
Submitted:
01 September 2023
Posted:
06 September 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Pulmonary fibrosis is a chronic, progressive, and irreversible lung disease characterized by fibrotic scarring in the lung parenchyma. This condition involves the excessive accumulation of extracellular matrix (EM) due to a persistently activated wound-repair response. The aberrant activation of myofibroblasts in the alveolar environment by Transforming Growth Factor beta (TGF-β) and other signaling molecules is considered a key event in the development and progression of fibrosis. A primary target of TGF-β signaling in fibrosis is Collagen Triple Helix Repeat Containing 1 (CTHRC1), a secreted glycoprotein that plays a pivotal role in extracellular matrix deposition. CTHRC1 is transcriptionally regulated by TGF-β and inhibits both TGF-β and canonical Wnt signaling pathways. This dual function suggests that CTHRC1 is vital in regulating tissue remodeling during wound repair.
In this review, we will highlight recent studies suggesting that CTHRC1 can serve as a diagnostic and prognostic biomarker for fibrosis in idiopathic pulmonary fibrosis, rheumatoid arthritis-interstitial lung disease, systemic sclerosis, and post-COVID lung fibrosis. Notably, the expression of CTHRC1 is responsive to antifibrotic drugs such as pirfenidone indicating its potential as a therapeutic target. Collectively, these findings suggest that CTHRC1 may present new opportunities for the diagnosis, stratification, and treatment of patients with lung fibrosis.
Keywords:
Biomarker
; CTHRC1
; extracellular matrix
; interstitial lung disease
; idiopathic pulmonary fibrosis
; rheumatoid arthritis
; post-COVID
; pirfenidone
1. Introduction
Diffuse parenchymal lung diseases (DPLDs) encompass a diverse array of disorders (Figure 1), primarily characterized by progressive fibrosis of the pulmonary architecture, frequently culminating in respiratory failure [1,2]. Despite the varied etiologies of these pulmonary disorders, the majority exhibit aberrant fibrotic proliferation and infiltration of inflammatory cells as principal clinical manifestations, impacting the alveolar walls of the lungs. These pathological aberrations are predominantly observed in the lung interstitium, hence the alternate designation of interstitial lung diseases (ILDs) [3].
Idiopathic pulmonary fibrosis (IPF) is the most prevalent fibrotic ILD, characterized by the radiographic and histopathological pattern of usual interstitial pneumonia (UIP) in the absence of an identifiable etiology or association with a known cause of pulmonary fibrosis [4]. IPF is chronic and irreversible and frequently results in respiratory failure and mortality. IPF exhibits a higher incidence in males than females and is more prominent among individuals aged 60 years and above [5]. In contrast, other ILDs typically present at a younger mean age (20 to 60 years) and display an equal distribution between the sexes [5,6].
For classification and therapeutic considerations, ILDs are usually assigned to distinct disease categories (Figure 1), primarily based on the presence of an underlying medical condition (e.g., pulmonary fibrosis associated with rheumatoid arthritis), an inciting factor (e.g., pneumoconiosis), or the absence of an identifiable etiology (e.g., IPF) [7,8,9,10].
Symptoms of ILDs often overlap with those of other respiratory diseases, such as chronic obstructive pulmonary disease (COPD), asthma, or even heart failure. This makes it difficult to diagnose and stratify patients based solely on clinical presentation. Advances in molecular diagnostics and identification of disease biomarkers may provide a more accurate diagnosis and classification and may lead to additional treatment options [11]. In addition, biomarkers may improve the monitoring of treatment and outcomes. Recent research has highlighted the role of CTHRC1, a secreted protein involved in extracellular matrix (ECM) and tissue remodeling, as a biomarker for lung fibrosis and, together with co-expressed proteins, as a marker for disease diagnosis and the monitoring of pirfenidone treatment [12,13,14,15].
This review will focus on the pathophysiological characteristics of pulmonary fibrosis and its association with rheumatoid arthritis, as well as the potential role of CTHRC1 as a biomarker for both diseases.
2. Epidemiology
Despite the relatively low incidence of specific fibrosing ILDs, these conditions collectively impact a significant number of patients. Current estimates indicate 76.0 ILD cases per 100,000 individuals in Europe and 74.3 cases per 100,000 in the United States [16]. The most prevalent fibrotic ILDs include sarcoidosis, connective tissue disease (CTD)-associated ILDs, and IPF, with prevalence estimates of 30.2, 12.1, and 8.2 cases per 100,000 individuals [2,16].
3. Pathogenesis of lung fibrosis
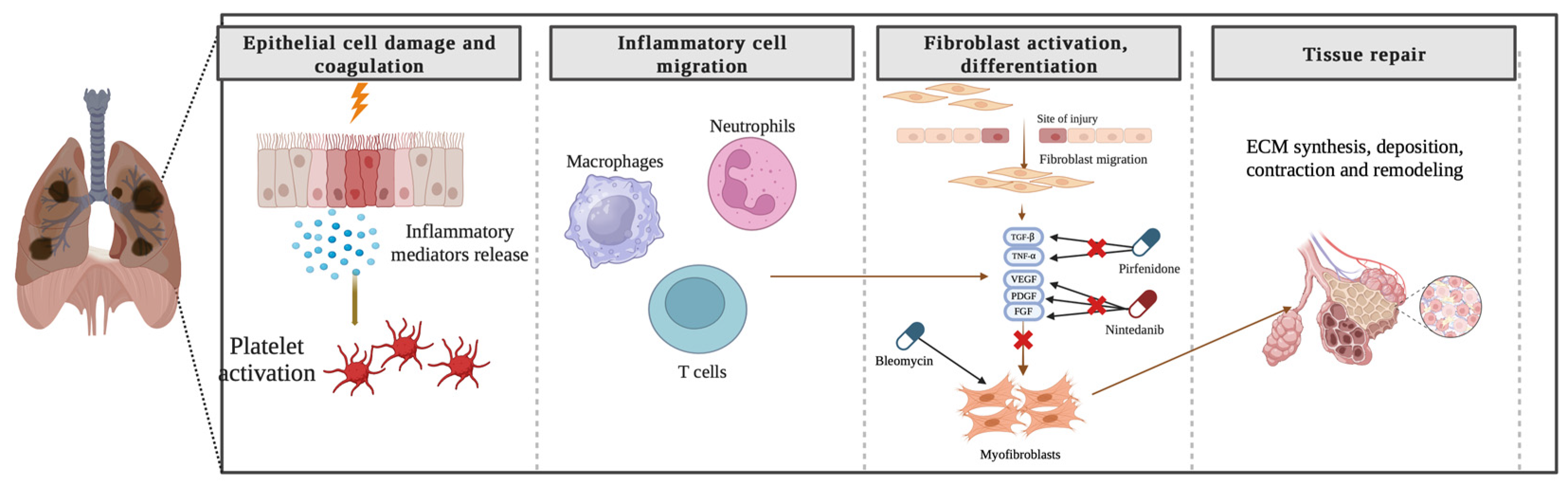
The precise pathogenesis of fibrosing ILDs remains incompletely understood. In the context of pulmonary fibrosis, diverse disease-specific triggers initiate abnormal cascades of inflammatory responses, culminating in the synthesis, deposition, contraction, and remodeling of ECM components and the formation of fibrotic tissue (Figure 2). Numerous aspects of the etiology of specific diseases and the criteria demarcating normal wound healing from fibrotic progression remain obscure. Notwithstanding the myriad etiologies underlying pulmonary fibrosis, the advanced stages exhibit shared pathophysiological mechanisms [20,21].
Genetic studies have identified both common and rare susceptibility loci associated with pulmonary fibrosis [22]. For instance, a common polymorphism in the MUC5B promoter, which is implicated in airway clearance and microbial host defense, has been correlated with an increased risk of IPF, rheumatoid arthritis with ILD (RA-ILD) [23], and chronic hypersensitivity pneumonitis (CHP) [24] but not with systemic sclerosis-ILD (SSc-ILD), sarcoidosis, or antisynthetase syndrome. Patients with IPF, RA-ILD, and CHP exhibit telomere shortening and alterations in the telomere-related genes TERT, TERC, RTEL1, and PARN [23,24]. Certain rare genetic variations, including genetic mutations affecting telomeres, are strongly associated with progressive disease [23].
In addition to shared genetic risk factors, the early stages of various ILDs exhibit heterogeneous and overlapping molecular pathways [7]. In IPF, an unidentified disruption of alveolar epithelial cell integrity might trigger pathology through interactions between endothelial cells and myofibroblasts [6]. In sarcoidosis, only a small fraction of patients develops fibrosis in response to granulomatous inflammation caused by a putative, persistent, and yet-to-be-identified etiology [25].
In a substantial proportion of SSc-ILD patients, an amalgamation of inflammatory responses, endothelial dysfunction, and vasculopathy contributes to pulmonary fibrosis, ultimately determining the disease outcome [20]. Investigation into specific cases has indicated that varied inflammatory responses may foster a profibrotic environment and cytokine milieu characterized by transforming growth factor, connective tissue growth factor, platelet-derived growth factor, and the activation of Wnt and Hedgehog signaling pathways. The complex interplay of signaling processes may initiate and sustain fibroblast activation and differentiation into myofibroblasts, leading to fibrosis development, including pathologic collagen deposition and scar formation [20].
4. CTHRC1—structural characteristics and expression
CTHRC1 is a secreted, highly conserved ECM glycoprotein that is pivotal in orchestrating vascular remodeling and tissue repair processes while promoting cell migration [26,27]. CTHRC1 encompasses an NH2-terminal peptide, a short collagen triple helix repeat comprising 36 amino acids, and a COOH-terminal globular domain [26]. Secreted CTHRC1 (30 kDa) observed in higher-order complexes exhibits a considerably increased molecular weight in comparison to cellular CTHRC1 (26 kDa) [28].
CTHRC1 is transiently expressed during adventitial remodeling and primarily associates with myofibroblasts. Under normal conditions, CTHRC1 is undetectable in healthy arteries and is predominantly expressed in various mesenchymal-derived cells during growth and injury-induced tissue repair [27,28]. During skeletal development, abundant CTHRC1 expression is observed in cartilage primordia, growth plate cartilage, bone matrix, and periosteum. In other tissues, CTHRC1 expression overlaps considerably with interstitial collagens and transforming growth factor-β (TGF-β) family members, indicating substantial collagen synthesis in the presence of TGF-β. Furthermore, the activation of TGF-β signaling may promote the proliferation and migration of vascular smooth muscle cells, resulting in neointimal hyperplasia [29,30]. Pathological expression of CTHRC1 has also been detected in the matrix of calcifying atherosclerotic plaques and mineralized bone tissues [31].
Elevated CTHRC1 expression has been implicated in the pathophysiology of numerous cancerous and autoimmune disorders. Accordingly, CTHRC1 was identified as a novel oncogene aberrantly overexpressed in various malignant tumors, exhibiting associations with bone metastasis and unfavorable patient outcomes [32,33,34,35]. CTHRC1 gene expression is also upregulated in RA, lupus erythematosus, muscular dystrophies, and lung, cardiac, and liver fibrosis [15,36,37,38,39,40,41,42]. In contrast, CTHRC1 silencing protects against asthmatic airway remodeling and inflammation both in vitro and in vivo [43].
5. CTHRC1 plays a role in several signaling pathways
CTHRC1 enhances collagen promoter activity by binding to ligands, potentially playing a role in vascular remodeling through the regulation of collagen matrix deposition and cell migration [44]. CTHRC1 directly or indirectly regulates and affects several signaling pathways, such as TGF-β, Wnt, integrin β/FAK, Src/FAK, MEK/ERK, PI3K/AKT/ERK, HIF-1α, and PKC-δ/ERK. The Cthrc1 transcript is induced by both TGF-β and BMP-4, and its inhibition of TGF-β sensitive reporters, along with the observed reduction in phospho-Smad2/3 levels in vivo, collectively support the evidence that these proteins could function as antagonists to preserve equilibrium among extracellular matrix components [44]. In addition, CTHRC1 promotes accelerated wound repair in mice by regulating the TGF-β and Notch pathways [45]. Similarly, CTHRC1 may also contribute to IL-1β-induced apoptosis of chondrocytes by activating the JNK1/2 pathway [46]. CTHRC1-expressing synoviocytes exerted an anti-inflammatory effect in a collagen antibody-induced arthritis model [47]. CTHRC1 can also contribute to fat and glycogen synthesis [27,48]. Moreover, CTHRC1 has been identified as a positive regulator of bone homeostasis that increases bone mass by promoting osteoblastic bone formation.
6. CTHRC1 as a biomarker for RA
CTHRC1 has recently been reported as a potential biomarker for RA diagnosis [36]. Initially, CTHRC1 was linked to RA pathogenesis in mice via a Cthrc1 gene polymorphism, which affected arthritis progression in mouse models. In mice, the Cthrc1 gene is located within the proteoglycan-induced arthritis 8 (Pgia8) locus on chromosome 15, and this locus controls PGIA severity in a sex-specific manner [49,50]. Within the Pgia8 locus, Cthrc1 strongly correlated with arthritis severity corresponding to the levels of the proinflammatory cytokines IL-6 and IL-1β [50].
To assess the impact of CTHRC1 on RA in humans, CTHRC1 levels were analyzed in plasma. The results showed a significant increase in individuals with established RA compared to healthy individuals [36]. This elevation was positively associated with RA disease markers, such as RF, anti-citrullinated protein antibodies (ACPA), and C-reactive protein (CRP) [36]. A similar study independently identified CTHRC1 as a diagnostic marker for RA [51]. Together, the findings in mouse models and the observations in patients indicate that CTHRC1 may be a potential biomarker for RA diagnosis. CTHRC1 serum levels may also be a candidate diagnostic biomarker for inflammatory conditions and fibrosis.
In RA patients, in addition to increased serum levels, immunohistochemistry analysis revealed highly elevated CTHRC1 expression at the invasive edge of the pannus [52]. When closely analyzed, two subtypes of fibroblast-like synoviocytes (FLSs) contributed to high expression in isolated tissues from RA patients. One subtype was characterized by the CD34+ CDH11+ THY1/CD90- phenotype and was proposed to recruit monocytes via IL-6, CXCL12, and CCL2. Another phenotype included the expression of CD34− CDH11+ THY1/CD90+, and this subtype was strongly linked with RA pathology [53]. In addition, both subsets also expressed several other genes related to migration and invasion, including TWIST1, POSTN, LOXL2, PDGFRB, and MMP14 [54,55], and these activities were confirmed in vitro. Therefore, CTHRC1 may contribute to synovial destruction by modulating the migration and invasion of various FLS populations, resulting in an abnormal immune response in RA.
Many studies have revealed CTHRC1 expression in FLSs in RA, and CTHRC1 can participate in the Wnt or TGF-β signaling pathways. Upregulated expression of these signaling pathways is observed in RA [56], is associated with chronic activation of FLSs and drives a localized immune response. This elevated production is associated with WNT5A expression, which is detected in FLSs and identified as the primary driver of cytokine production during inflammation [57,58]. Similarly, circulating WNT5A was associated with lung fibrosis associated with RA [59].
7. Common pathogenic pathways in RA-ILD and pulmonary fibrosis
The precise etiology of RA-associated lung fibrosis is not yet known. A complex interplay between genetic and environmental factors may contribute to disease pathogenesis. Individuals with specific, shared HLA-DRB1 epitopes show enhanced susceptibility [60,61]. Environmental risk factors, such as tobacco smoking, may further enhance immunogenicity in these individuals by triggering smoking-induced citrullination of lung proteins [62,63,64]. Increased citrullination can be a direct target for ACPAs and T-cell-mediated immunity, resulting in immune factors targeting the lung. However, elevated protein citrullination has also been observed in non-smoking individuals, suggesting the potential involvement of additional environmental factors, including particulate matter, silica, atmospheric pollution, and alterations in the pulmonary microbiome [65,66]. These environmental triggers may induce aberrant gene expression and subsequently activate the immune system, leading to the generation of circulating autoantibodies [67]. This process may, in turn, instigate pro-inflammatory cascades, culminating in persistent pulmonary inflammation [67,68].
Lung inflammation may contribute to early RA-related autoimmunity. Multiple mechanisms have been proposed through which mucosal inflammation progresses to joint inflammation [65]. These include commonly shared autoantigens, epitope spreading to a joint-specific antigen and circulating immune complexes that may be responsible for the onset of articular disease. For example, five shared citrullinated target peptides were identified between the lung and synovial tissue, two of which are citrullinated-vimentin peptides [69,70]. These shared autoantigens may provide a direct link between multifactorial diseases involving the lung and joints.
Among RA-associated autoantibodies, increased levels of ACPA, rheumatoid factor (RF), anti-peptidyl arginine deiminases (anti-PADs), and anti-carbamylated protein antibodies are observed three to five years prior to the appearance of clinically apparent inflammatory arthritis (IA) [71,72,73]. These circulating autoantibodies point to an extra-articular site as the origin of autoimmunity. The mucosal origin of autoimmunity in pre-RA can be supported by the responses of IgA-related autoimmunity, particularly IgA-ACPA responses [74]. To date, among mucosal sites, the lung has been extensively studied due to the high number of reports of lung diseases preceding joint destruction in RA [75]. In addition, organized lymphoid areas in the lungs, named inducible bronchus-associated lymphatic tissue (iBALT), are implicated in RA-related autoimmunity [76]. Other proposed autoimmunity triggers include mucosal inflammation in the gut and/or gingiva, and more research is needed to fully understand the contribution of these sites to autoimmunity in RA [77,78,79].
The concept of a shared pathogenic mechanism underlying the development of RA-ILD and IPF is supported by phenotypic similarities and shared environmental risk factors. The discovery of an excess of uncommon genetic variations found in RA-ILD patients linked to familial pulmonary fibrosis provides further support [80]. Notably, there was an excess of variants in genes involved in telomere maintenance (TERT, PARN, RETL1) and in the gene SFTPC, which encodes a protein involved in surfactant homeostasis, with an odds ratio (OR) of 3.17 when compared to controls [80]. Although these findings point to a shared genetic background between RA-ILD and IPF, at least for rare variants, one of the main limitations was the absence of patients with RA without ILD in the control group. Thus, these findings do not rule out the possibility that these mutations impact the genetic predisposition to RA. In addition, a functional MUC5B rs35705950 promoter variation, which is a significant risk factor for IPF (OR 8.3; 95% CI, 5.8-11.9; P4.6 × 10−31) [81], was recently discovered to be a risk factor for RA-ILD but was not connected to RA without ILD. Importantly, intense MUC5B staining was observed in the hyperplastic alveolar epithelium in fibrotic regions of lung biopsies from individuals with RA-ILD, comparable to what was observed in IPF [23]. This finding clearly indicates that RA-ILD and IPF have the same genetic architecture [23,81].
8. Role of CTHRC1 in pulmonary fibrosis
The fibrotic environment in the lung might be due to chronic Wnt/β-catenin signaling that induces cellular senescence in lung epithelial cells. This chronic increase in Wnt/β-catenin signaling might contribute to the senescence of alveolar type II cells and cell reprogramming, thus leading to further progenitor cell dysfunction and impaired lung repair [82].
In a murine model of bleomycin-induced lung fibrosis, CTHRC1 attenuated fibrotic tissue formation and preserved lung function [41], suggesting that the regulatory activity of CTHRC1 may inhibit TGF-β [44] and indirectly canonical Wnt signaling [28]. In a Cthrc1 knockout murine model, bleomycin administration resulted in increased immune response and decreased lung function, most likely due to the rapid accumulation of collagen in the lung [41]. These observations underscore the significant role that CTHRC1 plays in modulating lung matrix biology.
Analogous clinical manifestations are observed in patients with lung fibrosis, including breathing problems that result in severe dyspnea in the later stage of the disease. In patients, these signs and symptoms are typically preceded by months of undetected early disease progression [83]. Several studies have consistently reported that the invasive phenotype of fibroblasts drives pulmonary fibrosis in the bleomycin-induced murine model [84,85]. Significantly, CTHRC1 levels were elevated in these pathological fibroblasts. One study identified a fibroblast subset populating the fibrotic lungs of both mice and humans, characterized by high levels of type 1 collagen production, particularly COL1A1 and COL3A1, and increased expression of CTHRC1 [15]. Importantly, these COL1A1+ and CTHRC1+ cells were designated as ‘pathological fibroblasts,’ given their enhanced migratory capacity to colonize the lung and their localization at the edge of fibrotic processes [15]. Another study corroborated these results and further showed that inhibition of the CTHRC1/HIF-1α axis by NEDD4L-induced β-catenin ubiquitination impedes the initiation and progression of interstitial lung fibrosis [86].
Similarly, CTHRC1 has emerged as a marker of activated fibroblasts driving the development of SSc. Previous studies implicated the TGF-β-responsive genes periostin (POSTN) and cartilage oligomeric matrix protein (COMP) as biomarkers of activated TGF-β signaling and showed that POSTN and COMP exhibit heightened expression in the skin, manifest correlation with modified Rodnan skin score (mRSS), and demonstrate predictive value concerning disease progression [87,88]. In both dermal tissues from SSc patients and skin tissues from mice with bleomycin-induced fibrosis, CTHRC1 expression was elevated along with POSTN and other pathologic ECM components [89,90]. In the bleomycin-induced murine model of dermal fibrosis, recombinant CTHRC1 inhibited TGF-β-stimulated collagen deposition by fibroblasts and reduced fibrotic changes [91].
Taken together, the elevated expression of CTHRC1 in a subset of pathological fibroblasts driving fibrosis development suggests that it might be a key player in driving fibrotic processes by contributing to abnormal tissue repair, ECM remodeling, and myofibroblast activation. The identification of CTHRC1 as a marker for pathologic fibroblasts highlights its potential as a diagnostic biomarker for fibrotic diseases, including IPF, RA-ILD, and SSc. Targeting CTHRC1 and the signaling pathways it influences could offer novel therapeutic strategies to mitigate fibrosis progression and improve patient outcomes.
9. CTHRC1: A Potential Prognostic Marker for Severe Lung Complications in COVID-19 Patients
The coronavirus disease 2019 (COVID-19) pandemic due to the SARS-CoV-2 virus caused a range of symptoms in affected individuals, from asymptomatic cases to severe conditions that resulted in lung fibrosis and potentially fatal respiratory failure [92]. Lung tissue affected by COVID-19 showed a considerable increase in the number of cells from the monocyte-macrophage lineage [71,93], and these were predominantly located in the extravascular lung tissue, which is primarily ‘interstitial’ rather than ‘alveolar’ [71,73]. These macrophage populations were described as extremely active and expressed inflammatory markers as well as genes linked with tissue repair and fibrogenesis [14,94].
Patients experiencing long-haul COVID display enduring disruptions in immune responses even 8 months post-infection. This is evident through persistent increases in activated CD14+CD16+ monocytes and plasmacytoid dendritic cells compared to controls. Additionally, sustained elevation in type I (IFNβ) and type III (IFNλ1) interferons persists, which, along with other factors such as pentraxin 3, IFNγ, IFNλ2/3, and IL-6, forms a combination indicative of long-haul COVID, with accuracies reaching 78.5% to 81.6% [95]. These elements, often tied to acute severe disease, imply a delayed or ineffective resolution of inflammation in these patients. Conversely, overly intense inflammatory responses may precipitate irreversible lung fibrosis, causing significant respiratory function impairment. Markers, such as lipocalin-2, matrix metalloprotease-7, and hepatocyte growth factor, closely align with inflammation severity and impaired pulmonary function [96]. The capacity of SARS-CoV-2 to alter immune homeostasis mechanisms impacting tissue inflammation likely underlies persistent lung injuries. The decline in alveolar macrophages, integral for lung integrity, in severe COVID-19 may stem from damage to alveolar type II cells, which also express ACE2 receptors that the virus targets [97,98,99]. In COVID-19 fatalities, lung analyses have unveiled inflammation-associated AT2 cell states that hinder proper regeneration, coupled with pathogenic fibroblasts expressing CTHRC1, possibly driving rapid pulmonary fibrosis progression [100]. TGF-β and epithelium-derived IL-6 could be implicated in this fibrotic process [14,15,101]. Macrophages have been implicated in the immunopathology observed in fatal COVID-19 cases, and profibrotic macrophages, particularly those involving interleukin 1 beta (IL-1β), can hinder epithelial repair [14,102]. Importantly, a subpopulation of CTHRC1+ pathological fibroblasts was found to be increased in lungs affected by COVID-19 [15,93] and associated with the progression of pulmonary fibrosis in COVID-19 patients [14,100,103]. Researchers identified four fibroblast clusters—adventitial, alveolar, intermediate pathological, and pathological. Remarkably, the latter two cell clusters exhibited substantial expansion in COVID-19 lungs compared to controls and were characterized by the expression of CTHRC1, COL1A1, and COL3A1 [14,100,103]. These fibroblasts have previously been identified as pathologic drivers of fibrosis in patients with IPF [15]. These cells produce the highest levels of type 1 collagen and have an enhanced capacity to migrate and colonize the lung [15]. The increased risk of developing fibrosis could be attributed to the emergence of these CTHRC1+ fibroblast populations, as well as their demonstrated close relationship to macrophages [71,93].
A recent study discovered overexpression of profibrotic genes, including collagen and POSTN, in COVID-19 patients. This study further confirmed a significant increase in the expression of CTHRC1, a marker for myofibroblasts, colocalizing with regions of high alpha-smooth muscle expression [104,105,106]. Importantly, pulmonary fibrosis can develop and persist even after a patient fully recovers from COVID-19, emphasizing the need for diagnostic biomarkers and long-term treatment options to slow the progression of the disease [107].
Overall, these findings suggest that CTHRC1 may serve as a diagnostic biomarker for patient stratification. Additionally, targeting CTHRC1 expression may represent a potential therapeutic avenue for slowing the progression of fibrosis in patients who experience severe COVID-19 symptoms.
10. Targeting CTHRC1: Therapeutic options for lung fibrosis
Numerous compounds with diverse mechanisms of action are currently being evaluated in clinical trials as potential anti-fibrotic drugs [108]. These drugs target specific pathways and molecules that play critical roles in the development and progression of fibrosis. The drugs are often categorized based on their primary targets, and their efficacy is being assessed across various fibrotic diseases. TGF-β is a central player in fibrosis, and several drug categories are designed to modulate its effects. Pirfenidone, an FDA-approved drug for IPF, inhibits TGF-β synthesis and activation [109]. It has shown effectiveness in reducing lung function decline and mortality in IPF patients [13,110].
Nintedanib, a receptor tyrosine kinase inhibitor, has shown benefits in reducing fibrosis and disease progression in IPF [109]. In a mouse model of RA-ILD, nintedanib was found to inhibit the development of lung fibrosis [111,112]. Nintedanib has been investigated in two randomized, double-blind placebo-controlled trials, one of patients with SSc-ILD and one of patients with progressive fibrosing ILD. Both studies showed that nintedanib reduced the decline in forced vital capacity (FVC) [113,114]. ZSP1603, which blocks PDGFRs, FGFRs, and VEGFR2, exhibits anti-fibrotic effects in preclinical models [115]. In addition, Pamrevlumab, an anti-CTGF antibody, has demonstrated efficacy in early-phase trials for IPF [116,117].
The antifibrotic properties of pirfenidone were evaluated in a double-blind, randomized placebo-controlled trial in patients with non-IPF progressive fibrotic ILDs. The study suggested that pirfenidone added to conventional therapy reduced the decline in FVC [118]. This finding may be linked to pirfenidone significantly inhibiting CTHRC1 expression in the bleomycin-induced lung fibrosis model in mice [12]. Since pathological CTHRC1+ fibroblasts were strongly involved in ensuing fibrosis progression in COVID-19 patients, these findings raised the possibility of including pirfenidone within a standard treatment protocol to improve the outcome of post-COVID lung fibrosis patients. Interestingly, recent studies suggest that treating COVID-19 patients with pulmonary fibrosis using pirfenidone, particularly in the early stages of fibrosis, can improve patient outcomes, thus offering a promising therapeutic approach [119,120,121].
11. Conclusion
The information discussed in this review highlights the potential of CTHRC1 as a biomarker for the early detection of pulmonary fibrosis and as a possible target for the development of novel therapeutic strategies. Recent studies have underscored the importance of CTHRC1 in relation to RA-associated pulmonary fibrosis and its regulatory role in maintaining the integrity of the lung matrix. The potential disruption of CTHRC1-related processes might contribute to the abnormal proliferation of fibroblasts and their transformation into invasive phenotypes. In addition, other research has indicated the potential involvement of CTHRC1 in ECM remodeling in different tissues, including hepatic and cardiac fibrosis. The current understanding of CTHRC1 is consistent with its modulatory effects on fibroblast proliferation mediated by the TGF-β and canonical Wnt signaling pathways, as well as EMT. Additional research is necessary to clarify the more specific downstream actions of CTHRC1, aiming to identify potential strategies for therapeutic targeting to manage the progression of RA-related pulmonary fibrotic disease. Furthermore, CTHRC1 alone or as part of a pro-fibrotic gene expression signature could serve as a marker that could aid in the development of more effective diagnostic tools, preventive measures, or targeted therapeutic interventions for the progression of fibrosis and inflammation in DPLDs. CTHRC1 could offer new avenues for the early stratification of patients allowing more effective targeted treatment options.
Author Contributions
Conceptualization, Z.M. and J.K.; writing—original draft preparation, Z.M.; writing—review and editing, Z.M., A.A., and J. K; figures, Z.M. and J. K; project administration, A.A. and J.K.; funding acquisition, J.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a Collaborative Research Program Grant, grant number 021220CRP1722, and a Faculty Development Competitive Research Project Grant, grant number 021220FD2951 (both awarded by Nazarbayev University to JK).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Acknowledgments
We would like to thank Dr. Esther van de Vosse for help with article editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tomassetti, S.; Ravaglia, C.; Poletti, V. Diffuse parenchymal lung disease. Eur. Respir. Rev. 2017, 26, 170004. [Google Scholar] [CrossRef] [PubMed]
- Wijsenbeek, M.; Suzuki, A.; Maher, T.M. Interstitial lung diseases. Lancet 2022, 400, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Podolanczuk, A.J.; Wong, A.W.; Saito, S.; Lasky, J.A.; Ryerson, C.J.; Eickelberg, O. Update in Interstitial Lung Disease 2020. Am. J. Respir. Crit. Care Med. 2021, 203, 1343–1352. [Google Scholar] [CrossRef]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 515–546. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis — A Common Pathway to Organ Injury and Failure. New Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Wolters, P.J.; Blackwell, T.S.; Eickelberg, O.; E Loyd, J.; Kaminski, N.; Jenkins, G.; Maher, T.M.; Molina-Molina, M.; Noble, P.W.; Raghu, G.; et al. Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir. Med. 2018, 6, 154–160. [Google Scholar] [CrossRef]
- Wells, A.U.; Brown, K.K.; Flaherty, K.R.; Kolb, M.; Thannickal, V.J. What’s in a name? That which we call IPF, by any other name would act the same. Eur. Respir. J. 2018, 51, 1800692. [Google Scholar] [CrossRef]
- Gochuico, B.R.; Avila, N.A.; Chow, C.K.; Novero, L.J.; Wu, H.-P.; Ren, P.; MacDonald, S.D.; Travis, W.D.; Stylianou, M.P.; Rosas, I.O. Progressive Preclinical Interstitial Lung Disease in Rheumatoid Arthritis. Arch. Intern. Med. 2008, 168, 159–166. [Google Scholar] [CrossRef]
- Brown, K.K. Roger S. Mitchell Lecture. Rheumatoid Lung Disease. Proc. Am. Thorac. Soc. 2007, 4, 443–448. [Google Scholar] [CrossRef]
- Lurje, I.; Gaisa, N.T.; Weiskirchen, R.; Tacke, F. Mechanisms of organ fibrosis: Emerging concepts and implications for novel treatment strategies. Mol. Asp. Med. 2023, 92, 101191. [Google Scholar] [CrossRef]
- Bauer, Y.; Tedrow, J.; de Bernard, S.; Birker-Robaczewska, M.; Gibson, K.F.; Guardela, B.J.; Hess, P.; Klenk, A.; Lindell, K.O.; Poirey, S.; et al. A Novel Genomic Signature with Translational Significance for Human Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Togo, S.; Kadoya, K.; Tulafu, M.; Namba, Y.; Iwai, M.; Watanabe, J.; Nagahama, K.; Okabe, T.; Hidayat, M.; et al. Pirfenidone attenuates lung fibrotic fibroblast responses to transforming growth factor-β1. Respir. Res. 2019, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 2021, 595, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Tsukui, T.; Sun, K.-H.; Wetter, J.B.; Wilson-Kanamori, J.R.; Hazelwood, L.A.; Henderson, N.C.; Adams, T.S.; Schupp, J.C.; Poli, S.D.; Rosas, I.O.; et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Duchemann, B.; Annesi-Maesano, I.; de Naurois, C.J.; Sanyal, S.; Brillet, P.-Y.; Brauner, M.; Kambouchner, M.; Huynh, S.; Naccache, J.M.; Borie, R.; et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur. Respir. J. 2017, 50, 1602419. [Google Scholar] [CrossRef]
- Nannini, C.; Ryu, J.H.; Matteson, E.L. Lung disease in rheumatoid arthritis. Curr. Opin. Rheumatol. 2008, 20, 340–346. [Google Scholar] [CrossRef]
- Ascherman, D.P. Interstitial Lung Disease in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2010, 12, 363–369. [Google Scholar] [CrossRef]
- Kadura, S.; Raghu, G. Rheumatoid arthritis-interstitial lung disease: manifestations and current concepts in pathogenesis and management. Eur. Respir. Rev. 2021, 30, 210011. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Györfi, A.-H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Meyer, K.C. Pulmonary fibrosis, part I: Epidemiology, pathogenesis, and diagnosis. Expert Rev. Respir. Med. 2017, 11, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Adegunsoye, A.; Vij, R.; Noth, I. Integrating Genomics Into Management of Fibrotic Interstitial Lung Disease. Chest 2019, 155, 1026–1040. [Google Scholar] [CrossRef] [PubMed]
- Juge, P.-A.; Lee, J.S.; Ebstein, E.; Furukawa, H.; Dobrinskikh, E.; Gazal, S.; Kannengiesser, C.; Ottaviani, S.; Oka, S.; Tohma, S.; et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. N. Engl. J. Med. 2018, 379, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Torgerson, D.G.; Oldham, J.M.; Adegunsoye, A.; Liu, S.; Li, J.; Elicker, B.M.; Henry, T.S.; Golden, J.A.; Jones, K.D.; et al. Rare Protein-Altering Telomere-related Gene Variants in Patients with Chronic Hypersensitivity Pneumonitis. Am. J. Respir. Crit. Care Med. 2019, 200, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, J.; Grutters, J.C.; Arkema, E.V.; Saketkoo, L.A.; Moller, D.R.; Müller-Quernheim, J. Sarcoidosis. Nat. Rev. Dis. Prim. 2019, 5, 45. [Google Scholar] [CrossRef]
- Pyagay, P.; Heroult, M.; Wang, Q.; Lehnert, W.; Belden, J.; Liaw, L.; Friesel, R.E.; Lindner, V. Collagen Triple Helix Repeat Containing 1, a Novel Secreted Protein in Injured and Diseased Arteries, Inhibits Collagen Expression and Promotes Cell Migration. Circ. Res. 2005, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Stohn, J.P.; Perreault, N.G.; Wang, Q.; Liaw, L.; Lindner, V. Cthrc1, a Novel Circulating Hormone Regulating Metabolism. PLOS ONE 2012, 7, e47142. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Nishimura, O.; Misaki, K.; Nishita, M.; Minami, Y.; Yonemura, S.; Tarui, H.; Sasaki, H. Cthrc1 Selectively Activates the Planar Cell Polarity Pathway of Wnt Signaling by Stabilizing the Wnt-Receptor Complex. Dev. Cell 2008, 15, 23–36. [Google Scholar] [CrossRef]
- Smith, J.D.; Bryant, S.R.; Couper, L.L.; Vary, C.P.H.; Gotwals, P.J.; Koteliansky, V.E.; Lindner, V. Soluble Transforming Growth Factor-β Type II Receptor Inhibits Negative Remodeling, Fibroblast Transdifferentiation, and Intimal Lesion Formation But Not Endothelial Growth. Circ. Res. 1999, 84, 1212–1222. [Google Scholar] [CrossRef]
- Bryant, S.R.; Bjercke, R.J.; Erichsen, D.A.; Rege, A.; Lindner, V. Vascular Remodeling in Response to Altered Blood Flow Is Mediated by Fibroblast Growth Factor-2. Circ. Res. 1999, 84, 323–328. [Google Scholar] [CrossRef]
- Durmus, T.; LeClair, R.J.; Park, K.-S.; Terzic, A.; Yoon, J.K.; Lindner, V. Expression analysis of the novel gene collagen triple helix repeat containing-1 (Cthrc1). Gene Expr. Patterns 2006, 6, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lee, M.; Yu, G.; Lee, H.; Han, X.; Kim, D. CTHRC1 activates pro-tumorigenic signaling pathways in hepatocellular carcinoma. Oncotarget 2017, 8, 105238–105250. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Ren, F.; Xu, M.; Tan, C.; Weng, W.; Huang, Z.; Sheng, W.; Huang, D. CTHRC1 overexpression predicts poor survival and enhances epithelial-mesenchymal transition in colorectal cancer. Cancer Med. 2018, 7, 5643–5654. [Google Scholar] [CrossRef]
- Liu, J.; Li, W.; Liu, S.; Zheng, X.; Shi, L.; Zhang, W.; Yang, H. Knockdown of Collagen Triple Helix Repeat Containing 1 (CTHRC1) Inhibits Epithelial-Mesenchymal Transition and Cellular Migration in Glioblastoma Cells. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2017, 25, 225–232. [Google Scholar] [CrossRef]
- Jiang, N.; Cui, Y.; Liu, J.; Zhu, X.; Wu, H.; Yang, Z.; Ke, Z. Multidimensional Roles of Collagen Triple Helix Repeat Containing 1 (CTHRC1) in Malignant Cancers. J. Cancer 2016, 7, 2213–2220. [Google Scholar] [CrossRef]
- Myngbay, A.; Bexeitov, Y.; Adilbayeva, A.; Assylbekov, Z.; Yevstratenko, B.P.; Aitzhanova, R.M.; Matkarimov, B.; Adarichev, V.A.; Kunz, J. CTHRC1: A New Candidate Biomarker for Improved Rheumatoid Arthritis Diagnosis. Front. Immunol. 2019, 10, 1353. [Google Scholar] [CrossRef]
- Myngbay, A.; Manarbek, L.; Ludbrook, S.; Kunz, J. The Role of Collagen Triple Helix Repeat-Containing 1 Protein (CTHRC1) in Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 2426. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, Q.; Sun, H. Collagen triple helix repeat containing-1: a novel biomarker associated with disease activity in Systemic lupus erythematosus. Lupus 2018, 27, 2076–2085. [Google Scholar] [CrossRef]
- Spector, I.; Zilberstein, Y.; Lavy, A.; Genin, O.; Barzilai-Tutsch, H.; Bodanovsky, A.; Halevy, O.; Pines, M. The Involvement of Collagen Triple Helix Repeat Containing 1 in Muscular Dystrophies. Am. J. Pathol. 2013, 182, 905–916. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Ma, M.; Jiang, S.; Zhang, X.; Zhang, Y.; Yang, X.; Xu, C.; Tian, G.; Li, Q.; et al. Autocrine CTHRC1 activates hepatic stellate cells and promotes liver fibrosis by activating TGF-β signaling. EBioMedicine 2019, 40, 43–55. [Google Scholar] [CrossRef]
- Binks, A.P.; Beyer, M.; Miller, R.; LeClair, R.J. Cthrc1 lowers pulmonary collagen associated with bleomycin-induced fibrosis and protects lung function. Physiol. Rep. 2017, 5, e13115. [Google Scholar] [CrossRef]
- El-Mallah, R.; Farrag, D.A.; Safwat, N.A. Potential value of collagen triple helix repeat containing-1 (CTHRC1) in systemic lupus erythematosus (SLE) patients with arthritis detected clinically or by musculoskeletal ultrasound. Egypt. Rheumatol. 2023, 45, 197–202. [Google Scholar] [CrossRef]
- Feng, Y.; Hu, J.; Liu, F.; Shang, Y. Collagen Triple Helix Repeat Containing 1 Deficiency Protects Against Airway Remodeling and Inflammation in Asthma Models In Vivo and In Vitro. Inflammation 2023, 46, 925–940. [Google Scholar] [CrossRef] [PubMed]
- LeClair, R.; Lindner, V. The Role of Collagen Triple Helix Repeat Containing 1 in Injured Arteries, Collagen Expression, and Transforming Growth Factor β Signaling. Trends Cardiovasc. Med. 2007, 17, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Zheng, J.-H.; Xia, Z.-H.; Qian, J.; Deng, C.-L.; Yang, S.-L. CTHRC1 promotes wound repair by increasing M2 macrophages via regulating the TGF-β and notch pathways. BioMedicine 2019, 113, 108594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yin, Z.; Zhang, F.; Cao, K.; Sun, H. CTHRC1 mediates IL-1β-induced apoptosis in chondrocytes via JNK1/2 signaling. Int. J. Mol. Med. 2018, 41, 2270–2278. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.-R.; Stohn, J.P.; Wang, Q.; Nagano, K.; Baron, R.; Bouxsein, M.L.; Rosen, C.J.; Adarichev, V.A.; Lindner, V. Inhibition of osteoclast differentiation and collagen antibody-induced arthritis by CTHRC1. Bone 2017, 97, 153–167. [Google Scholar] [CrossRef]
- Stohn, J.P.; Wang, Q.; Siviski, M.E.; Kennedy, K.; Jin, Y.-R.; Kacer, D.; DeMambro, V.; Liaw, L.; Vary, C.P.; Rosen, C.J.; et al. Cthrc1 controls adipose tissue formation, body composition, and physical activity. Obesity 2015, 23, 1633–1642. [Google Scholar] [CrossRef]
- Adarichev, V.A.; Nesterovitch, A.B.; Bárdos, T.; Biesczat, D.; Chandrasekaran, R.; Vermes, C.; Mikecz, K.; Finnegan, A.; Glant, T.T. Sex effect on clinical and immunologic quantitative trait loci in a murine model of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 1708–1720. [Google Scholar] [CrossRef]
- Kudryavtseva, E.; Forde, T.S.; Pucker, A.D.; Adarichev, V.A. Wnt signaling genes of murine chromosome 15 are involved in sex-affected pathways of inflammatory arthritis. Arthritis Rheum. 2011, 64, 1057–1068. [Google Scholar] [CrossRef]
- Hu, T.; Liu, Y.; Tan, L.; Huang, J.; Yu, J.; Wu, Y.; Pei, Z.; Zhang, X.; Li, J.; Song, L.; et al. Value of serum collagen triple helix repeat containing-1(CTHRC1) and 14-3-3η protein compared to anti-CCP antibodies and anti-MCV antibodies in the diagnosis of rheumatoid arthritis. Br. J. Biomed. Sci. 2020, 78, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Shekhani, M.T.; Forde, T.S.; Adilbayeva, A.; Ramez, M.; Myngbay, A.; Bexeitov, Y.; Lindner, V.; Adarichev, V.A. Collagen triple helix repeat containing 1 is a new promigratory marker of arthritic pannus. Arthritis Res. Ther. 2016, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Li, X.; Zhu, H.; Luo, H. Single-Cell Sequencing in Rheumatic Diseases: New Insights from the Perspective of the Cell Type. Aging Dis. 2022, 13, 1633–1651. [Google Scholar] [CrossRef]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Abuwarwar, M.H.; Knoblich, K.; Fletcher, A.L. A pathogenic hierarchy for synovial fibroblasts in rheumatoid arthritis. Ann. Transl. Med. 2018, 6, S75–S75. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Nawata, M.; Wakitani, S. Expression Profiles and Functional Analyses of Wnt-Related Genes in Human Joint Disorders. Am. J. Pathol. 2005, 167, 97–105. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.; Kim, D.W.; Ha, Y.; Ihm, M.H.; Kim, H.; Song, K.; Lee, I. Wnt5a Induces Endothelial Inflammation via β-Catenin–Independent Signaling. J. Immunol. 2010, 185, 1274–1282. [Google Scholar] [CrossRef]
- Rauner, M.; Stein, N.; Winzer, M.; Goettsch, C.; Zwerina, J.; Schett, G.; Distler, J.H.; Albers, J.; Schulze, J.; Schinke, T.; et al. WNT5A is induced by inflammatory mediators in bone marrow stromal cells and regulates cytokine and chemokine production. J. Bone Miner. Res. 2011, 27, 575–585. [Google Scholar] [CrossRef]
- Yu, M.; Guo, Y.; Zhang, P.; Xue, J.; Yang, J.; Cai, Q.; You, X.; Ma, J.; Yang, D.; Jia, Y.; et al. Increased circulating Wnt5a protein in patients with rheumatoid arthritis-associated interstitial pneumonia (RA-ILD). Immunobiology 2019, 224, 551–559. [Google Scholar] [CrossRef]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and environmental risk factors for rheumatoid arthritis. Best Pr. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef]
- Reynisdottir, G.; Karimi, R.; Joshua, V.; Olsen, H.; Hensvold, A.H.; Harju, A.; Engström, M.; Grunewald, J.; Nyren, S.; Eklund, A.; et al. Structural Changes and Antibody Enrichment in the Lungs Are Early Features of Anti-Citrullinated Protein Antibody-Positive Rheumatoid Arthritis. Arthritis Rheumatol. 2013, 66, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, H.; Kusaba, T.; Yamaguchi, M. [Cause of death in autopsied RA patients]. Ryumachi. [Rheumatism] 1993, 33. [Google Scholar]
- Kelly, C.A.; Saravanan, V.; Nisar, M.; Arthanari, S.; Woodhead, F.A.; Price-Forbes, A.N.; Dawson, J.; Sathi, N.; Ahmad, Y.; Koduri, G.; et al. Rheumatoid arthritis-related interstitial lung disease: associations, prognostic factors and physiological and radiological characteristics--a large multicentre UK study. Rheumatol. 2014, 53, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Koga, Y.; Sugimoto, M. Different risk factors between interstitial lung disease and airway disease in rheumatoid arthritis. Respir. Med. 2012, 106, 1591–1599. [Google Scholar] [CrossRef]
- Catrina, A.I.; Ytterberg, A.J.; Reynisdottir, G.; Malmström, V.; Klareskog, L. Lungs, joints and immunity against citrullinated proteins in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 645–653. [Google Scholar] [CrossRef]
- Sparks, J.A.; Karlson, E.W. The Roles of Cigarette Smoking and the Lung in the Transitions Between Phases of Preclinical Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2016, 18, 1–17. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Toews, G.B.; White, E.S.; Iii, J.P.L.; Martinez, F.J. Mechanisms of Pulmonary Fibrosis. Annu. Rev. Med. 2004, 55, 395–417. [Google Scholar] [CrossRef]
- Larsen, J.M.; Steen-Jensen, D.B.; Laursen, J.M.; Søndergaard, J.N.; Musavian, H.S.; Butt, T.M.; Brix, S. Divergent pro-inflammatory profile of human dendritic cells in response to commensal and pathogenic bacteria associated with the airway microbiota. PLoS ONE 2012, 7, e31976. [Google Scholar] [CrossRef]
- Van Steendam, K.; Tilleman, K.; De Ceuleneer, M.; De Keyser, F.; Elewaut, D.; Deforce, D. Citrullinated vimentin as an important antigen in immune complexes from synovial fluid of rheumatoid arthritis patients with antibodies against citrullinated proteins. Arthritis Res. Ther. 2010, 12, R132–R132. [Google Scholar] [CrossRef]
- Van Steendam, K.; Tilleman, K.; Deforce, D. The relevance of citrullinated vimentin in the production of antibodies against citrullinated proteins and the pathogenesis of rheumatoid arthritis. Rheumatol. 2011, 50, 830–837. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Dorward, D.A.; Russell, C.D.; Um, I.H.; Elshani, M.; Armstrong, S.D.; Penrice-Randal, R.; Millar, T.; Lerpiniere, C.E.B.; Tagliavini, G.; Hartley, C.S.; et al. Tissue-Specific Immunopathology in Fatal COVID-19. Am. J. Respir. Crit. Care Med. 2021, 203, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Barra, L.; Scinocca, M.; Saunders, S.; Bhayana, R.; Rohekar, S.; Racapé, M.; Coles, R.; Cairns, E.; Bell, D.A. Anti-Citrullinated Protein Antibodies in Unaffected First-Degree Relatives of Rheumatoid Arthritis Patients. Arthritis Rheum. 2013, 65, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Mikuls, T.R.; Payne, J.B.; Deane, K.D.; Thiele, G.M. Autoimmunity of the lung and oral mucosa in a multisystem inflammatory disease: The spark that lights the fire in rheumatoid arthritis? J. Allergy Clin. Immunol. 2016, 137, 28–34. [Google Scholar] [CrossRef]
- Rangel-Moreno, J.; Hartson, L.; Navarro, C.; Gaxiola, M.; Selman, M.; Randall, T.D. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J. Clin. Investig. 2006, 116, 3183–3194. [Google Scholar] [CrossRef]
- Ossipova, E.; Cerqueira, C.F.; Reed, E.; Kharlamova, N.; Israelsson, L.; Holmdahl, R.; Nandakumar, K.S.; Engström, M.; Harre, U.; Schett, G.; et al. Affinity purified anti-citrullinated protein/peptide antibodies target antigens expressed in the rheumatoid joint. Thromb. Haemost. 2014, 16, R167–11. [Google Scholar] [CrossRef]
- Janssen, K.M.J.; de Smit, M.J.; Brouwer, E.; Kok, F.A.C.d.; Kraan, J.; Altenburg, J.; Verheul, M.K.; A Trouw, L.; van Winkelhoff, A.J.; Vissink, A.; et al. Rheumatoid arthritis–associated autoantibodies in non–rheumatoid arthritis patients with mucosal inflammation: a case–control study. Arthritis Res. Ther. 2015, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Demoruelle, M.K.; Deane, K.D.; Holers, V.M. When and where does inflammation begin in rheumatoid arthritis? Curr. Opin. Rheumatol. 2014, 26, 64–71. [Google Scholar] [CrossRef]
- Juge, P.-A.; Borie, R.; Kannengiesser, C.; Gazal, S.; Revy, P.; Wemeau-Stervinou, L.; Debray, M.-P.; Ottaviani, S.; Marchand-Adam, S.; Nathan, N.; et al. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1602314. [Google Scholar] [CrossRef]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A CommonMUC5BPromoter Polymorphism and Pulmonary Fibrosis. New Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef]
- Lehmann, M.; Hu, Q.; Hu, Y.; Hafner, K.; Costa, R.; Berg, A.v.D.; Königshoff, M. Chronic WNT/β-catenin signaling induces cellular senescence in lung epithelial cells. Cell. Signal. 2020, 70, 109588–109588. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical Course and Prediction of Survival in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; A Karimi-Shah, B.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2007, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Tsukui, T.; Ueha, S.; Abe, J.; Hashimoto, S.-I.; Shichino, S.; Shimaoka, T.; Shand, F.H.; Arakawa, Y.; Oshima, K.; Hattori, M.; et al. Qualitative Rather than Quantitative Changes Are Hallmarks of Fibroblasts in Bleomycin-Induced Pulmonary Fibrosis. Am. J. Pathol. 2013, 183, 758–773. [Google Scholar] [CrossRef]
- Chen, L.; Yang, Y.; Yan, H.; Peng, X.; Zou, J. NEDD4L-induced β-catenin ubiquitination suppresses the formation and progression of interstitial pulmonary fibrosis via inhibiting the CTHRC1/HIF-1α axis. Int. J. Biol. Sci. 2021, 17, 3320–3330. [Google Scholar] [CrossRef]
- Hesselstrand, R.; Andréasson, K.; Wuttge, D.M.; Bozovic, G.; Scheja, A.; Saxne, T. Increased serum COMP predicts mortality in SSc: results from a longitudinal study of interstitial lung disease. Rheumatology 2012, 51, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Kanaoka, M.; Yamaguchi, Y.; Komitsu, N.; Feghali-Bostwick, C.A.; Ogawa, M.; Arima, K.; Izuhara, K.; Aihara, M. Pro-fibrotic phenotype of human skin fibroblasts induced by periostin via modulating TGF-β signaling. J. Dermatol. Sci. 2018, 90, 199–208. [Google Scholar] [CrossRef]
- Gur, C.; Wang, S.-Y.; Sheban, F.; Zada, M.; Li, B.; Kharouf, F.; Peleg, H.; Aamar, S.; Yalin, A.; Kirschenbaum, D.; et al. LGR5 expressing skin fibroblasts define a major cellular hub perturbed in scleroderma. Cell 2022, 185, 1373–1388. [Google Scholar] [CrossRef]
- Valenzi, E.; Bulik, M.; Tabib, T.; Morse, C.; Sembrat, J.; Bittar, H.T.; Rojas, M.; Lafyatis, R. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Rheumatology 2019, 78, 1379–1387. [Google Scholar] [CrossRef]
- Shen, Z.; Su, T.; Chen, J.; Xie, Z.; Li, J. Collagen triple helix repeat containing-1 exerts antifibrotic effects on human skin fibroblast and bleomycin-induced dermal fibrosis models. Ann. Transl. Med. 2021, 9, 801–801. [Google Scholar] [CrossRef]
- Mehandru, S.; Merad, M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022, 23, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Rendeiro, A.F.; Ravichandran, H.; Bram, Y.; Chandar, V.; Kim, J.; Meydan, C.; Park, J.; Foox, J.; Hether, T.; Warren, S.; et al. The spatial landscape of lung pathology during COVID-19 progression. Nature 2021, 593, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Dolby, H.W.; Potey, P.; Wilder-Smith, A.B.; Clohisey, S.; E Millar, J.; Baillie, J.K.; A Dorward, D.; Lucas, C.D.; Russell, C.D. Histological Evidence of Pulmonary Microthrombosis and Vasculitis in Life-Threatening Respiratory Virus Diseases. Open Forum Infect. Dis. 2020, 8, ofaa640. [Google Scholar] [CrossRef] [PubMed]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J.; et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.J.; Coutavas, E.; Pine, A.B.; Lee, A.I.; Yu, V.L.; Shallow, M.K.; Giovacchini, C.X.; Mathews, A.M.; Stephenson, B.; Que, L.G.; et al. Immunofibrotic drivers of impaired lung function in postacute sequelae of SARS-CoV-2 infection. J. Clin. Investig. 2021, 6. [Google Scholar] [CrossRef]
- Schneider, C.; Nobs, S.P.; Kurrer, M.; Rehrauer, H.; Thiele, C.; Kopf, M. Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat. Immunol. 2014, 15, 1026–1037. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035. [Google Scholar] [CrossRef]
- Hou, Y.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446. [Google Scholar] [CrossRef]
- Pérez-Mies, B.; Caniego-Casas, T.; Bardi, T.; Carretero-Barrio, I.; Benito, A.; García-Cosío, M.; González-García, I.; Pizarro, D.; Rosas, M.; Cristóbal, E.; et al. Progression to lung fibrosis in severe COVID-19 patients: A morphological and transcriptomic study in postmortem samples. Front. Med. 2022, 9, 976759. [Google Scholar] [CrossRef]
- Coker, R.K.; Laurent, G.J.; Jeffery, P.K.; du Bois, R.M.; Black, C.M.; McAnulty, R.J. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax 2001, 56, 549–556. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Sikkema, L.; Ramírez-Suástegui, C.; Strobl, D.C.; Gillett, T.E.; Zappia, L.; Madissoon, E.; Markov, N.S.; Zaragosi, L.-E.; Ji, Y.; Ansari, M.; et al. An integrated cell atlas of the lung in health and disease. Nat. Med. 2023, 29, 1563–1577. [Google Scholar] [CrossRef] [PubMed]
- Jyothula, S.S.; Peters, A.; Liang, Y.; Bi, W.; Shivshankar, P.; Yau, S.; Garcha, P.S.; Yuan, X.; Akkanti, B.; Collum, S.; et al. Fulminant lung fibrosis in non-resolvable COVID-19 requiring transplantation. EBioMedicine 2022, 86. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, M.; Ramachandran, P. Immunology of human fibrosis. Nat. Immunol. 2023, 24, 1423–1433. [Google Scholar] [CrossRef]
- Li, A.; Chen, J.-Y.; Hsu, C.-L.; Oyang, Y.-J.; Huang, H.-C.; Juan, H.-F. A Single-Cell Network-Based Drug Repositioning Strategy for Post-COVID-19 Pulmonary Fibrosis. Pharmaceutics 2022, 14, 971. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, L.; Wang, M.; Zhou, S.; Lu, Y.; Cui, H.; Racanelli, A.C.; Zhang, L.; Ye, T.; Ding, B.; et al. Targeting fibrosis: mechanisms and clinical trials. Signal Transduct. Target. Ther. 2022, 7, 1–21. [Google Scholar] [CrossRef]
- Amati, F.; Stainer, A.; Polelli, V.; Mantero, M.; Gramegna, A.; Blasi, F.; Aliberti, S. Efficacy of Pirfenidone and Nintedanib in Interstitial Lung Diseases Other than Idiopathic Pulmonary Fibrosis: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 7849. [Google Scholar] [CrossRef]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 476–486. [Google Scholar] [CrossRef]
- Redente, E.F.; Aguilar, M.A.; Black, B.P.; Edelman, B.L.; Bahadur, A.N.; Humphries, S.M.; Lynch, D.A.; Wollin, L.; Riches, D.W.H. Nintedanib reduces pulmonary fibrosis in a model of rheumatoid arthritis-associated interstitial lung disease. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L998–L1009. [Google Scholar] [CrossRef] [PubMed]
- Deterding, R.; Young, L.R.; DeBoer, E.M.; Warburton, D.; Cunningham, S.; Schwerk, N.; Flaherty, K.R.; Brown, K.K.; Dumistracel, M.; Erhardt, E.; et al. Nintedanib in children and adolescents with fibrosing interstitial lung diseases. Eur. Respir. J. 2022, 61, 2201512. [Google Scholar] [CrossRef] [PubMed]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. New Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. New Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef]
- Liu, Z.-W.; Zhao, M.-Y.; Su, X.-L.; Ye, T.-H.; Zhuang, Y.-J.; Zhang, Y.; Zhang, Z.-Z.; Yang, J.-L.; Chen, L.-J.; Long, C.-F.; et al. The antifibrotic effect and mechanism of a novel tyrosine kinase inhibitor, ZSP1603, in preclinical models of pulmonary fibrosis. . 2020, 24, 1481–1491. [Google Scholar]
- Sgalla, G.; Franciosa, C.; Simonetti, J.; Richeldi, L. Pamrevlumab for the treatment of idiopathic pulmonary fibrosis. Expert Opin. Investig. Drugs 2020, 29, 771–777. [Google Scholar] [CrossRef]
- Wells, A.U. Pamrevlumab in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2019, 8, 2–3. [Google Scholar] [CrossRef]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 476–486. [Google Scholar] [CrossRef]
- Malik, B.; Abdelazeem, B.; Ghatol, A. Pulmonary Fibrosis After COVID-19 Pneumonia. Cureus 2021, 13. [Google Scholar] [CrossRef]
- Boshra, M.S.; Warda, A.E.A.; Sayed, M.A.; Elkomy, M.H.; Alotaibi, N.H.; Mohsen, M.; Sarhan, R.M. Effect of Pirfenidone on Risk of Pulmonary Fibrosis in COVID-19 Patients Experiencing Cytokine Storm. Healthcare 2022, 10, 2387. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, D.; Kong, X.; Wei, C.; LvQiu, S.; Wang, L.; Lin, Y.; Yin, Z.; Zhou, Z.; Luo, H. Case Report: Pirfenidone in the Treatment of Post-COVID-19 Pulmonary Fibrosis. Front. Med. 2022, 9, 925703. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The causes and contributing factors of diffuse parenchymal lung diseases (DPLDs). Created with BioRender.com.
Figure 1.
The causes and contributing factors of diffuse parenchymal lung diseases (DPLDs). Created with BioRender.com.
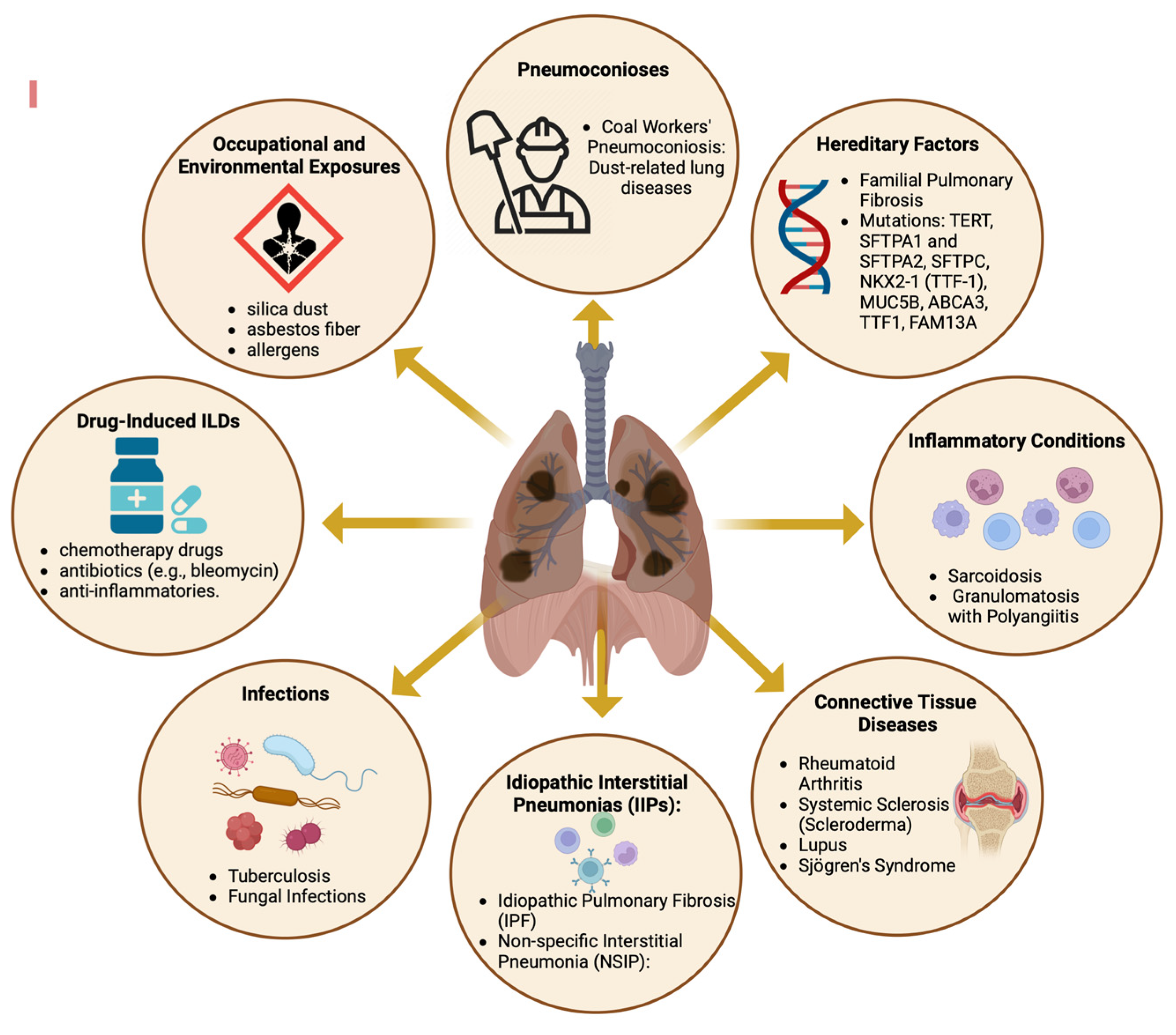
Figure 2.
Pathogenesis of Pulmonary Fibrosis: Unraveling the Cascade of Inflammatory Responses and Fibrotic Remodeling. Created with BioRender.com.
Figure 2.
Pathogenesis of Pulmonary Fibrosis: Unraveling the Cascade of Inflammatory Responses and Fibrotic Remodeling. Created with BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



