
Submitted:
08 September 2023
Posted:
11 September 2023
You are already at the latest version
Abstract
Rhabdomyosarcoma (RMS) is the most common soft tissue malignancy in children and adolescents. Respecting the age of the patients and the tumor aggressiveness, investigation of the molecular mechanisms of RMS tumorigenesis is directed toward the identification of novel therapeutic targets. To contribute to a better understanding of the molecular pathology of RMS, we investigated ankyrin repeat domain 1 (ANKRD1), designated as a potential marker for differential diagnostics. In this study, we used three RMS cell lines (SJRH30, RD, and HS-729) to assess its expression profile, intracellular localization, and turnover. They express wild-type ANKRD1, as judged by the sequencing of the open reading frame. Each cell line expressed a different amount of ANKRD1 protein, although the transcript level was similar. According to Western blot analysis, ANKRD1 protein was expressed at detectable levels in the SJRH30 and RD cells (SJRH30>RD), but not in the HS-729, even after immunoprecipitation. Immunocytochemistry revealed nuclear and cytoplasmic localization of ANKRD1 in all examined cell lines. Moreover, the punctate pattern of ANKRD1 staining in the nuclei of RD and HS-729 cells overlapped with coilin, indicating its association with Cajal bodies. We have shown that RMS cells are not able to overexpress ANKRD1 protein, which can be attributed to its proteasomal degradation. The unsuccessful attempt to overexpress ANKRD1 in RMS cells indicates the possibility that its overexpression may be lethal for RMS cells and opens a window for therapeutic targeting.
Keywords:
rhabdomyosarcoma
; ANKRD1
; Cajal bodies
; proteasomal degradation
1. Introduction
Rhabdomyosarcoma (RMS), the most prevalent soft tissue malignancy in children and adolescents, arises from abnormal differentiation of skeletal muscle precursor cells [1]. Traditionally classified on morphological features, RMS comprises three primary subtypes (embryonal, alveolar, and pleomorphic), driven by distinct molecular mechanisms, presenting unique challenges for therapeutic interventions. Hence, recent advancements in molecular biology and genetics have facilitated improvements in RMS diagnostics, classification, patient risk stratification, and management strategies [2]. Moreover, elucidating the molecular biology of RMS holds promise for identifying novel therapeutic targets [3].
The diagnostics of RMS relies on immunohistochemical detection of muscle markers like desmin, myoglobin, myogenin, and MyoD. However, there is a need for new markers as the conventional ones are not always sensitive and specific enough to ensure an accurate diagnosis [4]. Among potential muscle-specific markers, the ankyrin repeat domain 1 (ANKRD1) has emerged for differential diagnostics of RMS, due to its higher expression in RMS compared to other sarcomas [5]. Despite that, its role and regulation in RMS remain unclear.
ANKRD1, (also known as cardiac ankyrin repeat protein, CARP), a muscle ankyrin repeat protein (MARP) family member, is a highly conserved, multifunctional protein [6]. This mechanical stress-inducible protein is involved in cardiac and skeletal muscle remodeling [7]. It participates in cardiogenesis, mechanosensing, regulation of gene expression, and intracellular signaling [6,8-11]. ANKRD1 is mostly expressed in cardiomyocytes, both in the nucleus (role of transcriptional regulator) and at the sarcomeric I-band (role in sarcomere organization). It is able to translocate from the cytoplasm to the nucleus in response to mechanical stimuli, contributing to the transformation of biomechanical stress signals to the regulation of gene expression.
Changes in ANKRD1 expression have been observed in various tumors, including RMS and ovarian carcinoma [12,13]. Altered ANKRD1 expression has prognostic implications in ovarian and renal clear cell carcinoma and has been associated with chemotherapy resistance in ovarian and lung carcinoma [14-16]. Furthermore, ANKRD1 inhibition sensitizes ovarian cancer cells to endoplasmic reticulum stress-induced apoptosis [13], while ANKRD1 upregulation promotes osteosarcoma cell proliferation and invasion [17]. In contrast, ANKRD1 upregulation suppresses colony formation in hepatocellular, lung, colon, and prostate cancer cell lines [18,19]. The YAP/TAZ oncogene complex and tumor suppressor p53 are involved in the transcriptional regulation of ANKRD1 expression [20,21,22].
Considering the significance of ANKRD1 in carcinogenesis, its association with chemotherapy resistance in various tumor types, and its role in normal muscle cell differentiation, which is disrupted in RMS, we aimed to further investigate this potential diagnostic marker by examining its expression at the mRNA and protein level, as well as its intracellular localization and protein turnover, in cell lines representing three main subtypes of RMS.
2. Materials and Methods
2.1. Cell culture, treatments, and transfection
Human rhabdomyosarcoma cell lines: RD (ATCC CCL-136), HS-729 (ATCC HTB-153) and SJRH30 (ATCC CRL-2061), and HeLa (ATCC CCL-2) cells were used in this study. Cells were cultured in Dulbecco’s Modified Eagle’s Medium - high glucose (Sigma-Aldrich, USA) supplemented with 10% of heat-inactivated fetal bovine serum (Gibco, Thermo Fisher Scientific, USA) and antibiotic-antimycotic mixture (Sigma-Aldrich, USA). Cells were maintained in a humidified environment with 5% CO2 at 37°C and subcultured twice a week. Cellular treatments included incubation with 10 µM proteasome inhibitor, MG132 (Merck KGaA, Germany) for 4 and 8 h. Transient transfections were performed at 90% confluence, with the pCMV-Tag2B expression vector containing ANKRD1 or ANKRD2 open reading frame, using Lipofectamine 2000 Reagent (Invitrogen, Thermo Fisher Scientific, USA) according to the manufacturer’s instructions.
2.2. RT-PCR and quantitative PCR
Total RNA was extracted from cells using the TRIzol reagent (Thermo Fisher Scientific, USA), according to the manufacturer’s protocol. RNA concentration and purity were determined by spectrophotometry using the BioSpec-nano (Shimadzu, Japan). Complementary DNA (cDNA) was synthesized from 2 μg of total RNA using random hexamer primers and High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, USA), according to the manufacturer’s instructions. ANKRD1 and GAPDH fragments were amplified using Native Taq DNA polymerase (EURx, Poland), cDNA as a template, and primers, listed in Table 1. The quantity of cDNA in each reaction corresponded to 100 ng of total RNA. The PCR reaction was performed in the 2720 Thermal Cycler (Applied Biosystems, USA). GAPDH was used as a cDNA quality control.
Quantitative PCR reactions were carried out in technical triplicate for each sample and for three biological replicates of each cell line on a 7900HT Fast Real-Time System (Applied Biosystems, USA) using TaqMan Gene Expression Assay (Applied Biosystems, USA). The GAPDH transcript was used as an internal reference to normalize the mRNA levels in different samples. The data were analyzed using the 2−∆Ct method. The results are presented as mean ± standard deviation.
2.3. Sequencing of the ANKRD1 open reading frame
ANKRD1 open reading frame was amplified using the cDNA templates, Phusion High-Fidelity PCR Master Mix (Thermo Fisher Scientific, USA), and primers given in Table 1. The PCR reaction was performed in the Mastercycler gradient (Eppendorf, Germany). Generated fragments were sequenced using the BigDyeTM Terminator Version 3.1 Ready Reaction Kit (Applied Biosystems, USA) according to the manufacturer’s protocol, on a 3130 Genetic Analyzer (Applied Biosystems, USA) with primers listed in Table 1.
2.4. Western blot
Total cellular proteins were extracted as previously described [23], separated by SDS-PAGE, and blotted onto the PVDF membrane (Immobilon P, Millipore, USA). The anti-ANKRD1 antibody against full-length ANKRD1, generated by the procedure reported in Kojic et al. [24] was used at 1:750 dilution; rabbit polyclonal anti-GAPDH antibody (Guangzhou Dingguo Biology, China) was used at 1:2000 dilution; mouse monoclonal anti-FLAG antibody (Sigma-Aldrich, USA), and rabbit polyclonal anti-p21 antibody (Santa Cruz Biotechnology, USA) were used at 1:1000 dilution.
2.5. Immunoprecipitation
For ANKRD1 protein enrichment, RMS cell extracts containing 0.5 or 1 mg of total proteins were incubated overnight at 4°C with anti-ANKRD1 antibody. The following day, the antibody-antigen complex was pulled out of the mixture using the Pierce Protein A/G UltraLink Resin (Thermo Fisher Scientific, USA). Resins were then washed with phosphate-buffered saline (PBS), resuspended in 30 µl of coIP buffer (125mM Tris-HCl pH 6.8, 4% SDS, 20% glycerol, 100mM DTT, bromophenol blue), incubated for 5 min at 95°C and centrifuged for 1 min at 2000 rpm. The supernatants were analyzed by Western blot.
2.6. Immunocytochemistry and microscopy
After seeding on coverslips and reaching 80% confluence, cells were fixed in 4% paraformaldehyde, permeabilized with 0.5% TritonX in PBS, and incubated overnight at 4°C in blocking solution (3% bovine serum albumin in PBS). Coverslips were then incubated with primary antibodies, diluted in 1% bovine serum albumin in PBS, for 3 h at room temperature, washed with PBS, and incubated with secondary antibodies labeled with Alexa Fluor 488 or Alexa Fluor 568 (Invitrogen, Thermo Fisher Scientific, USA) for 1 h. Primary antibodies were: anti-ANKRD1 (mouse, 1:100 dilution), anti-coilin (rabbit, 1:300 dilution, Cell Signaling Technology, USA), and anti-PML (goat, 1:100 dilution, Santa Cruz Biotechnology, USA). Nuclei were stained with DAPI (100 ng/ml) before mounting with Dako fluorescence medium (Agilent Technologies, Denmark). Confocal imaging and image manipulation were performed using the Leica SP8 system and Leica Application Suite X (Leica, Germany).
2.7. Statistical analysis
Statistical analysis was conducted using SPSS software v.21.0 (IBM SPSS Statistics, USA). The normal distribution and variance homogeneity of the data were verified by the Shapiro–Wilk and the Levene’s test, respectively. One-way ANOVA was employed for the comparison of the ANKRD1 transcript levels between the RMS cell lines. Results are presented as mean±SD, and p-value ≤ 0.05 was considered significant.
3. Results
3.1. ANKRD1 transcript and protein are differently expressed in RMS cell lines
We investigated if there are differences in the ANKRD1 expression level and primary structure between RMS cells. ANKRD1 transcript was detected in all tested cell lines (Figure 1, left panel) and the quantitative analysis of transcript levels using qPCR showed no significant differences among them. Subsequent sequencing of the ANKRD1 open reading frame demonstrated that all cell lines express wild-type ANKRD1.
In contrast to the mRNA, Western blot analysis demonstrated different expression levels of ANKRD1 protein among the RMS cell lines (Figure 2). Initially, we analyzed 50 μg of total proteins per cell line and ANKRD1 was only detected in SJRH30 (Figure 2A). Subsequently, we conducted immunoprecipitation using the anti-ANKRD1 antibody. In the RD cell line, a band of the expected molecular weight was observed, indicating a lower expression level of ANKRD1 in this cell line compared to SJRH30 (Figure 2B). However, the immunoprecipitation experiment failed to detect ANKRD1 in the HS-729 cell extracts, even when 1 mg of total proteins was used as an input (Figure S1B). Ponceau S staining of the membrane confirmed equal loading and protein transfer across samples (Figure S1B). Additionally, we tested the possibility that ANKRD1 protein was pelleted after the centrifugation step during the preparation of protein extracts. Western blot confirmed that the absence of signal was not due to the sequestration of ANKRD1 protein in the pellet (Figure S2).
3.2. ANKRD1 is localized in the Cajal bodies of RMS cells’ nuclei
Immunofluorescence staining of RMS cell lines revealed the presence of ANKRD1 in both the cytoplasm and the nucleus (Figure 3), with dominant nuclear localization. The absence of staining in the control sample (without primary antibody) confirmed the specificity of the ANKRD1 immunostaining (Figure S3). Moreover, distinct nuclear structures containing ANKRD1 were observed in RD and HS-729 cells (Figure 3, insets).
To identify these nuclear structures, we performed co-staining of RMS cells for ANKRD1 and promyelocytic leukemia protein, a marker of PML bodies. However, no colocalization was detected (Figure S4) implying that ANKRD1 does not localize to PML bodies. Next, we tested if ANKRD1 is present in Cajal bodies. The immunofluorescence staining of RMS cells for ANKRD1 and Cajal body marker, coilin, revealed the overlapping signals (Figure 4) in RD and HS-729 cell lines, but not in SJRH30.
3.3. ANKRD1 is prone to proteasomal degradation in RMS cells
To test if ANKRD1 is subjected to proteasomal degradation in RMS cell lines we treated the cells with a proteasome inhibitor, MG132. The effectiveness of MG132 treatment was validated by monitoring the level of p21 protein, a known target of proteasomal degradation in RMS cells [25]. As expected, Western blot analysis showed elevated level of p21 protein in cells treated with MG132, confirming the successful inhibition of proteasomal activity. At the same time, an increased level of ANKRD1 protein in cells treated with MG132 for 4 and 8 h compared to the vehicle-treated control cells was revealed, indicating the inhibition of its proteasomal degradation (Figure 5).
3.4. RMS cells do not tolerate overexpression of ANKRD1
To study the function of ANKRD1 in RMS cells we tried to overexpress it by transfecting the cells with an expression vector carrying the ANKRD1 open reading frame. Since we could not detect ANKRD1 overexpression after several attempts, we designed an experiment to find out why it was not possible. We transfected RMS cell lines and HeLa cells with constructs ANKRD1/pCMV-Tag2B and ANKRD2/pCMV-Tag2B to express FLAG-tagged ANKRD1 and ANKRD2, a closely related protein sharing structural motifs with ANKRD1, including PEST sequences. While ANKRD2 overexpression was successful in all used RMS cell lines, ANKRD1 expression remained low (SJRH30 and HS-729) or undetectable (RD). Conversely, ANKRD1 overexpression was achieved in HeLa cells, indicating that the inability to express ANKRD1 ectopically is a specific feature of RMS cells.
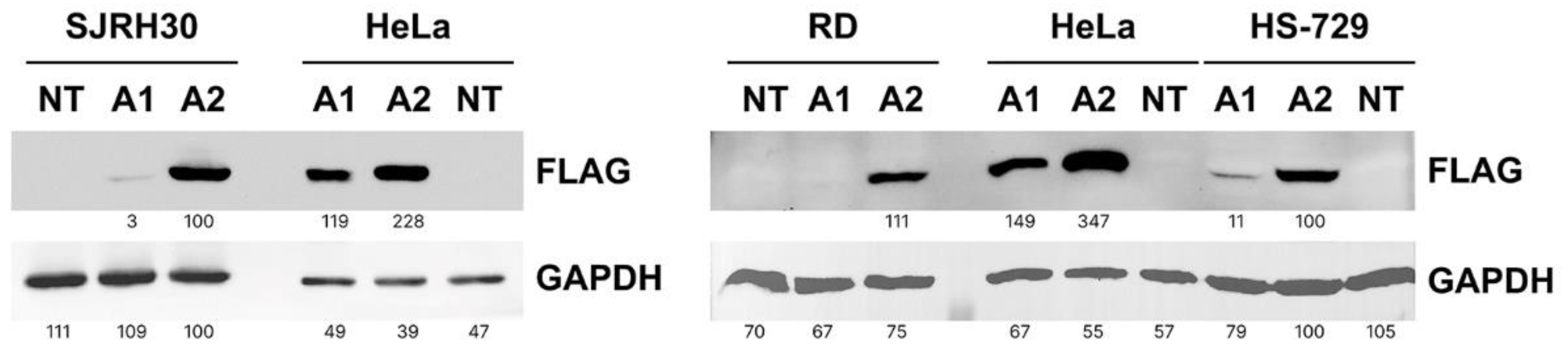
Figure 6.
RMS cell lines failed to overexpress ANKRD1 protein. RMS and HeLa cells were transfected with the pCMV-Tag2B vector carrying the ANKRD1 (A1) or ANKRD2 (A2) open reading frame to induce their overexpression. The non-transfected control (NT) was included as a baseline. Protein levels were assessed by Western blot, with GAPDH serving as the loading control. 50 µg of RMS and 10 µg of HeLa protein extracts was loaded on the gel. Representative blots are shown. Numbers represent the ratio between the values of densitometric analysis of the bands of anti-FLAG against those of anti-GAPDH.
Figure 6.
RMS cell lines failed to overexpress ANKRD1 protein. RMS and HeLa cells were transfected with the pCMV-Tag2B vector carrying the ANKRD1 (A1) or ANKRD2 (A2) open reading frame to induce their overexpression. The non-transfected control (NT) was included as a baseline. Protein levels were assessed by Western blot, with GAPDH serving as the loading control. 50 µg of RMS and 10 µg of HeLa protein extracts was loaded on the gel. Representative blots are shown. Numbers represent the ratio between the values of densitometric analysis of the bands of anti-FLAG against those of anti-GAPDH.
4. Discussion
ANKRD1 had been essentially investigated in muscle cells, where numerous reports had suggested its involvement in muscle differentiation under physiological and stress conditions, as well as in mechanosignaling pathways in mature muscle cells [6,8-10]. Parallel studies have reported an increased ANKRD1 expression in cells and tissues originating from certain primary tumors, such as rhabdomyosarcoma [10] and ovarian carcinoma [14]. Transcriptomic data from the Human Protein Atlas states increased ANKRD1 transcript level in cell lines derived from several tumor types, including rhabdomyosarcoma, testis, liver, ovarian, and pancreatic cancer. Additionally, ANKRD1 was suggested to play a role in cancer development since it is implicated in the progression of cancers, including those affecting ovaries, breast, and pancreas [13,26,27], and in the chemoresistance of ovarian and lung cancer [14,15]. In RMS, ANKRD1 was previously investigated as a potential diagnostic marker. Ishiguro et al. [5] reported upregulation of ANKRD1 in RMS compared to other sarcoma types, indicating its potential utility in differential diagnostics.
Driven by many findings suggesting the role of ANKRD1 in molecular mechanisms of tumorigenesis, we performed this in vitro study to examine its expression profile and regulation in cell lines derived from three RMS subtypes: RD (embryonal), SJRH30 (alveolar), and HS-729 (pleomorphic).
In RD and SJRH30 cell lines, we verified the ANKRD1 transcript expression as reported in Human Protein Atlas, and additionally detected a similar ANKRD1 mRNA level in HS-729 cells. In contrast to the similar mRNA levels, the ANKRD1 protein exhibited differential expression among these cell lines, suggesting different regulatory mechanisms controlling its protein level in RMS subtypes. Western blot analysis revealed detectable ANKRD1 protein levels in SJRH30 and RD cells (SJRH30 > RD), while in HS-729 cells the corresponding signal was not observed, even after immunoprecipitation. However, ANKRD1 was detected in HS-729 cells by immunofluorescence. This could be attributed to the differences in sensitivity or antibody affinities in two methods.
The fast protein turnover rate by proteasomal degradation could explain the discrepancy between ANKRD1 mRNA and protein levels and different amounts of endogenous ANKRD1 detected in protein extracts derived from different batches of cells, since proteasomal inhibition led to an increase in the ANKRD1 protein level in RMS cell lines. Similarly to other proteins containing PEST sequences that serve as proteolytic signals, ANKRD1 possesses a short intracellular half-life due to rapid degradation in 26S proteasomes [28,29]. This mechanism could also contribute to our inability to achieve ANKRD1 overexpression in RMS cells. However, in line with previous evidence [30], the overexpression of ANKRD2, a closely related protein sharing structural motifs with ANKRD1, including PEST sequences, was achieved. Additionally, we attained ANKRD1 overexpression in HeLa cells, despite its short half-life due to the proteasomal degradation in this cell line [28]. Our results imply that resistance to excessive ANKRD1 levels is a specific feature of RMS cells and mechanisms for regulation of its protein level in RMS cells warrant further exploration. For example, it would be worth investigating if ANKRD1 is implicated in growth inhibition of RMS cells triggered by the loss of proteasomal activity induced by bortezomib treatment [31,32].
Similar cases of cell lines exhibiting intolerance to gene overexpression have been reported. Lee et al. [33] demonstrated that mouse embryonic cell lines do not tolerate SUMO1 overexpression, adapting by reducing SUMO1 levels. Analogous observations include findings on glucocorticoid receptor expression in small-cell lung cancer cell lines [34] and research on SIRT6 overexpression in cancer cell lines [35]. The list goes on, including many pro-apoptotic and tumor suppressor proteins, like Bax, caspase-9, Beclin1, and p21 [36,37,38,39]. Additionally, the role of ANKRD1 in apoptosis modulation in various cancer types with its differential pro- and anti-apoptotic effects [,13,17,18,19,27], further confounds the understanding of its function in RMS cells. The function of ANKRD1 in tumor cells, particularly in apoptosis, should be considered in regard to p53. In addition to their interaction at protein level, we have previously shown that ANKRD1 upregulates p53 activity [20]. Furthermore, overexpressed ANKRD1 triggers the transcriptional activation of p53, inducing cellular apoptosis in cardiomyocytes. [40]. Our speculation that overexpression of ANKRD1 might be involved in induction of apoptotic death of RMS cells via activation of p53 remains to be clarified.
In muscle cells, ANKRD1 has dual, cytoplasmic and nuclear localization. It participates in sarcomere mechanosensing by translocation to the nucleus upon mechanical stimuli where it participates in the regulation of stress response genes [6,11]. In RMS cells we also observed both cytoplasmic and nuclear localization of ANKRD1 in RMS cells, which is in alignment with the findings of Ishiguro et al. [10]. However, inconsistently with their results, we detected higher ANKRD1 abundance in the nucleus compared to the cytoplasm. This could arise from differences in sample types (tissue sections vs. cell lines) or in the use of different antibodies (with different epitopes and sensitivity). Nuclear localization of ANKRD1 in RMS and RMS cell lines suggests its regulatory role. Accordingly, the next step would be to identify transcriptional targets downstream of ANKRD1 specific to RMS.
We detected ANKRD1 in structures resembling nuclear bodies in nuclei of RD and HS-729 cells. To identify these structures, the first hint was to explore the localization of ANKRD1 in PML bodies, since another MARP family member ANKRD2 is known to co-localize with PML protein in the PML bodies of human myoblasts [41]. However, there was no co-localization of ANKRD1 with a marker of PML bodies. Then we tested if ANKRD1 is localized to Cajal bodies. There was co-localization with coilin, resident protein of Cajal bodies, membraneless organelles implicated in the assembly of spliceosomal snRNPs [42], and in the maintenance of telomeres notably in cancer cells [43]. Although having many known protein partners, ANKRD1’s interactions with Cajal bodies-resident proteins like fibrillarin, SMN, and coilin have not been reported. Furthermore, there is no evidence that ANKRD1 could bind to RNA molecules. However, the reported interaction of ANKRD1 with YB-1 [44], an RNA-binding protein suggested as a target for RMS therapy [45], introduces intriguing prospects. Although rare, YB-1's presence in Cajal bodies [46] and its role as a component of spliceosomes [47,48] further emphasize the need for further research into the interactions of ANKRD1 with YB-1 and related proteins in Cajal bodies. Another possible direction would be to investigate if ANKRD1, as a downstream target of the Hippo pathway [19] involved in the responses to oxidative stress [49], has a role in the redox homeostasis driven by Cajal bodies in cancer cells [50].
Although the presence of ANKRD1 within the nucleoli fibrillar center is documented in various cell lines (proteinatlas.org), our study does not report ANKRD1 in nuclear structures other than Cajal bodies. Given the dynamic relationship between Cajal bodies and nucleoli in ribosome biogenesis, telomerase localization, RNA interference, and stress responses [51], translocation of ANKRD1 between Cajal bodies and nucleoli might present a topic for further investigation.
5. Conclusions
Our investigation provides novel insights into the expression and regulation of ANKRD1 in RMS cells. The disparity between ANKRD1 protein and mRNA levels, RMS cell resistance to ANKRD1 overexpression, and ANKRD1 localization within the Cajal bodies underline the unique attributes of RMS cells in regard to ANKRD1. These findings collectively establish a groundwork for future studies aiming to unravel the involvement of ANKRD1 in RMS pathogenesis, potentially leading to the identification of novel therapeutic targets.
Supplementary Materials
Table S1: Ct values ± SD for ANKRD1 and GAPDH amplicons obtained in the qPCR analysis; Figure S1: Original uncropped blots and membranes of Figure 2; Figure S2: ANKRD1 is not sequestered in the pellet during the protein extraction procedure; Figure S3: Control immunostaining of RMS cells; Figure S4: ANKRD1 is not localized to PML nuclear bodies of RMS cells; Figure S5: Original uncropped blots and membranes of Figure 5; Figure S6: Original uncropped blots and membranes of Figure 6.
Author Contributions
Conceptualization, E.M., J.J. and S.K.; validation, E.M., V.C., J.J. and S.K.; formal analysis, E.M. and M.N.; investigation, E.M.; resources, A.B. and S.K.; writing—original draft preparation, E.M.; writing—review and editing, M.N., V.C., A.B., S.K. and J.J.; visualization, E.M.; supervision, J.J. and S.K.; project administration, S.K.; funding acquisition, S.K. All authors have read and agreed to the published version of the manuscript.”.
Funding
This research was supported by Ministry of Science, Technological Development and Innovation of the Republic of Serbia (project number 451-03-9/2021-14/200042, 451-03-68/2022-14/200042, 451-03-47/2023-01/200042).
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Saab:, R.; Spunt, S.L.; Skapek, S.X. Myogenesis and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Curr Top Dev Biol 2011, 94, 197–234. [Google Scholar] [CrossRef] [PubMed]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat Rev Dis Primers 2019, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.A.; Barr, F.G. Molecular diagnostics in the management of rhabdomyosarcoma. Expert Rev Mol Diagn 2017, 17, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Dziuba, I.; Kurzawa, P.; Dopierała, M.; Larque, A.B.; Januszkiewicz-Lewandowska, D. Rhabdomyosarcoma in children - current pathologic and molecular classification. Pol J Pathol 2018, 69, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, N.; Motoi, T.; Araki, N.; Ito, H.; Moriyama, M.; Yoshida, H. Expression of cardiac ankyrin repeat protein, CARP, in malignant tumors: diagnostic use of CARP protein immunostaining in rhabdomyosarcoma. Hum Pathol 2008, 39, 1673–1679. [Google Scholar] [CrossRef]
- Miller, M.K.; Bang, M.L.; Witt, C.C.; Labeit, D.; Trombitas, C.; Watanabe, K.; Granzier, H.; McElhinny, A.S.; Gregorio, C.C.; Labeit, S. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J Mol Biol 2003, 333, 951–964. [Google Scholar] [CrossRef]
- Ling, S.S.M.; Chen, Y.T.; Wang, J.; Richards, A.M.; Liew, O.W. Ankyrin Repeat Domain 1 Protein: A Functionally Pleiotropic Protein with Cardiac Biomarker Potential. Int J Mol Sci 2017, 18, 1362. [Google Scholar] [CrossRef]
- Jeyaseelan, R.; Poizat, C.; Baker, R.K.; Abdishoo, S.; Isterabadi, L.B.; Lyons, G.E.; Kedes, L. A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J Biol Chem 1997, 272, 22800–22808. [Google Scholar] [CrossRef]
- Kuo, H.; Chen, J.; Ruiz-Lozano, P.; Zou, Y.; Nemer, M.; Chien, K.R. Control of segmental expression of the cardiac-restricted ankyrin repeat protein gene by distinct regulatory pathways in murine cardiogenesis. Development 1999, 126, 4223–4234. [Google Scholar] [CrossRef]
- Ishiguro, N.; Baba, T.; Ishida, T.; Takeuchi, K.; Osaki, M.; Araki, N.; Okada, E.; Takahashi, S.; Saito, M.; Watanabe, M.; et al. Carp, a cardiac ankyrin-repeated protein, and its new homologue, Arpp, are differentially expressed in heart, skeletal muscle, and rhabdomyosarcomas. Am J Pathol 2002, 160, 1767–1778. [Google Scholar] [CrossRef]
- Bang, M.L.; Mudry, R.E.; McElhinny, A.S.; Trombitás, K.; Geach, A.J.; Yamasaki, R.; Sorimachi, H.; Granzier, H.; Gregorio, C.C.; Labeit, S. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J Cell Biol 2001, 153, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Kojic, S.; Radojkovic, D.; Faulkner, G. Muscle ankyrin repeat proteins: their role in striated muscle function in health and disease. Crit Rev Clin Lab Sci 2011, 48, 269–294. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Henderson, B.R.; Emmanuel, C.; Harnett, P.R.; deFazio, A. Inhibition of ANKRD1 sensitizes human ovarian cancer cells to endoplasmic reticulum stress-induced apoptosis. Oncogene 2015, 34, 485–495. [Google Scholar] [CrossRef]
- Scurr, L.L.; Guminski, A.D.; Chiew, Y.E.; Balleine, R.L.; Sharma, R.; Lei, Y.; Pryor, K.; Wain, G.V.; Brand, A.; Byth, K.; et al. Ankyrin repeat domain 1, ANKRD1, a novel determinant of cisplatin sensitivity expressed in ovarian cancer. Clin Cancer Res 2008, 14, 6924–6932. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Seike, M.; Chiba, M.; Takahashi, S.; Nakamichi, S.; Matsumoto, M.; Takeuchi, S.; Minegishi, Y.; Noro, R.; Kunugi, S.; et al. Ankyrin Repeat Domain 1 Overexpression is Associated with Common Resistance to Afatinib and Osimertinib in EGFR-mutant Lung Cancer. Sci Rep 2018, 8, 14896. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Q.; Tang, Y.; Liu, Z.; Sun, G.; Lu, Z.; Chen, Y. Identification and validation of a cigarette smoke-related five-gene signature as a prognostic biomarker in kidney renal clear cell carcinoma. Sci Rep 2022, 12, 2189. [Google Scholar] [CrossRef]
- Yin, P.; Tong, C. LncRNA RGMB-AS1 up-regulates ANKRD1 Through Competitively Sponging miR-3614-5p to Promote OSA Cell Proliferation and Invasion. Arch Med Res 2022, 53, 131–137. [Google Scholar] [CrossRef]
- Park, J.H.; Liu, L.; Kim, I.H.; Kim, J.H.; You, K.R.; Kim, D.G. Identification of the genes involved in enhanced fenretinide-induced apoptosis by parthenolide in human hepatoma cells. Cancer Res 2005, 65, 2804–2814. [Google Scholar] [CrossRef]
- Jiménez, A.P.; Traum, A.; Boettger, T.; Hackstein, H.; Richter, A.M.; Dammann, R.H. The tumor suppressor RASSF1A induces the YAP1 target gene. Oncotarget 2017, 8, 88437–88452. [Google Scholar] [CrossRef]
- Kojic, S.; Nestorovic, A.; Rakicevic, L.; Belgrano, A.; Stankovic, M.; Divac, A.; Faulkner, G. A novel role for cardiac ankyrin repeat protein Ankrd1/CARP as a co-activator of the p53 tumor suppressor protein. Arch Biochem Biophys 2010, 502, 60–67. [Google Scholar] [CrossRef]
- Tang, J.; Tian, Z.; Liao, X.; Wu, G. SOX13/TRIM11/YAP axis promotes the proliferation, migration and chemoresistance of anaplastic thyroid cancer. Int J Biol Sci 2021, 17, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhou, J.; Tang, J.; Zhou, F.; He, Z.; Liu, T. MINDY1 promotes bladder cancer progression by stabilizing YAP. Cancer Cell Int 2021, 21, 395. [Google Scholar] [CrossRef] [PubMed]
- Jasnic-Savovic, J.; Nestorovic, A.; Savic, S.; Karasek, S.; Vitulo, N.; Valle, G.; Faulkner, G.; Radojkovic, D.; Kojic, S. Profiling of skeletal muscle Ankrd2 protein in human cardiac tissue and neonatal rat cardiomyocytes. Histochem Cell Biol 2015, 143, 583–597. [Google Scholar] [CrossRef]
- Kojic, S.; Medeot, E.; Faulkner, G. Characterization of Antibodies Directed Against the Ankrd2 Human Muscle Protein. Archive of Biological Science 2009, 61, 9. [Google Scholar] [CrossRef]
- Phillips, D.C.; Hunt, J.T.; Moneypenny, C.G.; Maclean, K.H.; McKenzie, P.P.; Harris, L.C.; Houghton, J.A. Ceramide-induced G2 arrest in rhabdomyosarcoma (RMS) cells requires p21Cip1/Waf1 induction and is prevented by MDM2 overexpression. Cell Death Differ 2007, 14, 1780–1791. [Google Scholar] [CrossRef]
- Kikuchi, M.; Yamashita, K.; Waraya, M.; Minatani, N.; Ushiku, H.; Kojo, K.; Ema, A.; Kosaka, Y.; Katoh, H.; Sengoku, N.; et al. Epigenetic regulation of ZEB1-RAB25/ESRP1 axis plays a critical role in phenylbutyrate treatment-resistant breast cancer. Oncotarget 2016, 7, 1741–1753. [Google Scholar] [CrossRef]
- Hui, B.; Ji, H.; Xu, Y.; Wang, J.; Ma, Z.; Zhang, C.; Wang, K.; Zhou, Y. RREB1-induced upregulation of the lncRNA AGAP2-AS1 regulates the proliferation and migration of pancreatic cancer partly through suppressing ANKRD1 and ANGPTL4. Cell Death Dis 2019, 10, 207. [Google Scholar] [CrossRef]
- Badi, I.; Cinquetti, R.; Frascoli, M.; Parolini, C.; Chiesa, G.; Taramelli, R.; Acquati, F. Intracellular ANKRD1 protein levels are regulated by 26S proteasome-mediated degradation. FEBS Lett 2009, 583, 2486–2492. [Google Scholar] [CrossRef]
- Samaras, S.E.; Chen, B.; Koch, S.R.; Sawyer, D.B.; Lim, C.C.; Davidson, J.M. 26S proteasome regulation of Ankrd1/CARP in adult rat ventricular myocytes and human microvascular endothelial cells. Biochem Biophys Res Commun 2012, 425, 830–835. [Google Scholar] [CrossRef]
- Piazzi, M.; Kojic, S.; Capanni, C.; Stamenkovic, N.; Bavelloni, A.; Marin, O.; Lattanzi, G.; Blalock, W.; Cenni, V. Ectopic Expression of Ankrd2 Affects Proliferation, Motility and Clonogenic Potential of Human Osteosarcoma Cells. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Bersani, F.; Taulli, R.; Accornero, P.; Morotti, A.; Miretti, S.; Crepaldi, T.; Ponzetto, C. Bortezomib-mediated proteasome inhibition as a potential strategy for the treatment of rhabdomyosarcoma. Eur J Cancer 2008, 44, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Péron, J.; Polivka, V.; Chabaud, S.; Poupart, M.; Ceruse, P.; Ramade, A.; Girodet, D.; Zrounba, P.; Fayette, J. An effective and well-tolerated strategy in recurrent and/or metastatic head and neck cancer: successive lines of active chemotherapeutic agents. BMC Cancer 2014, 14, 504. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Zhu, Y.; Sabo, Y.; Goff, S.P. Embryonic Cells Redistribute SUMO1 upon Forced SUMO1 Overexpression. mBio 2019, 10. [Google Scholar] [CrossRef]
- Sommer, P.; Le Rouzic, P.; Gillingham, H.; Berry, A.; Kayahara, M.; Huynh, T.; White, A.; Ray, D.W. Glucocorticoid receptor overexpression exerts an antisurvival effect on human small cell lung cancer cells. Oncogene 2007, 26, 7111–7121. [Google Scholar] [CrossRef]
- Van Meter, M.; Mao, Z.; Gorbunova, V.; Seluanov, A. SIRT6 overexpression induces massive apoptosis in cancer cells but not in normal cells. Cell Cycle 2011, 10, 3153–3158. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Chen, S.T.; Tafani, M.; Snyder, J.W.; Farber, J.L. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J Biol Chem 1998, 273, 7770–7775. [Google Scholar] [CrossRef]
- Druskovic, M.; Suput, D.; Milisav, I. Overexpression of caspase-9 triggers its activation and apoptosis in vitro. Croat Med J 2006, 47, 832–840. [Google Scholar]
- Zhu, J.; Cai, Y.; Xu, K.; Ren, X.; Sun, J.; Lu, S.; Chen, J.; Xu, P. Beclin1 overexpression suppresses tumor cell proliferation and survival via an autophagy-dependent pathway in human synovial sarcoma cells. Oncol Rep 2018, 40, 1927–1936. [Google Scholar] [CrossRef]
- Mansour, M.A.; Rahman, M.; Ayad, A.A.; Warrington, A.E.; Burns, T.C. P21 Overexpression Promotes Cell Death and Induces Senescence in Human Glioblastoma. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Shen, L.; Chen, C.; Wei, X.; Li, X.; Luo, G.; Zhang, J.; Bin, J.; Huang, X.; Cao, S.; Li, G.; et al. Overexpression of ankyrin repeat domain 1 enhances cardiomyocyte apoptosis by promoting p53 activation and mitochondrial dysfunction in rodents. Clin Sci (Lond) 2015, 128, 665–678. [Google Scholar] [CrossRef]
- Kojic, S.; Medeot, E.; Guccione, E.; Krmac, H.; Zara, I.; Martinelli, V.; Valle, G.; Faulkner, G. The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle. J Mol Biol 2004, 339, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Staněk, D. Cajal bodies and snRNPs - friends with benefits. RNA Biol 2017, 14, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Machyna, M.; Heyn, P.; Neugebauer, K.M. Cajal bodies: where form meets function. Wiley Interdiscip Rev RNA 2013, 4, 17–34. [Google Scholar] [CrossRef]
- Zou, Y.; Evans, S.; Chen, J.; Kuo, H.C.; Harvey, R.P.; Chien, K.R. CARP, a cardiac ankyrin repeat protein, is downstream in the Nkx2-5 homeobox gene pathway. Development 1997, 124, 793–804. [Google Scholar] [CrossRef]
- Oda, Y.; Kohashi, K.; Yamamoto, H.; Tamiya, S.; Kohno, K.; Kuwano, M.; Iwamoto, Y.; Tajiri, T.; Taguchi, T.; Tsuneyoshi, M. Different expression profiles of Y-box-binding protein-1 and multidrug resistance-associated proteins between alveolar and embryonal rhabdomyosarcoma. Cancer Sci 2008, 99, 726–732. [Google Scholar] [CrossRef]
- Bogolyubova, I.O.; Lyabin, D.N.; Bogolyubov, D.S.; Ovchinnikov, L.P. Immunocytochemical study of YB-1 nuclear distribution in different cell types. Tissue Cell 2014, 46, 457–461. [Google Scholar] [CrossRef]
- Chansky, H.A.; Hu, M.; Hickstein, D.D.; Yang, L. Oncogenic TLS/ERG and EWS/Fli-1 fusion proteins inhibit RNA splicing mediated by YB-1 protein. Cancer Res 2001, 61, 3586–3590. [Google Scholar]
- Rapp, T.B.; Yang, L.; Conrad, E.U.; Mandahl, N.; Chansky, H.A. RNA splicing mediated by YB-1 is inhibited by TLS/CHOP in human myxoid liposarcoma cells. J Orthop Res 2002, 20, 723–729. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, L.; Li, X.; Xu, W.; Yang, S.; Song, J.; Zhang, W.; Zhan, J.; Luo, J.; Zhang, H. Oxidative stress-CBP axis modulates MOB1 acetylation and activates the Hippo signaling pathway. Nucleic Acids Res 2022, 50, 3817–3834. [Google Scholar] [CrossRef]
- Beneventi, G.; Munita, R.; Cao Thi Ngoc, P.; Madej, M.; Cieśla, M.; Muthukumar, S.; Krogh, N.; Nielsen, H.; Swaminathan, V.; Bellodi, C. The small Cajal body-specific RNA 15 (SCARNA15) directs p53 and redox homeostasis via selective splicing in cancer cells. NAR Cancer 2021, 3, zcab026. [Google Scholar] [CrossRef]
- Trinkle-Mulcahy, L.; Sleeman, J.E. The Cajal body and the nucleolus: "In a relationship" or "It's complicated"? RNA Biol 2017, 14, 739–751. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Ankyrin repeat domain 1 (ANKRD1) gene expression in human rhabdomyosarcoma (RMS) cell lines. (A) ANKRD1 transcript fragments from all three RMS cell lines were amplified by RT-PCR. GAPDH was used as the reference gene. A reaction without cDNA served as a negative control (ctrl). Three biological replicates (1-3) were analyzed per cell line. (B) The relative quantification of the ANKRD1 mRNA in RMS cell lines by qPCR. The expression level was calculated using the 2-ΔCt method and normalized to the endogenous reference gene GAPDH. Values were multiplied by 1000 for easier comparison. The data are presented as the mean ± SD of three biological replicates for each cell line. The average Ct values ± SD are listed in Table S1.
Figure 1.
Ankyrin repeat domain 1 (ANKRD1) gene expression in human rhabdomyosarcoma (RMS) cell lines. (A) ANKRD1 transcript fragments from all three RMS cell lines were amplified by RT-PCR. GAPDH was used as the reference gene. A reaction without cDNA served as a negative control (ctrl). Three biological replicates (1-3) were analyzed per cell line. (B) The relative quantification of the ANKRD1 mRNA in RMS cell lines by qPCR. The expression level was calculated using the 2-ΔCt method and normalized to the endogenous reference gene GAPDH. Values were multiplied by 1000 for easier comparison. The data are presented as the mean ± SD of three biological replicates for each cell line. The average Ct values ± SD are listed in Table S1.
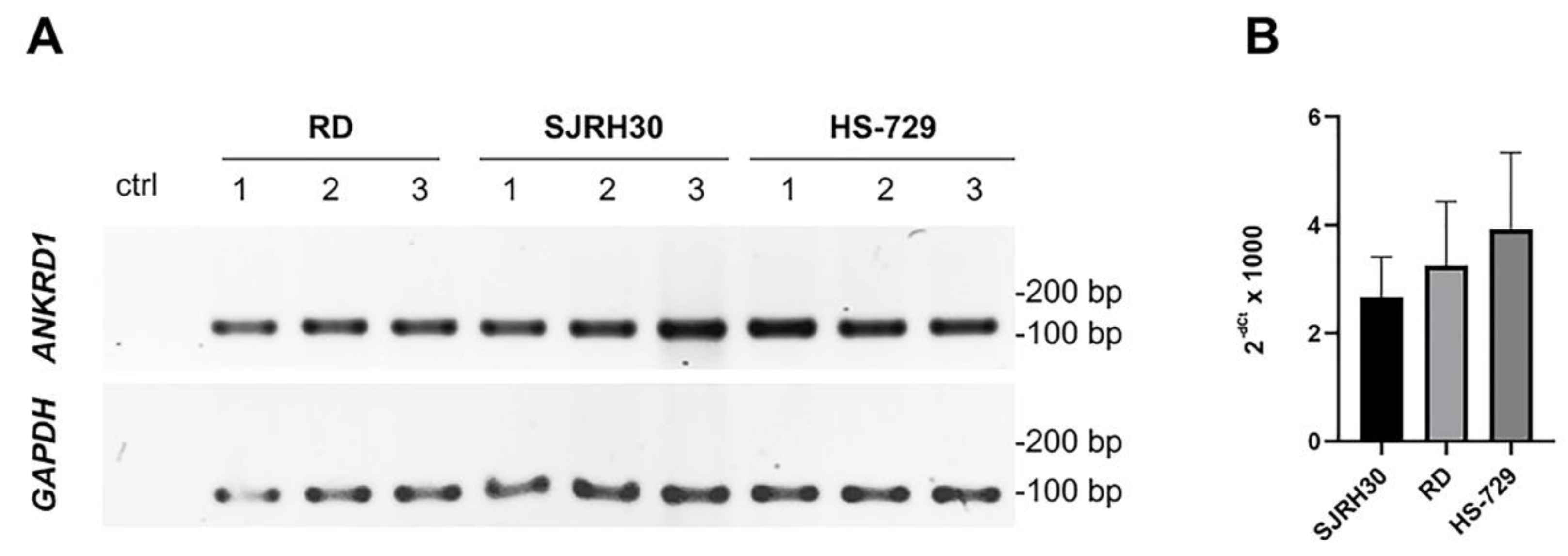
Figure 2.
ANKRD1 protein expression in RMS cell lines. (A) The detection of ANKRD1 protein in 50 µg of total cellular protein extracts of RMS cells. A positive signal, corresponding to the band of expected molecular weight of approximately 38 kDa, was observed in the extract derived from SJRH30 cells. The presented image is representative of one out of three independent experiments. Numbers represent the ratio between the values of densitometric analysis of the bands of anti-ANKRD1 against those that correspond to actin bands on Ponceau S stained membrane. (B) The ANKRD1 protein was selectively enriched via immunoprecipitation procedure using the anti-ANKRD1 antibody and 0.5 mg of total proteins isolated from RMS cells as an input. The displayed image represents one out of two independent experiments. *: 15% input for RD and SJRH30 and 10% input for HS-729. Numbers represent the values of densitometric analysis of the bands detected with anti-ANKRD1 antibody.
Figure 2.
ANKRD1 protein expression in RMS cell lines. (A) The detection of ANKRD1 protein in 50 µg of total cellular protein extracts of RMS cells. A positive signal, corresponding to the band of expected molecular weight of approximately 38 kDa, was observed in the extract derived from SJRH30 cells. The presented image is representative of one out of three independent experiments. Numbers represent the ratio between the values of densitometric analysis of the bands of anti-ANKRD1 against those that correspond to actin bands on Ponceau S stained membrane. (B) The ANKRD1 protein was selectively enriched via immunoprecipitation procedure using the anti-ANKRD1 antibody and 0.5 mg of total proteins isolated from RMS cells as an input. The displayed image represents one out of two independent experiments. *: 15% input for RD and SJRH30 and 10% input for HS-729. Numbers represent the values of densitometric analysis of the bands detected with anti-ANKRD1 antibody.
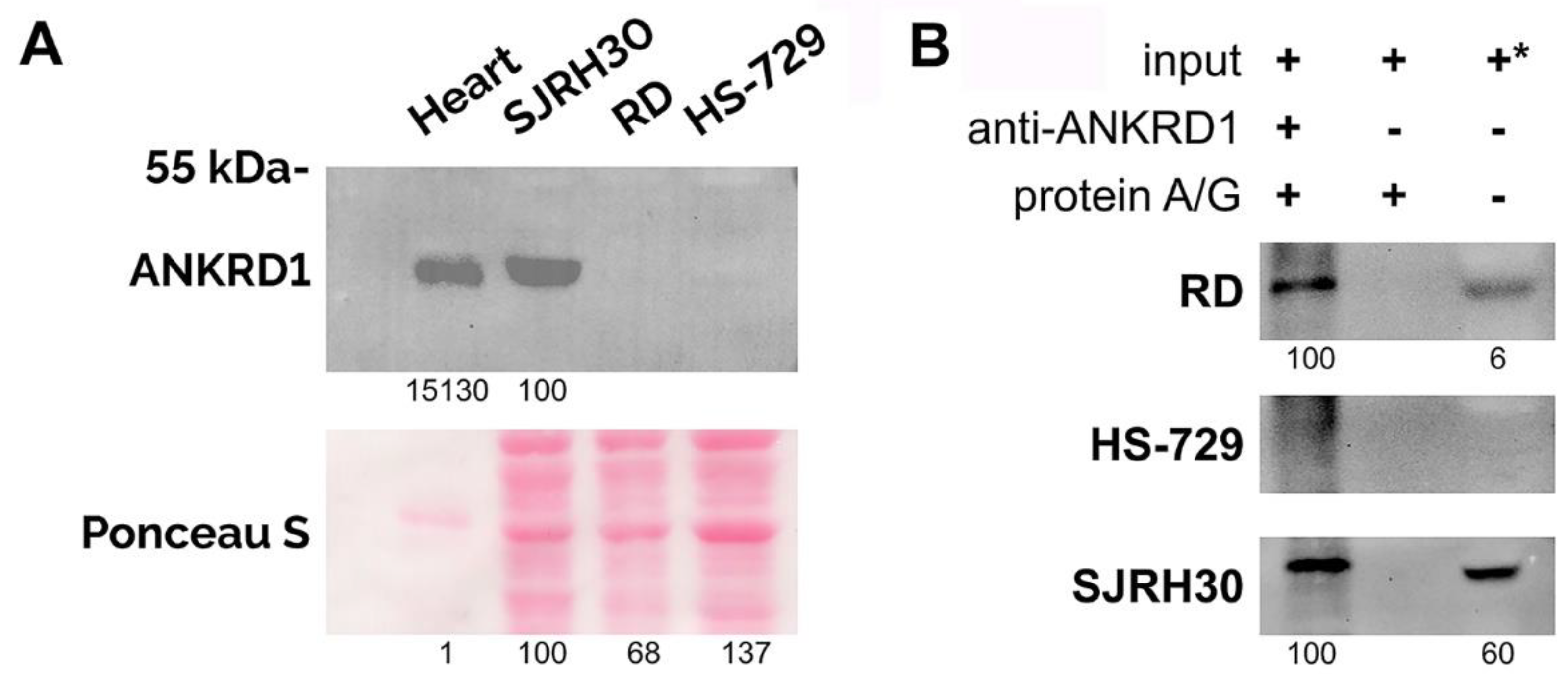
Figure 3.
Intracellular localization of ANKRD1 in RMS cells. Immunofluorescence staining revealed nuclear and cytoplasmic localization of ANKRD1 (green) in RMS cells. Nuclei were labeled with DAPI (blue). White arrows point to nuclear structures exhibiting high ANKRD1 content (insets). The signal intensity is not comparable between images. Scale bar = 10 μm.
Figure 3.
Intracellular localization of ANKRD1 in RMS cells. Immunofluorescence staining revealed nuclear and cytoplasmic localization of ANKRD1 (green) in RMS cells. Nuclei were labeled with DAPI (blue). White arrows point to nuclear structures exhibiting high ANKRD1 content (insets). The signal intensity is not comparable between images. Scale bar = 10 μm.
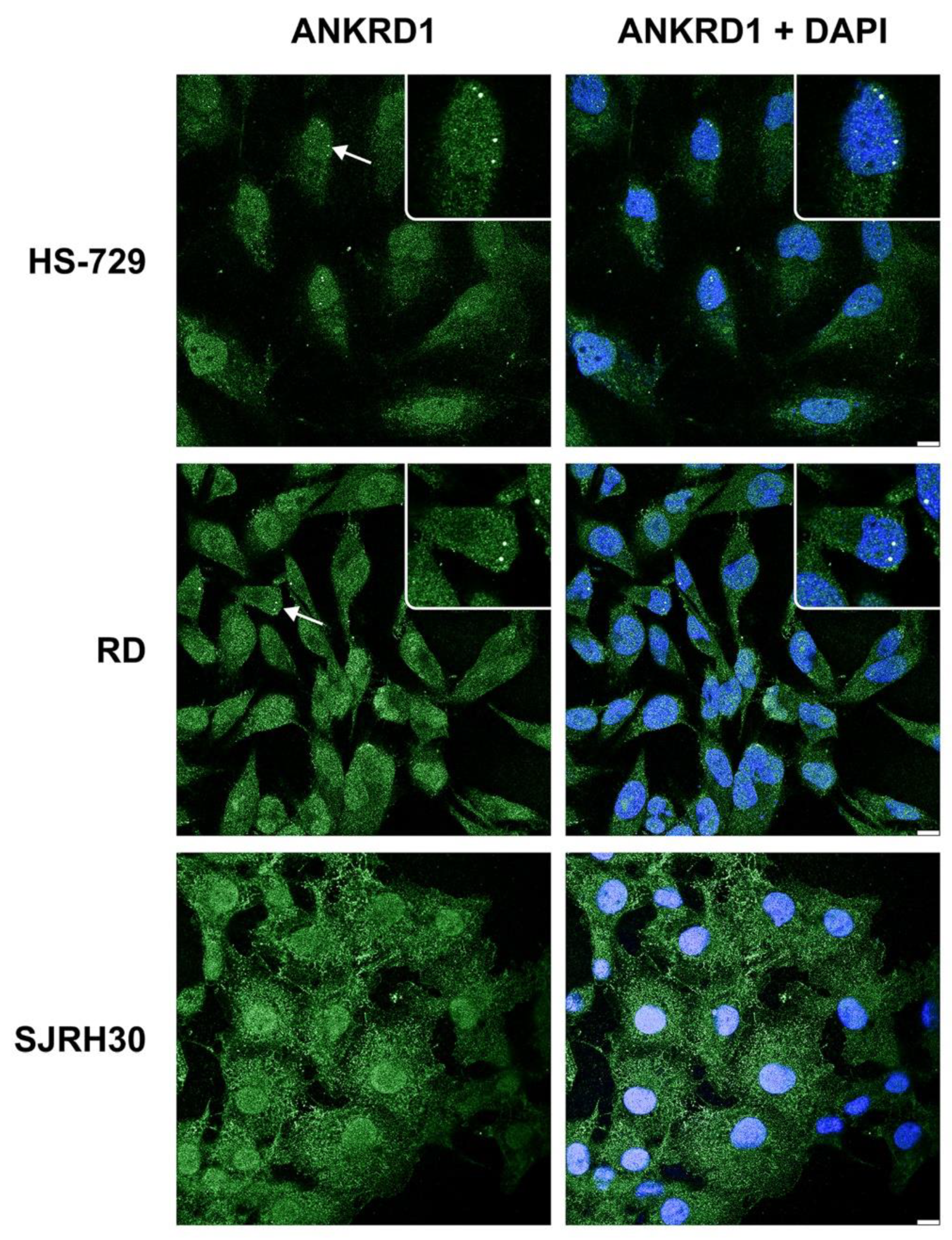
Figure 4.
Localization of ANKRD1 in Cajal bodies of RD and HS-729 cells. The immunofluorescence staining of ANKRD1 (green) and the Cajal body marker, coilin (red). Nuclei were stained with DAPI (blue). Scale bar = 5 µm.
Figure 4.
Localization of ANKRD1 in Cajal bodies of RD and HS-729 cells. The immunofluorescence staining of ANKRD1 (green) and the Cajal body marker, coilin (red). Nuclei were stained with DAPI (blue). Scale bar = 5 µm.
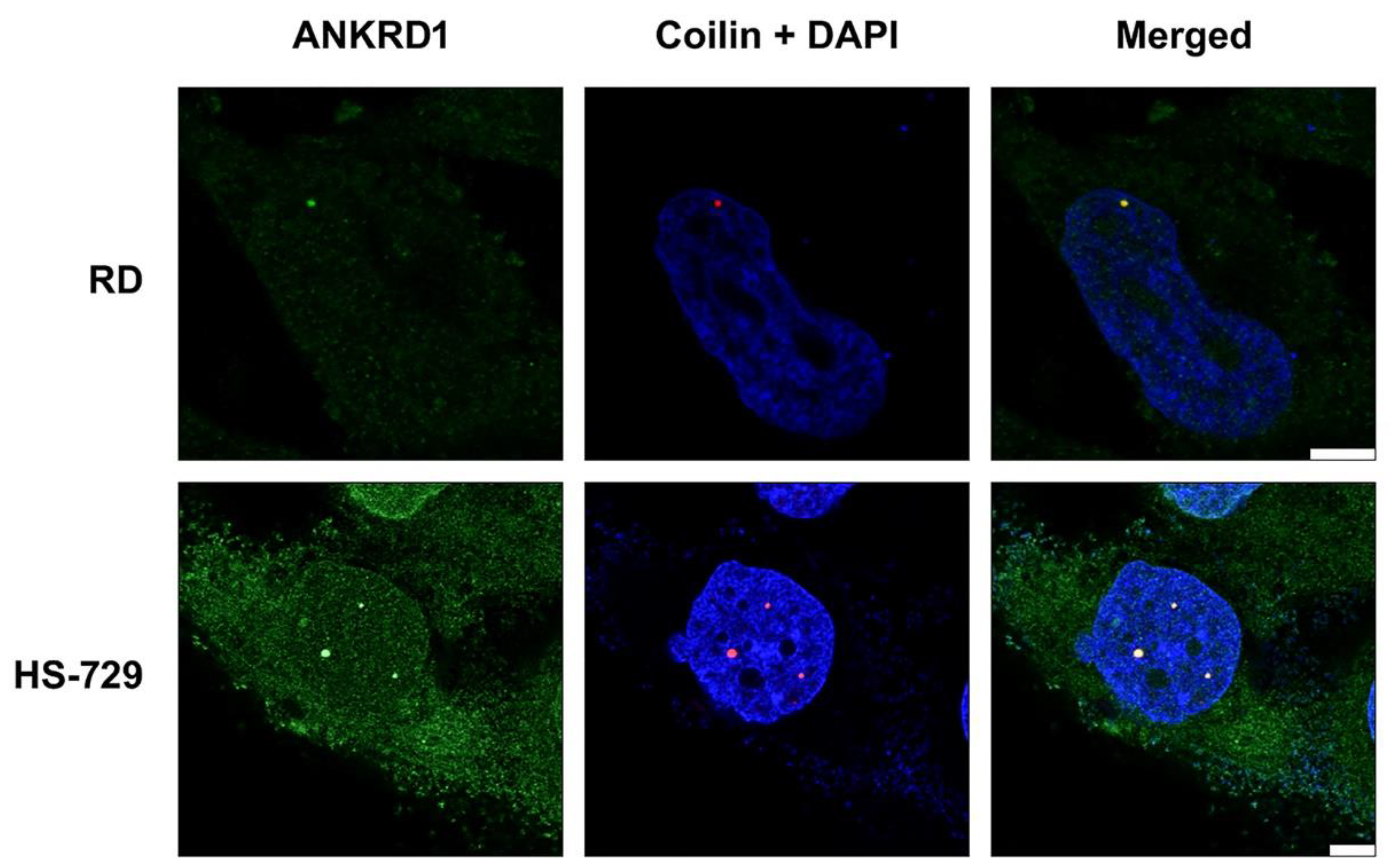
Figure 5.
ANKRD1 protein is prone to proteasomal degradation in RMS cells. Treatment of RMS cells with the 10 µM proteasome inhibitor MG132 for 4 and 8 h resulted in the increase of ANKRD1 protein level in comparison to the vehicle-treated control (-k). The effectiveness of the treatment was confirmed by increased p21 protein level. GAPDH was detected as a loading control. 60 µg of protein extracts was loaded on the gel. One representative out of two independent experiments is shown. Numbers represent the ratio between the values of densitometric analysis of the bands of anti-ANKRD1 and anti-p21 against those of anti-GAPDH.
Figure 5.
ANKRD1 protein is prone to proteasomal degradation in RMS cells. Treatment of RMS cells with the 10 µM proteasome inhibitor MG132 for 4 and 8 h resulted in the increase of ANKRD1 protein level in comparison to the vehicle-treated control (-k). The effectiveness of the treatment was confirmed by increased p21 protein level. GAPDH was detected as a loading control. 60 µg of protein extracts was loaded on the gel. One representative out of two independent experiments is shown. Numbers represent the ratio between the values of densitometric analysis of the bands of anti-ANKRD1 and anti-p21 against those of anti-GAPDH.
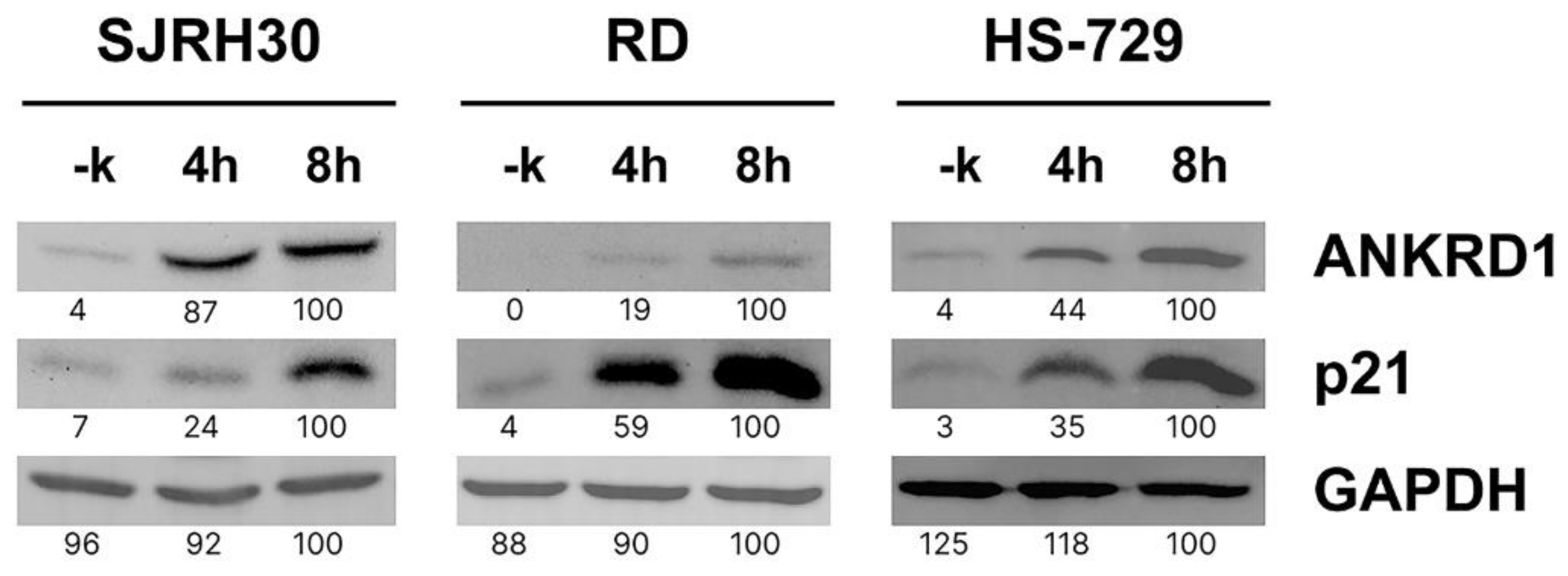

Table 1.
List of primers.
| Gene | Primer sequence (5’-3’) | Direction | Application | Position within ORF (bp) |
|---|---|---|---|---|
| GAPDH | GTGAAGGTCGGAGTCAACG | Forward | RT-PCR | 10-28 |
| GAPDH | TGAGGTCAATGAAGGGGTC | Reverse | RT-PCR | 103-121 |
| ANKRD1 | AGTAGAGGAACTGGTCACTGG | Forward | RT-PCR, Sequencing | 15-35 |
| ANKRD1 | TGGGCTAGAAGTGTCTTCAGAT | Reverse | RT-PCR, Sequencing | 131-152 |
| ANKRD1 | ACGCCAAAGACAGAGAAGGA | Forward | Sequencing | 737-756 |
| ANKRD1 | GCTGCAGCGATGATGGTACTGAAAGTA | Forward | Sequencing, ORF amplification | 1-18 |
| ANKRD1 | TAGGATCCTCAGAATGTAGCTATGCG | Reverse | Sequencing, ORF amplification | 943-960 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



