
Submitted:
01 September 2023
Posted:
04 September 2023
You are already at the latest version
Abstract
The synthesis, anticancer and antioxidant activity of a series of indole-derived hybrid chalcones are reported here. First, using well known Claisen-Schmidt condensation method a set of 29 chalcones has been designed, synthesized, and consequently characterized. Subsequently, screening for the antiproliferative activity of the synthesized hybrid chalcones was performed on the on five cancer (HCT116, HeLa, Jurkat, MDA-MB-231 and MCF7) and two non-cancer (MCF-10A and Bj-5ta) cell lines. Chalcone 18c bearing 1-methoxyindole and catechol structural features, exhibited selective activity against cancer cell lines with IC50 values of 8.0 ± 1.4 µM (Jurkat) and 18.2 ± 2.9 µM (HCT116) and showed no toxicity to non-cancer cells. Furthermore, antioxidant activity was evaluated using tree different methods. The in vitro studies of radical scavenging activity utilizing DPPH radicals, as well as FRAP method, demonstrated the strong activity of catechol derivatives 18a-c. According to the ABTS radical scavenging assay, the 3-methoxy-4-hydroxy substituted chalcones 19a–c were slightly more favorable. In general, a series of 3,4-dihydroxychalcone derivatives showed properties as a lead compound for both antioxidant and anticancer activity.
Keywords:
indole chalcone
; antiproliferative activity
; antioxidant activity
; 2-fluoro substitution
; hydroxy substitution
1. Introduction
Chalcones are a group of naturally occurring compounds with diverse biological effects and serve as promising starting point for drug design. They possess the structure of 1,3-diphenylprop-2-en-1-one (1) and are open-chain precursors for the biosynthesis of flavonoids and isoflavonoids, which are predominantly as polyphenolic compounds. Chalcone-containing plants have been extensively utilized in traditional medicine. Additionally, they also serve as lead compounds in the development of new drugs [1], with some of them already undergoing clinical trials or being in use as drugs [2].
Replacing phenyl in the structure of chalcones with various heterocycles, numerous hybrid chalcones with significant biological effects have been obtained [3]. Indole, a potent pharmacodynamic nucleus found in various natural products such as signal molecules (neurotransmitter serotonin, auxin indole-3-acetic acid, indole phytoalexins) as well as many drugs (sunitinib, indomethacin, vincristine, panobinostat) [4] proved to be a suitable heterocycle in the development of hybrid chalcones. One important indole chalcone is MOMIPP (2), which effectively reduces the growth and viability of temozolomide-resistant glioblastoma and doxorubicin-resistant breast cancer cells at low micromolar concentrations. This compound was found to act by a non-apoptotic mechanism of cell death - metuosis and may serve as a prototype drug against cancer that is resistant to common forms of cell death (for example, apoptosis) [5]. Later, PIKFYVE, a class III phosphoinositide (PI) kinase, was identified as the protein target responsible for the activity, allowing a better understanding of the mechanism of metuose-inducing chalcones [6].
In our recent study among the 19 investigated chalcones with 1-methoxyindole and 2-alkoxyindole skeleton, four inhibit the proliferation of colorectal cancer cells HCT-116 with IC50 values <8 μM and display low cytotoxicity to fibroblast cell line 3T3 and most of them showed activity against human leukemic T cell lymphoma Jurkat with IC50 below 15 μM. The study also demonstrated the binding interaction of selected chalcones with CT DNA and BSA [7]. The chalcone L1 (3) was subsequently studied [8] in a human cervical carcinoma (HeLa) cell model. The role of L1 as an effective agent with antiproliferative activity supported by G2/M cell cycle arrest and apoptosis has been demonstrated. Moreover, it has been proved that L1 is involved in modulating Transforming Growth Factor-β1 (TGF-β) signal transduction through Smad proteins and it also modulates other signaling pathways including Akt, JNK, p38 MAPK, and Erk1/2. The involvement of L1 in epithelial-to-mesenchymal transition was demonstrated by the regulation of N-cadherin, E-cadherin, and MMP-9 levels. The potential role of the chalcone L1 as a modulator of the anti-tumor microenvironment was also suggested [8].
Our previous study clearly demonstrated the effectiveness of fluorinated derivatives [7]. Consequently, we are committed to developing and exploring new fluorinated derivatives of chalcones with an indole core. Our determination is supported by the fact that over last thirty years, fluoro-pharmaceuticals make up 20% of all approved medicines [9]. Many reviews have been published that analyzed the database of fluorinated drugs to provide reliable insights into drug discovery [9,10,11]. In recent years, while the retreat of small molecule drugs has been observed at the expense of the rising star of biologics, the number of fluorine-containing drugs remains comparable to biologics. The number of fluorinated pharmaceuticals is expected to increase in the future in parallel with advances in fluorinated functionalization methodologies. The largest group of fluorinated drugs are drugs containing Ar-F (45%), and the most common are drugs with monofluorinated groups (67%)[9]. The high prevalence of fluorinated drugs can be attributed to several factors [9] influencing the absorption, distribution, metabolism and excretion (ADME) of candidate drugs: i) the smallest atom (H) can be replaced by the second smallest atom (F) without causing drastic changes in the parent structure; ii) the C-F bond, one of the strongest carbon bonds, often enhances the metabolic stability of fluorinated drugs; iii) fluorine, being the most electronegative element (3.98), induces bond polarization, thereby altering the lipophilicity/hydrophilicity balance of the compound and consequently changing the pKa values compared to the parent molecules. iv), additionally, fluorine acts as a weak hydrogen bond acceptor and can serve as a bioisoster of the hydroxyl group (OH)[9]. Halogenation is a commonly employed and successful method of derivatizing chalcones in the development of prototype drug structures. A series of 2-fluoro, 4-fluoro, and 2,5-difluoro-substituted chalcone derivatives were synthesized and evaluated against five cancer cell lines (IC50 in the range of 0.029–0.729 µM), with compound 4 identified as the most promising compound towards the development of new therapeutic agents, specifically for the treatment of kidney cancer in humans [12]. In another study, 2-fluoro-4'-aminochalcone 5 was among the most active compounds investigated and induced apoptosis rather than necrosis in both cells, increasing p53 expression in ER-positive cells (MCF-7 line)[13].
As mentioned earlier, chalcones are natural substances widely distributed in vegetables, tea and other plants. Hydroxyl and methoxy function are the groups most often used by nature in the formation of chalcones [14]. Significant findings have been revealed in the latest examination of drugs that contain hydroxyl groups, making up a substantial 37% of all medications [15]. Natural products make up a quarter of all medicines sold, while the majority are of synthetic origin. Up to 69% of natural products contain hydroxyl functions, while for synthetics it is only 23%. It is common for natural products to exhibit multiple hydroxyl functions, while most synthetic drugs contain at most one OH group. It was found that the probability drug containing hydroxyl functions is much higher for drugs derived from natural products compared to synthetic drugs origin. Many natural chalcones bearing hydroxyl groups have a wide spectrum of biological activities [16]. Licochalcon A (6), isolated from the root of Glycyrrhiza glabra (liquorice) or Glycyrrhiza inflata, exhibits prominent anticancer effects and has also been found to inhibit the efflux of antineoplastic drugs from cancer cells [16]. Its potential anticancer properties have been shown in various types of cancer cells such as gastric, ovarian, breast, glioma, bladder, liver cancer cells [17]. Studies show that licochalcone A (6) induces apoptosis of U87 glioma cells, nasopharyngeal cancer cells, epithelial ovarian carcinoma cells, and bladder cancer cells.
The 2,4,3´,4´-tetrahydroxychalcone, butein (8) inhibits the growth of different breast cancer cells but the molecular mechanisms of butein-induced apoptosis remain unclear. While butein has been reported to induce ROS generation in triple-negative MDA-MB- 231 breast cancer cells [18], other study has described reduction of ROS level leading to apoptosis, while not affecting butein-resistant HER2+ (HCC-1419, SKBR-3, and HCC-2218) cells [19]. Naturally occurring polyhydroxychalcones as brossochalcone A, butein (8) and xanthohumol (7), the main prenylchalcone in hops and beer, appear to be even more potent antioxidants then α-tocopherol [20,21,22].
In a constant attempt to identify potential candidates with antitumor properties, indole-hybrid chalcones reported by our research group have been successfully synthesized in our laboratory [7,8,23]. In continuation of this effort in the existing research, new indole chalcones were designed, synthesized and investigated for antiproliferative as well as antioxidant activity. The indole core is preserved in all compounds as an active pharmacophoric fragment and benzene ring is substituted with fluoro or hydroxy group (Figure 1).
2. Results and Discussion
2.1. Chemistry
The investigated chalcones 11-14 and 17-19 were synthesized using either base (50% aq. KOH or piperidine) or acid catalyzed (SOCl2 in anhydrous ethanol) Claisen–Schmidt reactions according to Scheme 1. The 1,3-diarylpropenones with a 3-indol-3-yl moiety 11a-c – 14a-c, were prepared by condensation of 1-H (9a), 1-methyl (9b [24]) or 1-methoxy (9c) substituted indole-3-carboxaldehyde [25,26] with equimolar amounts of different acetophenones (2-fluoro- (10a)/4-trifluoromethyl- (10b)/2-hydroxy- (10c) or 4-hydroxy- (10d)), Scheme 1. When the reaction time was extended to 24h during the KOH catalysed condensation of 1-methoxyindole-3-carboxaldehyde (9c) with 2-fluoroacetophenone 10a, in addition to the condensation reaction, a nucleophilic substitution occurred on the indole nucleus to position 2, resulting in the formation of 2-alkoxychalcones 11d-k. The binding of the alkoxy group (depending on the solvent used) occurred with the simultaneous cleavage of the methoxy group from the indole nitrogen. This nucleophilic substitution was previously described by Somei [27], and we recently employed it in the synthesis of similar 2-alkoxyindol-3-yl-4-fluorophenylprop-2-en-1-ones [7].
The reaction of 1H (15a), 1-methyl (15b [28]) or 1-methoxy-3-acetylindole (15c [29]) with 4-hydroxybenzaldehyde (16a), 3,4-dihydroxybenzaldehyde (16b) or vanillin (16c) resulted in formation of 1-(indol-3-yl)-1,3-diarylpropenones 17a-c, 18a-c and 19a-c with yields ranging from 55 to 81% (Scheme 2).
The desired 1,3-diarylpropenones were isolated, purified and characterized using NMR, IR, and HR-MS. The 1H NMR spectra exhibited two diagnostic doublet signals corresponding to H-2 and H-3 protons of 1,3-diarylpropenones, resonating between chemical shift values of 7.02–7.82 ppm and 7.84–8.21 ppm (for 3-(indol-3-yl)prop-2-en-1-ones) and within a narrow range of 7.47–7.64 ppm for both signals (for 1-(indol-3-yl)prop-2-en-1-ones). The coupling constant, J, for these doublets, ranged from 15.2 to 15.8 Hz, Table 1. Such large coupling constant values indicate the trans geometry at the double bond of the propenone linkage. For compounds 17b,c; 18c and 19b,c, a strong splitting was observed, where Δν/J is less than one. To determine the correct chemical shift positions for peaks H-2 and H-3, we calculated the weighted average of the positions of the two lines, weighted by the intensities of the two lines [30]. Even in the case of compound 18b, we observed that the H-2 and H-3 protons are isochronous and the interaction constants could not be determined. The 13C NMR spectra of prepared chalcones displayed signals at the farthest downfield ranging from 193.1 to 183.1 ppm, assignable to C1 of propenone linkage. The carbonyl atom with 2-hydroxyphenyl substitution (chalcones 13a-c) was shifted downfield to 193.1-193.0 ppm, and C-1 with indol-3-yl substitution (chalcones 17-19) was shifted upfield to 183.1-183.8 ppm. Signals for C-1, C-2 and C-3 were detected as doublets with JC,F in the range of 2.3-0.9 Hz for 2-fluorophenyl chalcones 11a-k. Complete assignment of all 1H and 13C resonances was performed using 2D NMR experiments. 1H and 13C NMR spectra of synthesized compounds are presented in Supplementary materials: Figures S1–S52.
2.2. Antiproliferative Activity
Screening for antiproliferative activity of the synthesized hybrid chalcones was performed on the HCT116 (human colorectal carcinoma), HeLa (human cervical adenocarcinoma), Jurkat (human acute T-lymphoblastic leukemia), MDA-MB-231 (human mammary gland adenocarcinoma) and MCF7 (human breast adenocarcinoma) cell lines. Compounds were evaluated for selective cytotoxicity against cancer cells compared to normal cells, utilizing two cell lines: MCF-10A (human mammary epithelial cells) and Bj-5ta (immortalized foreskin fibroblasts). The IC50 values are listed in Table 2. Values less than 10 μM are typed in bold. Each value represents the mean ± SD of three independent experiments.
The results indicate that the 2-fluoroderivatives 11a-k exhibit moderate activity against cancer cells, with the highest efficacy observed against Jurkat leukemic cells. Chalcones having N-H, N-methoxy and 2-ethoxy substituents on the indole moiety 11a, 11c and 11e exhibited an activity of less than 8.3 μM on this cell line. Chalcone with 2-propoxy group 11f significantly suppressed the proliferation of breast cancer cells (34 and 37.3 μM for MDA-MB- 231 and MCF-7) with minimal effect on non-cancer mammary epithelial cells MCF-10A. The mode of cell death was recently evaluated by our group [32] using flow cytometry, Western blot, and fluorescence microscopy. Chalcone 11f was found to induce cell cycle arrest in the G2/M phase and apoptosis. This was associated with the release of cytochrome c, increased activity of caspase 3 and caspase 7, PARP cleavage, decreased mitochondrial membrane potential, and the activation of the DNA damage response system. Chalcone 11f was shown to initiate autophagy as a defence mechanism in treated cells trying to escape the harmful effects induced by the chalcone [32].
Among the 4-trifluoromethyl chalcones 12a-c, selective activity against HCT116 and MCF-7 cells was observed, with an IC50 of 12.3 and 20.5 μM for the N-methyl derivative 12b, and 59.1 and 43.4 μM for unsubstituted derivative 12a. Minimal activity was observed against non-cancer cells MCF-10A and Bj-5ta (> 100 μM). Such desired selectivity was not observed for the compound with a 1-methoxyindole core 12c, even on non-cancer MCF-10A cells a higher toxicity was observed than on cancer cells. In a recent study, Lagu et al [19] synthesized and evaluated the antibacterial and antifungal activity of fluorinated chalcones bearing trifluoromethyl and trifluoromethoxy substituents. Compounds bearing the indole ring 12a were found to exhibit the greatest antimicrobial activity compared to standard drugs without showing cytotoxicity on a normal human liver cell line (L02). In recent years, the trend has been to develop multi-targeted drugs to fight cancer and microbial infections commonly seen in immunocompromised patients undergoing chemotherapy. Compounds containing a trifluoromethylphenyl group are undoubtedly an excellent choice for designing multi-targeted drugs.
Another evaluated group are hydroxylated chalcones 13-14a-c, 17-19a-c. The results of the antiproliferative activity of the investigated compounds align with the findings that synthetic compounds based on natural substances have a higher likelihood of succeeding as drugs [15]. Natural chalcones often contain a hydroxy group in position 4, as well as 3,4-dihydroxy substitution is relatively common in chalcones. On the Jurkat cell line, 4-hydroxyphenyl and 3,4-dihydroxyphenyl chalcones 14c and 18c, both bearing a methoxy group on the indole nitrogen, exhibited IC50 values of 7.3 and 8.0 µM, proving to be the most effective. In addition, chalcone 18c also demonstrated activity against HCT116 with an IC50 of 18.5 µM and showed no toxicity against non-cancer cells at the investigated concentrations. Plants from the family Cruciferae (Brassicaceace) produce natural antimicrobial substances called indole phytoalexins, often with the methoxy group attached to the nitrogen in the indole nucleus. In addition to antimicrobial activity, antiproliferative and chemopreventive activity have also been demonstrated for these compounds [33]. The suitability of the methoxy group as a substituent on indole nitrogen for the development of indole hybrid chalcones with antiproliferative effect was demonstrated in our previous studies [7,8]. Most hydroxylated chalcones evaluated exhibited moderate activity and lacked selectivity. Some, including 14b, 17a, 17b, 18a, and 19a-c, were more toxic to non-cancer cells than to cancer cells.
2.3. Antioxidant Activity
In addition to anticancer activity, series of indole-chalcone hybrids were tested for antioxidant activity. Chalcones are precursors for flavonoids and isoflavonoids with well known antioxidant properties. Also chalcones and chalcones derivatives, especially, hydroxychalcones, have showed high capacity for the scavenging of free radicals [34,35]. Within this study, as seen in Table 3, the set of synthetized hybrid chalcones were assessed for antioxidant activity using tree different in vitro methods, namely DPPH (2,2-diphenyl-1-picrylhydrazyl) radical-scavenging, ABTS (2,2´-azinobis-(3- ethylbenzothiazoline-6-sulfonic acid) radical-scavenging, and ferric reducing antioxidant power assay (FRAP). In addition to chalcones, the activities of natural polyphenols caffeic acid and p-coumaric acid were also determined in same concentration of 1 mM and results expressed as µmol equivalent of gallic acid per mmol sample. Generally, in vitro antioxidant assays are relatively simple to perform, but the reaction mechanism is complex and depends on several factors [36,37]. In the case of FRAP, the mechanism is due to the single electron transfer (SET), but the ABTS and DPPH methods its assumed to be mixed SET and hydrogen atom transfer (HAT) mechanism [36,38].
DPPH free radical scavenging is simply and an accepted mechanism for screening the antioxidant activity. According to the DPPH assay, the synthesized chalcones exhibited activity ranging from weak to strong. The series of synthesized compounds 11a-c, 13a-c, 14a-c, and 17a-c showed only mild scavenging activity against DPPH radical. The most potent structure was series of compound 18a-c with activity ranging from 520.1 ± 6.9 to 589.1 ± 8.9 µmol GAE/mmol. This series almost reached the activity of caffeic acid (690.7 ± 36.6 GAE/mmol) and represents about half of that of gallic acid.
The ABTS test proved to be more sensitive also for the compound 11a-c, 13 a-c, 14 a-c, and 17 a-c. Additionally, there was not such a significant difference in the activities between individual series of chalcones and almost all revealed considerable reducing ability. On the other hand, chalcones series 19a-c with 4-hydroxy-3-methoxyphenyl function group showed slightly stronger ABTS radical scavenging than series 18a-c with 3,4-dihydroxyl moiety. The structural feature of the 4-hydroxy-3-methoxyphenyl unit is also found in natural curcumin and synthetic derivatives with potent antioxidant activity. [39]
Consequently, reducing power according to FRAP assay was also used to measure the total antioxidant power of newly synthesized hybrid chalcones. In this method, the first set of synthesized compounds 11a-c, 13a-c, 14a-c, and 17a-c showed poor to moderate reducing activity similar to the DPPH test. The resting set was more effective, where the best reducing ability carried compound 18a-c with comparable activity to caffeic acid followed by chalcones 19a-c.
Generally, it was observed that the free radicals' scavenging activity and ferric reducing capacity is mainly related to the number and position of hydroxyl substituents. The presence of two hydroxyl groups arranged in the catechol moiety of derivatives 18a-c revealed the main influence on the antioxidant activity. This finding is also demonstrated by the improved activity of caffeic acid-bearing 3,4-dihydroxy groups against p-coumaric acid with only one. Moreover, it is consistent with the literature, where cinnamic acid substituted in the aromatic ring with two hydroxy groups (i.e., caffeic acid) had a higher antioxidant capacity compared to monohydroxycinnamic acid (p-coumaric acid) measured in DPHH, ABTS, CUPRAC and FRAP assays [40]. Also, naturally occurring 3,4-dihydoxy chalcone broussochalcone A inhibited iron-induced lipid peroxidation, DPPH radical, and exerted potent inhibitory effects on NO production [41]. A possible mechanism starts with the abstraction of the H atom of the 4-hydroxy group by the DPPH radical, then an additional H atom at the 3-OH position is abstracted to form a quinone structure [20].
According to the published QSAR analysis of a series of 25 chalcones [42], the spatial, structural and lipophilic properties of the compounds were shown to determine their antioxidant properties. Among 1-H, 1-methyl- and 1-methoxy substituted indole derivatives, the activity was higher in favor of unsubstituted derivatives. The isomerism does not play as important a role as the number of substituents, because within isomeric chalcones 14a-c and 17a-c was no significant difference in activity. Also, the position of a hydroxyl group (2-OH or 4-OH) is not as important as the number of oxygen atoms in the molecule, since both the 2-alkoxy on the indole (11f) and the 3-methoxy on the benzene ring also contribute (19a-c) to the antioxidant activity. The effect of the fluorine atom can be analyzed by comparing compounds 11a-c and compound 13a-c where a minor decline in activity was reported by compounds 11a-c containing electron-withdrawing fluorine instead of the H-bond donor hydroxyl group. This finding was also observed in other series of synthetic chalcones [35].
Moreover, the correlations obtained quantitatively confirm the parallelism between the DPPH scavenging activity and reducing power (R=0,941).
3. Materials and Methods
3.1. Chemistry
3.1.1. General Method and Materials
All chemicals and solvents used in the synthesis were obtained commercially and were used without further purification, unless otherwise specified. Reactions were magnetically stirred and monitored by thin-layer chromatography (TLC). TLC was performed on TLC aluminium sheets Silica gel 60 F254 and visualized with UV fluorescence. Column chromatographic purification was performed using Silica 60A Particle size 40-63 micron (Davisil®) with the indicated eluent. Melting points were measured on digital melting point apparatus (Electrothermal) using open glass capillaries and are uncorrected.
NMR spectra were recorded on a VNMRS spectrometer (Varian) operating at 600 MHz for 1H and 150 MHz for 13C at 299.15 K. Chemical shifts (δ in ppm) are given from internal solvent, DMSO-d6.
The infrared spectra were recorded on IRAffinity-1 FTIR Spectrophotometer (Shimadzu) in the range 4000−500 cm-1 using the KBr method (1mg sample and 150 mg KBr) or with Avatar FT−IR 6700 spectrometer in the range 4000−400 cm-1 with 64 repetitions for a single spectrum using ATR (attenuated total reflectance) technique and obtained data were analyzed using Omnic 8.2.0.387 (2010) software.
High-resolution fragmentation spectra were obtained with the use of Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), according to the procedure mentioned in our previous publication [43]. Data processing: To verify the structure of the studied molecules (11a-k and 12a-c, 13a-c, 14a-c, 18c, and 19c) the precursor ions were exhaustively fragmented using different fragmentation techniques (CID, HCD) at multiple collision energies. The acquired collections of fragmentation spectra at each fragmentation level (MS2 to MS4) enabled the generation comprehensive spectral tree of information (see Figure S53 in Supplementary information). The measured data were manually processed with Mass Frontier™ 8.0 software (Thermo Scientific™ Bratislava, Slovakia) in the Curator module. This module uses advanced algorithms to detect incompatibility between the declared structure precursor and the product MSn fragmentation spectra. The fragments for each structure were obtained by using Mass Frontier™ 8.0 software in the Fragments and Mechanisms module. This module enables the prediction of in silico fragmentation and recommends a comprehensive fragmentation pathway based on a set of general ionization, fragmentation, and rearrangement rules. For the prediction of possible fragments, information from the HighChem Fragmentation Library™ with around 227,000 mechanisms was also used. This library is based on the collection of all available scientific journals for mass spectrometry. The Fragments and Mechanisms tool automatically generates fragments from a user-supplied molecular structure. If the in-silico generated fragments of a given compound agree with observed fragments, the ion peaks in the MSn spectra can be annotated. This curation process is performed for every spectrum every collision energy, every fragmentation type, every MSn level, and for each precursor ion. The obtained data was of a high-quality level. Because of this comprehensive data, which contained high-resolution MS/MS and multi-stage MSn spectra, acquired at various collision energies using different fragmentation techniques, and because the unequivocal structure was confirmed (sub-3-ppm mass errors), these compounds were added to a high-quality mzCloud™ commercially available spectral library (https://www.mzcloud.org). It is a highly valued spectral library, for the identification of small molecules using tandem mass spectrometry. The content of the mzCloud™ spectral library is primarily used to identify an unknown compound through a sub-structural search, where the experimental fragmentation spectrum is searched against the mzCloud™ mass spectral database. For prepared compounds, mzCloud™ ID are 11a (11251), 11b (11252), 11c (11232), 11d (11233), 11e (11234), 11f (11235), 11g (11248), 11 h (11249), 11i (11250), 11j (11253), 11k (11266), 12a (11211), 12b (11212), 12c (11210), 13a (11214), 13b (11215), 13c (11213), 14a (11197), 14b (11198), 14c (11196), 18c (11230) and 19c (11231).
3.1.2. General Procedure A) Acid-Catalyzed Claisen-Schmidt Condensation
To a solution of acetophenone or the corresponding acetylindole (1 mmol) and the appropriate arylaldehyde (1 mmol) in anhydrous ethanol (10 mL), molecular sieves were added, and then SOCl2 (2 mmol) was slowly added. The course of the reaction was monitored using TLC. After completion of the reaction, the molecular sieves were filtered out, and the reaction mixture was poured into water, followed by extraction with EtOAc. The organic layer was washed with brine, dried over Na2SO4, evaporated and the product was isolated through chromatography and crystallisation.
3.1.3. General Procedure B1) Base-Catalyzed Claisen-Schmidt Condensation (50% aq. KOH)
To a stirred solution of acetophenone or the corresponding acetylindole (0.5 mmol) in solvent (5 mL) 50% KOH in H2O (0.5 mL) was added, followed by the appropriate arylaldehyde (0.5 mmol). The reaction mixture was stirred at room temperature. After completion of the reaction, the reaction mixture was cooled to 0-10°C, acidified with 1M HCl to pH 4. Then, the precipitated product was filtered, washed with H2O and cold alcohol, dried and crystallized. When the product did not precipitate, the mixture was extracted with EtOAc, and the extract was washed with brine. After drying over anhydrous Na2SO4, the filtrate was evaporated to dryness, and the product was purified by column chromatography on SiO2 and then crystalized to yield the chalcones.
3.1.4. General Procedure B2) Base-Catalyzed Claisen-Schmidt Condensation (Piperidine)
Arylaldehyde (1 mmol) and acetophenone (1 mmol) were dissolved in anhydrous ethanol (5 mL), and piperidine (2 mmol) was added. The reaction mixture was heated at 60–70 °C, and the course of the reaction was monitored by TLC. After completion of the reaction, the mixture was cooled to room temperature. Then, the precipitated product was filtered, washed with ethanol, dried and either crystallized or chromatographed on SiO2.
3.1.5. Synthesis and Characterisation of Compounds 11 - 14, 17 - 19
(2E)-1-(2''-Fluorophenyl)-3-(1'H-indol-3'-yl)prop-2-en-1-one (11a)
Procedure A: 1.5 h, 23%; Procedure B2: 27 h, 15%; Rf 0.39 (hexane/acetone 2:1); yellow crystals; mp 103–105.8 °C (CH2Cl2/hexane); 1H NMR (600 MHz, DMSO-d6): δ 11.97 (s, 1H, NH), 8.08 (s, 1H, H-2'), 7.92 (dd, 1H, J 7.1, 1.2 Hz, H-4'), 7.91 (dd, 1H, J 15.7, 1.2 Hz, H-3), 7.75 (td, 1H, J 7.6, 1.7 Hz, H-6''), 7.63 (dddd, 1H, J 8.3, 7.2, 5.2, 1.8 Hz, H-4''), 7.51 – 7.48 (m, 1H, H-7'), 7.39 – 7.34 (m, 2H, H-3'', H-5''), 7.29 (dd, 1H, J 15.7, 2.3 Hz, H-2), 7.25 (td, 1H, J 7.4, 1.3 Hz, H-6'), 7.22 (td, 1H, J 7.4, 1.3 Hz, H-5'); 13C NMR (150 MHz, DMSO-d6): δ 188.2 (d, JCF 2.1 Hz, C-1), 160.0 (d, JCF 249.8 Hz, C-2''), 140.0 (C-3), 137.6 (C-7'a), 134.1 (C-2'), 133.4 (d, JCF 8.7 Hz, C-4''), 130.3 (d, JCF 3.0 Hz, C-6''), 127.8 (d, JCF 14.0 Hz, C-1''), 124.9 (C-3'a), 124.8 (d, JCF 3.2 Hz, C-5''), 122.9 (C-6'), 121.4 (C-5'), 120.1 (C-4'), 119.5 (d, JCF 4.9 Hz, C-2), 116.5 (d, JCF 22.6 Hz, C-3''), 112.6 (C-7'), 112.4 (C-3'); IR (KBr): νmax 3266, 3036, 1609, 1597, 1570, 1508, 1486, 1413, 1313, 1242, 1197, 1162, 1069, 831, 771, 745 cm−1; HRMS: m/z [M+H]+: 266.09757 for C17H13FNO (calcd. 266.09778).
(2E)-1-(2''-Fluorophenyl)-3-(1'-metyl-1'H-indol-3'-yl)prop-2-en-1-one (11b)
Procedure B1: EtOH, 27 h, 57%; Rf 0.49 (hexane/acetone 2:1); yellow crystals; mp 117–120 °C (CH2Cl2/hexane); 1H and 13C NMR data identical with literature [23]; IR (KBr): νmax 3104, 1646, 1609, 1585, 1560, 1528, 1448, 1375, 1285, 1078, 1025, 975, 776, 740 cm−1; HRMS: m/z [M+H]+: 280.11374 for C18H15FNO (calcd. 280.11322).
(2E)-1-(2''-Fluorophenyl)-3-(2'-methoxy-1'H-indol-3'-yl)prop-2-en-1-one (11d)
Procedure B1: MeOH, 24 h, 35%; Rf 0.48 (EtOAc/hexane 2:1); yellow crystals; mp 172–173 °C (acetone/hexane); 1H NMR (DMSO-d6, 600 MHz): δ 12.17 (s, 1H, NH), 7.84 (dd, 1H, J 15.4, 1.5 Hz, H-3), 7.70 (td, 1H, J 7.6, 1.8 Hz, H-6''), 7.66 (d, 1H, J 7.6 Hz, H-4'), 7,59 (dddd, 1H, J 8.3, 7.2, 5.2, 1.8 Hz, H-4''), 7.36 – 7.32 (m, 3H, H-7', H-5'', H-3''), 7.17 (td, 1H, J 7.5, 1.2 Hz, H-6'), 7.12 (td, 1H, J 7.7, 1.2 Hz, H-5'), 7.02 (dd, 2H, J 15.4, 2.2 Hz, H-2), 4.14 (s, 3H, OCH3); 13C (DMSO-d6, 150 MHz): δ 187.4 (d, JCF 2.1 Hz, C-1), 159.8 (d, JCF 249.0 Hz, C-2''), 157.7 (C-2'), 136.8 (C-3), 132.9 (d, JCF 8.5 Hz, C-4''), 132.1 (C-7'a), 130.2 (d, JCF 3.2 Hz, C-6''), 128.2 (d, JCF 14.5 Hz, C-1''), 125.1 (C-3'a), 124.7 (d, JCF 3.3 Hz, C-5''), 121.8 (C-5'), 121.2 (C-6'), 118.5 (C-4'), 116.4 (d, JCF 22.9 Hz, C-3''), 115.7 (d, JCF 5.0 Hz, C-2), 111.6 (C-7'), 93.4 (C-3'), 58.8 (OCH3); IR (KBr): νmax 3146, 2938, 1612, 1588, 1560, 1547, 1491, 1474, 1455, 1445, 1434, 1358, 1319, 1285, 1265, 1230, 1211, 1166, 1075, 828, 760 cm−1; HRMS: m/z [M+H]+: 296.10846 for C18H15 FNO2 (calcd. 296.10813).
(2E)-3-(2'-ethoxy-1'H-indol-3'-yl)-1-(2''-fluorophenyl)prop-2-en-1-one (11e)
Procedure B1: EtOH, 24 h at 60 °C, 31%; Rf 0.38 (hexane/acetone 2:1); yellow crystals; mp 170–171 °C (acetone/hexane); 1H (DMSO-d6, 600 MHz ): δ 12.11 (s, 1H, NH), 7.88 (dd, 1H, J 15.4, 1.5 Hz, H-3), 7.73 (td, 1H, J 7.6, 1.8 Hz, H-6''), 7.66 (d, 1H, J 7.6 Hz, H-4'), 7.59 (dddd, 1H, J 8.3, 7.2, 5.2, 1.8 Hz, H-4''), 7.36 – 7.32 (m, 3H, H-7', H-5'', H-3''), 7.16 (td, 1H, J 7.7, 1.2 Hz, H-6'), 7.11 (td, 1H, J 7.6, 1.2 Hz, H-5'), 7.07 (dd, 1H J 15.4, 2.3 Hz, H-2), 4.45 (q, 2H, J 7.0 Hz, CH2CH3), 1.44 (t, 3H, J 7.0, CH2CH3); 13C NMR (150 MHz, DMSO-d6): δ 187.1 (d, JCF 2.2 Hz, C-1), 160.0 (d, JCF 249.2 Hz, C-2''), 156.9 (C-2'), 136.8 (C-3), 133.0 (d, JCF 8.7 Hz, C-4''), 132.1 (C-7'a), 130.3 (d, JCF 3.2 Hz, C-6''), 128.2 (d, JCF 14.2 Hz, C-1''), 125.0 (C-3'a), 124.7 (d, JCF 3.3 Hz, C-5''), 121.8 (C-5'), 121.2 (C-6'), 118.4 (C-4'), 116.4 (d, JCF 23.0 Hz, C-3''), 115.6 (d, JCF 5.5 Hz, C-2), 111.5 (C-7'), 93.9 (C-3'), 67.6 (OCH2 CH3), 14.7 (OCH2CH3); IR (KBr): νmax 3179, 2984, 2938, 1611, 1543, 1486, 1386, 1345, 1284, 1262, 1227, 1125, 1099, 1075, 1055, 1008, 828, 774, 747 cm−1; HR-MS: m/z [M+H]+: 310.12396 for C19H17FNO2 (calcd. 310.12378).
(2E)-1-(2''-Fluorophenyl)-3-(2'-propoxy-1'H-indol-3'-yl)prop-2-en-1-one (11f)
Procedure B1: n-PrOH, 24 h, 33%; Rf 0.50 (hexane/acetone 2:1); orange crystals; mp 142–144 °C (acetone/hexane); 1H NMR (DMSO-d6, 600 MHz): δ 12.11 (s, 1H, NH), 7.88 (dd, 1H, J 15.4, 1.5 Hz, H-3), 7.71 (td, 1H, J 7.6, 1.8 Hz, H-6''), 7.66 (d, 1H, J 7.8 Hz, H-4'), 7.59 (tdd, 1H, J 8.1, 5.2, 1.8 Hz, H-4''), 7.35 – 7.32 (m, 3H, H-7', H-5'', H-3''), 7.16 (td, 1H, J 7.5, 1.1 Hz, H-6'), 7.11 (td, 1H, J 7.7, 1.1 Hz, H-5'), 7.06 (dd, 1H, J 15.4, 2.2 Hz, H-2), 4.36 (t, 2H, J 6.5 Hz, OCH2CH2CH3), 1.82 (qt, 2H, J 7.4, 6.5 Hz, OCH2CH2CH3), 1.01 (t, 3H, J 7.4 Hz, OCH2CH2CH3); 13C NMR (DMSO-d6, 150 MHz): δ 187.3 (d, JCF 2.2 Hz, C-1), 159.9 (d, JCF 249.1 Hz, C-2''), 156.9 (C-2'), 136.9 (C-3), 133.0 (d, JCF 8.7 Hz, C-4''), 132.0 (C-7'a), 130.3 (d, JCF 3.2 Hz, C-6''), 128.2 (d, JCF 14.4 Hz, C-1''), 125,0 (C-3'a), 124.7 (d, JCF 3.2 Hz, C-5''), 121.8 (C-5'), 121.2 (C-6'), 118.4 (C-4'), 116.4 (d, JCF 23.0 Hz, C-3''), 115.7 (d, JCF 5.5 Hz, C-2), 111.5 (C-7'), 93.8 (C-3'), 73.0 (OCH2CH2CH3), 22.1 (OCH2CH2CH3), 10.0 (OCH2CH2CH3). IR (KBr): νmax 1624, 1607, 1579, 1561, 1517, 1484, 1460, 1339, 1280, 1229, 1211, 1203, 1065 cm−1; HRMS: m/z [M+H]+: 324.13989 for C20H19FNO2 (calcd. 324.13943).
(2E)-1-(4''-Fluorophenyl)-3-(2'-isopropoxy-1'H-indol-3'-yl)prop-2-en-1-one (11g)
Procedure B1: iso-PrOH, 24 h, 19%; Rf 0.31 (hexane/acetone 2:1) and (hexane/EtOAc 2:1); orange-brown powder; mp 150–152 °C (acetone/hexane); 1H (DMSO-d6, 600 MHz): δ 12.05 (s, 1H, NH), 7.87 (dd, 1H, J 15.6, 1.5 Hz, H-3), 7.73 (td, 1H, J 7.6, 1.8 Hz, H-6''), 7.67 (d, 1H, J 7.8 Hz, H-4'), 7.60 (dddd, 1H, J 8.3, 7.3, 5.2, 1.8 Hz, H-4''), 7.36 – 7.32 (m, 3H, H-7', H-5'', H-3''), 7.16 (td, 1H, J 7.6, 1.1 Hz, H-6'), 7,12 (td, 1H, J 7.6, 1.1 Hz, H-5'), 7.09 (dd, 1H, J 15.6, 2.3 Hz, H-2), 4,91 (septet, 1H, J 6.1 Hz, OCH(CH3)2), 1.40 (d, 6H, J 6.0, OCH(CH3)2); 13C NMR (DMSO-d6, 150 MHz): δ 187.2 (d, JCF 2.1 Hz, C-1), 160.0 (d, JCF 249.1 Hz, C-2''), 155.9 (C-2'), 136.9 (C-3), 133.1 (d, JCF 8.6 Hz, C-4''), 132.2 (C-7'a), 130.3 (d, JCF 3.0 Hz, C-6''), 128.2 (d, JCF 14.6 Hz, C-1''), 124.8 (C-3'a), 124.7 (d, JCF 3.0 Hz, C-5''), 121.7 (C-5'), 121.4 (C-6'), 118.5 (C-4'), 116.4 (d, JCF 23.0 Hz, C-3''), 115.8 (d, JCF 6.0 Hz, C-2), 111.5 (C-7'), 95.2 (C-3'), 75.7 (OCH(CH3)2), 22.1 (OCH(CH3)2); IR (KBr): νmax 3179, 2984, 2938, 1611, 1543, 1486, 1386, 1345, 1284, 1262, 1227, 1125, 1099, 1075, 1055, 1008, 828, 774, 747 cm−1; HRMS: m/z [M+H]+: 324.13986 for C20H19FNO2 (calcd. 324.13943).
(2E)-3-(2'-butoxy-1'H-indol-3'-yl)-1-(2''-fluorophenyl)prop-2-en-1-one (11h)
Procedure B1: n-BuOH, 24 h, 33%; Rf 0.47 (hexane/acetone 2:1) and (hexane/EtOAc 2:1); dark-orange crystals; mp 141–143 °C (acetone/hexane); 1H (DMSO-d6, 600 MHz): δ 12.11 (s,1H, NH), 7,86 (dd, 1H, J 15.4, 1.5 Hz, H-3), 7.71 (td, 1H, J 7.5, 1.7 Hz, H-6''), 7.66 (d, 1H, J 7.6 Hz, H-4'), 7.59 (dddd, 1H, J 8.3, 7.2, 5.2, 1.8 Hz, H-4''), 7.35 – 7.31 (m, 3H, H-7', H-5'', H-3''), 7.16 (td, 1H, J 7.6, 1.2 Hz, H-6'), 7.11 (td, 1H, J 7.6, 1.2 Hz, H-5'), 7.05 (dd, 1H, J 15.4, 2.2 Hz, H-2), 4.39 (t, 2H, J 6.4 Hz, CH2CH2CH2CH3), 1.81 – 1.76 (m, 2H, OCH2CH2CH2CH3), 1.50 – 1.44 (m, 2H, OCH2CH2CH2CH3), 0.95 (t, 3H, J 7.4 Hz, OCH2CH2CH2CH3); 13C NMR (DMSO-d6, 150 MHz): δ 187.4 (d, JCF 2.0 Hz, C-1), 159.9 (d, JCF 249.1 Hz, C-2''), 156.9 (C-2'), 136.9 (C-3), 132.9 (d, JCF 8.7 Hz, C-4''), 132.0 (C-7'a), 130.2 (d, JCF 3.1 Hz, C-6''), 128.2 (d, JCF 14.4 Hz, C-1''), 125.0 (C-3'a), 124.7 (d, JCF 3.2 Hz, C-5''), 121.8 (C-5'), 121.2 (C-6'), 118.4 (C-4'), 116.3 (d, JCF 23.0 Hz, C-3''), 115.8 (d, JCF 4.9 Hz, C-2), 111.5 (C-7'), 93.9 (C-3'), 71.4 (OCH2CH2CH2CH3), 30.6 (OCH2CH2CH2CH3), 18.5 (OCH2CH2CH2CH3), 13.5 (OCH2CH2CH2CH3); IR (KBr): νmax 3052, 2958, 2872, 1622, 1607, 1560, 1524, 1482, 1344, 1280, 1228, 1071, 1015, 982, 775, 739 cm−1; HRMS: m/z [M+H]+: 338.15570 for C21H21FNO2 (calcd. 338.15508).
(2E)-3-(2'-isobutoxy-1'H-indol-3'-yl)-1-(4''-fluorophenyl)prop-2-en-1-one (11i)
Procedure B1: iso-BuOH, 24 h, 27%; Rf 0.48 (hexane/acetone 2:1); dark-orange crystals; mp 157–159 °C (acetone/hexane); 1H NMR (DMSO-d6, 600 MHz): δ 12.11 (s, 1H, NH), 7.88 (dd, 1H, J 15.4, 1.4, H-3), 7.70 (td, 1H, J 7.4, 1.5 Hz, H-6''), 7.66 (d, 1H, J 7.7 Hz, H-4'), 7.59 (dddd, 1H, J 8.1, 7.2, 5.3, 1.8 Hz, H-4''), 7.35 – 7.31 (m, 3H, H-7', H-5'', H-3''), 7.16 (td, 1H, J 7.6, 1.0 Hz, H-6'), 7.11 (td, 1H, J 7.6, 1.1 Hz, H-5'), 7.06 (dd, 1H, J 15.4, 2.1 Hz, H-2), 4.18 (d, 2H, J 6.5 Hz, OCH2CH(CH3)2), 2.16 – 2.07 (m, 1H, OCH2CH(CH3)2), 1.01 (d, J 6.4 Hz, OCH2CH(CH3)2); 13C NMR (DMSO-d6, 150 MHz): δ 187.4 (d, JCF 2.0 Hz, C-1), 159.9 (d, JCF 248.9 C-2''), 156.9 (C-2'), 137.0 (C-3), 132.9 (d, JCF 8.6 Hz, C-4''), 132.0 (C-7'a), 130.2 (d, JCF 3.2 Hz, C-6''), 128.2 (d, JCF 14.5 Hz, C-1''), 125.1 (C-3'a), 124.7 (d, JCF 3.3 Hz, C-5''), 121.8 (C-5'), 121.2 (C-6'), 118.3 (C-4'), 116.3 (d, JCF 22.0 Hz, C-3''), 115.8 (d, JCF 5.1 Hz, C-2), 111.5 (C-7'), 93.7 (C-3'), 77.3 (OCH2CH(CH3)2), 27.9 (OCH2CH(CH3)2), 18.6 (OCH2CH(CH3)2); IR (KBr): νmax 3120, 2960, 2872, 1622, 1609, 1558, 1524, 1483, 1459, 1346, 1280, 1230, 1203, 1072, 1017, 829, 775 cm−1; HRMS: m/z [M+H]+: 338.15530 for C21H21FNO2 (calcd. 338.15508).
(2E)-1-(2-fluorophenyl)-3-{2-[(1-methoxypropan-2-yl)oxy]-1H-indol-3-yl}prop-2-en-1-one (11j)
Procedure B1: 1-methoxypropan-2-ol, 24 h, 32 %; Rf 0.24 (hexane/EtOAc 2:1); orange oil; 1H NMR (DMSO-d6, 600 MHz): δ 12.06 (s, 1H, NH), 7.87 (dd, 1H, J 15.5, 1.4, H-3), 7.73 (td, 1H, J 7.6, 1.8 Hz, H-6''), 7.67 (d, 1H, J 7.7 Hz, H-4'), 7.60 (dddd, 1H, J 8.2, 7.6, 5.2, 1.8 Hz, H-4''), 7.37 – 7.31 (m, 3H, H-7', H-5'', H-3''), 7.16 (td, 1H, J 7.5, 1.2 Hz, H-6'), 7.12 (td, 1H, J 7.6, 1.2 Hz, H-5'), 7.09 (dd, 1H, J 15.5, 2.2 Hz, H-2), 4.91 (tk, 1H, J 4.8, 6.3 Hz, OCH(CH3)(CH2OCH3)), 3.58 (d, 2H, J 4.8, OCH(CH3)(CH2OCH3)), 3,30 (s, 3H, OCH(CH3)(CH2OCH3)), 1.35 (d, 3H, J 6.3 Hz, OCH(CH3)(CH2OCH3)); 13C NMR (DMSO-d6, 150 MHz): δ 187.7 (d, JCF 2.2 Hz, C-1), 160.4 (d, JCF 249.3 C-2''), 156.5 (C-2'), 137.4 (C-3), 133.5 (d, JCF 8.7 Hz, C-4''), 132.6 (C-7'a), 130.7 (d, JCF 3.1 Hz, C-6''), 128.6 (d, JCF 14.2 Hz, C-1''), 125.2 (C-3'a), 125.1 (d, JCF 3.3 Hz, C-5''), 122.1 (C-5'), 121.8 (C-6'), 119.0 (C-4'), 116.8 (d, JCF 23.0 Hz, C-3''), 116.4 (d, JCF 5.9 Hz, C-2), 111.9 (C-7'), 95.6 (C-3'), 78.4 (OCH(CH3)(CH2OCH3)), 75.1 (OCH(CH3)(CH2OCH3)), 59.0 (OCH(CH3)(CH2OCH3)), 17.2 (OCH(CH3)(CH2OCH3)); IR: νmax 3191, 2927,1622, 1715, 1607, 1519, 1448, 1335,1275, 1202, 1100, 1060, 1008, 971, 845, 827, 738 cm−1; HRMS: m/z [M+H]+: 354.15040 for C21H20FNO3 (calcd. 354.15000).
(2E)-1-(2-fluorophenyl)-3-[2-(2-hydroxyethoxy)-1H-indol-3-yl]prop-2-en-1-one(11k)
Procedure B1: ethan-1,2-diol, 24 h, 23%; Rf 0.72 (hexane/acetone 1:1) and (hexane/acetone 2:1); orange crystals; mp 140–143 °C (acetone/hexane); 1H NMR (DMSO-d6, 600 MHz): δ 12.07 (s, 1H, NH), 7.94 (dd, 1H, J 15.5, 1.3, H-3), 7.74 (td, 1H, J 7.6, 1.8 Hz, H-6''), 7.66 (d, 1H, J 7.8 Hz, H-4'), 7.59 (dddd, 1H, J 8.5, 7.1, 5.1, 1.8 Hz, H-4''), 7.36 – 7.32 (m, 3H, H-7', H-5'', H-3''), 7.16 (td, 1H, J 7.5, 1.2 Hz, H-5'), 7.11 (td, 1H, J 7.6, 1.2 Hz, H-6'), 7.08 (dd, 1H, J 15.5, 2.3 Hz, H-2), 5.14 (s, 1H, OH), 4.43 – 4.40 (m, 2H, CH2), 3.82 – 3.79 (m, 2H, CH2); 13C NMR (DMSO-d6, 150 MHz): δ 187.0 (d, JCF 2.3 Hz, C-1), 160.0 (d, JCF 249.4 C-2''), 157.2 (C-2'), 136.8 (C-3), 133.0 (d, JCF 8.6 Hz, C-4''), 132.1 (C-7'a), 130.3 (d, JCF 3.1 Hz, C-6''), 128.3 (d, JCF 14.1 Hz, C-1''), 124.9 (C-3'a), 124.7 (d, JCF 3.3 Hz, C-5''), 121.8 (C-6'), 121.2 (C-5'), 118.5 (C-4'), 116.4 (d, JCF 23.0 Hz, C-3''), 115.5 (d, JCF 5.6 Hz, C-2), 111.5 (C-7'), 93.9 (C-3'), 73.4 (CH2), 59.4 (CH2); IR: νmax 3054, 1603;1552, 1520, 1479, 1367, 1338, 1275, 1228, 1068, 1051, 1017, 884, 842, 738 cm-1; HRMS: m/z [M+H]+: 326.11877 for C19H17FNO3 (calcd. 326.11870).
(2E)-3-(1H-indol-3-yl)-1-(4-trifluoromethylphenyl) prop-2-en-1-one (12a)
Procedure B2: r.t., 24 h, 20%; Rf 0.39 (hexane/EtOAc 2:1); yellow crystals; m.p. 195–197 °C (CH2Cl2/hexane); m.p. 62 °C [44]; 1H NMR (DMSO-d6, 600 MHz): δ 11.99 (s, 1H, NH), 8.29 (d, 2H, J 8.1 Hz, H-2″,6″), 8.16 (s, 1H, H-2′), 8.11 (d, 1H, J 15.4 Hz, H-3), 8.11-8.09 (m, 1H, H-4′), 7.92 (d, 2H, J 8.1 Hz, H-3″,5″), 7.63 (d, 1H, J 15.4 Hz, H-2), 7.52-7.49 (m, 1H, H-7′), 7.26 (ddd, 1H, J 8.6, 7.1, 1.5 Hz, H-6′), 7.24 (ddd, 1H, J 8.5, 7.1, 1.4 Hz, H-5′); 13C NMR (150 MHz, DMSO-d6): δ 188.2 (C-1), 141.9 (C-1″), 140.3 (C-3), 137.6 (C-7′a), 134.1 (C-2′), 131.7 (q, JCF 32.3 Hz, C-4″), 128.9 (C-2″,6″), 125.6 (q, JCF 3.6 Hz, C-3″,5″), 125.1 (C-3′a), 124.0 (q, JCF 272.8 Hz, CF3), 122.9 (C-6′), 121.3 (C-5′), 120.5 (C-4′), 115.1 (C-2) 112.8 (C-3′), 112.5 (C-7′); IR: νmax 3218, 1630, 1583, 1537, 1512, 1433, 1322, 1242,1228,1108, 1064, 1011, 815, 740 cm-1; HRMS m/z: [M+H]+: 316.09442 for C18H12F3NO (calc. 316.09438).
(2E)-3-(1-methyl-1H-indol-3-yl)-1-(4-trifluoromethylphenyl) prop-2-en-1-one (12b)
Mechanochemical synthesis [23] Rf 0.53 (hexane/EtOAc 2:1); yellow crystals; m.p. 182–184 °C; m.p. 160–162 °C [45]; 1H and 13C NMR data identical with literature (2); IR: νmax 3110, 1648, 1548, 1524, 1460, 1372, 1330,1319,1107, 1027, 810, 734 cm-1; HRMS m/z: [M+H]+: 330.11038 for C19H14F3NO (calc. 330.11003).
(2E)-3-(1-methoxy-1H-indol-3-yl)-1-(4-trifluoromethylphenyl) prop-2-en-1-one (12c)
Procedure B1: 20 min., 25%; Procedure B2: r.t., 24 h, 30%; Rf 0.58 (hexane/EtOAc 2:1); yellow crystals; m.p. 113–115 °C; 1H NMR (400 MHz, DMSO-d6): δ 8.55 (s, 1H, H-2'), 8.30 (d, 2H, J 8.1 Hz, H-2'',6''), 8.15 (dt, 1H, J 8.0, 0.9 Hz, H-4'), 8.02 (d, 1H, J 15.4 Hz, H-3), 7.93 (d, 2H, J 8.1 Hz, H-3'',5''), 7.69 (d, 1H, J 15.4 Hz, H-2), 7.59 (dt, 1H, J 8.2, 0.9 Hz, H-7'), 7.38 (ddd, 1H, J 8.2, 7.1, 1.0 Hz, H-6'), 7.31 (ddd, 1H, J 8.1, 7.1, 1.1 Hz, H-5'), 4.17 (s, 3H, OCH3); 13C NMR (100 MHz, DMSO-d6): δ 188.6 (C-1), 142.1 (C-1''), 139.2 (C-3), 132.9 (C-7'a), 130.2 (C-2'), 132.4 (q, JCF 31.8 Hz, C-4''), 129.4 (C-2'',6''), 126.1 (q, JCF 3.7 Hz, C-3'',5''), 122.4 (C-3'a), 124.4 (q, JCF 272.6 Hz, CF3), 124.2 (C-6'), 122.6 (C-5'), 121.2 (C-4'), 117.0 (C-2), 109.6 (C-7'), 109.0 (C-3'), 67.2 (OCH3); IR: νmax 3102, 2949, 1655, 1584, 1562, 1556, 1512, 1368, 1317, 1274, 1245, 1212, 1105, 953, 804, 730 cm-1; HRMS m/z: [M+H]+: 346.10530 for C19H14F3NO2 (calc. 346.10494).
(2E)-1-(2-hydroxyphenyl)-3-(1H-indol-3-yl)prop-2-en-1-one (13a)
Procedure B2: 10.5 h, 57%; Rf 0.39 (hexane/EtOAc 2:1); orange-yellow crystals; m.p. 179.5–181 °C (EtOAc/hexane), 181–185 °C [46]; 165 °C [47]; 1H (600 MHz, DMSO-d6): δ 13.16 (s, 1H, OH), 12.07 (s, 1H, NH), 8.25 (dd, 1H, J 8.1, 1.6 Hz, H-6''), 8.21 (s, 1H, H-2'), 8.21 (d, 1H, J 15.2 Hz, H-3), 8.15-8.12 (m, 1H, H-4'), 7.77 (d, 1H, J 15.2 Hz, H-2), 7.56 (ddd, 1H, J 8.3, 7.1, 1.6 Hz, H-4''), 7.52-7.50 (m, 1H, H-7'), 7.27 (dd, 1H, J 7.1, 1.7 Hz, H-6'), 7.25 (dd, 1H, J 7.1, 1.6 Hz, H-5'), 7.01 (ddd, 1H, 8.1, 7.1, 1.2 Hz, H-5''), 6.98 (dd, 1H, J 8.3, 1.1 Hz, H-3''); 13C NMR (150 MHz, DMSO-d6): δ 193.1 (C-1), 162.2 (C-2''), 140.4 (C-3), 137.6 (C-7a'), 135.7 (C-4''), 134.4 (C-2'), 130.3 (C-6''), 125.1 (C-3a'), 123.0 (C-6'), 121.5 (C-5'), 120.52 (C-1''), 120.5 (C-4'), 119.0 (C-5''), 117.7 (C-3''), 113.9 (C-2), 113.0 (C-3'), 112.6 (C-7'); IR: (KBr) νmax 3299, 3092, 1631, 1577, 1547, 1484, 1438, 1373, 1344, 1293, 1249, 1202, 1153, 1108, 1032, 966, 830, 762, 740 cm−1; HRMS: m/z [M + H]+: 264.101912 for C17H13NO2 (calcd. 264.10220).
(2E)-1-(2-hydroxyphenyl)-3-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (13b)
Procedure B: 7 h; 90%; 15% [48]; Rf 0.37 (hexane/EtOAc 2:1); yellow crystals; m.p. 205–207 °C (MeCN), 208–209 °C [48]; 1H (600 MHz, DMSO-d6): δ 13.16 (s, 1H, OH), 8.24 (dd, 1H, J 8.1, 1.6 Hz, H-6''), 8,20 (s, 1H, H-2'), 8.16 (d, 1H, J 15.2 Hz, H-3), 8.15 (d, 1H, J 8.0 Hz, H-4'), 7.75 (d, 1H, J 15.2 Hz, H-2), 7.59 (d, 1H, J 8.0 Hz, H-7'), 7.54 (ddd, 1H, J 8.3, 7.1, 1.6 Hz, H-4''), 7.34 (ddd, 1H, J 8.0, 7.0, 1.1 Hz H-6'), 7.30 (ddd, 1H, J 8.0, 7.1, 1.1 Hz, H-5'), 7.01 (ddd, 1H, J 8.1, 7.1, 1.2 Hz, H-5''), 6.98 (dd, 1H, 8.3, 1.2 Hz, H-3''), 3.88 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 193.0 (C-1), 162.2 (C-2''), 139.7 (C-3), 138.1 (C-7a'), 137.7 (C-2'), 135.7 (C-4''), 130.3 (C-6''), 125.6 (C-3a'), 123.0 (C-6'), 121.8 (C-5'), 120.6 (C-4'), 120.5 (C-1''), 119.0 (C-5''), 117.7 (C-3''), 113.9 (C-2), 112.0 (C-3'), 111.0 (C-7'), 33.2 (N-CH3); IR: (KBr) νmax 3097, 3058, 2909, 2833, 1632, 1573, 1548, 1528, 1487, 1393, 1346, 1298, 1262, 1205, 1135, 1075, 1029, 842, 773, 755 cm-1; HRMS: m/z [M + H]+: 278.11768 for C18H15NO2 (calcd. 278.11765).
(2E)-1-(2-hydroxyphenyl)-3-(1-methoxy-1H-indol-3-yl)prop-2-en-1-one (13c)
Procedure B (50 °C): 3.5 h; 53%; Rf 0.62 (hexane/EtOAc 2:1); light-orange crystals; m.p. 139–140 °C (CH2Cl2/hexane); 1H (600 MHz, DMSO-d6): δ 13.0 (s, 1H, OH), 8.59 (s, 1H, H-2'), 8.24 (dd, 1H, J 8.1, 1.6 Hz, H-6''), 8.17 (d, 1H, J 7.9 Hz, H-4'), 8.11 (d, 1H, J 15.3 Hz, H-3), 7.82 (d, 1H, J 15.3 Hz, H-2), 7,60 (d, 1H, J 8.1 Hz, H-7'), 7.55 (ddd, 1H, J 8.3, 7.1, 1.6 Hz, H-4''), 7.38 (t, 1H, J 7.6 Hz H-6'), 7.32 (t, 1H, J 7.6 Hz, H-5'), 7.02 (dd, 1H, J 8.1, 7.1 Hz, H-5''), 6.99 (d, 1H, 8.3 Hz, H-3''), 4.18 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6): δ 193.1 (C-1), 162.1 (C-2''), 138.7 (C-3), 135.8 (C-4''), 132.5 (C-7a'), 130.4 (C-6''), 129.9 (C-2'), 123.8 (C-6'), 122.3 (C-5'), 122.1 (C-3a'), 120.8 (C-4'), 120.5 (C-1''), 119.0 (C-5''), 117.8 (C-3''), 115.7 (C-2), 109.2 (C-7'), 108.7 (C-3'), 66.8 (OCH3); IR: (KBr) νmax 3462, 3103, 1632, 1560, 1488, 1377, 1352, 1298, 1261, 1204, 1156, 1034, 953, 840, 762, 724 cm-1; HRMS: m/z [M + H]+: 294.11292 for C18H15NO3 (calcd. 294.11247).
(2E)-1-(4-hydroxyphenyl)-3-(1H-indol-3-yl)prop-2-en-1-one (14a)
Procedure A: 1 h, 18%; 65% [49]; Rf 0.22 (hexane/acetone 2:1); light-yellow crystals; m.p. 218 °C d (acetone/hexane); 147–148 °C [49]; 1H (600 MHz, DMSO-d6): δ 11.84 (s, 1H, OH), 10.28 (s, 1H, NH), 8.08 – 8.06 (m, 1H, H-4'), 8.07 (s, 1H, H-2'), 8.04 (d, 2H, J 8.7 Hz, H-2'', H-6''), 7.99 (d, 1H, J 15.5 Hz, H-3), 7.64 (d, 1H, J 15.5 Hz, H-2), 7.49 – 7.48 (m, 1H, H-7'), 7.25-7.21 (m, 2H, H-5', H-6'), 6.90 (d, 2H, J 8.7 Hz, H-3'', H-5''); 13C NMR (150 MHz, DMSO-d6): δ 187.0 (C-1), 161.7 (C-4''), 137.7 (C-3), 137.5 (C-7a'), 132.6 (C-2'), 130.6 (C-2'', C-6''), 129.8 (C-1''), 125.2 (C-3a'), 122.6 (C-6'), 121.0 (C-5'), 120.3 (C-4'), 115.4 (C-2), 115.3 (C-3'', C-5''), 112.8 (C-3'), 112.4 (C-7'); IR: (KBr) νmax 3414, 3293, 3092, 1642, 1604, 1557, 1443, 1348, 1274, 1230, 1167, 1039, 818, 737 cm-1; HRMS: m/z [M + H]+: 264.10193 for C17H13NO2 (calcd. 264.10191).
(2E)-1-(4-hydroxyphenyl)-3-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (14b)
Procedure A: 1.5 h, 12%; Rf 0.29 (hexane/acetone 2:1); light-yellow crystals; m.p. 244 °C d (acetone/hexane); 1H (600 MHz, DMSO-d6): δ 10.29 (s, 1H, OH), 8.08 (dt, 1H, J 8.0, 1.1 Hz, H-4'), 8.05 (s, 1H, H-2'), 8.03 (d, 2H, J 8.7 Hz, H-2'', H-6'', 7.94 (d, 1H, J 15.5 Hz, H-3), 7.62 (d, 1H, J 15.5 Hz, H-2), 7.55 (dt, 1H, J 8.1, 1.1 Hz, H-7'), 7.31 (ddd, 1H, J 8.1, 7.0, 1.1 Hz, H-6'), 7.27 (ddd, 1H, J 8.0, 7.0, 1.1 Hz, H-5'), 6.90 (d, 2H, J 8.7 Hz, H-3'', H-5''), 3.85 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 187.0 (C-1), 161.6 (C-4''), 138.0 (C-7a'), 137.0 (C-3), 136.0 (C-2'), 130.6 (C-2'', C-6''), 129.8 (C-1''), 125.6 (C-3a'), 122.7 (C-6'), 121.3 (C-5'), 120.5 (C-4'), 115.4 (C-2), 115.3 (C-3'', C-5''), 111.8 (C-3'), 110.8 (C-7'), 33.0 (CH3); IR (KBr): νmax 3097, 1626, 1587, 1528, 1508, 1473, 1387, 1276, 1225, 1165, 1077, 1050, 973, 816, 745 cm-1; HRMS: m/z [M + H]+: 278.11752 for C18H15NO2 (calcd. 278.11756).
(2E)-1-(4-hydroxyphenyl)-3-(1-methoxy-1H-indol-3-yl)prop-2-en-1-one (14c)
Procedure A: 4.5 h, 28%; Rf 0.33 (hexane/acetone 2:1); orange crystals; m.p. 182–184 °C (acetone/hexane); 1H (600 MHz, DMSO-d6): δ 10.32 (s, 1H, OH), 8.46 (s, 1H, H-2'), 8.11 (dt, 1H, J 8.0, 1.0 Hz, H-4'), 8.05 (d, 2H, J 8.7 Hz, H-2'', H-6''), 7.91 (d, 1H, J 15.5 Hz, H-3), 7.69 (d, 1H, J 15.5 Hz, H-2), 7.57 (dt, 1H, J 8.1, 1.0 Hz, H-7'), 7.36 (ddd, 1H, J 8.1, 7.1, 1.0 Hz, H-6'), 7.27 (ddd, 1H, J 8.0, 7.1, 1.0 Hz, H-5'), 6.91 (d, 2H, J 8.7 Hz, H-3'', H-5''), 4.15 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6): δ 186.9 (C-1), 161.7 (C-4''), 136.1 (C-3), 132.4 (C-7a'), 130.8 (C-2'', C-6''), 129.6 (C-1''), 128.5 (C-2'), 123.5 (C-6'), 122.0 (C-3a'), 121.8 (C-5'), 120.7 (C-4'), 117.0 (C-2), 115.3 (C-3'', C-5''), 109.0 (C-3'), 108.7 (C-7'), 66.6 (OCH3); IR: (KBr) νmax 3241, 1643, 1591, 1555, 1507, 1327, 1287, 1211, 1166, 1046, 952, 838, 742 cm-1; HRMS: m/z [M + H]+: 294.11282 for C18H15NO3 (calcd. 294.11247).
(2E)-3-(4-hydroxyphenyl)-1-(1H-indol-3-yl)prop-2-en-1-one (17a)
Procedure A: 7 h, 49%; 55% [49]; Rf = 0.27 (hexane/acetone 2:1); light-orange crystals; m.p. 197–200 °C d (acetone/hexane); 213–215 °C [49]; 1H (600 MHz, DMSO-d6): δ 12.03 (d, 1H, J 3.1 Hz, NH), 9.92 (s, 1H, OH), 8.66 (d, 1H, J 3.1 Hz, H-2'), 8.33 (ddd, 1H, J 7.9, 1.5, 0.8 Hz, H-4'), 7.70 – 7.67 (m, 2H, H-2'', H-6''), 7.63 (d, 1H, J 15.4 Hz, H-3), 7.55 (d, 1H, J 15.4 Hz, H-2), 7.48 (dt, 1H, J 7.9, 1.0 Hz, H-7'), 7.23 (ddd, 1H, J 7.9, 7.0, 1.5 Hz, H-6'), 7.20 (ddd, J 8.0, 6.9, 1.0 Hz, 1H, H-5'), 6.85 – 6.81 (m, 2H, H-3'', H-5''); 13C NMR (150 MHz, DMSO-d6): δ 183.8 (C-1), 159.3 (C-4''), 139.8 (C-3), 136.8 (C-7a'), 134.2 (C-2'), 130.3 (C-2'', C-6''), 126.3 (C-1''), 125.9 (C-3a'), 123.0 (C-6'), 121.8 (C-5'), 121.7 (C-4'), 121.3 (C-2), 117.8 (C-3'), 115.7 (C-3'', C-5''), 112.1 (C-7'); IR: νmax 3493, 3120, 1633, 1604, 1584, 1512, 1442, 1238, 1156, 970. 822, 746 cm-1.
(2E)-3-(4-hydroxyphenyl)-1-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (17b)
Procedure A: 7 h, 55%; Rf 0.33 (hexane/acetone 2:1); light-red crystals; m.p. 236–239 °C (acetone/hexane); 1H (600 MHz, DMSO-d6): δ 9.94 (s, 1H OH), 8.67 (s, 1H, H-2'), 8.34 (dt, 1H, J 7.9, 1.0 Hz, H-4'), 7.69 – 7.65 (m, 2H, H-2'', H-6''), 7.56 (dt, 1H, J 8.1, 0.9 Hz, H-7'), 7.56 (d, 1H, J 15.8 Hz, H-3), 7.56 (d, 1H, J 15.8 Hz, H-2), 7.30 (ddd, 1H, J 8.1, 7.1, 1.3 Hz, H-6'), 7.25 (ddd, 1H, J 7.9, 7.1, 1.0 Hz, H-5'), 6.86 – 6.83 (m, 2H, H-3'', H-5''), 3.90 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 183.3 (C-1), 159.3 (C-4''), 139.8 (C-3), 137.8 (C-2'), 137.4 (C-7a')130.2 (C-2'', C-6''), 126.3 (C-3a'), 126.2 (C-1''), 123.0 (C-6'), 122.0 (C-5'), 121.9 (C-4'), 121.2 (C-2), 116.6 (C-3'), 115.7 (C-3'', C-5''), 110.6 (C-7'), 33.3 (CH3); IR: νmax 3227, 3115, 1634, 1604, 1556, 1523, 1510, 1463, 1428, 1371, 1267, 1217, 1087, 988, 969, 829, 749, cm-1.
(2E)-3-(4-hydroxyphenyl)-1-(1-methoxy-1H-indol-3-yl)prop-2-en-1-one (17c)
Procedure A: 5 h, 76%; Rf 0.38 (hexane/acetone 2:1); light-yellow crystals; m.p. 213–215 °C (acetone/hexane); 1H (600 MHz, DMSO-d6): δ 9.97 (s, 1H, OH), 9.06 (s, 1H, H-2'), 8.38 (dt, 1H, J 8.0, 1.0 Hz, H-4'), 7.71 – 7.67 (m, 2H, H-2'', H-6''), 7.59 (d, 1H, J 15.5 Hz, H-3), 7.58 (dt, 1H, J 8.1, 0.9 Hz, H-7'), 7.57 (d, 1H, J 15.5 Hz, H-2), 7.36 (ddd, 1H, J 8.1, 7.1, 1.1 Hz, H-6'), 7.29 (ddd, 1H, J 8.0, 7.1, 1.0 Hz, H-5'), 6.86 – 6.83 (m, 2H, H-3'', H-5''), 4.21 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6): δ 183.4 (C-1), 159.5 (C-4''), 140.4 (C-3), 132.1 (C-7a'), 131.0 (C-2'), 130.4 (C-2'', C-6''), 126.1 (C-1''), 123.9 (C-6'), 122.8 (C-5'), 122.5 (C-3a'), 122.2 (C-4'), 120.8 (C-2), 115.7 (C-3'', C-5''), 113.0 (C-3'), 108.8 (C-7'), 66.9 (OCH3); IR: νmax 3103, 1633, 1607, 1586, 1510, 1388, 1368, 1325, 1275, 1236, 1192, 1167, 1065, 975, 949, 815, 739, 715 cm-1.
(2E)-3-(3,4-dihydroxyphenyl)-1-(1H-indol-3-yl)prop-2-en-1-one (18a)
Procedure A: 3 h, 80%; Rf 0.11 (hexane/acetone 2:1); yellow-green crystals; m.p. 212–215 °C d (acetone/hexane); 1H (600 MHz, DMSO-d6): δ 12.01 (bs, 1H, OH), 9.26 (bs, 1H, OH), 8.65 (s, 1H, H-2'), 8.32 (ddd, 1H, J 7.9, 1.4, 0.9 Hz, H-4'), 7.54 (d, 1H, J 15.5 Hz, H-3), 7.48 (dt, 1H, J 7.9, 1.0 Hz, H-7'), 7.47 (d, 1H, J 15.5 Hz, H-2), 7.24 – 7.21 (m, 1H, H-6'), 7.23 (d, 1H, J 2.1 Hz, H-2''), 7.19 (ddd, 1H, J 8.1, 7.0, 1.1, H-5'), 7.14 (dd, 1H, 8.1, 2.1 Hz), 6.80 (d, 1H, J 8.1 Hz, H-5''); 13C NMR (150 MHz, DMSO-d6): δ 183.8 (C-1), 147.8 (C-4''), 145.5 (C-3''), 140.3 (C-3), 136. 8 (C-7'a), 134.1 (C-2'), 126. 8 (C-1''), 125.9 (C-3'a), 122.9 (C-6'), 121.8 (C-5'), 121.6 (C-4'), 121.2 (C-6''), 121.2 (C-2), 117.8 (C-3'), 115.7 (C-5''), 115.4 (C-2''), 112.1 (C-7'); IR: νmax 3150, 1632, 1607, 1583, 1514, 1440, 1290, 1236, 1150, 1099, 984, 958, 840, 808, 749 cm-1.
(2E)-3-(3,4-dihydroxyphenyl)-1-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (18b)
Procedure A: 4 h, 81%; Rf 0.15 (hexane/acetone 2:1); yellow-green crystals; m.p. 210–214 °C d (acetone/hexane); 1H (600 MHz, DMSO-d6): δ 9.30 (s, 2H, OH), 8.67 (s, 1H, H-2'), 8.33 (dt, 1H, J 7.9, 1.0 Hz, H-4'), 7.56 (dt, 1H, J 8.1, 0.9 Hz, H-7'), 7.47 (s, 2H, H-3, H-2), 7.30 (ddd, 1H, J 8.1, 7.1, 1.3 Hz, H-6'), 7.25 (ddd, 1H, J 8.0, 7.1, 1.1 Hz, H-5'), 7.22 (d, 1H, J 2.1 Hz, H-2''), 7.11 (dd, 1H, J 8.2, 2.1 Hz, H-6''), 6.80 (d, 1H, J = 8.1 Hz, H-5''), 3.90 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6): δ 183.3 (C-1), 147.8 (C-4''), 145.6 (C-3''), 140.4 (C-3), 137.8 (C-2'), 137.4 (C-7'a), 126.7 (C-1''), 126.3 (C-3'a), 123.0 (C-6'), 122.0 (C-5'), 121.9 (C-4'), 121.2 (C-6''), 121.1 (C-2), 116.6 (C-3'), 115.7 (C-5''), 115.2 (C-2''), 110.6 (C-7'), 33.3 (CH3); IR: νmax 3383, 2951, 2715, 1623, 1598, 1518, 1438, 1368, 1276, 1185, 1084, 966, 769 cm-1.
(2E)-3-(3,4-dihydroxyphenyl)-1-(1-methoxy-1H-indol-3-yl)prop-2-en-1-one (18c)
Procedure A: 3 h, 74%; Rf 0.21 (hexane/acetone 2:1); brown-grey crystals; m.p. 206–209 °C d (acetone/hexane); 1H (600 MHz, DMSO-d6): δ 9.75-8.90 (bs, 2H, OH), 9.07 (s, 1H, H-2'), 8.38 (dt, 1H, J 7.9, 0.9 Hz, H-4'), 7.58 (dt, 1H, J 8.1, 0.9 Hz, H-7'), 7.51 (d, 1H, J 15.5 Hz, H-2), 7.50 (d, 1H, J 15.5 Hz, H-3), 7.35 (ddd, 1H, J 8.1, 7.1, 1.0 Hz, H-6'), 7,29 (ddd, 1H, J 7.9, 7.1, 1.0 Hz, H-5'), 7.25 (d, 1H, J 2.1 Hz, H-2''), 7.14 (dd, 1H, J 8.1, 2.1 Hz, H-6''), 6.81 (d, 1H, J 8.1 Hz, H-5''), 4.21 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6): δ 183.4 (C-1), 148.0 (C-4''), 145.6 (C-3''), 140.9 (C-3), 132.1 (C-7'a), 131.0 (C-2'), 126.7 (C-1''), 123.9 (C-6'), 122.7 (C-5'), 122.5 (C-3'a), 122.2 (C-4'), 121.4 (C-6''), 120.8 (C-2), 115.6 (C-5''), 115.4 (C-2''), 113.0 (C-3'), 108.8 (C-7'), 66.9 (OCH3); IR (KBr): νmax 3526, 3441, 3118 do 2560, 1636, 1508, 1450, 1370, 1326, 1283, 1253, 1205, 1113, 1063, 977, 801, 738 cm-1; HRMS: m/z [M+H]+: 310.10764 for C18H15 NO4 (calcd. 310.10738).
(2E)-3-(4-hydroxy-3-methoxyphenyl)-1-(1H-indol-3-yl)prop-2-en-1-one (19a)
Procedure A: 4 h, 79%; 75% [50]; Rf 0.20 (hexane/ acetone 2:1); ligth-orange crystals; m.p. 200–203 °C (acetone/hexane); 215 °C [50]; 1H (600 MHz, DMSO-d6): δ 12.05 (bs, 1H, NH), 9.51 (bs, 1H, OH), 8.69 (s s, 1H, H-2'), 8.33 (dt, 1H, J 7.8, 1.0 Hz, H-4'), 7.64 (d, 1H, J 15.4 Hz, H-2), 7.55 (d, 1H, J 15.4 Hz, H-3), 7.49 (dt, 1H, J 7.9, 1.0 Hz, H-7'), 7.46 (d, 1H, J 1.8 Hz, H-2''), 7.25 - 7.22 (m, 1H, H-6'), 7.23 (dd, 1H, J 8.1, 1.8 Hz, H-6''), 7.20 (ddd, 1H, J 8.1, 7.1, 1.2 Hz, H-5'), 6.83 (d, 1H, J 8.1 Hz, H-5''), 3.88 (s, 3H, OCH3); 13C NMR (150 MHz, DMSO-d6): δ 183.8 (C-1), 148.8 (C-4''), 147.9 (C-3''), 140.2 (C-3), 136.8 (C-7'a), 134.2 (C-2'), 126.7 (C-1''), 125.9 (C-3'a), 123.1 (C-6''), 123.0 (C-6'), 121.8 (C-5'), 121.7 (C-4'), 121.4 (C-2), 117.8 (C-3'), 115.5 (C-5''), 112.1 (C-7'), 111.4 (C-2''), 55.8 (OCH3); IR: νmax 3544, 3115, 1638, 1504, 1427, 1279, 1261, 1149, 1137, 975, 745 cm-1.
(2E)-3-(4-hydroxy-3-methoxyphenyl)-1-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (19b)
Procedure A: 4.5 h, 81%; Rf 0.30 (hexane/ acetone 2:1); red crystals; m.p. 170–172 °C (acetone/hexane); 1H (600 MHz, DMSO-d6): δ 9.54 (bs, 1H, OH), 8.67 (s, 1H, H-2'), 8.34 (dd, 1H, J 7.9, 1.0 Hz, H-4'), 7.56 (d, 1H, J 15.6 Hz, H-2), 7.58 – 7.56 (m, 1H, H-7'), 7.55 (d, 1H, J 15.6 Hz, H-3), 7.42 (d, 1H, J 1.8 Hz, H-2''), 7.31 (ddd, 1H, J 8.1, 7.1, 1.0 Hz, H-6'), 7.25 (ddd, 1H, J 8.0, 7.1, 1.0 Hz, H-5'), 7.23 (dd, 1H, J 8.1, 1.8 Hz, H-6''), 6.84 (d, 1H, J 8.1 Hz, H-5''), 3.91 (s, 3H, N-CH3), 3.88 (s, 3H, C-OCH3); 13C NMR (150 MHz, DMSO-d6): δ 183.3 (C-1), 148.9 (C-4''), 147.9 (C-3''), 140.3 (C-3), 137.8 (C-2'), 137.5 (C-7'a), 126.7 (C-1''), 126.4 (C-3'a), 123.0 (C-6'), 122.96 (C-6''), 122.0 (C-5'), 121.9 (C-4'), 121.3 (C-2), 116.6 (C-3'), 115.6 (C-5''), 111.5 (C-2''), 110.6 (C-7'), 55.8 (C-OCH3), 33.3 (N-CH3); IR νmax 3514, 3118, 1616, 1579, 1512, 1400, 1372, 1267, 1207, 1123, 1081, 964, 740 cm-1.
(2E)-3-(4-hydroxy-3-methoxyphenyl)-1-(1-methoxy-1H-indol-3-yl)prop-2-en-1-one (19c)
Procedure A: 3 h, 61%; Rf 0.12 (hexane/EtOAc 2:1); ligth-yellow crystals; m.p. 155–156 °C (acetone/hexane); 1H NMR (600 MHz, DMSO-d6): δ 9.57 (bs, 1H, OH), 9.07 (s, 1H, H-2'), 8.39 (dd, 1H, J 8.1, 0.9 Hz, H-4'), 7.59 (d, 1H, J 15.5 Hz, H-2), 7.60 – 7.58 (m, 1H, H-7'), 7.58 (d, 1H, J 15.5 Hz, H-3), 7.46 (d, 1H, J 1.8 Hz, H-2''), 7.36 (ddd, 1H, J 8.1, 7.1, 1.0 Hz, H-6'), 7.30 (ddd, 1H, J 8.0, 7.1, 0.9 Hz, H-5'), 7.25 (dd, 1H, J 8.1, 1.8 Hz, H-6''), 6.84 (d, 1H, J 8.1 Hz, H-5''), 4.22 (s, 3H, N-OCH3), 3.89 (s, 3H, C-OCH3); 13C NMR (150 MHz, DMSO-d6): δ 183.3 (C-1), 149.1 (C-4''), 147.9 (C-3''), 140.9 (C-3), 132.1 (C-7'a), 131.0 (C-2'), 126.6 (C-1''), 123.9 (C-6'), 123.3 (C-6''), 122.8 (C-5'), 122.5 (C-3'a), 122.2 (C-4'), 120.9 (C-2), 115.6 (C-5''), 113.0 (C-3'), 111.5 (C-2''), 108.9 (C-7'), 66.9 (N-OCH3), 55.9 (C-OCH3); IR (KBr): νmax 3526, 3423, 3255, 1642, 1586, 1519, 1453, 1377, 1329, 1284, 1201, 1126, 1065, 1031, 976, 842, 807, 738 cm-1; HR-MS: m/z: [M+H]+ 324.12323 for C19H17NO4 (calcd. 324.12303).
3.2. Antiproliferative Activity Studies
3.2.1. Cell Cultures
In the experiments evaluating the biological activity of the synthesized chalcones, various tumor cell lines were used. Cell lines MDA-MB-231 (human breast adenocarcinoma), HeLa (human cervical adenocarcinoma), HCT116 (human colorectal carcinoma), and Jurkat (human T lymphocyte leukemia) were cultured in RPMI 1640 medium (Biosera, Kansas City, MO, USA). MCF-7 (human breast adenocarcinoma) cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM). Media were enriched with 1% HyCloneTM antibiotic/antimycotic solution containing penicillin, streptomycin, and amphotericin B (Merck, Darmstadt, Germany), and 10% fetal bovine serum (FBS; Gibco, Thermo Scientific, Rockford, IL, USA). The non-tumor cell line MCF-10A (human mammary epithelial cells) was cultured in DMEM F12 medium (high-glucose Dulbecco's Modified Eagle's Medium F12, Biosera, Kansas City, MO, USA), supplemented with antibiotic/antimycotic solution, insulin (final concentration of 10 µg/mL), 10% fetal bovine serum, EGF (final concentration of 20 ng/mL), and hydrocortisone (final concentration of 0.5 µg/mL) (Merck, Darmstadt, Germany). Human fibroblast cells, Bj-5ta (immortalized foreskin fibroblasts), were cultured in a mixture of DMEM and M199 media in a 4:1 ratio and supplemented with 10% FBS and hygromycin B (final concentration of 0.01 mg/mL). Cells were maintained in a humidified atmosphere containing 5% CO2 at 37 °C. Cell viability was higher than 95% for each experiment.
3.2.2. MTT Assay
To evaluate the antiproliferative/cytotoxic activity and inhibition of the metabolism of the tested compounds against cell lines, a colorimetric test of the metabolic activity of MTT (3-(4,5-di-methylthiazol-2-yl)2,5-diphenyltetrazolium bromide) was used (Sigma-Aldrich Chemie, Steinheim, Germany). Tested cells were seeded on 96-well culture plates at a density (5 × 103/well) and cultured in the respective culture medium for 24 hours. Tested chalcones were added to the cells at concentrations of 10, 50 and 100 μmol/L and incubated for 72 hours. After incubation, 100 µL of 10% MTT solution (5 mg/mL, Sigma-Aldrich Chemie, Steinheim, Germany) was added to each well. After 4 hours at 37 °C in a 5% CO2 atmosphere, MTT precipitated to insoluble formazan and 100 µL of 10% SDS (sodium dodecyl sulfate) was added to each well. After dissolving the crystals after 12 hours, the absorbance was measured at a wavelength of 540 nm using the automated Cytation™ 3 Cell Imaging Multi-Mode Reader (Biotek, Winooski, VT, USA). IC50 values, determined as half-inhibitory concentrations versus control cells, were calculated using the statistical predictive function Trend.
3.3 Antiproliferative Activity Studies
3.3.1. DPPH Radical Scavenging Activity
The DPPH-scavenging activities of the compounds, based on the decolorization of the stable purple DPPH free radical measured at 517 nm[51], were estimated with slight modification [52]. 50 µL of sample stock solution (1 mmol.L-1) or standard gallic acid (0.05-1.5 mmol.L-1) was mixed with 2.0 mL of DPPH solution (0.1 mmol.L-1). At the same time, 50 µL of methanol was used instead of the sample as a blank. After 30 min of incubation in the dark, absorbance was measured. The inhibition of radical was calculated as percentage of the blank according to the formula (%) = (1-Asample /Ablank)×100 and then the antioxidant activity was expressed as micromoles of gallic acid equivalents per millimole of sample (µmol GAE/mmol).
3.3.2. ABTS Radical Scavenging Activity
The ABTS radical scavenging ability of the compounds was determined by the decrease of absorbance of radical cation (ABTS•+) at 734 nm. The ABTS•+ was generated by reacting equal volumes of ABTS (7 mmol.L-1) in H2O with oxidizing agent K2S2O8 (2.45 mmol.L-1) for 12-16 h in the dark at room temperature and subsequently diluted with methanol to obtain absorbance of 0.76 ± 0,01 at 734 nm. Then, 50 µL of sample stock solution (1 mmol.L-1) or standard gallic acid (0.05 – 1.5 mmol.L-1) was added to ABTS solution (2 mL) and mixed. The absorbance of the sample was read at 734 nm after a 30 min of incubation at room temperature. The inhibition of radical was calculated as percentage of the blank according to the formula (%) = (1-Asample /Ablank)×100, and then the antioxidant activity was expressed as micromoles of gallic acid equivalents per millimole of sample (µmol GAE/mmol).
3.3.3. Ferric Reducing Antioxidant Power (FRAP)
The FRAP method is based on measurement of intense blue coloured Frap-Fe2+ complex formation having an absorption maximum at 595 nm after reduction of Fe3+ to Fe2+ ions in complex by sample [53]. The FRAP reagent contained 10 mmol.L-1 of TPTZ (2,4,6-tris(2-pyridyl)-s-triazine) solution in 40 mmol.L-1 HCl, 20 mM FeCl3, and 0.3 M acetate buffer at pH 3.6 in ratio 1:1:10. Briefly, 50 µL of sample stock solution (1 mmol.L-1) or standard gallic acid (0.05 – 1.5 mmol.L-1) were mixed with 2 ml of FRAP reagent and incubated for 30 min in the dark. The antioxidant activity was expressed as micromoles of gallic acid equivalents per millimole of sample (µmol GAE/mmol).
3.3.4. Statistical Analysis
The experimental results were performed in triplicate. The data were recorded asmean ± standard deviation and analyzed by one-way ANOVA followed by Tukey’s t test. Statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA, USA). Results were considered significantly different when p < 0.05.
4. Conclusions
In summary, we reported here the synthesis of a novel series of hybrid chalcones bearing unsubstituted, N-metyl- or N-methoxy- indole pharmacophore and benzene ring with OH, F, CF3 substituents. Antiproliferative activity screening indicates that the 2-fluoroderivatives 11a-k exhibit moderate and selective activity against cancer cell lines, with the highest efficacy observed against Jurkat leukemic cells, even N-H, N-methoxy and 2-ethoxy chalcones 11a, 11c, 11e exhibited an activity of less than 8.3 μM on this cell line. Another 2-fluro-chalcone with 2-propoxy group 11f significantly suppressed the proliferation of breast cancer cells with minimal effect on non-cancer mammary epithelial cells MCF-10A. The compound (2E)-3-(3,4-dihydroxyphenyl)-1-(1-methoxy-1H-indol-3-yl)prop-2-en-1-one (18c) from a series of hydroxylated chalcones demonstrated remarkable effectiveness against Jurkat leukemia cells and a human colon cancer cell line HCT116, with IC50 values of 8.0 ± 1.4 and 18.2 ± 2.9 μM, respectively. This structure contains a 1-methoxyindole nucleus, which shows great potential for the development of prototype anticancer drugs. Additionally, 1-methoxychalcone 14c exhibited impressive efficacy against Jurkat leukemia cells with an IC50 value of 7.3 ± 0.1 μM. These findings confirm the suitability of integrating the 1-methoxyindole nucleus into the design of novel and effective cancer treatments. Compounds 18a-c and 19a-c possess the highest antioxidant potential as determined by the in vitro DPPH, ABTS and FRAP methods.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1-S52 1H NMR and 13C NMR Spectral Data of 11a, 11d-f, 12a-c, 13a-c, 14a-c, 17a-c, 18a-c, 19a-c; Figure S53 HR-MS fragmentation spectra for compound 11a
Author Contributions
Conceptualization, Z.K. and J.M.; methodology, Z.K., R.M., A.S. and S.B.; validation, Z.K., R.M. A.S. and S.B.; experiments, Z.K., R.M., A.S., M.K. and S.B.; data curation, Z.K., R.M., A.S.; M.V. and S.B.; writing—original draft preparation, Z.K., R.M., A.S., M.K., S.B. and J.M.; writing—review and editing, Z.K., R.M., A.S., M.K., M.V. and J.M.; supervision, Z.K., J.M.; funding acquisition, A.S. M.V. and J.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by the Grant Agency of the Ministry of the Education, Science, Research and Sport of the Slovak Republic (VEGA 1/0539/21, VEGA 1/0513/21, VEGA 2/0112/22) and the Slovak Research and Development Agency under contract No. APVV-16–0446. Moreover, this publication is the result of the project implementation: “Open scientific community for modern interdisciplinary research in medicine (OPENMED)”, ITMS2014+: 313011V455, supported by the Operational Programme Integrated Infrastructure, funded by the ERDF.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gomes, M.N.; Muratov, E.N.; Pereira, M.; Peixoto, J.C.; Rosseto, L.P.; Cravo, P.V.L.; Andrade, C.H.; Neves, B.J. Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules 2017, 22, 1210. [CrossRef]
- McCluskey, A.; Russell, C. Chalcones: Potential Anticancer Agents. In Translational Research in Cancer; Http://dx.doi.org/10.5772/intechopen.9144, D., Ed.; 2021; pp. 1–25 ISBN 0000957720.
- Gao, F.; Huang, G.; Xiao, J. Chalcone Hybrids as Potential Anticancer Agents: Current Development, Mechanism of Action, and Structure-Activity Relationship. Med. Res. Rev. 2020, 40, 2049–2084. [CrossRef]
- Sravanthi, T. V.; Manju, S.L. Indoles - A Promising Scaffold for Drug Development. Eur. J. Pharm. Sci. 2016, 91, 1–10. [CrossRef]
- Robinson, M.W.; Overmeyer, J.H.; Young, A.M.; Erhardt, P.W.; Maltese, W.A. Synthesis and Evaluation of Indole-Based Chalcones as Inducers of Methuosis, a Novel Type of Non-Apoptotic Cell Death. J Med Chem. 2012, 55, 1940–1956. [CrossRef]
- Cho, H.; Geno, E.; Patoor, M.; Reid, A.; Mcdonald, R.; Hild, M.; Jenkins, J.L. Indolyl-Pyridinyl-Propenone-Induced Methuosis through the Inhibition of PIKFYVE. ACS Omega 2018, 3, 6097–6103. [CrossRef]
- Kudličková, Z.; Takáč, P.; Sabolová, D.; Vilková, M.; Baláž, M.; Béres, T.; Mojžiš, J. Novel 1-Methoxyindole- and 2-Alkoxyindole-Based Chalcones: Design, Synthesis, Characterization, Antiproliferative Activity and DNA, BSA Binding Interactions. Med. Chem. Res. 2021, 30, 897–912. [CrossRef]
- Kuruc, T.; Kello, M.; Petrova, K.; Kudlickova, Z.; Kubatka, P.; Mojzis, J. The Newly Synthetized Chalcone L1 Is Involved in the Cell Growth Inhibition, Induction of Apoptosis and Suppression of Epithelial-to-Mesenchymal Transition of HeLa Cells. Molecules 2021, 26, 1–27. [CrossRef]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [CrossRef]
- Yu, Y.; Liu, A.; Dhawan, G.; Mei, H.; Zhang, W.; Izawa, K.; Soloshonok, V.A.; Han, J. Fluorine-Containing Pharmaceuticals Approved by the FDA in 2020: Synthesis and Biological Activity. Chinese Chem. Lett. 2021, 32, 3342–3354. [CrossRef]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. - A Eur. J. 2019, 25, 11797–11819. [CrossRef]
- Burmaoglu, S.; Algul, O.; Anil, D.A.; Gobek, A.; Duran, G.G.; Ersan, R.H.; Duran, N. Synthesis and Anti-Proliferative Activity of Fluoro-Substituted Chalcones. Bioorganic Med. Chem. Lett. 2016, 26, 3172–3176. [CrossRef]
- dos Santos, M.B.; Bertholin Anselmo, D.; de Oliveira, J.G.; Jardim-Perassi, B. V.; Alves Monteiro, D.; Silva, G.; Gomes, E.; Lucia Fachin, A.; Marins, M.; de Campos Zuccari, D.A.P.; et al. Antiproliferative Activity and P53 Upregulation Effects of Chalcones on Human Breast Cancer Cells. J. Enzyme Inhib. Med. Chem. 2019, 34, 1093–1099. [CrossRef]
- Rozmer, Z.; Perjési, P. Naturally Occurring Chalcones and Their Biological Activities. Phytochem. Rev. 2016, 15, 87–120. [CrossRef]
- Cramer, J.; Sager, C.P.; Ernst, B. Hydroxyl Groups in Synthetic and Natural-Product-Derived Therapeutics: A Perspective on a Common Functional Group. J. Med. Chem. 2019, 62, 8915–8930. [CrossRef]
- Constantinescu, T.; Lungu, C.N. Anticancer Activity of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2021, 22, 1–33. [CrossRef]
- Deng, N.; Qiao, M.; Li, Y.; Liang, F.; Li, J.; Liu, Y. Anticancer Effects of Licochalcones: A Review of the Mechanisms. Front. Pharmacol. 2023, 14, 1–10. [CrossRef]
- Yang, L.H.; Ho, Y.J.; Lin, J.F.; Yeh, C.W.; Kao, S.H.; Hsu, L.S. Butein Inhibits the Proliferation of Breast Cancer Cells through Generation of Reactive Oxygen Species and Modulation of ERK and P38 Activities. Mol. Med. Rep. 2012, 6, 1126–1132. [CrossRef]
- Cho, S.-G.; Woo, S.-M.; Ko, S.-G. Butein Suppresses Breast Cancer Growth by Reducing a Production of Intracellular Reactive Oxygen Species. J. Exp. Clin. Cancer Res. 2014, 33, 51. [CrossRef]
- Chen, W.J.; Song, J.R.; Guo, P.; Wen, Z.Y. Butein, a More Effective Antioxidant than α-Tocopherol. J. Mol. Struct. THEOCHEM 2006, 763, 161–164. [CrossRef]
- Miranda, C.L.; Stevens, J.F.; Ivanov, V.; Mccall, M.; Frei, B.; Deinzer, M.L.; Buhler, D.R. Antioxidant and Prooxidant Actions of Prenylated and Nonprenylated Chalcones and Flavanones in Vitro. J. Agric. Food Chem. 2000, 2000, 3876–3884.
- Cai, Y.Z.; Mei Sun; Jie Xing; Luo, Q.; Corke, H. Structure-Radical Scavenging Activity Relationships of Phenolic Compounds from Traditional Chinese Medicinal Plants. Life Sci. 2006, 78, 2872–2888. [CrossRef]
- Kudličková, Z.; Stahorský, M.; Michalková, R.; Vilková, M.; Baláž, M. Mechanochemical Synthesis of Indolyl Chalcones with Antiproliferative Activity. Green Chem. Lett. Rev. 2022, 15, 474–482. [CrossRef]
- Suzdalev, K.F.; Babakova, M.N. Synthesis of Analogues of Indole Alkaloids from Sea Sponges – Aplysinopsins by the Reaction of Amines with (4Z)-4-[(1H-Indol-3-Yl)- Methylene]-1,3-Oxazol-5(4H)-Ones. J. Heterocycl. Chem. 2015, 46, 1200–1206. [CrossRef]
- Somei, M.; Kawasaki, T.; Kodama, A.; Nishida, T.; Shimizu, K. Preparation of 1-Hydroxyindole Derivatives and a New Route to 2-Substituted Indoles. Heterocycles 1991, 32, 221. [CrossRef]
- Acheson, B.R.M.; Hunt, P.G.; Littlewood, D.M.; Murrer, B.A.; Rosenberg, H.E. The Synthesis, Reactions, and Spectra of 1-Acetoxy-, 1-Hydroxy-, and 1-Methoxy-Indoles. J. Chem. Soc., Perkin Trans. 1 1978, 1117–1125. [CrossRef]
- Somei, M.; Nakajou, M.; Teramoto, T.; Tanimoto, A.; Yamada, F. [NUCLEOPHILIC SUBSTITUTION REACTION OF 3-ACETYL-I-METHOXY- INWLE AND ITS APPLICATION FOR THE SYNTHESIS OF NOVEL 2-SUBSTITUTED METHYL 2,3-DIHYDRO-l-METHYL3-0X05H-PYRIDO (4,3-b)INDOLE-4-CARBOXYLATES. 1999, 51, 1949–1956.
- Venkatanarayana, M.; K. Dubey, P. Novel and Simple Methodology for the Synthesis of 3-Acetylindoles and Their N-Alkyl Derivatives Using TBAB as Phase Transfer Catalyst. Lett. Org. Chem. 2011, 8, 656–662. [CrossRef]
- Somei, M.; Nakajou, M.; Teramoto, T.; Tanimoto, A.; Yamada, F. Nucleophilic Substitution Reaction of 3-Acetyl-1-Methoxyindole and Its Application for the Synthesis of Novel 2-Substituted Methyl 2,3-Dihydro-1-Methyl-3-Oxo-5H-Pyrido-[4,3-b]Indole-4-Carboxylates. Heterocycles 1999, 51, 1949–1956. [CrossRef]
- Jacobsen, N.E. NMR Data Interpretation Explained : Understanding 1D and 2D NMR Spectra of Organic Compounds and Natural Products; John Wiley & Sons, Inc., Hoboken, New Jersey, 2016; ISBN 9781118370223.
- Kudličková, Z.; Stahorský, M.; Michalková, R.; Vilková, M.; Baláž, M. Mechanochemical Synthesis of Indolyl Chalcones with Antiproliferative Activity. Green Chem. Lett. Rev. 2022, 15, 474–482. [CrossRef]
- Michalkova, R.; Kello, M.; Kudlickova, Z.; Gazdova, M.; Mirossay, L.; Mojzisova, G.; Mojzis, J. Programmed Cell Death Alterations Mediated by Synthetic Indole Chalcone Resulted in Cell Cycle Arrest , DNA Damage , Apoptosis and Signaling Pathway Modulations in Breast Cancer Model. Pharmaceutics 2022, 14, 503. [CrossRef]
- Chripkova, M.; Zigo, F.; Mojzis, J. Antiproliferative Effect of Indole Phytoalexins. Molecules 2016, 21, 6–8. [CrossRef]
- Gacche, R.N.; Dhole, N.A.; Kamble, S.G.; Bandgar, B.P. In-Vitro Evaluation of Selected Chalcones for Antioxidant Activity. J. Enzyme Inhib. Med. Chem. 2008, 23, 28–31. [CrossRef]
- Chu, J.; Guo, C.L. Design and Discovery of Some Novel Chalcones as Antioxidant and Anti-Inflammatory Agents via Attenuating NF-ΚB. Arch. Pharm. (Weinheim). 2016, 349, 63–70. [CrossRef]
- Przybylski, P.; Konopko, A.; Łętowski, P.; Jodko-Piórecka, K.; Litwinienko, G. Concentration-Dependent HAT/ET Mechanism of the Reaction of Phenols with 2,2-Diphenyl-1-Picrylhydrazyl (Dpph˙) in Methanol. RSC Adv. 2022, 12, 8131–8136. [CrossRef]
- Wang, G.; Xue, Y.; An, L.; Zheng, Y.; Dou, Y.; Zhang, L.; Liu, Y. Theoretical Study on the Structural and Antioxidant Properties of Some Recently Synthesised 2,4,5-Trimethoxy Chalcones. Food Chem. 2015, 171, 89–97. [CrossRef]
- Munteanu, I.G.; Apetrei, C. Analytical Methods Used in Determining Antioxidant Activity: A Review. Int. J. Mol. Sci. 2021, 22. [CrossRef]
- Jung, J.C.; Lee, Y.; Min, D.; Jung, M.; Oh, S. Practical Synthesis of Chalcone Derivatives and Their Biological Activities. Molecules 2017, 22, 1–11. [CrossRef]
- Kalinowska, M.; Płońska, A.; Trusiak, M.; Gołębiewska, E.; Gorlewska-Pietluszenko, A. Comparing the Extraction Methods, Chemical Composition, Phenolic Contents and Antioxidant Activity of Edible Oils from Cannabis Sativa and Silybum Marianu Seeds. Sci. Rep. 2022, 12, 1–16. [CrossRef]
- Cheng, Z.J.; Lin, C.N.; Hwang, T.L.; Teng, C.M. Broussochalcone A, a Potent Antioxidant and Effective Suppressor of Inducible Nitric Oxide Synthase in Lipopolysaccharide-Activated Macrophages. Biochem. Pharmacol. 2001, 61, 939–946. [CrossRef]
- Sivakumar, P.M.; Prabhakar, P.K.; Doble, M. Synthesis, Antioxidant Evaluation, and Quantitative Structure-Activity Relationship Studies of Chalcones. Med. Chem. Res. 2011, 20, 482–492. [CrossRef]
- Garberová, M.; Potočňák, I.; Tvrdoňová, M.; Bago-Pilátová, M.; Bekešová, S.; Kudličková, Z.; Samoľová, E.; Kešeľáková, A.; Elečko, J.; Vilková, M. Spectral, Structural, and Pharmacological Studies of Perillaldehyde and Myrtenal Based Benzohydrazides. J. Mol. Struct. 2023, 1271. [CrossRef]
- Lagu, S.B.; Yejella, R.P.; Bhandare, R.R.; Shaik, A.B. Design, Synthesis, and Antibacterial and Antifungal Activities of Novel Trifluoromethyl and Trifluoromethoxy Substituted Chalcone Derivatives. Pharmaceuticals 2020, 13, 1–16. [CrossRef]
- Yao, Y.; Huang, T.; Wang, Y.; Wang, L.; Feng, S.; Cheng, W.; Yang, L.; Duan, Y. Angiogenesis and Anti-Leukaemia Activity of Novel Indole Derivatives as Potent Colchicine Binding Site Inhibitors. J. Enzyme Inhib. Med. Chem. 2022, 37, 652–665. [CrossRef]
- Venturella, P.; Bellino, A.; Piozzi, F. Synthesis of Indolylchalcones and Indolylchrom Indolylchromonols. Farm. Ed. Sci. 1971, 26, 591–596.
- Yesuthangam, Y.; Pandian, S.; Venkatesan, K.; Gandhidasan, R.; Murugesan, R. Photogeneration of Reactive Oxygen Species and Photoinduced Plasmid DNA Cleavage by Novel Synthetic Chalcones. J. Photochem. Photobiol. B Biol. 2011, 102, 200–208. [CrossRef]
- Chang, M.Y.; Chen, K.T.; Tsai, Y.L.; Chen, H.Y. One-Pot Access to 2-Aryl-3-(Arylmethyl)Chromones. Synth. 2020, 52, 861–872. [CrossRef]
- Kumar, D.; Kumar, N.M.; Akamatsu, K.; Kusaka, E.; Harada, H.; Ito, T. Synthesis and Biological Evaluation of Indolyl Chalcones as Antitumor Agents. Bioorganic Med. Chem. Lett. 2010, 20, 3916–3919. [CrossRef]
- Rani, P.; Srivastava, V.K.; Kumar, A. Synthesis and Antiinflammatory Activity of Heterocyclic Indole Derivatives. Eur. J. Med. Chem. 2004, 39, 449–452. [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a Free Radical Method to Evaluate Antioxidant Activity. LWT - Food Sci. Technol. 1995, 28, 25–30. [CrossRef]
- Tvrdoňová, M.; Borovská, B.; Salayová, A.; Rončák, R.; Michalčin, P.; Bednáriková, Z.; Gažová, Z. Design and Synthesis of Novel Carbohydrate-Amino Acid Hybrids and Their Antioxidant and Anti-β-Amyloid Aggregation Activity. Bioorg. Chem. 2023, 137, 106636. [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of “Antioxidant Power”: The FRAP Assay. Anal. Biochem. 1996, 239, 70–76. [CrossRef]
Figure 1.
Structure of chalcones.
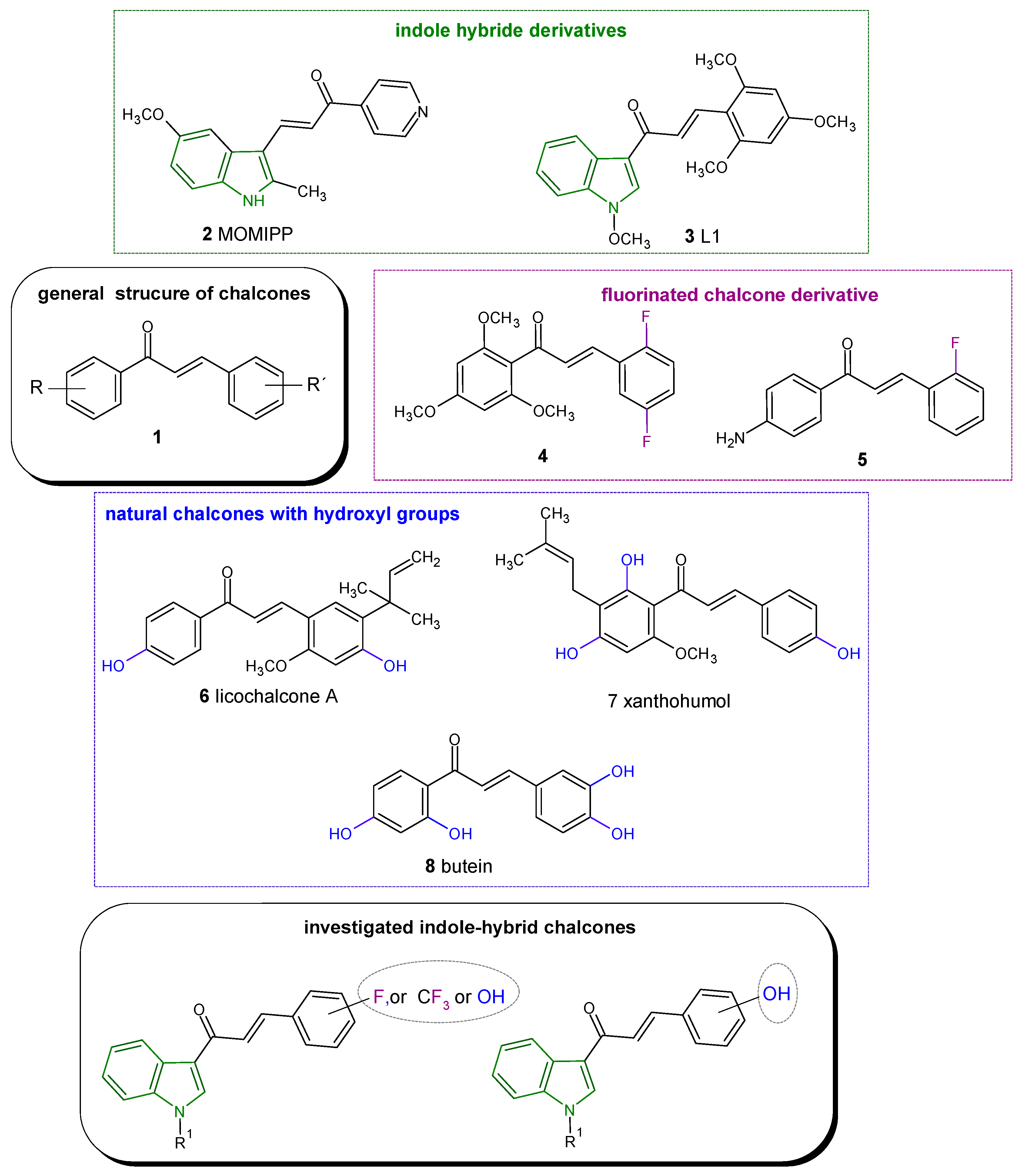
Scheme 1.
The synthetic pathway for the synthesis of the arylindolylpropenones 11a-k, 12a-c, 13a-c, 14a-c.
Scheme 1.
The synthetic pathway for the synthesis of the arylindolylpropenones 11a-k, 12a-c, 13a-c, 14a-c.
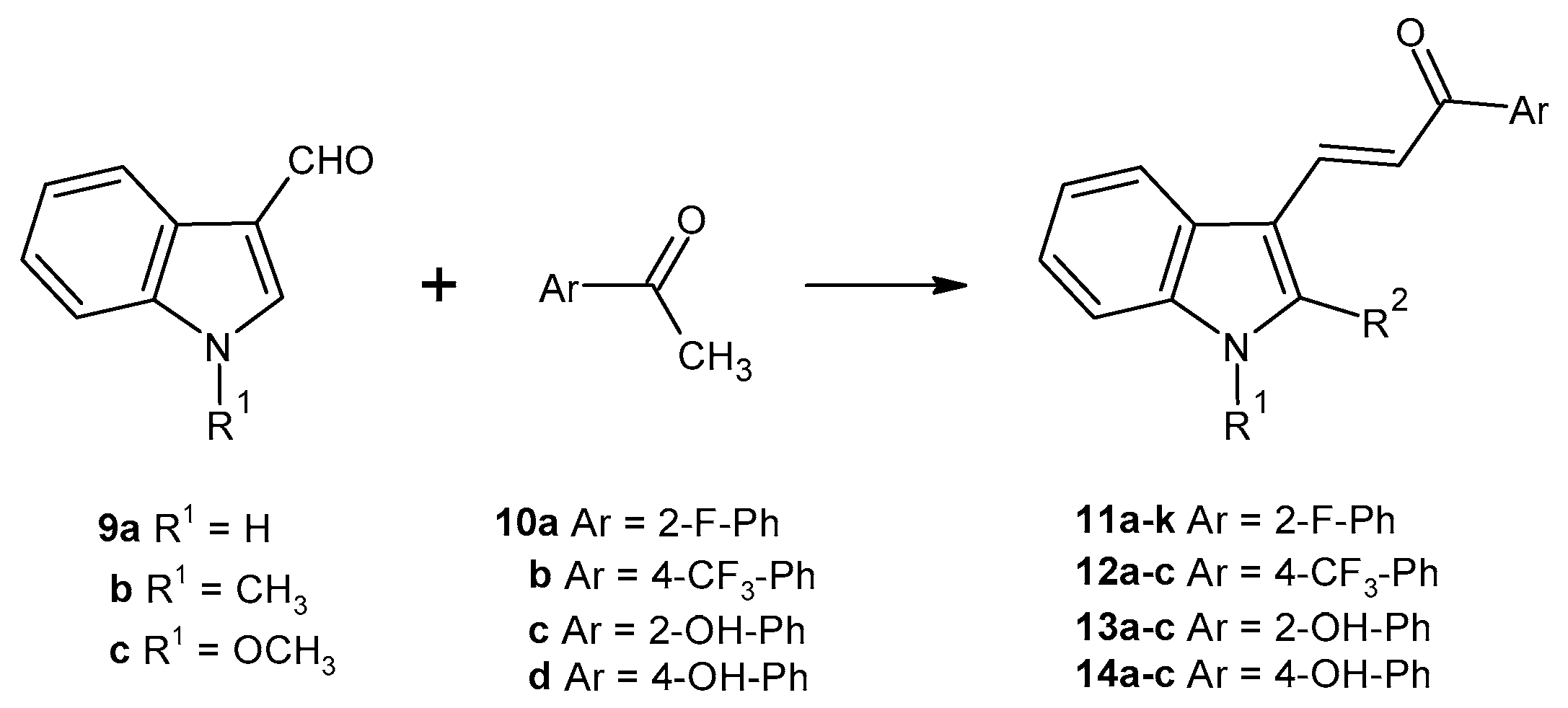
Scheme 2.
The synthetic pathway for the synthesis of the arylindolylpropenones 17a-c, 18a-c, 19a-c.

Table 1.
The chemical shifts (ppm) and coupling constants J ( Hz) of chalcones.
| comp. | Ar | R1 | R2 | H-2 (J) | H-3 (J) | C-1 | C-2 | C-3 |
|---|---|---|---|---|---|---|---|---|
| 11a | 2-F-Ph | H | H | 7.29 (15.7, 2.3) | 7.91 (15.7, 1.2) | 188.2 | 119.5 | 140.0 |
| 11b | CH3 | 7.25 (15.7, 2.3) | 7.88 (15.7, 1.4) | 188.1 | 119.4 | 139.3 | ||
| 11c | OCH3 | 7.33 (15.8, 2.2) | 7.84 (15.7, 0.9) | 188.3 | 120.9 | 138.5 | ||
| 11d | H | OCH3 | 7.02 (15.4, 2.2) | 7.84 (15.4, 1.5) | 187.4 | 115.7 | 136.8 | |
| 11e | OCH2CH3 | 7.07 (15.4, 2.3) | 7.88 (15.4, 1.5) | 187.1 | 115.6 | 136.8 | ||
| 11f | OCH2CH2CH3 | 7.06 (15.4, 2.2) | 7.88 (15.4, 1.5) | 187.3 | 115.7 | 136.9 | ||
| 11g | OCH(CH3)2 | 7.09 (15.6, 2.3) | 7.87 (15.6, 1.5) | 187.2 | 115.8 | 136.9 | ||
| 11h | OCH2CH2 CH2CH3 | 7.05 (15.4, 2.2) | 7.86 (15.4, 1.5) | 187.4 | 115.8 | 136.9 | ||
| 11i | OCH2CH(CH3)2 | 7.06 (15.4, 2.1) | 7.88 (15.4, 1.4) | 187.4 | 115.8 | 137.0 | ||
| 11j | OCHCH3CH2 OCH3 | 7.09 (15.5, 2.2) | 7.87 (15.5, 1.4) | 187.7 | 116.4 | 137.4 | ||
| 11k | OCH2CH2OH | 7.08 (15.5, 2.3) | 7.94 (15.5, 1.3) | 187.0 | 115.5 | 136.8 | ||
| 12a | 4-CF3-Ph | H | H | 7.63 (15.4) | 8.11 (15.4) | 188.2 | 115.1 | 140.3 |
| 12b | CH3 | 7.63 (15.4) | 8.06 (15.4) | 188.0 | 114.9 | 139.7 | ||
| 12c | OCH3 | 7.69 (15.4) | 8.02 (15.4) | 188.6 | 117.0 | 139.2 | ||
| 13a | 2-OH-Ph | H | H | 7.77 (15.2) | 8.21 (15.2) | 193.1 | 113.9 | 140.4 |
| 13b | CH3 | 7.75 (15.2) | 8.16 (15.2) | 193.0 | 113.9 | 139.7 | ||
| 13c | OCH3 | 7.82 (15.3) | 8.11 (15.3) | 193.1 | 115.7 | 138.7 | ||
| 14a | 4-OH-Ph | H | H | 7.64 (15.5) | 7.99 (15.5) | 187.0 | 115.4 | 137.7 |
| 14b | CH3 | 7.62 (15.5) | 7.94 (15.5) | 187.0 | 115.4 | 137.0 | ||
| 14c | OCH3 | 7.69 (15.5) | 7.91 (15.5) | 186.9 | 117.0 | 136.1 | ||
| 17a | 4-OH-Ph | H | 7.55 (15.4 | 7.63 (15.4) | 183.8 | 121.3 | 139.8 | |
| 17b | CH3 | 7.56 (15.8 | 7.56 (15.8) | 183.3 | 121.2 | 139.8 | ||
| 17c | OCH3 | 7.57 (15.5 | 7.59 (15.5) | 183.4 | 120.8 | 140.4 | ||
| 18a | 3,4-diOH-Ph | H | 7.47 (15.5) | 7.54 (15.5) | 183.8 | 121.2 | 140.3 | |
| 18b | CH3 | 7.47 | 7.47 | 183.3 | 121.1 | 140.4 | ||
| 18c | OCH3 | 7.50 (15.5) | 7.51 (15.5) | 183.4 | 120.8 | 140.9 | ||
| 19a | 3-OCH3-4-OH-Ph | H | 7.64 (15.4) | 7.55 (15.4) | 183.1 | 121.4 | 140.2 | |
| 19b | CH3 | 7.56 (15.6) | 7.55 (15.6) | 183.3 | 121.3 | 140.3 | ||
| 19c | OCH3 | 7.59 (15.5) | 7.58 (15.5) | 183.3 | 120.9 | 140.9 |
Table 2.
IC50 (μM) ± SD of tested compounds in different cell lines after 72 h incubation.
| comp. | Ar | R1 | R2 | Cell line, IC50 (µM) ± SD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MDA-MB-231 | HCT116 | Jurkat | Hela | MCF-7 | MCF-10A | Bj-5ta | ||||
| 11a | 2-F-Ph | H | H | 51.4 ± 3.2 | 31.8 ± 2.1 | 7.6 ± 0.4 | 41.7 ± 0.2 | 37.1 ± 0.4 | 89.1 ± 2.4 | NT |
| 11b[31] | CH3 | > 100 | 41.5 ± 1.7 | 32.4 ± 1.2 | 57.9 ± 0.4 | 47.5 ± 4.3 | > 100 | NT | ||
| 11c[7] | OCH3 | 21.1 ± 3.9 | 37.9 ± 2.7 | 5.9 ± 1.0 | 19.2 ± 5.7 | 20.0 ± 7.9 | 67 ± 2.9 | NT | ||
| 11d | H | OCH3 | 95.5 ± 2.7 | 45.5 ± 2.4 | 11.4 ± 1.9 | > 100 | 32 ± 1.2 | 98 ± 1.3 | NT | |
| 11e | OCH2CH3 | 72.2 ± 5.4 | 40.1 ± 0.6 | 8.3 ± 0.2 | 75.2 ± 4.1 | 30.3 ± 2.2 | 94 ± 4.6 | NT | ||
| 11f[32] | OCH2CH2CH3 | 34.0 ± 3.0 | 34.4 ± 1.1 | 37.7 ± 1.1 | 45.3 ± 0.7 | 37.3 ± 1.5 | > 100 | NT | ||
| 11g | OCH(CH3)2 | 33.3 ± 0.3 | 31.7 ± 1.7 | 36.7 ± 4.9 | 34.2 ± 0.1 | 17.1 ± 2.3 | 41.8 ± 1.5 | NT | ||
| 11h | OCH2CH2 CH2CH3 | 36.3 ± 2.1 | 31 ± 2.9 | 35.9 ± 1.8 | 48.2 ± 1.0 | 13.8 ± 3.5 | 65.5 ± 3.7 | NT | ||
| 11i | OCH2CH(CH3)2 | 42.9 ± 0.4 | 33.8 ± 0.1 | 40.1 ± 3.2 | 54.6 ± 3.6 | 30.7 ± 0.6 | 93.3 ± 3.4 | NT | ||
| 11j | OCHCH3CH2 OCH3 | 51.2 ± 4.1 | 32.4 ± 1.3 | 40.4 ± 2.1 | 50.0 ± 1.8 | 26.2 ± 0.5 | 76.5 ± 1.1 | NT | ||
| 11k | OCH2CH2OH | 64.5 ± 4.7 | 39.4 ± 1.5 | 37.8 ± 0.9 | 52.9 ± 1.6 | 38.1 ± 3.3 | 94.7 ± 2.6 | NT | ||
| 12a | 4-CF3-Ph | H | H | > 100 | 59.1 ± 1.1 | > 100 | 68.1 ± 3.3 | 43.3 ± 0.6 | > 100 | > 100 |
| 12b[31] | CH3 | > 100 | 12.3 ± 0.2 | > 100 | 32.8 ± 0.4 | 20.5 ± 5.1 | > 100 | > 100 | ||
| 12c | OCH3 | 81.3 ± 1.2 | 55.3 ± 1.7 | 39.3 ± 1.2 | 98 ± 2.8 | 86 ± 3.5 | 38.4 ± 3.1 | > 100 | ||
| 13a | 2-OH-Ph | H | H | 92.7 ± 1.8 | 40.5 ± 3.1 | 25.9 ± 2.4 | 39.6 ± 3.5 | 44.7 ± 2.6 | 55.4 ± 3.6 | 81.5 ± 0.2 |
| 13b | CH3 | 82 ± 3.2 | 34.7 ± 0.2 | > 100 | 42.8 ± 2.5 | 29.7 ± 2.3 | > 100 | > 100 | ||
| 13c | OCH3 | > 100 | 54.0 ± 2.2 | 42 ± 1.8 | 73.9 ± 3.4 | 53.4 ± 2.5 | > 100 | > 100 | ||
| 14a | 4-OH-Ph | H | H | 66 ± 2.5 | 36.8 ± 0.5 | 33.9 ± 2.5 | 39.9 ± 2.6 | 42.2 ± 0.7 | 51.5 ± 1.7 | 61 ± 0.6 |
| 14b | CH3 | > 100 | 49.3 ± 1.3 | > 100 | 93 ± 2.6 | 82.3 ± 1.8 | 54 ± 2.1 | > 100 | ||
| 14c | OCH3 | 82.2 ± 3.1 | 34.5 ± 0.7 | 7.3 ± 0.1 | 34.6 ± 0.3 | 52.7 ± 2.6 | 59 ± 0.2 | 76.4 ± 1.7 | ||
| 17a | 4-OH-Ph | H | 39.6 ± 2.1 | NT | NT | 31.4 ± 2.0 | 47.9 ± 0.2 | 28.6 ± 0.5 | NT | |
| 17b | CH3 | NT | > 100 | NT | NT | 50.3 ± 0.6 | 22.8 ± 0.4 | NT | ||
| 17c | OCH3 | > 100 | NT | NT | 39.8 ± 1.6 | > 100 | > 100 | NT | ||
| 18a | 3,4-diOH-Ph | H | 39.3 ± 1.9 | 29.8 ± 0.7 | NT | NT | 42.3 ± 3.3 | 11.5 ± 1.4 | NT | |
| 18b | CH3 | NT | 7.1 ± 0.7 | NT | 6.7 ± 0.8 | 37.4 ± 2.2 | 7.8 ± 0.3 | NT | ||
| 18c | OCH3 | > 100 | 18.2 ± 2.9 | 8.0 ± 1.4 | > 100 | > 100 | > 100 | > 100 | ||
| 19a | 3-OCH3-4-OH-Ph | H | NT | 62.7 ± 2.9 | NT | 25.5 ± 1.6 | NT | 24.7 ± 2.8 | NT | |
| 19b | CH3 | NT | NT | NT | 34.9 ± 1.3 | 69.8 ± 1.1 | 28.9 ± 0.8 | NT | ||
| 19a | OCH3 | 45.3 ± 0.5 | 35.3 ± 0.6 | 34.0 ± 0.3 | 41.4 ± 2.5 | 43.1 ± 1.6 | 40.2 ± 1.2 | 66.6 ± 0.1 | ||
Table 3.
Antioxidant activity.
|
Compond |
DPPH µmol GAE/mmol |
ABTS µmol GAE/mmol |
FRAP µmol GAE/mmol |
|---|---|---|---|
| 11a | 20.5 (±4.0) | 150.6 (±4.7) | 62.2 (±9.9) |
| 11b | 18.9 (±2.3) | 5.7 (±2.7) | 47.2 (±1.0) |
| 11c | 23.2 (±11.9) | 41.4 (±6.7) | 27.4 (±0.9) |
| 11f | 227.3 (±11.0) | 175.1 (±10.4) | 183.5 (±0.9) |
| 13a | 14.2 (±0.4) | 292.4 (±12.4) | 125.2 (±9.4) |
| 13b | 7.6 (±3.8) | 116.8 (±5.8) | 41.2 (±0.7) |
| 13c | 30.3 (±15.4) | 214.6 (±28.4) | 43.4 (±1.8) |
| 14a | 12.3 (±2.3) | 194.8 (±21.4) | 105.5 (±3.4) |
| 14b | 7.3 (±3.1) | 84.6 (±2.7) | 114.3 (±4.2) |
| 14c | 21.1 (±5.8) | 75.9 (±5.3) | 42.0 (±2.0) |
| 17a | 16.0 (±1.1) | 227.6 (±5.4) | 161.6 (±5.7) |
| 17b | 20.7 (±6.6) | 218.9 (±5.6) | 156.2 (±1.1) |
| 17c | 13.6 (±0.9) | 209.0 (±6.6) | 134.1 (±6.7) |
| 18a | 520.1 (±6.9) | 401.2 (±39.2) | 480.9 (±28.8) |
| 18b | 589.1 (±8.9) | 487.3 (±43.9) | 564.8 (±2.2) |
| 18c | 551.1 (±14.9) | 298.1 (±21.0) | 432.0 (±21.4) |
| 19a | 223.5 (±11.2) | 564.7 (±74.5) | 307.2 (±11.9) |
| 19b | 225.6 (±14.7) | 410.7 (±28.9) | 262.2 (±14.2) |
| 19c | 207.7 (±8.6) | 456.0 (±24.6) | 296.2 (±10.4) |
| p-coumaric acid | 23.0 (±3.1) | 278.8 (±11.1) | 105.2 (±4.2) |
| caffeic acid | 690.7 (±36.6) | 586.0 (±32.8) | 500.0 (±33.5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



