
Submitted:
11 September 2023
Posted:
13 September 2023
You are already at the latest version
Abstract
The structures of Ag complexes with dimethyl amino phenyl substituted phtalocyanine m[dmaphPcAg]q of various charges q and in two lowest spin states m were optimized using the B3LYP method within the D4h symmetry group and its subgroups. The most stable reaction intermediate in the supposed photoinitiation reaction is 3[dmaphPcAg]-. Group-theoretical analysis of the optimized structures and of their electron states reveals two symmetry-descent mechanisms. Stable structures of maximal symmetry of complexes 1[dmaphPcAg]+, 3[dmaphPcAg]+, 2[dmaphPcAg]0 and 4[dmaphPcAg]2- correspond to the D4 group as a consequence of the pseudo-Jahn-Teller effect within unstable D4h structure. Complexes 4[dmaphPcAg]0, 1[dmaphPcAg]-, 3[dmaphPcAg]- and 2[dmaphPcAg]2- with double degenerate electron ground states in D4h symmetry structures undergo a symmetry descent to stable structures corresponding to maximal D2 symmetry not because of a simple Jahn-Teller effect but due to hidden pseudo-Jahn-Teller effect (strong vibronic interaction between excited electron states). The reduction of the neutral photoinitiator causes symmetry descent to its anionic intermediate because of vibronic interactions that must significantly affect the polymerization reactions.
Keywords:
DFT
; TD-DFT
; symmetry descent
; electron states
; vibronic interactions
1. Introduction
New photosensitive systems with improved efficiency for initiating free-radical and/or cationic polymerizations under light activation are developed using sophisticated chemical treatments. Very recently, Breloy et al. [1] synthesized dimethyl amino substituted phtalocyanine dmaphPcH2 and its Ag(II) complex 2[dmaphPcAg]0 (Figure 1). Photoexcitation of [dmaphPcAg]0 in the presence and absence of an iodonium salt produced acidic and radical species that initiated cationic and free-radical polymerizations, respectively. The photoinitiator properties were investigated by spectroscopic and quantum-chemical methods. The polymerization kinetics in laminate and under air was studied. In the first step, irradiation (385 and 405 nm) caused a reduction of Ag(II) to Ag(I) and nitrogen-centered radicals were formed. Subsequently, Ag nanoparticles and carbon-centered radicals developed. Intermediates [dmaphPc]-, Ag0 nanoparticles, and [dmaphPcAg]q complexes, q = -1 → + 1, are supposed within the proposed reaction mechanism. DFT calculations indicate a D4 → D2 symmetry descent in [dmaphPcAg]q complexes which could be ascribed to the Jahn-Teller (JT) effect. The aim of our recent study is to shed more light on this problem and to verify the above explanation using a group theoretical analysis of the DFT optimized structures of [dmaphPcAg]q complexes within the highest possible D4h symmetry group and its subgroups.
2. Theoretical background
According to the Jahn-Teller theorem [2], any nonlinear configuration of atomic nuclei in a degenerate electron state is unstable. Hence, there is at least one such configuration of lower symmetry, where the above degeneracy is removed. In other words, the multidimensional representation in the high-symmetric (HS) structure is split into nondegenerate representations in the low-symmetric (LS) structure.
The JT active coordinate QJT describes the above mentioned symmetry descent. If the potential energy surface E = f(Qi) of N atoms is a function of 3N - 6 independent nuclear coordinates Qi, for the JT ‘unstable’ HS structure the following relation for any QJT is valid
The HS → LS geometry change described by QJT is connected with an energy decrease which is denoted as the JT stabilization energy EJT
EJT = EHS – ELS
An analogous symmetry descent and energy decrease for pseudodegenerate electron states is known as the pseudo-Jahn-Teller (PJT) effect. It can be observed for a sufficiently strong vibronic interaction and relatively small energy difference Δij between the interacting electron states Ψi and Ψj [3].
The (P)JT effects are usually related to the ground state Ψ0 (and some of the excited states Ψj). Nevertheless, excited states can undergo to these effects as well. In some cases the corresponding vibronic interaction can be so strong that the lower surface penetrates the potential surface of the ground electron state corresponding the HS structure and becomes the lowest state in the LS structure (i.e. its ground electron state). These consequences of the strong JT and PJT vibronic interactions within excited electron states are known as the hidden JT (HJT) and hidden PJT (HPJT) effects, respectively [4].
The (P)JT potential surfaces can be described by an analytical function based on perturbation theory. However, the search for their extremal points corresponding to the ‘stable’ or ‘unstable’ (P)JT structures is too complicated for large molecular systems. In such cases, a group-theoretical treatment must be used.
The epikernel principle method [5,6] is based on the QJT symmetry in the HS structure. A nonzero value of the integral
for the interacting electron states Ψi and Ψj and the full-symmetric Hamilton operator demands that the direct product
where Γi , ΛJT, and Γj are representations of Ψi, QJT, and Ψj, respectively (the asterisk denotes a complex conjugate value), must contain a full-symmetric representation. Alternatively
In the case of the JT effect and the degenerate states Ψi = Ψj , i.e. Γi = Γj, we obtain the even stronger condition
where […]+ denotes the symmetric direct product.
Γi* ⊗ ΛJT ⊗ Γj
ΛJT ∈ Γi* ⊗ Γj
ΛJT ∈ [Γi ⊗ Γi]+
The epikernel principle states that the extrema of a JT energy surface correspond to the kernel K(G, ΛJT) or epikernel E(G, ΛJT) subgroups of the HS parent group G. Kernels contain the symmetry operations of G that leave the ΛJT representation invariant. The symmetry operations of epikernels leave invariant only some components of the degenerate ΛJT representation.
The epikernel principle was originally formulated for the systems in degenerate electron states only [5,6], but it has been successfully extended (except Eq. (6)) to pseudodegenerate electron states as well [7]. Its drawback is in the restriction to JT active coordinates that have been derived within perturbation theory for the linear Taylor expansion of the perturbation only. This method might offer incomplete results in some cases (e.g., in systems with C5 rotations) [7].
The method of step-by-step symmetry descent [8,9] is based on consecutive splitting of a degenerate electron state within a JT symmetry descent. The corresponding multidimensional representation can be split within symmetry descent to an immediate subgroup of the parent HS group G. If the structure corresponding to this subgroup is in a nondegenerate electron state, it is JT stable and the symmetry descent stops. If the structure corresponding to an immediate subgroup of G is in a degenerate electron state described by a multidimensional representation (a JT unstable structure), the symmetry descent continues, and the whole procedure is repeated. As every group can have several immediate subgroups, various symmetry descent paths are possible and the JT stable structures can correspond to various LS symmetry groups. The only condition is its nondegenerate electron state (i.e. one-dimensional representation) obtained by splitting the degenerate electron state (i.e., multidimensional representation) of the HS structure of the parent group G.
3. Results
The highest possible structures of [dmaphPcAg]q complexes are of the D4h symmetry group with side phenyl groups perpendicular to the central phtalocyanine plane (Figure 2). Because of the great number of possible mutual orientations of dimethyl amino phenyl groups in less symmetric structures, we restricted our study to the stable structures of maximal symmetry group only. Another restriction is implied by the inability of standard DFT methods to optimize the atomic configurations in degenerate electron states [10]. The results of the geometry optimizations of the m[dmaphPcAg]q complexes with total charges q = +1 → -2 in the two lowest spin states m are presented in Table 1. It is interesting that the [dmaphPcAg]q complexes in the triplet spin state are more stable than these ones with the same charge in the singlet spin state. 3[dmaphPcAg]- of D2 symmetry is even the most stable complex under study. It must be mentioned that the use of an unrestricted ‘broken symmetry’ treatment [11] results in zero spin populations and does not reduce the energy of the systems studied. Therefore, only the restricted Kohn-Sham formalism has been used in subsequent TD-DFT calculations of our complexes in singlet ground states.
Further inspection of Table 1 shows that the studied complexes can be divided into two categories:
- i)
- Category I contains complexes 1[dmaphPcAg]+, 3[dmaphPcAg]+, 2[dmaphPcAg]0 and 4[dmaphPcAg]2- with optimized structures of D4h (unstable) and D4 (stable) symmetry groups.
- ii)
- Category II contains complexes 4[dmaphPcAg]0, 1[dmaphPcAg]-, 3[dmaphPcAg]- and 2[dmaphPcAg]2- where only their stable optimized structures of the D2 symmetry group were found, while the D4h optimized structures were absent. This can be explained by the electron configurations of the D4h complexes in Table 2. We may conclude that the category II complexes should contain partially occupied eg molecular orbitals, which implies Eg or Eu ground electron states. Hence, they are not accessible by standard DFT methods.
3.1. Category I complexes
The optimized D4h structures of these complexes are unstable due to several imaginary vibrations of Λim representations (Table 1) that coincide with the JT active coordinates. The JT stabilization energies EJT correspond to D4h → D4 symmetry decrease. Stable D4 structures with equally rotated phenyl rings (Figure 3) and without any imaginary vibration correspond to the K(D4h,Λim) kernel subgroups for the coordinate of the a1u representation. As indicated by its wavenumber νim, the energies of the corresponding vibrations are comparable with the highest energy ones in all category I complexes. The optimized structures of D2d and C4v symmetries are not stable (not presented), and hence only the D4 ones fulfill the condition of the stable structure of the highest symmetry group.
For known ground state Γ0 and JT coordinate Λim representations the Eqs. (4) and (5) enable to determine the excited state representations Γexc (see Table 1) interacting with the ground electron state in D4h structures of our complexes. The comparison of the TD-DFT calculated electron states in the corresponding D4h and D4 structures (Table 3) shows that the energy difference Eexc between the PJT interacting states increases after symmetry descent as a consequence of their vibronic interaction. This confirms the correct assignment of the corresponding states in both groups because the standard treatment based on similarity of their oscillator strengths f is hardly usable in our cases. The ground states in the D4h structures also correspond to those in their D4 subgroups.
We can see (Table 3) that the PJT active excited states in the D4h structures are relatively high. This indicates that their excitation energies are less important for possible vibronic interactions than the energies of JT active coordinates.
3.2. Category II complexes
As mentioned above, the ground state of the D4h structures of these complexes is double degenerate (Eg or Eu representations) and only the stable D2 structures (Figure 4) were obtained by geometry optimizations. Possible representations of the corresponding JT active coordinates can be obtained by the symmetric direct products for the HS group
[Eg ⊗ Eg]+ = [Eu ⊗ Eu]+ = A1g ⊕ B1g ⊕ B2g
The full-symmetric a1g coordinate does not change the symmetry of the structure (i.e. cannot be JT active) and the kernels
do not explain the existence of the optimized D2 structure. Therefore, the method of the epikernel principle [5,6] cannot explain the D4h → D2 symmetry descent.
K(D4h, b1g) = D2h(C2’)
K(D4h,b2g) = D2h(C2”)
The method of step-by-step symmetry descent [8,9] for the structures of the D4h symmetry group in double degenerate electron states is based on the scheme in Figure 5. Except D2h, all immediate subgroups of the D4h group (i. e. D4, D2d, C4v and C4h) are JT unstable because of preserved electron degeneracy. After the removal of the C4 axis from the D4 group we obtain its immediate subgroup D2, which is JT stable because the degeneracy is removed here. Alternatively, the D2 group can be obtained by symmetry descent through JT unstable D2d group with preserved electron degeneracy. The structures of the C4 and S4 symmetry groups preserve electron degeneracy and therefore cannot be stable. We have not found any stable structure of D2h, C2v, or C2h symmetry groups by geometry optimization. Therefore, the stable D2 structures meet the condition of maximal groups for category II complexes.
During the symmetry descent from D4h to D2 group the double degenerate representation is split into the nondegenerate B2 and B3 ones
Eg or Eu (D4h) → E (D4 or D2d) → B2 ⊕ B3 (D2)
None of these representations corresponds to the calculated ground electron state of the stable D2 structures. (Table 4). This discrepancy can be explained by the above-mentioned HPJT effect and the ground state of the D2 structure corresponds to the lower PJT interacting excited state in its supergroup. In our case, the situation is complicated by the fact that D2 is not an immediate subgroup of the D4h group. Therefore, three possible two-step symmetry descent paths are possible:
Based on group-subgroup relations we can assign the electron state representations of the D2 symmetry group to the corresponding D4, D2h or D2d ones despite there can be several alternatives. The same holds for the D4h to D4, D2h, or D2d symmetry descent. For known representations of ground states and JT active coordinates, it might be possible to determine the HPJT excited state representations according to Eq. (4). For the two-step symmetry descent there are too many possibilities where almost all excited-state representations (except two-dimensional) might be involved. Therefore, we shall not deal with this problem.
4. Methods
We have performed geometry optimization of the complexes m[dmaphPcAg]q with charges q = +1 → -2 in the two lowest spin states (singlet to quartet) with spin multiplicities m within the D4h symmetry and its subgroups. In agreement with our previous study [1], B3LYP hybrid functional [12], GD3 dispersion correction [13], cc-pVDZ-PP pseudopotential and basis set for Ag [14] and cc-pVDZ basis sets for remaining atoms [15] were used. The optimized structures were tested on the number of imaginary vibrations. Excited states (up to 50) were calculated for every optimized structure using time-dependent DFT (TD-DFT) treatment [16,17] analogously to our previous studies [1,18,19]. All calculations were performed with Gaussian16 (Revision B.01) software [20]. MOLDRAW (https://www.moldraw.software.informer.com, accessed on 9 September 2019) software [21] was used for visualization and geometry modification purposes. Finally, a group-theoretical analysis of the obtained results was carried out using the methods of the epikernel principle [5,6,7] and of step-by-step symmetry descent [8,9].
5. Conclusions
To shed more light on the photopolymerization action of the Ag(II) complex with dimethylamino phenyl-substituted phtalocyanine 2[dmaphPcAg]0, we have performed a quantum-chemical model study of possible reaction intermediates [dmaphPcAg]q with charges q = +1 → -2 with the aim to explain the possible role of the JT effect. Group-theoretical analysis of the obtained results shows that the complexes under study are of two categories.
Stable structures of maximal symmetry of 1[dmaphPcAg]+, 3[dmaphPcAg]+, 2[dmaphPcAg]0 and 4[dmaphPcAg]2- complexes (category I) correspond to the D4 group as a consequence of the PJT effect within the unstable D4h structure. On the other hand, complexes 4[dmaphPcAg]0, 1[dmaphPcAg]-, 3[dmaphPcAg]- and 2[dmaphPcAg]2- (category II) with double degenerate electron ground states in (JT unstable) D4h symmetry structures undergo a symmetry descent to stable structures corresponding to maximal D2 symmetry not because of a simple JT effect, but due to HPJT effect. The most stable reaction intermediate in the supposed photoinitiation reaction [1] is surprisingly 3[dmaphPcAg]- (a singlet electron state was expected). Therefore, the reduction of 2[dmaphPcAg]0 photoinitiator (D4 symmetry) to 3[dmaphPcAg]- intermediate (D2 symmetry) must be significantly affected by vibronic interactions (PJT and HPJT effects). Their influence on reaction rates and thermodynamics must be investigated in solutions.
The presented study shows how group-theoretical treatment can be profitable by solving chemical problems for large molecules. The method of epikernel principle [5,6,7] is restricted to JT active coordinates based on perturbation theory and thus grants incomplete results but can also be used for the PJT effect. The method of step-by-step symmetry descent [8,9] based on splitting degenerate electron states during symmetry descent is more universal but not suitable for the PJT effect. We have shown the usefulness of the combination of both methods. Further theoretical studies in this field are welcome.
Author Contributions
Investigation, writing—original draft preparation, writing—review and editing, M.B. All authors have read and agreed to the published version of the manuscript.
Funding
The work has been supported by the Slovak Research and Development Agency (no. APVV-19-0087), by the Slovak Scientific Grant Agency VEGA (no. 1/0139/20), and by Ministry of Education, Science, Research and Sport of the Slovak Republic within the scheme “Excellent research teams”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Acknowledgments
The authors thank the HPC center at the Slovak University of Technology in Bratislava, which is a part of the Slovak Infrastructure of High Performance Computing (SIVVP project ITMS 26230120002 funded by the European Region Development Funds) for the computational time and resources made available.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Sample Availability
Not applicable.
References
- Breloy, L.; Alcay, Y.; Yilmaz, I.; Breza, M.; Bourgon, J.; Brezová, V.; Yagci, Y.; Versace, D.-L. Dimethyl amino phenyl substituted silver phthalocyanine as a UV- and visible-light absorbing photoinitiator: in situ preparation of silver/polymer nanocomposites. Polym. Chem. 2021, 12, 1273–1285. [Google Scholar] [CrossRef]
- Jahn, H. A.; Teller, E. Stability of polyatomic molecules in degenerate electronic states. I. Orbital degeneracy. Proc. R. Soc. London A 1937, 161, 220–235. [Google Scholar]
- Bersuker, I. B. Pseudo-Jahn–Teller Effect—A Two-State Paradigm in Formation, Deformation, and Transformation of Molecular Systems and Solids. Chem. Rev. 2013, 113, 1351–1390. [Google Scholar] [CrossRef]
- Bersuker I.B. Recent Developments in the Jahn–Teller Effect Theory. In The Jahn-Teller Effect. Fundamentals and Implications for Physics and Chemistry; Köppel, H., Yarkony, D.R., Barentzen, H., Eds.; Springer, 2009; pp. 3–24. ISBN 978-3642034312.
- Ceulemans, A.; Beyens, D.; Vanquickenborne, L. G. Symmetry aspects of Jahn-Teller activity: structure and reactivity. J. Am. Chem. Soc. 1984, 106, 5824–5837. [Google Scholar] [CrossRef]
- Ceulemans, A.; Vanquickenborne, L. G. The Epikernel Principle. Struct. Bonding 1989, 71, 125–159. [Google Scholar]
- Breza, M. Group-Theoretical Treatment of Pseudo-Jahn-Teller Systems. In Vibronic Interactions and the Jahn-Teller Effect: Theory and Application. (Prog. Theor. Chem. Phys. B 23); Atanasov, M., Daul, C., Tregenna-Piggott, P.L.W., Eds.; Springer: Dordrecht, The Netherlands; Berlin/Heidelberg, Germany; London, UK; New York, NY, USA, 2012; pp. 59–82. ISBN 1567-7354.
- Pelikán, P.; Breza, M. Classification of the possible symmetries of the Jahn—Teller systems. Chem. Papers 1984, 39, 255–270. [Google Scholar]
- Breza, M. Group-Theoretical Analysis of Jahn-Teller Systems. In The Jahn-Teller Effect. Fundamentals and Implications for Physics and Chemistry; Köppel, H., Yarkony, D.R., Barentzen, H., Eds.; Springer, 2009; pp. 51–76. ISBN 978-3642034312.
- Kaplan, I. G. Problems in DFT with the total spin and degenerate states. Int. J. Quantum Chem. 2007, 107, 2595–2603. [Google Scholar] [CrossRef]
- Schurkus, H.F.; Chan, G. K.-L.; Chen, G.-T.; Cheng, H.-P.; Stanton, J.F. Theoretical prediction of magnetic exchange coupling constants from broken-symmetry coupled cluster calculations J. Chem. Phys. 2020, 152, 234115. [Google Scholar] [CrossRef] [PubMed]
- Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys., 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys., 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Dunning Jr., T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys., 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M. J.; Mennucci, B.; Tomasi, J.; Cammi, R.; Barone, V. Geometries and properties of excited states in the gas phase and in solution: theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 2006, 124, 94107. [Google Scholar] [CrossRef]
- Štellerová, D.; Lukeš, V.; Breza, M. How does pseudo-Jahn-Teller effect induce the photoprotective potential of curcumin? Molecules 2023, 28, 2946. [Google Scholar] [CrossRef] [PubMed]
- Štellerová, D.; Lukeš, V.; Breza, M. On the Potential Role of (Pseudo-) Jahn-Teller effect in Membrane Transport Processes: Enniatin B and Beauvericin. Molecules 2023, 28, 6264. [Google Scholar] [CrossRef] [PubMed]
- Frisch, G.W.M.J.; Schlegel, B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Ugliengo, P. MOLDRAW: A Program to Display and Manipulate Molecular and Crystal Structures, University Torino, Torino. 2012. Available online: https://www.moldraw.software.informer.com (accessed on 9 September 2019).
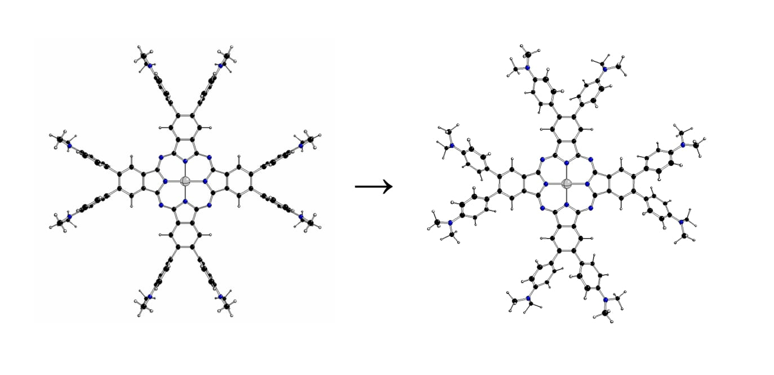
Figure 1.
Structure of 2[dmaphPcAg]0 [1].
Figure 1.
Structure of 2[dmaphPcAg]0 [1].
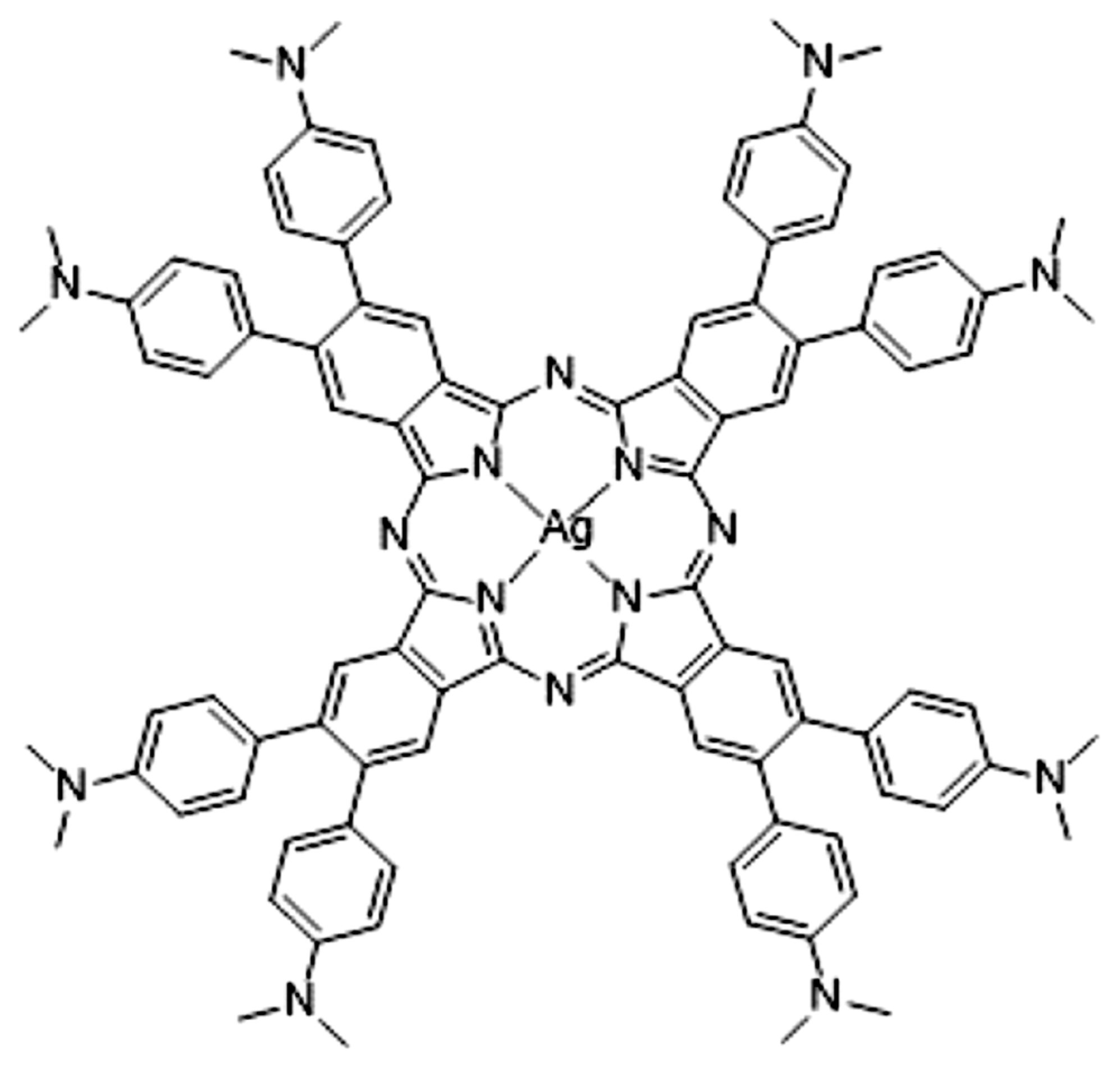
Figure 2.
Optimized D4h structure of 2[dmaphPcAg]0 (C – black, N – blue, H – white, Ag – grey).
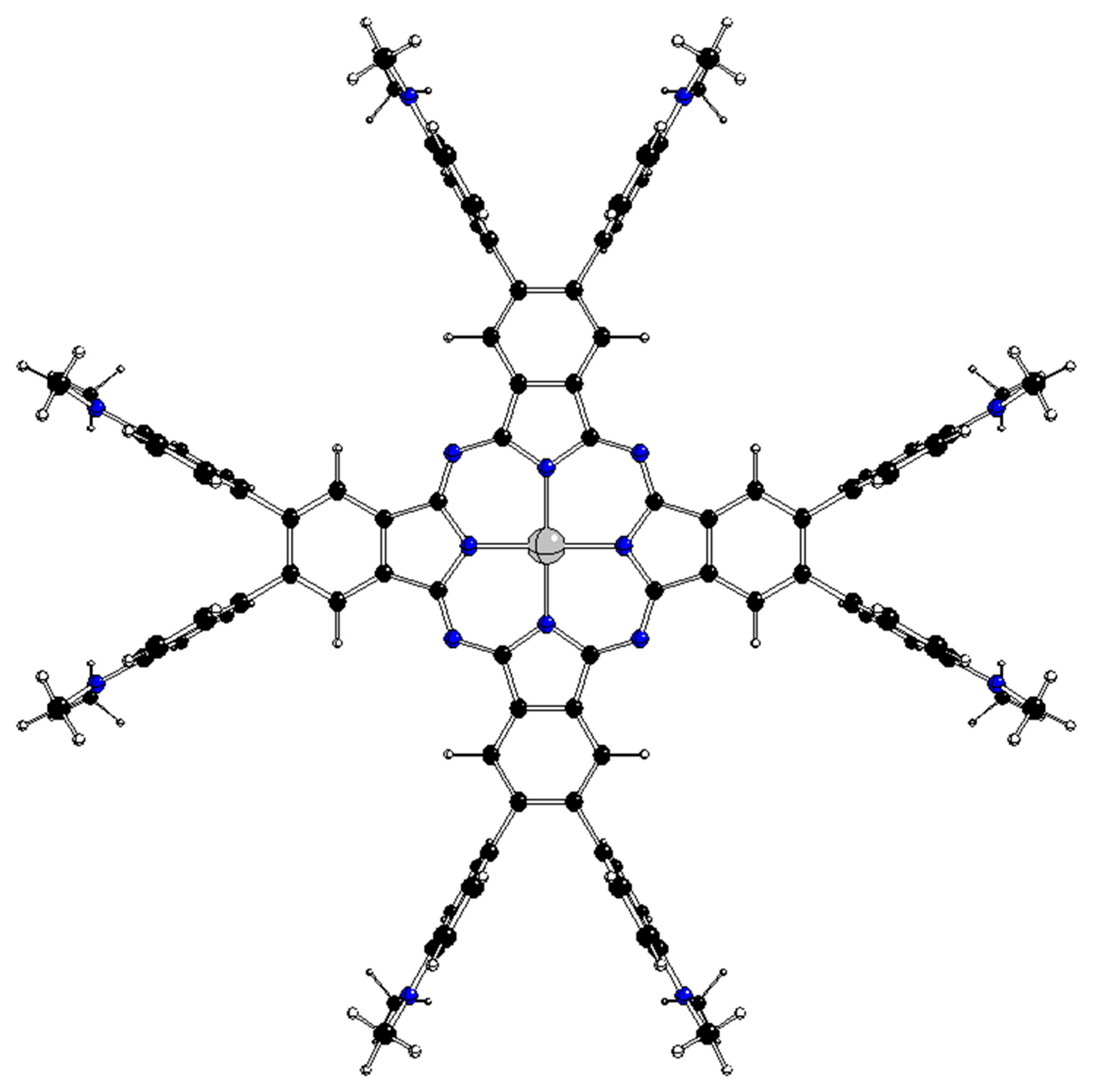
Figure 3.
Optimized D4 structure of 2[dmaphPcAg]0 (see Fig. 2 for atom notation).
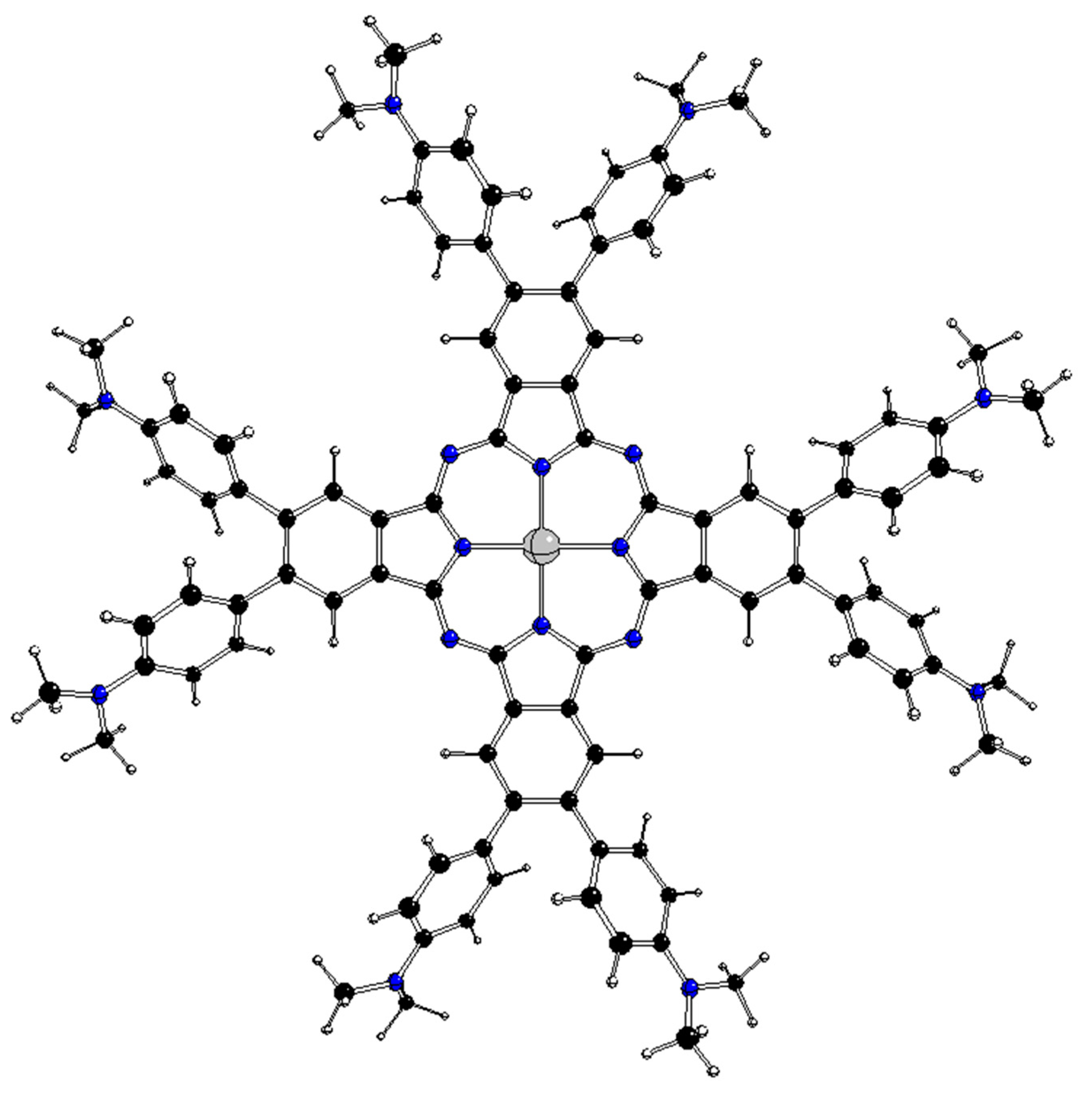
Figure 4.
Optimized D2 structure of 1[dmaphPcAg]- (see Figure 2 for atom notation).
Figure 4.
Optimized D2 structure of 1[dmaphPcAg]- (see Figure 2 for atom notation).
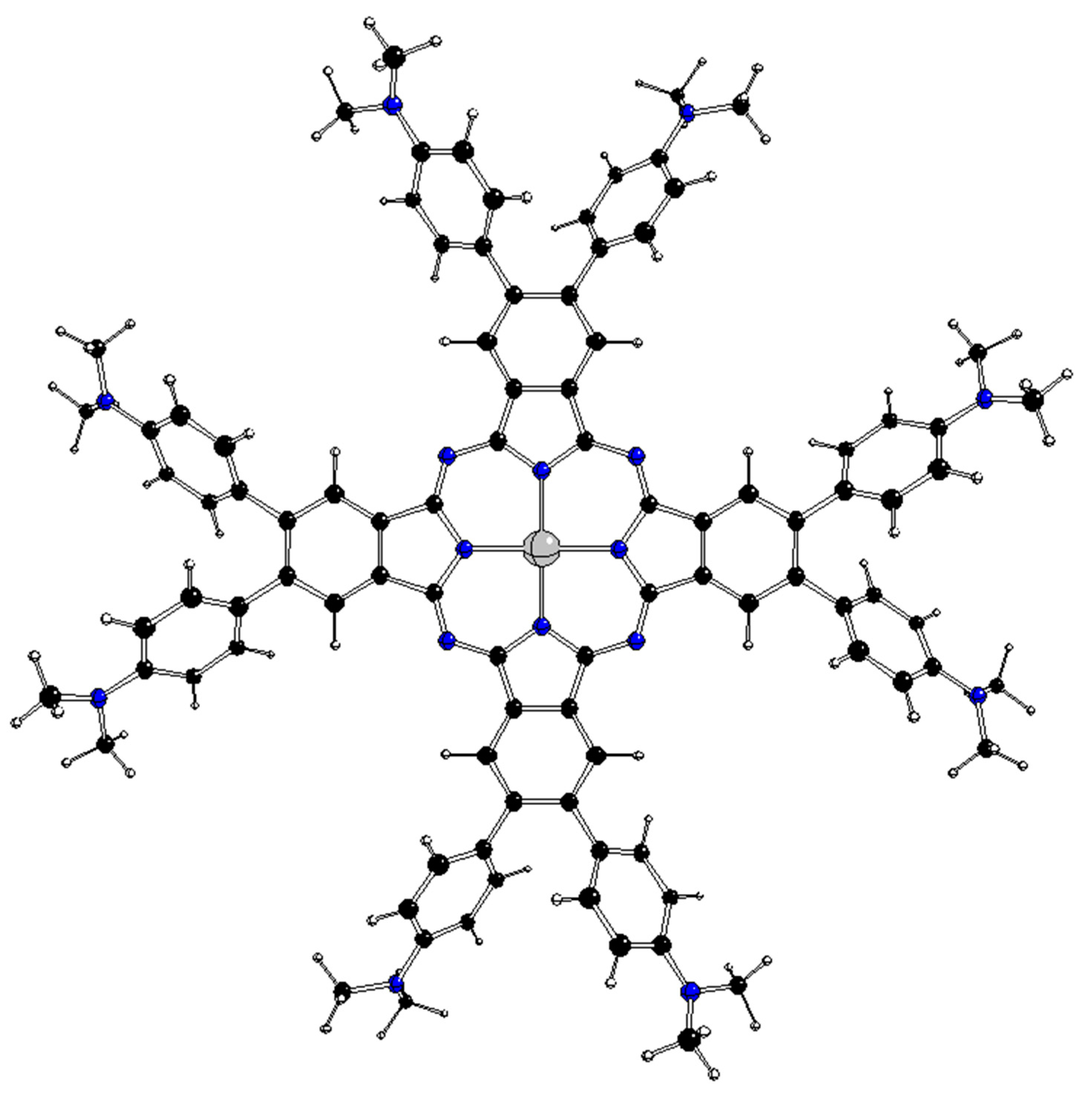
Figure 5.
Possible symmetry descent paths of D4h structures in double degenerate electron states [9]. The top and bottom lines of individual rectangles denote symmetry groups and the corresponding irreducible representations, respectively.
Figure 5.
Possible symmetry descent paths of D4h structures in double degenerate electron states [9]. The top and bottom lines of individual rectangles denote symmetry groups and the corresponding irreducible representations, respectively.
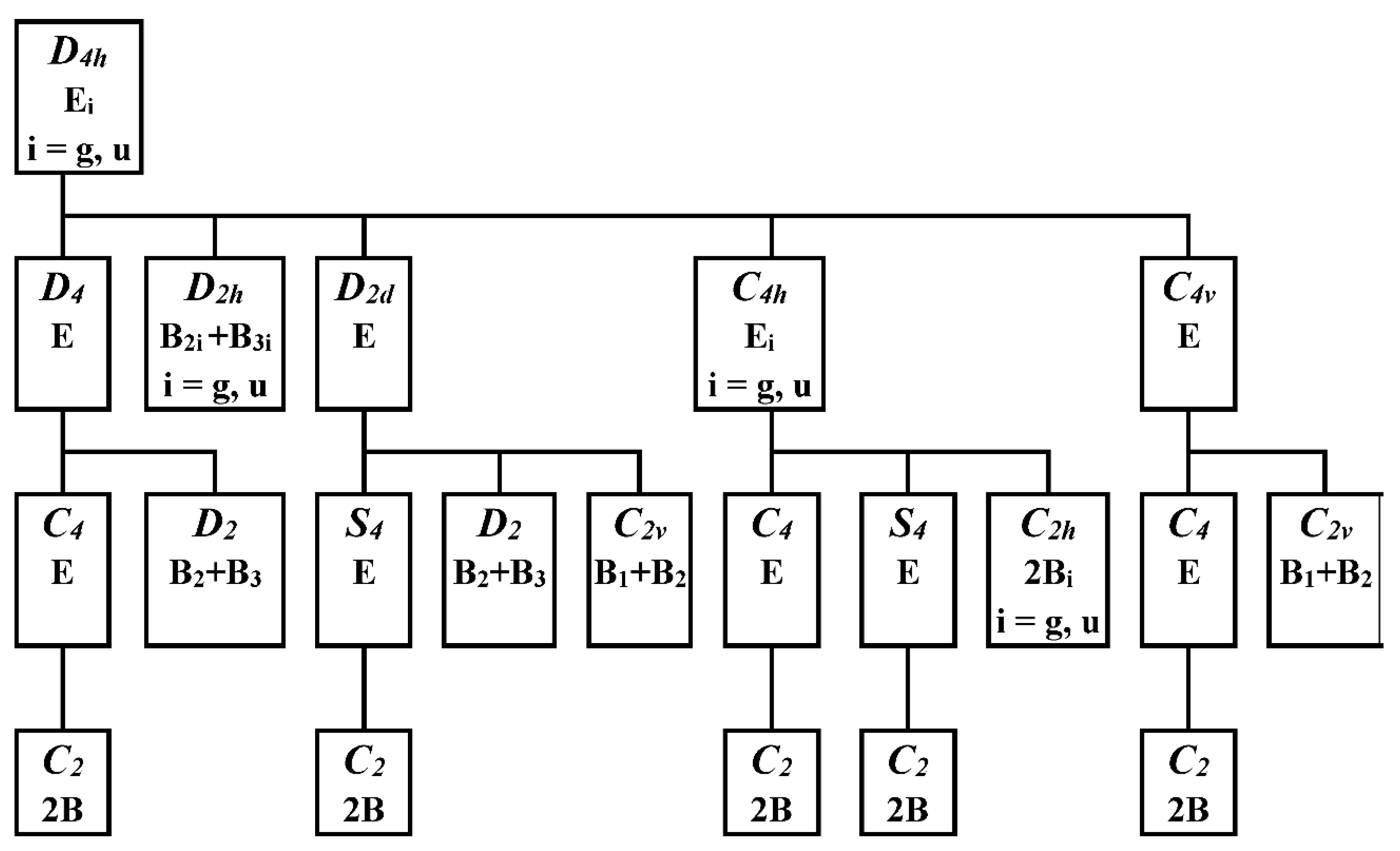

Table 1.
Charge q, spin multiplicity m, symmetry group G, representation of the ground electron state Γ0, DFT energy EDFT, JT stabilization energy EJT, representations Λim and wavenumbers νim of imaginary vibrations, kernel K(D4h,Λim) and epikernel K(D4h,Λim) subgroups of D4h, and representations of relevant PJT excited states Γexc of m[dmaphPc]q complexes under study (the preserved symmetry elements in the kernel and epikernel subgroups are in parentheses). The most stable structure is shown in bold.
Table 1.
Charge q, spin multiplicity m, symmetry group G, representation of the ground electron state Γ0, DFT energy EDFT, JT stabilization energy EJT, representations Λim and wavenumbers νim of imaginary vibrations, kernel K(D4h,Λim) and epikernel K(D4h,Λim) subgroups of D4h, and representations of relevant PJT excited states Γexc of m[dmaphPc]q complexes under study (the preserved symmetry elements in the kernel and epikernel subgroups are in parentheses). The most stable structure is shown in bold.
| q | m | G | Γ0 | EDFT [Hartree] | EJT [eV] | Λim | νim [cm-1] | K(D4h,Λim) | E(D4h,Λim) | Γexc |
|---|---|---|---|---|---|---|---|---|---|---|
| +1 | 1 | D4h | 1A1g | -4734.55067 | - | b1u | -48 | D2d(C2’) | B1u | |
| 2eg | -47, -31 | C1 | C2h(C2’), C2h(C2”) | Eg | ||||||
| a1u | -46 | D4 | A1u | |||||||
| a2u | -31 | C4v | A2u | |||||||
| b2u | -31 | D2d(C2”) | B2u | |||||||
| +1 | 1 | D4 | 1A1 | -4734.58352 | 0.894 | - | - | |||
| +1 | 3 | D4h | 3B1u | -4734.54698 | - | b1u | -18 | D2d(C2’) | A1g | |
| eg | -18 | C1 | C2h(C2’), C2h(C2”) | Eu | ||||||
| a1u | -18 | D4 | B1g | |||||||
| +1 | 3 | D4 | 3B1 | -4734.58550 | 1.048 | - | - | |||
| 0 | 2 | D4h | 2B1g | -4734.74708 | - | b1u | -42 | D2d(C2’) | A1u | |
| eg | -42 | C1 | C2h(C2’), C2h(C2”) | Eg | ||||||
| a1u | -41 | D4 | B1u | |||||||
| a2u | -23 | C4v | B2u | |||||||
| 3b2u | -23(3×) | D2d(C2”) | A2u | |||||||
| 0 | 2 | D4 | 2B1 | -4734.77283 | 0.701 | - | - | |||
| 0 | 4 | D2 | 4B2 | -4734.73113 | ? | - | - | |||
| -1 | 1 | D2 | 1A | -4734.79846 | ? | - | - | |||
| -1 | 3 | D2 | 3B2 | -4734.83355 | ? | - | - | |||
| -2 | 2 | D2 | 2B1 | -4734.80520 | ? | - | - | |||
| -2 | 4 | D4h | 4B1u | -4734.77355 | - | b1u | -47 | D2d(C2’) | A1g | |
| 2eg | -46,-27 | C1 | C2h(C2’), C2h(C2”) | Eu | ||||||
| a1u | -45 | D4 | B1g | |||||||
| a2u | -27 | C4v | B2g | |||||||
| b2u | -27 | D2d(C2”) | A2g | |||||||
| -2 | 4 | D4 | 4B1 | -4734.80581 | 0.878 | - | - |
Table 2.
Charge q, spin multiplicity m, electron configuration, and ground electron state representation Γ0 of studied m[dmaphPc]q complexes of D4h, symmetry group.
Table 2.
Charge q, spin multiplicity m, electron configuration, and ground electron state representation Γ0 of studied m[dmaphPc]q complexes of D4h, symmetry group.
| q | m | Electron configuration | Γ0 |
|---|---|---|---|
| +1 | 1 | …(b2g)2(eu)4(a2g)2(b1g)0(eg)0(b1u)0… | 1A1g |
| +1 | 3 | α: …(b1g)1(a2g)1(eu)2(b2g)1(eg)0(b1u)0…$$$β: …(eu)2(b2g)1(a1u)0(eg)0(b1g)0(b1u)0… | 3B1u |
| 0 | 2 | α:…(eu)2(b2g)1(b1g)1(a1u)1(eg)0(b1u)0…$$$β:…(eu)2(b2g)1(a1u)1(eg)0(b1g)0(b1u)0… | 2B1g |
| 0 | 4 | ? | 4Eg or 4Eu |
| -1 | 1 | ? | 1Eg or 1Eu |
| -1 | 3 | ? | 3Eg or 3Eu |
| -2 | 2 | ? | 2Eg or 2Eu |
| -2 | 4 | α:…(eg)2(b2g)1(a1u)1(b1g)1(eg)2(b2u)0…$$$β:…(eg)2(a2u)1(a1u)1(eg)0(a2u)0(b2u)0… | 4B1u |
Table 3.
Charge q, spin multiplicity m, symmetry group G, ground electron state representation Γ0, representations Γexc, excitation energies Eexc and oscillator strengths f of the low excited electron states of the studied m[dmaphPc]q complexes in D4h and D4 symmetry groups. The excited states that interact with the ground states are in bold.
Table 3.
Charge q, spin multiplicity m, symmetry group G, ground electron state representation Γ0, representations Γexc, excitation energies Eexc and oscillator strengths f of the low excited electron states of the studied m[dmaphPc]q complexes in D4h and D4 symmetry groups. The excited states that interact with the ground states are in bold.
| q | m | G | Γ0 | Γexc | Eexc [eV] | f | G | Γ0 | Γexc | Eexc [eV] | f |
|---|---|---|---|---|---|---|---|---|---|---|---|
| +1 | 1 | D4h | 1A1g | 11A2g | 0.156 | 0.000 | D4 | 1A1 | 11B1 | 0.167 | 0.000 |
| 11Eu | 0.158 | 0.002 | 11B2 | 0.255 | 0.000 | ||||||
| 11B2g | 0.159 | 0.000 | 11E | 0.259 | 0.000 | ||||||
| 11B1g | 0.349 | 0.000 | 11A2 | 0.267 | 0.000 | ||||||
| 21Eu | 0.353 | 0.081 | 21E | 0.631 | 0.014 | ||||||
| 11B1u | 0.377 | 0.000 | 11A1 | 0.642 | 0.000 | ||||||
| 11A1g | 0.487 | 0.000 | 21B1 | 0.858 | 0.000 | ||||||
| 11Eg | 1.010 | 0.000 | 31E | 1.187 | 0.050 | ||||||
| 11A1u | 1.010 | 0.000 | 21A1 | 1.192 | 0.001 | ||||||
| 11B1u | 1.010 | 0.000 | 21A2 | 1.200 | 0.000 | ||||||
| +1 | 3 | D4h | 3B1u | 13B2g | 0.011 | 0.000 | D4 | 3B1 | 13E | 0.188 | 0.007 |
| 13A2g | 0.015 | 0.001 | 13B2 | 0.190 | 0.000 | ||||||
| 13Eu | 0.016 | 0.001 | 13A2 | 0.195 | 0.000 | ||||||
| 13A1g | 0.235 | 0.000 | 13A1 | 0.600 | 0.000 | ||||||
| 23Eu | 0.235 | 0.014 | 23E | 0.617 | 0.298 | ||||||
| 13B2u | 0.239 | 0.000 | 13B1 | 0.779 | 0.000 | ||||||
| 23Eu | 1.255 | 0.000 | 33E | 1.244 | 0.001 | ||||||
| 33Eu | 1.327 | 0.003 | 43E | 1.391 | 0.033 | ||||||
| 13Eg | 1.493 | 0.008 | 23B1 | 1.400 | 0.000 | ||||||
| 0 | 2 | D4h | 2B1g | 12Eu | 1.195 | 0.000 | D4 | 2B1 | 12E | 1.157 | 0.000 |
| 12Eg | 1.306 | 0.000 | 22E | 1.305 | 0.000 | ||||||
| 12B1u | 2.014 | 0.000 | 32E | 1.901 | 0.624 | ||||||
| 22Eu | 2.050 | 0.710 | 12B2 | 1.942 | 0.000 | ||||||
| 22Eg | 2.140 | 0.000 | 12A2 | 1.944 | 0.000 | ||||||
| 12B2u | 2.140 | 0.000 | 42E | 1.956 | 0.004 | ||||||
| 12A2u | 2.140 | 0.000 | 22A2 | 1.969 | 0.000 | ||||||
| 32Eg | 2.163 | 0.000 | 12A1 | 2.055 | 0.000 | ||||||
| 22B2u | 2.164 | 0.000 | 32A2 | 2.056 | 0.000 | ||||||
| 22A2u | 2.164 | 0.000 | 12B1 | 2.059 | 0.001 | ||||||
| -2 | 4 | D4h | 4B1u | 14Eg | 0.957 | 0.000 | D4 | 4B1 | 14E | 0.888 | 0.191 |
| 14A2u | 0.976 | 0.000 | 14B1 | 1.356 | 0.000 | ||||||
| 14B2u | 0.978 | 0.000 | 24E | 1.360 | 0.010 | ||||||
| 24Eg | 1.025 | 0.000 | 14A1 | 1.360 | 0.000 | ||||||
| 14A1u | 1.042 | 0.000 | 34E | 1.396 | 0.010 | ||||||
| 14B1u | 1.048 | 0.000 | 14A2 | 1.415 | 0.000 | ||||||
| 34Eg | 1.118 | 0.110 | 14B2 | 1.417 | 0.000 | ||||||
| 14A1g | 1.495 | 0.000 | 44E | 1.436 | 0.012 | ||||||
| 14B1g | 1.495 | 0.000 | 24B2 | 1.472 | 0.001 | ||||||
| 14Eu | 1.496 | 0.000 | 24A2 | 1.481 | 0.000 | ||||||
| 24A1u | 1.501 | 0.000 | 34B1 | 1.523 | 0.000 |
Table 4.
Charge q, spin multiplicity m, representations of the ground electron state Γ0 and of the excited states Γexc, excitation energies Eexc and oscillator strengths f of low excited electron states of stable m[dmaphPc]q complexes under study in the D2 symmetry group. The irreducible representations obtained by splitting the two-dimensional representations of the degenerate ground state in D4h structures are in bold.
Table 4.
Charge q, spin multiplicity m, representations of the ground electron state Γ0 and of the excited states Γexc, excitation energies Eexc and oscillator strengths f of low excited electron states of stable m[dmaphPc]q complexes under study in the D2 symmetry group. The irreducible representations obtained by splitting the two-dimensional representations of the degenerate ground state in D4h structures are in bold.
| q | m | Γ0 | Γexc | Eexc [eV] | f | q | m | Γ0 | Γexc | Eexc [eV] | f |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 4 | 4B2 | 14B1 | 0.349 | 0.000 | -1 | 1 | 1A | 11B2 | 0.523 | 0.000 |
| 14B3 | 0.814 | 0.012 | 11B1 | 0.539 | 0.000 | ||||||
| 24B1 | 0.829 | 0.000 | 11B3 | 0.815 | 0.000 | ||||||
| 14B2 | 0.927 | 0.006 | 21B2 | 1.337 | 0.023 | ||||||
| 34B1 | 0.948 | 0.000 | 21B3 | 1.478 | 0.028 | ||||||
| 24B3 | 1.253 | 0.000 | 31B2 | 1.551 | 0.005 | ||||||
| 24B2 | 1.254 | 0.213 | 41B2 | 1.793 | 0.809 | ||||||
| 14A | 1.291 | 0.000 | 31B3 | 1.965 | 0.018 | ||||||
| 34B3 | 1.356 | 0.164 | 11A | 2.056 | 0.000 | ||||||
| 24A | 1.423 | 0.000 | 41B3 | 2.097 | 0.011 | ||||||
| -1 | 3 | 3B2 | 13B1 | 0.272 | 0.000 | -2 | 2 | 2B1 | 12B1 | 0.048 | 0.000 |
| 13B3 | 1.256 | 0.000 | 22B1 | 0.377 | 0.000 | ||||||
| 13B2 | 1.350 | 0.021 | 12B2 | 0.900 | 0.007 | ||||||
| 23B3 | 1.356 | 0.014 | 22B2 | 1.095 | 0.283 | ||||||
| 23B2 | 1.796 | 0.792 | 12B3 | 1.112 | 0.306 | ||||||
| 33B3 | 1.835 | 0.007 | 32B2 | 1.168 | 0.000 | ||||||
| 43B3 | 1.987 | 0.012 | 12A | 1.343 | 0.000 | ||||||
| 13A | 1.993 | 0.000 | 42B2 | 1.348 | 0.002 | ||||||
| 23B1 | 2.000 | 0.000 | 22A | 1.362 | 0.000 | ||||||
| 23A | 2.092 | 0.000 | 22B3 | 1.367 | 0.002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



