Submitted:
01 September 2023
Posted:
04 September 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The analysis reported here is unique in that it is the first study of the original data from the Pfizer/BioNTech BNT162b2 mRNA vaccine clinical trial (CA4591001) to be carried out by a group unaffiliated with the trial sponsor. Our study is a forensic analysis of the 38 trial subjects who died between July 27, 2020, the start of Phase 2/3 of the clinical trial, and March 13, 2021, the data end date of their 6-Month Interim Report. Phase 2/3 of the trial involved 44,060 subjects who were equally distributed into two groups and received Dose 1 of either the BNT162b2 mRNA vaccinated or the Placebo control (0.9% normal saline). At Week 20, when the BNT162b2 mRNA vaccine received Emergency Use Authorization from the U.S. FDA, subjects in the placebo arm were given the option to be BNT162b2 vaccinated. All but a few accepted. Surprisingly, a comparison of the number of subject deaths per week during the 33 Weeks of this study found no significant difference between the number of deaths in the vaccinated versus placebo arms for the first 20 weeks of the trial, the placebo-controlled portion of the trial. After Week 20, as subjects in the Placebo were unblinded and vaccinated, deaths among this still unvaccinated cohort of this group slowed and eventually plateaued. Deaths in the BNT162b2 vaccinated subjects continued at the same rate. Our analysis revealed inconsistencies between the subject data listed in the 6-Month Interim Report and publications authored by Pfizer/BioNTech trial site administrators. Most importantly, we found evidence of an over 3.7-fold increase in number of deaths due to cardiovascular events in BNT162b2 vaccinated subjects compared to Placebo controls. This significant adverse event signal was not reported by Pfizer/BioNTech. Potential sources of these data inconsistencies are identified.
Keywords:
BNT162b2 vaccine
; Pfizer/BioNTech
; cardiovascular events
; COVID-19
Introduction
The first human cases of a “novel” Coronavirus respiratory disease called COVID-19 were reported in Wuhan, People’s Republic of China in December 2019 . There is now significant evidence that suggests the virus was circulating in the United States as early as Fall of 2019 [1,2,3]. On January 30, 2020, the World Health Organization (W.H.O.) declared the COVID-19 outbreak a Public Health Emergency of International Concern and, on March 11, 2020, declared the outbreak a pandemic. In the United States, then Secretary of Health and Human Services, Alexander Azar, issued a Public Health Emergency Declaration under the PREP Act for medical countermeasures against COVID-19 on March 10, 2020. Thus, the race to develop a vaccine against COVID-19 began. The Public Readiness and Emergency Preparedness Act (PREP Act) passed the United States Congress and was signed into law by President George W. Bush in December 2005. It is a controversial tort liability shield intended to protect vaccine manufacturers from financial risk in the event of a declared public health emergency to be determined by the Secretary of Health and Human Services. Thus, the careful yet time-consuming processes normally followed to develop a safe and effective vaccine – foundational animal laboratory studies, the establishment of sound manufacturing and distribution standards, and a thorough review by regulatory agencies and medical review boards – could be waived.
Multiple pharmaceutical corporations jumped onboard to accept the challenge to develop, manufacture, animal test, and conduct massive human trials, in what was referred to as Operation Warp Speed. On December 10, 2020, a mere 9 months after HHS Secretary Azar’s Declaration and less than 6 months from the start of human trials, the U.S. Food and Drug Administration (FDA) made the highly controversial decision to grant Pfizer/BioNTech Emergency Use Authorization for their experimental BNT162b2 mRNA vaccine. Unfortunately, the evidence that this experimental product was “safe and effective” and “prevented transmission and serious illness”, was cloaked in secrecy and lacked even minimal transparency until June 2022.
BioNTech is a German biotechnology company that develops and manufactures active immunotherapies for patient-specific treatment of cancer and rare/orphan diseases as well as techniques for targeted protein replacement. In early 2020, BioNTech partnered with Pfizer, Inc to carry out a clinical trial to determine the efficacy and safety of BioNTech’s novel BNT162b2 mRNA SARS2-CoV vaccine. Pfizer/BioNTech applied to the U.S. FDA for a collaborative multi-national clinical trial entitled, “A Phase 1/2/3 Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of RNA Vaccine Candidates Against COVID-19 in Healthy Individuals” [4]. The application was approved by the FDA. Subject enrollment for Phase 1 of the 3-Phase trial began in April 2020, which was a small trial to determine the optimal dosing level of the vaccine. Phase 2/3 trial, the safety and efficacy phase involving over 43,548 subjects, began on July 27, 2020.
On November 20, 2020 Pfizer/BioNTech submitted to the U.S. FDA an Application for Emergency Use Authorization (EUA) for an Unapproved Product Review Memorandum [5]. The Application described the clinical trial results to the data cutoff data of November 14, 2020. The FDA made a copy of the EUA Application available on their website on December 11, 2020. This was the first opportunity for the general public and medical professionals to evaluate the clinical trial data supporting the safety and efficacy of their BNT162b2 mRNA vaccine. Polack et al. [6] published a journal article on December 10, 2020 entitled, “Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine,” summarizing these same findings. The authors of Polack et al. [6] consisted of the site administrators of the 153 clinical trial sites in over 7 different countries. Dr. Fernando P. Polack was Principal Investigator and site administrator of the trial site in Argentina and Dr. Stephen J. Thomas, the lead co-author, was the chief Principal Investigator of Clinical Trial CA4591001. Thus, the authors of these publications were intimately aware of the trial’s findings. On September 15, 2021, the same group of site administrators published another journal article entitled, “Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine through 6 months” [7]. With the knowledge and approval of the U.S. FDA, NONE of the original clinical trial data was to be made available for study by the world’s medical research community for 75 years. The health outcomes of the 44,060 subjects taking part in the trial were considered too sensitive for public review.
Public Health and Medical Professionals for Transparency (PHMPT), a non-profit alliance of over 80 public health officers and medical researchers, filled a FOIA lawsuit in the U.S. District Court, Fort Worth, Texas in September 2021 to obtain and disseminate the original clinical trial data upon which the FDA relied when it licensed Pfizer’s (Comirnaty) COVID-19 mRNA vaccine. To quote Dr. Aaron Kheriaty, one of the US physicians leading this court filing, “A group of us were concerned about the trial design, the shortened duration of the clinical trial, and the patchwork system that was in place for the post-marketing surveillance of adverse events.” The PHMPT case was approved. Over the objections of the FDA, a Federal Court Judge ordered the expedited release of Pfizer’s clinical trial data and documents at the rate of 55,000 per month. Data release began early in June 2022 to the Public Health and Medical Professionals for Transparency Documents site and was projected to take 8 months to complete. Unfortunately, it is taking much longer than estimated and documents continue to be downloaded to this site. The overwhelming size and complexity of these documents stimulated the formation of the DailyClout Pfizer/BioNTech Document analysis volunteers, a group of about 3,500 medical professionals, scientists, data analysts, statisticians, lawyers, and more, who have offered up their time and skills to analyze the Pfizer/BioNTech clinical trial documents. Team 3 is a subset of these volunteers dedicated to data investigation.
This report focuses on the 38 trial subjects listed in the Pfizer/BioNTech 6-month Interim Report [8] who died between the start of the trial on July 27, 2020 and March 13, 2021, the data end date of the 6-Month Interim Report. Our analysis revealed important inconsistencies between the subject data listed in the 6-Month Interim Report and the publicly available publications on this data submitted by Pfizer/BioNTech to the FDA: Pfizer/BioNTech’s FDA Application for Emergency Use Authorization [5], Polack et al. [6], and Thomas et al. [7]. Most alarming, we found evidence of an over 3.7-fold increase in number of deaths due to cardiovascular events in BNT162b2 vaccinated subjects that Pfizer/BioNTech did not report. Had this information been known at critical time points, it might have been sufficient to question the safety of the BNT162b2 mRNA vaccine, delay EUA approval of the vaccine, and alter recommendations made to the public during its worldwide roll-out.
Methods and Materials
The original Pfizer/BioNTech documents are available at Public Health and Medical Professionals for Transparency (PHMPT) website (https://phmpt.org/pfizers-documents/). The following documents were downloaded from this site and were main sources of data for our analysis.
- 6-Month Interim Report (16.2.7.4.1 Listing of Adverse Events – All Subjects ≥16 Years of Age) [8]
- Randomization Scheme and Actual Vaccine Received (16.1.7.1 Listing of Randomization Scheme and Actual Vaccine Received – All Subjects ≥16 Years of Age) [9]
- Listing of Discontinued Subjects (16.2.1.1 Listing of Subjects Discontinued From Vaccination and/or From the Study – All Subjects ≥16 Years of Age) [10]
- 6-Month Summary of Clinical Safety (2.7.4 Summary of Clinical Safety [11]
Documents 16.2.7.4.1 Listing of Adverse Events [8] and 16.1.7.1 Listing of Randomization Scheme and Actual Vaccine Received [9] were converted from PDF to Excel files and merged into a single Excel Pivot Table file. This allowed duplicate entries for a particular Subject ID to be removed and enabled searching for specific Preferred Terms for an Adverse Event. Thus, in a single searchable file listing all Subjects exhibiting an Adverse Event, we could determine the type of dose received (BNT162b2 mRNA vaccine or Placebo), the date that each Dose was administered, the date of onset of the Adverse Event, the Preferred Term for the Adverse Event, the Study Site physician’s diagnosis of the Adverse Event, and the decision of Pfizer’s safety physician whether the event was related to the trial.
Additional information came from the following.
- Pfizer/BioNTech Clinical Trial C4591001 – A Phase 1/2/3, Placebo-controlled, Randomized, Observer-blind, Dose-finding Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of SARS-COV-2 RNA Vaccine Candidates Against COVID-19 in Healthy Individuals (https://clinicaltrials.gov/ct2/show/NCT04816643)
- Emergency Use Authorization (EUA) for an Unapproved Product [5]
- Analysis Data Reviewer Guide – BLA Analysis for Participants ≥16 Years of Age, BioNTech SE and PFIZER INC, Study CA4591001 [12]
We also used the Abstractor search tool available on the DailyClout website (https://vaccines.shinyapps.io/abstractor/) to search for Clinical Reports Forms, Narratives, and other documents specific to a particular Subject ID’s, Preferred Terms, or clinical investigational data.
Results
On July 1, 2022, Pfizer/BioNTech released their report on the adverse events that occurred during the first 33-week period of the clinical trial, July 27 to March 13, 2021 entitled, 16.2.7.4.1 Listing of Adverse Events – All Subjects ≥16 Years of Age [8]. Section 16.2.7.7 of this document, found on pages 3640 – 3642, is a “Listing of Deaths – All Subjects ≥16 Years of Age”. Thirty-eight (38) subjects are reported as having died during this initial period. This document provides their Subject ID, Sex and Age at Death, and the Date of Death as well as the Primary Cause of Death for all 38 deceased subjects and a Secondary Cause of Death for 8 individuals.
We determined the vaccination status (BNT1626b2 mRNA vaccine or Placebo) of each deceased subject and the date that they received the first injection (Dose 1) using document 16.1.7.1 Listing of Randomization Scheme and Actual Vaccine Received – All Subjects ≥16 Years of Age [9]. To facilitate working with these two PDF files, they were converted to Excel files and merged into searchable pivot table format.
Overview of the initial 33 weeks of the trial. Phase 2/3 of the Pfizer/BioNTech BNT162b2 mRNA vaccine clinical trial began on July 27, 2020. Starting on this date, subjects who were deemed eligible by the screening process were randomized equally into the vaccinated or control arms of the clinical trial and received Dose 1 of either BNT162b2 mRNA vaccine or 0.9% normal saline Placebo, respectively. Almost all subjects randomized subjects had received Dose 2 by November 14, 2020 (Week 16). During this vaccination period to Week 16 and the initial weeks of the follow-up period to Week 20, subjects were followed for the occurrence of any adverse events (AE) and returned to the trial site for scheduled check-ups. This period of the trial is referred to by Pfizer/BioNTech as the “Blinded Placebo-controlled Period” and includes events from July 27 to December 10, 2020. In addition to July 27, 2020, four other important landmark dates are noteworthy.
- November 14, 2020 (end of Week 16) was the data cutoff date for Pfizer/BioNTech’s application to the US Food and Drug Administration (FDA) for Emergency Use Authorization (EUA) for their BNT162b2 mRNA vaccine [5].
- The application was submitted to the FDA on November 20, 2020 and included all data submitted to Pfizer/BioNTech from the 153 clinical trial sites through November 14, 2020. Data was collected several times each week from the trial sites. Since November 14, 2020 was a Saturday, we can assume that the data reported in the November 20th application, one week later, was completely up to date.
- December 10, 2020 (end of Week 20) Pfizer/BioNTech reported their results to the FDA’s Vaccines and Related Biological Products Advisory Committee (VRBPAC). The briefing documents [13] and a video of this meeting can be found (https://www.youtube.com/watch?v=owveMJBTc2I).
- December 11, 2020 began what Pfizer/BioNTech refers to as the “Open-label” or “Unblinded” Period. Their EUA application was approved by the FDA on December 11, 2020. The FDA also approved their request to unblind all subjects in the clinical trial. Unblinding means that they were permitted to inform all subjects whether they had received the BNT162b2 mRNA vaccine or the Placebo in Doses 1 and 2. Unblinded Placebo subjects were offered the BNT162b2 mRNA vaccine, Doses 3 and 4. All subjects of all vaccine status continued to be followed for 24 months. Required follow-up appointments were scheduled and, if needed, subjects were seen for emergency medical care. Trial site investigators were notified of hospitalizations and deaths. Deaths were immediately reported to Pfizer/BioNTech via an electronic reporting system.
- The period from December 11, 2020 to January 24, 2021 is referred to as the “Open-label Follow Up Period”. No explanation is given for the choice of January 24, 2021 but this date became evident from our analysis of the data reported in Thomas et al. [7].
- January 25, 2021 begins what Pfizer refers to as the “Open-label Observational Period”, which ended at the March 13, 2021 data cutoff date of the 6-Month Interim Report [8].
Flow charts showing the numbers of subjects at different stages of the trial are shown in Polack et al. [6] and Thomas et al. [7]. We found that the numbers reported were often not internally consistent within the published article and with numbers we determined based on the Listing of Discontinued Subjects [10]. Nevertheless, it is important to keep in mind that the number of Phase 2/3 subjects that were randomized and received Dose 1 were 22,030 BNT162b2 vaccinated and 22,030 Placebo for a total of 44,060 subjects. This number of doses could not be administered to all participants on the same day nor could return visits, whether scheduled or not, happen on the same day. Instead, all visits occurred over the course of weeks during the periods outlined above. Moreover, of the 20,794 subjects who originally received the placebo and were still trial subjects on December 11, 2020, only 19,685 were vaccinated after unblinding. Administration of the BNT162b2 vaccine to these individuals stretched over Weeks 20 to 33.
Deaths during 6-month safety period. Figure 1 plots the number of subject deaths per week over the period covered in Pfizer/BioNTech’s 6-Month Interim Report [8] as reported in Section 16.2.7.7. This document was generated on April 1, 2021 and thus should have accurate listings for the Date of Death. Important trial landmarks dates discussed above are marked in Figure 1. Week 1 started on Monday July 27, 2020, the date that subjects began to receive Dose 1. November 14, 2020, the EUA application data cutoff, was at the end of Week 16. December 11, 2020, the date the Pfizer/BioNTech EUA was approved, was the Friday of Week 20. March 13, 2021, the data cutoff for the 6-Month Interim Report [8], fell on the Saturday of Week 33. This 33 Week period was divided into 3 block, as described above and shown on Figure 1: the Blinded Placebo-controlled Period (July 27 to December 10, 2020); the Open-label Follow-up Period (December 11, 2020 to January 24, 2021); and the Open-label Observation Period (January 25 to March 13, 2021). The importance of these time periods will become clear later in this report.
The number of subject deaths in the BNT162b2 vaccinated and Placebo arms of the trial are plotted separately in Figure 1. Figure 1 also presents a plot of the cumulative number of deaths in trial each arm, as determined at the end of each week. The first Placebo subject death occurred in Week 5 and the first BNT162b2 vaccinated subject death occurred in Week 7. Very few deaths occurred in the first 12 weeks of the trial, probably because new subjects were still entering the trial.
Two things stand out from the results in Figure 1. First, the total number of deaths during this 6-month safety period is remarkably low. Only 38 individuals died from among the approximate 44,060 number of subjects in the trial pool. Second, the plots of the cumulative number of deaths in both arms of the trial appear to overlie one another until about Week 20 after which the cumulative plots diverge. Following Week 20, the number of deaths in the Placebo arm of the trial slows down and plateaus by about Week 30 while the number of deaths among BNT162b2 vaccinated subjects continues to increase at the same rate. This decreased rate in the Placebo arm likely results from the decrease in the numbers of unvaccinated Placebo subjects remaining in this arm of the trial due to the unblinding and vaccination process that occurred after December 11.
Causes of subject deaths. Table 1 details the information on the 38 deceased subjects shown in Figure 1. The data sources for Table 1 were the same as for Figure 1, the 6-Month Interim Report [8] and the Listing of Randomization Scheme and Actual Vaccine Received [9]. BNT162b2 vaccinated and Placebo subjects are listed separately. Within each grouping, subjects are listed according to their date of death. The two Unblinded Placebo subjects who died after receiving at least one dose of BNT162b2 vaccine are listed with the BNT162b2 vaccinated group and are highlighted in light gray. Table 1 includes the Subject ID assigned at the time of randomization for each deceased trial participant as well as their sex, age at death, and date of death. The data in Table 1 can be confirmed and original copies of the CRFs obtained using the DailyClout Abstractor and searching with the Subject ID.
Table 1 also lists the primary cause of death, and secondary cause of death for some, as given in the 6-Month Interim Report [8]. Investigators at each trial site were responsible for reporting all subject medical information to Pfizer/BioNTech for inclusion in the subjects Clinical Report Files (CRF). Deaths were to be reported immediately. Pfizer/BioNTech used a list of Preferred Terms that is based on the MedDRA coding dictionary, a standardized listing medical terminology for safety monitoring studies. The list includes 1,519 different Preferred Terms but, surprisingly, Death is not one of them. Often the Preferred Term used were vague and duplicative, which contributed to confusion regarding diagnoses. As we will show below, the lack of specificity in the terminology allowed the investigators to avoid requiring an autopsy to clarify the true cause of death, particularly in cases where myocardial infarction was a possibility. Myocardial infarction is a specific hypoxic irreversible injury to cardiac muscle tissue. In many of these 38 cases, the documentation provided in the CRF did not adequately support the cause of death diagnosis or did not allow one to rule out the possibility of a cardiovascular event with an autopsy. Frequent communications between Pfizer/BioNTech physicians and trial site medical staff are obvious in the CRFs, which were often quite lengthy some well over 400 to 900 pages.
Pfizer/BioNTech made available all 38 CRFs for those subjects who died. In general, our review of the CRFs found them to be lacking in detail and extremely difficult to interpret and develop a good timeline of events. Often, a subject’s pre-trial clinical history was absent. Absent also were results of the extensive array of medical testing carried out at the pre-trial screening and at other regularly scheduled visits. These test results include complete blood counts, metabolic tests, pregnancy tests, COVID-19 tests, a comprehensive list of active medications, and more, and would have clarified the subjects overall medical status. More detailed clinical data on the trial subjects exists but is still being withheld. Given the limitations of what has been provided, we determined that the information in the CRFs was frequently insufficient to support the investigator’s conclusions regarding the cause of death. In the more glaring cases, we indicated such in Table 1 with a superscript of 1 and 2. Interestingly, many of these concerns were also voiced by the Pfizer/BioNTech physician responsible for the dialog with the site medical managers suggesting that this critical interchange was often less than ideal even for internal review.
Working with what was available in the CRF and through DailyClout’s Abstractor, we evaluated each CRF. Our overall comments and concerns regarding the diagnosis of the causes of death are indicated in Table 1. The finding from this evaluation were particularly revealing and brief reports on several subjects are presented below. Two subjects, #11271112 and 10841266, are especially important because their CRF indicated that cardiovascular events likely contributed to their death, something that was not mentioned in Pfizer/BioNTech’s listing of their cause of death. Subjects #12291083 and #10971023 should also have been excluded from this list of 38 deceased subjects because they did not meet eligibility requirements at the time of randomization. Subject #10841470 had serious protocol deviations after randomization (see below). Because these 3 subjects are included in Pfizer/BioNTech’s list of 38 deaths, we did not remove them in our analysis.
Subject # 10841266 was a 77-year-old male with a history of severe vascular disease, gangrene, and multiple toe amputations likely related to diabetes and other comorbidities. He received a single dose of BNT162b2 vaccine after which he developed cholecystitis, had surgery, became septic, and died of multi-organ failure. No autopsy report is mentioned in the CRF, which is unfortunate because the CRF describes some confusion as to the primary cause of death. Emphysematous cholecystitis is a deadly bacterial form of gall bladder infection, which further increased the subject’s risks related to his severe diabetes. This infection started the cascade of events leading to death. The NSTEMI, a non-ST elevation myocardial infarction, first reported on November 23 was likely part of the cascade of organ failure. On November 23, the subject was hospitalized with elevated troponin levels and a suspected NSTEMI. Elevated troponin levels were confirmed on December 1 but on December 2 the entry into the CRF indicated that the NSTEMI was not considered a SAE, serious adverse event. This case had significant back and forth between the trial site and Pfizer/BioNTech regarding the primary cause of death and whether or not the subject had an NSTEMI in the hospital in addition to other reported issues. It appears that “sepsis” was his immediate cause of death but the NSTEMI should be listed as a contributing factor, at least as a secondary cause of death.
Subject # 10841470 is an obese 65-year-old Hispanic male with a medical history including pulmonary fibrosis and hypertension. He was in the Placebo arm of the trial and received Doses 1 and 2 on September 30 and October 21, 2020, respectively. On December 23, 2020, the subject received Dose 1 of the Moderna mRNA vaccine. This Protocol Deviation was reported in his CRF after the subject reported symptoms of COVID-19 on December 28, 2020 and was admitted to the hospital on December 31, 2020. While hospitalized, he became hypoxic and was intubated on January 2, 2021. He received monoclonal antibodies as part of his treatment in the hospital. Despite these efforts, the subject continued to deteriorate, lapsed into multisystem organ failure, and ultimately died on January 11, 2021. Subject #10841470 was in the List of Discontinued Subjects [10] as a “Death” and in the 6-Month Interim Report [8] as a Placebo death with COVID-19 as the secondary cause of death. This is a misrepresentation of the subject’s clinical information. The subject should have been discontinued from the Pfizer/BioNTech clinical trial because the “subject received non-study COVID-19 vaccine”.
Subject #11271112 was a 53-year-old Native American male with COPD and history of “stress related myocardial infarction”. The subject died suddenly of “cardiopulmonary arrest” on December 4, 2020, less than two months after Dose 2 of the BNT162b2 vaccine. On December 18, after the subject’s death, the trial site medical monitor listed the cause of death as “cardiopulmonary arrest related to myocardial infarction”. On December 19, Pfizer/BioNTech informed the trial site that multiple causes of death cannot be entered into the CRF and requested that “related to myocardial infarction” be deleted. The medical monitor refused to change the wording of the entry. On January 5, 2021, Pfizer/BioNTech overrode the trial site and changed the cause of death to “cardiopulmonary arrest” and chose not to list “myocardial infarction” as a secondary cause of death, which was an option that could have been used to deal with such conflicting conclusions. It is not clear why a specific diagnosis of an AESI was later changed to something undefined. Without the critical autopsy report to either confirm or deny the on-site medical monitor’s diagnosis, we felt it most appropriate to include this subject in the cardiovascular signal event group.
Subject #11621327 was found dead shortly after receiving Dose 1 of the BNT162b2 vaccine on September 10th. His body was found at home (with lividity) on the 13th of September when the police performed a welfare check. “According to the medical examiner, the probable cause of death was progression of atherosclerotic disease.” The cause of death listed in the 6-Month Interim Report [8] is “Arteriosclerosis”. However, there were multiple queries in the CRF about the cause of death being ascribed to atherosclerosis. Atherosclerosis was not documented in the CRF as a comorbidity of the patient. The subject’s CRF is only 127 pages in length and does not include the pre-screening portion of comorbidities, the section of the CRF that would have provided evidence on whether the subject had a history of atherosclerosis. Moreover, if an autopsy had been done, progression of atherosclerosis would have been documented but autopsy results were not provided or available. Based only on the medical documentation in the CRF, there is no basis for ascribing the subject’s death to advanced atherosclerosis or concluding that the death was unrelated to the vaccine. The following statement taken from the interim narrative document for the corresponding subject, in our opinion, is unfounded. “In the opinion of the investigator, there was no reasonable possibility that the arteriosclerosis was related to the study intervention, concomitant medications, or clinical trial procedures, but rather it was related to suspected underlying disease.” Pfizer/BioNTech concurred with the investigator’s causality assessment. It is likely that the subject died within a day or two of vaccination. This was a clear indication that his death could have been related to the BNT162b2 vaccine and this should not have been ruled out without a more rigorous investigation. In our opinion, this diagnosis was premature and an egregious misjudgment of the evidence at hand.
Subject #12291083 received the Placebo and died 76 days after Dose 1. The primary cause of death was first diagnosed as Diabetes mellites based on the subject’s medical history. This diagnosis was revised several times, despite the presence of very high blood-glucose levels, until finally settling on COVID-19 pneumonia as a secondary cause of death. The subject was HIV positive with a HIV RNA load of 50 copies per ml, which is just over the acceptable limit for inclusion in the trial. The subject should not have been randomized and approved as a trial participant.
Subject ID #12314987 was a 47-year-old male with a history of hypertension, obesity, and a smoker for 27 years. He received the Placebo and died 82 days after Dose 1. At an unscheduled visit he presented with abdominal pain, vomiting, and back pain at 9 PM on December 5, 2020 and died in the hospital at 7 AM the next morning, December 6, 2020. No record of an autopsy is available and the family was not responsive to inquiry. The cause of death was deemed "non traumatic cardiorespiratory arrest" but given the subject’s medical history, a firmer diagnosis should have been aggressively pursued.
Subject ID #12315324 received the Placebo and died 136 days after Dose 1. The primary cause of death was listed as “Multiple organ dysfunction syndrome” but the symptoms support a diagnosis of COVID-19. It appears that the otherwise healthy subject was hospitalized with COVID-19 symptoms. The patient required mechanical ventilation due to lobar pneumonia in the ICU and was documented as having acute kidney failure requiring dialysis. Other than vasopressors, there is no record of any other medication that the patient received as part of their hospital care.
In conclusion, we had no choice but to accept the cause of death diagnoses listed in the 6-Month Interim Report [8] as accurate, with the exception of subjects #11271112 and #10841266. Based on our medical expertise and in the interest of simplifying the search for potential safety signals among these 38 deceased subjects, we grouped the terms myocardial infarction, cardiac arrest, sudden cardiac death, cardiac failure congestive, and arteriosclerosis under the umbrella term “cardiovascular events”. Subjects diagnosed to have died as a result of a cardiovascular event are indicated with an asterisk* in Table 1. In the two exceptional cases, subjects #11271112 and #10841266, Table 1 still lists the cause of death as determined by Pfizer/BioNTech but in our opinion myocardial infarction could not be excluded as a cause of death. Therefore, subjects #11271112 and #10841266 were included in our “cardiovascular events” group, as indicated by the asterisk* next to the diagnosis of the cause of death.
Discrepancies in reports on subject deaths. Comparison of the data plotted in Figure 1 to the results reported in the Pfizer/BioNTech EUA application [5], Polack et al. [6], and Thomas et al. [7] revealed several discrepancies. Discrepancies among these various data sources are particularly disconcerting. The data with which we are working comes directly from Pfizer/BioNTech’s 6-Month Interim Report [8] on Clinical Trial C4591001 in the section entitled “Listing of Deaths – All Subjects ≥16 Years of Age”. As such, it should be entirely consistent with data presented in the other Pfizer/BioNTech documents and published reports. These discrepancies are illustrated in Table 2.
Table 2 compares the results reported by Pfizer/BioNTech, Polack et al. [6], and Thomas et al. [7] (left column) to the data from our analysis of the 6-Month Interim Report [8] (right column). The data is reported by time periods as shown in Figure 1: Blinded Placebo-controlled Period to EUA application Data Collection Cutoff (July 27 to November 14, 2020), the Blinded Placebo-controlled and Open-label Follow-up Period (July 27 to January 24, 2021), and the Open-label Observational Period to Data Collection Cutoff of the 6-Month Interim Report (January 25 to March 13, 2021). The basis for selecting January 24, 2021 as the end of the Open-label Follow-up Period is unclear and not explained in Thomas et al. [7]. The last section of Table 2 is a summary of the full 6-Month Period (July 27, 2020 to March 13, 2021). It should be noted that both Polack et al. [6] and Thomas et al. [7] have internal inconsistencies between the number of deaths reported in their flow charts and the number reported the text of the manuscript. These inconsistencies do not appear to have been identified by reviewers of either manuscript.
Data reported in the EUA application (July 27 to November 14, 2020). The first section of Table 2 compares Pfizer/BioNTech’s published data to the data reported here in Figure 1 and Table 1. The first 16 Weeks are the most important period of the clinical trial because the decision as to whether to approve the BNT162b2 vaccine rested entirely on these results. Pfizer/BioNTech’s EUA application [5] and Polack et al. [6], which was published on December 10, 2020 and updated on December 16th, reported that only 6 trial participants died prior to November 14, 2020: 2 in the vaccinated arm of the trial and 4 in the placebo arm. Based on comments on the cause of death in Polack et al. [6], we determined the Subject IDs of these 6 subjects. These are marked with a superscript # in Table 1. In contrast, our findings in Figure 1 and Table 1 show 11 deaths in total prior to November 14 (Week 16), 6 subjects in the vaccinated arm and 5 in the placebo arm. A careful review of the date of death of the 6 deceased subjects reported by Polack et al. [6] shows that they include only 2 of the 6 vaccinated and 4 of the 5 Placebo subjects whose date of death we found to be before November 14. This is the first discrepancy noted in our analysis.
Table 2 compares the results reported by Pfizer/BioNTech, Polack et al. [6], and Thomas et al. [7] (left column) to the data from our analysis of the 6-Month Interim Report [8] (right column). The data is reported by time periods as shown in Figure 1: Blinded Placebo-controlled
Period to EUA application Data Collection Cutoff (July 27 to November 14, 2020), the Blinded Placebo-controlled and Open-label Follow-up Period (July 27 to January 24, 2021), and the Open-label Observational Period to Data Collection Cutoff of the 6-Month Interim Report (January 25 to March 13, 2021). The basis for selecting January 24, 2021 as the end of the Open-label Follow-up Period is unclear and not explained in Thomas et al. [7]. The last section of Table 2 is a summary of the full 6-Month Period (July 27, 2020 to March 13, 2021). It should be noted that both Polack et al. [6] and Thomas et al. [7] have internal inconsistencies between the number of deaths reported in their flow charts and the number reported the text of the manuscript. These inconsistencies do not appear to have been identified by reviewers of either manuscript.
Data reported in the EUA application (July 27 to November 14, 2020). The first section of Table 2 compares Pfizer/BioNTech’s published data to the data reported here in Figure 1 and Table 1. The first 16 Weeks are the most important period of the clinical trial because the decision as to whether to approve the BNT162b2 vaccine rested entirely on these results. Pfizer/BioNTech’s EUA application [5] and Polack et al. [6], which was published on December 10, 2020 and updated on December 16th, reported that only 6 trial participants died prior to November 14, 2020: 2 in the vaccinated arm of the trial and 4 in the placebo arm. Based on comments on the cause of death in Polack et al. [6], we determined the Subject IDs of these 6 subjects. These are marked with a superscript # in Table 1. In contrast, our findings in Figure 1 and Table 1 show 11 deaths in total prior to November 14 (Week 16), 6 subjects in the vaccinated arm and 5 in the placebo arm. A careful review of the date of death of the 6 deceased subjects reported by Polack et al. [6] shows that they include only 2 of the 6 vaccinated and 4 of the 5 Placebo subjects whose date of death we found to be before November 14. This is the first discrepancy noted in our analysis.
Of the 6 deceased subjects reported by Pfizer/BioNTech, Table 1 indicates that 1 of the BNT162b2 vaccinated subjects and 2 of the Placebo subjects died of a cardiovascular event. In Polack et al. [6] and the Pfizer/BioNTech EUA application [5], it is stated that the trial investigators did not consider any of these deaths to be related to the vaccine. By comparison, our analysis of the 11 deaths observed in the Blinded Placebo-controlled period shows that about half were due to cardiovascular events: 4 in the BNT162b2 vaccinated and 2 in the Placebo arms. While the numbers are small, they represent a 2-fold increase in cardiovascular events in BNT162b2 vaccinated subjects. This should have alerted Pfizer/BioNTech to the possibility that cardiovascular events could be a vaccine-related signal event. It did not because information on 5 subjects who died prior to November 14 had not been reported to the FDA in the EUA application. Long gaps exist between the actual date of death and the date that this was officially recorded in the subject’s CRF (Table 3).
December 10, 2020 presentation to the FDA. On December 10th, Pfizer/BioNTech presented their evidence supporting their request for Emergency Use Authorization of their BNT162b2 mRNA vaccine [5] to the FDA’s Vaccines and Related Biological Products Advisory Committee (VRBPAC). This presentation took place 25 days after the November 14th data cutoff date for the EUA application. It was an opportunity for Pfizer/BioNTech to update their results to December 10th. Instead, Pfizer/BioNTech representatives reported the exact same results as those that appeared in the EUA application 25 days prior. CA4591001 was an ongoing clinical trial. It would not have been unusual to find additional deaths during this time period. In fact, our analysis of the 6-Month Interim Report [8] indicates that 6 more subjects died between November 14 and December 10, 2020 bringing the actual total number of deaths to 17 between July 27th and December 10th. Sixteen (16) of these 17 subjects, 8 BNT162b2 vaccinated and 8 Placebo, were known to Pfizer/BioNTech by December 10th. This is the second discrepancy noted in our analysis.
A careful review of the CRFs for each of the 38 deceased subjects revealed that the date of death recorded in the 6-Month Interim Report was not officially recorded in the subject’s CRF for several days, sometimes weeks. We decided to explore a possible pattern in the delay. Table 3 groups the 38 deceased subjects based on whether they received the only the Placebo or the BNT162b2 vaccine, either originally or after unblinding. The 6 subjects reported in the Pfizer/BioNTech EUA application [5] and in Polack et al. [6] are indicated by superscript #. Subjects whose listing is highlighted in gray are those whose death was not discussed at the FDA’s VRBPAC meeting but whose death was in fact known to Pfizer/BioNTech on December 10th, the date of their presentation. Based on the information in Table 3, Pfizer/BioNTech knew of 10 more subjects who died between November 14 and December 10th bringing their total number of officially recorded deaths to 16 (see Table 3 rows shaded in gray or marked with #). (Subject #10881126’s death on December 1 was not officially recorded in the CRF until 72 days later on February 11, 2021 and is not included.) These 16 deaths known to Pfizer/BioNTech were equally distributed to both arms of the trial, 8 in the BNT162b2 vaccinated arm and 8 in the Placebo arm. The causes of death are not. Based on our determination of the number of the cardiovascular events as reported in Table 1, the number of deaths related to a cardiovascular event is 5 in the BNT162b2 vaccinated arm and 2 Placebo arm, a 2.5-fold increase in the cardiovascular signal in BNT162b2 vaccinated subjects. If instead we use “cardiac arrest”, the only cardiovascular event reported by Pfizer/BioNTech by this date, the numbers are 2 in the BNT162b2 vaccinated arm and 2 Placebo arm, that is, balanced between the trial arms.
Pfizer/BioNTech should have voluntarily made known any new information that could contribute to the FDA’s decision. It was factually misleading for them not to do so. On the other hand, everyone at the VRBPAC meeting should have realized that the data from November 14th was outdated. Surprisingly, no members of VRBPAC requested an update on adverse events that occurred between the EUA data cutoff date (November 14) and the date of this meeting (December 10) (https://www.youtube.com/watch?v=owveMJBTc2I). Nor did they request more information on the causes of death and evaluate the deceased subjects CRFs independently. Sixteen deceased subjects is a manageable number and this was a critical point in the approval process. It appears that the FDA decision to approve the Pfizer/BioNTech EUA was based solely on 16 Weeks of data, data that was a misrepresentation of the full story that was not evaluated with a critical eye.
Blinded Placebo-controlled and Open-label Follow-up periods. This section of Table 2 reports on the first 26 Weeks of the trial. Here, the numbers and causes of subject deaths reported in the Pfizer/BioNTech 6-Month Summary Clinical Safety [11] and Thomas et al. [7] are compared to those reported in the Pfizer/BioNTech 6-Month Interim Report. The Summary Clinical Safety [11] was submitted to the FDA on May 5, 2021 and includes data up to March 13, 2021. Table 7 of this report lists the number of subjects who died in each arm of the trial from receipt of Dose 1 to the Unblinding Date (not defined) and Table 16 gives the cause of death. The information in Table 16 is reproduced in Table S4 of Thomas et al. [7] with no updating despite the fact that Thomas et al. [7] was published on September 15, 2021. Conclusions reported in both documents are identical: 15 deaths in the BNT162b2 group and 14 deaths in the placebo group during the blinded placebo-controlled period for a total of 29 deaths.
Section 2.7.4.2.4.2.2.1.1 of the Summary Clinical Safety [11] reports that two deaths occurred among the subset of 200 HIV-positive Phase 2/3 participants, one from each trial arm, and both were withdrawn from the study. Subject IDs for these participants were #11561160 and #12291083. Thomas et al. [7] appears to have included these 2 subjects in the flow chart figure that show the disposition of subjects during the trial. It appears that only one of these subjects was excluded to arrive at the final number of 29 deaths reported in the text of the article. Our analysis of the CRF for subject #12291083 indicates that this individual was ineligible for randomization (discussed above) but, because subject #12291083 was retained in the list of 38 deceased subjects, we did not exclude this individual from our accounting in Table 2. No information is available on why subject #11561160 might also have been excluded or if these were the excluded subjects. The disposition of the HIV positive subjects in Thomas et al. [7] and how Thomas et al. [7] arrives at the total of 29 subject deaths is not presented clearly. This confusion represents an internal inconsistency in their data and could explain one of the difference between our data showing total number of 30 subject deaths and that of Thomas et al. [7].
Of the 30 deaths during this first 26 Week period of the trial, we found a total of 13 deaths due to a cardiovascular event, 10 in the BNT162b2 vaccinated group and 3 in the Placebo. Cardiovascular events clearly constitute an adverse event safety signal for the BNT162b2 vaccine. Surprisingly, this signal was not mentioned by Pfizer/BioNTech. Thomas et al. [7] states, “No new serious adverse events were considered by the investigators to be related to BNT162b2 after the data cutoff date of the previous report” and “No new safety signals were observed during the longer follow-up period.” Since 3 of the 6 subjects that Pfizer/BioNTech reported to have died prior to November 14 died of cardiovascular events (myocardial infarction and arteriosclerosis), Thomas et al. [7] presumably did not consider deaths after that date to be due to a “new” adverse events. The number of total subject deaths and the imbalance in the causes of death is the third discrepancy noted in our analysis.
Open-label Observational Period to March 13, 2021. Thomas et al. [7] and Pfizer/BioNTech in the Summary Clinical Safety [11] report 3 deaths in the BNT162b2 group and 2 in the unblinded BNT162b2 vaccinated originally Placebo group in the trial period we entitled “Open-label Observational Period”, as shown in the third section of Table 2. We show a total of 8 subjects who died in this period: 3 in the BNT162b2 group, 2 in the unblinded BNT162b2 vaccinated original Placebo group, and 3 in the original Placebo group that were never vaccinated. It is not clear why Pfizer/BioNTech excludes this last group of subjects. Of the 8 deaths we report during Weeks 27-33, we found 1 cardiovascular event in the BNT162b2 vaccinated group and none in the Placebo arm. Thomas et al. [7] says that the “Causes of death were balanced between the BNT162b2 and Placebo groups”. Again, the number of total subject deaths during this final period of the 6-Month Interim Report and the small imbalance in the cardiovascular deaths is the fourth discrepancy noted in our analysis.
Summary of subject deaths. The last section of Table 2 provides a full accounting of the deaths that occurred over the first 33 weeks of the Pfizer/BioNTech CA4591001 clinical trial. Pfizer/BioNTech account for a total of 34 subjects who died during the 6-month follow-up period, 20 subjects who received BNT162b2 vaccine and 14 who were in the placebo control group. As discussed above, four of the 38 deaths listed in the 6-Month Interim Report are not included in their calculations: possibly the 2 HIV positive subjects, #12291083 and 11561160, and 2 Placebo subjects who died after January 24, 2021. Our results account for all 38 subject deaths: 21 deaths in the BNT162b2 vaccinated subjects and 17 in the Placebo. Three of the 38 should not have been listed in the 6-Month Interim Report, which would have brought that number to 35 subject deaths. Subjects #12291083 (Placebo) and #10971023 (BNT162b2) did not meet eligibility requirements and should have been excluded before randomization. Subject #10841470 (Placebo) received a received non-study COVID-19 vaccine. Interestingly, COVID-19 is given as the cause of death of both of these Placebo subjects.
Of the 38 deaths reported in the 6-Month Interim Report [8], the foundational document of our forensic analysis, we revealed that 14 subjects died from a cardiovascular event, over one-third of all deaths (36.8%). Of these 14, 11 were from the BNT162b2 vaccinated trial arm and 3 from the Placebo-only trial arm. This represents a 3.7-fold increase in cardiovascular events in subjects who received the BNT162b2 vaccine. Thomas et al. [7] and Pfizer/BioNTech’s Summary Clinical Safety [11] do not identify or remark on this clear serious adverse event signal.
Sources of the data discrepancies. The data discrepancies described above are critical to understanding why the cardiovascular safety signal was not reported to the public in a timely fashion. Table 3 provides an explanation. Our analysis of the data in Table 3 discussed above, showed that Pfizer/BioNTech used the date that the death was officially recorded in the CRF to determine which time period to report the death NOT the actual date of death, although both were available to them. According to the CA4591001 Protocol, Pfizer/BioNTech was to be notified of a subject death immediately. We do not have access to records that would have confirmed that the trial sites were diligent regarding death notification but the existence of other steps in the death notification process are alluded to in the CRFs that could have played a role in delaying entries into the CRF. Preliminary databases, such as a Death Details Form, are suggested in interactions logged into the CRFs. We have not been given access to any of these. Completion of the Death Details Form, and perhaps other requirements, appears to be partly computerized and automatic. There may be other steps as well. In addition, a member of the Pfizer/BioNTech pharmacovigilance leadership team was needed to review and approve entries into the CRF, particularly regarding the cause of death and whether event was “vaccine related”.
To identify possible patterns in this recordkeeping delay, we determined the delay in officially recording the death of all 38 deceased subjects reported in Table 3 during July 27, 2020 to March 13, 2021. We divided this into two time periods comparing the recording delays prior to December 11, 2020, the date the EUA was approved by the FDA, to those after EUA approval. All told, Table 3 shows that this official notification process contributed to the recordkeeping delays in recording data in the CRFs. For example, of the 5 deceased subjects missing from Polack et al. [6] but included in our list of 11 subjects who died prior to November 14th, Pfizer/BioNTech could justify their exclusion based on the delay in officially recording their death in the CRF. What other patterns became clear when all 38 deceased subjects were studied?
Of the 8 BNT162b2 vaccinated subjects that should have been reported to the VRBPAC on December 10th, the median reporting delay was 18 days (average of 17.5 days). Among the 8 Placebo subjects, the median delay was 5 days (average of 5.9 days). When the recording delay after December 11 is analyzed, we found a dramatic decrease in both arms of the trial. The median delay in the BNT162b2 arm of the trial was 7 days (average 9.8 days) and in the Placebo arm the delay was 3 days (average of 15.9 days). The median is a better measure of the delay because a small number of outliers such as 50 and 72 skew the average.
These results are a clear demonstration that the long official recording delays are not distributed equally between the two arms of the trial but are clustered in the BNT162b2 vaccinated arm, particularly before FDA approval of the EUA. Once the EUA was approved, Pfizer/BioNTech reported the date of death in a timelier fashion, although delays were still longer among vaccinated subjects. Instead of using the actual date of death, Pfizer/BioNTech based critical aspects of their analysis on when that death was officially recorded in the CRF. Had the recording delays been comparable in both arms of the trial, this technicality might not have had an impact on the analysis. Unfortunately, that was not the case and Table 3 demonstrates this.
Discussion
This study is the first analysis of the original trial data from the Pfizer/BioNTech BNT162b2 mRNA vaccine clinical trial (CA4591001) carried out by a group unaffiliated with the trial sponsor. The small number of deaths reported in Pfizer/BioNTech’s initial 6-Month Interim Report [8] allowed us to carry out an in-depth study at a level of detail that would not otherwise have been possible on such a large dataset. As such, it is best described as a forensic analysis of these 38 deaths. We reveal that reports on the BNT162b2 mRNA vaccine clinical trial done in public forums by Pfizer/BioNTech and their representatives were presented in a manner that obscured the actual trial results. Flawed data analysis and reporting errors by Pfizer/BioNTech played a role in the handling of the COVID-19 pandemic.
38 Deaths is a surprisingly low number. Given the large number of participants in the clinical trial, upwards of 44,060 subjects, the expected number of deaths would have been expected to be greater than the 38 reported in the 6-Month Interim Report [8]. In 2018, the year prior to the appearance of SARS2-CoV19, the United Nations estimates the all-cause all-ages death rate at 7.546 per 1,000 persons per year which would correspond to an expected number of 210.5 deaths during a 33 week period in 2018 (https://www.macrotrends.net/countries/WLD/world/death-rate). Thirty-eight (38) deaths is less than 20% of this expected number. Clinical trial CA4591001 clinical trial excluded people younger than 15 years and older than 85, age groups that normally include the largest percentage of deaths under normal circumstances. Nevertheless, it would be surprising if 80% of the deaths during 2020 would be in these excluded age groups. The 6 deaths of the 37,066 subjects in the safety population reported by Pfizer/BioNTech on November 14th is also unexpectedly lower than the expected 85.6 deaths during the 16-week period (16.6%). The COVID-19 infection rate was at a peak in the summer/fall of 2020 and preferentially impacted older individuals. Nonetheless, the pandemic cannot explain this rather low death rate among clinical trial subjects. It should be noted that, to our knowledge, at no time did any members of any international medical health regulatory agencies or medical literature reviewers, who evaluated the Pfizer/BioNTech trial data comment on this finding and request an explanation.
One possible explanation for the low number of subject deaths lies in the large number of “Discontinued Subjects” in CA4591001, 4.2% of the randomized subjects. The most disturbing of the reasons for discontinuation was “Lost to Follow-up”. These were subjects who did not show up for scheduled visits or other required protocol tasks. Apparently, trial site staff made attempts to contact these individuals via phone and certified mail or via an emergency contact but eventually after multiple attempts the effort was abandoned. Based on the List of Discontinued Subjects file [10], we found 395 unique subjects listed as “Lost to Follow-up” during the period of 6-Month Interim Report: 178 in the BNT162b2 vaccine and 217 in the Placebo arms. Of these, 203 (99 in the BNT162b2 vaccine and 104 in the Placebo arms) were lost prior to November 14, 2020, the data cutoff date for the Pfizer/BioNTech EUA application and 192 (79 in the BNT162b2 vaccine and 113 in the Placebo arms) after that date up to March 13, 2021. These are not insignificant numbers and could easily account for the low number of deaths reported in this safety period of the trial.
Rate of death is not decreased by BNT162b2 vaccination. Figure 1 clearly shows that the plots of the cumulative numbers of death in both the BNT162b2 vaccinated and Placebo arms of the trial overlie each other for about the first 20 Weeks (July 27, 2020 to December 11, 2020). This is an entirely unexpected finding. During the fall of 2020 the spread of COVID-19 was at its peak. To state that the BNT162b2 vaccine saved lives, Pfizer/BioNTech should have shown a reduction in all-cause mortality due to a decrease in COVID-19 mortality in the vaccinated arm of the trial. Figure 1 does not support any such claim for Weeks 1 – 20 and, in fact, speaks against this conclusion in the weeks following Week 20 in which the Placebo cumulative plot is distinctly below that of the BNT162b2 vaccinated. Week 20 is the point at which unblinding and BNT162b2 vaccination of the Placebo arm began. It is likely that the slowed rate of increase in the number of Placebo deaths and the plateau at Week 30 result from the gradual reduction in the size of the Placebo group. If the BNT162b2 vaccine were 95% effective, as claimed, the plots would have been reversed.
Any intervention whether it is a drug or a therapeutic procedure may have unrecognized risks and adverse side effects that may negate any positive effect. This is the basis for carrying out a placebo-controlled randomized trial. As is clear in this data, random deaths occurred in the Placebo (control) arm of this study. The Placebo arm is required to get the baseline numbers for the randomized subject population. Without this control, it is impossible to say with confidence that the treatment under study is having a positive, negative, or no effect at all. It is entirely inappropriate and poor scientific policy to compare a test subject population to the population at large. By approving the unblinding and vaccination of subjects in the Placebo arm of this study, the FDA ended all semblance of placebo-controlled clinical trial on December 11, 2020.
Causes of death are unbalanced between the two arms of the trial. The data in Table 1 and Table 2 show that, despite the finding that the all-cause mortality in both arms of the trial are similar, the causes of death are not balanced. We found that 14 of the 38 deaths, well over one-third of the deaths (36.7%), were the result of cardiovascular events, a 3.7-fold increase in deaths due to cardiovascular events in the treatment arm of the clinical trial. Moreover, the increased number of deaths due to cardiovascular events more than accounts for the difference between the number of deaths in the BNT162b2 arm (21 deaths) compared to the number in the Placebo arm (17 deaths). Given the fact that deaths due to cardiovascular events had been occurring from at least Week 5, why was the unbalance not reported by the sponsors of the trial prior to Week 20? Several factors contributed.
In summary, the CRF system used by Pfizer/BioNTech for the CA4591001 clinical trial did not conform with accepted industry standards and probably contributed to confusions regarding the cause of death of trial subjects [8]. The diagnoses listed in Table 1 were often not evidence-based and the CRFs lacked transparency, were not user friendly, and did not appear to provide a complete “chain of custody” of the responses between Pfizer/BioNTech and the trial site medical monitors. These issues become particularly relevant with regard to subjects 11271112 and 10841266, whose cause of death should have been attributed, at least in part, to myocardial infarction or progression of a pre-existing MI condition. Trial coordinators were dealing with only 38 deaths, the most serious of serious adverse events (SAEs). Their paramount should have been to determine the true cause of death.
In a placebo-controlled randomized clinical trial, causality is determined on a total statistical basis rather than by the commercial sponsor of the trail on a case-by-case basis, as was done in this trial by the sponsor Pfizer/BioNTech. The various oversight boards and the FDA’s VRBPAC relied on Pfizer/BioNTech to identify and report any safety signals. Due diligence was not done to confirm the trial sponsor’s data evaluation. The “Related to Vaccination” category should not have been included in the 6-Month Interim Report [8] or any of Pfizer/BioNTech’s published reports. Whether an adverse event is related to the treatment under investigation should have been determined by the regulatory agency overseeing the trial. Since CA4591001 was an on-going study, the appropriate time to do that would have been on the day Pfizer/BioNTech made its presentation to the FDA VRBPAC, December 10, 2020. This was not done nor was Pfizer/BioNTech required to update their trial data to December 10th.
Our analysis (Table 2) shows that discrepancies in the numbers of deaths reported are observed at two critical time points in the study – November 14th, the data cutoff date for the EUA application, and December 10th, the date of Pfizer/BioNTech’s presentation to the FDA VRBAC – had the effect of obscuring a 3-fold increase in the number of deaths due to cardiovascular events, a critically important safety signal that the FDA wanted highlighted as an Adverse Event of Special Interest (AESI) in the CA4591001 protocol (https://clinicaltrials.gov/ct2/show/NCT04816643).
We found that Pfizer/BioNTech used unnecessarily confusing terminology in their reports. Their list of Preferred Terms had more categories than warranted in light of the lack of rigor of the CRF medical information. Additionally, rather than simply giving exact start and ending dates for a time period, Pfizer/BioNTech used vague terms such as “Open-label observational period” or “After the Unblinding”. This was particularly relevant during the analysis of the “Open-label follow-up period” and “Open-label observational period” and likely contributed to the loss of 3 Placebo subjects by Thomas et al. [7]. Titles of tables found in Pfizer/BioNTech’s Summary Clinical Safety 6-month report [11] do more to confuse the reader than to clarify the data reported therein. Two typical examples follow: “Table 13: Number (%) of Subjects Reporting at Least 1 Adverse Event From Dose 1 to 6 Months After Dose 2, by System Organ Class and Preferred Term – Subjects With at Least 6 Months of Follow-up Time After Dose 2 – Phase 2/3 Subjects ≥16 Years of Age (Subjects Who Originally Received BNT162b2) – Safety Population” and “Table 19: Incidence Rates of at Least 1 Serious Adverse Event From Unblinding Date to Data Cutoff Date (13MAR2021), by System Organ Class and Preferred Term – Open-Label Follow-up Period – Subjects Who Originally Received BNT162b2 – Phase 2/3 Subjects ≥16 Years of Age – Safety Population”.
All told, these techniques served to obfuscate the true evidence being revealed by the CA4591001 clinical trial. The CRF format used by Pfizer/BioNTech was not up to normally expected standards and not transparently maintained. A subject’s true date of death was not recorded in their CRF in a timely fashion for all subjects regardless of their treatment arm. Oversight of the clinical trial’s commercial sponsor by the regulatory agencies also was lacking. Moreover, the medical literature publications on the clinical trial were not reviewed and edited with a critical eye or, possibly, without appropriate access to the underlying data.
This report shines light on very serious flaws in the processes used by federal agencies such as the FDA, CDC, and NIH in the development and safety/efficacy evaluation of new drugs. In our opinion, the flaws were made possible by a series of Congressional legislations and amendments that date back decades: the Bayh–Dole Act of 1980 on Patent and Trademark Law Amendments Act, the Project Bioshield Act of 2004, the 2005 Public Readiness and Emergency Preparedness (PREP) Act, and the 2016 21st Century Cures Act. These laws allowed Pfizer/BioNTech, the manufacturing and development corporations of the BNT162b2 mRNA vaccine, to retain full control of the trial’s original data while waiving all liability considerations. With FDA approval, Pfizer/BioNTech was allowed to block access to the original source data by medical and scientific research experts with no conflicts of interest in the trial vaccine. Information on 44,060 subjects was collected, monitored, evaluated, stored, and analyzed by Pfizer/BioNTech personnel. A review of the C4591001 protocol (https://clinicaltrials.gov/ct2/show/NCT04816643) should have made it obvious that the data from this trial would be massive involving a database of potentially millions of medical reports, clinical test results, scheduled and unscheduled visit reports, and more, all of which had to be organized, evaluated, and reported in an extremely short time window. Everything was handled by Pfizer/BioNTech personnel who also authored the reports that were submitted to the FDA and other international medical regulatory agencies, who were given only days to complete their evaluation.
Some aspects of this particular clinical trial review were unique. Progress of clinical trials is usually monitored by a Data Safety Monitoring Board (DSMB), a small group of independent experts whose role it to review the safety and efficacy data during the course of the trial and provide advice on whether to continue, modify, or terminate the study. But this was not the case for this clinical trial. Because the Pfizer/BioNTech trial was not funded by the U.S. government, Pfizer/BioNTech was allowed to establish its own DSMB. Thus, the Pfizer/BioNTech DSMB members could not be considered independent and without conflicting interests. The FDA had only a matter of days, November 20 – December 11, to review this massive data set of information to make their decisions regarding safety. Most likely the FDA and its VRBPAC relied far too heavily on summarized reports from Pfizer/BioNTech on their massive data bank of information and on the rigor and thoroughness of the oversight provided by Pfizer/BioNTech’s DSMB. Had it not been for the successful court case brought by the Public Health and Medical Professionals for Transparency, no one outside of the Pfizer and BioNTech corporations would have had the opportunity to investigate the original data generated by this clinical trial and none of the discrepancies reported here would have been revealed.
In summary, the decision to approve the BNT162b2 mRNA vaccine by the U.S. FDA and other international regulatory agencies was not an informed decision based on an unbiased, thorough, and transparent evaluation of the evidence intended to demonstrate that this vaccine met the criteria that it was a “safe and effective” means of controlling the COVID-19 pandemic.
In the past 50 years, the US has undertaken several mass immunization programs to control viral epidemics. In 1976, 362 cases of Guillain–Barré Syndrome occurred in the 6 weeks following the swine influenza vaccination of 45 million persons, an 8.8-fold increase above normal background rates [14]. The NY Times reported that the vaccination program was finally halted after the deaths of 3 elderly patients in 9 states, all of whom died with heart disease soon after receiving the same vaccine lot [15,16]. Guillain–Barré Syndrome is a very rare disorder and thus is easily recognized as a safety signal. Death and heart attacks are far more common adverse events. As such, they are not easily recognizable as warning signals and require extremely large numbers of trial subjects and longer follow-up periods to be identified. Had the FDA been aware of the cardiovascular event signal observed here, regulators might have given second thoughts regarding safety problems with the mRNA vaccine, as was seen in the 1976 swine flu vaccine debacle.
Despite evidence of the validity of the early warning signals and other adverse events reported in the post-marketing of the mRNA vaccines, this novel type of vaccine platform has not been removed from the market and has even been approved for children as young as 6 months. Physicians are still told to recommend mRNA vaccines without ever having had the opportunity to independently evaluate their safety and efficacy. At the very least, now is the time to inform physicians and other medical professionals of the dangers of the mRNA vaccines so that they can better advise their patients and help them to evaluate their individual personal risk versus benefit when deciding whether to be vaccinated. This would return healthcare decisions back to individuals and their medical providers where it belongs.
References
- Basavaraju SV, Patton ME, Grimm K, Rasheed MAU, Lester S, Mills L, et al. Serologic Testing of US Blood Donations to Identify Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)–Reactive Antibodies: December 2019–January 2020. Clinical Infectious Diseases 2021, 72, e1004–e1009. [Google Scholar] [CrossRef] [PubMed]
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- A Phase 1/2/3 Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of RNA Vaccine Candidates Against COVID-19 in Healthy Individuals. Available online: https://cdn.pfizer.com/pfizercom/2020-11/C4591001_Clinical_Protocol_Nov2020.pdf.
- Emergency Use Authorization for an Unapproved Product Review Memorandum. Available online: https://archive.org/details/emergency-use-authorization-eua-for-an-unapproved-product-review-memorandum.
- Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Thomas SJ, Moreira ED, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months. N Engl J Med 2021, 385, 1761–1773. [Google Scholar] [CrossRef] [PubMed]
- 6-Month Interim Report of Adverse Events CA4591001. Available online: https://pdata0916.s3.us-east-2.amazonaws.com/pdocs/070122/125742_S1_M5_5351_c4591001-interim-mth6-adverse-events.zip#page=3646.
- 16.1.7.1 Listing of Randomization Scheme and Actual Vaccine Received – All Subjects ≥16 Years of Age. Available online: https://phmpt.org/wp-content/uploads/2022/05/125742_S1_M5_5351_c4591001-interim-mth6-randomization-sensitive.pdf#page=4377.
- 16.2.1.1 Listing of Subjects Discontinued From Vaccination and/or From the Study – All Subjects ≥16 Years of Age. Available online: https://phmpt.org/wp-content/uploads/2022/07/125742_S1_M5_5351_c4591001-interim-mth6-discontinued-patients.pdf#page=233.
- 2.7.4 STN Summary Clinical Safety. Available online: https://phmpt.org/wp-content/uploads/2021/12/STN-125742_0_0-Section-2.7.4-summary-clin-safety.pdf#page=345.
- Analysis Data Reviewer Guide BLA Analysis for Participants ≥16 Years of Age BioNTech SE and PFIZER INC. Study C4591001. Available online: https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_c4591001-A-adrg.pdf#page=85.
- Pfizer-BioNTech COVID-19 Vaccine FDA Briefing Document VRBPAC December 10, 2020 meeting. Available online: https://archive.org/details/vrbpac-12.17.20-meeting-briefing-document-fda-0.
- Nelson, KE. Invited Commentary: Influenza Vaccine and Guillain-Barre Syndrome--Is There a Risk? American Journal of Epidemiology 2012, 175, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, H. Swine Flu Fiasco. New York Times 1976.
- Schmeck, HM. Swine Flu Program Halted in 9 States As 3 Die After Shots. New York Times 1976:1.
Figure 1.
Weekly subject deaths during the initial 33 weeks of Pfizer/BioNTech Clinical Trial CA4591001. The 38 subjects who died are shown in order of their date of death during the 33 weeks starting Monday, July 27, 2020 and ending Friday, March 13, 2021. The number of subject deaths per week are presented in a bar graph: solid bars, BNT162b2 vaccinated subjects; gray bars, Placebo subjects; hatched bars, Unblinded Placebo BNT162b2 vaccinated subjects. The cumulative number of deaths in each trial arm is shown as a linear graph: solid line, BNT162b2 vaccinated subjects; dotted line, Placebo subjects. The Unblinded Placebo BNT162b2 vaccinated subjects are included with the BNT162b2 vaccinated subjects. The three periods of this part of the clinical trial are as follows: Blinded Placebo-controlled period, July 27 – December 10, 2020; Open-label Follow-up period, December 11, 2020 – January 24, 2021; Open-label Observation period, January 25 – March 13, 2021.
Figure 1.
Weekly subject deaths during the initial 33 weeks of Pfizer/BioNTech Clinical Trial CA4591001. The 38 subjects who died are shown in order of their date of death during the 33 weeks starting Monday, July 27, 2020 and ending Friday, March 13, 2021. The number of subject deaths per week are presented in a bar graph: solid bars, BNT162b2 vaccinated subjects; gray bars, Placebo subjects; hatched bars, Unblinded Placebo BNT162b2 vaccinated subjects. The cumulative number of deaths in each trial arm is shown as a linear graph: solid line, BNT162b2 vaccinated subjects; dotted line, Placebo subjects. The Unblinded Placebo BNT162b2 vaccinated subjects are included with the BNT162b2 vaccinated subjects. The three periods of this part of the clinical trial are as follows: Blinded Placebo-controlled period, July 27 – December 10, 2020; Open-label Follow-up period, December 11, 2020 – January 24, 2021; Open-label Observation period, January 25 – March 13, 2021.
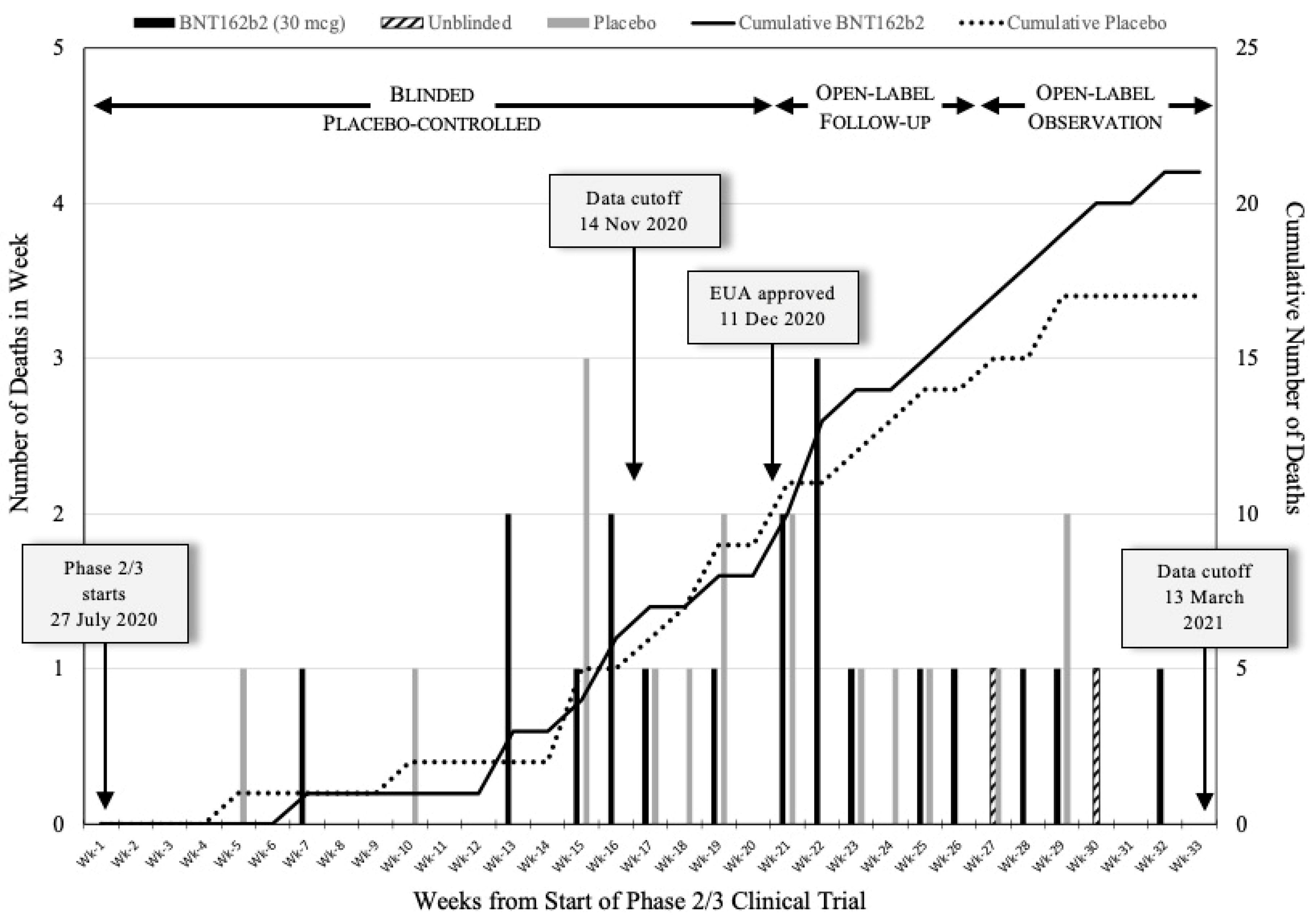

Table 1.
Cause of death of Pfizer/BioNTech clinical trial subjects.
| Subject ID | Sex | Age at Death | Date of Death | Days Post Dose 1 | Primary Cause of Death (Secondary Cause of Death) |
|
|---|---|---|---|---|---|---|
| BNT162b2 mRNA vaccinated subjects (21 Subjects) | ||||||
| 1# | 11621327 | M | 60 | 13Sept2020 | 4 | *Arteriosclerosis1 |
| 2 | 11141050 | F | 64 | 19Oct2020 | 63 | *Sudden cardiac death1 |
| 3# | 10071101 | F | 56 | 21Oct2020 | 84 | *Cardiac arrest |
| 4 | 11201050 | F | 58 | 07Nov2020 | 36 | *Cardiac arrest1 |
| 5 | 11521497 | M | 72 | 11Nov2020 | 96 | Shigella sepsis |
| 6 | 10891073 | F | 63 | 12Nov2020 | 99 | Chronic obstructive pulmonary disease |
| 7 | 10391010 | M | 85 | 18Nov2020 | 90 | *Arteriosclerosis (Hypertensive heart disease)1 |
| 8 | 11271112 | M | 53 | 04Dec2020 | 107 | *Cardio-respiratory arrest3 |
| 9 | 11361102 | M | 76 | 19Dec2020 | 52 | *Cardiac arrest2 |
| 10 | 10211127 | M | 54 | 19Dec2020 | 111 | *Cardiac failure - congestive |
| 11 | 10971023 | F | 87 | 21Dec2020 | 120 | Septic shock4 |
| 12 | 11561160 | F | 62 | 24Dec2020 | 95 | Road traffic accident |
| 13 | 12521010 | M | 81 | 26Dec2020 | 132 | COVID-19 pneumonia |
| 14 | 11401117 | M | 59 | 29Dec2020 | 158 | *Cardiac arrest2 |
| 15 | 10841266 | M | 77 | 12Jan2021 | 144 | *Sepsis3 (Emphysematous cholecycystis) |
| 16 | 11201266 | M | 51 | 19Jan2021 | 132 | Lung cancer metastatic |
| 17 | 11351033 | M | 67 | 29Jan2021 | 178 (5) | Suicide2 |
| 18 | 11291166 | F | 79 | 03Feb2021 | 149 | *Myocardial infarction1,2 |
| 19 | 10361140 | M | 64 | 10Feb2021 | 112 | Road traffic accident |
| 20 | 11311204 | M | 84 | 15Feb2021 | 147 (26) | Cardio-pulmonary arrest (Cerebrovascular accident) |
| 21 | 10881139 | M | 83 | 06Mar2021 | 143 | Metastases to lung (Pancreatic carcinoma metastatic) |
| Placebo subjects (17 Subjects) | ||||||
| 1# | 11521085 | F | 42 | 26Aug2020 | 8 | Death (Undetermined causes)1 |
| 2# | 12313972 | F | 61 | 28Sept2020 | 35 | Hemorrhagic stroke |
| 3 | 11561124 | M | 53 | 02Nov2020 | 54 | Overdose |
| 4# | 10661350 | M | 58 | 03Nov2020 | 16 | *Myocardial infarction |
| 5# | 10811194 | F | 51 | 04Nov2020 | 56 | *Myocardial infarction1,2 |
| 6 | 11681083 | M | 65 | 18Nov2020 | 86 | Aortic rupture |
| 7 | 11281009 | M | 66 | 28Nov2020 | 121 | Pneumonia1 |
| 8 | 10881126 | M | 66 | 01Dec2020 | 93 | *Cardiac arrest2 |
| 9 | 12314987 | M | 47 | 06Dec2020 | 101 | Cardio-respiratory arrest1,2 |
| 10 | 10191146 | M | 67 | 17Dec2020 | 108 | Metastases to liver (Biliary cancer metastatic) |
| 11 | 10941112 | F | 57 | 18Dec2020 | 102 | Acute respiratory failure (COVID-19 pneumonia) |
| 12 | 10891088 | F | 82 | 30Dec2020 | 146 | Dementia |
| 13 | 12291083 | F | 56 | 05Jan2021 | 97 | Diabetes mellites4 (COVID-19 pneumonia) |
| 14 | 10841470 | M | 65 | 11Jan2021 | 104 | Multi-organ dysfunction4 syndrome (COVID-19) |
| 15 | 12315324 | F | 59 | 31Jan2021 | 156 | Multiple organ dysfunction syndrome |
| 16 | 12071055 | M | 65 | 09Feb2021 | 97 | Bacterial pneumonia |
| 17 | 10271191 | F | 68 | 13Feb2021 | 156 | Respiratory failure (COVID-19) |
Table 1: Cause of death of Pfizer/BioNTech clinical trial subjects. The 38 subjects who died during the period July 27, 2020 to March 13, 2021 are listed separately according to their Clinical Trial arm, BNT162b2 vaccinated or Placebo, and numbered in order of their date of death after receiving their first trial dose (Dose 1). Rows for Subjects 11351033 and 11311204 are shaded in gray to indicate that these subjects were Unblinded Placebo subjects, from the original Placebo arm but BNT162b2 vaccinated after the unblinding. In parentheses, are the number of days these subjects died after they received Dose 3, the BNT162b2 vaccine dose. #Indicates those subjects included in the EUA application and Polack et al. [6]. *Indicates that the cause of death diagnosis was considered a Cardiovascular event. 1CRF does not provide sufficient clinical data to support diagnosis. 2CRF is incomplete; needs autopsy results to confirm diagnosis. 3CRF supports “Cardiovascular event” as the underlying cause of death. 4Subject did not meet criteria for Randomization or had a Protocol Deviation. .
Table 2.
Comparison of subject deaths reported during periods of the Pfizer/BioNTech BNT162b2 mRNA vaccine clinical trial CA491001.
Table 2.
Comparison of subject deaths reported during periods of the Pfizer/BioNTech BNT162b2 mRNA vaccine clinical trial CA491001.
| PFIZER/BIONTECH PUBLICATIONS DATA |
PFIZER/BIONTECH 6-MONTH INTERIM REPORT DATA |
|
|---|---|---|
| BLINDED PLACEBO-CONTROLLED PERIOD TO EUA APPLICATION DATA COLLECTION CUTOFF: July 27 to November 14, 2020 | ||
6 Deaths
|
11 Deaths
|
|
| BLINDED PLACEBO-CONTROLLED PERIOD AND OPEN-LABEL FOLLOW-UP PERIOD: July 27, 2020 to January 24, 2021 | ||
29 Deaths
|
30 Deaths
|
|
| OPEN-LABEL OBSERVATIONAL PERIOD TO DATA COLLECTION CUTOFF OF 6-MONTH INTERIM REPORT: January 25, 2021 to March 13, 2021 | ||
5 Deaths
Conclusion: “Causes of death were balanced between BNT162b2 and Placebo groups.” (REF) |
8 Deaths
|
|
| SUMMARY OF DEATHS IN 6-MONTH REPORTING PERIOD: July 27, 2020 to March 13, 2021 | ||
34 Deaths: 18 BNT162b2 vs 16 Placebo
Conclusion: No summary of causes of death for all deceased subjects presented. |
38 Deaths: 21 BNT162b2 vs 17 Placebo
|
|
Table 2: Comparison of subject deaths reported during periods of the Pfizer/BioNTech BNT162b2 mRNA vaccine clinical trial CA491001. The lefthand column presents data taken from the following publications: Pfizer/BioNTech EUA application [5], Polack et al. [6], and Thomas et al. [7]. The righthand column presents data from the Pfizer/BioNTech 6-Month Interim Report on Adverse Events [8]. Cardiovascular event numbers are based on our analysis of subject CRFs as presented in Table 1. Conclusion statements are taken from the texts of Pfizer/BioNTech EUA application [5], Polack et al. [6], and Thomas et al. [7]. The table is divided into 4 different time periods of the 6-Month Interim Report: Blinded Placebo-controlled Period to EUA application Data Collection Cutoff (July 27 to November 14, 2020); Blinded Placebo-controlled and Open-label Follow-up Period (July 27 to January 24, 2021); Open-label Observational Period to Data Collection Cutoff of the 6-Month Interim Report (January 25 to March 13, 2021); and a summary of the 6-Month Interim Report (July 27, 2020 to March 13, 2021).
Table 3.
Delay in recording subject death.
| Period | Subject ID | Date of Death | Officially Recorded Date (from CRF) |
Delay Recording Death (Days) |
|---|---|---|---|---|
| BNT162b2 mRNA vaccinated subjects (Subjects with available CRF) | ||||
| #P-C | 11621327 | 13Sept2020 | 24Sept2020 | 11 |
| P-C | 11141050 | 19Oct2020 | 25Nov2020 | 37 |
| #P-C | 10071101 | 21Oct2020 | 5Nov2020 | 15 |
| P-C | 11201050 | 07Nov2020 | 3Dec2020 | 26 |
| P-C | 11521497 | 11Nov2020 | 18Nov2020 | 7 |
| P-C | 10891073 | 12Nov2020 | 4Dec2020 | 22 |
| P-C | 10391010 | 18Nov2020 | 9Dec2020 | 21 |
| P-C | 11271112 | 04Dec2020 | 05Dec2020 | 1 |
| O-L, F | 11361102 | 19Dec2020 | 22Jan2021 | 34 |
| O-L, F | 10211127 | 19Dec2020 | 30Dec2020 | 11 |
| O-L, F | 10971023 | 21Dec2020 | 28Dec2020 | 7 |
| O-L, F | 11561160 | 24Dec2020 | 14Jan2021 | 21 |
| O-L, F | 12521010 | 26Dec2020 | 29Dec2020 | 3 |
| O-L, F | 11401117 | 29Dec2020 | 05Jan2021 | 7 |
| O-L, F | 10841266 | 12Jan2021 | 15Jan2021 | 3 |
| O-L, F | 11201266 | 19Jan2021 | 25Jan2021 | 6 |
| O-L, O | 11351033 | 29Jan2021 | 24Feb2021 | 26 |
| O-L, O | 11291166 | 03Feb2021 | 5Feb2021 | 2 |
| O-L, O | 10361140 | 10Feb2021 | 22Feb2021 | 12 |
| O-L, O | 11311204 | 15Feb2021 | 18Feb2021 | 3 |
| O-L, O | 10881139 | 06Mar2021 | 08Mar2021 | 2 |
| Placebo subjects (Subjects with available CRF) | ||||
| #P-C | 11521085 | 26Aug2020 | 27Aug2020 | 1 |
| #P-C | 12313972 | 28Sept2020 | 1Oct2020 | 3 |
| P-C | 11561124 | 02Nov2020 | 19Nov2020 | 17 |
| #P-C | 10661350 | 03Nov2020 | 10Nov2020 | 7 |
| #P-C | 10811194 | 04Nov2020 | 11Nov2020 | 7 |
| O-L, F | 11681083 | 18Nov2020 | 19Nov2020 | 1 |
| O-L, F | 11281009 | 28Nov2020 | 8Dec2020 | 10 |
| O-L, F | 10881126 | 01Dec2020 | 11Feb2021 | 72 |
| O-L, F | 12314987 | 06Dec2020 | 7Dec2020 | 1 |
| O-L, F | 10191146 | 17Dec2020 | 5Feb2021 | 50 |
| O-L, F | 10941112 | 18Dec2020 | 21Dec2020 | 3 |
| O-L, F | 10891088 | 30Dec2020 | 04Jan2021 | 5 |
| O-L, F | 12291083 | 05Jan2021 | 5Jan2021 | 0 |
| O-L, F | 10841470 | 11Jan2021 | 19Jan2021 | 8 |
| O-L, O | 12315324 | 31Jan2021 | 3Feb2021 | 3 |
| O-L, O | 12071055 | 09Feb2021 | 11Feb2021 | 2 |
| O-L, O | 10271191 | 13Feb2021 | 16Feb2021 | 3 |
Table 3: Delay in recording subject death. Subjects who died during the period July 27, 2020 to March 13, 2021 are listed. Subjects receiving BNT162b2 vaccine are listed separately from those who received the Placebo and in order of the true date of death. #Indicates those subjects included whose death was reported in the Pfizer/BioNTech EUA application [5] and Polack et al. [6]. The rows shaded in gray highlight those individuals whose death was officially recorded in their CRF between November 14 and December 10, 2020 indicating that Pfizer/BioNTech knew the subject died during this time period. Periods of the trial: P-C is Placebo-controlled, Blinded period; O-L, F is the Open-label Follow-up period (December 11, 2020 to January 24, 2021); O-L, O is the Open-label Observational period (January 25 to March 13, 2021).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



