
Submitted:
10 September 2023
Posted:
12 September 2023
You are already at the latest version
Abstract
The cancer stem cell hypothesis suggests that neoplastic cells with stem characteristics hierarchically regulate tumor generation and its high cellular heterogeneity. These cells have been detected in all cancer types, and specific signaling pathways give the regulation of self-renewal and differentiation. In prostate cancer, androgen receptor signaling has been extensively studied, and in non-stem cells, it promotes cell proliferation and tumor progression, but in the cancer stem cell population, it negatively regulates processes such as self-renewal. However, in other types of cancer, such as breast and glioblastoma, the androgen receptor seems to favor the maintenance of cancer stem cells, suggesting that androgen signaling has different effects depending on the tumor context. This review discusses the role of androgen receptor in maintaining cancer stem cells by regulating proliferation, self-renewal, and differentiation, as well as the possible signaling pathways involved in these processes.
Keywords:
Cancer Stem Cell
; Androgen Receptor
; Prostate Cancer
; Breast Cancer
; Glioblastoma
1. Introduction
Cell replacements exist in any body system as part of homeostasis, a process driven by the generation and proliferation of new cells. Typically, cell generation is regulated by a small pool of undifferentiated cells with the ability to divide asymmetrically to generate specialized cells while maintaining the original progeny. These cells are called stem cells and have two fundamental characteristics: the ability to self-renew and high differentiation potential [1].
Stem cells can be classified based on several criteria, including potency and origin. There are four distinct types of stem cells based on their potency: totipotent, pluripotent, multipotent, and unipotent [2]. Also, can be classified based on their origin: embryonic stem cells (ESC), which have pluripotency characteristics and are capable of generating cell lineages from the three germ layers; fetal and adult stem cells, which usually display multipotency capabilities and their progeny are linked to specific tissues [3]. Interestingly, cancerous cell subpopulations with characteristics similar to stem cells have been found in all cancers. These cells are called cancer stem cells (CSCs) and are characterized by being undifferentiated cells with self-renewal capacity and highly tumorigenic potential. They can also generate more differentiated cancer cells and express a phenotype like cells present in the tissue [4].
CSCs were first described in the 1990s in patients with acute myeloid leukemia using the CD34+/CD38- cell markers, which they share with hematopoietic stem cells. In xenograft models in immunodeficient mice, CSCs repopulated all blood lineages [5]. Subsequently, CSCs were discovered in solid tumors such as breast [6], brain [7], prostate [8], and colon tumors [9]. It is currently postulated that tumors are generated by CSCs, which maintain themselves by self-renewal and are highly resistant to conventional therapies. In addition, CSCs generate progeny in a hierarchical manner due to their ability to generate more differentiated cells, which explains the high tumor heterogeneity [10]. Several signaling pathways, such as Wnt/β-catenin, TGF-β/Smad, Notch, and HedgeHog, have the ability to modulate CSCs self-renewal and proliferation [11]. Similarly, signaling mediated by sex hormone receptors has been associated with the maintenance of different populations of CSCs [12].
Androgens belong to the group of sex hormones and participate in the appearance and maintenance of male sexual characteristics. These hormones play a key role in the generation and differentiation of the prostate, as well as in the neoplasms of this organ [13]. Testosterone is the most representative androgen, exerting its primary function through the androgen receptor (AR), which functions as a ligand-activated transcription factor. Due to the action of the enzyme 5α reductase (5-αR), testosterone is metabolized into dihydrotestosterone (DHT), the most potent endogenous androgen [14]. To perform their physiological role, androgens cross the cell membrane by simple diffusion to interact with the AR in the cytoplasm, causing conformational changes that allow it to dissociate from chaperone proteins and translocate to the nucleus. Within the nucleus, AR forms homodimers and acts as a transcription factor, binding to specific DNA sequences in the promoter regions called androgen response elements (AREs) (Figure 1a) [15]. AR activation also regulates cellular processes through non-genomic signaling due to direct interaction in the cytoplasm with the PI3K/AKT and MAPKs pathways. Interestingly, AR expression has been found in CSCs from different types of tumors, such as breast, ovarian, and glioblastoma, demonstrating its potential to positively regulate the expression of genes associated with stemness, as well as the proliferation and self-renewal [16,17,18]. In prostate cancer (PC), AR is preferentially expressed in cells with a higher degree of differentiation, negatively regulating the maintenance of CSCs [19]. Current evidence suggests that AR can positively and negatively regulate processes that maintain CSCs, depending on the tumor context (Figure 1b). This review focuses on the role of androgen signaling in the self-renewal and maintenance of CSCs.
2. AR Signaling in Prostate Cancer
PC is the second most frequently diagnosed cancer in men, with a highly diverse incidence worldwide [20]. Similar to the healthy prostate, androgens play a crucial role in the development and progression of PC. Depending on the stage and grade of the cancer, treatment options may include active surveillance, radical prostatectomy, radiation, chemotherapy, and hormone therapy. In the case of advanced PC, androgen deprivation therapy (ADT) is usually recommended in the therapeutic scheme. To decrease androgen signaling, drugs that inhibit testosterone synthesis or act as AR antagonists are often used [21]. Despite the benefits of ADT, CP eventually develops resistance to treatment, termed castration-resistant prostate cancer (CRPC). [21,22]. When T or DHT activates the AR in PC cells, it exerts its role as a transcription factor, facilitating the expression of genes that promote cell proliferation and regulating signaling pathways that inhibit apoptosis. In CRPC, several hypotheses suggest how the AR promotes cell growth and tumor progression, even without testosterone or DHT presence [23]. These mechanisms include changes in the AR expression, such as its amplification and overexpression in tumor cells, the expression of splicing variants that are constitutively active, and point mutations that can modulate its activation by other ligands [24]. Androgen receptor splice variant 7 (AR-V7) is one of the most studied variants in PC, which, by having the LBD truncated, exerts its role as a transcription factor independently of activation by testosterone or DHT [25]. AR-V7 activity increases the expression of transcription factors that regulate cell cycle entry, cell growth, and proliferation. In fact, it has been pointed out as an oncological modification that promotes therapeutic resistance [26].
2.1. The Role of AR in PC Tumor Growth
Cell proliferation can be regulated by the direct interaction of AR with components of the AKT and MAPK signaling pathways, triggering cellular responses in shorter periods of time compared to the genomic pathway in PC [27,28]. Activation of the AR by its ligand positively regulates the AKT pathway through direct interaction with the p85α subunit of the PI3K protein, causing AKT phosphorylation at residues S473 and T308. Androgen-mediated activation of AKT promotes cell survival through phosphorylation of the proapoptotic proteins Bad and FKHR [27]. Similarly, AKT can positively modulate AR activation to promote cell growth. Just as activation of the AR by its ligands promotes AKT phosphorylation, activation of AKT by the Her2 signaling pathway promotes AR phosphorylation at residues S213 and S79. Thus, AKT positively modulates the AR in an androgen-independent manner [29]. Various growth factors, such as EGF and IL-8, also promote AR phosphorylation and activation under androgen-free conditions [30]. These molecules modulate the activation of the MAPK signaling cascade, causing Src kinase to phosphorylate tyrosine residues in the AR, promoting its transcriptional activity and the expression of genes that promote cell growth [30].
AR activity can modulate other signaling pathways that promote cell survival. Apoptosis is vital in cell viability and resistance to current therapeutic options. Evidence suggests that AR activation may play a protective role in conjunction with the transcription factor FOXO3a promoting FLICE-inhibitory protein (FLIP, inhibitor of death receptor-mediated apoptosis) expression in LNCaP cells [31], promoting a protective role for tumor cells. Similarly, TGF-β/SMAD is another signaling pathway that regulates cell growth and apoptosis in PC [32]. Androgen signaling has an essential role in regulating the activation of the TGF-β pathway because AR suppresses the transcriptional activity of SMAD3, the main effector of the TGF-β pathway. Thus, negatively regulating its antitumor activity and promoting cell survival [33,34]
AR activity also promotes tumor malignancy by regulating invasion and cell migration. In LNCaP and LAPC-4 cell lines, treatments with the synthetic androgen R-1881 increased the expression of the metalloprotease MMP-2 in an AR-dependent manner [35]. Another essential protein for cell invasion is ezrin. Treatments with R-1881 also positively regulated its expression in LNCaP cells. Similarly, AR mediates the phosphorylation of residues Thr-567 and Tyr-353, consequently promoting cell invasion in matrigel assays [36]. Congruently, androgen signaling participates in PC epithelial-mesenchymal transition (EMT), enhancing cell migration and metastasis. Both the silencing and blocking of the AR with antagonists decreased the ability of cells to migrate and invade, a process mediated by inhibition in the expression of Slug, Snail, MMP-2, B- catenin, and vimentin, essential proteins that induce EMT in the C4-2B cell line [37]. All these data suggest that, in addition to effects on cell proliferation and death, AR activity promotes tumor malignancy by increasing migratory and invasive capacities in PC.
2.2. AR Expression in Prostate Cancer Stem Cells
PC stem cells (PCSCs) were isolated in 2005 by Collins et al., showing a CD44+/α2β1hi/CD133+ phenotype, representing only 0.1% of the cells in the tumor [38]. They also have a high capacity for proliferation and self-renewal. Currently, for the study of PCSCs, classic markers such as CD133, CD44, or α6 integrin are demonstrated in the laboratory [8,39]. Due to their properties, PCSCs are considered to play a fundamental role in the development of CRPC. The Wnt signaling pathway is important in the regulation of PCSCs, as well as in the progression towards CRPC. SRFP1 is a modulator of Wnt pathway activation, and its presence is associated with increased expression of pluripotency genes such as NANOG, SOX2, and OCT4, as well as increased self-renewal capacity [40]. Even though androgen signaling plays a fundamental role in the progression of PC by promoting cell growth, migration, and invasion, most PCSCs have an AR- phenotype [38,41,42]. Interestingly, studies carried out in LNCaP cell lines, LAP4 and LAP9, demonstrated that androgen depletion in the medium increased the population of cells with stem characteristics, showing a CD44+/CD24- phenotype, as well as a high tumorigenic capacity [43].
PCSCs can reconstitute the original tumor, generating basal, luminal, and neuroendocrine epithelial cells [41]. These stem cells have an AR- phenotype, but once the differentiation process is triggered, the receptor begins to be expressed, suggesting that androgen signaling favors differentiation processes over self-renewal [41]. Functional assessment in suspension cultures demonstrated that AR silencing promoted PCSCs self-renewal, increasing the number of spheres with a larger size. In contrast, AR overexpression decreased PCSCs self-renewal, showing a lower capacity in generating spheres [44]. Interestingly, bicalutamide decreased and increased the proliferation of LNCaP-CD133- and LNCaP-CD133+ cells in vivo, respectively, suggesting AR activation has differential effects in stem and non-stem cells [42].
AR activation has also been shown to downregulate the expression of proteins that promote stemness characteristics in PC cells. STAT3 protein maintains pluripotency in different stem cells, promoting self-renewal processes. Schroeder et al. demonstrated that AR loss increases the expression of STAT3, allowing the generation of stemness characteristics [45]. Additionally, pharmacological inhibition of the AR promoted the expression of stemness factors such as Sox2 and CD44 [45].
AR activation can negatively modulate effector proteins of signaling pathways involved in self-renewal and proliferation, such as the Akt and Wnt pathways. Indeed, the LNCaP and C4-2 cell lines negatively regulated the expression of Akt, Wnt-1 and c-myc, as well as the antiapoptotic protein Bcl-2 [44]. These data suggest that AR downregulates stemming characteristics in both cell models. Vummidi et al. reported that the AR- phenotype of PCSCs is mediated by the MDM2 ubiquitin ligase, which continuously degrades AR and supports the maintenance of stemness. The loss of MDM2 induced the AR re-expression and the differentiation towards luminal cell [46]. Therefore, AR signaling could negatively regulate stem characteristics in PC.
3. AR Signaling in Breast Cancer
Breast cancer (BC) is the most frequent cancer in women and the second cause of cancer death among women worldwide [47]. BC is classified according to the expression profile of the estrogen receptor α (ERα), the progesterone receptor (PR), and the human epidermal growth factor receptor (HER2). Hence, tumors that express the estrogen receptor (ER) are denominated as luminal A and luminal B. Luminal A tumors express ERα and PR but not HER2, whereas luminal B tumors express ERα, PR, and HER2, and show a higher proliferation rate than luminal A tumors; HER2-enriched tumors, lack ERα and PR expression but have high HER2 expression; and triple-negative breast cancer (TNBC) tumors do not express any of the previously mentioned receptors [47]. Interestingly, AR is expressed in 60-80% of mammary gland tumors, mainly among Luminal A neoplasias [48].
The effects of AR on cell proliferation and tumor progression are unclear since its action depends on the co-expression of ERα in BC. In ERα positive tumors, AR expression has been associated with a longer life expectancy, having smaller and lower-grade tumors [49]. However, a high AR:ER ratio is associated with the development of resistance to treatment, which suggests that the expression level of both receptors could influence tumor growth [50]. In the case of tumors that do not express ERα, AR exerts mainly a protumoral activity [51,52].
3.1. Action of Androgens in ERα- and ERα+ Tumors
In neoplasias lacking ERα, androgens stimulate cell proliferation and tumor growth. Experimentally, AR can regulate the Wnt signaling pathway, which plays an essential role in cell proliferation [52,53]. Moreover, AR interacts with β-catenin to modulate gene expression, enhancing the transcription of the CMYC oncogene, and promoting cell growth processes [52]. Similarly, in the MDA-MB-453 cell line, AR activation through DHT promotes WNT7B transcription, an essential mediator in the Wnt activation. In this model, transcriptional complexes comprising AR, β-catenin, and FOXA1 promoted HER3 expression [53]. HER3 can form heterodimers in HER2+ BC cells, driving cell proliferation via the PI3K/AKT pathway [54]. Bicalutamide treatment reduces HER3 expression and AKT phosphorylation [53]. Similarly, enzalutamide treatment and AR silencing in SKBR3 and HCC1954 cell lines decreased HER2 phosphorylation without affecting HER2 and HER3 protein content. This suggests that AR exerts non-genomic action in HER2+ overexpressing cells, regulating downstream pathways, such as PI3K and ERK [51]. Notably, inhibiting the PI3K pathway leads to decreased AR expression [55].
In TNBC cells, activation of the G-protein-coupled estrogen receptor (GPER) exerts antitumor activity [56]. In MDA-MB-231 and Hs578T cell lines, DHT promotes cell growth and viability while suppressing GPER expression at both transcript and protein levels. Notably, GPER activation inhibited AR-mediated cell growth [57]. In addition to promoting cell proliferation in ERα- cancer cells, there are reports of its participation in cell migration. Once the AR is activated, it can produce complexes with Src kinase and PI3K, which generate structural changes that promote cell motility and invasion [58]. Overall, in ERα- BC cells, AR activity promotes malignant processes by regulating essential signaling pathways in cell proliferation, as well as in cell migration and invasion processes.
In BC tumors that express ERα, estrogens play an important role in cell proliferation and tumor growth. Certainly, several of these tumors co-express other sex hormone receptors, such as PR and AR. ERα promotes gene expression in signaling pathways that promote cell growth in cancer cells [49]. The AR activation is associated with antiproliferative activity in these tumors. Androgen administration decreased cell growth in in vitro models. DHT treatments in the MCF-7 cell line decreased cell proliferation due to reduced cyclin D1 expression provoked by repression in the CCND1 gene transcription mediated by AR [59]. Similar results were observed in aromatase expression, which is repressed by AR [60], causing less estrogen synthesis from testosterone. In this cellular model, the repressor action on AR transcription was mediated by DAX1 [59,60].
AR expression has been associated with a longer life expectancy, having smaller and lower-grade tumors. AR exerts antiestrogenic activity due to the competition with the ERα for their hormone response elements in the promoter region of their target genes. Thus, AR inhibits the gene expression that promotes cell proliferation regulated by the ERα genes [61]. Also, AR can bind to the AREs sequences in the ERβ promoter, inducing its expression and provoking an antitumoral activity [62]. These data suggest that the antitumoral activity of AR in ER+ tumors could be regulating ERα and ERβ activity.
3.2. Regulation of Breast Cancer Stem Cells by AR
BC stem cells (BCSCs) were the first cancer stem cells reported in solid tumors. For its isolation, Al-Hajj et al. identified a cell population in mammary tumors that expressed the CD44+/CD24- phenotype, with a very high tumorigenic capacity, inducing tumors with 200 cells inoculated in immunosuppressed mice [6]. Sex hormone receptors, such as ER, PR, and AR, are also expressed in this population. In TNBC cells, enzalutamide decreased the ability to generate colonies on soft agar, suggesting lower growth under anchorage-free conditions. In addition, the drug decreased cell viability and increased apoptosis and necrosis in in vivo models [63]. A higher expression of AR was also found in a study with cells in forced suspension to increase the population of BCSCs [64]. AR silencing decreased both the ALDH+ cell population from 55 to 40% in cells of the SUM159PT line, as well as the generation of three-dimensional structures that allow the enrichment of BCSC, called mammospheres. These results were also observed with enzalutamide. In the MDA453 cell line, AR silencing increased the percentage of CD24+ cells, indicative of a differentiated phenotype, and reduced the efficiency of mammospheres derivation [64]. Fernandez et al. determined that AR is a positive modulator of the transcription factor RUNX1 at transcript and protein levels and its transcriptional activity is related to the expression of genes associated with stemness such as KLF4, OCT4, and SOX4 in TNBC [65]. A study by Rosas et al. demonstrated that activation of the TGF-β signaling pathway increased AR expression under anchorage-independent conditions. Consistently, AR interacted with TGFB1 and SMAD3 regulatory sequences. Inhibition of both TGF-β and AR decreased survival in anchorage-independent conditions [66]. In summary, the experimental evidence suggests that the activity of AR in cancer stem cells present in TNBC tumors positively regulates the self-renewal and the expression of markers associated with stemness; however, it is necessary to evaluate the actions of AR in ER-positive tumors to clarify if it has a role in the maintenance of BCSCs, evaluating the expression of stem genes, self-renewal and tumorigenesis.
4. AR Signaling in Glioblastoma
Gliomas are tumors of the central nervous system (CNS) of glial origin. Glioblastoma (GB) is the most frequent and lethal, with an incidence of 3.2 per 100,000 inhabitants, more frequently in males than in females, with a ratio of 3:2 [67]. This tumor rarely metastasizes outside the CNS; however, it is highly infiltrative in the brain parenchyma, generating diffuse tumors with indefinite borders. The higher incidence in men than women suggests that sex hormones play a role in the prevalence of this cancer. There is evidence that patients with GB have a higher serum testosterone concentration than healthy subjects, regardless of gender [68]. Consistent with these data, tumor tissue samples from GB patients showed increased AR expression compared to peripheral normal brain tissue. Indeed, AR expression has also been found in human GB cell lines A172, LN18, LN229, M059, T98G, U87MG, U118MG, and U138MG [69].
Several studies suggest that AR activation in GB cells can occur in both androgen-dependent and independent mechanisms. In vitro studies showed that testosterone or DHT treatments promote cell proliferation, migration, and invasion of human GB-derived cell lines through AR activation [70,71]. Concordantly, the activation of the AR by DHT in the U87-MG cell line hindered the antiproliferative effects produced by the TGF-β signaling pathway. These effects resulted from the AR binding to SMAD3, the primary effector of this pathway, which prevented SMAD3 translocation into the nucleus and its subsequent activity [69]. In the U87 cell line, androgen-independent AR activation was observed, wherein EGFR signaling induced AR phosphorylation and its translocation to the nucleus to execute its functions [72]. Additionally, the presence of the splicing variant AR-v7 was reported. This variant lacks the ligand-binding domain, leading to the constitutive activation of the AR in an androgen-independent manner, thereby promoting tumor growth [73]. All these data suggest that AR activation, either in an androgen-dependent or independent manner, promotes GB cell proliferation and plays a critical role in its progression.
4.1. AR Activity in the Maintenance of GSCs
Glioma stem cells (GSCs) have been identified in GB, displaying highly proliferative and the ability to self-renew, generate new tumors, and differentiate into cells of neuroglial lineages. The pioneering work of Sheila Singh and her colleagues involved isolating GSCs through the membrane marker CD133 [7]. Subsequently, different markers have been utilized for their study and isolation, including CD15 [74], α6-integrin [75], Sox2 [76], Oct4 [77], Nanog [78], and ALDH1A3 [79], among others. There is evidence that mutations in neural stem cells (NSCs) can give rise to GSCs, making NSCs a potential origin for GSC [80]. In physiological contexts, NSCs participate in developing the CNS during embryogenesis and neurogenesis in adult mammals [81]. Both adult and embryonic NSCs express AR. Recent evidence suggests that testosterone and DHT promote the proliferation and self-renewal of NSCs generated from the H1 and H9 human embryonic stem cell lines. Private androgens facilitated the differentiation of NSCs into excitatory neurons in an organoid model [82]. DHT treatments also enhanced the self-renewal of neural progenitor cells derived from mouse embryos, leading to an increase in the number of neurospheres in the culture and the expression of the stemness marker ALDH1A3. Interestingly, a more pronounced limitation to neuronal differentiation was observed in the presence of DHT [83]. These findings suggest that androgens promote NSCs proliferation and self-renewal while limiting their differentiation capabilities. Similar trends were noticed in GSCs. The utilization of bicalutamide and enzalutamide reduced the ability of human GB-derived cells to generate neurospheres and the proportion of CD133+ cells within the culture [18]. Congruently, treatment with the 5α-reductase inhibitor, finasteride, decreased sphere formation in the U373 and T98G cell lines [84]. The decline in the number and size of neurospheres implies that AR activity regulates GSC self-renewal. Additionally, the inhibition of AR activity reduced the expression of stemness markers such as NANOG, OCT4, and SOX2 [18,84]. In BC models, androgen signaling has been linked to the Wnt pathway, demonstrating the AR interaction with β-catenin to regulate gene expression. Finasteride treatments in GSCs lowered β-catenin expression, suggesting that androgens could regulate Wnt signaling similar to that reported in BC, thus contributing to stem cell maintenance [84]. Currently, experimental evidence suggests that AR activation is a crucial player in maintaining stem cells within GB, fostering the expression of stemness factors and self-renewal. However, more studies are required to validate these observations. Like BCSC, AR in GB seems to support stemness contrasting with the observed role in PCSC (Table 1).
5. Conclusions
Cancer stem cells are a highly researched topic in oncology due to their remarkable ability to sustain tumor growth through self-renewal and differentiation. These cells resist conventional therapies and are closely linked to tumor relapse. While the discovery of cancer stem cells is not recent, an effective strategy to inhibit their growth in several cancer types remains elusive. Although AR signaling has been extensively studied in PC, its analysis is gaining importance in other types of cancer. Despite its pivotal role in tumor maintenance, AR expression is often absent in PCSCs. On the contrary, its expression in this cell population correlates with reduced proliferation and self-renewal. In contrast, AR plays a positive role in maintaining stemness in BC and GB stem cells. To explain these divergent actions, the critical consideration is the specific tumor context. The AR has the ability to regulate different signaling pathways, which may exert pro- or anti-tumor effects. For instance, in the PI3K/AKT pathway, AR activation through the non-genomic pathway promotes AKT phosphorylation, activating effector proteins that positively regulate cell proliferation and survival. On the other hand, genomic AR activity negatively impacts AKT function. The interaction of the AR with other determinant signaling pathways in the maintenance of CSCs such as Wnt/β-catenin, MAPK, and β-TGF has also been documented. In BC, the ARs action is influenced by the expression of sex hormones receptors, particularly ERα. Understanding such interactions prompts the need to delineate the expression profile of sex hormone receptors across various cancer models. Moreover, differences in the expression of stemness-associated genes, such as SOX2, NANOG, OCT4, among others, exist among different CSCs types. Despite our current knowledge, extensive studies on various types of cancer are essential to elucidate the regulatory role of RA in the maintenance of stemness.
Author Contributions
Conceptualization and primary draft was prepared by J.C.Q. and I.C.-A.; comments and revisions were made by N.F.D., M.R.-D., and I.C.-A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by DGAPA-PAPIIT, grant number: IN201823, UNAM, México and by a scholarship to Juan Carlos Quintero from Consejo Nacional de Humanidades, Ciencia y Tecnología (CONAHCYT), México.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The figure featured in this article was created using Biorender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006, 441, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008, 132, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Rossi F, Noren H, Jove R, Beljanski V, Grinnemo KH. Differences and similarities between cancer and somatic stem cells: Therapeutic implications. Stem Cell Res Ther. 2020, 11, 489. [Google Scholar] [CrossRef]
- Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauß A, Blaudszun AR, Yevsa T, Fricke S, Kossatz-Boehlert U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumor-initiating cells. Nature. 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. The side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005, 65, 6207–6219. [Google Scholar] [CrossRef]
- O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumor growth in immunodeficient mice. Nature. 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003, 3, 895–902. [Google Scholar] [CrossRef]
- Borlongan MC, Wang H. Profiling and targeting cancer stem cell signaling pathways for cancer therapeutics. Front Cell Dev Biol. 2023, 11, 1125174. [Google Scholar]
- Lee J, Troike K, Fodor R, Lathia JD. Unexplored Functions of Sex Hormones in Glioblastoma Cancer Stem Cells. Endocrinology. 2022, 163, bqac002. [Google Scholar] [CrossRef] [PubMed]
- Banerjee PP, Banerjee S, Brown TR, Zirkin BR. Androgen action in prostate function and disease. Am J Clin Exp Urol. 2018, 6, 62–77. [Google Scholar]
- Batista RL, Mendonca BB. Integrative and Analytical Review of the 5-Alpha-Reductase Type 2 Deficiency Worldwide. Appl Clin Genet. 2020, 13, 83–96. [Google Scholar] [CrossRef]
- Jin HJ, Kim J, Yu J. Androgen receptor genomic regulation. Transl Androl Urol. 2013, 2, 157–177. [Google Scholar]
- Ling K, Jiang L, Liang S, Kwong J, Yang L, Li Y, PingYin, Deng Q, Liang Z. Nanog interaction with the androgen receptor signaling axis induce ovarian cancer stem cell regulation: Studies based on the CRISPR/Cas9 system. J Ovarian Res. 2018, 11, 36.
- Riaz N, Idress R, Habib S, Azam I, Lalani EM. Expression of Androgen Receptor and Cancer Stem Cell Markers (CD44+/CD24- and ALDH1+): Prognostic Implications in Invasive Breast Cancer. Transl Oncol. 2018, 11, 920–929. [Google Scholar] [CrossRef]
- Zhao N, Wang F, Ahmed S, Liu K, Zhang C, Cathcart SJ, DiMaio DJ, Punsoni M, Guan B, Zhou P, Wang S, Batra SK, Bronich T, Hei TK, Lin C, Zhang C. Androgen Receptor, Although Not a Specific Marker For, Is a Novel Target to Suppress Glioma Stem Cells as a Therapeutic Strategy for Glioblastoma. Front Oncol. 2021, 21, 616625. [Google Scholar]
- Verma S, Shankar E, Kalayci FNC, Mukunda A, Alassfar M, Singh V, Chan ER, MacLennan GT, Gupta S. Androgen Deprivation Induces Transcriptional Reprogramming in Prostate Cancer Cells to Develop Stem Cell-Like Characteristics. Int J Mol Sci. 2020, 21, 9568. [Google Scholar] [CrossRef]
- Bergengren O, Pekala KR, Matsoukas K, Fainberg J, Mungovan SF, Bratt O, Bray F, Brawley O, Luckenbaugh AN, Mucci L, Morgan TM, Carlsson SV. 2022 Update on Prostate Cancer Epidemiology and Risk Factors-A Systematic Review. Eur Urol. 2023, 84, 191–206. [Google Scholar] [CrossRef]
- Yu EM, Aragon-Ching JB. Advances with androgen deprivation therapy for prostate cancer. Expert Opin Pharmacother. 2022, 23, 1015–1033. [Google Scholar] [CrossRef] [PubMed]
- Morote J, Aguilar A, Planas J, Trilla E. Definition of Castrate Resistant Prostate Cancer: New Insights. Biomedicines. 2022, 10, 689. [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Fujita K, Nonomura N. Role of Androgen Receptor in Prostate Cancer: A Review. World J Mens Health. 2019, 37, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. Development of AR-V7 as a putative treatment selection marker for metastatic castration-resistant prostate cancer. Asian J Androl. 2016, 18, 580–585. [Google Scholar] [CrossRef]
- Konda P, Viswanathan SR. How splicing confers treatment resistance in prostate cancer. Elife. 2022, 11, e82070. [Google Scholar] [CrossRef]
- Baron S, Manin M, Beaudoin C, Leotoing L, Communal Y, Veyssiere G, Morel L. Androgen receptor mediates non-genomic activation of phosphatidylinositol 3-OH kinase in androgen-sensitive epithelial cells. J Biol Chem. 2004, 279, 14579–14586. [Google Scholar] [CrossRef]
- Dahiya V, Bagchi G. Non-canonical androgen signaling pathways and implications in prostate cancer. Biochim Biophys Acta Mol Cell Res. 2022, 1869, 119357. [Google Scholar]
- Wen Y, Hu MC, Makino K, Spohn B, Bartholomeusz G, Yan DH, Hung MC. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000, 60, 6841–6845. [Google Scholar]
- Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, Njar VC, Brodie AM, Yu LR, Veenstra TD, Chen H, Qiu Y. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006, 10, 309–319. [Google Scholar] [CrossRef]
- Cornforth AN, Davis JS, Khanifar E, Nastiuk KL, Krolewski JJ. FOXO3a mediates the androgen-dependent regulation of FLIP and contributes to TRAIL-induced apoptosis of LNCaP cells. Oncogene. 2008, 27, 4422–4433. [Google Scholar] [CrossRef] [PubMed]
- Brodin G, ten Dijke P, Funa K, Heldin CH, Landström M. Increased smad expression and activation are associated with apoptosis in normal and malignant prostate after castration. Cancer Res. 1999, 59, 2731–2738. [Google Scholar]
- Wang H, Song K, Sponseller TL, Danielpour D. Novel function of androgen receptor-associated protein 55/Hic-5 as a negative regulator of Smad3 signaling. J Biol Chem. 2005, 280, 5154–5162. [Google Scholar] [CrossRef]
- Song K, Wang H, Krebs TL, Wang B, Kelley TJ, Danielpour D. DHT selectively reverses Smad3-mediated/TGF-beta-induced responses through transcriptional down-regulation of Smad3 in prostate epithelial cells. Mol Endocrinol. 2010, 24, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Liao X, Thrasher JB, Pelling J, Holzbeierlein J, Sang QX, Li B. Androgen stimulates matrix metalloproteinase-2 expression in human prostate cancer. Endocrinology. 2003, 144, 1656–1663. [Google Scholar] [CrossRef]
- Chuan YC, Pang ST, Cedazo-Minguez A, Norstedt G, Pousette A, Flores-Morales A. Androgen induction of prostate cancer cell invasion is mediated by ezrin. J Biol Chem. 2006, 281, 29938–29948. [Google Scholar] [CrossRef]
- Lin CY, Jan YJ, Kuo LK, Wang BJ, Huo C, Jiang SS, Chen SC, Kuo YY, Chang CR, Chuu CP. Elevation of androgen receptor promotes prostate cancer metastasis by induction of epithelial-mesenchymal transition and reduction of KAT5. Cancer Sci. 2018, 109, 3564–3574. [Google Scholar] [CrossRef]
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Patrawala L, Calhoun-Davis T, Schneider-Broussard R, Tang DG. Hierarchical organization of prostate cancer cells in xenograft tumors: The CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells. Cancer Res. 2007, 67, 6796–6805. [Google Scholar] [CrossRef]
- Losada-García A, Salido-Guadarrama I, Cortes-Ramirez SA, Cruz-Burgos M, Morales-Pacheco M, Vazquez-Santillan K, Rodriguez-Martinez G, González-Ramírez I, Gonzalez-Covarrubias V, Perez-Plascencia C, Rodríguez-Dorantes M. SFRP1 induces a stem cell phenotype in prostate cancer cells. Front Cell Dev Biol. 2023, 11, 1096923. [Google Scholar]
- Gu G, Yuan J, Wills M, Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007, 67, 4807–4815. [Google Scholar] [CrossRef] [PubMed]
- Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, Calhoun-Davis T, Li H, Palapattu GS, Pang S, Lin K, Huang J, Ivanov I, Li W, Suraneni MV, Tang DG. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012, 10, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Seiler D, Zheng J, Liu G, Wang S, Yamashiro J, Reiter RE, Huang J, Zeng G. Enrichment of putative prostate cancer stem cells after androgen deprivation: Upregulation of pluripotency transactivators concurs with resistance to androgen deprivation in LNCaP cell lines. Prostate. 2013, 73, 1378–1390. [Google Scholar] [CrossRef]
- Lee SO, Ma Z, Yeh CR, Luo J, Lin TH, Lai KP, Yamashita S, Liang L, Tian J, Li L, Jiang Q, Huang CK, Niu Y, Yeh S, Chang C. New therapy targeting differential androgen receptor signaling in prostate cancer stem/progenitor vs. non-stem/progenitor cells. J Mol Cell Biol. 2013, 5, 14–26. [Google Scholar] [CrossRef]
- Schroeder A, Herrmann A, Cherryholmes G, Kowolik C, Buettner R, Pal S, Yu H, Müller-Newen G, Jove R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef]
- Vummidi Giridhar P, Williams K, VonHandorf AP, Deford PL, Kasper S. Constant Degradation of the Androgen Receptor by MDM2 Conserves Prostate Cancer Stem Cell Integrity. Cancer Res. 2019, 79, 1124–1137.
- Łukasiewicz S, Czeczelewski M, Forma A, Baj J, Sitarz R, Stanisławek A. Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies-An Updated Review. Cancers (Basel). 2021, 13, 4287. [Google Scholar] [CrossRef] [PubMed]
- Kensler KH, Regan MM, Heng YJ, Baker GM, Pyle ME, Schnitt SJ, Hazra A, Kammler R, Thürlimann B, Colleoni M, Viale G, Brown M, Tamimi RM. Prognostic and predictive value of androgen receptor expression in postmenopausal women with estrogen receptor-positive breast cancer: Results from the Breast International Group Trial 1-98. Breast Cancer Res. 2019, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Hwang KT, Kim YA, Kim J, Park JH, Choi IS, Hwang KR, Chai YJ, Park JH. Influence of Androgen Receptor on the Prognosis of Breast Cancer. J Clin Med. 2020, 9, 1083. [Google Scholar] [CrossRef]
- Cochrane DR, Bernales S, Jacobsen BM, Cittelly DM, Howe EN, D'Amato NC, Spoelstra NS, Edgerton SM, Jean A, Guerrero J, Gómez F, Medicherla S, Alfaro IE, McCullagh E, Jedlicka P, Torkko KC, Thor AD, Elias AD, Protter AA, Richer JK. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014, 22, R7. [Google Scholar]
- He L, Du Z, Xiong X, Ma H, Zhu Z, Gao H, Cao J, Li T, Li H, Yang K, Chen G, Richer JK, Gu H. Targeting Androgen Receptor in Treating HER2 Positive Breast Cancer. Sci Rep. 2017, 7, 14584. [Google Scholar] [CrossRef] [PubMed]
- Huang R, Han J, Liang X, Sun S, Jiang Y, Xia B, Niu M, Li D, Zhang J, Wang S, Wei W, Liu Q, Zheng W, Zhang G, Song Y, Panga D. Androgen Receptor Expression and Bicalutamide Antagonize Androgen Receptor Inhibit β-Catenin Transcription Complex in Estrogen Receptor-Negative Breast Cancer. Cell Physiol Biochem. 2017, 43, 12–2225. [Google Scholar]
- Ni M, Chen Y, Lim E, Wimberly H, Bailey ST, Imai Y, Rimm DL, Liu XS, Brown M. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell. 2011, 20, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Claus J, Patel G, Autore F, Colomba A, Weitsman G, Soliman TN, Roberts S, Zanetti-Domingues LC, Hirsch M, Collu F, George R, Ortiz-Zapater E, Barber PR, Vojnovic B, Yarden Y, Martin-Fernandez ML, Cameron A, Fraternali F, Ng T, Parker PJ. Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface. Elife. 2018, 7, e32271. [Google Scholar] [CrossRef]
- Cuenca-López MD, Montero JC, Morales JC, Prat A, Pandiella A, Ocana A. Phospho-kinase profile of triple negative breast cancer and androgen receptor signaling. BMC Cancer. 2014, 14, 302. [Google Scholar]
- Chen ZJ, Wei W, Jiang GM, Liu H, Wei WD, Yang X, Wu YM, Liu H, Wong CK, Du J, Wang HS. Activation of GPER suppresses epithelial mesenchymal transition of triple negative breast cancer cells via NF-κB signals. Mol Oncol. 2016, 10, 775–788. [Google Scholar] [CrossRef]
- Shen Y, Yang F, Zhang W, Song W, Liu Y, Guan X. The Androgen Receptor Promotes Cellular Proliferation by Suppression of G-Protein Coupled Estrogen Receptor Signaling in Triple-Negative Breast Cancer. Cell Physiol Biochem. 2017, 43, 2047–2061. [Google Scholar] [CrossRef]
- Giovannelli P, Di Donato M, Auricchio F, Castoria G, Migliaccio A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells Through AR/Src/PI3-K Complex Assembly. Sci Rep. 2019, 9, 4490. [Google Scholar] [CrossRef]
- Lanzino M, Maris P, Sirianni R, Barone I, Casaburi I, Chimento A, Giordano C, Morelli C, Sisci D, Rizza P, Bonofiglio D, Catalano S, Andò S. DAX-1, as an androgen-target gene, inhibits aromatase expression: A novel mechanism blocking estrogen-dependent breast cancer cell proliferation. Cell Death Dis. 2013, 4, e724. [Google Scholar] [CrossRef]
- Lanzino M, Sisci D, Morelli C, Garofalo C, Catalano S, Casaburi I, Capparelli C, Giordano C, Giordano F, Maggiolini M, Andò S. Inhibition of cyclin D1 expression by androgen receptor in breast cancer cells--identification of a novel androgen response element. Nucleic Acids Res. 2010, 38, 5351–5365. [Google Scholar] [CrossRef]
- Peters AA, Buchanan G, Ricciardelli C, Bianco-Miotto T, Centenera MM, Harris JM, Jindal S, Segara D, Jia L, Moore NL, Henshall SM, Birrell SN, Coetzee GA, Sutherland RL, Butler LM, Tilley WD. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res. 2009, 69, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Anestis A, Sarantis P, Theocharis S, Zoi I, Tryfonopoulos D, Korogiannos A, Koumarianou A, Xingi E, Thomaidou D, Kontos M, Papavassiliou AG, Karamouzis MV. Estrogen receptor beta increases sensitivity to enzalutamide in androgen receptor-positive triple-negative breast cancer. J Cancer Res Clin Oncol. 2019, 145, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Barton VN, D'Amato NC, Gordon MA, Lind HT, Spoelstra NS, Babbs BL, Heinz RE, Elias A, Jedlicka P, Jacobsen BM, Richer JK. Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. 2015, 14, 769–778. [Google Scholar] [CrossRef]
- Barton VN, Christenson JL, Gordon MA, Greene LI, Rogers TJ, Butterfield K, Babbs B, Spoelstra NS, D'Amato NC, Elias A, Richer JK. Androgen Receptor Supports an Anchorage-Independent, Cancer Stem Cell-like Population in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 3455–3466. [Google Scholar] [CrossRef] [PubMed]
- Fernández NB, Sosa SM, Roberts JT, Recouvreux MS, Rocha-Viegas L, Christenson JL, Spoelstra NS, Couto FL, Raimondi AR, Richer JK, Rubinstein N. RUNX1 Is Regulated by Androgen Receptor to Promote Cancer Stem Markers and Chemotherapy Resistance in Triple Negative Breast Cancer. Cells. 2023, 12, 444. [Google Scholar] [CrossRef] [PubMed]
- 66. Rosas E, Roberts JT, O'Neill KI, Christenson JL, Williams MM, Hanamura T, Spoelstra NS, Vahrenkamp JM, Gertz J, Richer JK. A Positive Feedback Loop Between TGFβ and Androgen Receptor Supports Triple-negative Breast Cancer Anoikis Resistance. Endocrinology, 2021; 162, bqaa226.
- Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, Batchelor TT, Bindra RS, Chang SM, Chiocca EA, Cloughesy TF, DeGroot JF, Galanis E, Gilbert MR, Hegi ME, Horbinski C, Huang RY, Lassman AB, Le Rhun E, Lim M, Mehta MP, Mellinghoff IK, Minniti G, Nathanson D, Platten M, Preusser M, Roth P, Sanson M, Schiff D, Short SC, Taphoorn MJB, Tonn JC, Tsang J, Verhaak RGW, von Deimling A, Wick W, Zadeh G, Reardon DA, Aldape KD, van den Bent MJ. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Bao D, Cheng C, Lan X, Xing R, Chen Z, Zhao H, Sun J, Wang Y, Niu C, Zhang B, Fang S. Regulation of p53wt glioma cell proliferation by androgen receptor-mediated inhibition of small VCP/p97-interacting protein expression. Oncotarget. 2017, 8, 23142–23154. [Google Scholar] [CrossRef]
- Yu X, Jiang Y, Wei W, Cong P, Ding Y, Xiang L, Wu K. Androgen receptor signaling regulates growth of glioblastoma multiforme in men. Tumour Biol. 2015, 36, 67–72.
- Rodríguez-Lozano DC, Piña-Medina AG, Hansberg-Pastor V, Bello-Alvarez C, Camacho-Arroyo I. Testosterone Promotes Glioblastoma Cell Proliferation, Migration, and Invasion Through Androgen Receptor Activation. Front Endocrinol. 2019, 10, 16. [Google Scholar] [CrossRef]
- Rodríguez-Lozano DC, Velázquez-Vázquez DE, Del Moral-Morales A, Camacho-Arroyo I. Dihydrotestosterone Induces Proliferation, Migration, and Invasion of Human Glioblastoma Cell Lines. Onco Targets Ther. 2020, 13, 8813–8823. [Google Scholar] [CrossRef]
- Zalcman N, Gutreiman M, Shahar T, Weller M, Lavon I. Androgen Receptor Activation in Glioblastoma Can Be Achieved by Ligand-Independent Signaling through EGFR-A Potential Therapeutic Target. Int J Mol Sci. 2021, 22, 10954. [Google Scholar] [CrossRef] [PubMed]
- Zalcman N, Canello T, Ovadia H, Charbit H, Zelikovitch B, Mordechai A, Fellig Y, Rabani S, Shahar T, Lossos A, Lavon I. Androgen receptor: A potential therapeutic target for glioblastoma. Oncotarget. 2018, 9, 19980–19993. [Google Scholar] [CrossRef] [PubMed]
- Read TA, Fogarty MP, Markant SL, McLendon RE, Wei Z, Ellison DW, Febbo PG, Wechsler-Reya RJ. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, Rich JN. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010, 6, 421–432. [Google Scholar] [CrossRef]
- Alonso MM, Diez-Valle R, Manterola L, Rubio A, Liu D, Cortes-Santiago N, Urquiza L, Jauregi P, Lopez de Munain A, Sampson N, Aramburu A, Tejada-Solís S, Vicente C, Odero MD, Bandrés E, García-Foncillas J, Idoate MA, Lang FF, Fueyo J, Gomez-Manzano C. Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PLoS ONE. 2011, 6, e26740. [Google Scholar]
- Du Z, Jia D, Liu S, Wang F, Li G, Zhang Y, Cao X, Ling EA, Hao A. Oct4 is expressed in human gliomas and promotes colony formation in glioma cells. Glia. 2009, 57, 724–733. [Google Scholar] [CrossRef]
- Niu CS, Li DX, Liu YH, Fu XM, Tang SF, Li J. Expression of NANOG in human gliomas and its relationship with undifferentiated glioma cells. Oncol Rep. 2011, 26, 593–601. [Google Scholar]
- Rasper M, Schäfer A, Piontek G, Teufel J, Brockhoff G, Ringel F, Heindl S, Zimmer C, Schlegel J. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro Oncol. 2010, 12, 1024–1033. [Google Scholar] [CrossRef]
- Loras A, Gonzalez-Bonet LG, Gutierrez-Arroyo JL, Martinez-Cadenas C, Marques-Torrejon MA. Neural Stem Cells as Potential Glioblastoma Cells of Origin. Life (Basel). 2023, 13, 905. [Google Scholar]
- Matsubara S, Matsuda T, Nakashima K. Regulation of Adult Mammalian Neural Stem Cells and Neurogenesis by Cell Extrinsic and Intrinsic Factors. Cells. 2021, 10, 1145. [Google Scholar] [CrossRef]
- Kelava I, Chiaradia I, Pellegrini L, Kalinka AT, Lancaster MA. Androgens increase excitatory neurogenic potential in human brain organoids. Nature. 2022, 602, 112–116. [Google Scholar] [CrossRef] [PubMed]
- La Rosa P, Bartoli G, Farioli Vecchioli S, Cesari E, Pagliarini V, Sette C. Androgen Receptor signaling promotes the neural progenitor cell pool in the developing cortex. J Neurochem. 2021, 157, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Kim HJ, Kim TJ, Kim YG, Seong C, Cho JH, Kim W, Lee KH, Kim DY. Antioxidant and Antiproliferative Activity of Finasteride against Glioblastoma Cells. Pharmaceutics. 2021, 13, 1410. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Androgen receptor signaling in cancer stem cells. A) Testosterone, due to its hydrophobic characteristics, crosses the cell membrane ①. Once in the cytoplasm, it can be metabolized into DHT or directly bind to the AR ③. The interaction of AR with its ligand generates conformational changes that release it from heat shock proteins ④ that keep it stable in the cytoplasm and protect it from degradation. Once the AR is bound to its ligand, it can be phosphorylated ⑤ and subsequently form a homodimer ⑥, which can translocate to the nucleus and recognize specific gene sequences ⑦. B) The activated AR in CSCs may promote the expression of genes associated with stemness. However, its effects on self-renewal, differentiation, and tumorigenic capacity seem to depend on the tumor context. Created with Biorender.com.
Figure 1.
Androgen receptor signaling in cancer stem cells. A) Testosterone, due to its hydrophobic characteristics, crosses the cell membrane ①. Once in the cytoplasm, it can be metabolized into DHT or directly bind to the AR ③. The interaction of AR with its ligand generates conformational changes that release it from heat shock proteins ④ that keep it stable in the cytoplasm and protect it from degradation. Once the AR is bound to its ligand, it can be phosphorylated ⑤ and subsequently form a homodimer ⑥, which can translocate to the nucleus and recognize specific gene sequences ⑦. B) The activated AR in CSCs may promote the expression of genes associated with stemness. However, its effects on self-renewal, differentiation, and tumorigenic capacity seem to depend on the tumor context. Created with Biorender.com.
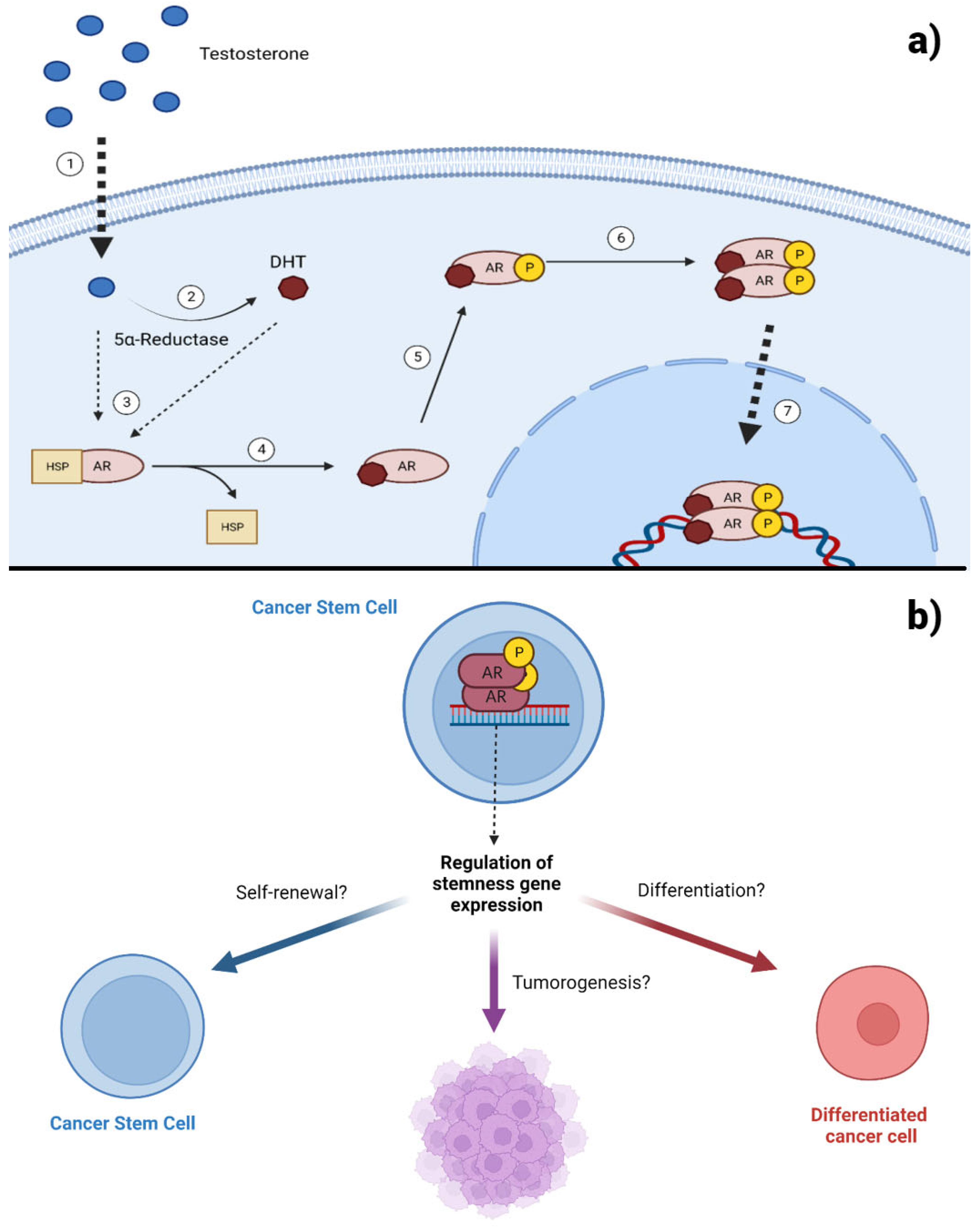

Table 1.
Effects of AR signaling on stemness maintenance.
| Cancer Stem Cell | AR effect on stemness | References |
|---|---|---|
| Prostate Cancer Stem Cell | Decreases self-renewal capacity Decreases expression of stemness factors Increases cell differentiation |
[42,44,45] |
| Breast Cancer Stem Cell | Promotes self-renewal Promotes expression of stemness factors |
[63,64,65] |
| Glioma Stem Cell | Promotes self-renewal Promotes expression of stemness factors |
[18,84] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



