
Submitted:
01 September 2023
Posted:
05 September 2023
You are already at the latest version
Abstract
The integrated stress response is a signaling network comprised of four branches, each of which senses different cellular stressors, converging on the phosphorylation of eIF2α to depress global translation and initiate recovery. One of these branches is composed of GCN2, which senses cellular amino acid insufficiency and participates in the maintenance of amino acid homeostasis. Previous studies have shown that GCN2 is a viable cancer target when amino acid stress is provoked by inhibiting an additional target. In this light, we screened a combination of drugs to identify biologically active compounds which synergize with the GCN2 inhibitor TAP20. First, a panel of 25 compounds was assayed in six cancer cell lines for drug sensitivity. Each compound was then combined with TAP20 at concentrations below their IC50, and the impact on cell growth was assessed. Selected strongly synergistic combinations were further characterised using synergy analyses and matrix-dependent invasion assays. Inhibitors of proteostasis, of the MEK-ERK pathway and pan-CDK inhibitors flavopiridol and seliciclib were found to potently synergize with TAP20 in two of the tested cell lines. Among their common CDK targets is CDK7, which was selectively targeted with THZ-1 and found to synergize with TAP20. Finally, these combinations were found to be partially synergistic when assessed using matrix-dependent invasion assays. However, we found that TAP20 alone was sufficient to restrict invasion at concentrations well below its growth-inhibitory IC50. We conclude that GCN2 can be targeted for treating cancers by polytherapy or even monotherapy.
Keywords:
integrated stress response
; amino acid
; PERK
; cyclin-dependent kinases
; cell cycle
; amino acid transport
1. Introduction
The integrated stress response (ISR) pathway allows cells to adapt to stressful situations such as amino acid depletion, endoplasmic reticulum (ER) stress, viral infection and heme depletion [1]. The ISR is comprised of four different protein kinases that respond to the different stressors and have as their only known target the eukaryotic translation initiation factor eIF2α. They are general control nonderepressible 2 (GCN2), which respond to amino acid depletion by using uncharged tRNAs as a sensor of amino acid imbalances; protein kinase R-like endoplasmic reticulum kinase (PERK), which senses ER-stress [2]; protein kinase R (PKR), which detects double-stranded RNA [3]; and heme-regulated inhibitor, which responds to stresses associated with hemoglobin production [4]. In the context of this study, we focused on GCN2. Phosphorylation of eIF2α reduces overall protein biosynthesis and at the same time allows the translation of specific mRNAs with upstream open-reading frames, in particular ATF4 [5]. ATF4 is a general activating transcription factor, which promotes the synthesis of genes involved in amino acid biosynthesis, transport, autophagy and antioxidant responses [6,7]. These mechanisms can restore amino acid homeostasis by improving the provision of amino acids and enhancing the recycling of proteins and cellular materials. In cancer cells rapid growth is thought to cause amino acid depletion and ER stress [8], which has drawn attention to the role of GCN2 and PERK in cancerous cell growth [9,10]. In vitro, however, cancer cell lines are often unstressed and require the inhibition of other components of amino acid homeostasis to elicit the ISR [11-13]. Elements of amino acid homeostasis are frequently explored for cancer therapy. Asparaginase, for instance, is used to treat acute lymphocytic leukemia [14]. Inhibition of amino acid transporter LAT1 [15] is explored to treat a variety of solid tumors. Bortezomib is a proteasome inhibitor that affects protein recycling [16], while rapamycin-derived mTORC1 inhibitors were explored in a large variety of clinical trials [17].
Activation of GCN2 initiates a rescue mechanism for cancer cells to deal with cellular stresses elicited by other anticancer drugs, which can thwart the expected effects of targeting amino acid homeostasis [13,16,17]. Thus pairing a drug, which targets amino acid homeostasis with a GCN2 inhibitor could improve its efficacy. Anticancer drug pairs can be beneficial over monotherapies as they can suppress the emergence of drug resistance and achieve sustained remission. Tumours frequently consist of a heterogenic population of malignant cells. The diversity of oncogenic profiles within a malignancy is driven by the genomic instability that is intrinsic to cancer cells and by different selection pressures arising from the immediate microenvironment of various subpopulations [18]. One way to target diverse populations of cells is combination therapy. A substantial combination screening effort by the US National Cancer Institute, involving 5000 FDA-approved anticancer drugs in 60 human cancer cell lines, discovered two promising drug pairings: clofarabine with bortezomib, and nilotinib with paclitaxel. Both combinations are in Phase I clinical trials (NCT02211755 and NCT02379416) [19].
Several GCN2 inhibitors have been developed and characterised for kinase selectivity and pharmacological properties. Nakamura et al. [14] reported the discovery of GCN2iB with an IC50 at 2.4 nM. GCN2iB also inhibits MAP2K5, STK10 and ZAK with slightly lower potencies but has a pharmacokinetic profile which makes it suitable for in vivo studies. GZD824 (olverembatinib) is a broad-spectrum Bcr-Abl inhibitor that is orally bioavailable and recently repurposed as a GCN2 inhibitor; however, it is highly promiscuous in kinase assays [20]. Lastly, triazolo[4,5-d]pyrimidines (TAP) constitute an entire class of GCN2 inhibitors [21,22]. Among these, compound 20 (TAP20) demonstrated superior potency and selectivity. In a cell-free assay, TAP20 achieved an IC50 of 17 nM against GCN2. It was similarly potent against GSK3α/β, and, to a lesser extent, CDK9/cyclinD1.
In this study we explored synergistic relationships between 25 experimental and approved drugs and the GCN2 inhibitor TAP20 using several cell lines.
2. Materials and Methods
Cell lines: All cell lines were procured from the American Type Culture Collection (ATCC) and maintained in DMEM/F-12 with 10% heat-inactivated FBS, 2 mM glutamine and 0.1% penicillin/streptomycin. Cells were stored in a humidified atmosphere with 5% CO2 in an incubator set to 37 °C in 25 cm2 or 75 cm2 culture flasks. Once confluent, cells were trypsinised using 0.25% Trypsin-EDTA (Gibco). Experiments were conducted using cells passaged 5–20 times.
Chemicals and antibodies: Drugs and chemicals were purchased from several sources. TAP20 was synthesised and provided by Merck KGaA (Darmstadt, Germany). YH16899 was purchased from AOBIOUS (Gloucester, MA, USA); thapsigargin from Thermo Fisher Scientific (Waltham, MA, USA), Bay876 from Sigma-Aldrich (St Louis, MO, USA) and all other compounds from MedChemExpress (Monmouth Junction, NJ, USA). Bumetanide and 5-fluorouracil stocks were dissolved in water and all other compounds were dissolved in DMSO. Cetuximab was purchased from MedChemExpress and all other antibodies were from Cell Signaling Technology (Danvers, MA, USA).
Proliferation assay: Cell growth was monitored using the IncuCyte system (Essen Biosciences). To increase throughput, cell confluence was measured only at the end of the assay. Briefly, cells were seeded in 96-well flat-bottom plates at 3000 cells/well in growth medium and allowed to attach overnight. The next day, the growth medium from each well was replaced with 300 µL of growth medium with or without treatment. Cells were stored in the cell incubator (5% CO2 humidified atmosphere; 37°C) for a predefined period before being scanned by the IncuCyte system. This period was determined based on the shortest time required for the untreated control cells to reach >90% confluence. This ranged from four to ten days and depended on the cell line. Confluence was recorded as the mean from two images taken for each well. Peripheral wells were excluded from the analysis.
IC50 curves were plotted from data obtained in this assay using the logistic growth equation:
Where A1 is the maximal confluence, A2 is the minimal confluence, x is the inhibitor’s concentration, x0 is the inhibitor’s concentration at half maximal effect and p is the power value.
To evaluate synergism among drug pairings, the coefficient of drug interaction [23] (CDI) was calculated using the following equation:
Where AB is the confluence of cells treated with the drug combination and A and B are the confluence values of cells exposed to each drug alone. CDI scores of 1 indicate an additive effect, those above 1 indicate antagonism and those below 1 indicate synergism.
Synergy analysis: Synergy diagrams were generated from data produced in the assay described above. A matrix of seven concentrations per drug was arrayed using MDA-MB-231 and HPAF-II cells seeded across three 96-well plates for each experiment. Drug concentrations were incremented by values of 1, 2 and 4 per order of magnitude. As above, peripheral wells were excluded from analysis and each plate contained untreated controls to which the confluence values of treated cells were normalised. Normalised data were then converted to synergy scores using SynergyFinder [23] and the Loewe equation.
Western blotting: Intracellular proteins were prepared for SDS-PAGE by washing cells grown almost to full confluence in 35 mm dishes with PBS (pH 7.4) and adding 100–150 μL of RIPA lysis and extraction buffer (Thermo Fisher Scientific; Waltham, MA, USA) containing cOmplete Mini Protease Inhibitor Cocktail (Roche; Basel, Switzerland). The lysate was collected using a cell scraper and transferred to a microcentrifuge tube. After 5 min of mixing and 5 min of centrifugation at top speed, 10 μL of the supernatant was sampled and diluted at least ten-fold in water for the Bradford assay (reagent obtained from Sigma-Aldrich; St Louis, MO, USA) to measure protein concentration and to normalise the protein mass added to the wells of an SDS-PAGE gel. Lysates were then mixed with 4×Bolt LDS and 10×Bolt Sample Reducing Agent (Life Technologies) and water up to 50 µL. A volume of the mixture, equivalent to 20–30 µg protein, was then loaded into the wells of a precast NuPAGE 4-12% Bis-Tris 10- or 15-well gel fitted to a Mini Gel Tank (Life Technologies) filled with MOPS-SDS running buffer (Thermo Fisher; Waltham, MA, USA). Sample lanes were flanked with wells loaded with either SeeBlue Plus2 or Novex Sharp pre-stained protein standards (Invitrogen; Waltham, MA, USA). Proteins were separated for approximately 1 h at 150 V, after which proteins were transferred onto a 0.45-micron Amersham Protran nitrocellulose membrane (GE Healthcare; Chicago, IL, USA) in an ice-cold wet transfer system (Bio-Rad; Hercules, CA, USA) containing Towbin buffer (192 mM NaCl, 25 mM Tris base, 20% methanol; pH 8.3) for 2 h at 100 V. The blot was then incubated in 5 % (w/v) skim milk powder in PBS-Tween (0.1% Tween-20; pH 7.4) for 1 h at room temperature or overnight at 4°C. In between blocking and antibody solutions, blots were always washed thrice with PBS-Tween for 5 min at a time. Primary antibodies were always incubated with blots overnight, while secondary antibodies were incubated for 2–6 h. To visualise the proteins, either the SuperSignal West Pico PLUS or the SuperSignal West Femto Maximum Sensitivity chemiluminescent substrates (Thermo Scientific; Waltham, MA, USA) were added dropwise onto the blots. Either the ChemiDoc MP (Bio-Rad) or Fusion Solo 2 (Vilber) imaging systems were used to visualise chemiluminescence.
All blots were probed first for the protein of interest and lastly for a housekeeping protein. β-Actin was probed as loading control. To remove protein-bound antibodies from the previous immunodetection, blots were incubated at 65°C in stripping buffer (2% w/v SDS; 62.5 mM Tris; and 0.7% v/v 2-mercaptoethanol; pH 6.8) for 10 min with agitation. The blots were then washed with PBS-Tween and incubated for at least 2 h with 5% skim milk in PBS-Tween.
Matrix-dependent invasion assay: To investigate drug-pair effects on matrix-dependent invasion, 143B osteosarcoma cells were used in a Cultrex 3D spheroid invasion system (R&D Systems; Minneapolis, MN, USA) as they readily form the necessary spheroids unlike the six cell lines used in the synergy screen. Initially, 143B were seeded in 1 × spheroid formation medium (ECM) (3000 cells per well in 50 μl) into low-adhesion, round-bottom 96-well plates to induce formation of spheroids. The plates were centrifuged at 300 × g for 3 min to position cells at the bottom of the well. Working on ice, 50 μl invasion matrix was added to the spheroids after 72 h. Subsequently, plates were centrifuged at 300 × g at 4°C to position the spheroid at the bottom of their wells. Gel formation was initiated by incubation at 37°C for 1 h followed by addition of 100 μl of pre-warmed culture medium. Plates were then incubated at 37°C/5% CO2 in a humidified incubator for 3-6 days, and images were taken with a Leica M205FA stereomicroscope. Invasion perimeter area was measured using the Leica Application Suite, version 4.12.
3. Results
We reasoned that current cancer drugs and experimental anticancer compounds might cause stress to cell lines, which was counteracted by GCN2 activation. Blocking GCN2 with TAP20 would reduce the cellular response flexibility, thereby reducing cellular fitness.
We selected 24 experimental compounds with well-known targets (Table 1). The reported target IC50 values are all in the nanomolar range typical for modern pharmaceutical compounds. To establish a baseline for a potential synergistic effect of TAP20, we determined the IC50 of 25 candidate drugs (Table 2) for cell growth using a range of cell lines that have a variety of oncogenic signatures [24]. Growth IC50-values can be higher than the target IC50-values due to membrane permeability, drug-resistance mechanisms and cellular reliance on the function of the target.
To determine synergy with TAP20, candidate drugs were adjusted to a concentration where inhibition was just observable (i.e. < IC50, Table 3). In this range, synergy is readily observed as growth rates are especially sensitive to reduced fitness. In general, we did not employ concentrations >10 µM due to insolubility or increased non-specific drug interactions. The dose-response curves for all cell lines can be found in supplementary Fig S1.
As a last preparation, we determined the response of cell lines to TAP20. TAP20 alone typically does not inhibit growth when cell lines are cultured in replete growth media because they experience little stress, which would initiate activation of GCN2. To quantify the action of TAP20 on GCN2 we starved the cell lines for 24 h in the presence of different concentrations of TAP20 (Figure 1). In our hands, the expression of ATF4 was the most reliable indicator of GCN2 activation and we used western blotting to determine ATF4 expression at different doses of TAP20. Except for Panc 02.03 cells, ATF4 expression was suppressed by TAP20 concentrations > 1 µM. The IC50-values were MDA-MB-231 = 0.54 µM, HPAFII = 0.56 µM, SKOV3 = 0.29 µM. For all synergy tests, TAP20 was used at 3 µM to ensure that the ISR could not be activated via GCN2.
In the synergy experiments, control cells were grown to confluency and the growth compared to that of the treated cells. A coefficient of drug interaction was calculated and used to compare the experiments (Table 4). A score <1 (green in Table 4) indicates a synergistic relationship, a score of 1 an additive effect (yellow) and scores >1 indicate an antagonistic relationship (red). The unprocessed growth data for each combination are shown in supplementary Figure S2. Several trends were observed. First, instances of drug synergy were not consistently observed across all cell lines. We observed a variety of synergistic relations in MDA-MB-231, HPAFII and SKOV3 cells, while these were mostly additive in MDA-MB-468 and Panc02.03 cells. We observed only a limited number of synergistic relations in Ovcar3 cells. Second, rapamycin was antagonistic to TAP20. This was expected because inhibition of mTORC1 is known to activate GCN2 and enhance phosphorylation of eIF2α [25].
Numerous synergistic relationships between TAP20 and the candidate drugs could be identified and grouped into several cellular processes. Some of the relationships can be readily rationalized. V-9302 for instance is an inhibitor of amino acid transporters LAT1 and SNAT2 [26]. Both transporters play a key role in the maintenance of cytosolic amino acid levels. Inhibition of LAT1 has been shown in the past to activate GCN2 [27]. Similarly, we expected inhibitors of amino acid homeostasis to synergize with TAP20. Halofuginone and YH16889 interfere with prolyl-tRNA synthetase and lysyl-tRNA synthetase, respectively, and are therefore mechanistically close to the tRNA-binding function of GCN2. CB-5083 inhibits p97 ATPase, a protein which is involved in unfolding of ubiquitinated proteins and membrane proteins [28]. The unfolded proteins are then transferred to the proteasome for degradation, a process that replenishes cellular amino acid pools. Another inhibitor of proteostasis is bortezomib which blocks the proteasome itself, although synergy was observed in different cell lines compared to CB-5083. Thapsigargin, an inhibitor of the SERCA ATPase, induces ER stress which is an independent activator of GCN2 through the protein kinase PERK. However, thapsigargin only synergised with TAP20 in HPAFII and in SKOV3 cells.
A direct link between the MEK-ERK MAPK signalling pathway and the ISR has been demonstrated previously in HepG2 cells [29]. Consistently, we found a strong synergy between TAP20 and ERK inhibitor SCH772984. The MEK inhibitor selumetinib, which inhibits the MEK-ERK pathway upstream of ERK had a similar effect, although not in HPAFII cells.
The most striking synergy we identified was between elements of cell-cycle regulation and TAP20. This was exemplified by inhibitors of cyclin-dependent kinases, such as flavopiridol, seliciclib and THZ-1 [30] and danusertib an inhibitor of aurora kinases [31]. While flavopiridol and seliciclib are non-specific CDK inhibitors, THZ-1 is a more selective CDK-7 inhibitor.
To further explore the scope of these synergistic relationships we performed growth experiments exploring a matrix of different drug concentrations and tested the synergistic effect on matrix-dependent cell invasion. Due to low invasiveness of the cell lines used in the growth assays, we performed the cell invasion assay with 143B osteosarcoma cells, while the synergy analysis was made using MDA-MB-231 cells and HPAF2 cells, which showed the strongest responses in this study.
Combining TAP20 with p97-ATPase inhibitor CB5083 showed a synergistic effect as shown by growth inhibition and calculation of synergy scores using Synergyfinder [23]. In HPAFII cells the optimal effect was achieved at 1 µM CB5083 and >1 µM of TAP20 (Figure 2).
Although TAP20 alone had little effect on growth in all cell lines, it dramatically reduced matrix-dependent invasion of 143B cells at 1–3 µM, but to evaluate synergistic effects, a lower concentration, 0.75 µM, was used (Figure 2). In agreement with the growth experiments, spheroid size was reduced in the presence of both inhibitors (compare the growth area of the control with that of TAP20/CB5083).
The highly synergistic relationship between the CDK inhibitor flavopiridol and TAP20 was confirmed in the synergy analysis yielding scores of up to 40 in MDA-MB-231 and HPAFII cells (Figure 3). Optimal synergy was achieved with relatively low concentrations of both compounds (MDA-MB-231 TAP20: 0.2 µM; flavopiridol: 0.04 µM (Figure 3 a, b); HPAFII TAP20 1 µM, flavopiridol 0.03 µM (Figure 3 c, d)). Synergy disappeared at higher drug concentrations in MDA-MB-231 cells (Figure 3 b). As shown in Figure 2, TAP20 (0.75 µM) had a significant effect on matrix-dependent invasion of 143B cells alone, which was similar to the effect of 50 nM flavopiridol (Figure 3 e). This suggests that invasion generates cellular stress, which the cells ameliorate by activation of GCN2. Combination of both drugs significantly potentiated the effect. In the presence of both drugs hardly any cell protrusions from the original spheroid were observed (Figure 3 f).
This result was confirmed using the non-specific CDK inhibitor seliciclib (Figure 4). Inhibition of growth of MDA-MB-231 cells and synergy markedly increased when both drugs were applied at higher concentrations (Figure 4 a, b). Synergy was confined to a narrow range of concentrations. At 10 µM, seliciclib significantly reduced matrix-dependent invasion on its own, but in combination with TAP20 (1 µM), no outgrowth was observed compared to tumor spheroids in the absence of matrix (Figure 4 c, d).
Recently, CDK7 has been implicated in the ISR [32]. To investigate whether this isoform might be underlying the responses observed for flavopiridol and seliciclib, we used THZ-1, a specific CDK7 inhibitor. While THZ-1 showed a strong synergistic action in MDA-MB-231 (Figure 5 a, b) and HPAFII cells (Figure 5 c, d) it was ineffective alone or in combination with TAP20 to suppress growth or invasion of 143B cells (Figure 5 e, f).
We also analysed the MEK and ERK inhibitors selumetinib (Figure 6) and SCH772984 (Figure 7) in detail. Both compounds inhibited growth at low concentrations (Figure 6a and Figure 7a) and showed high synergy scores at low concentrations (SCH772984 0.4 µM, selumetinib 2 µM). However, synergy was generally low for SCH772984, which was a very effective drug independent of TAP20. At 100 nM selumetinib not only reduced invasion but also the size of the tumor spheroids (Figure 6 c, d), suggesting that cells were dying during the treatment. A similar pattern was observed with SCH772984 (Figure 7). Spheroids were markedly smaller than control spheroids grown in the absence of matrix (Figure 7 c, d)
In summary, we have identified key cellular processes inhibition of which causes nutrient stress and requires metabolic flexibility, which is afforded by activation of GCN2. This can be abrogated by concurrent inhibition of GCN2. Combination of drugs is an important strategy to overcome drug resistance and to inhibit processes that require significant pathway flexibility such as metastasis.
4. Discussion
While cancer cell lines experience little nutritional stress in replete growth media, cancer cells in the microenvironment of the tumor are more likely to experience nutrient stress [33,34]. Evidence for this is the close association of cancer stem cells with blood vessels [35]. the ISR is one of the cellular programs to deal with stress and to elicit responses that optimize cellular fitness [1,36]. Recycling of amino acids through lysosomal and proteasomal protein degradation is a key element of amino acid homeostasis [37]. Consistently, we found synergy with the proteasomal inhibitor bortezomib, the p97-ATPase inhibitor CB-5083 and the amino acid transport inhibitor V-9302. We did not detect synergy with the PERK inhibitor AMG PERK44, suggesting that our chosen cancer cell lines do not experience significant ER stress. It is tempting to speculate that AMG PERK44 may synergize well with CB-5083 or thapsigargin, both of which cause ER stress.
A synergistic relationship between TAP20 and inhibitors of cyclin-dependent kinases may appear surprising at first sight. However, before undergoing division, cells must ensure that sufficient amino acids are available for abundant protein synthesis to duplicate cellular content. Accordingly, amino acid deprivation stops cell cycle progression by stabilizing the mRNA of endogenous cyclin-dependent kinase inhibitors p22 and p27 [29].
CDK-7 is an unusual cyclin-dependent kinase, as it appears to activate other CDKs instead of directly regulating steps of the cell cycle. In addition, it has a role as a transcriptional activator [38]. It allows RNA polymerase II to initiate transcription and facilitates capping of mRNAs [39]. A direct link between CDK-7 and expression of the amino acid transporter SNAT2 was identified by Stretton et al. [32]. Upregulation of SNAT2 mRNA in response to amino acid limitation depended on CDK-7 activity. While RNA transcription and execution of cellular programs are generally associated, activation of GCN2 silences CAP-dependent transcription allowing mRNAs with uORFs, such as SNAT2, to be translated by a CAP-independent mechanism [40]. Sirtuin 6 was recently identified as a link between CDK-7 and the ISR [41]. Sirtuin 6 binds to ATF4, stabilizing the latter and preventing its degradation. Low expression levels of sirtuin 6 causes cells to become sensitive to CDK7 inhibition. CDK-7 was found to enhance transcription of more than 2000 genes, including MYC [41]. Thus, inhibition of CDK-7 will cause cellular stress, which elicits the ISR.
Related to cell cycle progression, Piecyk et al., found synergy between oxaliplatin and GCN2 inhibition in colon cancer tumoroids [42]. This was based on GCN2 sustaining ribosomal RNA transcription in nutrient-rich conditions, which synergised with inhibition of RNA polymerase I.
Notably, inhibitors of proteostasis, such as CB-5083 and bortezomib have a significant influence on cell-cycle progression as rapid breakdown of cell-cycle proteins is required at certain stages of the cell cycle [28,43]. This may contribute to the synergy observed in this study.
The observed synergy with the MEK-ERK pathway is harder to explain. In the absence of stress, growth-promoting signals lead to Akt-dependent and or Erk-dependent phosphorylation of the tuberous sclerosis-2 (TSC2), which in turn causes the release of the TSC complex from the lysosome resulting in mTORC1 activation [44]. Thus, we might have expected an antagonistic relationship similar to rapamycin. However, a direct link between the MEK-ERK MAPK signalling pathway and the ISR has been demonstrated previously in HepG2 cells [29]. In these cells, an ERK inhibitor prevented increased transcription of ATF4 and of the amino acid response target gene SNAT2 [45]. The study further showed that phosphorylation of eIF2α was also blocked by inhibition of ERK without ERK directly phosphorylating eIF2α. The interaction is interdependent, as lack of GCN2 prevents phosphorylation of ERK in response to amino acid limitation. Moreover, there is also a direct link between the activity of amino acid transporter SNAT2 and ERK1/2 activity [46]. As a result, blocking the MEK-ERK axis could increase nutrient stress, which can be aggravated by simultaneously blocking GCN2.
Our results highlight the mutual relationship between nutrient stress and the ability of cancer cells to adaptively respond to stressful situations using the GCN2-ATF4 pathway. While blocking the GCN2-ATF4 axis alone may have limited efficacy to reduce cancer cell growth, its blockade in conjunction with stress-inducing drug regimens could be a promising strategy to reduce cancer cell growth and invasion.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Response of six cell lines to experimental anticancer drugs; Figure S2: Growth data for combinations shown in Table 4.
Author Contributions
G.G. and A.B. performed experiments, analyzed data and prepared data for publication. S.B. designed the study and wrote the publication. F.R. helped with microscopy and measuring the spheroid periphery. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a research contract provided by Merck KgAa.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors thank Ralph Lindemann, Ansgar Wegner, Dirk Wienke and Dieter Dorsch (Merck KGaA, Darmstadt, Germany) for fruitful discussions on the mechanism and use of the GCN2 inhibitors and for providing a collaborative atmosphere for the project.
Conflicts of Interest
This research was funded by a research contract provided by Merck KgAa.
Appendix A
Dose-Response curves of cancer growth inhibitors on cell lines used in this study.
Appendix B
Synergy between TAP20 and experimental and approved cancer growth inhibitors.
References
- Tian, X.; Zhang, S.; Zhou, L.; Seyhan, A.A.; Hernandez Borrero, L.; Zhang, Y.; El-Deiry, W.S. Targeting the Integrated Stress Response in Cancer Therapy. Front Pharmacol 2021, 12, 747837. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.H.; Petryshyn, R.; London, I.M. Characterization of double-stranded-RNA-activated kinase that phosphorylates alpha subunit of eukaryotic initiation factor 2 (eIF-2 alpha) in reticulocyte lysates. Proc Natl Acad Sci U S A 1980, 77, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; London, I.M. Regulation of protein synthesis by heme-regulated eIF-2 alpha kinase. Trends Biochem Sci 1995, 20, 105–108. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 2003, 11, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Cordova, R.A.; Misra, J.; Amin, P.H.; Klunk, A.J.; Damayanti, N.P.; Carlson, K.R.; Elmendorf, A.J.; Kim, H.G.; Mirek, E.T.; Elzey, B.D.; et al. GCN2 eIF2 kinase promotes prostate cancer by maintaining amino acid homeostasis. Elife 2022, 11. [Google Scholar] [CrossRef]
- Tameire, F.; Verginadis, I.; Leli, N.M.; Polte, C.; Conn, C.S.; Ojha, R.; Salas Salinas, C.; Chinga, F.; Monroy, A.M.; Fu, W.; et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat Cell Biol 2019, 21, 889–899. [Google Scholar] [CrossRef]
- Gold, L.T.; Masson, G.R. GCN2: roles in tumour development and progression. Biochem Soc Trans 2022, 50, 737–745. [Google Scholar] [CrossRef]
- Jin, Y.; Saatcioglu, F. Targeting the Unfolded Protein Response in Hormone-Regulated Cancers. Trends Cancer 2020, 6, 160–171. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wek, R.C. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem 2005, 280, 14189–14202. [Google Scholar] [CrossRef]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [PubMed]
- Broer, A.; Rahimi, F.; Broer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J Biol Chem 2016, 291, 13194–13205. [Google Scholar] [CrossRef]
- Nakamura, A.; Nambu, T.; Ebara, S.; Hasegawa, Y.; Toyoshima, K.; Tsuchiya, Y.; Tomita, D.; Fujimoto, J.; Kurasawa, O.; Takahara, C.; et al. Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc Natl Acad Sci U S A 2018, 115, E7776–E7785. [Google Scholar] [CrossRef] [PubMed]
- Okano, N.; Kawai, K.; Yamauchi, Y.; Kobayashi, T.; Naruge, D.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phaseⅠstudy of JPH203 in patients with advanced solid tumors. Journal of Clinical Oncology 2018, 36, 419–419. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of therapy with bortezomib in solid tumors: a review based on 32 clinical trials. Future Oncol 2014, 10, 1795–1807. [Google Scholar] [CrossRef]
- Lane, H.A.; Breuleux, M. Optimal targeting of the mTORC1 kinase in human cancer. Curr Opin Cell Biol 2009, 21, 219–229. [Google Scholar] [CrossRef]
- Ables, G.P.; Hens, J.R.; Nichenametla, S.N. Methionine restriction beyond life-span extension. Ann N Y Acad Sci 2016, 1363, 68–79. [Google Scholar] [CrossRef]
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D.; et al. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res 2017, 77, 3564–3576. [Google Scholar] [CrossRef]
- Kato, Y.; Kunimasa, K.; Takahashi, M.; Harada, A.; Nagasawa, I.; Osawa, M.; Sugimoto, Y.; Tomida, A. GZD824 Inhibits GCN2 and Sensitizes Cancer Cells to Amino Acid Starvation Stress. Mol Pharmacol 2020, 98, 669–676. [Google Scholar] [CrossRef]
- Dorsch, D.; Hölzemann, G.; Calderini, M.; Wegener, A.; Pöschke, O. Triazolo[4,5-d]pyrimidine derivatives for the treatment of diseases such as cancer. 2014.
- Broer, A.; Gauthier-Coles, G.; Rahimi, F.; van Geldermalsen, M.; Dorsch, D.; Wegener, A.; Holst, J.; Broer, S. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J Biol Chem 2019, 294, 4012–4026. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Wang, W.; Aldahdooh, J.; Malyutina, A.; Shadbahr, T.; Tanoli, Z.; Pessia, A.; Tang, J. SynergyFinder Plus: Toward Better Interpretation and Annotation of Drug Combination Screening Datasets. Genomics Proteomics Bioinformatics 2022. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, R.; Brown, K.R.; Suarez, F.; Sayad, A.; Karamboulas, K.; Krzyzanowski, P.M.; Sircoulomb, F.; Medrano, M.; Fedyshyn, Y.; Koh, J.L.Y.; et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov 2012, 2, 172–189. [Google Scholar] [CrossRef]
- Wengrod, J.; Wang, D.; Weiss, S.; Zhong, H.; Osman, I.; Gardner, L.B. Phosphorylation of eIF2α triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma. Sci Signal 2015, 8, ra27. [Google Scholar] [CrossRef]
- Broer, A.; Fairweather, S.; Broer, S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front Pharmacol 2018, 9, 785. [Google Scholar] [CrossRef]
- Cormerais, Y.; Giuliano, S.; LeFloch, R.; Front, B.; Durivault, J.; Tambutte, E.; Massard, P.A.; de la Ballina, L.R.; Endou, H.; Wempe, M.F.; et al. Genetic Disruption of the Multifunctional CD98/LAT1 Complex Demonstrates the Key Role of Essential Amino Acid Transport in the Control of mTORC1 and Tumor Growth. Cancer Res 2016, 76, 4481–4492. [Google Scholar] [CrossRef]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol 2012, 14, 117–123. [Google Scholar] [CrossRef]
- Leung-Pineda, V.; Pan, Y.; Chen, H.; Kilberg, M.S. Induction of p21 and p27 expression by amino acid deprivation of HepG2 human hepatoma cells involves mRNA stabilization. Biochem J 2004, 379, 79–88. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Meulenbeld, H.J.; Mathijssen, R.H.; Verweij, J.; de Wit, R.; de Jonge, M.J. Danusertib, an aurora kinase inhibitor. Expert Opin Investig Drugs 2012, 21, 383–393. [Google Scholar] [CrossRef]
- Stretton, C.; Lipina, C.; Hyde, R.; Cwiklinski, E.; Hoffmann, T.M.; Taylor, P.M.; Hundal, H.S. CDK7 is a component of the integrated stress response regulating SNAT2 (SLC38A2)/System A adaptation in response to cellular amino acid deprivation. Biochim Biophys Acta Mol Cell Res 2019, 1866, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Finicle, B.T.; Jayashankar, V.; Edinger, A.L. Nutrient scavenging in cancer. Nat Rev Cancer 2018, 18, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Gouirand, V.; Guillaumond, F.; Vasseur, S. Influence of the Tumor Microenvironment on Cancer Cells Metabolic Reprogramming. Front Oncol 2018, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Nofal, M.; Wang, T.; Yang, L.; Jankowski, C.S.R.; Hsin-Jung Li, S.; Han, S.; Parsons, L.; Frese, A.N.; Gitai, Z.; Anthony, T.G.; et al. GCN2 adapts protein synthesis to scavenging-dependent growth. Cell Syst 2022, 13, 158–172.e159. [Google Scholar] [CrossRef]
- Broer, S.; Broer, A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem J 2017, 474, 1935–1963. [Google Scholar] [CrossRef]
- Nigg, E.A. Cyclin-dependent kinase 7: at the cross-roads of transcription, DNA repair and cell cycle control? Curr Opin Cell Biol 1996, 8, 312–317. [Google Scholar] [CrossRef]
- Fisher, R.P. Cdk7: a kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription 2019, 10, 47–56. [Google Scholar] [CrossRef]
- Gaccioli, F.; Huang, C.C.; Wang, C.; Bevilacqua, E.; Franchi-Gazzola, R.; Gazzola, G.C.; Bussolati, O.; Snider, M.D.; Hatzoglou, M. Amino acid starvation induces the SNAT2 neutral amino acid transporter by a mechanism that involves eukaryotic initiation factor 2alpha phosphorylation and cap-independent translation. J Biol Chem 2006, 281, 17929–17940. [Google Scholar] [CrossRef]
- Kartha, N.; Gianopulos, J.E.; Schrank, Z.; Cavender, S.M.; Dobersch, S.; Kynnap, B.D.; Wallace-Povirk, A.; Wladyka, C.L.; Santana, J.F.; Kim, J.C.; et al. Sirtuin 6 is required for the integrated stress response and resistance to inhibition of transcriptional cyclin-dependent kinases. Sci Transl Med 2023, 15, eabn9674. [Google Scholar] [CrossRef]
- Piecyk, M.; Triki, M.; Laval, P.A.; Duret, C.; Fauvre, J.; Cussonneau, L.; Machon, C.; Guitton, J.; Rama, N.; Gibert, B.; et al. The stress sensor GCN2 differentially controls ribosome biogenesis in colon cancer according to the nutritional context. Mol Oncol 2023. [Google Scholar] [CrossRef]
- King, R.W.; Deshaies, R.J.; Peters, J.M.; Kirschner, M.W. How proteolysis drives the cell cycle. Science 1996, 274, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Cormerais, Y.; Vucetic, M.; Parks, S.K.; Pouyssegur, J. Amino Acid Transporters Are a Vital Focal Point in the Control of mTORC1 Signaling and Cancer. Int J Mol Sci 2020, 22. [Google Scholar] [CrossRef] [PubMed]
- Thiaville, M.M.; Pan, Y.X.; Gjymishka, A.; Zhong, C.; Kaufman, R.J.; Kilberg, M.S. MEK signaling is required for phosphorylation of eIF2alpha following amino acid limitation of HepG2 human hepatoma cells. J Biol Chem 2008, 283, 10848–10857. [Google Scholar] [CrossRef] [PubMed]
- Franchi-Gazzola, R.; Visigalli, R.; Bussolati, O.; Dall’Asta, V.; Gazzola, G.C. Adaptive increase of amino acid transport system A requires ERK1/2 activation. J Biol Chem 1999, 274, 28922–28928. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Inhibition of GCN2 by TAP20 in different cell lines as ascertained by ATF4 expression. Cell lines were incubated in amino acid-free media for 24 h in the absence or presence of increasing concentrations of TAP20. ATF4 expression was evaluated by immunoblotting; β-actin was used as the loading control. In some cell lines, ATF4 immunodetection revealed two bands of which the lower band was used for quantification. In all cell lines, ATF4 expression was strongly inhibited with > 1µM TAP20.
Figure 1.
Inhibition of GCN2 by TAP20 in different cell lines as ascertained by ATF4 expression. Cell lines were incubated in amino acid-free media for 24 h in the absence or presence of increasing concentrations of TAP20. ATF4 expression was evaluated by immunoblotting; β-actin was used as the loading control. In some cell lines, ATF4 immunodetection revealed two bands of which the lower band was used for quantification. In all cell lines, ATF4 expression was strongly inhibited with > 1µM TAP20.
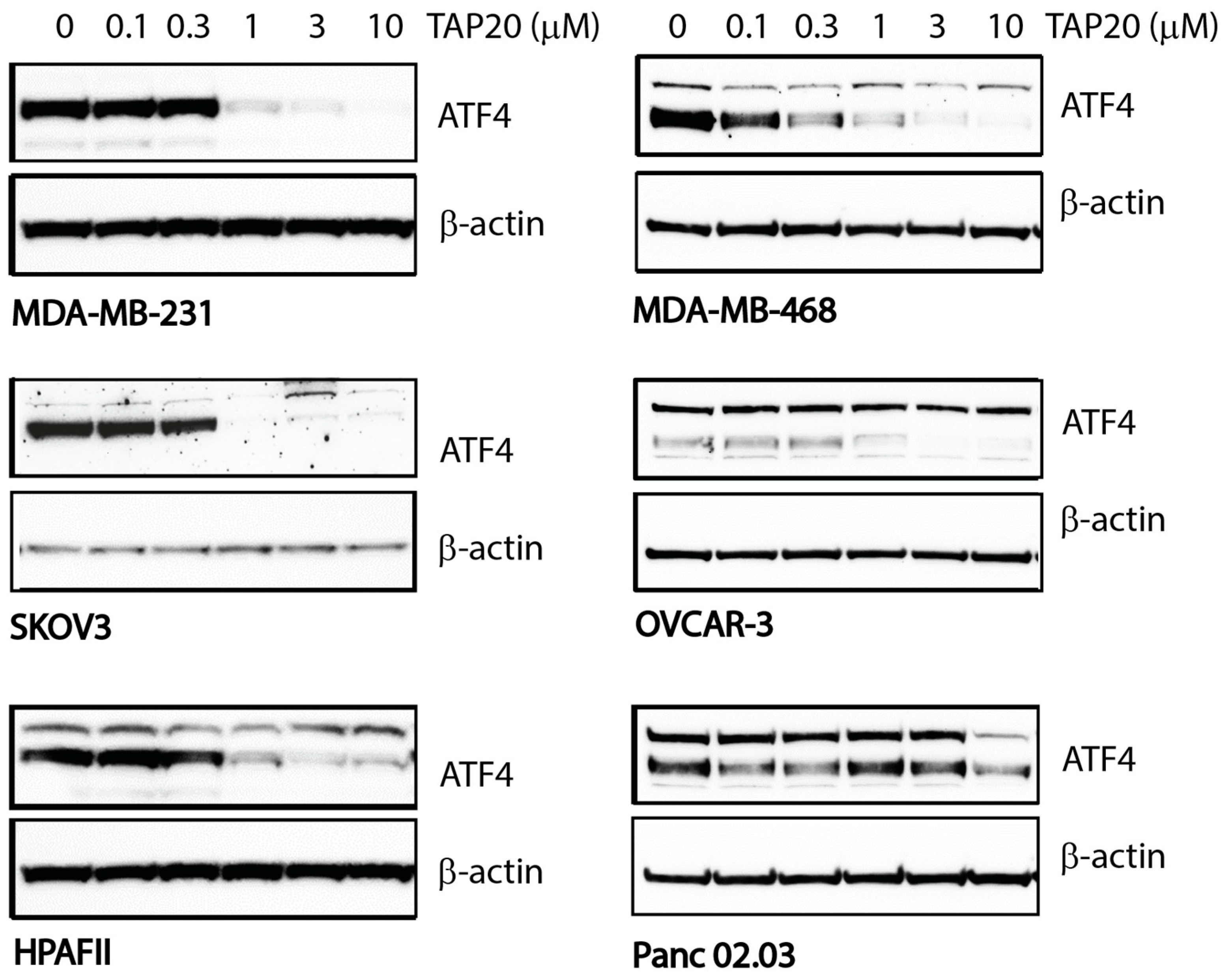
Figure 2.
Effect of TAP20 and of CB5083 on cell growth and matrix-dependent invasion. For synergy analysis HPAFII cells were grown in the presence of different concentrations of TAP20 and CB5083 (n = 3). Growth is expressed as a percentage of the uninhibited control (a). Synergy scores for the experiment are shown in (b). (c, d) 143B osteosarcoma cells were grown into spheroids before adding the extracellular matrix for invasion. Invasion areas were determined by microscopy (n = 6). (c) Quantitative evaluation of outgrowth over eight days. (d) Examples of spheroid formation in the presence or absence of inhibitors. Outgrowths with or without matrix are shown as controls.
Figure 2.
Effect of TAP20 and of CB5083 on cell growth and matrix-dependent invasion. For synergy analysis HPAFII cells were grown in the presence of different concentrations of TAP20 and CB5083 (n = 3). Growth is expressed as a percentage of the uninhibited control (a). Synergy scores for the experiment are shown in (b). (c, d) 143B osteosarcoma cells were grown into spheroids before adding the extracellular matrix for invasion. Invasion areas were determined by microscopy (n = 6). (c) Quantitative evaluation of outgrowth over eight days. (d) Examples of spheroid formation in the presence or absence of inhibitors. Outgrowths with or without matrix are shown as controls.
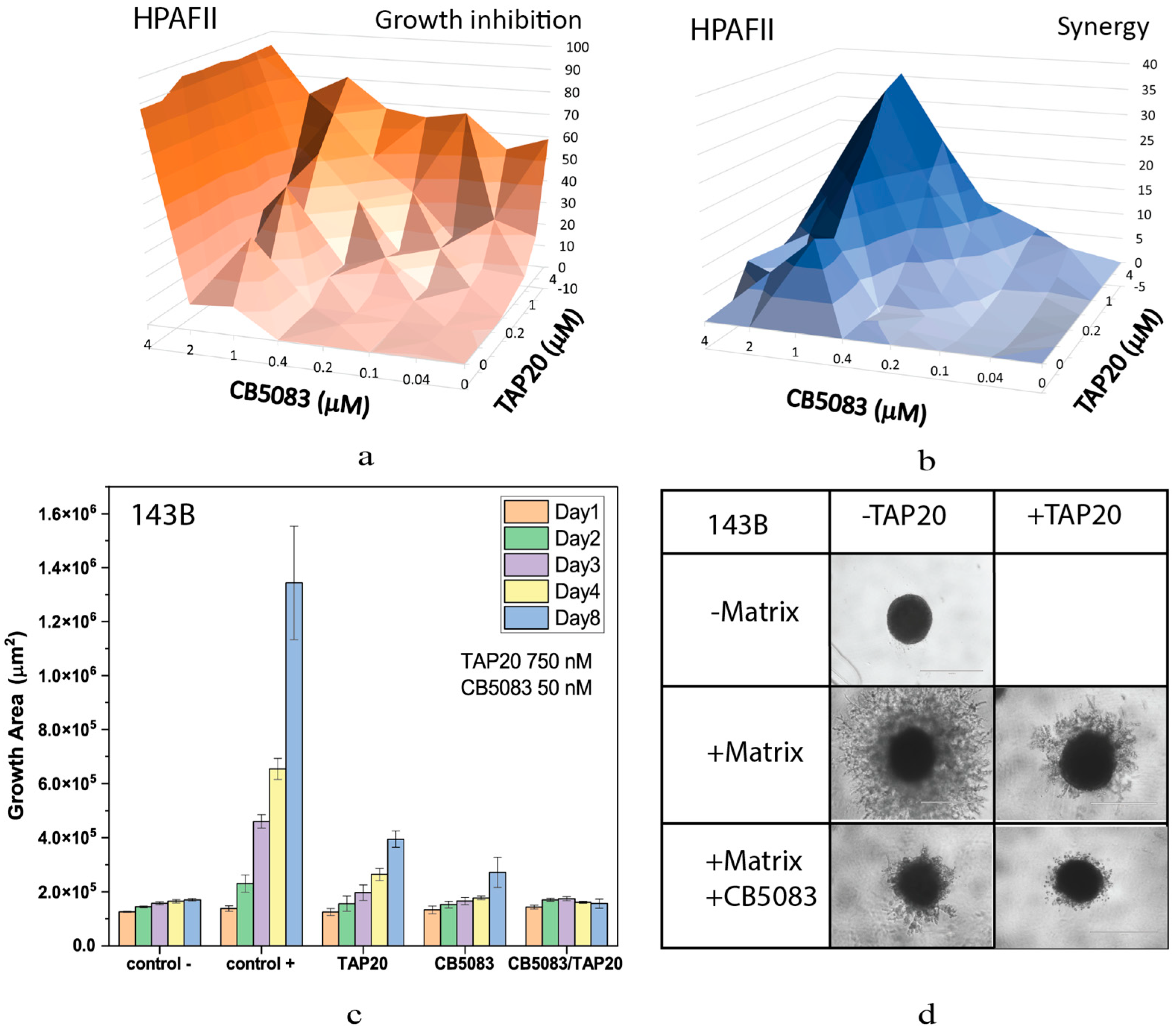
Figure 3.
Synergy analysis of TAP20 and flavopiridol and their effects on matrix-dependent invasion. For the synergy analysis, MDA-MB-231 (a, b) and HPAFII (c, d) cells were grown in the presence of different combined concentrations of TAP20 and flavopiridol (n = 3). Growth inhibition data (a, c) were used to calculate synergy scores (b, d). For matrix-dependent invasion, 143B osteosarcoma cells were grown to spheroids before adding the extracellular matrix for invasion (n = 6). Invasion areas were determined by microscopy. (e) Quantitative evaluation of outgrowth over eight days. (f) Examples of the effects flavopiridol and TAP20 on invasion. Controls are the same as in Figure 2).
Figure 3.
Synergy analysis of TAP20 and flavopiridol and their effects on matrix-dependent invasion. For the synergy analysis, MDA-MB-231 (a, b) and HPAFII (c, d) cells were grown in the presence of different combined concentrations of TAP20 and flavopiridol (n = 3). Growth inhibition data (a, c) were used to calculate synergy scores (b, d). For matrix-dependent invasion, 143B osteosarcoma cells were grown to spheroids before adding the extracellular matrix for invasion (n = 6). Invasion areas were determined by microscopy. (e) Quantitative evaluation of outgrowth over eight days. (f) Examples of the effects flavopiridol and TAP20 on invasion. Controls are the same as in Figure 2).
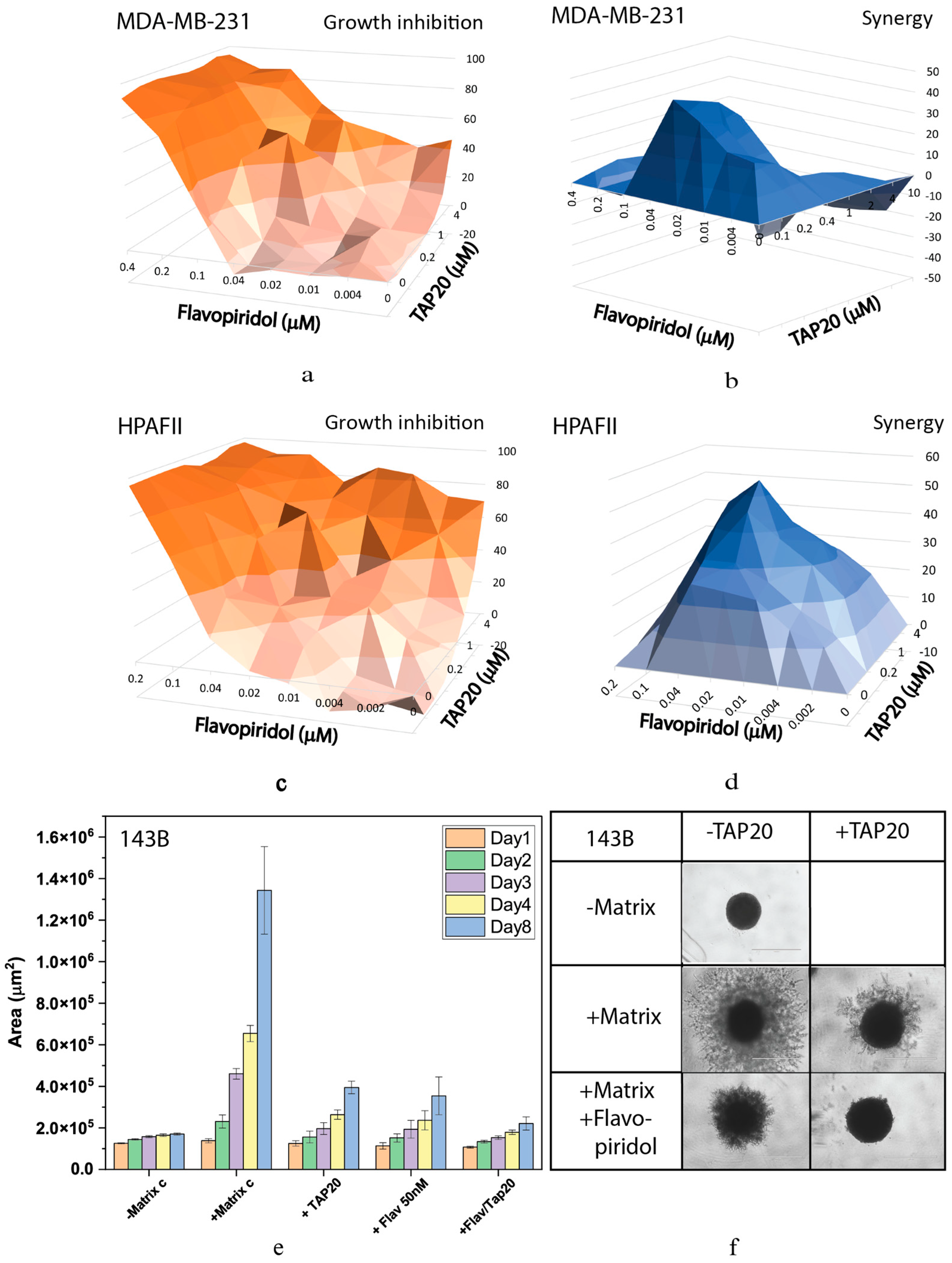
Figure 4.
Synergy analysis of TAP20 and seliclib and their effects on matrix-dependent invasion. For the synergy analysis, MDA-MB-231 cells were grown in the presence of different combined concentrations of TAP20 and seliciclib (n = 3). Growth data (a) were used to calculate synergy scores (b). For matrix-dependent invasion, 143B osteosarcoma cells were grown to spheroids before the extracellular matrix was added for invasion (n = 6). Invasion areas were determined by microscopy. (c) Quantitative evaluation of outgrowth over eight days. (d) Examples of the effects flavopiridol and TAP20 on invasion. Controls are the same as in Figure 2.
Figure 4.
Synergy analysis of TAP20 and seliclib and their effects on matrix-dependent invasion. For the synergy analysis, MDA-MB-231 cells were grown in the presence of different combined concentrations of TAP20 and seliciclib (n = 3). Growth data (a) were used to calculate synergy scores (b). For matrix-dependent invasion, 143B osteosarcoma cells were grown to spheroids before the extracellular matrix was added for invasion (n = 6). Invasion areas were determined by microscopy. (c) Quantitative evaluation of outgrowth over eight days. (d) Examples of the effects flavopiridol and TAP20 on invasion. Controls are the same as in Figure 2.
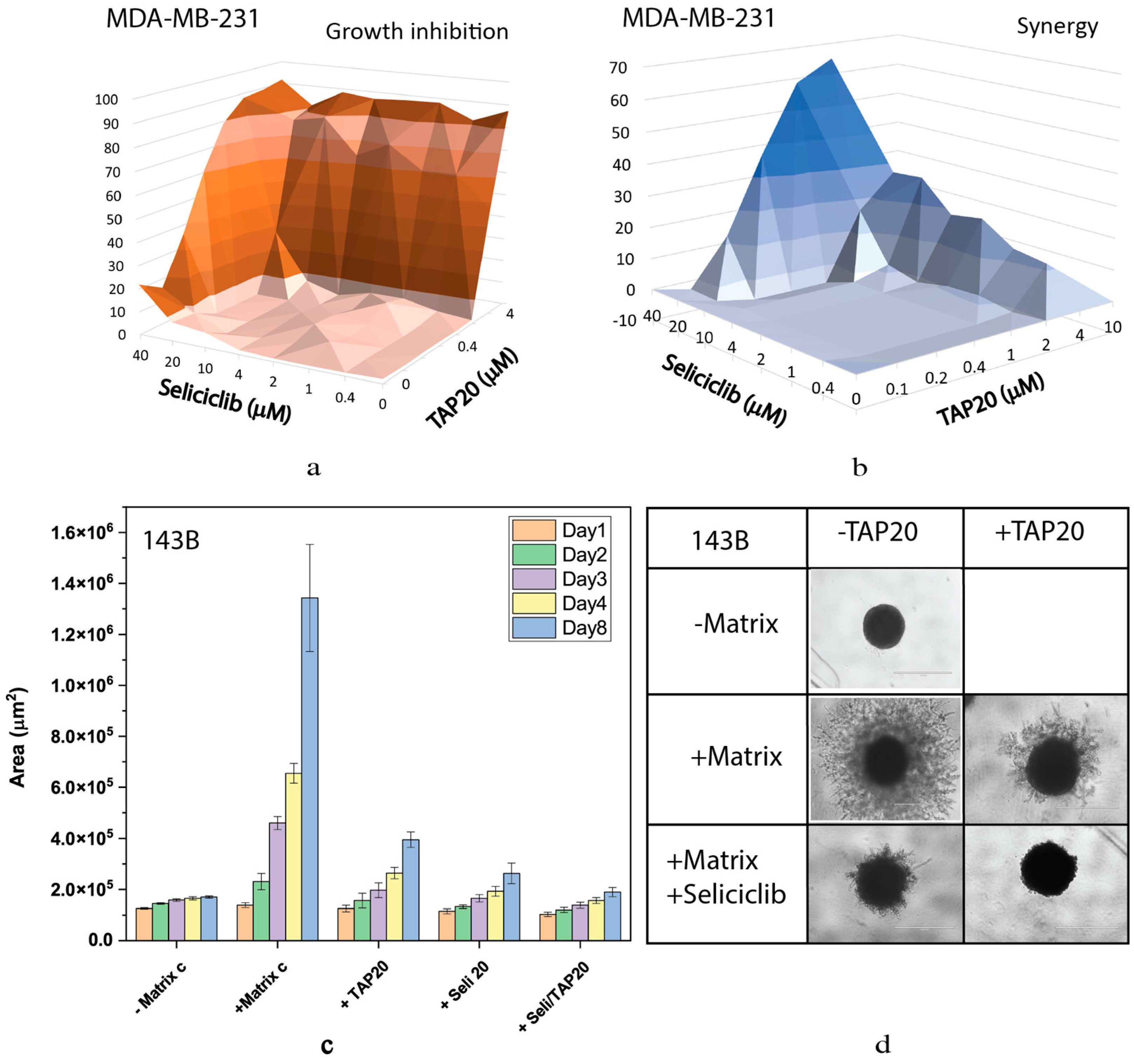
Figure 5.
Synergy analysis of TAP20 and THZ-1 and their effect on matrix-dependent invasion. For the synergy analysis, MDA-MB-231 (a, b) and HPAFII (c, d) cells were grown in the presence of different combined concentrations of TAP20 and THZ-1 (n = 3). For matrix-dependent invasion, 143B osteosarcoma cells were grown to spheres before adding the extracellular matrix for invasion (n = 6). Growth and invasion areas were determined by microscopy. (e) Growth of spheroids in the absence of matrix in the presence of different drug combinations. (f) Outgrowth into the matrix over seven days in the presence of the same drug combinations.
Figure 5.
Synergy analysis of TAP20 and THZ-1 and their effect on matrix-dependent invasion. For the synergy analysis, MDA-MB-231 (a, b) and HPAFII (c, d) cells were grown in the presence of different combined concentrations of TAP20 and THZ-1 (n = 3). For matrix-dependent invasion, 143B osteosarcoma cells were grown to spheres before adding the extracellular matrix for invasion (n = 6). Growth and invasion areas were determined by microscopy. (e) Growth of spheroids in the absence of matrix in the presence of different drug combinations. (f) Outgrowth into the matrix over seven days in the presence of the same drug combinations.
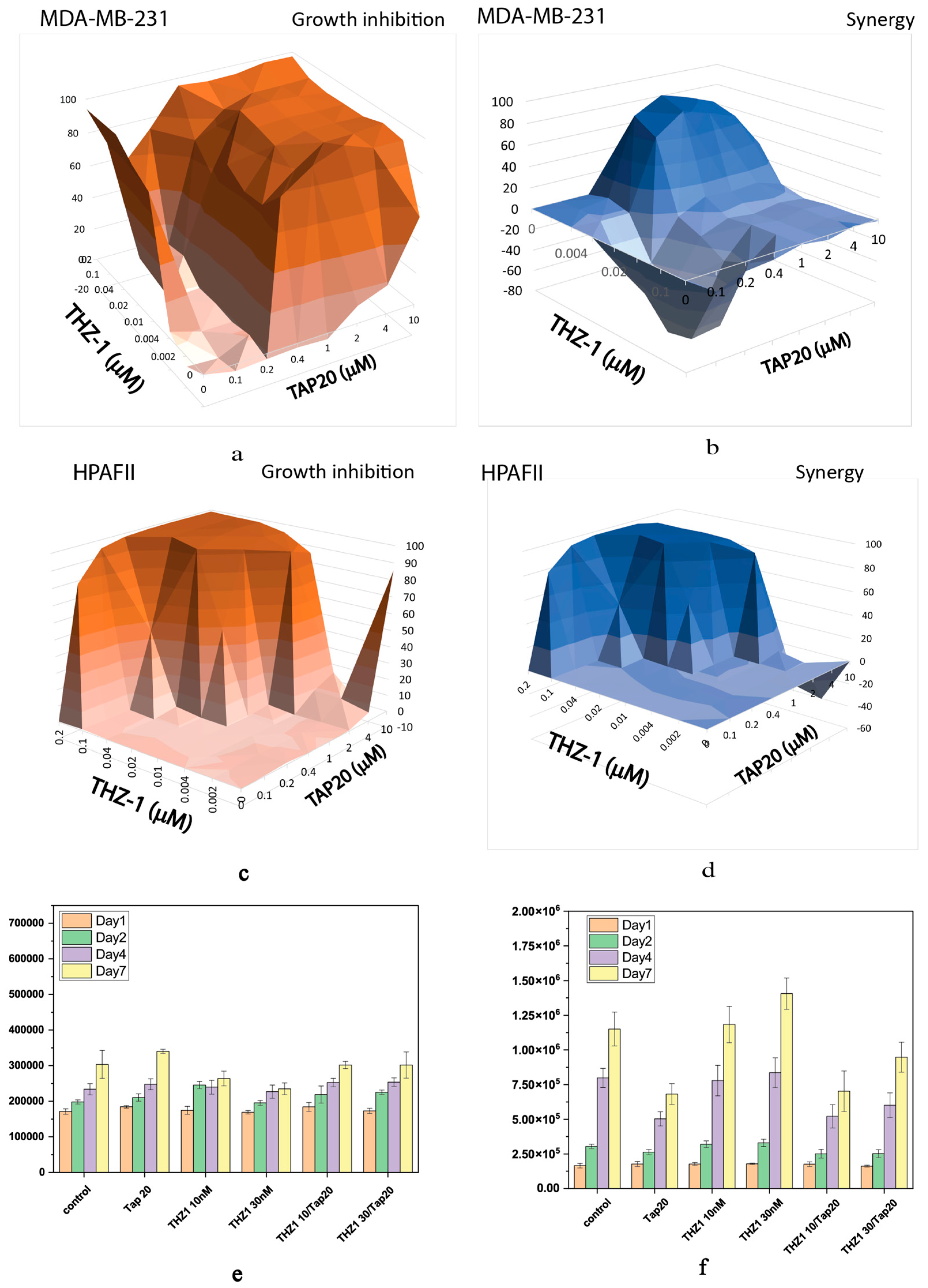
Figure 6.
Synergy analysis of TAP20 and selumetinib and their effects on matrix-dependent invasion. For the synergy analysis, MDA-MB-231 cells were grown in the presence of different combined concentrations of TAP20 and selumetinib (n = 3). (a) Inhibition of growth and (b) synergy score for different combinations of selumetinib and TAP20. For matrix-dependent invasion 143B osteosarcoma cells were grown to spheroids before adding the extracellular matrix for invasion (n = 6). Invasion areas were determined by microscopy. (c) Quantitative evaluation of outgrowth. (d) Example of the effects selumetinib and TAP20 on invasion. Controls are the same as in Figure 2.
Figure 6.
Synergy analysis of TAP20 and selumetinib and their effects on matrix-dependent invasion. For the synergy analysis, MDA-MB-231 cells were grown in the presence of different combined concentrations of TAP20 and selumetinib (n = 3). (a) Inhibition of growth and (b) synergy score for different combinations of selumetinib and TAP20. For matrix-dependent invasion 143B osteosarcoma cells were grown to spheroids before adding the extracellular matrix for invasion (n = 6). Invasion areas were determined by microscopy. (c) Quantitative evaluation of outgrowth. (d) Example of the effects selumetinib and TAP20 on invasion. Controls are the same as in Figure 2.
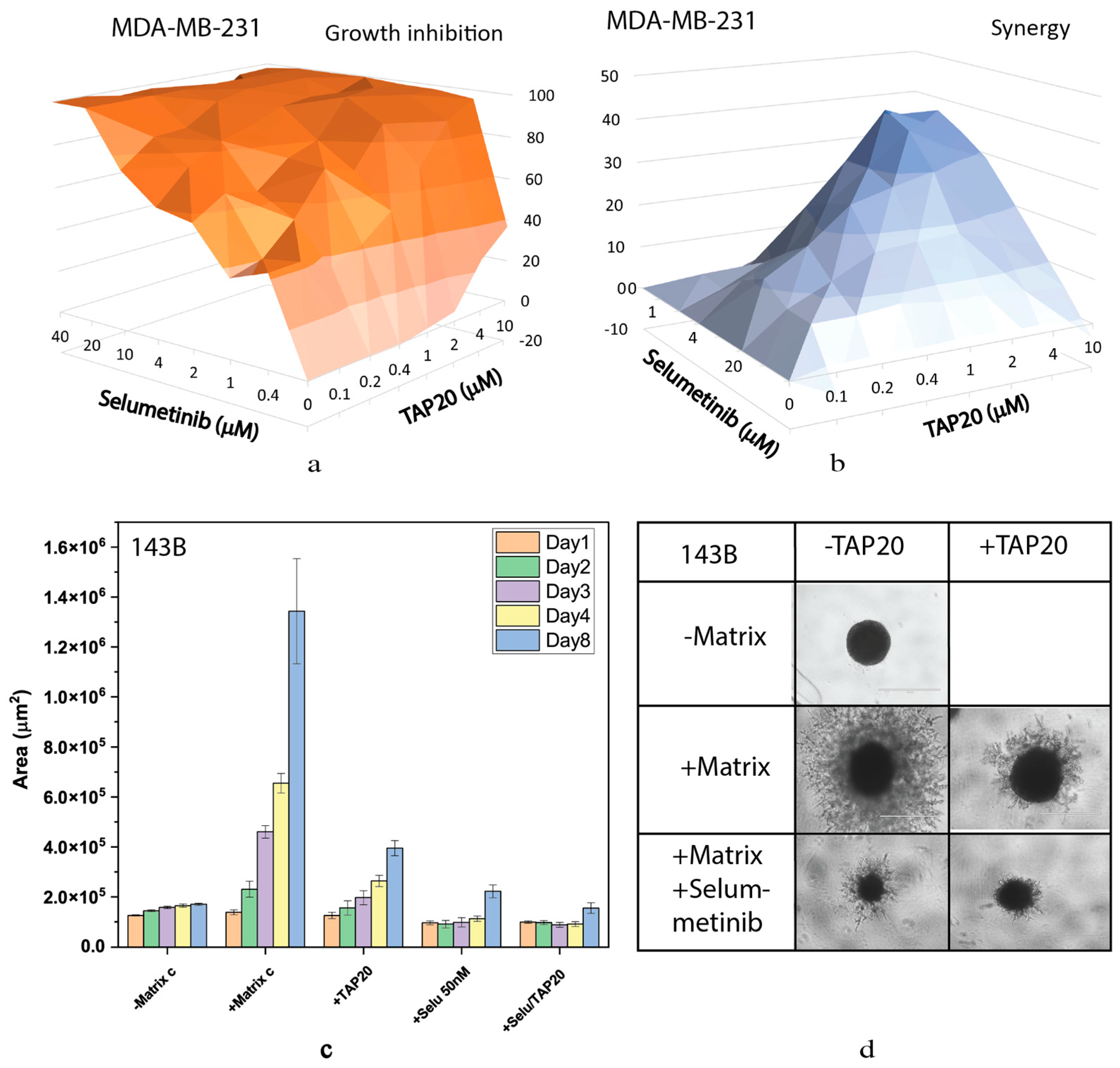
Figure 7.
Synergy between TAP20 and SCH772984 and their effects on matrix-dependent invasion. For the synergy analysis, MDA-MB-231 cells were grown in the presence of different combined concentrations of TAP20 and SCH772984 (n = 3). (a) Inhibition of growth by different combinations of TAP20 and SCH772984. (b) Synergy scores for the same combinations. For matrix-dependent invasion (c, d) 143B osteosarcoma cells were grown to spheres before adding the extracellular matrix for invasion (n = 6). Invasion areas were determined by microscopy. Outgrowths with or without matrix are shown as controls. (c) Quantitative evaluation of outgrowths. (d) Examples of the effects SCH772894 and TAP20 on invasion. Outgrowths with or without matrix are shown as controls.
Figure 7.
Synergy between TAP20 and SCH772984 and their effects on matrix-dependent invasion. For the synergy analysis, MDA-MB-231 cells were grown in the presence of different combined concentrations of TAP20 and SCH772984 (n = 3). (a) Inhibition of growth by different combinations of TAP20 and SCH772984. (b) Synergy scores for the same combinations. For matrix-dependent invasion (c, d) 143B osteosarcoma cells were grown to spheres before adding the extracellular matrix for invasion (n = 6). Invasion areas were determined by microscopy. Outgrowths with or without matrix are shown as controls. (c) Quantitative evaluation of outgrowths. (d) Examples of the effects SCH772894 and TAP20 on invasion. Outgrowths with or without matrix are shown as controls.
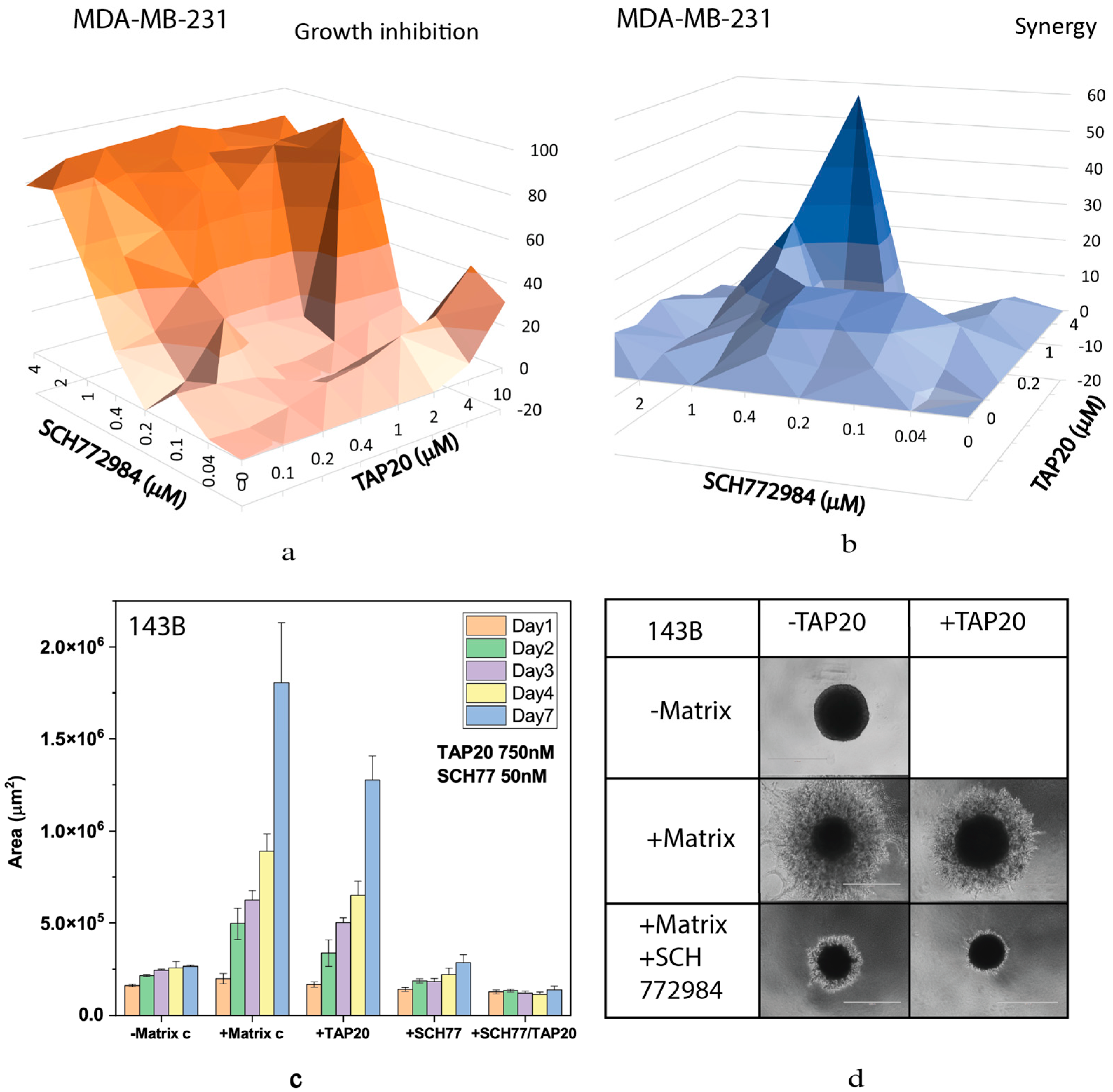

Table 1.
Compounds used in this study.
| Compound | Target | IC50 |
|---|---|---|
| Agerafenib | BRAFV600E | 14 nM |
| AMG PERK44 | PERK | 6 nM |
| AZD3965 | MCT1 | 1.6 nM |
| Bay876 | GLUT1 | 2 nM |
| Bortezomib | 20S proteasome | 0.6 nM |
| Bumetanide | NKCC1 | 680 nM |
| CB-839 | Glutaminase 1 | 25 nM |
| CB-5083 | p97AAA ATPase | 11 nM |
| Cetuximab | EGFR | 0.2 nM |
| Danusertib | Aurora-kinases | 13–79 nM |
| GSK2837808A | LDH-A | 2.6 nM |
| GZD824 | Bcr-Abl | 0.32 nM |
| Flavopiridol | CDK (non-specific) | 6–300 nM |
| 5-Fluorouracil | Thymidylate synthase | Irr1 |
| Halofuginone | Prolyl-tRNA synthetase | 18 nM |
| NCT-503 | PHGDH | 2500 nM |
| PLX8394 | BRAF/BRAFV600E | 14 nM/5 nM |
| Rapamycin | FKBP12 | 0.1 nM |
| SCH772984 | Erk1/2 | 4 nM/1 nM |
| Seliciclib | CDK (non-specific) | 200–800 nM |
| Selumetinib | MEK1/2 | 14 nM |
| Thapsigargin | SERCA | 0.4 nM |
| THZ-1 | CDK7 | Irr1 |
| V-9302 | SNAT2/LAT1 | n.d. |
| YH16899 | KRS-67LR interaction | 8600 nM |
1 irr: irreversible inhibitor.
Table 2.
Sensitivity of cell lines to compounds used in this study. Inhibition of growth is shown as IC50 (μM). n.i. = no inhibition, n.d. = not determined. For each cell line n = 12–18 independent biological replicates were analysed.
Table 2.
Sensitivity of cell lines to compounds used in this study. Inhibition of growth is shown as IC50 (μM). n.i. = no inhibition, n.d. = not determined. For each cell line n = 12–18 independent biological replicates were analysed.
| Compound | MDA-MB-231 | MDA-MB-468 | HPAFII | Panc02.03 | SKOV3 | OVCAR3 | ||
|---|---|---|---|---|---|---|---|---|
| Agerafenib | 4.3 | 4.3 | 1.6 | 3.7 | 4.7 | 6.5 | ||
| AMG PERK44 | >10 | >10 | 9 | >10 | >10 | >10 | ||
| AZD3965 | >10 | >10 | >10 | >10 | >10 | >10 | ||
| Bay876 | 10 | 10 | 0.25 | 0.15 | 1.4 | 0.16 | ||
| Bortezomib | 0.002 | 0.003 | 0.003 | 0.004 | 0.004 | 0.002 | ||
| Bumetanide | n.i. | n.d. | n.i. | n.d. | n.i. | n.d. | ||
| CB-839 | 10 | n.d. | >10 | n.d. | >10 | n.d. | ||
| CB-5083 | 0.76 | 0.36 | 0.19 | 0.12 | 1.2 | 0.36 | ||
| Cetuximab | >10 | n.d. | >10 | n.d. | >10 | n.d. | ||
| Danusertib | <0.1 | <0.1 | 0.8 | 0.36 | 2.2 | <0.1 | ||
| GSK2837808A | >10 | n.d. | >10 | n.d. | >10 | n.d. | ||
| GZD824 | 1 | 1.3 | <0.1 | 0.31 | 1.6 | 1.8 | ||
| Flavopiridol | 0.1 | 0.04 | <0.1 | 4 | 0.22 | 1.7 | ||
| 5-Fluorouracil | >10 | >10 | >10 | 8.9 | >10 | 1.7 | ||
| Halofuginone | 0.032 | 0.054 | 0.038 | 0.057 | 0.019 | 0.058 | ||
| NCT-503 | >10 | >10 | >10 | >10 | >10 | >10 | ||
| PLX8394 | 10 | 10 | 4.5 | 10 | 6 | 3.6 | ||
| Rapamycin | >10 | n.d. | >10 | n.d. | >10 | n.d. | ||
| SCH772984 | 1 | 1 | 0.45 | 0.4 | 8.4 | 5 | ||
| Seliciclib | >10 | >10 | 9.5 | 6.3 | >10 | 7.4 | ||
| Selumetinib | 10 | 6.3 | <0.1 | 0.88 | 10 | 1.4 | ||
| Thapsigargin | <0.1 | 0.005 | <0.1 | 0.004 | 0.52 | <0.003 | ||
| V-9302 | 1.1 | 1.1 | 1.1 | 2 | 2.6 | 1.1 | ||
| YH16899 | >10 | n.d. | >10 | n.d. | >10 | n.d |
Table 3.
Compound concentrations used to detect synergy with TAP20. All concentrations are given as μM.
Table 3.
Compound concentrations used to detect synergy with TAP20. All concentrations are given as μM.
| Compound | MDA-MB-231 | MDA-MB-468 | HPAFII | Panc02.03 | SKOV3 | OVCAR3 | ||
|---|---|---|---|---|---|---|---|---|
| Agerafenib | 3 | 3 | 1 | 0.3 | 3 | 3 | ||
| AMG PERK44 | 10 | 10 | 10 | 10 | 10 | 10 | ||
| AZD3965 | 10 | 10 | 10 | 10 | 10 | 3 | ||
| Bay876 | 10 | 3 | 0.1 | 0.03 | 1 | 0.1 | ||
| Bortezomib | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | ||
| Bumetanide | 10 | 10 | 10 | 10 | 10 | 10 | ||
| CB-839 | 10 | 10 | 10 | 10 | 10 | 10 | ||
| CB-5083 | 1 | 0.3 | 0.1 | 0.1 | 1 | 0.3 | ||
| Cetuximab | 0.033 | 0.033 | 0.033 | 0.033 | 0.033 | 0.033 | ||
| Danusertib | 0.01 | 0.03 | 0.3 | 0.1 | 1 | 0.01 | ||
| GSK2837808A | 10 | 10 | 10 | 10 | 10 | 10 | ||
| GZD824 | 0.3 | 0.3 | 0.01 | 1 | 0.1 | 0.1 | ||
| Flavopiridol | 0.1 | 0.03 | 0.03 | 3 | 0.1 | 1 | ||
| 5-Fluorouracil | 10 | 3 | 3 | 3 | 10 | 1 | ||
| Halofuginone | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | ||
| NCT-503 | 10 | 10 | 10 | 10 | 10 | 10 | ||
| PLX8394 | 10 | 3 | 3 | 3 | 3 | 1 | ||
| Rapamycin | 10 | 10 | 10 | 10 | 10 | 10 | ||
| SCH772984 | 1 | 0.3 | 0.3 | 0.1 | 3 | 0.1 | ||
| Seliciclib | 10 | 3 | 3 | 3 | 10 | 3 | ||
| Selumetinib | 10 | 3 | 0.03 | 0.3 | 10 | 0.3 | ||
| Thapsigargin | 0.01 | 0.001 | 0.003 | 0.001 | 0.1 | 0.001 | ||
| V-9302 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| YH16899 | 10 | 10 | 10 | 10 | 10 | 10 |
Table 4.
Synergy between TAP20 and experimental compounds. Values of coefficient of drug interaction (CDI) are shown. Antagonism (>1), additive (1), synergistic (<1). Bold values are statistically significant (p<0.05). For each combination n = 18 replicates were analysed, except MDA-MB-231/THZ-1 (n = 6) and SKOV3/THZ-1 (n = 12).
Table 4.
Synergy between TAP20 and experimental compounds. Values of coefficient of drug interaction (CDI) are shown. Antagonism (>1), additive (1), synergistic (<1). Bold values are statistically significant (p<0.05). For each combination n = 18 replicates were analysed, except MDA-MB-231/THZ-1 (n = 6) and SKOV3/THZ-1 (n = 12).
| Compound | MDA-MB-231 | MDA-MB-468 | HPAFII | Panc02.03 | SKOV3 | OVCAR3 |
|---|---|---|---|---|---|---|
| Agerafenib | 1.21 | 0.96 | 0.97 | 1.07 | 1.49 | 1.57 |
| AMG PERK44 | 0.88 | 0.94 | 0.66 | 0.97 | 1.07 | 1.3 |
| AZD3965 | 0.77 | 0.79 | 0.76 | 1.05 | 0.96 | 0.84 |
| Bay876 | 0.97 | 0.63 | 0.73 | 0.73 | 0.87 | 0.72 |
| Bortezomib | 0.79 | 0.82 | 0.62 | 0.83 | 0.89 | 0.91 |
| Bumetanide | 0.83 | 0.81 | 0.86 | 0.99 | 0.97 | 0.93 |
| CB-839 | 0.86 | 0.9 | 0.89 | 1.03 | 0.97 | 0.97 |
| CB-5083 | 0.85 | 0.89 | 0.51 | 0.96 | 1.17 | 0.81 |
| Cetuximab | 1.11 | 0.77 | 1.06 | 0.97 | 0.99 | 0.91 |
| Danusertib | 0.85 | 0.65 | 0.23 | 0.67 | 0.65 | 1.53 |
| GSK2837808A | 0.85 | 0.87 | 0.78 | 1.03 | 0.99 | 0.99 |
| GZD824 | 0.82 | 0.86 | 0.87 | 0.91 | 1.5 | 1.1 |
| Flavopiridol | 0.24 | 0.96 | 0.4 | 1.06 | 0.8 | 0.56 |
| 5-Fluorouracil | 0.7 | 0.95 | 0.71 | 0.87 | 0.94 | 0.51 |
| Halofuginone | 0.78 | 0.88 | 0.53 | 0.82 | 0.73 | 0.93 |
| NCT-503 | 0.68 | 0.88 | 0.74 | 0.86 | 0.95 | 1.11 |
| PLX8394 | 0.91 | 0.6 | 0.88 | 0.88 | 1.16 | 0.97 |
| Rapamycin | 1.29 | 1.05 | 1.56 | 0.97 | 1.57 | 2.38 |
| SCH772984 | 0.23 | 1.4 | 0.29 | 0.86 | 0.53 | 0.88 |
| Seliciclib | 0.47 | 0.85 | 0.51 | 0.96 | 0.87 | 0.88 |
| Selumetinib | 0.3 | 0.63 | 1.21 | 0.7 | 0.71 | 0.63 |
| Thapsigargin | 0.87 | 0.95 | 0.57 | 1.01 | 0.69 | 0.87 |
| THZ-1 | 0.71 | n.d. | 0.12 | n.d. | 0.4 | n.d. |
| V-9302 | 0.63 | 0.96 | 0.44 | 0.84 | 0.93 | 0.77 |
| YH16899 | 0.9 | 0.75 | 0.55 | 0.94 | 0.93 | 0.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



