
Submitted:
01 September 2023
Posted:
04 September 2023
You are already at the latest version
Abstract
The protein TRPM8, a non-selective, calcium (Ca2+) permeable ion channel is implicated in several pathological conditions, including neuropathic pain states. In our previous research endeavors, we have identified β–lactam derivatives with high hydrophobic character that exhibit potent and selective TRPM8 antagonist activity. This work describes the synthesis of novel derivatives featuring C-terminal amides and diversely substituted N’-terminal monobenzyl groups, in an attempt to increase the total polar surface area (TPSA) in this family of compounds. The primary goal was to assess the influence of these substituents on the inhibition of menthol-induced cellular Ca2+ entry, thereby establishing critical structure-activity relationships. While the substitution of the tert-butyl ester by isobutyl amide moieties improved the antagonist activity, none of the N’-monobencyl derivatives, regardless of the substituent on the phenyl ring, achieved the activity of the model dibenzyl compound. The antagonist potency of the most effective compounds was subsequently verified using Patch-Clamp electrophysiology experiments. Furthermore, we evaluated the selectivity of one of these compounds against other members of the TRP ion channel family and some receptors connected to peripheral pain pathways. This compound demonstrated specificity for TRPM8 channels. To better comprehend the potential mode of interaction, we conducted docking experiments to uncover plausible binding sites on the functionally active tetrameric protein, which come upon four main poses by the pore zone, differents from those described for other known antagonists. Finally, in vivo experiments, involving a couple of selected compounds, revealed significant antinociceptive activity within a mice model of cold allodynia induced by oxaliplatin.
Keywords:
TRPM8 channel
; β–lactams
; antagonists
; docking
; antinociceptive activity
1. Introduction
Somatosensory neurons are capable of perceiving changes in external temperature through thermosensitive transient receptor potential (TRP) ion channels [1,2]. These thermoTRP channels play pivotal roles in human physiology [3,4], but they are also implicated in human pathology [5,6,7], making them important targets for therapeutic interventions [8,9]. Among the thermoTRP family, TRPM8 stands out as a notable member. Initially identified as a marker for prostate cancer [10], TRPM8 was subsequently recognized as a sensory ion channel, crucial for cold sensing and pain transmission [11,12,13,14,15]. This cationic channel, with a particular preference for Ca2+ permeability, operates through multimodal activation mechanisms encompassing cold temperatures ( > 15 < 28 ºC), voltage changes, osmolarity alterations, and cooling compounds of natural origin, such as menthol and icilin [16,17].
Numerous experimental findings have correlated the expression, function and mutation of TRPM8 and various pathological conditions [18,19]. Thus, this channel is involved in pain perception and modulation, and TRPM8 inhibition and channel desensitization (by agonists pressure) have been shown to alleviate both acute and chronic pain conditions [13]. For instance, painful baldder hypersensity and peripheral neuropaty resulting from chemotherapy have been linked to elevated TRPM8 expression and/or function [19,20,21,22]. Notably, studies using TRPM8 agonists suggest their potential for treating patients with neuropathic ocular pain, while TRPM8 blockade has proven effective as well in alleviating pain associated with severe dry eye disease [23]. In pulmonary diseases, TRPM8 channels have been related to asthma and chonic obstructive disease (COPD). Accordingly, downregulation of TRPM8 expression has been connected with enhanced vasoreactivity in pulmonary hypertension in mice [24]. Additionally, certain TRPM8 and TRPA1 polymorphisms have been implicated in the pathogenesis of COPD [25]. Moreover, it has been observed that exposure to cold air triggers airway inflammatory responses and alters TRPM8 expression. Interestingly, knockdown of TRPM8 has been shown to mitigate respiratory hypersensitivity in asthmatic individuals [26].
The altered expression of TRPM8 channels also underlies the mechanisms driving tumor progression in various types of cancers, including prostate, pancreas, breast, colon, and lung. However, the impact on expression varies based on the tumor type and stage of advancement, with contrasting patterns of overexpression or downregulation, depending on the specific context [27].
Since its discovery in 2001, a wide array of chemically diverse compounds have been identified as modulators (agonists and antagonists) of the TRPM8 channel [8,9,28], with the most important contribution from several pharmaceutical companies. Notably, among these efforts, compounds like PF-05105679 and AMG333, both orally active TRPM8 antagonists, advanced into clinical trials with the aim of exploring their potential for treating cold pain hypersensitivity and relieving migraine symptoms, respectively [29,30]. However, these promising compounds failed to progress beyond phase I trials [31]. Volunteers participating in the trials reported significant side effects, including intense sensations of heat in the hands, arms, and mouth [32], which could likely be attributed to insufficient selectivity against heat-activated TRP channels. Other notable drawbacks of these clinical candidates included dysesthesia, paresthesia, and dysgeusia. These limitations might be connected to the pivotal role of the TRPM8 channel in regulating core body temperature, as well as other less understood physiological functions [33,34]. This context could potentially restrict the use of TRPM8 modulators as oral or systemic medications. Nonetheless, the prospect of utilizing these modulators as locally administered or topical treatments for TRPM8-related conditions remains largely underexplored.
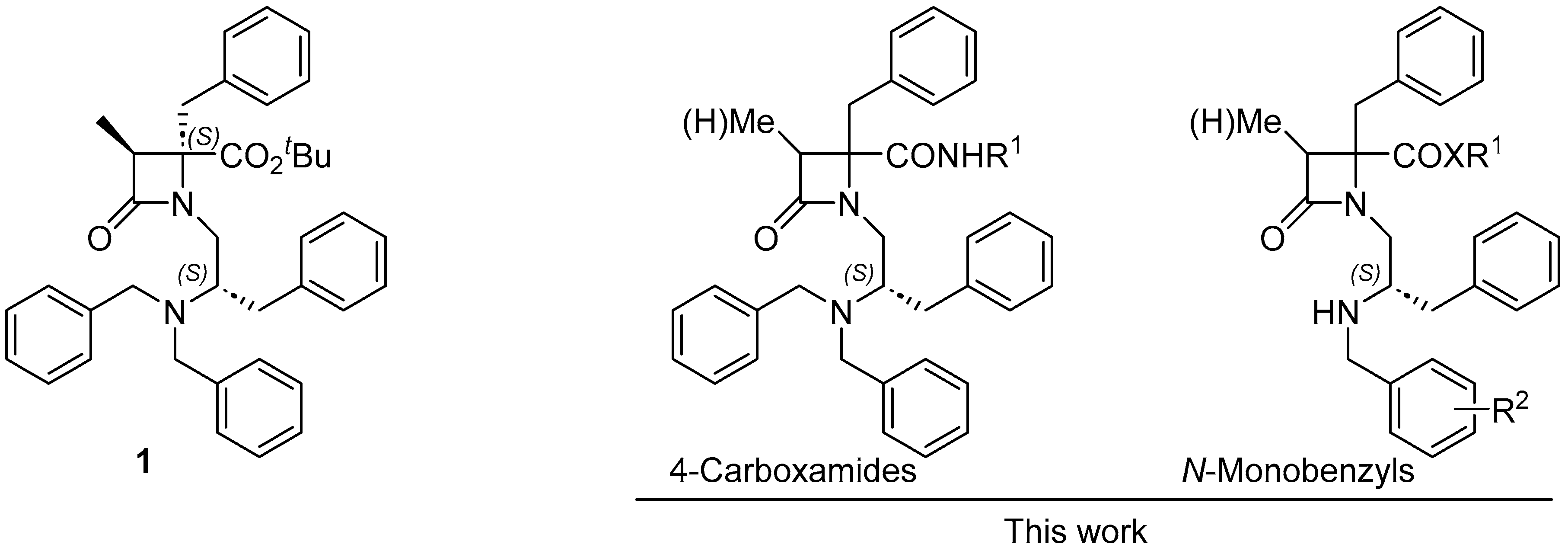
In previous research, our team has identified several β–lactam derivatives exhibiting potent and selective TRPM8 antagonist activity [35,36]. Furthermore, some of these derivatives have demonstrated antinociceptive properties in various animal models [37,38]. In the present study, our focus lies in deepening our understanding of the structure-activity relationships within this compound family. Specifically, we investigate the introduction of different amides at the C-terminus and explore the potential of various substitutions in N-monobenzyl derivatives (Figure 1). We were particularly interested in enhancing the total polar surface area (TPSA) of the previously identified N,N-dibenzyl derivative 1, to reduced absorption capabilities, and thereby to open up possibilities for investigating local or topical applications.
Figure 1.
Structure of model compond 1 and analogues prepared and studied in this work.
2. Results and Discussion
2.1. 1,4,4-Trisubstituted β–lactam Derivatives
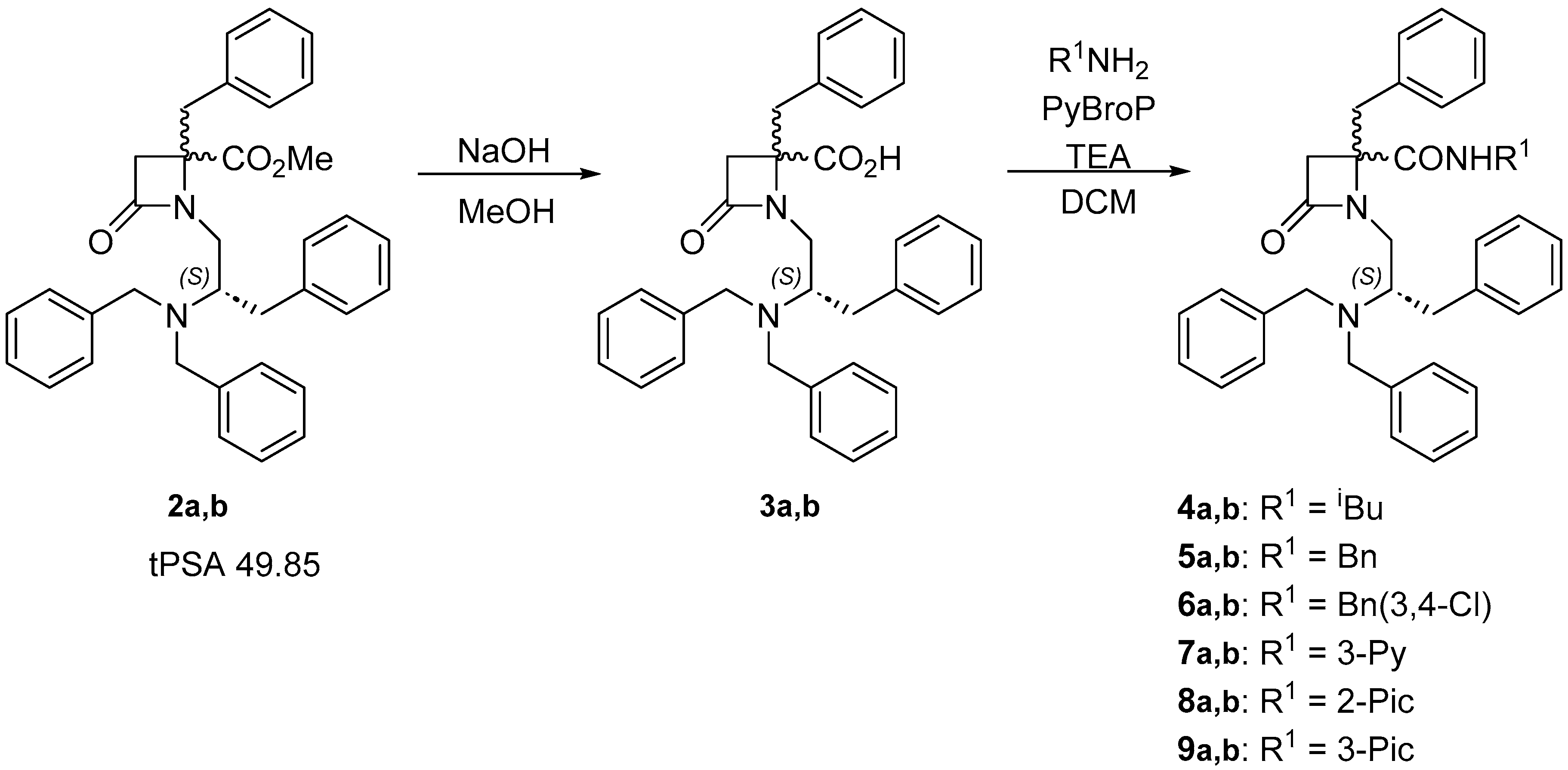
For the first exploratory structure-activity relationships, we decided to prepare and evaluate diastereoisomeric 1,4,4-trisubstituted β–lactam derivatives, which can be more easily prepared than enantiomerically pure analogues. In previous works, we have demonstrated that both 4-S- and 4-R-β-lactams rendered TRPM8 antagonists [37], therefore we expected thet 4-R,S-diastereoisomeric mixtures could be active and allow us to discern among important substituents. Firstly, we proposed the incorporation of different amides in the 4-carboxylate group, including structural diversity with an alkyl, a benzyl, a pyridine group and other related heterocyclic systems, able to slightly increase the TPSA values (Table S1). The six amines selected for the formation of the corresponding amides were isobutylamine, benzylamine, 3,4-dichlorobenzylamine, 2- and 3-picolines (2-, 3-Pic) and 3-aminopyridine (3-Py). In a previous publication on Asp-derived β-lactams, amide derivatives from 4-pyridine and 4-picoline showed lower activity than the corresponding 3-aminopyridyl analogues [35]. Therefore, the 4-picolyl derivative was discarded in this case, while the 2-picoline derivative was prepared, since it had not been explored previously. In addition, pyridyl- and picolyl-derived amides incorporate two protonable nitrogens, which could increase aqueous solubility of final molecules. The 3,4-dichlorobenzylamine was selected because this substitution is present in a kappa opioid agonist with antagonist-like properties in a mouse model related to TRPM8 activation by icilin [39]. Saponification of β–lactam 2a,b (see supporting information for synthetic details), afforded the carboxylic acid intermediate 3ab (a:b = 1:1.8), which was then coupled with the selected amines, in the presence of PyBroP, to allow the preparation of amides 4a,b-9a,b (Scheme 1). These amides were isolated in different diastereoisomeric ratios (Table S2), most probably due to different conversion of the diasteromeric acids 3a and 3b or to the enrichment in a particular diastereoisomer during the purification process.
Scheme 1.
Synthetic procedure for the preparation of amides 4a,b-9a,b from β-lactam 2a,b.
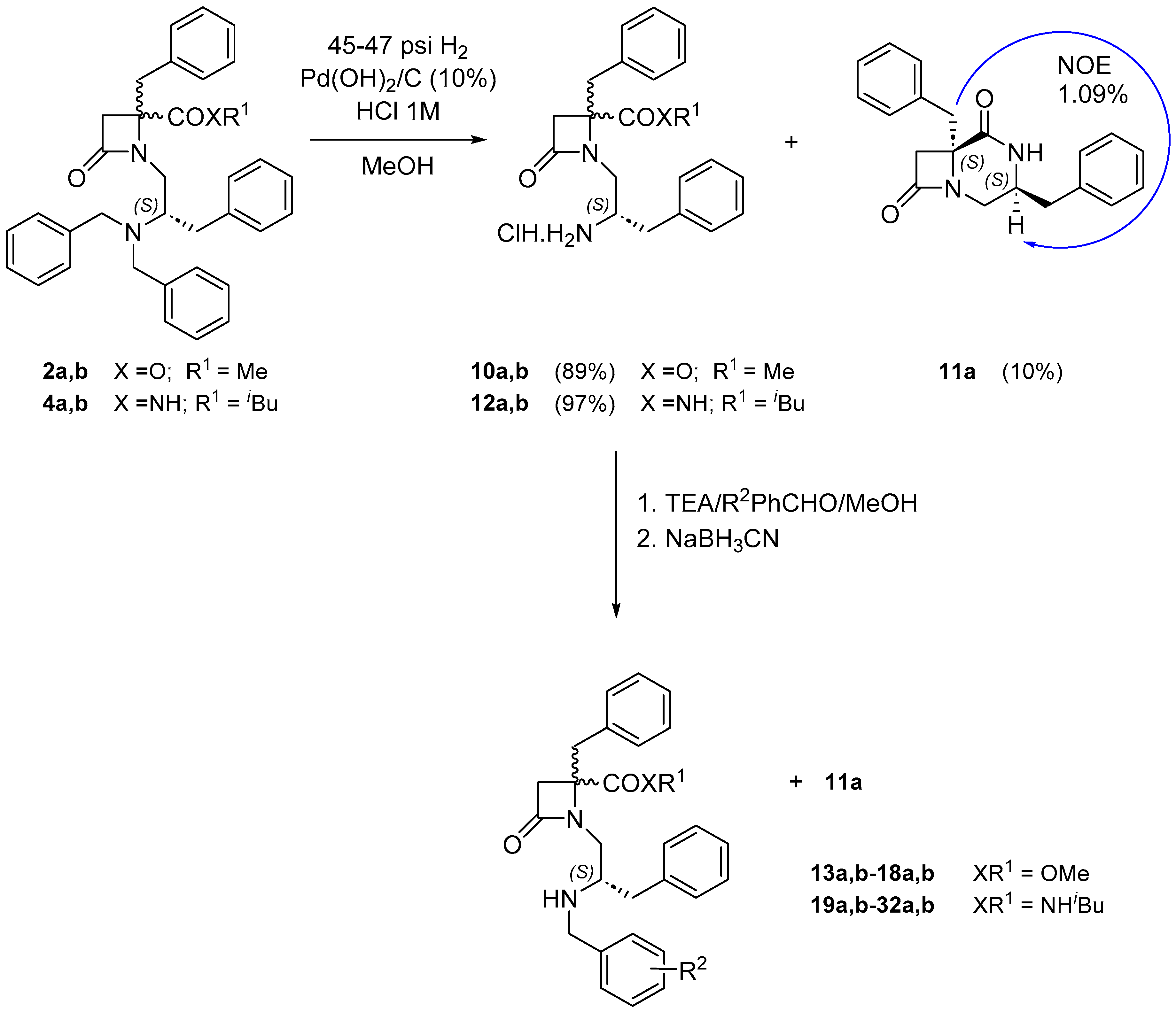
Another way to increase TPSA values is the removal of one of the N-benzyl groups, while the incorporation of different substitutions on the remaining N-benzyl moiety could serve to fine-tune the antagonist activity. To this end, the hydrogenolysis of β-lactam 2a,b, using Pd(OH)2/C as catalyst in the presence of HCl, gave rise to the primary amine derivative 10a,b in its hydrochloride form (1:2.5, a:b diastereoisomer ratio). The slight enrichement in te major diastereoisomer (b) compared to the starting material is due to the formation of the (3S,6S)-3,6-dibenzyl-1,4-diazabicyclo[4.2.0]octane-5,8-dione 11a (configuration determined through NOE experiments) from minor isomer 10a (Scheme 2). This secondary product was also formed during the reductive amination reaction of compound 10a,b with aldehydes to the corresponding monobenzyl derivatives, complicating the purification steps. To avoid this issue, most reactions in this section were performed with the isobutylamide derivative 12a,b, synthesized from 4a,b. Differently substituted benzaldehydes, containing alkyl or benzyl groups, and electron atrancting or withdrawing groups, and 2-naphtaldehyde were used for the N-monobezyl derivatives preparation (Scheme 2, Table S2). Following some of the activity initial results, detailed later on, most substituents on the benzyl group are at position 3 of the phenyl ring, but some 4-substituted analogues were also prepared to verify preliminary outcomes. Thus, amine derivative 10a,b was treated with the corresponding benzaldehyde, to form the corresponding intermediate imines, which were subsequently reduced with NaBH3CN to afford β-lactam N-monobenzylated derivatives 13a,b-17a,b (Scheme 2, Table S2). Similarly, the reaction of 12a,b with different aldehydes, containing an aliphatic (Me), two aromatic (Ph, Nf), all halides (F, Cl, Br, I), donor groups (OMe, OPh, OBn) and electron acceptors (CN, NO2) substituents afforded the corresponding monobenzyl derivatives 18a,b-31a,b (Scheme 2, Table S3).
Scheme 2.
Hydrogenolysis of β-lactams 2a,b and 4a,b and reductive amination to N-monobenzylamines.
The diastereomeric 1,4,4-trisubstitutes β-lactams were evaluated as modulators of the TRPM8 channel by calcium microfluorography assays. These assays use a HEK293 cell line that stably overexpressed rat TRPM8 channel (rTRPM8). Menthol was used as the agonist during these experiments, whereas AMTB and model compound 1 served as prototype antagonists for comparison.
Within these assays, β-lactam 2a,b, choosen as a pivotal intermediate, displayed submicromolar potency as an antagonists of the TRPM8 channel (Table 1). Among the amide series, the most favorable IC50 values were achieved with compounds 4a,b and 5a,b, sugesting that the methyl ester of 2a,b can be substituted by an aliphatic (4a,b) or benzylic (5a,b) amide, with only marginal loss of activity (Table 1). In contrast, the introduction of two chlorine atoms into the benzylamide group (6a,b) causes a notable reduction in antagonist activity. The amide derived from 2-picoline (8a,b) exhibited micromolar potency, while the 3-aminopyridyl (7a,b) and 3-picolyl (9a,b) derivatives demonstrated lesser potency. When comparing these results to the model antagonist AMTB, it becomes apparent that the 4-CO2Me derivative 2a,b and its amide counterparts 4a,b and 5a,b exhibit antagonist activity one order of magnitude more potent.

Table 1.
Antagonist activity of amides 4a,b-9a,b in rTRPM8 channels (Ca2+ microfluorography assay).
| Compound | R1 | a:b | IC50 (μM) | 95% confidential intervals |
|---|---|---|---|---|
| 2a,b | 1:1.8 | 0.15 ± 1.23 | 0.095 to 0.22 | |
| 4a,b | iBu | 1:1.6 | 0.71 ± 1.20 | 0.49 to 1.03 |
| 5a,b | Bn | 1:2.6 | 0.71 ± 1.27 | 0.43 to 1.16 |
| 6a | (3,4-Cl)Bn | ND | _ | |
| 6a,b | (3,4-Cl)Bn | 1:1.4 | ND | _ |
| 7a,b | 3-Py | 1:1.4 | 11.62 ± 1.30 | 6.73 to 20.06 |
| 8a,b | 2-Pic | 1:1.8 | 1.99 ± 1.26 | 1.24 to 3.17 |
| 9a,b | 3-Pic | 1:1.2 | 10.69 ± 1.38 | 5.52 to 20.73 |
| 1 | 1.06 ± 1.21 | 0.72 to 1.55 | ||
| AMTB | 7.3 ± 1.5 | 8.82 to 7.85 |
Within the monobenzyl series, all compounds exibit IC50 values in the micromolar range, displaying slight variation across most of them (1.10 to 12.1 μM), and lower potency in comparison to the dibenzyl model compounds 2a,b and 4a,b. The importance of having at least one benzyl-type group at the N-terminal amino moiety is demonstrated by the substantial decline in antagonist activity observed in 11a (with less than 50% inhibition at 50 μM), although the conformational restriction could also contribute to the lower activity.
Regarding the substituents on the benzyl group, placement at meta (3-) position seems slightly preferred over para (4-) positioning. This observation is drawn from the comparison between compounds 14a,b vs 15a,b, as well as between 20a,b and 21a,b vs 30a,b and 31a,b, respectively. Large ubstituents around position 3, such as Ph (20a,b), Cl (22a,b), Br (16a,b and 23a,b), I (24a,b), and the condensed 2-naphthyl moiety (17a,b and 32a,b), appear to enhance activity. Generally, strong electron-donating groups like OMe (25a,b) and OBn (27a,b), which result in an increased TPSA up to 70 A2 (Table S1), exhibit slightly lower potency than their corresponding unsubstituted analogue (13a,b). On the contrary, a distinct behavior is noted between the two analogues bearing strong electron-withdrawing groups (CN and NO2). While the nitrile-bearing derivative 28a,b ranks as the less effective compound in this series (IC50 = 12.1 µM), the 3-NO2 analog 29a,b maintains the one-digit micromolar antagonist activity akin to the unsubstituted counterpart. The calculated TPSA values of these latter compounds have increased by more than 25 and 55 units, respectively, when compared to the unsubstituted analogue 13a,b (as shown in Table S1). However, compound 29a,b wasn not pursued further due to the potential interference posed by the NO2 group, which is signaled as a Pan-Assay Interference Compound (PAIN) alert [40].
All these observations on structure-activity relationships must be approached with the required caution, considering the variations in the ratio of diastereomers among different compounds. In general, the N-monobenzylated derivatives exhibit a reduced activity ranging between one and two orders of magnitude when compared to the corresponding N,N-dibenzylated model compounds. The introduction of different substituents on the phenyl ring does not result in significant impacts on activity, independently of the substituent size and electronis character. Even large substituents (such as Ph or Br) on the N-monobenzyl moiety failed to compensate for the absence of the second benzyl group.
Table 2.
Antagonist activity of N-monobenzyl derivatives 13a,b-32a,b in rTRPM8 channels (Ca2+ microfluorography assays).
Table 2.
Antagonist activity of N-monobenzyl derivatives 13a,b-32a,b in rTRPM8 channels (Ca2+ microfluorography assays).
| Compound | XR1 | R2 | a:b | IC50 (μM) | 95% confidential intervals |
|---|---|---|---|---|---|
| 2a,b | OMe | - | 1:1.8 | 0.15 ± 1.23 | 0.095 to 0.22 |
| 4a,b | NHiBu | - | 1:1.6 | 0.71 ± 1.20 | 0.49 to 1.03 |
| 11a | - | - | - | ND | ND |
| 13a,b | OMe | H | 1:3.6 | 3.95 ± 1.20 | 2.66 to 5.85 |
| 14a,b | OMe | 3-Me | 1:3.6 | 3.58 ± 1.41 | 1.74 to 7.35 |
| 15a,b | OMe | 4-Me | 1:5.6 | 3.90 ± 1.47 | 1.74 to 8.74 |
| 16a,b | OMe | 3-Br | 1:2 | 1.10 ± 1.38 | 0.56 to 2.16 |
| 17a,b | OMe | (2-Nph)CH2 | 1:4 | 2.60 ± 1.23 | 1.69 to 3.98 |
| 18a,b | OMe | 4-OPr | 1:9.5 | 8.90 ± 1.40 | 4.37 to 18.10 |
| 19a,b | NHiBu | 3-Me | 1:1.5 | 3.97 ± 1.31 | 2.28 to 6.93 |
| 20a,b | NHiBu | 3-Ph | 1:1.1 | 2.03 ± 1.32 | 1.15 to 3.60 |
| 21a,b | NHiBu | 3-F | 1:2.6 | 5.15 ± 1.46 | 2.37 to 11.22 |
| 22a,b | NHiBu | 3-Cl | 1:2.4 | 2.57 ± 1.32 | 1.45 to 4.53 |
| 23a,b | NHiBu | 3-Br | 1:2.9 | 3.30 ± 1.36 | 1.73 to 6.29 |
| 24a,b | NHiBu | 3-I | 1:2.7 | 3.83 ± 1.28 | 2.33 to 6.29 |
| 25a,b | NHiBu | 3-OMe | 1:2.5 | 4.48 ± 1.25 | 2.83 to 7.09 |
| 26a,b | NHiBu | 3-OPh | 1:5.7 | 8.22 ± 1.30 | 4.79 to 14.08 |
| 27a,b | NHiBu | 3-OBn | 1:1.5 | 3.27 ± 1.37 | 1.72 to 6.20 |
| 28a,b | NHiBu | 3-CN | 1:1.1 | 12.10 ± 1.31 | 6.99 to 20.94 |
| 29a,b | NHiBu | 3-NO2 | 1.2:1 | 2.12 ± 1.28 | 1.28 to 3.50 |
| 30a,b | NHiBu | 4-Ph | 1:1.9 | 4.57 ± 1.22 | 3.07 to 6.82 |
| 31a,b | NHiBu | 4-F | 1.1:1 | 8.32 ± 1.30 | 4.87 to 14.22 |
| 32a,b | NHiBu | 3,4-Ph (2-Nph) | 1:1.1 | 1.81 ± 1.21 | 1.22 to 2.68 |
| 1 | 1.06 ± 1.21 | 0.72 to 1.55 | |||
| AMTB | 7.30 ± 1.50 | 8.82 to 7.85 |
2.2. 1,3,4,4-Tetrasubstituted, Enantiopure β–lactam Derivatives
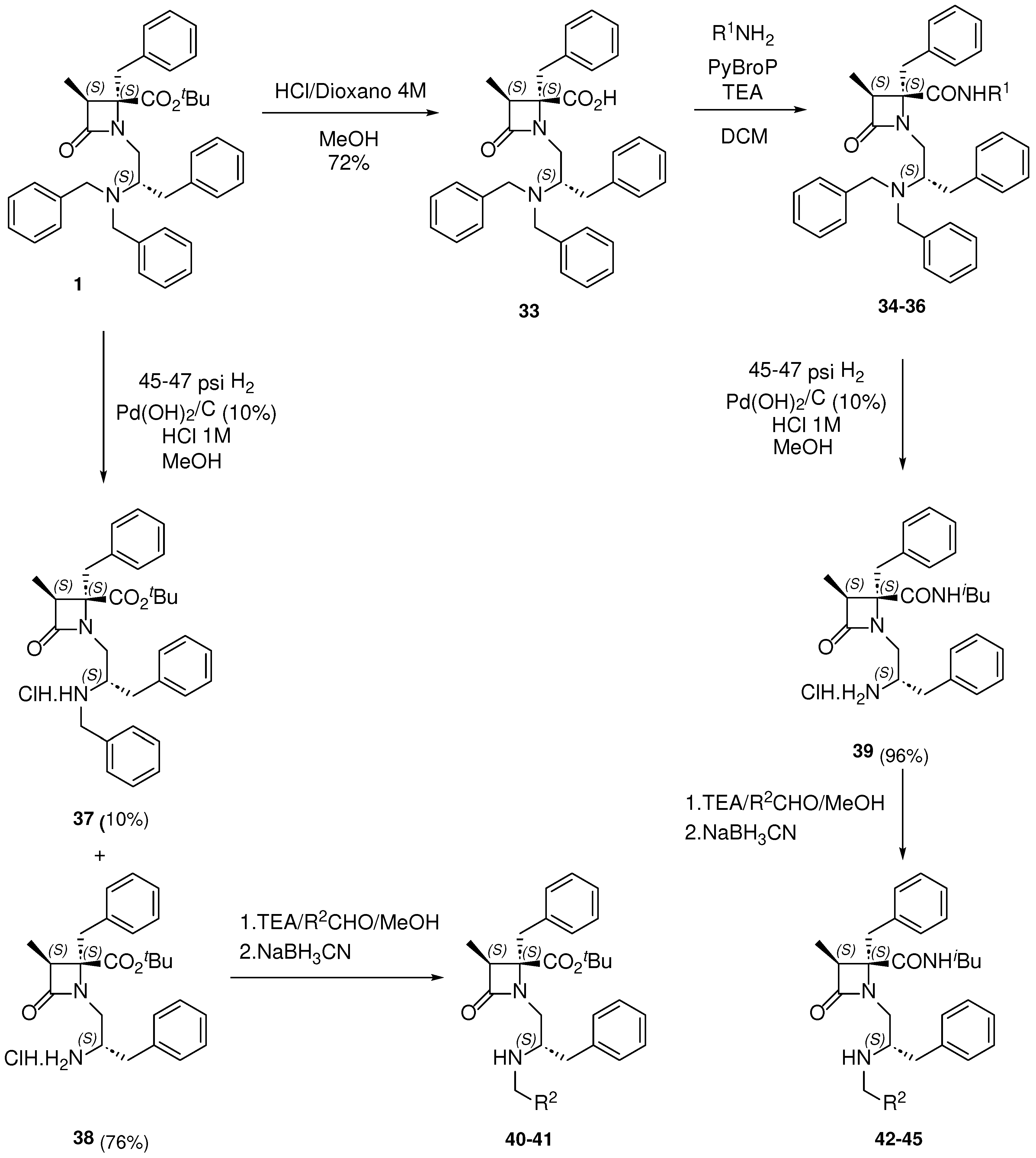
The challenges in making precise comparisons among compounds of the previous 1,4,4-trisubstituted series, due to the variability in the diastereoisomeric ratio and the requirement to study pure isomers for further pharmaceutical development, prompted us to transition to a new series of diastereo- and enantiopure β-lactam derivatives. In this newly designed series, the most promising substituents identified earlier were incorporated onto a 1,3,4,4-tetrasubstituted β–lactam scaffold. These enantiopure β–lactams can be synthesized using chiral 2-chloropropanoic acid derivatives, as previously reported [38,41].
Staring from compound 1, the tert-butyl ester underwent acidic hydrolysis to yield the free carboxylic acid intermediate 33. This intermediate was subsequently coupled to isobutyl, bencyl and 2-picolyl amines to generate the corresponding amides, 34-36, with very good yield (Scheme 3). Hydrogenolysis of compound 1, using Pd(OH)2 as catalyst, resulted in a mixture of the monobenzyl derivative 37 and the fully deprotected amino analogue 38, which were separated using column chromatography. A similar reaction, starting from compound 34, rendered the free amino derivative 39 in almost quantitative yield. Further trestments of compounds 38 and 39 with differently substituted benzaldehydes, followed by reduction of the resulting imine intermediates with NaBH3CN, led to the synthesis of the expected N-monobenzyl derivatives 40-45 (Scheme 3, Table S3).
Scheme 3.
Preparation of enantiopure β–lactam derivatives.
The initial biological evaluation of this series of 1,3,4,4-tetrasubstituted β-lactams was performed by calcium microfluorography assays using the indicated HEK293 cell line overexpressing rTRPM8 channels. As outlined in Table 3, submicromolar IC50 values were achieved for the three prepared amide derivatives, with their antagonist activities ranking as follows: 34 (R1 = iBu) > 35 (R1 = Bn) > 36 (R1 = CH2(2-Pic)). These levels of potency surpass that of the OtBu model compound 1 and are one order of magnitude higher than the reference antagonist AMTB. In line with previous findings, most of the N-monobenzylic compounds displayed reduced potency compared to their dibenzyl analogues. Furthermore, substitution at the 3-position of the benzyl aromatic ring appears to be slightly favored over the 4-position (comparison of 40 vs 41). Compounds with 3-Ph (42), 3-Br (44) and 2-Nf (45) exhibited similar micromolar activities. Unfortunately, our efforts to increase the TPSA values by the incorporation of additional heteroatoms in amides and N-monobenzyl derivatives resulted mostly in lower potencies compared to model compound 1. Interestingly, amides 34 and 35 improved the TRPM8 antagonist activity, although the increases in TPSA values are tiny for these compounds (Table S1).
Table 3.
Antagonist activity of enantiopure β–lactam derivatives at rTRPM8 channels (Ca2+ microfluorography assays).
Table 3.
Antagonist activity of enantiopure β–lactam derivatives at rTRPM8 channels (Ca2+ microfluorography assays).
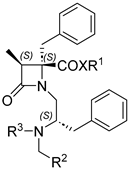 | |||||
|---|---|---|---|---|---|
| Compound | XR1 | R2 | R3 | IC50 (µM) | 95% Confidence Intervals |
| 1 | OtBu | 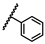 |
Bn | 1.06 ± 1.21 | 0.72 to 1.55 |
| 34 | NHiBu | 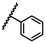 |
Bn | 0.30 ± 1.26 | 0.19 to 0.50 |
| 35 | NHBn | 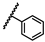 |
Bn | 0.49 ± 1.27 | 0.30 to 0.79 |
| 36 | NHCH2(2-Pic) | 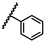 |
Bn | 0.84 ± 1.32 | 0.48 to 1.49 |
| 37 | OtBu | 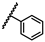 |
H | 3.06 ± 1.25 | 2.67 to 3.51 |
| 40 | OtBu | 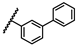 |
H | 6.36 ± 1.41 | 3.08 to 13.17 |
| 41 | OtBu | 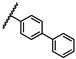 |
H | 9.26 ± 1.41 | 4.44 to 19.35 |
| 42 | NHiBu | 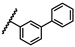 |
H | 2.19 ± 1.36 | 1.15 to 4.17 |
| 43 | NHiBu | 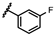 |
H | 4.03 ± 1.35 | 2.14 to 7.60 |
| 44 | NHiBu | 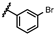 |
H | 3.27 ± 1.31 | 1.86 to 5.75 |
| 45 | NHiBu | 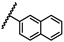 |
H | 3.56 ± 1.40 | 1.74 to 7.31 |
| AMTB | 7.3 ± 1.50 | 6.82 to 7.85 | |||
2.3. Activity in hTRPM8 Channels and Electrophysiology Assays
To further characterize this family of compounds, calcium microfluorography assays were conducted using the HEK293 cell line, which expresses the human isoform of the TRPM8 channel. Amide derivatives 34 and 35 were selected, and their outcomes were compared with those of the model compound 1 (Table 4). These new β–lactams exhibit a somewhat diminished antagonist activity, as evidenced by higher IC50 values, when evaluated in hTRPM8 as compared to rTRPM8. This behaviour is similar to those observed for other TRPM8 antagonists [42].
Table 4.
Antagonist activity of β-lactams 34 and 35 in hTRPM8 (Ca2+ microfluorommetry) and electrophysiology in rTRPM8 channels (Patch-clamp assays).
Table 4.
Antagonist activity of β-lactams 34 and 35 in hTRPM8 (Ca2+ microfluorommetry) and electrophysiology in rTRPM8 channels (Patch-clamp assays).
| Compound | Ca2+ Microfluorography Assays | Patch-Clamp Assay | ||||
|---|---|---|---|---|---|---|
|
rTRPM8 IC50 (µM) |
95% Confidence Intervals |
hTRPM8 IC50 (µM) |
95% Confidence Intervals |
rTRPM8 IC50 (µM) |
95% Confidence Intervals | |
| 1 | 1.06 ± 1.21 | 0.72 to 1.55 | 1.74 ± 1.19 | 1.23 to 2.45 | 0.60 ± 1.66 | 0.20 to 1.76 |
| 34 | 0.30 ± 1.26 | 0.19 to 0.50 | 2.58 ± 1.24 | 1.65 to 4.02 | 0.16 ± 0.82 | 0.10 to 0.271 |
| 35 | 0.49 ± 1.27 | 0.30 to 0.79 | 3.33 ± 1.28 | 2.03 to 5.48 | 0.48 ± 1.33 | 0.18 to 1.23 |
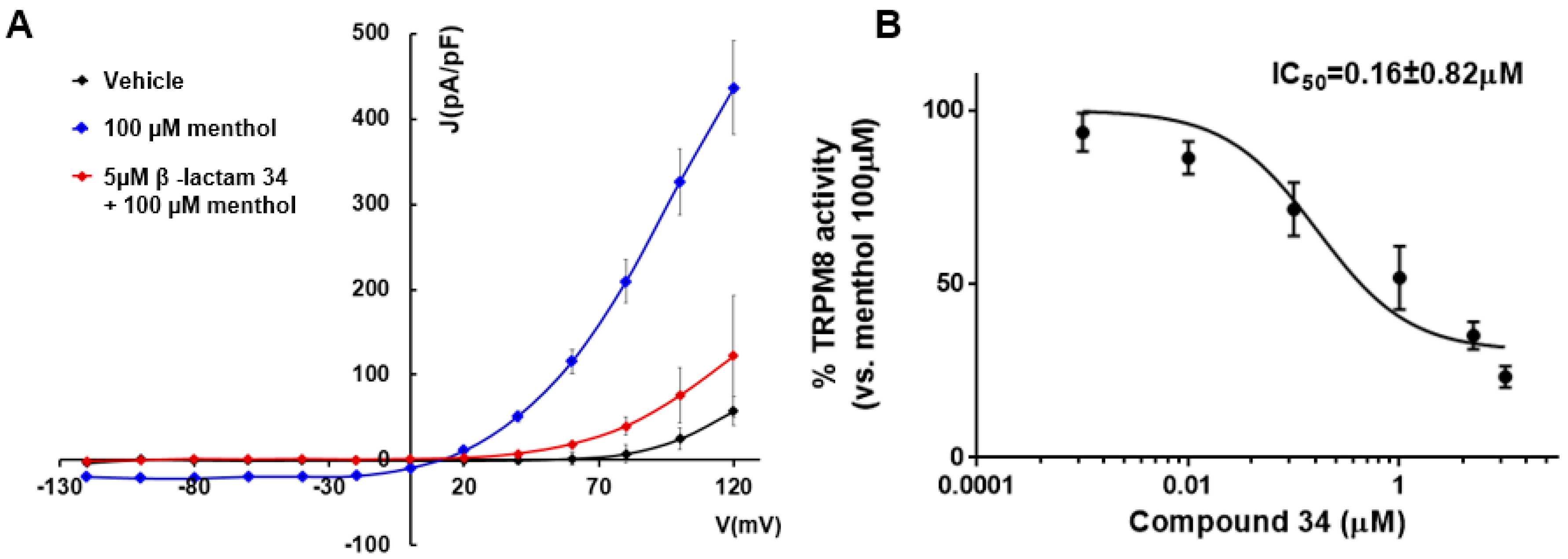
To confirm the antagonistic activity of the most potent compounds, electrophysiology studies were conducted using the Patch-Clamp technique on single HEK293 cells expressing the rat TRPM8 channel (Table 4, Figure 2). As depicted in Figure 2A, perfusion with 100 µM menthol (shown in blue) resulted in a strongly outward rectifying ion current. This is evident by a negligible current at negative potentials and a linear intensification of the ohmic current at positive voltages. When applying compound 34 at a concentration of 5 µM, a noticeable reduction in menthol-induced activity at depolarizing voltages was observed (indicated by the red line), compared to menthol alone. Dose-response relationships for both tested compounds were established through triplicate experiments at various concentrations, applying a holding voltage of −60 mV (as shown in Figure 2B). These compounds effectively blocked the TRPM8-mediated and menthol-induced responses. In these assays, both derivatives displayed higher potency than the model β–lactam 1, being the the isopropylamide derivative 34 the most potent, with a efficacy in the low nanomolar range (Table 4).
Figure 2.
Compound 34 blocks TRPM8-mediated responses evoked by menthol in rTRPM8-expressing HEK293 cells. (A) IV curves obtained after exposure to vehicle solution (green trace), 100 µM menthol (blue trace) and 100 µM menthol + 10 µM 34 (red trace). Peak current data were expressed as pA/pF (to allow comparison among different size cells). Each point is the mean ± SEM of n = 15. (B) Concentration/response curves for rTRPM8 current blockade by 34, at a holding voltage of −60 mV. The solid line represents fits of the experimental data to the following equation: Y = Bottom + (Top-Bottom)/(1 + 10^((X-LogIC50))) with a standard slope of −1.0 (Hill coefficient) and a restriction of the minimum (Top = 100). The fitted value for IC50 was 0.16 ± 0.82. Each point is the mean ± SEM of n = 15.
Figure 2.
Compound 34 blocks TRPM8-mediated responses evoked by menthol in rTRPM8-expressing HEK293 cells. (A) IV curves obtained after exposure to vehicle solution (green trace), 100 µM menthol (blue trace) and 100 µM menthol + 10 µM 34 (red trace). Peak current data were expressed as pA/pF (to allow comparison among different size cells). Each point is the mean ± SEM of n = 15. (B) Concentration/response curves for rTRPM8 current blockade by 34, at a holding voltage of −60 mV. The solid line represents fits of the experimental data to the following equation: Y = Bottom + (Top-Bottom)/(1 + 10^((X-LogIC50))) with a standard slope of −1.0 (Hill coefficient) and a restriction of the minimum (Top = 100). The fitted value for IC50 was 0.16 ± 0.82. Each point is the mean ± SEM of n = 15.
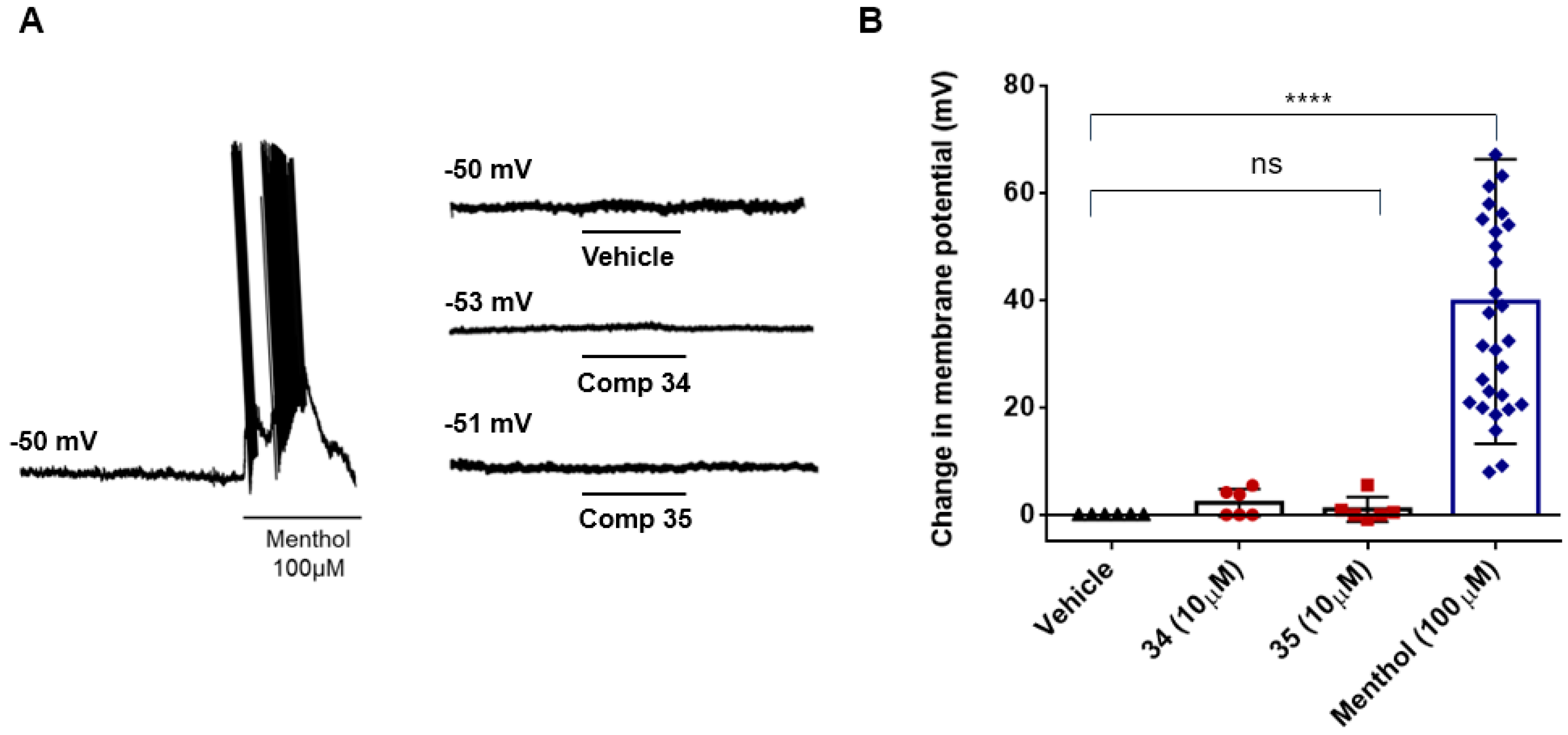
We also investigated the effect of β-lactams derivatives on the resting membrane potential (RMP) of DRG sensory neurons. Under current clamp conditions in patch-clamp assays, small sensory neurons displayed a RMP of ~-50 mV (Figure 3A, upper left trace). Exposure of the neurons to 10 µM of compound 34 or 35 did not alter the RMP of these neurons (Figure 3A, lower right traces and Figure 3B). As control to ensure that neurons were able to exhibit action potential firing they were exposed to 100 µM menthol (Figure 3A, right trace, and Figure 3B).
Figure 3.
β-lactams derivatives 34 and 35 do not affect the membrane resting potential of sensory neurons. (A) Representatives recordings of resting membrane potential measured under current-clamp configuration. The traces show the action potentials (APs) firings from DRG neuron elicited by 100 μM Menthol (left trace) or in the presence of vehicle (0.5% DMSO) or the β-lactams derivatives (left traces). (B) Changes in the Vm in the absence and in the presence of 10 μM of of the β-lactams derivatives. Data were analyzed by one-way ANOVA followed by Bonferroni post hoc test for multiple comparisons (ns = no significance, **** = 0.0001) and given as mean ± SEM; n ≥ 40 cells from 4 cultures.
Figure 3.
β-lactams derivatives 34 and 35 do not affect the membrane resting potential of sensory neurons. (A) Representatives recordings of resting membrane potential measured under current-clamp configuration. The traces show the action potentials (APs) firings from DRG neuron elicited by 100 μM Menthol (left trace) or in the presence of vehicle (0.5% DMSO) or the β-lactams derivatives (left traces). (B) Changes in the Vm in the absence and in the presence of 10 μM of of the β-lactams derivatives. Data were analyzed by one-way ANOVA followed by Bonferroni post hoc test for multiple comparisons (ns = no significance, **** = 0.0001) and given as mean ± SEM; n ≥ 40 cells from 4 cultures.
2.4. Activity in Other TRP Channels and Pain-Related Peripheral Receptors
To asess the selectivity for the TRPM8 channel, the effect of compound 34 was examined on different ion channels and receptors implcated in pain-related processes. First, some TRP channels that play a role in temperature integration and nociception were selected [31,43], including hTRPV1, hTRPV3, hTRPA1 and TRPM3. Additionally, the evaluation encompassed the hASIC3 channel, with widespread distribution within the peripheral nervous system and contributions to excitability of primary sensory neurons [44]. In each instance, the compound was subjected to evaluation at a consistent concentration of 10 µM. As outlined in Table 5, it is evident that compound 34 did not exhibit noteworthy modulatory effects on any of the assessed TRP and ASIC3 channels. Importantly, amide derivative 34 did not show activation on the TRPV1 channel, which was about 30% in the model compound 1 [38]. Compound 35 underwent evaluation as a potential antagonist for the TRPM3 channel, revealing a non-significant degree of inhibition (4.1 ± 10.8) in the Ca2+ influx induced by pregnenolone sulfate.
Table 5.
Functional activity of the selected β-lactam 34 (10 μM) in Ca2+ microfluorography assays of TRPV1, TRPV3, TRPA1, TRPM3 and ASIC3 channels and percentages of inhibition of specific binding at CGRPR, CB2 and M3 receptors.
Table 5.
Functional activity of the selected β-lactam 34 (10 μM) in Ca2+ microfluorography assays of TRPV1, TRPV3, TRPA1, TRPM3 and ASIC3 channels and percentages of inhibition of specific binding at CGRPR, CB2 and M3 receptors.
| Compound | % Channel Inhibition at 10 μM | % Binding at 10 μM | ||||||
|---|---|---|---|---|---|---|---|---|
| hTRPV1 | hTRPV3 | hTRPA1 | TRPM3 | ASIC3 | hCGRPR | hCB2 | hM3 | |
| 1 | 19.4 ± 1.7 | −6.3 ± 5.9 | 4.2 ± 1.1 | ND | 6.0 ± 0.9 | −1.0 ± 4.3 | 7.3 ± 0.3 | −4.1 ± 3.1 |
| 34 | 8.9 ± 2.9 | 17.3 ± 5.7 | 2.0 ± 3.4 | 6.9 ± 10.7 | 2.2 ± 9.6 | −11.9 ± 5.2 | 7.0 ± 2.1 | 5.2 ± 3.2 |
hTRPV1: transient receptor potential vanilloid, type 1. hTRPV3: transient receptor potential vanilloid, type 3. hTRPA1: transient receptor potential ankirin, type 1. ASIC3: acid sensing ion cannel, subunit 3. hCGRPR: human calcitonin gen-related peptide receptor. hCB2: human cannabinoid receptor, subtype 2. hM3: human muscarin receptor, subtype 3. In all cases, data is from two experiments in duplicate. Ago.: assay for agonist activity. Antago.: assay for antagonist activity (see methods). Agonists used for activation: TRPV1 (Capsaicin, 30 nM), TRPV3 (2-Aminoethoxydiphenyl borate, 2-APB. 30 µM), TRPA1 (Allylisothiocyanate, 10 µM), TRPM3 (pregnenolone sulfate, 50 µM), ASIC3 (Buffer, pH 5.5). Reference antagonists: TRPV1 (Capsazepin, IC50 1.3·10−7), TRPV3 (Ruthenium red, IC50 2.5·10−7 M), TRPA1 (Ruthenium red, 10 µM), TRPM3 (isosakuranetin, 10 µM; IC50 50 nM), ASIC3 (Amiloride. 1 mM). Radioligand for hCGRPR: [125]hGCRPα, agonist hGCRPα (1 µM). Radioligand for hCB2: [3H]WIN 55212-2, agonist WIN 55212-2 (5 µM). Radioligand for hM3: [3H]4-DMAP, agonist 4-DMAP (1 µM).
Furthermore, the investigation extended to measuring the binding affinity of compound 34 to calcitonin gene-related peptide hCGRPR, cannabinoid hCB2 and muscarinic hM3 receptors, all relevant to different pain conditions [45,46,47,48,49]. As shown in Table 5, this compound show inability to displace the radioligands for hCB2, hM3, and hCGRPR receptors.
2.5. Antinociceptive Activity in a Mouse Model of Chemotherapy-Induced Cold Allodynia
It is established that treatments with the chemotherapeutic drug oxaliplatin (OXA) induce painful peripheral neuropathy, known as CIPN, which becomes more pronounced in cold conditions [50]. The TRPM8 channel has been implicated in mouse models of CIPN pain induced by OXA [20,51]. In fact, the OXA-induced peripheral neuropathy mouse model is being used for the pharmacological characterizion of TRPM8 antagonists. For this peripheral neuropathy assay, mice were subcutaneously (s.c.) injected with OXA was subcutaneously (s.c.) injected at a dose of 6 mg/kg dose on days 1, 3 and 5 to male mice, to induce incresed sensitivity to cold, which can be monitored through the acetone drop test.
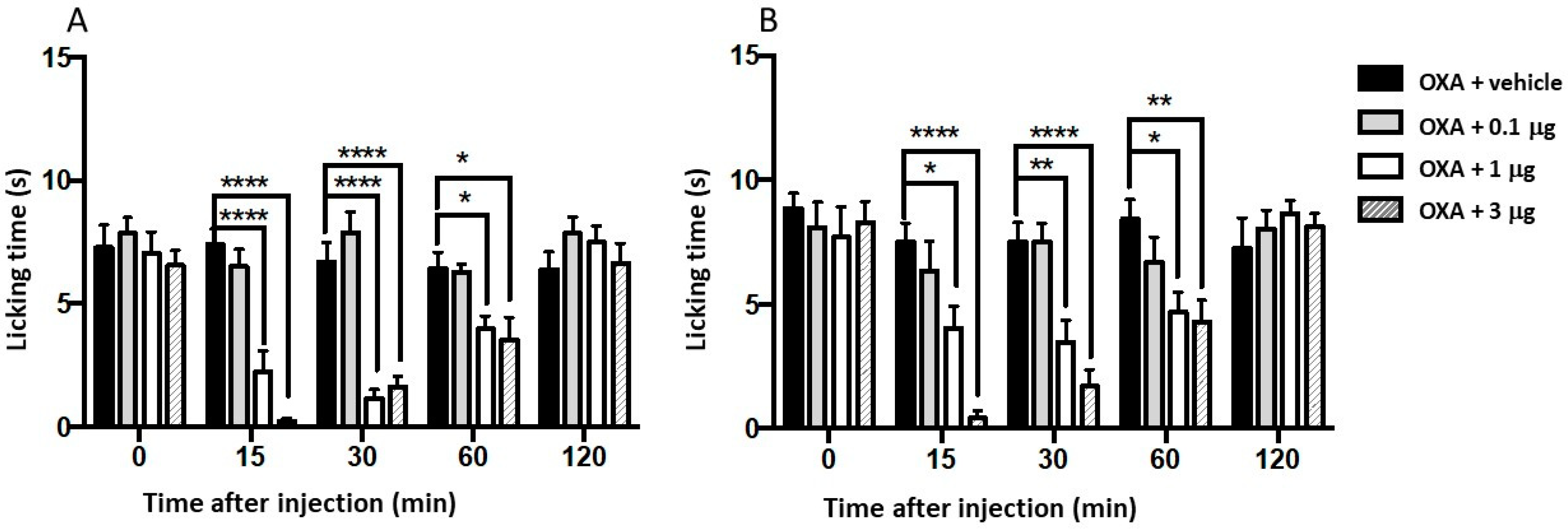
Consecuently, compounds 34 and 35 were assessed for their ability to alleviate OXA-induced cold hypersensitivity after intraplantar administration (i.pl., 3, 1 and 0.1 μg). As depicted in Figure 4A,B, both compounds, 34 and 35 exhibited a dose-dependent and significant reduction the cold-induced paw licking. This effect was observed 15 min post-administration and sustained up to 60 min. These findings parallel those reported for a biphenylamide N-spiro[4.5]decan-8-yl derivative which demonstrated antiallodynic activity lasting up to 60 minutes at a 1µg dose, despite possessing higher nanomolar potency on rat TRPM8.[52] Notably, compounds 34 and 35 displayed greater potency and a longer lasting action compared to another group of TRPM8 antagonists with an imidazo[10,50:1,6]pyrido[3,4-b]indole-1,3(2H)-dione central scaffold.[53] The superior in vivo activity of this β–lactam family might be attributed to their distinct mode of interaction, as suggested by modeling studies.
Figure 4.
Effects of compounds 34 (A) and 35 (B) on the oxaliplatin (OXP)-induced cold hypersensitivity (drop acetone test). Peripheral neuropathy was induced in male mice by injecting OXA (6 mg/kg, s.c.) on days 1, 3 and 5. Control animals were treated with vehicle. Compounds 34 (A) and 35 (B) (3 µg/i.pl., 1 µg/i.pl. and 0.1 µg/i.pl.) were administered to the OXP-treated animals, and the time-course of cold hypersensitivity was measured. Data are given ± SEM (n = 5). Statistical analysis, comparing with oxaliplatin-treated mice, was performed by two-way ANOVA followed by post hoc Bonferroni test by multiple comparisons: **** p < 0.0001.
Figure 4.
Effects of compounds 34 (A) and 35 (B) on the oxaliplatin (OXP)-induced cold hypersensitivity (drop acetone test). Peripheral neuropathy was induced in male mice by injecting OXA (6 mg/kg, s.c.) on days 1, 3 and 5. Control animals were treated with vehicle. Compounds 34 (A) and 35 (B) (3 µg/i.pl., 1 µg/i.pl. and 0.1 µg/i.pl.) were administered to the OXP-treated animals, and the time-course of cold hypersensitivity was measured. Data are given ± SEM (n = 5). Statistical analysis, comparing with oxaliplatin-treated mice, was performed by two-way ANOVA followed by post hoc Bonferroni test by multiple comparisons: **** p < 0.0001.
2.6. Insights into the Mode of Interaction of Selected β–lactams with the TRPM8 Channel
To shed light on the mechanism by which β–lactam amides and monobenzyls interact with the TRPM8 channel, we select compounds 34, 35, and 37, which represent the most potent TRPM8 antagonists within their respective series. For our docking studies, we employed software tools in Yassara suite of programs, and the interactions were analyzed with Yasara and Protein-Ligand Interaction Profiler (PLIP). Docking simulations were performed on a model of rat TRPM8 channel generated from the cryoelectron microscopy (cryo-EM) structure of Ficedula albicollis TRPM8 (PDB code 6BPQ) [54].
Initially, we used the TRPM8 structure encompassing transmembrane helices, as well as the internal and extracellular loops of the indicated tetrameric channel. As outlined in Table 6, the most prominent binding solutions for the three compounds consistently aligned them within or close to the pore channel. Curiously, these calculations did not provided any solution in which compounds 34, 35, or 37 bind to the menthol binding pocket [55].
Table 6.
Percentage of main docking solutions (and binding energy estimation in kcal/mol) obtained in molecular docking studies using Yasara. rTRPM8 with extracellular loops.
Table 6.
Percentage of main docking solutions (and binding energy estimation in kcal/mol) obtained in molecular docking studies using Yasara. rTRPM8 with extracellular loops.
| Subsite | Channel Location | 34 | 35 | 37 |
|---|---|---|---|---|
| 1 | Pore, external tower | 20.0 (9.09) | 14.4 (7.82) | 28.2 (9.85) |
| 2 | Pore, high S3-S4, S6 | 13.8 (8.51) | 16.4 (10.11) | 7.4 (7.40) |
| 3 | Inner pore, S5S6, S5 loops | 29.3 (9.81) | 24.6 (9.40) | 31.8 (7.62) |
| 4 | Pore, internal mouth | 6 .0 (11.21) | 7.6 (10.68) | 13.9 (8.49) |
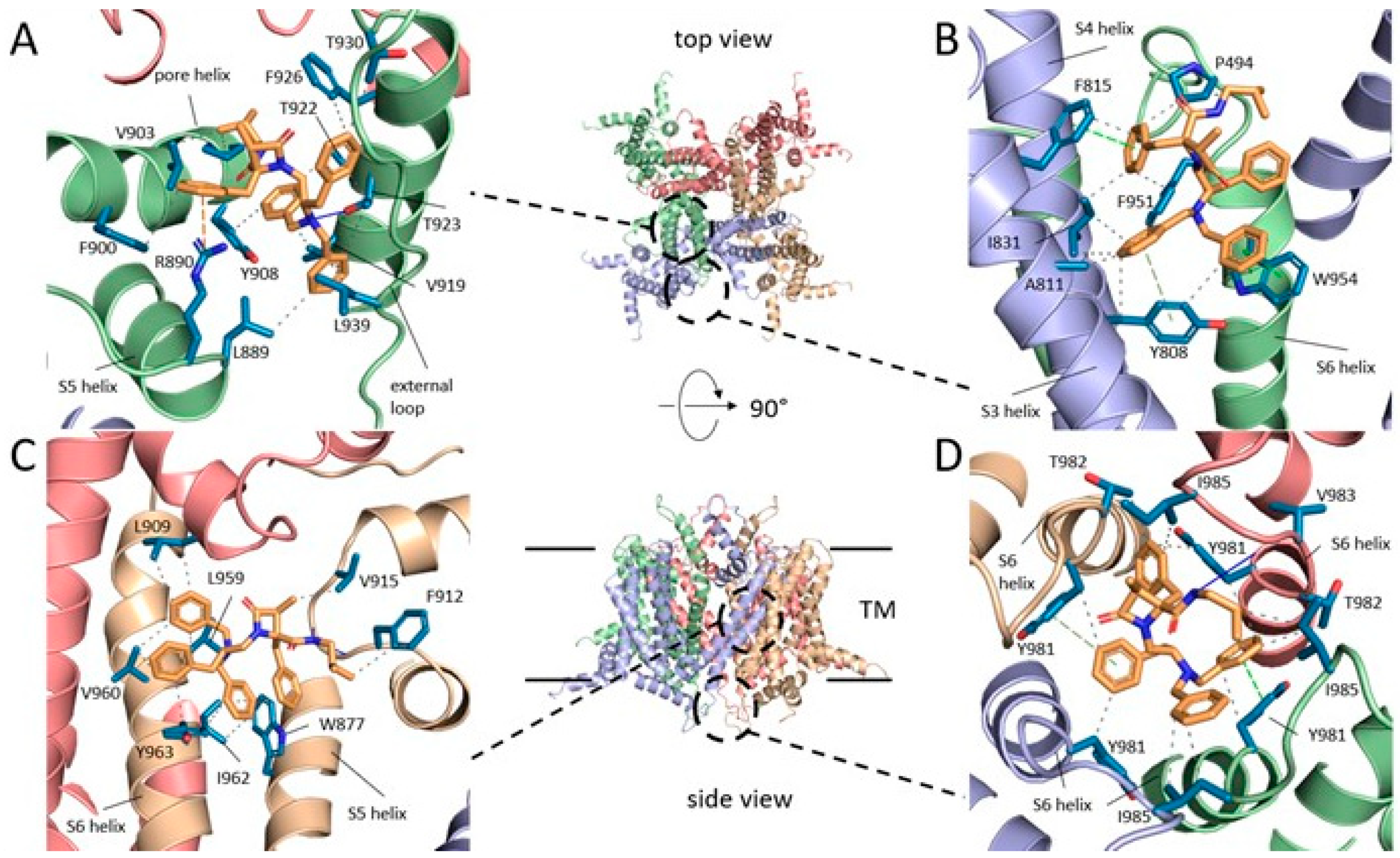
Upon analyzing the most populated solutions for these compounds, we identified four primary binding subsites within the TRPM8 tetrameric protein. Progressing from the top (extracellular) to the bottom (intracellular) regions, we observed that 14 to 28% of the solutions were clustered around the tower formed by the external loops (Subsite 1), being the second subsite most populated and that of the better biding energy in the case of monobenzyl derivative 37. A second subsite (Subsite 2), involving transmembrane helices S3 and S4 of a protein subunit and S6 of an adjacent monomer, was found to be more populated for the dibenzyl amide derivatives 34 and 35 in comparison to the monobenzyl analogue 37. Subsite 3, bounded by interactions with transmembrane helices S5 and S6 from a single monomer, along with certain loops near the pore helix on the same monomer and a neighboring protein subunit, emerged as the most populated binding site for all three compounds. Subsite 4, located within the cytoplasmic pore loops, accumulated a greater number of solutions for the smaller monobenzyl derivative 37 when compared to the dibenzyl analogues 34 and 35. However, for the latter compounds, subsite 4 corresponded to the binding pocket with the better binding energy.
To investigate whether the extracellular loops in the previous model play a role in guiding the distribution of β–lactam derivatives into cavities along the pore zone, as previously hypothesized for related analogs [36,38], an additional structure of the channel was incorporated into the modeling studies (compounds 34 and 37). This alternate structure retained the transmembrane helices and inner pore loops but excluded the extracellular loops. Within this framework, many different solutions emerged, but subsites 2, 3 and 4 remained the most frequently populated solutions, with an increased preference for subsite 4 in the case of the smaller compound 37 (see SM for details). Once more, no interactions with the menthol binding site were observed.
The stabilizing interactions among the external loops of the channel (Subsite 1) and amide derivatives 34 and 35 are characterized by hydrophobic contacts with L889, V903, V919, T922, T923, F926, K937, and L939, among others, from a single protein monomer (Figure 4A). Using PLIP program, both compounds establishes a π–cation interconnection, between R890 and the Ph ring of the 4-Bn group in 34, and between K937 and one of the aromatic rings of the N-Bn2 moiety in 35. Calculations conducted with Yasara identified some π–π interactions involving F900 and/or F926. Furthermore, both software programs indicate that the CO group of the β–lactam ring in 35 engages in a hydrogen-bond with NH/NH2 protons of the R890 guanidino group.
Within subsite 2 (Figure 4B), amides 34 and 35 exhibit two π–π interactions involving F815 and W954 of the protein and different phenyl groups of the small-molecule. Additionally, different hydrophobic contacts are observed, mainly delimited by Y808 (TM S3), and F815, I832 and I831 (TM S4) of a monomer, and by P949 and F951 of TM S6 of an adjacent protein unit (Figure 4B).
Deeper within the pore channel, subsite 3 corresponds with the most populated docking solutions for both compounds 34 and 35. This subsite entails two transmembrane helices from a protein subunit, namely S5 and S6, and two loops positioned at the exit of the pore helix, one from the same monomer, and the other from a contiguous protein in the tetramer and its S5. At this subsite, both software programs identify an H-bond involving the α–CO of G913 residue, acting as the acceptor, while the 4-CONH amide donates its proton to the interaction (identified in both compounds, 34 and 35). In addition, several hydrophobic contacts occur among different parts of the indicated small-molecules (NBn2, 2’-Bn, 3-Me, and iBu or Bn amide group) and different protein residues. These include interactions with L909 and L915 of the S5-S6 connector, at the end of pore-helix (two subunits), and L959, V960, I962 and Y963 of transmembrane S6 (Figure 4C). Residues W877 or F874 from S5 are also implicated in the complex stabilizing interactions.
At the pore internal mouth (Subsite 4), amide compounds 34 and 35 interconnect with residues spanning all four protein units. The hydrophobic interactions are primarily enabled by Y981 (4 subunits), I985 (3), T982 (1) and M978 (1) residues. However, both compounds adopt different poses within the channel and establish a singular H-bond, between the α–CO of V983 and the 4-CONH proton in 34 (Figure 4D), while the 4-CO of the β–lactam connects with the α–NH proton of residue I985 in 35. Additionally, the aromatic side-chains of Tyr981 residues are able of engaging π–π stacking and T-edge interactions with the aromatic rings of either 2´-Bn and N-Bn2 in 34, or with that of 4-Bn in 35.
The monobenzyl derivative 37 has binding interactions with the same four subsites as its dibenzyl analogue 34. However, the population of solutions is increased at subsites 1, 3 and 4. Residues of the protein implicated in the hydrophobic interconnections with 37 are similar to those above described for the dibenzyl compound at pockets 2-4. However, a different pose was identified at subsite 1, where Y924 residues from all four protein subunits enclose the molecules with the assistance of D920 from a single monomer. Moreover, a π–π stacking interaction is formed between one of these Y924 residues and the phenyl ring at 2´-position of 37. This compound also engages in two H-bonds, involving the 2’-NH and the CO of T982, and the CO of the β–lactam ring and the α–NH of one of the Y981 residues.
The recently resolved cryo-EM structures of TRPM8 complexed with known antagonists, like AMTB and TC-I2014, situated these compounds within a well-defined pocket, formed by the lower part of the TM S1-S4 helices and the TRP domain [56,57]. It encompasses a quite big box that includes a combination of hydrophobic and charged residues. As indicated above, our docking studies to identify potential interaction points of our β–lactams within the TRPM8 tetramer did not lead to any results directed to this precise pocket, thus suggesting a different mode of action for this series of compounds.
Figure 5.
Main subsites for the interaction of compound 34 with TRPM8 channel including extracellular loops. Top and side views of the TRPM8 channels indicate the approximate localization of the docking subsites. Subsite 1 (A); Subsite 2 (B); Subsite 3 (C); Subsite 4 (D). Interactions are drawn with dashed lines and colored gray for hydrophobic, green for π-stacking, and orange for cation-pi interactions. Hydrogen bonds are drawn with continuous blue lines.
Figure 5.
Main subsites for the interaction of compound 34 with TRPM8 channel including extracellular loops. Top and side views of the TRPM8 channels indicate the approximate localization of the docking subsites. Subsite 1 (A); Subsite 2 (B); Subsite 3 (C); Subsite 4 (D). Interactions are drawn with dashed lines and colored gray for hydrophobic, green for π-stacking, and orange for cation-pi interactions. Hydrogen bonds are drawn with continuous blue lines.
3. Materials and Methods
3.1. Synthesis
General procedures, synthesis of key inermediates and specific details on the characterization of diastereomeric β–lactams are gathered in the Supplementary Materials.
General method for the synthesis of substituted 4-carboxamides. To a solution of the β-lactam 4-carboxylate (0.188 mmol) in dry DCM (5 mL) was added PyBrOP (0.225 mmol, 0.105 g), TEA (0.225 mmol, 0.031 mL) and the corresponding amine (0.225 mmol). The reaction was stirred at rt, and after the starting product disappears, the solvent was evaporated to dryness. The reaction mixture was dissolved in EtOAc, washed with 0.1 M HCl, NaHCO3 (10%) and saturated NaCl solution. The organic phase was dried over anhydrous Na2SO4, filtered and evaporated to dryness. The resulting residue was purified on a silica gel column, using the eluent system indicated in each case.
General procedure for the synthesis of 2’-N-monobenzyl derivatives. To a solution of the corresponding 2’-NH2 β-lactam derivative (0.225 mmol) in MeOH (4 mL) was added TEA (0.225 mmol, 0.031 mL) and the corresponding aldehyde (0.337 mmol). The reaction mixture was stirred for 1.5 h at rt. Then, NaBH4 (0.450 mmol, 0.017 g) was added at 0 ºC and stirred at rt for additional 24 h, and finally the solvent was evaporated to dryness. The organic residue was dissolved in EtOAc and washed with H2O and saturated NaCl solution successively. The organic phase was dried over anhydrous Na2SO4, filtered and evaporated to dryness. The resulting residue was purified on a silica gel column, using the eluent system indicated in each case.
4S-Benzyl-4-[N-(iso-butyl)-carbamoyl]-3S-methyl-1-[(2’S-dibenzylamino-3’- phenyl)prop-1’-yl]-2-oxoazetidine (34). Syrup. Yield: 86% (From 33 and iso-butylamine). Eluent: 20 to 30% of EtOAc in hexane. HPLC: tR = 7.54 min (gradient from 15% to 95% of A in 10 min). [α]D = -25.50 (c 1, CH3Cl). 1H-NMR (400 MHz, CDCl3): δ 7.75 - 6.89 (m, 20H, Ar), 6.08 (t, J = 6.5 Hz, 1H, NH), 3.72 (s, 4H, NCH2), 3.72 (dd, J = 14.3, 6.7 Hz, 1H, H1’), 3.25 (p, J = 6.9 Hz, 1H, H2’), 3.18 (dd, J = 14.3, 3.8 Hz, 1H, H1’), 3.16 (d, J = 14.5 Hz, 1H, 4-CH2), 3.16 (m, 1H, H3), 3.02 (d, J = 14.6 Hz, 1H, 4-CH2), 2.95 (dd, J = 13.3, 6.7 Hz, 1H, CH2, iBu), 2.84 (dd, J = 13.8, 6.6 Hz, 1H, H3’), 2.82 (dd, J = 13.4, 6.6 Hz, 1H, CH2, iBu), 2.61 (dd, J = 13.8, 7.1 Hz, 1H, H3’), 1.46 (m, 1H, CH, iBu), 1.18 (d, J = 7.5 Hz, 3H, 3-CH3), 0.75 (d, J = 6.7 Hz, 3H, CH3, iBu), 0.71 (d, J = 6.7 Hz, 3H, CH3, iBu). 13C-NMR (75 MHz, CDCl3): δ 171.7 (C2), 170.3 (4-CONH), 139.5, 139.2, 135.9, 130.0, 129.3, 129.1, 129.0, 128.5, 128.5, 127.4, 127.3, 126.3 (Ar), 69.4 (C4), 60.0 (C2’), 54.0 (C3), 53.2 (NCH2), 47.4 (CH2, iBu), 42.8 (C1’), 40.2 (4-CH2), 36.0 (C3’), 28.4 (CH, iBu), 20.4 (CH3, iBu), 20.3 (CH3, iBu), 10.4 (3-CH3). MS(ES)+: 588.51 [M + H]+. Exact mass calculated for C39H45N3O2: 587.35118, found 587.35311.
4S-Benzyl-4-[N-(benzyl)-carbamoyl]-3S-methyl-1-[(2’S-dibenzylamino-3’-phenyl)prop-1’-yl]-2-oxoazetidine (35). Syrup. Yield: 74% (From 33 and benzylamine). Eluent: 20% to 30% of EtOAc in hexane. HPLC: tR = 7.60 min (gradient from 15% to 95% of A in 10 min). [α]D = - 41.10 (c 1, CH3Cl). 1H-NMR (400 MHz, CDCl3): δ 7.45 (m, 2H, Ar, NH), 7.31 - 7.13 (m, 18H, Ar), 7.09 (m, 2H, Ar), 7.01 (m, 2H, Ar), 6.89 (m, 2H, Ar), 4.53 (dd, J = 14.6, 6.4 Hz, 1H, NHCH2, Bn), 4.15 (dd, J = 14.6, 6.3 Hz, 1H, NHCH2, Bn), 3.77 (m, 1H, H1’), 3.64 (d, J = 14.4 Hz, 2H, NCH2), 3.58 (d, J = 14.4 Hz, 2H, NCH2), 3.37 (d, J = 14.6 Hz, 1H, 4-CH2), 3.23 (q, J = 7.3 Hz, 1H, H3), 3.17 (m, 1H, H1’), 3.14 (m, 1H, H2’), 3.11 (d, J = 14.7 Hz, 1H, 4-CH2), 2.66 (dd, J = 13.7, 5.6 Hz, 1H, H3’), 2.44 (dd, J = 13.6, 8.2 Hz, 1H, H3’), 1.23 (d, J = 7.5 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 172.2 (C2), 170.7 (4-CONH), 139.2, 138.6, 138.1, 135.6, 130.0, 129.2, 129.1, 128.8, 128.7, 128.6, 128.5, 128.1, 127.6, 127.4, 127.3, 126.3 (Ar), 69.7 (C4), 60.3 (C2’), 54.5 (C3), 52.8 (NCH2), 43.9 (NHCH2, Bn), 42.6 (C1’), 39.8 (4-CH2), 35.9 (C3’), 10.2 (3-CH3). MS(ES)+: 622.50 [M + H]+. Exact mass calculated for C42H43N3O2: 621.33553, found 621.33767.
4S-Benzyl-4-[N-(3’’-pyridyl)methylcarbamoyl]-3S-methyl-1-[(2’S-dibenzylamino-3’-phenyl)prop-1’-yl]-2-oxoazetidine (36). Syrup. Yield: 73% (From 33 and 3-picoline). Eluent: 10% of EtOAc in DCM. HPLC: tR = 5.93 min (gradient from 15% to 95% of A in 10 min). [α]D = - 39.71 (c 1, CH3Cl). 1H-NMR (400 MHz, CDCl3): δ 8.44 (dd, J = 4.9, 1.7 Hz, 1H, H4’’), 8.22 (d, J = 2.3 Hz, 1H, H2’’), 7.64 (t, J = 5.9 Hz, 1H, NH), 7.33 (dt, J = 7.8, 2.0 Hz, 1H, H6’’), 7.23 - 7.00 (m, 16H, H5’’, Ar), 6.86 (m, 2H, Ar), 6.75 (m, 2H, Ar), 6.60 (m, 1H, Ar), 4.32 (dd, J = 14.7, 6.1 Hz, 1H, NHCH2), 4.08 (dd, J = 14.8, 5.8 Hz, 1H, NHCH2), 3.88 (d, J = 13.8 Hz, 2H, NCH2), 3.88 (m, 1H, H1’) , 3.75 (d, J = 13.9 Hz, 2H, NCH2), 3.37 (d, J = 14.7 Hz, 1H, 4-CH2), 3.23 (q, J = 7.4 Hz, 1H, H3), 3.13 (d, J = 14.6 Hz, 1H, 4-CH2), 3.12 (m, 2H, H1’, H2’), 2.75 (dd, J = 13.6, 4.3 Hz, 1H, H3’), 2.40 (dd, J = 13.5, 8.6 Hz, 1H, H3‘), 1.20 (d, J = 7.6 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 172.2 (C2), 171.3 (4-CONH), 149.6, 149.0, 139.0, 138.5, 136.1, 135.5, 133.8, 130.0, 129.4, 129.0, 128.9, 128.7, 128.7, 127.6, 127.3, 126.5, 123.5 (Ar), 69.7 (C4), 60.7(C2’), 54.7 (C3), 53.0 (NCH2), 42.4 (C1’), 41.4 (NHCH2), 39.4 (4-CH2), 35.5 (C3’), 10.2 (3-CH3). MS(ES)+: 623.44 [M + H]+. Exact mass calculated for C41H42N4O2: 622.33078, found 622.33184.
4S-Benzyl-4-(terc-butoxy)carbonyl-3S-methyl-1-[(2’S-N-benzylamino-3’-phenyl) prop-1’-yl]-2-oxoazetidine (37). Syrup. Yield: 10% (From 1). Eluent: 3% to 9% of MeOH in DCM. HPLC: tR = 4.74 min (gradient from 15% to 95% of A en 5 min). [α]D = + 25.50 (c 1, CH3Cl). 1H-NMR (400 MHz, CDCl3): δ 7.34 - 7.20 (m, 11H, Ar), 7.14 (m, 4H, Ar), 3.83 (d, J = 13.2 Hz, 1H, NHCH2), 3.76 (d, J = 13.2 Hz, 1H, NHCH2), 3.44 (d, J = 14.5 Hz, 1H, 4-CH2), 3.30 (m, 1H, H2’), 3.20 (dd, J = 14.1, 7.8 Hz, 1H, H1’), 3.09 (q, J = 7.7 Hz, 1H, H3), 3.07 (d, J = 14.5 Hz, 1H, 4-CH2), 3.02 (dd, J = 14.1, 4.6 Hz, 1H, H1’), 2.84 (dd, J = 13.9, 6.1 Hz, 1H, H3’), 2.75 (dd, J = 13.9, 6.9 Hz, 1H, H3’), 2.33 (s ancho, 1H, NH), 1.39 (s, 9H, CH3, tBu), 1.21 (d, J = 7.5 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 170.6 (COO), 170.1 (C2), 138.7, 135.5, 130.1, 129.5, 128.7, 128.5, 128.5, 128.5, 127.4, 127.0, 126.4 (Ar), 83.2 (C, tBu), 68.5 (C4), 57.7 (C2’), 53.4 (C3), 51.3 (NCH2), 47.4 (C1’), 41.0 (4-CH2), 39.1 (C3’), 28.1 (CH3, tBu), 10.6 (3-CH3). MS(ES)+: 499.42 [M + H]+. Exact mass calculated for C38H42N2O3: 498.2882, found 498.2881.
4S-Benzyl-4-(terc-butoxy)carbonyl-3S-methyl-1-[(2’S-N-(3’’-biphenyl)methyl amino-3’-phenyl) prop-1’-yl]-2-oxoazetidine (40). Syrup. Yield: 77% (From 38 and biphenyl-3-carboxaldehyde). Eluent: 9% to 100% of EtOAc in hexane. HPLC: tR = 7.43 min (gradient from 15% to 95% of A in 10 min). [α]D = + 25.50 (c 1, CH3Cl). 1H-NMR (400 MHz, CDCl3): δ 7.56 (m, 2H, Ar), 7.43 (m, 4H, Ar), 7.33 (m, 2H, Ar), 7.21 (m, 7H, Ar), 7.11 (m, 4H, Ar), 3.82 (d, J = 13.2 Hz, 1H, NHCH2), 3.77 (d, J = 13.2 Hz, 1H, NHCH2), 3.40 (d, J = 14.5 Hz, 1H, 4-CH2), 3.28 (m, 1H, H2’), 3.15 (q, J = 7.6 Hz, 1H, H3), 3.05 (d, J = 14.5 Hz, 1H, 4-CH2), 3.04 (m, 2H, H1’), 2.78 (dd, J = 13.9, 6.5 Hz, 1H, H3’), 2.71 (dd, J = 13.9, 6.7 Hz, 1H, H3’), 1.66 (s ancho, 1H, NH), 1.34 (s, 9H, CH3, tBu), 1.14 (d, J = 7.5 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 170.5 (COO), 170.2 (C2), 141.3, 139.0, 135.6, 130.1, 129.5, 128.9, 128.8, 128.7, 128.5, 127.4, 127.3, 127.3, 127.3, 127.2, 126.3, 125.7 (Ar), 83.2 (C, tBu), 68.5 (C4), 57.6 (C2’), 53.5 (C3), 51.5 (NCH2), 47.6 (C1’), 41.0 (4-CH2), 39.5 (C3’), 28.1 (CH3, tBu), 10.6 (3-CH3). MS(ES)+: 575.36 [M + H]+. Exact mass calculated for C38H42N2O3: 574.31954, found 574.31961.
4S-Benzyl-4-terc-butoxycarbonyl-3S-methyl-1-[(2’S-N-(4’’-biphenyl)methylamino-3’- phenyl)prop-1’-yl]-2-oxoazetidine (41). Syrup. Yield: 68% (From 38 and 4-biphenyl-carboxaldehyde). Eluent: 9% to 100% of EtOAc in hexane. HPLC: tR = 7.44 min (gradient from 15% to 95% of A in 10 min). [α]D = - 17.99 (c 1, CH3Cl). 1H-NMR (400 MHz, CDCl3): δ 7.56 (m, 2H, Ar), 7.50 (m, 2H, Ar), 7.42 (m, (2H, Ar), 7.35 - 7.18 (m, 9H, Ar), 7.12 (m, 4H, Ar), 3.80 (d, J = 13.2 Hz, 1H, NHCH2), 3.74 (d, J = 13.1 Hz, 1H, NHCH2), 3.41 (d, J = 14.5 Hz, 1H, 4-CH2), 3.28 (m, 1H, H2’), 3.15 (q, J = 7.6 Hz, 1H, H3), 3.06 (d, J = 14.6 Hz, 1H, 4-CH2), 3.05 (m, 2H, H1’), 2.79 (dd, J = 13.9, 6.5 Hz, 1H, H3’), 2.71 (dd, J = 13.9, 6.7 Hz, 1H, H3’), 1.64 (s ancho, 1H, NH), 1.36 (s, 9H, CH3, tBu), 1.19 (d, J = 7.5 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 170.6 (COO), 170.2 (C2), 141.3, 134.0, 139.8, 139.0, 135.6, 130.1, 129.6, 129.5, 128.8, 128.8, 128.7, 128.5, 127.4, 127.2, 127.2, 126.3 (Ar), 83.2 (C, tBu), 68.5 (C4), 57.7 (C2’), 53.5 (C3), 51.1 (NCH2), 47.7 (C1’), 41.02 (4-CH2), 39.6 (C3’), 28.2 (CH3, tBu), 10.7 (3-CH3). MS(ES)+: 575.28 [M + H]+. Exact mass calculated for C38H42N2O3: 574.31954, found 574.32059.
4S-Benzyl-4-[N-(iso-butyl)-carbamoyl]-3S-methyl-1-[(2’S-(3’’-phenylbencil)amino-3’- phenyl)prop-1’-yl]-2-oxoazetidine (42). Syrup. Yield: 40% (From 39 and 3-phenylbenzaldehyde). Eluent: 25% of EtOAc in hexane. HPLC: tR = 7.35 min (gradient from 15% to 95% of A in 10 min). [α]D = - 82.17 (c 1, CH3Cl). 1H-NMR (300 MHz, CDCl3): δ 10.22 (t, J = 5.5 Hz, 1H, 4-CONH), 7.47 (m, 5H, Ar), 7.42 - 7.21 (m, 10H, Ar), 7.02 (m, 3H, Ar), 6.78 (dt, J = 7.6, 1.4 Hz, 1H, Ar), 4.07 (d, J = 14.2 Hz, 1H, 4-CH2), 3.64 (dd, J = 14.9, 6.3 Hz, 1H, H1’), 3.40 (d, J = 12.8 Hz, 1H, NHCH2), 3.32 (d, J = 12.8 Hz, 1H, NHCH2), 3.14 (q, J = 7.6 Hz, 1H, H3), 3.08 (m, 2H, CH2, iBu, H1’), 2.98 (d, J = 14.3 Hz, 1H, 4-CH2), 2.73 (dd, J = 12.6, 2.6 Hz, 1H, H3’), 2.67 - 2.50 (m, 3H, H2’, H3’, NHCH2), 1.62 (s ancho, 1H, NH), 1.58 (m, 2H, NH, CH, iBu), 1.26 (d, J = 7.5 Hz, 3H, 3-CH3), 0.70 (d, J = 6.6 Hz, 3H, CH3, iBu), 0.69 (d, J = 6.6 Hz, 3H, CH3, iBu’). 13C-NMR (75 MHz, CDCl3): δ 173.5 (4-CONH), 170.8 (C2), 141.7, 140.8, 139.0, 137.9, 137.0, 130.3, 129.1, 129.0, 129.0, 128.9, 128.8, 127.6, 127.4, 127.2, 126.9, 126.9, 126.9, 126.2 (Ar), 70.6 (C4), 56.3 (C2’), 55.8 (C3), 47.8 (NCH2), 47.1 (CH2, iBu), 42.7 (C1’), 41.7 (4-CH2), 38.3 (C3’), 28.1 (CH, iBu), 20.4 (CH3, iBu), 10.9 (3-CH3). MS(ES)+: 574.58 [M + H]+. Exact mass calculated for C38H43N3O2: 573.33553, found 573.33686.
4S-Benzyl-4-[N-(iso-butyl)carbamoyl]-3S-methyl-1-[(2’S-(3’’-fluorobenzyl)amino-3’- phenyl)prop-1’-yl]-2-oxoazetidine (43). Syrup. Yield: 53% (From 39 and 3-fluorobenzaldehyde). Eluent: 25% of EtOAc in hexane. HPLC: tR = 6.82 min (gradient from 15% to 95% of A in 10 min). [α]D = - 42.44 (c 1, CH3Cl). 1H-NMR (300 MHz, CDCl3): δ 9.99 (s ancho, 1H, 4-CONH), 7.30 (m, 8H, Ar), 7.16 (m, 1H, Ar), 6.96 (m, 2H, Ar), 6.88 (m, 1H, Ar), 6.58 (d, J = 7.6 Hz, 1H, Ar), 6.48 (d, J = 9.7 Hz, 1H, Ar), 4.06 (d, J = 14.2 Hz, 1H, 4-CH2), 3.61 (dd, J = 14.9, 6.5 Hz, 1H, H1’), 3.36 (d, J = 13.0 Hz, 1H, NHCH2), 3.29 (d, J = 13.1 Hz, 1H, NHCH2), 3.14 (q, J = 7.2 Hz, 1H, H3), 3.12 (m, 2H, H1’, CH2, iBu), 2.97 (d, J = 14.3 Hz, 1H, 4-CH2), 2.72 - 2.49 (m, 4H, H2’, CH2, iBu,H3’), 1.68 (s ancho, 1H, NH), 1.60 (m, 2H, NH, CH, iBu), 1.26 (d, J = 7.4 Hz, 3H, 3-CH3), 0.74 (d, J = 6.7 Hz, 3H, CH3, iBu), 0.73 (d, J = 6.7 Hz, 3H, CH3, iBu). 13C-NMR (75 MHz, CDCl3): δ 173.4 (4-CONH), 170.7 (C2), 162.9 (d, J = 24.8 Hz, C3’’), 141.0 (d, J = 7.3 Hz, C1’’), 137.7, 136.9, 130.3, 130.2 (d, J = 8.7 Hz, C5’’), 129.0, 128.9, 128.8, 127.3, 127.0, 123.6 (d, J = 3.0 Hz, C6’’), 114.9 (d, J = 21.4 Hz, C2’’), 114.4 (d, J = 21.0 Hz, C4’’) (Ar), 70.5 (C4), 56.1 (C2’), 55.8 (C3), 47.2 (CH2, iBu), 47.0 (NCH2), 42.6 (C1’), 41.6 (4-CH2), 38.1 (C3’), 28.2 (CH, iBu), 20.4 (CH3, iBu), 10.7 (3-CH3). MS(ES)+: 516.55 [M + H]+, 518.54 [M + 2]+. Exact mass calculated for C32H38FN3O2: 515.29481, found 515.2949.
4S-Benzyl-4-[N-(iso-butyl)carbamoyl]-3S-methyl-1-[(2’S-(3’’-bromobenzyl)amino-3’-phenyl)prop-1’-yl]-2-oxoazetidine (44). Syrup. Yield: 43% (From 39 and 3-bromobenzaldehyde). Eluent: 9% to 14% of EtOAc in DCM. HPLC: tR = 7.04 min (gradient from 15% to 95% of A in 10 min). [α]D = - 88.58 (c 1, CH3Cl). 1H-NMR (300 MHz, CDCl3): δ 9.98 (t, J = 5.4 Hz, 1H, 4-CONH), 7.37 - 7.19 (m, 8H, Ar), 7.06 (t, J = 7.8 Hz, 1H, Ar), 7.00 - 6.91 (m, 4H, Ar), 6.70 (dt, J = 7.6, 1.3 Hz, 1H, Ar), 4.08 (d, J = 14.3 Hz, 1H, 4-CH2), 3.61 (dd, J = 14.9, 6.6 Hz, 1H, H1’), 3.32 (d, J = 13.1 Hz, 1H, NHCH2), 3.24 (d, J = 13.0 Hz, 1H, NHCH2), 3.14 (q, J = 7.3 Hz, 1H, H3), 3.13 (m, 1H, CH2, iBu), 3.03 (dd, J = 15.1, 2.8 Hz, 1H, H1’), 2.96 (d, J = 14.3 Hz, 1H, 4-CH2), 2.74 - 2.46 (m, 4H, H2’, H3’, CH2, iBu), 1.58 (m, 2H, NH, CH, iBu), 1.55(s ancho, 1H, NH), 1.27 (d, J = 7.5 Hz, 3H, 3-CH3), 0.75 (d, J = 6.6 Hz, 3H, CH3, iBu), 0.75 (d, J = 6.7 Hz, 3H, CH3, iBu). 13C-NMR (75 MHz, CDCl3): δ 173.4 (4-CONH), 170.7 (C2), 140.9, 137.7, 137.0, 131.2, 130.6, 130.3, 130.2, 129.1, 128.9, 128.9, 127.4, 127.1, 126.6, 122.8 (Ar), 70.6 (C4), 56.0 (C2’), 56.0 (C3), 47.2 (CH2, iBu), 46.9 (NCH2), 42.5 (C1’), 41.7 (4-CH2), 38.2 (C3’), 28.2 (CH, iBu), 20.5 (CH3, iBu), 10.8 (3-CH3). MS(ES)+: 576.49 [M + H]+, 578.56 [M + 2]+. Exact mass calculated for C32H38BrN3O2: 575.21474, found 575.21447.
4S-Benzyl-4-[N-(iso-butyl)-carbamoyl]-3S-methyl-1-[(2’S-(2’’-naphtylmethyl) amino-3’-phenyl)prop-1’-yl]-2-oxoazetidine (45). Syrup. Yield: 41% (From 39 and 2-naphtaldehyde). Eluent: 33% of EtOAc in DCM. HPLC: tR = 7.35 min (gradient from 15% to 95% of A in 10 min). [α]D = - 62.25 (c 1, CH3Cl). 1H-NMR (300 MHz, CDCl3): δ 10.10 (s ancho, 1H, 4-CONH), 7.78 (d, J = 8.1 Hz, 1H, Ar), 7.71 (d, J = 8.3 Hz, 1H, Ar), 7.43 - 7.19 (m, 12H, Ar), 6.97 (m, 3H, Ar), 4.11 (d, J = 14.2 Hz, 1H, 4-CH2), 3.85 (d, J = 11.8 Hz, 1H, NHCH2), 3.79 (dd, J = 14.9, 6.9 Hz, 1H, H1’), 3.64 (d, J = 12.0 Hz, 1H, NHCH2), 3.17 (q, J = 7.2 Hz, 1H, H3) 3.15 (m, 1H, H1’), 3.00 (d, J = 14.2 Hz, 1H, 4-CH2), 2.79 - 2.52 (m, 4H, H3’, CH2, iBu, H2’), 2.31 (m, 1H, CH2, iBu), 1.50 (s ancho, 1H, NH), 1.28 (d, J = 7.5 Hz, 3H, 3-CH3), 1.13 (m, 1H, CH, iBu), 0.45 (d, J = 6.6 Hz, 3H, CH3, iBu), 0.40 (d, J = 6.6 Hz, 3H, CH3, iBu). 13C-NMR (75 MHz, CDCl3): δ 173.6 (4-CONH), 170.6 (C2), 137.8, 137.1, 134.3, 133.8, 131.5, 130.4, 129.0, 128.8, 128.3, 127.4, 127.0, 126.8, 126.5, 125.8, 125.4, 122.8 (Ar), 70.8 (C4), 57.0 (C2’), 56.0 (C3), 46.9 (CH2, iBu), 45.5 (NHCH2), 42.9 (C1’), 41.8 (4-CH2), 38.0 (C3’), 27.7 (CH, iBu), 20.2 (CH3, iBu), 10.8 (3-CH3). MS(ES)+: 548.63 [M + H]+. Exact mass calculated for C36H41N3O2: 547.31988, found 547.32163.
3.2. Molecular Modeling
The docking protocol used the algorithm AutoDockLGA [58] and the force filed AMBER03 [59], implemented in Yasara software version 22.5.22 [60]. Interaction analysis were done with PLIP, a fully automated protein-ligand interaction server [61]. Figures were drawn with PyMol [The PyMOL Molecular Graphics System, Version 2.6 Schrödinger, LLC.). Retrieved from http://www.pymol.org/pymol]. Detailed interaction can be seen in Supplementary Materials.
3.3. Biological Assays
4. Conclusions
Two novel series of β–lactam derivarives, featuring N-susbstituted 4-carboxamides and/or 2’-N’-monobenzyl groups, have been prepared and analyzed on their capacity to inhibit the Ca2+ entry triggered by menthol in TRPM8 channels. Structure-activity relationships unveiled that N’-monobenzyl derivatives, regardeless the size and the electronic character of the substituents on the phenyl ring, failed to competing with the antagonist activity demonstrated by the corresponding N’,N’-dibenzyl analogues. However, the 4-isobutyl- and 4-benzylcarboxamides, having a minute increase of 3 units in the TPSA, exhibited enhanced inhibitory potency against the agonist-induced TRPM8 activation when contrasted with the OtBu analogue. Unfortunately, the introduction of an extra nitrogen atom within the N-benzyl moiety, leading to increased TPSA values, visibly compromised the activity. The most potent compounds within the 4-carboxamide series, 34 and 35, confirmed their activity in electrophysiology assays, show selectivity for the TRPM8 channel, and displayed antiallodynic activity in a model of oxaliplatin-induced peripheral neuropathy. Ongoing research within our group to further explore this family of TRPM8 antagonists is directed to enhance TPSA values while maintaining the antagonist activity.
Supplementary Materials
Author Contributions
Conceptualization, R.G.-M.; investigation, C.M.-E., M.Á.B., J.A.M., A.M.-P., J.d.A.-L., S.G.-R., S.K. and G.F.-B.; writing—original draft, C.M.-E. and R.G.-M.; writing—review & editing, A.F.-M., A.F.-C. and R.G.-M.; supervision, G.F.-B., T.V., A.F.-C. and R.G.-M.; funding acquisition, A.F.-M., A.F.-C. and R.G.-M.. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MICINN, grant numbers RTI2018-097189-B-C21 (to A.F.-M. and A.F.-C.), RTI2018-097189-B-C22 (to R.G.-M.), PID2021-126423OB-C21 (to A.F.-M. and A.F.-C.) and PID2021-126423OB-C21 (to R.G.-M.), Generalitat Valenciana, PROMETEO/2021/031 (to A.F.-M.) and Comunidad de Madrid, IND2017/BMD7673 (to R.G.-M.).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of Miguel Hernández University (UMH.IBM.AFM.02.18—11 October 2018).
Informed Consent Statement
Not applicable.
Acknowledgments
We would like to thank Aída García and Aída Batalla for her support in the economic management of the projects. Part of this work was awarded with the Menarini Prize within the XXI call of Prizes for Young Reserchears (to Cristina Martín-Escura), organized by the Spanish Society of Medicinal Chemistry (SEQT).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kashio, M.; Tominaga, M. TRP channels in thermosensation. Curr. Opin. Neurobiol. 2022, 75, 102591. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M. Temperature sensing. Igaku no Ayumi 2019, 270, 998–1003. [Google Scholar]
- Yue, L.; Xu, H. TRP channels in health and disease at a glance. J. Cell Sci. 2021, 134, jcs258372. [Google Scholar] [CrossRef]
- Guibert, C.; Ducret, T.; Savineau, J.-P. Expression and physiological roles of TRP channels in smooth muscle cells. Adv. Exp. Med. Biol. 2011, 704, 687–706. [Google Scholar] [PubMed]
- Voets, T.; Vriens, J.; Vennekens, R. Targeting TRP Channels - Valuable Alternatives to Combat Pain, Lower Urinary Tract Disorders, and Type 2 Diabetes? Trends Pharmacol. Sci. 2019, 40, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cobos, J.C.; Zhang, X.; Motiani, R.K.; Harmon, K.E.; Trebak, M. TRPs to cardiovascular disease. In Proceedings of the TRP Channels Drug Discovery; Springer, 2012; Vol. 2; pp. 3–40. [Google Scholar]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef]
- Perez de Vega, M.J.; Gomez-Monterrey, I.; Ferrer-Montiel, A.; Gonzalez-Muniz, R. Transient Receptor Potential Melastatin 8 Channel (TRPM8) Modulation: Cool Entryway for Treating Pain and Cancer. J. Med. Chem. 2016, 59, 10006–10029. [Google Scholar] [CrossRef]
- Izquierdo, C.; Martin-Martinez, M.; Gomez-Monterrey, I.; Gonzalez-Muniz, R. TRPM8 Channels: Advances in Structural Studies and Pharmacological Modulation. Int. J. Mol. Sci. 2021, 22, 8502. [Google Scholar] [CrossRef]
- Zhang, L.; Barritt, G.J. TRPM8 in prostate cancer cells: a potential diagnostic and prognostic marker with a secretory function? Endocr. Relat. Cancer 2006, 13, 27–38. [Google Scholar] [CrossRef]
- Bautista, D.M.; Siemens, J.; Glazer, J.M.; Tsuruda, P.R.; Basbaum, A.I.; Stucky, C.L.; Jordt, S.E.; Julius, D. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 2007, 448, 204–208. [Google Scholar] [CrossRef]
- Dhaka, A.; Earley, T.J.; Watson, J.; Patapoutian, A. Visualizing cold spots: TRPM8-expressing sensory neurons and their projections. J. Neurosci. 2008, 28, 566–575. [Google Scholar] [CrossRef]
- De Caro, C.; Cristiano, C.; Avagliano, C.; Bertamino, A.; Ostacolo, C.; Campiglia, P.; Gomez-Monterrey, I.; La Rana, G.; Gualillo, O.; Calignano, A.; et al. Characterization of new TRPM8 modulators in pain perception. Int. J. Mol. Sci. 2019, 20, 5544. [Google Scholar] [CrossRef]
- Soeda, M.; Ohka, S.; Nishizawa, D.; Hasegawa, J.; Nakayama, K.; Ebata, Y.; Ikeda, K.; Soeda, M.; Fukuda, K.-I.; Ichinohe, T. Cold pain sensitivity is associated with single-nucleotide polymorphisms of PAR2/F2RL1 and TRPM8. Mol. Pain 2021, 17, 17448069211002008. [Google Scholar] [CrossRef]
- Pertusa, M.; Solorza, J.; Madrid, R. Molecular determinants of TRPM8 function: key clues for a cool modulation. Front. Pharmacol. 2023, 14, 1213337. [Google Scholar] [CrossRef] [PubMed]
- Voets, T.; Owsianik, G.; Nilius, B. TRPM8. Handb. Exp. Pharmacol. 2007, 179, 329–344. [Google Scholar]
- Almaraz, L.; Manenschijn, J.-A.; de la Pena, E.; Viana, F. TRPM8. Handb. Exp. Pharmacol. 2014, 222, 547–579. [Google Scholar]
- Liu, Y.; Mikrani, R.; He, Y.; Faran Ashraf Baig, M.M.; Abbas, M.; Naveed, M.; Tang, M.; Zhang, Q.; Li, C.; Zhou, X. TRPM8 channels: A review of distribution and clinical role. Eur. J. Pharmacol. 2020, 882, 173312. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, N.; Fujita, T. The TRPM8 channel as a potential therapeutic target for bladder hypersensitive disorders. J. Smooth Muscle Res. 2022, 58, 11–21. [Google Scholar] [CrossRef]
- Wu, B.; Su, X.; Zhang, W.; Zhang, Y.-H.; Feng, X.; Ji, Y.-H.; Tan, Z.-Y. Oxaliplatin depolarizes the IB4- dorsal root ganglion neurons to drive the development of neuropathic pain through TRPM8 in mice. Front. Mol. Neurosci. 2021, 14, 690858. [Google Scholar] [CrossRef] [PubMed]
- Aierken, A.; Xie, Y.-K.; Dong, W.; Apaer, A.; Lin, J.-J.; Zhao, Z.; Yang, S.; Xu, Z.-Z.; Yang, F. Rational Design of a Modality-Specific Inhibitor of TRPM8 Channel against Oxaliplatin-Induced Cold Allodynia. Adv. Sci. 2021, 8, 2101717. [Google Scholar] [CrossRef]
- Luyts, N.; Daniluk, J.; Freitas, A.C.N.; Bazeli, B.; Janssens, A.; Mulier, M.; Everaerts, W.; Voets, T. Inhibition of TRPM8 by the urinary tract analgesic drug phenazopyridine. Eur. J. Pharmacol. 2023, 942, 175512. [Google Scholar] [CrossRef] [PubMed]
- Fakih, D.; Baudouin, C.; Goazigo, A.R.-L.; Parsadaniantz, S.M. TRPM8: a therapeutic target for neuroinflammatory symptoms induced by severe dry eye disease. Int. J. Mol. Sci. 2020, 21, 8756. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-R.; Liu, Q.; Chen, G.-Y.; Hu, Y.; Sham, J.S.K.; Lin, M.-J. Down-Regulation of TRPM8 in Pulmonary Arteries of Pulmonary Hypertensive Rats. Cell. Physiol. Biochem. 2013, 31, 892–904. [Google Scholar] [CrossRef]
- Naumov, D.E.; Kotova, O.O.; Gassan, D.A.; Sugaylo, I.Y.; Afanas’eva, E.Y.; Sheludko, E.G.; Perelman, J.M. Effect of TRPM8 and TRPA1 Polymorphisms on COPD Predisposition and Lung Function in COPD Patients. J. Pers. Med. 2021, 11. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Q.; Hua, L.; Pan, J. Inhibition of transient receptor potential melastatin 8 alleviates airway inflammation and remodeling in a murine model of asthma with cold air stimulus. Acta Biochim. Biophys. Sin. 2018, 50, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Szallasi, A. ThermoTRP Channel Expression in Cancers: Implications for Diagnosis and Prognosis (Practical Approach by a Pathologist). Int. J. Mol. Sci. 2023, 24, 9098. [Google Scholar] [CrossRef]
- Gonzalez-Muniz, R.; Bonache, M.A.; Martin-Escura, C.; Gomez-Monterrey, I. Recent progress in TRPM8 modulation: an update. Int. J. Mol. Sci. 2019, 20, 2618. [Google Scholar] [CrossRef]
- Horne, D.B.; Biswas, K.; Brown, J.; Bartberger, M.D.; Clarine, J.; Davis, C.D.; Gore, V.K.; Harried, S.; Horner, M.; Kaller, M.R.; et al. Discovery of TRPM8 Antagonist (S)-6-(((3-Fluoro-4-(trifluoromethoxy)phenyl)(3-fluoropyridin-2-yl)methyl)carbamoyl)nicotinic Acid (AMG 333), a Clinical Candidate for the Treatment of Migraine. J. Med. Chem. 2018, 61, 8186–8201. [Google Scholar] [CrossRef]
- Andrews, M.D.; Af Forselles, K.; Beaumont, K.; Galan, S.R.G.; Glossop, P.A.; Grenie, M.; Jessiman, A.; Kenyon, A.S.; Lunn, G.; Maw, G.; et al. Discovery of a selective TRPM8 antagonist with clinical efficacy in cold-related pain. ACS Med. Chem. Lett. 2015, 6, 419–424. [Google Scholar] [CrossRef]
- Fernandez-Carvajal, A.; Gonzalez-Muniz, R.; Fernandez-Ballester, G.; Ferrer-Montiel, A. Investigational drugs in early phase clinical trials targeting thermotransient receptor potential (thermoTRP) channels. Expert Opin. Investig. Drugs 2020, 29, 1209–1222. [Google Scholar] [CrossRef]
- Gosset, J.R.; Beaumont, K.; Matsuura, T.; Winchester, W.; Attkins, N.; Glatt, S.; Lightbown, I.; Ulrich, K.; Roberts, S.; Harris, J.; et al. A cross-species translational pharmacokinetic-pharmacodynamic evaluation of core body temperature reduction by the TRPM8 blocker PF-05105679. Eur. J. Pharm. Sci. 2017, 109S, S161–S167. [Google Scholar] [CrossRef] [PubMed]
- Gavva, N.R.; Davis, C.; Lehto, S.G.; Rao, S.; Wang, W.; Zhu, D.X.D. Transient receptor potential melastatin 8 (TRPM8) channels are involved in body temperature regulation. Mol. Pain 2012, 8, 115. [Google Scholar] [CrossRef] [PubMed]
- Thapa, D.; Barrett, B.; Argunhan, F.; Brain, S.D. Influence of Cold-TRP Receptors on Cold-Influenced Behaviour. Pharmaceuticals 2022, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- de la Torre-Martinez, R.; Bonache, M.A.; Llabres-Campaner, P.J.; Balsera, B.; Fernandez-Carvajal, A.; Fernandez-Ballester, G.; Ferrer-Montiel, A.; Perez de Vega, M.J.; Gonzalez-Muniz, R. Synthesis, high-throughput screening and pharmacological characterization of β-lactam derivatives as TRPM8 antagonists. Sci. Rep. 2017, 7, 10766. [Google Scholar] [CrossRef] [PubMed]
- Bonache, M.Á.; Llabrés, P.J.; Martín-Escura, C.; De la Torre-Martínez, R.; Medina-Peris, A.; Butrón, L.; Gómez-Monterrey, I.; Roa, A.M.; Fernández-Ballester, G.; Ferrer-Montiel, A.; et al. Phenylalanine-derived β-lactam trpm8 modulators. Configuration effect on the antagonist activity. Int. J. Mol. Sci. 2021, 22, 2370. [Google Scholar] [CrossRef] [PubMed]
- Bonache, M.A.; Martin-Escura, C.; de la Torre Martinez, R.; Medina, A.; Gonzalez-Rodriguez, S.; Francesch, A.; Cuevas, C.; Roa, A.M.; Fernandez-Ballester, G.; Ferrer-Montiel, A.; et al. Highly functionalized β-lactams and 2-ketopiperazines as TRPM8 antagonists with antiallodynic activity. Sci. Rep. 2020, 10, 14154. [Google Scholar] [CrossRef]
- Martin-Escura, C.; Medina-Peris, A.; Spear, L.A.; de la Torre Martinez, R.; Olivos-Ore, L.A.; Barahona, M.V.; Gonzalez-Rodriguez, S.; Fernandez-Ballester, G.; Fernandez-Carvajal, A.; Artalejo, A.R.; et al. β-Lactam TRPM8 Antagonist RGM8-51 Displays Antinociceptive Activity in Different Animal Models. Int. J. Mol. Sci. 2022, 23, 2692. [Google Scholar] [CrossRef]
- Werkheiser, J.L.; Rawls, S.M.; Cowan, A. Mu and kappa opioid receptor agonists antagonize icilin-induced wet-dog shaking in rats. Eur. J. Pharmacol. 2006, 547, 101–105. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Perez-Faginas, P.; O’Reilly, F.; O’Byrne, A.; Garcia-Aparicio, C.; Martin-Martinez, M.; Perez de Vega, M. J.; Garcia-Lopez, M. T.; Gonzalez-Muniz, R. Exceptional Stereoselectivity in the Synthesis of 1,3,4-Trisubstituted 4-Carboxy β-Lactam Derivatives from Amino Acids. Org. Lett. 2007, 9, 1593–1596. [Google Scholar] [CrossRef]
- Journigan, V.B.; Alarcón-Alarcón, D.; Feng, Z.; Wang, Y.; Liang, T.; Dawley, D.C.; Amin, A.R.M.R.; Montano, C.; Van Horn, W.D.; Xie, X.Q.; et al. Structural and in Vitro Functional Characterization of a Menthyl TRPM8 Antagonist Indicates Species-Dependent Regulation. ACS Med. Chem. Lett. 2021, 12, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Vangeel, L.; Benoit, M.; Miron, Y.; Miller, P.E.; De Clercq, K.; Chaltin, P.; Verfaillie, C.; Vriens, J.; Voets, T. Functional expression and pharmacological modulation of TRPM3 in human sensory neurons. Br. J. Pharmacol. 2020, 177, 2683–2695. [Google Scholar] [CrossRef] [PubMed]
- Li, W.G.; Xu, T. Le ASIC3 channels in multimodal sensory perception. ACS Chem. Neurosci. 2011, 2, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Khan, S.; Kim, Y.S. Insight into Pain Modulation: Nociceptors Sensitization and Therapeutic Targets. Curr. Drug Targets 2019, 20, 775–788. [Google Scholar] [CrossRef]
- Yang, C.; Yamaki, S.; Jung, T.; Kim, B.; Huyhn, R.; McKemy, D.D. Endogenous inflammatory mediators produced by injury activate TRPV1 and TRPA1 nociceptors to induce sexually dimorphic cold pain that is dependent on TRPM8 and GFRa3. J. Neurosci. 2023, 43, 2803–2814. [Google Scholar] [CrossRef]
- Chen, S.R.; Chen, H.; Yuan, W.X.; Wess, J.; Pan, H.L. Dynamic control of glutamatergic synaptic input in the spinal cord by muscarinic receptor subtypes defined using knockout mice. J. Biol. Chem. 2010, 285, 40427–40437. [Google Scholar] [CrossRef]
- Lee, J.H.; Go, D.; Kim, W.; Lee, G.; Bae, H.; Quan, F.S.; Kim, S.K. Involvement of spinal muscarinic and serotonergic receptors in the anti-allodynic effect of electroacupuncture in rats with oxaliplatin-induced neuropathic pain. Korean J. Physiol. Pharmacol. 2016, 20, 407–414. [Google Scholar] [CrossRef]
- Camilleri, M. Toward an effective peripheral visceral analgesic: responding to the national opioid crisis. Am. J. Physiol. 2018, 314, G637. [Google Scholar] [CrossRef]
- Beijers, A.J.M.; Jongen, J.L.M.; Vreugdenhil, G. Chemotherapy-induced neurotoxicity: The value of neuroprotective strategies. Neth. J. Med. 2012, 70, 18–25. [Google Scholar]
- Rimola, V.; Osthues, T.; Koenigs, V.; Geisslinger, G.; Sisignano, M. Oxaliplatin causes transient changes in TRPM8 channel activity. Int. J. Mol. Sci. 2021, 22, 4962. [Google Scholar] [CrossRef]
- Journigan, V.B.; Feng, Z.; Rahman, S.; Wang, Y.; Amin, A.R.M.R.; Heffner, C.E.; Bachtel, N.; Wang, S.; Gonzalez-Rodriguez, S.; Fernández-Carvajal, A.; et al. Structure-Based Design of Novel Biphenyl Amide Antagonists of Human Transient Receptor Potential Cation Channel Subfamily M Member 8 Channels with Potential Implications in the Treatment of Sensory Neuropathies. ACS Chem. Neurosci. 2020, 11, 268–290. [Google Scholar] [CrossRef] [PubMed]
- Bertamino, A.; Ostacolo, C.; Medina, A.; Di Sarno, V.; Lauro, G.; Ciaglia, T.; Vestuto, V.; Pepe, G.; Basilicata, M.G.; Musella, S.; et al. Exploration of TRPM8 Binding Sites by β-Carboline-Based Antagonists and Their In Vitro Characterization and In Vivo Analgesic Activities. J. Med. Chem. 2020, 63, 9672–9694. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S.-Y. Structure of the cold- and menthol-sensing ion channel TRPM8. Science. 2018, 359, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S. Structure of the cold- and menthol-sensing ion channel. Science. 2017, 359, 237–241. [Google Scholar] [CrossRef]
- Diver, M.M.; Cheng, Y.; Julius, D. Structural insights into TRPM8 inhibition and desensitization. Science. 2019, 365, 1434–1440. [Google Scholar] [CrossRef]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.-Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science. 2019, 363, eaav9334. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock and AutoDockTools: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Ozvoldik, K.; Stockner, T.; Rammner, B.; Krieger, E. Assembly of Biomolecular Gigastructures and Visualization with the Vulkan Graphics API. J. Chem. Inf. Model. 2021, 61, 5293–5303. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443. [Google Scholar] [CrossRef]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.-D.; Benoit, M.; Xue, F.-Q.; Janssens, A.; Kerselaers, S.; et al. TRPM3 Is a Nociceptor Channel Involved in the Detection of Noxious Heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Muniz, R.; Perez Ma. Jesus, de V.; Bonache de Marcos, M.A.; Ferrer Montiel, A.; Fernandez Carvajal, A.; De la Torr, e. R. Heterocyclic compounds as TRPM8 channel antagonists and uses thereof. WO 2017005950 2017, WO 2017005950.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



