
Submitted:
08 September 2023
Posted:
12 September 2023
You are already at the latest version
Abstract
Metastasis is a critical step in the process of carcinogenesis and a vast majority of cancer related mortalities result from metastat-ic disease that is resistant to current therapies. Cell migration and invasion are the first steps of the metastasis process, which mainly occurs by two important biological mechanisms i.e., cytoskeletal remodelling and Epithelial to Mesenchymal Transition (EMT). Akt (also known as Protein Kinase B) is a central signalling molecule of the PI3K-Akt signalling pathway. Aberrant acti-vation of this pathway has been identified in a wide range of cancers. Several studies have revealed that Akt actively engages with the migratory process in motile cells, including metastatic cancer cells. The downstream signalling mechanism of Akt in cell migration depends upon the tumour type, sites, and intracellular localisation of activated Akt. In this review, we focus on the role of Akt in the regulation of two events that control cell migration and invasion in various cancers including head and neck squamous cell carcinoma (HNSCC) and the status of PI3K-Akt pathway inhibitors in clinical trials in HNSCC.
Keywords:
Akt
; cancer
; metastasis
; HNSCC
; EMT
; cytoskeletal remodelling
1. Introduction
The primary reason for cancer-related deaths is metastatic disease [1]. The spreading of tumour cells from the primary lesion is the main cause for the mortality and morbidity of cancer patients, whether it exists at the time of diagnosis, progresses during treatment, or happens at the time of disease relapse [2]. The metastasis process involve a series of sequential, interconnected steps including: separation of tumour cells from the primary lesion and invasion of neighbouring, healthy connective tissue, intravasation into the blood and lymphatic vessels, circulation through the blood vessels (circulating tumour cells) to other tissues in the body, extravasation from the blood vessel into the new tissue, growth in specific distant organs and building a secondary tumour [3,4,5] (Figure 1).
Many of the metastatic stages are dependent on tumour cell migration and invasion, which allows the cells to change tissue location. Tumour cells employ similar mechanism of migration to spread to other tissues to those that happen in non-tumour cells during physiological events such as wound healing, angiogenesis, inflammatory immune responses and embryonic morphogenesis [6]. However, tumour cell migration has shown to be stimulated by diverse promigratory factors ignoring stop signals, including tumour cell-derived autocrine factors and the soluble factors present at secondary sites [7,8]. Due to this imbalance of signals, cancer cells become unceasingly migratory and invasive, causing tumour expansion across tissue boundaries and hence the development of cancer metastases [4,7].
Cell migration through tissues results from highly integrated multistep cellular events [6,9,10]. First, the moving cell polarises, elongates and extends protrusions in the way of migration reacting to a migration-promoting agents. There are two types of protrusions, which can be spike-like filopodia, or large and broad lamellipodia. Protrusions are typically guided by actin polymerisation and are stabilised by adhering to the extracellular matrix or adjacent cells via related transmembrane receptors [11]. Consequently, forward extension of a lamellipodium and retraction of the trailing edge causes the translocation of the cell body [7,11]. Reorganisation of the actin cytoskeleton is the most important processes of cell motility and is vital for most types of cell migration [12]. In the process of cell migration, the actin cytoskeleton is dynamically remodeled, and this reorganisation creates the physical force essential for cell migration [13].
Variable experimental behaviour and histological patterns of tumour cells suggest that tumour cells can utilise different cellular and molecular modes of migration based on cell-type specific autonomous mechanisms and reactive mechanisms stimulated by the local microenvironments [14,15]. Tumour cells are detected as both single cells and organized collective sheets in malignant cancer patients, indicating that cancer cells exhibit the plasticity to switch between single and collective cell migration. Studies on single cell migration have founded the cellular and molecular basis providing a significant understanding into the spreading of tumours whose cells migrate constitutively as single cells such as leukaemia or lymphomas, after separation from cohesive lesions through the epithelial to mesenchymal transition (EMT) [10,16]. Collective cell migration occurs when the junctions between cells are retained over extended periods of time, so cells are adherent to their neighbours. The efficiency of the metastatic process is increased by the transition to single cell migration. However, circulating grouped tumour cells detected in the patient peripheral blood samples suggests that the intravasation process can also be enacted by a cell cluster [17,18]. Cell migration is the first step to invasion. The extracellular matrix is degraded by invasive cells via proteolysis before entering neighbouring tissues [7,19].
Highly integrated multistep cellular events lead to cell migration and invasion through tissues that are regulated by various cell signalling pathways, including PI3K-Akt signalling pathway. The serine/threonine kinase Akt is also known as protein kinase B (PKB). It was originally discovered as a proto-oncogene. Akt plays a significant regulatory role in various cellular activities including cell survival, cell migration and invasion progression, insulin metabolism, protein synthesis and thus became a focus of major attention. The Akt signalling pathway is activated by receptor tyrosine kinases (RTK), cytokine receptors, G-protein coupled receptors, integrins, B and T cells receptors and other stimuli that stimulates the production of phosphatidylinositol 3,4,5, triphosphates (PIP3) through phosphoinositide 3-kinase (PI3K) [20]. The PI3 kinases are a set of lipid kinases that phosphorylate the membrane phospholipid, phosphatidylinositol 4,5 biphosphate (PIP2), generating phosphatidylinositol 3,4,5, triphosphates (PIP3). PIP3 controls a range of effector molecules including the Akt group of oncogenic kinases termed Akt1, Akt2 and Akt3. Activation of Akt1, a 60 kDa kinase, depends on PI3K [21]. Increase of cellular PIP3 by PI3K eventually allows the activation of Akt1 by phosphorylation at Thr308 and Ser473 residues [22]. This activation is completed by structural modification stimulated by PI3K-dependent kinase-1 (PDK-1)-dependent phosphorylation at Thr308 and stabilisation by mTORC2 or DNA-PK (DNA-activated protein kinase) dependent phosphorylation at Ser473 [23,24,25]. A third phosphorylation site on Akt1 has been identified at Thr450 [26]. This site is referred to as the turn phosphorylation site and is controlled by mTORC2 activity [27,28]. Activation of the three Akt isoforms plays a pivotal role in fundamental cellular functions by phosphorylating a variety of substrates (Figure 2).
There are frequent alterations of the PI3K-Akt pathway in various types of human cancers. Amplification of the PIK3C gene encoding PI3K or the Akt gene lead to the constitutive activation of the PI3K-Akt pathway. PTEN (phosphatase and tensin homologue deleted on chromosome 10) can inhibit the Akt activation and mutation in the PTEN gene also causes the constitutive activation of Akt [29,30,31]. Recent evidence has also suggested that Akt plays an important role in cancer cell migration and invasion [32,33]. This review focuses on the regulatory roles of Akt in cancer cell metastasis including head and neck cancer emphasising cell migration. This review also briefly updates the current status of PI3K-Akt inhibitors in clinical trials in HNSCC.
2. Akt in Cytoskeletal rearrangements
The cytoskeleton is the supporting structure of cells which is composed of a filamentous network of micro filaments such as actin, myosin; intermediate filaments such as vimentin, keratin and microtubules such as tubulin [34]. The main purpose of the cytoskeleton is to maintenance of cellular structure, intracellular transport and supporting cell division. Cytoskeletal rearrangements occur in various physiological and pathological events such as cell movement, wound healing and cancer metastasis [35]. Cellular motility either in physiological events or in pathological conditions is driven by cytoskeletal remodelling, initiated by various signalling pathways. The synergistic effect of all the three basic elements-filamentous actin, microtubules and the intermediate filament vimentin is the potential basis for a cell to migrate [32]. Wide-ranging studies have focused on how stabilisation of intracellular filaments and dynamic polymerisation control cell migration [13,36]. Akt can phosphorylates a diverse group of key factors associated with the skeletal filaments.
Growth of the vascular network is essential for the spread of cancer cells. Angiogenesis is the process whereby new vessels are formed and involved in the supply of nutrients, oxygen and immune cells and also the removal of waste products [37]. Angiogenic factors plays huge role in neoplastic vascularisation, thus, increasingly getting attention. Vascular endothelial cell migration is a vital step for angiogenesis. Vascular endothelial growth factor (VEGF) activates Akt and stimulates the migration of endothelial cells by increasing actin polymerisation. Abrogated Akt activity by expression of a kinase-dead mutant inhibits actin bundle formation and blocked cell migration. This effect is enhanced when myristylated Akt is expressed [38], demonstrating that Akt is a critical mediator of VEGF-induced endothelial cell migration through actin reorganisation. Data also suggests that eNOS activation via phosphorylation of Ser-1177 by Akt is necessary and sufficient for VEGF-mediated EC migration [39,40].
In Chicken embryonic fibroblasts (CEF), PI3K-transduced migratory signal was blocked by inhibiting Akt activity. PI3K also activated p70S6K1 via Rac and induced actin filament remodelling and cell migration in CEF cells. This study confirms that the activation of PI3K activity alone is adequate to remodel actin filaments to increase cell migration through the activation of Akt and p70S6K1 in CEF cells [41]. Another study suggested that overexpression of the integrin-linked kinase (ILK) pathway is sufficient to stimulate PI3K-dependent Rac1 activation. Blocking of Akt, p70S6K1 or Rac1, inhibited the effect of ILK on actin filaments, hence blocked cell migration, implying a regulatory role for the PI3K/Akt/p70S6K1/Rac1 signalling pathway in response to ILK [42]. In ovarian cancer, p70S6K1, downstream of the PI3K/Akt pathway stimulated the rapid activation of Rac1 and cdc42 and their downstream effector molecule p21 activated kinase (PAK1) [43]. In neutrophils, activation of G-protein coupled receptors results in F-actin polymerisation and cytoskeleton contraction through PIP3 signalling. This pattern of actin reorganisation ensures pseudopod extension in human neutrophils during chemoattractant stimulation, which is dependent on Akt activity [44]. Breast cancer cell migration and invasion often occurs in an Akt dependent which is characterised by increased filopodia production. A specific Akt inhibitor named API-2 (Akt phosphorylation inhibitor 2) blocks breast cancer cell migration by blocking filopodia formation [45]. These observations of Akt activation and its role, suggest that Akt can potentially regulate cell migration through direct modulation of actin.
Other studies have shown that actin preferentially binds to phosphorylated Akt at pseudopodia with enriched bundles [46,47]. Another study further confirmed that Akt can phosphorylate actin and therefore cortical reorganisation of actin associated with cell migration is strongly dependent on Akt activation [48]. Studies with HeLa cells revealed that Akt phosphorylates PAK1, a protein which belongs to the p21-activated serine/threonine kinase family and facilitates its binding with the non-catalytic region of the tyrosine kinase adaptor protein (Nck) promoting chemotaxis [49]. This effect of Akt through PAK1 may be mediated by enhanced myosin 2 assembly and polarity [50].
The actin-rich structure of highly motile cells like invadopodia, filopodia and pseudopodia needs to be stabilised to function properly. Actin-associated proteins are responsible for stabilising this actin structure by blocking the degradation of newly formed actin filaments [51]. ALE (the Akt phosphorylation enhancer), also termed the ‘girder’ of actin filaments (Girdin) is one of the best examples of this type of protein. APE/Girdin provides the integrity of the actin meshwork (actin filament) at the leading edge of migrating cells. Reduction of APE/Girdin destabilises the actin bundles, triggering the ablation of stress fibres and actin structure. This results in the loss of directional migratory ability and establishes the vital activity of APE/Girdin in the regulation of cell migration. Enomoto et al proved that APE/Girdin is phosphorylated by Akt on Serine1416 (S1416) [52]. Upon stimulation by EGF, S1416 phosphorylation initiates the translocation of APE/Girdin, regulates actin reconstructions and Akt controlled cell motility in cancer-associated fibroblasts, fibroblasts, breast cancer and oesophageal squamous cell carcinoma cells [53,54,55,56,57]. Akt has also been shown to promote actin reorganisation and cell motility mediated by the mechano-protein and Akt substrate ANKRD2 (Ankyrin repeat domain protein 2, also known as ARPP) [58].
Actin-associated structural (cross-linker) protein, filamin A, is phosphorylated by Akt on residue S2152 [59,60,61]. In turn, phosphorylated filamin A mediates caveolin-1 induced cancer cell migration through the IGF signalling pathway [62,63]. Akt has been shown to phosphorylate NHE1 (sodium-hydrogen exchanger isoform 1), a key mediator of stress fibre disassembly on S648 and suggested to be critical for the growth factor-induced cytoskeletal rearrangements that favour cell migration and invasion [64]. Other studies have demonstrated the migration of different cell types by modulation of the cytoskeleton through NHE1, although the role of Akt was not elucidated [65,66,67,68]. A study in fibroblasts demonstrated that the Akt pathway is necessary for the translocation of NHE1 to the leading edge and actin nucleation at the lamellipodium that supports directional cell migration [69].
Extensive studies have been carried out to investigate the role of intermediate filaments in cell motility [70,71]. The most abundant intermediate protein that maintains normal cell and tissue integrity is called vimentin, a type 3 filamentous protein. It is phosphorylated by Akt1 on residue S39, stabilised and thereby regulates cancer cell invasion in aggressive sarcoma [72]. It has also been shown that vimentin is highly expressed in breast cancer lung metastases [73,74], however the specific mechanism to control cell migration by some Akt substrates are still undefined. Such as, S-phase kinase-associated protein 2 (skp2), a component of E3 ligase, is phosphorylated by Akt on S72 residue, stimulates Skp-2 dependent ligase activity and induce cell migration [75,76]. Akt also promotes cell migration by regulating microtubule dynamics through Akt/GSK3 beta axis-dependent activation of microtubule binding protein, APC (adenomatous polyposis coli) [77,78,79].
Akt interacts with promigratory proteins, in addition to targeting cytoskeletal proteins, thus facilitating crosstalk between associated signalling axes. The VEGFR/eNOS signalling pathway-controlled cell migration is dependent on Akt -mediated phosphorylation on S1177 [39]. Accumulating evidence has indicated the importance of Nitric Oxide (NO) in pathological conditions, especially in malignant tumours [80,81]. Furthermore, VEGFR signalling often cooperates with the G-protein coupled receptor, sphingosine-1-phosphate receptor 1 (SIPR1, also known as endothelial differentiation gene 1, EDG-1). SIP/SIPR1 activation leads to the phosphor-activation of VEGFR which phosphorylates Src kinase, consequently activating the PI3K/Akt/eNOS axis [82]. Akt-mediated phosphorylation of SIPR1 on T236 further enhanced their activity, stimulates cortical actin assembly, angiogenesis and chemotaxis [83,84]. Thus, Akt plays a vital role in regulating VEGFR and the SIP/SIPR1 signalling pathway and actively regulates cell migration. EphA2 (Ephrin receptor tyrosine kinase A2), a member of the largest tyrosine kinase family, is also phosphorylated by Akt on S897 residue. In human brain cancer cells, S897 phosphorylation in EphA2 is responsible for cell migration and invasion through dendritic actin cytoskeletal rearrangements and lamellipodia formation [85,86]. Scientists have shown that EphA2 recruits Ephexin4 (a guanine nucleotide exchange factor for the small GTPase, RhoG) upon phosphorylation of S896 and promotes breast and colorectal cancer cell migration and anoikis resistance [87].
It is now well established that membrane redistribution of integrin by various signalling pathways is a critical mediator of cellular movement. The ANK repeat and pleckstrin homology domain-containing protein 1 (ACAP 1) is a GTPase activating protein (GAP) for ADP ribosylation factor 6 (ARF6) known to participate in integrin beta recycling. ACAP1 is phosphorylated by Akt on S554 and stimulates integrin recycling and therefore promotes cell migration [88]. Another GTPase activation protein, RhoGAP22 is shown to be phosphorylated by Akt on S16, upon stimulation by insulin or possibly PDGF and this plays a vital role in regulating cell migration, leading to modulation of Rac1 activity [89]. Various studies have established the role of the mammalian targets of rapamycin complex 1 (mTORC1) in the cell migration and relationship with Akt [90,91]. Akt regulates mTORC1 activation and in turn, activates the phosphorylation of p70S6K1 (S6K1) and inhibits eukaryotic initiation factor 4E binding protein (4EBP1). It is suggested that tuberous sclerosis complex 1/2 (TSC 1/2), a tumour suppressor gene inhibits S6K1 and activates 4EBP1, facilitated by inhibiting mTORC1. Akt-mediated phosphorylation of TSC2 is also destabilises the complex and activates mTORC1 [92]. In the single cell motility assay, IGF-1-stimulated cell motility was inhibited by down-regulation of S6K1 using lentiviral and ectopic expression of constitutively hypophosphorylated 4EBP1 [93]. S6K1 regulates cell motility, which might be related to regulating phosphorylation of focal adhesion kinase (FAK), paxillin, p130cas, and F-actin organisation (or lamellipodia formation) [94]. Furthermore, mTORC1 mediates phosphorylation of ERK1/2 (extracellular signal related kinase) on T202 through direct and indirect regulation of PP2A (protein phosphatase 2A). Inhibition of PP2A activates ERK1/2 and promotes motility in a number of transformed and cancer cells [95,96,97,98,99].
Several studies have also demonstrated that transforming growth factor beta 1 (TGFβ1) enhances human chondrosarcoma and lung cancer cells migration through the PI3K/Akt signalling pathway. Akt phosphorylated IKKαβ (IkB kinase) which activates IkBα and p65 on S536 residue. This causes NFkB to dissociate from IkBα and hence activate β1 and αvβ3 integrin, promoting human lung cancer and chondrosarcoma cell migration [100,101]. Abrogation of mTOR signalling leads to the lack of functional mTORC1 in human trophoblast cells. mTORC1 regulates JAK/STAT signalling pathway and contributes to the invasiveness of trophoblast cells by regulating matrix remodelling enzymes such as MMP9 (matrix metalloproteinase), MMP2, uPA (urokinase plasminogen activator) and PAI-1 (plasminogen activator inhibitor) [8,102]. The opposing role of Akt in cell migration has also been discussed in different studies. Akt phosphorylates kidney ankyrin repeat-containing protein (Kank), which consequently leads to a negative regulation of stress fibre assembly and RhoA activation, attenuating cell migration [103]. An actin binding protein, paladin, phosphorylated by Akt1 on S507 inhibits breast cancer cell migration by disrupting F-actin bundles [104]. On the other hand, Akt2 contributes to paladin stability independent of S507 phosphorylation [105]. Similarly, Akt phosphorylates TSC2 (tuberous sclerosis complex), a Rho GTPase regulator that inhibits breast cancer cell migration due to impaired F-actin assembly [106].
3. Akt in EMT
Epithelial cells are tightly connected to their adjacent cells via E-cadherin and with actin filaments via α- or β-catenin. Epithelial tumour cells must break these intercellular junctions before migrating as single cell and invade stromal tissues. Epithelial tumour cells undergo a process named as epithelial to mesenchymal transition (EMT), to facilitate the invasion as single cell. EMT process can be induced either by extracellular growth factors, for example EGF, TGF-α and β, FGF, or by intracellular cues, such as oncogenic Ras [107,108]. Epithelial cells gain a mesenchymal phenotype by losing their polarity and cell-cell contacts during EMT. Functional loss of E-Cadherin and downregulation of epithelial cell markers such as Cytokeratins and ZO-1, and the overexpression of mesenchymal or fibroblast cell markers such as N-cadherin, vimentin and fibronectin are the main characteristics of EMT (Figure 3) [109,110].
EMT is reversible and sometimes, cells undergo the reciprocal mesenchymal to epithelial transition (MET). During the development process, EMT plays an essential role in the development of various tissues and organs such as the heart, neural crest, heart, peripheral nervous and musculoskeletal systems. Only a small number of cells in adult organisms have the ability to go through EMT process in specific physiological or pathological events such as wound healing. Nevertheless, tumour cells often gain the ability to reactivate the EMT process during metastasis, that enhance the migration and invasion capacity of cancer cells [110,111]. A number of studies have reported that Akt is frequently activated in human carcinomas [112,113,114,115,116]. Akt2 has been shown to be activated in ovarian carcinoma affecting epithelial cell morphology, tumourigenicity, cell motility and invasiveness, which is characterised by the loss of histological features of epithelial differentiation [117]. Akt shown to undergo EMT was first published in 2003 where squamous cell carcinoma cells, overexpressing activated mutant of Akt, were shown to undergo EMT and downregulate E-cadherin [118]. Loss of E-cadherin and relocalisation of β-catenin from the membrane to the nucleus is frequently detected in tumour cells undergoing EMT [119,120]. Several transcription factors have been recognized that can induce and maintain EMT process, including Snail, Twist and Zeb. The definitive molecular signalling mechanisms of normal regulation of these transcription factors, are still uncertain however they are apparently deregulated in many invasive cancers [109,121]. Evidence suggests a strong relationship between Akt and EMT-inducing transcription factors. Snail is phosphorylated by GSK3β (glycogen synthase kinase 3 beta) in normal epithelial cells but is very unstable and hardly detectable. Expression of Snail in epithelial cells strongly induces morphological changes associated with enhanced migratory capacity [122,123]. Phosphorylated Akt downregulates GSK3β by phosphorylating the S9 residue. GSK3β activates β-catenin and Snail that leads to their ubiquitination and degradation. Abrogated GSK3β on the other hand, causes the stabilisation and nuclear accumulation of β-catenin and Snail. Nuclear Snail suppresses transcription of the CDH1 gene encoding E-cadherin to stimulate the EMT process. Abrogated GSK3β stabilised the transcription factor, Snail and increases the expression of vimentin, N-cadherin and MMP-9. Nuclear β-catenin stimulates the transcription of cMYC and the cyclin D1 gene, that plays a vital role in the EMT process. This is possibly consistent with invasive cancers, where increased Akt phosphorylation leads to downregulation of GSK3β and Snail overexpression [124,125,126,127]. A recent study also suggested that the activation of the Akt/GSK3β/Snail pathway induced by Collagen type X1 α1 (COL11A1) plays a major role in the progression of pancreatic ductal cancer by facilitating EMT [128].
Y-box binding protein-1 (YB-1), a transcription/translation regulatory protein, is reported to be activated by Akt and translocated to the nucleus. Nuclear YB-1 thus phosphorylates Snail and decreases E-cadherin expression, which in turn induces EMT in invasive breast carcinoma [129]. Furthermore, upregulated Snail could in turn, increase Akt activity. Snail increases the binding of Akt2 to the E-cadherin (CDH1) promoter and Akt2 interference unexpectedly inhibits Snail repression of the CDH1 gene [130]. Akt2 could also be activated by another EMT-inducer, Twist, in invasive breast cancer cells [131]. Inhibition of Akt also downregulates Twist in cancer cells [77]. Furthermore, Akt phosphorylates and activates Twist1, which in turn enhances the phosphorylation of Akt because of increased TGFβ signalling in human breast cancer [132,133,134]. Data also suggests that the polycomb group protein named B lymphoma Mo-MLV insertion region 1 homolog (Bmi1) is a downstream target of Twist1 and is crucial for EMT and cancer metastasis [135]. Akt can phosphorylate Bmi1 directly in high grade prostate tumours [136]. Promotion of Akt activity by Bmi1 was also found to promote EMT by blocking GSK3β-mediated degradation of Snail in HNSCC and breast cancer [137,138]. Twist and Bmi1 also mediate suppression of a micro-RNA, miR let-7i, which results in NEDD9 and DOCK3 overexpression and promotes mesenchymal motility in HNSCC, melanoma and breast cancer via Rac1 [139,140,141]. In many cases breast cancer metastasis may be under the control of balance between Akt1 and Akt2 and their link with MiR-200/Zeb/E-cadherin axis [142,143]. Taken together, numerous studies establish the significant interaction between Akt and EMT inducer-associated signalling. This synergistic interaction has serious adverse pathological effects: 1) it sustains upregulation of PI3K/Akt signalling, which increases further the anti-apoptotic potential of cancer cells 2) it induces pro-invasive/metastatic gene expression, and 3) it halts the stress-induced cell cycle arrest in cancer cells [32,110,125]. Figure 4 below, illustrates the role of Akt in regulating downstream signalling molecules that in turn regulate cytoskeletal remodelling and EMT events in cancer cells.
4. Akt in HNSCC metastasis
Head and Neck Squamous Cell Carcinoma (HNSCC) denotes epithelial tumours that develop in the oral cavity, pharynx, larynx and nasal cavity. The main risk factors of HNSCC are alcohol and tobacco use and HPV infection [144,145]. It is the seventh most common cancer worldwide, with more than 887,000 cases and 450,000 deaths every year (accumulation of different head and neck cancer sites) [146]. It has recently been shown that Akt is persistently activated in the vast majority of HNSCC cases. Active forms of Akt (phosphorylated) can readily be detected in both experimental and human HNSCCs and HNSCC-derived cell lines [147,148,149]. Akt can be phosphorylated, hence activated by different growth factors, chemokines, integrins etc. and their respective receptors, ras mutations, PI3Ka gene amplification, overexpression, or activating mutations. Akt can also be activated by aberrant PTEN activity due to genetic alterations or reduced expression in HNSCC [150,151]. Akt activation is an early event in HNSCC progression which can be identified in as many as 50% of tongue preneoplastic lesions [152]. Akt activation also represents an independent prognostic marker of poor clinical outcome in both tongue and oropharyngeal HNSCCs [152,153] and is linked with the conversion of a potentially malignant oral lesion to OSCC (oral squamous cell carcinoma) [154].
Akt is known to induce morphological changes associated with EMT, loss of cell-cell adhesion and increased motility and invasion in HNSCC [109]. Oral carcinoma cells, of epithelial origin, ectopically express a mesenchyme-specific transcription factor (HMGA2) at the invasive front, which has a significant impact on tumour progression and patient survival [155]. However, the definitive evidence that EMT was induced by Akt was provided by a study in which oral squamous cell carcinoma cell lines overexpressing activated mutant Akt were shown to undergo EMT and down-regulate E-cadherin [118]. Snail and SIP1 exhibit an inverse correlation with E-cadherin expression levels in oral carcinoma cells [156,157]. OSCC clone with stable Snail overexpression displayed spindle morphology, amplified expression of vimentin and reduced expression of E-cadherin [158]. Julien et al reported that phosphorylation of NF-κB by Akt stimulate Snail expression and induces EMT in OSCC [159]. Bmi1 was found to bind with the promoter of the Akt inhibitor, PTEN and thus promoted Akt activity and in turn EMT by blocking GSK3β-mediated degradation of Snail. Interestingly, Bmi1 binds to the E-cadherin promoter but depends on Snail for E-cadherin repression. Thus, Bmi1 was found to be a player in EMT by activation of Akt, stabilisation of Snail and repression of E-cadherin in HNSCC [125,138]. Increased Twist expression is associated with downregulation of E-cadherin and may also influence the Akt pathway through an unclear mechanism in nasopharyngeal carcinoma cells [160]. Another study showed that pAkt inhibition could induce mesenchymal to epithelial transition (MET) though interaction between Twist and pAkt during EMT in OSCC [77]. SDF-1/CXCR4 system can also induce EMT via activation of the PI3k-Akt signalling pathway, resulting in lymph node metastasis of OSCC [161]. NOTCH1-inactivating mutations are observed in around 30% of HNSCC cases which activates cell proliferation and EMT though the induction of the EGFR/PI3K/Akt axis [162,163].
Research from our group suggested that VEGFA stimulated OSCC and oral cancer- associated fibroblast cell migration and can be inhibited by a specific PI3kinase and mTORC2 inhibitor. Addition of VEGF also caused increased Akt phosphorylation at both T308? and S473 residues. The phosphorylation of Akt was found to vary according to the concentration of VEGF, cell types, incubation time and assay format [148]. Although it has been suggested that differential phosphorylation of Akt at these two sites may modulate the substrate selectivity of Akt, a clear picture of this is yet to emerge [23]. In another study we also found, nuclear localisation of pAkt T308 both in VEGF-induced migrated oral carcinoma cells and VEGF-positive head and neck cancer tissue, while pAkt S473 was mainly localised in the cytoplasm. Vasco et al showed that the localisation of phosphorylated Akt varies between two forms of thyroid cancer, but nuclear localisation is linked with tumour invasion in both subtypes [164]. Akt has been reported to be abundant in the nucleus in many cancer cells yet, the mechanism of translocation, biological importance and activity has not yet been established [165]. Published data from our group also revealed that EGF (Epidermal Growth Factor), TGFα (Transforming Growth Factor α), TGFβ1(Transforming Growth Factor β1) and NGF (Nerve Growth Factor) can stimulate head and neck cancer cell migration and a specific PI3k/Akt pathway inhibitor such as PI103 or MK2206 can effectively block growth factor-induced cell migration [166,167]. Study from our research group also suggested that receptor tyrosine kinase inhibitors such as Gefitinib and Erlotinib inhibited the migration of head and neck cancer cells by inhibiting both Akt and MAPK phosphorylation [168]. Cetuximab, a monoclonal antibody, targeting EGFR is the only FDA approved targeted therapy for the treatment of recurrent/metastatic head and neck cancer, in combination with radiation therapy or as a single agent in patients who have had prior platinum-based therapy. The response rate, as a single agent, is only 13% and the patients who respond initially eventually develop resistance [169]. Evidence showed that an EGFR mutation at S493R inhibits Cetuximab-binding with the receptor but does not block EGF or TGFα binding. EGF or TGFα may therefore activate the downstream PI3K/Akt signalling pathway. Cetuximab resistance can also be mediated by the activation of the Akt signalling pathway in an alternative way, such as the overexpression of other growth factors (TGFβ, VEGF and NGF) and their associated receptors by the tumour cells and/or the tumour microenvironment [170]. A recent study also demonstrated that increased Akt 1/2/3 phosphorylation to be the characteristics for acquired Cetuximab resistance in head and neck squamous cell carcinoma [171]. The growth of HNSCC is maintained by a population of specialized cells, cancer stem cells (CSCs) which possess unlimited self-renewal potential and induce tumour regrowth, if not eliminated by therapy. Given their self-renewal properties, CSCs are thought to play a key role in tumour growth and metastasis, but also in recurrence making CSC-related gene and protein expression a promising biomarker candidate and therapeutic target [172]. Evidence suggests that HNSCC metastasis is associated with Bmi1-positive CSCs, which are responsible for tumour invasion, drug resistance and lymph node metastasis. Migration/invasion abilities, cancer stemness and EMT phenotype of HNSCC CSCs are maintained by the Twist/Bmi1/Akt/β-catenin signalling pathway [173,174]. So, targeting the Akt pathway in HNSCC CSCs could be an innovative way to treat cancer whilst avoiding drug resistance. A few recent clinical trials using an Akt inhibitor or PI3K inhibitor alone to treat late stage or recurrent head and neck cancer did not show promising outcomes (Table 1).
It is worth noting here that activated receptor tyrosine kinases activate not only the PI3K-Akt signalling pathway, but also other pathways including the MAPK and SMAD pathways. Signalling pathways are activated in a context-dependent manner and crosstalk among each other. Hence, targeted inhibition of one pathway downstream of the receptors may not affect other pathways and that adds complexity to therapeutic targeting. A recent study suggested that the combination of an Akt inhibitor and Cetuximab might be a favourable novel therapeutic strategy to overcome acquired Cetuximab resistance in HNSCC patients [171].
5. Conclusions
Extensive studies have demonstrated that activation of Akt by phosphorylation of different amino acid residues determines substrate selectivity and thus exerts different biological activity in different cell types. Three highly homologous Akt isoforms have non-overlapping and opposing functions in different cancer types. As Akt is the central signalling node that incorporates cell membrane, cytoplasmic and nuclear signals regulating cell fate, analysing Akt isoforms and cell type specific signalling pathways and targeting them will contribute to personalised targeted HNSCC therapy. So, carefully designing a clinical study using a combination of a PI3K-Akt pathway inhibitor and another signalling molecule inhibitor or receptor inhibitor during the early stages of HNSCC might result in an expected positive outcome.
Author Contributions
Writing—original draft preparation, MI.; writing—review and editing, SJ, IE.; visualization, MI.; supervision, SJ, IE. All authors have read and agreed to the published version of the manuscript.
Funding
The author would like to thank SORSAS (Scottish Overseas Research Student Award Scheme) and Anonymous trust for funding the project.
Institutional Review Board Statement
Not Applicable
Informed Consent Statement
Not Applicable
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The author would like to thank Jacqui Cox for her excellent technical work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dillekås H, Rogers MS, Straume O. Are 90% of deaths from cancer caused by metastases? Cancer medicine. 2019;8(12):5574-6.
- Palmer TD, Ashby WJ, Lewis JD, Zijlstra A. Targeting tumor cell motility to prevent metastasis. Adv Drug Deliv Rev. 2011;63(8):568-81.
- Leber MF, Efferth T. Molecular principles of cancer invasion and metastasis (review). Int J Oncol. 2009;34(4):881-95.
- Robert, J. Biology of cancer metastasis. Bulletin du cancer. 2013. [CrossRef]
- Woodhouse EC, Chuaqui RF, Liotta LA. General mechanisms of metastasis. Cancer. 1997;80(8 Suppl):1529-37.
- Friedl P, Brocker EB. The biology of cell locomotion within three-dimensional extracellular matrix. Cell Mol Life Sci. 2000;57(1):41-64.
- Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3(5):362-74. [CrossRef]
- Zhou H, Huang S. Role of mTOR Signaling in Tumor Cell Motility, Invasion and Metastasis. Curr Protein Pept Sci. 2011;12(1):30-42.
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84(3):359-69.
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell Migration: Integrating Signals from Front to Back. Science. 2003;302(5651):1704-9.
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84(3):371-9.
- Schmidt A, Hall MN. Signaling to the actin cytoskeleton. Annual review of cell and developmental biology. 1998;14:305-38. [CrossRef]
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112(4):453-65.
- Ahmed H, Ghoshal A, Jones S, Ellis I, Islam M. Head and Neck Cancer Metastasis and the Effect of the Local Soluble Factors, from the Microenvironment, on Signalling Pathways: Is It All about the Akt? Cancers (Basel). 2020;12(8). [CrossRef]
- Ellis IR, Islam MR, Aljorani L, Jones SJ. Fibronectin: the N-terminal region and its role in cell migration- implications for disease and healing. In: Beattie J, editor. Fibronectin: Current Concepts in Structure, Function and Pathology. Protein Biochemistry, Synthesis, Structure and Cellular Functions. New York: Nova Science publishers; 2012. p. 35-69.
- Friedl, P. Prespecification and plasticity: shifting mechanisms of cell migration. Curr Opin Cell Biol. 2004;16(1):14-23. [CrossRef]
- Inaki M, Vishnu S, Cliffe A, Rorth P. Effective guidance of collective migration based on differences in cell states. Proc Natl Acad Sci U S A. 2012;109(6):2027-32. [CrossRef]
- Rorth, P. Whence directionality: guidance mechanisms in solitary and collective cell migration. Dev Cell. 2011;20(1):9-18. [CrossRef]
- Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14(8):777-83.
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261-74.
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes & development. 1999;13(22):2905-27. [CrossRef]
- Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Current Opinion in Genetics & Development. 1998;8(1):55-62. [CrossRef]
- Bozulic L, Hemmings BA. PIKKing on PKB: regulation of PKB activity by phosphorylation. Current Opinion in Cell Biology. 2009;21(2):256-61.
- Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt Hydrophobic Motif Ser-473 Kinase as DNA-dependent Protein Kinase. Journal of Biological Chemistry. 2004;279(39):41189-96. [CrossRef]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098-101. [CrossRef]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17(3):313-25. [CrossRef]
- Hart JR, Vogt PK. Phosphorylation of AKT: a mutational analysis. Oncotarget. 2011;2(6):467-76.
- Ikenoue T, Inoki K, Yang Q, Zhou X, Guan K-L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27(14):1919-31. doi: http://www.nature.com/ emboj/journal/v27/ n14/ suppinfo/emboj2008119a_S1.html.
- De Marco C, Rinaldo N, Bruni P, Malzoni C, Zullo F, Fabiani F, et al. Multiple genetic alterations within the PI3K pathway are responsible for AKT activation in patients with ovarian carcinoma. PLoS One. 2013;8(2):e55362. [CrossRef]
- Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K, Sahin A, Liu S, Barrera JA, et al. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Molecular cancer therapeutics. 2011;10(6):1093-101.
- Wu R, Baker SJ, Hu TC, Norman KM, Fearon ER, Cho KR. Type I to Type II Ovarian Carcinoma Progression: Mutant Trp53 or Pik3ca Confers a More Aggressive Tumor Phenotype in a Mouse Model of Ovarian Cancer. Am J Pathol. 2013;182(4):1391-9.
- Xue G, Hemmings BA. PKB/Akt-Dependent Regulation of Cell Motility. J Natl Cancer Inst. 2013;105(6):393-404. [CrossRef]
- Yoeli-Lerner M, Toker A. Akt/PKB Signaling in Cancer: A Function in Cell Motility and Invasion. Cell Cycle. 2006;5(6):603-5.
- Frixione, E. Recurring views on the structure and function of the cytoskeleton: a 300-year epic. Cell motility and the cytoskeleton. 2000;46(2):73-94. [CrossRef]
- Bonello T, Coombes J, Schevzov G, Gunning P, Stehn J. Therapeutic Targeting of the Actin Cytoskeleton in Cancer. In: Kavallaris M, editor. Cytoskeleton and Human Disease. New York: Humana Press; 2012. p. 181-200.
- Bugyi B, Carlier MF. Control of actin filament treadmilling in cell motility. Annual review of biophysics. 2010;39:449-70. [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1(1):27-31.
- Morales-Ruiz M, Fulton D, Sowa G, Languino LR, Fujio Y, Walsh K, et al. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ Res. 2000;86(8):892-6.
- Dimmeler S, Dernbach E, Zeiher AM. Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett. 2000;477(3):258-62.
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601-5. [CrossRef]
- Qian Y, Corum L, Meng Q, Blenis J, Zheng JZ, Shi X, et al. PI3K induced actin filament remodeling through Akt and p70S6K1: implication of essential role in cell migration. Am J Physiol Cell Physiol. 2004;286(1):C153-63. [CrossRef]
- Qian Y, Zhong X, Flynn DC, Zheng JZ, Qiao M, Wu C, et al. ILK mediates actin filament rearrangements and cell migration and invasion through PI3K/Akt/Rac1 signaling. Oncogene. 2005;24(19):3154-65. [CrossRef]
- Ip CKM, Cheung ANY, Ngan HYS, Wong AST. p70 S6 kinase in the control of actin cytoskeleton dynamics and directed migration of ovarian cancer cells. Oncogene. 2011;30(21):2420-32. [CrossRef]
- Chodniewicz D, Zhelev DV. Chemoattractant receptor-stimulated F-actin polymerization in the human neutrophil is signaled by 2 distinct pathways. Blood. 2003;101(3):1181-4. [CrossRef]
- Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, et al. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64(13):4394-9. [CrossRef]
- Amiri A, Noei F, Jeganathan S, Kulkarni G, Pinke DE, Lee JM. eEF1A2 activates Akt and stimulates Akt-dependent actin remodeling, invasion and migration. Oncogene. 2007;26(21):3027-40. [CrossRef]
- Cenni V, Sirri A, Riccio M, Lattanzi G, Santi S, de Pol A, et al. Targeting of the Akt/PKB kinase to the actin skeleton. Cell Mol Life Sci. 2003;60(12):2710-20. [CrossRef]
- Ho YP, Kuo CW, Hsu YT, Huang YS, Yew LP, Huang WF, et al. beta-Actin is a downstream effector of the PI3K/AKT signaling pathway in myeloma cells. Mol Cell Biochem. 2011;348(1-2):129-39. [CrossRef]
- Zhou GL, Zhuo Y, King CC, Fryer BH, Bokoch GM, Field J. Akt phosphorylation of serine 21 on Pak1 modulates Nck binding and cell migration. Mol Cell Biol. 2003;23(22):8058-69.
- Chung CY, Potikyan G, Firtel RA. Control of cell polarity and chemotaxis by Akt/PKB and PI3 kinase through the regulation of PAKa. Mol Cell. 2001;7(5):937-47.
- Chhabra ES, Higgs HN. The many faces of actin: matching assembly factors with cellular structures. nature cell biology. 2007;9(10):1110-21.
- Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, et al. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell. 2005;9(3):389-402. [CrossRef]
- Jiang P, Enomoto A, Jijiwa M, Kato T, Hasegawa T, Ishida M, et al. An actin-binding protein Girdin regulates the motility of breast cancer cells. Cancer Res. 2008;68(5):1310-8. [CrossRef]
- Natsume A, Kato T, Kinjo S, Enomoto A, Toda H, Shimato S, et al. Girdin maintains the stemness of glioblastoma stem cells. Oncogene. 2012;31(22):2715-24. [CrossRef]
- Weng L, Enomoto A, Ishida-Takagishi M, Asai N, Takahashi M. Girding for migratory cues: roles of the Akt substrate Girdin in cancer progression and angiogenesis. Cancer Science. 2010;101(4):836-42. [CrossRef]
- Shibata T, Matsuo Y, Shamoto T, Hirokawa T, Tsuboi K, Takahashi H, et al. Girdin, a regulator of cell motility, is a potential prognostic marker for esophageal squamous cell carcinoma. Oncol Rep. 2013;29(6):2127-32. [CrossRef]
- Yamamura Y, Asai N, Enomoto A, Kato T, Mii S, Kondo Y, et al. Akt–Girdin Signaling in Cancer-Associated Fibroblasts Contributes to Tumor Progression. Cancer Research. 2015;75(5):813-23. [CrossRef]
- Cenni V, Bavelloni A, Beretti F, Tagliavini F, Manzoli L, Lattanzi G, et al. Ankrd2/ARPP is a novel Akt2 specific substrate and regulates myogenic differentiation upon cellular exposure to H(2)O(2). Mol Biol Cell. 2011;22(16):2946-56. [CrossRef]
- Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6(11):1034-8. [CrossRef]
- Ravid D, Chuderland D, Landsman L, Lavie Y, Reich R, Liscovitch M. Filamin A is a novel caveolin-1-dependent target in IGF-I-stimulated cancer cell migration. Exp Cell Res. 2008;314(15):2762-73. [CrossRef]
- Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, et al. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2(2):138-45. [CrossRef]
- Ravid D, Maor S, Werner H, Liscovitch M. Caveolin-1 inhibits cell detachment-induced p53 activation and anoikis by upregulation of insulin-like growth factor-I receptors and signaling. Oncogene. 2005;24(8):1338-47. [CrossRef]
- Nallapalli RK, Ibrahim MX, Zhou AX, Bandaru S, Sunkara SN, Redfors B, et al. Targeting filamin A reduces K-RAS-induced lung adenocarcinomas and endothelial response to tumor growth in mice. Mol Cancer. 2012;11:50. [CrossRef]
- Meima ME, Webb BA, Witkowska HE, Barber DL. The sodium-hydrogen exchanger NHE1 is an Akt substrate necessary for actin filament reorganization by growth factors. J Biol Chem. 2009;284(39):26666-75. [CrossRef]
- Denker SP, Barber DL. Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J Cell Biol. 2002;159(6):1087-96. [CrossRef]
- Martin C, Pedersen SF, Schwab A, Stock C. Intracellular pH gradients in migrating cells. Am J Physiol Cell Physiol. 2011;300(3):C490-5. [CrossRef]
- Stock C, Schwab A. Role of the Na/H exchanger NHE1 in cell migration. Acta physiologica (Oxford, England). 2006;187(1-2):149-57. [CrossRef]
- Stuwe L, Muller M, Fabian A, Waning J, Mally S, Noel J, et al. pH dependence of melanoma cell migration: protons extruded by NHE1 dominate protons of the bulk solution. The Journal of physiology. 2007;585(Pt 2):351-60. [CrossRef]
- Clement DL, Mally S, Stock C, Lethan M, Satir P, Schwab A, et al. PDGFRalpha signaling in the primary cilium regulates NHE1-dependent fibroblast migration via coordinated differential activity of MEK1/2-ERK1/2-p90RSK and AKT signaling pathways. J Cell Sci. 2013;126(Pt 4):953-65. [CrossRef]
- Chang L, Goldman RD. Intermediate filaments mediate cytoskeletal crosstalk. Nat Rev Mol Cell Biol. 2004;5(8):601-13. [CrossRef]
- Helfand BT, Chang L, Goldman RD. Intermediate filaments are dynamic and motile elements of cellular architecture. J Cell Sci. 2004;117(Pt 2):133-41. [CrossRef]
- Zhu QS, Rosenblatt K, Huang KL, Lahat G, Brobey R, Bolshakov S, et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene. 2011;30(4):457-70. [CrossRef]
- Lahat G, Zhu QS, Huang KL, Wang S, Bolshakov S, Liu J, et al. Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS One. 2010;5(4):e10105. [CrossRef]
- Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68(18):3033-46. [CrossRef]
- Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol. 2009;11(4):397-408. [CrossRef]
- Lin HK, Wang G, Chen Z, Teruya-Feldstein J, Liu Y, Chan CH, et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat Cell Biol. 2009;11(4):420-32. [CrossRef]
- Hong KO, Kim JH, Hong JS, Yoon HJ, Lee JI, Hong SP, et al. Inhibition of Akt activity induces the mesenchymal-to-epithelial reverting transition with restoring E-cadherin expression in KB and KOSCC-25B oral squamous cell carcinoma cells. Journal of experimental & clinical cancer research : CR. 2009;28:28. [CrossRef]
- McPhee TR, McDonald PC, Oloumi A, Dedhar S. Integrin-linked kinase regulates E-cadherin expression through PARP-1. Developmental dynamics : an official publication of the American Association of Anatomists. 2008;237(10):2737-47. [CrossRef]
- Onishi K, Higuchi M, Asakura T, Masuyama N, Gotoh Y. The PI3K-Akt pathway promotes microtubule stabilization in migrating fibroblasts. Genes to cells : devoted to molecular & cellular mechanisms. 2007;12(4):535-46. [CrossRef]
- Ridnour LA, Barasch KM, Windhausen AN, Dorsey TH, Lizardo MM, Yfantis HG, et al. Nitric oxide synthase and breast cancer: role of TIMP-1 in NO-mediated Akt activation. PLoS One. 2012;7(9):e44081. [CrossRef]
- Xu W, Liu LZ, Loizidou M, Ahmed M, Charles IG. The role of nitric oxide in cancer. Cell Res. 2002;12(5-6):311-20. [CrossRef]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4(5):397-407. [CrossRef]
- Lee MJ, Thangada S, Paik JH, Sapkota GP, Ancellin N, Chae SS, et al. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Mol Cell. 2001;8(3):693-704. [CrossRef]
- Ozaki H, Hla T, Lee MJ. Sphingosine-1-phosphate signaling in endothelial activation. Journal of atherosclerosis and thrombosis. 2003;10(3):125-31.
- Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16(1):9-20. [CrossRef]
- Pasquale, EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10(3):165-80. [CrossRef]
- Kawai H, Kobayashi M, Hiramoto-Yamaki N, Harada K, Negishi M, Katoh H. Ephexin4-mediated promotion of cell migration and anoikis resistance is regulated by serine 897 phosphorylation of EphA2. FEBS Open Bio. 2013;3(0):78-82. [CrossRef]
- Li J, Ballif BA, Powelka AM, Dai J, Gygi SP, Hsu VW. Phosphorylation of ACAP1 by Akt regulates the stimulation-dependent recycling of integrin beta1 to control cell migration. Dev Cell. 2005;9(5):663-73. [CrossRef]
- Rowland AF, Larance M, Hughes WE, James DE. Identification of RhoGAP22 as an Akt-dependent regulator of cell motility in response to insulin. Mol Cell Biol. 2011;31(23):4789-800. [CrossRef]
- Berven LA, Willard FS, Crouch MF. Role of the p70(S6K) pathway in regulating the actin cytoskeleton and cell migration. Exp Cell Res. 2004;296(2):183-95. [CrossRef]
- Sakakibara K, Liu B, Hollenbeck S, Kent KC. Rapamycin inhibits fibronectin-induced migration of the human arterial smooth muscle line (E47) through the mammalian target of rapamycin. Am J Physiol Heart Circ Physiol. 2005;288(6):H2861-8. [CrossRef]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648-57. [CrossRef]
- Liu, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25(53):7029-40. [CrossRef]
- Liu L, Chen L, Chung J, Huang S. Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene. 2008;27(37):4998-5010. [CrossRef]
- Benefield J, Meisinger J, Petruzzelli GJ, Young MR. Endothelial cell response to human head and neck squamous cell carcinomas involves downregulation of protein phosphatases-1/2A, cytoskeletal depolymerization and increased motility. Invasion & metastasis. 1997;17(4):210-20.
- Jackson JL, Young MR. Protein phosphatase-2A regulates protein tyrosine phosphatase activity in Lewis lung carcinoma tumor variants. Clin Exp Metastasis. 2003;20(4):357-64.
- Li Y, Wang X, Yue P, Tao H, Ramalingam SS, Owonikoko TK, et al. Protein phosphatase 2A and DNA-dependent protein kinase are involved in mediating rapamycin-induced Akt phosphorylation. J Biol Chem. 2013. [CrossRef]
- Liu L, Chen L, Luo Y, Chen W, Zhou H, Xu B, et al. Rapamycin inhibits IGF-1 stimulated cell motility through PP2A pathway. PLoS One. 2010;5(5):e10578. [CrossRef]
- Wlodarski P, Grajkowska W, Lojek M, Rainko K, Jozwiak J. Activation of Akt and Erk pathways in medulloblastoma. Folia neuropathologica / Association of Polish Neuropathologists and Medical Research Centre, Polish Academy of Sciences. 2006;44(3):214-20.
- Fong Y-C, Hsu S-F, Wu C-L, Li T-M, Kao S-T, Tsai F-J, et al. Transforming growth factor-β1 increases cell migration and β1 integrin up-regulation in human lung cancer cells. Lung Cancer. 2009;64(1):13-21. [CrossRef]
- Yeh Y-Y, Chiao C-C, Kuo W-Y, Hsiao Y-C, Chen Y-J, Wei Y-Y, et al. TGF-β1 increases motility and αvβ3 integrin up-regulation via PI3K, Akt and NF-κB-dependent pathway in human chondrosarcoma cells. Biochemical Pharmacology. 2008;75(6):1292-301. [CrossRef]
- Busch S, Renaud SJ, Schleussner E, Graham CH, Markert UR. mTOR mediates human trophoblast invasion through regulation of matrix-remodeling enzymes and is associated with serine phosphorylation of STAT3. Exp Cell Res. 2009;315(10):1724-33. [CrossRef]
- Kakinuma N, Roy BC, Zhu Y, Wang Y, Kiyama R. Kank regulates RhoA-dependent formation of actin stress fibers and cell migration via 14-3-3 in PI3K-Akt signaling. J Cell Biol. 2008;181(3):537-49. [CrossRef]
- Chin YR, Toker A. The Actin-Bundling Protein Palladin Is an Akt1-Specific Substrate that Regulates Breast Cancer Cell Migration. Molecular Cell. 2010;38(3):333-44. [CrossRef]
- Chin YR, Toker A. Akt2 regulates expression of the actin-bundling protein palladin. FEBS Lett. 2010;584(23):4769-74. [CrossRef]
- Liu, Radisky DC, Nelson CM, Zhang H, Fata JE, Roth RA, et al. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc Natl Acad Sci U S A. 2006;103(11):4134-9. [CrossRef]
- Nieto, MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annual review of cell and developmental biology. 2011;27:347-76. [CrossRef]
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818-29. [CrossRef]
- Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene. 2005;24(50):7443-54. [CrossRef]
- Zheng H, Kang Y. Multilayer control of the EMT master regulators. Oncogene. 2013. [CrossRef]
- Thompson EW, Williams ED. EMT and MET in carcinoma--clinical observations, regulatory pathways and new models. Clin Exp Metastasis. 2008;25(6):591-2. [CrossRef]
- Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Advances in cancer research. 2005;94:29-86. [CrossRef]
- Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer treatment reviews. 2004;30(2):193-204. [CrossRef]
- Ringel MD, Hayre N, Saito J, Saunier B, Schuppert F, Burch H, et al. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001;61(16):6105-11.
- Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A. 2001;98(20):10983-5. [CrossRef]
- Wu HT, Ko SY, Fong JH, Chang KW, Liu TY, Kao SY. Expression of phosphorylated Akt in oral carcinogenesis and its induction by nicotine and alkaline stimulation. J Oral Pathol Med. 2009;38(2):206-13.
- Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64(4):280-5.
- Grille SJ, Bellacosa A, Upson J, Klein-Szanto AJ, Van Roy F, Lee-Kwon W, et al. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer research. 2003;63(9):2172-8.
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420-8. [CrossRef]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871-90. [CrossRef]
- Bellacosa A, Larue L. PI3K/AKT Pathway and the Epithelial–Mesenchymal Transition. In: Thomas-Tikhonenko A, editor. Cancer Genome and Tumor Microenvironment. New York: Springer Science+Business Media; 2010. p. 11-31.
- Katoh M, Katoh M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol Ther. 2006;5(9):1059-64.
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6(10):931-40. [CrossRef]
- Ha G-H, Park J-S, Breuer E-KY. TACC3 promotes epithelial–mesenchymal transition (EMT) through the activation of PI3K/Akt and ERK signaling pathways. Cancer Letters. 2013;332(0):63-73. [CrossRef]
- Smith A, Teknos TN, Pan Q. Epithelial to mesenchymal transition in head and neck squamous cell carcinoma. Oral Oncol. 2013;49(4):287-92. [CrossRef]
- Wang H, Wang H-S, Zhou B-H, Li C-L, Zhang F, Wang X-F, et al. Epithelial–Mesenchymal Transition (EMT) Induced by TNF-α Requires AKT/GSK-3β-Mediated Stabilization of Snail in Colorectal Cancer. PLOS ONE. 2013;8(2):e56664. [CrossRef]
- Wu K, Fan J, Zhang L, Ning Z, Zeng J, Zhou J, et al. PI3K/Akt to GSK3β/β-catenin signaling cascade coordinates cell colonization for bladder cancer bone metastasis through regulating ZEB1 transcription. Cellular Signalling. 2012;24(12):2273-82. [CrossRef]
- Wang H, Zhou H, Ni H, Shen X. COL11A1-Driven pithelialMesenchymal Transition and Stemness of Pancreatic Cancer Cells Induce Cell Migration and Invasion by Modulating the AKT/GSK-3β/Snail Pathway. Biomolecules. 2022;12(3):391.
- Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell. 2009;15(5):402-15. [CrossRef]
- Villagrasa P, Diaz VM, Vinas-Castells R, Peiro S, Del Valle-Perez B, Dave N, et al. Akt2 interacts with Snail1 in the E-cadherin promoter. Oncogene. 2012;31(36):4022-33. [CrossRef]
- Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007;67(5):1979-87. [CrossRef]
- Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D, et al. Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-beta signaling axes. Cancer Discov. 2012;2(3):248-59. [CrossRef]
- Yao K, Ye PP, Tan J, Tang XJ, Shen Tu XC. Involvement of PI3K/Akt pathway in TGF-beta2-mediated epithelial mesenchymal transition in human lens epithelial cells. Ophthalmic research. 2008;40(2):69-76. [CrossRef]
- Yokoyama K, Kimoto K, Itoh Y, Nakatsuka K, Matsuo N, Yoshioka H, et al. The PI3K/Akt pathway mediates the expression of type I collagen induced by TGF-beta2 in human retinal pigment epithelial cells. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2012;250(1):15-23. [CrossRef]
- Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010;12(10):982-92. [CrossRef]
- Nacerddine K, Beaudry JB, Ginjala V, Westerman B, Mattiroli F, Song JY, et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J Clin Invest. 2012;122(5):1920-32. [CrossRef]
- Guo BH, Feng Y, Zhang R, Xu LH, Li MZ, Kung HF, et al. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol Cancer. 2011;10(1):10. [CrossRef]
- Song LB, Li J, Liao WT, Feng Y, Yu CP, Hu LJ, et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest. 2009;119(12):3626-36. [CrossRef]
- Ahn J, Sanz-Moreno V, Marshall CJ. The metastasis gene NEDD9 product acts through integrin beta3 and Src to promote mesenchymal motility and inhibit amoeboid motility. J Cell Sci. 2012;125(Pt 7):1814-26. [CrossRef]
- Sanz-Moreno, V. Tumour invasion: a new twist on Rac-driven mesenchymal migration. Curr Biol. 2012;22(11):R449-51. [CrossRef]
- Yang W-H, Lan H-Y, Huang C-H, Tai S-K, Tzeng C-H, Kao S-Y, et al. RAC1 activation mediates Twist1-induced cancer cell migration. Nat Cell Biol. 2012;14(4):366-74. doi: http://www.nature.com/ncb/ journal/ v14/n4/ abs/ ncb2455.html#supplementary-information.
- Hill L, Browne G, Tulchinsky E. ZEB/miR-200 feedback loop: at the crossroads of signal transduction in cancer. Int J Cancer. 2013;132(4):745-54. [CrossRef]
- Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, et al. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal. 2009;2(92):ra62. [CrossRef]
- Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92(9):709-20.
- Neville BW, Day TA. Oral cancer and precancerous lesions. CA: a cancer journal for clinicians. 2002;52(4):195-215.
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018;68(6):394-424. [CrossRef]
- Islam MR, Ellis IR, Macluskey M, Cochrane L, Jones SJ. Activation of Akt at T308 and S473 in alcohol, tobacco and HPV-induced HNSCC: is there evidence to support a prognostic or diagnostic role? Exp Hematol Oncol. 2014;3(1):25. [CrossRef]
- Islam MR, Jones SJ, Macluskey M, Ellis IR. Is there a pAkt between VEGF and oral cancer cell migration? Cellular Signalling. 2014;26(6):1294-302. [CrossRef]
- Amornphimoltham P, Sriuranpong V, Patel V, Benavides F, Conti CJ, Sauk J, et al. Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clin Cancer Res. 2004;10(12 Pt 1):4029-37. [CrossRef]
- Amornphimoltham P, Patel V, Molinolo A, Gutkind JS. Head and Neck Cancer and PI3K/Akt/mTOR Signaling Network: Novel Molecular Targeted Therapy. In: Glick AB, Van Waes C, editors. Signaling Pathways in Squamous Cancer. Springer Science+Business Media, LLC; 2011. p. 407-30.
- Marquard FE, Jücker M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem Pharmacol. 2020;172:113729. [CrossRef]
- Massarelli E, Liu DD, Lee JJ, El-Naggar AK, Lo Muzio L, Staibano S, et al. Akt activation correlates with adverse outcome in tongue cancer. Cancer. 2005;104(11):2430-6. [CrossRef]
- Yu Z, Weinberger PM, Sasaki C, Egleston BL, Speier WFt, Haffty B, et al. Phosphorylation of Akt (Ser473) predicts poor clinical outcome in oropharyngeal squamous cell cancer. Cancer Epidemiol Biomarkers Prev. 2007;16(3):553-8. [CrossRef]
- Pontes HA, de Aquino Xavier FC, da Silva TS, Fonseca FP, Paiva HB, Pontes FS, et al. Metallothionein and p-Akt proteins in oral dysplasia and in oral squamous cell carcinoma: an immunohistochemical study. J Oral Pathol Med. 2009;38(8):644-50. [CrossRef]
- Miyazawa J, Mitoro A, Kawashiri S, Chada KK, Imai K. Expression of Mesenchyme-Specific Gene HMGA2 in Squamous Cell Carcinomas of the Oral Cavity. Cancer Research. 2004;64(6):2024-9.
- Maeda G, Chiba T, Okazaki M, Satoh T, Taya Y, Aoba T, et al. Expression of SIP1 in oral squamous cell carcinomas: implications for E-cadherin expression and tumor progression. International journal of oncology. 2005;27(6):1535-41.
- Yokoyama K, Kamata N, Hayashi E, Hoteiya T, Ueda N, Fujimoto R, et al. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncology. 2001;37(1):65-71.
- Taki M, Kamata N, Yokoyama K, Fujimoto R, Tsutsumi S, Nagayama M. Down-regulation of Wnt-4 and up-regulation of Wnt-5a expression by epithelial-mesenchymal transition in human squamous carcinoma cells. Cancer Science. 2003;94(7):593-7.
- Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, et al. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26(53):7445-56. [CrossRef]
- Zhang X, Wang Q, Ling MT, Wong YC, Leung SC, Wang X. Anti-apoptotic role of TWIST and its association with Akt pathway in mediating taxol resistance in nasopharyngeal carcinoma cells. Int J Cancer. 2007;120(9):1891-8. [CrossRef]
- Onoue T, Uchida D, Begum NM, Tomizuka Y, Yoshida H, Sato M. Epithelial-mesenchymal transition induced by the stromal cell-derived factor-1/CXCR4 system in oral squamous cell carcinoma cells. International Journal of Oncology. 2006;29(5):1133-8.
- Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157-60. [CrossRef]
- Zheng Y, Wang Z, Xiong X, Zhong Y, Zhang W, Dong Y, et al. Membrane-tethered Notch1 exhibits oncogenic property via activation of EGFR–PI3K–AKT pathway in oral squamous cell carcinoma. Journal of Cellular Physiology. 2019;234(5):5940-52. [CrossRef]
- Vasko V, Saji M, Hardy E, Kruhlak M, Larin A, Savchenko V, et al. Akt activation and localisation correlate with tumour invasion and oncogene expression in thyroid cancer. J Med Genet. 2004;41(3):161-70.
- Wang R, Brattain MG. AKT can be activated in the nucleus. Cell Signal. 2006;18(10):1722-31.
- Alkhadar H, Macluskey M, White S, Ellis I. Nerve growth factor-induced migration in oral and salivary gland tumour cells utilises the PI3K/Akt signalling pathway: Is there a link to perineural invasion? J Oral Pathol Med. 2020;49(3):227-34. [CrossRef]
- Islam M, Alghamdi A, Sriramula P, Shalgm B, Jones S, Ellis I. Is it all just an Akt - you'd be SMAD to believe it! Role of TGFβ1 in oral cancer metastasis. Science Repository (Dental Oral Biology and Craniofacial Research). 2018;1(3). [CrossRef]
- Thwe AM, Mossey P, Ellis IR. Effect of tyrosine kinase inhibitors on cell migration and epithelial-to-mesenchymal transition in Asian head and neck cancer cell lines. J Oral Pathol Med. 2021;50(10):1031-9. [CrossRef]
- Khattri A, Sheikh N, Acharya R, Tan Y-HC, Kochanny S, Lingen MW, et al. Mechanism of acquired resistance to cetuximab in head and neck cancer. Journal of Clinical Oncology. 2018;36(15_suppl):e18061-e. [CrossRef]
- Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18(2):221-3. [CrossRef]
- Zaryouh H, De Pauw I, Baysal H, Pauwels P, Peeters M, Vermorken JB, et al. The Role of Akt in Acquired Cetuximab Resistant Head and Neck Squamous Cell Carcinoma: An In Vitro Study on a Novel Combination Strategy. Frontiers in Oncology. 2021;11(3658). [CrossRef]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105-11. [CrossRef]
- Chen D, Wu M, Li Y, Chang I, Yuan Q, Ekimyan-Salvo M, et al. Targeting BMI1(+) Cancer Stem Cells Overcomes Chemoresistance and Inhibits Metastases in Squamous Cell Carcinoma. Cell Stem Cell. 2017;20(5):621-34.e6. [CrossRef]
- Lai YJ, Yu WN, Kuo SC, Ho CT, Hung CM, Way TD, et al. CSC-3436 inhibits TWIST-induced epithelial-mesenchymal transition via the suppression of Twist/Bmi1/Akt pathway in head and neck squamous cell carcinoma. J Cell Physiol. 2019;234(6):9118-29. [CrossRef]
- Institute NC: Akt Inhibitor MK2206 in Treating Patients With Recurrent or Metastatic Head and Neck Cancer. https://ClinicalTrials.gov/show/NCT01349933 (2011). Accessed. 04 April.
- Ma BB, Goh BC, Lim WT, Hui EP, Tan EH, Lopes Gde L, et al. Multicenter phase II study of the AKT inhibitor MK-2206 in recurrent or metastatic nasopharyngeal carcinoma from patients in the mayo phase II consortium and the cancer therapeutics research group (MC1079). Invest New Drugs. 2015;33(4):985-91. [CrossRef]
- Ho AL, Foster NR, Meyers JP, Vasudeva SD, Katabi N, Antonescu CR, et al. Alliance A091104: A phase II trial of MK-2206 in patients (pts) with progressive, recurrent/metastatic adenoid cystic carcinoma. Journal of Clinical Oncology. 2015;33(15_suppl):6039-. [CrossRef]
- University Y: An Open Label, Single Arm, Multicenter Phase II Study of BYL719 in Patients With Recurrent or Metastatic Squamous Cell Carcinoma of Head and Neck Who Failed to Respond to Platinum-based Therapy. (2016). Accessed.
- Kim HR, Kang HN, Yun MR, Lim SM, Kim CG, Ahn M-J, et al. Clinical trials outcomes of combined BKM120 and cetuximab compared to BKM120 in recurrent and/or metastatic squamous cell carcinoma of head and neck (R/M-SCCHN). Journal of Clinical Oncology. 2015;33(15_suppl):6049-. [CrossRef]
- Rodon J, Curigliano G, Delord J-P, Harb W, Azaro A, Han Y, et al. A Phase Ib, open-label, dose-finding study of alpelisib in combination with paclitaxel in patients with advanced solid tumors. Oncotarget. 2018;9(60):31709-18. [CrossRef]
- Institute D-FC, Pharmaceuticals N: Phase Ib Study of BKM120 With Cisplatin and XRT in High Risk Locally Advanced Squamous Cell Cancer of Head and Neck. (2014). Accessed.
- Hospital SNU, Group KCS, Hospital CNU: Korean Cancer Study Group: Translational bIomarker Driven UMbrella Project for Head and Neck (TRIUMPH), Esophageal Squamous Cell Carcinoma- Part 1 (HNSCC). (2017). Accessed.
- Chicago Uo, Institute NC: PI3K Inhibitor BKM120 and Cetuximab in Treating Patients With Recurrent or Metastatic Head and Neck Cancer. (2013). Accessed.
Figure 1.
Metastasis cascade. Tumour cells proliferate uncontrollably and eventually lose their adhesive phenotype. The motile cells migrate and invade into surrounding tissues and intravasate to lymphatic and blood vessels. Circulating tumour cells then extravasate, enter into another tissue and form micro-metastases at the secondary site.
Figure 1.
Metastasis cascade. Tumour cells proliferate uncontrollably and eventually lose their adhesive phenotype. The motile cells migrate and invade into surrounding tissues and intravasate to lymphatic and blood vessels. Circulating tumour cells then extravasate, enter into another tissue and form micro-metastases at the secondary site.
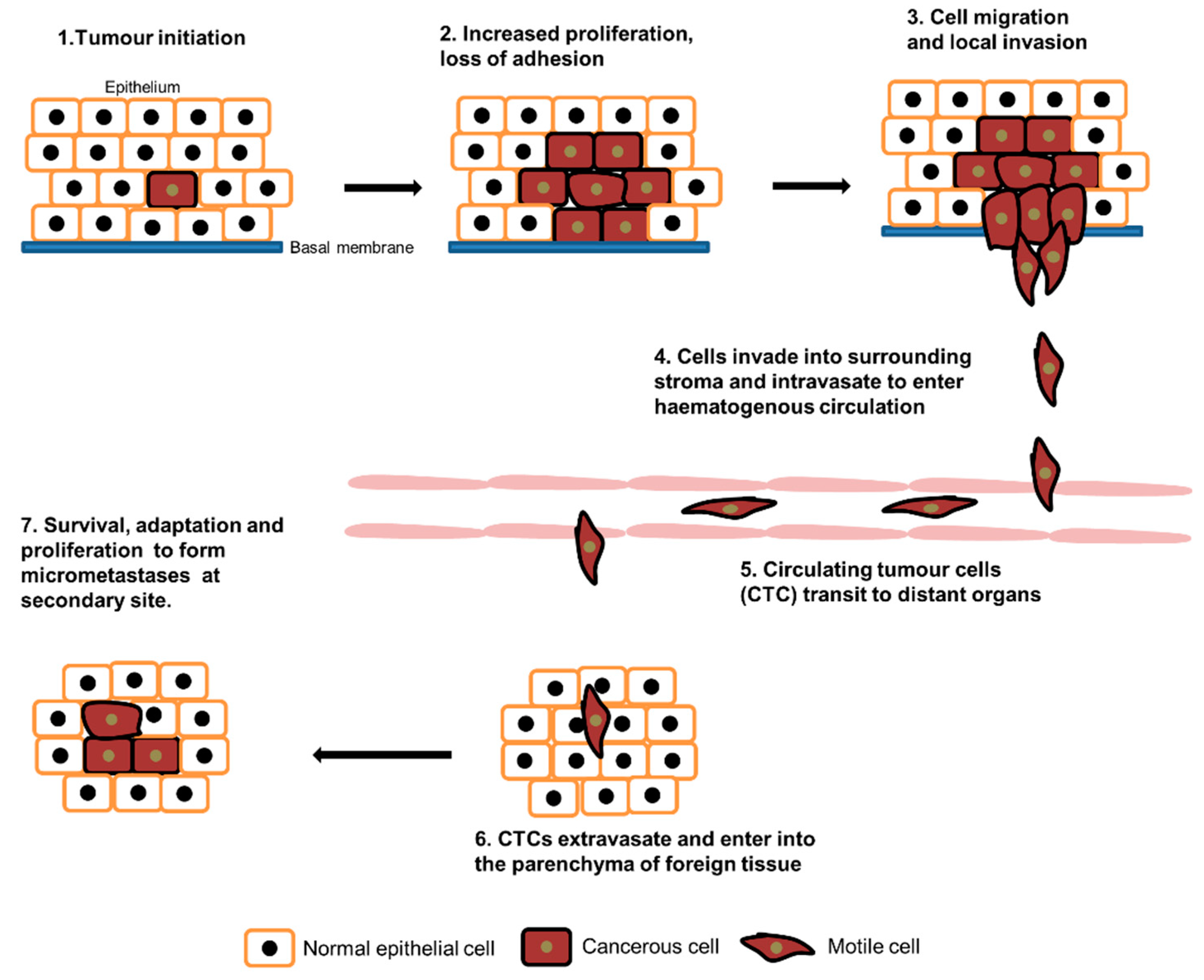
Figure 2.
PI3K-Akt signalling pathway. Upon ligand binding, conformational changes occur in the receptor tyrosine kinase (RTK), the PI3kinases are then activated by RTK and translocate to the plasma membrane. Activated PI3K then convert PIP2 to PIP3. Pleckstrin homology (PH) domain containing protein, Akt then translocate to the membrane, bind to PIP3 and phosphorylate at Threonine 308 residue by PDK1. Akt translocate back to cytoplasm and phosphorylate further at Serine 473 and Threonine 450 residues by mTORC2. Activated Akt is responsible for initiating various cellular activities such as proliferation, protein synthesis, autophagy, cell survival etc.
Figure 2.
PI3K-Akt signalling pathway. Upon ligand binding, conformational changes occur in the receptor tyrosine kinase (RTK), the PI3kinases are then activated by RTK and translocate to the plasma membrane. Activated PI3K then convert PIP2 to PIP3. Pleckstrin homology (PH) domain containing protein, Akt then translocate to the membrane, bind to PIP3 and phosphorylate at Threonine 308 residue by PDK1. Akt translocate back to cytoplasm and phosphorylate further at Serine 473 and Threonine 450 residues by mTORC2. Activated Akt is responsible for initiating various cellular activities such as proliferation, protein synthesis, autophagy, cell survival etc.
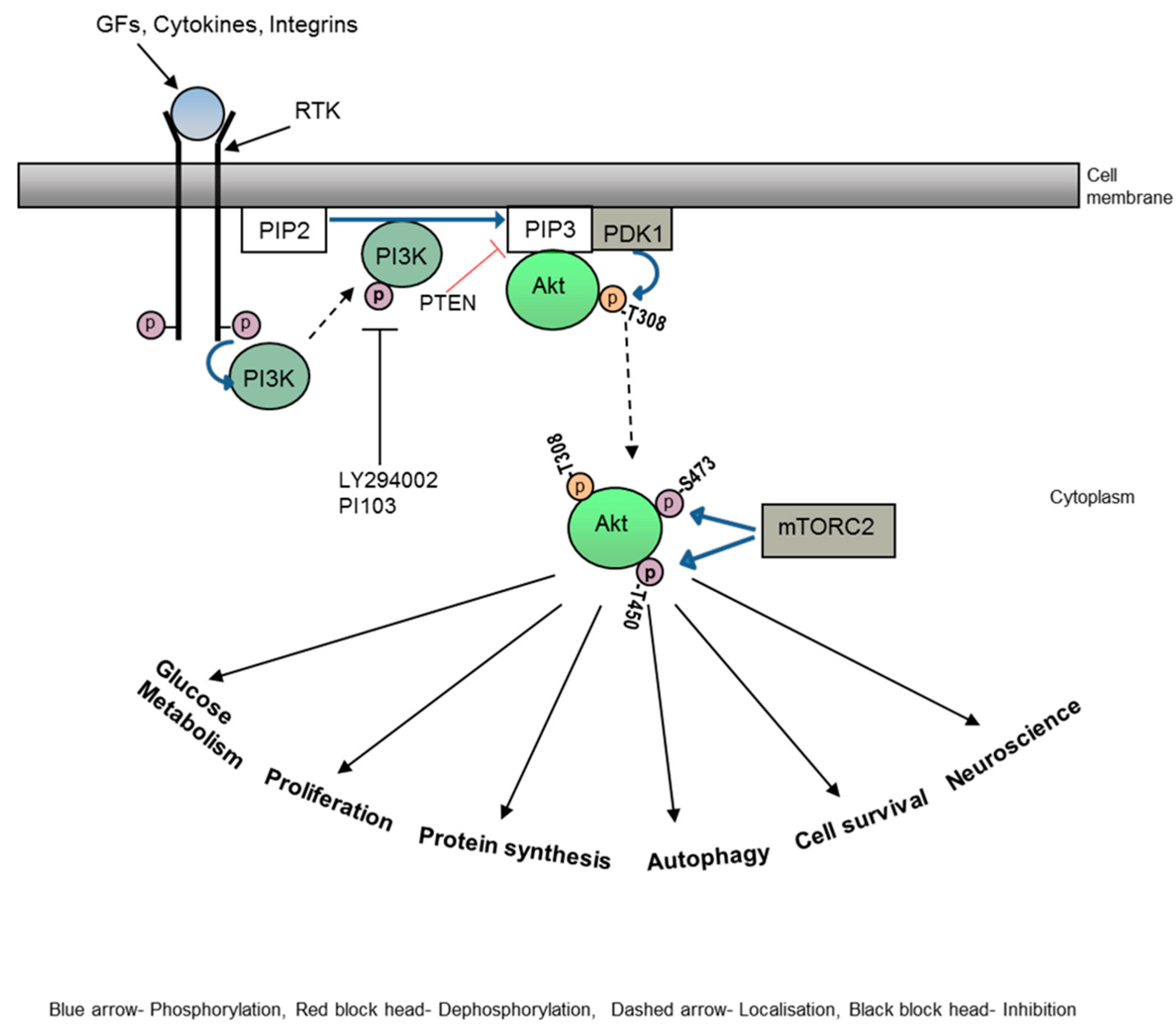
Figure 3.
Epithelial to Mesenchymal Transition (EMT) and associated biological markers. When appropriate signalling pathways are switched on, non-polarised, cobblestone shaped epithelial cells lose their cell-cell contacts and change to mesenchymal type motile cells. Extracellular matrix degradation enzymes, MMPs, then degrade the ECM and cells migrate through the basal lamina. This event can be detected at a molecular level by a reduction in levels of epithelial markers such as E-cadherin, b-catenin, cytokeratin etc. and higher levels of mesenchymal markers such as vimentin, Snail and Twist etc.
Figure 3.
Epithelial to Mesenchymal Transition (EMT) and associated biological markers. When appropriate signalling pathways are switched on, non-polarised, cobblestone shaped epithelial cells lose their cell-cell contacts and change to mesenchymal type motile cells. Extracellular matrix degradation enzymes, MMPs, then degrade the ECM and cells migrate through the basal lamina. This event can be detected at a molecular level by a reduction in levels of epithelial markers such as E-cadherin, b-catenin, cytokeratin etc. and higher levels of mesenchymal markers such as vimentin, Snail and Twist etc.
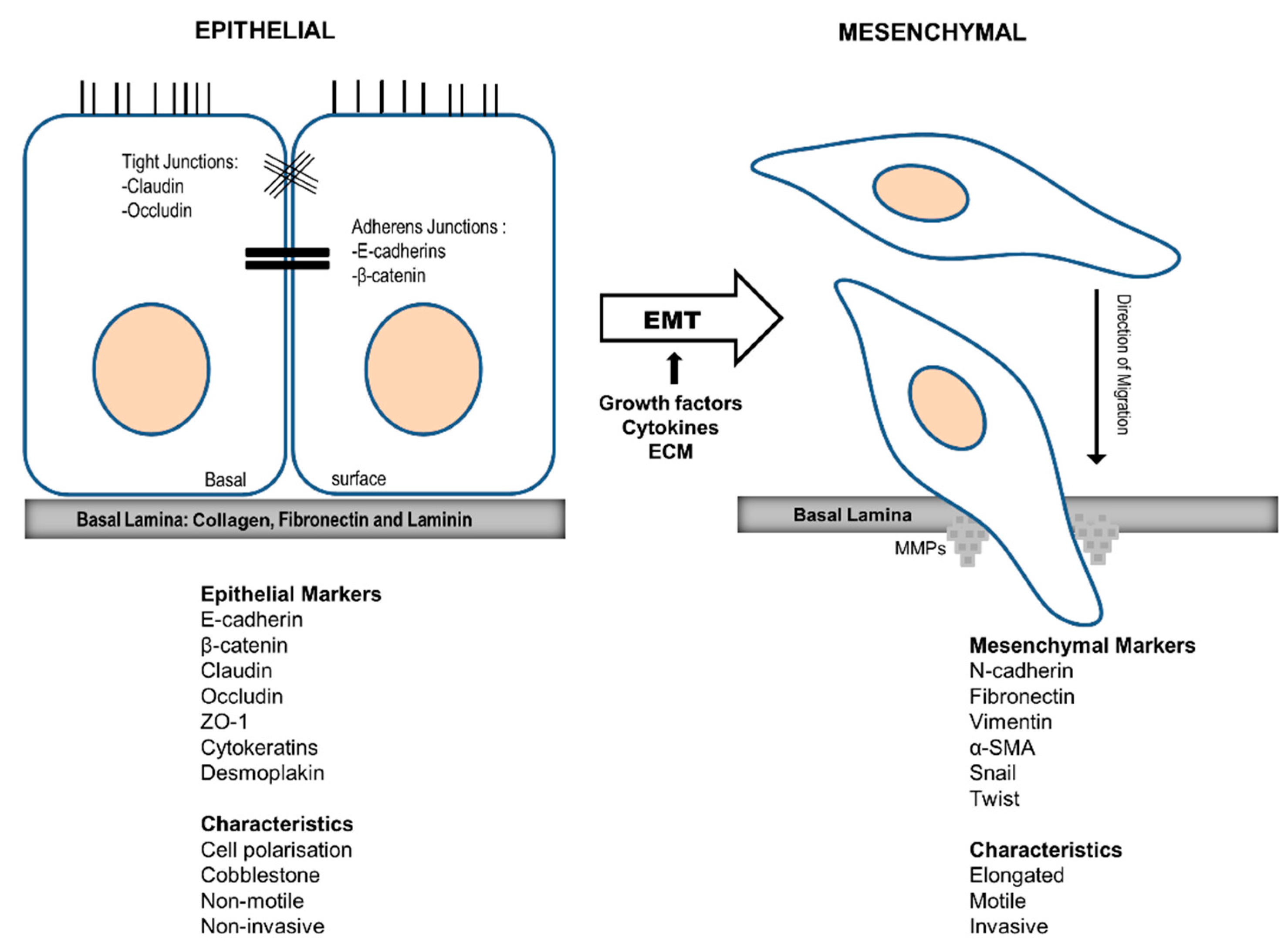
Figure 4.
The role of Akt in the metastasis process. Cellular motility or migration is the first step of the tumour cell metastatic process. Cytoskeletal remodelling and/or epithelial to mesenchymal transition are the two cellular events that are responsible for cell migration. Akt plays significant roles in cellular migration by controlling various downstream substrates which regulate these two events. The function of Akt has been found to be cell type, tumour type and site dependent.
Figure 4.
The role of Akt in the metastasis process. Cellular motility or migration is the first step of the tumour cell metastatic process. Cytoskeletal remodelling and/or epithelial to mesenchymal transition are the two cellular events that are responsible for cell migration. Akt plays significant roles in cellular migration by controlling various downstream substrates which regulate these two events. The function of Akt has been found to be cell type, tumour type and site dependent.
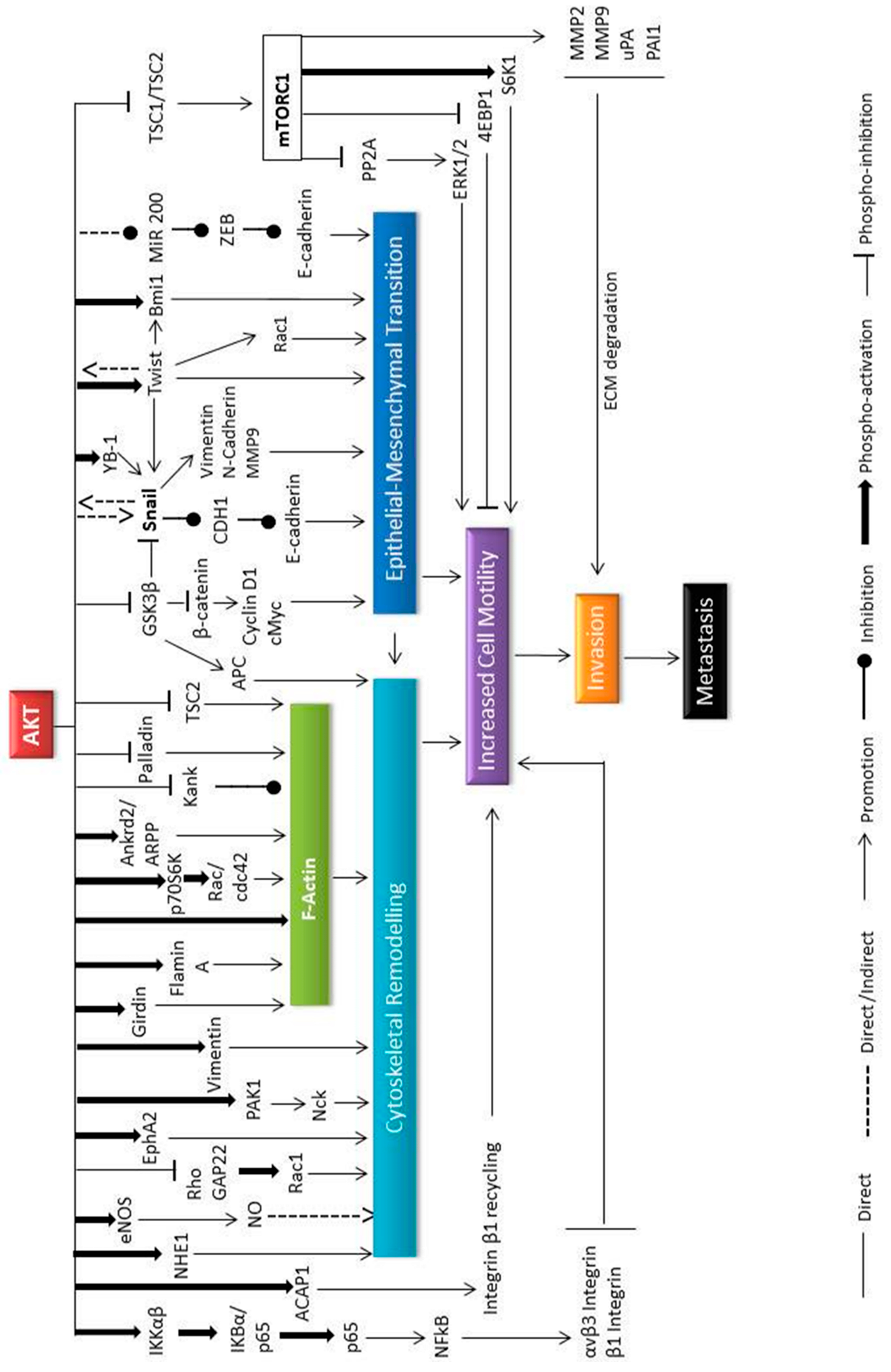

Table 1.
Recently completed clinical trials of PI3k/Akt pathway inhibitors in HNSCC.
| Trial identifier | Phase | Stage/type of HNSCC | Inhibitor | Combination | Result | Ref |
| NCT01349933 | II | IV/recurrent NPC | MK2206 (Akt inhibitor) | None | CR-0%, PR-4.8%, stable disease 52.4%, OS-10 months, PFS-3.5 months | [175] |
| NCT01370070 | II | Recurrent NPC | MK2206 | None | CR-0%, PR-5%, Stable disease-52%, OS-10 months, PFS-3.5 months | [176] |
| NCT01604772 | II | IV/recurrent ADCC | MK2206 | None | CR/PR-0%, Stable disease-81%, PFS-9.7 months, OS-18 months | [177] |
| NCT02145312 | II | Recurrent/metastatic HNSCC | BYL719/ Alpelisib (PI3K inhibitor) | None | Not published | [178] |
| NCT01527877 | II | Recurrent/metastatic HNSCC | BKM120/ Buparlisib (PI3K inhibitor) | None | RR-3%, Stable disease-49%, PFS-63 days, OS-143 days | [179] |
| NCT02021751 | Ib | Recurrent/metastatic HNSCC | BYL719 | Paclitaxel | Challenging safety profile, dose expansion phase was not initiated | [180] |
| NCT02113878 | Ib | Locally advanced HNSCC | BKM120 | Cisplatin/RT | Not published yet | [181] |
| NCT03292250 | II | HNSCC | BYL719 | Poziotinib (EGFR inhibitor) | Not published yet | [182] |
| NCT01816984 | I/II | Recurrent/metastatic HNSCC | BKM120 | Cetuximab | OS-9.3 months, PFS-2 months, RR-8-9% | [183] |
RR=Response rate, CR-Complete Response, PR-Partial response, OS-Overall survival, PFS-Progression free survival, NPC- Nasopharyngeal cancer, ADCC-Adenoid Cystic Carcinoma.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



