
Submitted:
30 August 2023
Posted:
06 September 2023
You are already at the latest version
Abstract
The aim of this systematic review was to describe clinical and genetic features of syndromes showing oligodontia as a sign. The review was performed according to the PRISMA 2020 checklist guidelines, and the search was conducted using PubMed, Scopus, Lilacs, Web of science, Livivo and EMBASE and supplemented by a gray literature search on Google Scholar and ProQuest, applying key terms relevant to the research questions. The systematic review identified 49 types of syndromes in 91 studies, and the most common was hypohidrotic ectodermal dysplasia, which was reported in 24 patients in 22 studies. Other commonest syndromes that reported oligodontia included Axenfeld-Rieger syndrome, Witkop’s syndrome, Ellis-van Creveld syndrome, blepharocheilodontic syndrome and oculo-facio-cardio-dental syndrome. The X-linked mode of inheritance was the most reported (n=14 studies), followed by the autosomal dominant (n=11 studies). The review describes the main syndromes that may have oligodontia as a clinical sign and reinforce the need of oro-dental-facial examining for adequate diagnosis and treatment of the affected patients. Molecular analysis in order to better understand the occurrence of oligodontia is imperative.
Keywords:
tooth agenesis
; oligodontia
; syndrome
; systematic review
1. Introduction
Tooth agenesis is defined as the absence of teeth from the normal series by a failure to develop, and encompasses hypodontia, oligodontia, and anodontia [1]. The absence of up to five teeth is classified as hypodontia, the congenital absence of six or more teeth is defined as oligodontia, and anodontia refers to the complete absence of all teeth from the normal series [1]. Tooth development is regulated by a series of signaling pathways, and genetic mutations in specific genes have been described as the cause of such defects [2,3]. Besides, environmental factors such as trauma, infections, toxins and dietary deficiencies have been implicated and could interact with the genetic factors as a complex and multifactorial disease [4,5].
The occurrence of oligodontia can be observed as an isolated trait (non-syndromic oligodontia) or accompanying other features as part of a syndrome [6,7,8,9]. In patients with non-syndromic oligodontia, the congenitally missing teeth are the only apparent clinical finding [10], and studies show that these cases are rare, with a prevalence ranging from 0.16% to 0.36%, depending on the population studied [11]. However, some patients can display mild phenotypes or the clinical expression of phenotypes other than oligodontia can only appear late in life, and the misdiagnosis of non-syndromic oligodontia can occur [12]. Oligodontia is a significant and integral diagnostic feature for many syndromes, but other concurrent dental anomalies may occur, including microdontia, short roots, dental impactions, delayed formation of teeth, delayed eruption, transposition of canines and premolars, taurodontism, and enamel hypoplasia [2,13,14]. Patients with oligodontia present serious deficiencies in their quality of life due to decreased masticatory function, phonetic ability and maxillofacial aesthetics [15,16]. As oligodontia can be a clinical manifestation of a large and heterogenous group of syndromes with multiple signs and symptoms, this systematic review aims summarize the available literature concerning the presence of oligodontia in syndromes, emphasizing the phenotype and the molecular etiology, in order to assist in diagnosis and management of the patients.
2. Materials and Methods
2.1. Protocol and Registration
This systematic review was performed according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) checklist [17], and its protocol was recorded in the International Prospective Register of Systematic Reviews (PROSPERO) database, under registration number CRD42020190814.
2.2. Eligibility Criteria
The PICOS approach was used to formulate the question for this study: P—participants (syndromic patients with oligodontia), I—intervention (none), C—comparison (none), O—outcomes (frequency and types of syndromes associated with oligodontia, pattern of missing teeth and frequency of pathogenic variants), S—study (case reports and case series).
The criteria for the exclusion of articles were: (1) Non-syndromic cases; (2) Studies that did not report or report unclear dental X-rays; (3) Studies that did not report representative cases of oligodontia; (4) Studies that report cases of hypodontia or anodontia; (5) Studies that do not include pattern of tooth agenesis; (6) Reviews, letters, conference abstract, personal opinions, and in vitro or in vivo animal studies; (7) Full-text copy not available; (8) Duplicated data from other study; (9) Articles that were not in Roman alphabet; (10) Studies with absence of clinical information.
2.3. Study Selection
The selection process of the studies was performed using an individual search in each bibliographic database: PubMed, Scopus, Lilacs, Web of science, Livivo and EMBASE. A gray literature search was conducted using Google Scholar and ProQuest. The research was performed in December 2022. However, a second literature search was performed using the same terms on August 8th, 2021, retrieving articles published between January and August 2023. The search strategy can be assessed in Supplementary Table S1. The duplicate references were removed by reference manager software (EndNote X7, Thomson Reuters). All references were transferred and worked on the Rayyan (Rayyan, Qatar Computing Research Institute, Qatar Foundation), developed specifically to expedite the initial screening of abstracts and titles [18].
The course of the research was established in two distinct phases. In the first phase, two authors (NLC and KKMR) independently read all titles and abstracts, taking into account the eligibility criteria initially defined. In cases of no consensus, a third author (ACA) was involved, who determined which articles would be included in the second phase. The second phase was carried out by the same authors, who performed a full-text reading of the screened articles. Disagreements were solved by discussion involving the third author (ACA).
2.4. Data Collection Process and Data Items
The data collection process was carried out by the two authors (NLC and KKMR) who selected the articles initially in the two phases, in which the necessary and relevant information of each study was collected. The information was checked by a third author (ACA).
2.5. Risk of Bias within Studies
The risk-of-bias was assessed by two authors (NLC and KKMR) using the Joanna Briggs Institute Critical Appraisal Tools for Studies Reporting Prevalence Data for Use in Systematic Reviews—referred to Case Reports [19]. The authors scored each item as “yes,” “no,” “unclear,” or “not applicable” when assessing the quality of each included study. Decisions about scoring were discussed by all reviewers, a study was characterized as having a high risk of bias when it reached a “yes” score of up to 49%, moderate when 50% to 69%, and low when >70%.
2.6. Interaction Analysis
The functional relevance of identified genes was further investigated with STRING, 11.0 version (search tool for the retrieval of interacting genes, http://string-db.org), which provides a P value after applying false discovery rate for correction of multiple testing.
3. Results
The searches conducted in the 6 databases resulted in 2,569 scientific articles. After removal of duplicates, 1,772 articles were totalled. The gray literature search resulted in 81 articles. After reading the titles and abstracts in the first phase, 288 articles were selected for the next phase. At the end of reading the full articles (second phase), 91 articles were included for the qualitative synthesis. The studies excluded in the second phase are listed in the Supplementary Table S2. The review process is schematized in a flowchart depicted on Figure 1.
The main data of each selected article are summarized in Supplementary Table S3. Together, the studies reported 106 patients, ages ranging from 3 to 75 years old, with oligodontia as a clinical feature of different types of syndromes. Risk-of-bias assessment in each study is reported in Supplementary Table S4.
The syndromes more frequently identified, in decreasing order, were hypohidrotic ectodermal dysplasia (HED) (24 patients in 22 studies), Axenfeld-Rieger syndrome (ARS) (8 patients in 5 studies), Witkop’s syndrome (6 patients in 5 studies), Ellis-van Creveld syndrome (EVCS) (5 patients in 4 studies), blepharocheilodontic syndrome (BCDS) (5 patients in 2 studies), oculo-facio-cardio-dental syndrome (4 patients in 4 studies), incontinentia pigmenti (3 patients in 3 studies), Hallermann-Streiff syndrome (3 patients in 3 studies), polycistic ovarian syndrome (3 patients in 2 studies), Down syndrome (2 patients in 2 studies), Carvajal syndrome (2 patients in 2 studies), Carpenter syndrome (2 patients in 2 studies) and Kabuki syndrome (2 patients in 2 studies). Other uncommon syndromes listed in Supplementary Table S3 were reported in 1 patient each.
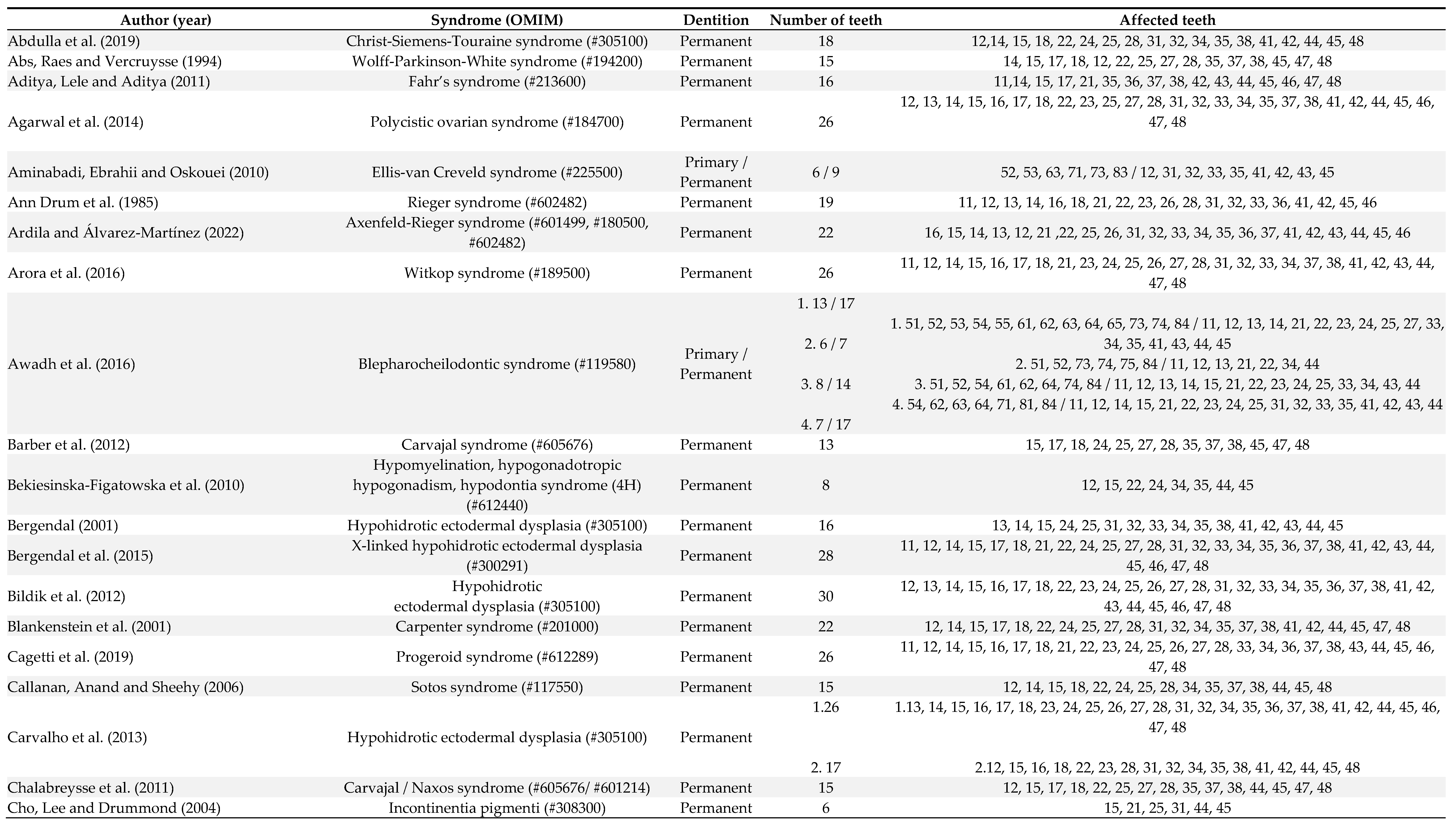
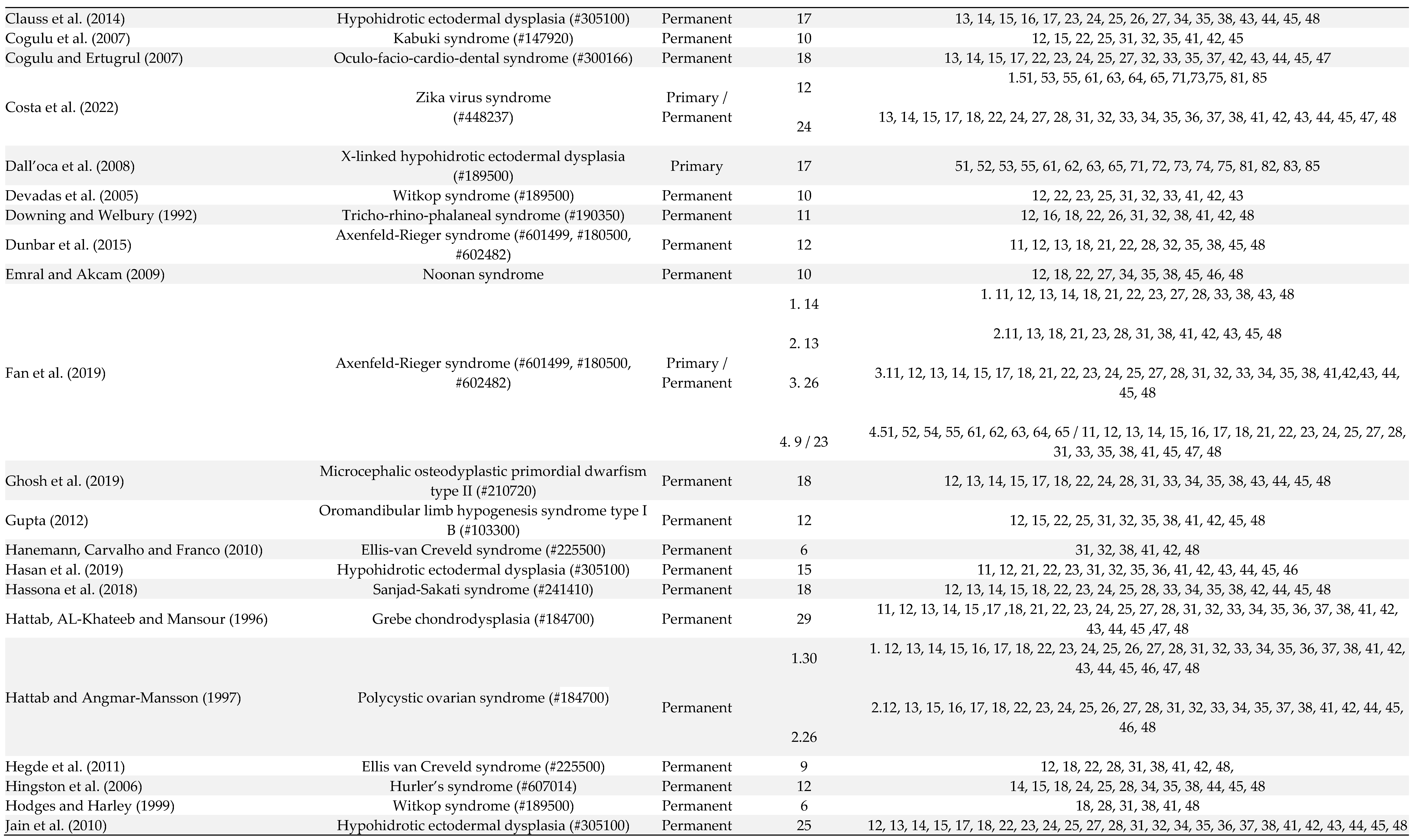
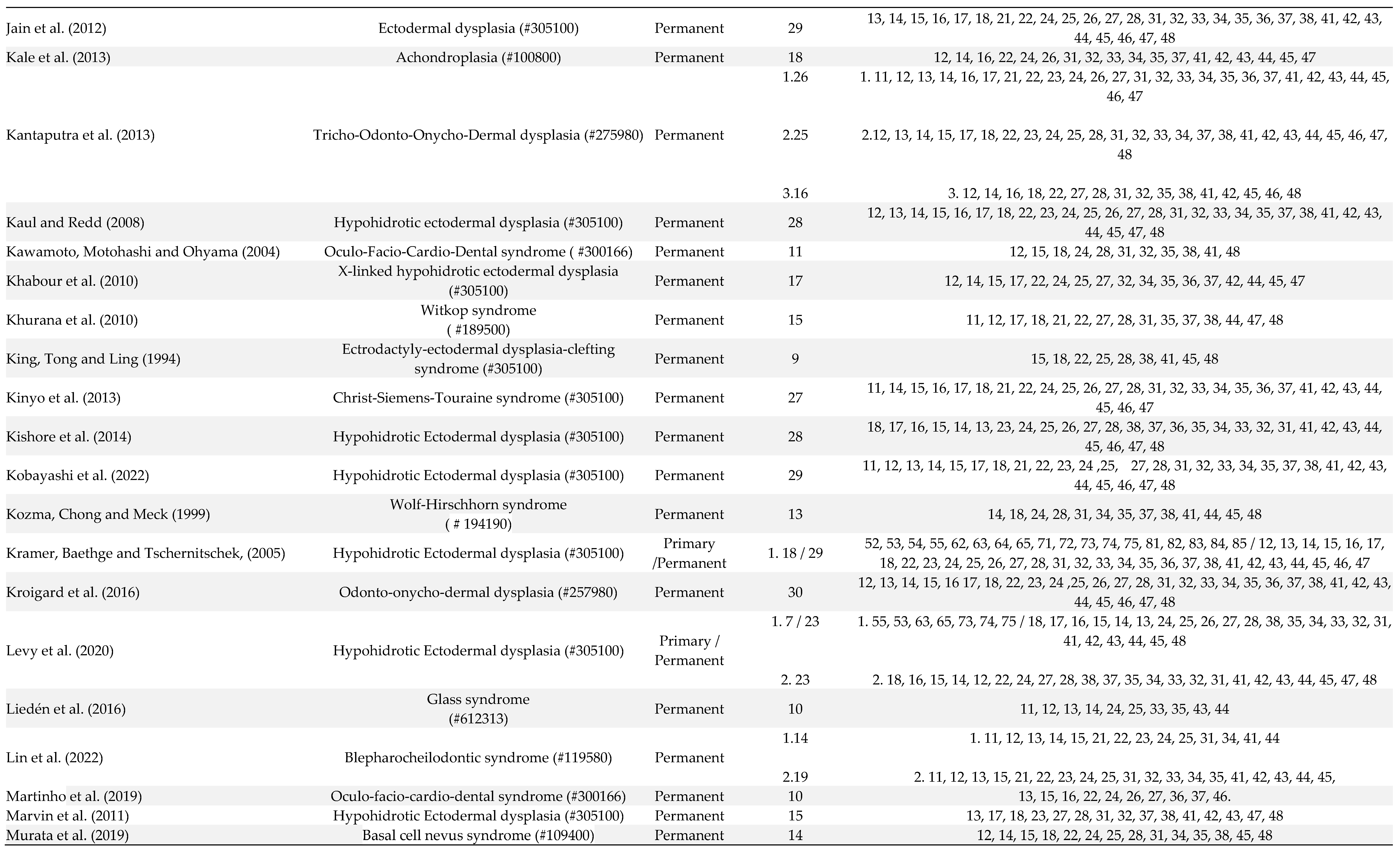
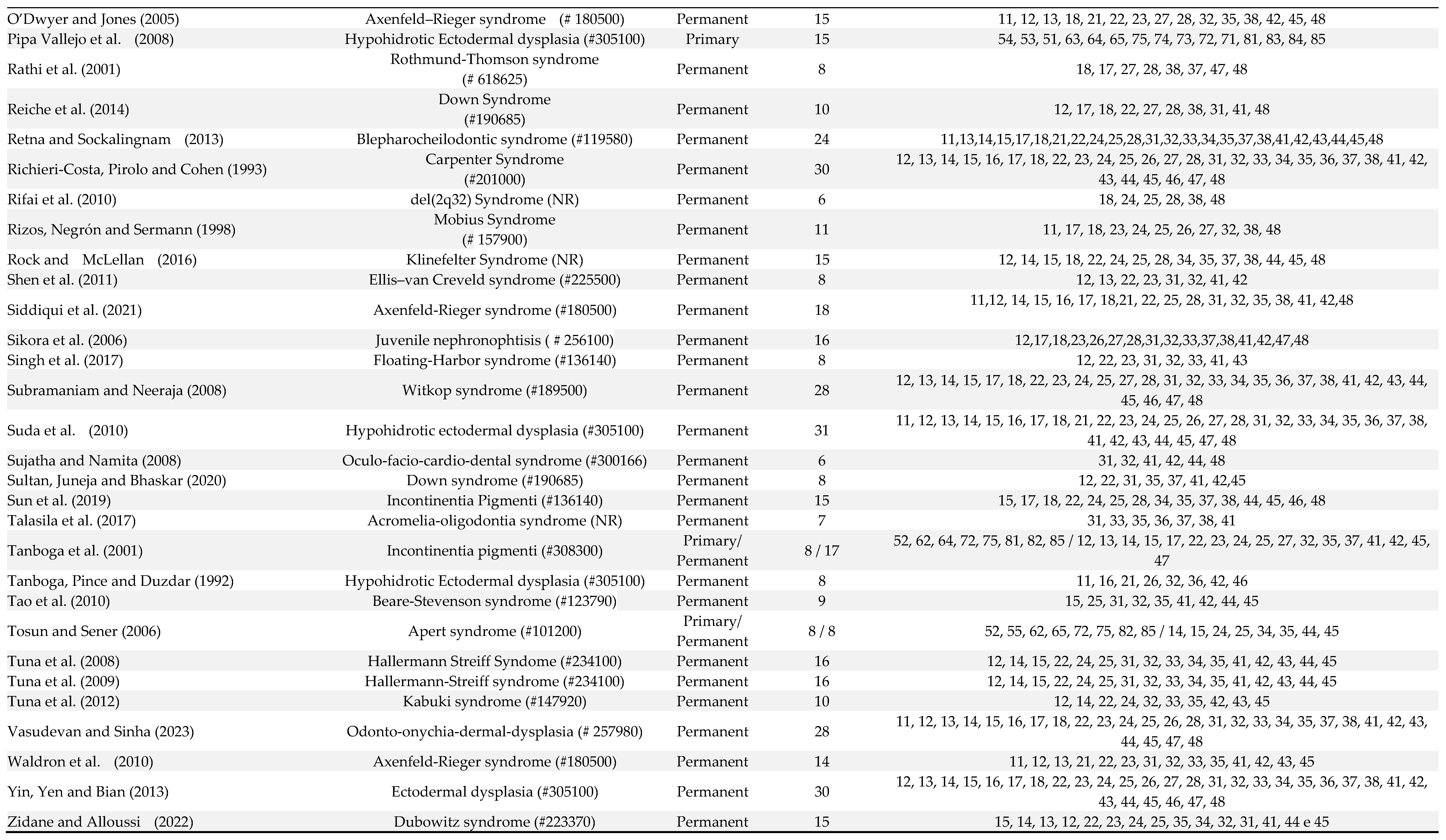
Out of 106 patients, 10 patients were affected by oligodontia in both primary and permanent dentitions, 3 had oligodontia only in the primary dentition, and in the other cases, oligodontia was exclusively observed in the permanent dentition (Table 1). The number of missing teeth ranged from 6 to 13 in the primary dentition and from 6 to 30 in the permanent dentition. In the deciduous dentition, the absence of the first molars, lateral and central incisors was observed in 76.9% of patients, and the second molars and canines were absent in 61.5% of patients (Table 1). In the permanent dentition, lateral incisors (76.2% of patients), first molars (75.5% of patients), central incisors (69.7% of patients), premolars (66% of patients), third molars (60% of patients) and second molars (40% of patients) were the more affected teeth (Table 1).
The genetic profile of the different studies is summarized in Supplementary Table S3. HED was the most found syndrome and the X-linked mode of inheritance was the most common for this syndrome. The most reported mode of inheritance for oculo-facio-cardio-dental syndrome, incontinentia pigmenti and Christ-Siemens-Touraine syndrome was X-linked. The mode of inheritance for Carvajal syndrome, Noonan syndrome, ARS, Witkop’s syndrome, Apert syndrome, BCDS and Kabuki syndrome was the autosomal dominant, whereas microcephalic osteodyplastic primordial dwarfism type II, tricho-odonto-onycho-dermal dysplasia, Rothmund-Thomson syndrome and EVCS were reported under autosomal recessive (Supplementary Table S3).
Eight studies reported mutations in EDA or WNT10A in HED, Christ-Siemens-Touraine syndrome, tricho-odonto-onycho-dermal dysplasia and odonto-onycho-dermal dysplasia. MSX1 was related twice in cases of Witkop’s syndrome. Others studies have reported mutations in DSP (Carvajal/Naxos syndrome), PITX2 (ARS), PCNT (microcephalic osteofysplastic dwarfism type 2), TBCE (Sanjad-Sakati syndrome), EDARADD (HED), LEF1 (HED), AXIN2 (HED), PTCH1 (basal cell nevus syndrome), FZD7 (del(2q32) syndrome), EVC2 (EVCS), NPHP1 (juvenile nephronophtisis), SRCAP (Floating-Harbor syndrome), BCOR (oculo-facio-cardio-dental syndrome), IKBKG (incontinentia pigmenti), and FGFR2 (Beare-Stevenson syndrome). Together, these genes participate of 227 biological processes and 20 pathways. The most significant biological processes were odontogenesis (GO:0042476, P = 9.55e-11), animal organ morphogenesis (GO:0009887, P = 6.51e-07) and epithelium development (GO:0060429, P = 2.90e-06), and the pathways were of basal cell carcinoma (hsa05217, P = 2.34e-07), pathways in cancer (hsa05200, P = 6.77e-06) and pathways of the gastric cancer (hsa05226, P = 6.77e-06) (Supplementary Table S5 and S6). The networks included 18 predicted interactions (Figure 2).
4. Discussion
Within the realm of syndromes characterized by oligodontia, questions arise regarding the consistency of this phenotype across cases, its varying expressiveness, diagnostic utility, and the specific teeth most affected. This review sought to comprehensively address these queries by collating pertinent information from a diverse array of syndromes exhibiting oligodontia within their clinical spectrum. The exploration commenced by surveying an extensive expanse of literature, transcending temporal constraints, leading to the identification of 49 distinct syndromes cataloged within the Online Mendelian Inheritance in Man. Among these, HED emerged as the most frequent. The hallmark trifecta of HED, involving hair, teeth and sweat gland anomalies [22], was evident in the affected patients. The spectrum of dental agenesis in HED spans mainly oligodontia, but reports of hypodontia and even anodontia is found in the literature, with a predilection for the mandible. This remarkable variability necessitates close attention for correct diagnosis [23,24]. Notably, the distinctive conical shape of anterior teeth, when present, offers a diagnostic clue. Furthermore, the potential confluence of maxillary retrusion, sagittal jaw underdevelopment, jaw displacement, and craniofacial alterations underlines the complex interplay of factors characterizing this syndrome [23,25].
Among the most commom syndromes idetifieded in the systematic review was EVCS, an autosomal recessive skeletal dysplasia that encompasses an intriguing spectrum of oral manifestations. Alongside limb abnormalities, the oral phenotype includes occlusion irregularities, labiogingival adhesions, hypertrophied labiogingival frenulum, accessory frenula, serrated incisal margins, dental transposition, diastemas, conical teeth, enamel hypoplasia, and congenital absence of serevral teeth [26,27]. The propensity for premature eruption or exfoliation accentuates the complexity of dental anomalies associated with EVCS. Oculo-facio-cardio-dental syndrome, a rare multi-systemic anomaly more prevalent in females, demonstrates an intricate interplay between congenital cataracts, facial dysmorphisms, dental anomalies like radiculomegaly and oligodontia, and congenital heart defects [28,29]. Witkop syndrome is an uncommon genetic disorder inherited in an autosomal dominant pattern, attributed to mutations occurring in MSX1. This gene holds significance in the formation of the teeth, nails, hair follicles and various other anatomical structures of ectoderma origin. Consequently, the syndrome is distinguished by two primary features: dental agenesis, mainly oligodontia, but the absence of up to 5 teeths (hypodontia) is also reported, and nail dysplasia. Conical-shaped teeth and teeth with narrow crowns are common dental features of Witkop syndrome [30]. ARS is a condition characterized by ocular dysgenesis affecting the anterior segment, along with concurrent systemic anomalies involving the teeth, heart, craniofacial structure and abdominal wall. It is frequently associated with a 6p25 distal microdeletion. However, diverse manifestations of this syndrome may also show connections with other genetic loci like 4q25 or 13q14. Various genes, including FOXC1, FOXC2, and FKHL7, are implicated in the context of ARS [31,32]. BCDS is an uncommon autosomal dominant disorder characterized by congenital facial clefting, oligodontia, euryblepharon, lagophthalmos, and ectropion. While the extent of its expression can differ, the prevalent features often include cleft lip and/or palate, ectropion and lagophthalmos [33].
Interrogating the consistency of oligodontia across syndromic cases is of paramount importance. The range of dental presentations, spanning from hypodontia to anodontia, underscores the variable expressiveness within these syndromes. Consequently, the manifestation of oligodontia should be viewed as a continuum instead of an absolute trait. This variable expressivity poses challenges in diagnosis and underscores the importance of considering broader phenotypic traits in conjunction with dental anomalies. Regarding the most affected teeth, patterns emerged from the collated data. In deciduous dentition, absence of first molars, lateral and central incisors was observed in the majority of patients, with second molars and canines affected in a significant proportion. In permanent dentition, lateral incisors, first molars, central incisors, premolars, third molars, and second molars exhibited the highest susceptibility to oligodontia. These patterns may offer clues for diagnosis and genetic assessment. However, as previously reported, there are several mechanisms that are involved in tooth development and other tissues of the body, establishing very heterogeneous phenotypes in affected individuals. Thus, radiographic and molecular diagnosis may be necessary. Analysis of dental radiographs is an important part of the diagnostic process in daily clinical practice, and interpretation by an expert includes teeth detection and numbering [34,35,36]. In the reading of the articles included, panoramic radiographs with poor quality were evidenced, which can affect the diagnosis and consequent interpretation of the case report. The detailed radiographic report of the observed alterations was also absent in most studies.
In some situations, in the differential diagnosis process, sequencing analysis is useful, and further exploration of the identified mutations can assist in the interpretation of the phenotypes (genotype-phenotype correlation). The important role of genetics has been increasingly recognized in recent years with regard to the understanding of dental anomalies such as tooth agenesis [37]. However, many of the included studies in this systematic review did not perform molecular analysis. Only 13 studies [38,51] performed genetic analysis and reported the genetic variants associated with the syndromes. These genes can be grouped in two groups. These genes can be grouped in two major groups. One with crucial roles at multiple stages of tooth development, skin and sweat glands (EDA, EDARADD, WNT10a, MSX1, DSP, LEF1, EVC2, PITX2, FGFR2, and AXIN2), which are involved in the signal pathway essential for ectodermal structure development [52,53], and the other with genes that intermedia the function and developmental cellular (PCNT, TBCE, PTCH1, TBCE, FZD7, SRCAP, NPHP1, IKBKG, and BCOR). Interestingly, some of the identified genes are also associated with non-syndromic oligodontia, but in these cases, the mutations cause reduced expression, decreased receptor-binding affinity or altered signaling-intensity of mutated protein, whereas the mutations associated with syndromic oligodontia are characterized by a more intense impact on protein function [54].
The teeth in patients with oligodontia frequently are affected by dental anomalies, including reduction in size, assuming a conoid shape, and delayed eruption [52,53,54,55,56,57,58], indicating a control by similar genetic mechanisms [55]. Corroborating with previous literature [37], our findings demonstrated that the permanent dentition is more frequently affected by oligodontia than primary dentition. Out of 106 patients, 68 patients showed bilateral agenesis of maxillary lateral incisors and 13 patients showed unilateral absence of the second mandibular premolar. Clinical studies have described that bilateral agenesis of the maxillary lateral incisors occurred more often than unilateral agenesis, and unilateral agenesis of the second mandibular premolar is more common than bilateral one [59].
The study has limitations, which are the result of use of different terminology to define oligodontia, including severe hypodontia or partial anodontia. The unavailability of radiographies to the correct diagnosis of oligodontia was alarming, reducing the sample size. This study was also limited by the fact that many studies did not perform genetic tests, precluding a more complete phenotype-genotype correlation. In addition, cases of very rare disorders may not be reported in the literature, limiting our map for syndrome with oligodontia.
5. Conclusion
Phenotype identification can reduce uncertainty in the diagnosis when the phenotype is highly predictive for a specific syndrome. Based on this review, during the diagnosis process of patients with oligodontia, clinians and geneticists should be aware of the most common syndromes with oligodontia as clinical sign, including HED, ARS, Witkop’s syndrome, EVCS, BCDS and oculo-facio-cardio-dental syndrome.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
N. L. Castilho: Data extraction, methodology, writing-original draft. K. K. M. Resende: Data extraction, methodology, writing-original draft. J. A. dos Santos: Data extraction, methodology. R. A. Machado: Data extraction, methodology, supervision, writing-original draft. R. D. Coletta: Conceptualization, supervision, writing-review and editing. E. N. S. Guerra: Conceptualization, methodology. A. C. Acevedo: Conceptualization, data extraction, methodology. H. Martelli-Junior: Conceptualization, project administration, validation, data curation, writing-review and editing.
Funding
The study was supported by the Minas Gerais State Research Foundation-FAPEMIG, Brazil, National Council for Scientific and Technological Development—CNPq, Brazil and the Coordination for the Improvement of Higher Education Personnel, CAPES, Brazil.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict interest.
References
- De La Dure-Molla, M.; Fournier, B.P.; Manzanares, M.C.; Acevedo, A.C.; Hennekam, R.C.; Friedlander, L.; Boy-Lefèvre, M.; Kerner, S.; Toupenay, S.; Garrec, P.; et al. Elements of morphology: Standard terminology for the teeth and classifying genetic dental disorders. Am. J. Med Genet. Part A 2019, 179, 1913–1981. [Google Scholar] [CrossRef] [PubMed]
- Weide, Y.S.-V.; Beemer, F.; Faber, J.; Bosman, F. Symptomatology of patients with oligodontia. J. Oral Rehabilitation 1994, 21, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, Y.; Zheng, J. [Progress in genetic research on tooth agenesis associated with Wnt/beta-catenin signaling pathway]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2021, 38, 506–509. [Google Scholar] [PubMed]
- Al-Ani, A.H.; Antoun, J.S.; Thomson, W.M.; Merriman, T.R.; Farella, M. Hypodontia: An Update on Its Etiology, Classification, and Clinical Management. BioMed Res. Int. 2017, 2017, 9378325. [Google Scholar] [CrossRef] [PubMed]
- Brook, H. Multilevel complex interactions between genetic, epigenetic and environmental factors in the aetiology of anomalies of dental development. Archives of Oral Biology 2009, 54, S3–S17. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, P.; Arte, S.; Pirinen, S.; Peltonen, L.; Thesleff, I. Gene defect in hypodontia: exclusion of MSX1 and MSX2 as candidate genes. Hum. Genet. 1995, 96, 305–308. [Google Scholar] [CrossRef]
- Gorlin RJ. Syndromes of head and neck (2nd ed.), Oxford, USA. 2001.
- Vieira, A. Oral Clefts and Syndromic Forms of Tooth Agenesis as Models for Genetics of Isolated Tooth Agenesis. J. Dent. Res. 2003, 82, 162–165. [Google Scholar] [CrossRef]
- Baba, R.; Sato, A.; Arai, K. Consecutive tooth agenesis patterns in non-syndromic oligodontia. Odontology 2021, 110, 183–192. [Google Scholar] [CrossRef]
- Ritwik, P.; Partsson, K.K. Diagnosis of Tooth Agenesis in Childhood and Risk for Neoplasms in Adulthood. The Ochsner Journal 2018, 18, 345–350. [Google Scholar] [CrossRef]
- Moses, J.; Gurunathan, D.; Rangeeth, B.N.; Kannan, K.S. Non-syndromic oligodontia of primary and permanent dentition: 5 years follow up- a rare case report. Journal of clinical and diagnostic research 2013, 7, 776–779. [Google Scholar] [CrossRef]
- Ross, J.N.; Ruigrok, L.C.; Fennis, W.M.M.; Cuen, M.S.; Rosenberg, A.J.W.P.; van Nunen, A.B.; et al. Gastrointestinal symptoms in patients with isolated oligodontia and a Wnt gene mutation. Oral Diseases 2023, 29, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Adaimy, L.; Chouery, E.; Mégarbané, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; de Mazancourt, P.; Mégarbané, A. Mutation in WNT10A Is Associated with an Autosomal Recessive Ectodermal Dysplasia: The Odonto-onycho-dermal Dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef] [PubMed]
- De Coster, P.J.; Marks, L.A.; Martens, L.C.; Huysseune, A. Dental agenesis: genetic and clinical perspectives. J. Oral Pathol. Med. 2008, 38, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nordgarden, H.; Jensen, J.; Storhaug, K. Oligodontia is associated with extra-oral ectodermal symptoms and low whole salivary flow rates. Oral Diseases 2001, 7, 226–232. [Google Scholar] [PubMed]
- Dhamo, B.; Kuijpers, M.A.R.; Balk-Leurs, I.; Boxum, C.; Wolvius, E.B.; Ongkosuwito, E.M. Disturbances of dental development distinguish patients with oligodontia-ectodermal dysplasia from isolated oligodontia. Orthod. Craniofacial Res. 2017, 21, 48–56. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. PLOS Med. 2021, 18, e1003583. [Google Scholar] [CrossRef] [PubMed]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A web and mobile app for systematic reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef]
- Moola, S.; Munn, Z.; Tufanaru, C.; Aromataris, E.; Sears, K.; Sfetcu, R.; et al. Systematic reviews of etiology and risk. In: Aromataris E, Munn Z, editors. Joanna Briggs Institute reviewer’s manual. Adelaide (Australia): Joanna Briggs Institute. Chap 7. 2017.
- Galluccio, G.; Castellano, M.; La Monaca, C. Genetic basis of non-syndromic anomalies of human tooth number. Arch. Oral Biol. 2012, 57, 918–930. [Google Scholar] [CrossRef]
- Øgaard, B.; Krogstad, O. Craniofacial structure and soft tissue profile in patients with severe hypodontia. Am. J. Orthod. Dentofac. Orthop. 1995, 108, 472–477. [Google Scholar] [CrossRef]
- Zeng, B.; Lu, H.; Xiao, X.; Zhou, L.; Lu, J.; Zhu, L.; et al. Novel EDA mutation in X-linked hypohidrotic ectodermal dysplasia and genotype-phenotype correlation. Oral Diseases 2015, 21, 994–1000. [Google Scholar] [CrossRef]
- Moura, E.; Rotenberg, I.S.; Pimpão, C.T. X-Linked Hypohidrotic Ectodermal Dysplasia—General Features and Dental Abnormalities in Affected Dogs Compared With Human Dental Abnormalities. Top. Companion Anim. Med. 2019, 35, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Reali, J.; Mendoza-Ramos, M.I.; Garrido-Guerrero, E.; Méndez-Catalá, C.F.; Méndez-Cruz, A.R.; Pozo-Molina, G. Hypohidrotic ectodermal dysplasia: clinical and molecular review. Int. J. Dermatol. 2018, 57, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Lexner, M.; Bardow, A.; Juncker, I.; Jensen, L.; Almer, L.; Kreiborg, S.; Hertz, J. X-linked hypohidrotic ectodermal dysplasia. Genetic and dental findings in 67 Danish patients from 19 families. Clin. Genet. 2008, 74, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Baujat, G.; Le Merrer, M. Ellis-van Creveld syndrome. Orphanet Journal of Rare Diseases 2007, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Susami, T.; Kuroda, T.; Yoshimasu, H.; Suzuki, R. Ellis-van Creveld syndrome: craniofacial morphology and multidisciplinary treatment. The Cleft Palate-Craniofacial Journal 1999, 36, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Cogulu, D.; Oncag, O.; Celen, E.; Ozkinay, F. Kabuki Syndrome with additional dental findings: a case report. Journal of Dentistry for Children 2008, 75, 185–187. [Google Scholar] [PubMed]
- Sujatha, R. Barriers in Career Growth of Women Managers: An Indian Scenario. Asia Pac. Bus. Rev. 2008, 4, 108–116. [Google Scholar] [CrossRef]
- Hodges, S.; Harley, K. Witkop tooth and nail syndrome: report of two cases in a family. Int. J. Paediatr. Dent. 2001, 9, 207–211. [Google Scholar] [CrossRef]
- Seifi, M.; Walter, M.A. Axenfeld-Rieger syndrome. Clin Genet. 2018, 93, 1123–1130. [Google Scholar] [CrossRef]
- Smith, R.S.; Zabaleta, A.; Kume, T.; Savinova, O.V.; Kidson, S.H.; Martin, J.E.; Nishimura, D.Y.; Alward, W.L.M.; Hogan, B.L.M.; John, S.W.M. Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum. Mol. Genet. 2000, 9, 1021–1032. [Google Scholar] [CrossRef]
- Yen, M.T.; Lucci, L.M.; Anderson, R.L. Management of eyelid anomalies associated with Blepharo-cheilo-dontic syndrome. Am. J. Ophthalmol. 2001, 132, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Tuzoff, D.V.; Tuzova, L.N.; Bornstein, M.M.; Krasnov, A.S.; Kharchenko, M.A.; Nikolenko, S.I.; Sveshnikov, M.M.; Bednenko, G.B. Tooth detection and numbering in panoramic radiographs using convolutional neural networks. Dentomaxillofac. Radiol. 2019, 48, 20180051. [Google Scholar] [CrossRef] [PubMed]
- Román, J.C.M.; Fretes, V.R.; Adorno, C.G.; Silva, R.G.; Noguera, J.L.V.; Legal-Ayala, H.; Mello-Román, J.D.; Torres, R.D.E.; Facon, J. Panoramic Dental Radiography Image Enhancement Using Multiscale Mathematical Morphology. Sensors 2021, 21, 3110. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.C.; Line, S.R. The genetics of amelogenesis imperfecta: a review of the literature. Journal of applied oral science 2005, 13, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Vastardis, H. The genetics of human tooth agenesis: new discoveries for understanding dental anomalies. Am. J. Orthod. Dentofac. Orthop. 2000, 117, 650–655. [Google Scholar] [CrossRef]
- Chalabreysse, L.; Senni, F.; Bruyère, P.; Aime, B.; Ollagnier, C.; Bozio, A.; et al. A new hypo/oligodontia syndrome: Carvajal/Naxos syndrome secondary to desmoplakin-dominant mutations. Journal of Dental Research 2011, 90, 58–64. [Google Scholar] [CrossRef]
- Clauss, F.; Waltmann, E.; Barriere, P.; Hadj-Rabia, S.; Manière, M.-C.; Schmittbuhl, M. Dento-maxillo-facial phenotype and implants-based oral rehabilitation in Ectodermal Dysplasia with WNT10A gene mutation: Report of a case and literature review. J. Cranio-Maxillofacial Surg. 2014, 42, e346–e351. [Google Scholar] [CrossRef]
- Fan, Z.; Sun, S.; Liu, H.; Yu, M.; Liu, Z.; Wong, S.W.; et al. Novel PITX2 mutations identified in Axenfeld-Rieger syndrome and the pattern of PITX2-related tooth agenesis. Oral Diseases 2019, 25, 2010–2019. [Google Scholar] [CrossRef]
- Ghosh, S.; Garg, M.; Gupta, S.; Choudhary, M.; Chandra, M. Microcephalic osteodyplastic primordial dwarfism type II: case report with unique oral findings and a new mutation in the pericentrin gene. Oral Surgery, Oral Med. Oral Pathol. Oral Radiol. 2019, 129, e204–e211. [Google Scholar] [CrossRef]
- Kantaputra, P.; Kaewgahya, M.; Hatsadaloi, A.; Vogel, P.; Kawasaki, K.; Ohazama, A.; Cairns, J.K. GREMLIN 2 Mutations and Dental Anomalies. J. Dent. Res. 2015, 94, 1646–1652. [Google Scholar] [CrossRef]
- Khabour, O.; Mesmar, F.; Al-Tamimi, F.; Al-Batayneh, O.; Owais, A. Missense mutation of the EDA gene in a Jordanian family with X-linked hypohidrotic ectodermal dysplasia: phenotypic appearance and speech problems. Genet. Mol. Res. 2010, 9, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Khurana, V.K.; Gupta, R.K.; Kumar, L.P. Witkop syndrome: A case report of an affected family. Dermatol. Online J. 2012, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Kinyó, A.; Vályi, P.; Farkas, K.; Nagy, N.; Gergely, B.; Tripolszki, K.; Török, D.; Bata-Csörgő, Z.; Kemény, L.; Széll, M. A newly identified missense mutation of the EDA1 gene in a Hungarian patient with Christ–Siemens–Touraine syndrome. Arch. Dermatol. Res. 2013, 306, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Krøigård, A.B.; Thomassen, M.; Lænkholm, A.-V.; Kruse, T.A.; Larsen, M.J. Evaluation of Nine Somatic Variant Callers for Detection of Somatic Mutations in Exome and Targeted Deep Sequencing Data. PLOS ONE 2016, 11, e0151664. [Google Scholar] [CrossRef]
- Marvin, M.L.; Mazzoni, S.M.; Herron, C.M.; Edwards, S.; Gruber, S.B.; Petty, E.M. AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am. J. Med Genet. Part A 2011, 155, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Kurosaka, H.; Ohata, Y.; Aikawa, T.; Takahata, S.; Fujii, K.; Miyashita, T.; Morita, C.; Inubushi, T.; Kubota, T.; et al. A novel PTCH1 mutation in basal cell nevus syndrome with rare craniofacial features. Hum. Genome Var. 2019, 6, 16. [Google Scholar] [CrossRef]
- Shen, W.; Han, D.; Zhang, J.; Zhao, H.; Feng, H. Two novel heterozygous mutations of EVC2 cause a mild phenotype of Ellis-van Creveld syndrome in a Chinese family. Am. J. Med Genet. Part A 2011, 155, 2131–2136. [Google Scholar] [CrossRef]
- Yin, W.; Ye, X.; Bian, Z. The Second Deletion Mutation in Exon 8 of EDA Gene in an XLHED Pedigree. Dermatology 2013, 226, 105–110. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Bongkochwilawan, C.; Kaewgahya, M.; Ohazama, A.; Kayserili, H.; Erdem, A.P.; et al. Enamel-Renal-Gingival syndrome, hypodontia, and a novel FAM20A mutation. American journal of medical genetics Part A 2014, 164A, 2124–2128. [Google Scholar] [CrossRef]
- Zeng, B.; Zhao, Q.; Li, S.; Lu, H.; Lu, J.; Ma, L.; Zhao, W.; Yu, D. Novel EDA or EDAR Mutations Identified in Patients with X-Linked Hypohidrotic Ectodermal Dysplasia or Non-Syndromic Tooth Agenesis. Genes 2017, 8, 259. [Google Scholar] [CrossRef]
- Andreoni, F.; Sgattoni, C.; Bencardino, D.; Simonetti, O.; Forabosco, A.; Magnani, M. Missense mutations in EDA and EDAR genes cause dominant syndromic tooth agenesis. Mol. Genet. Genom. Med. 2020, 9, e1555. [Google Scholar] [CrossRef] [PubMed]
- Mues, G.; Tardivel, A.; Willen, L.; Kapadia, H.; Seaman, R.; Frazier-Bowers, S.; Schneider, P.; D'Souza, R.N. Functional analysis of Ectodysplasin-A mutations causing selective tooth agenesis. Eur. J. Hum. Genet. 2009, 18, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Peck, J.; Douglas, G.; Wu, C.H.; Burbelo, P.D. Human RhoGAP domain-containing proteins: structure, function and evolutionary relationships. FEBS Lett. 2002, 528, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, P. Genetic basis of Tooth agenesis. Journal of Experimental Zoology Part B: Molecular and Developmental Evolution 2009, 312, 320–342. [Google Scholar] [CrossRef] [PubMed]
- Der Weide, Y.S.-V.; Steen, W.H.; Bosman, F. Distribution of missing teeth and tooth morphology in patients with oligodontia. ASDC J. Dent. Child. 1992, 59, 133–140. [Google Scholar]
- Van Der Weide, Y.S.; Prahl-Andersen, B.; Bosman, F. Tooth formation in patients with oligodontia. Angle Orthod. 1993, 63, 31–37. [Google Scholar] [CrossRef]
- Polder, B.J.; Hof, M.A.V.; Van der Linden, F.P.G.M.; Kuijpers-Jagtman, A.M. A meta-analysis of the prevalence of dental agenesis of permanent teeth. Community Dent. Oral Epidemiology 2004, 32, 217–226. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Flow diagram of literature search and selection criteria adapted from Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA).
Figure 1.
Flow diagram of literature search and selection criteria adapted from Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA).
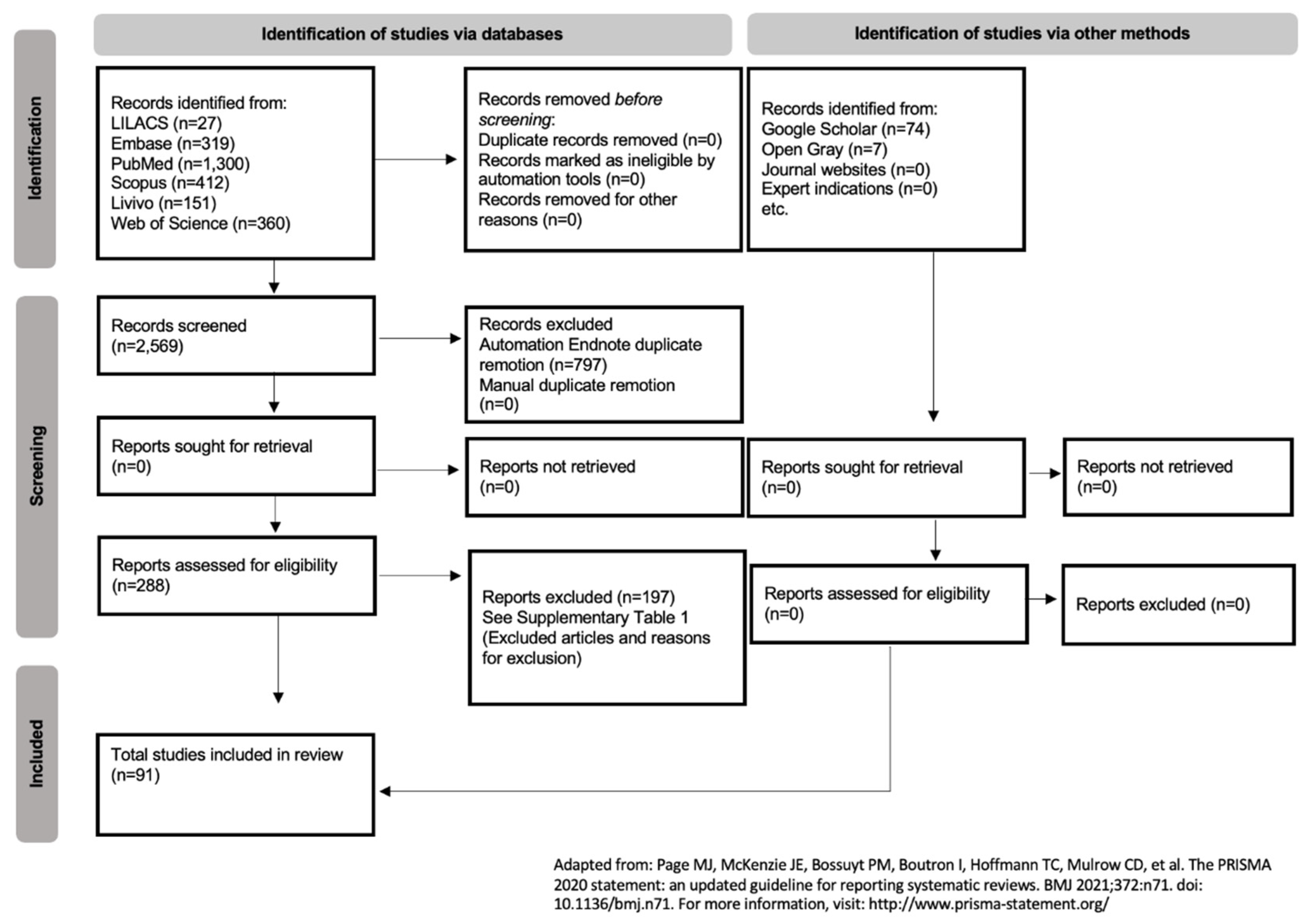
Figure 2.
Protein-protein interaction network with the genes associated with syndromes with oligodontia. Two nodes of interactions, involving AXIN2, EDA, EDARADD, EVC2, FGFR2, FZD7, IKBKG, LEF1, MSX1, PTCH1, PITX2 and WNT10A (P < 1.0e-16), and between PCNT and NPHP1 (P = 0.02), were identified. Different colors represent different levels of evidence of connection between proteins. Light blue represents curated databases, purple experimental evidence, green gene neighborhood, red gene fusions, blue gene co-occurrence, light green evidence from text mining, black co-expression, and violet protein homology. This analysis had an average confidence score of 0.601, suggesting a low rate for false-positive interactions.
Figure 2.
Protein-protein interaction network with the genes associated with syndromes with oligodontia. Two nodes of interactions, involving AXIN2, EDA, EDARADD, EVC2, FGFR2, FZD7, IKBKG, LEF1, MSX1, PTCH1, PITX2 and WNT10A (P < 1.0e-16), and between PCNT and NPHP1 (P = 0.02), were identified. Different colors represent different levels of evidence of connection between proteins. Light blue represents curated databases, purple experimental evidence, green gene neighborhood, red gene fusions, blue gene co-occurrence, light green evidence from text mining, black co-expression, and violet protein homology. This analysis had an average confidence score of 0.601, suggesting a low rate for false-positive interactions.
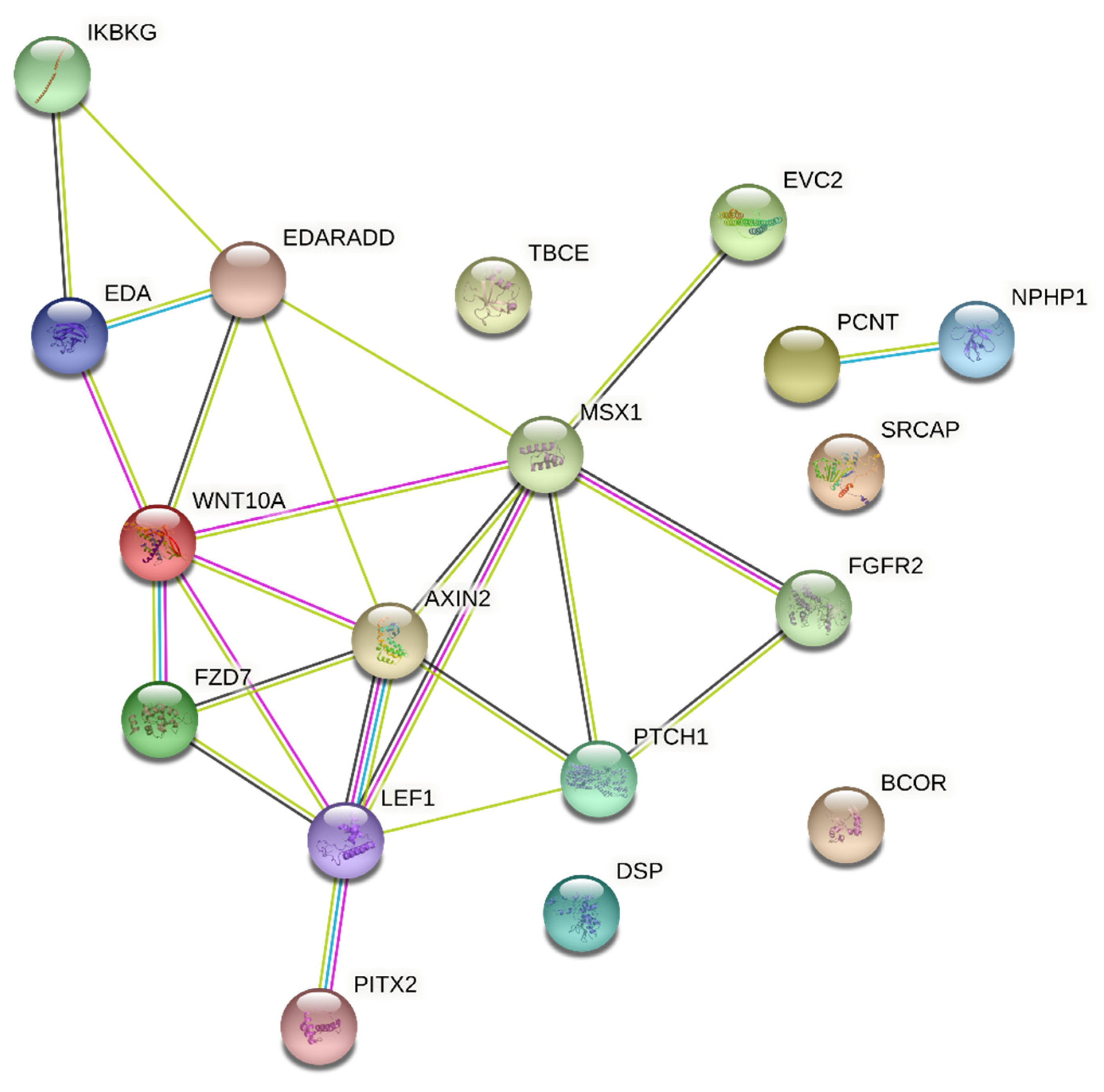

Table 1.
Manifestations of oligodontia in primary and permanent dentition of patients in the included articles number of studies (91).
Table 1.
Manifestations of oligodontia in primary and permanent dentition of patients in the included articles number of studies (91).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



