
Submitted:
03 September 2023
Posted:
05 September 2023
You are already at the latest version
Abstract
Cancer metastasis accounts for approximately 90% of cancer deaths, and elucidating markers in metastasis is the first step in its prevention. To characterize metastasis marker genes of breast cancer (MGs), XGBoost models that classify metastasis status were trained with gene expression profiles from TCGA. Then, a metastasis score (MS) was assigned to each gene by calculating the inner product between the feature importance and AUC performance of the models. As a result, the 54, 202, and 357 genes with the highest MS were characterized as MGs by empirical P-value cutoffs of 0.001, 0.005, and 0.01, respectively. The three sets of MGs were compared with those from existing metastasis marker databases, which provided significant results in most comparisons. We noticed that the set of MGs with the median EP cutoff showed better performance than the other two sets, suggesting the importance of the cutoff used in determining MGs. They were also significantly enriched in biological processes associated to breast cancer metastasis. The MGs that could not be identified by statistical analysis (e.g., GOLM1, ELAVL1, UBP1, and AZGP1) as well as the MGs with the highest MS (e.g., ZNF676, FAM163B, LDOC2, IRF1, and STK40) were verified via the literature. Additionally, we checked how close the MGs are located to each other in the protein–protein interaction networks. We expect that the characterized markers will help understand and prevent breast cancer metastasis.
Keywords:
metastasis marker
; gene expression
; machine learning
; XGBoost
; breast cancer
; feature importance.
1. Introduction
Cancer metastasis is one of the main causes of cancer mortality, accounting for approximately 90% of cancer deaths [1]. Metastatic cancers go through four steps (i.e., detachment, migration, invasion, and adhesion) and show different characteristics from primary cancers, which makes the treatment of metastasis much more challenging [2]. Drugs chosen to treat primary cancers are almost never effective against metastatic cancers [3]. Therefore, it is important to prevent primary cancer from progressing to the metastatic stages.
The identification of genes that play key roles in metastasis will be the beginning of its prevention. In many previous studies, differentially expressed genes (DEGs) were selected and utilized as the main strategy to identify metastasis markers. For example, Chen’s group determined 97 DEGs between primary lung cancers and lung cancer metastasized to the brain, and their involved biological functions and signaling mechanisms were identified [4]. In addition, 664 DEGs were identified by analyzing transcriptome profiling in matched breast cancer and lymph node metastatic tissues of seven patients [5]. Wei’s group elucidated 472 DEGs involved in the metastasis of renal cell carcinoma by examining the expression profiling for renal cell carcinoma patients with and without metastasis [6].
Utilizing machine learning models could also be a good alternative to the DEG approaches for characterizing metastasis markers. This is because cancer metastasis, as well as cancer itself, is intricately related to multiple biological events and numerous factors, and a machine learning model is able to deal with multiple factors in a combinatorial manner [3]. However, to date, few machine learning models have been developed for characterizing metastasis markers. One of them is Metri’s work, which identified genes that discriminate metastatic from primary melanoma with AdaBoost machine learning models [7]. Wei’s group constructed support vector machines to identify marker genes associated with metastasis for cutaneous melanoma based on expression profiles [8]. In addition, Burton and colleagues compared seven kinds of machine learning models that predict metastasis outcome in breast cancer patients [9]. While exploring several related works, we noticed one of the challenges of using a machine learning approach is the difficulty of determining a specific number of metastasis markers [3].
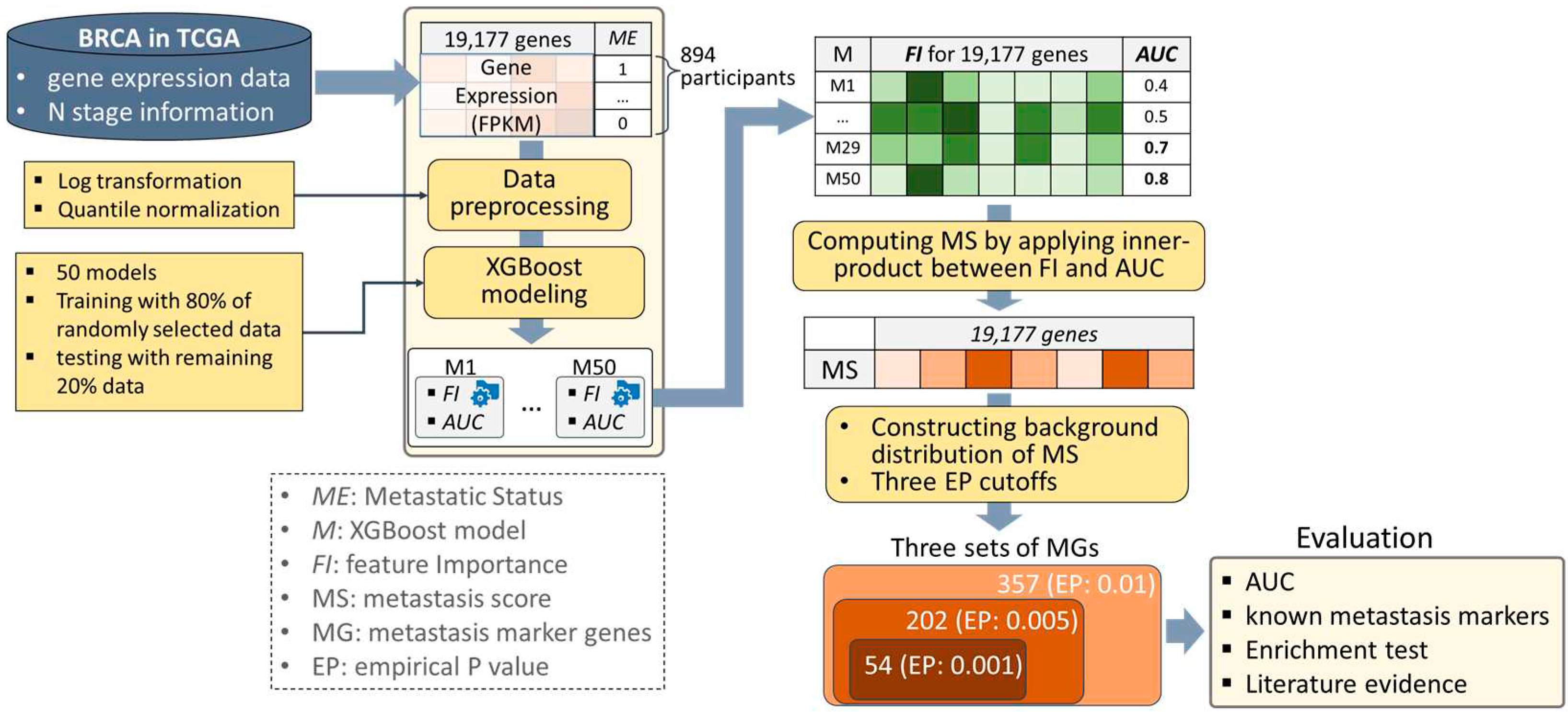
In this study, we devised an algorithm that specifies a set of significant metastasis markers based on the trained machine learning models. Specifically, a scoring function was designed that calculates the inner product between the feature importance and AUC performance of the trained models. In this study, breast cancer was selected as the cancer to be analyzed, which is the most diagnosed cancer in the world [10] and has the most samples in the TCGA database. The eXtreme Gradient Boosting (XGBoost) models were trained with expression profiles of breast cancer from TCGA (BRCA), and a metastatic score (MS) was assigned to each gene by applying the devised scoring function. Then, their significance was determined using an empirical P-value (EP) that was obtained by comparing it to the background distribution of the MS. As a result, three sets of MGs were characterized with three kinds of EP cutoffs, which are 0.001, 0.005, and 0.01. The results were evaluated in four ways, including 1) measuring AUCs of the models built using only the characterized MGs, 2) comparing them with known metastasis markers, 3) performing an enrichment test on processes associated with metastasis, and 4) exploring evidence from the literature. The strategy overview is depicted in Figure 1.
Figure 1.
A strategy overview. We prepared gene expression and metastasis information of the 894 breast cancer (BRCA) participants obtained from TCGA. After two kinds of data preprocessing steps, XGBoost models classifying metastatic status were trained 50 times, which produced a matrix consisting of feature importance (FI) as well as AUC performance of the 50 models. A metastasis score (MS) was assigned to each gene by calculating the inner product between FI and AUC, and their significance was determined by empirical P-value (EP) with background distribution of MS. Three sets of MGs were determined by three different EP cutoffs (i.e., 0.001, 0.005, and 0.01), and they were evaluated in four ways, including measuring AUCs, comparing them with known metastasis markers, performing enrichment test on processes associated with metastasis, and exploring evidence in the literature.
Figure 1.
A strategy overview. We prepared gene expression and metastasis information of the 894 breast cancer (BRCA) participants obtained from TCGA. After two kinds of data preprocessing steps, XGBoost models classifying metastatic status were trained 50 times, which produced a matrix consisting of feature importance (FI) as well as AUC performance of the 50 models. A metastasis score (MS) was assigned to each gene by calculating the inner product between FI and AUC, and their significance was determined by empirical P-value (EP) with background distribution of MS. Three sets of MGs were determined by three different EP cutoffs (i.e., 0.001, 0.005, and 0.01), and they were evaluated in four ways, including measuring AUCs, comparing them with known metastasis markers, performing enrichment test on processes associated with metastasis, and exploring evidence in the literature.
2. Materials and Methods
2.1. Data Preparation
Each sample of breast cancer in the TCGA database (BRCA) is given ‘ajcc_pathologic_m’ information, indicating metastasis to other organs [11]. However, only 22 samples from the breast cancer dataset (i.e., 2% of the total samples) were classified as having a metastasis status, which is too small to use in this study. Thus, we decided to use ‘ajcc_pathologic_n’ information instead, which indicates whether cancer is metastatic in nearby lymph nodes. There are four kinds of N stages in ‘ajcc_pathologic_n’ information, i.e., N0, N1, N2, and N3. N0 indicates that the cancer has not spread to nearby lymph nodes, and N1, N2, and N3 indicate that the cancer has spread to nearby lymph nodes, where higher numbers indicate a higher number of lymph nodes affected by cancer. N1 is also called the micrometastasis stage, and N2 and N3 are called the macrometastasis stages [12]. In this study, N0 is referred to as M0, i.e., nonmetastatic status, and N1, N2, and N3 are referred to as M1, i.e., metastatic status. Next, FPKM expression profiles for all BRCA samples were collected and processed by using the TCGAbiolinks R package [13], and they were integrated with the metastatic information.
The expression profiles of more than 40k RNAs in the TCGA database include not only coding genes but also noncoding RNAs, and too many features in machine learning not only increase computational efforts but also degrade performance due to noise and redundancy [14,15]. Thus, we decided to use 19,177 genes reported in the Cancer Cell Line Encyclopedia (CCLE) [16] as features of machine learning. As a result, we obtained expression profiles of 19,177 genes from 894 (333 M0 and 561 M1) samples.
2.2. Data Preprocessing
The gene expression matrix was preprocessed using the following four techniques sequentially. First, gene expression was averaged per gene by mapping the ensemble IDs to gene symbols. Second, gene expression was averaged per participant by mapping the TCGA barcodes to participants. Third, log transformation was performed on every expression value to minimize outlier effects. Fourth, quantile normalization was applied to allow for an equal expression distribution for each participant (see Figure S1 for the boxplots of the preprocessed expressions).
2.3. XGBoost Modeling
Out of the various machine learning models, we decided to use eXtreme Gradient Boosting (XGBoost) to predict metastatic status, which is an ensemble model that has been intensively employed and has outstanding performance in biology fields [17,18]. An ensemble model combines several base models that retain their good performance individually and exhibit diversities, and the XGBoost model uses a gradient boosting algorithm that trains the base model to reduce residuals passed on from the previous base model [19]. The XGBoost models were established with the Python XGBoost package (https://xgboost.readthedocs.io/en/stable/) with their default parameters.
When an XGBoost model is being trained, feature importance (FI) scores are generated. An FI for a certain feature presents the amount of decrease in performance when it is perturbed, which is assigned to every feature while a model is being trained. A feature with a high FI indicates that it plays an important role in discriminating the class. When an XGBoost model is tested, the Area Under the ROC Curve (AUC) is generated, which is one of the most used performance metrics in machine learning approaches [20].
2.4. Characterizing Metastasis Marker Genes
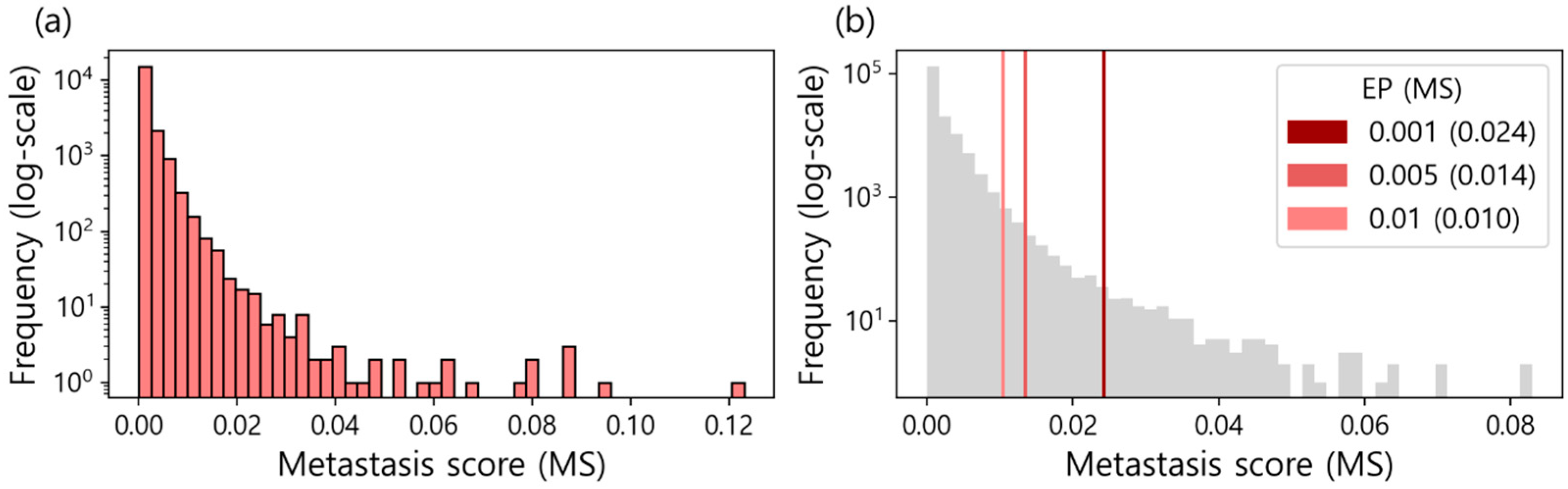
In this study, metastasis marker genes (MGs) are considered as genes with the highest FI in the trained XGBoost models classifying metastasis status. It is because that a high FI indicates that a gene plays an important role in discriminating metastasis status. To compute FIs, the 50 XGBoost models were constructed, each of which was trained with 80% of the randomly selected data and tested with the remaining 20% of the data. Here, we noticed that the AUCs of the 50 models varied from 0.494 to 0.692 (Figure S2). We believe that the FI of a model with a high AUC should receive a higher score than the FI of a model with a low AUC, even for the same FI. Thus, a scoring function to generate metastasis score (MS) was designed, as shown in Algorithm 1, which calculates the inner product between the FIs and AUCs of the 50 models. Here, the AUC is used as the weight of the FI. By applying the scoring function, we generated a set of MSn, where n=1 to 19,177. The detailed results are presented in Table S1, and their distribution is described in Figure 2a.
<Algorithm 1>
, and
where
Significance cutoffs of the MS to determine MGs were not available. Thus, MGs were determined with an empirical P-value (EP) that was obtained by constructing the background distribution. To this end, <algorithm 1> was performed ten times on the data with the shuffled metastasis status, allowing the background distribution to consist of 191,770 MSs (Figure 2b). For characterizing MGs, we decided to use three kinds of EPs (i.e., 0.001, 0.005, and 0.01) as significance cutoffs, whose corresponding MSs are 0.024, 0.014, and 0.010, respectively (Table S2 and Figure 2b).
Figure 2.
(a) The distribution of metastasis score (MS) for 19,177 genes. (b) The background distribution of MS. It was constructed by training XGB models on the data with the shuffled metastasis status. The three kinds of empirical P-value (EP) cutoffs (i.e., 0.001, 0.005, and 0.01) were used to characterize metastasis marker genes (MGs).
Figure 2.
(a) The distribution of metastasis score (MS) for 19,177 genes. (b) The background distribution of MS. It was constructed by training XGB models on the data with the shuffled metastasis status. The three kinds of empirical P-value (EP) cutoffs (i.e., 0.001, 0.005, and 0.01) were used to characterize metastasis marker genes (MGs).
3. Results and Evaluations
3.1. Metastasis Marker Genes
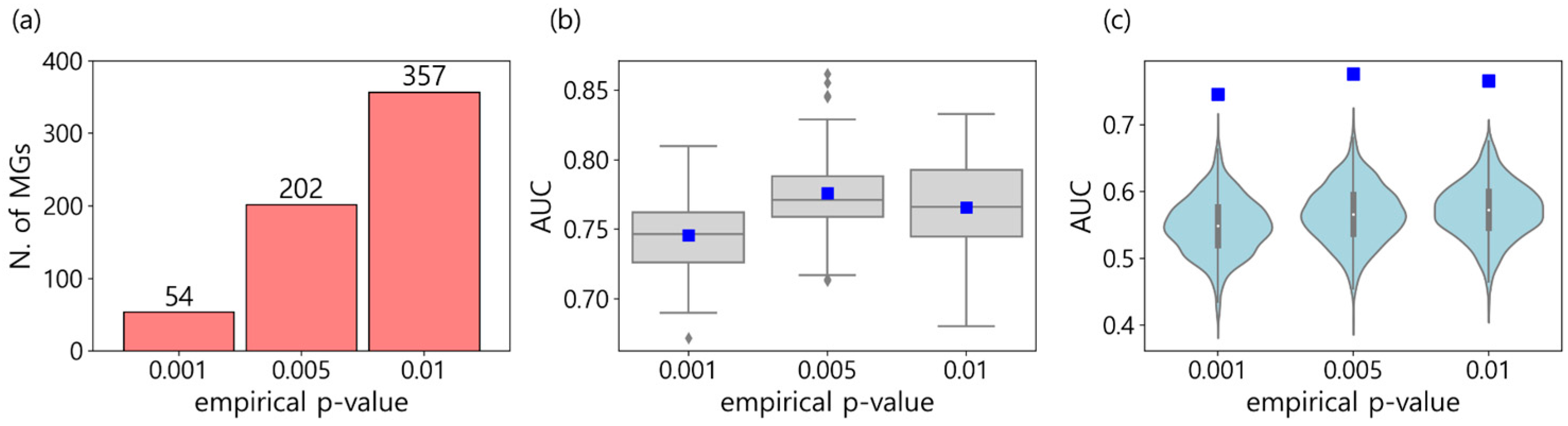
As a result, three sets containing 54, 202, and 357 MGs were characterized by EP cutoffs of 0.001, 0.005, and 0.01 (Figure 3a and Table S2). To evaluate the performance of the MGs, XGB models were trained by using only the MGs of each set. For each set, the 50 XGB models were generated with 80% of the randomly sampled training data, and their AUCs are depicted in Figure 3b as a box plot. The mean AUCs were 0.746, 0.776, and 0.766 for each set of MGs with EPs of 0.001, 0.005, and 0.01. We noticed that all of these AUCs are higher than 0.593, which is the mean AUC obtained by using all 19,177 genes from the CCLE (Figure S2). In addition, the models using 202 genes (EP cutoff: 0.005) performed better than the models using 357 genes (EP cutoff: 0.01), which include the 202 genes with an EP cutoff of 0.005. This is consistent with the assertion that too many features in machine learning not only increase computational efforts but also degrade performance due to noise and redundancy [14,15].
Furthermore, we investigated how significant the AUCs of the MGs were when compared to those of randomly selected genes. To do this, for each of the three sets of MGs, 1,000 XGBoost models were constructed with randomly selected genes, using as many as the corresponding MGs. Their AUCs are depicted as boxplots in Figure 3c. We noticed that the AUC of the MGs was located at the top in all three comparisons, which indicates that the MGs are not randomly selected but have more capabilities in classifying the two kinds of metastasis statuses.
Figure 3.
(a) The number of metastasis marker genes (MGs) for three empirical P-value (EP) cutoffs. The list of genes for each set is depicted in Table s2. (b) The AUC boxplots of the XGB models trained with only the characterized MGs in each of the three sets. The mean AUC is presented as a blue square (i.e., 0.746, 0.776, and 0.766 for EP cutoff of 0.001, 0.005, and 0.01). (c) The AUC distributions of the XGB models trained with randomly selected genes numbering as many as the characterized MGs in each of the three sets. Blue squares are mean AUCs in Figure 3b.
Figure 3.
(a) The number of metastasis marker genes (MGs) for three empirical P-value (EP) cutoffs. The list of genes for each set is depicted in Table s2. (b) The AUC boxplots of the XGB models trained with only the characterized MGs in each of the three sets. The mean AUC is presented as a blue square (i.e., 0.746, 0.776, and 0.766 for EP cutoff of 0.001, 0.005, and 0.01). (c) The AUC distributions of the XGB models trained with randomly selected genes numbering as many as the characterized MGs in each of the three sets. Blue squares are mean AUCs in Figure 3b.
3.2. Comparing with known metastasis markers
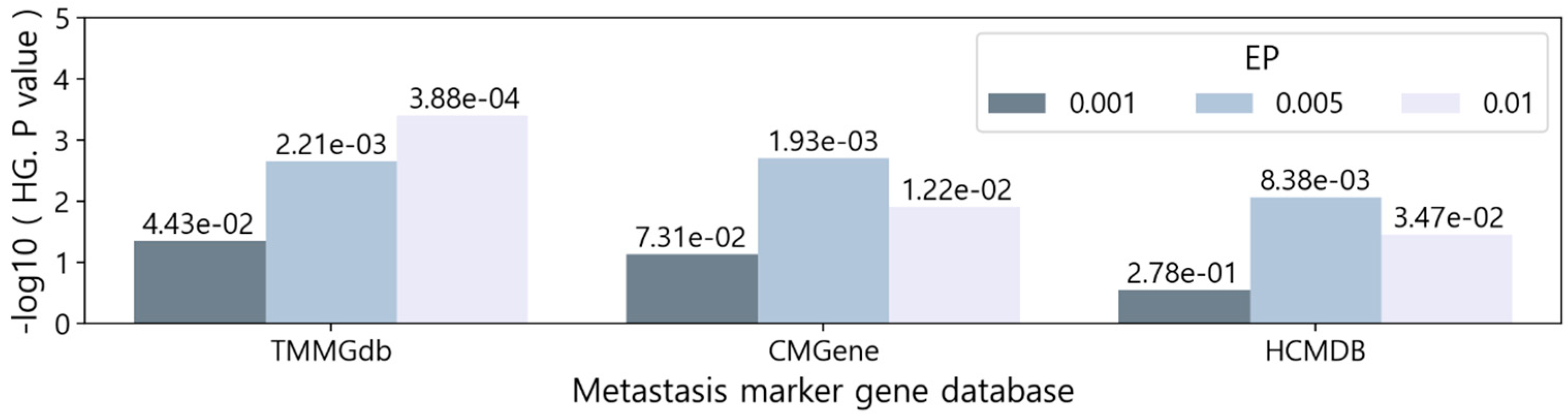
We evaluated the characterized MGs by comparing them with known metastasis markers. To do this, we obtained access to three metastasis marker databases, which are the Tumor Metastasis Mechanism-associated Gene Database (TMMGdb [21]), Cancer Metastasis-related Genes database (CMGene [22]), and Human Cancer Metastasis Database (HCMDB [23]). The TMMGdb contains 3.2K genes collected with the text-mining tool BioBERT, taking into account the terms of metastatic subprocesses. The CMGene database includes 2K genes integrated by applying a series of text-mining techniques followed by manual curation. The HCMDB contains 1.9K genes obtained by collecting metastasis-related expression profiles and analyzing them. The gene lists provided by the three databases are presented in Table S3.
The three sets of MGs were statistically compared to the genes in each of the three databases by applying hypergeometric tests. As a result, seven of the nine comparisons produced significant results (p-value < 0.05), and there were three significant comparisons with a stricter p-value cutoff (p-value < 0.005) (Figure 4 and Table S4). On average, the level of significance was high in the order of EP 0.005, 0.01, and 0.001, which is the same order as the AUC result in Figure 3b.
Figure 4.
Hypergeometric test (HG) results between the characterized MGs and the genes in the three metastasis marker gene databases. EP: empirical P-value.
Figure 4.
Hypergeometric test (HG) results between the characterized MGs and the genes in the three metastasis marker gene databases. EP: empirical P-value.
3.3. Enrichment tests on metastasis-related processes
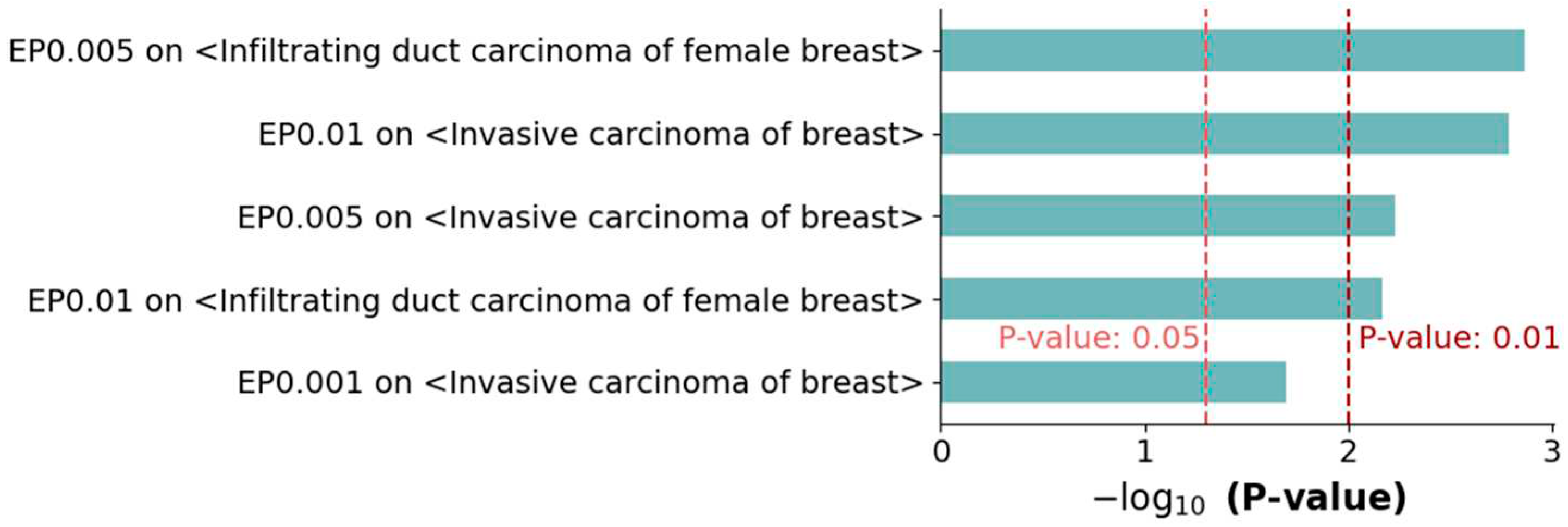
DisGeNET is a discovery platform containing one of the largest publicly available collections of genes associated with human diseases, which integrates data from GWAS catalogs, animal models, and the scientific literature. DisGeNET contains 1,134,942 gene–disease associations that have been identified between 21,671 genes and 30,170 diseases [24]. For each of the three sets of MGs, the MGs were evaluated by performing enrichment tests on the two breast cancer metastatic terms in DisGeNET, i.e., infiltrating duct carcinoma of the female breast and invasive carcinoma of the breast. As a result, five of the six comparisons presented with significant consequences (p-value < 0.05) (Figure 5 and Table S5), and four comparisons showed more significant results (p-value <0.01). Similar to the previous results, the set of MGs with an EP of 0.005 showed better performance than the other two sets.
Figure 5.
Enrichment tests on metastatic terms in the DisGeNET database. The two breast cancer metastasis-related terms in DisGeNET, infiltrating duct carcinoma of female breast and invasive carcinoma of breast, were compared to the three sets of MGs. This produced significant results in five out of six enrichment tests (P-value < 0.05).
Figure 5.
Enrichment tests on metastatic terms in the DisGeNET database. The two breast cancer metastasis-related terms in DisGeNET, infiltrating duct carcinoma of female breast and invasive carcinoma of breast, were compared to the three sets of MGs. This produced significant results in five out of six enrichment tests (P-value < 0.05).
3.4. Literature Evidence
3.4.1. Metastasis marker genes with the highest Metastasis score
The gene with the first highest MS is ZNF676, which is closely associated with the PRMT1 gene that is involved in breast cancer metastasis [25]. The gene with the second highest MS is FAM163B, which has not yet been elucidated, but its paralog FAM163A (also known as NDSP) is associated with an increased risk for the development of cancer metastasis in bone marrow [26]. The gene with the third highest MS is LDOC2, whose function is a tumor suppressor that inhibits proliferation and metastasis [27]. The LDOC2 gene regulates WNT5A expression, which promotes breast cancer cell migration [28]. The gene with the fifth highest MS is IRF1, which plays a dual role in the process of the epithelial-to-mesenchymal transition (EMT). In more detail, the suppression of IRF1 in mammary epithelial cells increases the expression of mesenchymal factors; however, conversely, the inhibition of IRF1 during a TGFβ-induced EMT prevents a mesenchymal transition [29]. The gene with the eighth highest MS is STK40, whose depletion decreases cell viability and colony formation in triple-negative breast cancers (TNBCs). The knockdown of STK40 also delays tumor growth in in vivo experiments [30].
3.4.2. Metastasis marker genes not identified by statistical analysis
The 202 MGs identified with an EP cutoff of 0.005 produced the best performance in the multiple evaluations among the three sets of MGs. Among them, we noticed that the 75 genes failed to show statistical significance when a T-test was performed (P-value>0.1), which means that they could not be revealed by statistical analysis (refer to Table S6). We examined the literature evidence showing that they are also associated with breast cancer metastasis. For example, GOLM1 (EP: 0.0005, T-test P-value: 0.103) induces the EMT and promotes the proliferation, migration, and invasion of breast cancer cells. Also, the overexpressing of GOLM1 markedly promotes the metastasis of breast cancer cells in vivo [31]. ELAVL1 (EP: 0.0016, T-test P-value: 0.805) was found to be modulated by MUC16, which promotes triple-negative breast cancer lung metastasis [32]. UBP1 (EP: 0.0034, T-test P-value: 0.546) consists of the CP2 transcription factor with TFCP2, which is known to be essential for the EMT process [33]. AZGP1 (EP: 0.0014, T-test P-value: 0.209) is known to reduce cell proliferation and promote invasion, and it also found to be a blocker of the EMT induced by the TGFbeta1-ERK2 pathway [34].
4. Discussion
We paid attention to the 75 MGs presented in Section 3.4.2, which were not statistically significant but were identified as the MGs. One of the reasons for being characterized as MGs despite this small difference might be their biological interactions in complex molecular networks, which are combinatorial rather than individual. Thus, we checked how close the MGs are located to each other in the protein–protein interaction networks. To this end, protein–protein interaction (PPI) networks were constructed from the BIOGRID database [35] by integrating protein interactions associated with ‘affinity chromatography technology’ or the ‘two hybrid’ detection method, resulting in 597,215 interactions among 19,160 nodes. Then, for each of the 75 MGs, the number of adjacent MGs was calculated for the PPI network. As a result, 15 of the 75 genes we found to be involved in one or more of the adjacent MGs, such as ELAVL1 (n: 14), KRT38 (n: 5), and UBP1 (n:3). Considering that the average number of adjacent MGs is 0.54, we can say that they have many adjacent MGs. The detailed information is depicted in Table S6. For some of them, the association with breast cancer metastasis was validated with the literature evidence presented in Section 3.4.2.
The set of MGs with an EP of 0.005, which is the middle value of the three EP cutoffs, showed a higher AUC score as well as better performance in all evaluations performed in this study than the other two sets. This means that, in machine learning approaches, the selection of an appropriate number of features will produce significant results. This suggests how important the cutoff decision is when selecting a specific number of features.
In this study, XGBoost modeling was employed to characterize a set of breast cancer metastasis markers (MGs). A metastasis score was assigned to each gene by calculating the inner product between the FIs and AUCs of the trained models. Then, three sets of MGs were characterized by applying three empirical P-value (EP) cutoffs, and they were evaluated in several different ways. We noticed that the characterized MGs contain genes that could not be detected by T-tests, and we confirmed that they are also associated with breast cancer metastasis by verifying this information with that already published in the literature. We expect that the results of this study will be of great help in elucidating the mechanism of metastasis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Boxplots of the preprocessed gene expressions for randomly selected participants (left) and genes (right); Figure S2: The box plots of the 50 AUCs obtained from the 50 trained XGBoost models; Table S1: Metastasis scores computed with feature importance (FI) and AUC of the 50 XGB models; Table S2: The metastasis marker genes (MGs) determined by empirical P-value; Table S3: Lists of metastasis-associated genes obtained from TMMGdb, GMGene, and HCMDB databases; Table S4: Hypergeometric tests with the three metastasis marker databases (TMMGdb, CMGene, and HCMDB); Table S5: GSEA results applied to the breast metastatic-associated terms from the DisGeNET database; Table S6: T-test results and the number of adjacent MGs for the 202 MGs identified with EP cutoff of 0.005.
Author Contributions
Conceptualization, J.J.; methodology, J.J. and S.Y.; software, J.J.; validation, S.Y.; investigation, S.Y.; resources, J.J.; writing—original draft preparation, J.J.; writing—review and editing, J.J. and S.Y.; visualization, J.J. and S.Y.; supervision, S.Y.; project administration, S.Y.; funding acquisition, J.J. and S.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (NRF-2022R1C1C1008823) and by a grant from the Ministry of Food and Drug Safety given in 2021 (21162MFDS045).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article or Supplementary Materials. Python implementations are available at https://github.com/jmjung83/breast_cancer_metastasis_marker.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Dillekas, H., M.S. Rogers, and O. Straume, Are 90% of deaths from cancer caused by metastases? Cancer Med, 2019. 8(12): p. 5574–5576. [CrossRef]
- Guan, X., Cancer metastases: Challenges and opportunities. Acta Pharm Sin B, 2015. 5(5): p. 402–18. [CrossRef]
- Albaradei, S., et al., Machine learning and deep learning methods that use omics data for metastasis prediction. Comput. Struct. Biotechnol. J., 2021. 19: p. 5008–5018. [CrossRef]
- Chen, C., et al., Screening and evaluation of the role of immune genes of brain metastasis in lung adenocarcinoma progression based on the TCGA and GEO databases. J Thorac Dis, 2021. 13(8): p. 5016–5034. [CrossRef]
- Kim, G.E., et al., Differentially expressed genes in matched normal, cancer, and lymph node metastases predict clinical outcomes in patients with breast cancer. Appl Immunohistochem Mol Morphol, 2020. 28(2): p. 111–122. [CrossRef]
- Wei, W., et al., Identification of key genes involved in the metastasis of clear cell renal cell carcinoma. Oncol Lett, 2019. 17(5): p. 4321–4328. [CrossRef]
- Metri, R., et al., Identification of a gene signature for discriminating metastatic from primary melanoma using a molecular interaction network approach. Sci Rep, 2017. 7(1): p. 17314. [CrossRef]
- Wei, D., A multigene support vector machine predictor for metastasis of cutaneous melanoma. Mol Med Rep, 2018. 17(2): p. 2907–2914. [CrossRef]
- Burton, M., et al., Gene expression profiles for predicting metastasis in breast cancer: A cross-study comparison of classification methods. Sci. World J., 2012. 2012: p. 380495. [CrossRef]
- Tamar, G. and T. Vasil, THE BURDEN OF BREAST CANCER IN TBILISI IN 2015-2019. European journal of biomedical and life sciences, 2021(4): p. 27-33.
- Tomczak, K., P. Czerwińska, and M. Wiznerowicz, Review the cancer genome atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol., 2015. 2015(1): p. 68–77. [CrossRef]
- Liu, J., et al., An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell, 2018. 173(2): p. 400–416.e11.
- Colaprico, A., et al., TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic acids research, 2016. 44(8): p. e71-e71. [CrossRef]
- Abawajy, J., A. Darem, and A.A. Alhashmi, Feature subset selection for malware detection in smart IoT platforms. Sensors (Basel), 2021. 21(4): p. 1374. [CrossRef]
- Hughes, G., On the mean accuracy of statistical pattern recognizers. IEEE Trans. Inf. Theory, 1968. 14(1): p. 55–63. [CrossRef]
- Barretina, J., et al., The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 2012. 483(7391): p. 603-607. [CrossRef]
- Li, Y., et al., Putative biomarkers for predicting tumor sample purity based on gene expression data. BMC Genom., 2019. 20(1): p. 1021. [CrossRef]
- Pellegrino, E., et al., Machine learning random forest for predicting oncosomatic variant NGS analysis. Sci Rep, 2021. 11(1): p. 21820. [CrossRef]
- Chen, T. and C. Guestrin, Xgboost: A scalable tree boosting system, in Proceedings of the 22nd ACM SIGKDD international conference on knowledge discovery and data mining. 2016, Association for Computing Machinery: San Francisco, California, USA. p. 785–794.
- Bradley, A.P., The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern recognition, 1997. 30(7): p. 1145-1159. [CrossRef]
- Liu, H.-C., et al., TMMGdb-Tumor Metastasis Mechanism-associated Gene Database. Current Bioinformatics, 2023. 18(1): p. 63-75. [CrossRef]
- Liu, Y., et al., CMGene: A literature-based database and knowledge resource for cancer metastasis genes. Journal of Genetics and Genomics, 2017. 44(5): p. 277-279. [CrossRef]
- Zheng, G., et al., HCMDB: The human cancer metastasis database. Nucleic Acids Res, 2018. 46(D1): p. D950–955. [CrossRef]
- Piñero, J., et al., The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res, 2020. 48(D1): p. D845–855. [CrossRef]
- Papatsirou, M., et al., Identification of novel circular RNAs of the human protein arginine methyltransferase 1 (PRMT1) gene, expressed in breast cancer cells. Genes, 2022. 13(7): p. 1133. [CrossRef]
- Vasudevan, S.A., et al., Neuroblastoma-derived secretory protein is a novel secreted factor overexpressed in neuroblastoma. Molecular cancer therapeutics, 2009. 8(8): p. 2478-2489. [CrossRef]
- Keenan, A.B., et al., ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic acids research, 2019. 47(W1): p. W212-W224. [CrossRef]
- Yong, B.-C., et al., LDOC1 regulates Wnt5a expression and osteosarcoma cell metastasis and is correlated with the survival of osteosarcoma patients. Tumor Biology, 2017. 39(2): p. 1010428317691188. [CrossRef]
- Meyer-Schaller, N., et al., A dual role of Irf1 in maintaining epithelial identity but also enabling EMT and metastasis formation of breast cancer cells. Oncogene, 2020. 39(24): p. 4728-4740. [CrossRef]
- Maubant, S., et al., LRP5 regulates the expression of STK40, a new potential target in triple-negative breast cancers. Oncotarget, 2018. 9(32): p. 22586. [CrossRef]
- Zhang, R., et al., Golgi membrane protein 1 (GOLM1) promotes growth and metastasis of breast cancer cells via regulating matrix metalloproteinase-13 (MMP13). Medical science monitor: international medical journal of experimental and clinical research, 2019. 25: p. 847. [CrossRef]
- Chaudhary, S., et al., MUC16 promotes triple-negative breast cancer lung metastasis by modulating RNA-binding protein ELAVL1/HUR. Breast Cancer Research, 2023. 25(1): p. 1-15. [CrossRef]
- Zhao, Y., et al., A feedback loop comprising EGF/TGFα sustains TFCP2-mediated breast cancer progression. Cancer research, 2020. 80(11): p. 2217-2229. [CrossRef]
- Xu, M.-Y., et al., AZGP1 suppresses epithelial-to-mesenchymal transition and hepatic carcinogenesis by blocking TGFβ1-ERK2 pathways. Cancer letters, 2016. 374(2): p. 241-249. [CrossRef]
- Oughtred, R., et al., The BioGRID interaction database: 2019 update. Nucleic acids research, 2019. 47(D1): p. D529-D541. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



