
Submitted:
06 September 2023
Posted:
11 September 2023
Read the latest preprint version here
Abstract
Virgin Argan Oil (VAO) is extracted from the fruits of the Moroccan endemic species argan [Argania spinosa (L.) Skeels]. It is known for its richness in polyunsaturated fatty acids and its unique composition in tocopherols conferring a panoply of pharmacological properties, documented in several studies. The aim of this study was to investigate the potential protective effect of VAO against oxidative and nitrosative stress induced in cells of the model species Tetrahymena pyriformis. As a comparison, well-known Virgin Olive Oil (VOO) from Olea europaea L. was used instead. Both oils were subjected to a preliminary analysis of phytochemicals and properties of interest. Oxidative stress in T. pyriformis cells was induced by hydrogen peroxide (H2O2) at 350µM, whereas sodium nitroprusside (SNP) nitrosative stress was induced using a 1mM concentration. Neither of these concentrations caused relevant changes in cell viability. The level of reactive oxygen species (ROS) was evaluated in the cell cultures by using H2-DCFDA dye. The activity and cell localization of the antioxidant enzymes catalase (CAT), superoxide dismutase (SOD) and glutathione peroxidase (GPX) was evaluated as well as the levels of glutathione (GSH) and the malondialdehyde (MDA) generated after lipid peroxidation. Finally, the presence and localization of intracellular lipid droplets was assessed by using Nile red. The treatment of cultures with H2O2 produced significant increases in the activity of CAT, SOD and GPX and in the level of GSH, as also did the SNP treatment. MDA level was increased by both the treatments. VAO and VOO treatment were found to protect T. pyriformis from oxidative stress and increased cells defense in nitrosative stress. In another hand VAO and VOO decreased significantly MDA level increased by H2O2 treatment and failed after SNP treatment. The quantification of fluorescence signal obtained from immunolocalization of antioxidant enzymes confirmed the results obtained after the evaluation of their activities. Interestingly the level of ROS and the number of lipid droplets increased by H2O2 treatment was significantly decreased by VAO and VOO co-treatments. VAO and VOO represent strong antioxidants, playing an important role in protecting cells against oxidative stress.
Keywords:
Argania spinosa Skeels (L.)
; oxidative stress
; nitrosative stress
; Tetrahymena pyriformis
; virgin argan oil
; virgin olive oil
1. Introduction
The argan tree [Argania spinosa (L.) Skeels] of the family Sapotaceae is an endemic tree mainly located in the South-Western region of Morocco, where the argan forest covers about 8000 km2 [1]. From the fruit of this specie, berber women extract what is one of the world’s most expensive vegetable oils. Edible argan oil is a cold-pressed oil prepared from the roasted kernels contained in the argan fruits, whereas unroasted kernels are used to prepare argan oil destined for cosmetics, which is used as moisturizing and to repair various skin conditions, slow down the appearance of wrinkles and prevent hair loss [2].
Chemical analysis of argan oil revealed a unique composition in terms of mono-unsaturated and polyunsaturated fatty acids. Unsaturated fatty acids are the major component (80%) of argan oil, principally oleic and linoleic acids (44.8% and 33.7%, respectively) [3]. Unsaponifiable compounds represent a minor fraction of 1% of total oil constituents only. They comprise tocopherols (α, β, δ, and especially γ-tocopherol), phenols (ferulic, syringic, and vanillic acids), sterols (schottenol and spinasterol), carotenoids, triterpene alcohols, xanthophyls, and squalene [4]. These compounds are the reason why argan oil is considered an important and powerful antioxidant source [5,6,7] and an inti-inflammatory agent [8,9]. Moreover, argan oil exerts potential actions on risk factors for cardiovascular diseases, such as hyperlipidemia, hypercholesterolemia, and hypertension [3,10]. In his study, [11] suggest the efficacy of argan oil supplementation in lowering plasma concentrations of total cholesterol, LDL and triglycerides as well as increasing HDL. Indeed, oleic acid is directly responsible for the reduction of blood pressure, through regulation of membrane lipid structure [12].
It is now clear that Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS) contribute to the appearance and/or progression of several human diseases. Oxidative/nitrosative stress can be defined as a disturbance in the pro-oxidant/antioxidant balance in favor of the former, leading to potential damages [13]. These damages are not only the result of the elevation of the level of oxidation. They can also result from failure of repair or neutralization systems [14]. ROS/RNS are products of normal metabolic processes in all aerobic organisms, with mitochondria as the major responsible for most of the reactive oxygen species via oxidative phosphorylation [15]. When ROS and RNS are present at certain physiological concentration, they play important roles in cell signaling and maintenance of the functioning of the body. When excessively produced, and because of their high reactivity with multiple macromolecules (proteins, DNA, lipids, sugars…), ROS/RNS may lead to serious damage in the body. Cells responses to oxidative/nitrosative stress range from prevention of cell division, senescence and necrosis, to apoptosis [16]. According to several studies [17,18,19], the interactions between RNS and ROS promote the apoptotic process by generating powerful oxidants. To cope with the oxidative and nitrosative stress, cells had to develop defense strategies to maintain the intracellular redox homeostasis; these strategies can be divided to two principal mechanisms: enzymatic and non-enzymatic systems [20]. The enzymatic systems involve superoxide dismutase (SOD, considered as the primary defense of cells against the oxidative injury caused by superoxide) [21], catalase and glutathione peroxidase, which hydrolyze H2O2 to H2O and O2. Non-enzymatic defenses include redox-active cellular low-molecular-mass compounds such as glutathione (GSH), flavonoids, carotenoids, thiols and the radical-scavenging vitamins E and C [22]. These antioxidants can be synthesized in vivo or taken in from the diet [23]. Thus, several edible oils have demonstrated health-promoting properties due to their antioxidant capacity, which is transferred to humans and animal systems through their intake. Such properties are highly remarkable, and have been widely reported in the virgin olive oil [24,25,26].
In order to investigate the harmful effect of oxidative/nitrosative stress many type of cells and animals have been used as models: mice [27,28,29], rats [30,31] and even humans in clinical studies [32,33]. Simple biological organisms like the protozoa T. pyriformis have been extensively used as model organisms in biomedical research for decades for its many advantages especially its ability to grow on simple media, cost-effective laboratory handling and also short generation time of about 2 hours [34], which has made of it a model of choice for in vitro rapid bioassays. The purpose of the present study was to investigate the potential beneficial effects of argan oil on preventing damages caused by oxidative/nitrosative stress induced on T. pyriformis. For this purpose, olive virgin oil (VOO) was used in most experiments in a parallel way, as a comparison.
2. Materials and Methods
2.1. Preparation of oil extracts
Edible virgin argan oil (VAO) and virgin olive oil (VOO) were provided by associations in south Morocco, who used traditional processes for oils extraction.
The oils were exposed to methanol to separate the polar and non-polar fraction following the method described by [35]. For this purpose, mixtures of methanol:oil (1:1) were prepared and vigorously stirred 1 h and then centrifuged at 2500 g for 5 m in order to separate the two phases: the methanolic fraction (MF) and the lipidic fraction (LF). Whole oils (total fraction: TO) were also used in the study to compare the different fractions with the non-fractioned oils.
2.2. Determination of total phenolics content (TPC)
The content of phenolic compounds in the different extracts was estimated using the Folin-Ciocalteu method according to [36]. Briefly, 2.5 mL of Folin’s reagent (diluted 10 times) was added to 500 μL of the samples corresponding to the MFs, LFs and TOs, and to the standards (prepared in methanol) with suitable dilutions, together with 2 mL of sodium carbonate (7.5%). After 15 minutes of incubation in a water bath at 45 °C, the absorbance was measured at 765 nm against a blank without extract. The quantification of total polyphenols was calculated from the regression equation of the calibration range established with gallic acid under the same conditions as the sample. The results were expressed in mg of gallic acid equivalent per gram of dry weight of plant material (mg EAG/g of dry weight).
2.3. Determination of total flavonoids content (TFC)
The determination of total flavonoids was carried out by the colorimetric method of aluminum chloride described by [37]. The concentration of flavonoids was deduced from a calibration curve established with quercetin; results were expressed as micrograms of quercetin equivalent per gram of dry weight of plant material (mgEQ/g of dry weight).
2.4. Determinations of antioxidant capacity
2.4.1. DPPH radical scavenging assay
In order to evaluate the anti-radical activity of the MFs, LFs fractions and TOs, the method of 2,2-diphenyl-1-picrylhydrazyl radical (DPPH•) was used according to the protocol described by [38]. Samples (0.3 mL) were mixed with a solution of 0.2 mM DPPH in methanol (2.7 mL). The mixture was shaken vigorously and allowed to stand for 1 h before the absorbance was measured at 517 nm. Radical-scavenging activity was calculated as the following percentage: [(As-Ai)/As]×100 (As = absorbance of DPPH alone, Ai = absorbance of DPPH in the presence of MFs, LFs or TOs). A calibration curve generated using ascorbic acid was used as the reference.
2.4.2. Ferric reducing antioxidant power (FRAP)
The ferric reducing ability was determined according to the method described by [39]. FRAP is based on the reduction at low pH of a colorless ferric complex (Fe3+-tripyridyl-triazine) to a blue-colored ferrous complex (Fe2+-tripyridyl-triazine) by the antioxidants. One milliliter of the MFs, LFs or TOs were mixed with 2.5 mL sodium phosphate buffer (0.2 M, pH 6.6) and 2.5 mL of 1% potassium ferricyanide (K3Fe(CN)6) solution. The mixture was incubated at 50 °C for 20 m. Then, trichloroacetic acid (10%, 2.5 mL) was added to the mixture. Afterwards, 2.5 mL of this solution was mixed with 2.5 mL distilled water and 0.5 mL of 0.1% FeCl3, and the absorbance was followed at 700 nm. Ascorbic acid was used in order to generate a calibration curve. Results were expressed as mg ascorbic acid equivalent per gram of dry weight of plant material.
2.4.3. ABTS radical scavenging assay
The ABTS test was measured using the decolorization assay described by [40]. This method is based on the ability of antioxidant molecules to quench the stable bisradical cation [2,2′-azinobis (3-ethylbenzothiazoline-6-sulfonic acid) diammonium] ABTS•+. In this study, the ABTS•+ solution was prepared by reaction of a 7 mM aqueous ABTS•+ solution and 2.45 mM potassium-persulfate solution. After storage in the dark for 16 h, the radical cation solution was further diluted with 5 mM phosphate-buffered saline (PBS, pH 7.4) until the initial absorbance value of 0.7 ± 0.05 at 734 nm was reached. Then, 160 μL of ABTS⋅+ solution was added to 40 μL of MFs, LFs fractions or TOs. Absorbance was measured at 734 nm after 10 min and ABTS radical scavenging activity (%) for each concentration relative to a blank absorbance was calculated using the following equation:
where A control represents the initial concentration of the ABTS⋅+ and A sample is the absorbance of the remaining concentration of ABTS⋅+ in the presence of the sample. Trolox was used as a positive control.
ABTS (%) = (A control-A sample)/(A control) × 100
2.5. Cell culture
Ciliate Tetrahymena pyriformis, wild strain (E, ATCC 30005), was used in this study. Tetrahymena pyriformis cells were aseptically cultured at 25 oC in a PPYG medium [1.5% (w/v) peptone, 0.25% (w/v) yeast extract and 0.2% (w/v)] glucose supplemented with penicillin G (100 U/mL) and streptomycin (100 µg/mL) to prevent bacterial growth in the culture medium. Log-phase culture was used for the different experiments. The cells were used in a final concentration of 1×104. cells/mL.
2.6. Cell treatments and viability assay
Cells were maintained in a PPYG medium supplemented with a panel of concentrations of H2O2 (50, 150, 250, 350, 450 and 550 µM) and sodium nitroprusside (SNP) (0.4, 0.6, 0.8, 1.0, 1.2 and 1.4 mM) in order to determine the 50% inhibitory concentrations (IC50). To evaluate the protective effects of olive and argan oil against hydrogen peroxide- and SNP-induced cytotoxicity, the cultures of T. pyriformis cells were supplemented with non-toxic concentrations of each one of the oils dissolved in ethanol 0.15% at different concentrations [0.25, 0.5, 0.75, 1.25 and 1.5% (v/v) oil in ethanol 0.15%]. Cells treated with ethanol only (0.15%) were used as the control. To determine the adequate concentration of ethanol used as the vehicle for oils (0.15%), prior experiments were carried out by adding ethanol at different concentrations (0.1, 0.15, 0.3, 0.6, 1.2 and 2.4%] to the culture medium, and assessing cell viability. All treatments were maintained for 24h.
In order to assess T. pyriformis viability, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test was performed according to the protocol described by [41]. Cells were incubated with MTT at a final concentration of 0.05 mg/ml for 3 hours at 37 °C. Afterwards, the cell suspension was centrifuged at 4000 g for 5 min, the supernatant was discarded and the formazan crystals in the pellet were dissolved by the addition of 500 µL of DMSO. The optical densities were revealed by spectrophotometry at 540 nm. The viability of T. pyriformis cells was calculated as percentage compared to the control following the formula: % Viability = (OD experiment/OD control) × 100%.
2.7. Localization and quantification of cellular ROS by Fluorescence Microscopy (FM)
After 24 hours of treatment, T. pyriformis cells were collected by centrifugation at 4000 g for 10 min then incubated with DCFH2-DA (2’,7’-dichlorodihydrofluorescein diacetate) (Calbiochem) to a final concentration of 10µM prepared in MES (2-[N-morpholino] ethanesulfonic acid)-KCl buffer (5 µM KCl, 50 µM CaCl2, 10 mM MES, pH 6.15) for 30 min at 37 °C. Cells were subjected to a wash step in fresh buffer for 15 min, stained with Hoechst dye and then imaged in a Nikon Eclipse Ti-U inverted microscope fitted with a CoolLed pI-300White epifluorescence excitation device, by using sequentially the ultraviolet and then the blue ranges of excitation. Negative controls were treated with MES-KCl buffer only. The intensity of the green fluorescence in each cell was quantified by image J software using cells treated as above but omitting the fluorophore as the control. Both average and standard deviation were calculated after measurement of a minimum of three images containing multiple cells, corresponding to three independent experiments.
2.8. Detection of intracellular lipid droplets by Nile Red staining
Nile Red is a fluorescent probe used to reveal intracellular neutral lipid accumulation in the form of lipid droplets. After 24h treatment, T. pyriformis cells were collected by centrifugation at 4000 g for 10 min, washed twice with PBS to eliminate the culture medium residues, and then fixed with 3% formaldehyde in PBS for 30 min. After three washes with PBS cells were incubated with Nile red dye at a final concentration of 10 µg/ml for 30min at room temperature in the dark [42]. Afterwards, cells were washed in fresh buffer, stained with Hoechst dye and imaged in a Nikon Eclipse Ti-U inverted microscope fitted with a CoolLed pI-300White epifluorescence excitation device, by using sequentially the ultraviolet and then the green ranges of excitation. The intensity of the green fluorescence in each cell was quantified by image J software using cells treated as above but omitting the fluorophore as the control. Both average and standard deviation were calculated after measurement of a minimum of three images containing multiple cells, corresponding to three independent experiments.
2.9. Preparation of cell extracts
T. pyriformis cells were harvested by centrifugation at 4000 g for 10 min at 4 °C, and then washed before been suspended in 1 mL of phosphate buffer (50 mM, pH 7.4). The mixture was homogenized using an ULTRA TURRAX homogenizer (Sigma) for 20 min at 4 °C followed by sonication of the sample (80 W, 60s). The homogenate was then centrifuged at 4000 g for 10 min at 4 °C. The supernatant obtained represents the crude protein extract.
2.10. Protein assay
The protein content was measured according to the method described by [43], using bovine serum albumin as standard.
2.11. Total superoxide dismutase (SOD) activity measurement
The activity of SOD was quantified according to the method of [44]. This method involves the inhibition of the photochemical reduction of nitroblue tetrazolium (NBT) in the presence of riboflavin. One unit of SOD activity is defined as the amount of enzyme required to inhibit NBT reduction by 50%. Briefly, 3 mL of the reaction mixture contained 50 mM sodium phosphate (pH 7.8), 13 mM methionine, 75 mM NBT, 2 mM riboflavin, 100 mM EDTA, and 200 mL of the enzyme extract. The change in absorbance of the sample was then recorded at 560 nm after the production of blue formazan.
2.12. Catalase (CAT) activity measurement
Catalase activity was determined by measuring the kinetics of H2O2 disappearance at 240 nm according to the method described by [45]. In a 3 mL quartz tank, 0.05 mL of the homogenate was added to 1.95 mL of 50 mM phosphate buffer and 1 mL of H2O2 (0.019 M). The optical density was followed at 240 nm. The catalase activity was expressed as µmol/min/mg of protein.
2.13. Glutathione peroxidase (GPX) activity measurement
The activity of glutathione peroxidase (GPX) was determined using the method of [46]. Briefly, 1 mL of the homogenate, 0.2 mL of phosphate buffer (0.1 M pH 7.4), 0.2 mL of GSH (4 mM) and 0.4 mL of H2O2 (5 mM) was incubated at 37 °C for 1 min and the reaction stopped by the addition of 0.5 mL TCA (5%, w/v). After centrifugation at 1500g for 5 min, aliquot (0.2 mL) from supernatant was mixed with 0.5 mL of phosphate buffer (0.1 M pH 7.4) and 0.5 mL DTNB (10 mM) and absorbance recorded at 412 nm. GPX activity was expressed as μmol of GSH consumed/min/mg protein.
2.14. Determination of the level of glutathione (GSH)
Glutathione level was evaluated based on the method described by [47]. The reaction mixture contained 200μL of TCA (5%) and 400 μL of the protein extract. After centrifugation at 12,000 g for 10 minutes, 50 μL of the supernatant was removed and added to 100 μL of DTNB (6 mM) and to 850μL phosphate buffer (50 mM, pH 8). The optical density was measured after 5 minutes at 412 nm. Results of glutathione assay were expressed as µmol/mg of protein.
2.15. Lipid peroxidation measurement
Lipid peroxidation was determined by the measurement of malondialdehyde (MDA) generated by the thiobarbituric acid (TBA) reaction as described by [48]. For analysis, 0.5 ml of the homogenate was mixed with 0.5 ml of trichloroacetic acid (TCA, 20%) and 1 ml of thiobarbituric acid (TBA, 0.67%), and heated at 100 °C for 15 minutes. After cooling the mixture, 4 ml of n-butanol was added and a centrifugation at 3000 g for 15 minutes was performed. The absorbance of the upper phase containing the pink pigment was read at 532 nm. MDA levels were determined by using an extinction coefficient for MDA–TBA complex of 1.56×105 M-1 cm-1, and expressed as µmol/mg of protein.
2.16. FM Immunolocalization of SOD, CAT and GPX
After treatments for 24 hours, cells were collected by centrifugation at 4000 g for 10 min at 4 °C and washed twice with HEPES buffer, then fixed with 4% (w/v) paraformaldehyde and 0.2% (v/v) glutaraldehyde diluted in HEPES buffer before being embedded in agarose (6%). After that, cells were re-suspended in cacodylate buffer (0.03M, pH 7.2) in ice and washed three times 30 min each. Samples were dehydrated in ethanol series and embedded in Unicryl resin at −20 °C using ultraviolet light. Semithin sections were obtained with a Reichert-Jung Ultracut E microtome using a glass knife. Sections were placed on Biobond-coated slides and used for inmunolocalization of superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX). Cells permeabilization was performed by incubating cells with PBS-Triton X 100 (0.1%), then slides were washed with PBS three times 5 min each. Afterwards slides were incubated with 1% BSA, 22.52 mg/mL glycine in PBST (PBS+ 0.1% Tween 20) for 30 min to block unspecific binding of the antibodies. Slides were then incubated with one of the following antibodies: rabbit anti-SOD primary antibody (Agrisera) solution (1:1000 dilution in blocking buffer), rabbit anti-CAT primary antibody (Agrisera) solution (1:1000 dilution in blocking buffer), or rabbit anti-GPX primary antibody (Agrisera) solution (1:1500 dilution in blocking buffer) in a humidified chamber for 1 h at room temperature or overnight at 4 °C. The slides were washed three times in PBS for 5 min. The secondary antibody (1:1000 solution of goat anti-rabbit IgG antibody conjugated to DyLight 488) (Agrisera) was added in 1% BSA for 1h at room temperature in the dark and washed three times with PBS for 5 min. The slides were then stained with Hoechst dye and then imaged in a Nikon Eclipse Ti-U inverted microscope fitted with a CoolLed pI-300White epifluorescence excitation device, by sequentially using the ultraviolet and then the blue ranges of excitation. Negative control sections were treated as above but using normal rabbit preimmune serum instead of the different antibodies.
2.17. Statistical Analysis
Data were statistically analyzed with GraphPad Prism 8.0 soft-ware (GraphPad Software, San Diego, CA, USA). They were expressed as mean values ± standard deviations (SD) from at least three separate experiments and compared with an ANOVA test followed by a Tukey’s test, which provide multiple comparisons and allows to assess any interactions. MTT test and data from phytochemical screening tests were compared with a student’s t-test. A p-value less than 0.05 was considered statistically significant.
3. Results
3.1. Total polyphenols and flavonoids content
Table 1 shows several quantitative data regarding chemical characteristics of the oils analyzed. Significant differences between the MF and LF fractions extracted from VAO and VOO, oils and the non-fractioned oils (TO) were detected. In terms of total polyphenol content, the values ranged from 80.64 ± 1.32 to 28.31 ± 4.6 mg EAG/g of oil). We noticed that MF always displayed the highest value when compared to LF and TO, and this for the two oils, with the MF of VOO showing the highest value, followed by the MF of VAO. Table 1 also show similar trends for LFs and TOs than with MFs, with the LF and TO of VOO, significantly higher than LF and TO of VAO.
3.2. Antioxidant activity and free radical scavenging capacity
The antioxidant activity and free radical scavenging capacities of the MF, LF and TF of VOO and CO were evaluated using the DPPH, ABTS and FRAP assay. The results varied from an oil to another and among the fractions in each oil (Table 1).
3.2.1. DPPH• radical scavenging activity
DPPH• radical scavenging activity was quantified in terms of percentage inhibition of a pre-formed free radical by antioxidants in each sample (Table 1). The percentage inhibition of the DPPH• radical by the oils and oils fractions varied from 55.35%± 1.71 to 31.67% ± 0.62. The MF of VOO showed the highest value, followed by TO from the same oil, both inhibiting the radical over the half. In the next places came the MF and TF of VAO that exhibited values of inhibition significantly lower than the MF and TF of VOO, respectively.
3.2.2. FRAP assay
In order to determine the ferric reducing activity of our samples the FRAP assay was performed. The results (Table 1) show a variation between the oils and oil fractions in term of ferric reducing capacities. VAO MF and TF showed ferric reducing activity very close to those of MF and TF of VOO (less than 8% and 9.5% inferior, respectively). Oppositely, ferric reducing activity of VAO LF was even slightly higher than that of VOO LF (1% higher).
3.2.3. ABTS•+ radical cation scavenging activity
In ABTS assay the antioxidant activity of the oil fractions was quantified in terms of percentage inhibition of the ABTS•+ radical cations by antioxidants in each sample (Table 1). Within the oils and oils fractions the TF of VAO fraction displayed the highest ABTS•+ scavenging capacity, that reached (55.09%) compared to the TF of VOO. Oppositely, the MF fraction of VAO showed a significant decrease (39.26%) compared to the MF of VOO.
3.2.4. Determination of potential cytotoxic effects of the treatments
In order to determine the potential cytotoxicity of H2O2, SNP, ethanol and the two oils (VAO, VOO) on T. pyriformis, MTT viability tests were performed after 24h incubation with different concentrations of the components, and the results were expressed as a percentage of the viability displayed by a control culture (untreated) (Figure 1). Viability of T. pyriformis cells treated with H2O2 produced statistically relevant reductions of viability even at low concentrations like 50 µM. Viability was reduced by 50% when a concentration of 350µM H2O2 was used, and reached a minimum of 12.88% at the highest concentration tested (550µM) (Figure 1A).
SNP (Figure 1B) lowered significantly viability starting at a concentration at 0.8 mM. Such reduction reached 50% at 1mM, whilst the highest concentration tested (1.4mM SNP) allowed 14.32% viability only. Ethanol (Figure 1C), although significantly lowered viability at concentrations higher than 1.2% (v/v) (82.87% viability), was used in the present study at much lower concentrations (0.15%) as a vehicle for oils. At such concentration, viability was not significantly different than untreated controls.
The treatment with the two oils (Figure 1D–F) had no pronounced effect on viability even reaching 1% concentration. Nevertheless, VOO stimulated viability at low concentration over the control (112.23%). High viability levels were maintained in the cultures (e.g., 66% in the case of VOO) even with a concentration of 1.5% VOO. These results suggest that VAO and VOO at low concentrations had no toxic effects on T. pyriformis cells.
3.2.5. Fluorescence detection of ROS by H2-DCFDA in challenged cultures
To assess the putative anti-oxidative activity of VAO and VOO, we examined the generation of intracellular ROS by using a H2-DCFDA staining assay of T. pyriformis cells in culture. Treatments used include VOO, VAO and the combined treatment VOO-H2O2 and VAO-H2O2 (Figure 2A,C). The presence of green fluorescence revealed the cellular distribution of ROS in a quantitative manner. ROS were broadly present in most cellular compartments (Figure 2B). One-way ANOVA multiple comparison Tukey test analysis of FM quantification data showed that the treatment with H2O2 (Figure 2C) significantly increased ROS generation by control cells. Also, the treatment with either VOO or VAO significantly lowered the amounts of ROS generated by the cells in comparison with the control cells, independently of whether H2O2 was added to the culture (Figure 2C). No statistical differences were detected between the effects of VOO or VAO themselves.
3.2.6. Effects of oxidative treatments on the presence of lipids and lipid droplets
Nile red (9-diethylamino-5H-benzo[a]phenoxazine-5-one), has been widely used as a vital stain for the detection of intracellular lipids and lipid droplets/lipid bodies by fluorescence microscopy and flow cytofluorometry. We report here the use of Nile red as a rapid and inexpensive method to estimate cellular lipids of T. pyriformis by the direct application of the dye to fixed cells without any extraction or purification.
To assess the potential effects of the different components assayed here on the presence of lipid components in T. pyriformis cells in culture, we used Nile red after the oxidative induction treatments, which included the combined treatment VOO-H2O2 and VAO-H2O2, as well as additional controls consisting of VOO and VAO oils (Figure 3A). FM observations show both diffuse orange fluorescence in the cytoplasm of T. pyriformis cells, as well as abundant discrete signals corresponding to lipid droplets (Figure 3B). Statistical analysis of fluorescence intensity data showed that the treatment with H2O2 significantly increased overall lipid staining in cells treated with H2O2 alone, in comparison with the control group (Figure 3C). When VAO or VOO were added to the culture medium, the intensity of green fluorescence remained, or was restored to levels statistically similar to that of control cultured cells in the absence of H2O2, independently of whether the oils were co-incubated or not with H2O2. The effect of VAO was even stronger than that of VOO, as fluorescence with VOO in the presence of H2O2 did not statistically differ of the control cells treated with H2O2 (Figure 3C).
3.2.7. Effects of nitrosative treatments on the presence of lipids and lipid droplets
Nile red was used again to assess the potential effects of nitrosative induction treatments, which included the combined treatment VOO-SNP and VAO-SNP, as well as additional controls consisting of VOO and VAO oils (Figure 4A). As shown before, FM observations show both diffuse orange fluorescence in the cytoplasm of T. pyriformis cells, as well as abundant discrete signals corresponding to lipid droplets (Figure 4B). Statistical analysis of fluorescence intensity data show that the treatments with SNP, or with VOO or VAO oils either alone or in combination with SNP do not significantly alter overall lipid staining of control cells (Figure 4C). However, cells treated with VOO and SNP display a significantly higher level of fluorescence than cells treated just with VOO. An analogous relationship was detected after comparing cells treated with VAO in combination SNP and just VAO (Figure 4C).
3.2.8. Effect of oxidative stress treatment on the activity of key antioxidant enzymes and substrates
The first line of defense against oxidative stress include different enzymes like superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX), which represent key factors in cell defense strategy against oxidation process, together with antioxidants like glutathione. Also, assessment of oxidation products like malondialdehyde may help to identify the effects of the treatments on the T. pyriformis cells.
Figure 5 shows the effects of VOO and VAO on the specific activities of several antioxidant defense enzymes in H2O2-stimulated T. pyriformis cells. Treatment with 350 µM of H2O2 significantly increased enzyme activities like SOD (1470.35±220.21 vs. 3110.82±158.88 IU/mg of proteins p≤0.02), catalase (1592.29±432.47 vs. 669.19±168.35 IU/mg of proteins p≤0.05), and GPX (56.16±2.59 vs. 17.30±0.16 IU/mg of proteins p≤0.002). GSH content was also significantly increased (56.80±1.95 vs. 19.49±0.25 µmoles/mg of protein p≤0.001), as well as the presence of MDA (209.76±8.51 vs. 41.88±3.71 µmoles/mg of protein p≤0.001) in control cells treated with H2O2 alone compared to untreated control cells. Noteworthy, the treatment of the cells with either VOO or VAO significantly lowered the enhancement of the activity of the enzymes SOD, CAT and GPX and the levels of GSH and MDA caused by H2O2, which became again statistically similar (with the exception of catalase activity), to the basal levels displayed by control cells without the treatment with H2O2. In most cases, the treatment with the oils further diminished the level of antioxidant activity or the presence of GSH or MDA to levels significantly lower than the basal ones present in untreated control cells. Several small differences were detected between VOO and VAO in their capacity of eliciting these effects (Figure 5).
3.2.9. Effect of nitrosative stress treatment on the activity of key antioxidant enzymes and substrates
Different results were observed when cells were treated with the nitric oxide (NO) donor, sodium nitroprusside (SNP) instead of H2O2. The results (Figure 6) show that SNP induces a decrease in catalase (230,27±33.31 vs. 669.191±168.35 IU/mg of protein p≤0.03), SOD (749.94±96.11 vs. 1470.35±220.21 IU/mg of protein p≤0.01) and GPX (6.25±0.11 vs. 17.30±0.16 IU/mg of protein p≤0.001) activities, as well as in GSH (6.03±0.19 vs. 19.49±0.25 µmol/mg of protein p≤ 0.0006) and MDA. The cotreatment SNP-VOO and SNP-VAO increased significantly the level of SOD (332 and 322.51% p≤0.004) compared to SNP-treated group, whereas oils cotreatments conveyed no significant effect on catalase rate. Finally, the treatment SNP-VOO exhibited a significant increase in GPX level, (205.72% p≤0.03) compared to SNP-treated group.
3.2.10. Immunocytochemical evidence of the effects of oxidative and nitrosative stress treatments on the presence of key antioxidant enzymes
In order to determine whether the changes observed in the activity of the three key antioxidant enzymes SOD, CAT and GPX may become a consequence of a divergence in the level or localization of such enzymes, immunolocalization of the three enzymes was performed in the cultures using FM immunofluorescence approaches.
Conspicuous changes in the level of fluorescence occur concomitantly with the treatments. Thus, cell treatment with H2O2 produced a significant increase of the presence of SOD enzyme of 367.27% (p≤0.004), which becomes restored to a level similar to that of the H2O2-untreated cells when H2O2 was added to the culture concomitantly with either VOO or VAO (Figure 7A,C). Treatment with SNP itself did not alter significantly the level of SOD enzyme (Figure 7A,D); however, the joint treatment of SNP with either VOO or VAO significantly enhanced the levels of the immunodetected SOD compared to the control untreated cells, and compared to the cultures treated with the oils (VOO or VAO) individually added to the culture, which did not alter the levels of SOD in comparison with the control untreated cells. (Figure 7A,C,D). Significant difference was detected in the effect of the addition of VAO to the cultures compared with the addition of VOO regarding the levels of immunodetected CAT (Figure 8D).
The treatments with H2O2, SNP and VOO and VAO produced significant changes in the presence of the enzyme in the T. pyriformis cells. Similarly to SOD, cell treatment with H2O2 produced a significant increase of the presence of CAT enzyme of 215.5% p≤0.03, which became restored to a level similar to that of the H2O2-untreated cells when H2O2 was added to the culture concomitantly with either VOO or VAO (Figure 8A,C). Treatment with SNP itself did not alter significantly the level of SOD enzyme (Figure 8A,D). In this case, the joint treatment of SNP with either VOO or VAO did not modify the levels of the immunodetected CAT compared to the control untreated cells, as neither did the treatments of the cultures with the oils (VOO or VAO) individually added to the culture. (Fig 8A, C-D). Significant difference was detected in the effect of the addition of VAO to the cultures compared with the addition of VOO regarding the levels of immunodetected CAT (Figure 8D).
Several of the treatments assayed produced significant changes in the presence of the GPX enzyme in the T. pyriformis cells. Cell treatment with H2O2 produced a significant increase of the presence of GPX enzyme of 365.31 p≤0.03, which became restored to a level similar to that of the H2O2-untreated cells when H2O2 was added to the culture concomitantly with VOO (Figure 9A,C). Such restoration, although visible was not statistically significant for H2O2 when cultured concomitantly with VAO (Figure 9A,C). Treatment with SNP itself did not alter significantly the level of GPX enzyme (Figure 9A,D). In this case, the joint treatment of SNP with VOO did modify the levels of the immunodetected GPX compared to the control untreated cells, but this effect was not observed with the joint treatment of SNP with VAO. Our observations also detected that VAO treatment joint with H2O2 was statistically different to the VOO treatment joint with H2O2 regarding the levels of immunodetected CAT (Figure 9D).
4. Discussion
Overall, our data concerning the chemical composition of the oils used here are in good agreement with those present in the literature available. Thus, phenolic total amount of olive oil varies from 100 to 1000 mg/Kg oil [49,50]. They are also consistent with those observations made by [51,52], regarding virgin argan oil, which presents lower polyphenol content than virgin olive oil but a higher content than other edible vegetable oils.
For flavonoids determination assay, differences in the amount of these compounds were detected between the methanolic fractions of VOO, which were significantly higher than those of VAO and TF. This profile was similar in the case of flavonoids present in the TF fractions of the two oils. However, such differences where less relevant in the flavonoids present in the LF fractions, which were higher in VOO than in VAO and TF, but were only statistically significant in the later. Phenolic total amount and flavonoids composition of oils varies depending on cultivars, place of origin, extraction methods and storage conditions [53,54].
Both oils used in the present work have a good antioxidant capacity, based on the different determinations carried out, well within the figures described in the literature. Thus, the results of the ABTS radical scavenging assay fall well within the range of 0.19 and 0.87 mmol Trolox/kg described by [51] as regards to DPPH anti radical scavenging capacity for argan virgin oils. FRAP is basically a hydrophilic antioxidant assay that does not respond well to lipophilic antioxidants [55], therefore explaining the results obtained with the LF fractions of the oils. Enhanced radical scavenging effect of VAO observed here can be explained by its high total tocopherol content that in argan oils have been described to range from 427.0 mg/kg to 654.0 mg/kg [4,52,56]. The two oils used here are therefore representative of these oils.
Our results show that the addition of the oils may help to control or mitigate oxidative stress conditions artificially triggered in T. pyriformis cell cultures at physiological levels not limiting cell viability. Such effects were assessed in a large number of key parameters. Thus, H2O2 is detected by the H2-DCFDA fluorochrome, and also promotes generation of other ROS which cause oxidative stress and cytotoxicity and may lead to cell death [57,58,59,60]. The ROS scavenging effect observed here after concomitant treatment with VOO and VAO likely reflects the effects of the presence of antioxidant components in both oils. Thus, olive oil contains numerous antioxidant components, which include an unsaponifiable fraction, monounsaturated and polyunsaturated fatty acids, and a fraction composed of natural antioxidants such as carotenoids, phytosterols, flavonoids, α-tocopherol, and other phenolic compounds (reviewed by [61]. These authors indicate that olive tree polyphenols are major antioxidant components in VOO, and include flavonols, lignans and glycosides in particular. Olive glycosides are iridoids (monoterpenes derived from geraniol), whose cleavage produces secoiridoids and iridoid derivatives, which are mainly found in glycated form in the olive, as also in other Oleaceae plants and in general in the plant subclass Asteridae [62].
Several of these secoiridoids display slight additional chemical modifications and receive specific names, like the most abundant one in VOO (oleuropein in its glucose-bound form), elenolic acid, oleacein, oleocanthal, or ligstroside aglycone. Additional polyphenols include verbascoside, the caffeoylrhamnosylglucoside of HT, 1-acetoxypinoresinol and pinoresinol (two lignans) [61,63,64].
Referred to VAO, unique composition and properties is characterized by its richness in minor compounds such as tocopherols, sterols (schottenol and spinasterol), phenols (ferulic, syringic and vanillic acid) and vitamin E, with potential ROS scavenger properties [65,66,67]. Moreover, there is a large number of studies providing evidence that the radical scavenging activity of VOO is strictly correlated with its polyphenol content [68,69,70,71].
A previous work [72] describes several effects of argan oil on T. pyriformis cultures treated with H2O2, which include several markers in common with the present one. The present work additionally stablishes a valuable comparison between both oils (VAO and VOO), with different origins and well valuated in the food marker.
Additional markers have been assessed in our present work. Thus, the use of fluorescence detection of ROS by H2-DCFDA in challenged cultures of T. pyriformis represents a quick and simple procedure to identify the presence of ROS (mainly H2O2) in numerous biological systems [73]. Fluorescence measurements are sensitive enough to clearly make visible the changes in ROS concentrations either as increment of fluorescence in response to the H2O2 treatment, or as lowered fluorescence generated in the cells after co-cultivation with both oils.
Fluorescence labeling of lipids and lipid bodies by Nile red was also able to dissect the response of the cultures to the different conditions assayed. Lipid droplets consist of a core of neutral lipid, including triacylglycerol and cholesteryl ester, surrounded by a phospholipid monolayer [74]. We report here the use of Nile red as a rapid and inexpensive method to estimate cellular lipids of T. pyriformis by the direct application of the dye to fixed cells without any extraction or purification. Oxidative stress trigger intracellular lipid droplets accumulation [75,76,77]. Lipid droplets may serve as energy reservoirs, sources of lipid for membrane biosynthesis, or storage sites for potentially toxic lipid species [78]. In this work, quantification of Nile red fluorescence allows to identify the balancing effects of both oils on the increased fluorescence caused by H2O2, and is even able to identify differences between both oils.
Complementary to the previous work on T. pyriformis carried out by [72], the present work also explores the effects of nitrosative stress on T. pyriformis cultures, by testing a broad range of concentrations of the NO donor sodium nitroprusside (SNP) (0.4, 0.6, 0.8, 1.0, 1.2 and 1.4 mM). NO is a free radical indispensable for cell signaling [79], that when produced at high concentrations can be reduced or oxidized and thereby leads to nitrosative stress. SNP had been used by many authors to induce nitrosative stress in different cell models [80,81,82].
The use of nitrosative stress conditions in the cultures, also produced an increase in the levels of fluorescence by Nile red, similar to that observed when oxidative stress conditions were applied. Such increase could be caused by the presence of cooperative (oxidative/nitrosative) pathways, However, the fact that co-cultivation with neither VOO nor VAO was unable to revert such increase of lipids, and that both co-incubations even increased the level of Nile red fluorescence account for the presence of a different mechanism, not so far identified clearly in the literature according to our knowledge. In a human T-lymphocyte cell line (Jurkat), palmitic acid (PA) treatment has been shown to increase neutral lipid content in a dose-dependent manner, as assessed by Nile Red [83]. The added PA was incorporated into all lipid classes analyzed (triglycerides, cholesterol ester, phospholipids, and free fatty acids). The authors of this study additionally observed that the PA treatment resulted in nitrosative stress by inducing the production of reactive nitrogen species (RNS), for example, NO. A similar effect might be triggered here by the addition of the oils to the cultures, as they contain certain amounts of PA. Further analyses should be necessary in order to dissect the nature of the induced effects in this case.
As we have seen here, and has been widely reported, H2O2 is an agent known for its capacity to induce oxidative stress in T. pyriformis and many other cell model [84,85,86,87]. The elevation of the activity of antioxidant enzymes may be observed here as a sign of the existence of strong oxidative processes that the cells try to contest by the increase in CAT, SOD and GPX activities. These results are in accordance with those of [72,88] and also with [89] in the case of the enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH, EC 1.2.1.12). Interestingly, this effect was significantly and completely reversed by the co-treatments with VAO and VOO. Such oils played an important role in enhancing cells ability to cope with the oxidative stress induced by H2O2 through the presence of a high level of antioxidant molecules in their composition. Their effect was reflected here by the significant decrease of the activity and level of the different enzymes and the GSH and MDA rate in the co-treated groups. Such VAO and VOO capacities in monitoring CAT, SOD and GPX activities under oxidative stress have been widely demonstrated in other studies carried out in rats and humans [90,91,92].
The induction of nitrosative stress by SNP produced an increase in the activities of the antioxidant enzymes analyzed here (CAT, SOD, GPX) as well as in the levels of GSH and MDA. Again, it is difficult to attribute this effect to a particular mechanism. There is a biologically active interplay between ROS and RNS signals that modulates cellular responses to environmental stimuli. Several authors have described a number of candidate mechanisms through which ROS, antioxidant molecules and RNS cross-talk. They include redox-based post-translational protein modifications, the generation of ONOO− through the reaction of O2·− with NO, the reaction between reduced glutathione (GSH) and nitric oxide (NO) to form GSNO and others [89,93,94,95]. Thus, the production of NO from SNP can mimic and even trigger the induction of an oxidative condition, with a concomitant increase of antioxidant activities as described by [96], in the same way that we have detected in the present work with lipids and at the enzymatic level.
However, we have shown here that co-cultivation with either VOO or VAO in the presence of SNP not even failed to revert the levels of antioxidant enzymes and molecules to those of the control, but even enhanced such levels. This fact could be attributed to the absence of specific anti-nitrosative components in VOO/VAO oils, in a way in which continued NO signaling occurs as the result of the SNP treatment, and concomitant oxidative condition therefore continues.
Such hypothesis must be confirmed in future studies through additional assays, which might include treatments with NO scavengers, additional NO donors like GSNO, and/or monitoring of NO levels [90]. At this last regard, the use of quantitative fluorochromes, chemiluminescence, electrochemical or highly resolutive and quantitative methods like EPR (electron paramagnetic resonance) combined with NO spin traps would be of application [97].
Immunofluorescence localization of antioxidant enzymes is a powerful way to analyze the specific localization of these proteins as well as to characterize their presence in a quantitative way, among other advantages like for example determining their potential co-localization [98]. It has been used in numerous organisms, including plants, animals, invertebrates, parasites, bacteria and human tissues [99,100,101,102]. In the present work, antibodies with a recognized cross-reactivity among species were used, showing in all cases a subcellular distribution compatible with that cited in the literature for these enzymes. Thus,
The immunocytochemical evidence of the presence of the enzymes obtained here by means of quantification of the immunofluorescence clearly parallels the changes in enzyme activities described before through enzyme activity assays. Therefore, the changes in SOD, CAT and GPX in response to the treatments seem to be a consequence of modifications in the levels of the corresponding enzymes, more than to inductory or inhibitory aspects of their activity. Immunocytochemical methods also confirm the previous observations concerning the antioxidant properties of both VOO and VAO in co-incubation experiments, together with the likely absence of specific anti-nitrosative compounds hypothesized for these oils.
In this context, continued treatment with NO donor + OVO/OAO may conduct to the formation of nitro fatty-acids, considered of high relevance at both physiological and nutritional level because of their regulatory and anti-inflammatory actions [103,104]. Such potential interactions must be also monitored in the future.
5. Conclusions
Virgin Argan Oil and Virgin Olive Oils used in this work shown a large proportion of polyphenols and flavonoids, together with high antioxidant and free radical scavenging capacity, comparable with those displayed in commercial oils analyzed in the literature.
The conditions used in the present work to induce both oxidative and nitrosative stress do not compromise viability of the T. pyriformis cultures, as neither did the treatments with the oils or the vehicles used to generate oil extracts.
Both fluorescent probes H2-DCFDA and Nile Red have been successfully used to monitor the presence of ROS and their effect on lipid/lipid bodies, respectively, and have served to determine the effectivity of the stress treatments applied and the capacity of both oils to modulate the induced stress.
Both oxidative and nitrosative inductions caused a relevant increase in the activities of different antioxidant enzymes and oxidative molecule markers, which was reverted by the application of oils concomitantly to the stress agent. However, nitrosative stress induction was not reverted by such co-cultivation. The reasons and the consequences of this lack of response in the presence of the oils has to be further analyzed, although several hypotheses focus into the absence of anti-nitrosative components in the oils, or the involvement of complex interactions between the pathways regulating the levels of both ROS and RNS in cell systems.
Immunolocalization of the antioxidant enzymes analyzed here confirms previous observations in a quantitative manner, pointing to transcriptional and translational mechanisms of control more than to the action of postraduccional modifications or changes in enzyme activity, although such modifications cannot be discarded.
Author Contributions
Conceptualization, SE, JDA, BN; investigation SE, ELC, JDA; analysis of results, SE, ELC, SEK, REK, AE, KM, PA, MCM, KS, GL, AJC, BN, JDA; original draft preparation, SE, ELC, AJC, JDA; funding acquisition, JDA, BN; writing—review and editing, SE, JDA. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by research projects BFU-2016-77243-P and PID2020-113324GB-100 of the Spanish Ministry of Science, Innovation and Universities (MICIIN)/State Research Agency (AGE) and research projects P18-RT-1577, CA8313, AT17_5247 y PYC20 RE 009 CSIC. EEZ and UMA20-FEDERJA-029 of the Junta de Andalucía. These projects are partially co-funded by the European Regional Development Fund (ERDF)/European Union (EU).
Acknowledgments
The authors would like to acknowledge the support of the Confocal and Transmission Electron Microscopy (CTEM) facility of the EEZ-CSIC.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Charrouf, Z.; Guillaume, D. Argan oil: Occurrence, composition and impact on human health. European Journal of Lipid Science and Technology 2008, 110(7), 632–636. [Google Scholar] [CrossRef]
- Boucetta, K. Q.; Charrouf, Z.; Derouiche, A., Rahali; Y., Bensouda, Y. Skin hydration in postmenopausal women : argan oil benefit with oral and/or topical use. Prz Menopauzalny 2014, 13(5), 280–288. [CrossRef]
- Cherki, M.; Berrougui, H.; Drissi, A.; Adlouni, A.; Khalil, A. Argan oil : Which benefits on cardiovascular diseases ? Pharmacological Research 2006, Issue 1, 1–5. [Google Scholar] [CrossRef]
- Khallouki, F.; Younos, C.; Soulimani, R.; Oster, T.; Charrouf, Z.; Spiegelhalder, B.; Bartsch, H.; Owen, R.W. Consumption of argan oil (Morocco) with its unique profile of fatty acids, tocopherols, squalene, sterols and phenolic compounds should confer valuable cancer chemopreventive effects. European Journal of Cancer Prevention 2003, 12(1), 67–75. [Google Scholar] [CrossRef] [PubMed]
- Bakour, M.; Soulo, N.; Hammas, N.; Fatemi, H.E.L.; Aboulghazi, A.; Taroq, A.; Abdellaoui, A.; Al-waili, N. The antioxidant content and protective effect of argan oil and Syzygium aromaticum essential oil in hydrogen peroxide-induced biochemical and histological changes. Int. J. Mol. Sci. 2018, 19(2), 610. [Google Scholar] [CrossRef]
- Drissi, A.; Girona, J., Cherki, M.; Godàs, G.; Derouiche; A., El Messal, M.; Saile, R.; Kettani, A.; Solà, R., Masana, L.; Adlouni, A. Evidence of hypolipemiant and antioxidant properties of argan oil derived from the argan tree (Argania spinosa). Clinical Nutrition 2004, 23(5), 1159–1166. [CrossRef]
- Haimeur, A., Messaouri, H., Ulmann, L.; Mimouni, V.; Masrar, A.; Chraibi, A., Tremblin, G.; Meskini, N. Argan oil prevents prothrombotic complications by lowering lipid levels and platelet aggregation, enhancing oxidative status in dyslipidemic patients from the area of Rabat (Morocco). Lipids in Health and Disease 2013, 12(1), 1–9. [CrossRef]
- Kamal, R.; Kharbach, M., Vander, Y.; Zohra, H.; Ghchime, R.; Bouklouze, A.; Cherrah, Y.; Alaoui, K. In vivo anti - inflammatory response and bioactive compounds’ profile of polyphenolic extracts from edible Argan oil (Argania spinosa L .), obtained by two extraction methods. Journal of food biochemistry 2019, 43(12), e13066. [CrossRef]
- Menni, H.B.; Belarbi, M.; Menni, D. B.; Bendiab, H. , Kherraf, Y.; Ksouri, R.; Djebli, N.; Visioli, F. Anti-inflammatory activity of argan oil and its minor components. International Journal of Food Sciences and Nutrition 2019, 0(0), 1–8. [Google Scholar] [CrossRef]
- El Abbassi, A.; Khalid, N.; Zbakh, H.; Ahmad, A. Physicochemical Characteristics, Nutritional Properties, and Health Benefits of Argan Oil: A Review. Critical Reviews in Food Science and Nutrition 2014, 54(11), 1401–1414. [Google Scholar] [CrossRef]
- Ursoniu, S.; Serban, M.C. (2017). The impact of argan oil on plasma lipids in humans: Systematic review and meta - analysis of randomized controlled trials. Phytotherapy Research 2017, 32(3), 377-383. [CrossRef]
- Teres, S.; Coblijn, G.B.; Benet, M.; Alvarez, R.; Bressani, R., Halver, J. Oleic acid content is responsible for the reduction in blood pressure induced by olive oil. 105(37). Proceedings of the National Academy of Sciences 2008, 105(37), 13811-13816. [CrossRef]
- Sies, H. Physiological Society Symposium : Impaired Endothelial and Smooth Muscle Cell Function in Oxidative Stress Reaction of Nitric Oxide With Superoxide : Experimental Physiology 1997, 13(December 1995), 305–316. 19 December.
- Liguori, L. , Russo, G., Curcio, F.; Bulli, G.; Aran, L.; Della-morte, D.; Testa, G.; Cacciatore, F., Bonaduce, D.; Abete, P. Oxidative stress, aging, and diseases. Clinical interventions in aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Holzerová, E.; Prokisch, H. Mitochondria: Much ado about nothing? How dangerous is reactive oxygen species production? International Journal of Biochemistry and Cell Biology 2015, 63, 16–20. [Google Scholar] [CrossRef]
- Tang, S.Y.; Whiteman, M.; Peng, Z.F.; Jenner, A.; Yong, E.L.; Halliwell, B. Characterization of antioxidant and antiglycation properties and isolation of active ingredients from traditional chinese medicines. Free Radical Biology and Medicine 2004, 36(12), 1575–1587. [Google Scholar] [CrossRef]
- Kobayashi, N.; DeLano, F.A., Schmid-Schönbein, G.W. Oxidative stress promotes endothelial cell apoptosis and loss of microvessels in the spontaneously hypertensive rats. Arteriosclerosis, Thrombosis, and Vascular Biology 2005, 25(10), 2114–2121. [CrossRef]
- Xu, H.; Li, C.; Mozziconacci, O.; Zhu, R.; Xu, Y.; Tang, Y.; Chen, R.; Huang, Y., Holzbeierlein, J.M.; Christian Schöneich, J.H.; Li, B. Xanthine oxidase-mediated oxidative stress promotes cancer cell-specific apoptosis. Free Rad. Biol. Med. 2019, Aug 1;139:70-79. [CrossRef]
- Zhang, J., Zheng, S., Wang, S., Liu, Q., & Xu, S. Cadmium-induced oxidative stress promotes apoptosis and necrosis through the regulation of the miR-216a-PI3K/AKT axis in common carp lymphocytes and antagonized by selenium. Chemosphere 2020, 258, 127341. [CrossRef]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous non-enzymatic antioxidants in the human body. Advances in Medical Sciences 2018, 63(1), 68–78. [Google Scholar] [CrossRef] [PubMed]
- Younus, H. Therapeutic potentials of superoxide dismutase. International Journal of Health Sciences 2018, 12(3), 88–93. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5969776/pdf/IJHS-12-88.pdf.
- Adwas, A.A.; Elsayed, A.; Azab, A.E.; Quwaydir, F.A. Oxidative stress and antioxidant mechanisms in human body. J. J. Appl. Biotechnol. Bioeng 2019, 6(1), 43–47. [Google Scholar] [CrossRef]
- Kanarek, N.; Petrova, B.; Sabatini, D.M. Dietary modifications for enhanced cancer therapy. Nature 2020, 579(7800), 507–517. [Google Scholar] [CrossRef] [PubMed]
- Biel, S.; Mesa, M.D.; de la Torre, R.; Espejo, J.A., Fernández-Navarro, J.R.; Fitó, M.; Sánchez-Rodriguez, E.; Rosa, C.; Marchal, C.; Alche, J.D.; Expósito, M.; Brenes, M.; Gandul, B.; Calleja, M.A.; Covas, M.I. The NUTRAOLEOUM Study, a randomized controlled trial, for achieving nutritional added value for olive oils. BMC complementary and alternative medicine 2016, 16(1), 1-9. [CrossRef]
- Sanchez-Rodriguez, E.; Lima-Cabello, E.; Biel-Glesson, S.; Fernandez-Navarro, J.R.; Calleja, M.A.; Roca, M.; Espejo Calvo, J.A.; Extremera, B.G., Florido, M.S.; de la Torre, R., Fito, M.; Covas, M.I.; Alché, J.D.; Martinez de Victoria, E.; Gil, A.; Mesa, M. D. Effects of virgin olive oils differing in their bioactive compound contents on metabolic syndrome and endothelial functional risk biomarkers in healthy adults: a randomized double-blind controlled trial. Nutrients 2018, 10(5), 626. pii: E626. E: 626. pii. [CrossRef]
- Sanchez-Rodriguez, E.; Biel-Glesson, S.; Fernandez-Navarro, J.R.; Calleja, M.A.; Espejo-Calvo, J.A.; Gil-Extremera, B.; de la Torre, R.; Fito, M.; Covas, M.I.; Vílchez, P.; Alché, J.D.; Martinez de Victoria, E.; Gil, A.; Mesa, M. D. Effects of virgin olive oils differing in their bioactive compound contents on biomarkers of oxidative stress and inflammation in healthy adults: A randomized double-blind controlled trial. Nutrients 2019, 11(3), 561. [Google Scholar] [CrossRef]
- Hamilton, R.T.; Walsh, M.E.; Van Remmen, H. Mouse models of oxidative stress indicate a role for modulating healthy aging. J Clin Exp Pathol 2012, ; Suppl 4. [CrossRef]
- Navarro, A., Gomez, C., López-Cepero, J. M., Boveris, A. Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. American journal of physiology-regulatory, integrative and comparative physiology 2004, 286(3), R505-R511.
- Yamauchi, Y.; Matsuno, T.; Omata, K.; Satoh, T. Relationship between hyposalivation and oxidative stress in aging mice. Journal of Clinical Biochemistry and Nutrition 2017, 61(1), 40–46. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, E.; Roy, A.; Royapuram Veeraragavan, G.; Magesh, A.; Varikalam Sleeba, A.; Arivarasu, L.; Marimuthu Parasuraman, B. β-Sitosterol attenuates carbon tetrachloride–induced oxidative stress and chronic liver injury in rats. Naunyn-Schmiedeberg’s Archives of Pharmacology 2020, 393, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Samarghandian, S.; Farkhondeh, T.; Samini, F.; Borji, A. (2016). Protective effects of carvacrol against oxidative stress induced by chronic stress in rat’s brain, liver, and kidney. Biochemistry research international 2016. [CrossRef]
- Marrocco, I.; Altieri, F.; Peluso, I. (2017). Review Article Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxidative medicine and cellular longevity 201. 2017. [CrossRef]
- Usharani, P.; Merugu, P.L.; Nutalapati, C. (2019). Evaluation of the effects of a standardized aqueous extract of Phyllanthus emblica fruits on endothelial dysfunction, oxidative stress, systemic inflammation and lipid profile in subjects with metabolic syndrome : a randomised, double blind, placebo controlled clinical study. BMC complementary and alternative medicine 2019, 19, 1–8. [Google Scholar] [CrossRef]
- Cassidy-Hanley, D. M. Tetrahymena in the Laboratory: Strain Resources, Methods for Culture, Maintenance, and Storage. In: Methods in Cell Biology Volume 109, 2012, Pages 237-276. [CrossRef]
- Espín, J.C., Soler-Rivas, C.; Wichers, H.J. Characterization of the total free radical scavenger capacity of vegetable oils and oil fractions using 2,2-diphenyl-1-picrylhydrazyl radical. Journal of Agricultural and Food Chemistry 2000, 48(3), 648–656. [CrossRef]
- Osorio-Esquivel, O.; Álvarez, V.B.; Dorantes-Álvarez, L.; Giusti, M.M. Phenolics, betacyanins and antioxidant activity in Opuntia joconostle fruits. Food Research International 2011, 44(7), 2160–2168. [Google Scholar] [CrossRef]
- Dehpour, A.A.; Ebrahimzadeh, M.A.; Fazel, N.S.; Mohammad, N.S. Antioxidant activity of the methanol extract of Ferula assafoetida and its essential oil composition. Grasas y aceites 2009, 60(4), 405–412. [Google Scholar]
- Huang, B.; Ke, H.; He, J.; Ban, X. , Zeng, H.; Wang, Y. Extracts of Halenia elliptica exhibit antioxidant properties in vitro and in vivo. Extracts of Halenia elliptica exhibit antioxidant properties in vitro and in vivo. Food and Chemical Toxicology 2011, 49(1), 185–190. [Google Scholar] [CrossRef]
- Oyaizu, M. Studies on products of browning reaction antioxidative activities of products of browning reaction prepared from glucosamine. The Japanese journal of nutrition and dietetics 1986, 44(6), 307–315. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free radical biology and medicine 1999, 26(9-10), 1231-1237. 1231. [CrossRef]
- Lizard, G.; Gueldry, S.; Deckert, V.; Gambert, P.; Lagrost, L. Evaluation of the cytotoxic effects of some oxysterols and of cholesterol on endothelial cell growth: methodological aspects. Pathologie-biologie 1997, 45(4), 281-290. /: 281-290. https.
- Dürichen, H. , Siegmund, L.; Burmester, A.; Fischer, M.S.; Wöstemeyer, J. Ingestion and digestion studies in Tetrahymena pyriformis based on chemically modified microparticles. European Journal of Protistology 2016, 52, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.; Rosebrough, N. , Farr, A.L.; Randall, R. Protein measurement with the Folin phenol reagent. Protein measurement with the Folin phenol reagent. Journal of biological chemistry 1951, 193(1), 265–275. [Google Scholar] [CrossRef]
- Paoletti, F.; Aldinucci, D.; Mocali, A.; Caparrini, A. A sensitive spectrophotometric method for the determination of superoxide dismutase activity in tissue extracts. Analytical biochemistry 1986, 154(2), 536–541. [Google Scholar] [CrossRef] [PubMed]
- Aebi, H. Catalase in vitro. Metods in Enzymology 1984, 105(1947), 121–126. [Google Scholar] [CrossRef]
- Gonzler, W.A. The term glutathione peroxidase (glutathione: H202 oxidoreductase, EC 1. 11. 1.9) is reserved for the selenoprotein catalyzing the reaction: Heal. San Fr 1984, 105, 114–120. [Google Scholar]
- Ellman, G.L. Tissue sulfhydryl groups. Archives of biochemistry and biophysics 1959, 82(1), 70–77. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for Lipid Peroxides in Animal Tissues Thiobarbituric Acid Reaction. Analytical biochemistry 1979, 95(2), 351–358. [Google Scholar] [CrossRef]
- Houshia, O.J.; Qutit, A., Zaid, O., Shqair, H., Zaid, M. Determination of Total Polyphenolic Antioxidants Contents in West-Bank Olive Oil. Journal of Natural Sciences Research 2014, 4(15), 71–76.
- Nenadis, N.; Mastralexi, A.; Tsimidou, M.Z. Physicochemical characteristics and antioxidant potential of the Greek PDO and PGI Virgin Olive Oils (VOOs). European journal of lipid science and technology 2019, 121(3), 1800172. [Google Scholar] [CrossRef]
- Marfil, R.; Giménez, R., Martínez, O.; Bouzas, P.R.; Rufián-Henares, J.A.; Mesías, M.; Cabrera-Vique, C. Determination of polyphenols, tocopherols, and antioxidant capacity in virgin argan oil (Argania spinosa, Skeels). European Journal of Lipid Science and Technology 2011, 113(7), 886–893. [CrossRef]
- Zarrouk, A.; Martine, L.; Grégoire, S.; Nury, T.; Meddeb, W.; Camus, E.; Badreddine, A; Durand, Ph.; Namsi, A.; Yammine, A.; Nasser, B.; Mejri, M.; Bretillon, L.; Mackrill, J.J.; Cherkaoui-Malki, M.; Hammami, M.; Lizard, G.; Lizard, G. Profile of fatty acids, tocopherols, phytosterols and polyphenols in mediterranean oils (argan oils, olive oils, milk thistle seed oils and nigella seed oil) and evaluation of their antioxidant and cytoprotective activities. Current pharmaceutical design 2019, 25(15), 1791-1805. 1791. [CrossRef]
- Saffar, S.; Jafari, S.M.; Bahrami, A. Evaluation of changes in the quality of extracted oil from olive fruits stored under different temperatures and time intervals Scientific Reports 2019, 9(1), 19688. [CrossRef]
- Bongiorno, D.; Di Stefano, V.; Indelicato, S.; Avellone, G.; Ceraulo, L. Bio-phenols determination in olive oils: Recent mass spectrometry approaches. Mass Spectrometry Reviews 2021, e21744. [Google Scholar] [CrossRef]
- Çelik, S.E.; Özyürek, M.; Güçlü, K.; Apak, R. Solvent effects on the antioxidant capacity of lipophilic and hydrophilic antioxidants measured by CUPRAC, ABTS/persulphate and FRAP methods. Talanta 2010, 81(4–5), 1300–1309. [CrossRef]
- Cayuela, J.A.; Rada, M.; Pérez-camino, M.C., Benaissa, M; Abdelaziz, E., Guinda, Á. Characterization of artisanally and semiautomatically extracted argan oils from Morocco. haracterization of artisanally and semiautomatically extracted argan oils from Morocco 2008, 110(12), 1159–1166. [CrossRef]
- Ahamed, M.; Akhtar, M.J.; Alaizeri, Z.A.M.; Alhadlaq, H.A. TiO2 nanoparticles potentiated the cytotoxicity, oxidative stress and apoptosis response of cadmium in two different human cells. Environmental Science and Pollution Research 2020, 27(10), 10425–10435. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.H.; Martinez, L.J.; Coleman, M.C.; Spitz, D.R.; Weintraub, N.L.; Kader, K.N. Mechanisms of H2O2-induced oxidative stress in endothelial cells. Free Radical Biology and Medicine 2006, 40(12), 2206–2213. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.S.; Dutta, S.; Raha, S. Hydrogen peroxide-induced apoptosis-like cell death in Entamoeba histolytica. Parasitology International 2010, 59, 166–172. [Google Scholar] [CrossRef]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxidants & redox signaling 2007, 9(1), 49-89. [CrossRef]
- Kiritsakis, A.K.; Kiritsakis, K.A.; Tsitsipas, C.K. A review of the evolution in the research of antioxidants in olives and olive oil during the last four decades. Journal of Food Bioactives 2020, 11. [Google Scholar] [CrossRef]
- Dinda, B.; Dubnath, S.; Harigaya, Y. Naturally occurring iridoids. A review, part 1. Chem. Pharm. Bull. 2007, 55, 159–222. [Google Scholar] [CrossRef]
- Fabiani, R.; Rosignoli, P.; de Bartolomeo, A.; Fuccelli, R.; Servili, M.; Montedoro, G.F.; Morozzi, G. Oxidative DNA damage is prevented by extracts of olive oil, hydroxytyrosol, and other olive phenolic compounds in human blood mononuclear cells and HL60 cells. J. Nutr. 2008, 138, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Tundis, R.; Loizzo, M.; Menichini, F.; Statti, G.; Menichini, F. Biological and pharmacological activities of iridoids: Recent developments. Mini Rev. Med. Chem. 2008, 8, 399–420. [Google Scholar] [CrossRef]
- Midaoui, A.; El Haddad, Y.; Filali-Zegzouti, Y.; Couture, R. Argan Oil as an Effective Nutri-Therapeutic Agent in Metabolic Syndrome: A Preclinical Study. International Journal of Molecular Sciences 2017, 18(11), 2492. [Google Scholar] [CrossRef]
- Orabi, S.H.; Allam, T.S.; Shawky, S.M.; El-Aziz Tahoun, A.; Khalifa, H.K.; Almeer, R., Abdel-Daim, M.M.; Borai El-Borai, N.; Mousa, A.A.; Eg, A.A.M. The Antioxidant, Anti-Apoptotic, and Proliferative Potency of Argan Oil against Betamethasone-Induced Oxidative Renal Damage in Rats. Biology, 2020, 352. [CrossRef]
- Şekeroğlu, Z.A.; Aydın, B. , Şekeroğlu, V. Argan oil reduces oxidative stress, genetic damage and emperipolesis in rats treated with acrylamide. Biomedicine and Pharmacotherapy 2017, 94, 873–879. [Google Scholar] [CrossRef]
- Haman, N.; Longo, E.; Schiraldi, A. , Scampicchio, M. Radical scavenging activity of lipophilic antioxidants and extra-virgin olive oil by isothermal calorimetry. Thermochimica Acta 2017, 658, 1–6. [Google Scholar] [CrossRef]
- Paradiso, V.M.; Flamminii, F.; Pittia, P.; Caponio, F.; Mattia, C.Di. (2020). Radical Scavenging Activity of Olive Oil Phenolic Antioxidants in Oil or Water Phase during the Oxidation of O/W Emulsions: An Oxidomics Approach. Antioxidants 2020, 9(10), 996. [CrossRef]
- Quintero-Flórez, A.; Pereira-Caro, G.; Sánchez-Quezada, C.; Moreno-Rojas, M.; Gaforio, J.J.; Jimenez, A.; Beltrán, G. Effect of olive cultivar on bioaccessibility and antioxidant activity of phenolic fraction of virgin olive oil. European Journal of Nutrition 2018, 57, 1925–1946. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M., Caruso, F.; Kwok, L.; Lee, G.; Caruso, A.; Gionfra, F.; Candelotti, E.; Belli, S.L.; Molasky, N.; Raley-Susman, K.M.; Leone, S.; Filipský, T.; Tofani, D.; Pedersen, J.; Incerpi, S. (2017). Protection by extra virgin olive oil against oxidative stress in vitro and in vivo. Chemical and biological studies on the health benefits due to a major component of the Mediterranean diet. PLoS One, 12(12), e0189341. [CrossRef]
- Cadi, R.; Mounaji, K.; Amraoui, F.; Soukri, A. Protective and antioxidant potential of the argan oil on induced oxidative stress in Tetrahymena pyriformis. Journal of Medicinal Plants Research 2013, 7(27), 1961–1968. [Google Scholar] [CrossRef]
- Zafra, A.; Rodríguez-García, M.I.; Alché, J.D.D. Cellular localization of ROS and NO in olive reproductive tissues during flower development. BMC Plant Biology 2020, 10(1), 1–14. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Brown, D.A. Fluorescent Detection of Lipid Droplets and Associated Proteins. Current Protocols in Cell Biology 2007, 35(1), 1–11. [Google Scholar] [CrossRef]
- Cruz, A.L.S.; Barreto, E.D.A.; Bozza, P.T. Lipid droplets : platforms with multiple functions in cancer hallmarks. Cell Death and Disease 2020. [CrossRef]
- Lee, S.; Zhang, J.; Choi, A.M.K.; Kim, H.P. Mitochondrial Dysfunction Induces Formation of Lipid Droplets as a Generalized Response to Stress. Oxidative medicine and cellular longevity 2013. [CrossRef]
- Lee, J.; Homma, T.; Kurahashi, T.; Kang, E.S.; Fujii, J. Biochemical and Biophysical Research Communications Oxidative stress triggers lipid droplet accumulation in primary cultured hepatocytes by activating fatty acid synthesis. Biochemical and Biophysical Research Communications 2015, 1–7. [Google Scholar] [CrossRef]
- Petan, T.; Jarc, E.; Jusovic, M. (2018). Lipid Droplets in Cancer : Guardians of Fat in a Stressful World. Molecules 2018, 23(8), 194111–15. [CrossRef]
- Del Rio, L.A., Sandalio, L.M.; Corpas, F.J., Palma, M., Barroso, J.B.. Reactive Oxygen Species and Reactive Nitrogen Species in Peroxisomes . Production , Scavenging , and Role. Plant physiology 2006, 141(2), 330-335. [CrossRef]
- Chen, R.; Tai, Y.; Chen, T.; Lin, T. Propofol protects against nitrosative stress-induced apoptotic insults to cerebrovascular endothelial cells via an intrinsic mitochondrial mechanism. Surgery 2013, 154(1), 58–68. [Google Scholar] [CrossRef]
- Chen, T.; Wu, G.; Hsu, C.; Fong, T.; Chen, R. Chemico-Biological Interactions Nitrosative stress induces osteoblast apoptosis through downregulating MAPK-mediated NF B/AP-1 activation and subsequent Bcl-X L expression. Chemico-Biological Interactions 2010, 184(3), 359–365. [CrossRef]
- Rogstam, A.; Larsson, J.T.; Kjelgaard, P.; Wachenfeldt, C. Von. (2007). Mechanisms of Adaptation to Nitrosative Stress in Bacillus subtilis. ( 189(8), 3063–3071. [CrossRef]
- Takahashi, H.K.; Cambiaghi, T.D.; Luchessi, A.D.; Hirabara, S.M.; Vinolo, M.A.R.; Newsholme, P.; Curi, R. Activation of survival and apoptotic signaling pathways in lymphocytes exposed to palmitic acid. Journal of cellular physiology 2012, 227(1), 339–350. [Google Scholar] [CrossRef]
- Guenaou, I.; Hmimid, F.; Azzahra, F.; Errami, A.; Santé, L.; Environnement, E., Des, F.; Ain, S.; Hassan, U., Casablanca, I. I. De. (2021). Comparative Biochemistry and Physiology , Part C Cytoprotective effect of ethyl acetate fraction from Ephedra fragilis on H2O2 - induced oxidative damage in Tetrahymena pyriformis. Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology 2021, 239, 108899. [CrossRef]
- Salla, S.; Sunkara, R.; Ogutu, S.; Walker, L.T., Verghese, M. Antioxidant activity of papaya seed extracts against H2O2 induced oxidative stress in HepG2 cells. LWT - Food Science and Technology 2015. [CrossRef]
- Zhang, L.; Liu, Y.; Li, J.Y.; Li, L.Z.; Zhang, Y.L.; Gong, H.Y.; Cui, Y. Protective Effect of Rosamultin against H2O2 -Induced Oxidative Stress and Apoptosis in H9c2 Cardiomyocytes. Oxidative Medicine and Cellular Longevity 2018. [CrossRef] [PubMed]
- Zwiewka, M.; Bielach, A.; Tamizhselvan, P.; Madhavan, S.; Dobrev, P.; Vankova, R.; Ryad, E.E.; Tan, S. Root Adaptation to H2O2 -Induced Oxidative Stress by ARF-GEF BEN1- and Cytoskeleton-Mediated PIN2 Trafficking. Plant and Cell Physiology 2019, February 2019, 255–273. [Google Scholar] [CrossRef]
- Grina, F.; Abdellatif, R.; Talal, S.; Nasser, B. (2020). Could the Fucus Spiralis Algal Extract Prevent the Oxidative Stress in Could the Fucus Spiralis Algal Extract Prevent the Oxidative Stress in Tetrahymena Pyriformis Model ? Biointerface Research in Applied Chemistry 2020, 11(2), 8978–8995. [Google Scholar] [CrossRef]
- Fourrat, L.; Iddar, A.; Valverde, F.; Serrano, A.; Soukri, A. Effects of Oxidative and Nitrosative Stress on Tetrahymena pyriformis Glyceraldehyde-3-Phosphate Dehydrogenase. Journal of eukaryotic microbiology 2007, 54(4), 338–346. [Google Scholar] [CrossRef] [PubMed]
- Ayd, B. (2017). Effects of argan oil on the mitochondrial function, antioxidant system and the activity of NADPH- generating enzymes in acrylamide treated rat brain. Biomedicine & pharmacotherapy 2017, 87, 476–481. [Google Scholar] [CrossRef]
- Er, R.; Aydın, B.; Şekeroğlu, V.; Atlı Şekeroğlu, Z. Protective effect of Argan oil on mitochondrial function and oxidative stress against acrylamide-induced liver and kidney injury in rats. Biomarkers 2020, 25(6), 458–467. [Google Scholar] [CrossRef]
- lavarasi, K., Kiruthiga, P.V.; Pandian, S.K.; Devi, K. P. Chemosphere Hydroxytyrosol , the phenolic compound of olive oil protects human PBMC against oxidative stress and DNA damage mediated by 2 , 3 , 7 , 8-TCDD. Chemosphere 2011, 84(7), 888–893. Chemosphere. [CrossRef]
- Molassiotis, A.; Fotopoulos, V. Oxidative and nitrosative signaling in plants: two branches in the same tree?. Plant Signaling & Behavior 2011,, 6(2), 210-214. [CrossRef]
- Corpas, F.J.; Alché, J.D.; Barroso, J. B. Current overview of S-nitrosoglutathione (GSNO) in higher plants. Frontiers in plant science 2013, 4, 126. [Google Scholar] [CrossRef]
- Chaki, M.; Begara-Morales, J.C.; Valderrama, R.; Aranda-Caño, L.; Barroso, J.B. New Insights into the Functional Role of Nitric Oxide and Reactive Oxygen Species in Plant Response to Biotic and Abiotic Stress Conditions. In: Plant Growth and Stress Physiology. Plant in Challenging Environments, vol 3. Gupta, D.K., Palma, J.M. (eds) Springer, Plant Growth and Stress Physiology. Plant in Challenging Environments, vol 3. 2021 Cham. 215-235. [CrossRef]
- Bączek-Kwinta, R. Nitric Oxide and Reactive Oxygen Species Interactions in Plant Tolerance and Adaptation to Stress Factors. In: Biotic and Abiotic Stress Tolerance in Plants. Vats, S. (eds) Springer, 2018, Singapore. 239-256. [CrossRef]
- Goshi, E.; Zhou, G.; He, Q. Nitric oxide detection methods in vitro and in vivo. Medical gas research 2019, 9(4), 192. [Google Scholar] [CrossRef]
- Crouch, R. K.; Thaete, L.G. Immunolocalization of CuZn SOD. In Handbook Methods For Oxygen Radical Research 271-276. CRC Press. 2018.
- Michonneau, P.; Fleurat-Lessard, P.; Roblin, G.; Béré, E. CuZn-superoxide dismutase is differentially modified in localization and expression by three abiotic stresses in miniature rose bushes. Micron 2023, 103524. [Google Scholar] [CrossRef] [PubMed]
- Dvořák, P.; Krasylenko, Y.; Ovečka, M.; Basheer, J.; Zapletalová, V.; Šamaj, J.; Takáč, T. In vivo light-sheet microscopy resolves localisation patterns of FSD1, a superoxide dismutase with function in root development and osmoprotection. Plant, Cell & Environment 2021, 44(1), 68-87. [CrossRef]
- Lohana, P.; Suryaprawira, A.; Woods, E.L.; Dally, J.; Gait-Carr, E.; Alaidaroos, N.Y.; Heard, Ch.M.; Lee, K.Y.; Ruge, F.; Farrier, J.N.; Enoch, S.; Caley, M.P.; Peake, M.A.; Davies, L.C.; Giles, P.J.; Thomas, D.W.; Stephens, Ph.; Moseley, R. Role of enzymic antioxidants in mediating oxidative stress and contrasting wound healing capabilities in oral mucosal/skin fibroblasts and tissues. Antioxidants 2023, 12(7), 1374. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzewska, A.K.; Ocłoń, E. , Kucharski, M.; Sechman, A. Effect of in vitro sodium fluoride treatment on CAT, SOD and Nrf mRNA expression and immunolocalisation in chicken (Gallus domesticus) embryonic gonads. Theriogenology 2020, 157, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Mata-Pérez, C.; Sánchez-Calvo, B.; Padilla-Serrano, M.N.; Begara-Morales, J.C.; Luque, F.; Melguizo, M.; Jiménez-Ruiz, J.; Fierro-Risco, J.; Peñas-Sanjuán, A.; Valderrama, R.; Corpas, F.J.; Barroso, J.B. Nitro-fatty acids in plant signaling: nitro-linolenic acid induces the molecular chaperone network in Arabidopsis. Plant Physiol, 170:686-670. [CrossRef]
- Mata-Pérez C, Sánchez-Calvo B, Padilla M.N.; Begara-Morales, J.C.; Valderrama, R.; Corpas, F.J.; Barroso, J.B (2017) Nitro-fatty acids in plant signaling: new key mediators of nitric oxide metabolism. Redox Biol 2016, 11:554–561. [CrossRef]
Figure 1.
Effect of H2O2 (A), SNP (B), ethanol (C), VOO (D) and VAO (E) on T. pyriformis viability measured by the MTT test. The values were normalized against the control. The protozoan cells were maintained in PPYG medium supplemented with H2O2 [50, 150, 250, 350, 450 and 550 µM], SNP [0.4, 0.6, 0.8, 1, 1.2 and 1.4 mM] oils [0.25, 0.5, 0.75, 1.25 and 1.5% in ethanol] and ethanol [0.1, 0.15, 0.3, 0.6, 1.2 and 2.4%] for 24h. Prior to incubation with MTT at 0.05 mg/ml. All values are means ± SD (n = 3). An asterisk indicates values that were significantly different (P ≤0.05) from control group.
Figure 1.
Effect of H2O2 (A), SNP (B), ethanol (C), VOO (D) and VAO (E) on T. pyriformis viability measured by the MTT test. The values were normalized against the control. The protozoan cells were maintained in PPYG medium supplemented with H2O2 [50, 150, 250, 350, 450 and 550 µM], SNP [0.4, 0.6, 0.8, 1, 1.2 and 1.4 mM] oils [0.25, 0.5, 0.75, 1.25 and 1.5% in ethanol] and ethanol [0.1, 0.15, 0.3, 0.6, 1.2 and 2.4%] for 24h. Prior to incubation with MTT at 0.05 mg/ml. All values are means ± SD (n = 3). An asterisk indicates values that were significantly different (P ≤0.05) from control group.
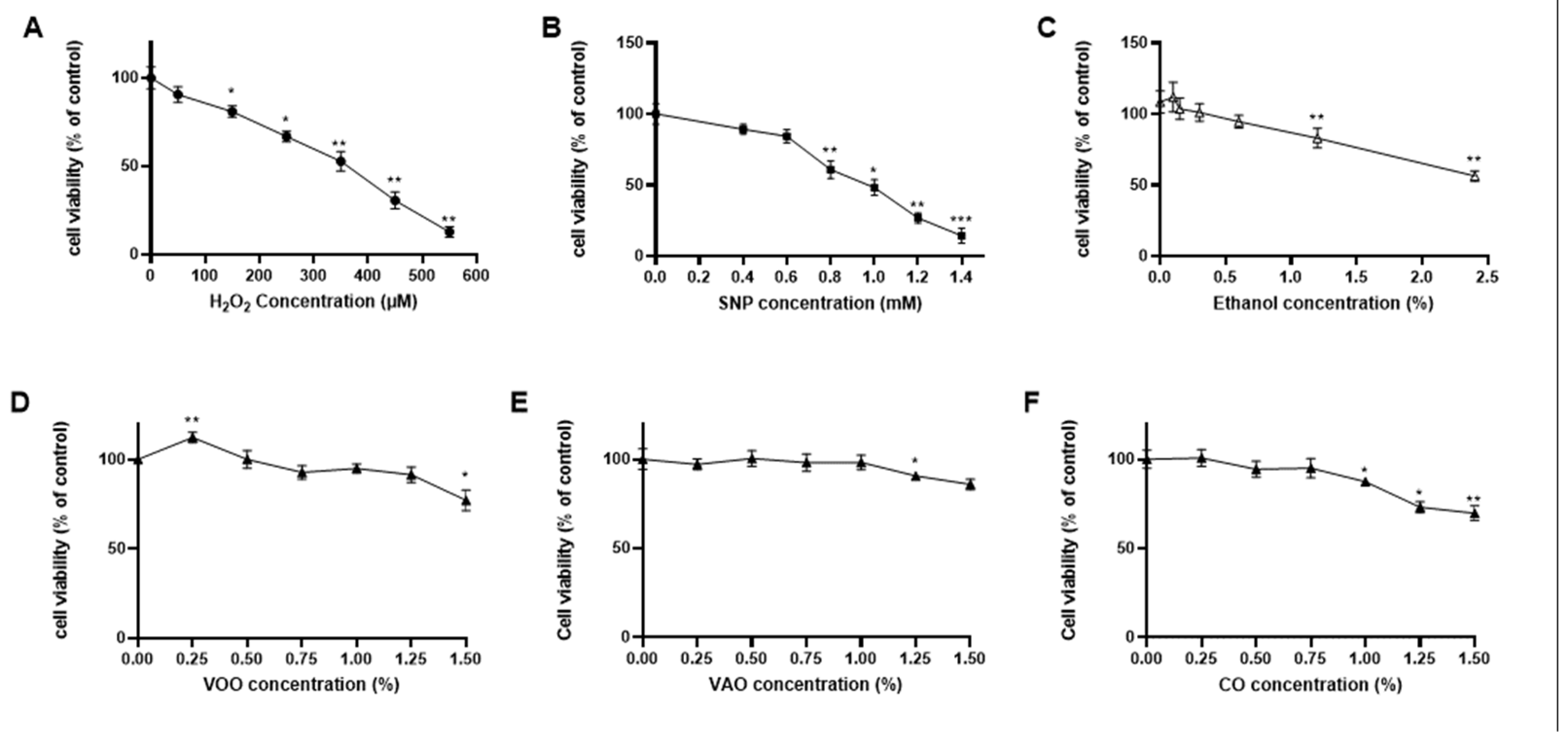
Figure 2.
(A): FM images of T. pyriformis cells exposed to the different treatments, and stained with H2-DCFDA ROS staining dye. Left panel in (A) shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in (A) shows identical images captured after blue excitation (H2-DCFDA). The intensity of the green fluorescent staining differs among the treatments applied (control, H2O2, H2O2 + VOO, H2O2 + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in (A). Fluorescence is present in most cell compartments. (C): Quantification of green fluorescence intensity as percentage of intensity compared to the control culture (100%). Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison versus control, b: comparison versus H2O2. Scale bars represent 50 µm.
Figure 2.
(A): FM images of T. pyriformis cells exposed to the different treatments, and stained with H2-DCFDA ROS staining dye. Left panel in (A) shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in (A) shows identical images captured after blue excitation (H2-DCFDA). The intensity of the green fluorescent staining differs among the treatments applied (control, H2O2, H2O2 + VOO, H2O2 + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in (A). Fluorescence is present in most cell compartments. (C): Quantification of green fluorescence intensity as percentage of intensity compared to the control culture (100%). Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison versus control, b: comparison versus H2O2. Scale bars represent 50 µm.
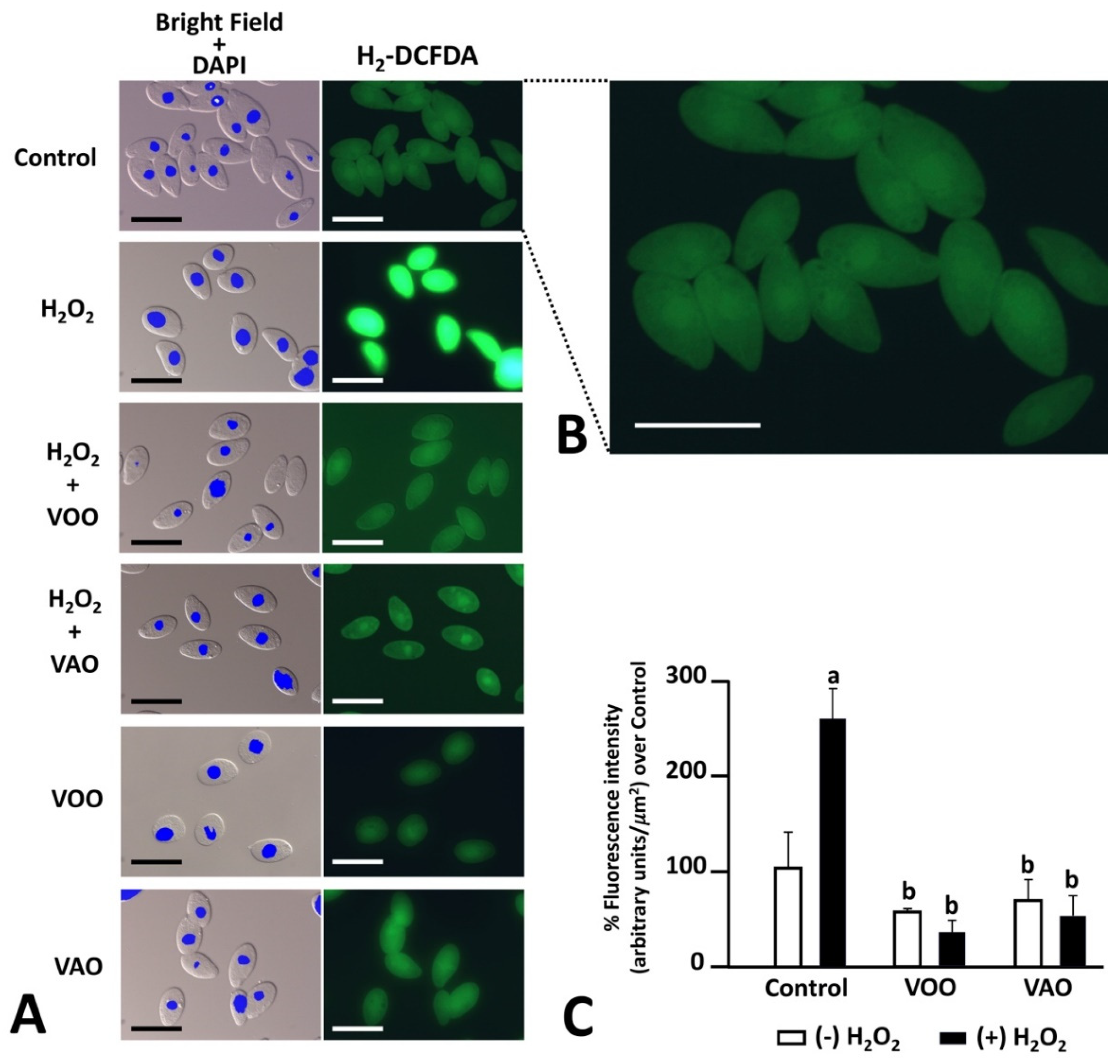
Figure 3.
(A): FM images of T. pyriformis cells exposed to the different oxidative treatments, and stained with Nile red staining dye. Left panel in (A) shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in (A) shows identical images captured after green excitation. The intensity of the orange fluorescent staining differs among the treatments applied (control, H2O2, H2O2 + VOO, H2O2 + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in A. Fluorescence is present in the form of diffuse orange cytoplasmic staining (squares) together with discrete orange spots likely representing lipid droplets (white arrows). (C): Quantification of orange fluorescence intensity as percentage of intensity compared to the control culture (100%). Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison versus control, b: comparison versus H2O2, Scale bars represent 50 µm.
Figure 3.
(A): FM images of T. pyriformis cells exposed to the different oxidative treatments, and stained with Nile red staining dye. Left panel in (A) shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in (A) shows identical images captured after green excitation. The intensity of the orange fluorescent staining differs among the treatments applied (control, H2O2, H2O2 + VOO, H2O2 + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in A. Fluorescence is present in the form of diffuse orange cytoplasmic staining (squares) together with discrete orange spots likely representing lipid droplets (white arrows). (C): Quantification of orange fluorescence intensity as percentage of intensity compared to the control culture (100%). Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison versus control, b: comparison versus H2O2, Scale bars represent 50 µm.
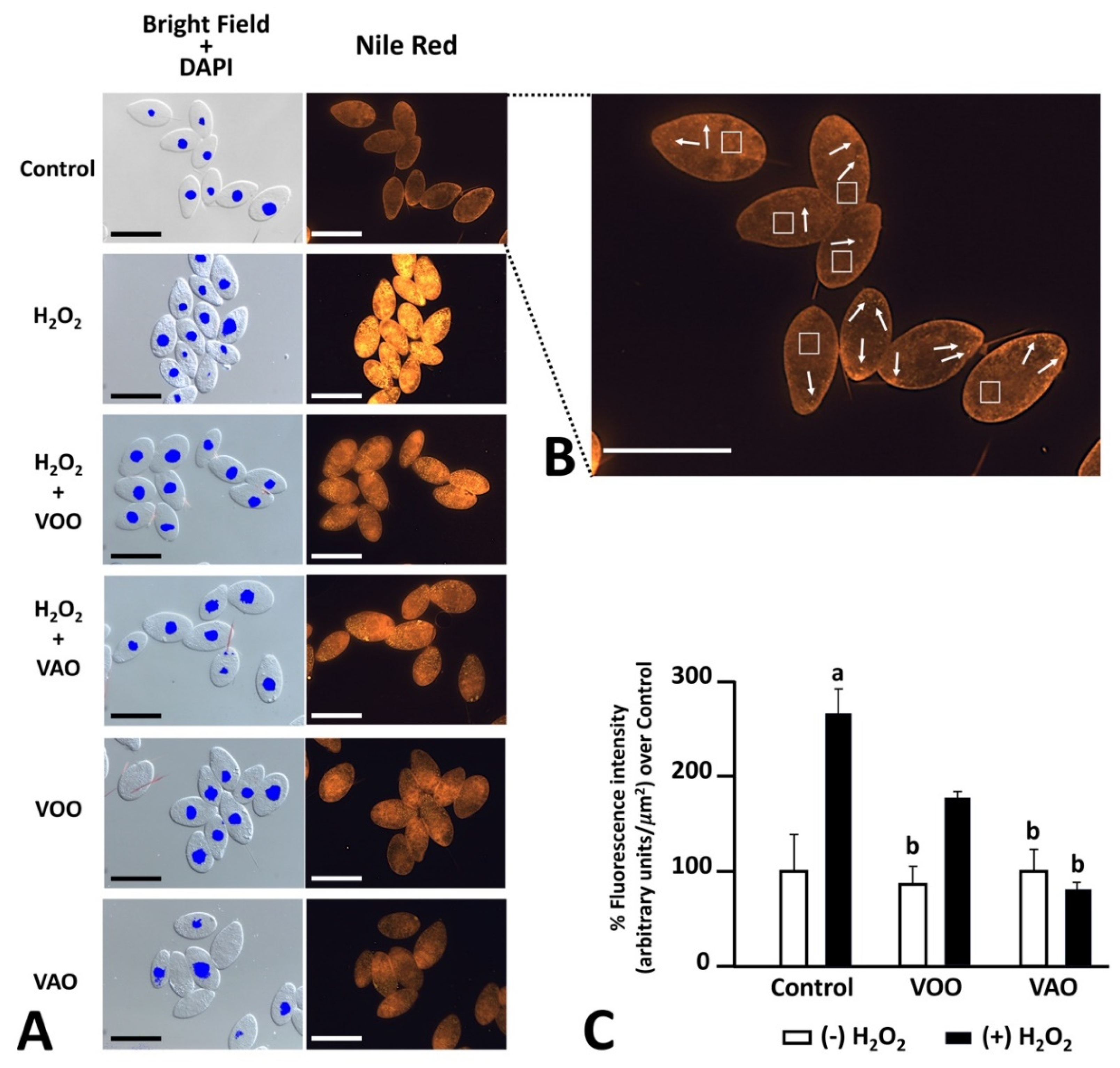
Figure 4.
(A): FM images of T. pyriformis cells exposed to the different nitrosative treatments, and stained with Nile red staining dye. Left panel in (A) shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in (A) shows identical images captured after green excitation. The intensity of the orange fluorescent staining differs among the treatments applied (control, SNP, SNP + VOO, SNP + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in A. Fluorescence is present in the form of diffuse orange cytoplasmic staining (white squares) together with discrete orange spots likely representing lipid droplets (white arrows). (C): Quantification of orange fluorescence intensity as percentage of intensity compared to the control culture (100%). Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, c: comparison versus VOO, d: comparison versus VAO. Scale bars represent 50 µm.
Figure 4.
(A): FM images of T. pyriformis cells exposed to the different nitrosative treatments, and stained with Nile red staining dye. Left panel in (A) shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in (A) shows identical images captured after green excitation. The intensity of the orange fluorescent staining differs among the treatments applied (control, SNP, SNP + VOO, SNP + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in A. Fluorescence is present in the form of diffuse orange cytoplasmic staining (white squares) together with discrete orange spots likely representing lipid droplets (white arrows). (C): Quantification of orange fluorescence intensity as percentage of intensity compared to the control culture (100%). Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, c: comparison versus VOO, d: comparison versus VAO. Scale bars represent 50 µm.
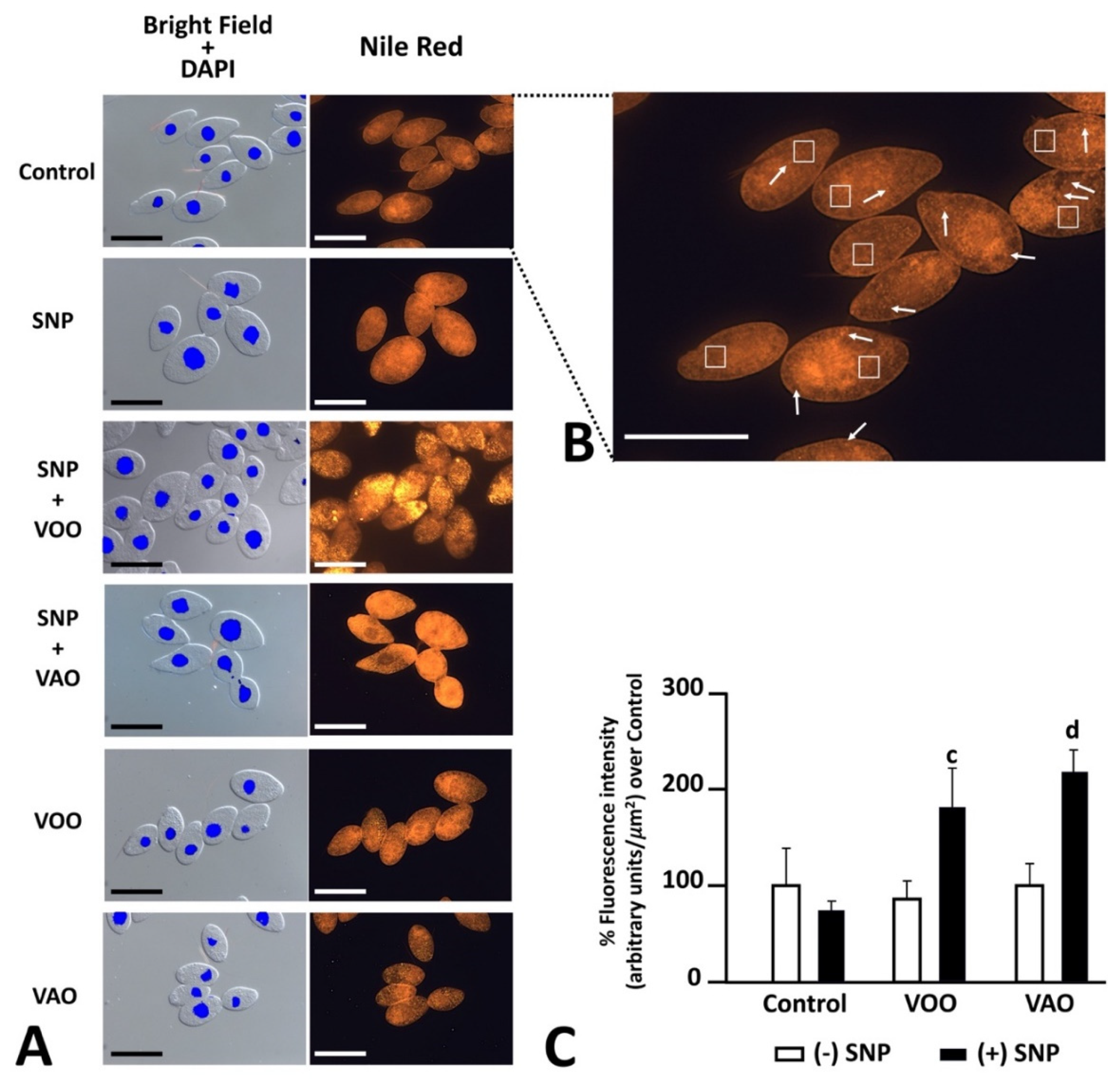
Figure 5.
Effect of VOO and VAO on key antioxidant enzymes and metabolites involved in the oxidative stress induced by H2O2 treatment of T. pyriformis cultures. The activity of SOD (A), CAT (B) and GPX (C) were evaluated, as well as the levels of GSH (D) and MDA (E). T. pyriformis cells were maintained in PPYG medium supplemented with VOO and VAO at 5%. H2O2 treatment was performed using a concentration of 350 µM for 24 h. All values are means ± SD (n=3). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison vs. control, b: comparison vs. H2O2, c: comparison vs. VOO, d: comparison vs. VAO, e: comparison vs. H2O2 + VOO, f: comparison vs. H2O2 + VAO, h: comparison vs. SNP +VOO.
Figure 5.
Effect of VOO and VAO on key antioxidant enzymes and metabolites involved in the oxidative stress induced by H2O2 treatment of T. pyriformis cultures. The activity of SOD (A), CAT (B) and GPX (C) were evaluated, as well as the levels of GSH (D) and MDA (E). T. pyriformis cells were maintained in PPYG medium supplemented with VOO and VAO at 5%. H2O2 treatment was performed using a concentration of 350 µM for 24 h. All values are means ± SD (n=3). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison vs. control, b: comparison vs. H2O2, c: comparison vs. VOO, d: comparison vs. VAO, e: comparison vs. H2O2 + VOO, f: comparison vs. H2O2 + VAO, h: comparison vs. SNP +VOO.
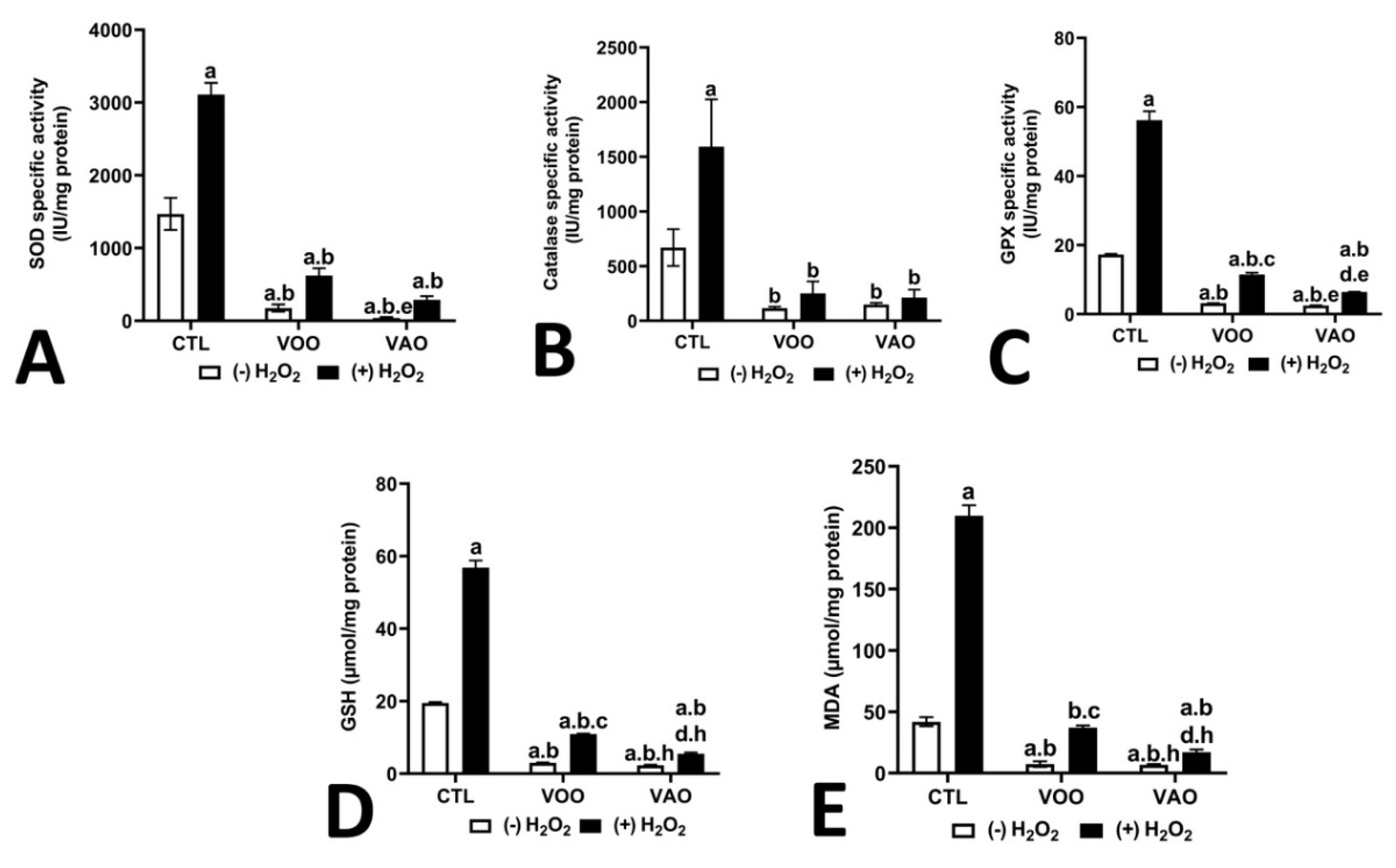
Figure 6.
Effect of VOO and VAO on key antioxidant enzymes and metabolites involved in the nitrosative stress induced by SNP treatment of T. pyriformis cultures. The activity of SOD (A), CAT (B) and GPX (C) were evaluated, as well as the levels of GSH (D) and MDA (E). T. pyriformis cells were maintained in PPYG medium supplemented with VOO and VAO at 5%. SNP treatment was performed using a concentration of 350 µM for 24 h. All values are means ± SD (n=3). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison vs. control, g: comparison vs. SNP, c: comparison vs. VOO, d: comparison vs. VAO, h: comparison vs. SNP + VOO.
Figure 6.
Effect of VOO and VAO on key antioxidant enzymes and metabolites involved in the nitrosative stress induced by SNP treatment of T. pyriformis cultures. The activity of SOD (A), CAT (B) and GPX (C) were evaluated, as well as the levels of GSH (D) and MDA (E). T. pyriformis cells were maintained in PPYG medium supplemented with VOO and VAO at 5%. SNP treatment was performed using a concentration of 350 µM for 24 h. All values are means ± SD (n=3). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison vs. control, g: comparison vs. SNP, c: comparison vs. VOO, d: comparison vs. VAO, h: comparison vs. SNP + VOO.

Figure 7.
(A): Immunofluorescence detection of SOD in T. pyriformis cells under different oxidative and nitrosative stress conditions of culture, and in the presence of the VOO and VAO oils. Left panel in A shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in A shows identical images captured after blue excitation. The intensity of the green fluorescent staining differs among the treatments applied (control, H2O2, H2O2 + VOO, H2O2 + VAO, SNP, SNP + VOO, SNP + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in A. Fluorescence is present in the form of diffuse green cytoplasmic staining (white squares) together with discrete green spots likely representing organelles (white arrows). (C,D): Quantification of green fluorescence intensity as percentage of intensity compared to the control culture (100%). (C) refers to oxidative vs. control conditions, whereas (D) refers to nitrosative vs. control conditions. Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison versus control, b: comparison versus H2O2, c: comparison vs. VOO, d: comparison vs. VAO, h: comparison vs. SNP + VOO. Scale bars represent 50 µm.
Figure 7.
(A): Immunofluorescence detection of SOD in T. pyriformis cells under different oxidative and nitrosative stress conditions of culture, and in the presence of the VOO and VAO oils. Left panel in A shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in A shows identical images captured after blue excitation. The intensity of the green fluorescent staining differs among the treatments applied (control, H2O2, H2O2 + VOO, H2O2 + VAO, SNP, SNP + VOO, SNP + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in A. Fluorescence is present in the form of diffuse green cytoplasmic staining (white squares) together with discrete green spots likely representing organelles (white arrows). (C,D): Quantification of green fluorescence intensity as percentage of intensity compared to the control culture (100%). (C) refers to oxidative vs. control conditions, whereas (D) refers to nitrosative vs. control conditions. Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison versus control, b: comparison versus H2O2, c: comparison vs. VOO, d: comparison vs. VAO, h: comparison vs. SNP + VOO. Scale bars represent 50 µm.
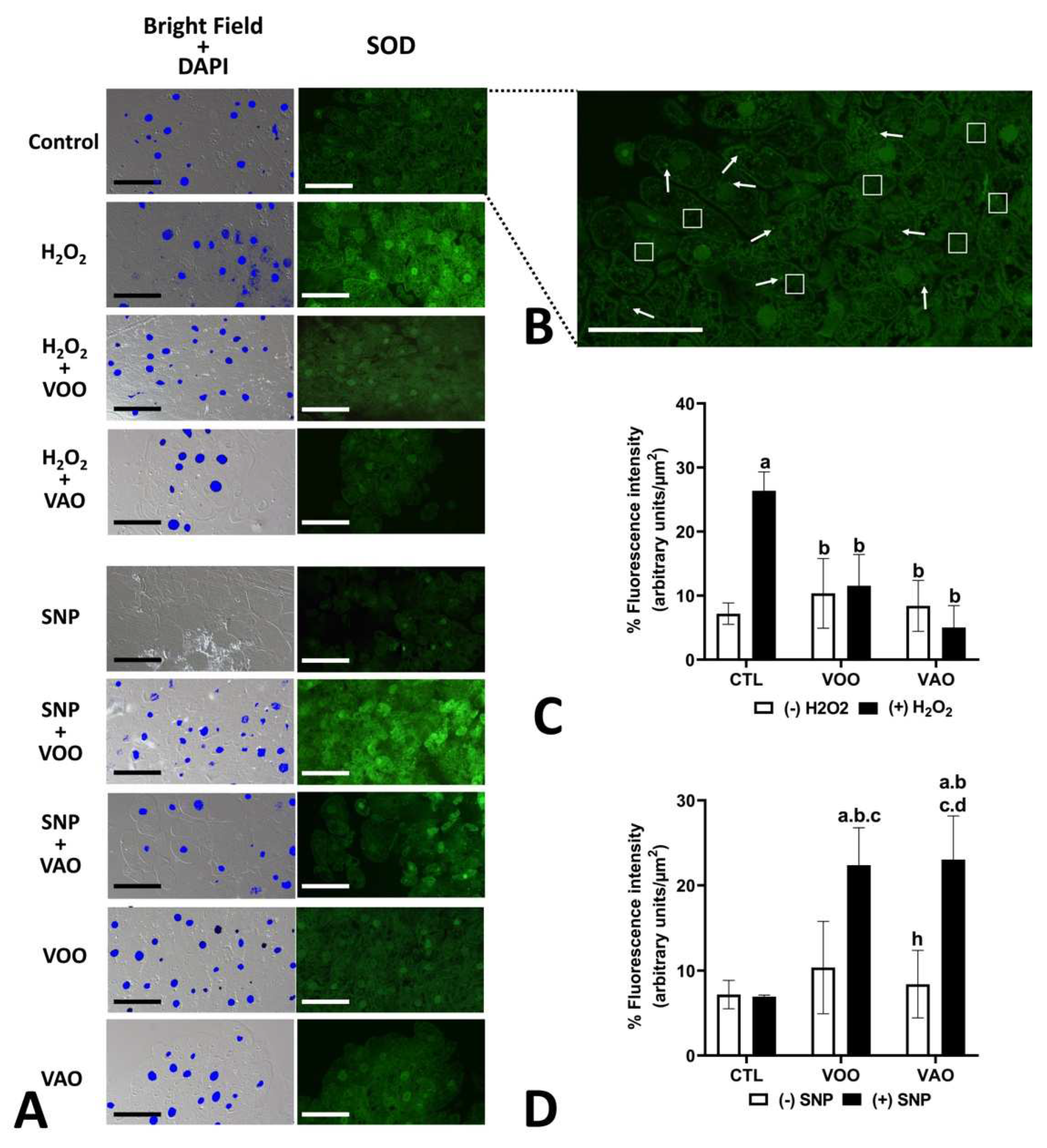
Figure 8.
(A): Immunofluorescence detection of CAT in T. pyriformis cells under different oxidative and nitrosative stress conditions of culture, and in the presence of the VOO and VAO oils. Left panel in (A) shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in (A) shows identical images captured after blue excitation. The intensity of the green fluorescent staining differs among the treatments applied (control, H2O2, H2O2 + VOO, H2O2 + VAO, SNP, SNP + VOO, SNP + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in (A). Fluorescence is present in the form of discrete green spots likely representing organelles (white arrows). (C,D): Quantification of green fluorescence intensity as percentage of intensity compared to the control culture (100%). (C) refers to oxidative vs. control conditions, whereas (D) refers to nitrosative vs. control conditions. Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison vs. control, b: comparison vs. H2O2, h: comparison vs. SNP + VOO. Scale bars represent 50 µm.
Figure 8.
(A): Immunofluorescence detection of CAT in T. pyriformis cells under different oxidative and nitrosative stress conditions of culture, and in the presence of the VOO and VAO oils. Left panel in (A) shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in (A) shows identical images captured after blue excitation. The intensity of the green fluorescent staining differs among the treatments applied (control, H2O2, H2O2 + VOO, H2O2 + VAO, SNP, SNP + VOO, SNP + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in (A). Fluorescence is present in the form of discrete green spots likely representing organelles (white arrows). (C,D): Quantification of green fluorescence intensity as percentage of intensity compared to the control culture (100%). (C) refers to oxidative vs. control conditions, whereas (D) refers to nitrosative vs. control conditions. Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison vs. control, b: comparison vs. H2O2, h: comparison vs. SNP + VOO. Scale bars represent 50 µm.

Figure 9.
(A): Immunofluorescence detection of GPX in T. pyriformis cells under different oxidative and nitrosative stress conditions of culture, and in the presence of the VOO and VAO oils. Left panel in (A) shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in (A) shows identical images captured after blue excitation. The intensity of the green fluorescent staining differs among the treatments applied (control, H2O2, H2O2 + VOO, H2O2 + VAO, SNP, SNP + VOO, SNP + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in A. Fluorescence is present in the form of diffuse green cytoplasmic staining (white squares) together with discrete green spots likely representing organelles (white arrows). (C,D): Quantification of green fluorescence intensity as percentage of intensity compared to the control culture (100%). (C) refers to oxidative vs. control conditions, whereas (D) refers to nitrosative vs. control conditions. Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison vs. control, b: comparison vs. H2O2, c: comparison vs. VOO, d: comparison vs. VAO, h: comparison vs. SNP + VOO, i: comparison vs. SNP + VAO. Scale bars represent 50 µm.
Figure 9.
(A): Immunofluorescence detection of GPX in T. pyriformis cells under different oxidative and nitrosative stress conditions of culture, and in the presence of the VOO and VAO oils. Left panel in (A) shows transmitted light images combined with DAPI detection of nuclei, whereas right panel in (A) shows identical images captured after blue excitation. The intensity of the green fluorescent staining differs among the treatments applied (control, H2O2, H2O2 + VOO, H2O2 + VAO, SNP, SNP + VOO, SNP + VAO, VOO and VAO). (B): Large magnification image of the control cells shown in A. Fluorescence is present in the form of diffuse green cytoplasmic staining (white squares) together with discrete green spots likely representing organelles (white arrows). (C,D): Quantification of green fluorescence intensity as percentage of intensity compared to the control culture (100%). (C) refers to oxidative vs. control conditions, whereas (D) refers to nitrosative vs. control conditions. Original measurement was performed as arbitrary units/µ2 using imageJ software. The values were expressed as mean±SD (n = 10). A multiple comparative analysis between the groups was carried out using a one-way ANOVA test followed by a Tukey’s test. A p-value less than 0.05 was considered statistically significant. The statistically significant differences between the groups are indicated by different letters, a: comparison vs. control, b: comparison vs. H2O2, c: comparison vs. VOO, d: comparison vs. VAO, h: comparison vs. SNP + VOO, i: comparison vs. SNP + VAO. Scale bars represent 50 µm.
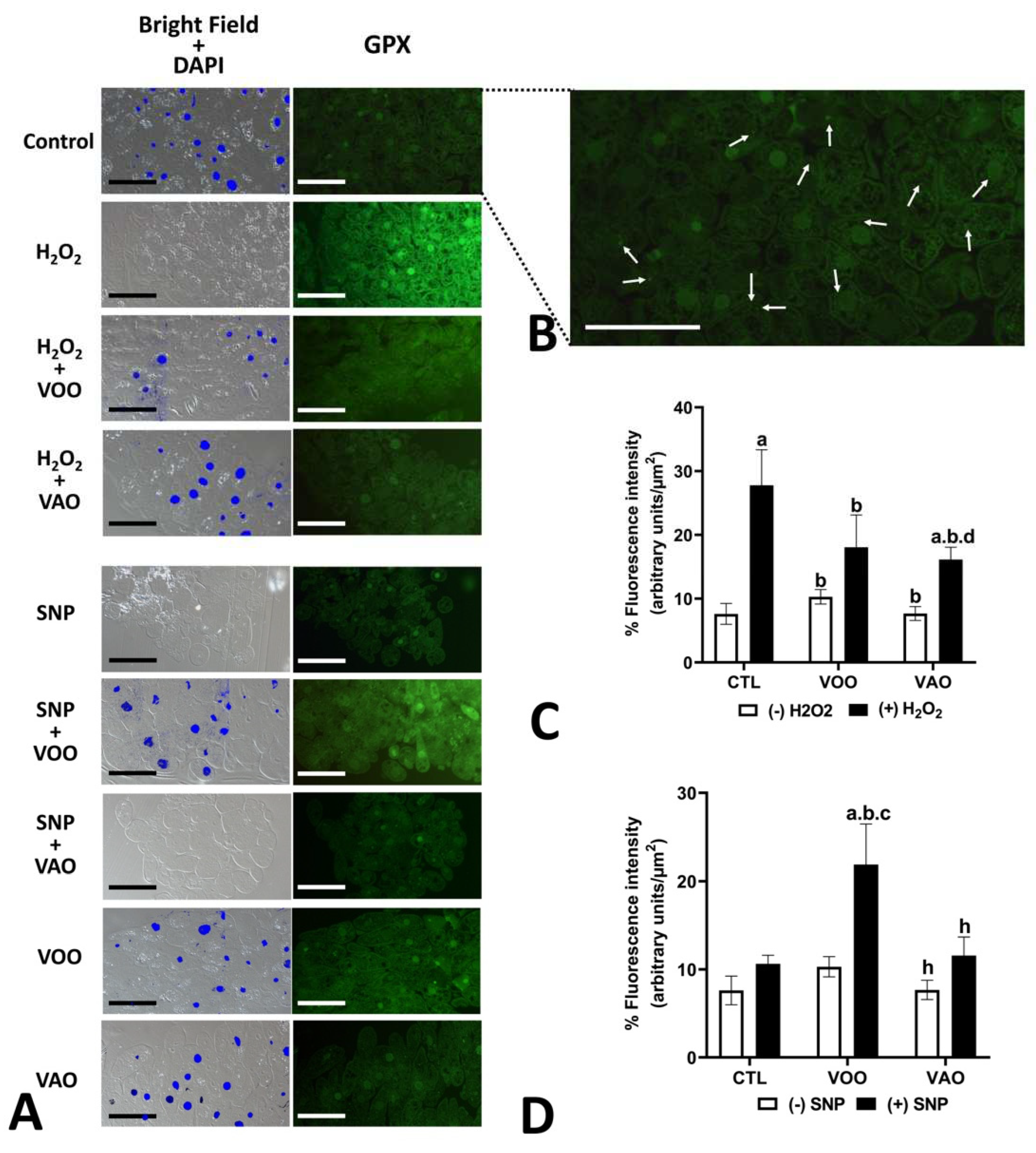

Table 1.
Total phenolic and flavonoids content, and antioxidants activities determined by DPPH, FRAP and ABTS of the Methanolic Fraction (MF), Lipidic Fraction (LF) and Total Oil (TO) from Virgin Olive Oil (VOO) and Virgin Argan Oil (VAO). All values are means ± standard deviations of data from three independent experiments. Means with different asterisks (*) are significantly different according to the student’s test compared to VOO.
Table 1.
Total phenolic and flavonoids content, and antioxidants activities determined by DPPH, FRAP and ABTS of the Methanolic Fraction (MF), Lipidic Fraction (LF) and Total Oil (TO) from Virgin Olive Oil (VOO) and Virgin Argan Oil (VAO). All values are means ± standard deviations of data from three independent experiments. Means with different asterisks (*) are significantly different according to the student’s test compared to VOO.
| Virgin Olive Oil (VOO) | Virgin Argan Oil (VAO) | |||||
|---|---|---|---|---|---|---|
| MF | LF | TO | MF | LF | TO | |
| Total polyphenols (mg EAG/g dry weight) | 80.64±1.32 | 50.73±1.63 | 66.72±1.36 | 76.20±2.33 | 31.10±1.69 *** |
50.85±1.51 ** |
| Flavonoids (mg EQ/mg of dry weight) | 169±9.24 | 100.5±9.78 | 139.36±5.63 | 151.18±8.53 ** |
72.72±8.41 | 103.48±4.2 * |
| DPPH (% of inhibition) | 55.35±1.71 | 43.76±1.03 | 54.43±0.99 | 47.27±0.68 ** |
39.33±0.88 | 42.95±1.03 ** |
| FRAP (mg EAA/g of the extract) | 71.65±4.33 | 32.99±2.36 | 55.68±3.76 | 66.76±1.61 |
33.31±2.76 | 51.65±2.58 |
| ABTS (% of inhibition) | 49.31±1.48 | 32.24±1.77 | 50.11±0.72 | 39.26±1.51 * |
26.50±0.82 | 55.09±2.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



