
Submitted:
07 September 2023
Posted:
11 September 2023
You are already at the latest version
Abstract
Potassium 6-oxo-7,13,16,22-tetraazatetracyclo[12.6.2.18,12.017,21]tricosa-1(20),8(23),9,11,14,16,18,21-octaen-2-yne-15-carboxylate was synthesized through a multi-step pathway starting from commercially available 3-iodo-1,2-phenylenediamine. Structure characterization of this new substituted macrocyclic quinoxaline compound was achieved by using 1H NMR, 13C NMR, and HRMS spectral analysis. This new macrocyclic derivative demonstrated submicromolar potency on both Pim-1 and Pim-2 isoforms, with an interesting selectivity profile against a selected panel of human kinases.

Keywords:
macrocycle
; quinoxaline
; Pim kinases
; kinase inhibitor
1. Introduction
Pim (Provirus Integration site for Moloney murine leukemia virus) kinases are a family of three constitutively active proto-oncogenic serine/threonine protein kinases (Pim-1, Pim-2 and Pim-3), regulating various cellular processes, including cell proliferation, survival and differentiation [1,2]. Because, they share a certain level of sequence homology, they can activate similar cellular pathways and can sometimes be considered as compensatory proteins [2,3]. However, these kinases present individual characteristics, especially in their tissue distribution [4]. Pim kinases are implicated in oncogenesis, particularly in tumor progression and metastasis, and are considered as important drivers of chemotherapy resistance [5]. Thus, these kinases are overexpressed in a large variety of tumors, with differences in their expression pattern according to the cancer type. Thereby, while Pim-1 and Pim-2 are commonly up-regulated in hematopoietic cancers [6,7,8,9,10], Pim-3 is mostly over-expressed in some solid cancers (e.g. prostate cancers) [11]. Finally, mice deficient for all Pim kinases displayed mild phenotypic modifications, including reduced body size and impaired responses to hematopoietic growth factors [12,13], demonstrating the interest of targeting these kinases in oncology. Moreover, crystal structures of Pim-1 and Pim-2 revealed unique particularities in comparison to others kinases, which can be exploited to develop selective Pim inhibitors [14,15].
In this context, and in the course of our drug discovery program on the development of new targeted antileukemic treatments, we previously identified the new quinoxaline lead compound 1, acting as a submicromolar dual Pim1/2 inhibitor (IC50 of 130 nM and 170 nM, on Pim-1 and Pim-2, respectively) (Figure 1), but displaying also micromolar inhibition of DYRK1A and GSK3β off-target mammalian kinases [16]. In the light of these results, we decided to prepare optimized analogues of compound 1, with an improved selectivity profile. Thus, taking into account our experience on the structure-activity relationships (SAR) in our previously described quinoxaline derivatives series [16,17], we used the quinoxaline-2-carboxylic acid scaffold as a template for the design and the synthesis of a new macrocyclic compound. SAR and molecular modeling studies highlighted the crucial role of the carboxylic acid moiety in position 2 for the Pim kinase inhibitory activity, establishing a key salt bridge with the catalytic lysine residue of Pim1/2 kinases at physiological pH. Moreover, macrocycles have been emerging as a valuable class of pharmacological agents over the past decade. Indeed, macrocyclization allows restriction of the conformational freedom observed in small molecules, permitting to optimize affinity and selectivity [18], and macrocyclic kinase inhibitors have reached advanced clinical trial, particularly in oncology [19,20]. In 2021, the Food and Drug Administration approved lorlatinib, the first macrocyclic kinase inhibitor in metastatic anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer. Here, we report the synthesis and structural identification of the potassium 6-oxo-7,13,16,22-tetraazatetracyclo[12.6.2.18,12.017,21]tricosa-1(20),8(23),9,11,14,16,18,21-octaen-2-yne-15-carboxylate 8. This original macrocyclic quinoxaline 8 was further evaluated on human Pim-1 and Pim-2 kinases and on a selected panel of human protein kinases, to determine its selectivity profile.
2. Results and Discussion
2.1. Potassium 6-oxo-7,13,16,22-tetraazatetracyclo[12.6.2.18,12.017,21]tricosa-1(20),8(23),9,11,14,16,18,21-octaen-2-yne-15-carboxylate
The synthesis of the potassium 6-oxo-7,13,16,22-tetraazatetracyclo[12.6.2.18,12.017,21]tricosa-1(20),8(23),9,11,14,16,18,21-octaen-2-yne-15-carboxylate 8 was performed as described in Scheme 1.
First, intermediate 3 was synthesized in two steps. The peptide coupling reaction between commercially available pent-4-ynoic acid and tert-butyl (3-aminophenyl)carbamate in N,N-dimethylformamide (DMF), using 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) and 2,6-lutidine, gave the carbamate intermediate 2, which was then deprotected, in a 10% trifluoroacetic acid (TFA) solution to provide the attempted trifluoroacetic salt 3.
The preparation of 3-chloro-5-iodoquinoxaline key intermediate 5a was then achieved in two steps according to literature procedures [16,17,21,22]. Briefly, commercial 3-iodo-1,2-phenylenediamine was condensed with diethyl 2-oxomalonate, in ethanol, using citric acid as catalyst to give ester 4a, which was separated from its 8-iodo isomer 4b by silica column chromatography. Chlorination in position 3 of esters 4a and 4b was realized, using DMF as a catalyst, in refluxing phosphorous oxychloride to give the corresponding esters 5a and 5b. Intermediate 5b, in contrast with its isomer 5a, gave a yellow single crystal, which was used for the 3D structural determination by X-ray crystallography (Figure 2), to identify the position of the iodo group on the quinoxaline scaffold in the solid state of this isomer, confirming the structure of each isomer.
Access to the macrocyclic quinoxaline 8 was then performed, using a three-step synthetic pathway. After treatment of trifluoroacetic salt 3 with N,N’-diisopropylethylamine (DIPEA), the resulting amine derivative was then engaged in a nucleophilic aromatic substitution reaction with quinoxaline 5a, in refluxing dry tetrahydrofuran (THF), to yield intermediate 6, which underwent an intramolecular Sonogashira cross-coupling reaction, using PdCl2(PPh3)2, and CuI, as catalysts, and triethylamine in refluxing THF, leading to the macrocyclic quinoxaline 7. Hydrolysis of ethyl ester 7 with potassium carbonate in 80% aqueous methanol was then performed to afford the macrocyclic potassium salt 8. The structure of this new macrocyclic quinoxaline derivative 8 was then confirmed by 1H NMR, and HRMS analysis (see Supplementary Materials).
2.2. Protein Kinase Assays
The ability of the macrocyclic quinoxaline 8 to inhibit the in vitro enzymatic activity of human Pim-1 and Pim-2, and of a selected panel of six human off-target protein kinases (comprising DYRK1A, CDK5/p25, CDK9/CyclinT, Haspin, CK1ε and GSK3β), was evaluated, using a luminescence-based kinase assay [23]. The commercially available pan-Pim protein kinase inhibitor, SGI-1776 [24], was used as a control for the in vitro studies. As shown in Table 1, compound 8 displayed a submicromolar activity on Pim-1 and Pim-2 (IC50 of 400 nM, and 100 nM, respectively) with the same level of activity on Pim-2 but a slightly decreased activity on Pim-1, in comparison to lead compound 1 and SGI-1776, the reference drug. However, the general selectivity profile of macrocycle 8 on the panel of human protein kinases studied was significantly improved in comparison to compound 1 and SGI-1776. Indeed, while compound 1 displayed low micromolar inhibition of DYRK1A and GSK3β, and SGI-1776 potently inhibited five of the six human kinases tested (IC50 values of 0.05 to 9.53 µM), compound 8 displayed an interesting selectivity profile against the six potential off-target kinases (CDK5/p25, CDK9/CyclinT, Haspin, CK1ε and GSK3β), with IC50 inhibition values > 10 µM in every case.
3. Materials and Methods
All solvents were anhydrous reagents from commercial sources. Unless otherwise noted, all chemicals and reagents were obtained commercially and used without purification. Melting points (m.p.) were determined on a Stuart capillary apparatus and are uncorrected. High-resolution mass spectra (HRMS) were performed on a Bruker maXis mass spectrometer by the SALSA platform from ICOA laboratory, in positive mode with an ESI source. NMR spectra were recorded at 400 MHz (1H), 101 MHz (13C) or 376 MHz (19F) on a Bruker Avance (400 MHz) spectrometer. The chemical shifts are reported in parts per million (ppm, δ) relative to residual deuterated solvent peaks. The abbreviations s = singlet, d = doublet, t = triplet, q = quadruplet, m = multiplet and bs = broad signal were used throughout.
3.1. Tert-butyl (3-(pent-4-ynamido)phenyl)carbamate (2)
To a solution of pent-4-ynoic acid (106 mg, 1.08 mmol) in dry N,N-dimethylformamide (DMF) (3 mL), under an argon atmosphere, were added 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluoro-phosphate (HATU) (680 mg, 1.79 mmol) in solution in dry DMF (3 mL) and 2,6-lutidine (207 µL, 1.79 mmol), and the resulting mixture was stirred magnetically at room temperature for 10 minutes. Tert-butyl (3-aminophenyl)carbamate (188 mg, 0.90 mmol) was then added and the mixture was stirred at room temperature for 24 h. The solvent was then removed under reduced pressure, and the residue was finally purified by silica column chromatography using cyclohexane with ethyl acetate gradient (0-60%) as eluent, to give compound 2 (214 mg, 69%) as a white powder, m.p. 130 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.91 (s, 1H), 9.32 (s, 1H), 7.78 (s, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.13 (dd, J = 8.4, 8.8 Hz, 1H), 7.02 (d, J = 8.8 Hz, 1H), 2.78 (t, J = 2.4 Hz, 1H), 2.50-2.40 (m, 4H), 1.47 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 169.2, 152.7, 139.8, 139.4, 128.7, 113.3, 113.0, 109.1, 83.7, 78.9, 71.4, 35.1, 28.1 (3 × C), 14.1.
3.2. 3-(Pent-4-ynamido)benzenaminium trifluoroacetic salt (3)
To a solution of compound 2 (1.20 g, 4.16 mmol,) in dichloromethane (DCM) (40 mL) was added dropwise a 10% trifluoroacetic acid (TFA) solution (4 mL) at 0 °C. The resulting mixture was stirred magnetically at 0 °C for 30 minutes, and then, at room temperature for 5 h. The solvent was then removed under reduced pressure, and the residue was finally purified by silica column chromatography using DCM with methanol (MeOH) gradient (0-15%) as eluent, to give compound 3 (1.19 g, 100%) as a brown oil. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 7.50 (s, 1H), 7.20 (dd, J = 8.4, 7.6 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 6.69 (d, J = 7.6 Hz, 1H), 4.25-3.00 (m, 3H), 2.79 (t, J = 2.4 Hz, 1H), 2.55-2.40 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 169.8, 140.2, 134.7, 130.0, 116.7, 116.3, 112.2, 83.6, 71.5, 35.2, 14.1. 19F NMR (376 MHz, DMSO-d6) δ -74.1 (s).
3.3. Ethyl 5-iodo-3-oxo-3,4-dihydroquinoxaline-2-carboxylate (4a) and ethyl 8-iodo-3-oxo-3,4-dihydroquinoxaline-2-carboxylate (4b)
A mixture of 3-iodo-1,2-phenylenediamine (1.00 g, 4.27 mmol), diethyl 2-oxomalonate (0.81 mL, 5.31 mmol) and citric acid (132 mg, 0.69 mmol) in ethanol (50 mL) was stirred magnetically at 50 °C for 3 h. Ethanol was then evaporated under reduced pressure, and the resulting residue was purified by silica column chromatography using cyclohexane with ethyl acetate gradient (0-70%) as eluent to give compound 4a (275 mg, 19%) and its 8-iodo isomer 4b (1.01 g, 69%) as yellow powders.
Compound 4a: m.p. 162 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.54 (bs, 1H), 8.21 (d, J = 6.0 Hz, 1H), 7.90 (d, J = 7.6 Hz, 1H), 7.24 (dd, J = 7.6, 6.0 Hz, 1H), 4.39 (q, J = 7.2 Hz, 2H), 1.33 (t, J = 7.2 Hz, 3H).
Compound 4b: m.p. 238 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.96 (bs, 1H), 7.89 (dd, J = 4.4, 2.4 Hz, 1H), 7.37-7.29 (m, 2H), 4.39 (qd, J = 7.2, 2.0 Hz, 2H), 1.33 (td, J = 7.2, 2.0 Hz, 3H).
3.4. Ethyl 3-chloro-5-iodoquinoxaline-2-carboxylate (5a)
Method A: into a dry three-neck round bottom flask was introduced compound 4a (100 mg, 0.29 mmol) in phosphorous oxychloride (0.96 mL) at ice bath temperature. The mixture was vigorously stirred magnetically at 0 °C for 5 min and DMF (42 µL) was then added at 0 °C and the reaction mixture was refluxed for 1 h. After cooling at 0 °C, the resulting mixture was neutralized with a 1 M sodium hydroxide aqueous solution, and extracted with ethyl acetate. The combined organic layers were washed with brine, and dried over MgSO4, filtered, and evaporated under reduced pressure to obtain derivative 5a (102 mg, 97%) as a yellow solid, m.p. 120 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.61 (dd, J = 7.6, 1.2 Hz, 1H), 8.23 (dd, J = 8.4, 1.2 Hz, 1H), 7.75 (dd, J = 8.4, 7.6 Hz, 1H), 4.50 (q, J = 7.2 Hz, 2H), 1.39 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 162.9, 144.8, 143.9, 143.0, 141.7, 139.7, 133.0, 129.9, 100.7, 62.8, 13.9.
3.5. Ethyl 3-chloro-8-iodoquinoxaline-2-carboxylate (5b)
The title compound was synthesized according to the general method A from compound 4b (100 mg, 0.29 mmol), phosphorous oxychloride (0.96 mL) and DMF (42 μL). Compound 5b was obtained (94 mg, 89%) as a yellow solid, m.p. 105 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.55 (dd, J = 7.2, 0.8 Hz, 1H), 8.10 (dd, J = 8.4, 0.8 Hz, 1H), 7.75 (dd, J = 8.4, 7.2 Hz, 1H), 4.52 (qd, J = 7.2, 1.6 Hz, 2H), 1.40 (td, J = 7.2, 1.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.4, 145.4, 144.1, 142.4, 142.2, 139.7, 134.8, 129.3, 102.9, 63.3, 14.4.
3.6. Ethyl 5-iodo-3-((3-(pent-4-ynamido)phenyl)amino)quinoxaline-2-carboxylate (6)
To a solution of trifluoroacetic salt 3 (160 mg, 0.56 mmol) and N,N’-diisopropylethylamine (DIPEA) (500 µL, 2.87 mmol) in dry tetrahydrofuran (THF) (3 mL), under an argon atmosphere, was added dropwise compound 5a (64 mg, 0.18 mmol) in solution in dry THF (1 mL). The resulting mixture was refluxed for 2 days. The solvent was then removed under reduced pressure, and the residue was finally purified by silica column chromatography using cyclohexane with ethyl acetate gradient (0-100%) as eluent to give intermediate 6 (37 mg, 41%) as a yellow powder, m.p. 127 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 9.99 (s, 1H), 8.56 (d, J = 8.4 Hz, 1H), 8.38 (dd, J = 7.6, 1.2 Hz, 1H), 8.02 (dd, J = 8.0, 1.2 Hz, 1H), 7.92 (s, 1H), 7.41-7.32 (m, 2H), 7.22 (d, J = 8.8 Hz, 1H), 4.51 (q, J = 7.2 Hz, 2H), 2.82 (t, J = 2.4 Hz, 1H), 2.59-2.50 (m, 4H), 1.43 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.5, 165.2, 148.6, 142.3, 142.1, 139.5, 139.1, 135.5, 133.3, 130.2, 129.3, 127.6, 115.0, 114.3, 110.8, 98.5, 83.7, 71.5, 62.5, 35.2, 14.1, 14.0.
3.7. Ethyl 6-oxo-7,13,16,22-tetraazatetracyclo[12.6.2.18,12.017,21]tricosa-1(20),8(23),9,11,14,16,18,21-octaen-2-yne-15-carboxylate (7)
Into a sealed tube were introduced bis(triphenylphosphine)palladium(II) dichloride (PdCl2(PPh3)2) (4 mg , 5.7 µmol), copper iodide (CuI) (0.3 mg, 1.6 µmol) and triethylamine (16 µL, 0.12 mmol) in dry THF (3 mL), under an argon atmosphere. Then, intermediate 6 (20 mg, 39 µmol) was added dropwise, and the reaction mixture was refluxed for 16 h. The solvent was then removed under reduced pressure, and the residue was finally purified by silica column chromatography using cyclohexane with ethyl acetate gradient (0-100%) as eluent to give the macrocycle 7 (1.2 mg, 8%), as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 9.60 (s, 1H), 9.48 (bs, 1H), 7.97 (dd, J = 8.4, 0.8 Hz, 1H), 7.87 (dd, J = 7.2, 0.8 Hz, 1H), 7.53 (dd, J = 8.4, 7.2 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.06 (dd, J = 8.0, 1.6 Hz, 1H), 6.79 (dd, J = 8.0, 1.6 Hz, 1H), 4.50 (q, J = 7.2 Hz, 2H), 2.95 (t, J = 5.6 Hz, 2H), 2.59 (t, J = 5.6 Hz, 2H), 1.42 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.7, 165.2, 147.9, 142.6, 139.6, 139.3, 135.4, 135.0, 132.5, 129.3, 129.0, 126.1, 120.4, 119.0, 118.6, 116.8, 95.5, 78.6, 62.4, 29.0, 16.2, 14.0. HRMS (ESI) m/z: [M+H]+ calcd for C22H19N4O3, 387.14572; found 387.14516.
3.8. Potassium 6-oxo-7,13,16,22-tetraazatetracyclo[12.6.2.18,12.017,21]tricosa-1(20),8(23),9,11,14,16,18,21-octaen-2-yne-15-carboxylate (8)
To ester 7 (1 mg, 2.6 µmol) in aqueous methanol (80%, 2 mL), potassium carbonate (0.36 mg, 2.6 µmol) was added and the reaction mixture was stirred magnetically at room temperature for 1 h. After cooling, MeOH was removed under reduced pressure, and the aqueous phase was washed with ethyl acetate, and evaporated under reduced pressure to yield macrocycle 8 (1 mg, 97%) as a yellow powder, m.p. > 350 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.29 (s, 1H), 9.62 (s, 1H), 9.61 (bs, 1H), 7.86 (dd, J = 8.0, 1.2 Hz, 1H), 7.66 (d, J = 7.2, 1.2 Hz, 1H), 7.38 (dd, J = 8.0, 7.2 Hz, 1H), 7.28 (t, J = 8.0 Hz, 1H), 6.86 (d, J = 8.0, 2.0 Hz, 1H), 6.70 (dd, J = 8.0, 2.0 Hz, 1H), 2.94 (t, J = 4.8 Hz, 2H), 2.65 (t, J = 4.8 Hz, 2H). HRMS (ESI) m/z: [M+2H]+ calcd for C20H15N4O3, 359.11442, found 359.11401.
3.9. X-Ray Data
The structure of compound 5b was established by X-ray crystallography (Figure 2). The yellow single crystal of 5b was obtained by slow evaporation from a methanol/chloroform solution (v/v : 20/80): triclinic, space group P-1, a = 6.6838(5) Å, b = 8.6592(6) Å, c = 10.5128 (7) Å, α = 79.521(2)°, β = 89.243(2)°, γ = 86.626(2)°, V = 597.26 (7)Å3, Z = 2, δ(calcd) = 2.016 Mg.m−3, FW = 362.54 for C11H8ClIN2O2, F(000) = 348. Full crystallographic results have been deposited at the Cambridge Crystallographic Data Centre (CCDC-2262892), UK, as supplementary material [25]. The data were corrected for Lorentz and polarization effects and for empirical absorption correction [26]. The structure was solved by direct methods Shelx 2013 [27] and refined using Shelx 2013 [27] suite of programs.
3.10. Protein Kinase Assays
4. Conclusions
Taking into account our previous studies, using the biological active quinoxaline-2-carboxylic acid scaffold, we designed and synthesized a new potassium 6-oxo-7,13,16,22-tetraazatetracyclo[12.6.2.18,12.017,21]tricosa-1(20),8(23),9,11,14,16,18,21-octaen-2-yne-15-carboxylate 8 and then evaluated its anti-Pim-1/2 kinase activity. This macrocyclic quinoxaline 8 exhibited submicromolar activity on Pim-1 and Pim-2, with a significantly improved selectivity profile on the panel of human protein kinases studied in comparison to lead compound 1 and the reference drug SGI-1776. This compound could therefore represent a new attractive candidate for extending further pharmacomodulation studies and pharmacological investigations.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, 1H NMR, 13C NMR, and HRMS.
Author Contributions
Conceptualization, C.D.-S. and C.L.; methodology, C.D.-S., C.B. M.-C.V.-M., and N.J.; investigation, C.B., S.L., C.L., C.D.-S., J.G., N.P., T.R. and S.B.; writing—original draft preparation, C.D.-S. and C.B.; writing—review and editing, C.D.-S.; supervision, C.D.-S.; project administration, C.D.-S.; funding acquisition, C.D.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by a grant from the Centre-Val de Loire Region and from the French National Research Agency under the program “Investissements d’avenir” Grant Agreement LabEx SynOrg (ANR-11-LABX-0029). The authors also thank the Cancéropôle Grand Ouest (3MC network—Marine Molecules, Metabolism and Cancer), GIS IBiSA (Infrastructures en Biologie Santé et Agronomie) and Biogenouest (Western France life science and environment core facility network) for supporting the KISSf screening facility. C. Blouet thanks the Centre-Val de Loire Region and the LabEx SynOrg for her PhD fellowship.
Acknowledgments
The authors would like to thank Cyril Colas from the "Fédération de Recherche" ICOA/CBM (FR2708)" for HRMS analysis, and Cécile Croix, from CEPR, for NMR spectrometry and technical support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mochizuki, T.; Kitanaka, C.; Noguchi, K.; Muramatsu, T.; Asai, A.; Kuchino, Y. Physical and Functional Interactions between Pim-1 Kinase and Cdc25A Phosphatase. Implications for the Pim-1-Mediated Activation of the c-Myc Signaling Pathway. J. Biol. Chem. 1999, 274, 18659–18666. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Kosan, C.; Xing, P.X.; Montenarh, M.; Hoffmann, I.; Möröy, T. The Oncogenic Serine/Threonine Kinase Pim-1 Directly Phosphorylates and Activates the G2/M Specific Phosphatase Cdc25C. Int. J. Biochem. Cell Biol. 2006, 38, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Theo Cuypers, H.; Selten, G.; Quint, W.; Zijlstra, M.; Maandag, E.R.; Boelens, W.; van Wezenbeek, P.; Melief, C.; Berns, A. Murine Leukemia Virus-Induced T-Cell Lymphomagenesis: Integration of Proviruses in a Distinct Chromosomal Region. Cell 1984, 37, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Maja Narlik-Grassow, Carmen Blanco-Aparicio, and A.C. The PIM Family of Serine/Threonine Kinases in Cancer. Med. Res. Rev. 2014, 34, 136–159. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.K.; Warfel, N.A. Targeting PIM Kinases to Overcome Therapeutic Resistance in Cancer. Mol. Cancer Ther. 2021, 20, 3–10. [Google Scholar] [CrossRef]
- Keane, N.A.; Reidy, M.; Natoni, A.; Raab, M.S.; O’Dwyer, M. Targeting the Pim Kinases in Multiple Myeloma. Blood Cancer J. 2015, 5(7), e325. [Google Scholar] [CrossRef]
- Amson, R.; Sigaux, F.; Przedborski, S.; Flandrin, G.; Givol, D.; Telerman, A. The Human Protooncogene Product P33pim Is Expressed during Fetal Hematopoiesis and in Diverse Leukemias. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 8857–8861. [Google Scholar] [CrossRef]
- Koblish, H.; Li, Y. long; Shin, N.; Hall, L.; Wang, Q.; Wang, K.; Covington, M.; Marando, C.; Bowman, K.; Boer, J.; et al. Preclinical Characterization of INCB053914, a Novel Pan-PIM Kinase Inhibitor, Alone and in Combination with Anticancer Agents, in Models of Hematologic Malignancies. PLoS One 2018, 13, 1–22. [Google Scholar] [CrossRef]
- Czardybon, W.; Windak, R.; Gołas, A.; Gałezowski, M.; Sabiniarz, A.; Dolata, I.; Salwińska, M.; Guzik, P.; Zawadzka, M.; Gabor-Worwa, E.; et al. A Novel, Dual Pan-PIM/FLT3 Inhibitor SEL24 Exhibits Broad Therapeutic Potential in Acute Myeloid Leukemia. Oncotarget 2018, 9, 16917–16931. [Google Scholar] [CrossRef]
- Bellon, M.; Nicot, C. Targeting Pim Kinases in Hematological Cancers: Molecular and Clinical Review. Mol. Cancer 2023, 22, 1–25. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, C.; Du, E.; Wang, A.; Yang, Y.; Guo, J.; Wang, A.; Zhang, Z.; Xu, Y. Pim-3 Is a Critical Risk Factor in Development and Prognosis of Prostate Cancer. Med. Sci. Monit. 2016, 22, 4254–4260. [Google Scholar] [CrossRef] [PubMed]
- Mikkers, H.; Nawijn, M.; Allen, J.; Brouwers, C.; Verhoeven, E.; Jonkers, J.; Berns, A. Mice Deficient for All PIM Kinases Display Reduced Body Size and Impaired Responses to Hematopoietic Growth Factors. Mol. Cell. Biol. 2004, 24, 6104–6115. [Google Scholar] [CrossRef] [PubMed]
- An, N.; Kraft, A.S.; Kang, Y. Abnormal Hematopoietic Phenotypes in Pim Kinase Triple Knockout Mice. J. Hematol. Oncol. 2013, 6, 1. [Google Scholar] [CrossRef]
- Kumar, A.; Mandiyan, V.; Suzuki, Y.; Zhang, C.; Rice, J.; Tsai, J.; Artis, D.R.; Ibrahim, P.; Bremer, R. Crystal Structures of Proto-Oncogene Kinase Pim1: A Target of Aberrant Somatic Hypermutations in Diffuse Large Cell Lymphoma. J. Mol. Biol. 2005, 348, 183–193. [Google Scholar] [CrossRef]
- Bullock, A.N.; Russo, S.; Amos, A.; Pagano, N.; Bregman, H.; Debreczeni, J.É.; Lee, W.H.; von Delft, F.; Meggers, E.; Knapp, S. Crystal Structure of the PIM2 Kinase in Complex with an Organoruthenium Inhibitor. PLoS One 2009, 4(10), e7112. [Google Scholar] [CrossRef] [PubMed]
- Oyallon, B.; Brachet-Botineau, M.; Logé, C.; Robert, T.; Bach, S.; Ibrahim, S.; Raoul, W.; Croix, C.; Berthelot, P.; Guillon, J.; et al. New Quinoxaline Derivatives as Dual Pim-1/2 Kinase Inhibitors: Design, Synthesis and Biological Evaluation. Molecules 2021, 26, 867. [Google Scholar] [CrossRef] [PubMed]
- Oyallon, B.; Brachet-Botineau, M.; Logé, C.; Bonnet, P.; Souab, M.; Robert, T.; Ruchaud, S.; Bach, S.; Berthelot, P.; Gouilleux, F.; et al. Structure-Based Design of Novel Quinoxaline-2-Carboxylic Acids and Analogues as Pim-1 Inhibitors. Eur. J. Med. Chem. 2018, 154, 101–109. [Google Scholar] [CrossRef]
- Liang, Y.; Fang, R.; Rao, Q. An Insight into the Medicinal Chemistry Perspective of Macrocyclic Derivatives with Antitumor Activity: A Systematic Review. Molecules 2022, 27(9), 2837. [Google Scholar] [CrossRef]
- Basit, S.; Ashraf, Z.; Lee, K.; Latif, M. First Macrocyclic 3rd-Generation ALK Inhibitor for Treatment of ALK/ROS1 Cancer: Clinical and Designing Strategy Update of Lorlatinib. Eur. J. Med. Chem. 2017, 134, 348–356. [Google Scholar] [CrossRef]
- Verstovsek, S.; Komrokji, R.S. A Comprehensive Review of Pacritinib in Myelofibrosis. Futur. Oncol. 2015, 11, 2819–2830. [Google Scholar] [CrossRef]
- Mahesh, R.; Dhar, A.K.; Sasank T.v.n.v., T.; Thirunavukkarasu, S.; Devadoss, T. Citric Acid: An Efficient and Green Catalyst for Rapid One Pot Synthesis of Quinoxaline Derivatives at Room Temperature. Chinese Chem. Lett. 2011, 22, 389–392. [Google Scholar] [CrossRef]
- Mahesh, R.; Devadoss, T.; Dhar, A.K.; Venkatesh, S.M.; Mundra, S.; Pandey, D.K.; Bhatt, S.; Jindal, A.K. Ligand-Based Design, Synthesis, and Pharmacological Evaluation of 3-Methoxyquinoxalin-2-Carboxamides as Structurally Novel Serotonin Type-3 Receptor Antagonists. Arch. Pharm. (Weinheim). 2012, 345, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A Bioluminescent and Homogeneous Adp Monitoring Assay for Kinases. Assay Drug Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Aparicio, C.; Carnero, A. Pim Kinases in Cancer: Diagnostic, Prognostic and Treatment Opportunities. Biochem. Pharmacol. 2013, 85, 629–643. [Google Scholar] [CrossRef]
- Supplementary X-ray crystallographic data: Cambridge Crystallographic Data Centre, University Chemical Lab, Lensfield Road, Cambridge, CB2 1EW, UK. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 15 May 2023).
- Sheldrick, G.M. (1996) SADABS, University of Göttingen, Germany.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
Figure 1.
Chemical structure of lead compound 1.
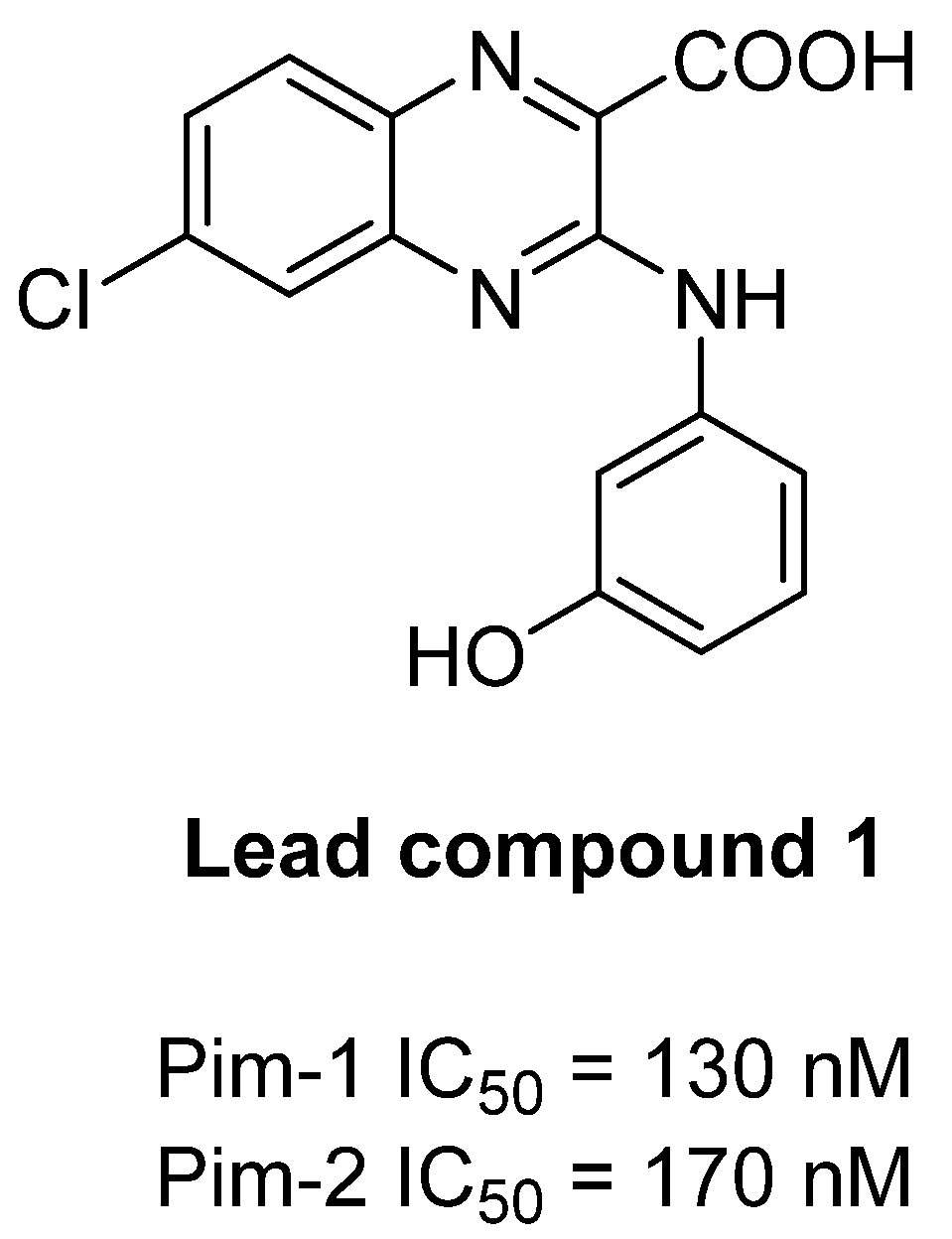
Scheme 1.
Synthesis of Potassium 6-oxo-7,13,16,22-tetraazatetracyclo[12.6.2.18,12.017,21]tricosa-1(20),8(23),9,11,14,16,18,21-octaen-2-yne-15-carboxylate (8).
Scheme 1.
Synthesis of Potassium 6-oxo-7,13,16,22-tetraazatetracyclo[12.6.2.18,12.017,21]tricosa-1(20),8(23),9,11,14,16,18,21-octaen-2-yne-15-carboxylate (8).
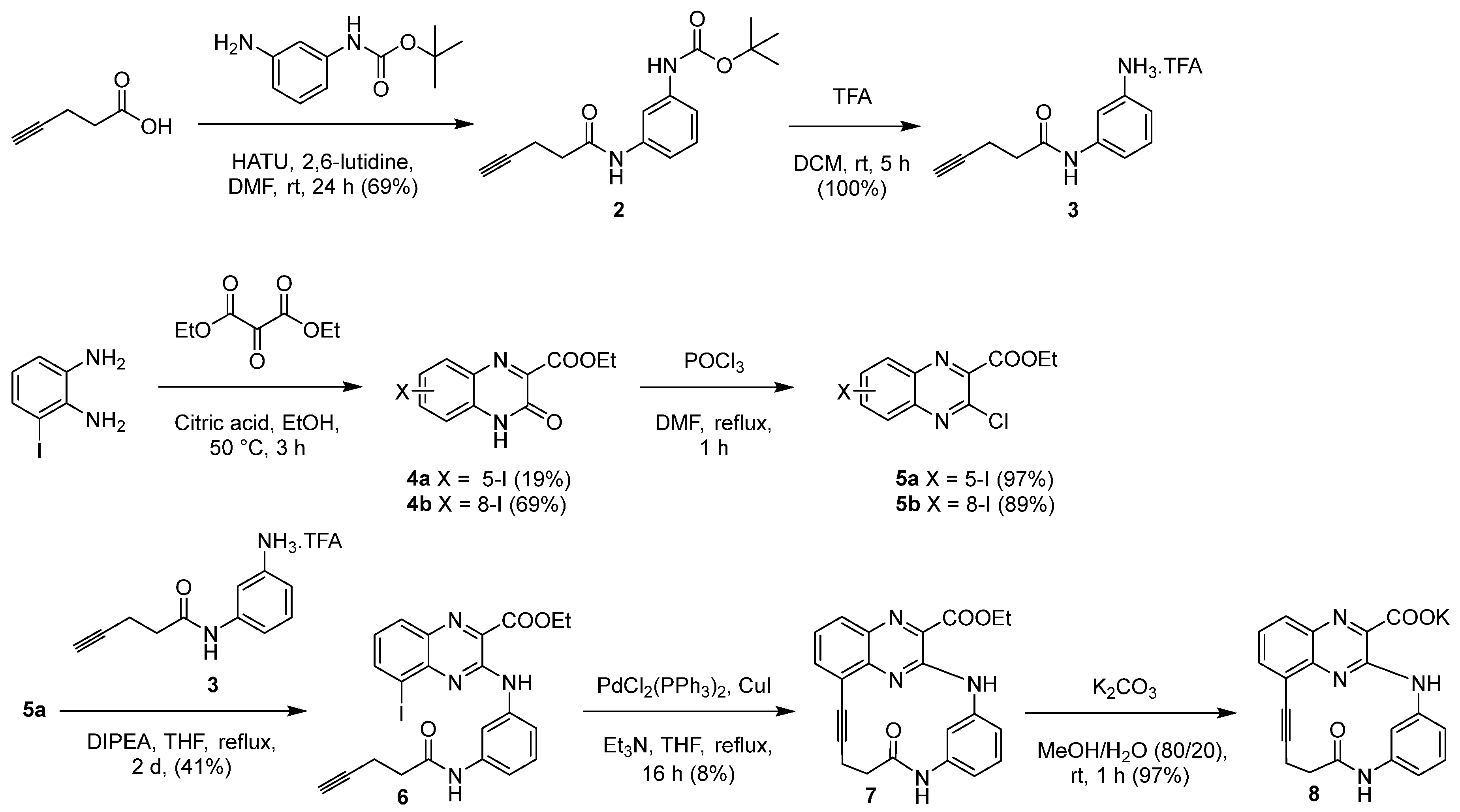
Figure 2.
The ORTEP (Oak Ridge Thermal Ellipsoid Plot) drawing of ethyl 3-chloro-8-iodoquinoxaline-2-carboxylate (5b) with thermal ellipsoids at 30% level.
Figure 2.
The ORTEP (Oak Ridge Thermal Ellipsoid Plot) drawing of ethyl 3-chloro-8-iodoquinoxaline-2-carboxylate (5b) with thermal ellipsoids at 30% level.

Table 1.
Kinase selectivity profile of compounds 1 and 8.
| Compound | Kinase enzymatic IC50 (µM) (a) | |||||||
| Pim-1 | Pim-2 | DYRK1A | CDK5/p25 |
CDK9/ CyclinT |
Haspin | CK1ε | GSK3β | |
| 1 | 0.13 | 0.17 | 2.58 | > 10 | > 10 | > 10 | > 10 | 2.80 |
| 8 | 0.40 | 0.10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
| SGI-1776 | 0.05 | 0.10 | 3.80 | 9.53 | 1.08 | 0.05 | 6.54 | > 10 |
a IC50 on disease-related kinase activity were calculated from dose-response curves. Each inhibitor concentration was tested in duplicate. All protein kinases used here are human. DYRK1A: dual specificity tyrosine phosphorylation regulated kinase 1A, CDK: cyclin-dependent kinase, Haspin: haploid germ cell-specific nuclear protein kinase, CK1: casein kinase 1, GSK3: glycogen synthase kinase 3.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



