
Submitted:
07 September 2023
Posted:
11 September 2023
You are already at the latest version
Abstract
Non-alcoholic steatohepatitis (NASH) is diagnosed by the occurrence of fat buildup, inflammation, and fibrotic changes within the liver. NASH has the capacity to proceed to the advancement of decompensated cirrhosis and carcinoma by inflammation and oxidative stress. Although Agrimonia pilosa has traditionally been used medicinal purposes, its effects on the liver remains incompletely elucidated. In this research, we assessed the therapeutic outcomes of Agrimonia pilosa extract (APE) on free fatty acid (FFA)-treated HepG2 cellular hepatic steatosis and in a choline-deficient, L-amino acid-defined, high-fat diet with 0.1% methionine (CDAHFD)-induced NASH mouse model. In FFA-treated cells, APE inhibited intracellular lipid accumulation, and regulated the mRNA and protein expressions of modulators of fatty acid (FA) synthesis as well as FA oxidation proteins, through down-regulation of the AMP-activated protein kinase/sirtuin 1 signaling pathway. In the CDAHFD-induced NASH mouse model, APE significantly enhanced liver histology and serum levels of liver damage. APE also significantly attenuated hepatic expression of mRNA and proteins related to FA synthesis, inflammation, and fibrosis. Furthermore, APE mitigated the expression levels of oxidative stress and endoplasmic reticulum stress. Overall, these discoveries indicate that Agrimonia pilosa has the potential to treat NASH by regulating hepatic lipid metabolism, alleviating inflammation, and mitigating oxidative stress.
Keywords:
non-alcoholic steatohepatitis
; Agrimonia pilosa
; lipid metabolism
; inflammation
; oxidative stress
; AMPK/SIRT1 signaling pathway
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is metabolic dysfunction that exerts its influence on approximately one-quarter of the global populace, thereby emerging as a significant worldwide health concern [1,2]. It is distinguished by the occurrence of fat formation within the liver without alcohol intake. The spectrum of these conditions encompasses a range from uncomplicated hepatic steatosis to non-alcoholic steatohepatitis (NASH), which represents an advanced manifestation of the disease distinguished by inflammatory and fibrotic changes within the liver. NASH has the potential to evolve into long-term liver conditions, encompassing compensated cirrhosis, decompensated cirrhosis, and uncorrectable hepatocellular carcinoma. [1,3]. Moreover, NASH exhibits a strong correlation with metabolic disorders, including obesity, dyslipidemia, insulin resistance, and chronic kidney dysfunction. [4,5,6,7,8,9]. Despite the urgent need for effective NAFLD treatments, there currently exist no approved pharmaceutical agents that specifically target this disorder. In recent years, various therapeutic agents, including pharmacological agents and natural compounds, have been assessed for their effectiveness in managing of NAFLD.
Several pharmacological agents, such as pioglitazone, vitamin E, and obeticholic acid, have exhibited encouraging outcomes undergoing clinical trials for the therapeutic intervention of NAFLD [10]. Pioglitazone, a thiazolidinedione, is an insulin sensitizer that has been documented efficacy in enhancing hepatic steatosis, lipid metabolism, and insulin resistance among individuals diagnosed with NAFLD. [11]. Vitamin E, a potent agent of antioxidant, has demonstrated its effectiveness in enhancing liver biochemical and histological parameters of the liver in patients with NASH [12]. Furthermore, obeticholic acid, a farnesoid X receptor agonist, has been reported to significantly enhance fibrotic liver lesions among individuals diagnosed with NASH [13]. However, these medicinal agents are subject to limitations, including potential adverse effects and an absence of extensive, long-term safety documentation.
It is crucial to develop a NASH treatment with minimal side effects that targets both inflammation and oxidative stress, as these factors are closely related to the progression and recurrence of the disease [14]. An overabundance of fat buildup within the liver can trigger an inflammatory reaction within Kupffer cells, inducing the expulsion of proinflammatory cytokines and activation of the immune system [15]. The ensuing inflammatory response may further amplify the production of reactive oxygen species (ROS), leading to a vicious cycle of inflammation and oxidative stress that can ultimately advance to more severe forms of NASH [16]. Therefore, natural compounds with anti-inflammatory and antioxidant properties are particularly promising for preventing and managing of NASH.
Agrimonia pilosa (A. pilosa), also known as hairy agrimony or Chinese agrimony, is a perennial herbaceous plant classified within the Rosaceae family. A. pilosa is native to Asia, including China, Korea, Japan, and Vietnam, and is commonly found growing on grassy slopes, in meadows, and in forests. A. pilosa has been used in traditional remedy for centuries as a treatment for various conditions such as fever, diarrhea, dysentery, liver and gallbladder disorders, and inflammatory diseases. The phytochemicals in A. pilosa include triterpenoids, phenolic acids, tannins, and flavonoids, which exhibit antioxidant and anti-inflammatory effects [17,18]. Recent scientific research has focused on investigating the potential of A. pilosa as a natural remedy for several diseases. Several studies have demonstrated that A. pilosa extract (APE) has anti-inflammatory properties in lipopolysaccharide-stimulated macrophages and alleviates the release of proinflammatory cytokines in a mouse model of croton oil-induced ear edema [19,20]. Another study showed that APE can ameliorate hyperglycemia and hepatic steatosis provoked by a lack of estrogen in a mouse model simulating postmenopausal metabolic syndrome [21]. However, the potential of A. pilosa in the treatment of NASH is not well understood.
In this study, we examined the therapeutic effects of A. pilosa on cellular hepatic steatosis in HepG2 cells exposed to free fatty acids (FFAs), and its potential in mitigating the development of NAFLD/NASH in a CDAHFD diet-induced NASH mouse model. Our findings offer valuable insights into the potential use of A. pilosa as a therapeutic agent for NAFLD/NASH, highlighting the importance of regulating hepatic immune responses and oxidative stress as key elements in the treatment of these diseases.
2. Materials and Methods
2.1. Preparation of Agrimonia pilosa Extract
Agrimonia pilosa extract (APE) was provided from BTC Corporation (Sangnok-gu, Ansan, Korea). The dried leaves of A. pilosa was extracted with 50% ethanol (Ducksan, Ansan, Republic of Korea) at 80℃ for 2 h twice. The first and second extracts were acquired after filtering, separately. The supernatants of two extractions were blended well. The complete extract was concentrated through vacuum evaporation and dehydrated to produce APE.
2.2. High-Performance Liquid Chromatography (HPLC) Analysis
The concentration of luteolin-7-O-glucuronide in APE was determined by HPLC using an Agilent Infinity 1260 series system with a diode array detector (Agilent Technologies, Palo Alto, CA, USA). The separation was conducted at 30℃ using a X Select HSS C18 column (4.6 X 250 mm, 5 μm, Waters, Milford, MA, USA). The mobile phase composition was as follows: A, 0.1% phosphoric acid in distilled water; B, Acetonitrile. The gradient elution conditions were as follows; 0-20 min, 10% B; 20-35 min, 10%-12% B; 35-60 min, 12%-12% B; 60-70 min, 12%-30% B; 70-72 min, 30%-80% B; 72-75 min, 80%-80% B; 75-76 min, 80%-10% B; 76-90 min, 10%-10% B. The flow rate was 1.6 mL/min, and the injection volume was 5 μL. The chromatograms were obtained at 350 nm. APE was standardized to obtain at least 0.55% luteolin-7-O-glucuronide (Figure 1).
2.3. Cell Culture
The human liver carcinoma cell line HepG2 cells were sourced from American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were cultured in Minimum Essential Medium with Earle’s Balanced Salts Medium (MEM/EBSS) (Hyclone, South Logan, UT, USA) with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 U/mL streptomycin (Gibco, MA, USA) in a CO2 incubator containing 5% CO2 at 37℃. All the experiments were conducted when the cells reached 80% confluence.
2.4. Preparation of Stock solution of Fatty acids
To prepare the FFA-BSA conjugate, we conjugated palmitic acid (PA) and oleic acid (OA) with fatty acid-free bovine serum albumin (BSA) according to protocol described below. We dissolved 0.0641 g PA and 79 μL OA in 2.5 mL 100% ethanol (Sigma-Aldrich, St. Louis, MO, USA) to acquire a FFA concentration of 100 mM FFA stock solution. Then, we conjugated these solutions with 20% fatty acid-free BSA in PBS at 70℃ for 1 h to obtain an ultimate FFA stock solution (10 mM). All stock solutions were stored at -20℃.
2.5. Cell Viability Assay
Cell viability was evaluated using the Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies, Japan). Briefly, HepG2 cells were initially seeded at a density of 1 × 105 cells/well in 96-well plates (SPL Life Science, South Korea) and incubated overnight at 37℃ for attachment. Subsequently, various concentrations of APE (ranging from 0 to 400 μg/mL) or 0.125 mM FFA in combination with varying APE concentrations were administered to the cells for 24 h. The cells were treated with 10 μL of CCK-8 solution per well and incubated for 2 h in a CO2 incubator. The absorbance of each well was then quantified at 450 nm using a microplate reader (Bio-Tek Instruments, Winooski, VT, USA).
2.6. Oil-Red O Staining
To visualize the accumulation of lipid droplets in HepG2 cells, the cells were initially seeded at a density of 2.5 × 105 cells/well in 12-well plates and incubated overnight. Subsequently, the cells were pre-treated with APE at the indicated concentrations for 2 h, followed by exposure to 0.125 mM FFA for 24 h. Cells were then fixed using 4% paraformaldehyde (PFA) at room temperature for 30 min. Following fixation, cells were subjected to a rinse with 60% isopropanol, followed by staining utilizing a diluted Oil-Red O working solution for 10 min. Lipid droplets were visualized on an inverted microscope. For quantification of the lipid droplets, they were extracted with isopropanol and their absorbance was subsequently determined at 500 nm.
2.7. Triglyceride (TG) Assay
TG levels were determined using TG Quantification Kit (MAK266, Sigma-Aldrich, St. Louis, MO, USA). Briefly, HepG2 cells were initially seeded at a density of 5 × 105 cells/well in 6-well plates and incubated overnight. Subsequently, the cells received a pre-treatment with APE at concentrations as indicated for 2 h, followed by exposure to 0.125 mM FFA for 24 h. After treatment, cells were lysed in 200 μL of Triglyceride Regent. The lysates were centrifuged at 10,000 × g for 10 min at 4℃, resulting in the collection of supernatants utilized for TG quantification. The supernatants of 10 μL were mixed with 50 μL of the Master Reaction Mix to each well, and incubated at 37℃ for 30 min. The absorbance of each well was then quantified at 570 nm using a microplate reader. TG levels were calculated based on a standard curve generated with Triglyceride standards.
2.8. Animals and Experimental Protocols
Male C57BL/6J mice (6-week-old) were purchased from Joongang Experimental Animal Co. (Seoul, South Korea). The animals were accommodated in separate, ventilated cages within a controlled laboratory environment (22-24℃, 40%-50% humidity) with a 12 h light/dark cycle. Following a 1 week period of acclimation, the mice were randomly distributed to seven groups (n = 8 mice in each group) as follows: Group 1, Control: Mice were fed with a normal diet (Teklad 2018S, Envigo, Madison, WI, USA); Group 2, NASH: fed with a CDAHFD (A06071302, Research Diets Inc., New Brunswick, NJ, USA); Groups 3, 4, and 5, APE 25, 50, and 100: fed a CDAHFD with oral administration of APE (25, 50, and 100 mg/kg per day); Group 6, Sily: fed a CDAHFD with oral administration of silymarin (100 mg/kg per day); Group 7, Lute: fed a CDAHFD with oral administration of luteolin-7-O-glucuronide (luteolin) (20 mg/kg per day) for 12 weeks. At the end of the 12-week experimental period, mice were anesthetized using the isoflurane, liver samples and blood from the portocaval vein were collected and stored at -75℃ until further analysis. All animal experiments were approved by the Kyungpook National University Institutional Animal Care and Use Committee (authorization no. KNU- 2021-0224).
2.9. Measurement of Biochemical Markers
To evaluate liver function and detect liver damage, the blood samples were obtained and centrifuged at 8,000 rpm for 30 min at 4℃ to isolate the serum. The serum levels of aspartate transaminase (AST), and alanine transaminase (ALT) were analyzed using a Fuji DRI-CHEM NX500i plasma biochemistry analyzer (Fujifilm Corporation, Tokyo, Japan).
2.10. Histological Analysis
The liver tissues were preserved in 10% neutral buffered formalin and then embedded in paraffin. The paraffin embedded liver samples were cut at a thickness of 3 μm, and then stained with hematoxylin and eosin (H&E), or Sirius Red to assess fibrosis using standard procedures. For H&E staining, the sections were first deparaffinized with xylene and gradually rehydrated using a series of alcohol solutions with varying concentrations. Subsequently, they were treated with hematoxylin for 4 minutes, and counterstained with eosin for 3 minutes. Afterward, the sections were subjected to dehydration using graded alcohols, clearing in xylene, and finally mounted with Canada balsam (Sigma-Aldrich, St. Louis, MO, USA). For Sirius Red staining, the sections were first deparaffinized with xylene and gradually rehydrated using a series of alcohol solutions with varying concentrations, and rinsed with distilled water. They were then exposed to a solution of 0.1% Sirius Red (Direct Red 80; Sigma-Aldrich, St. Louis, MO, USA) in saturated picric acid at room temperature for 1 h. The sections were subsequently rinsed twice in acidified water, dehydrated through graded alcohols, cleared in xylene, and mounted with Canada balsam. The microscopic examination of these liver sections was conducted using a Nikon ECLIPS 80i microscope. To quantify fibrosis, the areas of Sirius Red-stained regions were quantified in random 10x magnification fields of Sirius Red-stained sections using ImageJ software.
2.11. Immunohistochemical Staining (IHC)
Immunohistochemistry was carried out to analyze the expression of inflammation and fibrosis-related proteins in the liver tissue. Paraffin-embedded liver tissues were deparaffinized, and rehydrated using a series of alcohol solutions with varying concentrations, and rinsed with distilled water. The sections underwent incubation with a 3% hydrogen peroxide (H2O2) solution in methanol for a duration of 10 min. Antigen retrieval was facilitated by subjecting the sections to citrate buffer within a pressure cooker at 110℃ for 5 min. The sections were then performed with Histostatin Plus Broad Spectrum (Invitrogen, California, USA). The primary antibodies were used F4/80, Collagen Ⅰ (COLⅠ), Collagen Ⅴ (COLⅤ) (Abcam, Cambridge, UK). The sections were subjected to an overnight incubation with these primary antibodies at a temperature of 4℃. Following a thorough rinse with PBS, the sections were incubated with secondary antibody for 30 min. Then, the sections underwent an incubation step with streptavidin peroxidase conjugate solution lasting for 30 min. Finally, the sections were subjected to diaminobenzidine (DAB) substrate for 5 min and were counterstained briefly with hematoxylin for 10 s. After dehydration in graded alcohols and clearing in xylene, the sections were mounted with Canada balsam. Images were observed under a Nikon ECLIPS 80i microscope.
2.12. Real-time Polymerase Chain Reaction
Liver samples were homogenized using a FastPrep-24TM 5G Homogenizer (MP biomedicals, Solon, OH, USA) and total RNA was extracted from the HepG2 cells and liver samples using TRIzol reagent (Life Technologies, Grand Island, NY) following the manufacturer’s protocol. For the reverse transcription process, 2.5 μg of total RNA was converted into cDNA using SuperiorScript Ⅲ RT Master Mix (Enzynomics, South Korea), incubating at 25℃ for 10 min, and 42℃ for 60 min, and 85℃ for 5 min. Real-time polymerase chain reaction (Real-time PCR) was conducted using TOPrealTM qPCR 2X PreMIX (SYBR Green with low ROX) (Enzynomics, South Korea) on a qTOWER3G instrument (Analytik Jena, Germany). The thermal cycling conditions were as follows: 95℃ for 10 min; 40 cycles of 95℃ for 10 s, 60℃ for 15 s, 72℃ for 30 s; and 72℃ for 7 min. To ensure primer specificity, a melting curve analysis was carried out for each sequence-specific primer. Subsequent to data acquisition, relative gene expression levels were computed using the comparative Ct method (2-ΔΔCt) with β-actin as the reference gene. The results were indicated as the fold-change of gene expression in the experimental group compared to the control group. The primer sequences for target genes are shown in Table 1.
2.13. Western Blot
HepG2 cells and mouse liver samples were homogenized in RIPA buffer (Millipore, Bedford, MA, USA) adding the protease inhibitors (Roche, Swiss). The homogenates were centrifuged at 13,000 rpm for 30 min at 4℃, with subsequent collection of the supernatants. Equivalents of protein were loaded onto SDS-PAGE gels, ranging from 7.5% to 12%, and electrophoretically separated. The separated proteins were transferred onto PVDF membranes (Millipore, Bedford, MA, USA). After transfer, the membranes were blocked with 5% non-fat dry milk in Tris-buffered saline with 0.1% Tween-20 (TBST) at room temperature for 1 h. The blocking membranes were then incubated overnight at 4℃ with primary antibodies targeting specific proteins, including sterol regulatory element-binding protein 1c (SREBP-1c, MyBioSource, San Diego, CA, USA), CCAAT enhancer-binding protein alpha (C/EBPα, SantaCruz, CA, USA), carnitine palmitoyltransferase 1A (CPT1a, Abcam, Cambridge, UK), sirtuin 1 (SIRT1, Abcam, Cambridge, UK), phospho-AMP-activated protein kinase (p-AMPKα, Cell Signaling, Beverly, MA, USA), AMPKα (Cell Signaling, Beverly, MA, USA), cyclooxygenase 2 (COX-2, Abcam, Cambridge, UK), alpha-smooth muscle actin (α-SMA, Sigma-Aldrich, St. Louis, MO, USA), nuclear factor erythroid 2-related factor 2 (Nrf2, Santa Cruz, CA, USA), heme oxygenase-1 (HO-1, Cell Signaling, Beverly, MA, USA), β-actin (Cell Signaling, Beverly, MA, USA). Following thorough washing with TBST, the membranes underwent incubation with specific HRP-conjugated secondary antibodies at an 1 : 1000 dilution in TBST with 5% skim milk at room temperature for 1 h. After washing the secondary antibodies with TBST, protein bands were visualized utilizing the SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA) and were captured using the C-DiGit Blot Scanner (Li-COR, Corp., Lincoln, NE, USA).
2.14. Statistical Analysis
Data are expressed as the means ± standard deviation (SD). Statistical analysis was determined one-way analysis of variance (ANOVA) followed by Dunnett’s test. * p < 0.05, ** p < 0.01, *** p < 0.001, # p < 0.05, ## p < 0.01, and ### p < 0.001 were regarded as demonstrating a statistically significant distinction. Data were evaluated using Prism software Version 5.0 (GraphPad Prism Software, Inc.).
3. Results
3.1. Effect of APE on the Cell Viability in HepG2 cells
To investigate the impact of FFA and APE on the cell viability, HepG2 cells were evaluated at the different concentrations of APE either alone or in combination with 0.125 mM FFA for a duration of 24 h. These findings demonstrated that APE did not influence the cell viability at the treated concentrations up to 50 μg/mL. However, at a concentration of 200 μg/mL, cell viability was notably reduced, showing an approximately 50% decline (33.82 ± 5.22%; 200 μg/mL, 22.11 ± 1.46%; 400 μg/mL; p < 0.001; Figure 2a). In the presence of 0.125 mM FFA alone, cell viability decreased to 84.35% ± 0.93% of the control cells, attributed to FFA-induced lipotoxicity (p < 0.001). Cell viability decreased by approximately 50% or more when treated with both APE and FFA at APE concentrations ranging from 100 to 400 μg/mL (49.00% ± 7.11% at 100 μg/mL, 17.66% ± 2.62% at 200 μg/mL, 6.00% ± 0.81% at 400 μg/mL; p < 0.001; Figure 2b). Based on these results, the induction of cellular steatosis in HepG2 cells was carried out at a concentration of 0 to 50 ug/ml.
3.2. Effects of APE on Intracellular Lipid Accumulation in HepG2 cells
To assess the impact of APE on accumulation of lipid droplets, cells were initially pre-treated with the indicated concentrations of APE for 2 h, followed by a 24 h treatment with 0.125 mM FFA. Subsequently, staining of the cells was conducted using Oil-red O, and quantification was performed by measuring absorbance at 500 nm. The results of morphological observation after staining revealed a higher frequency of visible lipid droplets in HepG2 cells exposed to 0.125 mM FFA in comparison to the control cells. This finding provides evidence that treatment with FFA successfully induced hepatic steatosis in the cell model. However, APE treatment effectively decreased the visible lipid droplets, suggesting its potential as a therapeutic agent for hepatic steatosis (Figure 3a). Oil-red O dye absorption in APE-treated cells was also reduced dose-dependently compared to the FFA-treated cells (0.19 ± 0.018 nm; 12.5 μg/mL; p < 0.01, 0.11 ± 0.017 nm; 25 μg/mL, 0.04 ± 0.005 nm; 50 μg/mL; p < 0.001; Figure 3b). To analyze the effect of APE on intracellular lipid accumulation, cellular TG contents were quantified using a TG quantification kit. The intracellular TG content displayed a significant elevation in cells exposed to FFA (0.22 ± 0.01 μg/mg; p < 0.001), whereas APE treatment strongly suppressed TG levels between 12.5 and 50 μg/mL (0.16 ± 0.008 μg/mg; 12.5 μg/mL; p < 0.01, 0.14 ± 0.021 μg/mg; 25 μg/mL, 0.12 ± 0.008 μg/mg; 50 μg/mL; p < 0.001; Figure 3c). Collectively, these findings suggest that APE possesses the capacity to effectively mitigate intracellular lipid accumulation in 0.125 mM FFA-exposed HepG2 cells.
3.3. Effects of APE on the Expression of Lipid Metabolism-associated Genes and Proteins in HepG2 cells
The signaling pathways of AMPK and SIRT1 have important roles in regulating metabolism and energy homeostasis. The signaling pathways of AMPK/SIRT1 activate several regulators involved in fatty acid synthesis, including SREBP-1c, C/EBPα, fatty acid synthase (FAS), and peroxisome proliferator-activated receptor gamma (PPARγ), while suppressing enzymes that perform fatty acid oxidation, including CPT1a and PPARα [22]. To identify the underlying molecular mechanisms of lipid metabolism within HepG2 cells, we measured both the mRNA and protein expressions of these key regulators and enzymes involved in FA synthesis and FA oxidation. In HepG2 cells, exposure to FFA notably upregulated the mRNA expressions of genes linked to fatty acid synthesis. The mRNA expressions of C/EBPα and FAS in the FFA group were found to be increased by approximately 1.3-fold, and the mRNA expressions of PPARγ in the FFA group was found to be 1.66-fold higher than in the control group (1.30 ± 0.08-fold; FAS; p < 0.01, 1.36 ± 0.12-fold; C/EBPα, 1.66 ± 0.12-fold; PPARγ; p < 0.001). However, there was no substantial distinction in the mRNA expression of SREBP-1c in the FFA-treated cells. Interestingly, treatment with APE dose-dependently attenuated the upregulation of SREBP-1c, C/EBPα, FAS, and PPARγ. The mRNA expressions related to FA synthesis in the APE 50 μg/mL group were found to be reduced to a level similar to that of the control group (0.91 ± 0.038-fold; C/EBPα, 0.88 ± 0.008-fold; FAS, 0.96 ± 0.026-fold; PPARγ; p < 0.001; Figure 4a-4d). The mRNA expressions of the FA oxidation enzymes were significantly downregulated in FFA-treated cells in comparison to the control cells (0.85 ± 0.044-fold; CPT1a; p < 0.05, 0.55 ± 0.041-fold; PPARα; p < 0.01). However, APE treatment enhanced this decrease dose-dependently, and APE treatment at a high concentration (50 μg/mL) significantly elevated the mRNA expressions of CPT1a and PPARα by 1.3 and 1.1-fold than in the control group (1.30 ± 0.081-fold; CPT1a; p < 0.001, 1.09 ± 0.064-fold; PPARα; p < 0.01; Figure 4e and 4f). Western blot analysis revealed comparable results regarding the levels of protein expression. The protein expressions of SREBP-1c and C/EBPα were found to increase in FFA-treated cells, while APE treatment strongly alleviated this upregulation at a high concentration (50 μg/mL). In contrast, exposure to FFA resulted in a notable reduction in the protein expressions of CPT1a, SIRT1, and the phosphorylation of AMPKα. However, APE treatment strongly elevated the protein expressions of AMPK phosphorylation, SIRT1, and FA oxidation (Figure 4g). These findings indicate that APE can ameliorate hepatic lipid accumulation by inhibiting FA synthesis and inducing FA oxidation in FFA-mediated steatosis.
3.4. Effects of APE on the Body Weight and Liver Weight in CDAHFD-induced NASH mouse model
To evaluate the impact of APE on NASH induced by CDAHFD diet, mice were provided with a CDAHFD containing added vehicle, APE, silymarin or luteolin for 12 weeks. Changes in mouse body weight and liver tissues were observed. At the end of the 12-week experimental period, the average of final body weight in the CDAHFD-induced NASH group and control group was 26.0 ± 1.6 g and 28.2 ± 1.8 g, respectively. The average of final body weight in the NASH group exhibited a notable decrease against to that of the control group (p < 0.001). Administration of APE at a dosage of 100 mg/kg elevated slightly in the average final body weight compared with the NASH group (27.1 ± 1.4 g; Figure 5a). The macroscopic appearance of the liver in the NASH group showed noticeable alterations against with the control group, including a shift from reddish brown to yellowish brown coloration and an increase in overall liver size. These changes indicated the development of steatohepatitis induced by CDAHFD. These morphological alterations were remarkably attenuated in the APE, Sily, and Lute groups. Notably, administration of APE at a dosage of 100 mg/kg led to a morphological liver appearance that closely resembled appearance to the control group (Figure 5b). Furthermore, the average ratio of liver to body weight exhibited a statistically significant increase in the NASH group (4.605 ± 0.451%) than in the control group (3.260 ± 0.203%; p < 0.001). However, the average ratios of liver to body weight of the APE, Sily, and Lute groups were observed to be lower than that of the NASH group (4.058 ± 0.389%; Sily; p < 0.05; Figure 5c).
3.5. Effects of APE on Hepatic Steatosis and Serum Markers of Liver Injury in CDAHFD-induced NASH mice
To identify any protective effects of APE against hepatic steatosis, we conducted a histological examination of liver tissues and analyzed the serum concentrations of ALT and AST, which are often used as markers for liver function and injury. Microscopic examination of the H&E-stained liver sections of the NASH group exhibited diffuse steatohepatitis characterized by the presence of numerous lipid droplets, mild inflammation, and ballooning degeneration within hepatocytes. However, the lipid droplets in hepatocytes and immune cells were significantly reduced in the APE, Sily, and Lute groups. These results demonstrate that APE treatment can significantly inhibit hepatic steatosis and inflammation (Figure 6a).
The serum levels of ALT and AST were dramatically elevated by CDAHFD. The mean serum ALT level in the NASH group was 311.2 ± 66.7 U/L, whereas that in the control group was 26.0 ± 2.2 U/L (p < 0.001). Similarly, the mean serum AST level in the NASH group was 219.0 ± 55.7 U/L, whereas that in the control group was 38.8 ± 6.4 U/L (p < 0.001). However, in the treatment groups, the APE 100, Sily, and Lute groups showed significantly mitigated serum levels of ALT and AST compared to the serum levels of ALT and AST in the NASH group (p < 0.001). Among the treatment groups, the APE 100 group demonstrated a slightly more pronounced effect on the serum levels of both liver damage markers than the other groups (160.0 ± 49.1 U/L; ALT, 96.0 ± 18.7 U/L; AST; p < 0.001; Figure 6b and 6c). These results demonstrate that APE may have protective effects against hepatic steatosis and liver inflammation.
3.6. Effects of APE on Inflammation and Fibrosis in CDAHFD-induced NASH mice
To examine the effects of APE on liver inflammation and fibrosis in NASH, IHC along with Sirius Red staining was conducted to detect F4/80, COLⅠ, and COLⅤ. The results of IHC for F4/80 demonstrated that the control group had a low number of F4/80-positive cells, whereas the NASH group had a higher number of these cells, reflecting a greater degree of inflammation (1.35 ± 0.09%; CON, 3.66 ± 0.16%; NASH; p < 0.001). Conversely, treatment with APE, silymarin, and luteolin resulted in reduction in the quantity of F4/80-positive cells compared with the NASH group (1.46 ± 0.20%; Sily, 1.73 ± 0.11%; Lute; p < 0.001). Remarkably, the groups treated with APE exhibited a dose-dependent decrease in the quantity of F4/80-positive cells, with both the APE 50 and APE 100 groups showing significant reductions (1.81 ± 0.15%; APE 50, 1.39 ± 0.07%; APE 100; p < 0.001; Figure 7a and 7b). Sirius Red staining revealed that the NASH group showed an enormous increase in collagen deposition, indicating liver fibrosis, whereas the control group showed minimal collagen deposition in the liver (0.03 ± 0.01%; CON, 5.63 ± 0.39%; NASH; p < 0.001). Sirius Red staining revealed that the APE, Sily, and Lute groups exhibited a substantial reduction in collagen deposition compared with the NASH group (1.69 ± 0.002%; Sily, 2.63 ± 0.12%; Lute; p < 0.001). Administration of APE significantly decreased fibrotic staining in all of the APE groups (2.39 ± 0.16%; APE 25, 1.81 ± 0.16%; APE 50, 1.54 ± 0.03%; APE 100; p < 0.001). IHC results for COLⅠ and COLⅤ were similar pattern to the Sirius Red staining results. The NASH group showed a marked increase in COLⅠ and COLⅤ staining, whereas the control group exhibited minimal COLⅠ and COLⅤ staining. Administration of APE, silymarin, and luteolin led to a notable reduction in COLⅠ and COLⅤ staining. The overall level of fibrosis in the APE 100 group was comparable to that of the control group (Figure 7c and 7d). Histologically, these results indicate that APE can provide protection against liver inflammation and fibrosis in NASH.
3.7. Effects of APE on the Expression of Lipid Metabolism-associated Genes and Proteins in CDAHFD-induced NASH mice
To examine the molecular mechanisms of lipid metabolism in the mouse liver, we analyzed the mRNA and protein expression levels of enzymes involved in FA synthesis, FA oxidation, and very low-density lipoprotein processing. Our results showed that the NASH group exhibited elevated the mRNA expressions of SREBP-1c and PPARγ by 1.33-fold and 2.48-fold, respectively (2.48 ± 0.43-fold; PPARγ; p < 0.001). However, the mRNA expression of PPARγ experienced a significant reduction in the APE, Sily, and Lute groups in comparison to the NASH group (1.59 ± 0.17-fold; APE 25, 1.66 ± 0.16-fold; APE 50, 1.51 ± 0.31-fold; APE 100; p < 0.05, 1.45 ± 0.17-fold; Sily, 1.45 ± 0.24-fold; Lute; p < 0.01; Figure 8a and 8b). Conversely, the mRNA expressions of CPT1a and PPARα were found to be reduced by 0.73-fold and 0.65-fold in the NASH group (0.73 ± 0.07-fold; CPT1a, 0.65 ± 0.06-fold; PPARα). However, treatment with APE at a dose of 100 mg/kg led to a substantial elevation in the mRNA expressions of CPT1a and PPARα by 1.06-fold and 1.10-fold, respectively (1.06 ± 0.04-fold; CPT1a, 1.10 ± 0.19-fold; PPARα; p < 0.05; Figure 8c and 8d). In addition, the mRNA expression of genes related to VLDL secretion, including apolipoprotein B (ApoB) and microsomal triglyceride transfer protein (MTTP), were significantly reduced approximately 0.5-fold in the NASH group (0.48 ± 0.06-fold; ApoB, 0.50 ± 0.01-fold; MTTP; p < 0.01). Nonetheless, APE treatment led to gradually increase in the mRNA expressions of ApoB and MTTP. In particular, the mRNA expressions of VLDL secretion (ApoB and MTTP) in the APE 100 group were 0.94-fold and 1.01-fold, showing a tendency similar to those of the control group (0.94 ± 0.04-fold; ApoB; p < 0.05, 1.01 ± 0.24-fold; MTTP; p < 0.01; Figure 8e and 8f). Western blot analysis showed similar results for the protein expression levels. The NASH group demonstrated increased protein levels of SREBP-1c and C/EBPα in comparison to the control group, and APE treatment alleviated gradually the levels of these proteins. Conversely, the protein levels of CPT1a and SIRT1 and the phosphorylation of AMPKα were strongly decreased in the NASH group, which were restored following treatment with APE, silymarin, and luteolin. Moreover, APE treatment elevated the protein concentrations of SIRT1 and promoted the phosphorylation of AMPKα in a dose-dependent manner (Figure 8g). These results demonstrate that APE treatment can regulate lipid metabolism by suppressing lipid synthesis and promoting lipolysis.
3.8. Effects of APE on the Expression of Inflammatory Factors in CDAHFD-induced NASH mice
In the progression of NASH, inflammation is intricately linked to the pathogenesis and advancement of hepatic steatosis to steatohepatitis [23]. To evaluate whether APE regulates the immune response in the liver, we measured the mRNA and protein expression of various markers related to inflammation, including macrophage markers (F4/80 and YM-1), chemokines (monocyte chemoattractant protein 1 [MCP-1], and macrophage inflammatory protein-2 [MIP-2]), and proinflammatory cytokines (interleukin-6 [IL-6], tumor necrosis factor alpha [TNF-α], and COX-2). The mRNA expressions of macrophage markers involved in inflammation, namely, F4/80 and YM-1, were considerably increased by 11.34-fold and 39.91-fold in the NASH group (11.34 ± 1.24-fold; F4/80, 39.91 ± 6.89-fold; YM-1; p < 0.001), however, the mRNA expressions of macrophage markers were strongly reduced in APE-treated groups (3.04 ± 1.12-fold; APE 100 (F4/80), 12.10 ± 4.09-fold; APE 100 (MTTP); p < 0.001; Figure 9a and 9b). Furthermore, the mRNA expressions of the chemokines MCP-1 and MIP-2 were significantly higher in the NASH group by 72.11-fold and 4.60-fold than in the control group, respectively (72.11 ± 6.90-fold; MCP-1, 4.60 ± 0.36-fold; MIP-2; p < 0.001), however, APE treatment led to a significant decrease in their expressions in a dose-dependent manner (39.19 ± 2.49-fold; APE 100 (MCP-1); p < 0.001, 2.55 ± 0.07-fold; APE 100 (MIP-2); p < 0.01; Figure 9c and 9d). The mRNA expressions of the proinflammatory cytokines including IL-6, TNF-α, and COX-2 were significantly augmented in the NASH group compared with the control (2.29 ± 0.16-fold; IL-6; p < 0.01, 16.33 ± 1.36-fold; TNF-α, 4.77 ± 0.70-fold; COX-2; p < 0.001). The mRNA expressions of these cytokines exhibited a dose-dependent reduction in the APE-treated groups, with the APE 100 group showing the most significant decrease (0.79 ± 0.36; IL-6; p < 0.01, 3.76 ± 1.47; TNF-α; p < 0.001, 2.42 ± 0.71; COX-2; p < 0.01; Figure 9e-9g). Western blot analysis revealed similar results for COX-2 at the protein expression level. Among the treated groups, the APE 100 group demonstrated the most substantial decrease in COX-2 protein expression, exhibiting levels comparable to the control group (Figure 9h). These findings demonstrate that APE exerts anti-inflammatory effects in the liver by restricting the expulsion of chemokines and proinflammatory cytokines.
3.9. Effects of APE on the Expression of Fibrosis-related Proteins in CDAHFD-induced NASH mice
Hepatic fibrosis is caused by infiltration of immune cells due to chronic inflammation. Inflammation triggers the activation of hepatic stellate cells (HSCs), which impart to the formation of liver myofibroblasts in NAFLD models [24]. The frequency of activated HSCs expressing α-SMA and collagen type I alpha 1 chain (COL1A1) can be monitored to gauge the degree of liver fibrosis. However, α-SMA and COL1A1 positivity describes only a subpopulation of activated HSCs and myofibroblasts, so we also used other myofibroblast markers, such as desmin and transforming growth factor beta 1 (TGF-β1). To investigate the liver’s protective potential of APE against fibrosis, the abovementioned fibrosis markers were examined via real-time PCR and western blot analysis. Based on the mRNA levels of these markers, the NASH group was more fibrogenic than the control group (11.94 ± 1.15; α-SMA, 159.58 ± 5.20; Col1a1, 6.64 ± 0.07; Desmin, 10.42 ± 0.91; TGF-β1; p < 0.001). Treatment with APE again demonstrated a dose-dependent attenuation of elevated mRNA expression levels of these markers, indicating a significant reduction in fibrotic potential (1.98 ± 0.54; APE 100 (α-SMA), 26.21 ± 10.43; APE 100 (Col1a1); 2.19 ± 0.16; APE 100 (Desmin), 3.31 ± 0.34; APE 100 (TGF-β1); p < 0.001; Figure 10a-10d). Moreover, western blot analysis of α-SMA revealed that its expression level was significantly higher in the NASH group than in the control group. However, the administration of APE led to a dose-dependent reduction in the protein expression level of α-SMA (Figure 10e). These findings show that APE can attenuate NASH-related fibrosis and HSC and myofibroblast activation in the liver.
3.10. Effects of APE on Oxidative Stress and Endoplasmic Reticulum (ER) Stress in CDAHFD-induced NASH mice
The development from hepatic steatosis to NASH can be initiated by oxidative stress induced by the production of reactive oxygen species and endoplasmic reticulum (ER) stress. [25,26]. To assess whether APE prevents NASH-induced oxidative stress and ER stress, we conducted quantitative PCR and western blot analysis to assess the levels of stress markers that mediate NASH progression. The results revealed that the NASH group had significantly increased mRNA expression levels related to the cellular response to oxidative stress, including HO-1, NADPH oxidase 2 (NOX2), NOXA2, and NAD(P)H dehydrogenase quinone 1 (NQO1) (12.46 ± 0.53; HO-1, 15.55 ± 1.10; NOX2, 18.88 ± 1.01; NOXA2, 3.71 ± 0.39; NQO1; p < 0.001). This elevated mRNA expression was abrogated by APE treatment in a dose-dependent manner, with the most significant decrease observed in the APE 100 group (4.52 ± 0.42; HO-1, 3.66 ± 1.11; NOX2, 6.91 ± 0.03; NOXA2, 1.29 ± 0.11; NQO1; p < 0.001; Figure 11a-11d). Western blot analysis indicated that the protein levels of Nrf2 and HO-1 were also elevated in the NASH group compared with the control group, which were dose-dependently decreased by APE treatment (Figure 11e).
ER stress represents another pivotal factor in the pathogenesis and progression of NASH. Although we observed no significant changes in apoptotic response regulators G protein-coupled receptor 78 (GPR78) and X-box binding protein 1 (XBP1) expression, the mRNA expression level of growth arrest- and DNA damage-inducible gene 153 (GADD153) was increased in the NASH group (3.10 ± 0.14; p < 0.001). The APE, Sily, and Lute groups exhibited a significant reduction in the mRNA expression of GADD153 (1.19 ± 0.23; APE 100, 0.90 ± 0.22; Sily, 1.42 ± 0.04; Lute; p < 0.001) compared with the NASH group (Figure 11f-11h). Overall, these findings indicate that APE treatment can inhibit the pathogenesis and progression of NASH by suppressing oxidative stress and ER stress.
4. Discussion
NASH is a complex liver condition characterized by the accumulation of excess fat in liver cells, inflammation, and liver damage. [27]. NASH treatments have seen development efforts on a global scale. However, their limited success can be attributed to the lack of drugs capable of effectively and comprehensively regulating the diverse pathways involved in the progression of NASH. In particular, synthetic pharmaceutical agents are typically formulated to selectively obstruct a singular signaling pathway. Consequently, addressing the intricate task of mitigating NASH is expected to present an even greater level of complexity. Natural products such as herbs, plant extracts, and dietary supplements often contain bioactive compounds with hepatoprotective properties that can act on multiple targets and pathways involved in NASH pathogenesis. The natural compounds used for NASH treatment should possess the capacity to mitigate oxidative stress and inflammation, which are key drivers of NASH progression. Several natural compounds have been shown to possess antioxidant and anti-inflammatory properties, including polyphenols, such as resveratrol, curcumin, and quercetin, as well as omega-3 FAs found in fish oil [28,29,30,31]. These compounds have been studied extensively in animal models and some clinical trials, with promising results in terms of their ability to mitigate oxidative stress and inflammation as well as improve liver histology and function. One potential advantage of natural compounds over synthetic drugs is their relatively low toxicity and fewer side effects.
Therefore, our study tested the potential of APE as a candidate material for the treatment of NASH based on its strong antioxidant and anti-inflammatory effects. In the present study, we conducted experiments via FFA-induced cellular steatosis in HepG2 cells and a CDAHFD-induced NASH model in mice. Our findings revealed that A. pilosa exerted inhibitory effects on the accumulation of intracellular lipid droplets and TG in both FFA-treated HepG2 cells and the liver tissue of CDAHFD-induced NASH mice.
Furthermore, we aimed to evaluate the expression of genes related to lipid metabolism, specifically concerning the inhibition of fat globule accumulation and TG reduction following the administration of APE. Several cell signaling pathways are involved in the regulation of fat metabolism and contribute to the development and progression of NASH. Among these pathways, the SREBP pathway plays a prominent role in the regulation of lipogenesis. Transcription factors known as SREBPs control the expression of genes involved in FA and TG synthesis. Dysregulation of the SREBP pathway in NASH can lead to excessive lipogenesis and an augmented accumulation of TGs within hepatocytes [32,33]. Another significant pathway implicated in NASH is the PPARγ pathway. PPARγ, a nuclear receptor, plays a significant role in adipocyte differentiation and lipid metabolism. Activation of PPARγ promotes adipogenesis and facilitates lipid storage. Aberrant activation of PPARγ in NASH can contribute to generated lipogenesis and lipid droplets accumulation in the liver [34,35]. In addition, the AMPK pathway, serving as a key cellular energy sensor, regulates lipid metabolism. Activation of AMPK promotes lipolysis by phosphorylating and inhibiting key enzymes involved in lipogenesis while simultaneously activating enzymes responsible for TG breakdown. Impaired AMPK signaling in NASH disrupts lipolysis and contributes to the accumulation of TGs [36,37]. These pathways interact with and influence each other, collectively forming a complex network that governs lipid metabolism. Our study found that treatment with APE in FFA-induced hepatic steatosis reduced the expression levels of lipid metabolism-related genes, such as SREBP-1c and C/EBPα, and elevated the expression levels of FA oxidation-related genes, such as CPT1a and PPARα, via the AMPK/SIRT1 pathway. Furthermore, in line with our in vitro data, treatment of APE in a CDAHFD-induced NASH mouse model downregulated SREBP-1c expression, activated AMPK, and upregulated PPARα expression in the liver. These results demonstrate that AMPK/SIRT1 activation and subsequent lipid metabolism inhibition is a potential mechanism for the beneficial effect of A. pilosa on hepatic steatosis.
Considering the complex pathogenesis of NASH, we further investigated the effects of A. pilosa the expression of genes related to on inflammation, oxidative stress, ER stress, and liver fibrosis. Inflammation and oxidative stress play a crucial role in the development of NASH by triggering a cascade of events that lead to more severe liver diseases. In addition, inflammation and oxidative stress are known to contribute to ER stress by increasing the demand for protein folding and promoting the accumulation of misfolded proteins. In turn, ER stress can also activate the unfolded protein response and contribute to the development of inflammation and oxidative stress [38,39]. The interplay among inflammation, oxidative stress, and ER stress in NASH can lead to fibrosis, cirrhosis, and hepatocellular carcinoma. In the present study, we additionally conducted an examination of gene expression patterns associated with inflammation, oxidative stress, ER stress, and liver fibrosis to assess the potential inhibitory impact of A. pilosa on NASH. Notably, our findings revealed that treatment with APE strongly attenuated inflammation, as evidenced by the decreased expression of key macrophage, cytokine, and chemokine markers, including F4/80, MCP-1, IL-6, TNF-α, and COX-2. Moreover, A. pilosa demonstrated its ability to suppress hepatic oxidative stress and ER stress, as indicated by the lower expression levels of Nrf2, HO-1, and GADD 153. We also demonstrated that APE treatment led to a decrease in hepatic fibrosis markers, such as α-SMA, COL1A1, desmin, and TGF-β1. The above results indicate that A. pilosa contributes to the prevention of the advancement towards hepatic fibrosis through the suppression of lipid metabolism, attenuation of damage induced by various intracellular stressors, and inhibition of intrahepatic fat accumulation.
Overall, A. pilosa is expected to be more competitive than other plant extracts. These results are attributed to the influence of various active ingredients, as they exhibit higher efficacy compared to luteolin, one of the liver damage-inhibiting constituents present in the APE studied in this study.
5. Conclusions
In summary, our study provides evidence that A. pilosa possesses the potential to mitigate intracellular lipid accumulation in FFA-treated HepG2 cells, suggesting that A. pilosa exerts a protective effect against NASH by modulating lipid metabolism. Moreover, the introduction of A. pilosa in a CDAHFD-induced NASH mouse model resulted in substantial improvements, including reduced fat accumulation in hepatocytes, decreased levels of pro-inflammatory cytokines, lower inflammatory mediator expression, reduced oxidative stress, and diminished hepatic fibrosis. The results of the present study underscore the therapeutic potential of A. pilosa in addressing NASH-related complications. Overall, these findings highlight the promising role of A. pilosa in managing NASH and suggest its potential as a multifaceted therapeutic intervention.
Author Contributions
Min-Jeong Jo and Youngjae Ryu contributed equally to this study. Sang-Joon Park and Yongduk Kim conceived and designed the study. Jae Woo Lee and Yoon Hee Kim provided and analysed A. pilosa extract. Min-Jeong Jo, Youngjae Ryu, and Myung-Gi Seo carried out in vivo and in vitro experiments. Min-Jeong Jo, Youngjae Rye, Jin-Kyu Park, and Sang-Joon Park wrote and review the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by the 2021 Technology development program (Development of new products subject to purchase conditions, S3137537), funded by the Ministry of SMEs and Startups (MSS, Republic of Korea).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nature Reviews Gastroenterology & Hepatology 2013, 10, 686-690. [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (Baltimore, Md.) 2016, 64, 73-84. [CrossRef]
- Maurice, J.; Manousou, P. Non-alcoholic fatty liver disease. Clinical medicine journal 2018, 18, 245-250. [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology (Baltimore, Md.) 2010, 51, 679-689. [CrossRef]
- Milić, S.; Lulić, D.; Štimac, D. Non-alcoholic fatty liver disease and obesity: Biochemical, metabolic and clinical presentations. World journal of gastroenterology 2014, 20, 9330-9337. [CrossRef]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [CrossRef]
- Saponaro, C.; Gaggini, M.; Gastaldelli, A. Nonalcoholic fatty liver disease and type 2 diabetes: Common pathophysiologic mechanisms. Current diabetes reports 2015, 15, 607. [CrossRef]
- Zhang, Q.Q.; Lu, L.G. Nonalcoholic Fatty Liver Disease: Dyslipidemia, Risk for Cardiovascular Complications, and Treatment Strategy. Journal of clinical and translational hepatology 2015, 3, 78-84. [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD as a driver of chronic kidney disease. Journal of hepatology 2020, 72, 785-801. [CrossRef]
- Nassir, F. NAFLD: Mechanisms, Treatments, and Biomarkers. Biomolecules 2022, 12, 824. [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Annals of internal medicine 2016, 165, 305-315. [CrossRef]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of NAFLD. Diseases (Basel, Switzerland) 2018, 6, 86. [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (London, England) 2019, 394, 2184-2196. [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism: Clinical and experimental 2016, 65, 1038-1048. [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology (Baltimore, Md.) 2010, 52, 1836-1846. [CrossRef]
- Ucar, F.; Sezer, S.; Erdogan, S.; Akyol, S.; Armutcu, F.; Akyol, O. The relationship between oxidative stress and nonalcoholic fatty liver disease: Its effects on the development of nonalcoholic steatohepatitis. Redox report : Communications in free radical research 2013, 18, 127-133. [CrossRef]
- Jin, T.; Chi, L.; Ma, C. Agrimonia pilosa: A Phytochemical and Pharmacological Review. Evidence-based complementary and alternative medicine : eCAM 2022, 2022, 3742208. [CrossRef]
- Wen, S.; Zhang, X.; Wu, Y.; Yu, S.; Zhang, W.; Liu, D.; Yang, K.; Sun, J. Agrimonia pilosa Ledeb.: A review of its traditional uses, botany, phytochemistry, pharmacology, and toxicology. Heliyon 2022, 8, e09972. [CrossRef]
- Chen, L.; Teng, H.; Fang, T.; Xiao, J. Agrimonolide from Agrimonia pilosa suppresses inflammatory responses through down-regulation of COX-2/iNOS and inactivation of NF-κB in lipopolysaccharide-stimulated macrophages. Phytomedicine : International journal of phytotherapy and phytopharmacology 2016, 23, 846-855. [CrossRef]
- Feng, J.H.; Kim, H.Y.; Sim, S.M.; Zuo, G.L.; Jung, J.S.; Hwang, S.H.; Kwak, Y.G.; Kim, M.J.; Jo, J.H.; Kim, S.C.; et al. The Anti-Inflammatory and the Antinociceptive Effects of Mixed Agrimonia pilosa Ledeb. and Salvia miltiorrhiza Bunge Extract. Plants (Basel, Switzerland) 2021, 10, 1234. [CrossRef]
- Jang, H.H.; Bae, J.H.; Kim, M.J.; Park, M.Y.; Kim, H.R.; Lee, Y.M. Agrimonia pilosa Ledeb. Ameliorates Hyperglycemia and Hepatic Steatosis in Ovariectomized Rats Fed a High-Fat Diet. Nutrients 2020, 12, 1631. [CrossRef]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. The Journal of biological chemistry 2008, 283, 20015-20026. [CrossRef]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nature reviews. Gastroenterology & hepatology 2018, 15, 349-364. [CrossRef]
- Jiao, J.; Friedman, S.L.; Aloman, C. Hepatic fibrosis. Current opinion in gastroenterology 2009, 25, 223-229. [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants (Basel, Switzerland) 2021, 10, 174. [CrossRef]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free radical research 2015, 49, 1405-1418. [CrossRef]
- Neuschwander-Tetri, B.A. Non-alcoholic fatty liver disease. BMC medicine 2017, 15, 45. [CrossRef]
- Heebøll, S.; El-Houri, R.B.; Hellberg, Y.E.; Haldrup, D.; Pedersen, S.B.; Jessen, N.; Christensen, L.P.; Grønbaek, H. Effect of resveratrol on experimental non-alcoholic fatty liver disease depends on severity of pathology and timing of treatment. Journal of gastroenterology and hepatology 2016, 31, 668-675. [CrossRef]
- Afrin, R.; Arumugam, S.; Rahman, A.; Wahed, M.I.; Karuppagounder, V.; Harima, M.; Suzuki, H.; Miyashita, S.; Suzuki, K.; Yoneyama, H.; et al. Curcumin ameliorates liver damage and progression of NASH in NASH-HCC mouse model possibly by modulating HMGB1-NF-κB translocation. International immunopharmacology 2017, 44, 174-182. [CrossRef]
- Marcolin, E.; San-Miguel, B.; Vallejo, D.; Tieppo, J.; Marroni, N.; González-Gallego, J.; Tuñón, M.J. Quercetin treatment ameliorates inflammation and fibrosis in mice with nonalcoholic steatohepatitis. The Journal of nutrition 2012, 142, 1821-1828. [CrossRef]
- Caldwell, S. NASH Therapy: Omega 3 supplementation, vitamin E, insulin sensitizers and statin drugs. Clinical and molecular hepatology 2017, 23, 103-108. [CrossRef]
- Moslehi, A.; Hamidi-Zad, Z. Role of SREBPs in Liver Diseases: A Mini-review. Journal of clinical and translational hepatology 2018, 6, 332-338. [CrossRef]
- Ferré, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes, obesity & metabolism 2010, 12 Suppl 2, 83-92. [CrossRef]
- Liss, K.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65-74. [CrossRef]
- Skat-Rørdam, J.; Højland Ipsen, D.; Lykkesfeldt, J.; Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic & clinical pharmacology & toxicology 2019, 124, 528-537. [CrossRef]
- Strzyz, P. AMPK against NASH. Nature Reviews Molecular Cell Biology 2020, 21, 181-181. [CrossRef]
- Anggreini, P.; Kuncoro, H.; Sumiwi, S.A.; Levita, J. Role of the AMPK/SIRT1 pathway in non-alcoholic fatty liver disease (Review). Molecular medicine reports 2023, 27, 2. [CrossRef]
- Pomacu, M.M.; Trașcă, M.D.; Pădureanu, V.; Bugă, A.M.; Andrei, A.M.; Stănciulescu, E.C.; Baniță, I.M.; Rădulescu, D.; Pisoschi, C.G. Interrelation of inflammation and oxidative stress in liver cirrhosis. Experimental and therapeutic medicine 2021, 21, 602. [CrossRef]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d'Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Frontiers in cellular neuroscience 2014, 8, 213. [CrossRef]
Figure 1.
High-performance liquid chromatography with diode array detection (HPLC-DAD) chromatograms. Panel (a) represents the blank chromatogram. Panel (b) shows the chromatogram of the luteolin-7-O-glucuronide standard. Panel (c) displays the chromatogram of Agrimonia pilosa with excitation at 350 nm.
Figure 1.
High-performance liquid chromatography with diode array detection (HPLC-DAD) chromatograms. Panel (a) represents the blank chromatogram. Panel (b) shows the chromatogram of the luteolin-7-O-glucuronide standard. Panel (c) displays the chromatogram of Agrimonia pilosa with excitation at 350 nm.
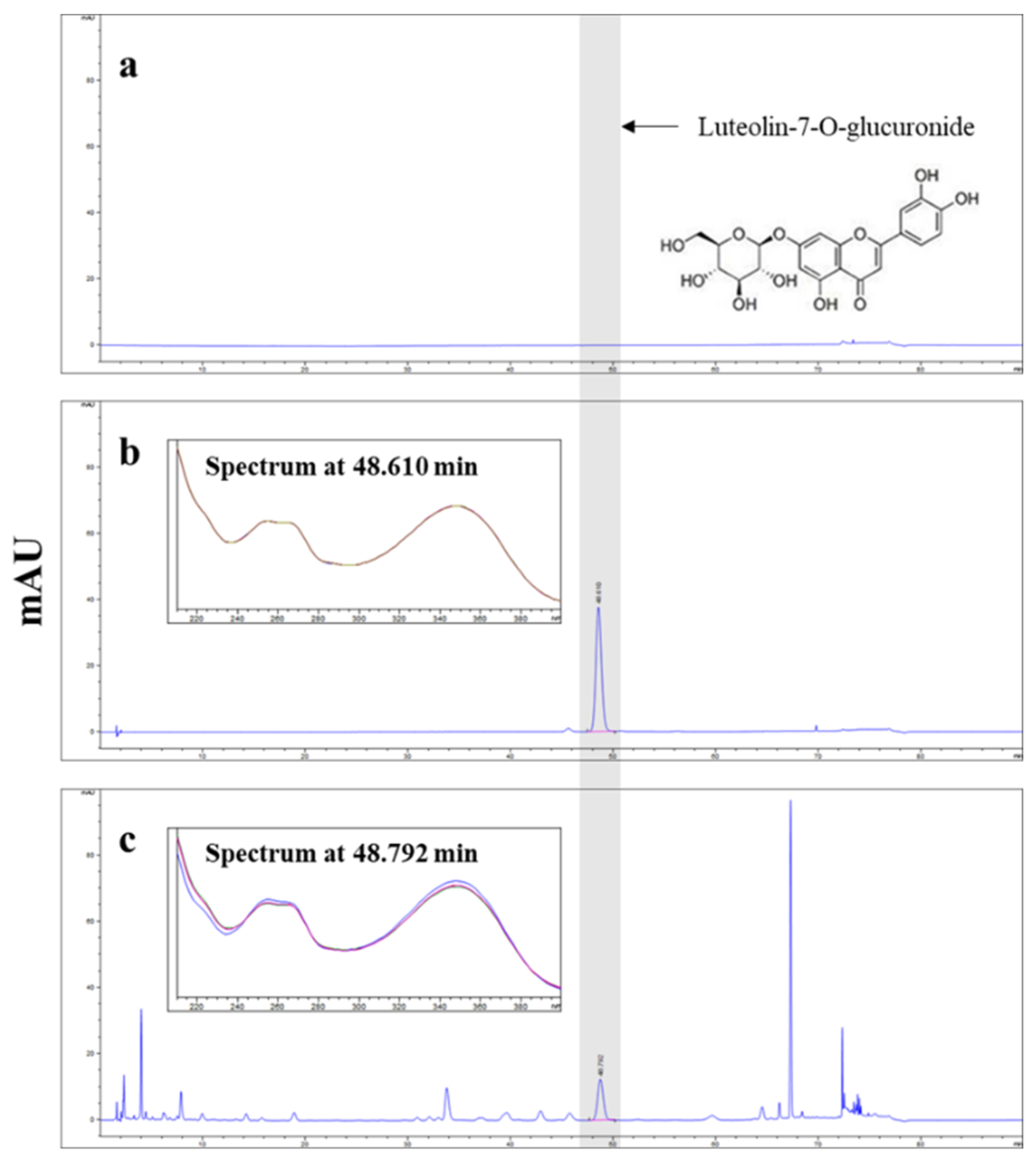
Figure 2.
Effects of APE on the cell viability of HepG2 cells. (a) HepG2 cells were treated with different concentrations of APE (0, 3.125, 6.25, 12.5, 25, 50, 100, 200, and 400 μg/mL) for 24 h. (b) Cells were exposed with 0.125 mM FFA and different concentrations of APE (0, 3.125, 6.25, 12.5, 25, 50, 100, 200, and 400 μg/mL) for 24 h. Cell viability was assessed through the utilization of the CCK-8 assay. The data are presented as the mean ± SD. * p < 0.05, *** p < 0.001 vs. the control group; ### p < 0.001 vs. the group treated with FFA alone.
Figure 2.
Effects of APE on the cell viability of HepG2 cells. (a) HepG2 cells were treated with different concentrations of APE (0, 3.125, 6.25, 12.5, 25, 50, 100, 200, and 400 μg/mL) for 24 h. (b) Cells were exposed with 0.125 mM FFA and different concentrations of APE (0, 3.125, 6.25, 12.5, 25, 50, 100, 200, and 400 μg/mL) for 24 h. Cell viability was assessed through the utilization of the CCK-8 assay. The data are presented as the mean ± SD. * p < 0.05, *** p < 0.001 vs. the control group; ### p < 0.001 vs. the group treated with FFA alone.
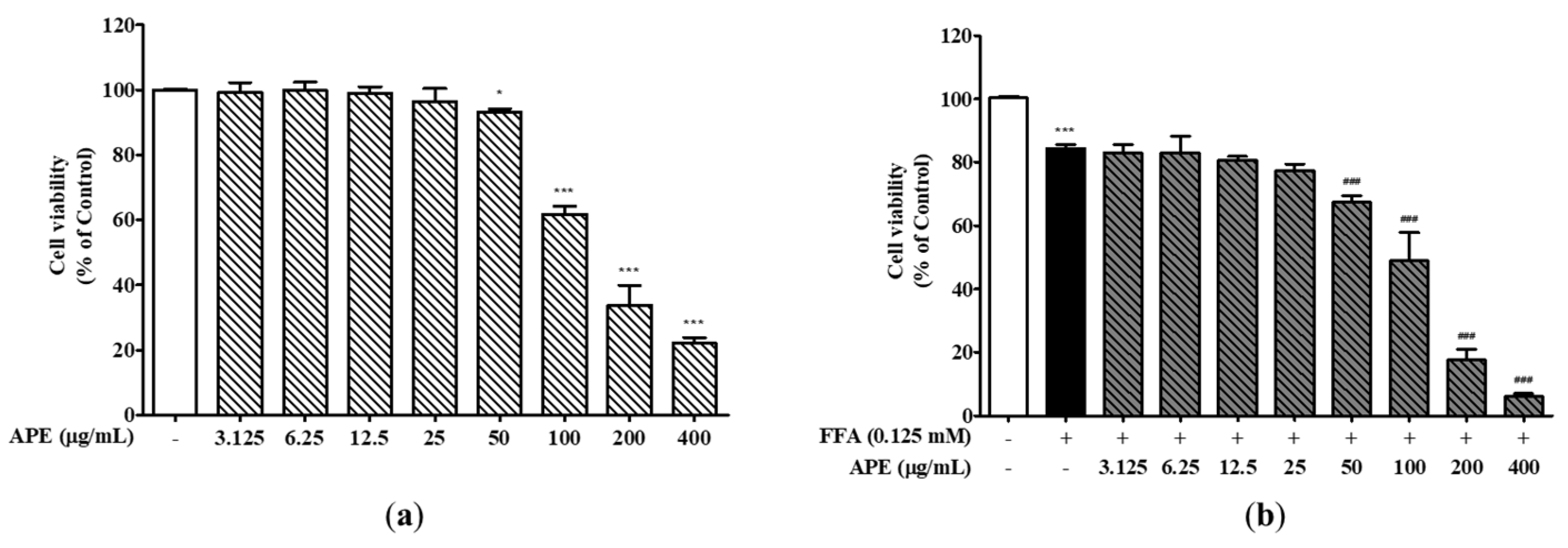
Figure 3.
Effects of APE on intracellular lipid accumulation in HepG2 cells. HepG2 cells were initially pre-treated with the indicated concentrations of APE (0, 6.25, 12.5, 25, 50 μg/mL) for 2 h, followed by a 24 h treatment with 0.125 mM FFA. Cells were stained with Oil-Red O; (a) accumulation of lipid droplets were visualized using a microscope (200x magnification), and (b) Oil-Red O absorption was quantified by assessing the optical density at 500 nm. (c) Intracellular TG contents were analyzed with a TG quantification kit. The data are presented as the mean ± SD. *** p < 0.001 vs. the control group; ## p < 0.01, ### p < 0.001 vs. the group treated with FFA alone.
Figure 3.
Effects of APE on intracellular lipid accumulation in HepG2 cells. HepG2 cells were initially pre-treated with the indicated concentrations of APE (0, 6.25, 12.5, 25, 50 μg/mL) for 2 h, followed by a 24 h treatment with 0.125 mM FFA. Cells were stained with Oil-Red O; (a) accumulation of lipid droplets were visualized using a microscope (200x magnification), and (b) Oil-Red O absorption was quantified by assessing the optical density at 500 nm. (c) Intracellular TG contents were analyzed with a TG quantification kit. The data are presented as the mean ± SD. *** p < 0.001 vs. the control group; ## p < 0.01, ### p < 0.001 vs. the group treated with FFA alone.
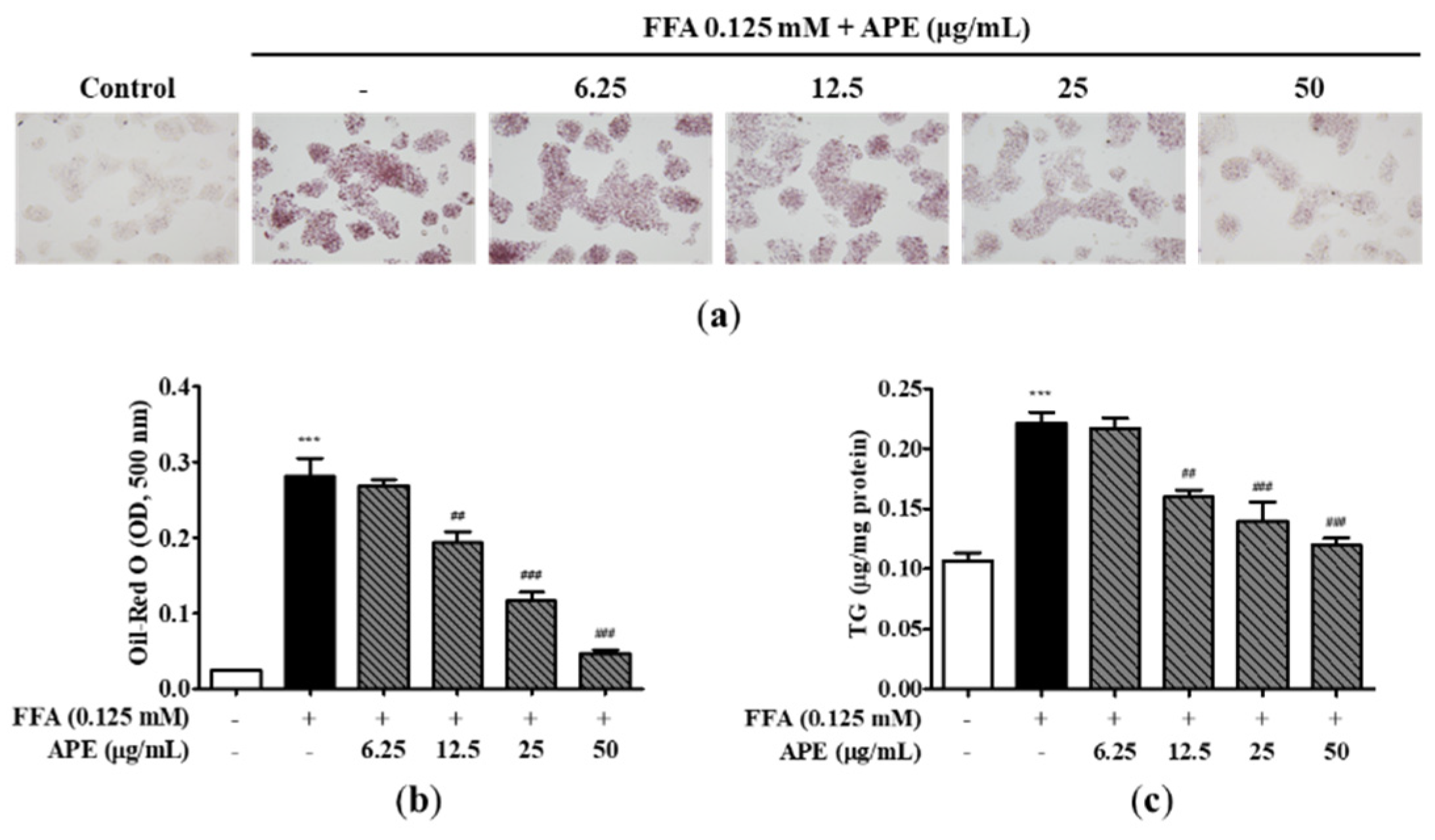
Figure 4.
Effect of APE on the expression of lipid metabolism-related genes and proteins in HepG2 cells. mRNA expression levels of (a) SREBP-1c, (b) C/EBPα, (c) FAS, (d) PPARγ, (e) CPT1a, and (f) PPARα were analyzed using quantitative real-time PCR and normalized to β-actin. (g) Protein expression levels of SREBP-1c, C/EBPα, CPT1a, SIRT1, phosphorylated-AMPKα, and AMPKα were determined using western blot analysis. The data are presented as the mean ± SD. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. the control group; # p < 0.05, ## p < 0.01, ### p < 0.001 vs. the group treated with FFA alone.
Figure 4.
Effect of APE on the expression of lipid metabolism-related genes and proteins in HepG2 cells. mRNA expression levels of (a) SREBP-1c, (b) C/EBPα, (c) FAS, (d) PPARγ, (e) CPT1a, and (f) PPARα were analyzed using quantitative real-time PCR and normalized to β-actin. (g) Protein expression levels of SREBP-1c, C/EBPα, CPT1a, SIRT1, phosphorylated-AMPKα, and AMPKα were determined using western blot analysis. The data are presented as the mean ± SD. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. the control group; # p < 0.05, ## p < 0.01, ### p < 0.001 vs. the group treated with FFA alone.
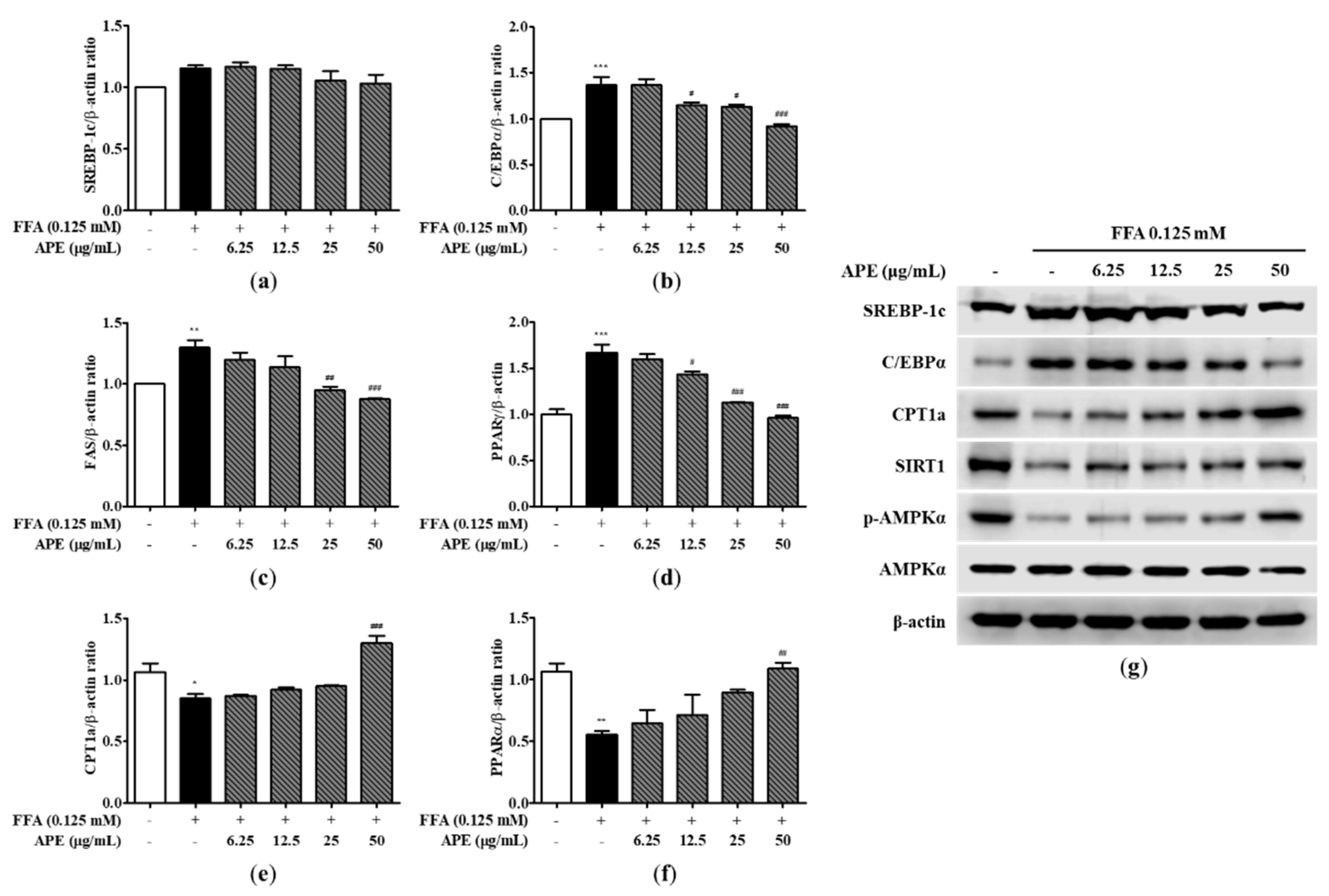
Figure 5.
Effects of APE on mouse body weight and liver weight in a CDAHFD-induced NASH mouse model. (a) Body weight of mice in the CON (normal diet and vehicle), NASH (CDAHFD and vehicle), APE (CDAHFD and 25, 50, 100 mg/kg of APE), Sily (CDAHFD and 100 mg/kg of silymarin), and Lute (CDAHFD and 20 mg/kg of luteolin) groups over the course of 12 weeks. (b) Representative morphology of liver tissues. (c) Ratios of liver weight to body weight. The data are presented as the mean ± SD. ** p < 0.01, *** p < 0.001 vs. the CON group; # p < 0.05 vs. the NASH group.
Figure 5.
Effects of APE on mouse body weight and liver weight in a CDAHFD-induced NASH mouse model. (a) Body weight of mice in the CON (normal diet and vehicle), NASH (CDAHFD and vehicle), APE (CDAHFD and 25, 50, 100 mg/kg of APE), Sily (CDAHFD and 100 mg/kg of silymarin), and Lute (CDAHFD and 20 mg/kg of luteolin) groups over the course of 12 weeks. (b) Representative morphology of liver tissues. (c) Ratios of liver weight to body weight. The data are presented as the mean ± SD. ** p < 0.01, *** p < 0.001 vs. the CON group; # p < 0.05 vs. the NASH group.
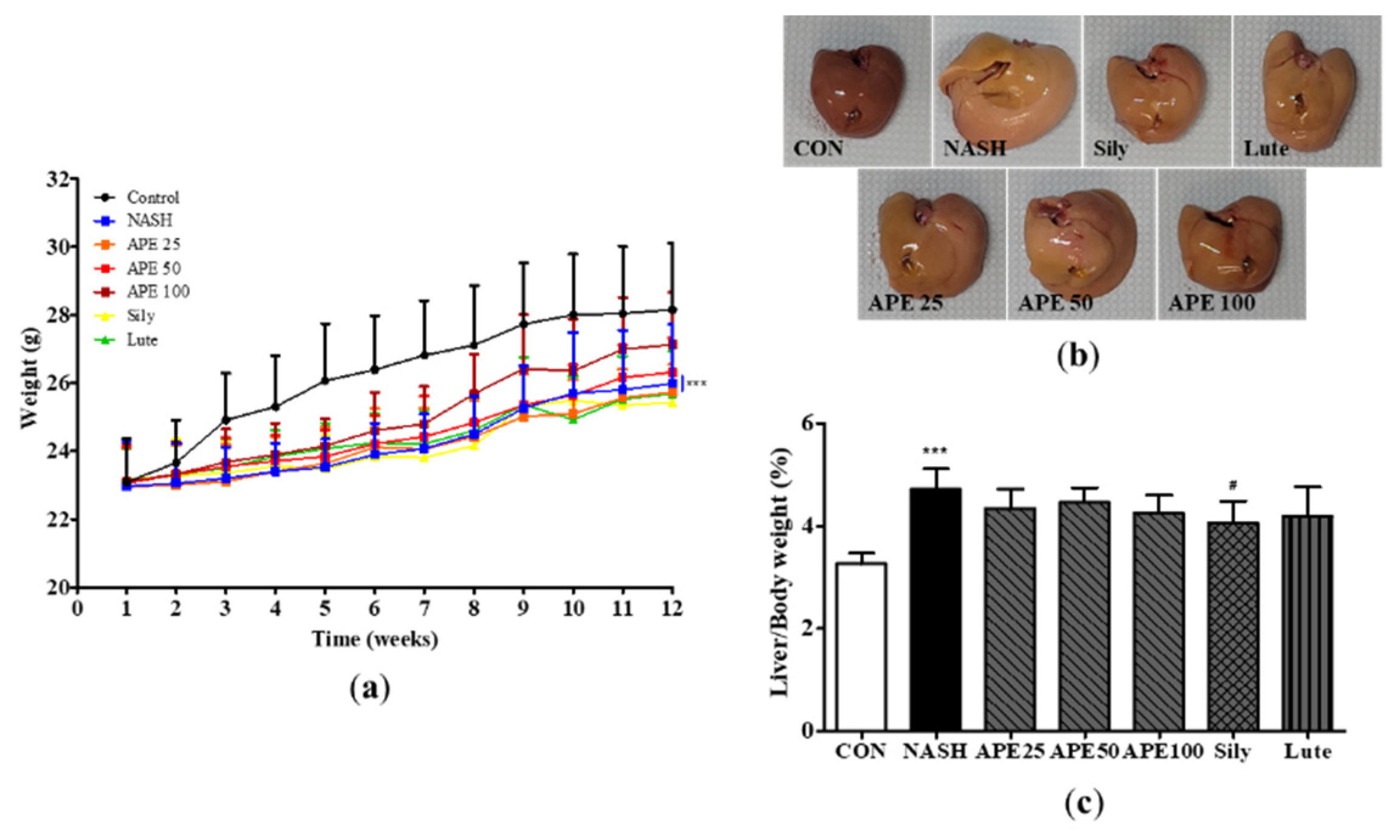
Figure 6.
Effect of APE on hepatic steatosis and serum markers of liver injury in a CDAHFD-induced NASH mouse model. (a) Representative histological images stained with H&E (200x magnification). Serum levels of (b) ALT and (c) AST. The data are presented as the mean ± SD. *** p < 0.001 vs. the CON group, # p < 0.05, ## p < 0.01, ### p < 0.001 vs. the NASH group.
Figure 6.
Effect of APE on hepatic steatosis and serum markers of liver injury in a CDAHFD-induced NASH mouse model. (a) Representative histological images stained with H&E (200x magnification). Serum levels of (b) ALT and (c) AST. The data are presented as the mean ± SD. *** p < 0.001 vs. the CON group, # p < 0.05, ## p < 0.01, ### p < 0.001 vs. the NASH group.
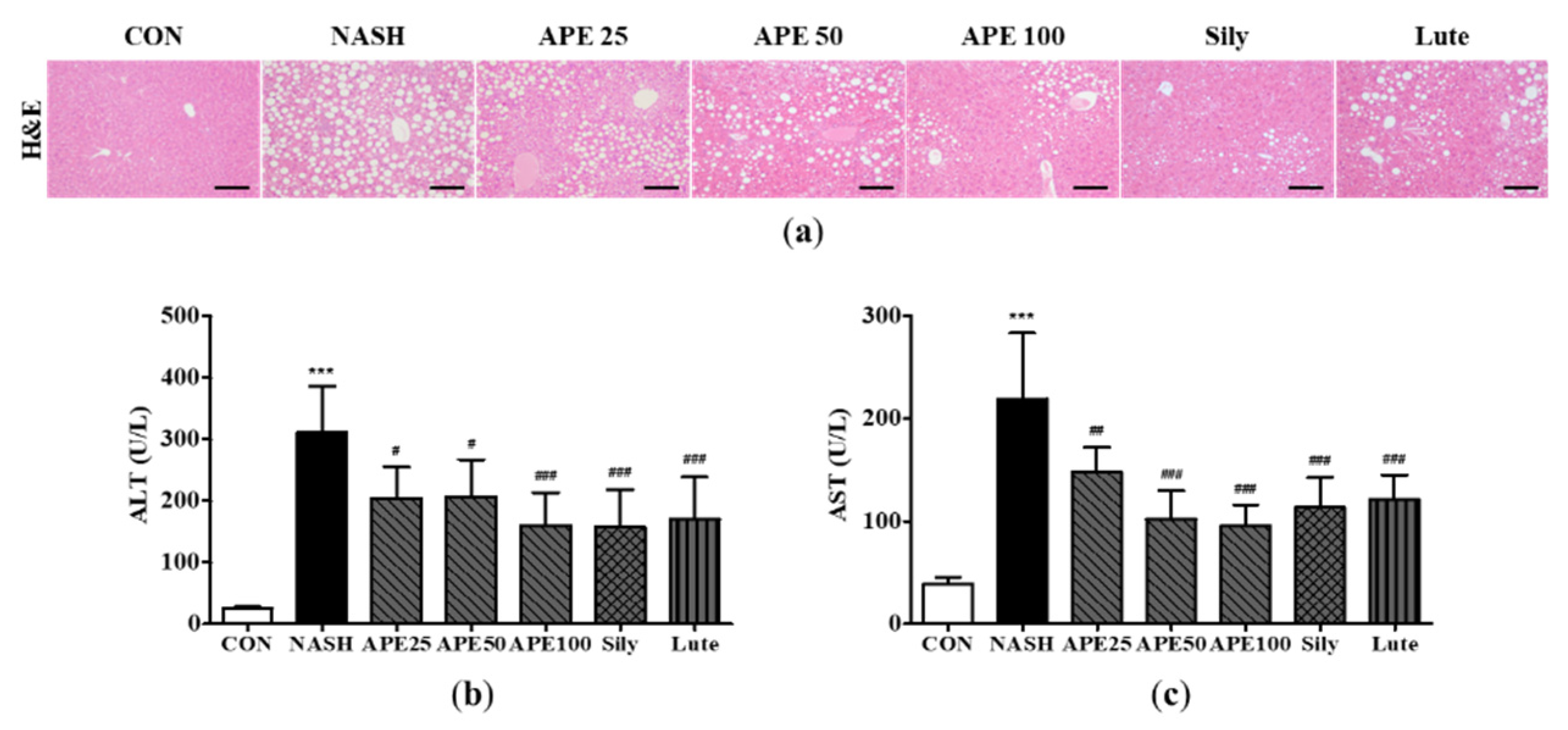
Figure 7.
Effect of APE on histopathology and immunohistochemistry of the liver in a CDAHFD-induced NASH mouse model. (a) Representative images of immunohistochemical staining for F4/80 (200x magnification). (b) Quantification of the positive areas of F4/80. (c) Representative images of Sirius Red staining and immunohistochemical staining of COLⅠ and COLⅤ (200x magnification). (d) Quantification of the positive areas of Sirius Red staining. The data are presented as the mean ± SD. *** p < 0.001 vs. the CON group, ### p < 0.001 vs. the NASH group.
Figure 7.
Effect of APE on histopathology and immunohistochemistry of the liver in a CDAHFD-induced NASH mouse model. (a) Representative images of immunohistochemical staining for F4/80 (200x magnification). (b) Quantification of the positive areas of F4/80. (c) Representative images of Sirius Red staining and immunohistochemical staining of COLⅠ and COLⅤ (200x magnification). (d) Quantification of the positive areas of Sirius Red staining. The data are presented as the mean ± SD. *** p < 0.001 vs. the CON group, ### p < 0.001 vs. the NASH group.
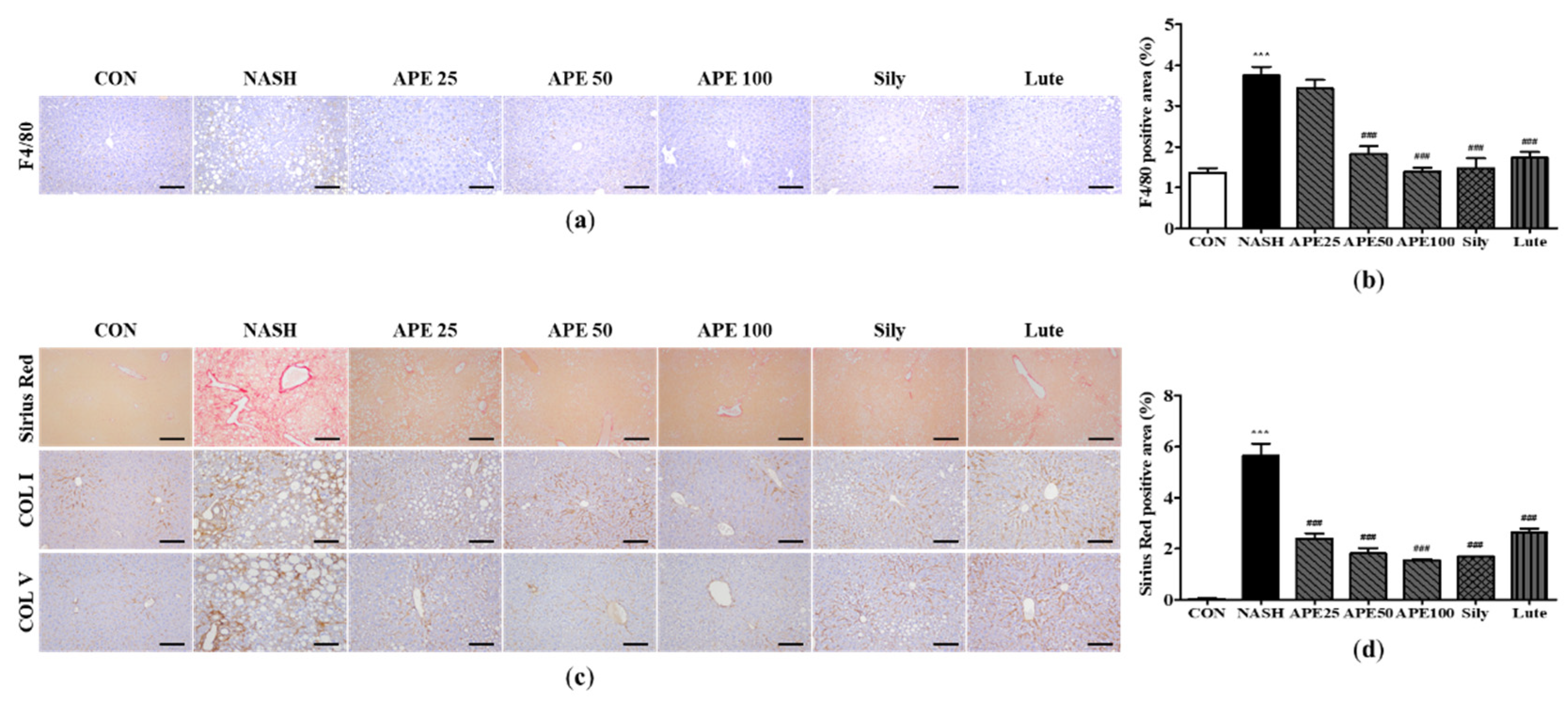
Figure 8.
Effects of APE on the expression of lipid metabolism-related genes and proteins in a CDAHFD-induced NASH mouse model. mRNA expression levels of (a) SREBP-1c, (b) PPARγ, (c) CPT1a, (d) PPARα, (e) ApoB, and (f) MTTP were analyzed via quantitative real-time PCR and normalized to β-actin. (g) Protein expression levels of SREBP-1c, C/EBPα, CPT1a, SIRT1, phosphorylated-AMPKα, and AMPKα were determined via western blot analysis. The data are presented as the mean ± SD. ** p < 0.01, *** p < 0.001 vs. the CON group, # p < 0.05, ## p < 0.01 vs. the NASH group.
Figure 8.
Effects of APE on the expression of lipid metabolism-related genes and proteins in a CDAHFD-induced NASH mouse model. mRNA expression levels of (a) SREBP-1c, (b) PPARγ, (c) CPT1a, (d) PPARα, (e) ApoB, and (f) MTTP were analyzed via quantitative real-time PCR and normalized to β-actin. (g) Protein expression levels of SREBP-1c, C/EBPα, CPT1a, SIRT1, phosphorylated-AMPKα, and AMPKα were determined via western blot analysis. The data are presented as the mean ± SD. ** p < 0.01, *** p < 0.001 vs. the CON group, # p < 0.05, ## p < 0.01 vs. the NASH group.
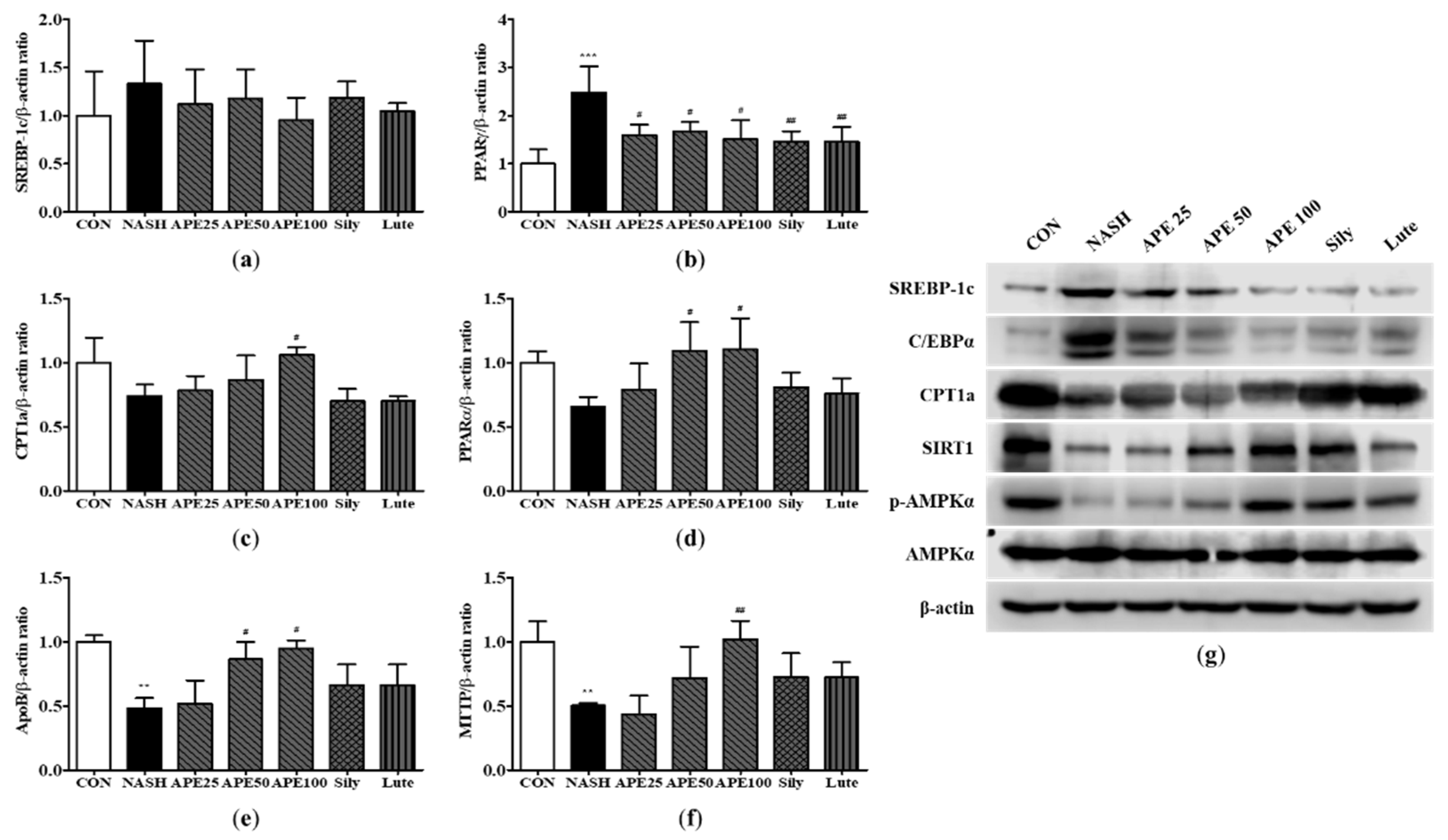
Figure 9.
Effects of APE on the expression of inflammatory factors in a CDAHFD-induced NASH mouse model. mRNA expression levels of (a) F4/80, (b) YM-1, (c) MCP-1, (d) MIP-2, (e) IL-6, (f) TNF-α, and (g) COX-2 were analyzed via quantitative real-time PCR and normalized to β-actin. (h) The protein expression level of COX-2 was determined via western blot analysis. The data are presented as the mean ± SD. ** p < 0.01, *** p < 0.001 vs. the CON group, # p < 0.05, ## p < 0.01, ### p < 0.001 vs. the NASH group.
Figure 9.
Effects of APE on the expression of inflammatory factors in a CDAHFD-induced NASH mouse model. mRNA expression levels of (a) F4/80, (b) YM-1, (c) MCP-1, (d) MIP-2, (e) IL-6, (f) TNF-α, and (g) COX-2 were analyzed via quantitative real-time PCR and normalized to β-actin. (h) The protein expression level of COX-2 was determined via western blot analysis. The data are presented as the mean ± SD. ** p < 0.01, *** p < 0.001 vs. the CON group, # p < 0.05, ## p < 0.01, ### p < 0.001 vs. the NASH group.
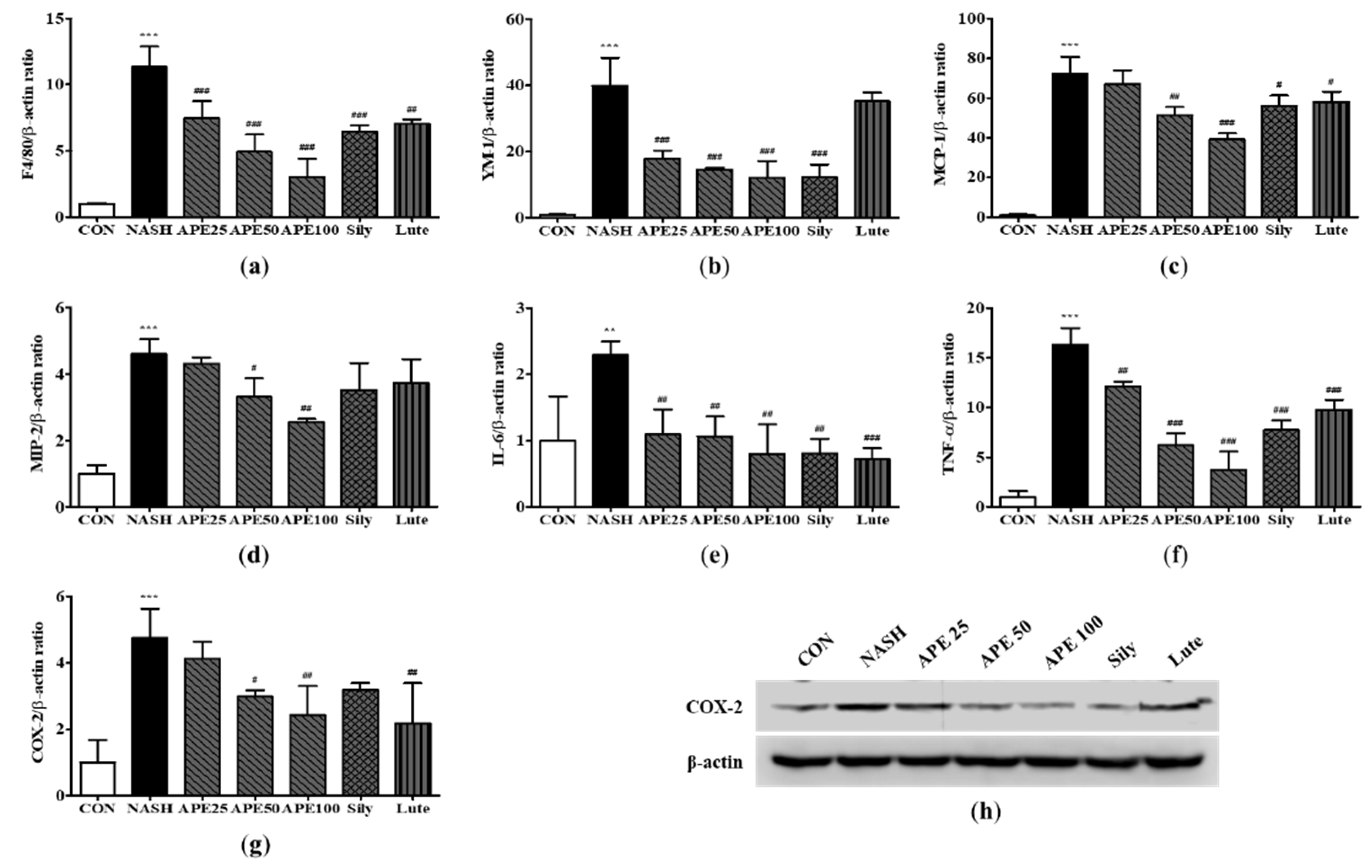
Figure 10.
Effects of APE on the expression of fibrosis-related proteins in a CDAHFD-induced NASH mouse model. mRNA expression levels of (a) α-SMA, (b) COL1A1, (c) desmin, and (d) TGF-β1 were analyzed via quantitative real-time PCR and normalized to β-actin. (e) Protein expression level of α-SMA was determined via western blot analysis. The data are presented as the mean ± SD. *** p < 0.001 vs. the CON group, ### p < 0.001 vs. the NASH group.
Figure 10.
Effects of APE on the expression of fibrosis-related proteins in a CDAHFD-induced NASH mouse model. mRNA expression levels of (a) α-SMA, (b) COL1A1, (c) desmin, and (d) TGF-β1 were analyzed via quantitative real-time PCR and normalized to β-actin. (e) Protein expression level of α-SMA was determined via western blot analysis. The data are presented as the mean ± SD. *** p < 0.001 vs. the CON group, ### p < 0.001 vs. the NASH group.
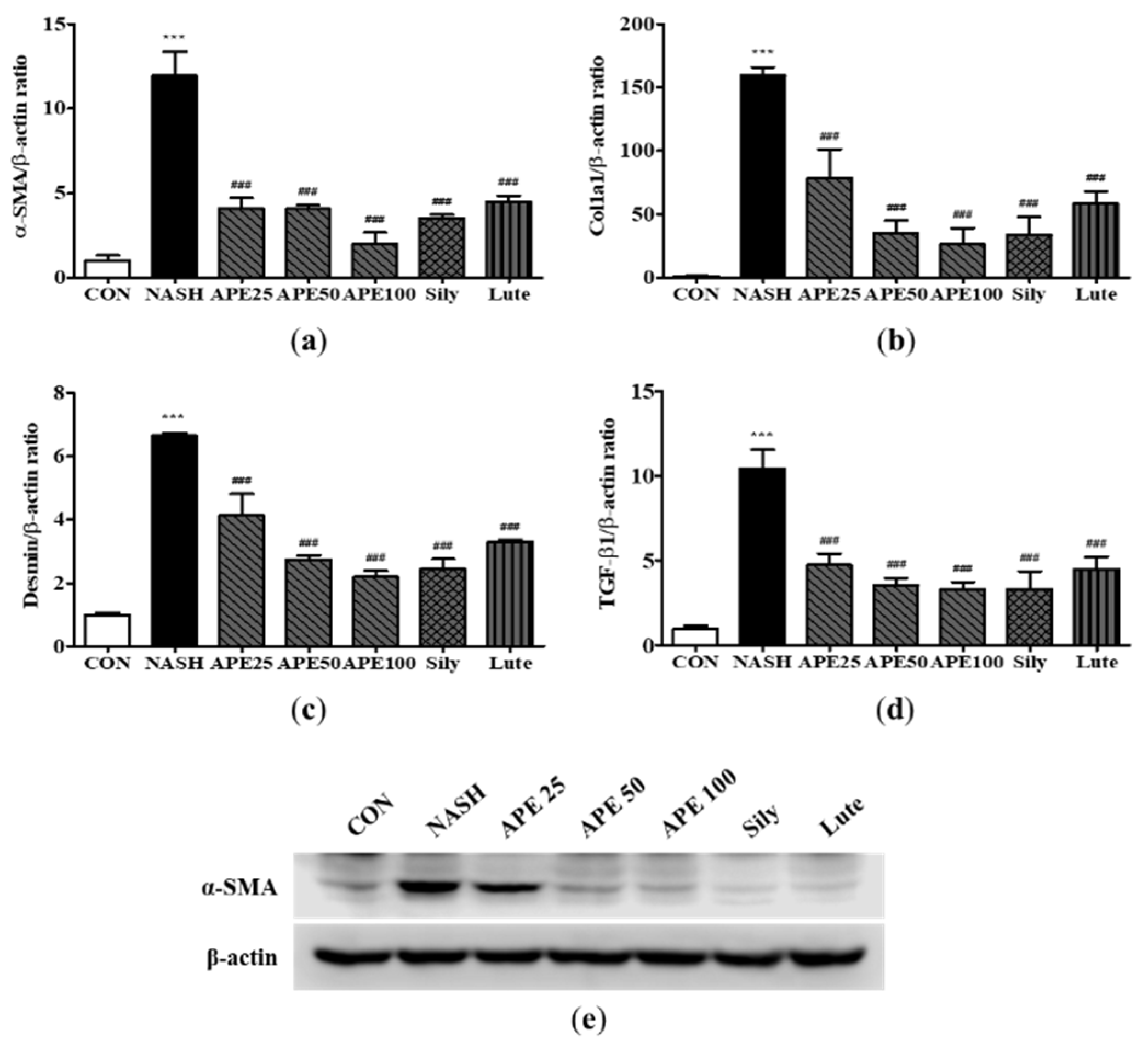
Figure 11.
Effect of APE on the expression of oxidative stress and ER stress in a CDAHFD-induced NASH mouse model. mRNA expression levels of (a) HO-1, (b) NOX2, (c) NOXA2, (d) NQO1, (f) GADD 153, (g) GPR-78, and (h) XBP-1 were analyzed via quantitative real-time PCR and normalized to β-actin. (e) Protein expression levels of Nrf2 and HO-1 were determined via western blot analysis. The data are presented as the mean ± SD. *** p < 0.001 vs. the CON group, # p < 0.05, ## p < 0.01, ### p < 0.001 vs. the NASH group.
Figure 11.
Effect of APE on the expression of oxidative stress and ER stress in a CDAHFD-induced NASH mouse model. mRNA expression levels of (a) HO-1, (b) NOX2, (c) NOXA2, (d) NQO1, (f) GADD 153, (g) GPR-78, and (h) XBP-1 were analyzed via quantitative real-time PCR and normalized to β-actin. (e) Protein expression levels of Nrf2 and HO-1 were determined via western blot analysis. The data are presented as the mean ± SD. *** p < 0.001 vs. the CON group, # p < 0.05, ## p < 0.01, ### p < 0.001 vs. the NASH group.
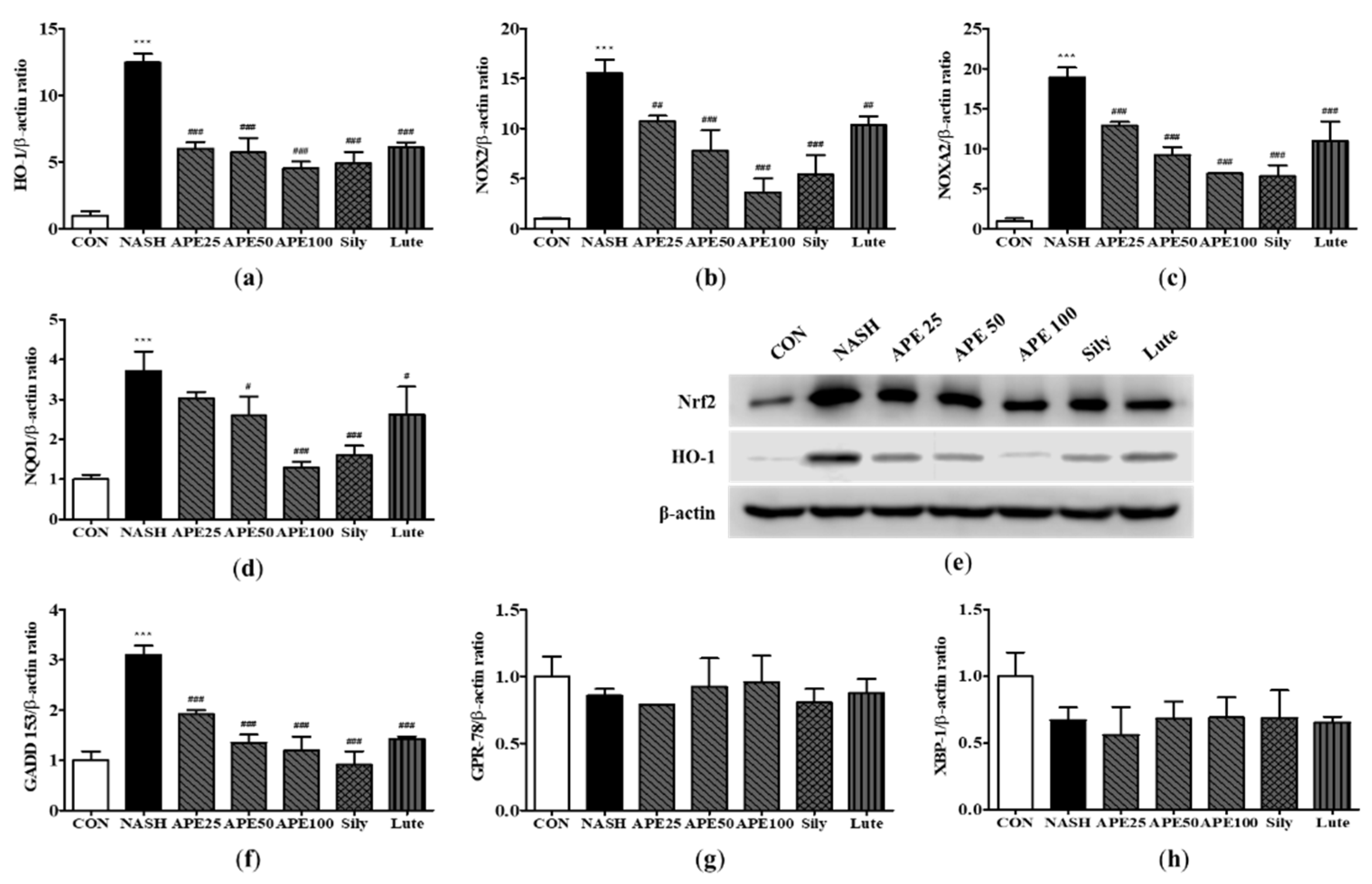

Table 1.
Real-time PCR primers used in this study.
| Genes | Forward primer | Reverse primer |
|---|---|---|
| human | ||
| hSREBP-1c | GCGCCTTGACAGGTGAAGTC | GCCAGGGAAGTCACTGTCTTG |
| hC/EBPα | TGGACAAGAACAGCAACGAGTA | ATTGTCACTGGTCAGCTCCAG |
| hFAS | CCCCTGATGAAGAAGGATCA | ACTCCACAGGTGGGAACAAG |
| hPPARγ | TGCAGGTGATCAAGAAGACG | AGTGCAACTGGAAGAAGGGA |
| hCPT1a | CCTCCGTAGCTGACTCGGTA | GGAGTGACCGTGAACTGAAA |
| hPPARα | ACGATTCGACTCAAGCTGGT | GTTGTGTGACATCCCGACAG |
| mouse | ||
| mSREBP-1c | GCTACCGGTCTTCTATCAATG | GCAAGAAGCGGATGTAGTC |
| mPPARγ | AGGCCGAGAAGGAGAAGCTGTTG | TGGCCACCTCTTTGCTCTGCTC |
| mCPT1a | TCCACCCTGAGGCATCTATT | ATGACCTCCTGGCATTCTCC |
| mPPARα | AGAGCCCCATCTGTCCTCTC | ACTGGTAGTCTGCAAAACCAAA |
| mApoB | GGACTGTCTGACTTCCATATTC | AAGACTTGCCACCCAAAG |
| mMTTP | TCTCACAGTACCCGTTCTT | TCTTCTCCGAGAGACATATCC |
| mF4/80 | CAACACTCTCGGAAGCTATTAT | GAATTCCTGGAGCACTCATC |
| mYM-1 | TCTGGTGAAGGAAATGCGTA | AATGATTCCTGCTCCTGTGG |
| mMCP-1 | TGCTTCTGGGCCTGCTGTTC | ACCTGCTGCTGGTGATCCTCT |
| mMIP-2 | CACTGCGCCCAGACAGAAGT | GGGCTTCAGGGTCAAGGCAA |
| mIL-6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| mTNF-α | GCTGAGCTCAAACCCTGGTA | AGTACTTGGGCAGATTGACCT |
| mCOX-2 | GCCTACTACAAGTGTTTCTTTTTGCA | CATTTTGTTTGATTGTTCACACCAT |
| mα-SMA | CTGACAGAGGCACCACTGAA | GAAGGAATAGCCACGCTCAG |
| mCol1a1 | TCCTCCAGGGATCCAACGA | GGCAGGCGGGAGGTCTT |
| mDesmin | TACACCTGCGAGATTGATGC | ACATCCAAGGCCATCTTCAC |
| mTGF-β1 | TTGCTTCAGCTCCACAGAGA | TGGTTGTAGAGGGCAAGGAC |
| mHO-1 | CACGCATATACCCGCTACCT | CCAGAGTGTTCATTCGAGCA |
| mNOX2 | TGGACGGCCCAACTGGGATA | CAAGGCTTCAGGGCCACACA |
| mNOXA2 | CCGAGCAGGCCTTCACCAAA | TCCCACGAAGCTGCGTCAAG |
| mNQO1 | TTCCCATTGCAGTGGTTTGG | CTGCTACGAGCACTCTCTCA |
| mGADD 153 | CTGGAAGCCTGGTATGAGGAT | CAGGGTCAAGAGTAGTGAAGGT |
| mGPR-78 | CGAAGGGATCATCTGCTATTAC | CTTCATAGTCCTGCCCATTG |
| mXBP-1 | TCCGCAGCACTCAGACTATG | ACAGGGTCCAACTTGTCCAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



