Submitted:
11 September 2023
Posted:
12 September 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Monoclonal antibodies (mAbs) are an important treatment option for COVID-19 caused by SARS-CoV-2, especially in immunosuppressed patients. However, this treatment option can become ineffective due to mutations in the SARS-CoV-2 genome, mainly in the receptor binding domain (RBD) of the spike (S) protein. In the present study 7950 SARS-CoV-2 positive samples from the Uppsala and Örebro regions of central Sweden collected between March 2022 and May 2023 were whole-genome sequenced using next-generation sequencing, mainly with the Nanopore sequencing method. Pango lineages were determined and all single nucleotide polymorphism (SNP) mutations that occurred in these samples were identified. The dominant sublineages changed over time and mutations conferring resistance to currently available mAbs became common. Notable ones are R346T and K444T mutations in the RBD that confer significant resistance against tixagevimab and cilgavimab mAbs. Further, mutations conferring a high-fold resistance to bebtelovimab, such as the K444T and V445P mutations, were also observed in the samples. This study highlights that resistance mutations have over time rendered currently available mAbs ineffective against SARS-CoV-2 in most patients. Therefore, there is a need for continued surveillance of resistance mutations and the development of new mAbs that target more conserved regions of the RBD.
Keywords:
SARS-CoV-2
; coronavirus
; monoclonal antibodies
; resistance
; nanopore
; Sweden
; whole-genome sequencing
; receptor binding domain
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a positive sense, single-stranded RNA virus of the Coronaviridae family, which causes coronavirus disease 2019 (COVID-19). SARS-CoV-2 has caused a global pandemic and was considered a public health emergency of international concern by the World Health Organisation (WHO) between 30 January 2020 and 05 May 2023[1]. The mRNA vaccines have contributed to reducing severe disease and hospitalization, but long term immunity remains difficult to reach due to immune escape[2]. Consequently, antiviral options such as Paxlovid[3] or monoclonal antibodies (mAbs) are necessary to further reduce the COVID-19 health burden. Hence, mAbs have become valuable in the treatment of COVID-19, especially for high-risk individuals such as immunocompromised patients. The mAbs developed against SARS-CoV-2 have been divided into four major classes: Classes I-IV, based on their epitope recognition and binding mode with the receptor binding domain (RBD) of the spike (S) protein[4,5]. The RBD lies between amino acids 333 and 527 in the S protein[6]. It interacts with angiotensin-converting enzyme 2 (ACE2) of the host cell, thereby playing a vital role in viral entry into the host cell[7]. Since November 9, 2020, mAbs have been authorized (for emergency use) for treating COVID-19 patients (Table 1).
RNA viruses, in general, tend to accumulate mutations in their genome at relatively high rates. Some of these mutations, either alone or together, can provide a selective advantage to the virus. SARS-CoV-2 started to diversify even in the first months after it was first detected[22] but was initially thought to be only slowly evolving due to the proofreading function of non-structural protein 14,[23] but throughout the pandemic a large number of mutations have occurred globally[23,24]. Some of the mutations in the genome of SARS-CoV-2 are known to confer significant resistance towards the licenced mAbs. Tracking such mutations helps physicians predict whether certain treatments may or may not work. Previous studies have found several mutations, mainly in the RBD of the S protein that confer significant resistance against mAbs, among them are R346T[25,26], 371F[27], K444T[25,26], V445A/P[25,26], G446S[26], L452R[28], N460K[25,26], E484A[26], F486S/V/P[25,26] and Q493R[28,29]. Table 2 lists these and additional mutations and their corresponding resistances. These mutations have appeared in separate lineages through convergent evolution.[26] Further, treatment with mAbs can lead to or accelerate the development of resistance in the patient being treated [30,31,32,33] and may also cause these mutations to spread into the larger population [31,32].
Studying the genomic differences (i.e., mutations) between different variants and sublineages of SARS-CoV-2 is also necessary for understanding why they cause different symptoms and severities. The Omicron variant was first detected in South Africa[34] and then became the world-dominant SARS-CoV-2 variant. The Delta variant, preceding Omicron, differs from the latter variant in several ways. Cell-cell fusion and infectivity of lung and gut cells were reduced in the Omicron lineages BA.1 and BA.2 compared to that of the Delta variant.[35,36] Further, the Delta and Omicron variants differ in entry pathways[36]. Hence, identifying the mutation(s) that caused these phenotypes is important to understand how future variants will behave and what threats they may pose. There are also observed phenotypic differences between Omicron sublineages, e.g. the BA.5 sublineages infect lung tissues more efficiently than the BA.2 sublineages, and the Spike protein of BA.4 and BA.5 can more efficiently fuse lung cells compared to that of BA.2[37].
There are also other important reasons for tracking mutations and circulating lineages of SARS-CoV-2. Tracking immune escape mutations is important to understand the immune protection status of vaccinated or previously infected individuals, and they aid in the development of appropriate vaccines/booster vaccines. Resistance to antivirals other than mAbs may also arise in individual patients and spread across the population.
During the SARS-CoV-2 pandemic, several laboratories throughout Sweden have conducted whole-genome sequencing for contact tracing and surveillance purposes. The variants of concern and S-protein mutations detected in the Uppsala region have already been described for the year 2021[38] but not for the period after that. In the present study, SARS-CoV-2 genomes from COVID-19-positive samples between March 2022 and May 2023 obtained from Uppsala and Örebro regions- representing a large part of central Sweden have been sequenced using next-generation sequencing methods, mainly with the Nanopore sequencing method. Using the sequence data from these samples, we describe the prevalence of Omicron variants and how their susceptibility towards mAbs has changed over time.
2. Materials and Methods
2.1. Samples collection
Samples positive for SARS-CoV-2 RNA, used in this study, were collected between March 2022 and May 2023 from individuals residing in Uppsala and Örebro regions, two of Sweden’s 21 regional councils, as part of their SARS-CoV-2 surveillance and tracing programs. The study was approved by the Swedish Ethical Review Authority) under the case numbers Dnr 2022-01249-01 and 2023-02272-02. Informed consent was not applicable. Samples received were either assisted nasopharyngeal-throat swabs, saliva samples (via self-collection kit) or a combined sampling of gargled water and self-collected nasal/nasopharyngeal swab. Further, a small number of bronchoalveolar lavage, blood and lung biopsy samples were also included.
2.2. Viral RNA extraction and sequencing
For samples from Örebro, sequencing was performed on three different laboratories: department of Laboratory Medicine, Örebro University Hospital, National Pandemic centre (NPC) or Public Health Agency in Sweden (PHAS).
In Örebro the sequences were generated using either a MiSeq (Illumina, U.S) instrument with the ARTIC V3 tiled amplicon enrichment protocol (400 bp amplicon) or with a GridION (Oxford Nanopore Technologies, United Kingdom) instrument using the Midnight protocol based on the ARTIC network (1 200 bp amplicons). Both described in detail by Koskela von Sydow et al.[39] Consensus sequences from Illumina data were generated with either Ridom SeqSphere+ version 8.3.1 – 8.5.1[40], or gms-artic version v2.0[41], Ridom Seqsphere+ was used with the following settings: samtools mpileup (-A -d 1000000 -B -Q 0, v1.12); ivar consensus (-q 20 -t 0.7 -m 10); ivar variants (-q 20 -m 10 -t 0.1). Gms-artic was used with the default settings and “--scheme nCoV-2019”, “–schemeVersion” “V3” or “V4.1”. Consensus sequences for the 1200 bp amplicons were generated with wf-artic version v0.3.9-v0.3.24[42] from epi2me-labs using the default settings and --scheme_”version V1200” or “Midnight-IDT/V1”.
For sequencing at the NPC, sequences were generated on the MGI DNBSEQ-G400 using an ultraplex amplicon approach and PE100 library construction[43]. In brief, raw sequencing data were filtered using FastP[44], followed by mapping towards the reference genome. Alignments were trimmed from primer sequence by use of iVar[45], and Variant Call Files were calculated using Freebayes[46]. Consensus sequences were generated based on major frequency bases. Low coverage areas (<30) were masked to N, and deletions (as defined by variant call) were masked, and then the sequence was collapsed at the point of deletion to keep relative genome coordinates.
At the PHAS, the sequences were generated by Genome Sequencer HiSeq (Illumina, U.S), sequence mode NovaSeq 6000 S4 PE150 XP. High quality reads were aligned to the SARS-CoV-2 reference genome (isolate Wuhan-Hu-1, MN908947) using the BWA-MEM v0.7.17-r1188 algorithm and consensus sequences were generated using consensusfixer v0.4[47] with at least 15 supporting reads at each position. Base positions which showed less than 15x coverage were filled with N’s.
For samples from Uppsala, RNA extraction, reverse transcription PCR (RT-PCR) and Nanopore sequencing were performed at the Division of Clinical Microbiology and Hospital Hygiene, Uppsala University Hospital, Sweden. RNA extraction was performed according to the manufacturer’s instructions using Chemagic™ 360 (PerkinElmer, U.S.) or eMAG® (bioMérieux, France) instruments. Samples positive for SARS-CoV-2 were detected with an in-house RT-qPCR method or with the BIOFIRE® Respiratory 2.1 plus Panel (bioMérieux, France). After extraction, RNA eluates were stored at -20℃. The Ct value for each sample, needed for correct dilution of the RNA, was acquired from the in-house RT-qPCR method.
Between the start of the study period and June 27, 2022, the Midnight protocol[48] was used alongside the NEBNext® ARTIC SARS-CoV-2 Companion Kit protocol version 1.0_1/21 (with a few modifications) for the library preparation and sequencing. After June 27, 2022, only the ARTIC protocol was used. The modifications to the protocol included 33 PCR cycles and the replacement of the PCR bead cleanup, by a 1:10 dilution of all PCR products as per the ARTIC nCoV-2019 v3 (LoCost) protocol[49,50]. Library preparation was performed with the NEBNext® ARTIC SARS-CoV-2 Companion, Native Barcoding Expansion 96 (Catalogue number: EXP-NBD196; Oxford Nanopore Technologies, United Kingdom) and Ligation Sequencing (Catalogue number: SQK-LSK109; Oxford Nanopore Technologies, United Kingdom) Kits. Between the start of the study period and June 27, 2022, ARTIC Network SARS-CoV-2 V3 primers were used. After this, VarSkip Short v2 primers (New England BioLabs, U.S.) and BA.2 Spike-in primers[51] (New England BioLabs, U.S.) were used. This switch was done to increase the quality of sequences since issues with the older primer sets had previously been described.[38,52] The DNA concentration of the Library was measured using the Qubit HS dsDNA assay kit (Thermo Fisher, U.S.).
Sequencing was performed with the R9.4.1 flow cell (catalogue number: FLO-MIN106D) on a GridION instrument (Oxford Nanopore Technologies, United Kingdom). To reduce the risk of cross-contamination between runs, flow cells were never reused. The MinKNOW software was used with the following settings: high-accuracy base calling, barcodes on both ends, minimum barcoding alignment score = 60, minimum mid-read barcoding alignment score = 50, and trim barcodes.
Analysis of sequence data was performed with Geneious Prime version 2021.1.1[53]. Primer sequences were trimmed using the Geneious prime BBDuk plugin version 38.84[54] with the following settings: trim = left end, kmer length = 21, maximum substitutions = 3, trim low quality (<10) from both ends, discard reads shorter than 50 bp and custom BBDuk options; rcomp = f and restrictleft = 32. Sequence alignment and mapping to the SARS-CoV-2 Wuhan-Hu-1 reference sequence (NCBI accession number: NC_045512.2) was performed using the Geneious prime minimap2 version 2.17[55] plugin using the following settings: data type = “Oxford Nanopore (more sensitive)”, include secondary alignments enabled, maximum secondary alignments per read = 5, minimum secondary to primary alignment score ratio = 0.8 and “remove existing trim regions from sequences” enabled. Consensus sequences were generated using the “Generate Consensus Sequence” function in Geneious Prime using the following settings: minimum coverage = 4 reads, minimum nucleotide frequency = 0.5 and “Trim to reference sequence” enabled.
Consensus sequences from both Örebro and Uppsala were uploaded in FASTA format to the Global Initiative on Sharing Avian Influenza Data (GISAID) database[56].
The sequences were assigned Pango lineages according to the Pango dynamic lineage nomenclature scheme[57] using the Geneious wrapper plugin for Pangolin[58] that runs the Phylogenetic Assignment of Named Global Outbreak Lineages (Pangolin) tool[59]. Unaliased Pango lineages used for grouping lineages were acquired using modified versions of R scripts contained in the PangoLineageTranslator tool[60].
To identify the mutations in the sequences, Coronapp[61] was used to find all mutations across the entire genome. Coronapp is annotation based, which we have found necessary to find all mutations in our sequences generated from Nanopore sequencing which occasionally have frameshifts.
3. Results
From the period between March 1, 2022, to May 18, 2023, 7950 SARS-CoV-2 positive samples were successfully whole-genome sequenced and analysed, which is presented in this study. Of these samples, the majority (i.e., 6710) were obtained during 2022 and 1240 were from 2023. Approximately half (i.e., 4478) of these samples were from the Uppsala region and 3472 from the Örebro region. The distribution of samples sequenced over time is shown in Figure 1.
The Pangolin tool was used to determine the Pango lineage of the samples and the weekly frequencies of different sublineages as determined from their Pango lineage were plotted against time. Figure 2 shows how the dominant sublineages in our data from central Sweden have changed throughout the pandemic. The Omicron BA.2 sublineage and its descendants dominated from the start of our study in March 2022 until the middle of May 2022 when it was replaced, mainly by the BA.5 sublineage and its descendants, but to some degree also by the BA.4 sublineage. The BA.5 and its descendants, including BE, BF and BQ sublineages, were the main sublineages between June 2022 and December 2022. After this, the situation was more dynamic, with a mix of BA.2 and BA.5 lineages. The most prominent of the sublineages that increased during this time were the BA.2.75 (including BN and CH sublineages) and BQ sublineages. The XBB sublineages began to take over and became the most common ones by late spring 2023. The XBB sublineage is the product of a recombination event between the two sublineages BJ.1 and BM.1.1.1 which are both descendants of BA.2.[62].The increase in BQ and XBB sublineages is important because of their known resistance to the avaiable mAbs[25]. The number of sequences assigned to the XBB.1.9.1 lineage increased towards the end of the studied period (Figure 2) and was the most common Pango lineage in May 2023 (Table A1), the last month of the studied period.
Among the amino acid (aa) mutations in the RBD that are involved in mAb resistance, several changed in their relative abundance throughout the studied period (Figure 3). The over time relative abundance of all aa substitutions in the S-protein of SARS-CoV-2 from samples collected during the period March 2022 to May 2023 can be seen in Figure S1.
Mutations in the SARS-CoV-2 Spike protein, especially at positions 346 and 444, mainly R346T and K444T, increased during the autumn of 2022 (Figure 4a), indicating an increase in infections that were resistant to mAb treatment with tixagevimab and cilgavimab. There was a simultaneous increase in the incidence of samples with double mutations at aa positions 346 and 444, and samples with single mutations at one of the two positions, showing that several different lineages increased in frequency at the same time but were conferring similar mAb resistance through different mutations. Mutations in position 444, mainly K444T, increased during the autumn of 2022 but decreased by spring 2023, mutations in position 445, mainly V445P, increased during spring of 2023 (Figure 4b). K444T and V445P are involved in resistance to bebtelovimab.
4. Discussion
Since the start of the COVID-19 pandemic, SARS-CoV-2 has evolved rapidly and still continues to evolve. The changes that have occurred during this time have affected many aspects of the virus and the COVID-19 disease outcome. Since virus transmission and COVID-19 symptoms/severities vary between different sublineages[35,36,37], it is important to have a good knowledge of sublineage(s) that are in circulation locally at particular time. This will benefit in treating patients effectively and also in implementing effective control measures to prevent further spread. Using SARS-CoV-2 sequences collected between March 2022 and May 2023 from individuals residing in the Uppsala and Örebro regions of central Sweden, the dynamics of various sublineages in the regions and aa mutations that are relevant regarding mAbs are reported in this study.
We found that the mutations that have occurred during the studied period, especially in the RBD, have a significant effect on the usefulness of available mAbs[25,26].
Treatment with bamlanvimab, regdanvimab, casirivimab/imdevimab or sotrovimab have been unlikely to be successful in the Uppsala and Örebro regions throughout the entire studied period. Mutations like L452R, E484A, E484K, F486V or Q493R, either alone or in combination, confers a high-fold resistance (>400 fold-change range) against bamlanvimab (Table 2) and these mutations have been present in most samples during the studied period (Figure 3). Some of the aforementioned mutations, L452R and Q493R, also confer high-fold resistance to regdanvimab. While Q493R, which confers the highest resistance to regdanvimab of all the aa mutations (as a single mutation) in the RBD of the S protein, became rare after the first half of 2022, other mutations such as S371F and E484A are very common and L452R was very common between June 2022 and March 2023 (Figure 3). The common E484A mutation also confers medium-fold resistance to the combination of casirivimab and imdevimab (Table 2). In addition, the mutations K444T, V445P and G446S which became common during the end of 2022 and the start of 2023 (Figure 3) also confer low- to medium-fold resistance to this mAb combination. The S371F mutation, on the other hand, confers medium fold-change resistance against sotrovimab. This left tixagevimab and cilgavimab as the only available option in the EU while the U.S. also had access to bebtelovimab.
During the second half of 2022, the number of samples with mutations in 346 or 444 increased rapidly, from being present in <10% of samples to being present in >90% of samples in just four months (Figure 4a). This increase was mainly due to the aa mutations R346T and K444T which were much more common than R346S, R346K, R346I, K444R or K444N (Figure 3). The simultaneous increase of mutations in S-protein aa 346, 444 or both shows that prevalence of mAb resistance increased by the spread of several different lineages carrying different sets of mutations with similar phenotypic effects. This taken together with the fact that both sublineages originating from BA.2 and sublineages originating from BA.5 spread at the same time and showed similar mutations indicates that parallel evolution was and had been taking place. The proportion of samples with either a mutation in 444 or 445 increased rapidly during the last months of 2022 and the beginning of 2023 (Figure 4b). The increase in aa mutations at position 445 was mainly because of an increase of V445P (Figure 3) which is present in all XBB lineages. Mutations in aa positions 346 and/or 444 have been shown to cause high-fold resistance against the mAb treatment with tixagevimab and cilgavimab[25] used in Evusheld which was the main mAb treatment available in Europe during this time. In the U.S., bebtelovimab was available and if it had been available in Uppsala and Örebro it could have been used to treat patients until the mutations K444T, and/or V445P, which cause resistance to bebtelovimab[25,26] also became very common (Figure 4b). However, targeted testing and whole-genome sequencing of SARS-CoV-2 samples from candidates for bebtelovimab treatment could have been performed for several months before the 445 aa changes became fixed in the SARS-CoV-2 population. This would have allowed more patients to be treated with mAbs.
The abundance of sublineages BN.1, BQ.1, CH.1, XBB and their descendants which are known for their high resistance against the mAbs that were available during 2022 and the first half of 2023[25] increased among our samples during the end of 2022 and had become very dominant by the start of 2023 (Figure 2). During 2023, sublineages with high resistance to mAbs remained dominant but the composition of sublineages changed. Descendants of XBB, and in turn BA.2, have taken over and very few samples sequenced by from April to May 2023 are descendants of BA.5. By the final week of this study, XBB.1.9.1 had become the most common sublineage (Table S1). This increase of XBB.1.9.1 as well as that of XBB.1.5 (Figure 2) is likely because of the mutation F486P which increased the ACE2 binding affinity and is a strong neutralising antibody evading mutation[63]. This suggests that future studies should focus on investigating the XBB sublineages and investigate how mutations with an XBB genetic background affects any new vaccines, mAbs or other antivirals. However, parallel evolution can take place in different distantly related sublineages and replacements with completely new sublineages has happened rapidly and repeatedly, so investigation of other sublineages is also well justified. Treatment with currently available mAbs is unlikely to be successful for most current SARS-CoV-2 patients, but some patients with chronic infections may still carry SARS-CoV-2 variants who are susceptible to available mAbs. If samples from these patients are whole-genome sequenced it could help determine which mAb would be effective for their treatment.
While currently available mAbs may be ineffective for treating most SARS-CoV-2 patients, new mAbs are being developed, such as AZD3152 by AstraZeneca which is in clinical trials with an estimated primary completion date of 7 September 2023[64]. This antibody targets a conserved region of the RBD[65] and is active against currently circulating sublineages that are resistant to other mAbs (Table 2).
The repeated events of evolved resistance to mAbs means that the development of new mAbs and other antiviral treatments remains important for patients infected with SARS-CoV-2. The rapid increase in resistance to tixagevimab and cilgavimab highlights that these changes need to be communicated quickly by labs and scientists to the public. So, the continued surveillance of SARS-CoV-2 through whole-genome sequencing is essential for understanding the evolution of the virus and for providing scientists, physicians, patients, decision makers and drug manufacturers with correct and updated information for curbing this infection in the population.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Heatmap showing the abundance of all amino acid mutations over time in the S protein of the SARS-CoV-2 from sequences collected between March 2022 and May 2023 from individuals residing in Uppsala and Örebro region. Time points are weekly. Mutations that were present in less than 10 samples have been excluded. Table S1: Number of sequences of each Pango lineage in May 2023.
Author Contributions
Conceptualization, J.H., N.P. and J.Le.; methodology, J.H and J.Le.; software, J.H. and R.K.; formal analysis, J.H.; investigation, J.H., F.W., P.M., P.E., R.K and J.Le; resources, J.Li, M.S., R.K.; data curation, J.H. and F.W.; writing—original draft preparation, J.H.; writing—review and editing, J.H., N.P., F.W., P.M., P.E., J.Li., M.S., R.K and J.Le.; visualization, J.H.; supervision, R.K. and J.Le.; funding acquisition, J.Le. All authors have read and agreed to the published version of the manuscript.
Funding
J.Le. received financial support from the Regional Research Council Mid Sweden (RFR-980115).
Data Availability Statement
Consensus sequences for all samples are available on GISAID as EPI_SET_230905gu, doi: 10.55876/gis8.230905gu.
Acknowledgments
We would like to thank the personnel at Clinical Microbiology and Hospital Hygiene at Uppsala University Hospital for their help with handling samples, RNA extraction, PCR and whole-genome sequencing.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- IHR Emergency Committee on Novel Coronavirus (2019-nCoV). Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-statement-on-ihr-emergency-committee-on-novel-coronavirus-(2019-ncov) (accessed on 22 August 2023).
- Lyke, K.E.; Atmar, R.L.; Islas, C.D.; Posavad, C.M.; Szydlo, D.; Paul Chourdhury, R.; Deming, M.E.; Eaton, A.; Jackson, L.A.; Branche, A.R.; et al. Rapid Decline in Vaccine-Boosted Neutralizing Antibodies against SARS-CoV-2 Omicron Variant. Cell Rep Med 2022, 3, 100679. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, L.B.; Tedla, N.; Bull, R.A. Broadly-Neutralizing Antibodies Against Emerging SARS-CoV-2 Variants. Frontiers in Immunology 2021, 12. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 Neutralizing Antibody Structures Inform Therapeutic Strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Pillay, T.S. Gene of the Month: The 2019-nCoV/SARS-CoV-2 Novel Coronavirus Spike Protein. Journal of Clinical Pathology 2020, 73, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the Receptor-Binding Domain (RBD) of 2019 Novel Coronavirus: Implication for Development of RBD Protein as a Viral Attachment Inhibitor and Vaccine. Cell Mol Immunol 2020, 17, 613–620. [Google Scholar] [CrossRef]
- Commissioner, O. of the Coronavirus (COVID-19) Update: FDA Revokes Emergency Use Authorization for Monoclonal Antibody Bamlanivimab. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-monoclonal-antibody-bamlanivimab (accessed on 22 August 2023).
- EMA EMA Issues Advice on Use of Antibody Combination (Bamlanivimab / Etesevimab). Available online: https://www.ema.europa.eu/en/news/ema-issues-advice-use-antibody-combination-bamlanivimab-etesevimab (accessed on 22 August 2023).
- EMA Bamlanivimab and Etesevimab for COVID-19: Withdrawn Application. Available online: https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/bamlanivimab-etesevimab-covid-19 (accessed on 22 August 2023).
- Emergency Use Authorization 094. Available online: https://pi.lilly.com/eua/bam-and-ete-eua-fda-authorization-letter.pdf (accessed on 22 August 2023).
- EMA Regkirona. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/regkirona (accessed on 22 August 2023).
- Commissioner, O. of the Coronavirus (COVID-19) Update: FDA Limits Use of Certain Monoclonal Antibodies to Treat COVID-19 Due to the Omicron Variant. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-limits-use-certain-monoclonal-antibodies-treat-covid-19-due-omicron (accessed on 22 August 2023).
- Commissioner, O. of the Coronavirus (COVID-19) Update: FDA Authorizes Additional Monoclonal Antibody for Treatment of COVID-19. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-additional-monoclonal-antibody-treatment-covid-19 (accessed on 22 August 2023).
- Commissioner, O. of the FDA Roundup: April 5, 2022. Available online: https://www.fda.gov/news-events/press-announcements/fda-roundup-april-5-2022 (accessed on 22 August 2023).
- EMA Xevudy. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/xevudy (accessed on 22 August 2023).
- Commissioner, O. of the Coronavirus (COVID-19) Update: FDA Authorizes New Long-Acting Monoclonal Antibodies for Pre-Exposure Prevention of COVID-19 in Certain Individuals. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-new-long-acting-monoclonal-antibodies-pre-exposure (accessed on 22 August 2023).
- Research, C. for D.E. and FDA Announces Evusheld Is Not Currently Authorized for Emergency Use in the U.S. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-announces-evusheld-not-currently-authorized-emergency-use-us (accessed on 22 August 2023).
- EMA Evusheld. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/evusheld (accessed on 22 August 2023).
- Commissioner, O. of the Coronavirus (COVID-19) Update: FDA Authorizes New Monoclonal Antibody for Treatment of COVID-19 That Retains Activity Against Omicron Variant. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-new-monoclonal-antibody-treatment-covid-19-retains (accessed on 22 August 2023).
- Research, C. for D.E. and FDA Announces Bebtelovimab Is Not Currently Authorized in Any US Region. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-announces-bebtelovimab-not-currently-authorized-any-us-region (accessed on 22 August 2023).
- Kaden, R. Early Phylogenetic Diversification of SARS-CoV-2: Determination of Variants and the Effect on Epidemiology, Immunology, and Diagnostics. Journal of Clinical Medicine 2020, 9, 1615. [Google Scholar] [CrossRef]
- McCormick, K.D.; Jacobs, J.L.; Mellors, J.W. The Emerging Plasticity of SARS-CoV-2. Science 2021, 371, 1306–1308. [Google Scholar] [CrossRef]
- Lennerstrand, J.; Palanisamy, N. Global Prevalence of Adaptive and Prolonged Infections’ Mutations in the Receptor-Binding Domain of the SARS-CoV-2 Spike Protein. Viruses 2021, 13, 1974. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming Antibody Evasion Properties of Rising SARS-CoV-2 BQ and XBB Subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 Humoral Immunity Induces Convergent Omicron RBD Evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Chan, J.F.-W.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody Evasion Properties of SARS-CoV-2 Omicron Sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Dingens, A.S.; Bloom, J.D. Complete Map of SARS-CoV-2 RBD Mutations That Escape the Monoclonal Antibody LY-CoV555 and Its Cocktail with LY-CoV016. Cell Rep Med 2021, 2, 100255. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Novazzi, F.; Genoni, A.; Dentali, F.; Gasperina, D.D.; Baj, A.; Maggi, F. Emergence of SARS-COV-2 Spike Protein Escape Mutation Q493R after Treatment for COVID-19. Emerg Infect Dis 2021, 27, 2728–2731. [Google Scholar] [CrossRef]
- Truffot, A.; Andréani, J.; Le Maréchal, M.; Caporossi, A.; Epaulard, O.; Germi, R.; Poignard, P.; Larrat, S. SARS-CoV-2 Variants in Immunocompromised Patient Given Antibody Monotherapy. Emerg Infect Dis 2021, 27, 2725–2728. [Google Scholar] [CrossRef]
- Casadevall, A.; Focosi, D. SARS-CoV-2 Variants Resistant to Monoclonal Antibodies in Immunocompromised Patients Constitute a Public Health Concern. J Clin Invest 2023, 133. [Google Scholar] [CrossRef]
- Gupta, A.; Konnova, A.; Smet, M.; Berkell, M.; Savoldi, A.; Morra, M.; Averbeke, V.V.; Winter, F.H.R.D.; Peserico, D.; Danese, E.; et al. Host Immunological Responses Facilitate Development of SARS-CoV-2 Mutations in Patients Receiving Monoclonal Antibody Treatments. J Clin Invest 2023, 133. [Google Scholar] [CrossRef]
- Vellas, C.; Trémeaux, P.; Bello, A.D.; Latour, J.; Jeanne, N.; Ranger, N.; Danet, C.; Martin-Blondel, G.; Delobel, P.; Kamar, N.; et al. Resistance Mutations in SARS-CoV-2 Omicron Variant in Patients Treated with Sotrovimab. Clinical Microbiology and Infection 2022, 28, 1297–1299. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.T.M.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 Usage by SARS-CoV-2 Omicron Impacts Infectivity and Fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Zhao, H.; Lu, L.; Peng, Z.; Chen, L.-L.; Meng, X.; Zhang, C.; Ip, J.D.; Chan, W.-M.; Chu, A.W.-H.; Chan, K.-H.; et al. SARS-CoV-2 Omicron Variant Shows Less Efficient Replication and Fusion Activity When Compared with Delta Variant in TMPRSS2-Expressed Cells. Emerging Microbes & Infections 2022, 11, 277–283. [Google Scholar] [CrossRef]
- Hoffmann, M.; Wong, L.-Y.R.; Arora, P.; Zhang, L.; Rocha, C.; Odle, A.; Nehlmeier, I.; Kempf, A.; Richter, A.; Halwe, N.J.; et al. Omicron Subvariant BA.5 Efficiently Infects Lung Cells. Nat Commun 2023, 14, 3500. [Google Scholar] [CrossRef] [PubMed]
- Mannsverk, S.; Bergholm, J.; Palanisamy, N.; Ellström, P.; Kaden, R.; Lindh, J.; Lennerstrand, J. SARS-CoV-2 Variants of Concern and Spike Protein Mutational Dynamics in a Swedish Cohort during 2021, Studied by Nanopore Sequencing. Virol J 2022, 19, 164. [Google Scholar] [CrossRef] [PubMed]
- Koskela von Sydow, A.; Lindqvist, C.M.; Asghar, N.; Johansson, M.; Sundqvist, M.; Mölling, P.; Stenmark, B. Comparison of SARS-CoV-2 Whole Genome Sequencing Using Tiled Amplicon Enrichment and Bait Hybridization. Sci Rep 2023, 13, 6461. [Google Scholar] [CrossRef]
- Jünemann, S.; Sedlazeck, F.J.; Prior, K.; Albersmeier, A.; John, U.; Kalinowski, J.; Mellmann, A.; Goesmann, A.; von Haeseler, A.; Stoye, J.; et al. Updating Benchtop Sequencing Performance Comparison. Nat Biotechnol 2013, 31, 294–296. [Google Scholar] [CrossRef]
- Gms-Artic. Available online: https://github.com/genomic-medicine-sweden/gms-artic (accessed on 8 September 2023).
- Epi2me-Labs/Wf-Artic: ARTIC SARS-CoV-2 Workflow and Reporting. Available online: https://github.com/epi2me-labs/wf-artic (accessed on 8 September 2023).
- National Sample Collection Stored at NPC – The COVID-19 Library | Karolinska Institutet. Available online: https://ki.se/en/mtc/national-sample-collection-stored-at-npc-the-covid-19-library (accessed on 8 September 2023).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An Amplicon-Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and iVar. Genome Biol 2019, 20, 8. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing 2012.
- Töpfer, A. Cbg-Ethz/ConsensusFixer: Computes a Consensus Sequence with Wobbles, Ambiguous Bases, and in-Frame Insertions, from a NGS Read Alignment. Available online: https://github.com/cbg-ethz/consensusfixer (accessed on 8 September 2023).
- Freed, N.E.; Vlková, M.; Faisal, M.B.; Silander, O.K. Rapid and Inexpensive Whole-Genome Sequencing of SARS-CoV-2 Using 1200 Bp Tiled Amplicons and Oxford Nanopore Rapid Barcoding. Biology Methods and Protocols 2020, 5, bpaa014. [Google Scholar] [CrossRef]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC Multiplex PCR Method for SARS-CoV-2 Genome Sequencing Using Nanopore. bioRxiv 2020, 2020.09.04.283077. [CrossRef]
- Quick, J. nCoV-2019 Sequencing Protocol v3 (LoCost). 2020. 2020. [CrossRef]
- Can Coverage Be Improved for the BA.2 Variant? | NEB. Available online: https://international.neb.com/faqs/2022/04/28/can-coverage-be-improved-for-the-ba2-variant (accessed on 22 August 2023).
- Davis, J.J.; Long, S.W.; Christensen, P.A.; Olsen, R.J.; Olson, R.; Shukla, M.; Subedi, S.; Stevens, R.; Musser, J.M. Analysis of the ARTIC Version 3 and Version 4 SARS-CoV-2 Primers and Their Impact on the Detection of the G142D Amino Acid Substitution in the Spike Protein. Microbiology Spectrum 2021, 9, e01803-21. [Google Scholar] [CrossRef]
- Geneious | Bioinformatics Software for Sequence Data Analysis. Available online: https://www.geneious.com/ (accessed on 22 August 2023).
- BBMap. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 22 August 2023).
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- GISAID - Gisaid.Org. Available online: https://gisaid.org/.
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat Microbiol 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Geneious Wrapper Plugin for Pangolin. Available online: https://github.com/clinical-genomics-uppsala/Geneious_pangolin_wrapper (accessed on 29 August 2023).
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, S. Pango Lineage Translator. Available online: https://github.com/sschmutz/PangoLineageTranslator (accessed on 22 August 2023).
- Mercatelli, D.; Triboli, L.; Fornasari, E.; Ray, F.; Giorgi, F.M. Coronapp: A Web Application to Annotate and Monitor SARS-CoV-2 Mutations. Journal of Medical Virology 2021, 93, 3238–3245. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological Characteristics of the SARS-CoV-2 XBB Variant Derived from Recombination of Two Omicron Subvariants. Nat Commun 2023, 14, 2800. [Google Scholar] [CrossRef]
- Yue, C.; Song, W.; Wang, L.; Jian, F.; Chen, X.; Gao, F.; Shen, Z.; Wang, Y.; Wang, X.; Cao, Y. ACE2 Binding and Antibody Evasion in Enhanced Transmissibility of XBB.1.5. The Lancet Infectious Diseases 2023, 23, 278–280. [Google Scholar] [CrossRef]
- AstraZeneca A Phase I/III Randomized, Double Blind Study to Evaluate the Safety, Efficacy and Neutralizing Activity of AZD5156/AZD3152 for Pre Exposure Prophylaxis of COVID 19 in Participants With Conditions Causing Immune Impairment. Sub-Study: Phase II Open Label Sub-Study to Evaluate the Safety, PK, and Neutralizing Activity of AZD3152 for Pre-Exposure Prophylaxis of COVID-19; clinicaltrials.gov, 2023.
- ECCMID Poster: P2636 The SARS-CoV-2 Monoclonal Antibody AZD3152 Potently Neutralises Historical and Currently Circulating Variants.
Figure 1.
Distribution of sequenced samples per sampling week.
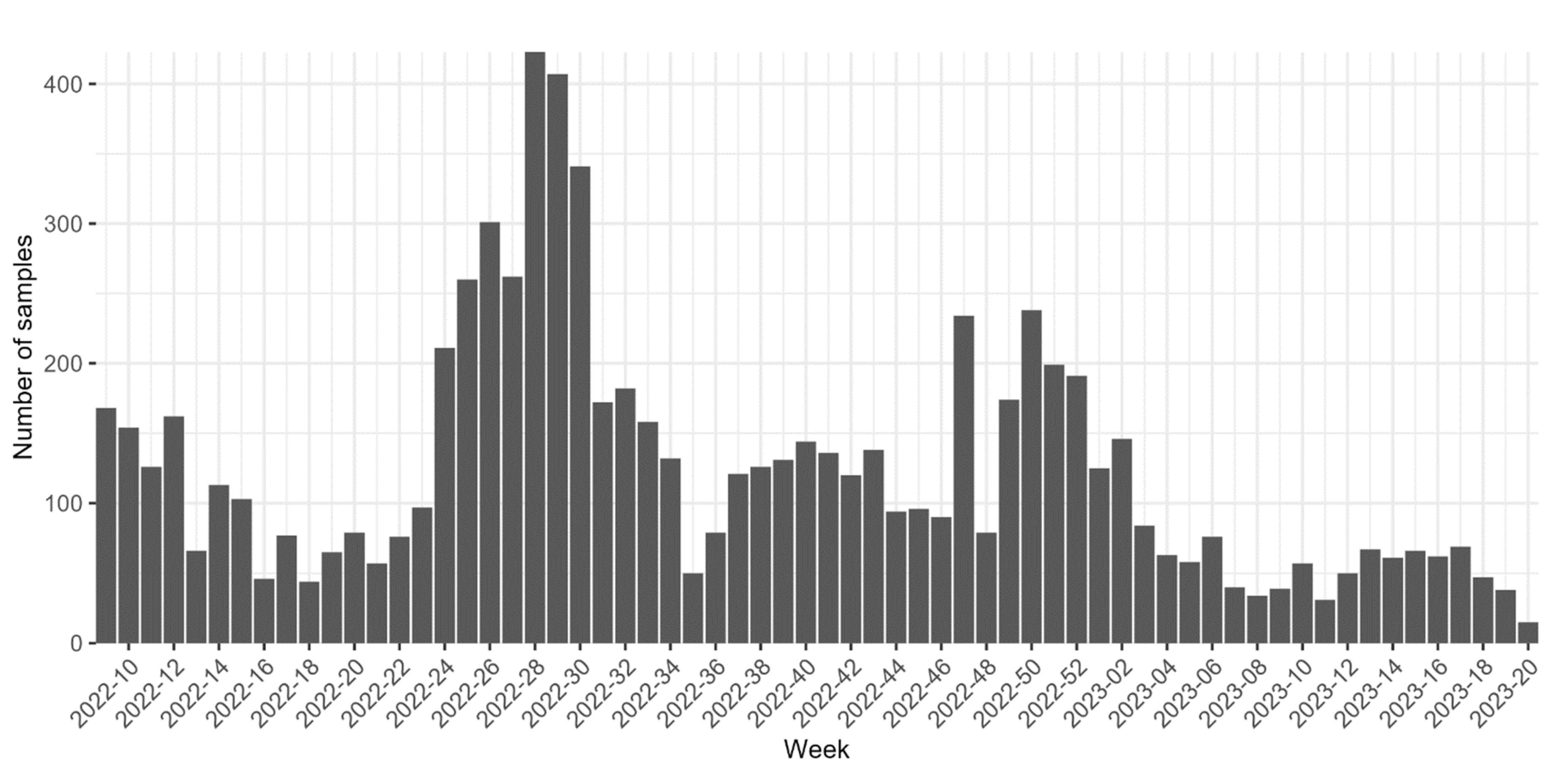
Figure 2.
Frequencies of different SARS-CoV-2 sublineages over time in Uppsala and Örebro region during the period March 2022 to May 2023 with weekly time points. Multiple Pango lineages have been grouped together into groups based on ancestry.
Figure 2.
Frequencies of different SARS-CoV-2 sublineages over time in Uppsala and Örebro region during the period March 2022 to May 2023 with weekly time points. Multiple Pango lineages have been grouped together into groups based on ancestry.
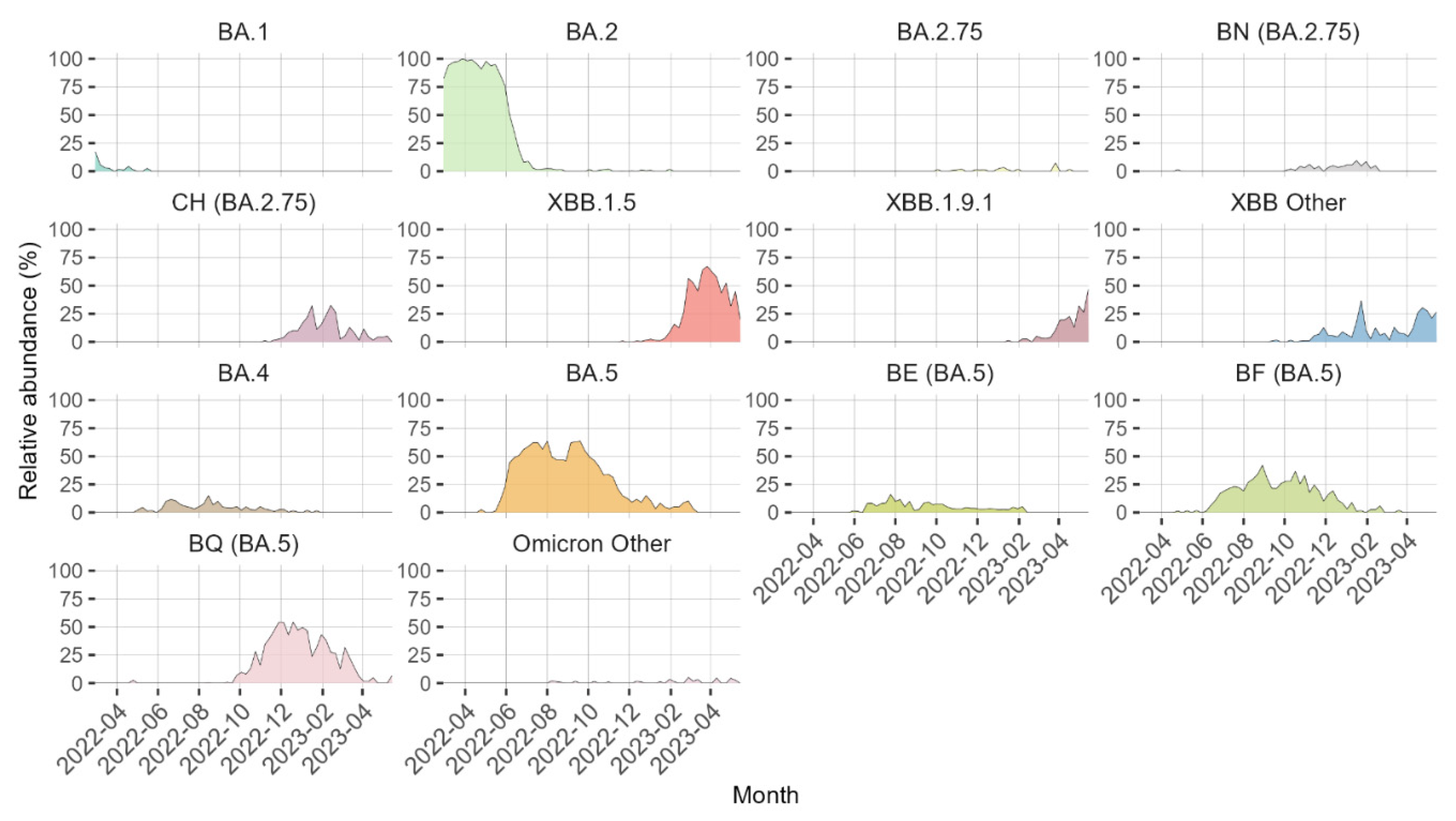
Figure 3.
Heatmap showing the relative abundance of amino acid mutations in the receptor binding domain (RBD) of the SARS-CoV-2 S-protein for each week between March 2022 to May 2023. Amino acid mutations that were present in less than 10 samples have been excluded.
Figure 3.
Heatmap showing the relative abundance of amino acid mutations in the receptor binding domain (RBD) of the SARS-CoV-2 S-protein for each week between March 2022 to May 2023. Amino acid mutations that were present in less than 10 samples have been excluded.
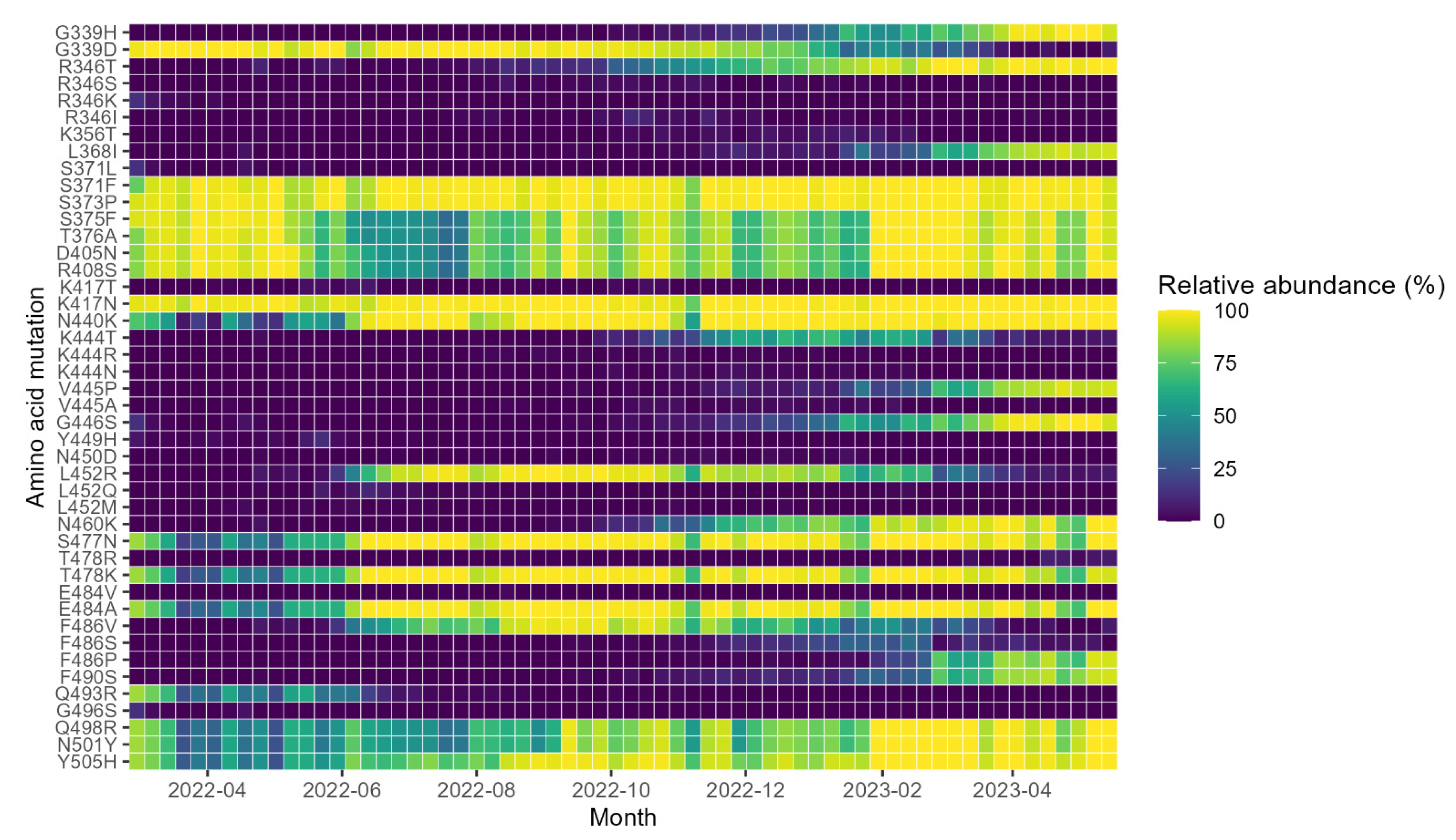
Figure 4.
Relative abundance per week from March 2022 to May 2023 of SARS-CoV-2 sequences containing double and single amino acid (aa) mutations in the S protein and. (a) Positions 346 and 444 (i.e., mainly R346T and K444T), relevant for resistance against tixagevimab and cilgavimab, and (b) positions 444 and 445 (i.e., mainly K444T and V445P), relevant for resistance against bebtelovimab.
Figure 4.
Relative abundance per week from March 2022 to May 2023 of SARS-CoV-2 sequences containing double and single amino acid (aa) mutations in the S protein and. (a) Positions 346 and 444 (i.e., mainly R346T and K444T), relevant for resistance against tixagevimab and cilgavimab, and (b) positions 444 and 445 (i.e., mainly K444T and V445P), relevant for resistance against bebtelovimab.
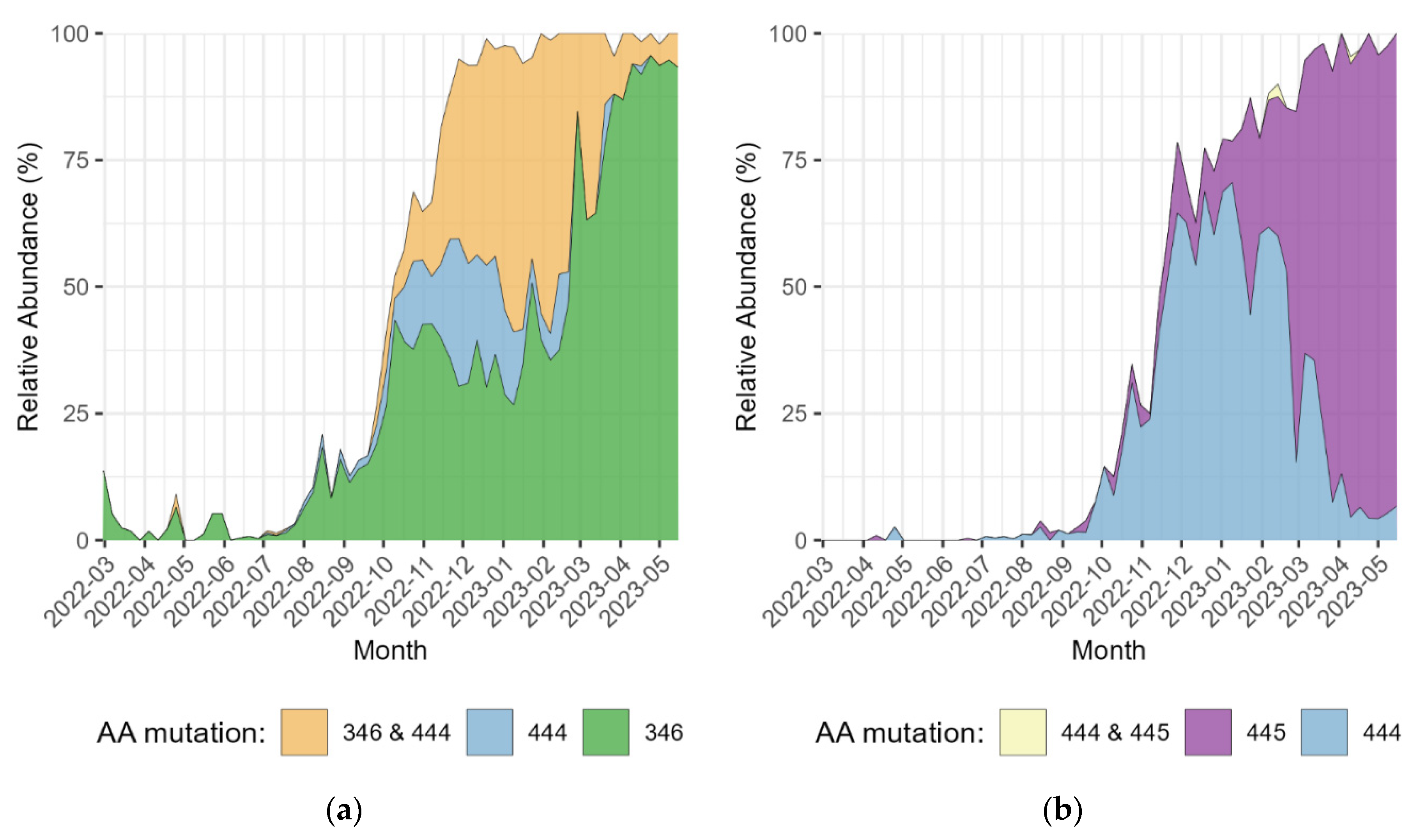

Table 1.
Monoclonal antibodies (mAbs) for the treatment of COVID-19 granted emergency use by the American Food and Drug Administration (FDA) or with a started European Medicines Agency (EMA) rolling review.
Table 1.
Monoclonal antibodies (mAbs) for the treatment of COVID-19 granted emergency use by the American Food and Drug Administration (FDA) or with a started European Medicines Agency (EMA) rolling review.
| Monoclonal antibody | Commercial name | Class | Granted emergency use by the FDA | Revised emergency use by the FDA | EMA Rolling Review Started | EMA Rolling Review Stopped |
|---|---|---|---|---|---|---|
| bamlanvimab | - | II | 09 November 2020[8] | 16 April 2021[8] | 11 March 2021[9] | 29 October 2021[10] |
| bamlanvimab & etesevimab | - | II & I | 09 February 2021[11] | - | 11 March 2021[9] | 29 October 2021[10] |
| regdanvimab | Regikrona | I | - | - | 12 November 2021[12] | - |
| casirivimab & imdevimab | Ronapreve/REGEN-COV | I & III | 21 November 2020[13] | 24 January 2022[13] | - | - |
| sotrovimab | Xevudy | III | 26 May 2021[14] | 05 April 2022[15] | 17 December 2021[16] | - |
| tixagevimab & cilgavimab | Evusheld | I & II | 08 December 2021[17] | 26 January 2023[18] | 25 March 2022[19] | - |
| bebtelovimab | - | III | 11 February 2022[20] | 30 November 2022[21] | - | - |
Table 2.
SARS-CoV-2 receptor binding domain (RBD) mutations and their fold-resistance towards currently available mAbs⁑.
Table 2.
SARS-CoV-2 receptor binding domain (RBD) mutations and their fold-resistance towards currently available mAbs⁑.
| Omicron lineage or mutants with a single RBD mutation | *Important resistance mutations in lineage | bamlanivimab | regdanvimab | casirivimab & imdevimab | sotrovimab | tixagevimab & cilgavimab | bebtelovimab | AZD3152 |
| BA 2 | S371F + T478K + E484A + Q493R | >1000 | >1000 | 387 | 21 | 8 | 1 | 0.6 |
| BA 2.75 | S371F + G446S + N460K + E484A | >1000 | 42 | >1000 | 12 | 24 | 3.1 | 1.9 |
| BA 4 | S371F + L452R + E484A + F486V | >1000 | >1000 | 25 | 22 | 25 | 1 | 0.2 |
| BA 5 | S371F + L452R + E484A + F486V | >1000 | >1000 | 25 | 22 | 25 | 1 | 0.2 |
| BE (BA.5) | S371F + L452R + E484A + F486V | - | - | - | - | - | - | - |
| BN (BA 2.75) | R346T + S371F + G446S + N460K + E484A | - | - | - | - | - | - | - |
| BQ (BA.5) | S371F + K444T + L452R + N460K + E484A + F486V | - | - | 200 | 26 | >1000 | 900 | 0.9 |
| CH (BA 2.75) | R346T + S371F + K444T + G446S + E484A | - | - | >1000 | 16 | >1000 | >1000 | - |
| XBB | R346T + S371F + V445P + G446S + N460K + E484A + F486S | >1000 | - | 200 | 14 | 738 | >1000 | 0.3 |
| XBB 1.5 | R346T + S371F + V445P + G446S + N460K + E484A + F486P | - | - | 751 | 18 | 867 | 475 | 0.2 |
| XBB 1.9.1 | R346T + S371F + V445P + G446S + N460K + E484A + F486P | - | - | - | - | - | - | - |
| R346T | R346T | - | - | - | 1.3 | - | - | - |
| S371F | S371F | 0.9 | 21 | 0.6 | 9.7 | 0.6 | 2.2 | - |
| K444T | K444T | - | - | 6.2 | - | - | >1000 | - |
| V445A | V445A | - | - | 4.7 | 3.4 | - | 83 | - |
| G446S | G446S | 1.2 | 1.1 | 4.5 | 1.6 | 2 | 2 | - |
| L452R | L452R | >1000 | 35 | 2 | 1.1 | 1 | 0.6 | - |
| N460K | N460K | 1.5 | - | 1.3 | 1.2 | 2 | 1.2 | - |
| E484A | E484A | 697 | 7.9 | 7.8 | 0.9 | 5.1 | 1.4 | - |
| E484K | E484K | >1000 | 1.4 | 1.5 | 0.4 | 3.6 | 0.7 | - |
| F486V | F486V | 490 | - | 2.7 | 1.1 | 9.5 | 1.5 | - |
| Q493R | Q493R | >1000 | 949 | - | 1.3 | 3.4 | 1 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



