methanol yield was only slightly reduced while selectivity reached 100% as no byproducts were detected, indicating optimal catalyst recyclability. Thermodynamic calculations predicted that two-dimensional CuO species form in the MOF channels, and over these Cu sites the DFT absorption energy of methane was found to be as low as ∼-0.9 kcal/mol. A DFT-based MTM reaction mechanism was also reported, according to which the interaction between CH and two distinct oxygen sites leads to the formation of CH and a OH site with an energy barrier of ∼27 kcal/mol. This step is followed by the exothermic production of methanol with a relatively low activation energy of ∼6 kcal/mol.
Lastly, in 2022 Lee and coworkers demonstrated that the copper-doped ZIF-7 (zinc benzimidazolate) system can hydroxylate methanol using HO as the oxidant [110]. PXRD analysis evidenced a phase transition of the catalyst to a square-planar dense structure upon copper insertion, while XAS analyses disclosed the presence of four-coordinated Cu(II) species. The reaction of methane with Cu/ZIF-7 and HO (0.5 M) in aqueous solution produced methanol as well as methyl hydroperoxide, hydroxymethyl hydroperoxide and carbon dioxide as overoxidation products, while a blank test performed with pristine ZIF-7 only yielded formic acid due to the presence of strongly acidic zinc sites. Through isotopic labeling catalytic tests, undesired oxidation processes of the benzimidazole ligands were identified as the source of the carbon dioxide product. The influence of reaction conditions was investigated, showing that CH can already be activated at 30 °C. Further, it was found that the total yields of methanol and of its overoxidation products increased as a function of temperature until a drastic reduction in selectivity was observed at 90 °C, while the maximum-yield temperature (70 °C) caused a deterioration of the framework. The catalyst performance was therefore determined to be optimal at 50 °C with an elevated methanol yield of 48 mol g. Increasing the concentration of hydrogen peroxide above 0.5 M increased the production of hydroxymethyl hydroperoxide and CO due to the augmented methanol overoxidation and ligand oxidation. To assess the reusability of the catalyst, the MTM process was repeated five times under optimized conditions demonstrating unaffected reactivity. Further, although the catalyst weight gradually decreased after each use, XAS and PXRD analyses showed no structural and electronic modifications of the Cu/ZIF-7 system. The reaction mechanisms leading to the formation of both methanol and methyl hydroperoxyde were studied through DFT calculations. Specifically, it was proposed that an initial dissociative reaction of HO to form HO and O-CuN as products occurs after the adsorption of HOO on the given copper site. A copper-oxyl species was then predicted to hydroxylate methane in a H-bond cleavage step, followed by radical rebound-driven methanol formation as observed in a number of iron- and copper-based systems [73,96]. The activation barrier for this step was determined to be ∼15.2 kcal/mol. Importantly, it was found that the reaction of O with the methyl radical produces CHOO, a stable intermediate that may abstract hydrogen from hydroxylated HO-CuN sites to form methyl hydroperoxide.
Table 2 summarizes the methane hydroxylation performance of the Cu-bearing MOFs illustrated in this Section.
5. Spectroscopic Characterization of the MOF-Based Methane Oxidation
To begin this Section, we observe that the development of next-generation catalysts to accomplish the efficient and selective MTM conversion is a highly complex and challenging process, where basic science, screening methodologies and engineering are all required to contribute in a synergic manner [111]. Nonetheless, while the rate and selectivity of the MTM reaction are the most sensitive descriptors of a given catalyst’s performance, these macroscopic criteria are not sufficient to deduce the underlying reaction mechanism and need to be complemented with additional microscopic information on the nature, structure, electronic properties and reactivity of the key reaction active sites and intermediates. It is for such information that one resorts to spectroscopic techniques, that when complemented with theoretical methods may shed light onto the active reaction mechanism and provide insights that are key to rationally design improved catalysts [111]. When considering the MTM reaction performed over iron and copper sites in MOFs, our attention primarily focuses on spectroscopic tools useful in heterogeneous catalysis, where surface properties are of great significance to drive the desired reaction pathways. In fact, the permanent porosity and very high surface areas exhibited by MOFs render these materials unique platforms for solid-gas catalytic reactions. The "ideal" spectroscopic technique to investigate the MTM reaction over MOFs would allow the identification of adsorbed molecules, while as well providing information on the structure, oxidation and spin states of the surface species, and on the intermolecular interactions established by adsorbed species (such as CH itself and the HO, NO or O oxidants) with the framework atoms. Further, the measurements should be possible over a wide range of pressures and temperatures. While such "ideal" spectroscopic probe does not exist as a single method alone, the judicious combination of diverse and complementary spectroscopic techniques operating in different energy ranges of the electromagnetic spectrum may often answer the majority of the experimental questions [111].
Some properties of individual spectroscopies relevant to heterogeneous catalysis and employed to investigate the MTM reaction over MOFs are summarized in Table 3. It is worth observing that for all spectroscopies there are restrictions on the nature and form of the investigated sample, as well as on the experimental conditions allowed by the given technique. For instance, ambient pressures are not accessible for XPS, because the mean free path of electrons is very limited even at pressures inferior to 10 atm, while EPR and Mössbauer spectroscopies require, respectively, unpaired electrons and Mössbauer-active nuclei, such as the Fe isotope.
As reported in Table 3, IR, Raman, NMR and EPR spectroscopies are more frequently employed to investigate the structural properties within molecules adsorbed on surfaces, offering also the possibility of making deductions about the adsorption configuration. Conversely, optical, Mössbauer and XPS techniques furnish direct insights into surface composition and on the local structural and electronic properties of the adsorption sites. Notably, XAS measurements in both the soft (0.1–3 keV) [112,113,114,115] and hard (3–100 keV) [116,117,118,119,120,121,122] energy ranges may provide at the same time information on the nature, structure and time evolution of the key intermediates with element-specificity and high sensitivity. It should be noted that the majority of works reported in the literature employed a combination of the above mentioned spectroscopic techniques, supported by DFT calculations, to characterize the catalytic active sites and the intermediate species formed while performing the MTM conversion over iron- and copper-bearing MOFs.
For instance, Gascon and coworkers [86] utilized Mössbauer, EPR and XAS spectroscopies to characterize the Fe species present in the MIL-53(Al,Fe) catalysts. From the EPR and Mössbauer experiments it was found that while the HTS samples mostly contain oligomeric species, the degree of dispersion observed in the ECS samples was higher. In particular, for the ECS samples the Mössbauer spectra recorded at -231.15 C (see Figure 12a) display a dominant doublet component with no magnetic hyperfine structure alongside a small component related to single Fe(III) atoms, supporting the view that the catalysts are comprised of a combination of isolated monomeric Fe(III) centers and antiferromagnetically coupled dimeric Fe(III)–Fe(III) sites [86]. Fe K-edge X-ray absorption near edge structure (XANES) spectra evidenced that octahedrally coordinated Fe(III) sites are present in all samples while an EXAFS analysis (Figure 12b) evidenced that each iron center is directly coordinated by two O atoms at 1.95–1.96 Å, which are associated to the shorter bridging Fe–-O–Fe bonds, and by two O atoms at 2.01–2.03 Å.
Bollini and coworkers [34] employed IR spectroscopy to demonstrate that solely Fe(II) centers are active towards the MTM conversion over MIL-100(Fe), using NO as a probe molecule selective to the MOF Fe(II) active centers. In fact, at sufficiently low NO pressures (below 5 kPa NO) Fe(II) open metal sites in MIL-100(Fe) have been shown to be saturated by NO with no indication of Fe(III)-nytrosyl species being formed [71]. Specifically, exposing the activated MIL-100 material to 0.5 kPa NO at 150 C resulted in a band at 1810 cm assigned to NO directly coordinated to the MOF Fe(II) sites. Further, the (Fe(II)–NO) and (Fe–OH) IR relative band areas were found to correlate linearly. This evidence supported the view that thermally treating the MOF to progressively higher temperatures generates larger Fe(II) site densities, with near complete dehydroxylation achieved at 250 C where one Fe(II) site per SBU is obtained.
Simons and coworkers [73] employed in situ XAS to track the structural and electronic variations of the Fe sites in PCN-250 both after thermal activation and after the NO + CH reaction. Figure 13a compares the XANES of the as-synthesized (black), thermally activated (red) and post-reaction (blue) materials. Overall, thermally activating the MOF in He to 240 C produced a shift of the XANES absorption edge energy to lower energies as well as a decrease in the white line intensity. In addition, a decrease in the fourier transform (FT) magnitude of the EXAFS spectrum at 2.03 Å was observed during activation (see Figure 13b), in agreement with the temperature-induced removal of HO and HO species directly coordinating the iron MOF centers. After performing the MTM reaction at 120 C, the XANES absorption edge shifted once again to higher energies while the magnitude of the FT at 2.04 Å increased, indicating that Fe(II) sites had been converted back to Fe(III) and likely coordinated by additional ligands. Fitting of the EXAFS data based on structural models in line with the XRD structure of PCN-250 [123] evidenced that activation from 125 to 240 C only negligibly affected the average Fe–O coordination number, from 5.8 ± 0.5 to 5.6 ± 0.5, while in the post-reaction catalyst the average Fe–O coordination number slightly increased again. This finding was consistent with the notion that apical HO ligands had been removed during activation and replaced by other axial ligands after the MTM conversion. Further, the authors resorted to in situ IR spectroscopy to investigate the mechanisms of CHOH protection during MTM reactive conditions. In the IR spectrum of PCN-250, activated by heating in vacuo to 240 C, there are solely bands belonging to the organic linker. Conversely, in the IR spectrum of the post-reaction MOF a band at 3682 cm and three new peaks appear (at 2904, 2876 and 2802 cm) which correlate with methanol formation quantified by means of H-NMR spectroscopy after washing the IR sample with DO. These bands are associated to specific C–H vibrations that belong to a methanol–derived species stabilized within the MOF pores and DFT calculations suggested they belong to a methoxy group coordinating a Fe(III) site within the PCN-250 SBU. The authors hypothesized that the formation of such stable methoxy species was due to CHOH molecules reacting with MOF surface hydroxyl groups (observed in PCN-250 using IR measurements) via water elimination. This was therefore proposed to constitute a pathway for the protection of gaseous CHOH additional to the radical–rebound mechanism. In fact, exposing PCN-250 bearing Fe(III)-OH groups to gas-phase CHOH resulted in a decrease of the band and in the formation of three new bands at 2904, 2876 and 2802 cm that were assigned to Fe(III)–OCH species [73].
An and coworkers [75] utilized a combination of diverse spectroscopic tools to establish the structure of the PMOF–RuFe(Cl) and PMOF–RuFe(OH) materials. PXRD patterns demonstrated that PMOF–RuFe(Cl) and PMOF–RuFe(OH) retain the UiO-67-type structure, while binding of Fe(III) ions to the MOF linker was supported by means of UV–Vis–near IR spectroscopy. XPS analyses of the active PMOF-RuFe(OH) species indicated that Fe(III)-OH species were present, Raman measurements excluded the formation of O–O bonds as in possible Fe–O adducts [75], while EPR spectra showed that both PMOF–RuFe(Cl) and PMOF–RuFe(OH) contained high-spin Fe(III) sites with highly rombic environments. The XANES spectrum of PMOF–RuFe(OH) displays a 1s→3d transition at 7114.8 eV which is less intense and at slightly higher energies if compared to the pre–edge peak in the XAS spectrum of the PMOF–RuFe(Cl) material (7114.4 eV). The EXAFS signal of PMOF–RuFe(OH) is consistent with a DFT–optimized [(bpy)Fe(OH)(HO)] complex, where the Fe–O and Fe–O bonds are equal to 1.78 ± 0.09 Å and 2.17 ± 0.09 Å, respectively [75]. Finally, a wavelet transform analysis of the EXAFS spectrum of PMOF–RuFe(OH) indicated that FeFe binuclear or cluster species are not present, as they do not contribute to second–shell scattering if compared to a FeO reference which exhibits Fe–Fe distances of ∼2.5 Å [75].
Rungtaweevoranit and coworkers [91] collected Fe Mössbauer spectra of the as–synthesized Fe/UiO-66 catalyst at -268.15 C and -193.15 C to characterize the MOF iron sites. These measurements supported the presence of both FeO nanoparticles and high-spin Fe(III) centers octahedrally coordinated by oxygen atoms in an approximately 1:1 ratio. Further, EPR analysis of the Fe/UiO-66 MOF identified two signals with g values of 2.01 and 4.24 attributed to –FeO and to an isolated Fe(III) species in a distorted octahedral coordination, respectively. The Fe K–edge XAS spectrum of Fe/UiO-66 possesses a pre-edge transition at 7114 eV, which is in the same region of that of FeO, suggesting that the MOF iron sites have a +3 oxidation state, while EXAFS data fits indicated that Fe is 6–coordinated and bound to HO and HO ligands. In addition, the XANES edge transition is located at 7127 eV, a value higher than typical Fe(III)-related absorption edges, supporting the view that the Fe sites are electron–deficient [91]. After pretreating the Fe/UiO-66 material and performing the MTM reaction, the iron first–shell coordination parameters remained basically unaffected while an increase in the FeFe backscattering coordination number (CN) was observed, likely due to the agglomeration of Fe sites into FeO nanoparticles. XPS analysis of the Fe 2p region indicated the presence of two components, the first one assigned to Fe(III) residing in FeO and the second one associated to the Fe(III) sites bound to electron–withdrawing ligands, a finding in agreement with the XAS data. To better track the evolution of the Fe sites during the MTM conversion, in situ DR UV–Vis spectroscopy was employed, evidencing that while the pre–reaction MOF samples contain a mixture of mono–, bi– and polynuclear Fe species, after the MTM reaction these isolated Fe species agglomerate into larger FeO nanoparticles. In situ IR measurements found that, while performing the MTM reaction, peaks arise attributable to surface methoxy and formate–related species (e.g. formate, formaldehyde and formic acid).
Ikuno and coworkers [78] performed Cu K-edge XAS measurements to determine the electronic and structural properties of the copper sites in the ALD-synthesized Cu-NU-1000 MOF. A linear combination fitting of the XANES spectrum of the pristine MOF (see Figure 14a) using CuO and Cu(OH) as Cu(I) and Cu(II) references, respectively, suggested that approximately 15% of Cu is present as Cu(I) and ∼85% as Cu(II). Further, there were only minor variations in the XANES and EXAFS spectra while activating the MOF in flowing O at 150 C (Figure 14b), while an increase in the Cu(I) characteristic 1s→4p transition was observed when exposing the activated MOF to CH, with an estimated ∼9% of Cu(I) being further reduced (Figure 14c). These evidences indicated that the majority of copper sites in the MOF retained the +2 oxidation state. The structural environment around the copper atoms was investigated by means of a Cu-EXAFS analysis. Specifically, the Cu-O bond was found to be shorter than in Cu(OH) suggesting the coexistence of multiple copper species in the MOF. Further, the EXAFS spectrum of Cu-Nu-1000 (Figure 14d, black curve) possesses a single scattering (SS) feature similarly to that of Cu(OH), supporting the view that small Cu clusters of a few Cu atoms (e.g., 2-4 atoms) or widely spaced sheets similar to Cu(OH) exist in the material. Subsequently, Cu(OH) was employed as a model to fit the EXAFS spectrum of Cu-NU-1000, evidencing that within the MOF Cu is coordinated to four first-shell O atoms with an average Cu-O distance of ∼1.94 Å and a square planar configuration, while two further O atoms are located at > 2.3 Å from the copper sites. In addition, the fit indicated Cu-Cu scattering at about 2.93 Å with an average CN equal to 1.3 ± 0.3, suggesting that copper may be present in Cu-NU-1000 as Cu-hydroxo dimers, trimers or tetramers [78]. To further support these hypotheses, the authors used DFT to optimize the geometry of a [Cu(OH)] fragment inserted and anchored by two hydroxyl groups of each MOF node, and found a reasonable agreement between the associated theoretical and experimental EXAFS spectra.
Baek and coworkers [76] utilized N K–edge XANES, Cu K–edge XAS, DR UV–Vis and resonance Raman spectroscopy measurements to obtain detailed structural and electronic information on the investigated three MOF–808–L–Cu catalysts, with the MOF–808–Bzz–Cu species being the most active towards MTM conversion (see Figure 15a). The anchoring of copper sites to N atoms part of the imidazole linkers was evidenced by N K–edge XANES spectra. In fact, as one may observe in Figure 15c, two absorption bands at 398.8 and 400.6 eV assignable to 1s→ transitions are present in the spectrum of MOF–808–Bzz, whereas upon metalation the peaks are shifted and their intensities modified, suggesting that the copper atoms are indeed coordinated by the N linker atoms. Since the ex situ N K–edge XANES spectra measured after each reaction step remain similar (Figure 15c), it could be concluded that Cu atoms are coordinated to N linker atoms during the entire MTM process.
The Cu K–edge XANES spectrum of the as–synthesized MOF–808–Bzz–Cu sample exhibited a weak 1s→3d adsorption peak at 8979 eV and a 1s→4p feature at 8984 eV attributable to Cu(II) and Cu(I) species, respectively, a mixture of which is present in the catalyst (Figure 15b). Pretreating the MOF in He at 150 C and subsequently exposing the material to 3% NO/He at the same temperature resulted in XANES modifications indicative of an initial Cu(II) to Cu(I) reduction, followed by the formation of active copper–oxygen species, respectively. After exposing the active MOF to CH, the Cu(I) peak at 8984 eV increased with a contextual decrease of the white line intensity, suggesting that Cu(II) is reduced to Cu(I) during the reaction. Further, flowing 3% steam/He at 150 C onto the catalyst in order to desorb methanol resulted in an intensity increase of the Cu(I) peak at 8984 eV, as shown in Figure 15c. Resonance Raman spectroscopy was used to identify the active copper species arising upon exposing the MOF to 3% NO/He. It was found that the Raman spectra of MOF–808–L–Cu samples oxygenated with either O or O display isotope–dependent Raman peaks at ∼560 cm and ∼640 cm, and at 545 cm and 630 cm, respectively, which are assigned to Cu–O bond vibrations in the core breathing mode of bis(-oxo) dicopper species (see Figure 15d). The presence of such dicopper species in the frameworks of all three MOF–808–L–Cu structures was also supported by DFT calculations and by EXAFS analyses.
Zheng and coworkers [77] employed in situ XAS, FTIR and EPR to probe the local variations of the active copper centers along the MTM reaction cycle in cation exchange-synthesized Cu-NU-1000. In particular, the authors investigated MOF catalysts with a Cu percentage in the 0.6-2.9 wt % range. XAS suggested that in the as-synthesized MOFs Cu exhibits a local structure similar to that of Cu in fully hydrated Cu(OH). After activating the MOF samples in O at 200 C, XANES and EXAFS spectra evidenced the removal of HO ligands coordinating the copper sites with a retention of the overall structure, nuclearity and oxidation state of copper in the pristine materials. In particular, within the MOF exhibiting highest Cu loading, mainly dinuclear copper species were found, identified predominantly as Cu oxyl-like and cupric hydroxide-like species, while the MOF with lowest Cu loading predominantly contained mononuclear Cu sites. DFT predictions, in agreement with the collected spectroscopic data, suggested that the dinuclear Cu species have one Cu atom anchored to two -OH groups of the Zr node and a second more distant Cu atom bridged to the first Cu center by means of two -OH groups [77].
Ren and coworkers [108] employed XPS to track the evolution of the Cu sites in CuCl@UiO-bpy, as well as in CuO@UiO-bpy before and after CH loading. From the analysis of the Cu 2p and Cu LMM XPS spectra, it was found that in the as-synthesized CuCl@UiO-bpy framework there is a relative content of Cu(II) and of Cu(I) equal to 84 and 16%, respectively. After activating the MOF in O, the Cu(II) increased to 94%, while after CH loading a partial reduction of Cu(II) was observed, with an increase in the relative Cu(I) content from 6% up to 11%. The nature of the CuO clusters located inside the MOF framework was assigned on the basis of DFT calculations, which suggested that the CuO clusters consist of 0.34% CuO, 80.85% CuO and 18.81% CuO in UiO-bpy after O activation, with the MTM conversion being most energetically favorable over CuO species.
Lastly, Lee and coworkers [110] leveraged XAS and XPS to characterize the Cu/ZIF-7 catalysts active towards the MTM reaction. The oxidation state of the Cu sites in the as-synthesized MOF was determined to be +2 from the energy positions of the main XAS absorption edge and of the pre-edge XAS transition, as well as from XPS data. Wavelet-transform Cu-EXAFS analyses indicated the absence of Cu-Cu bonds, suggesting that mononuclear Cu sites are dispersed within the zeolitic framework, while Cu-EXAFS fitting indicated a CuN first-shell coordination with identical Cu-N bond lengths equal to ∼1.97 Å.
6. Conclusions and perspectives
In summary, we have provided herein a brief overview of the state of the art concerning the application of MOFs with iron and copper active sites to accomplish the oxidation of methane to methanol, with a particular focus on the spectroscopic techniques employed by authors to unravel the electronic and structural properties of the MOF catalysts. Despite the field is still relatively young, a number of successful studies accomplishing the MTM reaction over iron- and copper-loaded MOFs in batch or flow reactors have been reported, and the combination of different spectroscopies, supported by theory, was proven essential to understand the mechanistic reaction details at a microscopic level. Nonetheless, it appears clear that the development of cost-efficient MOF catalysts for the MTM conversion still requires significant advances. For instance, the more widespread use of mild experimental conditions and of environmentally benigh oxidants (such as O or HO, which produces only HO as a byproduct) in the near future may allow one to access MTM conversion processes over MOFs that are more economically feasible. Further, the very high degree of tunability of MOF constituents may be further leveraged to design active site pockets of variable pore aperture sizes, in an effort to better control MTM selectivity [124], to incorporate carefully chosen linker functionalities that may act as reaction co-catalysts, such as N-heteroaromatic carboxylic acids [6,125], or to increase the hydrophobicity of the MOF inner cavities in order to favor the release of the hydrophylic methanol product. On the experimental side, synchrotron-based advanced techniques such as, among others, resonant and non-resonant X-ray emission spectroscopy (XES) may be employed in addition to XAS or to the laboratory spectroscopies explored to date, in order to further complement the MTM reaction picture. The theoretical treatment of MOF catalytic sites for the MTM reaction, so far mainly based on DFT methodologies, may also significantly benefit from the application of multireference (MR) approaches. Further, with the increasing use of data-driven theoretical models by the scientific community we expect that machine learning (ML) and artificial intelligence will provide a significant contribution to the efficient in silico screening and selection of promising, new MOF frameworks with desirable properties to accomplish the MTM conversion. In this regard, the availability of diverse spectroscopic data sets related to the given MOF catalyst key intermediates will be critical to train ML models.
In conclusion, MOFs with bioinspired iron and copper active sites provide unique opportunities to take on the challenge of efficiently converting methane to methanol, while the combination of advanced and complementary spectroscopic techniques allow access to the reaction mechanistic details which are required to rationally improve MOF catalysts for the MTM conversion.
Author Contributions
Conceptualization, Francesco Tavani, Alessandro Tofoni and Paola D’Angelo; writing—original draft preparation, Francesco Tavani and Alessandro Tofoni; writing—review and editing, Francesco Tavani and Alessandro Tofoni and Paola D’Angelo; supervision, Paola D’Angelo. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| GWP | Global Warming Potential |
| MTM | Methane To Methanol |
| BDE | Bond Dissociation Energy |
| MOF | Metal-Organic Frameworks |
| BDC | benzene-1,4-dicarboxylate |
| DOBDC | 2,5-2,5-dioxido-1,4-benzenedicarboxylate |
| BTC | benzene-1,3,5-tricarboxylate |
| ABTC | 3,3’,5,5’-azobenzene-tetracarboxylate |
| TBAPy | 1,3,6,8-tetrakis(p-benzoate)-pyrene |
| SBUs | Secondary Building Units |
| DFT | Density Functional Theory |
| sMMO | Soluble Methane Monooxygenase |
| NADH | Nicotinamide Adenine Dinucleotide |
| HTS | Hydrothermal Synthesis Samples |
| ECS | Electrochemical Synthesis Samples |
| LL | Low Loading |
| HL | High Loading |
| TOF | Turnover Frequency |
| TON | Turnover Number |
| bpy | 2,2’-bipyridine |
| Hbpydc | 2,2’-bipyridine-5,5’-dicarboxylic acid |
| XPS | X-ray Photoelectron Spectroscopy |
| EPR | Electron Paramagnetic Resonance |
| XAS | X-ray Absorption Spectroscopy |
| pMMO | Particulate Methane Monoxygenase |
| QM/MM | Quantum Mechanics/Molecular Mechanics |
| EDS | Energy-Dispersive Spectroscopy |
| XANES | X-ray Absorption Near Edge Structure |
| DRS | Diffuse Reflectance Spectroscopy |
| PXRD | Powder X-ray Diffraction |
| HAADF-STEM | High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy |
| Pair Distribution Function | |
| EXAFS | Extended X-ray Absorption Fine Structure |
| FT | Fourier Transform |
| CN | Coordination Number |
| SS | Single Scattering |
| MS | Multiple Scattering |
| XES | X-ray Emission Spectroscopy |
| MR | Multireference |
| ML | Machine Learning |
References
- Caballero, A.; Pérez, P.J. Methane as Raw Material in Synthetic Chemistry: the Final Frontier. Chem. Soc. Rev. 2013, 42, 8809–8820. [Google Scholar] [CrossRef] [PubMed]
- Bakkaloglu, S.; Cooper, J.; Hawkes, A. Methane Emissions Along Biomethane and Biogas Supply Chains are Underestimated. One Earth 2022, 5, 724–736. [Google Scholar] [CrossRef]
- MacDonald, G. Role of Methane Clathrates in Past and Future Climates. Clim. Change 1990, 16, 247–281. [Google Scholar] [CrossRef]
- Dummer, N.F.; Willock, D.J.; He, Q.; Howard, M.J.; Lewis, R.J.; Qi, G.; Taylor, S.H.; Xu, J.; Bethell, D.; Kiely, C.J.; Hutchings, G.J. Methane Oxidation to Methanol. Chem. Rev. 123, 6359–6411.
- Saunois, M.; Stavert, A.R.; Poulter, B.; Bousquet, P.; Canadell, J.G.; Jackson, R.B.; Raymond, P.A.; Dlugokencky, E.J.; Houweling, S.; Patra, P.K.; Ciais, P.; Arora, V.K.; Bastviken, D.; Bergamaschi, P.; Blake, D.R.; Brailsford, G.; Bruhwiler, L.; Carlson, K.M.; Carrol, M.; Castaldi, S.; Chandra, N.; Crevoisier, C.; Crill, P.M.; Covey, K.; Curry, C.L.; Etiope, G.; Frankenberg, C.; Gedney, N.; Hegglin, M.I.; Höglund-Isaksson, L.; Hugelius, G.; Ishizawa, M.; Ito, A.; Janssens-Maenhout, G.; Jensen, K.M.; Joos, F.; Kleinen, T.; Krummel, P.B.; Langenfelds, R.L.; Laruelle, G.G.; Liu, L.; Machida, T.; Maksyutov, S.; McDonald, K.C.; McNorton, J.; Miller, P.A.; Melton, J.R.; Morino, I.; Müller, J.; Murguia-Flores, F.; Naik, V.; Niwa, Y.; Noce, S.; O’Doherty, S.; Parker, R.J.; Peng, C.; Peng, S.; Peters, G.P.; Prigent, C.; Prinn, R.; Ramonet, M.; Regnier, P.; Riley, W.J.; Rosentreter, J.A.; Segers, A.; Simpson, I.J.; Shi, H.; Smith, S.J.; Steele, L.P.; Thornton, B.F.; Tian, H.; Tohjima, Y.; Tubiello, F.N.; Tsuruta, A.; Viovy, N.; Voulgarakis, A.; Weber, T.S.; van Weele, M.; van der Werf, G.R.; Weiss, R.F.; Worthy, D.; Wunch, D.; Yin, Y.; Yoshida, Y.; Zhang, W.; Zhang, Z.; Zhao, Y.; Zheng, B.; Zhu, Q.; Zhu, Q.; Zhuang, Q. The Global Methane Budget 2000–2017. Earth Syst. Sci. Data 2020, 12, 1561–1623. [Google Scholar] [CrossRef]
- Andrade, L.S.; Lima, H.H.; Silva, C.T.; Amorim, W.L.; Poco, J.G.; Lopez-Castillo, A.; Kirillova, M.V.; Carvalho, W.A.; Kirillov, A.M.; Mandelli, D. Metal–Organic Frameworks as Catalysts and Biocatalysts for Methane Oxidation: the Current State of the Art. Coord. Chem. Rev. 2023, 481, 215042. [Google Scholar] [CrossRef]
- Mar, K.A.; Unger, C.; Walderdorff, L.; Butler, T. Beyond CO2 Equivalence: the Impacts of Methane on Climate, Ecosystems, and Health. Environ. Sci. Policy 2022, 134, 127–136. [Google Scholar] [CrossRef]
- Yuliati, L.; Yoshida, H. Photocatalytic Conversion of Methane. Chem. Soc. Rev. 2008, 37, 1592–1602. [Google Scholar] [CrossRef]
- Schwarz, H. Chemistry with Methane: Concepts Rather than Recipes. Angew. Chem. Int. Ed. 2011, 50, 10096–10115. [Google Scholar] [CrossRef]
- Sher Shah, M.S.A.; Oh, C.; Park, H.; Hwang, Y.J.; Ma, M.; Park, J.H. Catalytic Oxidation of Methane to Oxygenated Products: Recent Advancements and Prospects for Electrocatalytic and Photocatalytic Conversion at Low Temperatures. Adv. Sci. 2020, 7, 2001946. [Google Scholar] [CrossRef]
- Innovation Outlook: Renewable Methanol; IRENA, 2021. https://www.irena.org/publications/2021/Jan/Innovation-Outlook-Renewable-Methanol. accessed: 2023-07-25.
- Methanol Price and Supply/Demand; Methanol Institute, 2022. https://www.methanol.org/methanol-price-supply-demand/. accessed: 2023-07-25.
- Acquarola, C.; Bhatelia, T.; Pareek, V.; Ao, M.; Shah, M.T. Optimized Process for Methanol Production via Bi-reforming Syngas. Ind. Eng. Chem. Res. 2022, 61, 5557–5567. [Google Scholar] [CrossRef]
- Bussche, K.; Froment, G. A Steady-State Kinetic Model for Methanol Synthesis and the Water Gas Shift Reaction on a Commercial Cu/ZnO/Al2O3Catalyst. J. Catal. 1996, 161, 1–10. [Google Scholar] [CrossRef]
- Latimer, A.A.; Kakekhani, A.; Kulkarni, A.R.; Nørskov, J.K. Direct Methane to Methanol: The Selectivity–Conversion Limit and Design Strategies. ACS Catal. 2018, 8, 6894–6907. [Google Scholar] [CrossRef]
- Liu, R.S.; Iwamoto, M.; Lunsford, J.H. Partial Oxidation of Methane by Nitrous Oxide over Molybdenum Oxide Supported on Silica. J. Chem. Soc., Chem. Commun. 1982, pp. 78–79.
- Khan, M.; Somorjai, G. A Kinetic Study of Partial Oxidation of Methane with Nitrous Oxide on a Molybdena-Silica Catalyst. J. Catal. 1985, 91, 263–271. [Google Scholar] [CrossRef]
- Suzuki, T.; Wada, K.; Shima, M.; Watanabe, Y. Photoinduced Partial Oxidation of Methane into Formaldehyde on Silica-Supported Molybdena. J. Chem. Soc., Chem. Commun. 1990, pp. 1059–1060. 1059.
- Zhang, X.; He, D.H.; Zhang, Q.J.; Xu, B.Q.; Zhu, Q.M. Methanol from Oxidation of Methane over MoOx/La-Co-Oxide Catalysts. In Natural Gas Conversion VII; Bao, X.; Xu, Y., Eds.; Elsevier, 2004; Vol. 147, Studies in Surface Science and Catalysis, pp. 541–546.
- Zhen, K.; Khan, M.; Mak, C.; Lewis, K.; Somorjai, G. Partial Oxidation of Methane with Nitrous Oxide over V2O5 SiO2 Catalyst. J. Catal. 1985, 94, 501–507. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Lin, L.; Chu, S.; Su, Y.; Song, W.; Wang, A.; Weckhuysen, B.M.; Luo, W. Highly Selective Oxidation of Methane into Methanol over Cu-Promoted Monomeric Fe/ZSM-5. ACS Catal. 2021, 11, 6684–6691. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Staykov, A.; Shiota, Y.; Yoshizawa, K. Direct Conversion of Methane to Methanol by Metal-Exchanged ZSM-5 Zeolite (Metal = Fe, Co, Ni, Cu). ACS Catal. 2016, 6, 8321–8331. [Google Scholar] [CrossRef]
- Starokon, E.V.; Parfenov, M.V.; Arzumanov, S.S.; Pirutko, L.V.; Stepanov, A.G.; Panov, G.I. Oxidation of Methane to Methanol on the Surface of FeZSM-5 Zeolite. J. Catal. 2013, 300, 47–54. [Google Scholar] [CrossRef]
- Wood, B.R.; Reimer, J.A.; Bell, A.T. Studies of N2O Adsorption and Decomposition on Fe–ZSM-5. J. Catal. 2002, 209, 151–158. [Google Scholar] [CrossRef]
- Alayon, E.M.; Nachtegaal, M.; Ranocchiari, M.; van Bokhoven, J.A. Catalytic Conversion of Methane to Methanol over Cu-Mordenite. Chem. Commun. 2012, 48, 404–406. [Google Scholar] [CrossRef]
- Knorpp, A.J.; Newton, M.A.; Pinar, A.B.; van Bokhoven, J.A. Conversion of Methane to Methanol on Copper Mordenite: Redox Mechanism of Isothermal and High-Temperature-Activation Procedure. Ind. Eng. Chem. Res. 2018, 57, 12036–12039. [Google Scholar] [CrossRef]
- Yousefzadeh, H.; Bozbag, S.E.; Sushkevich, V.; van Bokhoven, J.A.; Erkey, C. Stepwise Conversion of Methane to Methanol over Cu-Mordenite Prepared by Supercritical and Aqueous Ion Exchange Routes and Quantification of Active Cu Species by H2-TPR. Catal. Commun. 2023, 174, 106574. [Google Scholar] [CrossRef]
- Bozbag, S.E.; Alayon, E.M.C.; Pecháĉek, J.; Nachtegaal, M.; Ranocchiari, M.; van Bokhoven, J.A. Methane to Methanol over Copper Mordenite: Yield Improvement through Multiple Cycles and Different Synthesis Techniques. Catal. Sci. Technol. 2016, 6, 5011–5022. [Google Scholar] [CrossRef]
- Pappas, D.K.; Martini, A.; Dyballa, M.; Kvande, K.; Teketel, S.; Lomachenko, K.A.; Baran, R.; Glatzel, P.; Arstad, B.; Berlier, G.; Lamberti, C.; Bordiga, S.; Olsbye, U.; Svelle, S.; Beato, P.; Borfecchia, E. The Nuclearity of the Active Site for Methane to Methanol Conversion in Cu-Mordenite: A Quantitative Assessment. J. Am. Chem. Soc. 2018, 140, 15270–15278. [Google Scholar] [CrossRef]
- Cai, X.; Fang, S.; Hu, Y.H. Unprecedentedly High Efficiency for Photocatalytic Conversion of Methane to Methanol over Au-Pd/TiO2 - what is the Role of each Component in the System? J. Mater. Chem. A 2021, 9, 10796–10802. [Google Scholar] [CrossRef]
- Zhou, W.; Qiu, X.; Jiang, Y.; Fan, Y.; Wei, S.; Han, D.; Niu, L.; Tang, Z. Highly Selective Aerobic Oxidation of Methane to Methanol over Gold Decorated Zinc Oxide via Photocatalysis. J. Mater. Chem. A 2020, 8, 13277–13284. [Google Scholar] [CrossRef]
- Zecchina, A.; Rivallan, M.; Berlier, G.; Lamberti, C.; Ricchiardi, G. Structure and Nuclearity of Active Sites in Fe-Zeolites: Comparison with Iron Sites in Enzymes and Homogeneous Catalysts. Phys. Chem. Chem. Phys. 2007, 9, 3483–3499. [Google Scholar] [CrossRef]
- Zuo, H.; Xin, Q.; Meynen, V.; Klemm, E. Sensitivity of the Selective Oxidation of Methane Over Fe/ZSM-5 Zeolites in a Micro Fixed-Bed Reactor for the Catalyst Preparation Method. Appl. Catal. A: Gen. 2018, 566, 96–103. [Google Scholar] [CrossRef]
- Hall, J.N.; Bollini, P. Low-Temperature, Ambient Pressure Oxidation of Methane to Methanol Over Every Tri-Iron Node in a Metal–Organic Framework Material. Chem. Eur. J. 2020, 26, 16639–16643. [Google Scholar] [CrossRef]
- Shen, K.; Kumari, S.; Huang, Y.C.; Jang, J.; Sautet, P.; Morales-Guio, C.G. Electrochemical Oxidation of Methane to Methanol on Electrodeposited Transition Metal Oxides. J. Am. Chem. Soc. 2023, 145, 6927–6943. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, S.; Wang, S.; Zhang, Y.; Long, C.; Xie, J.; Fan, X.; Zhao, W.; Xu, P.; Fan, Y.; Cui, C.; Tang, Z. Enabling Specific Photocatalytic Methane Oxidation by Controlling Free Radical Type. J. Am. Chem. Soc. 2023, 145, 2698–2707. [Google Scholar] [CrossRef]
- Gan, Y.; Huang, M.; Yu, F.; Yu, X.; He, Z.; Liu, J.; Lin, T.; Dai, Y.; Niu, Q.; Zhong, L. Highly Selective Photocatalytic Methane Oxidation to Methanol Using CO2 as a Soft Oxidant. ACS Sustainable Chem. Eng. 2023, 11, 5537–5546. [Google Scholar] [CrossRef]
- Kaye, S.S.; Dailly, A.; Yaghi, O.M.; Long, J.R. Impact of Preparation and Handling on the Hydrogen Storage Properties of Zn4O(1,4-benzenedicarboxylate)3 (MOF-5). J. Am. Chem. Soc. 2007, 129, 14176–14177. [Google Scholar] [CrossRef] [PubMed]
- Bloch, E.D.; Murray, L.J.; Queen, W.L.; Chavan, S.; Maximoff, S.N.; Bigi, J.P.; Krishna, R.; Peterson, V.K.; Grandjean, F.; Long, G.J.; Smit, B.; Bordiga, S.; Brown, C.M.; Long, J.R. Selective Binding of O2 over N2 in a Redox-Active Metal-Organic Framework with Open Iron(II) Coordination Sites. J. Am. Chem. Soc. 2011, 133, 14814–14822. [Google Scholar] [CrossRef] [PubMed]
- Bara, D.; Meekel, E.G.; Pakamorė, I.; Wilson, C.; Ling, S.; Forgan, R.S. Exploring and Expanding the Fe-Terephthalate Metal-Organic framework Phase space by Coordination and Oxidation Modulation. Mater. Horiz. 2021, 8, 3377–3386. [Google Scholar] [CrossRef]
- Hendon, C.H.; Walsh, A. Chemical Principles Underpinning the Performance of the Metal-Organic Framework HKUST-1. Chem. Sci. 2015, 6, 3674–3683. [Google Scholar] [CrossRef]
- Quijia, C.R.; Lima, C.; Silva, C.; Alves, R.C.; Frem, R.; Chorilli, M. Application of MIL-100 (Fe) in Drug Delivery and Biomedicine. J. Drug. Deliv. Sci. Technol. 2021, 61, 102217. [Google Scholar] [CrossRef]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Yuan, S.; Sun, X.; Pang, J.; Lollar, C.; Qin, J.S.; Perry, Z.; Joseph, E.; Wang, X.; Fang, Y.; Bosch, M.; Sun, D.; Liu, D.; Zhou, H.C. PCN-250 Under Pressure: Sequential Phase Transformation and the Implications for MOF Densification. Joule 2017, 1, 806–815. [Google Scholar] [CrossRef]
- Chen, B.; Ockwig, N.W.; Millward, A.R.; Contreras, D.S.; Yaghi, O.M. High H2 Adsorption in a Microporous Metal–Organic Framework with Open Metal Sites. Angew. Chem. Int. Ed. 2005, 44, 4745–4749. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef]
- Stock, N.; Biswas, S. Synthesis of Metal-Organic Frameworks (MOFs): Routes to Various MOF Topologies, Morphologies, and Composites. Chem. Rev. 2012, 112, 933–969. [Google Scholar] [CrossRef] [PubMed]
- Rosen, A.S.; Notestein, J.M.; Snurr, R.Q. Structure–Activity Relationships That Identify Metal–Organic Framework Catalysts for Methane Activation. ACS Catal. 2019, 9, 3576–3587. [Google Scholar] [CrossRef]
- Rosen, A.S.; Notestein, J.M.; Snurr, R.Q. Identifying Promising Metal–Organic Frameworks for Heterogeneous Catalysis via High-Throughput Periodic Density Functional Theory. J. Comput. Chem. 2019, 40, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- Konnerth, H.; Matsagar, B.M.; Chen, S.S.; Prechtl, M.H.; Shieh, F.K.; Wu, K.C.W. Metal-Organic Framework (MOF)-Derived Catalysts for Fine Chemical Production. Coord. Chem. Rev. 2020, 416, 213319. [Google Scholar] [CrossRef]
- Maina, J.W.; Pozo-Gonzalo, C.; Kong, L.; Schütz, J.; Hill, M.; Dumée, L.F. Metal Organic Framework Based Catalysts for CO2 Conversion. Mater. Horiz. 2017, 4, 345–361. [Google Scholar] [CrossRef]
- Liu, M.; Wu, J.; Hou, H. Metal-Organic Framework (MOF)-Based Materials as Heterogeneous Catalysts for C-H Bond Activation. Chem. Eur. J. 2019, 25, 2935–2948. [Google Scholar] [CrossRef]
- Cui, W.G.; Zhang, G.Y.; Hu, T.L.; Bu, X.H. Metal-Organic Framework-Based Heterogeneous Catalysts for the Conversion of C1 Chemistry: CO, CO2 and CH4. Coord. Chem. Rev. 2019, 387, 79–120. [Google Scholar] [CrossRef]
- Bavykina, A.; Kolobov, N.; Khan, I.S.; Bau, J.A.; Ramirez, A.; Gascon, J. Metal–Organic Frameworks in Heterogeneous Catalysis: Recent Progress, New Trends, and Future Perspectives. Chem. Rev. 2020, 120, 8468–8535. [Google Scholar] [CrossRef]
- Li, Y.; Fu, Y.; Ni, B.; Ding, K.; Chen, W.; Wu, K.; Huang, X.; Zhang, Y. Effects of Ligand Functionalization on the Photocatalytic Properties of Titanium-Based MOF: a Density Functional Theory Study. AIP Adv. 2018, 8. [Google Scholar] [CrossRef]
- Barona, M.; Snurr, R.Q. Exploring the Tunability of Trimetallic MOF Nodes for Partial Oxidation of Methane to Methanol. ACS Appl. Mater. Interfaces 2020, 12, 28217–28231. [Google Scholar] [CrossRef]
- Xiao, D.J.; Bloch, E.D.; Mason, J.A.; Queen, W.L.; Hudson, M.R.; Planas, N.; Borycz, J.; Dzubak, A.L.; Verma, P.; Lee, K.; Bonino, F.; Crocellà, V.; Yano, J.; Bordiga, S.; Truhlar, D.G.; Gagliardi, L.; Brown, C.M.; Long, J.R. Oxidation of Ethane to Ethanol by N2O in a Metal-Organic Framework with Coordinatively Unsaturated Iron(II) Sites. Nat. Chem. 2014, 6, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Natarajan, S.; Mani, P.; Pankajakshan, A. Post-Synthetic Modification of Metal-Organic Frameworks Toward Applications. Adv. Funct. Mater. 2021, 31, 2006291. [Google Scholar] [CrossRef]
- Asgari, M.; Semino, R.; Schouwink, P.A.; Kochetygov, I.; Tarver, J.; Trukhina, O.; Krishna, R.; Brown, C.M.; Ceriotti, M.; Queen, W.L. Understanding How Ligand Functionalization Influences CO2 and N2 Adsorption in a Sodalite Metal–Organic Framework. Chem. Mater. 2020, 32, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, M.; Bury, W.; Karagiaridi, O.; Brown, Z.; Hupp, J.T.; Farha, O.K. Transmetalation: Routes to Metal Exchange within Metal-Organic Frameworks. J. Mater. Chem. A 2013, 1, 5453–5468. [Google Scholar] [CrossRef]
- Zheng, L.L.; Zhang, L.S.; Chen, Y.; Tian, L.; Jiang, X.H.; Chen, L.S.; Xing, Q.J.; Liu, X.Z.; Wu, D.S.; Zou, J.P. A New Strategy for the Fabrication of Covalent Organic Framework-Metal-Organic Framework Hybrids via In-Situ Functionalization of Ligands for Improved Hydrogen Evolution Reaction Activity. Chinese J. Catal. 2022, 43, 811–819. [Google Scholar] [CrossRef]
- Li, B.; Zhang, Y.; Ma, D.; Li, L.; Li, G.; Li, G.; Shi, Z.; Feng, S. A Strategy Toward Constructing a Bifunctionalized MOF Catalyst: Post-Synthetic Modification of MOFs on Organic Ligands and Coordinatively Unsaturated Metal Sites. Chem. Comm. 2012, 48, 6151–6153. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, X.; Kanchanakungwankul, S.; Lu, Z.; Noh, H.; Syed, Z.H.; Farha, O.K.; Truhlar, D.G.; Hupp, J.T. Unexpected “Spontaneous” Evolution of Catalytic, MOF-Supported Single Cu(II) Cations to Catalytic, MOF-Supported Cu(0) Nanoparticles. J. Am. Chem. Soc. 2020, 142, 21169–21177. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Y.; Zhao, Y.; Liu, C.j. Recent Progresses in the Size and Structure Control of MOF Supported Noble Metal Catalysts. Catal. Today 2016, 263, 61–68. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, G.; Chen, J.; Niu, H. Excellent Catalytic Performance of Ce–MOF with Abundant Oxygen Vacancies Supported Noble Metal Pt in the Oxidation of Toluene. Catalysts 2022, 12, 775. [Google Scholar] [CrossRef]
- Han, A.; Wang, B.; Kumar, A.; Qin, Y.; Jin, J.; Wang, X.; Yang, C.; Dong, B.; Jia, Y.; Liu, J.; Sun, X. Recent Advances for MOF-Derived Carbon-Supported Single-Atom Catalysts. Small Methods 2019, 3, 1800471. [Google Scholar] [CrossRef]
- Huang, H.; Shen, K.; Chen, F.; Li, Y. Metal-Organic Frameworks as a Good Platform for the Fabrication of Single-Atom Catalysts. ACS Catal. 2020, 10, 6579–6586. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, L.; Doyle-Davis, K.; Fu, X.; Luo, J.L.; Sun, X. Recent Advances in MOF-Derived Single Atom Catalysts for Electrochemical Applications. Adv. Energy Mater. 2020, 10, 2001561. [Google Scholar] [CrossRef]
- Li, Z.; Schweitzer, N.M.; League, A.B.; Bernales, V.; Peters, A.W.; Getsoian, A.B.; Wang, T.C.; Miller, J.T.; Vjunov, A.; Fulton, J.L.; Lercher, J.A.; Cramer, C.J.; Gagliardi, L.; Hupp, J.T.; Farha, O.K. Sintering-Resistant Single-Site Nickel Catalyst Supported by Metal–Organic Framework. J. Am. Chem. Soc. 2016, 138, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Vogiatzis, K.D.; Planas, N.; Borycz, J.; Xiao, D.J.; Long, J.R.; Gagliardi, L.; Truhlar, D.G. Mechanism of Oxidation of Ethane to Ethanol at Iron(IV)–Oxo Sites in Magnesium-Diluted Fe2(dobdc). J. Am. Chem. Soc. 2015, 137, 5770–5781. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.C.; Vitillo, J.G.; Babucci, M.; Hoffman, A.S.; Boubnov, A.; Beauvais, M.L.; Chen, Z.; Cramer, C.J.; Chapman, K.W.; Bare, S.R.; Gates, B.C.; Lu, C.C.; Gagliardi, L.; Bahn, A. Structure, Dynamics, and Reactivity for Light Alkane Oxidation of Fe(II) Sites Situated in the Nodes of a Metal–Organic Framework. J. Am. Chem. Soc. 2019, 141, 18142–18151. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.N.; Li, M.; Bollini, P. Light Alkane Oxidation over Well-Defined Active Sites in Metal-Organic Framework Materials. Catal. Sci. Technol. 2022, 12, 418–435. [Google Scholar] [CrossRef]
- Simons, M.C.; Prinslow, S.D.; Babucci, M.; Hoffman, A.S.; Hong, J.; Vitillo, J.G.; Bare, S.R.; Gates, B.C.; Lu, C.C.; Gagliardi, L.; Bahn, A. Beyond Radical Rebound: Methane Oxidation to Methanol Catalyzed by Iron Species in Metal–Organic Framework Nodes. J. Am. Chem. Soc. 2021, 143, 12165–12174. [Google Scholar] [CrossRef]
- Szécsényi, Á.; Li, G.; Gascon, J.; Pidko, E.A. Unraveling Reaction Networks Behind the Catalytic Oxidation of Methane with H2O2 Over a Mixed-Metal MIL-53(Al, Fe) MOF Catalyst. Chem. Sci. 2018, 9, 6765–6773. [Google Scholar] [CrossRef]
- An, B.; Li, Z.; Wang, Z.; Zeng, X.; Han, X.; Cheng, Y.; Sheveleva, A.M.; Zhang, Z.; Tuna, F.; McInnes, E.J.L.; Frogley, M.D.; Ramirez-Cuesta, A.J.; S. Natrajan, L.; Wang, C.; Lin, W.; Yang, S.; Schröder, M. Direct Photo-Oxidation of Methane to Methanol over a Mono-Iron Hydroxyl Site. Nat. Mater. 2022, 21, 932–938. [Google Scholar] [CrossRef]
- Baek, J.; Rungtaweevoranit, B.; Pei, X.; Park, M.; Fakra, S.C.; Liu, Y.S.; Matheu, R.; Alshmimri, S.A.; Alshehri, S.; Trickett, C.A.; Somorjai, G.; Yaghi, O.M. Bioinspired Metal–Organic Framework Catalysts for Selective Methane Oxidation to Methanol. J. Am. Chem. Soc. 2018, 140, 18208–18216. [Google Scholar] [CrossRef]
- Zheng, J.; Ye, J.; Ortuño, M.A.; Fulton, J.L.; Gutiérrez, O.Y.; Camaioni, D.M.; Motkuri, R.K.; Li, Z.; Webber, T.E.; Mehdi, B.L.; Browning, N.D.; Penn, R.L.; Farha, O.K.; Hupp, J.T.; Truhlar, D.G.; Cramer, C.J.; Lercher, J.A. Selective Methane Oxidation to Methanol on Cu-oxo Dimers stabilized by Zirconia Nodes of an NU-1000 Metal–Organic Framework. J. Am. Chem. Soc. 2019, 141, 9292–9304. [Google Scholar] [CrossRef] [PubMed]
- Ikuno, T.; Zheng, J.; Vjunov, A.; Sanchez-Sanchez, M.; Ortuño, M.A.; Pahls, D.R.; Fulton, J.L.; Camaioni, D.M.; Li, Z.; Ray, D.; Mehdi, B.L.; Browning, N.D.; Farha, O.K.; Hupp, J.T.; Cramer, C.J.; Gagliardi, L.; Lercher, J.A. Methane Oxidation to Methanol Catalyzed by Cu-oxo Clusters Stabilized in NU-1000 Metal–Organic Framework. J. Am. Chem. Soc. 2017, 139, 10294–10301. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Proshlyakov, Y.; D. , L.J.; Proshlyakov, D.A. Structure of the Key Species in the Enzymatic Oxidation of Methane to Methanol. Nature 2015, 518, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liang, A.D.; Lippard, S.J. Coupling Oxygen Consumption with Hydrocarbon Oxidation in Bacterial Multicomponent Monooxygenases. Acc. Chem. Res. 2015, 48, 2632–2639. [Google Scholar] [CrossRef]
- Wang, V.C.C.; Maji, S.; Chen, P.P.Y.; Lee, H.K.; Yu, S.S.F.; Chan, S.I. Alkane Oxidation: Methane Monooxygenases, Related Enzymes, and Their Biomimetics. Chem. Rev. 2017, 117, 8574–8621. [Google Scholar] [CrossRef]
- Baik, M.H.; Newcomb, M.; Friesner, R.A.; Lippard, S.J. Mechanistic Studies on the Hydroxylation of Methane by Methane Monooxygenase. Chem. Rev. 2003, 103, 2385–2420. [Google Scholar] [CrossRef]
- Liu, K.E.; Valentine, A.M.; Wang, D.; Huynh, B.H.; Edmondson, D.E.; Salifoglou, A.; Lippard, S.J. Kinetic and Spectroscopic Characterization of Intermediates and Component Interactions in Reactions of Methane Monooxygenase from Methylococcus Capsulatus (Bath). J. Am. Chem. Soc. 1995, 117, 10174–10185. [Google Scholar] [CrossRef]
- Jacobs, A.B.; Banerjee, R.; Deweese, D.E.; Braun, A.; Babicz, J.T.J.; Gee, L.B.; Sutherlin, K.D.; Böttger, L.H.; Yoda, Y.; Saito, M.; Kitao, S.; Kobayashi, Y.; Seto, M.; Tamasaku, K.; Lipscomb, J.D.; Park, K.; Solomon, E.I. Nuclear Resonance Vibrational Spectroscopic Definition of the Fe(IV)2 Intermediate Q in Methane Monooxygenase and Its Reactivity. J. Am. Chem. Soc. 2021, 143, 16007–16029. [Google Scholar] [CrossRef]
- Kazaryan, A.; Baerends, E.J. Ligand Field Effects and the High Spin-High Reactivity Correlation in the H Abstraction by Non-Heme Iron(IV)-Oxo Complexes: A DFT Frontier Orbital Perspective. ACS Catal. 2015, 5, 1475–1488. [Google Scholar] [CrossRef]
- Isolated Fe Sites in Metal Organic Frameworks Catalyze the Direct Conversion of Methane to Methanol. ACS Catal. 2018, 8, 5542–5548. [CrossRef]
- Loiseau, T.; Serre, C.; Huguenard, C.; Fink, G.; Taulelle, F.; Henry, M.; Bataille, T.; Férey, G. A Rationale for the Large Breathing of the Porous Aluminum Terephthalate (MIL-53) Upon Hydration. Chem. Eur. J. 2004, 10, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Férey, G.; Serre, C.; Mellot-Draznieks, C.; Millange, F.; Surblé, S.; Dutour, J.; Margiolaki, I. A Hybrid Solid with Giant Pores Prepared by a Combination of Targeted Chemistry, Simulation, and Powder Diffraction. Angew. Chem. Int. Ed. 2004, 43, 6296–6301. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Surblé, S.; Serre, C.; Hong, D.Y.; Seo, Y.K.; Chang, J.S.; Grenèche, J.M.; Margiolaki, I.; Férey, G. Synthesis and Catalytic Properties of MIL-100(Fe), an Iron(iii) Carboxylate with Large Pores. Chem. Commun. 2007, pp. 2820–2822.
- Colby, J.; Stirling, D.I.; Dalton, H. The Soluble Methane Mono-Oxygenase of Methylococcus Capsulatus (Bath). Its Ability to Oxygenate n-Alkanes, n-Alkenes, Ethers, and Alicyclic, Aromatic and Heterocyclic Compounds. Biochem. J. 1977, 165, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Rungtaweevoranit, B.; Abdel-Mageed, A.M.; Khemthong, P.; Eaimsumang, S.; Chakarawet, K.; Butburee, T.; Kunkel, B.; Wohlrab, S.; Chainok, K.; Phanthasri, J.; Wannapaiboon, S.; Youngjan, S.; Seehamongkol, T.; Impeng, S.; Faungnawakij, K. Structural Evolution of Iron-Loaded Metal-Organic Framework Catalysts for Continuous Gas-Phase Oxidation of Methane to Methanol. ACS Appl. Mater. Interfaces 2023, 15, 26700–26709. [Google Scholar] [CrossRef] [PubMed]
- Semrau, J.D.; DiSpirito, A.A.; Yoon, S. Methanotrophs and Copper. FEMS Microbiol. Rev. 2010, 34, 496–531. [Google Scholar] [CrossRef]
- Stanley, S.; Prior, S.; Leak, D.; Dalton, H. Copper Stress Underlies the Fundamental Change in Intracellular Location of Methane Mono-Oxygenase in Methane-Oxidizing Organisms: Studies in Batch and Continuous Cultures. Biotechnol. Lett. 1983, 5, 487–492. [Google Scholar] [CrossRef]
- Murrell, J.C.; McDonald, I.R.; Gilbert, B. Regulation of expression of methane monooxygenases by copper ions. Trends Microbiol. 2000, 8, 221–225. [Google Scholar] [CrossRef]
- Lawton, T.J.; Rosenzweig, A.C. Methane-Oxidizing Enzymes: an Upstream Problem in Biological Gas-to-Liquids Conversion. J. Am. Chem. Soc. 2016, 138, 9327–9340. [Google Scholar] [CrossRef]
- Yoshizawa, K.; Shiota, Y. Conversion of Methane to Methanol at the Mononuclear and Dinuclear Copper Sites of Particulate Methane Monooxygenase (pMMO): A DFT and QM/MM Study. J. Am. Chem. Soc. 2006, 128, 9873–9881. [Google Scholar] [CrossRef]
- Smith, S.M.; Rawat, S.; Telser, J.; Hoffman, B.M.; Stemmler, T.L.; Rosenzweig, A.C. Crystal Structure and Characterization of Particulate Methane Monooxygenase from Methylocystis Species Strain M. Biochemistry 2011, 50, 10231–10240. [Google Scholar] [CrossRef]
- Ross, M.O.; Rosenzweig, A.C. A Tale of Two Methane Monooxygenases. J. Biol. Inorg. Chem. 2017, 22, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Cutsail, G.E.; Ross, M.O.; Rosenzweig, A.C.; DeBeer, S. Towards a Unified Understanding of the Copper Sites in Particulate Methane Monooxygenase: an X-ray Absorption Spectroscopic Investigation. Chem. Sci. 2021, 12, 6194–6209. [Google Scholar] [CrossRef] [PubMed]
- Citek, C.; Herres-Pawlis, S.; Stack, T.D.P. Low Temperature Syntheses and Reactivity of Cu2O2 Active-Site Models. Acc. Chem. Res. 2015, 48, 2424–2433. [Google Scholar] [CrossRef]
- Woertink, J.S.; Smeets, P.J.; Groothaert, M.H.; Vance, M.A.; Sels, B.F.; Schoonheydt, R.A.; Solomon, E.I. A [Cu2O]2+ Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol. Proc. Nat. Ac. Sci. 2009, 106, 18908–18913. [Google Scholar] [CrossRef] [PubMed]
- Grundner, S.; Markovits, M.A.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-Site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef]
- Pappas, D.K.; Borfecchia, E.; Dyballa, M.; Pankin, I.A.; Lomachenko, K.A.; Martini, A.; Signorile, M.; Teketel, S.; Arstad, B.; Berlier, G.; Lamberti, C.; Bordiga, S.; Olsbye, U.; Lillerud, K.P.; Svelle, S.; Beato, P. Methane to Methanol: Structure-Activity Relationships for Cu-CHA. J. Am. Chem. Soc. 2017, 139, 14961–14975. [Google Scholar] [CrossRef]
- Platero-Prats, A.E.; Li, Z.; Gallington, L.C.; Peters, A.W.; Hupp, J.T.; Farha, O.K.; Chapman, K.W. Addressing the Characterisation Challenge to Understand Catalysis in MOFs: the Case of Nanoscale Cu Supported in NU-1000. Faraday Discuss. 2017, 201, 337–350. [Google Scholar] [CrossRef]
- Webber, T.E.; Desai, S.P.; Combs, R.L.; Bingham, S.; Lu, C.C.; Penn, R.L. Size Control of the MOF NU-1000 through Manipulation of the Modulator/Linker Competition. Cryst. Growth Des. 2020, 20, 2965–2972. [Google Scholar] [CrossRef]
- Tomkins, P.; Mansouri, A.; Bozbag, S.E.; Krumeich, F.; Park, M.B.; Alayon, E.M.C.; Ranocchiari, M.; van Bokhoven, J.A. Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature. Angew. Chem. Int. Ed. 2016, 128, 5557–5561. [Google Scholar] [CrossRef]
- Sheppard, T.; Hamill, C.; Goguet, A.; Rooney, D.; Thompson, J. A Low temperature, Isothermal Gas-Phase System for Conversion of Methane to Methanol Over Cu–ZSM-5. Chemical Commun. 2014, 50, 11053–11055. [Google Scholar] [CrossRef]
- Ren, M.; Shi, Q.; Mi, L.; Liang, W.; Yuan, M.; Wang, L.; Gao, Z.; Huang, W.; Huang, J.; Zuo, Z. Isothermal Conversion of Methane to Methanol over CuxOy@UiO-bpy. Mater. Today Sustain. 2021, 11-12, 100061. [Google Scholar] [CrossRef]
- Beznis, N.V.; Weckhuysen, B.M.; Bitter, J.H. Cu-ZSM-5 Zeolites for the Formation of Methanol from Methane and Oxygen: Probing the Active Sites and Spectator Species. Catal. Lett. 2010, 138, 14–22. [Google Scholar] [CrossRef]
- Lee, H.; Kwon, C.; Keum, C.; Kim, H.E.; Lee, H.; Han, B.; Lee, S.Y. Methane Partial Oxidation by Monomeric Cu Active Center Confined on ZIF-7. Chem. Eng. J. 2022, 450, 138472. [Google Scholar] [CrossRef]
- Delgass, W. Spectroscopy in Heterogeneous Catalysis; Elsevier, 2012.
- Braglia, L.; Tavani, F.; Mauri, S.; Edla, R.; Krizmancic, D.; Tofoni, A.; Colombo, V.; D’Angelo, P.; Torelli, P. Catching the Reversible Formation and Reactivity of Surface Defective Sites in Metal-Organic Frameworks: An Operando Ambient Pressure-NEXAFS Investigation. J. Phys. Chem. Lett. 2021, 12, 9182–9187. [Google Scholar] [CrossRef]
- Tavani, F.; Fracchia, M.; Tofoni, A.; Braglia, L.; Jouve, A.; Morandi, S.; Manzoli, M.; Torelli, P.; Ghigna, P.; D’Angelo, P. Structural and Mechanistic Insights into Low-Temperature CO Oxidation over a Prototypical High Entropy Oxide by Cu L-edge Operando Soft X-ray Absorption Spectroscopy. Phys. Chem. Chem. Phys. 2021, 23, 26575–26584. [Google Scholar] [CrossRef] [PubMed]
- Tavani, F.; Busato, M.; Braglia, L.; Mauri, S.; Torelli, P.; D’Angelo, P. Caught while Dissolving: Revealing the Interfacial Solvation of the Mg2+ Ions on the MgO Surface. ACS Appl. Mater. Interfaces 2022, 14, 38370–38378. [Google Scholar] [CrossRef] [PubMed]
- Tavani, F.; Busato, M.; Veclani, D.; Braglia, L.; Mauri, S.; Torelli, P.; D’Angelo, P. Investigating the High-Temperature Water/MgCl2 Interface through Ambient Pressure Soft X-ray Absorption Spectroscopy. ACS Appl. Mater. Interfaces 2023, 15, 26166–26174. [Google Scholar] [CrossRef] [PubMed]
- Capocasa, G.; Sessa, F.; Tavani, F.; Monte, M.; Olivo, G.; Pascarelli, S.; Lanzalunga, O.; Di Stefano, S.; D’Angelo, P. Coupled X-ray Absorption/UV-vis Monitoring of Fast Oxidation Reactions Involving a Nonheme Iron-Oxo Complex. J. Am. Chem. Soc. 2019, 141, 2299–2304. [Google Scholar] [CrossRef]
- Tavani, F.; Martini, A.; Capocasa, G.; Di Stefano, S.; Lanzalunga, O.; D’Angelo, P. Direct Mechanistic Evidence for a Non-Heme Complex Reaction Through a Multivariate XAS Analysis. Inorg. Chem. 2020, 59, 9979–9989. [Google Scholar] [CrossRef]
- Tavani, F.; Fracchia, M.; Pianta, N.; Ghigna, P.; Quartarone, E.; D’Angelo, P. Multivariate Curve Resolution Analysis of Operando XAS Data for the Investigation of the Lithiation Mechanisms in High Entropy Oxides. Chem. Phys. Lett. 2020, 760, 137968. [Google Scholar] [CrossRef]
- Tavani, F.; Capocasa, G.; Martini, A.; Sessa, F.; Di Stefano, S.; Lanzalunga, O.; D’Angelo, P. Direct Structural and Mechanistic Insights into Fast Bimolecular Chemical Reactions in Solution through a Coupled XAS/UV-Vis Multivariate Statistical Analysis. Dalton Trans. 2021, 50, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Tavani, F.; Capocasa, G.; Martini, A.; Sessa, F.; Di Stefano, S.; Lanzalunga, O.; D’Angelo, P. Activation of C-H Bonds by a Nonheme Iron(iv)-Oxo Complex: Mechanistic Evidence through a Coupled EDXAS/UV-Vis Multivariate Analysis. Phys. Chem. Chem. Phys. 2021, 23, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, D.; Tavani, F.; Di Berto Mancini, M.; Frateloreto, F.; Busato, M.; Oliveira De Souza, D.; Cenesi, F.; Lanzalunga, O.; Di Stefano, S.; D’Angelo, P. Two Faces of the Same Coin: Coupling X-Ray Absorption and NMR Spectroscopies to Investigate the Exchange Reaction Between Prototypical Cu Coordination Complexes. Chem. Eur. J. 2022, 28, e202103825. [Google Scholar] [CrossRef] [PubMed]
- Frateloreto, F.; Tavani, F.; Di Berto Mancini, M.; Del Giudice, D.; Isabelle, K.; Lanzalunga, O.; Di Stefano, S.; D’Angelo, P. Following a Silent Metal Ion: A Combined X-ray Absorption and Nuclear Magnetic Resonance Spectroscopic Study of the Zn2+ Cation Dissipative Translocation between Two Different Ligands. J. Phys. Chem. Lett. 2022, 13, 5522–5529. [Google Scholar] [CrossRef]
- Feng, D.; Wang, K.; Wei, Z.; Chen, Y.P.; Simon, C.M.; Arvapally, R.K.; Martin, R.L.; Bosch, M.; Liu, T.F.; Fordham, S.; Yuan, D.; Omary, M.A.; Haranczyk, M.; Smit, B.; Zhou, H.C. Kinetically Tuned Dimensional Augmentation as a Versatile Synthetic Route Towards Robust Metal-Organic Frameworks. Nat. Commun. 2014, 5, 5723. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Bols, M.L.; Rhoda, H.M.; Plessers, D.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Cage effects control the mechanism of methane hydroxylation in zeolites. Science 2021, 373, 327–331. [Google Scholar] [CrossRef]
- Kirillov, A.M.; Shul’pin, G.B. Pyrazinecarboxylic acid and analogs: Highly efficient co-catalysts in the metal-complex-catalyzed oxidation of organic compounds. Coord. Chem. Rev. 2013, 257, 732–754. [Google Scholar] [CrossRef]
Figure 2.
Proposed Fe(IV) diiron structure of compound Q in sMMO. The numbers denote amino acids in the side chains: H, histidine; E, glutamate. [Reprinted with permission from Ref.[81]. Copyright 2017, American Chemical Society].
Figure 2.
Proposed Fe(IV) diiron structure of compound Q in sMMO. The numbers denote amino acids in the side chains: H, histidine; E, glutamate. [Reprinted with permission from Ref.[81]. Copyright 2017, American Chemical Society].
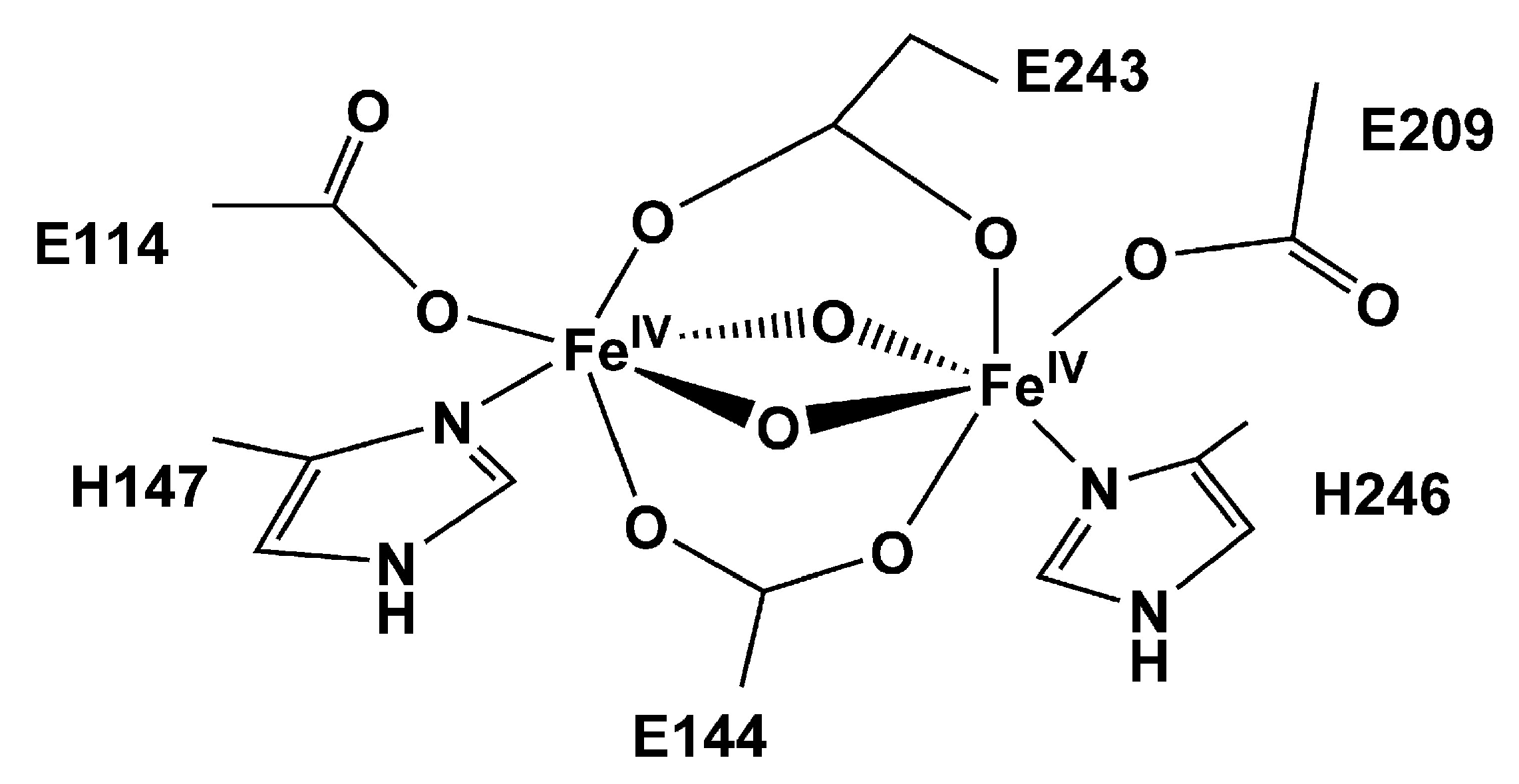
Figure 3.
Schematic depiction of the sMMO catalytic cycle. R represents the MMOR reductase component, while B is the regulatory MMOB component. [Reprinted with permission from Ref.[80]. Copyright 2015, American Chemical Society].
Figure 3.
Schematic depiction of the sMMO catalytic cycle. R represents the MMOR reductase component, while B is the regulatory MMOB component. [Reprinted with permission from Ref.[80]. Copyright 2015, American Chemical Society].
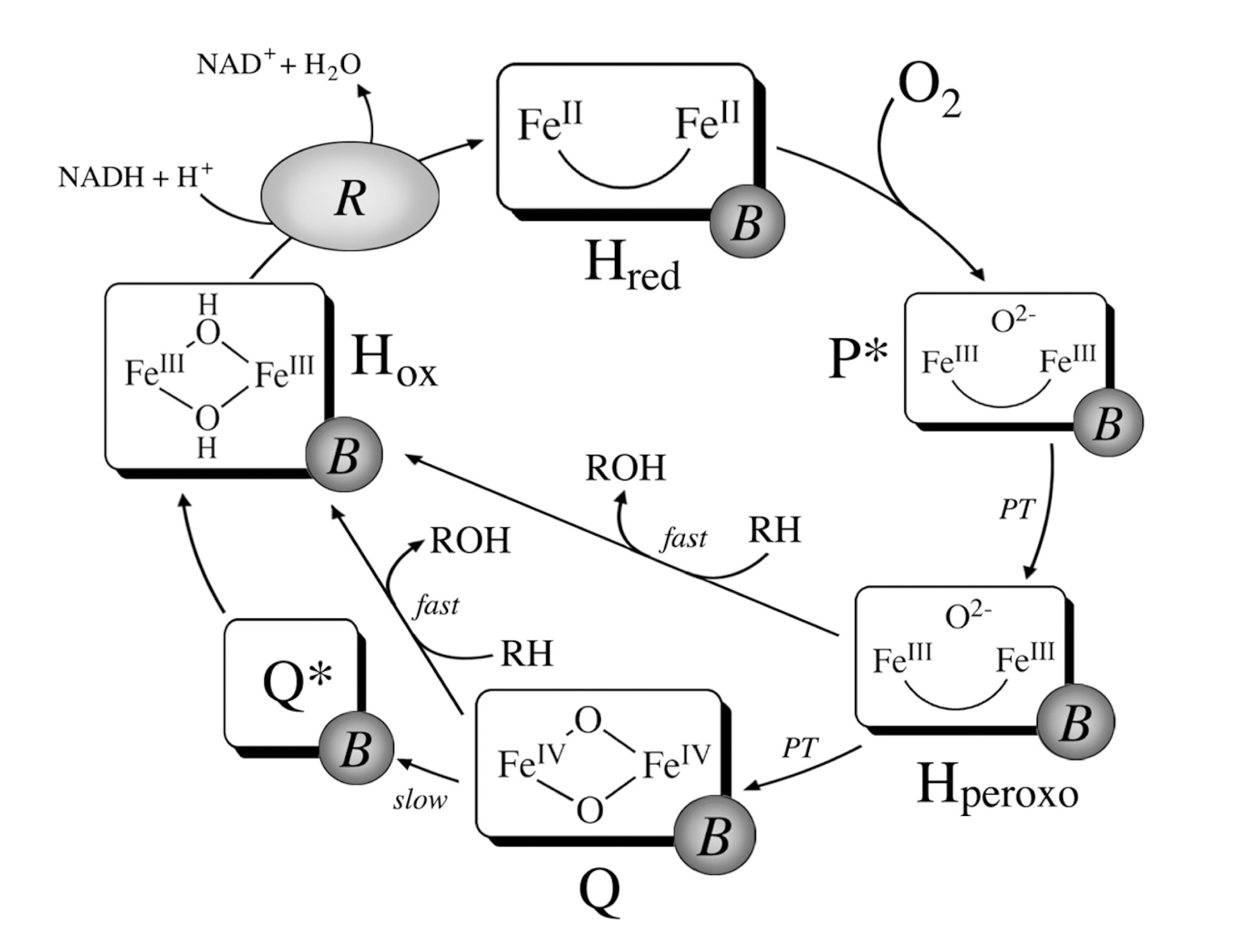
Figure 4.
(a) Depiction of the MIL-53(Al,Fe) crystal structure, along with a representation of an isolated Fe-Fe site within the MOF octahedral [AlO] chain. (b) Evolution of the oxidation products and of the associated turnover frequency (TOF) and turnover number (TON) values during the MTM conversion in the presence of low concentrations of the ECS-LL catalyst. [Adapted with permission from Ref.[86]. Copyright 2018, American Chemical Society].
Figure 4.
(a) Depiction of the MIL-53(Al,Fe) crystal structure, along with a representation of an isolated Fe-Fe site within the MOF octahedral [AlO] chain. (b) Evolution of the oxidation products and of the associated turnover frequency (TOF) and turnover number (TON) values during the MTM conversion in the presence of low concentrations of the ECS-LL catalyst. [Adapted with permission from Ref.[86]. Copyright 2018, American Chemical Society].
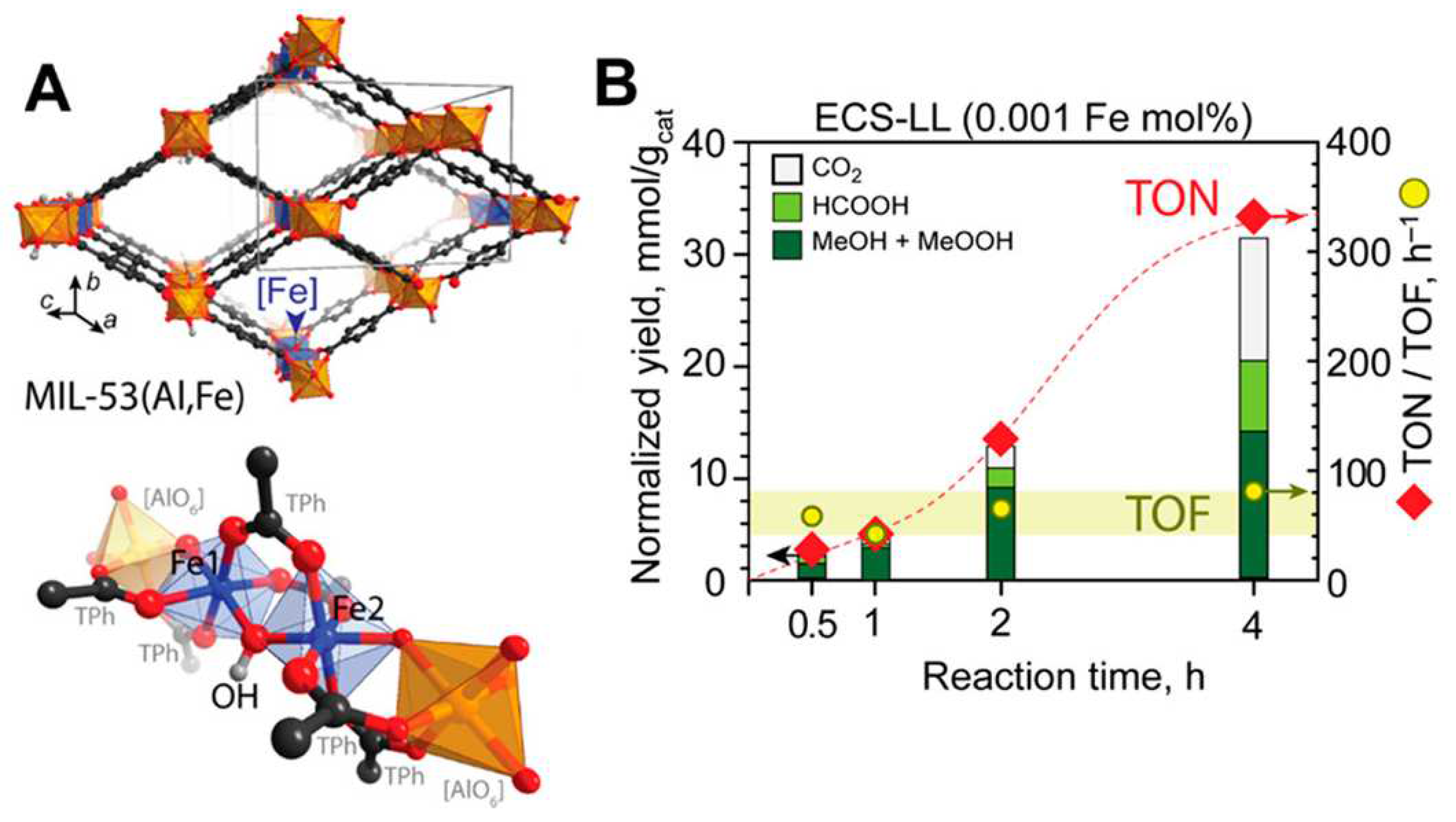
Figure 6.
On-stream MTM conversion conducted over the Fe/UiO-66 MOF. (a) Rates of CHOH and CO formation during the direct CH activation (10% CH, 5% O + 0.2% HO, balance Ar) at 5 bar and at 180 or 190 C. (b) Product selectivity towards the formation of methanol, expressed as [CHOH]/([CHOH] + [CO]). [Adapted with permission from Ref.[91]. Copyright 2023, American Chemical Society].
Figure 6.
On-stream MTM conversion conducted over the Fe/UiO-66 MOF. (a) Rates of CHOH and CO formation during the direct CH activation (10% CH, 5% O + 0.2% HO, balance Ar) at 5 bar and at 180 or 190 C. (b) Product selectivity towards the formation of methanol, expressed as [CHOH]/([CHOH] + [CO]). [Adapted with permission from Ref.[91]. Copyright 2023, American Chemical Society].
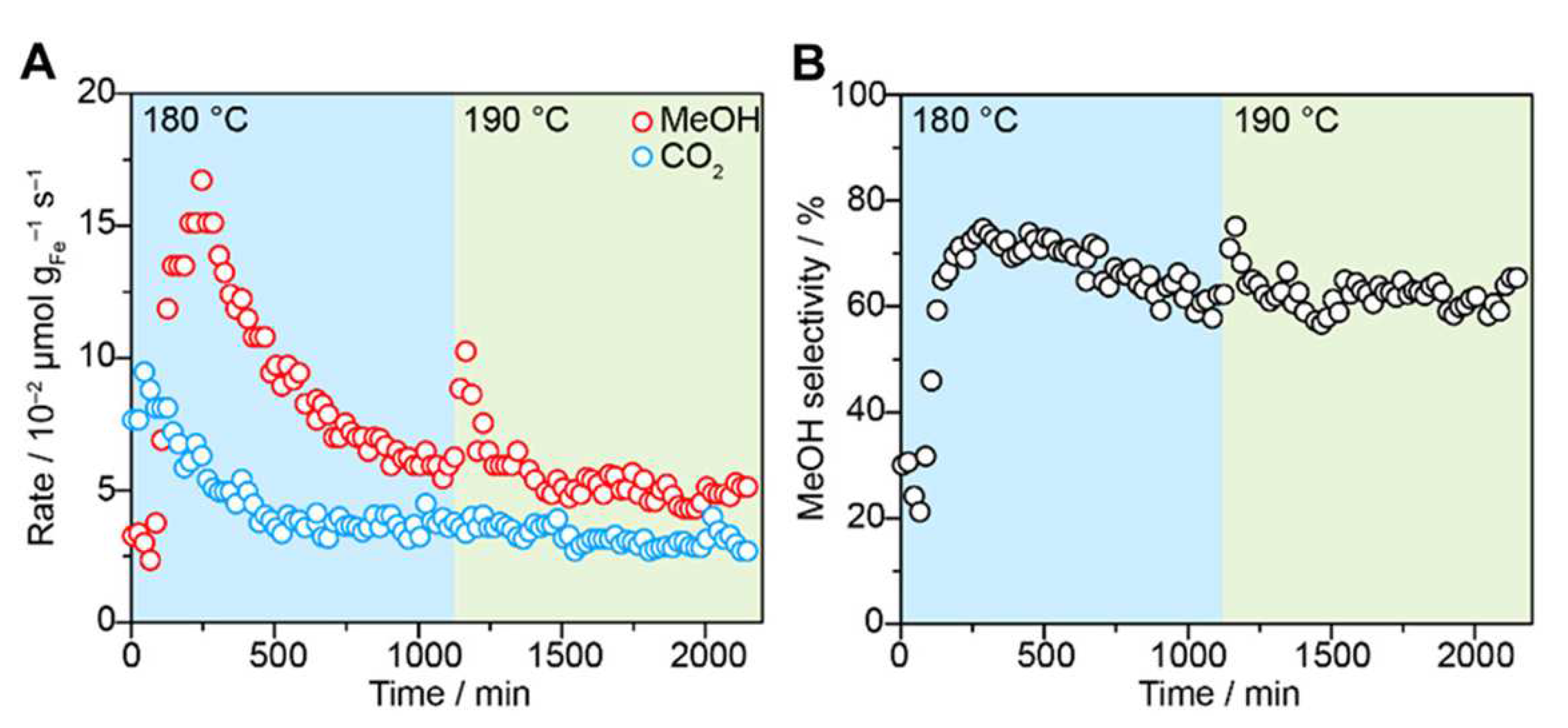
Figure 7.
QM/MM structures of the monomeric (a) and dimeric (b) copper active sites of pMMO optimized from crystallographic data. [Adapted with permission from Ref.[96]. Copyright 2006, American Chemical Society].
Figure 7.
QM/MM structures of the monomeric (a) and dimeric (b) copper active sites of pMMO optimized from crystallographic data. [Adapted with permission from Ref.[96]. Copyright 2006, American Chemical Society].
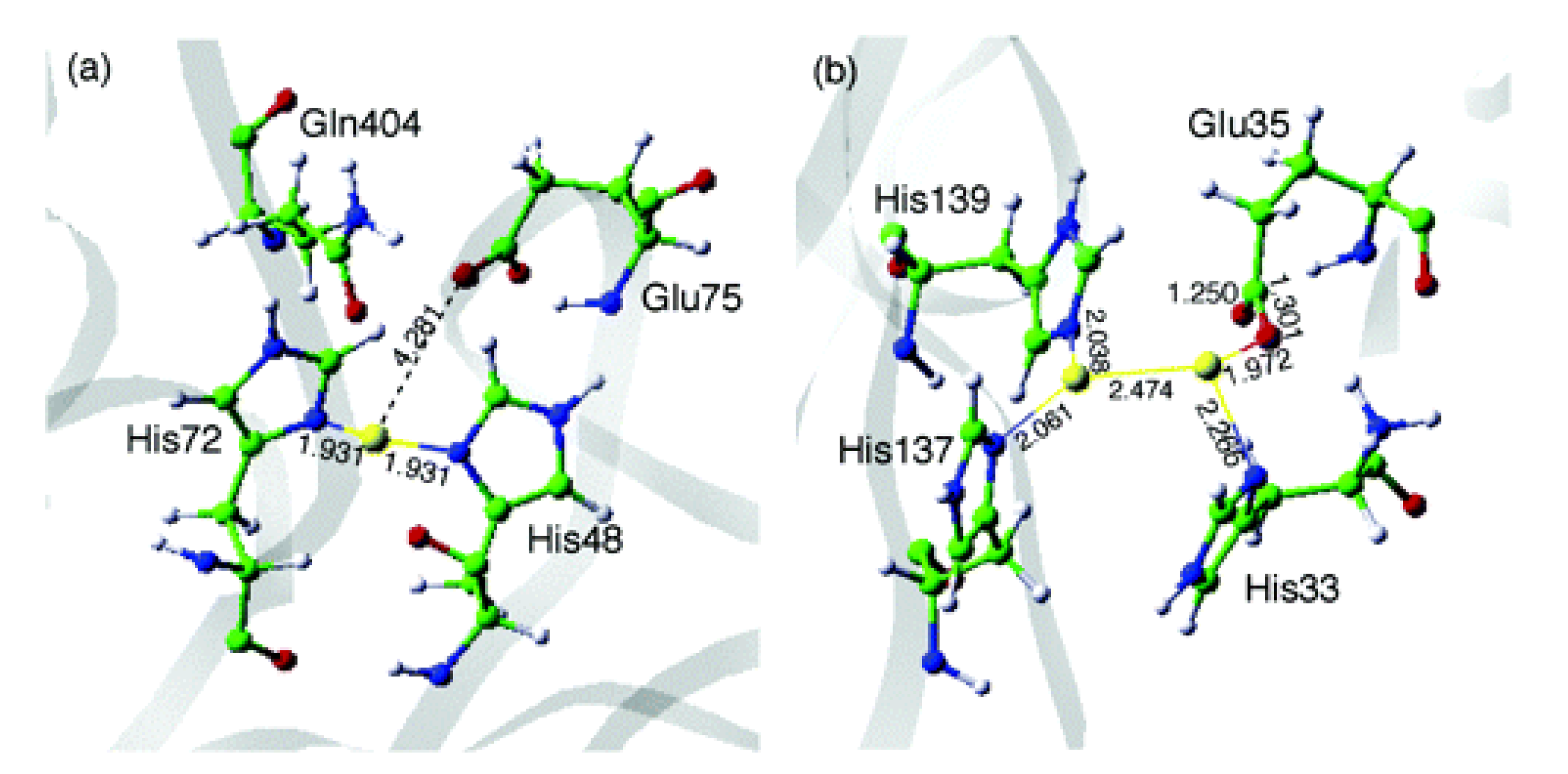
Figure 8.
Energy diagram for the MTM conversion over a proposed pMMO dimeric copper active site (energy units are expressed in kcal/mol). [Adapted with permission from Ref.[96]. Copyright 2006, American Chemical Society].
Figure 8.
Energy diagram for the MTM conversion over a proposed pMMO dimeric copper active site (energy units are expressed in kcal/mol). [Adapted with permission from Ref.[96]. Copyright 2006, American Chemical Society].
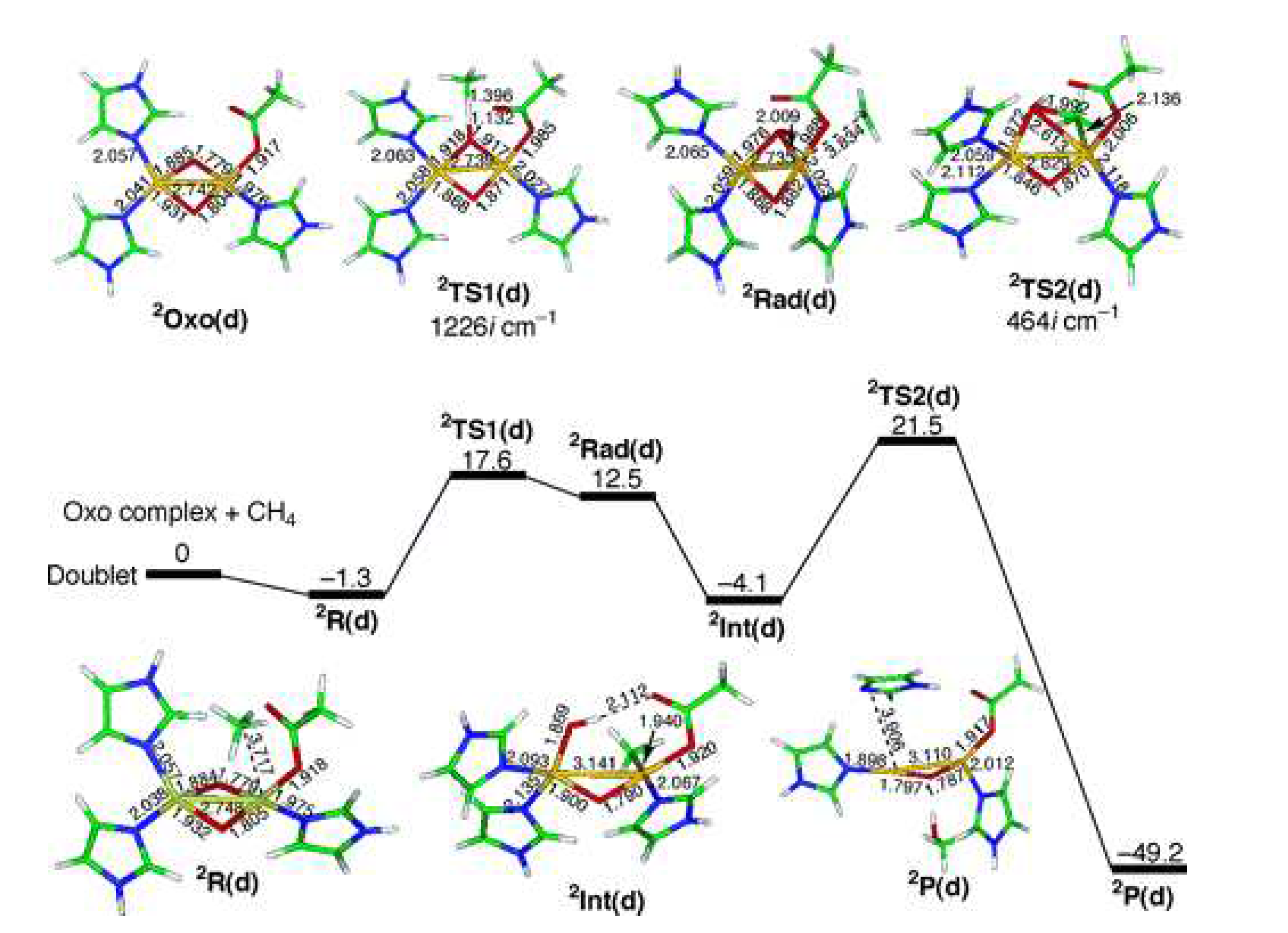
Figure 9.
Synthesis of bioinspired MOF-808-supported Cu catalysts. (a) MOF-808 structure. (b) Pseudohexagonal pore opening of MOF-808. (c) Replacement of formate groups with imidazole-containing ligands and subsequent metalation with Cu(I). [Adapted with permission from Ref.[76]. Copyright 2018, American Chemical Society].
Figure 9.
Synthesis of bioinspired MOF-808-supported Cu catalysts. (a) MOF-808 structure. (b) Pseudohexagonal pore opening of MOF-808. (c) Replacement of formate groups with imidazole-containing ligands and subsequent metalation with Cu(I). [Adapted with permission from Ref.[76]. Copyright 2018, American Chemical Society].
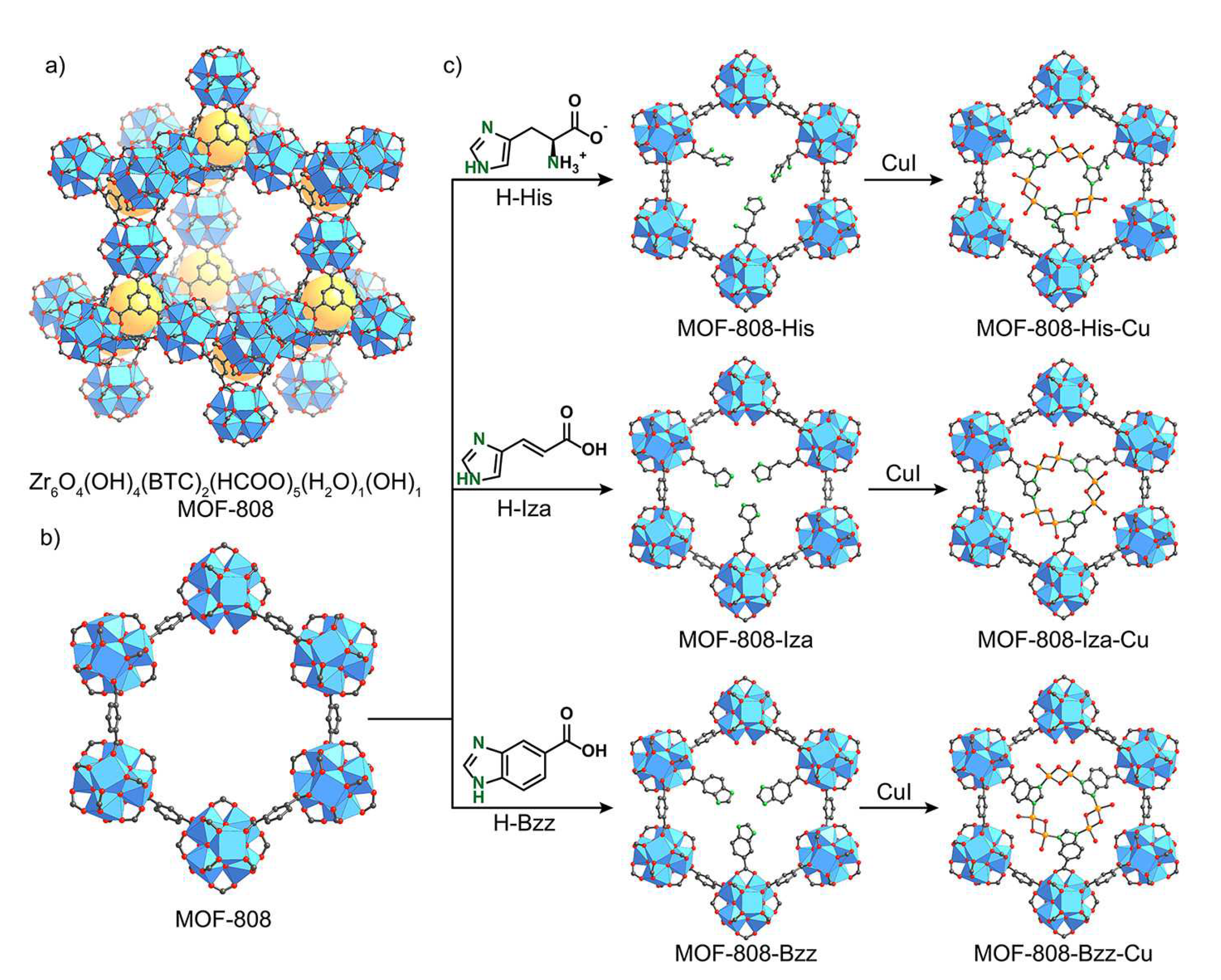
Figure 10.
Depiction of the NU-1000 (a) hexa-Zr nodes and (b) tetratopic organic linkers, as well as of the (c) MOF 3 nm-wide hexagonal pores. [Reprinted with permission from Ref.[105]. Copyright 2020, American Chemical Society].
Figure 10.
Depiction of the NU-1000 (a) hexa-Zr nodes and (b) tetratopic organic linkers, as well as of the (c) MOF 3 nm-wide hexagonal pores. [Reprinted with permission from Ref.[105]. Copyright 2020, American Chemical Society].
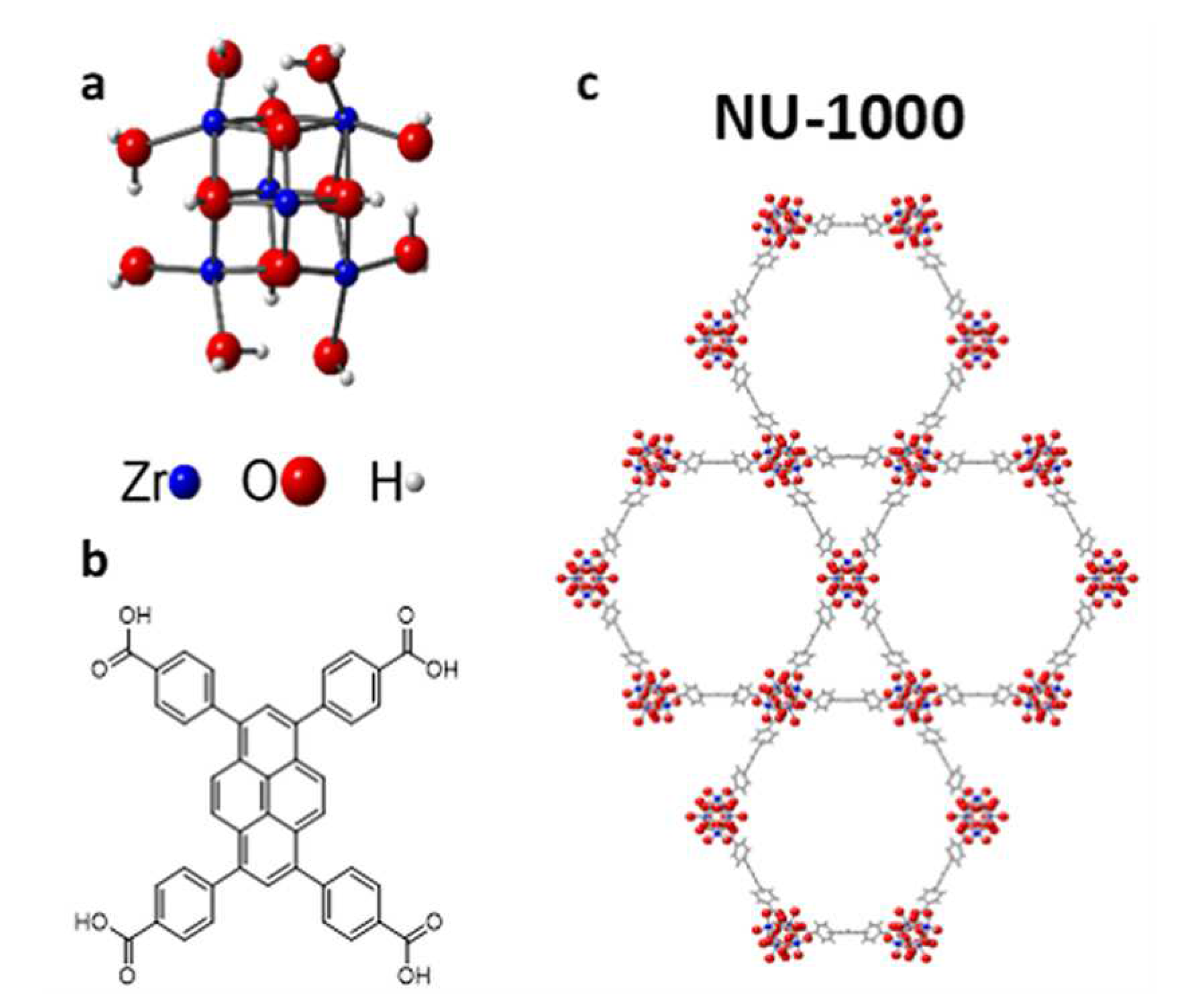
Figure 11.
Proposed DFT structures of the Cu-NU-1000 active sites, synthesized either by atomic layer deposition (a) or by metal exchange strategies (b). [(a) Reprinted with permission from Ref.[78]. Copyright 2017, American Chemical Society]. [(b) Reprinted with permission from Ref.[77]. Copyright 2019, Amerian Chemical Society].
Figure 11.
Proposed DFT structures of the Cu-NU-1000 active sites, synthesized either by atomic layer deposition (a) or by metal exchange strategies (b). [(a) Reprinted with permission from Ref.[78]. Copyright 2017, American Chemical Society]. [(b) Reprinted with permission from Ref.[77]. Copyright 2019, Amerian Chemical Society].
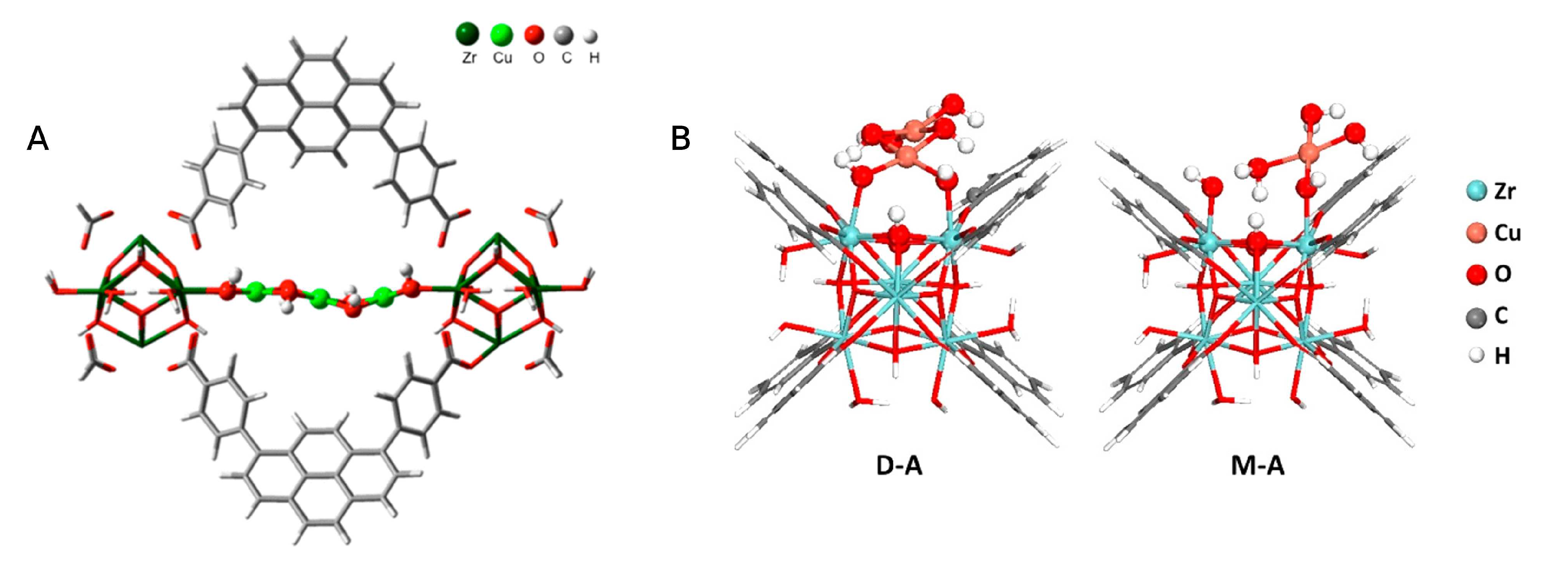
Figure 12.
(a) Mössbauer spectra of the HTS, ECS-HL and ECS-LL catalysts in zero field at -231.15 C (the experimental data are represented by black solid lines, and the corresponding fitted components are represented by colored lines). (b) Fourier transform of EXAFS Fe K-edge experimental (red) and fitted (black) data for the HTS-1, ECS-HL, and ECS-LL samples. [Adapted with permission from Ref.[86]. Copyright 2018, American Chemical Society].
Figure 12.
(a) Mössbauer spectra of the HTS, ECS-HL and ECS-LL catalysts in zero field at -231.15 C (the experimental data are represented by black solid lines, and the corresponding fitted components are represented by colored lines). (b) Fourier transform of EXAFS Fe K-edge experimental (red) and fitted (black) data for the HTS-1, ECS-HL, and ECS-LL samples. [Adapted with permission from Ref.[86]. Copyright 2018, American Chemical Society].
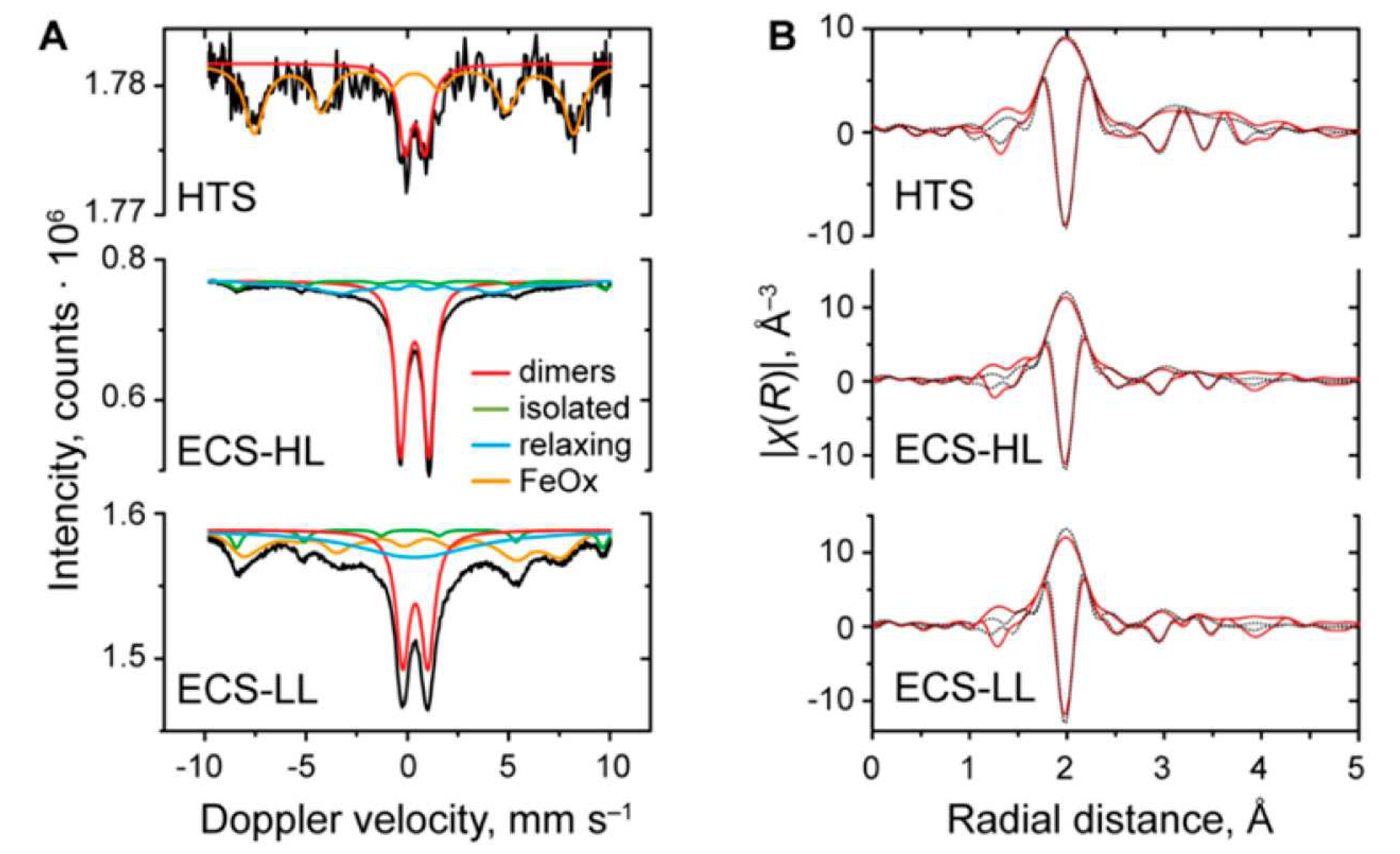
Figure 13.
Fe K-edge XANES (a) and EXAFS (b) spectra (k-weighted magnitude of the FT) characterizing PCN-250 in flowing He at 120 C (black curve), at 240 C (red curve), and after performing the MTM reaction with NO + CH at 120 C (blue curve). [Reprinted with permission from Ref.[73]. Copyright 2021, American Chemical Society].
Figure 13.
Fe K-edge XANES (a) and EXAFS (b) spectra (k-weighted magnitude of the FT) characterizing PCN-250 in flowing He at 120 C (black curve), at 240 C (red curve), and after performing the MTM reaction with NO + CH at 120 C (blue curve). [Reprinted with permission from Ref.[73]. Copyright 2021, American Chemical Society].
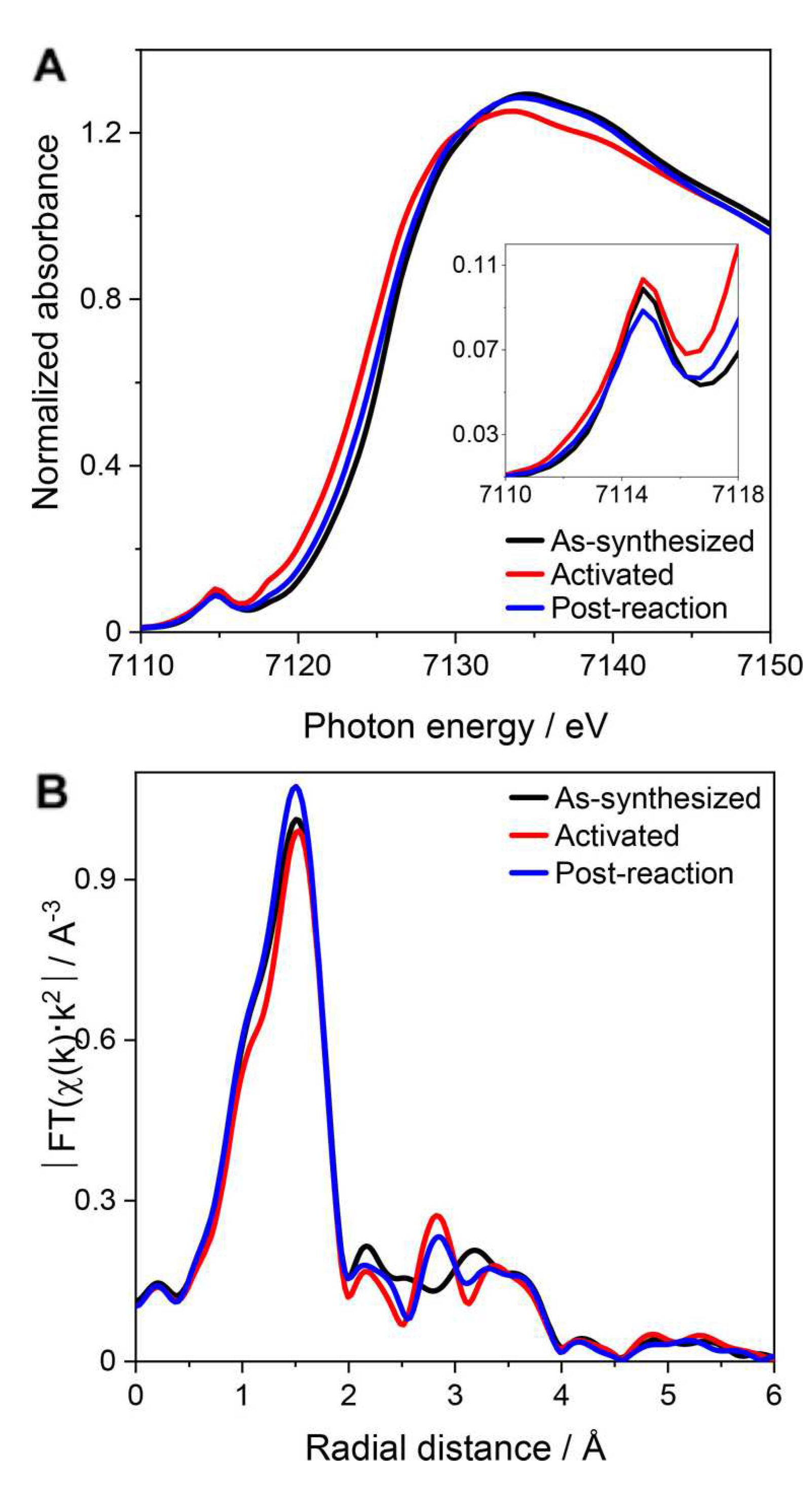
Figure 14.
(a–c) Normalized Cu-XANES spectra (a) and k-weighted Cu-EXAFS Img[(R)] spectra (c) of ALD-synthesized Cu-NU-1000 and of reference materials. In panel (c), arrows indicate the spectral regions most affected by Cu-O single scattering (SS) and multiple scattering (MS), as well as by Cu-Cu SS. (b–d) Normalized Cu-XANES spectra of Cu-NU-1000 during activation in oxygen (b) and methane loading (d) at 150 C. In panels (a), (b) and (d) the insets evidence the pre-edge feature at 8977-8978 eV due to the 1s→3d electronic transition for the distorted symmetry of Cu(II) as well as the 1s→4p transitions for Cu(I) and Cu(II) at 8982-8984 and 8985.5 eV, respectively. [Adapted with permission from Ref.[78]. Copyright 2017, American Chemical Society].
Figure 14.
(a–c) Normalized Cu-XANES spectra (a) and k-weighted Cu-EXAFS Img[(R)] spectra (c) of ALD-synthesized Cu-NU-1000 and of reference materials. In panel (c), arrows indicate the spectral regions most affected by Cu-O single scattering (SS) and multiple scattering (MS), as well as by Cu-Cu SS. (b–d) Normalized Cu-XANES spectra of Cu-NU-1000 during activation in oxygen (b) and methane loading (d) at 150 C. In panels (a), (b) and (d) the insets evidence the pre-edge feature at 8977-8978 eV due to the 1s→3d electronic transition for the distorted symmetry of Cu(II) as well as the 1s→4p transitions for Cu(I) and Cu(II) at 8982-8984 and 8985.5 eV, respectively. [Adapted with permission from Ref.[78]. Copyright 2017, American Chemical Society].
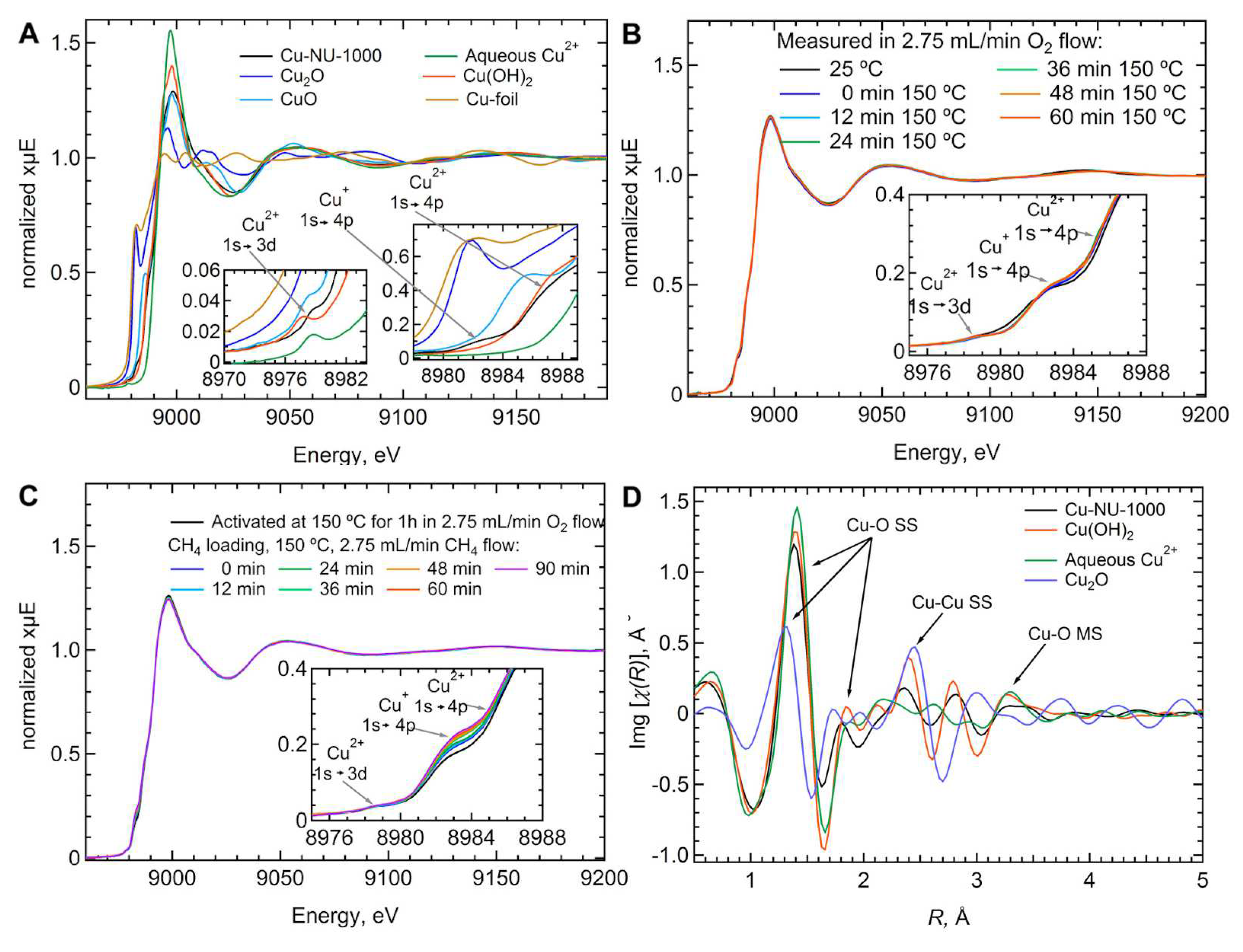
Figure 15.
(a) Average CHOH productivity of MOF-808-His-Cu, MOF-808-Iza-Cu and MOF-808-Bzz-Cu. (b) Ex situ Cu K–edge XANES spectra of MOF-808-Bzz-Cu collected after performing the reactions with He, 3% NO/He, CH, and 3% steam/He. (c) Ex situ N K–edge XANES spectra of MOF-808-Bzz, as-synthesized MOF-808-Bzz-Cu, and MOF-808-Bzz-Cu after performing the reactions with He, 3% NO/He, CH, and 3% steam/He. (d) Resonance Raman spectra of MOF-808-Bzz-Cu synthesized using O and O (the wavelength of the laser was 407 nm). [Reprinted with permission from Ref.[76]. Copyright 2018, American Chemical Society]
Figure 15.
(a) Average CHOH productivity of MOF-808-His-Cu, MOF-808-Iza-Cu and MOF-808-Bzz-Cu. (b) Ex situ Cu K–edge XANES spectra of MOF-808-Bzz-Cu collected after performing the reactions with He, 3% NO/He, CH, and 3% steam/He. (c) Ex situ N K–edge XANES spectra of MOF-808-Bzz, as-synthesized MOF-808-Bzz-Cu, and MOF-808-Bzz-Cu after performing the reactions with He, 3% NO/He, CH, and 3% steam/He. (d) Resonance Raman spectra of MOF-808-Bzz-Cu synthesized using O and O (the wavelength of the laser was 407 nm). [Reprinted with permission from Ref.[76]. Copyright 2018, American Chemical Society]
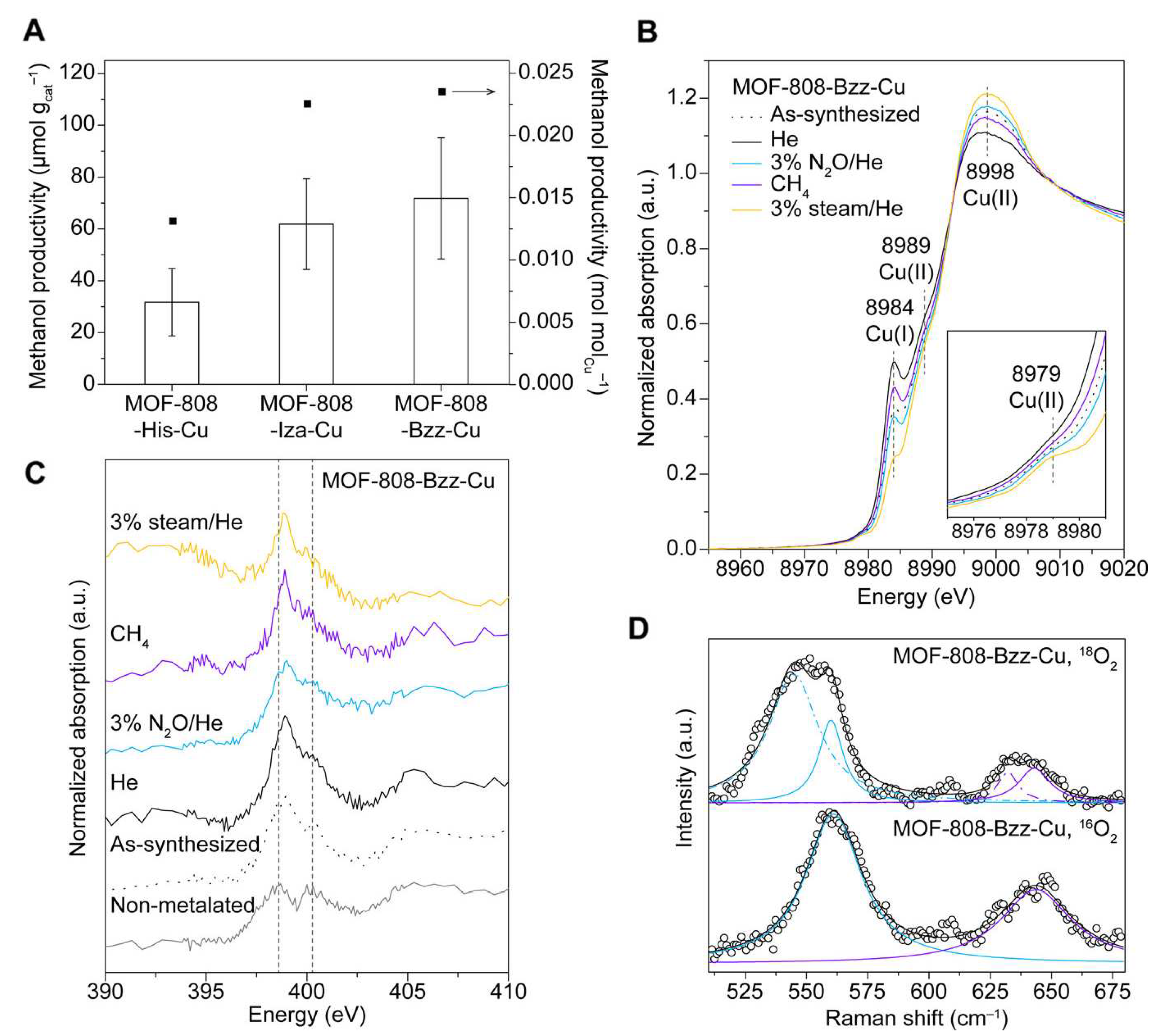

Table 1.
Summary of the MTM conversion performance for the Fe-containing MOFs illustrated in Section 3. *depending on Fe wt%
Table 1.
Summary of the MTM conversion performance for the Fe-containing MOFs illustrated in Section 3. *depending on Fe wt%
| MOF | Active site type | Oxidant | Methanol yield | Methanol selectivity | Reference |
|---|---|---|---|---|---|
| MIL-53(Al,Fe) | FeO SBU | HO | ∼10-30 molg* | ∼10-40* | [86] |
| MIL-100(Fe) | Fe--oxo trimer | NO | 0.34 mol(mol Fe) | ∼99% at 453 K | [34] |
| PCN-250 | Fe--oxo trimer | NO | 200 molg | 70% | [73] |
| PMOF-RuFe(OH) | mono-iron hydroxyl | O | 8.81±0.34 mmolgh | 100% | [75] |
| Fe-UiO-66 | mono-iron hydroxyl | O | 0.167 molgs | 62% | [91] |
Table 2.
Summary of the MTM conversion performance for the Cu-containing MOFs illustrated in Section 4.
Table 2.
Summary of the MTM conversion performance for the Cu-containing MOFs illustrated in Section 4.
| MOF | Active site type | Oxidant | Methanol yield (molg | Methanol selectivity | Reference |
|---|---|---|---|---|---|
| MOF-808-Bzz-Cu | histidine-supported Cu-oxo dimer | NO | 71.8 ± 23.4 | 100% | [76] |
| Cu-NU-1000 | Cu-oxo cluster | O | 17.7 | 45% (including DME) | [78] |
| Cu-2.9-NU-1000 | Cu-oxo dimer | O | 4.4 | 70% | [77] |
| Cu-UiO-bpy | CuO clusters | O | 24.3 | 88% | [108] |
| Cu-ZIF-7 | ligand-supported tetrahedral Cu | HO | 48 | 8% | [110] |
Table 3.
Comparison of different kinds of spectroscopic techniques useful for investigations in heterogeneous catalysis.
Table 3.
Comparison of different kinds of spectroscopic techniques useful for investigations in heterogeneous catalysis.
| Information mainly provided on: | |||||
|---|---|---|---|---|---|
| Spectroscopy | Energy (eV) | Kind of transition | Solid surface | Bond between adsorbed molecule and surface | Adsorbed molecule structure |
| Infrared | (50–2.5) × 10 | Vibrational | N | Y | Y |
| Raman | (50–0.6) × 10 | Vibrational | N | Y | Y |
| Visible/near infrared | 0.5–6.5 | Electronic, vibrational | Y | Y | Y |
| Mössbauer | 10–10 | Nuclear | Y | N | N |
| EPR | (14–3.8) × 10 | Nuclear spin | Y | N | Y |
| NMR | (2.5–8.3) × 10 | Nuclear spin | Y | Y | Y |
| XPS | (0.1–1500) | Bound electron to the continuum | Y | Y | Y |
| XAS | (0.1–100) × 10 | Bound electron to the continuum | Y | Y | Y |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



