Submitted:
05 September 2023
Posted:
11 September 2023
You are already at the latest version
Abstract
A full potential all-electron density functional method within generalized gradient approximation is used to investigate the electronic, elastic and transport properties of cubic copper sulphide and copper selenide. The electronic structure suggests metallic behavior with a zero energy band gap for both materials. Elastic property calculations suggest a stiff material with the bulk to shear modulus ratio of 0.35 and 0.44 for Cu2S and Cu2Se respectively. The transport properties were estimated using the Boltzmann transport approach. Seebeck coefficient, electrical conductivity, thermal conductivity, and power factor all suggest a potential p-type conductivity for Cu2S and n-type conductivity for Cu2Se.
Keywords:
CuS
; CuSe
; density functional theory
; electronic structure
; elastic constants
; transport properties
; power factor
1. Introduction
Copper based chalcogenides, especially the copper sulphides (CuS) and selenides (CuSe) have a potential of replacing some of the leading of the now becoming extinct silicon families of energy harvesting materials. These binary compounds are generated from the group IB transitional metals and group VIA non-metals. The three elements; copper, sulphur, and selenium are readily available from the earth’s crust. Uniquely designed to CuS and CuSe chalcogenides allow development of cost effective alongside minor environmental hazards energy compounds [1]. Literature studies show that both CuS and CuSe can exist in a variety of stoichiometries with crystal forms ranging from cubic to hexagonal phase [1,2,3,4,5,6,7,8]. Heating and cooling processes within the materials mostly bring about transition from one form to the other [2,3].
A CuSe configuration is a blended conductor, which displays diverse phase transitions from stable to metastable forms, also from low to high temperature forms. Particularly, Cu2Se undergoes a low temperature monoclinic phase to the high temperature face centered cubic (fcc) structured phase transition at 410 K [3]. On the other hand, the Cu2S undergoes two-phase transitions; one around 370 K and another at 700 K [4,5]. At 370 K, a transition from the monoclinic to hexagonal phase takes place coupled with hexagonal to cubic phase at 700 K. The intermediate phases, which include monoclinic and orthorhombic, can also be categorized as superionic due to them having fast mobile fluidic Cu1+ or Cu2+ ions within the focal Se lattice.
Namsani et al. [2] and Kim et al. [6], exposed that at room temperature, the CuSe are not well defined, but at high temperatures the cubic phase is dominant. The prevailing lattice originate from the Se ions with the Cu ions arbitrarily dispersed at different sites within this lattice matrix. Upon the cubic faced Se ions lattice, the Cu ions exhibit fluidic behaviour, which result in good thermoelectric character of the compound. Such a performance shows a cubic CuSe as a favourable material when coming to thermal and electronic parameter regulation. Even though the Se atoms lattice is well defined, the cubic CuSe still demonstrates inconsistencies on the thermos-physical and structural properties with increasing temperature. Such inconsistencies result in large coefficient of thermal expansion at high temperatures and a decrease in heat capacity towards the theoretical limit of liquids. Despite such reported anomalies, notable developments keep on being reported out of ongoing measurements and computations to further understand the material’s properties.
Coming to the CuS chalcocite, experiments further show Cu atoms being highly mobile in the temperature range 300 – 373 K because of its chemical stoichiometry [7]. Due to their elevated Cu atoms mobility, it is difficult to locate their positions, but instead refer to their statistical distributions. This explains the existence of a diverse crystal phases observed with this compound. A mixture of cubic and hexagonal chalcocite is present above 363 K, but as the temperature rises, the hexagonal phase continue to vanish until only cubic CuS chalcocite is observable above 410 K. On the same cause, the orthorhombic CuS transforms to a cubic chalcocite at temperatures above 346 K. On the other hand, Will et al. [4] recorded that below 346 K, the monoclinic chalcocite spontaneously transform into a cubic symmetry.
Coincidentally, in both CuS and CuSe chalcogenides, the cubic symmetry phase is considered the most stable, always evident at ambient and high temperatures, and are favored for thermo-electric transport applications [2,3,7]. Due to envisage potential of these exciting CuS and CuSe chalcogenides in a wide range of applications such as thermo-electric, solar cells, conductive fibers, optical filter and high-capacity cathode materials in lithium secondary batteries, further inquiry was deemed necessary [7,8]. In this article, the electronic structure, elastic and transport properties of the cubic Cu2S and Cu2Se phases are explored. To acquire such investigations, ab initio density functional theory (DFT) methods which apply all-electrons, full potential, linearized augmented plane waves approach have been used. The study may serve as an insight on how the electronic structure and elastic properties can be manipulated into effective thermo-electric energy based transport compounds.
2. Computational details
Computational studies of the electronic, elastic and transport properties of cubic copper sulphide (α-Cu2S) and selenide (α-Cu2Se) phases are presented. Calculations performed are based on the open source-Exciting package [9]. Density functional theory (DFT) using generalized gradient approximation (GGA) as the exchange-correlation functional energy is utilized [10,11]. A Perdew-Burke-Ernzerhof form applied in solids (PBE_sol) [11] is the chosen GGA. All-electrons, full-potential, linearized augmented plane waves on both cubic Cu2S and Cu2Se were exercised. For the electronic structure and elastic properties calculations, the cut-off energy of 500 eV with the forces tolerance of less than 0.02 eV/Å for each atom relaxation have been considered. A total energy convergence was also based on the convincing sampling mesh of the k-points and the expansion of the eigen-functions using the chosen size of the basis set. In this way, the calculation time scales linearly with respect to the k-points weighing and exponentially with respect to the basis set.
In both compounds, the interstitial bonding between Cu and S or Se atoms is described by the plane waves. Cu, S and Se atomic sites central potentials are using the muffin-tin basis function description [9]. To a good approximation, the working muffin-tin basis contains a sum of the radial eigen-functions and the derivative of the radial eigenfunctions with respect to energy all operating on the spherical harmonics component of the wavefunctions. This choice produces smooth and continuous muffin-tin basis function for the interstitial plane waves. The analysis of the electronic thermo-electric properties is carried using the Boltzmann theory [12]. This is because the calculations are based on the semi-classical approach on the solution of the linearized Boltzmann transport equation. The equations contain the derivative of the Fermi distribution function. The rigid body and relaxation time approximations are considered. A relaxation time of 1 x 10-15 s is imposed for the calculations of the Seebeck coefficient (S), the figure of merit (σS2), electrical conductivity per relaxation time (σ/τ), and thermal conductivity per relaxation time (κ/τ).
Cubic α-Cu2S and α-Cu2Se compounds possess the space group Fm-3m (No. 225) with a fluorite prototype structure. Ideally, Cu1+ is bonded equivalently to S2- or Se2- ions to create a mixture cell edge and corner sharing in a CuSe4 tetrahedron. Conversely, S2- or Se2- ion is bonded equally in a body centered cubic form to eight Cu1+ ions. The regular structure contains two highly symmetric positions where S2- (Se2-) and Cu1+ ions occupy localized positions: 4a (0.00 0.00 0.00) and 8c (0.25 0.25 0.25) respectively according to the Wyckoff table of coordinates [13]. The optimized lattice constants for α-Cu2S and α-Cu2Se used in the calculations are Bohr and Bohr and respectively. Here 1 Bohr = 1 atomic unit (a.u.) = 0.529 Å.
3. Results and discussion
All the electronic, elastic and transport properties of alpha Cu2S and Cu2Se were performed under room temperature. The electronic properties describe the state and behaviour of electrons in the material. For example, the electronic band structure and the density of state, which describes the state of the electrons in terms of their energies. Elastic properties determines the mechanical properties of the material. The transport properties assist with the understanding of various interactions in electronic systems such as the Seebeck coefficient, electrical conductivity, thermal conductivity and the power factor.
3.1. Electronic properties
The electronic structure of any material is important in order to understand the sub-atomic properties. This information is obtained from the calculated density of states (DOS) and band structure through the materials electron density. When doing the calculations, the energies were allowed to converge with respect to the k points density (9 × 9 × 9) for density of state, band structure and lattice calculations. In the process of structure optimization, all atoms were fully relaxed, and the energy convergence threshold is 1.0 × 10−5 eV. The force convergence threshold of each atom is 0.015 eV·Å −1.
Figure 1a,b shows the total energy against volume for the optimized structures of alpha type copper sulphide (α-Cu2S) (a) and copper selenide (α-Cu2Se) (b) respectively. The calculated lattice constants, volume and energy are displayed in Table 1. The lattice constants of Cu2S and Cu2Se were found to be 10.39 Bohr and 10.89 Bohr (5.59 Å and 5.50 Å) corresponding to the total energies of -3705.95 and -5735.90 eV respectively. Bharathi et al. [14] calculated the lattice constant of Cu2S and found it to be 5.60 Å, which differs with our results by 0.04 %. Sakuma et al. [15] calculated the lattice constant of cubic Cu2Se and found it to be 5.85 Å, which differs with our results by 0.36 %.
Figure 2(a) and (b) presents the calculated electronic band structure of the cubic α-Cu2S (a) and α-Cu2Se (b) symmetries respectively. As seen from the figures, there is an overlap of the bottom of the conduction and the top of the valence bands in the high symmetry k-points L to Γ. As such, both the α-Cu2S and α-Cu2Se compounds give rise to zero band gap, which is associated with metallic behaviour. The electronic density of states (DOS) results are displayed in Figure 3(a) and (b). In both compounds, the valence band area has notable two regions. A lower energy region which sweeps from -8.0 to -6.0 eV and an upper energy region which is from -3.5 to 0.0 eV (Fermi level). In both α-Cu2S and α-Cu2Se, the electron charge density is distributed in the same way in the lower and upper regions. The lower region is associated with the core p and s states of Cu and S (or Se) in α-Cu2S or (α-Cu2Se). The upper region is associated with a mixture of d, p and valence s states. As seen in Figure 3 (a) and (b), the 3d peaks are outstanding between -3.0 and -2.0 eV in the upper region of the valence band. For α-Cu2Se, the peaks are associated with a mixture of Cu and Se 3d states, whilst the peak on the α-Cu2S is associated with the lone Cu 3d states. Amalgamated results presented in Figure 3 (a) and (b) are in agreement with calculations of Råsander et al. [16] and tight binding model computations of Garba and Jacobs [17].
3.2. Elastic properties
Systematically, the elastic properties of alpha Cu2S and Cu2Se were also computed. Such calculations assist in understanding the stability, stiffness, ductility and anisotropy nature of the materials. Young’s modulus measures the stiffness of the materials, bulk modulus refers to the resistance to shape deformation, shear modulus reflects the resistance against the shear deformation and Poisson’s ratio predicts the ductility of the materials [18]. In the cubic crystal symmetry, there are three independent elastic constants, which are C11, C12, and C44. Theoretically, elastic constants can be calculated directly form the total ground state energy of the crystals as discussed by Stadler et al. [19] or using the relation between the stress () and strain () within a crystal as proposed by Nielsen and Martin [20]. In this article, elastic constants are calculated using the ElaStic@exciting [9] interface, which can be used to obtain full elastic constants of any crystal systems. In order to calculate the three elastic constants (C11, C12 and C44), bulk modulus (B0), Young’s modulus (Y), shear modulus (G) and the Poisson’s ratio (v), the following equation relations were considered [21]:
In Equation (1), C11 is associated with the longitudinal compression, whereas C12 is associated with the tranverse expansion.
In this equation (2), C44 directly extracts the shear modulus (G) on adjacent planes.
In equations (3) and (4), different operations on the bulk modulus (B0) and shear modulus (G) produce the Young’s modulus and Poisson’s ratio respectively.
Table 2 and Table 3 present the calculated elastic constants of α-Cu2S and α-Cu2Se respectively. In view of the fact that alpha Cu2S and Cu2Se are cubic materials, the elastic deformation stability needs to obey the following trend: ; ; and [3,21,22,23]. Values presented in Table 2 and Table 3, confirm that all conditions of cubic deformation stability are satisfied. Making use of the condition , a solid material can be predicted to obey either a homogeneous or an anisotropic elastic property. If, the material comply with homogeneous elasticity and if , the material conform to anisotropic elasticity. Reflecting on this condition, a conclusion that materials α-Cu2S and α-Cu2Se are mechanically stable and can also be viewed as belonging to homogeneous elastic media [22,23]. As portrayed in the tables, the bulk moduli of α-Cu2S and α-Cu2Se are much greater than the respective shear moduli, which leads to Poisson’s ratios of 0.35 and 0.44 respectively. Such positive values in the range of 0 – 0.5 suggest that both α-Cu2S and α-Cu2Se compounds can be stretched in one direction whilst they expand in other two directions which are perpendicular to the direction of compression [23]. Mott et al. [24], described that for incompressible materials, the bulk modulus () is typically large compared to the shear modulus () which leads to the Poisson’s ratio of about 0.5. This notion suggest the fact that both materials are incompressible, with α-Cu2Se more incompressible than α-Cu2S since α-Cu2Se Poisson’s ratio is very close to 0.5 [24]. Aimed at α-Cu2Se, the bulk, shear and Young’s modulus values are in agreement with the computational results [3] in compariosn with the experimental results [22]. In the case of α-Cu2S compound, only computational results are available for comparison. The Young’s moduli values suggest both α-Cu2S and α-Cu2Se materials as stiff [3].
3.3. Transport properties
The thermo-electric effect is a phenomenon, which explains the ability of certain materials to transform solar to electrical energy. In order to understand the thermo-electric properties of α-Cu2S and α-Cu2Se materials at 300 K, a presentation of the Seebeck coefficient, electrical and thermal conductivities per relaxation time as well as the power factor is made in Figure 4(a) – (d). Figure 4(a), displays the behaviour of the Seebeck coefficient (S) against the chemical potential (μ), which indicates two notable maxima peaks for both materials. Symmetrically, the positive maxima S values of Cu2S and Cu2Se are 150 μVK-1 and 125 μVK-1 respectively at µ = -0.05 eV whilst the negative minima of -100 μVK-1 and -220 μVK-1 respectively at μ = 0.05 eV. The calculated Seebeck coefficient provides a good consistency with the experimental outcomes of Byeon et al. [25], where the group studied Seebeck coefficients of Cu2Se and discovered two peaks with negative peaks possessing a larger magnitude than the positive peak. Theoretically, on the chemical potential abscissa of Figure 4(a), the positive and negative values suggest the electrons (n-type) and holes (p-type) dopants respectively [25]. So for α-Cu2S, the p-type dopants are more elevated compared to the n-type, but for α-Cu2Se, the n-type overcome the p-type dopants. This put forward the concept that α-Cu2S favours p-type conductivity whilst α-Cu2Se favours the n-type conductivity.
The electrical (σ/τ) and thermal (κ/τ) conductivities per relaxation time against the chemical potential (μ) appear in Figure 4 (b) and (c) respectively. Both curves display a similar behaviour relative to the changing μ and their turning points occur at the Fermi level. Around the Fermi level, the electrical conductivity suggests the metallic behaviour as the curve does not entirely touch the zero level of the σ for both materials [26]. The power factor (σS2) quantity describes how efficient a given thermo-electric material is [26]. In Figure 4(d), the observation of the visible active peaks are evident in the positive zone of both α-Cu2S and α-Cu2Se compounds, even though the peaks for α-Cu2S are somehow compromised. Such values suggest that the power factor is more inclined to the n-type conductivity for both α-Cu2S and α-Cu2Se. Mahan and Sofo [12] findings expressed that a good thermo-electric material is expected to have a large Seebeck coefficient, high electrical conductivity and a low thermal conductivity in order to acquire an enhanced figure of merit [12,26,27,28]. In this instance, for both α-Cu2S and α-Cu2Se, the S is having high positive values of 150 μVK-1 and 125 μVK-1 respectively and κ maximum values of 0.35 Wcm-1K-1 and 0.37 Wcm-1K-1 respectively are sufficiently small. A conclusion will be that the alpha Cu2S and Cu2Se satisfy the properties of being good thermo-electric materials.
Figure 5(a) and (b) show variation of the Seebeck coefficient with temperature at chemical potentials μ = -0.136 and 0.136 eV respectively for α-Cu2S and α-Cu2Se. According to Scheidemantel et al. [27], the argument is that the negative and positive chemical potentials are associated with the p- and n-type conductivities respectively. Considering Figure 5(a), for cubic Cu2S, the p-type conductivity reflects the highest S value in relation to the n-type conductivity. Furthermore, for p-type conductivity, the S increases continuously with the increasing temperature up to about 155 μVK-1 at 1200 K. Conversely, for the n-type conductivity, the S reaches a maximum of 100 μVK-1 around 600 K then decreases towards about 75 μVK-1 at 1200 K. Narjis et al. [29] and independently, Zhao et al. [30], pointed out that the positive values of the Seebeck coefficient indicate transport properties which are dominated by holes, suggesting p-type conductivity. On the other hand, the negative values of S indicate transport properties dominated by electrons, which suggest n-type conductivity materials. In this case, the suggestion that α-Cu2S is a p-type conductivity material holds. Looking at the Figure 5(b), which represent the α-Cu2Se, the n-type doping obtained high negative S value than the p-type value. In addition, on the n-type doping, S values decrease exponentially with the increasing temperature, which suggests α-Cu2Se as an n-type material.
Steadily, Figure 6(a) and (b) display electrical conductivity per relaxation time against temperature at constant chemical potentials μ = -0.136 and 0.136 eV of α-Cu2S and α-Cu2Se respectively. Compatible with the Seebeck coefficient’s performance, for the α-Cu2S, the p-type doping at μ = -0.136 eV demonstrates the highest electrical conductivity per relaxation time compared to the n-type at μ = 0.136 eV (Figure 6(a)). In a similar manner, in Figure 6(b), for the α-Cu2Se the n-type doping displays the highest electrical conductivity per relaxation time compared to the p-type. It must also be mentioned that in both instances of α-Cu2S and α-Cu2Se, the electrical conductivity per relaxation time at μ = -0.136 and 0.136 eV, increases with the increasing temperature which further suggest metallic character [31].
Advancing on, Figure 7(a) and (b) present the thermal conductivity per relaxation time against temperature at chemical potentials μ = -0.136 and 0.136 eV respectively for both α-Cu2S and α-Cu2Se. Previous studies by Jiang et al. [28] and Narjis et al. [29] clarified that a material with high electrical conductivity must have very low thermal conductivity. In accordance with this, Figure 7(a) shows that for α-Cu2S, the good thermal conductivity favours the p-type doping since it has the lowest thermal conductivity whereas, Figure 7(b) illustrates the good thermal conductivity of the α-Cu2Se is obtained along the n-type doping. At μ = -0.136 and 0.136 eV, the thermal conductivity per relaxation time increases exponentially with increasing temperature for both α-Cu2S and α-Cu2Se. In addition, Figure 7(a) and (b) illustrates that the minimum thermal conductivities per relaxation time of alpha Cu2S and Cu2Se are still very low at the values of about 0.02 and 0.05 Wcm-1K-1 at 300 K. Such a property further confirms that, the thermal conductivity behaviour of the α-Cu2S is in line with some previous related studies [28,29,30].
Lastly, Figure 8(a) and (b) illustrate the power factor against temperature at chemical potentials μ = -0.136 and 0.136 eV of α-Cu2S and α-Cu2Se respectively. The power factor is the product of the electrical conductivity and the square of the Seebeck coefficient. Hasan et al. [26] explained that if the maximum power factor was obtained at the hole-doping region (μ < 0), then it could be concluded that the material works better as a p-type thermo-electric. Similarly, when maximum power factor was obtained at the electron doping region (μ > 0), then the material works well as an n-type thermo-electric [26]. For α-Cu2S in Figure 8(a), the p-type doping suggest the high value of the power factor compared to the n-type doping. Moreover, the n-type doping demonstrates relatively constant power factor with the increasing temperature, though for the p-type doping, the power factor increases linearly as the temperature increases, which further confirms that indeed α-Cu2S works better as a p-type thermo-electric material. In Figure 8(b), which signify α-Cu2Se curves, the n-type suggest the highest value of the power factor related to the p-type doping. Also, on the p-type doping, the power factor demonstrates the highest value around 600 to 700 K, then lowers to 20 μWcm-1K2 at 1200 K, but for the n-type doping, the power factor increase with the increasing temperature. This also confirms that α-Cu2Se works better as an n-type thermo-electric material. Wrapping up, all the variations of the transport coefficients with temperature suggest α-Cu2S as a good p-type conductivity thermo-electric material whilst the α-Cu2Se being a good n-type conductivity thermo-electric material.
5. Conclusions
The electronic properties results are in agreement with previous studies, which show alpha copper sulphide and copper selenide having metallic character with zero energy band gap. This zero energy band gap is attributed to the overlap of the lowest valence and the highest conduction bands geometry between the symmetry points L and Γ on both materials. Elastic constants results demonstrate these chalcogenides as being mechanically stable with low shear moduli coupled with high bulk moduli. Such material composition is expected on thermo-electric property materials. Transport properties of the materials suggest a p-type conductivity for α-Cu2S and n-type conductivity for α-Cu2Se. The high positive (α-Cu2S) and negative (α-Cu2Se) values of the Seebeck coefficient, the high electrical conductivity, high thermal conductivity and the low thermal conductivity with respect to the chemical potentials suggest that, alpha Cu2S and Cu2Se are good thermo-electric transport material at the p-type doping and n-type doping respectively. This is further corroborated by the little change of the electrical conductivity with increasing temperature. Very small, suppressed thermal conductivity values with increasing temperature further support the findings. Such low shear and high bulk moduli materials have potential in thermo-electric transport applications. To quantify these results, a combination of such n-type and p-type conductivity materials suggest sustainable solar harvesting for prolonged electrical energy generation.
Author Contributions
Conceptualization, M.R. and T.M.; methodology, T.M.; formal analysis, M.R.; investigation, M.R.; resources, T.M.; data curation, M.R.; writing—original draft preparation, M.R.; writing—review and editing, T.M.; visualization, M.R.; supervision, T.M.; project administration, T.M.; funding acquisition, T.M. All authors have read and agreed to the published version of the manuscript.”.
Funding
This research was funded by the UNIVERSITY OF LIMPOPO.
Data Availability Statement
The data utilized in this article is available from the corresponding author on request.
Acknowledgments
The University of Limpopo (UL) and the Centre for High Performance Computing (CHPC) are thanked for computational facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ashfaq, A.; Tahir, S.; ur Rehman, U.; Ali, A.; Ashfaq, H.F.; Ahmad, W.; Mushtaq, S.; Saeed, R.; Haneef, M.; Khan, K.M.; Shabbir, K. Structural, morphological and thermoelectric properties of copper deficient and excessive Cu2-xS nanoparticles with (x=0-0.3). Surfaces and Interfaces 2022, 30, 101965. [Google Scholar] [CrossRef]
- Dioum, A.; Diakite, Y.I.; Malozovsky, Y.; Ayirizia, B.A.; Beye, A.C.; Bagayoko, D. First-principles investigation of electronic and related properties of cubic magnesium silicide (Mg2Si). Computation 2023, 11, 40. [Google Scholar] [CrossRef]
- Namsani, S. , Gahtori, B., Auluck, S.; Singh, J.K. An interaction potential to study the thermal structure evolution of a thermoelectric material: β-Cu2Se. Journal of computational chemistry 2017, 38, 2161–2170. [Google Scholar] [CrossRef]
- Potter, R.W. An electrochemical investigation of the system copper-sulfur. Economic Geology 1977, 72, 1524–1542. [Google Scholar] [CrossRef]
- Will, G.; Hinze, E.; Abdelrahman, A.R.M. Crystal structure analysis and refinement of digenite, Cu1. 8S, in the temperature range 20 to 500 °C under controlled sulfur partial pressure. European Journal of Mineralogy 2002, 14, 591–598. [Google Scholar] [CrossRef]
- Kim, H.; Ballikaya, S.; Chi, H.; Ahn, J.P.; Ahn, K.; Uher, C.; Kaviany, M. Ultralow thermal conductivity of β-Cu2Se by atomic fluidity and structure distortion. Acta Materialia 2015, 86, 247–253. [Google Scholar] [CrossRef]
- He, Y.; Day, T.; Zhang, T.; Liu, H.; Shi, X.; Chen, L.; Snyder, G.J. High thermoelectric performance in non-toxic earth-abundant copper sulfide. Advanced Materials 2014, 26, 3974–3978. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Q.; Ge, Z.H.; Feng, J. Synthesis and thermoelectric properties of copper sulfides via solution phase methods and spark plasma sintering. Crystals 2017, 7, 141. [Google Scholar] [CrossRef]
- Gulans, A.; Kontur, S.; Meisenbichler, C.; Nabok, D.; Pavone, P.; Rigamonti, S.; Sagmeister, S.; Werner, U.; Draxl, C. Exciting: a full-potential all-electron package implementing density-functional theory and many-body perturbation theory. Journal of Physics: Condensed Matter 2014, 26, 363202. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Physical review B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. arXiv:0707.2088v2 [cond-mat.other] 22 Oct 2007. arXiv:0707.2088v2 [cond-mat.other] 22 Oct 2007.
- Mahan, G.D.; Sofo, J.O. The best thermoelectric. Proc. Natl. Acad. Sci. USA 1996, 93, 7436–7439. [Google Scholar] [CrossRef]
- Aroyo, M.I.; Perez-Mato, J.M.; Capillas, C.; Kroumova, E.; Ivantchev, S.; Madariaga, G.; Kirov, A.; Wondratschek, H. Bilbao crystallogarphic server I: databases and crystallographic computing programs. Z. Krist. 2006, 221, 15–27. [Google Scholar]
- Bharathi, B.; Thanikaikarasan, S.; Ramesh, K. Structural, compositional, and optical properties of electrochemically deposited Cu2S thin films. International Journal of ChemTech Research 2014, 6, 1907–1909. [Google Scholar]
- Sakuma, T.; Sugiyama, K.; Matsubara, E.; Waseda, Y. Determination of the Crystal Structure of Superionic Phase of Cu2Se. Materials Transactions, JIM 1989, 30, 365–369. [Google Scholar] [CrossRef]
- Råsander, M.; Bergqvist, L.; Delin, A. Density functional theory study of the electronic structure of fluorite Cu2Se. Journal of Physics: Condensed Matter 2013, 25, 125503. [Google Scholar] [CrossRef] [PubMed]
- Garba, E.J.D.; Jacobs, R.L. The electronic structure of Cu2-xSe. Physica B+C 1986, 138, 253–260. [Google Scholar] [CrossRef]
- Zeng, X.; Peng, R.; Yu, Y.; Hu, Z.; Wen, Y.; Song, L. Pressure effect on elastic constants and related properties of Ti3Al intermetallic compound: a first-principles study. Materials 2018, 11, 2015. [Google Scholar] [CrossRef]
- Stadler, R.; Wolf, W.; Podloucky, R.; Kresse, G.; Furthmuller, J.; Hafner, J. Ab initio calculations of cohesive, elastic, and dynamical properties of CoSi2 by pseudopotential ans all-electrons techniques. Phys. Rev. B 1996, 54, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Martin, R.M. First-principles calculation of stress Phys Rev. Lett. 1983, 50, 697–700. [Google Scholar] [CrossRef]
- Jamal, M.; Asadabadi, S.J.; Ahmad, I.; Aliabad, H.R. Elastic constants of cubic crystals. Computational Materials Science 2014, 95, 592–599. [Google Scholar] [CrossRef]
- Hasan, S., Baral, K. and Ching, W.Y., 2019. Total bond order density as a quantum mechanical metric for materials design: Application to chalcogenide crystals. Preprint article 20 June 2019. 20 June.
- Zhang, J.; Zhang, C.; Zhu, T.; Yan, Y.; Su, X.; Tang, X. Mechanical properties and thermal stability of the high-thermoelectric-performance Cu2Se compound. ACS Applied Materials & Interfaces 2021, 13, 45736–45743. [Google Scholar]
- Mott, P.H. , Dorgan, J.R. and Roland, C.M. The bulk modulus and Poisson's ratio of “incompressible” materials. Journal of Sound and Vibration, 2008, 312, 572–575. [Google Scholar] [CrossRef]
- Byeon, D.; Sobota, R.; Delime-Codrin, K.; Choi, S.; Hirata, K.; Adachi, M.; Kiyama, M.; Matsuura, T.; Yamamoto, Y.; Matsunami, M.; Takeuchi, T. Discovery of colossal Seebeck effect in metallic Cu2Se. Nature communications 2019, 10, 72. [Google Scholar] [CrossRef]
- Hasan, S.; San, S.; Baral, K.; Li, N.; Rulis, P.; Ching, W.Y. First-Principles Calculations of Thermoelectric Transport Properties of Quaternary and Ternary Bulk Chalcogenide Crystals. Materials 2022, 15, 2843. [Google Scholar] [CrossRef] [PubMed]
- Scheidemantel, T.J.; Ambrosch-Draxl, C.; Thonhauser, T.; Badding, J.V.; Sofo, J.O. Transport coefficients from first-principles calculations. Physical Review B 2003, 68, 125210. [Google Scholar] [CrossRef]
- Jiang, Q.; Yan, H.; Khaliq, J.; Shen, Y.; Simpson, K.; Reece, M.J. Enhancement of thermoelectric properties by atomic-scale percolation in digenite CuxS. Journal of Materials Chemistry A 2014, 2, 9486–9489. [Google Scholar] [CrossRef]
- Narjis, A.; Outzourhit, A.; Aberkouks, A.; El Hasnaoui, M.; Nkhaili, L. Structural and thermoelectric properties of copper sulphide powders. Journal of Semiconductors 2018, 39, 122001. [Google Scholar] [CrossRef]
- Zhao, L.D.; Berardan, D.; Pei, Y.L.; Byl, C.; Pinsard-Gaudart, L.; Dragoe, N. Bi1− xSrxCuSeO oxyselenides as promising thermoelectric materials. Applied Physics Letters 2010, 97, 092118. [Google Scholar] [CrossRef]
- Yang, D.; Su, X.; Yan, Y.; He, J.; Uher, C.; Tang, X. Mechanochemical synthesis of high thermoelectric performance bulk Cu2X (X= S, Se) materials. APL Materials 2016, 4, 116110. [Google Scholar] [CrossRef]
Figure 1.
Change in total energy against volume. (a) cubic copper sulphide (α-Cu2S). (b) cubic copper selenide (α-Cu2Se).
Figure 1.
Change in total energy against volume. (a) cubic copper sulphide (α-Cu2S). (b) cubic copper selenide (α-Cu2Se).
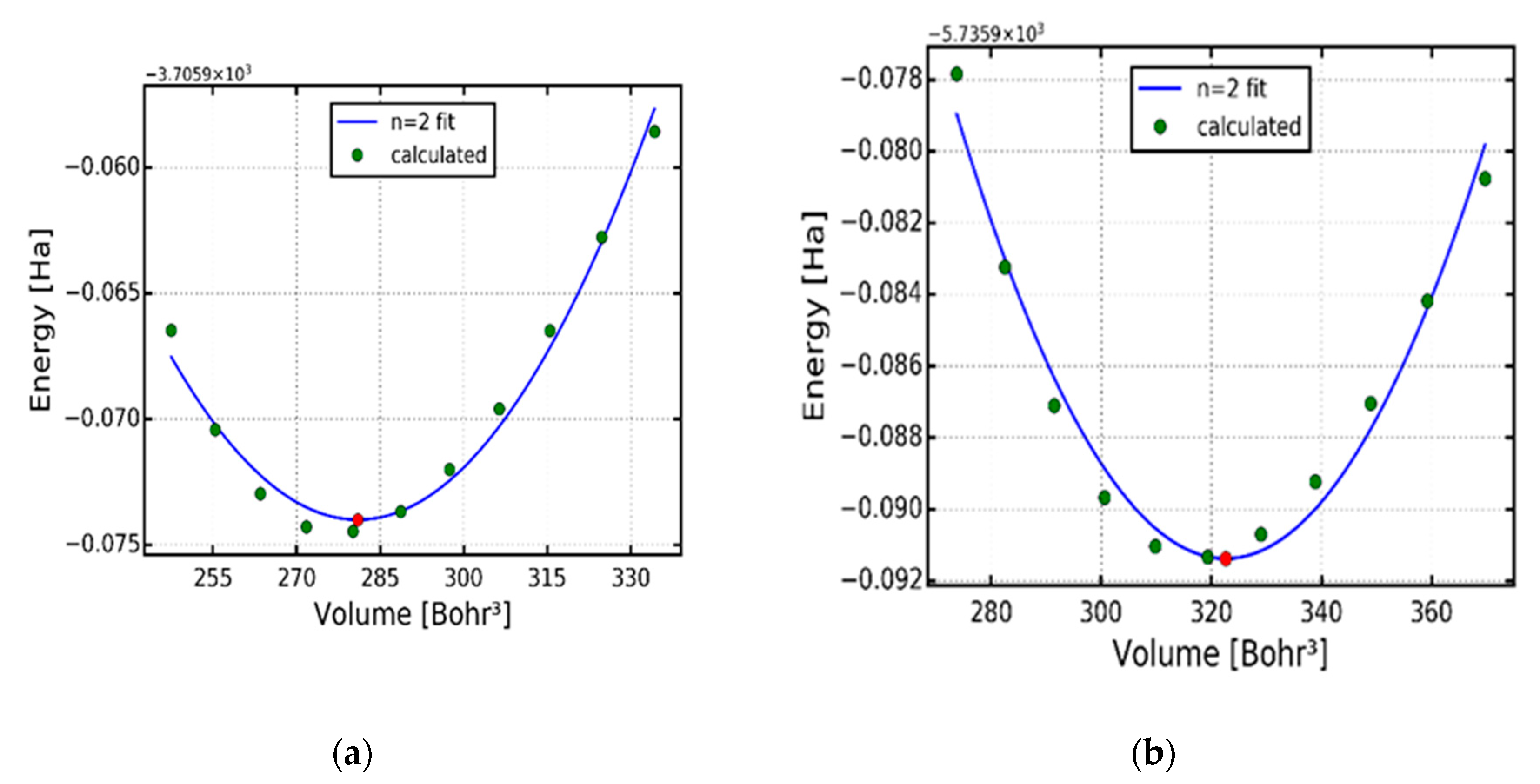
Figure 2.
Calculated electronic band structures. (a) cubic copper sulphide (α-Cu2S). (b) cubic copper selenide (α-Cu2Se).
Figure 2.
Calculated electronic band structures. (a) cubic copper sulphide (α-Cu2S). (b) cubic copper selenide (α-Cu2Se).
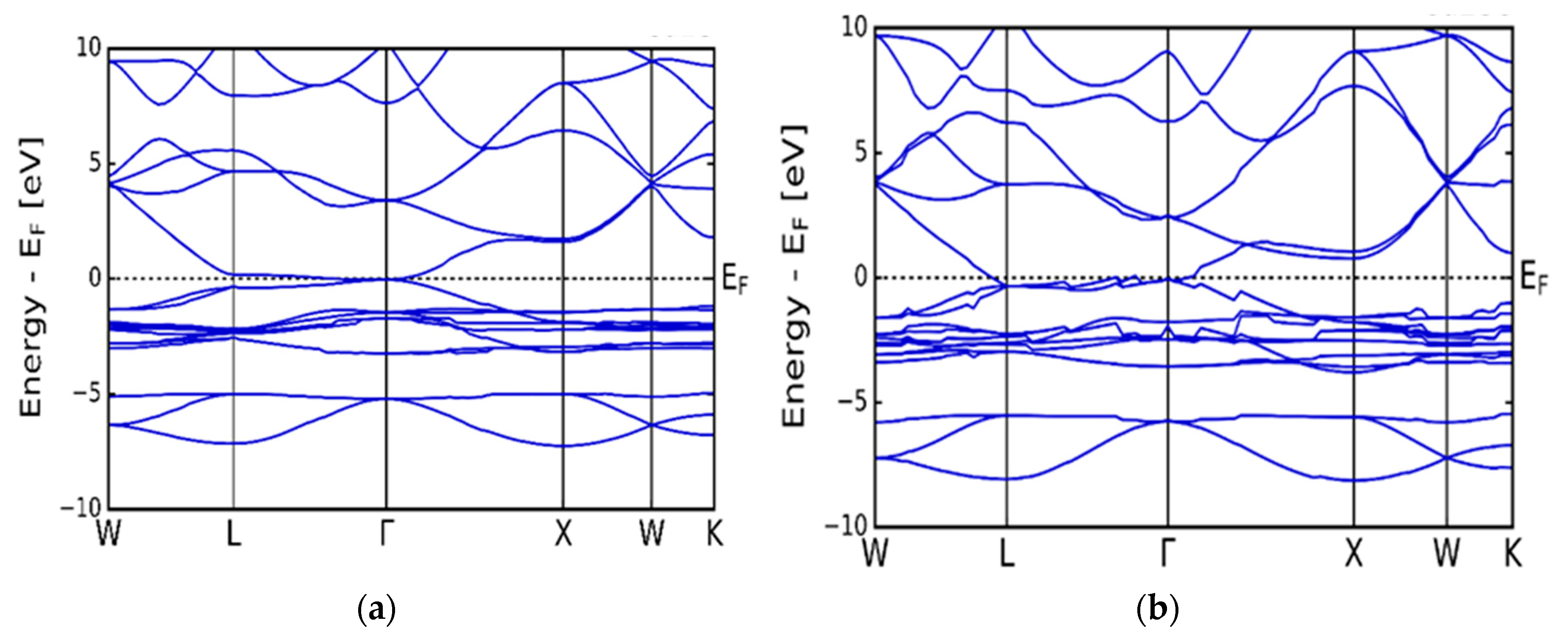
Figure 3.
Calculated electronic density of states (DOS). (a) cubic copper sulphide (α-Cu2S). (b) cubic copper selenide (α-Cu2Se).
Figure 3.
Calculated electronic density of states (DOS). (a) cubic copper sulphide (α-Cu2S). (b) cubic copper selenide (α-Cu2Se).
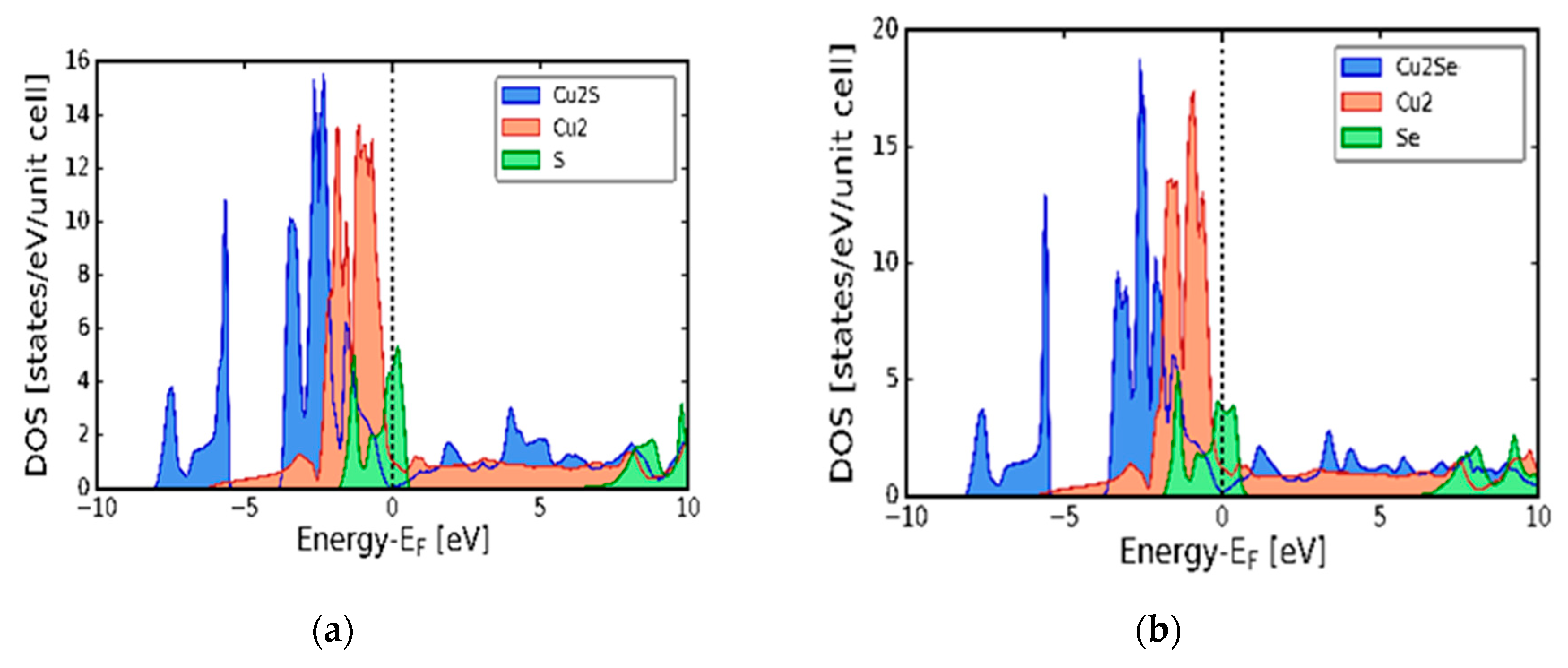
Figure 4.
Thermo-electric transport properties of cubic α-Cu2S and α-Cu2Se all at 300 K. (a) Variation of the Seebeck coefficient (S) against chemical potential (μ). (b) Variation of the electrical conductivity per relaxation time (σ/τ) against the chemical potential (μ). (c) Variation of the thermal conductivity per relaxation time (κ/τ) against the chemical potential (μ). (d) Variation of the Power factor (σS2) against the chemical potential (μ).
Figure 4.
Thermo-electric transport properties of cubic α-Cu2S and α-Cu2Se all at 300 K. (a) Variation of the Seebeck coefficient (S) against chemical potential (μ). (b) Variation of the electrical conductivity per relaxation time (σ/τ) against the chemical potential (μ). (c) Variation of the thermal conductivity per relaxation time (κ/τ) against the chemical potential (μ). (d) Variation of the Power factor (σS2) against the chemical potential (μ).
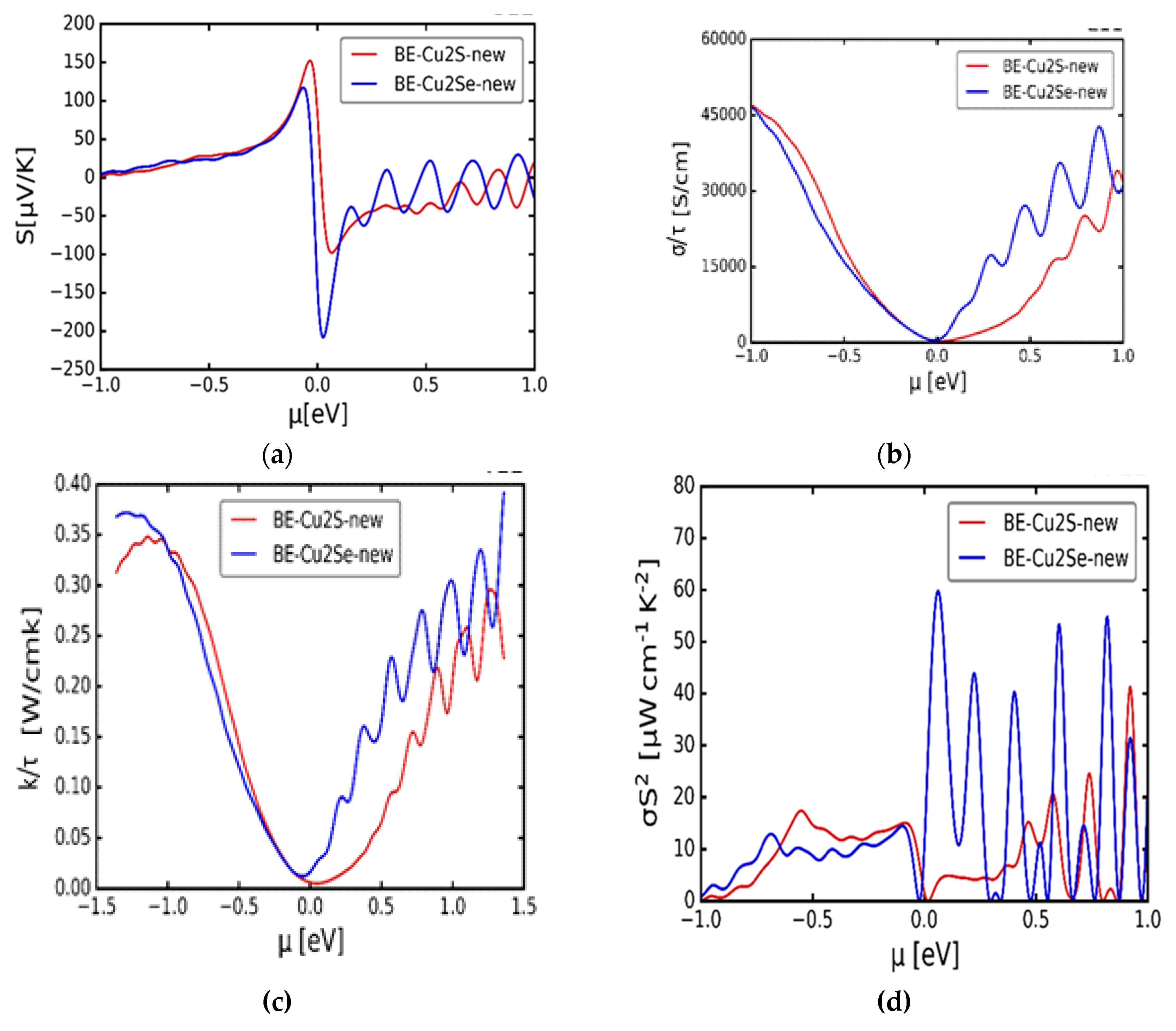
Figure 5.
Variation of the Seebeck coefficient (S) with temperature (T). (a) cubic α-Cu2S. (b) cubic α-Cu2Se.
Figure 5.
Variation of the Seebeck coefficient (S) with temperature (T). (a) cubic α-Cu2S. (b) cubic α-Cu2Se.
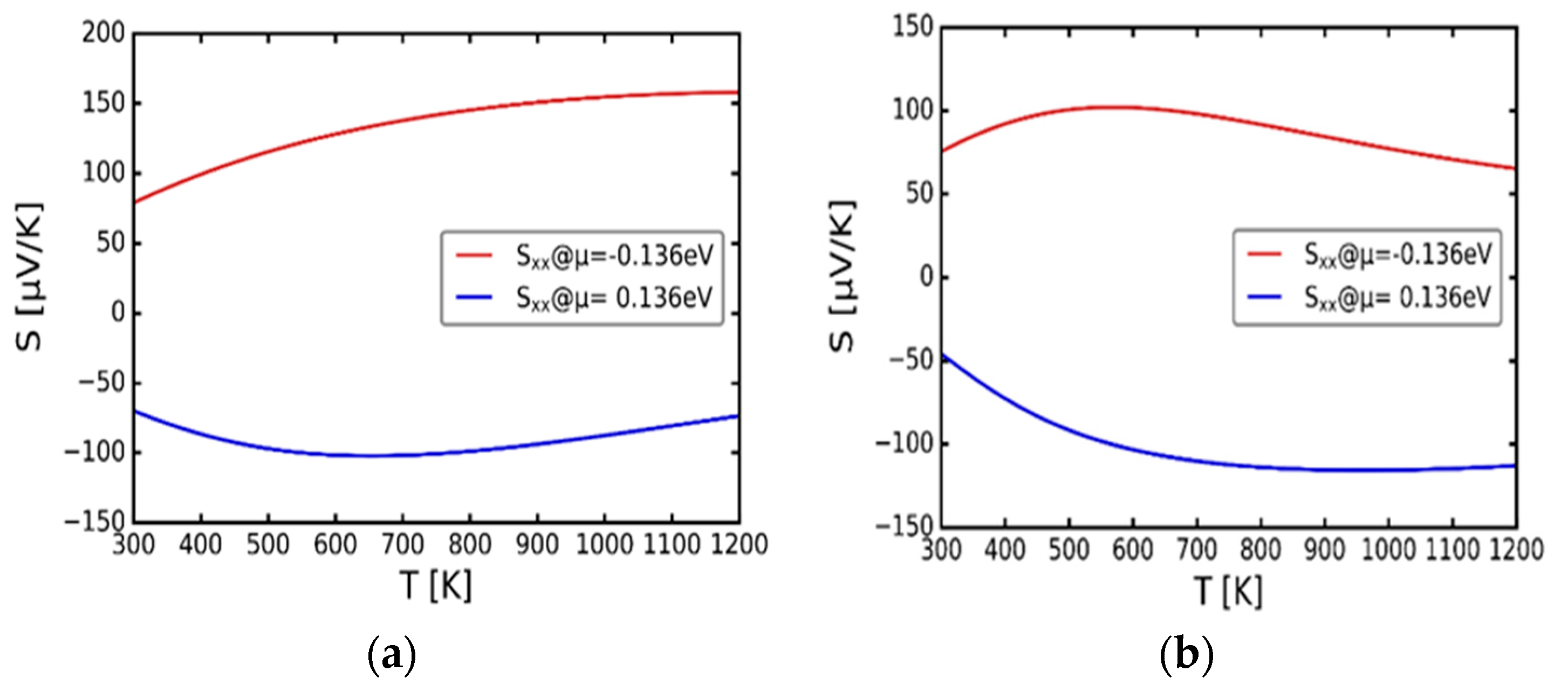
Figure 6.
Variation of the electrical conductivity per relaxation time (σ/τ) with temperature (T). (a) cubic α-Cu2S. (b) cubic α-Cu2Se.
Figure 6.
Variation of the electrical conductivity per relaxation time (σ/τ) with temperature (T). (a) cubic α-Cu2S. (b) cubic α-Cu2Se.
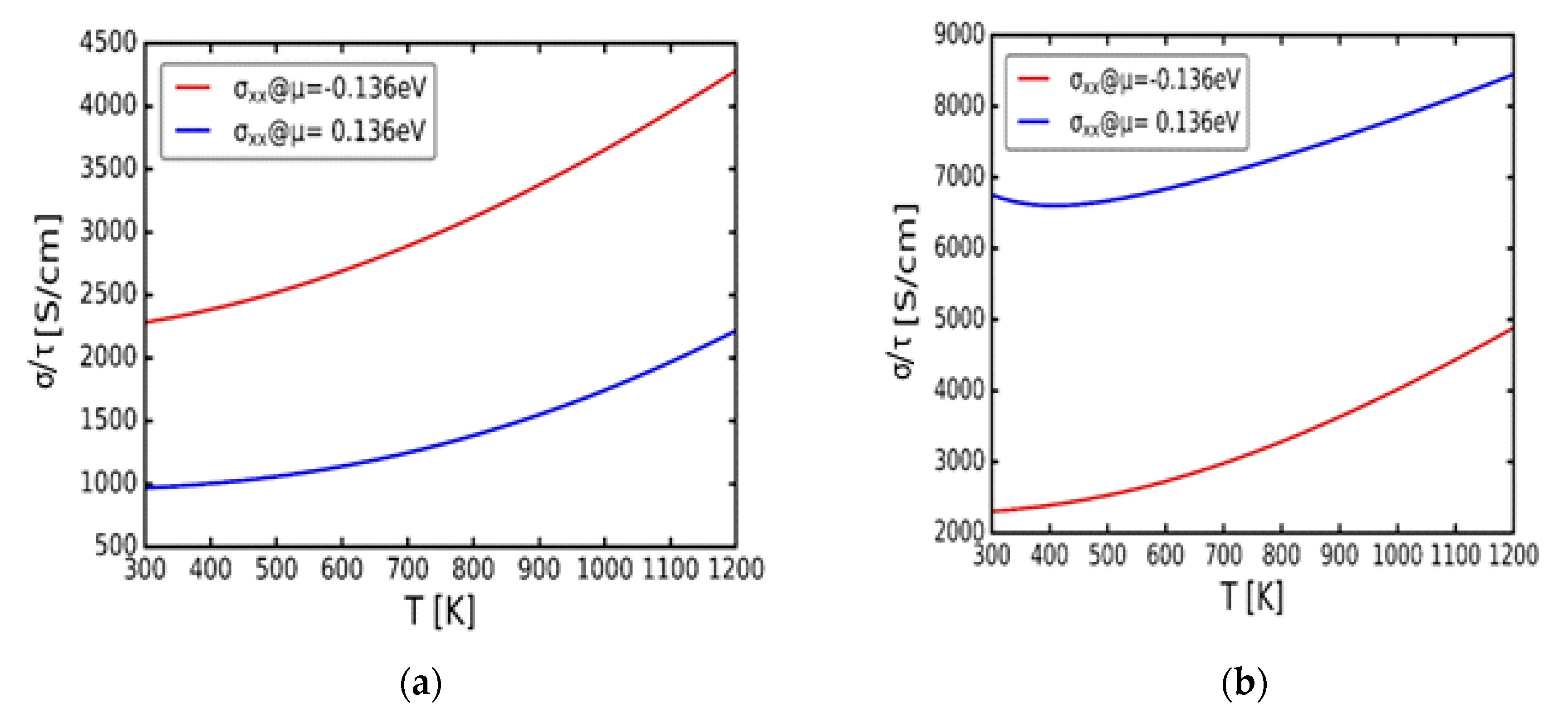
Figure 7.
Variation of the thermal conductivity per relaxation time (κ/τ) with temperature (T). (a) cubic α-Cu2S. (b) cubic α-Cu2Se.
Figure 7.
Variation of the thermal conductivity per relaxation time (κ/τ) with temperature (T). (a) cubic α-Cu2S. (b) cubic α-Cu2Se.
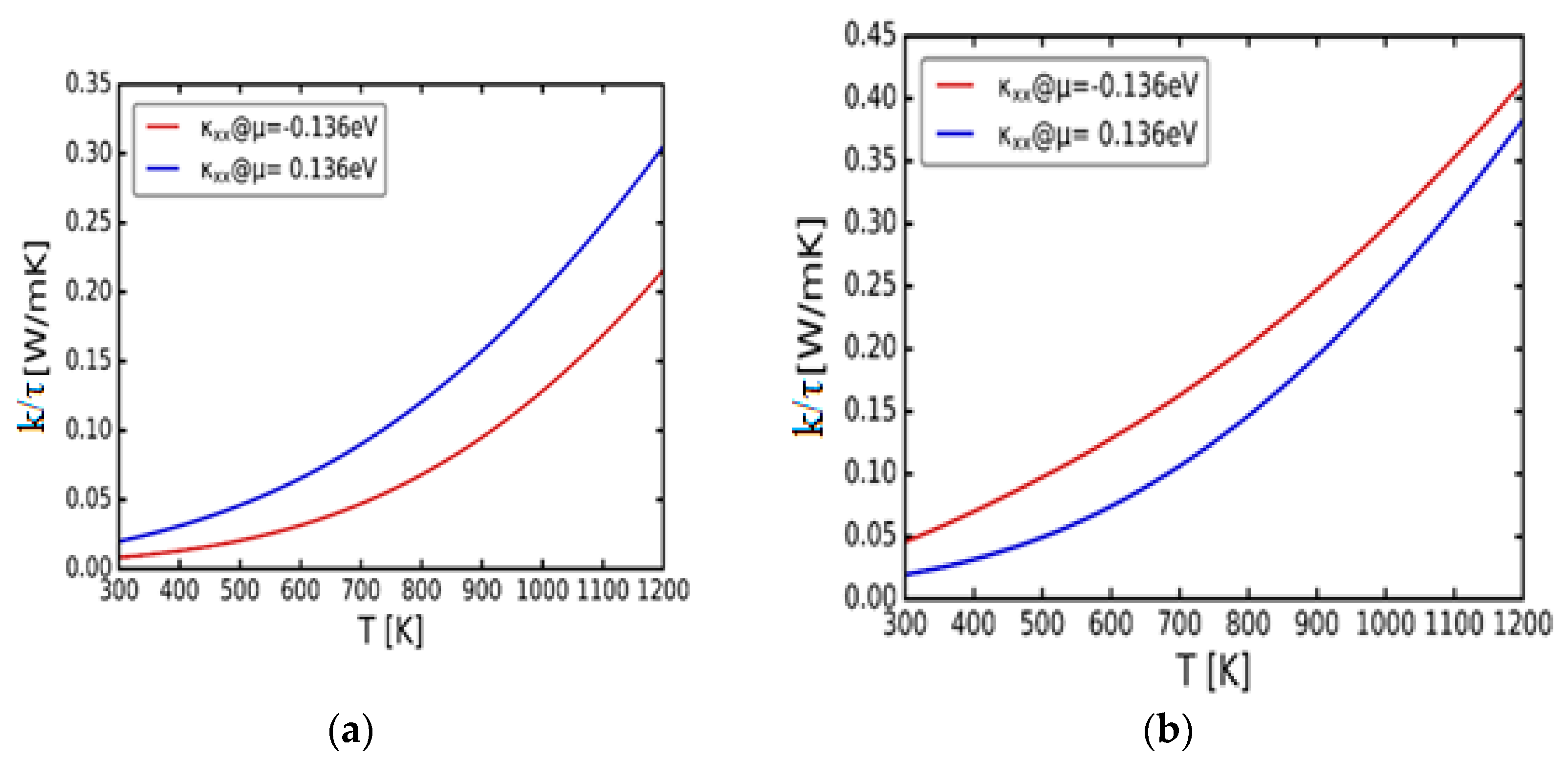
Figure 8.
Variation of the power factor (σS2) with temperature (T). (a) cubic α-Cu2S. (b) cubic α-Cu2Se.
Figure 8.
Variation of the power factor (σS2) with temperature (T). (a) cubic α-Cu2S. (b) cubic α-Cu2Se.
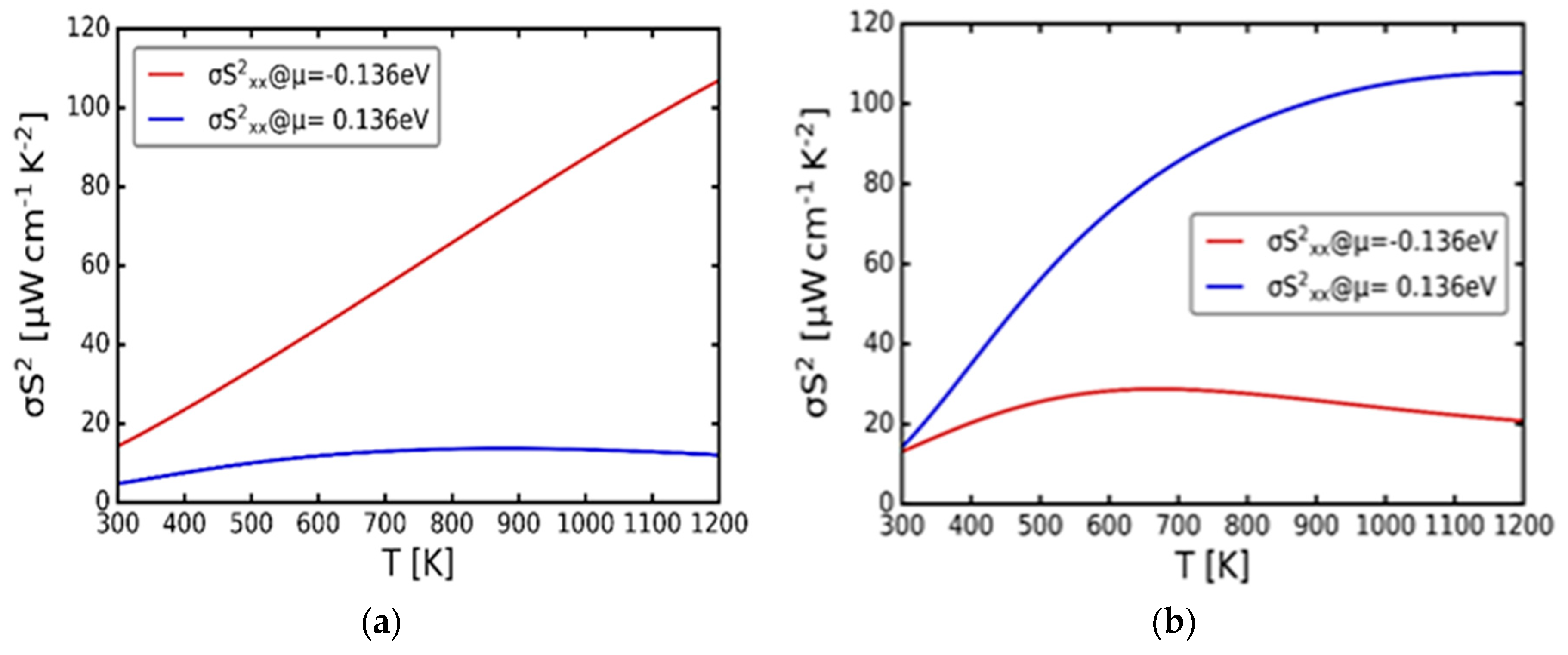

Table 1.
Minimum volume (V) in (Bohr3) units, lattice constant (a) in (Bohr) units and minimum energy (E) in (eV) units.
Table 1.
Minimum volume (V) in (Bohr3) units, lattice constant (a) in (Bohr) units and minimum energy (E) in (eV) units.
| Cu2S | Cu2Se | |
|---|---|---|
| (Bohr3) | 281.02 | 322.57 |
| (Bohr) | 10.39 | 10.89 |
| (eV) | -3705.95 | -5735.90 |
Table 2.
Elastic constants (C11, C12, C44), bulk modulus (B0), Young’s modulus (Y), shear modulus (G), all in GPa and Poisson’s ratio (ν) [22].
Table 2.
Elastic constants (C11, C12, C44), bulk modulus (B0), Young’s modulus (Y), shear modulus (G), all in GPa and Poisson’s ratio (ν) [22].
| α-Cu2S | C11 | C12 | C44 | B0 | Y | G | |
|---|---|---|---|---|---|---|---|
| This work (GPa) | 105.940 | 54,680 | 23.442 | 71.767 | 63.421 | 23.442 | 0.353 |
| Computational (GPa) | - | 80.70a | 65.413a | 23.692a | 0.360a | ||
[a]Ref [22].
Table 3.
Elastic constants (C11, C12, C44), bulk modulus (B0), Young’s modulus (Y), shear modulus (G), all in GPa and Poisson’s ratio (ν) [3,22,23].
| α-Cu2Se | C11 | C12 | C44 | B | Y | G | |
|---|---|---|---|---|---|---|---|
| This work (GPa) | 120.11 | 110.49 | 16.54 | 113.70 | 47.32 | 16.54 | 0.44 |
| Computational (GPa) | 121.298b | 73.419b | 74.101b | 89.276b | 53.57c | 20.58b | 0.300b |
| Experimental (GPa) | 72.80c | - | - | 45.70c | 34.20c | 13.60c | 0.390c |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



