
Submitted:
04 September 2023
Posted:
06 September 2023
You are already at the latest version
Abstract
A form of dementia distinct from healthy cognitive aging, Alzheimer's disease (AD) is a complex multi-stage disease that currently afflicts over 50 million people worldwide. Unfortunately, previous therapeutic strategies developed from murine models emulating different aspects of AD pathogenesis have failed. Consequently, researchers are now developing models that express several aspects of pathogenesis that better reflect the clinical situation in humans. As such, this review seeks to provide insight regarding current applications of mammalian models in AD research by addressing recent developments and characterizations of prominent transgenic models and their contributions to pathogenesis as well as discuss the advantages, limitations, and application of emerging models (hAβ-KI) that better capture genetic heterogeneity and mixed pathologies observed in the clinical situation.
Keywords:
Animal model
; Alzheimer’s Disease
; Amyloid β
; Tau Protein
; LOAD models
; Transgenic Murine Models
1. Introduction
Accounting for nearly 70% of all dementia cases with an average course of ten years, AD has the potential to overwhelm healthcare systems generating large care deficits. Unfortunately, such deficits are already prevalent in many healthcare systems requiring individuals to compensate (financial or otherwise) with limitations. For example, in 2017 and 2020, 16 and 11 million USA citizens provided 18.5 and 14 million (family members and health workers) hours of unpaid care, respectively [1]. However, even with continued contributions, by 2050, the USA AD care model will become untenable as predicted costs surpass $1 trillion/ annum. In addition to economic concerns, the negative impacts on other health sectors (mental) [2] are yet to be fully realized, with burnout and depression frequently reported among carers. Thus, the current directives for AD scientists and clinicians have shifted from developing end-stage treatments to ones that extend the early stages of AD, encouraging arresting actions and deficit reductions among patients and carers alike.
AD can present as early (<65 years) onset Alzheimer's disease (EOAD) or late (>65 years) onset Alzheimer's disease (LOAD). The disease itself has several stages and is characterized by three pathological hallmarks, extracellular amyloid beta (A) [3,4,5] plaques (dense and diffuse), intracellular neurofibrillary tangles (NFT), and cortical and hippocampal neurodegeneration [6,7,8,9,10]. The formation of extracellular A plaques starts with the sequential cleavage of amyloid precursor protein (APP) by a -secretase, -APP cleaving enzyme (BACE1), an aspartyl protease followed by γ-secretase cleavage (a protein complex composed of presenilin-1, nicastrin, APH-1, and PEN-2) [11,12,13,14,15,16,17] leading to the release of 3 protein fragments including Aβ peptides (Aβ40, Aβ42, extracellular space), the amyloid-intracellular domain (AICD) (cytoplasm) and soluble APP.
For the most part, the majority of Aβ peptides produced by γ-secretase end at amino acid 40 (Aβ40) and a minority at amino acid 42 (Aβ42). However, it was not until the discovery of familial mutations (1991) in the amyloid precursor protein (APP) and presenilin (PSEN1 and PSEN2) genes resulting in APP cleavage favouring the aggregation of amyloid-beta 42 (Aβ42), that the amyloid the cascade theory was proposed. In short, the initial theory followed a simple process: -amyloid deposition → tau phosphorylation and tangle formation → neuronal death. Since then [18], more than 300 mutations in APP, PSEN1 and PSEN2 genes have been identified [19]. Interestingly, the age of onset (AAO) and the severity of symptoms are markedly different between mutations. For example, carriers of pathogenic PSEN1 variants may present symptoms before 25 or after 60 years of age [20]. Moreover, numerous findings have emerged, suggesting tangle formations can precede amyloid deposition, and the degree of cognitive impairment correlates much better with NFT counts than amyloid plaque burden [21] challenging the original theory.
NFTs are composed of aggregated tau proteins, collectively called tauopathies, a major pathological feature of AD and other neurodegenerative conditions. Under normal conditions (homeostasis), tau is an unstructured axonal protein that binds and stabilizes microtubules [22]. The protein can present as a three-repeat (3R) or four-repeat (4R) isoform as exon 10 on the microtubule-associated protein tau gene (MAPT) is subject to alternative splicing [23]. The resultant isoforms can undergo oligomerization and hyperphosphorylation, contributing to the formation of intracellular NFT in the axons [24,25]. Aβ and tau accumulation in the brain promote several pathophysiological (breakdown of myelin sheath around neurons, astrocytic and microglial inflammation) changes affecting AD patients' cognitive function. In addition, Aβ and tau accumulation remains central to the histopathological post-mortem staging (Braak) systems, considered the gold standard in AD diagnosis.
These genetic, biochemical and neuropathological hallmarks warrant the creation and validation of AD animal models. Beyond the dominant forms of AD, mutations in apolipoprotein E (APOE) and the triggering receptor expressed on myeloid cells 2 (TREM2) are the most common genetic risk factors for LOAD.
Preclinical investigations using murine models are an essential part of the arsenal of tools to probe specific disease mechanisms and evaluate therapeutic efficacy and diagnostic strategies within a living organism. Of the 214 murine models registered between 1988-2022, 212 were transgenic, one naturally occurring, and one chromosomally induced [26]. When selecting a model from the register, matching the murine model (laboratory strain) to the experimental question (for ex., stage of the disease) under study remains the primary consideration [27]. The current plethora of available Tg models permits researchers to explore up to six pathological hallmarks, including plague and tangle formations, neuronal loss, synaptic loss, gliosis, cognitive impairment, and changes in long-term potentiation / long-term depression (LTP/LTD) but generally, not all in the same model. In this review, the advantages and limitations of prominent (based on the amyloid cascade hypothesis) Tg and non-transgenic models engineered to emulate the major pathological hallmarks of EOAD, then LOAD, are assessed. In addition, diagnostics (behavior, biomarkers, neuro imaging) used to evaluate the therapeutic potential of a selection of models are discussed.
PDAPP
The first amyloid model was presented in 1995 [28], expressing high levels of human APP with V717F Indiana mutation. These transgenic mice progressively developed pathological hallmarks of AD as the mutation increases the ratio of Aβ42/Aβ40. These Tg mice exhibit various features resembling human AD-like extracellular amyloid fibrils organized in plaques, degenerative subcellular changes, dystrophic neuritis, apoptosis, gliosis, and synaptic loss that spread progressively from the hippocampus to the cortex [28,29]. These mice also presented age-related memory loss (with highest at 12-15 months of age) which is the main feature in Alzheimer's patients [30,31,32]. Aβ plaques are observed at 6 months with no tau tangles. Synaptic loss presents after 8-9 months, whereas no neuronal loss is seen. Cognitive impairment by three months and gliosis was seen by 6 months [27].
Tg2576
In 1996 Karen Hsiao and colleagues developed the Tg2576 transgenic mouse. The model carries a double Swedish mutation K670N and M671L and is synomyous with early-onset familial AD [27,33]. Utilizing a prion promoter (PrP) that non-specifically drives hAPP gene the model can demonstrate up to a 5-fold increase in APP production, with amyloid pooling in the heart, kidney, and lungs in the first 3 months. Synaptic loss is observed within 4-5 months [34,35]. This is followed by reduced cerebral blood flow by 8 months, spatial working memory deficits by 5 months, and CAA by 9-10 months. Plaque formation is observed at 11-13 months in the temporal, frontal, entorhinal cortices, hippocampus, pre-subiculum and cerebellum. No profound cognitive impairment was seen [36] even though these mice can sometimes display hyperphosphorylated tau in the last months of life [27].
APP23
Sturchler-Pierrat et al. [37] described the APP23 mouse in 1997. Akin to the previous model, these mice model harbour the Swedish double-mutation on the hAPP gene and utilize a neuronal instead of a prion promoter to induce hAPP overexpression [37]. As a result, soluble Aβ blood levels remain stable in APP23 mice over time [38], presenting Aβ plaques, and spatial memory deficits earlier than Tg2576 mice at 3-6 months. Interestingly [39] when the APP23 APOE allele is silenced, vascular damage and oxidative stress occur; conversely, APOE4 overexpression in Tg2576 mice induces oxidative stress and vascular injury [40].
At six months, vascular abnormalities are observable in APP23, with integrin's overexpression on the platelets' surface, resulting in collagen and fibrinogen adhesion and vessel occlusion. These are accompanied by elevated IgG levels (1.2-fold) in neurons, blood vessels, and glial cells, which are absent in age-matched wild-type mice, implying that enhanced IgG synthesis may be unique to this model. A reduction in cerebral blood flow at 11 months is observed, with microhaemorrhage and CAA presentation around 18 months [41]. At the same time, other vascular irregularities, such as twisted or studded blood vessels with protrusions and reduced micro-vascular flow in the frontal and temporal cortices in APP23 mice, are evident [40]. Multiple studies indicate CAA and cerebral blood flow changes precede vascular injury [38,42]. Therefore, this model may be considered suitable for cerebral vascular investigations.
APP(V717I) & J20 models
Harbouring a common familial APP mutation (London V717I) this 1999 model also utilizes a neuronal Thy1 promoter to drive hAPP overexpression which leads to spatial memory deficits by 6 months, Aβ plaque formations by 10 months, CAA by 15 months, and micro haemorrhages by 25-30 months [42,43]. However, a recent clinical report [44] regarding the neuropathologic characterization of an APP V717I carrier, demonstrated that genetically determined ADAD can show significant phenotypic variability including widespread parenchymal beta-amyloid (Aβ) deposition, severe cerebral amyloid angiopathy (CAA) and predominant age-related TDP-43 proteinopathy (LATE).
In 2000, the J20 model carrying a hAPP gene with three mutations (Indiana (V717F) & Swedish double-mutation (K670N and M671L)) and a PDGF-β promoter [45] was introduced. J20 exhibits elevated soluble Aβ brain levels after 1 month, neuronal loss (3 months), spatial memory impairment (4months) and Aβ1-42 plaque morphologies (medium and diffuse) at 5-7months [46]. Micro haemorrhages, CAA and reduced flow ensue at 11 months, interestingly [47,48] the J20-hAPP mouse recently showed enhanced baseline blood volume and saturation of all vascular compartments under hyperoxic conditions suggesting it could be used to study potential treatments or early preventive actions such as saturated oxygen or moderate hypoxic conditioning.
TgCRND8 model
Akin to J20, the TgCRN8 model also harbours’ the hAPP gene with Swedish and Indiana mutations utilizing a prion promoter instead of PDGF-β resulting in hAPP overexpression in neuronal tissue [49,50]. TgCRND8 mice exhibit increases in the Aβ42/Aβ40 ratio, leading to accumulation of Aβ amyloid plaques, intraneuronal Aβ, soluble oligomeric Aβ (oAβ), and insoluble fibrillar Aβ (fAβ). TgCRND8 mice develop spatial deficits at 3 months followed by Aβ plaques at 5months, significant (>30%) neuronal loss at 6 months, and tauopathies between 7-12 months [51]. CAA and reduced cerebral blood flow develop later in J20mice than in TgCRND8 mice (11months vs. 6 months) due to impaired Aβ clearance [52]. From 12 months onwards, TgCRND8 mice develop capillary CAA. TgCRND8 mice exhibit a wide spectrum of including Aβ plaques, fibrils, and oligomers exhibiting greater neuronal toxicity than other models (Dutch (E693Q) that solely favour the overproduction of intraneuronal oligomers.
TgSwDI model
Introduced in 2004 [53] the TgSwDI mouse AD model harbours an hAPP gene segment with three mutations (Swedish (Lys670→Asn/ Met671→Leu), ((E693Q) Dutch Glu693→Gln), and Iowa (D694N (Asp694→Asn)) and the promoter Thy1.2[53]. Tg-SwDI mice express transgenic human AβPP only in the brain, but at levels below those of endogenous mouse AβPP. Moreover, the model shows that the overexpression of human AβPP is not necessary for the development of Aβ pathology in the mouse brain. Another unique characteristic of this model is the accumulations of numerous diffuse nonfibrillar plaques and fibrillar deposits in the parenchymal tissue. In TgSwDI mice, reduced cerebral blood flow, and memory deficits appear as early as 2-3 months, resulting from a robust accumulation of insoluble Aβ40 and Aβ42 [54]. In addition, further investigations suggest that said mutations drive Aβ deposition in the cerebrovasculature, leading to type 1 CAA and vascular fragility with cerebral haemorrhages at 6 months [55] followed by extensive deposition at year’s end.
Primarily fibrillar, the deposition is closely related with a severe inflammatory reaction, knocking out the APOE allele causes a shift from microvascular to parenchymal Aβ deposits without changing the overall Aβ burden indicating that APOE is the driving force for CAA and microhaemorrhages in this model [42]. Moreover [56] a longitudinal study demonstrated that APOE protein levels increased 3-fold in 9-month-old TgSwDI mice compared to 3-month-old wild type mice, indicating that CAA may also increase with age in these mice, suggesting that TgSwDI is well suited to therapeutic investigations regarding Aβ degradation and clearance.
5xFAD-
The 5xFAD double transgenic mouse was developed in 2006 containing 5 familial AD mutations: APP KM670/671NL (Swedish), APP I716V (Florida), APP V717I (London), PSEN1 M146L, PSEN1 L286V [57,58]. 5xFAD model shows an aggressive amyloid deposition accompanied by gliosis, synaptic and neuronal loss [59]. β- and γ- secretase are the two proteases that are responsible for cleaving Aβ from APP. APP is cut at the N terminus of Aβ domain by β- secretase to produce C99 (membrane bound fragment) and secreted APP ectodomain APPsβ. C99 is then cleaved by γ- secretase to generate C terminus of Aβ. Since the γ- secretase cleaving is not precise, Aβ peptide of 38-43 amino acid length are produced. A higher proportion of Aβ40 (40 amino acid) is produced under normal condition [57]. Intraneuronal Aβ depositions developed within 1.5 – 2 months of age with memory deficits in 4 months and neuronal loss at nine months [60] accompanied by cognitive and motor deficiencies, with observable tau tangles [61]. In addition, amyloid pathology in the spinal cord is observed at around 11 weeks in the lumbar regions and cervical extending along the cord by 19 weeks [62]. According to the study done by Oakley et al. in 2006, they showed that in 5XFAD mice model, before amyloid deposition starts intraneuronal accumulation of Aβ occurs mainly in the neurons present in the deep cortical layers and subiculum. Some plaques appear to be originating from the intraneuronal Aβ-containing neurons that have degenerated morphologies [57]. All the above features of the 5xFAD mouse make it a popular choice for AD research.
APPxPS1-
The APPxPS1 transgenic mouse model contain mutations: APP K670_M671delinsNL (Swedish) and PSEN1 L166P. This model shows a 3-fold higher expression of hAPP transgene than murine APP. Plaques deposition occurs in 1.5 months in cortex and 3-4 months in hippocampus [63]. Gliosis occurs at around 1.5 months of age with an increase in astrogliosis. Synaptic loss occurs almost after 4 weeks of plaque formation which continues for several months [64]. Cognitive impairment occurs around 7 months [65] but was also reported at 8 months by Radde et al. [63]. According to Gengler and colleagues, no detrimental effects on synaptic plasticity was found in young mice up to 5 months of age as plaque formation, Aβ production and level of inflammatory markers are relatively low. Impaired LTP in hippocampal CA1 region was found between 8-15 months of age [66]. No severe neuron loss is reported. According to Rupp et al., neuron loss occurred at 17 months of age in the dentate gyrus and other subregions with high neuronal density [67].
APP NL-F KI-
This transgenic mouse model was first reported in 2014 by Saito et al. and contained mutations: APP K670_M671delinsNL (Swedish) and APP I716F (Iberian). APP NL-F mice recapitulate AD pathologies like Aβ pathology, neuroinflammation, and memory impairment, all occurring age-dependently [68]. Amyloid plaques occurred at 6 months in the hippocampus and cortex region. Gliosis occurs at around the same age of 6 months, concurrent with plaque formation. Synaptic loss occurs at 9-12 months, memory impairment is seen at 18 months, and no tangle formation and no loss of neurons [26,68]. The Swedish mutation elevates the level of Aβ40 and Aβ42 whereas the Iberian mutation is responsible for increasing the ratio of Aβ42 to Aβ40 [68]. Two other related strains containing mutant APP with Swedish mutation alone (APPNL) or with Iberian, Arctic, and Swedish mutation (APPNL-G-F) were developed. APPNL show a less severe phenotype than APPNL-F, whereas APPNL-G-F shows a more severe phenotype, including aggressive Aβ deposition [26]. APPNL-F mice are a good candidate to study the effects of elevated Aβ against wild-type levels of APP.
A selection of prominent models utilized in A research is shown in Table 1.
| Model | Mutation | Model Background | Aβ Pathology | Other Pathologies and Impairments | References |
| Tg2576 | APP KM670/671NL (Swedish) | C57BL/6 x SJL | Plaques by 11-13 months | Synaptic loss by 4.5 monthsCognitive impairment by 6 months | [27], [33,34,35] |
| PDAPP | APP V717F (Indiana) | C57BL6 x DBA2 | Plaques at 6 months | Cognitive impairments at 3 monthsSynaptic loss by 8-9 monthsGliosis by 6 months | [28],[31], [29], [27] |
| 5xFAD (B6SJL) | APP KM670/671NL (Swedish), APP I716V (Florida), APP V717I (London), PSEN1 M146L, PSEN1 L286V | C57BL/6 x SJL | Plaques by 1.5 months | Gliosis at 2 monthsSynaptic and Neuronal loss at 4 and 6 monthsCognitive impairments by 4-5 months. | [27], [26] |
| 5xFAD (C57BL6) | APP KM670/671NL (Swedish), APP I716V (Florida), APP V717I (London), PSEN1 M146L, PSEN1 L286V | C57BL6 | Plaques by 1.5-2 months | Cognitive impairments between 3-6 months Synaptic and Neuronal loss at 6 and 12 months | [60], [59], [69], [62] |
| APPxPS1 | APP K670_M671delinsNL (Swedish) and PSEN1 L166P | C57BL/6 | Plaques by 1.5 months (cortex) and 3-4 months (hippocampus) | Cognitive impairment at 7 monthsGliosis at approx. 1.5 monthsSynaptic loss at 2-3 monthsNeuronal loss at 17 months | [63], [67], [64], [66], [65] |
| TREM2- BAC x 5xFAD | APP KM670/671NL (Swedish), APP I716V (Florida), APP V717I (London), PSEN1 M146L, PSEN1 L286V | TREM2-BAC: FVB/NJ;5xFAD: C57BL/6 x SJL | Plaques by 8.5 months | Neuronal and synaptic loss no data reportedGliosis by 7 months | [70], [71], [72], [73], [26] |
| APPNL-F | APP KM670/671NL (Swedish), APP I716F (Iberian) | C57BL/6 | Plaques by 6 months | Synaptic loss by 9-12 monthsGliosis in 6 months | [26], [68] |
| APPV717I | APP V717I (London) | C57BL/6 x FVB/N | Plaques by 10 months | Spatial memory deficits by 6 monthsCAA (15 months)Microhemorrhages (25-30 months) | [44], [43] |
| J20 | APP K670_M671delinsNL (Swedish), APP V717F (Indiana) | C57BL/6 | Plaques by 5-7 months | Neuronal loss, synaptic loss by 3 months, Cognitive impairments by 4 monthsTangles absent | [45], [46], [48], [47], [26] |
| TgCRND8 | APP K670_M671delinsNL (Swedish), APP V717F (Indiana) | Hybrid C3H/He-C57BL/6 | Plaques by 3 months | Cognitive impairment and gliosis by 3 months Tangles absent | [50], [51], [49], [52], [26] |
| Tg-SwDI | APP K670_M671delinsNL (Swedish), APP E693Q (Dutch), APP D694N (Iowa) | C57BL/6 | Plaques by 3 months | Cognitive impairment by 3 months,Gliosis by 6 months,Tangles absent | [26], [53], [54] |
Prominent Tau Mice Models
Abnormal hyperphosphorylation of tau protein (p-tau) is a key component of NFT especially, phosphorylation of specific Thr residues (Thr 181, 217, and 231) is a biomarker of AD pathology [74]. The tau protein can be either 3R or 4R depending on the presence of the repeat domain. Both these forms of tau are expressed equally in the adult brain but the 3R/4R ratio gets altered in most tauopathies [75].
The first model to showcase tauopathy expressed 4R (consisting of a group of neurodegenerative diseases which show cytoplasmic inclusions composed of tau protein with 4 microtubule-binding domain), the largest tau isoform and the most natural substrate for hyperphosphorylation but showed little to no NFT formation [76,77]. Transgenic mice that developed robust neuronal tauopathy were primarily based on transgenic overexpression of mutations that cause FTD with Parkinsonism linked to chromosome 17 (FTD-MAPT) [78,79,80]. Various important tau mice models have been summarized in Table 2 and are described below:
JNPL3 (P301L)-
Transgenic mice (JNPL3) expressing 4R tau with P301L missense mutation (Pro301→Leu) is a model of neurofibrillary and axonal deterioration, neuronal loss, behavioral deviations, and motor dysfunction [81,82]. Neurofibrillary tangles develop between 4.5 - 6.5 months in an age- or dose-dependent manner. Neuronal loss is observed from 9-10 months, and astrogliosis presents in the diencephalon, brainstem, and basal telencephalon after 10 months [82].
To evaluate the effect of P301L, Hutton et al. (2001) developed the Tau Microinjected Model at Mayo Clinic (Jacksonville, Florida). Central to the model is a mutant form of the microtubule-associated protein tau (MAPT) gene linked to chromosome 17 (FTDP-17), which encodes for tau resulting in protein dysfunction contributing to neurodegeneration [82,83].
The model was developed by microinjecting transgenic constructs containing the P301L mutation and mouse prion promotor into fertilized eggs of hybrid C57BL/6 x DBA2 x SW mice. In this regard, models can be homozygous (002508-M), hemizygous (001638-T), or the wild type (001638-W). Both homozygous and hemizygous microinjected mice displayed a late righting reflex (RR), and the motor functions were feeble within two weeks of the onset of signs [84].
hTau-
These mouse models were developed by crossing 8c mice (mice that express tau transgene derived from human PAC, H1 haplotype) [85] with tau exon 1 knockout mice [86,87]. As this model express all the isoforms of human tau, it is the best model to test the effect of drugs on human tau without murine tau intervention [86].
Hyperphosphorylated tau accumulates at 6 months and increases by 13-15 months with maximum accumulation in the hippocampal and neocortex compared to the brain stem and spinal cord [87,88]. hTau mice develop cognitive deficits and physiological impairment at 12 months, and significant neuronal death occurs after 14 months [89]. Early hyperphosphorylation of tau at Thr (181 and 231) and Ser (202 and 396) residues has been reported in the htau mice model with C57B1/6J strain. Hence, to analyze the efficacy of compounds on tauopathy, the above model and strain can be used at a young age [90,91].
rTg (tauP301L)-
The rTg(tauP301L)4510 mouse expresses the P301L mutation in tau (4R0N) associated with frontotemporal dementia and parkinsonism linked to chromosome 17 [92]. Since its initiation in 2005 [87,88], this mouse model has become popular, as it shows the phenotype of tau pathology. Pronounced neurodegeneration is observed in human tauopathies and provides researchers temporal control over mutant tau transgene expression. rTg4510 mice (‘r’ for regulatable) mutant tau is expressed approximately 13 folds higher than endogenous mouse tau. The degree of neurodegeneration and NFT pathology is aggressive [87,93]. Gender difference has also been reported in this model, with females showing more tau pathology and behavioural deficits than males [94,95]. Accumulation of tau pathology in the form of argyrophilic tangles occurs by 4 months in cortex and by 5 months in hippocampus [87,88]. This model develops neuronal loss. For example, Ramsden et al. (2005) and SantaCruz et al. (2005) reported 60% neuron loss in hippocampal CA1 region at 5.5 months [87,88] whereas Helboe et al. (2017) reported 43% neuron loss between 8-12 months [96]. Gliosis observed at 2.5 months of age [96]. Synaptic loss in dendritic spine at 8-9 months, motor impairments normal up to 6 months, and spatial memory retention impaired in 2.5-4 months [26]. Overexpression of tauP301L in forebrain of this mice is accepted as the cause of forebrain atrophy and other tauopathy phenotypes, a hypothesis stating that suppression of tauP301L expression by DOX (doxycycline) halts neuronal loss and improved memory function [88]. Mice expressing wild-type human Tau at levels equivalent to tauP301L in rTg4510 mice do not show the development of memory deficits or overt atrophy, claiming that the tauP301L mutant form of tau is the direct cause of these phenotypes in rTg4510 [97,98].
PS19 (tau P301S)-
PS19 mouse model was first developed and reported in 2007 by Lee and colleagues [99]. This widely used tau pathology model expresses the P301S mutant form of human MAPT. Cognitive decline and neurofibrillary tangles formation are observed at six months [26] with accumulation of latter in spinal cord, brain stem, hippocampus, neocortex, and amygdala. Synaptic loss and gliosis at 3 months. gliosis is observed mainly in the white matter of brain and spinal cord. Neuronal loss observed at 9-12 months in hippocampus and cortex, and severe neuronal loss by 12 months in amygdala and neocortex [26,99].
3xTg A composite model-
3xTg is a triple-transgenic mouse model developed in the year 2003 to demonstrate both Aβ and tau pathology. It contains mutagenic forms of the human MAPT gene segment (P301L), APP695 (KM670/671NL), and human presenilin-1 gene (M146V) all of which are controlled by the neuronal Thy1.2 promoter [100]. In the 3xTg model, Aβ plaques and tau pathology development starts at 6 months and mimics its development in the AD patient. The Aβ accumulates in the cortex and progresses to the hippocampus with age while tau pathology follows an inverse trend. Additionally, neurogenesis and cognitive dysfunction defects appear within 4 months [101] and gliosis after seven months with no neuronal loss [72,92]. Further investigations showed that neutrophils infiltrate the brain in 4–6-month-old 3xTg-AD mice, contributing to early cognitive decline. Aβ pathology is preceded by tau pathology with NFT at 12 months [27]. The age at CAA onset and microhaemorrhage extent are unknown for 3xTg-AD mice. Previous studies found the increased expression of RAGE (Receptors Advanced glycation end products) in 3xTg-AD transgenic mice [102], which in turn induced oxidative stress and expression of proinflammatory cytokines through various pathways [103].
Compared to the C57BL/6 model, 3xTg models show accelerated aging, mitochondrial function, and stem cell composition in the cortex and hippocampus region of the brain [104]. As the 3xTg model exhibits both important features of AD, it will be useful in pre-clinical studies to evaluate the effectiveness of compounds in ameliorating AD pathology.
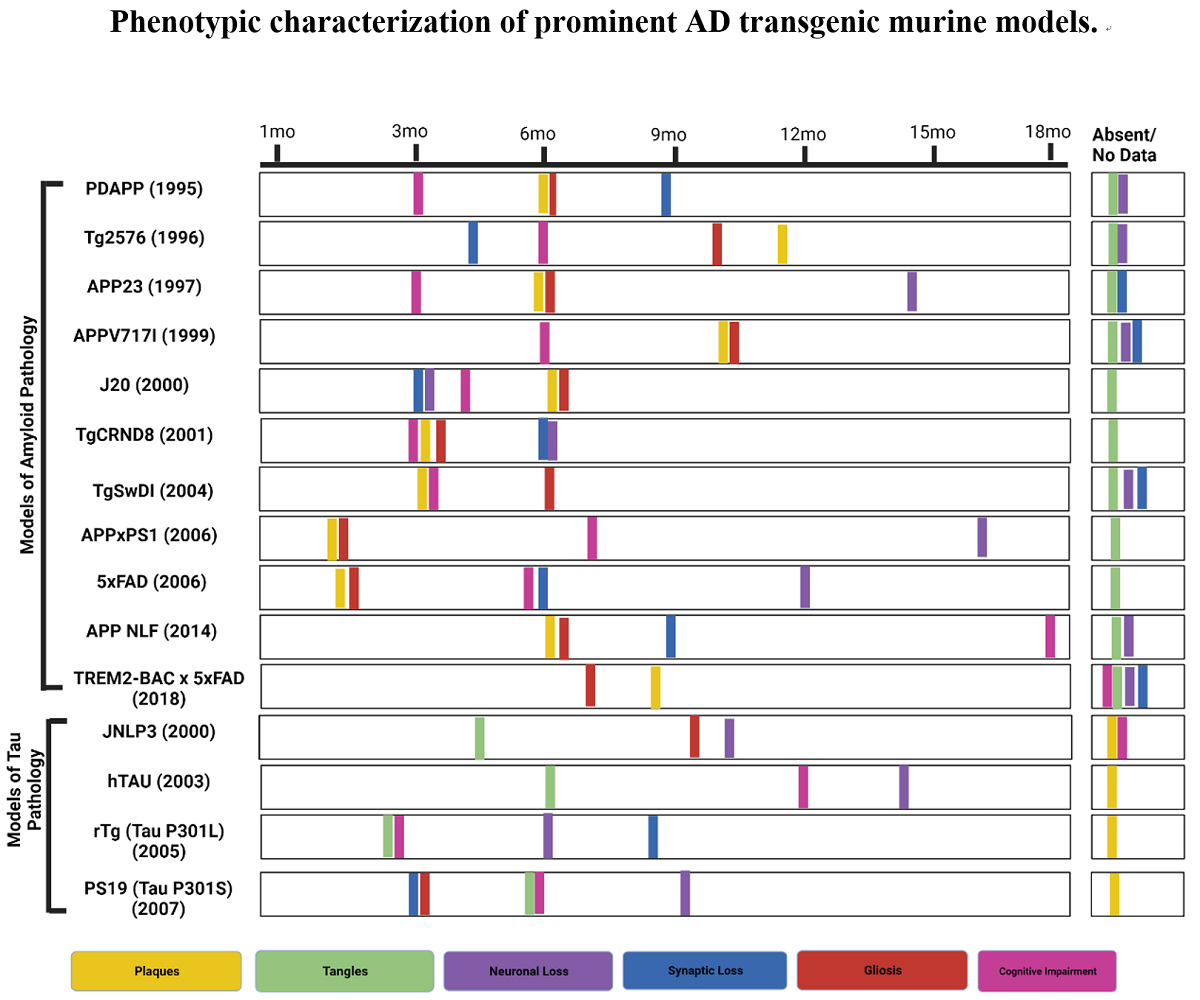
Figure 1.
Phenotypic characterization of prominent AD transgenic murine models. (Created with BioRender.com).
Figure 1.
Phenotypic characterization of prominent AD transgenic murine models. (Created with BioRender.com).
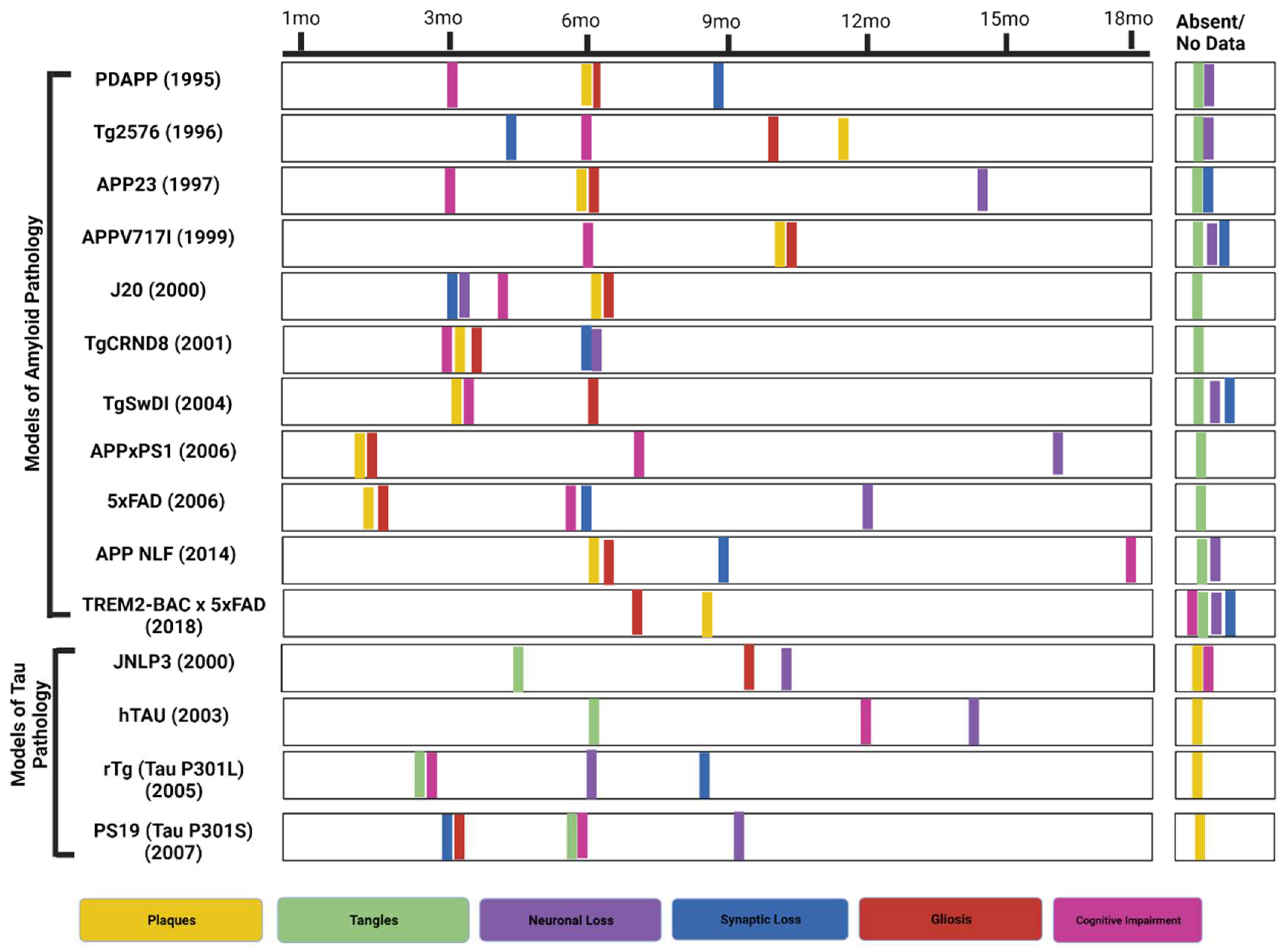

Table 2.
Summarizes of the some of the most popular Tau models currently in use.
| Model | Mutation | Model Background | Tau Pathology | Other Pathology | References |
|---|---|---|---|---|---|
| JNPL3 | MAPT P301L | C57BL/6, DBA/2, SW mixed background | NFTs develop from 4.5-6.5 months | Aβ plaques absentNeuronal loss at 9-10 monthsAstrogliosis by 10 months | [81], [82], [84], [83] |
| hTau | M.A.P.T.: knock out; M.A.P.T.: Transgenic | Cross between SW and B6D2F1 | Hyperphosphorylated tau at 6 months | Aβ plaques absentCognitive deficits at 12 monthsNeuronal death observed at 14 months | [87], [88], [89] |
| rTg (tau P301L) | MAPT P301L | Produced by crossing 129S6 (activator) to F.V.B. (responder) | Pretangles at 2.5 months. Argyrophilic tangle-like inclusions in the cortex by 4 months and in the hippocampus by 5.5 months | Aβ plaques absentNeuronal loss was observed at different periodsSynaptic loss at 8-9 monthsMotor impairments by 6 monthsMemory impairments by 2.5-4 months | [87], [88], [96], [26] |
| PS19 (tau P301S)- | MAPT P301S | (C57BL/6 x C3H)F1 | NFTs develop at 6 months | Cognitive deficits at 6 monthsSynaptic loss and gliosis at 3 monthsNeuronal loss at 9-12 months | [26], [99] |
LOAD Murine Models
Apolipoprotein E
Apolipoprotein E (APOE) (299 amino acids) is highly expressed by astrocytes, followed by microglial cells and neurons in the brain [8,9] and liver. There are three common APOE genetic variants ε2, ε3, and ε4, with (APOE) ε4 posing the greatest risk factor for LOAD [105,106]. Unlike animals, humans are the only known species that express multiple forms of apoE protein. Only 14% of the human population carries the apoE ε4 allele, and only 50% of AD patients are carriers [107]. Patients with a homozygous ε4 (APOE4/4) have a 30–55% risk of developing mild cognitive impairment (MCI) or AD by 85, compared to a 10–15% for those with the APOE 3/3 genotype. In other mammals, a similar version of human APOE ε4 protein is expressed [108]. ApoE ε4 leads to the development of AD via various mechanisms, including increased aggregation and decreased amyloid-polypeptide clearance; increased tau phosphorylation; reduced glucose metabolism, vascular and mitochondrial dysfunctions; network excitability; neurodevelopmental differences[109,110]. Very few mouse models have been studied for age-related alterations in AD, as existing models, along with behavioral experiments, have failed to show efficacy in clinical trials [111,112]. Studies related to genetics and the biology of LOAD have revealed that other pathways may contribute to disease pathogenesis [113].
TREM2- BAC x 5XFAD-
TREM2 receptor is expressed by microglia in the Brain and mediating microglial phagocytosis and inflammatory stimuli [70]. Several mouse models are available to study the effects of TREM2 in AD pathogenesis; including TREM2-BAC x 5XFAD [71], TREM2 Humanized (common variant) x 5XFAD [73], and TREM2 Humanized (R47H) x 5XFAD [72]. TREM2-BAC x 5xFAD model produces less cortical amyloid than the 5xFAD model in 7 months. No data on tau tangles, synaptic loss, and neuronal loss has been reported. Gliosis is observed at seven months of age [72]. The R47H variant results in 3 folds greater risk for developing AD. Mice carrying human TREM2 gene were crossed with 5XFAD to study the effects of R47H variant. In a recent study by Song et al. (2018), two transgenic mouse lines were generated, one with common variant (CV) and another with R47H variant into mouse TREM2 deficient mice and crossed with 5XFAD mouse model. The result showed that CV enhanced microglial activation and augment plaque associated microgliosis, indicating that R47H is a loss of function. Presence of soluble TREM2 on neuron cell bodies, on Aβ plaques and throughout brain tissues was found in CV but not in R47H variant mice. The study provided the evidence of in vivo loss of function of R47H implying a protective role of TREM2 [73].
Alternative Murine models
Senescence Accelerated Mouse (SAM, transgenic) and Senescence-Accelerated RAT (OXYS) (non-transgenic) models are some of the most popular occurring phenotypic murine models used in AD research [114,115,116,117]. SAM represent a series of inbred strains derived from the AKR/J strain, consisting of 9 senescence-prone strains (SAMP) and 4 senescence-resistant strains (SAMR) with the characteristic of accelerated and normal (controlled) ageing. Among SAMP strains, SAMP8 and SAMP10 mice exhibit numerous deficits /symptoms consistent with AD’s intermediate and later stages, such as learning and memory, emotional disorders, altered circadian rhythm, and specific biochemical, pharmacological, and pathological changes. For example, SAMP8 shows passive and active avoidance responses indicative of memory impairment and cognitive decline at 2 months, followed by tau hyperphosphorylation at 3 months, inflammation at 5 months, Aβ deposition at 6 months, synaptic degeneration at 8 months at and neuronal loss at 10 months [118]. Moreover, these strains manifest various pathobiological phenotypes spontaneously, suggesting they may have a significant role in investigating future drug candidates such as J147 and 6-chlorotacrine (huprine)-TPPU hybrids [119,120].
Sharing similar traits with the previous model, OXYS rats can elucidate the mechanisms of ageing and pathology and provide an objective evaluation of potential therapeutics and preventive actions [121]. For example, a recent study using said model assessed the association of cerebrovascular dysfunction with the development of AD-like pathology. As a result, the maximal density of blood vessels in prepubescent mice was observed on day 20, followed by a reduction in vessel density of hippocampal regions (СА1 and СА3) at 5 months and a complete reduction in all areas by 18 months [122].
The underlying cause or causes of AD, particularly LOAD, is unknown. In addition, finding a suitable model that spontaneously develops the underlying pathologies remain challenging. However, a long-lived (18 years) strictly herbivorous rodent (Octodon degus) found in the upper mountain regions of Chile [123] does spontaneously develop cognitive decline with the altered cholinergic transmission, hyperphosphorylated-tau, β-amyloid plaques, NFT, and neuroinflammation in the brain [124,125]. Moreover, said model performs coprophagy (faecal reingestion) and is susceptible to diabetes mellitus, a major risk factor for AD. Regarding drug development, its utility is still in the early stages. Although some data suggested that O. degus raised in captivity at lower altitudes cannot reproduce the AD hallmarks reported earlier [126] in a laboratory environment.
Limitations of murine models
Using an animal model gives us a clear insight into the pathology of a disease that is difficult to monitor in humans. For studying diseases such as AD or any other neurodegenerative disorders, animal models exhibit pathologies within months compared to years in humans. Using such models has yielded encouraging results, but their clinical value remains uncertain as the mice do not naturally develop neurodegeneration (which is interesting in itself), thus, the focus is mainly on familial AD (FAD) [127] and not on sporadic human AD (sAD). Likewise, there are considerable differences between the immune system of both mice and humans which most models fail to address. The effectiveness of a transgenic animal model in mimicking certain diseases relies on how they respond to behavioural assessments [72]. Until now, murine models were ineffective in exhibiting progressive loss of cholinergic neurons in AD [81]. However, different studies may provide different results using AD mouse models. Some newer models show physiological characteristics similar to human amyloid protein with quantifiable levels of amyloid plaques but still lack neurodegeneration, the main feature of human AD [128,129,130].
Furthermore, there is no consistency in the results when working with AD mouse models. Very few rodent models allow us to study the formation of plaques and tangles simultaneously as the presence of both is required to diagnose AD and to understand how the interaction between plaques and tangles contributes to the progression of Alzheimer's [11]. Evidence shows that tau increases Aβ toxicity leading to the understanding that both pathological characteristics are essential to replicate toxicity in human AD [11]. Mice have a shorter life span and a smaller and less developed prefrontal cortex, which can be a significant drawback for studying age-related neurodegenerative diseases like AD [127,131].
Although transgenic murine models remain the mainstay of AD laboratory research, some earlier models exhibiting overexpression are currently being phased out. The primary reason is that the models demonstrated incomplete pathologies such as widespread Aβ pathology without neuronal loss (strongest AD correlate), cognitive deficits [132], and Tau pathology. Conversely, other models have prioritized the overexpression of human tau proteins at the expense of Aβ pathology. Accordingly, these incomplete pathologies and the limited information provided have led to the emergence of newer models that better emulate the complete pathology of the disease, some of which will be addressed in later sections.
Evolutionarily closer models
The evolutionary distance between murine models and humans can be a limitation. Mammals phylogenetically closer to humans include canine and nonhuman primate models (NHP), which are functionally and neuroanatomically similar exhibiting larger brain sizes and providing larger sample volumes for biomarker studies [127]. Age is the greatest factor in the risk of AD, the course of ageing in canine and nonhuman primates provides more significance for life-span-related changes than that of mice. For example, aged dogs mainly accumulate Aß deposits with a similar amino acid sequence to the human protein which is an advantage over transgenic mouse models. But, Tau pathology, including aggregated intraneuronal topographies and pre-tangles is very limited in aged dogs [125,133,134]. Aß is also presented in canine’s CSF and the ratio of Aß42/Aß40 declines with age [125]. However, cerebrovascular changes linked to cognitive decline and mitochondrial dysfunction have been reported in canines [125]. Hence the aged dog model is suitable for studying the neuroprotective effect of drug molecules.
NHP are beneficial animal models for AD since they share genetic similarities with humans, have a developed prefrontal cortex, and exhibit age-related cognitive deficits [135]. Studies confirm amyloid plaque formation in aged monkeys and great apes [136,137,138]. Hyperphosphorylated tau has been reported in Macaca mulatta (rhesus monkey), Papio anubis (baboon), and saimiri sciureus (squirrel monkey) [139]. Diffuse Aß plaques and vascular amyloid were observed in the brain of Pongo pygmaeus (Orangutan) and Pan troglodytes (Chimpanzee) but they lacked NFT [140,141].
Many aged NHP species develop amyloid plaques without NFT or neuronal loss [142]. The utility of Macaca mulatta and the Macaca fascicularis (Philippine monkey) in Old-World Monkeys has been limited due to inconsistent demonstration of AD pathology [127,143,144]. The New World Monkeys (Marmosets, Capuchin monkeys, Night monkeys, Saki monkeys, and Spider monkeys) have proven useful for studying different aspects of human aging [145]. The fast aging (10-12 years) marmosets are particularly attractive as they have a well-developed platform and toolbox for gene editing, neurophysiological and cognitive studies, and neuropathology studies [146]. The literature proposes that NHPs are better models for non-pathological aging due to the absence of AD-like brain pathology and difference in amyloid biochemistry [127,144]. Animal models related to the amyloid cascade hypothesis are shown in Figure 2.
Emerging Models
The National Institute on Aging (NIA ) established Model Organism Development and Evolution for Late-onset Alzheimer’s Disease (MODEL-AD): 1) to develop new models of LOAD using humanized Aβ and tau to recapitulate human AD pathology 2) to identify novel genetic variants that increase the risk of LOAD, 3) to develop a robust preclinical testing line for testing newer therapeutics [113]. A knock-in model, hAβ-KI is developed which expresses humanized Aβ sequence within the murine APP gene. hAβ-KI mice models display age-dependent behavioural abnormalities and synaptic plasticity, but do not develop amyloid plaques [147]. hAβ-KI mice develop age-related insoluble Aβ, decreasing soluble Aβ, suggesting that this shift from soluble to insoluble Aβ may be playing a mechanistic role in the AD progression [148]. hAβ-KI mouse models produce human Aβ at physiological levels in cells which normally express APP, without the addition of any FAD mutations or over-expression of APP or its metabolites [113]. Amyloid plaques were not observed through 22 months of age, neither microgliosis nor astrogliosis was observed in 22 months old mice. Neuron number was the same in the hippocampal region of hAβ-KI and wild type mouse models. Cognitive impairment was observed at 10 months. Levels of pro-inflammatory cytokines increased in the hAβ-KI mouse model, with a concomitant decrease in the level of anti-inflammatory cytokines [147].
2. Behavioral Studies
The cognitive test is required to assess the ability to learn and think. In contrast, behavioral tests are required to determine an animal's ability to respond to a stimulus and to determine its sensory responses. As the AD pathology is severe in the entorhinal cortex and the hippocampal region of the medial temporal lobe [149], The medial temporal lobe is responsible for learning, memory, and emotional behaviour. The hippocampus, on the other hand, is responsible for spatial memory [150] and gets damaged at the early stages of AD; hence is considered essential to study the pathophysiology of the disease [151,152,153]. Therefore, NHP and rodent models are developed to measure hippocampal-dependent memory, which can showcase human memory [27,154,155].
Different types of cognitive-behavioural assays are performed which illustrate similar defects in human AD, namely, fear conditioning (FC) task studies the associative memory, Radial Arm Water Maze (RAWM) task studies the short-term memory affected early in the disease, and Morris Water Maze (MWM) task studies long term memory that is affected late in disease [153]. Analyzing animal behaviour has become an essential tool in translational neuroscience and for studying the physiological mechanism of a neurological disorder, modifications/changes induced by chemical treatment / genetic manipulations, and the efficacy of novel drugs that reverse the phenotypic conditions in the disease models. These behavioural-cognitive assays in mouse models can provide information regarding the validity and efficacy of the new drugs target/compounds, but the only limitation is that a few cognitive functions are unique to humans and cannot be measured in murine models [153].
3. Fluid Biomarkers
In line with the amyloid cascade theory, early AD diagnostics focused on changes in Aβ40, Aβ42, and total tau(t-tau) levels in cerebrospinal fluid (CSF). However, changes in these markers also occur in other neurological diseases and normal aging. Moreover, 25% of AD patients with mild cognitive impairment (MCI) have normal t-tau levels, while CSF t-tau is elevated in 50% of preclinical AD individuals [156]. Therefore, it has been suggested that elevated tau is due to increase production and neuronal plasticity, whereas elevations at the end stages of the disease can be attributed to dying neurons.
CSF sampling is invasive, and the physical burden on patients and murine models is high. However, the blood absorbs nearly 500 ml of CSF daily, providing a readily accessible sampling pool. Moreover, blood plasma is obtainable at fractional costs rendering it more suited to mass testing. However, t-tau CSF levels poorly correlate with plasma levels restricting its usage as a biomarker. In addition to total tau, other biomarkers spanning all three stages of AD include phosphorylated tau (p-tau) and amyloid beta oligomers (AO's)(Figure 3). The p-tau isoforms p-tau threonine 181, p-tau threonine 217, and p-tau threonine 231 plasma levels strongly correlate with CSF values and are considered AD early-mid stage (Braak 3–4) biomarkers and predictors of A- to A+ conversion in MCI patients [157].
Low molecular weight AβO is present at the beginning and later stages of the disease and can be found in blood, plasma, and CSF. Li et al. recently reported a highly specific imaging probe for soluble AO’s known as AN-SP, a fusion of aminonaphthalene-2-cyanoacrylate (ANCA) with a spiropyran (SP) [158]. Investigations regarding the specificity of ANSP for AOs performed on 8-month-old APP/PS1 transgenic and wildtype mice showed that AN-SP exhibited excellent colocalization with A oligomer-specific antibodies but not with conformational tau pathology. A summary of the current state of NIR imaging probes specific to A can be found in a recent review by Peng et al. [159]
4. Neuro Imaging in Murine Models
In addition to detecting changes in A and tau levels, a definitive AD diagnosis also requires a multi-layered (0.7 mm thickness/per layer) image of a patient's brain presenting significant NFT and plague deposition post-mortem. This can be achieved using various imaging modalities, including magnetic resonance imaging (MRI), near-infrared fluorescence (NIRF)g and positron emission tomography (PET).
MRI has been used to assess the metabolic profiles of APP, 3xTg-AD and hyperactive 5xFAD models. For example, [160] amyloid plaques as small as 35 μm have been detected in APP mouse models using magnetic resonance microimaging (MRMI) at a high magnetic field (9.4 T). Moreover, age-dependent reductions in metabolite concentrates (acetyl aspartate (NAA), glutamate and glutathione) and elevations in taurine (related to the degree of neuroprotection) [161] in the cerebral cortex of 19-month APP mice analogous to AD patients have been detected using single-voxel H(proton) magnetic resonance spectroscopy ((1)H MRS) at high magnetic field. Furthermore, in-vivo H-MRS found a significant reduction and elevation in taurine in 3xTg-AD mice compared to age-matched controls [162]. Moreover, a recent study using 3xTg-AD to evaluate the potential of scFv-h3D6 immunotherapy showed a partial recovery in brain volume reduction of the inflammatory marker, IL-6, compared to the controls at 12 months [163].
Magnetic resonance spectroscopy (MRS) has been used to identify biomarkers of AD progression in the hippocampus of 5xFAD mice [164]. H-MRS revealed decreased concentrations of Gamma-Aminobutyric Acid (GABA) at 9 and 10 months in the dorsal hippocampus and increased concentrations of myo-inositol (Myo). Several features of AD pathology are expressed by 5XFAD mice demonstrating its compatibility with studies reliant on brain imaging and pathogenic amyloid measurement. Studies have shown that cerebral metabolic alterations precede the clinical manifestation of AD symptoms. Distinct patterns of cerebral glucose metabolism also help differentiate AD from other neurodegenerative disease causes [164]. Traditionally cerebral metabolic rates of glucose have been measured utilizing the imaging tracers 2-[18F] fluoro-2-deoxy-D-glucose (FDG) and (18)F-AV-45 (florbetapir) in conjunction with PET to establish a proxy for neuronal activity, which has also been shown feasible for quantifying Aβ in 5XFAD mice. Moreover, a comprehensive evaluation of 5X FAD including whole brain imaging, autoradiography, blood plasma analysis (cytokines and A species) and numerous behavioral measurements, was reported by Oblak et al. [61] Interestingly, the authors concluded that 5XFAD was better suited to AD studies involving sex-based differences, immune dysfunction, and Aβ deposition than translational studies evaluating the efficacy of novel therapeutics targeting cognitive impairment.
5. Conclusion
The mammalian models developed for AD investigations exhibit various disease characteristics. However, none of these models can replicate all the pathophysiological features of human AD. Moreover, many transgenic murine models exhibit genetic drift (allelic drift), resulting in Jackson Laboratory (Jax) and the National Institutes of Health (NIH) sub strains, thus quality safeguards are necessary to monitor and maintain the genomic validity of the transgenic AD models of interest [165,166].
sAD constitutes 95% of the cases, but the models (Transgenic/NHP/Canine) on which anti-AD drug candidates are tested, utilize fAD partial models. This may account for the failure of drugs in clinical trials as the brain distribution of Aβ and tau is different in sAD and fAD [167]. Additionally, a few models express both NFT and amyloid plaques. The crosstalk between Aβ and NFT significantly increases the toxicity [168]; hence the models displaying pathological characteristics of both Aβ and tau are important to study these interactions during AD progression and therapeutic intervention. Drugs that lower Aβ levels have been the most desirable for drug development. For example, after many clinical trials, it has been shown that anti-Aβ antibody therapies or secretase inhibitor therapies lower Aβ levels in animals and humans. However, the safety concerns remain [169] and anti-amyloid immunization only arrests disease progression but does not improve cognition [170,171]. Thus, there is a need to develop next-generation animal models that will more precisely predict the efficacy of treatments enabling the translation of drugs from the lab to the clinic.
Author Contributions
Conceptualization, HS and SAA.; writing—original draft preparation, H.S. and.; writing—review and editing, JH., and SAA.; funding acquisition, SAA. All authors read and agreed to the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cummings, J.L. , et al., The costs of developing treatments for Alzheimer's disease: A retrospective exploration. Alzheimers Dement 2022, 18, 469–477. [Google Scholar] [CrossRef]
- Poon, C.H. , et al., Rodent models of amyloid-beta feature of alzheimer’s disease: Development and potential treatment implications. Aging and disease 2020, 11, 1235. [Google Scholar] [CrossRef] [PubMed]
- Therriault, J. , et al., Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nature Aging, 2022, 1-10.
- DeTure, M.A. and D.W. Dickson, The neuropathological diagnosis of Alzheimer’s disease. Molecular neurodegeneration 2019, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R. , et al., The development of amyloid β protein deposits in the aged brain. Science of aging knowledge environment 2006, 2006, re1–re1. [Google Scholar] [CrossRef]
- Jankowsky, J.L. and H. Zheng, Practical considerations for choosing a mouse model of Alzheimer’s disease. Molecular neurodegeneration 2017, 12, 1–22. [Google Scholar] [CrossRef]
- Wattmo, C. and A.K. Wallin, Early- versus late-onset Alzheimer's disease in clinical practice: cognitive and global outcomes over 3 years. Alzheimers Res Ther 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Verghese, P.B., J. M. Castellano, and D.M. Holtzman, Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol 2011, 10, 241–252. [Google Scholar] [CrossRef]
- Kim, J., J. M. Basak, and D.M. Holtzman, The role of apolipoprotein E in Alzheimer's disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Vassar, R. , BACE1: the beta-secretase enzyme in Alzheimer's disease. J Mol Neurosci 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Drummond, E. and T. Wisniewski, Alzheimer’s disease: experimental models and reality. Acta Neuropathologica 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Cole, S.L. and R. Vassar, The Alzheimer's disease beta-secretase enzyme, BACE1. Mol Neurodegener 2007, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H. , et al., The beta-Secretase BACE1 in Alzheimer's Disease. Biol Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I. , et al. , Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci 1999, 14, 419–427. [Google Scholar] [PubMed]
- Sinha, S. , et al., Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R. , et al., Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Lin, X. , et al., Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A 2000, 97, 1456–1460. [Google Scholar] [CrossRef]
- Goate, A. , et al., Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Petit, D. , et al., Aβ profiles generated by Alzheimer’s disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Molecular Psychiatry 2022, 27, 2821–2832. [Google Scholar] [CrossRef]
- Hardy, J. and D. Allsop, Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Selkoe, D.J. and J. Hardy, The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Mandelkow, E.-M. and E. Mandelkow, Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harbor perspectives in medicine 2012, 2, a006247. [Google Scholar] [CrossRef]
- Gendron, T.F. and L. Petrucelli, The role of tau in neurodegeneration. Molecular neurodegeneration 2009, 4, 1–19. [Google Scholar] [CrossRef]
- Ribeiro, F.M. , et al. , Animal models of neurodegenerative diseases. Revista brasileira de psiquiatria (Sao Paulo, Brazil : 1999) 2013, 35 (Suppl 2), S82–91. [Google Scholar] [CrossRef]
- KoSIK, K.S., C. L. Joachim, and D.J. Selkoe, Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proceedings of the National Academy of Sciences 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Research Models Alzheimer's Disease. Available from: https://www.alzforum.org/research-models/alzheimers-disease.
- Puzzo, D. , et al., Rodent models for Alzheimer's disease drug discovery. Expert opinion on drug discovery 2015, 10, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Games, D. , et al., Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Masliah, E. , et al., Comparison of Neurodegenerative Pathology in Transgenic Mice Overexpressing V717F \upbeta{}{-}{A}myloid Precursor Protein and Alzheimer's Disease. The Journal of Neuroscience 1996, 16, 5795–5811. [Google Scholar] [CrossRef] [PubMed]
- Chen, G. , et al., A learning deficit related to age and \upbeta{}{-}{a}myloid plaques in a mouse model of Alzheimer\textquotesingles disease. Nature 2000, 408, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Dodart, J.-C. , et al., Neuroanatomical Abnormalities in Behaviorally Characterized APPV717F Transgenic Mice. Neurobiology of Disease 2000, 7, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.F. , et al., Amyloid deposition in the hippocampus and entorhinal cortex: Quantitative analysis of a transgenic mouse model. Proceedings of the National Academy of Sciences 2003, 100, 4837–4842. [Google Scholar] [CrossRef]
- Hsiao, K. , et al., Correlative Memory Deficits, A$\upbeta$ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L. , et al., Impaired Spine Stability Underlies Plaque-Related Spine Loss in an Alzheimer\textquotesingles Disease Mouse Model. The American Journal of Pathology 2007, 171, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- IRIZARRY, M.I.C.H.A.E.L.C. , et al., {APPSW} Transgenic Mice Develop Age-related A$\upbeta$ Deposits and Neuropil Abnormalities, but no Neuronal Loss in {CA}1. Journal of Neuropathology and Experimental Neurology 1997, 56, 965–973. [Google Scholar] [CrossRef] [PubMed]
- King, D.L. and G.W. Arendash, Behavioral characterization of the Tg2576 transgenic model of Alzheimer\textquotesingles disease through 19 months. Physiology {\&}amp$\mathsemicolon$ Behavior 2002, 75, 627–642. [Google Scholar]
- Sturchler-Pierrat, C. , et al., Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Thal, D.R., W. S. Griffin, and H. Braak, Parenchymal and vascular Abeta-deposition and its effects on the degeneration of neurons and cognition in Alzheimer's disease. J Cell Mol Med 2008, 12, 1848–1862. [Google Scholar] [CrossRef]
- Reuter, B. , et al., Statin Therapy and the Development of Cerebral Amyloid Angiopathy--A Rodent in Vivo Approach. Int J Mol Sci 2016, 17. [Google Scholar]
- Fryer, J.D. , et al., Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci 2005, 25, 2803–2810. [Google Scholar] [CrossRef]
- Meyer, E.P. , et al., Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer's disease. Proc Natl Acad Sci U S A 2008, 105, 3587–3592. [Google Scholar] [CrossRef]
- Nehra, G., B. Bauer, and A.M. Hartz, Blood-brain barrier leakage in Alzheimer’s disease: from discovery to clinical relevance. Pharmacology & Therapeutics.
- Caroni, P. , Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods 1997, 71, 3–9. [Google Scholar] [CrossRef]
- Lloyd, G.M. , et al., Prominent amyloid plaque pathology and cerebral amyloid angiopathy in APP V717I (London) carrier - phenotypic variability in autosomal dominant Alzheimer's disease. Acta Neuropathol Commun 2020, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L. , et al., High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H. , et al., Global changes of phospholipids identified by MALDI imaging mass spectrometry in a mouse model of Alzheimer's disease. J Lipid Res 2016, 57, 36–45. [Google Scholar] [CrossRef]
- Ameen-Ali, K.E. , et al., The Time Course of Recognition Memory Impairment and Glial Pathology in the hAPP-J20 Mouse Model of Alzheimer's Disease. J Alzheimers Dis 2019, 68, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Shabir, O. , et al., Enhanced Cerebral Blood Volume under Normobaric Hyperoxia in the J20-hAPP Mouse Model of Alzheimer's Disease. Sci Rep 2020, 10, 7518. [Google Scholar] [CrossRef]
- Chishti, M.A. , et al., Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 2001, 276, 21562–21570. [Google Scholar] [CrossRef]
- Boy, J. , et al., Expression mapping of tetracycline-responsive prion protein promoter: digital atlasing for generating cell-specific disease models. Neuroimage 2006, 33, 449–462. [Google Scholar] [CrossRef]
- Cortes-Canteli, M. , et al., Fibrin deposited in the Alzheimer's disease brain promotes neuronal degeneration. Neurobiol Aging 2015, 36, 608–617. [Google Scholar] [CrossRef]
- Brautigam, H. , et al., The isotropic fractionator provides evidence for differential loss of hippocampal neurons in two mouse models of Alzheimer's disease. Mol Neurodegener 2012, 7, 58. [Google Scholar] [CrossRef]
- Davis, J. , et al., Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem 2004, 279, 20296–20306. [Google Scholar] [CrossRef]
- Davis, J. , et al., Deficient cerebral clearance of vasculotropic mutant Dutch/Iowa Double A beta in human A betaPP transgenic mice. Neurobiol Aging 2006, 27, 946–954. [Google Scholar] [CrossRef]
- Choi, S. , et al., Asymmetric dimethylarginine exacerbates cognitive dysfunction associated with cerebrovascular pathology. FASEB J 2020, 34, 6808–6823. [Google Scholar] [CrossRef]
- Searcy, J.L. , et al., Impact of age on the cerebrovascular proteomes of wild-type and Tg-SwDI mice. PLoS One 2014, 9, e89970. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H. , et al., Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. Journal of Neuroscience 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Forner, S. , et al., Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer’s disease. Scientific data 2021, 8, 270. [Google Scholar] [CrossRef]
- Jawhar, S. , et al., Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal A$\upbeta$ aggregation in the 5XFAD mouse model of Alzheimer{\textquotesingle}s disease. Neurobiology of Aging 2012, 33, 196–e29. [Google Scholar] [CrossRef]
- Oakley, H. , et al., Intraneuronal beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer\textquotesingles Disease Mutations: Potential Factors in Amyloid Plaque Formation. Journal of Neuroscience 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Oblak, A.L. , et al., Comprehensive evaluation of the 5XFAD mouse model for preclinical testing applications: a MODEL-AD study. Frontiers in aging neuroscience 2021, 13, 713726. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.-H. , et al., Axonal and myelinic pathology in 5xFAD Alzheimer's mouse spinal cord. PLOS ONE 2017, 12, e0188218. [Google Scholar] [CrossRef]
- Radde, R. , et al., Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 2006, 7, 940–946. [Google Scholar] [CrossRef]
- Bittner, T. , et al., Amyloid plaque formation precedes dendritic spine loss. Acta Neuropathol 2012, 124, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Serneels, L. , et al., gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science 2009, 324, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Gengler, S., A. Hamilton, and C. Holscher, Synaptic plasticity in the hippocampus of a APP/PS1 mouse model of Alzheimer's disease is impaired in old but not young mice. PLoS One 2010, 5, e9764. [Google Scholar] [CrossRef] [PubMed]
- Rupp, N.J. , et al., Early onset amyloid lesions lead to severe neuritic abnormalities and local, but not global neuron loss in APPPS1 transgenic mice. Neurobiol Aging 2011, 32, 2324–e1. [Google Scholar] [CrossRef]
- Saito, T. , et al., Single App knock-in mouse models of Alzheimer's disease. Nat Neurosci 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Forner, S. , et al., Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer's disease. Scientific Data 2021, 8. [Google Scholar] [CrossRef]
- Udeochu, J., F. A. Sayed, and L. Gan, TREM2 and Amyloid Beta: A Love-Hate Relationship. Neuron 2018, 97, 991–993. [Google Scholar] [CrossRef]
- Lee, C.Y.D. , et al., Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer's Disease Models. Neuron 2018, 97, 1032–1048. [Google Scholar] [CrossRef]
- Poon, C.H. , et al., Rodent Models of Amyloid-Beta Feature of Alzheimer's Disease: Development and Potential Treatment Implications. Aging and disease 2020, 11, 1235. [Google Scholar] [CrossRef]
- Song, W.M. , et al., Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. Journal of Experimental Medicine 2018, 215, 745–760. [Google Scholar] [CrossRef]
- Barthelemy, N.R. , et al., Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer's disease. J Exp Med 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M. and E.D. Roberson, Mouse models of Alzheimer's disease. Brain Res Bull 2012, 88, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R. , et al., Animal models in the study of Alzheimer's disease and Parkinson's disease: A historical perspective. Animal models and experimental medicine 2022, 5, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Probst, A. , et al., Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathologica 2000, 99, 469–481. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. and K.N. Green, Animal Models of Alzheimer Disease. Cold Spring Harbor Perspectives in Medicine 2012, 2, a006320–a006320. [Google Scholar] [CrossRef]
- Goedert, M., D. S. Eisenberg, and R.A. Crowther, Propagation of Tau Aggregates and Neurodegeneration. Annual review of neuroscience 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Dawson, T.M., T. E. Golde, and C. Lagier-Tourenne, Animal models of neurodegenerative diseases. Nature neuroscience 2018, 21, 1370–1379. [Google Scholar] [CrossRef]
- Lewis, J. , et al., Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nature genetics 2000, 25, 402–405. [Google Scholar] [CrossRef]
- Hutton, M. , Missense and splice site mutations in tau associated with FTDP-17: multiple pathogenic mechanisms. Neurology 2001, 56(11 Suppl 4), S21–5. [Google Scholar] [CrossRef]
- Duff, K. , et al., Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiology of disease 2000, 7, 87–98. [Google Scholar] [CrossRef]
- Hutton, M. , et al., Analysis of tauopathies with transgenic mice. Trends Mol Med 2001, 7, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, C. , et al., Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. Journal of Neurochemistry 2003, 86, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, C. , et al., Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. The Journal of neuroscience : the official journal of the Society for Neuroscience 2005, 25, 5446–5454. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, M. , et al. , Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). The Journal of neuroscience : the official journal of the Society for Neuroscience 2005, 25, 10637–10647. [Google Scholar]
- Santacruz, K. , et al. , Tau suppression in a neurodegenerative mouse model improves memory function. Science (New York, N.Y.) 2005, 309, 476–481. [Google Scholar]
- Tucker, K.L., M. Meyer, and Y.A. Barde, Neurotrophins are required for nerve growth during development. Nature neuroscience 2001, 4, 29–37. [Google Scholar] [CrossRef]
- Neddens, J. , et al., Constant Levels of Tau Phosphorylation in the Brain of htau Mice. Front Mol Neurosci 2020, 13, 136. [Google Scholar] [CrossRef]
- Wenger, K. , et al., Common mouse models of tauopathy reflect early but not late human disease. Mol Neurodegener 2023, 18, 10. [Google Scholar] [CrossRef]
- Billings, L.M. , et al., Intraneuronal Aβ causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef]
- Jankowsky, J.L. and H. Zheng, Practical considerations for choosing a mouse model of Alzheimer's disease. Mol Neurodegener 2017, 12, 89. [Google Scholar] [CrossRef]
- Yue, M. , et al., Sex difference in pathology and memory decline in rTg4510 mouse model of tauopathy. Neurobiol Aging 2011, 32, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Song, L. , et al., Analysis of tau post-translational modifications in rTg4510 mice, a model of tau pathology. Mol Neurodegener 2015, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Helboe, L. , et al., Early depletion of CA1 neurons and late neurodegeneration in a mouse tauopathy model. Brain Res 2017, 1665, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Hoover, B.R. , et al., Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef]
- Gamache, J. , et al., Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat Commun 2019, 10, 2479. [Google Scholar] [CrossRef]
- Yoshiyama, Y. , et al., Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Oddo, S. , et al., Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Rodriguez, J.J. , et al., Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS One 2008, 3, e2935. [Google Scholar] [CrossRef]
- Do, T.M. , et al. , Age-Dependent Regulation of the Blood-Brain Barrier Influx/Efflux Equilibrium of Amyloid-beta Peptide in a Mouse Model of Alzheimer's Disease (3xTg-AD). J Alzheimers Dis 2016, 49, 287–300. [Google Scholar]
- Origlia, N. , et al., MAPK, beta-amyloid and synaptic dysfunction: the role of RAGE. Expert Rev Neurother 2009, 9, 1635–1645. [Google Scholar] [CrossRef]
- Padmanabhan, P. and J. Götz, Clinical relevance of animal models in aging-related dementia research. Nature Aging 2023, 3, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Badea, A. , et al., Identifying Vulnerable Brain Networks in Mouse Models of Genetic Risk Factors for Late Onset Alzheimer's Disease. Front Neuroinform 2019, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E. , et al., Role of inflammatory molecules in the Alzheimer's disease progression and diagnosis. J Neurol Sci 2017, 376, 242–254. [Google Scholar] [CrossRef]
- Reiman, E.M. , et al., Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nature Communications 2020, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.K., K. Uchida, and H. Nakayama, White matter myelin loss in the brains of aged dogs. Experimental Gerontology 2012, 47, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y. and R.W. Mahley, Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer's diseases. Neurobiol Dis 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef]
- Liu, C.C. , et al., Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Bales, K.R. , The value and limitations of transgenic mouse models used in drug discovery for Alzheimer's disease: an update. Expert Opin Drug Discov 2012, 7, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Shineman, D.W. , et al., Accelerating drug discovery for Alzheimer's disease: best practices for preclinical animal studies. Alzheimers Res Ther 2011, 3, 28. [Google Scholar] [CrossRef]
- Oblak, A.L. , et al., Model organism development and evaluation for late-onset Alzheimer's disease: MODEL-AD. Alzheimers Dement (N Y) 2020, 6, e12110. [Google Scholar] [CrossRef]
- Yagi, H. , et al., Age-related deterioration of ability of acquisition in memory and learning in senescence accelerated mouse: SAM-P/8 as an animal model of disturbances in recent memory. Brain Res 1988, 474, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Tanisawa, K. , et al., Exome sequencing of senescence-accelerated mice (SAM) reveals deleterious mutations in degenerative disease-causing genes. BMC genomics 2013, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.A. , et al., Association of cerebrovascular dysfunction with the development of Alzheimer’s disease-like pathology in OXYS rats. BMC genomics 2018, 19, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Kolosova, N. , et al., Senescence-accelerated OXYS rats: A genetic model of premature aging and age-related diseases. Advances in Gerontology 2014, 4, 294–298. [Google Scholar] [CrossRef]
- Liu, B., J. Liu, and J.S. Shi, SAMP8 Mice as a Model of Age-Related Cognition Decline with Underlying Mechanisms in Alzheimer's Disease. J Alzheimers Dis 2020, 75, 385–395. [Google Scholar] [CrossRef]
- Kepchia, D. , et al., The Alzheimer's disease drug candidate J147 decreases blood plasma fatty acid levels via modulation of AMPK/ACC1 signaling in the liver. Biomed Pharmacother 2022, 147, 112648. [Google Scholar] [CrossRef]
- Codony, S. , et al., Discovery and In Vivo Proof of Concept of a Highly Potent Dual Inhibitor of Soluble Epoxide Hydrolase and Acetylcholinesterase for the Treatment of Alzheimer's Disease. J Med Chem 2022, 65, 4909–4925. [Google Scholar] [CrossRef]
- Kolosova, N.G. , et al., [The senescence-accelerated oxys rats--a genetic model of premature aging and age-dependent degenerative diseases]. Adv Gerontol 2014, 27, 336–340. [Google Scholar]
- Stefanova, N.A. , et al., Association of cerebrovascular dysfunction with the development of Alzheimer's disease-like pathology in OXYS rats. BMC Genomics 2018, 19 (Suppl 3). [Google Scholar] [CrossRef]
- Hurley, M.J. , et al., The long-lived Octodon degus as a rodent drug discovery model for Alzheimer's and other age-related diseases. Pharmacology & therapeutics 2018, 188, 36–44. [Google Scholar]
- Inestrosa, N.C. , et al., Human-like rodent amyloid-beta-peptide determines Alzheimer pathology in aged wild-type Octodon degu. Neurobiology of aging 2005, 26, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N. , et al., Accelerating Alzheimer's research through 'natural' animal models. Curr Opin Psychiatry 2015, 28, 155–164. [Google Scholar] [CrossRef]
- Steffen, J. , et al. , Revisiting rodent models: Octodon degus as Alzheimer's disease model? Acta neuropathologica communications 2016, 4, 91. [Google Scholar] [PubMed]
- Vitek, M.P. , et al., Translational animal models for Alzheimer\textquotesingles disease: An Alzheimer\textquotesingles Association Business Consortium Think Tank. Alzheimer{\textquotesingle}s {\&}amp$\mathsemicolon$ Dementia: Translational Research {\&}amp$\mathsemicolon$ Clinical Interventions 2020, 6. [Google Scholar]
- Bruce-Keller, A.J. , et al., Cognitive impairment in humanized APP\texttimesPS1 mice is linked to A\upbeta{}{1}{–}42 and NOX activation. Neurobiology of Disease 2011, 44, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. , et al., Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiology of disease 2007, 27, 301–311. [Google Scholar] [CrossRef]
- Xu, W. , et al., Cerebral microvascular rather than parenchymal amyloid-β protein pathology promotes early cognitive impairment in transgenic mice. Journal of Alzheimer's Disease 2014, 38, 621–632. [Google Scholar] [CrossRef]
- Fisher, E.M.C. and D.M. Bannerman, Mouse models of neurodegeneration: Know your question, know your mouse. Science translational medicine 2019, 11. [Google Scholar] [CrossRef]
- Sabbagh, J.J., J. W. Kinney, and J.L. Cummings, Animal systems in the development of treatments for Alzheimer's disease: challenges, methods, and implications. Neurobiol Aging 2013, 34, 169–183. [Google Scholar] [CrossRef]
- Cotman, C.W. and E. Head, The canine (dog) model of human aging and disease: dietary, environmental and immunotherapy approaches. Journal of Alzheimer's disease : JAD 2008, 15, 685–707. [Google Scholar] [CrossRef]
- Head, E. , A canine model of human aging and Alzheimer's disease. Biochim Biophys Acta 2013, 1832, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.L., T. M. Ballard, and C. Glavis-Bloom, Animal paradigms to assess cognition with translation to humans. Handbook of experimental pharmacology 2015, 228, 27–57. [Google Scholar] [PubMed]
- Perez, S.E. , et al., Alzheimer's disease pathology in the neocortex and hippocampus of the western lowland gorilla (Gorilla gorilla gorilla). Journal of Comparative Neurology 2013, 521, 4318–4338. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.E. , et al., Early Alzheimer's disease-type pathology in the frontal cortex of wild mountain gorillas (Gorilla beringei beringei). Neurobiol Aging 2016, 39, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Edler, M.K. , et al., Aged chimpanzees exhibit pathologic hallmarks of Alzheimer's disease. Neurobiol Aging 2017, 59, 107–120. [Google Scholar] [CrossRef]
- Oikawa, N., N. Kimura, and K. Yanagisawa, Alzheimer-type tau pathology in advanced aged nonhuman primate brains harboring substantial amyloid deposition. Brain Res 2010, 1315, 137–149. [Google Scholar] [CrossRef]
- Gearing, M. , et al., Neuropathology and apolipoprotein E profile of aged chimpanzees: implications for Alzheimer disease. Proc Natl Acad Sci U S A 1994, 91, 9382–9386. [Google Scholar] [CrossRef]
- Gearing, M. , et al., beta-Amyloid (A beta) deposition in the brains of aged orangutans. Neurobiol Aging 1997, 18, 139–146. [Google Scholar] [CrossRef]
- Heuer, E. , et al., Nonhuman primate models of Alzheimer-like cerebral proteopathy. Current pharmaceutical design 2012, 18, 1159–1169. [Google Scholar] [CrossRef]
- Kitt Cheryl, A. , et al., Evidence for Cholinergic Neurites in Senile Plaques. Science 1984, 226, 1443–1445. [Google Scholar] [CrossRef]
- Mufson, E.J. , et al., Apolipoprotein E-immunoreactivity in aged rhesus monkey cortex: Colocalization with amyloid plaques. Neurobiology of Aging 1994, 15, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Didier, E.S. , et al., Contributions of Nonhuman Primates to Research on Aging. Veterinary pathology 2016, 53, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Callejas, J.D., E. Fuchs, and C. Perez-Cruz, Evidence of Tau Hyperphosphorylation and Dystrophic Microglia in the Common Marmoset. Frontiers in Aging Neuroscience 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- hAβ-KI Alzforum. Available from: https://www.alzforum.org/research-models/hav-ki.
- Baglietto-Vargas, D. , et al., Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer's disease-like pathology. Nat Commun 2021, 12, 2421. [Google Scholar] [CrossRef]
- Serrano-Pozo, A. , et al., Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011, 1, a006189. [Google Scholar] [CrossRef]
- Squire, L.R., B. Knowlton, and G. Musen, The structure and organization of memory. Annu Rev Psychol 1993, 44, 453–495. [Google Scholar] [CrossRef]
- Bussière, T. , et al., Stereologic assessment of the total cortical volume occupied by amyloid deposits and its relationship with cognitive status in aging and Alzheimer’s disease. Neuroscience 2002, 112, 75–91. [Google Scholar] [CrossRef]
- Wang, L. , et al., Abnormalities of hippocampal surface structure in very mild dementia of the Alzheimer type. Neuroimage 2006, 30, 52–60. [Google Scholar] [CrossRef]
- Puzzo, D. , et al., Behavioral assays with mouse models of Alzheimer's disease: practical considerations and guidelines. Biochem Pharmacol 2014, 88, 450–467. [Google Scholar] [CrossRef]
- Clark, R.E. and L.R. Squire, Similarity in form and function of the hippocampus in rodents, monkeys, and humans. Proceedings of the National Academy of Sciences 2013, 110 (Suppl. S2), 10365–10370. [Google Scholar] [CrossRef]
- Watrous, A.J. , et al., A comparative study of human and rat hippocampal low-frequency oscillations during spatial navigation. Hippocampus 2013, 23, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Visser, P.J. , et al., Cerebrospinal fluid tau levels are associated with abnormal neuronal plasticity markers in Alzheimer’s disease. Molecular Neurodegeneration 2022, 17, 27. [Google Scholar] [CrossRef]
- An, S.S.A. and J.P. Hulme, Plasma amyloid-beta oligomer and phosphorylated tau: diagnostic tools for progressive Alzheimer’s disease. Neural Regeneration Research 2023, 18, 2391–2392. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. , et al., Dual-Modal NIR-Fluorophore Conjugated Magnetic Nanoparticle for Imaging Amyloid-beta Species In Vivo. Small 2018, 14, e1800901. [Google Scholar] [CrossRef]
- Peng, C. , et al., Versatile fluorescent probes for near-infrared imaging of amyloid-β species in Alzheimer's disease mouse model. Journal of Materials Chemistry B 2019, 7, 1986–1995. [Google Scholar] [CrossRef]
- Jack, C.R., Jr. , et al., Magnetic resonance imaging of Alzheimer's pathology in the brains of living transgenic mice: a new tool in Alzheimer's disease research. Neuroscientist 2007, 13, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J. , et al., Evaluation of the neuroprotective effect of taurine in Alzheimer’s disease using functional molecular imaging. Scientific Reports 2020, 10, 15551. [Google Scholar] [CrossRef] [PubMed]
- Chiquita, S. , et al., A longitudinal multimodal in vivo molecular imaging study of the 3xTg-AD mouse model shows progressive early hippocampal and taurine loss. Hum Mol Genet 2019, 28, 2174–2188. [Google Scholar] [CrossRef]
- Guell-Bosch, J. , et al., Progression of Alzheimer's disease and effect of scFv-h3D6 immunotherapy in the 3xTg-AD mouse model: An in vivo longitudinal study using Magnetic Resonance Imaging and Spectroscopy. NMR Biomed 2020, 33, e4263. [Google Scholar] [CrossRef]
- Mlynarik, V. , et al., Proton and phosphorus magnetic resonance spectroscopy of a mouse model of Alzheimer's disease. J Alzheimers Dis 2012, 31 (Suppl 3), S87–99. [Google Scholar] [CrossRef]
- Benavides, F. , et al., Genetic quality assurance and genetic monitoring of laboratory mice and rats: FELASA Working Group Report. Lab Anim 2020, 54, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Mekada, K. , et al., Development of SNP markers for C57BL/6N-derived mouse inbred strains. Exp Anim 2015, 64, 91–100. [Google Scholar] [CrossRef]
- Shinohara, M. , et al., Regional distribution of synaptic markers and APP correlate with distinct clinicopathological features in sporadic and familial Alzheimer's disease. Brain 2014, 137 Pt 5, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.M. , et al., Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol 2015, 129, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Hyde, L.A. , et al., Studies to investigate the in vivo therapeutic window of the γ-secretase inhibitor N2-[(2S)-2-(3, 5-difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6, 7-dihydro-5H-dibenzo [b, d] azepin-7-yl]-L-alaninamide (LY411, 575) in the CRND8 mouse. Journal of Pharmacology and Experimental Therapeutics 2006, 319, 1133–1143. [Google Scholar]
- Head, E. , et al., A Two-Year Study with Fibrillar β-Amyloid (Aβ) Immunization in Aged Canines: Effects on Cognitive Function and Brain Aβ. J. Neurosci. 2008, 28, 3555. [Google Scholar] [CrossRef]
- Davis, P.R. , et al., Aβ vaccination in combination with behavioral enrichment in aged beagles: effects on cognition, Aβ, and microhemorrhages. Neurobiology of aging 2017, 49, 86–99. [Google Scholar] [CrossRef]
Figure 2.
Illustrates the animal models used to replicate EOAD and LOAD (sporadic) pathologies over recent decades. (Created with BioRender.com).
Figure 2.
Illustrates the animal models used to replicate EOAD and LOAD (sporadic) pathologies over recent decades. (Created with BioRender.com).
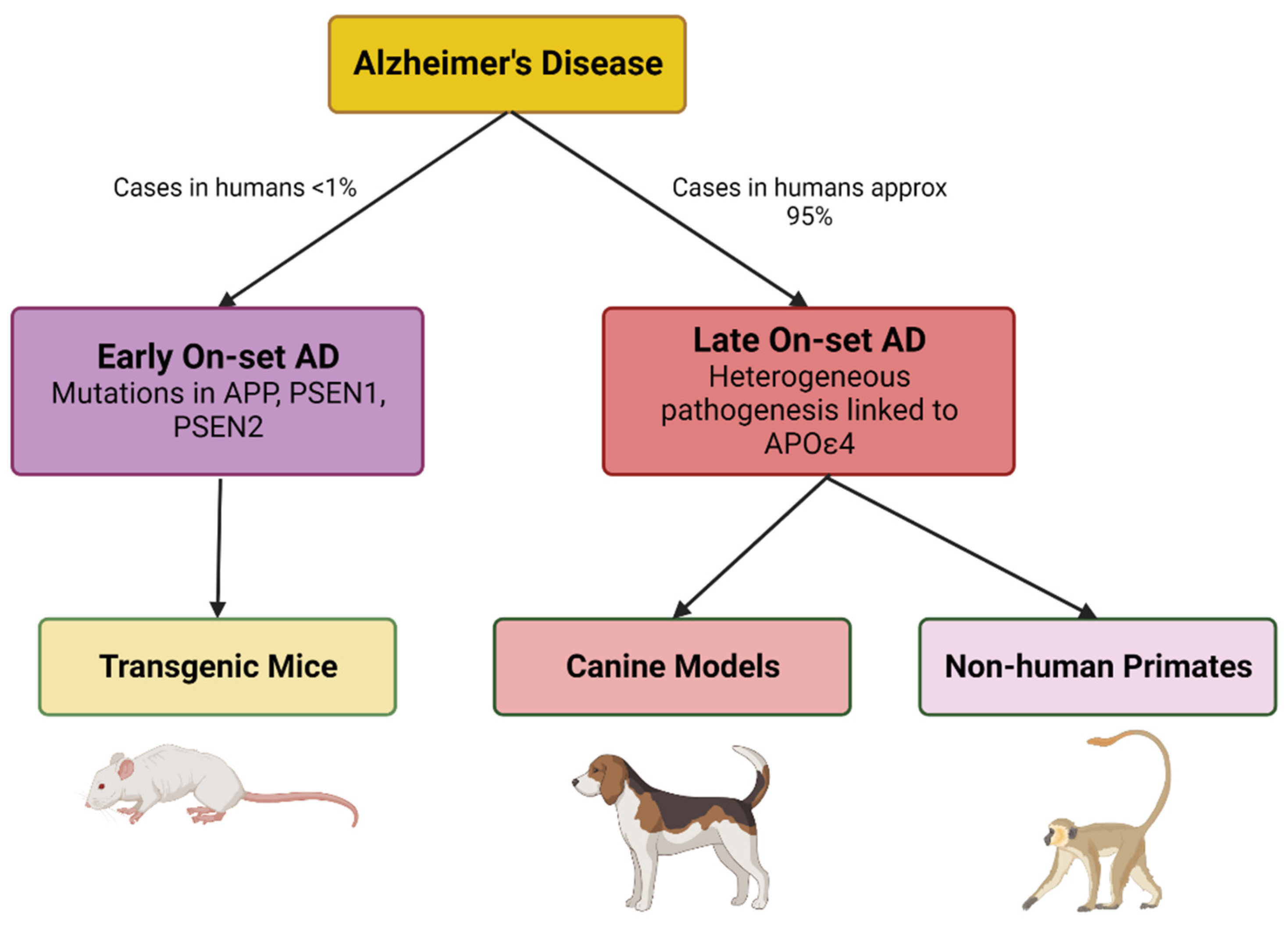
Figure 3.
Clinically relevant blood plasma biomarkers used in AD detection and Aβ: Amyloid-beta; CSF: cerebral spinal fluid; NCI: non-cognitive impairment; OAβ: oligomer amyloid beta; SCI: subjective cognitive impairment. Modified with permission [157].
Figure 3.
Clinically relevant blood plasma biomarkers used in AD detection and Aβ: Amyloid-beta; CSF: cerebral spinal fluid; NCI: non-cognitive impairment; OAβ: oligomer amyloid beta; SCI: subjective cognitive impairment. Modified with permission [157].
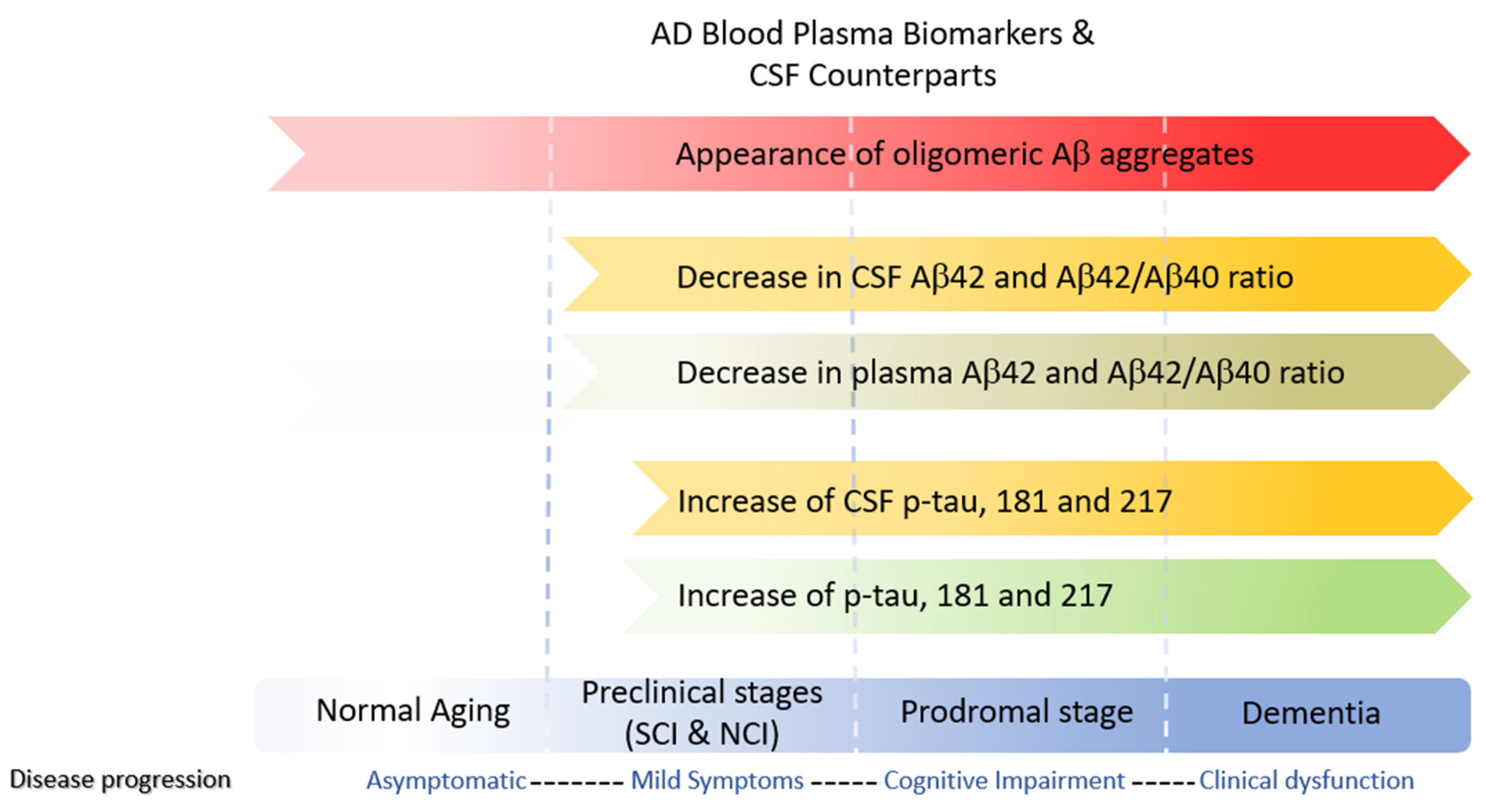
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



