
Submitted:
11 September 2023
Posted:
13 September 2023
You are already at the latest version
Abstract
Rhabdomyosarcoma is a rare cancer arising in skeletal muscle that typically impacts children and young adults. It is a worldwide challenge in child health as treatment outcomes for metastatic and recurrent disease still pose a major concern for both basic and clinical scientists. The treatment strategies for rhabdomyosarcoma include multi-agent chemotherapies after surgical resection with or without ionization radiotherapy. In this comprehensive review, we first provide a detailed clinical understanding of rhabdomyosarcoma including its classification and subtypes, diagnosis, and treatment strategies. Later we focus on chemotherapy strategies for this childhood sarcoma and discuss the impact of three mechanisms that are involved in chemotherapy response including apoptosis, macro-autophagy, and the unfolded protein response. Finally, we discuss in vivo mouse and zebrafish models, and in vitro 3-dimensional bioengineering models of Rhabdomyosarcoma to screen future therapeutic approaches and promote muscle regeneration.
Keywords:
Alveolar Rhabdomyosarcoma
; Apoptosis
; Autophagy
; Unfolded Protein Response
; Bioengineering
; Tumor Stiffness
1. Introduction
Rhabdomyosarcoma (RMS) is a pediatric soft tissue malignancy with poor survival rates for the high-risk and recurrent disease and involves the potential for significant morbidity associated with treatment. This review will characterize the clinical implications, methods of tumor differentiation and current chemotherapeutic agents that are involved in RMS management. Furthermore, we will discuss the roles of apoptosis, autophagy and the unfolded protein response (UPR) and their implications in RMS chemotherapy. As we focus on these cell death pathways, we will discuss the role of zebrafish and mouse models of RMS, and the relevance of tissue engineering strategies in RMS, emphasizing their importance in further understanding RMS and to direct future advances in the treatment. We are optimistic that this review will provide meaningful knowledge to guide further clinical advancements in RMS therapy in order to improve survival outcomes for the RMS cancer patients.
RMS, a cancer of skeletal muscle tissue is the most common pediatric soft tissue sarcoma. RMS is responsible for the 3% of all childhood malignant tumors and is the third most prevalent pediatric extracranial solid tumor [1,2]. For individuals under the age of 20, the incidence of RMS is approximately 4.5 patients per million in the United States, accounting for approximately 350 new cases each year with half of the diagnoses occurring in the patients under the age of 10 [1,2,3]. Treatment of RMS presents unique challenges when attempting local control due to the rarity of the disease and various anatomical sites in which the primary tumor can appear [3,4].
Patient survival rates depend upon several variables such as the tumor subtype, size, grade, primary site, as well as RMS disease stage and clinical group [4]. The overall 5-year survival rates for RMS in children have exceeded 70% [4,5,6,7,8,9,10,11]. However, despite advances in diagnostic and treatment methods over the past few decades, children with high-risk RMS and recurrent disease have 5-year survival rates of less than 30% and 17%, respectively [1,12,13]. Prognostic stratification is significant because 15-20% of children have diffused metastatic disease at the time of diagnosis [14,15]. Adults with RMS also experience poor outcomes, with 5-year survival rates ranging from 26.6%-61% [16,17,18]. Over the last three decades, there have been several national and international clinical trials which have resulted in refined treatment regimens based on the tumor stage and clinical group, leading to improved pediatric RMS survival rates [4,5,6,7,8,15,19]. In addition, advancements in molecular biology and next generation sequencing have allowed researchers and clinicians to further understand RMS pathogenesis and classification [1,4]. However, despite these developments, the cure rate for pediatric patients with metastatic or recurrent disease remains low and current RMS therapies continue to pose potential life-threatening toxicities, which can lead to lifelong morbidity [4].
2. RMS Subtypes
RMS is generally characterized into four main tumor subtypes, as recognized by the World Health Organization (WHO); Embryonal RMS (ERMS), Alveolar RMS (ARMS), Pleomorphic RMS (PRMS), and Sclerosing/Spindle RMS (Table 1) [1,3,4,20]. The most common subtypes are ERMS and ARMS, whereas spindle cell/sclerosing RMS and PRMS are considered rare. Primary RMS tumors tend to occur at three main anatomical regions including; the head and neck regions (35-40%), genitourinary system (25%), and the trunk/extremities (20%) [3]. Of the RMS tumors occurring in the head and neck region, 75% arise in the orbit of the eye [3,21].
ERMS is the most common subtype (~60-70% of cases) with bimodal distribution and peak incidence in the 0-4 and 14-18 age ranges [2,22]. In the patients with ERMS, the primary tumor is commonly located in the head and neck region, specifically the superior nasal quadrants and eye socket, as well as the genitourinary system where it is most often found in the bladder and prostate [23]. ERMS is associated with a loss of heterozygosity at the 11p15 locus in 80% of the cases which results in an altered insulin-like growth factor 2 (IGF2) gene [3,23]. Histologically, ERMS is composed of immature rhabdomyoblasts in a stroma-rich background and lacks the alveolar pattern seen in ARMS [24]. According to the Children’s Oncology Group Soft Tissue Sarcoma (COG-STS) Risk Stratification, low risk ERMS has the most favorable prognosis of the RMS subtypes with a 5-year survival of approximately 80-90% [25].
ARMS is the second most common subtype of RMS that tends to occur in late childhood/adolescence [2]. The ARMS primary tumor tends to arise on the trunk and extremities, but can also be located in the inferior orbit [23]. Histologically, ARMS is characterized by densely packed, small, round cells lining septations that resemble fetal alveoli [4]. Next generation DNA and RNA sequencing has allowed us to characterize 80% of the patients with ARMS as fusion-positive (FP); with 60% containing the PAX3-FOXO1 fusion onco-protein and 20% containing PAX7-FOXO1 fusion onco-protein (Table 1) [3]. Fusion status is clinically important as fusion-negative (FN) ARMS has molecular similarities to ERMS and clinical outcomes of children with FN ARMS are analogous to those of ERMS [26]. The ARMS is considered an intermediate/high-risk RMS subtype by the COG-STS Risk Stratification [12]. Intermediate-risk ARMS has an estimated 5-year survival rate of 65-73%, whereas high-risk lesions have a 5-year survival rate of less than 30% [25].
PRMS primarily occurs in adults between the ages of 40-70 with a peak incidence during the 6th decade of their life. PRMS is most often found in the lower extremities and can be subdivided into classic, round cell, and spindle cell subtypes based on the histological findings [4]. Immunohistochemistry and the presence of skeletal muscle proteins is used to differentiate PRMS from other high-grade soft tissue sarcomas found in adults [3,4]. In general, individuals diagnosed with PRMS have a worse prognosis relative to those with ERMS and ARMS due to unfavorable anatomic location of the primary tumor and a higher likelihood of being treated outside of a sarcoma specialized center [3]. In addition, PRMS is unique as it does not respond to chemotherapy, unlike ERMS and ARMS, and is often treated with radiation therapy with wide excision.
3. RMS Classification
The classification of RMS subtypes has changed over the last several years due to the advancements in nucleic acid sequencing [1]. Initially, RMS was divided into two main subtypes; ERMS and ARMS based on the light microscopy findings [27,28]. ARMS and ERMS both contained cells that resembled immature skeletal myoblasts distributed around an open central space [4,27,28]. RMS differs from other small round blue cell tumors such as Neuroblastoma and Ewings Sarcoma via immunohistochemical staining revealing muscle cell markers such as alpha-actin, MyoD1, Myogenin, and Desmin [3].
Recently, molecular biology approaches have further characterized RMS by the presence or absence of fusion proteins related to the balanced translocations between chromosomes 1 and 13 (t(1;13)) and chromosomes 2 and 13 (t(2;13)), which gave way to FP and FN classifications [1,4]. These fusion proteins are composed of paired box proteins PAX3 and PAX7, and Forkhead box protein O1 (FOXO1), which are transcribed yielding functional PAX3-FKHR and PAX7-FKHR transcription factors. Approximately, 60% of ARMS tumors can be characterized by t(1;13) (q35;q14) and the PAX3-FOXO1 fusion protein, while 20% of the ARMS tumors are characterized by t(2;13) (p36;q14) and the PAX7-FOXO1 fusion protein [1,3,4,22,23]. When comparing the historical subtype classification by microscopy and immunohistochemistry to the FP/FN classification, several studies found that 20% of FN ARMS tumors act more similarly to ERMS with regards to its prognosis and treatment, despite their histological differences. This shift in the classification of RMS subtypes creates unique challenges when comparing past and present literature due to the crossover in tumor subtypes between the microscopy-based classification in the early literature and more recent FN/FP classification of ARMS [4].
4. RMS Epidemiology
RMS accounts for approximately 4.5% of all cases of childhood cancer [29,30], with a bimodal distribution displaying peak incidence rates at 2-6 and 10-18 years of age as well as a slight male predominance [23,31]. The incidence of RMS is similar among countries around the world with the exception of East Asia. For example, the incidence of RMS is 4.5 cases per million (<20 years of age) in the United States and 4.9 cases per million (<15 years of age) in Sweden; however, in Japan, India and China, the incidence of RMS is 2 cases per million [23,32,33]. In the adult population, soft tissue sarcomas comprise less than 1% of all solid tumor malignancies, with RMS comprising less than 4% of adult soft tissue sarcomas [4,23,34]. From 1975-2020 there has been a stable incidence rate of RMS despite ongoing advances in the diagnosis and classification of RMS subtypes [35,36]. With regards to FP disease, PAX7-FOXO1 positive RMS tends to occur at a younger age than PAX3-FOXO1 positive RMS [22]. The overall risk of RMS is lower in Hispanics and is higher in those familial cancer syndromes; particularly Li-Fraumeni Syndrome (LFS) [4].
5. RMS Treatment
The mainstay of treatment for RMS involves multi agent systemic chemotherapy in order to eradicate disseminated disease, along with surgical resection of the primary tumor with or without addition of the ionizing radiation therapy for the control of local disease [4,37]. The timing of systemic chemotherapy remains controversial; however, most North American centers will administer chemotherapy in the neoadjuvant setting. Surgical resection has been shown to increase survival in Group I and II diseases, whereas those with Group III disease may experience increased morbidity without improved survival rate [4,15,38,39]. Complete surgical resection with circumferential margins greater than 0.5-1 cm is the preferred treatment method for the localized RMS [3,23]. Adequate negative margins are required unless the surgical excision threatens adjacent organs, leads to the loss of significant function, results in poor cosmesis, or is not technically feasible [23]. In the event that there are positive margins, patients may undergo radiation therapy or further surgical resection of the tumor. Re-excision of the recurrent RMS has been shown to increase 5 year survival rates from 8% to 37% compared to the patients without aggressive re-excision [40].
Currently approved chemotherapeutic agents for the treatment of RMS include cyclophosphamide, actinomycin-D, doxorubicin, etoposide, ifosfamide, irinotecan, melphalan, temsirolimus, vincristine and volasertib (Figure 1).
In the North America, chemotherapy for the pediatric patients consists of a backbone of vincristine, actinomycin D and cyclophosphamide (VAC) [1,21]. In comparison, in Europe VAC therapy is substituted for a regimen consisting of ifosfamide, vincristine, and actinomycin D (IVA), which has produced similar treatment outcomes [4,5]. There is no standardized chemotherapy regimen for the adult patients, with some studies suggest using a combination of ifosfamide, doxorubicin and vincristine, while others utilize pediatric regimens such as VAC [3,41]. When treating RMS, chemotherapy is typically administered in intervals over a 6 to 9 months period [4]. The Children’s Cancer Study Group A Trial in the 1960s and 70s reported up to 50-60% disease recurrence in the patients who didn’t receive chemotherapy [42]. This finding further emphasizes the importance of chemotherapy and its contribution to a successful multimodal curative treatment regimen. Though the chemotherapeutic regimen for the RMS treatment has remained unchanged over the last few decades, current research is still evaluating the efficacy of additional drugs such as doxorubicin, cisplatin and etoposide to the VAC therapy and the impact of variable chemotherapeutic dosing intensities [3,19]. Current literature fails to show a therapeutic advantage for higher doses of cyclophosphamide in children with intermediate risk tumors [43]. However, the COG does show improvement in the patients with disease relapse who use Irinotecan and Vincristine as part of their chemotherapy regimen [44].
Radiation therapy plays an important role in the treatment of patients with COG group II (microscopic residual) or group III (gross residual) diseases [3,45]. Dosing varies based on the patient’s clinical group, with patients in clinical group II typically receiving 40 Gy of radiotherapy, whereas those in group III typically receive 50 Gy [3]. The current literature is focused on balancing the effectiveness of radiotherapy in decreasing tumor size with the reduction of treatment side effects in young patients with RMS [3]. Advents such as the use of Intensity Modulated Radiation Therapy (IMRT) and Proton Beam Therapy (PBT) are currently being used to try and achieve this goal [3]. Notable side effects of radiotherapy include joint stiffness, soft tissue changes, appendicular skeletal growth problems and secondary malignancy.
Although the 5-year survival of the patients with low risk disease has approached 90%, children with metastatic disease have an overall survival rate of 25-30% at 3 years, despite the use of high dose of chemotherapy and stem cell rescue treatments [4,14,46,47]. Thus, there are several agents that are currently under investigation to improve treatments for this cohort with poor survival outcomes. For example, targeted therapeutic agents such as cixutumumab, crizotinib, pazopanib, sorafenib, and temsirolimus are currently being studied for their role in RMS treatment (Table 2). There are also various ongoing clinical trials for the chemotherapeutic agents such as vinorelbine and Trabectedin, and combination drugs such as mocetinostat and vinorelbine, dasatinib and ganitumab, and olaparib and temozolomide (Table 3). The most important chemotherapy medications for RMS are summarized in the following sections.
5.1. Temsirolimus
Temsirolimus is a second-generation analog of a natural product rapamycin – a macrolide antibiotic produced by the bacterium Streptomyces hygroscopicus [48]. The ester group of temsirolimus is hydrolyzed by cytochrome P450 CYP3A4 to its active metabolite, rapamycin [49]. Rapamycin was used as an immunosuppressant, but due to poor pharmacokinetic properties, the more polar analog temsirolimus has been developed [50]. This ester modification present in temsirolimus not only increases the solubility and bioavailability, but also decreases the immunosuppressive properties [51]. Following intravenously administration of temsirolimus, enzymatic hydrolysis occurs and rapamycin can be detected in the blood within 15 minutes, reaching a peak within 0.5-2 hr. Concomitant use of drugs that alter CYP3A4 activity results in drug serum level changes [52,53].
Temsirolimus was approved in 2007 for the treatment of advanced renal cell carcinoma. As a specific inhibitor of mammalian target of rapamycin (mTOR), it can also be used for the treatment of various tumors where mTOR is excessively activated [54]. mTOR is a serine-threonine kinase and plays an important role in the signal transduction process that leads to cell growth and tumor development. First, temsirolimus binds to the protein FKBP12, and this complex acts to inhibit mTOR [55,56]. Interestingly, even though temsirolimus binds to an allosteric modulator of mTOR, this binding inhibits the kinase activity of mTOR, which is required to initiate protein synthesis necessary for the cell cycle [57]. In addition, mTOR is involved in regulation of the Akt/PKB pathway [58], and plays an important role in controlling several factors that promote cell growth including vascular endothelial growth factor (VEGF), platelet-derived growth factor β (PDGF-β) and transforming growth factor (TGF) [59].
In 2012, temsirolimus was used in a phase II study in children and adolescents with high-grade glioma, neuroblastoma or RMS [60]. In this study, patients received temsirolimus (75 mg/m weekly) for twelve weeks, yet this treatment did not meet the primary objective efficacy threshold. However, some promising results have been published in a more recent study, where patients received temsirolimus treatment four the times over a period of 21-daystogether with vinorelbine and cyclophosphamide [44].
5.2. Vincristine
Several vinca alkaloids extracted from the leaves of Catharanthus roseus (periwinkle) are potent inhibitors of polymerization and cell division [63]. Targeting microtubules has been a promising strategy for the development of novel anticancer therapies since they play an important role in the mitosis process. Vinca alkaloids bind in the proximity of the single high-affinity site on the (+)-end of the tubules and decrease the uptake of guanosine-5`-triphosphate (GTP), which is essential for the tubule elongation [64,65]. There are three currently available vinca alkaloids: vincristine, vinblastine and vinorelbine. Vincristine, which acts by binding most tightly to the active site [66], is the least lipophilic of the three alkaloids and has the longest half-life, resulting in a greater anti-tumor efficacy [67,68]. Vincristine is composed of two polycyclic moieties: a dihydroindole nucleus (vindoline) and an indole nucleus (catharanthine), joined by a carbon-carbon bond. Both subunits are essential for the activity involved in tubulin binding [69,70,71]. Interestingly, vincristine binds to tubulin in a reversible manner at different sites compared to other inhibitors of tubulin, which makes this anticancer agent a good candidate for the combination therapy with possibility to produce synergistic effects [72]. In addition, vincristine is relatively safe and has very low bone marrow toxicity at standard therapeutic doses, which makes it popular in combination therapy along with other anticancer agents [73,74,75].
Originally, vincristine was formulated as a sulfate salt that has been approved to treat acute leukemia, and as a part of a multidrug regimen for Hodgkin’s and non-Hodgkin’s lymphomas. It can also be used for the treatment of gliomas, RMS, neuroblastoma, Wilm's tumor, and soft tissue cancers [76,77]. Vincristine has been commonly used to treat pediatric cancers due to its remarkable level of intrinsic sensitivity and better tolerance at therapeutic doses in children. Other uses have included treatment of several non-malignant hematologic disorders such as refractory autoimmune thrombocytopenia, hemolytic uremic syndrome and thrombotic thrombocytopenia purpura [63,78,79].
Since cellular mechanisms of resistance to vinca alkaloids have been observed in the clinical applications, combination therapies with other chemotherapeutic agents are preferred over monotherapy [80]. For the treatment of RMS, vincristine is used in combination with dactinomycin, or as a combination with dactinomycin and cyclophosphamide (VAC). In some cases, VAC uses alternating vincristine and irinotecan, also known as VAC/VI. Many multitarget approaches that include vincristine are currently in various phases of clinical trials (Table 3).

Table 3.
RMS Chemotherapies.
| Treatment | Clinical Trial Phase | Reference |
|---|---|---|
| Ifosfamide/vinorelbine | III | [480] |
| Ifosfamide/ doxorubicin | III | [481] |
| Vincristine, dactinomycin, and cyclophosphamide or vincristine, dactinomycin, and cyclophosphamide/vincristine and irinotecan | III | [482] |
| Trabectedin | II | [246] |
| Irinotecan or vincristine and irinotecan | II | [452] |
| Vincristine, doxorubicin, and cyclophosphamide/Ifosfamide and etoposide | II | [483] |
| Vincristine, irinotecan, and temozolomide | N/A | [484] |
| Vincristine and irinotecan + vincristine, doxorubicin, and cyclophosphamide/ ifosfamide and etoposide + temozolomide | II | [227] |
| Temozolomide + Irinotecan | Preclinical (Mouse models) | [485] |
5.3. Doxorubicin
Doxorubicin is a natural product that belongs to the antibiotic group of antineoplastic agents and was originally isolated from Streptomyces paucities [77,81]. These compounds target DNA function through several mechanisms, including alkylation, intercalation, and inhibiting enzymes crucial for the process of DNA replication [82]. Intercalation is a process wherein antineoplastics interact directly with DNA and insert between the base pairs of the double-stranded helix, forming noncovalent interactions with DNA bases [83]. The newly formed complex uncoils DNA resulting in incorrect replication, which is an important trigger for apoptosis [84]. In order to fit between the double-stranded DNA, all antibiotic antineoplastic compounds including doxorubicin, possess planar, aromatic moiety [85]. In the case of doxorubicin, this planar tricyclic system is called anthracyclinone, composed of aromatic rings B, C and D that can fit between the two DNA strands, orienting itself perpendicular to the long axis of DNA [86]. The interaction of antineoplastics and DNA base pairs occurs by the overlap of p-orbitals [87]. The drug-DNA complex is further stabilized through a combination of several noncovalent interactions, including van der Waals, and/or hydrogen bonds [88]. It has been speculated that these interactions will cause inhibition of normal DNA function. Moreover, it has been reported that doxorubicin binds better in the GC-rich regions [89,90]. However, these interactions of the anthracyclinone moiety alone are not sufficient to induce cell death in cancer cells. To complement intercalation, doxorubicin also inhibits topoisomerase II [91,92], an enzyme responsible for the proper DNA maintenance during replication process. The second part of the doxorubicin structure is the aminosugar, L-daunosamine. The role of the charged amino group in sugar is believed to stabilize the anthracyclinone-DNA complex, but several SAR studies have shown that structures lacking the aminosugar, have poor activity, thus it may also be involved in covalent binding to the DNA backbone [93,94]. It is believed that rings B, C and D of anthracyclinone moiety and the sugar portion are responsible for binding to the DNA, while ring A possesses the topoisomerase II inhibition activity [95].
As a final mechanism of action, doxorubicin has quinone moiety that participates in electron-transfer reactions and makes reactive oxygen species (ROS), including singlet oxygen, hydroxyl radicals, and peroxides. ROS are known to cause damage to DNA, RNA, proteins and lipids, which may eventually lead to the cell death. This mechanism is notably responsible for the peroxidation of myocardial lipids and therefore, cardiac toxicity of doxorubicin, which is the most important and severe complication [96].
5.4. Actinomycin D (Dactinomycin)
Actinomycin D also belongs to the group of antibiotic antineoplastics. It was first isolated from Streptomyces parvullus in 1940 [98]. As mentioned above, these drugs have intercalating properties and usually contain a flat aromatic moiety capable of slipping into the double helix of DNA and distort its structure. Actinomycin D has a planar, aromatic portion, known also as actinocin or phenoxazine system (which is accountable for yellow-red color of the drug), and two cyclic pentapeptides connected to this aromatic moiety [99]. Similar to doxorubicin, the phenoxazine system is capable of intercalating DNA, especially in the GC-rich regions [89]. Once the aromatic part is positioned between DNA base pairs, the cyclic pentapeptide moieties will form several important noncovalent interactions and stabilize the dactinomycin-DNA complex [100]. This binding of actinocin to DNA is thus much stronger compared to the binding of the anthracene moiety to DNA, which is present in doxorubicin [101]. As a result of forming this stable dactinomycin-DNA complex, topoisomerase II will be inhibited, which will in turn lead to improper DNA replication [102]. Previous SAR on the actinocin moiety showed that methyl groups are important for this activity [103]. Drugs, such as doxorubicin and dactinomycin are also known as topoisomerase II poisons, since they do not inhibit the enzyme directly [104].
It has also been revealed that using low doses of dactinomycin results in ribosomal stress, resulting in p53 stabilization and activation. The p53 protein is an important transcription factor that regulates multiple genes involved in cell cycle arrest, apoptosis, differentiation and even prevention of angiogenesis. Accordingly, administration of low doses of actinomycine D in combination with other antineoplastic agents is a promising cancer therapy [105,106,107,108].
The high affinity of dactinomycin for DNA, also results in a long half-life [109]. Interestingly, cancer cells that show resistance to vincristine, are also resistant to dactinomycin and doxorubicin [110].
This drug is the most effective therapy in the treatment of RMS and Wilms tumors in children. It is also used in several other carcinomas, such as Kaposi sarcoma, Ewing sarcoma, gestational trophoblastic tumors and testicular cancer [96].
5.5. Cyclophosphamide
Cyclophosphamide belongs to the group of alkylating agents and is a derivative of the first alkylating agent used as an anticancer therapeutic, the nitrogen mustard compound chlormethine [111]. The main characteristic of these antitumor drugs is their highly electrophilic nature and ability to form covalent bonds with nucleophilic groups present on the nucleic acid bases of DNA [112]. These agents can alkylate nucleophilic groups on non-tumor DNA and proteins as well, which leads to many adverse effects [113]. However, the fact that cancer cells divide faster than healthy cells, makes these drugs strong candidates in anticancer therapeutic approaches.
Cyclophosphamide is a prodrug metabolized into two parts in the liver by various CYP450 isoenzymes to yield phosphoramide mustard and acrolein [114,115]. Phosphoramide mustard is the active form and is capable of alkylating DNA [116], while acrolein is a highly reactive aldehyde responsible for many of the adverse effects [117]. The most common side effects of acrolein are toxicities related to the kidney and bladder [118]. Toxicity can be significantly reduced if cyclophosphamide is administered with Mesna, a sulfhydryl (-SH) containing agent, which will bind to acrolein and form a water-soluble adduct that can be excreted [119]. Other frequent side effects of cyclophosphamide are nausea, vomiting, alopecia, immunosuppression and gonadal damage, mostly due to its toxicity on rapidly proliferating tissues.
Cyclophosphamide is one of the most commonly used drugs in a wide variety of hematopoietic and solid tumors, some autoimmune diseases and in bone marrow transplants, as a single agent and also in combination chemotherapy [120].
5.6. Ifosfamide
Ifosfamide is also a nitrogen mustard derivative that was developed as a structural isomer of cyclophosphamide in the 1960s. It is used in adults and pediatrics as a single agent or in combination with other chemotherapeutic agents in the treatment of both hematological and non-hematological disease [121]. Ifosfamide is a prodrug activated by CYP P450 enzymatic system in the liver to form 4-hydroxyifosfamide. This conversion occurs at a slower rate compared to cyclophosphamide due to steric hindrance, which in turn requires higher doses to achieve the same anti-tumor effect [122]. 4-Hydroxyifosfamide is an unstable product which is in equilibrium with its tautomeric form aldoifosfamide. Aldoifosfamide is converted into the ultimate alkylating agent ifosforamide mustard and the biproduct acrolein, which is also cytotoxic and the main factor responsible for the urotoxic effects of ifosfamide. Ifosforamide mustard binds covalently to nucleophilic site of DNA through its alkyl groups leading to the block in DNA synthesis and cell apoptosis [123]. In addition, it upregulates ROS which results in DNA damage [124]. It has been shown that approximately 25% to 60% of Ifosfamide can be metabolized by dechloroethylation to produce chloroacetaldehyde, which causes glutathione depletion and cell death. Ifosfamide can also pass the blood-brain barrier and can therefore cause neurotoxicity ranging from mild somnolence and confusion to severe encephalopathy and coma [125,126]. Ifosfamide can also cause a greater degree of urotoxicity compared to cyclophosphamide, thus co-administration of mesna is recommended [127].
5.7. Melphalan
Melphalan is another alkylating agent and is commonly used in the treatment of multiple myeloma [130]. The mechanism is similar to other alkylating agents: it will alkylate the guanine base of DNA (N-7 of guanine is highly nucleophilic), which will prevent normal base pairing, and lead to inhibition of replication [131]. Melphalan was one of the first follow-up analogs of chlormethine and the methyl group of the parent drug is replaced with the amino acid L-phenylalanine, giving rise to the name L-phenylalanine mustard [132]. The amino acid substitution contributes to the increased transport into the cells by carrier proteins in membrane, increased stability, and oral administration [133]. The aromatic moiety next to the nitrogen is capable of stabilizing its lone electron pair by resonance, which decreases reactivity and severe side effects [98]. In addition, melphalan can also inhibit malignant cell growth via decreasing levels of Interleukin-6 (IL-6), a cytokine involved in tumor promotion. Moreover, melphalan is able to stimulate an immune response against cancer cells by creating an inflammatory medium [134]. This drug is available both orally and intravenously to treat a variety of solid cancers, including breast, colon and ovary, RMS, melanoma, neuroblastoma and Ewing’s sarcoma, as well as various hematologic malignancies. Although it is considered more patient-compliant, there is still a long-term risk of inducing secondary leukemia/myelodysplastic syndrome and other secondary cancers [135].
5.8. Etoposide
Etoposide is a semisynthetic derivative of podophyllotoxin of Podophyllum peltatum, also called the mandrake plant. Etoposide is a podophyllotoxin glycoside with a D-glucose derivative, and is structurally identical to the anticancer medication teniposide, with the exception of a methyl group (teniposide contains a thienyl group) [136]. Both of these chemicals were created with the goal of generating less lethal podophyllotoxin derivatives [137]. Etoposide is a crystalline powder that ranges in color from white to yellow-brown and dissolvable in organic solvents. It is utilized in the form of etoposide phosphate, which is more water-soluble than etoposide. Etoposide forms a ternary complex with DNA and the enzyme topoisomerase II, which aids in the relaxation of DNA negative or positive supercoils. Topoisomerase II often breaks one DNA double-strand, enabling another to pass through while re-ligating the damaged strands. Topoisomerase II is inhibited from re-ligating the broken DNA strands, enabling the topoisomerase II-induced DNA breaks to remain broken, as well as preventing the topoisomerase II molecule from leaving the area and relieving stress elsewhere [138]. Cancer cells rely on this enzyme more than healthy cells, because they proliferate at a higher pace. As a result, errors in DNA synthesis occur, resulting in the demise of cancer cells. Etoposide and etoposide phosphate intravenous and oral formulations and are often used in combination with other agents to treat a variety of malignancies, including lung and testicular cancer, sarcoma, lymphoma, leukemia, nonlymphocytic, glioblastoma multiforme, and rhabdomyosarcoma [139,140,141]. It is also used on occasion as part of a pre-transplant conditioning regimen for the blood stem cells and bone marrow [139].
Etoposide is well-known as an apoptotic pathway inducer; however, current studies show that it may also be implicated in autophagic pathways. Whether etoposide activation of autophagic mechanisms leads in cell death or has a pro-survival effect remains unknown [138]. In this context, it has been shown that etoposide increases autophagy-dependent ATP production in multiple glioma cells, which protects the cells and may lead to drug resistance [142]. This surge in ATP levels is prevented by pre-incubation with the autophagy inhibitor 3-methyladenine, siRNA-mediated down-regulation of Beclin-1, or the mitochondrial inhibitor oligomycin, but not by glucose restriction. By inhibiting autophagy-induced ATP production, all of these treatments promote non-apoptotic cell death [143].
Severe myelosuppression is the major but uncommon adverse reaction following administration of etoposide. Other side effects include allergic reactions, vomiting, diarrhea, bone marrow suppression, nausea, stomatitis, abdominal pain, fatigue, hypotension, peripheral neuropathy and hair loss [96].
5.9. Irinotecan
Irinotecan is an analogue of camptothecin (CPT), a natural product isolated from the bark and stem of Camptotheca acuminate [144]. CPT anticancer activity is linked to the inhibition of topoisomerase I. The lactone ring of CPT is extremely vulnerable to hydrolysis, and topoisomerase I is inhibited by reclosing the lactone ring, resulting in trapping a subset of topoisomerase-1-DNA complexes and preventing relegation of the DNA strand. Passive diffusion transports CPT into the cell, while lipophilicity promotes cellular uptake and increases intracellular accumulation. Because of better lactone partitioning into the red blood cells and subsequently reduced hydrolysis of the lactone, lipophilicity makes compounds more stable. CPT, particularly the carboxylate form of CPT, has high affinity for human serum albumin (HSA). As a result, the balance between the carboxylate form and the lactone ring is pushed in favor of the carboxylate. Improved activity might be achieved by reducing drug-HSA interactions [145,146].
Irinotecan is a prodrug that damages DNA by inhibiting topoisomerase and kills cells in S-phase. Lethal double-strand DNA breakage and cell death arise from the development of a cleavable drug-topoisomerase I-DNA complex. Its piperidine group at C10 site is hydrolyzed by carboxylesterases and butyrylcholinesterase to produce SN-38, which is more than 100-fold cytotoxic than irinotecan [96,147]. It was discovered and produced for the first time in 1983 in Japan, and it has since shown significant anticancer activity against a wide spectrum of cancers [148]. Irinotecan has shown activity against colorectal, gastric, esophageal, small-cell and non-small-cell lung cancers, lymphomas and leukemia, and central nervous system malignant gliomas [144,148]. In the United States, it was approved as a second-line therapy for metastatic colorectal cancer when 5-fluorouracil (5-FU) and leucovorin failed [149]. It was recently authorized for use as a first-line therapy of colorectal cancer in combination with 5-FU/leucovorin [150,151]. Irinotecan is being studied as an adjuvant treatment for node-positive colorectal cancer following resection. Similarly, phase II trials on advanced esophageal and gastric cancer showed significant response rates that were encouraging [152,153]. The combination of vincristine, irinotecan, and temozolomide (VIT) is frequently used to treat adolescents and children with relapsed RMS. A recent study has demonstrated that in the patients with first relapse RMS, VIT treatment in conjunction with sufficient local control is linked with some disease control and may be another viable alternative to give patients as salvage therapy [154].
SN-38 is metabolized and inactivated by glucuronidation to SN-38G and intrahepatic CYP450 enzymes and excreted mainly in the bile. However, SN-38G can be reactivated by β-glucuronidases to SN-38 in the intestine, which is related to the intestinal damage, mucositis and diarrhea complications, restored and reabsorbed [147,155].
5.10. Volasertib
Volasertib (BI 6727) is a potent dihydropteridinone derivative that inhibits Polo-Like Kinase-1 (PLK1) by acting as a small-molecule ATP-competitive kinase inhibitor [156]. It is a second-generation PLK1 inhibitor that is structurally similar to BI 2536 but has been chemically modified to increase its PLK1 activity and pharmacokinetic profile (i.e., large volume of distribution and long terminal half-life resulting in extensive penetration into the tissues and prolonged tumor exposure). PLK1 is a serine/threonine kinase that has a vital role in the cell cycle progression through mitosis as well as regulating DNA damage checkpoints. It is overexpressed in wide spectrum of cancers including Ewing sarcomas, medulloblastomas, non-small-cell lung cancer, breast cancer, and RMS, and its elevated level has been correlated with poor prognosis in some types of neoplasms making it a promising target in cancer therapy. Volasertib blocks cell cycle in prometaphase, also called polo arrest, and induces apoptosis. It inhibits PLK1 at sub-nanomolar doses (IC50 0.87 nM), however, it has also been shown to inhibit PLK2 and PLK3 at higher doses (IC50 5 and 56 nM, respectively) [157,158,159,160].
Volasertib has been clinically studied for years in various drug combinations in adult patients suffering acute myeloid leukemia and other solid malignancies, with mixed outcomes [161]. At low volasertib/BI 2536 dosages, the pre-clinical effects of volasertib in combination with vincristine in fusion-negative RMS models appear to be significant; however, the effects of fusion-positive RMS models with the volasertib/vincristine combination requires additional evaluation [161]. Given the broad usage of vincristine for the treatment of newly diagnosed and relapsed RMS, as well as the likely non-overlapping toxicities of volasertib and vincristine, the combination of these two drugs appears to be practical and has immediate clinical promise in both fusion genes negative and positive RMS. If limited proof-of-concept clinical testing confirms dosage and activity tolerance, more intense backbone chemotherapy and/or additional targeted medicines may be studied in the future, potentially improving RMS patients outcomes [161].
Overall, the treatment of low-risk tumors is evolving in an effort to decrease the burden of treatment by reserving intensive therapy for those with high risk or recurrent disease [4]. This approach to therapy has led to higher tumor recurrence rates in Europe (where this approach has been adopted), lower treatment associated side effects, and unchanged RMS survival rates in cases with low-risk tumors [4,7]. Genitourinary tumors are of particular concern when considering the side effects of treatment such as enuresis and sexual dysfunction, which are associated with local disease therapy (surgery and radiotherapy) [162]. Such side effects have led patients with high-risk RMS to unsuccessfully complete therapy due to attrition in addition to unplanned dose modifications outside of the protocol guidelines [163].
6. Apoptosis – General Considerations
Apoptosis, or programmed cell death, is one of the major mechanisms of cell death [164,165]. This process can occur either under physiological conditions, i.e., during development and differentiation of tissues, or as a result of prolonged stress induced by the environment of the cell [166,167,168]. Apoptosis is a strictly regulated process and can be distinguished from necrosis based on the characteristic morphological changes, such as chromatin condensation, fragmentation of DNA within the nucleus, or cell shrinkage [169,170]. Apoptosis can be induced in the cells either through the intrinsic mitochondrial pathway or the extrinsic death receptor pathways [171,172].
The intrinsic pathway involves the activity of Bcl-2 family proteins, located in the mitochondrial outer membrane. When the balance in their activity is tipped towards cell death, pro-apoptotic Bcl-2 proteins promote mitochondrial outer membrane permeability and subsequent release of cytochrome c, caspase activation, and apoptosis [173,174,175]. Conversely, the extrinsic pathway relies on stimulation of death receptors, such as Fas or TNFR (tumor necrosis factor receptor), by ligands [176,177,178]. This process is followed by the recruitment of adaptor proteins and initiator caspases – caspase 8 and 10, which form death-inducing signaling complex [179]. An overview of apoptosis signaling pathways is illustrated in Figure 2.
6.1. Avoidance of Apoptosis by RMS Cells
Resistance to the programmed cell death, which allows for a proliferative advantage, is a characteristic feature of the malignant cells. Defects in apoptosis often result in resistance to the cytotoxic therapies, as current conventional treatment relies on the neoplastic cells ability to undergo cell death in response to toxicity [180]. In most cancers, the avoidance of cell death occurs predominantly due to the overexpression of anti-apoptotic genes, or down-regulation of pro-apoptotic genes [181,182].
In fusion-positive (FP) RMS, the PAX3-FOXO1 and PAX7-FOXO1 fusion proteins function as drivers of oncogenesis by dysregulating multiple crucial cellular pathways. The fusion proteins drive the expression of other transcription factors such as MYCN and MYOD1, contributing to the RMS formation and progression [183]. Moreover, the fusion proteins drive the expression of receptor tyrosine kinases (RTKs). The overexpression or activation mutations of both genes encoding the RTKs or their downstream signaling effector genes are common in FP RMS [183]. This includes FGFR4 (fibroblast growth factor receptor 4), whose activating mutations are present in 7% of FP RMS patients triggering RAS and STAT signaling pathways that induce tumor growth [184]. Activation of the Ras/Raf/MEK/ERK, and JAK/STAT pathways can result in prevention of apoptosis through phosphorylation of Bim and Bad, which result in the loss of the ability to heterodimerize with survival proteins BCL-XL and BCL-2. Moreover, JAK/STAT signaling pathway can result in the overexpression of anti-apoptotic BCL-XL [185,186]. Taken together, these changes result in down-regulation of BAX/BAK effector proteins and apoptosis restriction. Additionally, the overexpression of FGFR4 tyrosine kinase in RMS cell lines induces its auto-phosphorylation and constitutive signaling that results in the prevention of apoptosis by targeting the IGF1R-PI3K-mTOR (Insulin growth factor 1 receptor/Phosphoinositide 3-kinase/mammalian target of rapamycin) pathway [187,188,189]. Additionally, knockdown of FGFR4 in RMS cell lines shows reduction in the cell proliferation and increase in apoptosis [190].
PDGFR (platelet-derived growth factor receptor) is another RTK driven by the fusion protein. Experimental data suggests that its overexpression regulates cancer cell stemness, differentiation, and apoptosis, with PDGFR inhibition resulting in an increase in apoptosis accompanied by the G2/M cell cycle arrest in RMS cell lines [191].
Other RTKs induced by the fusion protein and implicated in RMS progression can signal through the RAS-PI3K-AKT-mTOR and RAS-RAF-MAPK pathways [192,193]. Gene expression analyses reveal that over 50% of the patients with FP RMS carry mutations that impact the aforementioned pathways [194,195]. AKT serves as a member of pro-survival pathway, as its activity rescues cells from PTEN-mediated apoptosis [196]. The anti-apoptotic activity of AKT seems to be multifactorial, as it directly phosphorylates selected components of the apoptotic machinery. Phosphorylation of BAD by AKT prevents its dimerization with a member of the BCL-2 family – BCL-XL, restoring the latter anti-apoptotic function [197]. Moreover, through direct phosphorylation, AKT inhibits the activity of caspase 9 [198]. Finally, PAX-FOXO1 fusion protein can synergize with the loss of cyclin-dependent kinase inhibitor 2A (CDKN2A) or p53, functionally either indirectly through CDKN2A tumor suppressor gene loss, or TP53 promoter mutation [199].
An increasing body of evidence suggests that epigenetic regulation contributes to RMS development and progression [200]. In comparison with normal tissue, muscle-specific microRNAs (miRs) are down-regulated. These miRs are often involved in protecting the organism from malignant transformation, serving as antioncogenes. The inhibition of these specific miRs, such as miR-29, miR-450b-5p, miR-203, and miR-214, contributes to the enhanced tumorigenesis through diminished myogenic differentiation and inhibition of apoptosis [201,202]. While those miRs affect diverse molecular pathways, the effect is partly mediated by IGF1/AKT pathway, as transient transfection of miR-378a-3p in ARMS cell line induced apoptosis, impaired migration, and promoted myogenic differentiation [203].
6.2. Antineoplastic Agents Targeting the Apoptosis Pathway in RMS
There are several chemotherapeutic drugs that have been approved for the RMS treatment, which act through inducing cancer cell apoptosis. These treatment modalities initiate cell death pathway through diverse molecular mechanisms (e.g., through cell cycle blockade, interference with proliferation, or DNA damage) (Figure 2).
Alkylating agents, cyclophosphamide and ifosfamide, induce crosslinking between DNA strands (See Section 5 above). In the cell lines exposed to alkylating agents, a decrease in the DNA strands expression of the anti-apoptotic BCL-2 and an increase in pro-apoptotic BAX, caspase 3, and PARP expression have been observed [204,205]. Moreover, a dose-dependent inhibition of ERK1/2 and AKT phosphorylation was observed, suggesting that the changes in apoptosis-associated proteins is mediated by ERK/MAPK and PI3K/AKT signaling pathways [204].
While the exact mechanism through which etoposide leads to apoptosis is not fully understood, it seems to involve AKT regulation, whereby etoposide stimulates AKT to migrate into the mitochondria, enhancing its interaction with Smac, phosphorylating it at residue 67, which in turns leads to the enhancement of Smac interaction with X-chromosome linked IAP (XIAP) protein, which then upregulates the activity of caspase 3 [206,207]. During therapy with topoisomerase II inhibitor, caspase 2, 3, and 9 activation is observed, an effect which is partly mediated by BCL-2 [208]. Similarly, treatment with dactinomycin results in apoptosis in both a caspase-dependent and -independent manner. Dactinomycin treatment results in cell death through the activation of caspase 7 and 9, an affect which was only partly attenuated by caspase inhibition, suggesting the partial involvement of reactive oxygen species release, and upregulation of the apoptotic-inducing factor (AIF) expression [209].
As mentioned in the section 5 above, vincristine destabilizes microtubules through suppression of tubulin polymerization [210]. As a result, cells undergo arrest in the G2/M phase. Vincristine treatment also depletes the mitochondrial membrane potential, increasing the release of mitochondrial cytochrome c into the cytosol. Additionally, there is an observable increase in tBID, which in combination with lower concentrations of BCL-2 and BCL-XL, leads to the apoptosis through FADD-associated auto-cleavage and activation of procaspase-8 [210]. Another established mechanism through which the cell cycle becomes halted in the G2/M phase is the inhibition of pro-survival polo-like kinases (PLKs) [211]. Volasertib, a novel PLK inhibitor, induces apoptosis through caspase 3 activation [211].
Melphalan and temozolomide induce apoptosis in cancer cells through distinct molecular pathways. The former induces the cleavage of MCL-1, disrupting the MCL-1/BIM complex, which under normal conditions neutralizes the proapoptotic function of BIM and prevents the activation of death effectors [212]. The disappearance of MCL-1 allows for the release of BIM isoforms, which lead to further BAX activation and cytochrome c release. The mechanism through which temozolomide induces apoptosis remains unelucidated, but likely does not involve changes in MCL-1, BCL-2, BCL-XL, or BAX protein expression [212,213].
Irinotecan, a DNA topoisomerase I inhibitor increases intracellular BAX concentration. Moreover, it causes an increase in p53 and caspase 9 levels with accompanying decrease in the expression of BCL-XL [214]. Elevated p53 reinforces the induction of apoptosis by raising the expression of pro-apoptotic members of BCL-2 family and death receptors [215]. Nevertheless, a notable subset of patients exists, for whom the aforementioned treatment is ineffective by the means of rapidly acquired resistance. Therefore, significant efforts are placed to identify other, more efficacious therapeutic agents.
Temsirolimus is a derivative and prodrug of widely used immunosuppressant sirolimus, also known as rapamycin (See Section 5 for more details). Rapamycin and its derivatives act by inhibiting mTOR [216,217]. Blockage of this protein is followed by dysregulation of proliferation and hindrance of the cell growth [217]. Moreover, mTOR inhibition leads to the cell cycle arrest in the G1-phase and directs the cell towards apoptotic cell death [216]. This observation could be explained by a decrease in the mTOR downstream target p70S6K, which normally phosphorylates the pro-apoptotic BAD on serine 136, disrupting its ability to bind to BCL-XL and BCL-2 [218]. In RMS cells, mTOR inhibition can successfully abrogate tumor growth with a reduction in proliferation and invasiveness, as well as an induction of apoptosis through inhibition of BCL-2 expression [219]. The restriction of tumor growth is associated with the down-regulation of mTOR and Hedgehog (Hh) signaling, both of which are implicated in the pathogenesis of RMS. This implementation of molecular targeted therapy opens new avenues for the personalized therapy in the hope to improve therapeutic outcomes [219].
The addition of temsirolimus to the chemotherapeutic regimen is expected to enhance its efficacy, as mTOR inhibition presumably resensitizes previously chemoresistant cancer cells [220]. The clinical trial (NCT00106353) reported that this agent at the dose of 75 mg/m2/week prolongs stable disease. However, further evaluation of temsirolimus in combination with currently used therapy regimens is essential [221].
Some other examples of the molecular targeted therapies in RMS involve the use of vascular endothelial growth factor (VEGF) inhibitors. The expression of VEGF is indicative of poor prognosis in various solid tumors, including both ARMS and ERMS [222]. These observations strongly suggest that VEGF could be a suitable therapeutic target. Clinical trial NCT01222715, compared the efficacy of temsirolimus and VEGF-A inhibitor -bevacizumab, where 87 patients received the standard chemotherapy combined with one of the aforementioned agents. Temsirolimus was found to be more efficacious in terms of event-free survival between the two groups [223].
One of the targeted therapies that is currently being investigated in the context of RMS treatment (see Table 4) involves the use of IgG1 monoclonal antibody, cixutumumab, which is directed against the human insulin-like growth factor-1 receptor (IGF-1R) [224]. This therapeutic agent down-regulates PI3K and MAP signaling pathways, increasing caspase 3 and PARP cleavage [225]. The limited activity and acceptable toxicity of monotherapy supports the idea of including this antibody in the combined therapeutic regimens [226,227,228]. Other drug combinations involving cixutumumab with doxorubicin and temsirolimus are under scrutiny [226]. The preliminary results suggest that this antibody improves the outcomes of temsirolimus therapy [229]; however, the dependence of combined therapy on IGF-1R expression on cancer cells remains unclear [229,230].
Crizotinib and ceritinib, ALK (anaplastic lymphoma kinase) and ROS1 (c-ros oncogene 1) inhibitors, are other neoplastic drugs whose efficacy against RMS is under investigation [231,232]. ALK inhibition is a known mechanism of inducing apoptosis [233], and cancer cells (such as non-small-cell lung cancers (NSCLC) & RMS ) are often dependent on ALK and ROS1 function, providing a reasonable rationale for evaluating crizotinib and ceritinib in these cancers [234]. Nevertheless, studies characterizing the properties of ALK and ROS1 inhibitors failed to prove their efficacy as single agents against RMS [235,236,237]. However, the addition of ceritinib to another chemotherapeutic agent, especially kinase inhibitors such as dasatinib or sorafenib [238], improves the therapeutic outcomes [235,239]. Sorafenib in combination with PLKs inhibitors is under scrutiny in other types of cancers and primary results are promising [211]. Another kinase inhibitor, pazopanib is also under investigation [240]. According to the recent studies, it seems to be a promising therapeutic modality for the patients with refractory and relapsed sarcomas [241,242]. Similarly, regorafenib, does not improve progression-free survival in the treatment-refractory liposarcoma [243], but its combination with other agents in RMS treatment might lead to the superior results.
Trabectedin, which inhibits gene activation and blocks nucleotide excision repair, leads to the cell cycle arrest [244] and upregulation of BAX, BID, and caspase 3 transcripts [245]. While this agent failed to demonstrate sufficient activity as a single agent, it might become an element of a potent multi-drug regimen [246].
Another approach to influence the RMS apoptotic pathways is through inhibition of histone deacetylation. Posttranslational modifications of histones affect gene expression. Acetylation as one of these modifications, marks regions of the high transcriptional activity [247]. However, acetylation can be reversed by the histone deacetylases (HDACs) leading to the transcriptional repression. HDACs silence apoptosis inducers or tumor suppressor genes, contributing to oncogenesis [248,249]. HDAC inhibitors (HDACIs) are a promising group of therapeutic agents that are believed to restore physiological histone acetylation [248]. A study on HDACIs influence on apoptosis of RMS cells revealed that they, especially in combination with Bromodomain and Extra Terminal (BET) inhibitors, trigger the mitochondrial pathway of apoptosis [250,251]. Further analysis shows that BIM and BIF become upregulated, while BCL-XL and survivin are down-regulated [250,251,252,253]. HDACIs are also capable of cell cycle arrest in the M-phase [252]. One member of the HDACIs, entinostat, demonstrates synergistic anti-tumor activity if combined with vincristine [254], while another HDACI SAHA acts synergistically with doxorubicin [253] and has beneficial properties against RMS in both cell and mouse models [254].
The involvement of PARP proteins in the DNA repair prompted researchers to hypothesize that DNA breaks induced by radiotherapy would be more deadly to cancer cells if the therapy was combined with Olaparib, a PARP1-3 inhibitor [255,256]. This hypothesis was verified in a study on RMS cells, where combined exposure to PARP inhibitors and ionizing radiation elicited more robust cytotoxic effects than radiation alone [257].
Statins or 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA reductase) inhibitors are another group of the agents, whose antiproliferative properties might be beneficial during anti-RMS treatments. Statins are believed to activate the mitochondrial pathway of apoptosis. Recent research suggests that simvastatin activates caspases 3 and 9 [258]. Moreover, pretreatment with statins augments proapoptotic properties of other antineoplastic agents [258]. A summary of the available data on HMG-CoA reductase inhibitors suggests that impairing the Ras family GTPase signaling is crucial for the chemo-sensitizing effect [259,260,261,262]. Nevertheless, the clinical significance of statins as antineoplastic agents is still undetermined [259].
7. Autophagy Process
Autophagy is a Greek term that means self-digestion and was firstly proposed by Christian de Duve in 1963 [263]. In vivo, basal autophagy is constitutively active under normal conditions, and it can be further induced by physiological and environmental stressors such as DNA damage, reactive oxygen species (ROS), hypoxia, nutrient starvation, endoplasmic reticulum stress, adenosine triphosphate (ATP) deficiency, hormonal stimulation, and pharmacological treatment [264,265,266,267]. Based on the mechanism and morphology, autophagy is divided into three major types: microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy [268]. In microautophagy, small cargo substrates are directly delivered to the lysosome membrane without an autophagosome (a kind of double-membrane vesicle) and these substrates are digested by lysosome [269,270,271]. This type of autophagy cannot be activated by the stress or nutrient deprivation (Figure 3A) [272]. CMA has the most selective function and identifies unfolded substrate proteins containing a special recognition motif KFERQ (Lys-Phe-Glu-Arg-Gln) by chaperon proteins like HSP70 and HSPA8. LAMP2A (lysosomal CMA receptor) identifies these complexes containing chaperon and substrate proteins and transports them to the lysosome [273,274,275]. Like microautophagy, during CMA cytosolic components are not enveloped by a cytoplasmic membrane (Figure 3B) [276,277,278,279,280]. Macroautophagy is considered common autophagy [273]. This type of autophagy is evolutionarily conserved from yeast to mammals [273,281], and is defined by a process where cytoplasmic substrates are isolated by an autophagosome and then transported to the lysosome for digestion [273,282,283]. Generally, this process could be either selective or non-selective [284]. Although both selective and non-selective autophagy use the same mechanism for digesting substrates, in selective autophagy, special substrates such as mitochondria, ribosome and peroxisomes are targeted by autophagy receptors which contain an ATG8-interacting motif (AIM)/LC3-interacting region (LIR) to facilitate delivery to the autophagosome (Figure 3C) [285,286].
7.1. Targeting Autophagy to Increase the Effectiveness of Chemotherapy in Rhabdomyosarcoma
Autophagy has been observed to play both a survival role and a mode of cell death in cancer cells. This dual role of autophagy in cancer development has led to two different treatment strategies. The first approach involves the sensitization of cancer cells to chemo/radiotherapy through inhibition of the cytoprotective role of autophagy; while the second approach involves the induction of autophagic cell death in apoptosis-resistant cancer cells [287]. In this regard, combinatorial therapeutic strategies produce a synergistic or additive effect compared to monotherapies to overcome the resistance of tumor cells to cancer chemotherapeutic agents and enhance their response to anticancer compounds [288]. Rezaei Moghadam and colleagues have shown that autophagy inhibition increased temozolomide (TMZ)-induced extrinsic apoptosis in ARMS cell lines. Indeed, TMZ can activate autophagy flux in the ARMS cells by increasing the expression levels of autophagy proteins LC3-II, P62, and ATG5-12. Treatment of the ARMS cell line RH30 with the autophagy inhibitor Bafilomycin A1 significantly increased the anti-tumor effect of TMZ as a chemotherapy agent [289].
Statins are FDA-approved mevalonate (MEV) cascade inhibitors, more commonly known as cholesterol-lowering drugs, and widely used for the primary and secondary prevention of coronary artery disease [290,291,292]. Importantly, autophagy is induced and modulated in statin-induced cell death [293,294]. Shojaei et al., demonstrated that simvastatin enhances TMZ-induced apoptosis in human glioblastoma (GBM) cell lines. This investigation showed that simvastatin inhibited the TMZ-induced autophagic flux by blocking the fusion of autophagosomes and lysosomes [295]. Moreover, Werner et al., demonstrated that the combined application of doxorubicin and simvastatin had additive effects on activating the mitochondrial pathway of apoptosis in RMS cells compared to either drug alone [296].
Actinomycin D is an antibiotic that also has antitumor activity against malignancies, especially in RMS [297,298,299]. Wang and colleagues in their study depicted that the anti-Fas death receptor antibody/Actinomycin D (AF/AD) induced apoptosis and P38MAPK-mediated protective autophagy in human hepatocellular carcinoma Bel-7402 cells. They showed that adding the P38MAPK inhibitor SB203580 or the autophagy inhibitor 3-methyladenine (3-MA) to this combination could induce apoptosis in Bel-7402 cells [300].
Glutathione S-transferase P1 (GSTP1), a phase II detoxifying enzyme, is overexpressed in the tumor cells and contributes to multidrug resistance (MDR). Multidrug resistance in pediatric RMS is also associated with the GST family of genes. A combination of GST protein inhibitors OZO-H (4-phenyl-1,3,2-oxathiazolylium-5-oleate) or Etacrinic acid and cytotoxic drugs vincristine, doxorubicin, and topotecan modulates the drug sensitivity of alveolar RMS RH30 cells and embryonal RMS A204 cells and provides a noticeable additive effect on cell death [301]. This suggests a positive correlation between GST protein expression and soft tissue sarcoma resistance to Aariamycin, cisplatin and mitomycin C [302].
Autophagy function is dependent on lysosomal activity [303]; thus, inhibiting or modulating lysosomal activity could be a high-value target to improve chemosensitivity of RMS cells. Salerno et al., proved that blocking lysosomal acidification by the V-ATPase inhibitor Omeprazole, or by specific siRNA, considerably potentiated the cytotoxic effects of doxorubicin against an embryonal RMS cell line, but also mitigated the invasive potential of RMS cancer stem cells [304]. Ciclopirox olamine (CPX), a synthetic Hydroxypyridone derivative, is known to induce cell death in different cancer types including leukemia, breast cancer, and soft tissue sarcoma. Hongyu Zhou et al., found that CPX activates ROS-mediated JNK signaling pathway to provoke autophagy in human RMS (RH30 and RD) cells, and that inhibition of this autophagy response by chloroquine (CQ) exacerbates the anticancer effectiveness of CPX [305].
The ubiquitin-proteasome system (UPS) and the heat shock response (HSR) are two essential regulators for the cell homeostasis, as their inhibition has a great impact on the growth and survival of normal cells as well as the stress response and invasion of cancer cells. Peron et al., demonstrated that a combination of a lysosomal inhibitor (chloroquine), a proteasome inhibitor (bortezomib), and a competitive Hsp90 inhibitor (17-DMAG) sensitizes the alveolar and embryonal RMS cell lines (RH30 and RD) to anticancer drug-induced apoptosis [306]. Moreover, this sensitivity could be abrogated by the autophagy activator rapamycin, confirming that autophagy is a key resistance mechanism in RMS cells [306]. In agreement with these observations, disruption of autophagosome formation via shRNA sequence against ATG7 (shATG7) or by inhibition of both V-ATPase-dependent acidification and autophagosome-lysosome fusion using bafilomycin A1 can mitigate antitumor drug-induced autophagy and abolish the growth of embryonal (RD) and alveolar (RMS13) RMS cell lines [307].
SIRT1 and SIRT2 are deacetylase enzymes that belong to the mammalian Sirtuin (SIRT) family and are involved in various cellular processes such as metabolism [308], cell survival [309], differentiation [310], DNA repair [311], and pathogenesis of solid tumors and leukemias [312,313,314]. A study demonstrated that overexpression of SIRT1 and SIRT2 induced autophagic flux in human soft tissue sarcoma cell lines. Ma et al., further showed that pharmacological inhibition of Sirtuins with Tenovin-6 (Tv6), induced apoptosis and impaired autophagic flux in pediatric sarcoma cell lines, without impacting p53 acetylation. They indicated that using Tv6 or SIRT1 and SIRT2 siRNAs, has not only antiproliferative effects in the RMS cell lines (RD and RH30), but also an anti-expression effect on the protein level of LC3-II [315].
Temsirolimus is a specific pharmacological inhibitor of mTOR that has been well tolerated by patients with advanced solid tumors and melanoma in clinical phase I trial of the combinatorial therapy. This clinical study indicates that the combination of temsirolimus and hydroxychloroquine, as autophagy inhibitor, regulates autophagy in patients, and produces more synergistic antitumor activity [316].
The adenosine triphosphate (ATP)-binding cassette (ABC) transporters consists of a large superfamily of membrane proteins that transport substrates across the membranes by hydrolyzing ATP [317]. The ABCC subfamily constitutes 12 transporters and overexpression of these proteins causes chemotherapeutic drug resistance in tumor cells [318]. Among them, P-glycoprotein (P-gp/MDR1/ABCB1) is associated with resistance to the commonly used chemotherapeutic agents in RMS [319]. The inhibition of P-gp with different concentrations of Silibinin di-hemisuccinate (SDH), a flavonoid antioxidant, enhanced MTX-induced cytotoxicity in MTX-resistant human RMS (hRD) [320]. The cancer stem cells (CSC) in glioblastoma multiforme (GBM) display high levels of ABC transporters which are associated with chemo-resistance phenotype in GBM CSCs. A schematic overview of autophagy targeting in relation to RMS has been shown in Figure 4.
Collectively, these studies strongly suggest that specific and effective autophagy modulators could be a beneficial adjunct in combination cancer therapy. We summarized the available data collected from previous studies about the synergistic effect of autophagy inhibitors and other therapeutic agents on RMS in Table 5.
8. General Concepts of Unfolded Protein Response and its link to RMS
The Endoplasmic Reticulum (ER) is the cell ‘manufacturing and packaging plant’ playing important roles in the production, folding and post-transitional modification of proteins and biosynthesis of lipids. Given the importance of ER function, cells must constantly monitor ER health. Three ER anchored transmembrane receptors, Inositol Requiring Enzyme 1α (IRE1α), Protein Kinase R like endoplasmic reticulum kinase (PERK) and Activating Transcription Factor 6 (ATF6), survey the internal ER environment. Under non-stress conditions, each of these receptors is inactivated through binding of their N-terminus to the ER chaperone Glucose Regulated Protein 78 (Grp78) [321,322]. Accumulation of unfolded or misfolded proteins within the ER lumen, a condition known as ER stress, instigates Grp78 dissociation facilitating receptor activation [321,322]. IRE1α dimerizes and trans-autophosphorylates facilitating activation of its RNase activity [323,324,325]. Similar to IRE1α, PERK dimerizes and trans-autophoshorylates upon loss of Grp78 binding allowing it to acquire full catalytic activity [326]. In contrast to IRE1α and PERK, upon Grp78 dissociation, ATF6 translocate to the Golgi Apparatus where it is cleaved by Site 1 and Site 2 proteases forming ATF6N [327]. The collective signaling pathways downstream of IRE1α, PERK and ATF6 constitute the UPR. These pathways work in a cooperative, complimentary fashion to reduce the levels of unfolded proteins thereby restoring ER homeostasis [328].
IRE1α via its RNase activity splices XBP1 mRNA, which following relegation by RTCB, and translation produces a transcription factor referred to as spliced XBP1 or XBP1s [329,330]. XBP1s increases expression of genes encoding ER chaperone proteins and components of the ER-associated degradation machinery (ERAD) [331]. By doing so, IRE1 signaling helps to support the folding of those proteins that can be refolded while promoting the destruction of those proteins beyond repair. IRE1α RNase activity has also been linked to the degradation of selective mRNAs via a process referred to as Regulated IRE1 Dependent Decay (RIDD) [332,333]. Many mRNAs identified as RIDD targets encode ER targeted proteins. By facilitating their degradation, IRE1α avoids additional pressure being placed on an already stressed ER.
Similar to IRE1-RIDD signaling, PERK activation aids the resolution of ER stress by halting canonical cap dependent translation. PERK, via its kinase activity, phosphorylates Ser51 on eif2α [334]. Phosphorylation of eIF2α at Ser 51 blocks eIF2B-mediated exchange of GDP for GTP, thereby halting 5” cap-dependent translation. This translational block, while widespread, is not complete as genes with an upstream open reading frame or an internal ribosome entry site within their 5’ Untranslated Region (UTR) are selectively translated under these conditions [335]. Activating Transcription Factor 4 (ATF4) is one such example. ATF4 expression during the ER stress is linked to the regulation of adaptive genes including those involved in regulating oxidative stress, amino acid metabolism and ER chaperones [336,337]. PERK can also target and phosphorylate the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) [338]. Normally, NRF2 is retained in the cytoplasm through binding to Kelch like-ECH-associated protein 1 (KEAP1) [339]. PERK-mediated phosphorylation of NRF2 breaks the NRF2/KEAP1 interaction enabling nuclear translocation of NRF2, where it increases the expression of pro-survival genes [338]. The ATF6 signaling pathway both upregulates expression of genes encoding ER chaperones and supports IRE1 signaling by transcriptionally upregulating XBP1, thereby ensuring a plentiful pool of XBP1 for the IRE1-mediated splicing [329].
In addition to controlling the UPR directly, IRE1, PERK and ATF6 can stimulate and influence proteostasis through additional stress-induced pathways, in particular autophagy. Although a basally active process, levels of autophagy tend to increase during times of stress. IRE1, PERK and ATF6 signaling pathways have all been linked to the events such as upregulation of autophagy-related genes (ATG), repression of autophagy suppressive pathways such as mTORC1 signaling and disruption of Beclin 1/BCL-2 complexes which stimulate autophagy [340].
While the UPR is an adaptive process, unlike autophagy it is not meant to be constitutively activated. If ER stress is excessive or prolonged, UPR signaling transitions from a pro-survival to a pro-death pathway. Although ER stress-induced cell death and the mechanisms facilitating it have been extensively studied, exactly how and when a cell makes the decision to transition to death is still a matter of much debate. Regulation of BCL-2 family members leading to mitochondrial-mediated apoptosis was thought to be the predominant cell death pathway during ER stress, but recent studies have indicated a role for death receptors, in particular the Trail death receptor DR5 [341].
The fundamental role of UPR signaling in healthy cells is to provide cells with a means to survive during transient stress, but the situation in diseased cells, such as cancer cells, is much more complex. Unlike healthy cells, cancer cells have acquired the ability to sustain permanent activation of UPR mediators. Constitutive activation of IRE1 and PERK and their associated downstream pathways has been reported in multiple cancers including triple negative breast cancer, prostate cancer, lung cancer and more recently sarcomas including RMS (RMS) [342,343,344]. Sustained UPR signaling offers cancer cells a means to meet the protein folding demands instigated by the activation of oncogenes or loss of tumor suppressors. However, the impact of UPR signaling appears to be more extensive than simply aiding ER function, with IRE1 and PERK mediated pathways implicated in a range of pro-tumorigenic processes ranging from supporting metastasis to the development of chemoresistance [342].
Engaging pathways such as UPR, heat shock proteins, and autophagy are strategies cancer cells can exploit to maintain proteome integrity. Similar to many other cancers, RMS cells are known to have a high dependence upon proteostatic pathways including the UPR [344,345]. This presents a therapeutic opportunity, if these pathways can be impeded and proteostasis disrupted, cancer cells may engage death pathways. In RMS, chemical inhibitors of heat shock protein HSP70 have been shown to reduce RMS cell viability. The addition of MAL3-101 (HSP70 inhibitor) to RMS cell lines triggers cell death via a mechanism dependent upon UPR mediated induction of the pro-apoptotic transcription factor CHOP [346]. Subsequent studies demonstrated that while HSP70-based inhibition could elevate UPR signaling and cell death, RMS cells by increasing autophagy or ER associated degradation pathways developed resistance [347]. Combination with strategies to decrease autophagy, such as chloroquine addition, overcame HSP70 inhibitor resistance suggesting dual targeting of HSP70, and autophagy may be an effective combination [347].
While targeting HSP70 and autophagy may elevate UPR signaling to a point where it is untenable, an alternative way to also achieve this is by reducing basal UPR signaling. The recent development of small molecule inhibitors of UPR mediators offer the potential to selectively target and block these pathways. Inhibition of IRE1 RNase activity has been shown to exert beneficial effects as either a standalone treatment or in combination with chemotherapeutics in pre-clinical models of Triple Negative Breast Cancer (TNBC) and prostate cancer [348,349,350]. The status of basal UPR signaling pathways and outcome of UPR inhibition in RMS has not been extensively studied. However, McCarthy and colleagues recently reported constitutive activation of UPR mediators IRE1 and PERK in a panel of RMS cell lines encompassing both ARMS and ERMS subtypes (McCarthy N et al, 2020). Selective inhibition of IRE1 or PERK resulted in divergent outcomes with ARMS cells displaying a marked reduction in cell proliferation and long-term survival to IRE1 inhibition, whereas ERMS cell lines were more responsive to PERK inhibitors [343]. Further analysis demonstrated reduction in the cell proliferation, which was the consequence of cells transitioning into a non-proliferative senescent state [324]. Whether combination with senolytics is sufficient to trigger death of IRE1/PERK inhibitor-treated RMS cells is an interesting question to address in the future. An ever-expanding literature supports roles for the UPR mediators in pro-tumorigenic processes distinct from maintenance of cell viability. For example, IRE1 signaling has been linked to the tumor metabolism, epithelial to mesenchymal transition (EMT), angiogenesis and the development of chemoresistance [342]. Whether IRE1 or other UPR mediators also impact upon these processes in RMS remains unanswered.
Treatment for the RMS with a combination of vincristine, actinomycin-D, and cyclophosphamide (ie. VAC, see above for more details) is the favorable chemotherapy approach [39]. Although recent pre-clinical models and clinical trials assessing the efficacy of alternate chemotherapies associated with less toxic side effects, such as temozolomide (TMZ) have shown encouraging results [351] and while chemotherapeutics can be effective in inducing the death of RMS cells, relapse is a significant challenge. Alterations in proteostatic mechanisms such as autophagy or UPR are known to contribute to chemoresistance in many cancers. Induction of autophagy in response to TMZ has been reported in RH30 ARMs cells [352]. Combination with autophagy inhibitors increased TMZ induced cell death in RH30 ARMs cells suggesting the benefit of combining chemotherapeutics with autophagy inhibitors [352].
Current findings suggest that proteostatic pathways such as the UPR contribute to the progression of multiple cancers in diverse ways. As of yet, few studies have focused on UPR activation within the setting of RMS. Those that have suggested heightened, constitutive UPR signaling in RMS, but the functional consequences are on the whole unknown. Future studies examining the impact of the UPR upon RMS progression and responsiveness to chemotherapeutics are required.
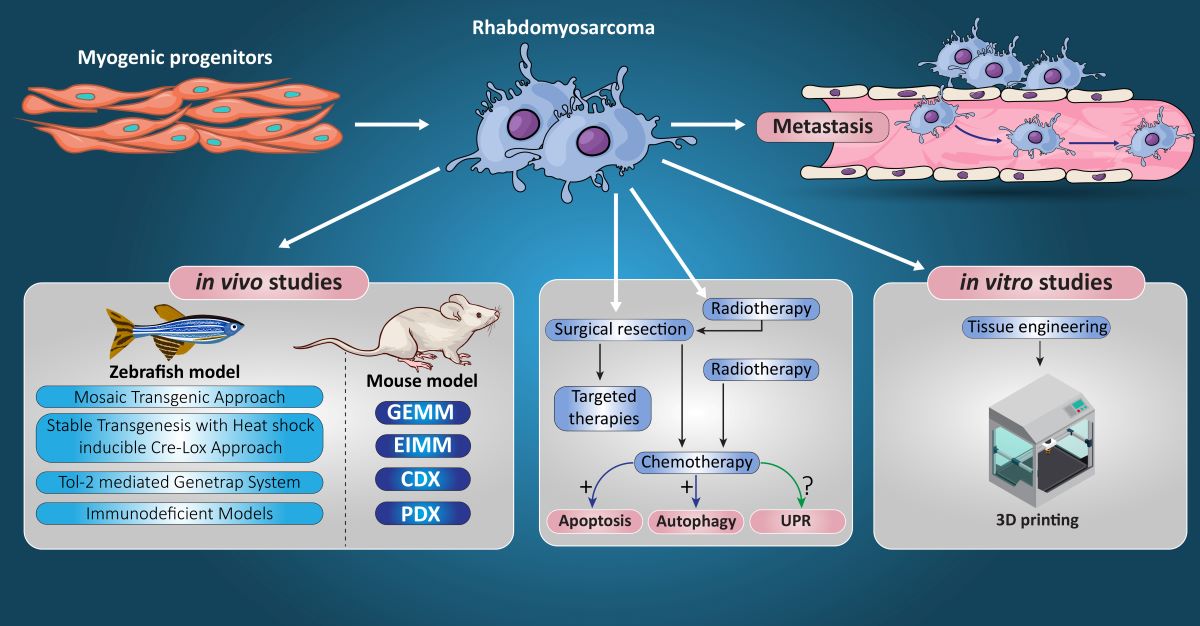
9. RMS In-Vivo Models
9.1. RMS In-Vivo Mouse Models
In general, there are four main groups of mouse models used for the RMS studies (Figure 5) including: 1) Cell-line derived Xenograft (CDXs); 2) Patient-derived xenografts (PDXs); 3) Environmental-induced mouse models (EIMMs); and 4) Genetically engineered mouse models (GEMMs). CDXs are xenograft models in which, specific cell lines are subcutaneously injected in immunocompromised mouse models to produce models that are the same as human tumor origin and are classified into two forms of orthotopic and heterotopic models [353]. These are used to simulate human cancer tissue, and are commonly used in pediatric RMS research [354]. In PDXs, primary tumor tissue is injected subcutaneously in immunocompromised mouse models, to obtain either the cells or tissue pieces (orthotopic or heterotopic) [355]. In GEMMs, specific genetic information (i.e.. typically from an oncogene or tumor suppressor gene) is used to produce the model [356]. According to the investigation purposes, different types of germlines or somatic mutations are used to produce different types of GEMMs [357]. In EIMMs, animals are exposed to the mutagens (like oxidative stress, aging or DNA methylation) to mimic the disease that confers different conclusions about the progress of the mutagenesis [354].
Each of the mentioned animal models is suitable for evaluating the specific types of therapeutic approaches (Figure 6). The CDX model is the commonly used mouse model for investigating the drug mechanism of action; however, these models may fail to recapitulate the disease phenotype. PDX models are adopted mostly for the translational sarcoma research including biomarker investigations, local therapies, targeted therapies, combinatorial chemotherapies, and radiotherapies [358]. To investigate specific research questions, mostly related to carcinogens, EIMM modeling could be used together with CDX mouse models [359]. GEMM models are mostly used in tumor microenvironment screening, tumorigenesis, tumor maintenance and some other applications related to the diagnostics and preclinical testing [357]. In the case of adolescent cancers, utilizing EIMMs is the most powerful approach than other types, while PDX and GEMM models are applied as complementary techniques [357]. Table 6 and Table 7 represent different types of the animal model approaches with their pros and cons.
9.1.1. Genetically Engineered Mouse Models (GEMMs)
There are four different strategies for the generation of GEMMs models (Figure 7) including: a) Spontaneous mutations by targeting the related gene [360], b) Chemical/radiation induced mutation via utilizing some external modulators [361], c) Retroviral transduction [362], and d) DNA microinjection.
In ERMS or ARMS, several genetic aberrations occur in specific and nonspecific nucleotide regions [362]. Due to the numerous variations in the RMS-related gene map, genetically engineered mouse models could be ideal candidates for the in vivo study of this disease [362]. Among different mutations, P53 pathway is one of the most common mutations used for mimicking the cancer case, due to the significant role of p53 from nuclear signaling to apoptosis [354,358]. Different types of models could be generated in this context including the inactivated or mutated p53 models [353], models with overexpression of p53 negative regulators like murine double minute 2 (MDM2) [353,363], and p53 null mice model (that could trigger the Pax3-FKHR chimeric factor to generate ARMS) [364],[365].
Additionally, Ras/Erk pathway, which has a close relation with p53, is also used for ERMS determination. Another study demonstrated the generation of an animal model with loss of p53 through Sonic Hedgehog (SHH) pathway. By this, they demonstrated the importance of local injection of mutagenic agents in the generation of different mutagenic patterns, especially in breeding GEMM models [357].
In the generation of GEMMs mouse models, the genetic background of RMS should also be well-known [357].
GEMM mouse models were used for the assessment of the therapeutic applications of RMS [366]. For instance, NODscid mice, a type of GEMMs mouse models known with a severe combined immune deficiency spontaneous mutation, were used for the evaluation of RMS treatment with vincristine (VCR)-loaded liposomes [366]. The results of this study showed prolonged circulation of the nanoformulation in blood and improved tumor accumulation into the targeted site that led to high therapeutic performances [366].
9.1.2. Environmental-induced Mouse Models (EIMMs)
EIMMs are generated by inducing natural-based mutation using carcinogenic agents and ionizing radiations [367]. While the childhood sarcoma is generated by the genetic variations, the adulthood sarcoma EIMM types are produced via utilizing harsh conditions (to mimic the environmental impacts) [358].
Heavy metals are the most common agents used for studying the toxic effect of environmental pollutant from the mutagenic and carcinogenic perspective. For instance, Gilman et al., confirmed that the intramuscular injection of cobalt and nickel to the rat animal models could lead to the RMS development [357,358]. Pyrrolizidine alkaloids are the other agents that are recognized to contribute the RMS formation [353]. The metabolite of these agents is dehydroretronecine, which is a type of natural toxin [368]. Benzenediazonium sulphate (BD) is another carcinogen agent that induces RMS formation in several mouse models. Swiss mice with subcutaneous injection of BD indicated the formation of RMS, fibrosarcomas, and osteosarcomas [368].
The effects of polycyclic aromatic hydrocarbons on RMS development, which are resulted from cigarette smoke, urban air, pollution or other additional external causes, could be monitored via utilizing Sprague-Dawley rats [364,367]. The CD-1 mice models are used to understand the ionizing radiation effects on RMS formation [361]. β-radiation-exposed mice models demonstrated p53 mutations that lead to RMS development [361]. Combinatorial studies are also performed to understand different co-locations and development patterns of RMS [369]. For instance, to understand the relation between the immune-dependency and tumor progression, immunocompetent mice models are used in which EIMMs models are used to determine the effects of external factors and different radiation types in tumor growth progression [370]. Despite a lot of research being done, there still remains not enough information about screening the effects of the environmentally-related parameters on RMS.
9.1.3. Cell-line Derived Xenograft Mouse Models (CDXs)
In comparison with other animal models, CDX mice are low cost and highly available models (near 70% among other animal models), which are feasible for in vitro tests [357]. However, only very aggressive types of RMS could grow in in vitro conditions and the adaptation of these cells with prolonged viability is required for the tumor stroma reflection. Thus, other types of animal models are generally used as complementary approach with CDX, especially for the therapeutic predictions [362].
In general, excess amounts of fetal calf serum (FCS) are used for the invitro cancer cell culturing for CDX mouse models [354]. Some of the common cell-lines derived mouse models used for the CDX studies are Rh30 (alveolar rhabdomyosarcoma—ARMS), A204 (embryonal rhabdomyosarcoma—ERMS), HS-SY-II (SySa), TC71 (Ewing Sarcoma—EwS) and KHOS (osteosarcoma—OS) [371]. Although, all of these cancer cells are not well-adapted in 2D cell culture conditions, they could decrease the activity in in vivo tests [357].
In a study, CDX and PDX models were used to indicate the therapeutical safety of antibody against B7-H3 receptor to treat solid tumor malignancies including RMS as pediatric cancer type [355]. Kendsersky et al., demonstrated that the CDX and PDX models should be used together for screening the therapeutical applications and using one type of animal models could not be enough for the investigation of such therapeutics effects [363].
9.1.4. Patient Derived Xenograft Mouse Models (PDXs)
In patient-derived animal models, tumors are extracted from the patients and directly inserted into the immunodeficient humanized mice. These models have various advantages aiding in the deeper understanding of actual cancer biology due to their ability to simulate the natural cancer progression. Mostly studied PDX models for RMS are related to musculoskeletal malignancies. For instance, Igarashi et al., generated successful animal models as orthotopic PDX models for the RMS study. However, collecting the samples and transplanting them into mice models are challenging [357,361,372]. Due to rarity of some sub-types of RMS, logistic challenging to access these samples is the most problematic case for this modeling approach [356].
Some models are generated and published as in the repositories or some data banks like https://www.europdx.eu/. These models obtained from the patient-derived tissues or primary cells inclusion into mouse models continue to differentiate to online platforms for the academic platforms and research organizations [356]. Despite the impressive success of the PDX animal models, necessary carefulness should be considered during the researches, due to the difficulties to have the actual patient samples and also their limited number, especially for the rare diseases like RMS [359].
Lu et al., studied patient-derived xenograft models in musculoskeletal malignancies by generating PDX mice models. In their study, the main aim was to struggle with the appropriate animal model usage due to the complexity and heterogeneity of musculoskeletal malignancies. The models were generated after receiving the samples from the patient as fresh tumor tissues and were preserved in fetal bovine serum (FBS) and transplanted through mice within 2 h for the highest yield. Soft tissue sarcoma was analyzed using PDX models and reliable results obtained in tumor growth with stable genomic alterations [373]. PDX model application has more successful outputs in the soft tissue sarcoma due to transplantation acceptance of the animals [353]. The stable gene alterations exist in PDX models lead to determine responsible genetic paths with a deeper understanding such as preserved genetic variations both in bone and soft tissue [373].
9.2. Zebrafish Models to Study RMS
In addition to mouse in vivo models, zebrafish models have been employed to study the development, histology, pathogenesis, tumor progression, metastasis and drug screening of the RMS [374]. Since ERMS is the most dominant type of RMS in humans, and it was revealed that fusion gene-negative ARMS are similar (both biologically and clinically) to ERMS, the initial models in zebrafish were mostly designed as ERMS subtype. ERMS is characterized by the mutations in the genes for RAS GTPases, MYOD1, and FGFR4. In fusion-positive RMS, the overexpression of fusion protein PAX3/PAX7-FOXO1, caused by the chromosomal translocation, leads to more aggressive type of RMS, or 85 % of ARMS [375,376,377]. These two RMS models can be recreated in the zebrafish either with genetic modifications or tumor cell transplantation [374].
To study RMS in vivo, zebrafish hold great advantages compared to other animal models. They are small and can produce numerous offspring from a single breeding, consuming less effort and are economically cheaper compared to the commonly used murine species [378]. A highly attractive trait is their ability to generate tumors with similar histological and genetic features to humans [379]. They also provide the opportunity for high-throughput drug screening by submersing larvae in a bath solution of the drug of choice, as well as transplanting primary tumors (obtained from the patient) into immunocompromised lines [375]. The translucent appearance of zebrafish provides the ability of imaging the tumor growth, shape, size and renewal, relapse over early larval stages via live in vivo confocal imaging in combination with the fluorescent reporter lines. Moreover, the use of zebrafish mutant lines lacking pigmentation altogether, such as the Casper line [380,381], allow highly tractable observations of tumor growth over larval to senescent stages of zebrafish development. This presents unique opportunities for tracking molecular markers, histogenesis and metastasis of cancer cells, as well as drug screening [375,382]. In particular, zebrafish ERMS has shown histological and molecular mechanisms similar to the human ERMS. This model uncovered the pathways that regulate the RMS growth, its propagation and self-renewal. Furthermore, transgenic zebrafish can be generated with the help of specific promoters added to the muscle for expressing the fluorescent proteins, which can be conditionally activated uniquely at different stages of muscle maturation, thus marking the state of the muscle cells [383].
Despite clear benefits of the zebrafish model, the zebrafish has several drawbacks. When the RMS model is generated via tumor transplantation, limited cell numbers are transferred to the larval fish, which are grown at 28°C. Furthermore, xenograft fish models are subjected to the drug screening experiments before the elimination of these cells via the immune system (usually around 10 days of their life), which prevents the visualization of drugs and cellular events during the tumor propagation and metastasis during the long term of experiments [384]. Thus, it seems that zebrafish models are most beneficial for the short-term treatment studies but may suffer for the longer-term experiments. In other words, their response against the therapeutic components could be stated when fishes are in the drug-container dish that makes it hard to estimate the drug uptake route. In this case, adult zebrafish, due to their unique features like low cost, optical clarity, good fecundity, and capability of performing high drug throughput and tumor progression studies, could be ideal candidates to be used as cell-transplantation models [380].
Currently, immunodeficient zebrafish models have been developed to eliminate the mentioned disadvantages of zebrafish for the xenograft RMS studies [380]. Based on this, in the following sections we will describe different methods used for the generation of RMS model in the zebrafish, including the mosaic transgenic approach, heat-shock inducible Cre-Lox and Tol2 mediated gene trap systems (Figure 8) and the immune compromised models for the tumor transplantation.
9.2.1. Mosaic Transgenic Approach
In this method, one or more genes were injected into zebrafish embryo at its one cell stage, to initiate the RMS tumor development in several parts of the fish. The most studied approach to generate ERMS in the zebrafish is to express constitutively active Kirsten rat sarcoma viral oncogene homolog (KRAS) (mutated version called KRASG12D) gene using a recombination activating 2 (rag2) promoters [375]. The rag2 is expressed in the progenitors of B and T cells, satellite muscle cells and myoblasts, but not in the multi-nucleated muscle fibers [374]. Once rag2-KRASG12D and rag2-Green Fluorescent Protein (GFP) constructs were linearized and co-injected into the embryos in one cell stage, the expression of KRASG12D was validated with GFP imaging at 10 days of post-fertilization, forming KRAS-driven ERMS [374].
For the RMS, labeling of more than one cell type and tumor niche were also achieved. Transgenic zebrafish expressing myogenic factor 5 (myf5)-GFP, myogenin-H2B-mRFP and mylpfa-lyn-Cyan was able to show the sub-population of developed ERMS tumors, indicating GFP fluorescence for tumor propagating cells (TPCs) and cyan fluorescence for differentiated cells. With this approach, tumor subpopulation and heterogeneity were labeled based on the activation of different promoters (myf5, myogenin-H2B and mylpfa) of different stages in the muscle development [385]. In another case, the expression of different promoters in the muscle development was achieved with the co-injection of rag2-KRASG12D: myogenin-H2B-RFP: mylz2-lyn-Cyan into mutant fish expressing myf5-GFP at one cell stage (Figure 9A). The heterogeneity in the tumor was labeled in ERMS cells differentiated early, mid and late phases at 16 days of their life (Figure 9B-C). After the serial cell engraftment to the syngeneic fish, only myf5-GFP-positive cells were transferred ERMS to the following recipient (Figure 9D). ERMS tumors hold similar histology in the primary tumor and the second recipient (Figure 9E-J) [386]. In addition to tumor growth, several other parameters were studied with the zebrafish KRAS-driven RMS tumors, which are outlined in Table 6.
Cadherin 15 (cdh15) promoter, which is expressed in the muscle satellite cells, was also utilized to generate KRAS-driven ERMS in the zebrafish. The cdh15-KRASG12D and mylz2-KRASG12D as well as rag2-KRASG12D were injected to double transgenic zebrafish embryo at single cell level. No histological difference was observed between rag2 and cdh15 promoters in KRASG12D expression, showing the potential of early muscle progenitor cells, which consist of mostly undifferentiated myoblast-like cells, to develop ERMS. Injection of the mylz2-KRASG12D into the fish resulted in the tumor propagation, which was similar to the mature skeletal muscle [387].
Once primary tumors were generated in the donor zebrafish successfully, cells can be harvested and transplanted to the syngeneic fish to study the tumor propagation and volume. In this assay, ERMS cells expressing fluorescence protein were collected from donor fish and sorted based on the fluorescence-activated cell sorting (FACS) technique. The sorted tumor cells were then injected to the recipient adult fish, either with intraperitoneal or intramuscular ways, imaged over weeks and used for the different analyses like cellular pathways, tumorigenesis and drug screening [388].
9.2.2. Stable Transgenesis with Heat-Shock Inducible Cre-LoxP Approach
The Cre-loxP approach is widely studied for gene manipulation composed of Cre recombinase enzyme and a pair of short nucleotide sequence called LoxP (5’-ATAACTTCGTATA-GCATACAT-TATACGAAGTTAT-3’). When flanking LoxP sites are recognized by the Cre recombinase, the enzyme cuts and recombines the LoxP sites, resulting in excision, insertion or inversion of genes located between two loxP sites. This strategy was applied in the zebrafish model for introducing KRAS-driven ERMS model. Transgenic fish expressing β-actin-LoxP-EGFP-STOP-LoxP-KRASG12D and heat-shock protein 70 (hsp70) with Cre (called hsp70-Cre) translates enhanced GFP (EFGP) ubiquitously (Figure 10). When Cre was encoded by the heat-shock treatment via hsp70-Cre, it recombines two LoxP sites, removing the EGFP and expressing KRASG12D, resulting in KRAS-driven ERMS tumor in zebrafish. Double transgenic zebrafish were able to express EGFP for 24 h (Figure 10B-C) and 44 days (Figure 10D-E) of post-fertilization without heat-shock. Upon heat treatment (37°C, 1 hr), the Cre-mediated excision was made for the expression of KRASG12D showing the tumor formation (Figure 10F-G). Surprisingly, non-heat-shocked transgenic zebrafish also formed tumors with low frequency, due to the hsp70 activation during the fish growth (Figure 10F-H) [389].
9.2.3. Tol-2 Mediated Gene Trap System
Gene trap technology provided by Tol2 transposon system was used to generate ARMS in the zebrafish, via expression of PAX3/FOXO1 oncogenic fusion protein, to study the in vivo development and tumorigenesis of ARMS. In this transgenesis system, synthetic transposase messenger RNA (mRNA) and Tol2 transposon, promoter and florescent protein containing transposon plasmid are co-injected into the fish embryos. The donor plasmid containing Tol2 was cut from the Tol2 sites, and inserted into zebrafish genome, creating stable transgenesis [390]. This model was used to study a novel target of PAX3/FOXO1, called HES3 transcription activator. To generate ARMS in zebrafish, human PAX3/FOXO1, linked with additional GFP or mCherry via viral 2A sequence, was added into the genome of the fish via Tol2 transposon-based system (Figure 11A). Fusion-positive tumors expressing PAX3/FOXO1 were observed for up to 19 weeks using florescence imaging and tracking the morphology. Several promoters were used to express PAX3/FOXO1 in zebrafish, among them CMV, β-actin and Ubiquitin promoters were able to generate Primitive neuroectodermal tumors (PNET), RMS and sarcoma, respectively (Figure 11B-D). It was also stated that PAX3-FOXO1 fusion and PAX3 alone exhibited different characteristics on the embryonal development of the fish. HER3 (zebrafish ortholog of HES3) expression was observed on the fish injected with the PAX3-FOXO1 vector. The expression of HES3 was linked with the pro-tumorigenic events in mammalian cells, which are also linked with the tumor progression and reduced survival in patients with the RMS tumors, thus, HES3 could be a novel target for the RMS treatment [391].
9.2.4. Immunodeficient/Compromised Zebrafish Models to Study RMS
Tumor grafting to adult fish is a very powerful tool to study tumor expansion, propagation, and recurrence. Using adult fish also provides injection of a higher number of cells. However, xenograft transplantation often fails due to the activation of immune response of the fish, resulting in loss of tumor cells. For the allograft transplantation or transferring zebrafish RMS tumor cells from one individual to another one with matching immunity, transgenic lines are required, which are from syngeneic background with more than four generations [392]. In this regard, several methods were developed to eliminate the engrafted cell rejection, for instance, using gamma rays could eliminate the immune rejection for about 20 days, whereby dexamethasone can eliminate it up to 30 days during which a sustained drug dose is required [392].
Transgenic zebrafish with compromised immune systems were generated via reduced B and T cells activity. In this approach, rag2E450fs homozygous AB strain mutant zebrafish were generated for the ERMS engraftment [392]. The rag2 promoter is expressed in satellite cells and myoblasts, as well as progenitors of T and B cells. To generate such mutant, gene inactivation and engineered zinc-finger nucleases were used to alter the rag2 When α-actin-RFP expressing zebrafish were used as donors, the immune compromised recipients were able to hold the engrafted cells, even if they were transformed to multinucleated muscle fibers; however, the wild type counters did not achieve fiber formation in the 30-day experiment. When myf5-GFP:mylpfa-mCherry double positive transgenic ERMS tumor were transplanted to the immune compromised recipients intraperitoneally, different cell types as well as tumor propagating cells were observed, which showed similar histological features to the donor zebrafish ERMS [392].
For further analysis of the dynamic and heterogeneity as well as the propagation of ERMS, rag2E450fs homozygous transparent Casper fish were utilized. The Casper zebrafish lack melanocytes and iridophores, making them translucent compared to the AB strain. The ERMS tumors growing in CG1 type zebrafish were successfully engrafted to the mutant Casper fish and it was revealed that the histology of the tumor was protected against the primary version. To image the dynamic of the ERMS tumors, KRAS-induced ERMS was generated in triple transgenic zebrafish with expressing myf5-GFP, myogenin-H2B-mRFP, and mylpfa-lyn-cyan (Figure 12A). Later, ERMS tumors were transplanted intramuscularly to the 3-month-old Casper fish with flk1-mCherry, rag2E450fs expressing (Figure 12B). Thus, tumor propagating cells were illustrated by the GFP florescence, while differentiated cells were labeled with the AmCyan (Figure 12C) [385].
The xenograft ERMS transplantation was achieved using the mutant Casper fish with the lack of T, B and natural killer cells. The immunocompromised Casper zebrafish were generated though inducing deficiency in protein kinase DNA-activated catalytic polypeptide (prkdc) and interlukin-2 receptor gamma a (il2rga) via crossing the mutant prkdcD3612fs/D3612fs and il2rgaY91fs/+ in adult Casper (roya9/a9 and nacrew2/w2 mutants). Homozygous inbreeds prkdcD3612fs/D3612fs and il2rgaY91fs/Y91fs were chosen as prkdc-/-, il2rga-/- fish. When GFP-expressing cancer cells were intraperitoneally injected, zebrafishe were able to hold the cells, and tumors were growing up to 4 weeks with a death ratio less than 15%. When RMS cells were transplanted to the immunodeficient zebrafish and mice, both recipients exhibited similar histological profiles. The mutant fish was also able to hold the patient-derived RMS tumor within 4 weeks at 37°C (Figure 13A). Translucent prkdc-/-, il2rga-/- Casper fish also provided the tracking of RMS cells at a single cell level over a week. When a drug cocktail including combination of temozolomide (TMZ) and olaparib was orally administrated to the EGFP-labeled human-RMS engrafted immunodeficient Casper fish, the tumor size decreased (Figure 13B-C). At the end of the drug treatment for 28 days, animals were sacrificed, and tumors were labeled with histology analysis via Hematoxylin and Eosin (H&E) (Figure 13C), Ki67 staining for the cell proliferation (Figure 13D) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay for the apoptosis (Figure 13E). According to Figure 13C, combination therapy showed elimination of tumor mass compared to TMZ or olaparib alone. The histological analysis showed the overall declines in cellularity in animals administrated with TMZ or olaparib alone and almost complete loss of cell proliferation after three cycles of combination therapy (Figure 13C-E) [380].
10. Tissue Engineering Basics
The engineering and manufacturing of replacement tissue is specific to the tissue engineering field. Recently, tissue engineering has obtained much attention in the field of medicine, as an alternative to grafts or transplants. Tissue engineering uses a patient’s own cells to generate a functional tissue or organ [393]. Overall, there are three models for tissue engineering, including acellular scaffolds, scaffold-free cell-only designs, and hybrid cellularized scaffolds. The emergence of 3D printing has made significant progress in the field of tissue engineering, as 3D printing allows the fabricated tissue to include multiple cell types [394,395,396], biomaterials [397,398] and growth factors [399]. A pressing issue remains in medical tissue engineering, notably how fabricated tissues are evaluated in terms of performance and compatibility. The gold standard has traditionally been the use of histological approaches to evaluate the structure of the engineered tissue. However, histology is an invasive semi-quantitative procedure that can destroy the tissue. Hence, it is vital to develop non-invasive imaging techniques, utilizing cooperation among biologists, physiologists and radiologists.
10.1. Application of 3D Printing in Muscles and Rhabdomyosarcoma Tissue Engineering and Treatment
In recent years, Additive Manufacturing (AM) or 3D printing has been widely used in different fields such as aerospace, automobile, construction, and medical science. In the medical setting, 3D printing can be used for three different purposes, including tissue engineering, implants, and surgical planning prototypes. A number of recent studies have highlighted the potential of 3D printing applications in medical tissue engineering [400,401,402], implants [403,404], and surgical planning prototypes [405,406] for the muscles and RMS modeling and treatment.
As a gold standard, surgery and resection of the tumor is the most reliable clinical treatment for RMS. Skeletal muscle has a robust ability to regenerate and remodel following injury largely due to the presence of muscle progenitor cells called satellite cells. However, if the resected damage to muscle is larger than the capacity of regeneration, or the size of the primary tumor is particularly large, the structural and functional deficits occur as fibrosis and sometimes fibro-fatty tissue infiltrate muscle tissue with the expansion of resident Fibro–adipogenic progenitors (FAPs). Thus, there are times when it is necessary to assist muscle regeneration with adequate functionality and structure at the tumor site. 3D printed scaffolds could help to create platforms with the complex microstructures to guide cell alignment and fusion, and consequently to regenerate replacement tissue for resected tissue [400,401,402]. Moreover, the application of either natural or engineered biomaterials in the printed scaffolds not only could increase tissue formation but also could improve the functionality of the regenerated tissue [407,408,409,410,411]. For example, Kim and Kim [410] used a collagen-based bio-ink for the skeletal muscle tissue regeneration. They produced 3D scaffolds made of C2C12 myoblast-laden bio-ink by using extrusion printing with uniaxially aligned topographical cues. In vitro testing revealed a high degree of cell-alignment and efficient differentiation. In another study, Kang et al., [412] developed an electroconductive C2C12-laden bio-ink composed of a phenol-rich gelatin (GHPA) and graphene oxide (GO) for the production of a 3D printed scaffold for muscle tissue regeneration. In vitro analysis showed that the myogenic differentiation of C2C12 myoblasts was spontaneously facilitated without the inclusion of myogenic differentiation-inducing factors. In recent work, Bilge et al., [413] used a 3D printed electroactive scaffold for the skeletal muscle tissue engineering. The scaffolds were made of poly(ɛ-caprolactone) (PCL) combined with carbonaceous material (CM) to incorporate the electrical conductivity. This group seeded the printed scaffolds with C2C12 myoblasts and subjected it to an electrical stimulation during in vitro test. Their results confirmed enhancement in myotube formation in electroactive scaffolds compared to non-conductive scaffolds. It was also observed that myotube formation and myotube maturity were significantly increased in the CM group following electrical stimulation (Figure 14) [413].
Although 3D printed scaffolds are useful in tissue engineering, they do have some limitations. Generally, implantation of the printed scaffolds, especially hydrogel-based ones, is a difficult process because they are not suturable and do not adhere properly to the host tissues [414,415]. Moreover, for the complex-shaped injuries, it is possible that the printed scaffold will not match the defect site exactly and consequently, a gap or overlap with the surrounding tissue will occur. Moreover, 3D printing of the scaffolds requires pre-processing operations, such as taking images or impressions of the injured area, creating a 3D model from the 2D images, and generating instructions for printing the desired shape, which are normally time-consuming processes. In addition, printing time varies based on the employed method, material, and complexity of the printed shape. In urgent cases, such as those resulting from traumatic injury, the initial surgical intervention should be done quickly after the accident [414]. Considering the time needed for preparing the scaffold, secondary surgery would be required to implant the scaffold into the injured area. To overcome this problem, some researchers have tried to develop a mobile bioprinter with in situ printing ability [414,416,417,418]. Russel et Al, [414] proposed a mobile extrusion-based bioprinter for in situ printing in the case of volumetric muscle loss (VML) (Figure 15).
This group employed a gelatin-based hydrogel, printed it directly into the defect area and cross-linked in situ. The suitability of the used materials was confirmed by several in vitro and in vivo tests. The results of hematoxylin and eosin (H&E) staining of the harvested samples from murine models with VML injuries showed adequate adhesion of the printed gel to the surrounding tissues. Moreover, there were no signs of rupturing in the structure of the gels, which confirmed its suitable mechanical properties (Figure 16) [414].
Another application of 3D printing in medical science is the production of patient-specific implants, and there are several reports of this approach used following treatment of RMS [403,404]. For instance, O’Sullivan et al., [404] produced a customized 3D printed eye cover for 18-year-old man with left maxillary ARMS utilizing a biocompatible material. They utilized a 3D scanner to map the surface of the patient and then created a 3D model of the implant. With the advantages of AM, the entire process from beginning to the end of printing was less than 72 hr. Thus, AM can assist to create custom-built implants for the patients in palliative care to meet rare and difficult clinical challenges [404].
As noted above, the main clinical treatment for RMS is tumor resection via surgery. Complete tumor resection is normally difficult to achieve and any error in the resection surgery may cause damage to the neighboring tissues and long-term sequelae. Thus, development of surgical skill is imperative. To this end, either physical models or surgical planning prototypes (i.e. phantoms), can be used. In the recent years, several studies have been done on the manufacturing phantoms using AM [405,419,420,421,422,423]. Among the AM methods that have been used in different studies, Material Jetting, stereolithography (SLA) and fused filament fabrication (FFF) are the most common methods used for producing phantoms [424]. The advantages and disadvantages of these methods are presented in Table 8.
The additively manufactured phantoms could be used for two different goals: visualization of the soft tissues and mimicking the modeled tissue for its shape and mechanical properties. In the first goal, the phantoms are used to give some insight about the tissue geometry to the surgeons before the operation. In this case, mechanical properties of the phantom are not important, and the accuracy of the tissue geometry is the only matter of fact [424]. However, for the second goal, the produced phantom should have both mechanical properties and geometrical parameters the same as the targeted tissue [405,424]. The FFF method is a cost-effective type of additive manufacturing, which is suitable for producing phantoms used for visualization applications, since the common material used in this method are rigid. However, most of the produced phantoms are mono-material and mono-color, which are not suitable to get a good insight from different parts of the tissues [405,406].
Recently, some researchers have combined the advantages of the AM with the other manufacturing methods to produce multilateral and multi-color phantoms with mechanical properties similar to the live soft tissues. Tejo-Oreto et al., [406] created a soft surgical planning prototype for a biliary tract RMS. To this end, they used Computed Tomography (CT) to obtain 2D images from liver that were then overlapped to reconstruct 3D model (image segmentation). To distinguish between different anatomical structures, divers parts of the prototypes are highlighted with different colors as follows: (1) red for the hepatic artery, (2) purple for the portal vein, (3) blue for the vena cava, (4) green for the gallbladder, and finally, (5) the brown color corresponds to the tumor (Figure 17A) [406]. According to the 3D models of the tissue that were created from the CT images, the inner parts of tissue and mold were designed to produce the main body of the tissue (Figure 17B). Since the surface quality of the inner parts was important, the inner parts were produced by the SLS, while the molds were produced by FFF. Then, the inner parts were located in their position inside the mold and phantom was produced using the casting method (Figure 17C). They suggested that 6%wt PVA (poly vinyl alcohol)/1%wt PHY (Phytagel)-1FT (Freeze-Thaw cycles) and 1%wt agarose have the highest similarity to the liver tissue in terms of the mechanical properties. They also have investigated the hardness and mechanical properties of different composites using Dynamic Mechanical Analysis (DMA) tests and Shore hardness tests to obtain the optimum combination for mimicking the liver tissue. The CT result showed that the produced phantom geometry had less than 1% difference with the designed model and the total cost to produce phantom was relatively lower than other technologies [406].
Besides two areas that phantoms can help for the skeletal muscle and RMS modeling and treatment, recently, phantoms have been used for the patient educations as well. Previously, to increase the interaction between the doctors and patients or even their families, CT or MRI images of the tissues or tumors used to be employed. Although it was fruitful, using 3D prototype will lead to better results in comparison with the 2D pictures and could aware patients about their treatment progress [425,426,427].
Additive manufacturing or 3D printing helped to resolve a lot of limitations such as reproducibility, accuracy, and precision in the manufacturing. However, 3D printed structures despite native tissues are not active in response to the external stimuli, i.e., dynamic 3D constructs [428,429]. In recent years, application of smart materials (stimuli-responsive material) in 3D printing, leads to four-dimensional (4D) printing as a new technology that can produce smart structures that are able to respond to external stimuli. Thus, the new dimension for 3D printed structures will be time [429]. Application of biocompatible smart material in 4D printing made it possible to use this technology in different fields of biomedical application such as tissue engineering [430,431,432,433,434,435,436,437,438], implants and medical devices [439,440], and soft robotics [441,442]. Some studies have been done in this field for engineering of different tissues like bone [430], neural conduits [431,432], vascular structures [433,434] and muscle tissue [435,436,437,438]. 4D printed scaffolds, which can undergo morphological changes in a pre-planned way, could be beneficial for the muscle tissue engineering and RMS treatment [438,443]. For instance, Constante et al., [443] have employed 4D printing to fabricate hollow scroll-like cellular structures with a specific orientation of myoblast. They have combined extrusion printing (methacrylated alginate) and melt electro writing (polycaprolactone) to fabricate the shape-morphing scaffold. The external stimuli in this study were changes in Ca2+ ions concentration in the surrounding medium since the methacrylated alginate gel has high sensitivity to this ion. First, they printed planar scaffold using methacrylated alginate gel by extrusion printing, then, added polycaprolactone fibers in the printed scaffold using melt electrowriting to add enough mechanical support to the whole structures of scaffold. After photo crosslinking of the hydrogel, the C2C12 mouse muscle cells were seeded in aqueous media into the scaffold. Then, by changing the Ca2+ ions concentration in the medium, the printed structure starts to fold to form a tubular structure with the enclosed cells. The results of in vitro study show that using a scrolled bilayer scaffold will help to increase the viability and proliferation of myoblasts cells. Moreover, it is possible to control the cells orientation very well by adding the patterned surface generated by PCL fibers, which is hard if not impossible to do on the hydrogel layer without fibers [443].
In summary, 3D printing has been used for the skeletal muscle and RMS modeling and treatment by manufacturing muscle tissue engineering scaffolds, patient-specific implants, and phantoms. In the case of tissue engineering scaffolds, in addition to microstructure design and printing techniques employed for the production of scaffolds, bio-ink materials are also extremely crucial for the improvement in their performances. The synthesis and development of bio-inks with higher bioactivity, biocompatibility, mechanical properties and electroconductivity will help to achieve this goal. In the case of surgical planning tools, development of new methods and material, which can mimic the modeled tissue while reducing the production cost, is desirable. Moreover, material that can mimic the skeletal muscle and the tumor mechanical behavior will help the surgeons to obtain better experience regarding the resection process before the main surgery.
There have been very limited studies about the application of 4D printing in the field of muscle and RMS modeling and treatment. It should be noted that this technology is still in its early stages. One of the important areas of research in 4D bioprinting is the development of smart bio-inks. Most of the available smart material are triggered by stimuli like changes in temperature and pH, that are not suitable for biomedical application [444]. In addition, development of new materials may need to advance the printing methods technologically as well. In the case of RMS and muscle tissue modeling and treatment, application of 4D printing could help in development of phantoms where their mechanical and rheological properties can change in response to the external stimuli. This can help the surgeons to obtain a better insight of the tissue or tumor behavior during the resection surgery.
One feature of RMS cells is their invasion and migration into the neighboring and distant tissues. Therefore, understanding the interactions involved in mediating metastasis of RMS cells is of importance. The experiments using 3D cell culture of RMS have shed some light on the cell-cell associations involved in tumor invasion [445]. To introduce tumor heterogeneity, malignant ERMS cells were co-cultured with normal human skeletal muscle myoblast (HSMM) using a cell sheet strategy. When the number of ERMS cells is lower compared to HSMM cells, cell sheet disruption occurs. However, sheets containing only ERMS or HSMM are intact. Further investigations revealed that malignant cells are able to interfere with HSMM cell alignment. Hence, ERMS cells negatively affect their surrounding tissues and cells due to their aggressive behavior. Furthermore, as muscles are affected by invasive ERMS cells, targeting them can be of importance in inhibiting cancer metastasis (Figure 18) [445].
The development of 3D culture systems also allows us to evaluate impact of chemotherapy and other therapeutic modalities in the treatment of RMS. Recently, we developed a 3D culture system for ARMS containing thermally cross-linked collagen disc and ARMS cells that have similar biochemical parameters of tumor extracellular matrix (ECM). This method is able to determine the potential of chemotherapeutic agents in ARMS suppression. Furthermore, we can evaluate apoptosis and autophagy induction in ARMS by chemotherapeutic agents in this model (Figure 19) [446].
11. Conclusion and Perspectives
Despite advances in RMS therapy, drug resistance and tumor recurrence continue to be long-standing clinical issues. Molecular research into RMS pathology has identified a strong link between genetic and epigenetic alterations and cell growth, proliferation, differentiation, and apoptosis. The identification of PAX-FKHR fusion genes has shifted our research focus with the goal elucidating pathways that lead not only to the chromosomal translocations, but also how these oncogenic fusion proteins alter the cell phenotype and can be exploited pharmacologically. This approach could allow for the identification of biomarkers that could be applied to individualize the targeted therapy and improve RMS prognosis.
The integration of new therapeutic agents into the currently recommended treatment regimens seems promising. The aforementioned studies have already led to multiple large-scale clinical trials. The upcoming results will show whether precise targeting of apoptosis can be successfully transferred into a clinical setup. However, numerous agents targeting CDK4–CDK6, MEK or TRK might have an even larger impact on the treatment efficaciousness. Moreover, the recent discoveries of microRNAs during RMS differentiation, along with the role of cell surface receptors preferentially expressed in the RMS cells may enhance personalized therapy through the use of antagomirs or monoclonal antibodies, respectively.
At the present time, there are few efficient therapeutic alternatives available for the RMS patients and the only standard treatment protocol for three subgroups of RMS including low, intermediate, and high risk is the VAC-triple therapy. The VAC regimen consists of an alkylating agent such as cyclophosphamide or ifosfamide along with vincristine and dactinomycin (actinomycin-D) [297,298,299,447]. Considerable research efforts have been made to improve the treatment outcome of pediatric metastatic RMS by adding one or more anticancer compounds to the standard VAC chemotherapy; however, to date, none of the new regimes have been more effective than the VAC protocol [448]. The IRS-IV study tested VAC (vincristine, dactinomycin, and cyclophosphamide) therapy compared to the VAC combined with vincristine, topotecan, and cyclophosphamide (VAC/VTC) in patients with intermediate-risk RMS. There were no significant differences in the effect of VAC against VAC/VTC between the risk groups [449]. Another study investigated VAC therapy compared to the vincristine and dactinomycin, ifosfamide (VAI) and vincristine, ifosfamide, and etoposide (VIE) in the patients with intermediate-risk RMS. This study showed that there was no significant difference in overall 3-year survival rate among patients who received VAI and VIE and those who received only VAC regimen [299]. In a clinical trial by the International Society of Pediatric Oncology (SIOP) in Europe, 457 patients aged 14 years with high-risk nonmetastatic soft tissue sarcoma were treated with either ifosfamide, vincristine, and dactinomycin (IVA), or IVA plus carboplatin, epirubicin, and etoposide (ICE) for 27 weeks. They reported no survival advantage but toxicity for this treatment protocol [450]. Other studies assessed VAC therapy followed by pre-administration of ifosfamide/etoposide (IE) [451], vincristine/melphalan (VM) [451], and ifosfamide/doxorubicin (ID) [451] in the patients with high-risk RMS. They found that the overall 3-year survival rate with the IE, ID, and VM-containing regimen was noticeably better than VAC regimen alone [451]. The Children's Oncology Group evaluated irinotecan alone and in combination with vincristine in intermediate-risk patients with RMS. They were unable to document any improvement in the survival rate in RMS patients treated with irinotecan plus vincristine versus irinotecan alone [452]. However, the vincristine, irinotecan, and temozolomide combination has shown synergistic antitumor activity against RMS, which is now the standard treatment protocol for children and adults with relapsed or refractory RMS in Europe [453]. The bottom line is that multidrug resistance (MDR) often occurs after prolonged chemotherapy, which in turn leads to refractory cancer and tumor recurrence. Therefore, proliferation-inhibiting and apoptosis-inducing in MDR tumor cells could be a new weapon for preventing the development of MDR in cancer therapy. Autophagy, a self-degradative process, generally arises during the treatment of multidrug-resistant tumors. In this regard, genetic and pharmacological autophagy inhibitors are used along with therapeutic agents in various malignancies including B cell lymphomas [454], colorectal cancer [455,456], myeloid leukemias [457], ovarian cancer [458], pancreatic cancer [459,460], renal cancer [461], bladder cancer [462,463], cervical carcinoma [464], and lung cancer [465,466,467], which in turn lead to the tumor growth impairment and therapeutic sensitivity improvement. On the other hand, as shown in Table 5, as a double-edged sword, autophagy may lead to the death of MDR cancer cells in which apoptosis pathways are inactive. Therefore, more investigations about the combination of autophagy modulators with therapeutic agents are urgently needed in the treatment of various cancer types. To date, numerous studies have been carried out by different combinations of autophagy inhibitors and activators, and chemotherapeutic drugs. Nevertheless, studies on the application of this treatment strategy for combatting the development of chemoresistance are very limited for RMS.
UPR is endogenously upregulated in RMS [343,344]. Thus, targeting UPR could be one the promising future therapeutic approaches in this rare childhood cancer. As an example, targeting IRE1-sXBP1, or PERK axis of UPR using MKC8866 or PERK inhibitors in combination with apoptosis inducing chemotherapy may improve the efficiency of chemotherapy in this deadly disease. On the other hand, the UPR is a regulator of both apoptosis and autophagy [328], therefore, targeting UPR could be a good strategy to potentiate the effect of chemotherapy compounds that affect these pathways.
One of the major challenges in cancer investigations, including RMS, is tumor stiffness during the tumor growth. As tumor cells grow, they remodel their environment, by altering the protein content and nearby cell type in their ECM [468]. This feature associated with the changes in microenvironment stiffness, results in altering the cellular behaviors. Besides, autophagy is also involved in ECM mechanotransduction, thus it was suggested that increasing autophagy is recorded in normal mammalian cells with the increased matrix stiffness [469]. This brings the idea of autophagy involvement in the mechanical regulation of cancer cells. We believe the need for modeling of 3D tumor environment with different ECM stiffness has emerged to mimic tumor environment to test the changes in autophagy, and then the chemotherapy response in RMS. Although there are animal models for the RMS including mice and zebrafish, it is hard to mimic the exact mechanical environment in animals. It was also stated that, measuring the tumor stiffness in the animals are hard, complex and complicated methods [470]. Unlike the disadvantages and complexities of the animal models, using 3D culturing techniques with hydrogels provides easy and better understanding of the studies related to the cell behavior and drug screening under different mechanical stressors in RMS models.
Author Contributions
Ali Zarrabi led the team for bioengineering, zebrafish and mouse model. Philip Kawalec, Micah Sommer prepared clinical part of the paper under David Perrin supervision. Mahboubeh Kavousi, Mohammadreza Bolandi, Mohsen Taheri, Bhavya Bhushan, Peyman Koleini prepared autophagy part under Joseph W Gordon and Saeid Ghavami supervision. Serap Sezen, Atefeh Zarepour prepared Zebra fish under Benjamin Lindsey supervision. Filip Majach, Jakub Rosik and Parvaneh Mehbod prepared apoptosis part under Marek J Los supervision. Saba Afifi, Mazaher Ahmadi prepared pharmacology and chemistry part of the paper under supervision of Stevan Pecic and Tayyebeh Madrakian. Mohsen Taheri prepared genetic related scientific content. Nilufer Cakir, , Kiavash Hushmandi, Ali Fallah, Bahattin Koc prepared bioengineering part under Ali Zarrabi supervision. Susan Logue prepared unfolded protein part. Parvaneh Mehrbod, Bhavaya Bhushan revised the final manuscript. Stevan Pecic, Joseph W Gordon, Saeid Ghavami led the whole team, finalized the final draft and did final copy edit of manuscript.
Funding
SG was supported by UCRP and CHRIM operating grant.
Data Availability Statement
N/A.
Acknowledgments
N/A.
Conflicts of Interest
The authors do not have any conflict of interest.
Abbreviation List
| 17-DMAG | 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin |
| 2D | Two-Dimensional |
| 3D | Three-Dimensional |
| 3-MA | 3-Methyladenine |
| 4D | Four-Dimensional |
| 5-FU | 5-Fluorouracil |
| AD | Actinomycin D |
| ADP | Adenosine diphosphate |
| ADR | Adriamycin |
| AF | Anti-Fas death receptor antibody |
| AIF | Apoptotic-inducing factor |
| AIM | ATG8-Interacting motif |
| ALK | Anaplastic lymphoma kinase |
| AM | Additive manufacturing |
| AMP | Adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| ARMS | Alveolar rhabdomyosarcoma |
| ATF4 | Transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| ATG | Autophagy related genes |
| ATP | Adenosine triphosphate |
| BAG3 | Bcl-2-associated athanogene 3 |
| BD | Benzenediazonium sulphate |
| cdh15 | Cadherin 15 |
| CDK | Cyclin-Dependent kinase |
| CDKN2A | Cyclin-Dependent kinase inhibitor 2A |
| CDXs | Cell-Line derived xenograft mouse models |
| CM | Carbonaceous material |
| CMA | Chaperone-mediated autophagy |
| CMP | Chaperone-mediated autophagy |
| COG-STS | Children’s oncology group soft tissue sarcoma |
| CPT | Camptothecin |
| CPX | Ciclopirox olamine |
| CQ | Chloroquine |
| CSC | Cancer stem cells |
| CT | Computed tomography |
| CYP450 | Cytochrome P450 |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMA | Dynamic mechanical analysis |
| DNA | Deoxyribonucleic acid |
| Dox | Doxorubicin |
| ECM | Extracellular matrix |
| EIMMs | Environmentally induced mouse models |
| EMT | Epithelial to mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERAD | ER associated degradation machinery |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| ERMS | Embryonal rhabdomyosarcoma |
| FACS | Fluorescence-Activated cell sorting |
| FADD | Fas-Associated protein with death domain |
| FAPs | Fibro-Adipogenic progenitors |
| FBS | Fetal bovine serum |
| FCS | fetal calf serum |
| FDA | Food and drug administration |
| FDG PET scan | Fluorodeoxyglucose (FDG)-positron emission tomography (PET) |
| FFF | Fused filament fabrication |
| FFS | Failure free survival |
| FG + | PAX3-FOXO1 fusion genes positive |
| FGFR4 | Fibroblast growth factor receptor 4 |
| FN | Fusion-negative |
| FOXO1 | Forkhead box protein O1 |
| FP | Fusion-positive |
| GBM | Glioblastoma multiforme |
| GDP | Guanosine diphosphate |
| GelMA | Gelatin-Methacryloyl |
| GEMMs | Genetically engineered mouse models |
| GFP | Green fluorescent protein |
| GHPA | Gelatin-hydroxyphenyl propionic acid |
| GO | Graphene oxide |
| Grp78 | ER chaperone glucose regulated protein 78 |
| GSK3 | Glycogen synthase kinase 3 |
| GSTP1 | Glutathione S-transferase P1 |
| GTP | Guanosine-5'-triphosphate |
| H&E | Hematoxylin and eosin |
| HAS | Human serum albumin |
| HDAC6 | Histone deacetylase 6 |
| HDACIs | Histone deacetylases inhibitors |
| HDACs | Histone deacetylases |
| Hgf | Hepatocyte growth factor |
| Hh | Hedgehog |
| HMG-CoA | β-Hydroxy β-methylglutaryl-CoA |
| hRD | Human rhabdomyosarcoma |
| HSMM | Human skeletal muscle myoblast |
| HSR | Heat shock response |
| ICE | Carboplatin, Epirubicin, and Etoposide |
| ICE | Carboplatin, Epirubicin, and Etoposide |
| ID | Ifosfamide/Doxorubicin |
| IE | Ifosfamide/Etoposide |
| IGF1R | Insulin growth factor 1 receptor |
| IGF2 | Insulin growth factor 2 |
| IgG1 | Immunoglobulin G1 |
| il2rga | Interlukin-2 receptor gamma a |
| IL-6 | Interleukin-6 |
| IMRT | Intensity modulated radiation therapy |
| IRE | Inositol requiring enzyme |
| IRE1α | Inositol requiring enzyme 1α |
| IRS | Intergroup rhabdomyosarcoma study |
| IVA | Ifosfamide, Vincristine, and Actinomycin D |
| JAK/STAT | Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway |
| KEAP1 | Kelch like-ECH-associated protein 1 |
| KRAS | the gene Kirsten rat sarcoma viral oncogene homolog |
| LAMP2A | Lysosomal chaperone-mediated autophagy receptor |
| LC3 | Light chain 3 |
| LFS | Li-Fraumeni syndrome |
| LIR | LC3-Interacting region |
| MAPK | Mitogen-Activated protein kinase |
| MDM2 | Murine double minute 2 |
| MDR | Multidrug resistance |
| MEV | Mevalonate |
| miRs | muscle-specific microRNAs |
| MMP9 | the Matrix metalloproteinase 9 |
| MRI | Magnetic resonance imaging |
| mRNA | messenger RNA |
| mTOR | Mammalian target of rapamycin |
| MTX | Methotrexate |
| myf5 | myogenic factor 5 |
| MyoD1 | Myogenic Differentiation 1 |
| NF1 | Neurofibromatosis type I |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NSCLCs | Non-Small cell lung cancers |
| OZO-H | 4-phenyl-1,3,2-oxathiazolylium-5-oleate |
| PARP | Poly (ADP-ribose) polymerase |
| PAS | Pre-autophagosomal structure |
| PBT | Proton beam therapy |
| PCL | Poly(ɛ-caprolactone) |
| PDGF-β | Platelet-Derived growth factor β |
| PDGFR | Platelet-Derived growth factor receptor |
| PDXs | Patient derived xenograft mouse models |
| PERK | Protein kinase R like endoplasmic reticulum kinase |
| P-gp | P-glycoprotein |
| PI3K | Phosphoinositide 3-kinase |
| PI3P | Phosphatidylinositol 3-phosphate |
| PLK1 | Polo-Like kinase-1 |
| PLKs | Polo-Like kinases |
| PNET | Primitive neuroectodermal tumors |
| PQC | Protein quality control system |
| prkdc | protein kinase DNA-activated catalytic polypeptide |
| PRMS | Pleomorphic rhabdomyosarcoma |
| PVA | Poly vinyl alcohol |
| RAC1 | Ras-related C3 botulinum toxin substrate 1 |
| rag2 | recombination activating 2 |
| RIDD | Regulated IRE1 dependent decay |
| RMS | Rhabdomyosarcoma |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| ROS1 | c-ros oncogene 1 |
| RTCB | RNA 2',3'-cyclic phosphate and 5'-OH ligase |
| RTKs | Receptor tyrosine kinases |
| S6K1 | ribosomal protein S6 kinase 1 |
| SAHA | Suberoylanilide hydroxamic acid |
| SAM | Syngeneic allograft model |
| SAR | Structure-Activity relationship |
| SDH | Silibinin di-hemisuccinate |
| shATG7 | shRNA sequence against ATG7 |
| SHH | Sonic hedgehog |
| SIOP | International Society of pediatric oncology |
| siRNA | short interfering RNA or silencing RNA |
| SIRT | the mammalian Sirtuin |
| SLA | Stereolithography |
| Smac | Second mitochondria-derived activator of caspase |
| ST80 | the cytoplasmic histone deacetylase 6 inhibitor ST80 |
| tBID | truncated BID |
| TGF | Transforming growth factor |
| TMZ | Temozolomide |
| TNBC | Triple negative breast cancer |
| TNFR | Tumor necrosis factor receptor |
| TPCs | Tumor propagating cells |
| TRAILR | TNF-related apoptosis-inducing ligand receptor |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| Tv6 | Tenovin-6 |
| ULK1/2 | Unc-51 Like Autophagy Activating Kinase 1/2 |
| UPR | the Unfolded protein response |
| UPS | the Ubiquitin-Proteasome system |
| UTR | Untranslated region |
| UV | Ultraviolet |
| VAC | Vincristine, Actinomycin D and Cyclophosphamide |
| VAI | Vincristine and Dactinomycin, Ifosfamide |
| V-ATPase | Vacuolar H+ ATPase |
| VEGF | Vascular endothelial growth factor |
| VI | Vincristine and Irinotecan |
| VIE | Vincristine, Ifosfamide, and etoposide |
| VIT | Vincristine, Irinotecan, and Temozolomide |
| VM | Vincristine/Melphalan |
| VML | Volumetric muscle loss |
| Vps | Vacuolar protein sorting |
| VTC | Vincristine, Topotecan, and Cyclophosphamide |
| WHO | the World health organization |
| XBP1 | X-box-binding protein 1 |
| XIAP | X-chromosome linked IAP protein |
References
- Shern, J.F.; Yohe, M.E.; Khan, J. Pediatric rhabdomyosarcoma. Crit Rev Oncog 2015, 20, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Ognjanovic, S.; Linabery, A.M.; Charbonneau, B.; Ross, J.A. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer 2009, 115, 4218–4226. [Google Scholar] [CrossRef]
- Ruiz-Mesa, C.; Goldberg, J.M.; Coronado Munoz, A.J.; Dumont, S.N.; Trent, J.C. Rhabdomyosarcoma in adults: new perspectives on therapy. Curr Treat Options Oncol 2015, 16, 27. [Google Scholar] [CrossRef]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; et al. Rhabdomyosarcoma. Nat Rev Dis Primers 2019, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Crist, W.M.; Anderson, J.R.; Meza, J.L.; Fryer, C.; Raney, R.B. ; Ruymann FB, et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol 2001, 19, 3091–3102. [Google Scholar] [PubMed]
- Arndt, C.A.; Stoner, J.A.; Hawkins, D.S.; Rodeberg, D.A.; Hayes-Jordan, A.A.; Paidas, C.N.; et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children's oncology group study D9803. J Clin Oncol 2009, 27, 5182–5188. [Google Scholar] [CrossRef]
- Stevens, M.C.; Rey, A.; Bouvet, N.; Ellershaw, C.; Flamant, F.; Habrand, J.L.; Marsden, H.B.; Martelli, H.; de Toledo, J.S.; Spicer, R.D.; et al. Treatment of Nonmetastatic Rhabdomyosarcoma in Childhood and Adolescence: Third Study of the International Society of Paediatric Oncology—SIOP Malignant Mesenchymal Tumor 89. J. Clin. Oncol. 2005, 23, 2618–2628. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, O.; Rey, A.; de Toledo, J.S.; Martelli, H.; Jenney, M.E.; Scopinaro, M.; Bergeron, C.; Merks, J.H.; Bouvet, N.; Ellershaw, C.; et al. Randomized Comparison of Intensified Six-Drug Versus Standard Three-Drug Chemotherapy for High-Risk Nonmetastatic Rhabdomyosarcoma and Other Chemotherapy-Sensitive Childhood Soft Tissue Sarcomas: Long-Term Results From the International Society of Pediatric Oncology MMT95 Study. J. Clin. Oncol. 2012, 30, 2457–2465. [Google Scholar] [CrossRef]
- Ferrari, A.; Casanova, M.; Collini, P.; Meazza, C.; Luksch, R.; Massimino, M.; Cefalo, G.; Terenziani, M.; Spreafico, F.; Catania, S.; et al. Adult-Type Soft Tissue Sarcomas in Pediatric-Age Patients: Experience at the Istituto Nazionale Tumori in Milan. J. Clin. Oncol. 2005, 23, 4021–4030. [Google Scholar] [CrossRef]
- Blakely, M.L.; Andrassy, R.J.; Raney, R.; Anderson, J.R.; Wiener, E.S.; Rodeberg, D.A.; Paidas, C.N.; E Lobe, T.; Crist, W.M. Prognostic factors and surgical treatment guidelines for children with rhabdomyosarcoma of the perineum or anus: A report of intergroup rhabdomyosarcoma studies I through IV, 1972 through 1997. J. Pediatr. Surg. 2003, 38, 347–353. [Google Scholar] [CrossRef]
- Crist, W.M.; Garnsey, L.; Beltangady, M.S.; Gehan, E.; Ruymann, F.; Webber, B.; Hays, D.M.; Wharam, M.; Maurer, H.M. Prognosis in children with rhabdomyosarcoma: a report of the intergroup rhabdomyosarcoma studies I and II. Intergroup Rhabdomyosarcoma Committee. J. Clin. Oncol. 1990, 8, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Anderson, J.R.; Crist, W.M.; Wharam, M.D.; Breitfeld, P.P.; Hawkins, D.; et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study Group. J Clin Oncol 1999, 17, 3487–3493. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Lyden, E.; Breitfeld, P.; Donaldson, S.S.; Wiener, E.; Parham, D.; et al. Two consecutive phase II window trials of irinotecan alone or in combination with vincristine for the treatment of metastatic rhabdomyosarcoma: the Children's Oncology Group. J Clin Oncol 2007, 25, 362–369. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Spunt, S.L.; Skapek, S.X. ; Committee COGSTS. Children's Oncology Group's 2013 blueprint for research: Soft tissue sarcomas. Pediatr Blood Cancer 2013, 60, 1001–1008. [Google Scholar] [PubMed]
- Crist, W.; Gehan, E.A.; Ragab, A.H.; Dickman, P.S.; Donaldson, S.S. ; Fryer C, et al. The third intergroup rhabdomyosarcoma study. J Clin Oncol 1995, 13, 610–630. [Google Scholar]
- Sultan, I.; Qaddoumi, I.; Yaser, S.; Rodriguez-Galindo, C.; Ferrari, A. Comparing Adult and Pediatric Rhabdomyosarcoma in the Surveillance, Epidemiology and End Results Program, 1973 to 2005: An Analysis of 2,600 Patients. J. Clin. Oncol. 2009, 27, 3391–3397. [Google Scholar] [CrossRef]
- Ferrari, A.; Dileo, P.; Casanova, M.; Bertulli, R.; Meazza, C. ; Gandola L, et al. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer 2003, 98, 571–80. [Google Scholar]
- Noujaim, J.; Thway, K.; Jones, R.L.; Miah, A.; Khabra, K.; Langer, R.; Kasper, B.; Judson, I.; Benson, C.; Kollàr, A. Adult Pleomorphic Rhabdomyosarcoma: A Multicentre Retrospective Study. Anticancer Res. 2015, 35, 6213–6217. [Google Scholar]
- Raney, R.B.; Maurer, H.M.; Anderson, J.R.; Andrassy, R.J.; Donaldson, S.S.; Qualman, S.J.; et al. The Intergroup Rhabdomyosarcoma Study Group (IRSG): Major lessons from the IRS-I through IRS-IV studies as background for the current IRS-V treatment protocols. Sarcoma 2001, 5, 9–15. [Google Scholar] [CrossRef]
- Kallen, M.E.; Hornick, J.L. The 2020 WHO Classification: What's new in soft tissue tumor pathology? Am J Surg Pathol 2022, 45, e1–e23. [Google Scholar] [CrossRef]
- Pappo, A.S.; Shapiro, D.N.; Crist, W.M.; Maurer, H.M. Biology and therapy of pediatric rhabdomyosarcoma. J. Clin. Oncol. 1995, 13, 2123–2139. [Google Scholar] [CrossRef]
- Kelly, K.M.; Womer, R.B.; Sorensen, P.H.; Xiong, Q.B.; Barr, F.G. Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J. Clin. Oncol. 1997, 15, 1831–1836. [Google Scholar] [CrossRef]
- Dasgupta, R.; Rodeberg, D.A. Update on rhabdomyosarcoma. Semin Pediatr Surg 2012, 21, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, E.R.; Teot, L.A.; Anderson, J.R.; Moore, J.; Bridge, J.A.; Barr, F.G.; et al. Dense pattern of embryonal rhabdomyosarcoma, a lesion easily confused with alveolar rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Am J Clin Pathol 2013, 140, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Malempati, S.; Hawkins, D.S. Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer 2012, 59, 5–10. [Google Scholar] [CrossRef]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol 2012, 30, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Enterline, H.T.; Horn, R.C. Alveolar Rhabdomyosarcoma. A Distinctive Tumor Type. Am. J. Clin. Pathol. 1958, 29, 356–366. [Google Scholar] [CrossRef]
- Patton, R.B.; Horn, R.C. Rhabdomyosarcoma: clinical and pathological features and comparison with human fetal and embryonal skeletal muscle. Surgery 1962, 52, 572–584. [Google Scholar] [PubMed]
- Toro, J.R.; Travis, L.B.; Wu, H.J.; Zhu, K.; Fletcher, C.D.; Devesa, S.S. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer 2006, 119, 2922–2930. [Google Scholar] [CrossRef]
- Merlino, G.; Helman, L.J. Rhabdomyosarcoma-working out the pathways. Oncogene 1999, 18, 5340–5348. [Google Scholar] [CrossRef]
- Miller, R.W.; Young JL, Jr.; Novakovic, B. Childhood cancer. Cancer 1995, 75, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Stiller, C.A.; Parkint, D.M. International variations in the incidence of childhood soft-tissue sarcomas. Paediatr. Périnat. Epidemiology 1994, 8, 107–119. [Google Scholar] [CrossRef]
- Lychou, S.E.; Gustafsson, G.G.; Ljungman, G.E. Higher rates of metastatic disease may explain the declining trend in Swedish paediatric rhabdomyosarcoma survival rates. Acta Paediatr. 2016, 105, 74–81. [Google Scholar] [CrossRef] [PubMed]
- A Furlong, M.; Mentzel, T.; Fanburg-Smith, J.C. Pleomorphic Rhabdomyosarcoma in Adults: A Clinicopathologic Study of 38 Cases with Emphasis on Morphologic Variants and Recent Skeletal Muscle-Specific Markers. Mod. Pathol. 2001, 14, 595–603. [Google Scholar] [CrossRef]
- Perez, E.A.; Kassira, N.; Cheung, M.C.; Koniaris, L.G.; Neville, H.L.; Sola, J.E. Rhabdomyosarcoma in children: a SEER population based study. J Surg Res 2011, 170, e243–e251. [Google Scholar] [CrossRef] [PubMed]
- Grufferman, S.; Ruymann, F.; Ognjanovic, S.; Erhardt, E.B.; Maurer, H.M. Prenatal X-ray exposure and rhabdomyosarcoma in children: a report from the children's oncology group. Cancer Epidemiol Biomarkers Prev 2009, 18, 1271–1276. [Google Scholar] [CrossRef]
- Little, D.J.; Ballo, M.T.; Zagars, G.K.; Pisters, P.W.; Patel, S.R.; El-Naggar, A.K.; et al. Adult rhabdomyosarcoma: outcome following multimodality treatment. Cancer 2002, 95, 377–88. [Google Scholar] [CrossRef]
- Maurer, H.M.; Beltangady, M.; Gehan, E.A.; Crist, W.; Hammond, D.; Hays, D.M.; et al. The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer 1988, 61, 209–20. [Google Scholar] [CrossRef]
- Maurer, H.M.; Gehan, E.A.; Beltangady, M.; Crist, W.; Dickman, P.S.; Donaldson, S.S.; et al. The Intergroup Rhabdomyosarcoma Study-II. Cancer 1993, 71, 1904–22. [Google Scholar] [CrossRef]
- Hayes-Jordan, A.; Doherty, D.K.; West, S.D.; Raney, R.B.; Blakely, M.L.; Cox CS, Jr.; et al. Outcome after surgical resection of recurrent rhabdomyosarcoma. J Pediatr Surg 2006, 41, 633–638. [Google Scholar] [CrossRef]
- Bramwell, V.H.C. Management of Advanced Adult Soft Tissue Sarcoma. Sarcoma 2003, 7, 43–55. [Google Scholar] [CrossRef]
- Heyn, R.M.; Holland, R.; Newton, W.A.; Tefft, M.; Breslow, N.; Hartmann, J.R. The role of combined chemotherapy in the treatment of rhabdomyosarcoma in children. Cancer 1974, 34, 2128–2145. [Google Scholar] [CrossRef]
- Spunt, S.L.; Smith, L.M.; Ruymann, F.B.; Qualman, S.J.; Donaldson, S.S.; Rodeberg, D.A.; et al. Cyclophosphamide dose intensification during induction therapy for intermediate-risk pediatric rhabdomyosarcoma is feasible but does not improve outcome: a report from the soft tissue sarcoma committee of the children's oncology group. Clin Cancer Res 2004, 10, 6072–6079. [Google Scholar] [CrossRef]
- Mascarenhas, L.; Chi, Y.-Y.; Hingorani, P.; Anderson, J.R.; Lyden, E.R.; Rodeberg, D.A.; Indelicato, D.J.; Kao, S.C.; Dasgupta, R.; Spunt, S.L.; et al. Randomized Phase II Trial of Bevacizumab or Temsirolimus in Combination With Chemotherapy for First Relapse Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 2866–2874. [Google Scholar] [CrossRef]
- Mandell, L.; Ghavimi, F.; Peretz, T.; LaQuaglia, M.; Exelby, P. Radiocurability of microscopic disease in childhood rhabdomyosarcoma with radiation doses less than 4,000 cGy. J. Clin. Oncol. 1990, 8, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, O.; Rey, A.; Lyden, E.; Bisogno, G.; Stevens, M.C.; Meyer, W.H.; Carli, M.; Anderson, J.R. Prognostic Factors in Metastatic Rhabdomyosarcomas: Results of a Pooled Analysis From United States and European Cooperative Groups. J. Clin. Oncol. 2008, 26, 2384–2389. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, G.; Ferrari, A.; Prete, A.; Messina, C.; Basso, E.; Cecchetto, G.; Indolfi, P.; Scarzello, G.; D’angelo, P.; De Sio, L.; et al. Sequential high-dose chemotherapy for children with metastatic rhabdomyosarcoma. Eur. J. Cancer 2009, 45, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Vardanyan, R. Chapter 10 - Classes of Piperidine-Based Drugs, in Piperidine-Based Drug Discovery, Vardanyan, R., Editor. 2017, Elsevier. p. 299-332.
- Cai, P.; Tsao, R.; Ruppen, M.E. In Vitro Metabolic Study of Temsirolimus: Preparation, Isolation, and Identification of the Metabolites. Drug Metab. Dispos. 2007, 35, 1554–1563. [Google Scholar] [CrossRef]
- Scholar, E. Temsirolimus, in xPharm: The Comprehensive Pharmacology Reference, Enna, S.J. and Bylund, D.B., Editors. 2009, Elsevier: New York. p. 1-4.
- Chan, F.; Samlowski, E.E.; Samlowski, W.E. Temsirolimus: A Review of its Use in the Treatment of Advanced Renal Cell Carcinoma. Clin. Med. Ther. 2009, 1, CMT–S2349. [Google Scholar] [CrossRef]
- Raymond, E.; Alexandre, J.; Faivre, S.; Vera, K.; Materman, E.; Boni, J.; Leister, C.; Korth-Bradley, J.; Hanauske, A.; Armand, J.-P. Safety and Pharmacokinetics of Escalated Doses of Weekly Intravenous Infusion of CCI-779, a Novel mTOR Inhibitor, in Patients With Cancer. J. Clin. Oncol. 2004, 22, 2336–2347. [Google Scholar] [CrossRef]
- Boni, J.P.; Hug, B.; Leister, C.; Sonnichsen, D. Intravenous Temsirolimus in Cancer Patients: Clinical Pharmacology and Dosing Considerations. Semin. Oncol. 2009, 36, S18–S25. [Google Scholar] [CrossRef] [PubMed]
- Proud CG. mTOR and its downstream targets, in Encyclopedia of Biological Chemistry (Second Edition), Lennarz, W.J. and Lane, M.D., Editors. 2013, Academic Press: Waltham. p. 194-199.
- Hausch, F.; Kozany, C.; Theodoropoulou, M.; Fabian, A.K. FKBPs and the Akt/mTOR pathway. Cell Cycle 2013, 12, 2366–70. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Buckner, J.C.; Erlichman, C.; Pollack, M.S.; Boni, J.P.; Dukart, G.; Marshall, B.; Speicher, L.; Moore, L.; Rowinsky, E.K. A Phase I and Pharmacokinetic Study of Temsirolimus (CCI-779) Administered Intravenously Daily for 5 Days Every 2 Weeks to Patients with Advanced Cancer. Clin. Cancer Res. 2006, 12, 5755–5763. [Google Scholar] [CrossRef]
- Chiarini, F.; Lonetti, A.; Teti, G.; Orsini, E.; Bressanin, D.; Cappellini, A.; Ricci, F.; Tazzari, P.L.; Ognibene, A.; Falconi, M.; et al. A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget 2012, 3, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Q.; Lindsley, C.W.; Cheng, G.Z.; Yang, H.; Nicosia, S.V. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene 2005, 24, 7482–7492. [Google Scholar] [CrossRef]
- Advani, S.H. Targeting mTOR pathway: A new concept in cancer therapy. Indian J. Med Paediatr. Oncol. 2010, 31, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Kieran, M.W.; Grupp, S.; Perek, D.; Clancy, J.; Krygowski, M.; Ananthakrishnan, R.; Boni, J.P.; Berkenblit, A.; Spunt, S.L. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur. J. Cancer 2012, 48, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Klümpen, H.J.; Beijnen, J.H.; Gurney, H.; Schellens, J.H. Inhibitors of mTOR. Oncologist 2010, 15, 1262–1269. [Google Scholar] [CrossRef]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA Approval Summary: Temsirolimus as Treatment for Advanced Renal Cell Carcinoma. Oncol. 2010, 15, 428–435. [Google Scholar] [CrossRef]
- Moudi, M.; Go, R.; Yien, C.Y.; Nazre, M. Vinca alkaloids. Int J Prev Med 2013, 4, 1231–1235. [Google Scholar]
- Pellegrini, F.; Budman, D.R. Review: tubulin function, action of antitubulin drugs, and new drug development. Cancer Invest 2005, 23, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Kerckhove, N.; Collin, A.; Condé, S.; Chaleteix, C.; Pezet, D.; Balayssac, D. Long-Term Effects, Pathophysiological Mechanisms, and Risk Factors of Chemotherapy-Induced Peripheral Neuropathies: A Comprehensive Literature Review. Front. Pharmacol. 2017, 8, 86. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule destabilising agents: far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Mittal, B.; Tulsyan, S.; Kumar, S.; Mittal, R.D.; Agarwal G. Chapter Four - Cytochrome P450 in cancer susceptibility and treatment, in Adv Clin Chem, Makowski, G.S., Editor. 2015, Elsevier. p. 77-139.
- Găman, A.M.; Egbuna, C.; Găman M-A. Chapter 6 - Natural bioactive lead compounds effective against haematological malignancies, in Phytochemicals as Lead Compounds for New Drug Discovery, Egbuna, C., Kumar, S., Ifemeje, J.C., Ezzat, S.M., and Kaliyaperumal, S., Editors. 2020, Elsevier. p. 95-115.
- Vardanyan, R.S. and V.J. Hruby, 28—Female Sex Hormones, in Synthesis of Essential Drugs, R.S. Vardanyan and V.J. Hruby, Editors. 2006, Elsevier: Amsterdam; pp. 365–379.
- Prakash, V.; Timasheff, S.N. Mechanism of interaction of vinca alkaloids with tubulin: catharanthine and vindoline. Biochemistry 1991, 30, 873–880. [Google Scholar] [CrossRef]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.-W.; Lai, M.-J.; Liou, J.-P.; Chang, Y.-L.; Wang, J.-C.; Pan, S.-L.; Teng, C.-M. The synergic effect of vincristine and vorinostat in leukemia in vitro and in vivo. J. Hematol. Oncol. 2015, 8, 1–15. [Google Scholar] [CrossRef]
- Khan, T.S.; Sundin, A.; Juhlin, C.; Wilander, E.; Öberg, K.; Eriksson, B. Vincristine, Cisplatin, Teniposide, and Cyclophosphamide Combination in the Treatment of Recurrent or Metastatic Adrenocortical Cancer. Med Oncol. 2004, 21, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Kamiya, O.; Hamajima, N.; Mizuno, H.; Kobayashi, M.; Hirabayashi, N.; Takeyama, H.; Kato, R.; Kawashima, K.; Nitta, M.; et al. Multi-drug Combination Therapy with Vincristine-Melphalan-Cyclophosphamide-Prednisolone Was More Effective than Cyclophosphamide-Prednisolone in Stage III Myeloma. Jpn. J. Cancer Res. 1990, 81, 1320–1327. [Google Scholar] [CrossRef]
- Nabors, L.B.; Surboeck, B.; Grisold W. Chapter 14 - Complications from pharmacotherapy, in Handbook of Clinical Neurology, Berger, M.S. and Weller, M., Editors. 2016, Elsevier. p. 235-250.
- Agrawal K. Vincristine, in xPharm: The Comprehensive Pharmacology Reference, Enna, S.J. and Bylund, D.B., Editors. 2007, Elsevier: New York. p. 1-4.
- Rassekh, S.R.; Ross CJD. Chapter 6 - Cancer Pharmacogenomics in Children, in Cancer Genomics, Dellaire, G., Berman, J.N., and Arceci, R.J., Editors. 2014, Academic Press: Boston. p. 77-92.
- Hong, W.K.; Holland, J.F.; American Association for Cancer, R. Microtubule-targeting natural products, in Holland-Frei Cancer Medicine 8. 2010, PMPH USA, Ltd: Shelton, Conn. p. 655-657.
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; et al. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg Med Chem Lett 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Agrawal, K. Doxorubicin, in xPharm: The Comprehensive Pharmacology Reference, Enna, S.J. and Bylund, D.B., Editors. 2007, Elsevier: New York. p. 1-5.
- Hena, S.; Znad, H. Chapter Six - Membrane bioreactor for pharmaceuticals and personal care products removal from wastewater, in Compr Anal Chem, Chormey, D.S., Bakırdere, S., Turan, N.B., and Engin, G.Ö., Editors. 2018, Elsevier. p. 201-256.
- Braña, M.F.; Cacho, M.; Gradillas, A.; de Pascual-Teresa, B.; Ramos, A. Intercalators as anticancer drugs. Curr Pharm Des 2001, 7, 1745–1780. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H. ; Klein TE, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics 2011, 21, 440–446. [Google Scholar] [PubMed]
- Gewirtz, D. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Madkour, LH. Chapter 7 - Classifications of DNA binding molecules—Drug interactions, in Nucleic Acids as Gene Anticancer Drug Delivery Therapy, Madkour, L.H., Editor. 2019, Academic Press. p. 87-101.
- Canals, A.; Purciolas, M.; Aymamí, J.; Coll, M. The anticancer agent ellipticine unwinds DNA by intercalative binding in an orientation parallel to base pairs. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005, 61, 1009–1012. [Google Scholar] [CrossRef]
- Kellogg, G.E.; Scarsdale, J.N.; Fornari, F.A. Identification and hydropathic characterization of structural features affecting sequence specificity for doxorubicin intercalation into DNA double-stranded polynucleotides. Nucleic Acids Res. 1998, 26, 4721–4732. [Google Scholar] [CrossRef]
- Yang, F.; Teves, S.S.; Kemp, C.J.; Henikoff, S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta 2014, 1845, 84–89. [Google Scholar] [CrossRef]
- Estève, P.-O.; Chin, H.G.; Pradhan, S. Molecular Mechanisms of Transactivation and Doxorubicin-mediated Repression of survivin Gene in Cancer Cells. J. Biol. Chem. 2007, 282, 2615–2625. [Google Scholar] [CrossRef]
- Taymaz-Nikerel, H.; Karabekmez, M.E.; Eraslan, S.; Kırdar, B. Doxorubicin induces an extensive transcriptional and metabolic rewiring in yeast cells. Sci. Rep. 2018, 8, 13672. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Piestrzeniewicz, M.K.; Wilmańska, D.; Szemraj, J.; Studzian, K.; Gniazdowski, M. Interactions of Novel Morpholine and Hexamethylene Derivatives of Anthracycline Antibiotics with DNA. Z Naturforsch C J Biosci 2004, 59, 739–748. [Google Scholar] [CrossRef]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as Topoisomerase II Poisons: From Early Studies to New Perspectives. Int. J. Mol. Sci. 2018, 19, 3480. [Google Scholar] [CrossRef]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem J 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Hong, W.K.; Holland, J.F.; American Association for Cancer, R. Drugs that target DNA topoisomerases, in Holland-Frei Cancer Medicine 8, editors, W.K.H.e.a., Editor. 2010, PMPH USA, Ltd: Shelton, Conn. p. 645-653.
- Martins-Teixeira, M.B.; Carvalho, I. Antitumour Anthracyclines: Progress and Perspectives. ChemMedChem 2020, 15, 933–948. [Google Scholar] [CrossRef] [PubMed]
- Kwok, K.K.; Vincent, E.C.; Gibson, JN. 36 - Antineoplastic Drugs, in Pharmacology and Therapeutics for Dentistry (Seventh Edition), Dowd, F.J., Johnson, B.S., and Mariotti, A.J., Editors. 2017, Mosby. p. 530-562.
- Avendaño, C.; Menéndez, JC. Chapter 4 - Anticancer Drugs Acting via Radical Species, Photosensitizers and Photodynamic Therapy of Cancer, in Medicinal Chemistry of Anticancer Drugs, Avendaño, C. and Menéndez, J.C., Editors. 2008, Elsevier: Amsterdam. p. 93-138.
- Selby, C.P.; Sancar, A. Noncovalent drug-DNA binding interactions that inhibit and stimulate (A)BC excinuclease. Biochemistry 1991, 30, 3841–3849. [Google Scholar] [CrossRef]
- Ijäs, H.; Shen, B.; Heuer-Jungemann, A.; Keller, A.; A Kostiainen, M.; Liedl, T.; A Ihalainen, J.; Linko, V. Unraveling the interaction between doxorubicin and DNA origami nanostructures for customizable chemotherapeutic drug release. Nucleic Acids Res. 2021, 49, 3048–3062. [Google Scholar] [CrossRef] [PubMed]
- Martin, SA. Chapter 6 - The DNA mismatch repair pathway, in DNA Repair in Cancer Therapy (Second Edition), Kelley, M.R. and Fishel, M.L., Editors. 2016, Academic Press: Boston. p. 151-177.
- Farquhar, D.; Cherif, A.; Bakina, E.; Nelson, J.A. Intensely Potent Doxorubicin Analogues: Structure−Activity Relationship. J. Med. Chem. 1998, 41, 965–972. [Google Scholar] [CrossRef]
- Vann, K.R.; Oviatt, A.A.; Osheroff, N. Topoisomerase II Poisons: Converting Essential Enzymes into Molecular Scissors. Biochemistry 2021, 60, 1630–1641. [Google Scholar] [CrossRef]
- Golomb, L.; Volarevic, S.; Oren, M. p53 and ribosome biogenesis stress: The essentials. FEBS Lett. 2014, 588, 2571–2579. [Google Scholar] [CrossRef]
- Ladds, M.J.G.W.; Laín, S. Small molecule activators of the p53 response. J. Mol. Cell Biol. 2019, 11, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-S.; Ho, D.-R.; Chen, F.-Y.; Chen, C.-R.; Ke, Y.-D.; Su, J.-G.J. AKT mediates actinomycin D-induced p53 expression. Oncotarget 2014, 5, 693–703. [Google Scholar] [CrossRef]
- van Leeuwen, I.M.; Higgins, M.; Campbell, J.; Brown, C.J.; McCarthy, A.R.; Pirrie, L.; et al. Mechanism-specific signatures for small-molecule p53 activators. Cell Cycle 2011, 10, 1590–8. [Google Scholar] [CrossRef] [PubMed]
- Veal, G.J.; Cole, M.; Errington, J.; Parry, A.; Hale, J.; Pearson, A.D.J.; et al. Pharmacokinetics of dactinomycin in a pediatric patient population: a United Kingdom Children's Cancer Study Group. Clin Cancer Res 2005, 11, 5893–5899. [Google Scholar] [CrossRef] [PubMed]
- Melguizo, C.; Prados, J.; E Fernández, J.; Vélez, C.; Alvarez, L.; Aránega, A. Actinomycin D causes multidrug resistance and differentiation in a human rhabdomyosarcoma cell line. Cell. Mol. Biol. 1994, 40, 137–145. [Google Scholar] [PubMed]
- Rider, BJ. Cyclophosphamide, in xPharm: The Comprehensive Pharmacology Reference, Enna, S.J. and Bylund, D.B., Editors. 2007, Elsevier: New York. p. 1-5.
- Ralhan, R.; Kaur, J. Alkylating agents and cancer therapy. Expert Opin Ther Pat 2007, 17, 1061–1075. [Google Scholar] [CrossRef]
- Konstantinov, S.M.; Berger, MR. Alkylating agents, in Encyclopedia of Molecular Pharmacology, Offermanns, S. and Rosenthal, W., Editors. 2008, Springer Berlin Heidelberg: Berlin, Heidelberg. p. 53-57.
- Huang, Z.; Roy, P.; Waxman, D.J. Role of human liver microsomal CYP3A4 and CYP2B6 in catalyzing N-dechloroethylation of cyclophosphamide and ifosfamide. Biochem. Pharmacol. 2000, 59, 961–972. [Google Scholar] [CrossRef]
- de Jonge, M.E.; Huitema, A.D.; Rodenhuis, S.; Beijnen, J.H. Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet 2005, 44, 1135–1164. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.A.; Chess-Williams, R.; McDermott, C. Novel insights into the mechanism of cyclophosphamide-induced bladder toxicity: chloroacetaldehyde’s contribution to urothelial dysfunction in vitro. Arch. Toxicol. 2019, 93, 3291–3303. [Google Scholar] [CrossRef] [PubMed]
- Moghe, A.; Ghare, S.; Lamoreau, B.; Mohammad, M.; Barve, S.; McClain, C.; Joshi-Barve, S. Molecular Mechanisms of Acrolein Toxicity: Relevance to Human Disease. Toxicol. Sci. 2015, 143, 242–255. [Google Scholar] [CrossRef]
- Monach, P.A.; Arnold, L.M.; Merkel, P.A. Incidence and prevention of bladder toxicity from cyclophosphamide in the treatment of rheumatic diseases: A data-driven review. Arthritis Rheum. 2010, 62, 9–21. [Google Scholar] [CrossRef]
- Luce, J.K.; Simons, J.A. Efficacy of mesna in preventing further cyclophosphamide-induced hemorrhagic cystitis. Med Pediatr Oncol 1988, 16, 372–374. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Giraud, B.; Hebert, G.; Deroussent, A.; Veal, G.J.; Vassal, G.; Paci, A. Oxazaphosphorines: new therapeutic strategies for an old class of drugs. Expert Opin. Drug Metab. Toxicol. 2010, 6, 919–938. [Google Scholar] [CrossRef] [PubMed]
- Sprangers, BEN.; Cosmai, L. Sprangers BEN.; Cosmai, L.; Porta C. 16 - Conventional chemotherapy, in Onco-Nephrology, Finkel, K.W., Perazella, M.A., and Cohen, E.P., Editors. 2020, Elsevier: Philadelphia. p. 127-153.e11.
- Sannu, A.; Radha, R.; Mathews, A.; Padmakumari Mony, R.; Prahladan, A.; James, F.V. Ifosfamide-induced malignancy of ureter and bladder. Cureus 2017, 9, e1594. [Google Scholar] [PubMed]
- Voelcker, G. Influence of the alkylating function of aldo-Ifosfamide on the anti-tumor activity. Anti-Cancer Drugs 2018, 29, 75–79. [Google Scholar] [CrossRef]
- Shin, Y.-J.; Kim, J.-Y.; Moon, J.-W.; You, R.-M.; Park, J.-Y.; Nam, J.-H. Fatal Ifosfamide-Induced Metabolic Encephalopathy in Patients with Recurrent Epithelial Ovarian Cancer: Report of Two Cases. Cancer Res. Treat. 2011, 43, 260–263. [Google Scholar] [CrossRef]
- Kataria, P.S.; Kendre, P.P.; Patel, A.A. Ifosfamide-induced Encephalopathy Precipitated by Aprepitant: A Rarely Manifested Side Effect of Drug Interaction. J. Pharmacol. Pharmacother. 2017, 8, 38–40. [Google Scholar] [CrossRef]
- Thirumaran, R.; Prendergast, G.C.; Gilman, P.B. Chapter 7 - Cytotoxic Chemotherapy in Clinical Treatment of Cancer, in Cancer Immunother, Prendergast, G.C. and Jaffee, E.M., Editors. 2007, Academic Press: Burlington. p. 101-116.
- Dechant, K.L.; Brogden, R.N.; Pilkington, T.; Faulds, D. Ifosfamide/mesna. A review of its antineoplastic activity, pharmacokinetic properties and therapeutic efficacy in cancer. Drugs 1991, 42, 428–467. [Google Scholar] [CrossRef]
- Kerbusch, T.; de Kraker, J.; Keizer, H.J.; van Putten, J.W.G.; Groen, H.J.M.; Jansen, R.L.H.; Schellens, J.H.M.; Beijnen, J.H. Clinical Pharmacokinetics and Pharmacodynamics of Ifosfamide and its Metabolites. Clin. Pharmacokinet. 2001, 40, 41–62. [Google Scholar] [CrossRef]
- Falco, P.; Bringhen, S.; Avonto, I.; Gay, F.; Morabito, F.; Boccadoro, M.; Palumbo, A. Melphalan and its role in the management of patients with multiple myeloma. Expert Rev. Anticancer. Ther. 2007, 7, 945–957. [Google Scholar] [CrossRef]
- Osborne, M.; Lawley, P. Alkylation of DNA by melphalan with special reference to adenine derivatives and adenine-guanine cross-linking. Chem. Interactions 1993, 89, 49–60. [Google Scholar] [CrossRef]
- Rider, BJ. Melphalan, in xPharm: The Comprehensive Pharmacology Reference, Enna, S.J. and Bylund, D.B., Editors. 2007, Elsevier: New York. p. 1-5.
- Vistica, D.T. Cytotoxicity as an indicator for transport mechanism. Evidence that melphalan is transported by two leucine-preferring carrier systems in the L1210 murine leukemia cell. Biochim. et Biophys. Acta (BBA) - Biomembr. 1979, 550, 309–317. [Google Scholar] [CrossRef]
- Esma, F.; Salvini, M.; Troia, R.; Boccadoro, M.; Larocca, A.; Pautasso, C. Melphalan hydrochloride for the treatment of multiple myeloma. Expert Opin Pharmacother 2017, 18, 1127–1136. [Google Scholar] [CrossRef]
- Sirohi, B.; Cunningham, D.; Powles, R.; Murphy, F.; Arkenau, T.; Norman, A.; et al. Long-term outcome of autologous stem-cell transplantation in relapsed or refractory Hodgkin's lymphoma. Ann Oncol 2008, 19, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Long, B.H.; Casazza, A.-M. Structure-activity relationships of VP-16 analogues. Cancer Chemother. Pharmacol. 1994, 34, S26–S31. [Google Scholar] [CrossRef]
- Montecucco, A.; Zanetta, F.; Biamonti, G. Molecular mechanisms of etoposide. EXCLI J 2015, 14, 95. [Google Scholar]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef]
- Sasada, S.; Kodaira, M.; Shimoi, T.; Shimomura, A.; Yunokawa, M.; Yonemori, K.; Shimizu, C.; Fujiwara, Y.; Tamura, K. Ifosfamide and Etoposide Chemotherapy in the Treatment of Recurrent/Refractory Rhabdomyosarcoma in Adults. Anticancer Res. 2016, 36, 2429–2432. [Google Scholar]
- Arndt, C.; Nascimento, A.; Schroeder, G.; Schomberg, P.; Neglia, J.; Sencer, S.; Silberman, T.; Moertel, C.; Tillisch, J.; Miser, J. Treatment of intermediate risk rhabdomyosarcoma and undifferentiated sarcoma with alternating cycles of vincristine/doxorubicin/cyclophosphamide and etoposide/ifosfamide. Eur. J. Cancer 1998, 34, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Katayama, M.; Kawaguchi, T.; Berger, M.S.; O Pieper, R. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007, 14, 548–558. [Google Scholar] [CrossRef]
- Yao, *!!! REPLACE !!!*; Xie, B.-S.; Zhao, H.-C.; Yao, S.-K.; Zhuo, D.-X.; Jin, B.; Lv, D.-C.; Wu, C.-L.; Ma, D.-L.; Gao, C.; et al. Autophagy inhibition enhances etoposide-induced cell death in human hepatoma G2 cells. Int. J. Mol. Med. 2011, 27, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.M. Fifteen years of irinotecan therapy for pediatric sarcoma: where to next? Clin Sarcoma Res 2015, 5, 20. [Google Scholar] [CrossRef]
- Adams, D.J.; Wahl, M.L.; Flowers, J.L.; Sen, B.; Colvin, M.; Dewhirst, M.W.; Manikumar, G.; Wani, M.C. Camptothecin analogs with enhanced activity against human breast cancer cells. II. Impact of the tumor pH gradient. Cancer Chemother. Pharmacol. 2006, 57, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Zunino, F.; Dallavalleb, S.; Laccabuea, D.; Berettaa, G.; Merlinib, L.; Pratesi, G. Current status and perspectives in the development of camptothecins. Curr Pharm Des 2002, 8, 2505–2520. [Google Scholar] [CrossRef]
- de Man, F.M.; Goey, A.K.L.; van Schaik, R.H.N.; Mathijssen, R.H.J.; Bins, S. Individualization of irinotecan treatment: A review of pharmacokinetics, pharmacodynamics, and pharmacogenetics. Clin Pharmacokinet 2018, 57, 1229–1254. [Google Scholar] [CrossRef]
- Xu, Y.; Villalona-Calero, M.A. Irinotecan: mechanisms of tumor resistance and novel strategies for modulating its activity. Ann. Oncol. 2002, 13, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Pyrhönen, S.; James, R.D.; Punt, C.J.; Hickish, T.F.; Heikkila, R.; Johannesen, T.B.; Starkhammar, H.; A Topham, C.; Awad, L.; et al. Randomised trial of irinotecan plus supportive care versus supportive care alone after fluorouracil failure for patients with metastatic colorectal cancer. Lancet 1998, 352, 1413–1418. [Google Scholar] [CrossRef]
- Saltz, L.B.; Cox, J.V.; Blanke, C.; Rosen, L.S.; Fehrenbacher, L.; Moore, M.J.; Maroun, J.A.; Ackland, S.P.; Locker, P.K.; Pirotta, N.; et al. Irinotecan plus Fluorouracil and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2000, 343, 905–914. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Cunningham, D.; Roth, A.D.; Navarro, M.; James, R.D.; Karasek, P.; Jandik, P.; Iveson, T.; Carmichael, J.; Alakl, M.; et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar] [CrossRef]
- Ilson, D.H.; Saltz, L.; Enzinger, P.; Huang, Y.; Kornblith, A.; Gollub, M.; O'Reilly, E.; Schwartz, G.; DeGroff, J.; Gonzalez, G.; et al. Phase II Trial of Weekly Irinotecan Plus Cisplatin in Advanced Esophageal Cancer. J. Clin. Oncol. 1999, 17, 3270–3275. [Google Scholar] [CrossRef]
- Ilson, D.H. Phase II trial of weekly irinotecan/cisplatin in advanced esophageal cancer. Oncology 2004, 18, 22–5. [Google Scholar] [CrossRef]
- Setty, B.A.; Stanek, J.R.; Mascarenhas, L.; Miller, A.; Bagatell, R.; Okcu, F.; et al. VIncristine, irinotecan, and temozolomide in children and adolescents with relapsed rhabdomyosarcoma. Pediatr Blood Cancer 2018, 65, 1–10. [Google Scholar] [CrossRef]
- Bailly, C. Irinotecan: 25 years of cancer treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, B.T.; Schöffski, P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015, 29, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Yen, C.-J.; Hsu, C.-H.; Su, W.-P.; Yeh, K.-H.; Lu, Y.-S.; Cheng, A.-L.; Huang, D.C.-L.; Fritsch, H.; Voss, F.; et al. A phase I study of two dosing schedules of volasertib (BI 6727), an intravenous polo-like kinase inhibitor, in patients with advanced solid malignancies. Br. J. Cancer 2014, 110, 2434–2440. [Google Scholar] [CrossRef]
- Rudolph, D.; Impagnatiello, M.A.; Blaukopf, C.; Sommer, C.; Gerlich, D.W.; Roth, M.; et al. Efficacy and mechanism of action of volasertib, a potent and selective inhibitor of Polo-like kinases, in preclinical models of acute myeloid leukemia. J Pharmacol Exp Ther 2015, 352, 579–589. [Google Scholar] [CrossRef]
- Janning, M.; Fiedler, W. Volasertib for the treatment of acute myeloid leukemia: a review of preclinical and clinical development. Futur. Oncol. 2014, 10, 1157–1165. [Google Scholar] [CrossRef]
- Gatz, S.A.; Aladowicz, E.; Casanova, M.; Chisholm, J.C.; Kearns, P.R.; Fulda, S.; Geoerger, B.; Schäfer, B.W.; Shipley, J.M. A Perspective on Polo-Like Kinase-1 Inhibition for the Treatment of Rhabdomyosarcomas. Front. Oncol. 2019, 9, 1271. [Google Scholar] [CrossRef]
- Macedo, A.; Ferreira, P.V.; Barroso, U.; Demarchi, G.T.; Garrone, G.; Liguori, R.; Caran, E.; Ortiz, V. Sexual function in teenagers after multimodal treatment of pelvic rhabdomyosarcoma: A preliminary report. J. Pediatr. Urol. 2010, 6, 605–608. [Google Scholar] [CrossRef]
- Gupta, A.A.; Chi, Y.Y.; Anderson, J.R.; Lyden, E.; Weigel, B.; Arndt, C.; et al. Patterns of chemotherapy-induced toxicities and outcome in children and adolescents with metastatic rhabdomyosarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 2017, 64, 1–11. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Siri, M.; Dastghaib, S.; Zamani, M.; Rahmani-Kukia, N.; Geraylow, K.R.; Fakher, S.; et al. Autophagy, unfolded protein response, and neuropilin-1 cross-talk in SARS-CoV-2 infection: What can be learned from other coronaviruses. Int J Mol Sci 2021, 22, 5992. [Google Scholar] [CrossRef] [PubMed]
- Meier, P.; Finch, A.; Evan, G. Apoptosis in development. Nature 2000, 407, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef]
- Behrouj, H.; Seghatoleslam, A.; Mokarram, P.; Ghavami, S. Effect of casein kinase 1alpha inhibition on autophagy flux and the AKT/phospho-beta-catenin (S552) axis in HCT116, a RAS-mutated colorectal cancer cell line. Can J Physiol Pharmacol 2021, 99, 284–293. [Google Scholar] [CrossRef]
- Häcker, G. The morphology of apoptosis. Cell Tissue Res 2000, 301, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Aftabi, S.; Moazeni-Roodi, A.; Sarani, H.; Wiechec, E.; Ghavami, S. Association of CASP8 polymorphisms and cancer susceptibility: A meta-analysis. Eur. J. Pharmacol. 2020, 881, 173201. [Google Scholar] [CrossRef]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular Mechanisms of Apoptosis and Roles in Cancer Development and Treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, I.; Vosoughi, A.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Sheikholeslami, K.; Sher, A.A.; Lockman, S.; Kroft, D.; Ganjibakhsh, M.; Nejati-Koshki, K.; Shojaei, S.; Ghavami, S.; Rastegar, M. Simvastatin Induces Apoptosis in Medulloblastoma Brain Tumor Cells via Mevalonate Cascade Prenylation Substrates. Cancers 2019, 11, 994. [Google Scholar] [CrossRef]
- Ghavami, S.; Cunnington, R.H.; Yeganeh, B.; Davies, J.J.; Rattan, S.G.; Bathe, K.; Kavosh, M.; Los, M.J.; Freed, D.H.; Klonisch, T.; et al. Autophagy regulates trans fatty acid-mediated apoptosis in primary cardiac myofibroblasts. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2012, 1823, 2274–2286. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Sharma, P.; Yeganeh, B.; Ojo, O.O.; Jha, A.; Mutawe, M.M.; Halayko, A.J. Airway mesenchymal cell death by mevalonate cascade inhibition: integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim Biophys Acta. 2014, 1843, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Karami, S.; Sarabandi, S.; Moazeni-Roodi, A.; Małecki, A.; Ghavami, S.; Wiechec, E. Association between PD-1 and PD-L1 Polymorphisms and the Risk of Cancer: A Meta-Analysis of Case-Control Studies. Cancers 2019, 11, 1150. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Moazeni-Roodi, A.; Ghavami, S. Association between CASP3 polymorphisms and overall cancer risk: A meta-analysis of case-control studies. J. Cell. Biochem. 2018, 120, 7199–7210. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.-J.; Yuan, J. Cleavage of BID by Caspase 8 Mediates the Mitochondrial Damage in the Fas Pathway of Apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Makin, G.; Dive, C. Apoptosis and cancer chemotherapy. Trends Cell Biol 2001, 11, S22–S26. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Wong, R.S.Y. Apoptosis in cancer: from pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Cao, L.; Yu, Y.; Bilke, S.; Walker, R.L.; Mayeenuddin, L.H.; Azorsa, D.O.; Yang, F.; Pineda, M.; Helman, L.J.; Meltzer, P.S. Genome-Wide Identification of PAX3-FKHR Binding Sites in Rhabdomyosarcoma Reveals Candidate Target Genes Important for Development and Cancer. Cancer Res 2010, 70, 6497–6508. [Google Scholar] [CrossRef]
- Taylor Vi, J.G.T.; Cheuk, A.T.; Tsang, P.S.; Chung, J.-Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.-R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 119, 3395–3407. [Google Scholar] [CrossRef]
- Packham, G.; White, E.L.; Eischen, C.M.; Yang, H.; Parganas, E.; Ihle, J.N.; et al. Selective regulation of Bcl-XL by a Jak kinase-dependent pathway is bypassed in murine hematopoietic malignancies. Genes Dev 1998, 12, 2475–2487. [Google Scholar] [CrossRef]
- A McCubrey, J.; Steelman, L.S.; Abrams, S.L.; E Bertrand, F.; E Ludwig, D.; Bäsecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia 2008, 22, 708–722. [Google Scholar] [CrossRef]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Taulli, R.; Scuoppo, C.; Bersani, F.; Accornero, P.; Forni, P.E.; Miretti, S.; Grinza, A.; Allegra, P.; Schmitt-Ney, M.; Crepaldi, T.; et al. Validation of Met as a Therapeutic Target in Alveolar and Embryonal Rhabdomyosarcoma. Cancer Res 2006, 66, 4742–4749. [Google Scholar] [CrossRef] [PubMed]
- Wachtel, M.; Rakic, J.; Okoniewski, M.; Bode, P.; Niggli, F.; Schäfer, B.W. FGFR4 signaling couples to Bim and not Bmf to discriminate subsets of alveolar rhabdomyosarcoma cells. Int. J. Cancer 2014, 135, 1543–1552. [Google Scholar] [CrossRef]
- Crose, L.E.; Etheridge, K.T.; Chen, C.; Belyea, B.; Talbot, L.J.; Bentley, R.C.; Linardic, C.M. FGFR4 Blockade Exerts Distinct Antitumorigenic Effects in Human Embryonal versus Alveolar Rhabdomyosarcoma. Clin. Cancer Res. 2012, 18, 3780–3790. [Google Scholar] [CrossRef]
- Ehnman, M.; Missiaglia, E.; Folestad, E.; Selfe, J.; Strell, C.; Thway, K.; Brodin, B.; Pietras, K.; Shipley, J.; Östman, A.; et al. Distinct Effects of Ligand-Induced PDGFRα and PDGFRβ Signaling in the Human Rhabdomyosarcoma Tumor Cell and Stroma Cell Compartments. Cancer Res 2013, 73, 2139–2149. [Google Scholar] [CrossRef]
- Shukla, N.; Ameur, N.; Yilmaz, I.; Nafa, K.; Lau, C.-Y.; Marchetti, A.; Borsu, L.; Barr, F.G.; Ladanyi, M. Oncogene Mutation Profiling of Pediatric Solid Tumors Reveals Significant Subsets of Embryonal Rhabdomyosarcoma and Neuroblastoma with Mutated Genes in Growth Signaling Pathways. Clin. Cancer Res. 2012, 18, 748–757. [Google Scholar] [CrossRef]
- Stewart, E.; McEvoy, J.; Wang, H.; Chen, X.; Honnell, V.; Ocarz, M.; Gordon, B.; Dapper, J.; Blankenship, K.; Yang, Y.; et al. Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell 2018, 34, 411–426. [Google Scholar] [CrossRef]
- Davicioni, E.; Finckenstein, F.G.; Shahbazian, V.; Buckley, J.D.; Triche, T.J.; Anderson, M.J. Identification of a PAX-FKHR Gene Expression Signature that Defines Molecular Classes and Determines the Prognosis of Alveolar Rhabdomyosarcomas. Cancer Res 2006, 66, 6936–6946. [Google Scholar] [CrossRef]
- Romualdi, C.; De Pittà, C.; Tombolan, L.; Bortoluzzi, S.; Sartori, F.; Rosolen, A.; et al. Defining the gene expression signature of rhabdomyosarcoma by meta-analysis. BMC Genomics 2006, 7, 287. [Google Scholar] [CrossRef]
- Li, J.; Simpson, L.; Takahashi, M.; Miliaresis, C.; Myers, M.P.; Tonks, N.; Parsons, R. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 1998, 58, 5667–5672. [Google Scholar] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; et al. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.; Arenkiel, B.R.; Coffin, C.M.; El-Bardeesy, N.; DePinho, R.A.; Capecchi, M.R. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Minerva Anestesiol. 2004, 18, 2614–2626. [Google Scholar] [CrossRef]
- Li, Z.; Yu, X.; Shen, J.; Liu, Y.; Chan, M.T.V.; Wu, W.K.K. MicroRNA dysregulation in rhabdomyosarcoma: a new player enters the game. Cell Prolif. 2015, 48, 511–516. [Google Scholar] [CrossRef]
- Huang, H.-j.; Liu, J.; Hua, H.; Li, S.-e.; Zhao, J.; Yue, S.; et al. MiR-214 and N-ras regulatory loop suppresses rhabdomyosarcoma cell growth and xenograft tumorigenesis. Oncotarget 2014, 5, 2161. [Google Scholar] [CrossRef]
- Diao, Y.; Guo, X.; Jiang, L.; Wang, G.; Zhang, C.; Wan, J.; Jin, Y.; Wu, Z. miR-203, a Tumor Suppressor Frequently Down-regulated by Promoter Hypermethylation in Rhabdomyosarcoma. J. Biol. Chem. 2014, 289, 529–539. [Google Scholar] [CrossRef]
- Megiorni, F.; Cialfi, S.; McDowell, H.P.; Felsani, A.; Camero, S.; Guffanti, A.; Pizer, B.; Clerico, A.; De Grazia, A.; Pizzuti, A.; et al. Deep Sequencing the MicroRNA Profile in Rhabdomyosarcoma Reveals Down-regulation of miR-378 Family Members. BMC Cancer 2014, 14, 880. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, Y.; Liu, J.; Wang, X.; Yan, Q. Cyclophosphamide-induced apoptosis in A431 cells is inhibited by fucosyltransferase IV. J. Cell. Biochem. 2011, 112, 1376–1383. [Google Scholar] [CrossRef]
- Becker, R.; Ritter, A.; Eichhorn, U.; Lips, J.; Bertram, B.; Wiessler, M.; Zdzienicka, M.Z.; Kaina, B. Induction of DNA breaks and apoptosis in crosslink-hypersensitive V79 cells by the cytostatic drug β-D-glucosyl-ifosfamide mustard. Br. J. Cancer 2002, 86, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Je, Y.-T.; Chun, K.-H. AKT is translocated to the mitochondria during etoposide-induced apoptosis of HeLa cells. Mol. Med. Rep. 2015, 12, 7577–7581. [Google Scholar] [CrossRef]
- Jeong, C.-H.; Chun, K.-S.; Kundu, J.; Park, B. Phosphorylation of Smac by Akt promotes the caspase-3 activation during etoposide-induced apoptosis in HeLa cells. Mol. Carcinog. 2015, 54, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-F.; Chen, C.-L.; Jan, M.-S.; Hsu, L.-J.; Wu, R.-H.; Fang, Y.-T.; Tang, M.-J.; Chang, W.-C.; Lin, Y.-S. Bcl-2 Rescues Ceramide- and Etoposide-induced Mitochondrial Apoptosis through Blockage of Caspase-2 Activation. J. Biol. Chem. 2005, 280, 23758–23765. [Google Scholar] [CrossRef]
- Wang, M.-J.; Liu, S.; Liu, Y.; Zheng, D. Actinomycin D enhances TRAIL-induced caspase-dependent and -independent apoptosis in SH-SY5Y neuroblastoma cells. Neurosci. Res. 2007, 59, 40–46. [Google Scholar] [CrossRef]
- Wang, L.-J.; Chiou, J.-T.; Lee, Y.-C.; Huang, C.-H.; Shi, Y.-J.; Chang, L.-S. SIRT3, PP2A and TTP protein stability in the presence of TNF-α on vincristine-induced apoptosis of leukaemia cells. J Cell Mol Med 2020, 24, 2552–2565. [Google Scholar] [CrossRef]
- Lin, S.-F.; Lin, J.-D.; Yeh, C.-N.; Huang, Y.-T.; Chou, T.-C.; Wong, R.J. Targeting PLKs as a therapeutic approach to well-differentiated thyroid cancer. Endocrine-Related Cancer 2019, 26, 727–738. [Google Scholar] [CrossRef]
- Gomez-Bougie, P.; Oliver, L.; Le Gouill, S.; Bataille, R.; Amiot, M. Melphalan-induced apoptosis in multiple myeloma cells is associated with a cleavage of Mcl-1 and Bim and a decrease in the Mcl-1/Bim complex. Oncogene 2005, 24, 8076–8079. [Google Scholar] [CrossRef]
- Moghadam, A.R.; da Silva Rosa, S.C.; Samiei, E.; Alizadeh, J.; Field, J.; Kawalec, P.; Thliveris, J.; Akbari, M.; Ghavami, S.; Gordon, J.W. Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells. Cell Death Discov. 2018, 4, 52. [Google Scholar] [CrossRef] [PubMed]
- Takeba, Y.; Sekine, S.; Kumai, T.; Matsumoto, N.; Nakaya, S.; Tsuzuki, Y.; Yanagida, Y.; Nakano, H.; Asakura, T.; Ohtsubo, T.; et al. Irinotecan-Induced Apoptosis Is Inhibited by Increased P-Glycoprotein Expression and Decreased p53 in Human Hepatocellular Carcinoma Cells. Biol. Pharm. Bull. 2007, 30, 1400–1406. [Google Scholar] [CrossRef]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by pOncogene 2003, 22, 9030–9040. [CrossRef]
- Hosoi, H.; Dilling, M.B.; Shikata, T.; Liu, L.N.; Shu, L.; A Ashmun, R.; Germain, G.S.; Abraham, R.T.; Houghton, P.J. Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res. 1999, 59, 886. [Google Scholar]
- Sehgal, S.N. Sirolimus: its discovery, biological properties, and mechanism of action. Transplant Proc 2003, 35, S7–S14. [Google Scholar] [CrossRef]
- Harada, H.; Andersen, J.S.; Mann, M.; Terada, N.; Korsmeyer, S.J. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci USA 2001, 98, 9666–9670. [Google Scholar] [CrossRef] [PubMed]
- Kaylani, S.Z.; Xu, J.; Srivastava, R.K.; Kopelovich, L.; Pressey, J.G.; Athar, M. Rapamycin targeting mTOR and hedgehog signaling pathways blocks human rhabdomyosarcoma growth in xenograft murine model. Biochem. Biophys. Res. Commun. 2013, 435, 557–561. [Google Scholar] [CrossRef]
- Trucco, M.M.; Meyer, C.F.; Thornton, K.A.; Shah, P.; Chen, A.R.; Wilky, B.A.; Carrera-Haro, M.A.; Boyer, L.C.; Ferreira, M.F.; Shafique, U.; et al. A phase II study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcomas. Clin. Sarcoma Res. 2018, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Kieran, M.W.; Grupp, S.; Perek, D.; Clancy, J.; Krygowski, M.; Ananthakrishnan, R.; Boni, J.P.; Berkenblit, A.; Spunt, S.L. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur. J. Cancer 2012, 48, 253–262. [Google Scholar] [CrossRef]
- Miyoshi, K.; Kohashi, K.; Fushimi, F.; Yamamoto, H.; Kishimoto, J.; Taguchi, T.; Iwamoto, Y.; Oda, Y. Close correlation between CXCR4 and VEGF expression and frequent CXCR7 expression in rhabdomyosarcoma. Hum. Pathol. 2014, 45, 1900–1909. [Google Scholar] [CrossRef]
- Mascarenhas, L.; Chi, Y.-Y.; Hingorani, P.; Anderson, J.R.; Lyden, E.R.; Rodeberg, D.A.; Indelicato, D.J.; Kao, S.C.; Dasgupta, R.; Spunt, S.L.; et al. Randomized Phase II Trial of Bevacizumab or Temsirolimus in Combination With Chemotherapy for First Relapse Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 2866–2874. [Google Scholar] [CrossRef]
- McKian, K.P.; Haluska, P. Cixutumumab. Expert Opin Investig Drugs 2009, 18, 1025–1033. [Google Scholar] [CrossRef]
- Attias-Geva, Z.; Bentov, I.; Ludwig, D.L.; Fishman, A.; Bruchim, I.; Werner, H. Insulin-like growth factor-I receptor (IGF-IR) targeting with monoclonal antibody cixutumumab (IMC-A12) inhibits IGF-I action in endometrial cancer cells. Eur. J. Cancer 2011, 47, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Chugh, R.; Griffith, K.; Davis, E.; Thomas, D.; Zavala, J.; Metko, G.; Brockstein, B.; Undevia, S.; Stadler, W.; Schuetze, S. Doxorubicin plus the IGF-1R antibody cixutumumab in soft tissue sarcoma: a phase I study using the TITE-CRM model. Ann. Oncol. 2015, 26, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Malempati, S.; Weigel, B.J.; Chi, Y.Y.; Tian, J.; Anderson, J.R.; Parham, D.M.; et al. The addition of cixutumumab or temozolomide to intensive multiagent chemotherapy is feasible but does not improve outcome for patients with metastatic rhabdomyosarcoma: a report from the Children’s Oncology Group. Cancer 2019, 125, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Weigel, B.; Malempati, S.; Reid, J.M.; Voss, S.D.; Cho, S.Y.; Chen, H.X.; et al. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children's Oncology Group. Pediatr Blood Cancer 2014, 61, 452–456. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Tap, W.D.; Qin, L.-X.; Livingston, M.B.; Undevia, S.D.; Chmielowski, B.; Agulnik, M.; Schuetze, S.M.; Reed, D.R.; Okuno, S.H.; et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2013, 14, 371–382. [Google Scholar] [CrossRef]
- Asmane, I.; Watkin, E.; Alberti, L.; Duc, A.; Marec-Berard, P.; Ray-Coquard, I.; Cassier, P.; Decouvelaere, A.-V.; Ranchère, D.; Kurtz, J.-E.; et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: A predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur. J. Cancer 2012, 48, 3027–3035. [Google Scholar] [CrossRef]
- Forde, P.M.; Rudin, C.M. Crizotinib in the treatment of non-small-cell lung cancer. Expert Opin. Pharmacother. 2012, 13, 1195–1201. [Google Scholar] [CrossRef]
- Sahu, A.; Prabhash, K.; Noronha, V.; Joshi, A.; Desai, S. Crizotinib: A comprehensive review. South Asian J Cancer 2013, 2, 91–97. [Google Scholar]
- Zhou, X.; Zhang, X.; Wu, Z.; Xu, X.; Guo, M.; Zhai, X.; Zuo, D.; Wu, Y. The novel ALK inhibitor ZX-29 induces apoptosis through inhibiting ALK and inducing ROS-mediated endoplasmic reticulum stress in Karpas299 cells. J. Biochem. Mol. Toxicol. 2021, 35, e22666. [Google Scholar] [CrossRef]
- Frentzel, J.; Sorrentino, D.; Giuriato, S. Targeting Autophagy in ALK-Associated Cancers. Cancers 2017, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- van Erp, A.E.M.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Fleuren, E.D.G.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Targeting anaplastic lymphoma kinase (ALK) in rhabdomyosarcoma (RMS) with the second-generation ALK inhibitor ceritinib. Target Oncol 2017, 12, 815–826. [Google Scholar] [CrossRef]
- Wierdl, M.; Tsurkan, L.; Chi, L.; Hatfield, M.J.; Tollemar, V.; Bradley, C.; Chen, X.; Qu, C.; Potter, P.M. Targeting ALK in pediatric RMS does not induce antitumor activity in vivo. Cancer Chemother. Pharmacol. 2018, 82, 251–263. [Google Scholar] [CrossRef]
- Schöffski, P.; Wozniak, A.; Leahy, M.G.; Aamdal, S.; Rutkowski, P.; Bauer, S.; Richter, S.; Grünwald, V.; Debiec-Rychter, M.; Sciot, R.; et al. The tyrosine kinase inhibitor crizotinib does not have clinically meaningful activity in heavily pre-treated patients with advanced alveolar rhabdomyosarcoma with FOXO rearrangement: European Organisation for Research and Treatment of Cancer phase 2 trial 90101 ‘CREATE’. Eur. J. Cancer 2018, 94, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Widemann, B.C.; Krailo, M.; Jayaprakash, N.; Fox, E.; Weigel, B.; et al. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: A report from the Children's Oncology Group. Pediatr Blood Cancer 2015, 62, 1562–1566. [Google Scholar] [CrossRef]
- Dolgikh, N.; Fulda, S. Rhabdomyosarcoma cells are susceptible to cell death by LDK378 alone or in combination with sorafenib independently of anaplastic lymphoma kinase status. Anti-Cancer Drugs 2017, 28, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.; Hingorani, P.; Gupta, A.A. Emerging Molecular-Targeted Therapies in Early-Phase Clinical Trials and Preclinical Models. Am. Soc. Clin. Oncol. Educ. Book 2013, 420–424. [Google Scholar] [CrossRef]
- Van Der Graaf, W.T.; Blay, J.-Y.; Chawla, S.P.; Kim, D.-W.; Bui-Nguyen, B.; Casali, P.G.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. The Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Sleijfer, S.; Ray-Coquard, I.; Papai, Z.; Le Cesne, A.; Scurr, M.; Schöffski, P.; et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European Organisation for Research and Treatment of Cancer–Soft Tissue and Bone Sarcoma Group (EORTC study 62043). J Clin Oncol 2009, 27, 3126–3132. [Google Scholar]
- Riedel, R.F.; Ballman, K.V.; Lu, Y.; Attia, S.; Loggers, E.T.; Ganjoo, K.N.; Livingston, M.B.; Chow, W.; Wright, J.; Ward, J.H.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase II Study of Regorafenib Versus Placebo in Advanced/Metastatic, Treatment-Refractory Liposarcoma: Results from the SARC024 Study. Oncol. 2020, 25, e1655–e1662. [Google Scholar] [CrossRef]
- Guan, Y.; Sakai, R.; Rinehart, K.L.; Wang, A.H. Molecular and crystal structures of ecteinascidins: potent antitumor compounds from the Caribbean tunicate Ecteinascidia turbinata. J. Biomol. Struct. Dyn. 1993, 10, 793–818. [Google Scholar]
- Banerjee, P.; Zhang, R.; Ivan, C.; Galletti, G.; Clise-Dwyer, K.; Barbaglio, F.; Scarfò, L.; Aracil, M.; Klein, C.; Wierda, W.; et al. Trabectedin Reveals a Strategy of Immunomodulation in Chronic Lymphocytic Leukemia. Cancer Immunol. Res. 2019, 7, 2036–2051. [Google Scholar] [CrossRef] [PubMed]
- Baruchel, S.; Pappo, A.; Krailo, M.; Baker, K.S.; Wu, B.; Villaluna, D.; et al. A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: a report from the Children’s Oncology Group. Eur J Cancer 2012, 48, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications – writers that read. Embo Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Enßle, J.C.; Boedicker, C.; Wanior, M.; Vogler, M.; Knapp, S.; Fulda, S. Co-targeting of BET proteins and HDACs as a novel approach to trigger apoptosis in rhabdomyosarcoma cells. Cancer Lett. 2018, 428, 160–172. [Google Scholar] [CrossRef]
- Laszig, S.; Boedicker, C.; Weiser, T.; Knapp, S.; Fulda, S. The novel dual BET/HDAC inhibitor TW09 mediates cell death by mitochondrial apoptosis in rhabdomyosarcoma cells. Cancer Lett. 2020, 486, 46–57. [Google Scholar] [CrossRef]
- Tomoyasu, C.; Kikuchi, K.; Kaneda, D.; Yagyu, S.; Miyachi, M.; Tsuchiya, K.; et al. OBP-801, a novel histone deacetylase inhibitor, induces M-phase arrest and apoptosis in rhabdomyosarcoma cells. Oncol Rep 2019, 41, 643–649. [Google Scholar] [CrossRef]
- Heinicke, U.; Fulda, S. Chemosensitization of rhabdomyosarcoma cells by the histone deacetylase inhibitor SAHA. Cancer Lett. 2014, 351, 50–58. [Google Scholar] [CrossRef]
- Bharathy, N.; Berlow, N.E.; Wang, E.; Abraham, J.; Settelmeyer, T.P.; Hooper, J.E.; et al. Preclinical rationale for entinostat in embryonal rhabdomyosarcoma. Skelet Muscle 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- de Haan, R.; van Werkhoven, E.; Heuvel, M.v.D.; Peulen, H.M.U.; Sonke, G.S.; Elkhuizen, P.; Brekel, M.W.M.v.D.; Tesselaar, M.E.T.; Vens, C.; Schellens, J.H.M.; et al. Study protocols of three parallel phase 1 trials combining radical radiotherapy with the PARP inhibitor olaparib. BMC Cancer 2019, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, P.; Lequesne, J.; Grellard, J.-M.; Dugué, A.; Coquan, E.; Brachet, P.-E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef]
- Camero, S.; Ceccarelli, S.; De Felice, F.; Marampon, F.; Mannarino, O.; Camicia, L.; Vescarelli, E.; Pontecorvi, P.; Pizer, B.; Shukla, R.; et al. PARP inhibitors affect growth, survival and radiation susceptibility of human alveolar and embryonal rhabdomyosarcoma cell lines. J. Cancer Res. Clin. Oncol. 2019, 145, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Sacher, J.; Hohenegger, M. Mutual amplification of apoptosis by statin-induced mitochondrial stress and doxorubicin toxicity in human rhabdomyosarcoma cells. Br. J. Pharmacol. 2004, 143, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Łos, M.J.; et al. Pleiotropic effects of statins: A focus on cancer. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2020, 1866, 165968. [Google Scholar] [CrossRef]
- Alizadeh, J.; Shojaei, S.; da Silva Rosa, S.; Moghadam, A.R.; Zeki, A.A.; Hashemi, M.; et al. Detection of small GTPase prenylation and GTP binding using membrane fractionation and GTPase-linked immunosorbent assay. J Visu Exp 2018, e57646. [Google Scholar]
- Yeganeh, B.; Wiechec, E.; Ande, S.R.; Sharma, P.; Moghadam, A.R.; Post, M.; et al. Targeting the mevalonate cascade as a new therapeutic approach in heart disease, cancer and pulmonary disease. Pharmacol Ther 2014, 143, 87–110. [Google Scholar] [CrossRef]
- Koohestanimobarhan, S.; Salami, S.; Imeni, V.; Mohammadi, Z.; Bayat, O. Lipophilic statins antagonistically alter the major epithelial-to-mesenchymal transition signaling pathways in breast cancer stem–like cells via inhibition of the mevalonate pathway. J. Cell. Biochem. 2019, 120, 2515–2531. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Ann Rew Physiol 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Musiwaro, P.; Smith, M.; Manifava, M.; Walker, S.A.; Ktistakis, N.T. Characteristics and requirements of basal autophagy in HEK 293 cells. Autophagy 2013, 9, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Habibzadeh, P.; Dastsooz, H.; Eshraghi, M.; Los, M.J.; Klionsky, D.J.; Ghavami, S. Autophagy: The potential link between SARS-CoV-2 and cancer. Cancers 2021, 13, 5721. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Siri, M.; Behrouj, H.; Dastghaib, S.; Zamani, M.; Likus, W.; Rezaie, S.; et al. Casein Kinase-1-Alpha Inhibitor (D4476) sensitizes microsatellite instable colorectal cancer cells to 5-Fluorouracil via authophagy fux inhibition. Arch Immunol Ther Exp (Warsz) 2021, 69, 26. [Google Scholar] [CrossRef]
- Hinton, M.; Eltayeb, E.; Ghavami, S.; Dakshinamurti, S. Effect of pulsatile stretch on unfolded protein response in a new model of the pulmonary hypertensive vascular wall. Biochem. Biophys. Rep. 2021, 27, 101080. [Google Scholar] [CrossRef]
- Wang, B.; Abraham, N.; Gao, G.; Yang, Q. Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 1–9. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 2014, 112, 24–49. [Google Scholar] [CrossRef]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; et al. Alzheimer's disease pathogenesis: Role of autophagy and mitophagy focusing in microglia. Int J Mol Sci 2021, 22, 3330. [Google Scholar] [CrossRef]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt signaling pathway: A focus on Wnt/beta-catenin signaling. Biochim Biophys Acta Mol Cell Res 2021, 1868, 118926. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Oh, J.E.; Lee, H.K. Autophagy in Innate Recognition of Pathogens and Adaptive Immunity. Yonsei Med J. 2012, 53, 241–247. [Google Scholar] [CrossRef]
- Hombach-Klonisch, S.; Natarajan, S.; Thanasupawat, T.; Medapati, M.; Pathak, A.; Ghavami, S.; Klonisch, T. Mechanisms of Therapeutic Resistance in Cancer (Stem) Cells with Emphasis on Thyroid Cancer Cells. Front. Endocrinol. 2014, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Mutawe, M.M.; Schaafsma, D.; Yeganeh, B.; Unruh, H.; Klonisch, T.; Halayko, A.J. Geranylgeranyl transferase 1 modulates autophagy and apoptosis in human airway smooth muscle. Am. J. Physiol. Cell. Mol. Physiol. 2012, 302, L420–L428. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Kochan, M.M.; Stewart, V.D.; Drewnik, D.A.; Hannila, S.S.; Ghavami, S. Inhibition of Autophagy Flux Promotes Secretion of Chondroitin Sulfate Proteoglycans in Primary Rat Astrocytes. Mol. Neurobiol. 2021, 58, 6077–6091. [Google Scholar] [CrossRef]
- Zeki, A.A.; Yeganeh, B.; Kenyon, N.J.; Post, M.; Ghavami, S. Autophagy in airway diseases: a new frontier in human asthma? Allergy 2016, 71, 5–14. [Google Scholar] [CrossRef]
- Alizadeh, J.; Glogowska, A.; Thliveris, J.; Kalantari, F.; Shojaei, S.; Hombach-Klonisch, S.; Klonisch, T.; Ghavami, S. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.-F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Zhou, S.; Zhao, L.; Kuang, M.; Zhang, B.; Liang, Z.; Yi, T.; Wei, Y.; Zhao, X. Autophagy in tumorigenesis and cancer therapy: Dr. Jekyll or Mr. Hyde? Cancer Lett. 2012, 323, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Hanif, E.A.M.; Chin, S.-F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, A.R. Targeting Rhabdomyosarcoma with Temozolomide: How Autophagy Regulates TMZ-induced Apoptosis in Rhabdomyosarcoma Cells. 2018: University of Manitoba.
- Alizadeh, J.; Zeki, A.A.; Mirzaei, N.; Tewary, S.; Moghadam, A.R.; Glogowska, A.; Nagakannan, P.; Eftekharpour, E.; Wiechec, E.; Gordon, J.W.; et al. Mevalonate Cascade Inhibition by Simvastatin Induces the Intrinsic Apoptosis Pathway via Depletion of Isoprenoids in Tumor Cells. Sci. Rep. 2017, 7, 44841. [Google Scholar] [CrossRef]
- Likus, W.; Siemianowicz, K.; Bieńk, K.; Pakuła, M.; Pathak, H.; Dutta, C.; et al. Could drugs inhibiting the mevalonate pathway also target cancer stem cells? Drug resist updat 2016, 25, 13–25. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Liu, P.-Y.; Liao, J.K. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Su, Z.; DeWitt, J.P.; Xie, L.; Chen, Y.; Li, X.; Han, L.; Li, D.; Xia, J.; Zhang, Y.; et al. Fluvastatin Prevents Lung Adenocarcinoma Bone Metastasis by Triggering Autophagy. EBioMedicine 2017, 19, 49–59. [Google Scholar] [CrossRef]
- Peng, X.; Li, W.; Yuan, L.; Mehta, R.G.; Kopelovich, L.; McCormick, D.L. Inhibition of Proliferation and Induction of Autophagy by Atorvastatin in PC3 Prostate Cancer Cells Correlate with Downregulation of Bcl2 and Upregulation of miR-182 and p21. PLOS ONE 2013, 8, e70442. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, I.; Vosoughi, A.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Sacher, J.; Hohenegger, M. Mutual amplification of apoptosis by statin-induced mitochondrial stress and doxorubicin toxicity in human rhabdomyosarcoma cells. Br. J. Pharmacol. 2004, 143, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Raney, R.B.; Walterhouse, D.O.; Meza, J.L.; Andrassy, R.J.; Breneman, J.C.; Crist, W.M.; et al. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. J Clin Oncol 2011, 29, 1312–1318. [Google Scholar] [PubMed]
- Walterhouse, D.O.; Pappo, A.S.; Meza, J.L.; Breneman, J.C.; Hayes-Jordan, A.; Parham, D.M.; et al. Reduction of cyclophosphamide dose for patients with subset 2 low-risk rhabdomyosarcoma is associated with an increased risk of recurrence: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Cancer 2017, 123, 2368–2375. [Google Scholar] [CrossRef]
- Crist, W.M.; Anderson, J.R.; Meza, J.L.; Fryer, C.; Raney, R.B.; Ruymann, F.B.; et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol 2001, 19, 3091–3102. [Google Scholar] [CrossRef]
- Wang, Y.; Xia, C.; Lv, Y.; Li, C.; Mei, Q.; Li, H.; Wang, H.; Li, S. Crosstalk Influence between P38MAPK and Autophagy on Mitochondria-Mediated Apoptosis Induced by Anti-Fas Antibody/Actinomycin D in Human Hepatoma Bel-7402 Cells. Molecules 2017, 22, 1705. [Google Scholar] [CrossRef]
- Seitz; Seitz, G. ; Bonin, M.; Fuchs, J.; Poths, S.; Ruck, P.; Warmann, S.W.; Armeanu-Ebinger, S. Inhibition of glutathione-S-transferase as a treatment strategy for multidrug resistance in childhood rhabdomyosarcoma. Int. J. Oncol. 2010, 36, 491–500. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Zhou, J.; Tan, S.-H.; Nicolas, V.; Bauvy, C.; Yang, N.-D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.-M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef]
- Salerno, M.; Avnet, S.; Bonuccelli, G.; Hosogi, S.; Granchi, D.; Baldini, N. Impairment of Lysosomal Activity as a Therapeutic Modality Targeting Cancer Stem Cells of Embryonal Rhabdomyosarcoma Cell Line RD. PLOS ONE 2014, 9, e110340–e110340. [Google Scholar] [CrossRef]
- Zhou, H.; Shen, T.; Shang, C.; Luo, Y.; Liu, L.; Yan, J.; Li, Y.; Huang, S. Ciclopirox induces autophagy through reactive oxygen species-mediated activation of JNK signaling pathway. Oncotarget 2014, 5, 10140–10150. [Google Scholar] [CrossRef] [PubMed]
- Peron, M.; Bonvini, P.; Rosolen, A. Effect of inhibition of the Ubiquitin-Proteasome System and Hsp90 on growth and survival of Rhabdomyosarcoma cells in vitro. BMC Cancer 2012, 12, 233–233. [Google Scholar] [CrossRef] [PubMed]
- Rapino, F.; Jung, M.; Fulda, S. BAG3 induction is required to mitigate proteotoxicity via selective autophagy following inhibition of constitutive protein degradation pathways. Oncogene 2014, 33, 1713–1724. [Google Scholar] [CrossRef] [PubMed]
- Sadeghabadi, Z.A.; Nourbakhsh, M.; Pasalar, P.; Emamgholipour, S.; Golestani, A.; Larijani, B.; Razzaghy-Azar, M. Reduced gene expression of sirtuins and active AMPK levels in children and adolescents with obesity and insulin resistance. Obes. Res. Clin. Pr. 2018, 12, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 Regulates Adipocyte Differentiation through FoxO1 Acetylation/Deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef]
- Ding, N.; Bonham, E.M.; Hannon, B.E.; Amick, T.R.; Baylin, S.B.; O'Hagan, H.M. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J. Mol. Cell Biol. 2016, 8, 244–254. [Google Scholar] [CrossRef]
- Wu, S.; Jiang, J.; Liu, J.; Wang, X.; Gan, Y.; Tang, Y. Meta-analysis of SIRT1 expression as a prognostic marker for overall survival in gastrointestinal cancer. Oncotarget 2017, 8, 62589–62599. [Google Scholar] [CrossRef]
- Grbesa, I.; Pajares, M.J.; Martínez-Terroba, E.; Agorreta, J.; Mikecin, A.-M.; Larráyoz, M.; Idoate, M.A.; Gall-Troselj, K.; Pio, R.; Montuenga, L.M. Expression of Sirtuin 1 and 2 Is Associated with Poor Prognosis in Non-Small Cell Lung Cancer Patients. PLOS ONE 2015, 10, e0124670–e0124670. [Google Scholar] [CrossRef]
- Chen, W.; Yuan, H.; Su, L. The emerging and diverse roles of sirtuins in cancer: a clinical perspective. OncoTargets Ther. 2013, 6, 1399–416. [Google Scholar] [CrossRef]
- Ma, L.; Maruwge, W.; Strambi, A.; D'Arcy, P.; Pellegrini, P.; Kis, L.; de Milito, A.; Lain, S.; Brodin, B. SIRT1 and SIRT2 inhibition impairs pediatric soft tissue sarcoma growth. Cell Death Dis. 2014, 5, e1483–e1483. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef]
- Jones, P.M.; George, A.M. The ABC transporter structure and mechanism: perspectives on recent research. Cell. Mol. Life Sci. 2004, 61, 682–699. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A Family of Drug Transporters: the Multidrug Resistance-Associated Proteins. JNCI J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Li, Z.; He, Z.; Qiu, J.; Zhou, S. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef]
- A Hussain, S.; Marouf, B.H. Silibinin improves the cytotoxicity of methotrexate in chemo resistant human rhabdomyosarcoma cell lines. Saudi Med J. 2013, 34, 1145–1150. [Google Scholar] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–56. [Google Scholar]
- Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039. [Google Scholar] [CrossRef]
- Sidrauski, C.; Walter, P. The Transmembrane Kinase Ire1p Is a Site-Specific Endonuclease That Initiates mRNA Splicing in the Unfolded Protein Response. Cell 1997, 90, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Schröder, M.; Kaufman, R.J. Ligand-independent Dimerization Activates the Stress Response Kinases IRE1 and PERK in the Lumen of the Endoplasmic Reticulum. J. Biol. Chem. 2000, 275, 24881–24885. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases that Process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Alizadeh, J.; Juarez, M.; Samali, A.; Halayko, A.J.; Kenyon, N.J.; et al. Autophagy, apoptosis, the unfolded protein response, and lung function in idiopathic pulmonary fibrosis. Cells 2021, 10, 1642. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, F.-X.; Wang, X. A Synthetic Biology Approach Identifies the Mammalian UPR RNA Ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Weissman, J.S. Decay of Endoplasmic Reticulum-Localized mRNAs During the Unfolded Protein Response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.S.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- B'Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef]
- McCarthy, N.; Dolgikh, N.; Logue, S.; Patterson, J.B.; Zeng, Q.; Gorman, A.M.; Samali, A.; Fulda, S. The IRE1 and PERK arms of the unfolded protein response promote survival of rhabdomyosarcoma cells. Cancer Lett. 2020, 490, 76–88. [Google Scholar] [CrossRef]
- Aghaei, M.; Nasimian, A.; Rahmati, M.; Kawalec, P.; Machaj, F.; Rosik, J.; Bhushan, B.; Bathaie, S.Z.; Azarpira, N.; Los, M.J.; et al. The Role of BiP and the IRE1α–XBP1 Axis in Rhabdomyosarcoma Pathology. Cancers 2021, 13, 4927. [Google Scholar] [CrossRef]
- Stewart, E.; McEvoy, J.; Wang, H.; Chen, X.; Honnell, V.; Ocarz, M.; Gordon, B.; Dapper, J.; Blankenship, K.; Yang, Y.; et al. Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell 2018, 34, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, A.J.; Guerriero, C.J.; Olivas, V.; Sayana, A.; Shue, J.; Flanagan, J.; Asthana, S.; Paton, A.W.; Paton, J.C.; Gestwicki, J.E.; et al. Combined chemical–genetic approach identifies cytosolic HSP70 dependence in rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 9015–9020. [Google Scholar] [CrossRef]
- Sannino, S.; Guerriero, C.J.; Sabnis, A.J.; Stolz, D.B.; Wallace, C.T.; Wipf, P.; et al. Compensatory increases of select proteostasis networks after Hsp70 inhibition in cancer cells. J Cell Sci 2018, 131, 217760. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J Clin Invest 2018, 128, 1283–1299. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mixon, B.A.; Eckrich, M.J.; Lowas, S.; Engel, M.E. Vincristine, Irinotecan, and Temozolomide for Treatment of Relapsed Alveolar Rhabdomyosarcoma. J. Pediatr. Hematol. 2013, 35, e163–e166. [Google Scholar] [CrossRef]
- Moghadam, A.R.; da Silva Rosa, S.C.; Samiei, E.; Alizadeh, J.; Field, J.; Kawalec, P.; Thliveris, J.; Akbari, M.; Ghavami, S.; Gordon, J.W. Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells. Cell Death Discov. 2019, 4, 52. [Google Scholar] [CrossRef]
- Manzella, G.; Schreck, L.D.; Breunis, W.B.; Molenaar, J.; Merks, H.; Barr, F.G.; Sun, W.; Römmele, M.; Zhang, L.; Tchinda, J.; et al. Phenotypic profiling with a living biobank of primary rhabdomyosarcoma unravels disease heterogeneity and AKT sensitivity. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical mouse solid tumour models: status quo, challenges and perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef]
- Kendsersky, N.M.; Lindsay, J.M.; Kolb, E.A.; Smith, M.A.; Teicher, B.A.; Erickson, S.W.; Earley, E.J.; Mosse, Y.P.; Martinez, D.; Pogoriler, J.; et al. The B7-H3–Targeting Antibody–Drug Conjugate m276-SL-PBD Is Potently Effective Against Pediatric Cancer Preclinical Solid Tumor Models. Clin. Cancer Res. 2021, 27, 2938–2946. [Google Scholar] [CrossRef] [PubMed]
- Risbridger, G.P.; Lawrence, M.G.; A Taylor, R. PDX: Moving Beyond Drug Screening to Versatile Models for Research Discovery. J. Endocr. Soc. 2020, 4. [Google Scholar] [CrossRef]
- Imle, R.; Kommoss, F.K.F.; Banito, A. Preclinical In Vivo Modeling of Pediatric Sarcoma—Promises and Limitations. J. Clin. Med. 2021, 10, 1578. [Google Scholar] [CrossRef]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Bhimani, J.; Ball, K.; Stebbing, J. Patient-derived xenograft models—the future of personalised cancer treatment. Br. J. Cancer 2020, 122, 601–602. [Google Scholar] [CrossRef]
- Onaciu, A.; Munteanu, R.; Munteanu, V.C.; Gulei, D.; Raduly, L.; Feder, R.I.; et al. Spontaneous and induced animal models for cancer research. Diagnostics 2020, 10:, 660. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.E.; Cantor, E.L.; Ehlen, M.S.; Banerjee, A.; Malempati, S.; Stenzel, P.; Woltjer, R.L.; Gandour-Edwards, R.; Goodwin, N.C.; Yang, Y.; et al. A Patient-Derived Xenograft Model of Parameningeal Embryonal Rhabdomyosarcoma for Preclinical Studies. Sarcoma 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Stewart, E.; Federico, S.; Karlstrom, A.; Shelat, A.; Sablauer, A.; Pappo, A.; Dyer, M.A. The Childhood Solid Tumor Network: A new resource for the developmental biology and oncology research communities. Dev. Biol. 2016, 411, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Jr, D.F.C.; Jacob, A.G.; Sanford, B.L.; Montes, M.; Goodwin, A.K.; Steiner, H.; Matsa, E.; Tapia-Santos, A.S.; Bebee, T.W.; Grieves, J.; et al. A novel mouse model of rhabdomyosarcoma underscores the dichotomy of MDM2-ALT1 function in vivo. Oncogene 2018, 37, 95–106. [Google Scholar] [CrossRef]
- Grunewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol Med 2020, 12, e11131. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Roveri, M.; Pfohl, A.; Jaaks, P.; Alijaj, N.; Leroux, J.-C.; Luciani, P.; Bernasconi, M. Prolonged circulation and increased tumor accumulation of liposomal vincristine in a mouse model of rhabdomyosarcoma. Nanomedicine 2017, 12, 1135–1151. [Google Scholar] [CrossRef]
- Kemp, C.J. Animal Models of Chemical Carcinogenesis: Driving Breakthroughs in Cancer Research for 100 Years. Cold Spring Harb. Protoc. 2015, 2015, 865–74. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Kovach, A.R.; DeMonia, M.; Slemmons, K.K.; Oristian, K.M.; Chen, C.; Linardic, C.M. Expression of oncogenic HRAS in human Rh28 and RMS-YM rhabdomyosarcoma cells leads to oncogene-induced senescence. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kawagoe, R.; Sasai, K.; Li, Y.; Russell, H.R.; Curran, T.; McKinnon, P.J. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 2007, 26, 6442–6447. [Google Scholar] [CrossRef]
- Gutierrez, W.R.; Scherer, A.; McGivney, G.R.; Brockman, Q.R.; Knepper-Adrian, V.; Laverty, E.A.; Roughton, G.A.; Dodd, R.D. Divergent immune landscapes of primary and syngeneic Kras-driven mouse tumor models. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Zanola, A.; Rossi, S.; Faggi, F.; Monti, E.; Fanzani, A. Rhabdomyosarcomas: an overview on the experimental animal models. J. Cell. Mol. Med. 2012, 16, 1377–1391. [Google Scholar] [CrossRef]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; et al. Genomic profiling of childhood tumor patient-derived xenograft models to enable rational clinical trial design. Cell Rep 2019, 29, 1675–1689. [Google Scholar] [CrossRef]
- Lu, W.; Chao, T.; Ruiqi, C.; Juan, S.; Zhihong, L. Patient-derived xenograft models in musculoskeletal malignancies. J. Transl. Med. 2018, 16, 1–16. [Google Scholar] [CrossRef]
- Chen, E.Y.; Langenau, D.M. Zebrafish Models of Rhabdomyosarcoma. Methods Cell Biol 2011, 105, 383–402. [Google Scholar] [CrossRef]
- Casey, M.J.; Stewart, R.A. Pediatric Cancer Models in Zebrafish. Trends Cancer 2020, 6, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, F.; Fahs, A.; Ghayad, S.E.; Saab, R. Signaling pathways in Rhabdomyosarcoma invasion and metastasis. Cancer Metastasis Rev. 2020, 39, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Zibat, A.; Missiaglia, E.; Rosenberger, A.; Pritchard-Jones, K.; Shipley, J.; Hahn, H.; Fulda, S. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene 2010, 29, 6323–6330. [Google Scholar] [CrossRef]
- Ali, S.; Champagne, D.L.; Spaink, H.P.; Richardson, M.K. Zebrafish embryos and larvae: A new generation of disease models and drug screens. Birth Defects Res. Part C: Embryo Today: Rev. 2011, 93, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Etchin, J.; Kanki, J.; Look, AT. Zebrafish as a model for the study of human cancer, in The Zebrafish: Disease Models and Chemical Screens Detrich, H.W., Weterfield, M., and Zon, L.I.B., Editors. 2011, Academic Press. p. 309-337.
- Yan, C.; Brunson, D.; Tang, Q.; Do, D.; Iftimia, N.A.; Moore, J.C.; Hayes, M.N.; Welker, A.M.; Garcia, E.G.; Dubash, T.D.; et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell 2019, 177, 1903–1914. [Google Scholar] [CrossRef]
- Heilmann, S.; Ratnakumar, K.; Langdon, E.M.; Kansler, E.R.; Kim, I.S.; Campbell, N.R.; Perry, E.B.; McMahon, A.J.; Kaufman, C.K.; van Rooijen, E.; et al. A Quantitative System for Studying Metastasis Using Transparent Zebrafish. Cancer Res 2015, 75, 4272–4282. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; DeRan, M.T.; Ignatius, M.S.; Grandinetti, K.B.; Clagg, R.; McCarthy, K.M.; et al. Glycogen synthase kinase 3 inhibitors induce the canonical WNT/beta-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc Natl Acad Sci USA 2014, 111, 5349–5354. [Google Scholar] [CrossRef] [PubMed]
- Ignatius, M.S.; Hayes, M.N.; Lobbardi, R.; Chen, E.Y.; McCarthy, K.M.; Sreenivas, P.; et al. The NOTCH1/SNAIL1/MEF2C pathway regulates growth and self-renewal in embryonal rhabdomyosarcoma. Cell Rep 2017, 19, 2304–2318. [Google Scholar] [CrossRef]
- Scientists see human cancer in zebrafish. Cancer Discov 2019, 9, 819–820. [CrossRef]
- Tang, Q.; Moore, J.C.; Ignatius, M.S.; Tenente, I.M.; Hayes, M.N.; Garcia, E.G.; Yordán, N.T.; Bourque, C.; He, S.N.; Blackburn, J.S.; et al. Imaging tumour cell heterogeneity following cell transplantation into optically clear immune-deficient zebrafish. Nat. Commun. 2016, 7, 10358. [Google Scholar] [CrossRef]
- Hayes, M.N.; Langenau, DM. Discovering novel oncogenic pathways and new therapies using zebrafish models of sarcoma, in Zebrafish, Detrich, H.W., Westerfield, M., and Zon, L., Editors. 2017, Academic Press. p. 525-561.
- Storer, N.Y.; White, R.M.; Uong, A.; Price, E.; Nielsen, G.P.; Langenau, D.M.; Zon, L.I. Zebrafish rhabdomyosarcoma reflects the developmental stage of oncogene expression during myogenesis. Development 2013, 140, 3040–3050. [Google Scholar] [CrossRef]
- Pham, T.Q.; Robinson, K.; Xu, L.; Pavlova, M.N.; Skapek, S.X.; Chen, E.Y. HDAC6 promotes growth, migration/invasion, and self-renewal of rhabdomyosarcoma. Oncogene 2021, 40, 578–591. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Langenau, D.M.; Keefe, M.D.; Kutok, J.L.; Neuberg, D.S.; Zon, L.I. Heat shock-inducible Cre/Lox approaches to induce diverse types of tumors and hyperplasia in transgenic zebrafish. Proc. Natl. Acad. Sci. USA 2007, 104, 9410–9415. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Pugach, E.K.; Hettmer, S.; Storer, N.Y.; Liu, J.; Wills, A.A.; DiBiase, A.; Chen, E.Y.; Ignatius, M.S.; Poss, K.D.; et al. A novel chemical screening strategy in zebrafish identifies common pathways in embryogenesis and rhabdomyosarcoma development. Development 2013, 140, 2354–2364. [Google Scholar] [CrossRef]
- Kendall, G.C.; Watson, S.; Xu, L.; LaVigne, C.A.; Murchison, W.; Rakheja, D.; et al. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. Elife 2018, 7, e33800. [Google Scholar] [CrossRef]
- Tenente, I.M.; Tang, Q.; Moore, J.C.; Langenau, D.M. Normal and malignant muscle cell transplantation into immune compromised adult zebrafish. J Vis Exp 2014, 52597. [Google Scholar]
- Rai, R.; Raval, R.; Khandeparker, R.V.; Chidrawar, S.K.; Khan, A.A.; Ganpat, M.S. Tissue engineering: Step ahead in maxillofacial reconstruction. J Int Oral Health 2015, 7, 138–142. [Google Scholar] [PubMed]
- Kang, H.-W.; Lee, S.J.; Ko, I.K.; Kengla, C.; Yoo, J.J.; Atala, A. A 3D bioprinting system to produce human-scale tissue constructs with structural integrity. Nat. Biotechnol. 2016, 34, 312–319. [Google Scholar] [CrossRef]
- Ma, X.; Qu, X.; Zhu, W.; Li, Y.-S.; Yuan, S.; Zhang, H.; et al. Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting. Proc Natl Acad Sci USA 2016, 113, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Qu, X.; Zhu, J.; Ma, X.; Patel, S.; Liu, J.; Wang, P.; Lai, C.S.E.; Gou, M.; Xu, Y.; et al. Direct 3D bioprinting of prevascularized tissue constructs with complex microarchitecture. Biomaterials 2017, 124, 106–115. [Google Scholar] [CrossRef]
- Jammalamadaka, U.; Tappa, K. Recent Advances in Biomaterials for 3D Printing and Tissue Engineering. J. Funct. Biomater. 2018, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Ma, X.; Zhu, W.; Wang, P.; Miller, K.L.; Stupin, J.; Koroleva-Maharajh, A.; Hairabedian, A.; Chen, S. Scanningless and continuous 3D bioprinting of human tissues with decellularized extracellular matrix. Biomaterials 2019, 194, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Berry, D.; Moran, A.; He, F.; Tam, T.; Chen, L.; Chen, S. Controlled Growth Factor Release in 3D-Printed Hydrogels. Adv. Healthc. Mater. 2020, 9, 1900977–e1900977. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Seol, Y.-J.; Ko, I.K.; Kang, H.-W.; Lee, Y.K.; Yoo, J.J.; Atala, A.; Lee, S.J. 3D Bioprinted Human Skeletal Muscle Constructs for Muscle Function Restoration. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Jun, Y.-J.; Kim, D.Y.; Yi, H.-G.; Chae, S.-H.; Kang, J.; Lee, J.; Gao, G.; Kong, J.-S.; Jang, J.; et al. A 3D cell printed muscle construct with tissue-derived bioink for the treatment of volumetric muscle loss. Biomaterials 2019, 206, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Mozetic, P.; Giannitelli, S.M.; Gori, M.; Trombetta, M.; Rainer, A. Engineering muscle cell alignment through 3D bioprinting. J Biom Mater Res A 2017, 105, 2582–2588. [Google Scholar] [CrossRef]
- Mendonça, G.; Edwards, S.P.; Mayers, C.A.; Meneghetti, P.C.; Liu, F. Digital Immediate Complete Denture for a Patient with Rhabdomyosarcoma: A Clinical Report. J. Prosthodont. 2021, 30, 196–201. [Google Scholar] [CrossRef]
- O’Sullivan, A.; Duffy, E.; O’Sullivan, K.; Cronin, U.; Lyons, E.; O’Sullivan, L.; et al. Bespoke 3D printed eye cover for teen with rhabdomyosarcoma. BMJ Support Palliat Care 2021, 2021, 002900. [Google Scholar] [CrossRef]
- Tejo-Otero, A.; Fenollosa-Artés, F.; Uceda, R.; Castellví-Fernández, A.; Lustig-Gainza, P.; Valls-Esteve, A.; et al. 3D printed prototype of a complex neuroblastoma for preoperative surgical planning. Ann 3D Print Med 2021, 2, 100014. [Google Scholar] [CrossRef]
- Tejo-Otero, A.; Lustig-Gainza, P.; Fenollosa-Artés, F.; Valls, A.; Krauel, L.; Buj-Corral, I. 3D printed soft surgical planning prototype for a biliary tract rhabdomyosarcoma. J Mech Behav Biomed Mater 2020, 109, 103844. [Google Scholar] [CrossRef]
- Shin, Y.C.; Kang, S.H.; Lee, J.H.; Kim, B.; Hong, S.W.; Han, D.-W. Three-dimensional graphene oxide-coated polyurethane foams beneficial to myogenesis. J. Biomater. Sci. Polym. Ed. 2018, 29, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Kim, C.; Song, S.-J.; Jun, S.; Kim, C.-S.; Hong, S.W.; Hyon, S.-H.; Han, D.-W.; Oh, J.-W. Ternary Aligned Nanofibers of RGD Peptide-Displaying M13 Bacteriophage/PLGA/Graphene Oxide for Facilitated Myogenesis. Nanotheranostics 2018, 2, 144–156. [Google Scholar] [CrossRef]
- Zhang, Z.; Klausen, L.H.; Chen, M.; Dong, M. Electroactive Scaffolds for Neurogenesis and Myogenesis: Graphene-Based Nanomaterials. Small 2018, 14, e1801983. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, G. A functional bioink and its application in myoblast alignment and differentiation. Chem. Eng. J. 2019, 366, 150–162. [Google Scholar] [CrossRef]
- Kim, Y.; Pagan-Diaz, G.; Gapinske, L.; Kim, Y.; Suh, J.; Solomon, E.; Harris, J.F.; Nam, S.; Bashir, R. Integration of Graphene Electrodes with 3D Skeletal Muscle Tissue Models. Adv. Heal. Mater. 2020, 9, 1901137–e1901137. [Google Scholar] [CrossRef]
- Kang, M.S.; Kang, J.I.; Le Thi, P.; Park, K.M.; Hong, S.W.; Choi, Y.S.; Han, D.-W.; Park, K.D. Three-Dimensional Printable Gelatin Hydrogels Incorporating Graphene Oxide to Enable Spontaneous Myogenic Differentiation. ACS Macro Lett. 2021, 10, 426–432. [Google Scholar] [CrossRef]
- Bilge, S.; Ergene, E.; Talak, E.; Gokyer, S.; Donar, Y.O.; Sınağ, A.; Huri, P.Y. Recycled algae-based carbon materials as electroconductive 3D printed skeletal muscle tissue engineering scaffolds. J. Mater. Sci. Mater. Med. 2021, 32, 1–15. [Google Scholar] [CrossRef]
- Russell, C.S.; Mostafavi, A.; Quint, J.P.; Panayi, A.C.; Baldino, K.; Williams, T.J.; Daubendiek, J.G.; Sánchez, V.H.; Bonick, Z.; Trujillo-Miranda, M.; et al. In Situ Printing of Adhesive Hydrogel Scaffolds for the Treatment of Skeletal Muscle Injuries. ACS Appl. Bio Mater. 2020, 3, 1568–1579. [Google Scholar] [CrossRef]
- Lee, V.K.; Lanzi, A.M.; Ngo, H.; Yoo, S.-S.; Vincent, P.A.; Dai, G. Generation of Multi-scale Vascular Network System Within 3D Hydrogel Using 3D Bio-printing Technology. Cell. Mol. Bioeng. 2014, 7, 460–472. [Google Scholar] [CrossRef]
- Hakimi, N.; Cheng, R.; Leng, L.; Sotoudehfar, M.; Ba, P.Q.; Bakhtyar, N.; Amini-Nik, S.; Jeschke, M.G.; Günther, A. Handheld skin printer: in situ formation of planar biomaterials and tissues. Lab a Chip 2018, 18, 1440–1451. [Google Scholar] [CrossRef]
- O’connell, C.D.; Di Bella, C.; Thompson, F.; Augustine, C.; Beirne, S.; Cornock, R.; Richards, C.J.; Chung, J.; Gambhir, S.; Yue, Z.; et al. Development of the Biopen: a handheld device for surgical printing of adipose stem cells at a chondral wound site. Biofabrication 2016, 8, 015019–015019. [Google Scholar] [CrossRef] [PubMed]
- Quint, J.P.; Mostafavi, A.; Endo, Y.; Panayi, A.; Russell, C.S.; Nourmahnad, A.; Wiseman, C.; Abbasi, L.; Samandari, M.; Sheikhi, A.; et al. In Vivo Printing of Nanoenabled Scaffolds for the Treatment of Skeletal Muscle Injuries. Adv. Heal. Mater. 2021, 10, 2002152–e2002152. [Google Scholar] [CrossRef] [PubMed]
- Chae, M.P.; Rozen, W.M.; McMenamin, P.G.; Findlay, M.W.; Spychal, R.T.; Hunter-Smith, D.J. Emerging applications of bedside 3D printing in plastic surgery. Front Surg 2015, 2, 25. [Google Scholar] [CrossRef]
- Krauel, L.; Fenollosa, F.; Riaza, L.; Pérez, M.; Tarrado, X.; Morales, A.; et al. Use of 3D prototypes for complex surgical oncologic cases. World J Surg 2016, 40, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Rebong, R.E.; Stewart, K.T.; Utreja, A.; Ghoneima, A.A. Accuracy of three-dimensional dental resin models created by fused deposition modeling, stereolithography, and Polyjet prototype technologies: A comparative study. Angle Orthod. 2018, 88, 363–369. [Google Scholar] [CrossRef]
- Adams, F.; Qiu, T.; Mark, A.; Fritz, B.; Kramer, L.; Schlager, D.; Wetterauer, U.; Miernik, A.; Fischer, P. Soft 3D-Printed Phantom of the Human Kidney with Collecting System. Ann. Biomed. Eng. 2017, 45, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Dini, D.; Baena, F.R.Y.; Forte, A.E. Composite hydrogel: A high fidelity soft tissue mimic for surgery. Mater. Des. 2018, 160, 886–894. [Google Scholar] [CrossRef]
- Tejo-Otero, A.; Buj-Corral, I.; Fenollosa-Artés, F. 3D Printing in Medicine for Preoperative Surgical Planning: A Review. Ann. Biomed. Eng. 2020, 48, 536–555. [Google Scholar] [CrossRef]
- van de Belt, T.H.; Nijmeijer, H.; Grim, D.; Engelen, L.J.; Vreeken, R.; van Gelder, M.M.; ter Laan, M. Patient-Specific Actual-Size Three-Dimensional Printed Models for Patient Education in Glioma Treatment: First Experiences. World Neurosurg. 2018, 117, e99–e105. [Google Scholar] [CrossRef]
- Biglino, G.; Koniordou, D.; Gasparini, M.; Capelli, C.; Leaver, L.-K.; Khambadkone, S.; Schievano, S.; Taylor, A.M.; Wray, J. Piloting the Use of Patient-Specific Cardiac Models as a Novel Tool to Facilitate Communication During Cinical Consultations. Pediatr. Cardiol. 2017, 38, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, J.-C.; Isotani, S.; Matsugasumi, T.; Duddalwar, V.; Hung, A.J.; Suer, E.; Baco, E.; Satkunasivam, R.; Djaladat, H.; Metcalfe, C.; et al. Personalized 3D printed model of kidney and tumor anatomy: a useful tool for patient education. World J. Urol. 2016, 34, 337–345. [Google Scholar] [CrossRef]
- Agarwal, T.; Hann, S.Y.; Chiesa, I.; Cui, H.; Celikkin, N.; Micalizzi, S.; Barbetta, A.; Costantini, M.; Esworthy, T.; Zhang, L.G.; et al. 4D printing in biomedical applications: emerging trends and technologies. J. Mater. Chem. B 2021, 9, 7608–7632. [Google Scholar] [CrossRef]
- Saska, S.; Pilatti, L.; Blay, A.; Shibli, J.A. Bioresorbable Polymers: Advanced Materials and 4D Printing for Tissue Engineering. Polymers 2021, 13, 563. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Zhang, P.; Liu, Y.; Lv, L.; Zhou, Y. Four-dimensional bioprinting: Current developments and applications in bone tissue engineering. Acta Biomater. 2020, 101, 26–42. [Google Scholar] [CrossRef]
- Apsite, I.; Constante, G.; Dulle, M.; Vogt, L.; Caspari, A.; Boccaccini, A.R.; Synytska, A.; Salehi, S.; Ionov, L. 4D Biofabrication of fibrous artificial nerve graft for neuron regeneration. Biofabrication 2020, 12, 035027. [Google Scholar] [CrossRef]
- Aigner, T.; Scheibel, T. Self-Rolling Refillable Tubular Enzyme Containers Made of Recombinant Spider Silk and Chitosan. ACS Appl. Mater. Interfaces 2019, 11, 15290–15297. [Google Scholar] [CrossRef]
- Kirillova, A.; Maxson, R.; Stoychev, G.; Gomillion, C.T.; Ionov, L. 4D biofabrication using shape-morphing hydrogels. Adv Mater 2017, 29, 1703443. [Google Scholar] [CrossRef]
- Zhang, L.; Xiang, Y.; Zhang, H.; Cheng, L.; Mao, X.; An, N.; et al. A Biomimetic 3D-Self-Forming Approach for Microvascular Scaffolds. Adv Sci 2020, 7, 1903553. [Google Scholar] [CrossRef] [PubMed]
- Stroganov, V.; Pant, J.; Stoychev, G.; Janke, A.; Jehnichen, D.; Fery, A.; Handa, H.; Ionov, L. 4D Biofabrication: 3D Cell Patterning Using Shape-Changing Films. Adv. Funct. Mater. 2018, 28. [Google Scholar] [CrossRef]
- Stroganov V, 4D Biofabrication using self-folding polymers. 2018, University of Bayreuth.
- Hendrikson, W.J.; Rouwkema, J.; Clementi, F.; A van Blitterswijk, C.; Farè, S.; Moroni, L. Towards 4D printed scaffolds for tissue engineering: exploiting 3D shape memory polymers to deliver time-controlled stimulus on cultured cells. Biofabrication 2017, 9, 031001. [Google Scholar] [CrossRef] [PubMed]
- Miao, S.; Nowicki, M.; Cui, H.; Lee, S.-J.; Zhou, X.; Mills, D.K.; Zhang, L.G. 4D anisotropic skeletal muscle tissue constructs fabricated by staircase effect strategy. Biofabrication 2019, 11, 035030–035030. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-Y.; Xie, H.; Xu, Z.-Y.; Li, L.; Jiang, M.-N.; Tang, L.; Yang, K.-K.; Wang, Y.-Z. 4D printing of shape memory aliphatic copolyester via UV-assisted FDM strategy for medical protective devices. Chem. Eng. J. 2020, 396, 125242. [Google Scholar] [CrossRef]
- Wan, X.; Wei, H.; Zhang, F.; Liu, Y.; Leng, J. 3D printing of shape memory poly(d,l-lactide-co-trimethylene carbonate) by direct ink writing for shape-changing structures. J Appl Polym Sci 2019, 136, 48177. [Google Scholar] [CrossRef]
- Podstawczyk, D.; Nizioł, M.; Szymczyk-Ziółkowska, P.; Fiedot-Toboła, M. Development of Thermoinks for 4D Direct Printing of Temperature-Induced Self-Rolling Hydrogel Actuators. Adv. Funct. Mater. 2021, 31, 2009664. [Google Scholar] [CrossRef]
- Nishiguchi, A.; Zhang, H.; Schweizerhof, S.; Schulte, M.F.; Mourran, A.; Moeller, M. 4D Printing of a Light-Driven Soft Actuator with Programmed Printing Density. ACS Appl. Mater. Interfaces 2020, 12, 12176–12185. [Google Scholar] [CrossRef]
- Constante, G.; Apsite, I.; Alkhamis, H.; Dulle, M.; Schwarzer, M.; Caspari, A.; Synytska, A.; Salehi, S.; Ionov, L. 4D Biofabrication Using a Combination of 3D Printing and Melt-Electrowriting of Shape-Morphing Polymers. ACS Appl. Mater. Interfaces 2021, 13, 12767–12776. [Google Scholar] [CrossRef]
- Miao, S.; Castro, N.; Nowicki, M.; Xia, L.; Cui, H.; Zhou, X.; Zhu, W.; Lee, S.-J.; Sarkar, K.; Vozzi, G.; et al. 4D printing of polymeric materials for tissue and organ regeneration. Mater. Today 2017, 20, 577–591. [Google Scholar] [CrossRef]
- Li, M.; Nagamori, E.; Kino-Oka, M. Disruption of myoblast alignment by highly motile rhabdomyosarcoma cell in tissue structure. J. Biosci. Bioeng. 2017, 123, 259–264. [Google Scholar] [CrossRef]
- Stefanek, E.; Samiei, E.; Kavoosi, M.; Esmaeillou, M.; Geraylow, K.R.; Emami, A.; Ashrafizadeh, M.; Perrin, D.; Gordon, J.W.; Akbari, M.; et al. A bioengineering method for modeling alveolar Rhabdomyosarcoma and assessing chemotherapy responses. MethodsX 2021, 8, 101473. [Google Scholar] [CrossRef]
- Roll, W.; Dirksen, U.; Weckesser, M. Update PET in der Pädiatrischen Onkologie. Der Nukl. 2018, 41, 211–221. [Google Scholar] [CrossRef]
- Oberlin, O.; Rey, A.; Lyden, E.; Bisogno, G.; Stevens, M.C.; Meyer, W.H.; Carli, M.; Anderson, J.R. Prognostic Factors in Metastatic Rhabdomyosarcomas: Results of a Pooled Analysis From United States and European Cooperative Groups. J. Clin. Oncol. 2008, 26, 2384–2389. [Google Scholar] [CrossRef]
- Arndt, C.A.; Stoner, J.A.; Hawkins, D.S.; Rodeberg, D.A.; Hayes-Jordan, A.A.; Paidas, C.N.; et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children's oncology group study D9803. J Clin Oncol 2009, 27, 5182. [Google Scholar] [CrossRef] [PubMed]
- Van Winkle, P.; Angiolillo, A.; Krailo, M.; Cheung, Y.K.; Anderson, B.; Davenport, V.; et al. Ifosfamide, carboplatin, and etoposide (ICE) reinduction chemotherapy in a large cohort of children and adolescents with recurrent/refractory sarcoma: the Children's Cancer Group (CCG) experience. Pediatr Blood Cancer 2005, 44, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Breitfeld, P.P.; Lyden, E.; Raney, R.B.; Teot, L.A.; Wharam, M.; Lobe, T.; Crist, W.M.; Maurer, H.M.; Donaldson, S.S.; Ruymann, F.B. Ifosfamide and Etoposide Are Superior to Vincristine and Melphalan for Pediatric Metastatic Rhabdomyosarcoma When Administered With Irradiation and Combination Chemotherapy: A Report From the Intergroup Rhabdomyosarcoma Study Group. J. Pediatr. Hematol. 2001, 23, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Lyden, E.; Breitfeld, P.; Donaldson, S.S.; Wiener, E.; Parham, D.; et al. Two consecutive phase II window trials of irinotecan alone or in combination with vincristine for the treatment of metastatic rhabdomyosarcoma: the Children's Oncology Group. J Clin Oncol 2007, 25, 362–369. [Google Scholar] [CrossRef]
- Defachelles, A.-S.; Bogart, E.; Casanova, M.; Merks, J.H.M.; Bisogno, G.; Calareso, G.; Melcon, S.G.; Gatz, S.A.; Le Deley, M.-C.; McHugh, K.; et al. Randomized Phase II Trial of Vincristine-Irinotecan With or Without Temozolomide, in Children and Adults With Relapsed or Refractory Rhabdomyosarcoma: A European Paediatric Soft Tissue Sarcoma Study Group and Innovative Therapies for Children With Cancer Trial. J. Clin. Oncol. 2021, 39, 2979–2990. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370–370. [Google Scholar] [CrossRef]
- Yang, H.-Z.; Ma, Y.; Zhou, Y.; Xu, L.-M.; Chen, X.-J.; Ding, W.-B.; Zou, H.-B. Autophagy contributes to the enrichment and survival of colorectal cancer stem cells under oxaliplatin treatment. Cancer Lett. 2015, 361, 128–136. [Google Scholar] [CrossRef]
- Han, W.; Sun, J.; Feng, L.; Wang, K.; Li, D.; Pan, Q.; Chen, Y.; Jin, W.; Wang, X.; Pan, H.; et al. Autophagy Inhibition Enhances Daunorubicin-Induced Apoptosis in K562 Cells. PLOS ONE 2011, 6, e28491. [Google Scholar] [CrossRef]
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-C.; Wang, H.-C.; Hou, Y.-C.; Tung, H.-L.; Chiu, T.-J.; Shan, Y.-S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 2015, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Bläuer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur. J. Cancer 2014, 50, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Si, S.; Schoen, S.; Chen, J.; Jin, X.-B.; Wu, G. Suppression of autophagy enhances preferential toxicity of paclitaxel to folliculin-deficient renal cancer cells. J. Exp. Clin. Cancer Res. 2013, 32, 99–99. [Google Scholar] [CrossRef]
- Quan, Y.; Lei, H.; Wahafu, W.; Liu, Y.; Ping, H.; Zhang, X. Inhibition of autophagy enhances the anticancer effect of enzalutamide on bladder cancer. Biomed. Pharmacother. 2019, 120, 109490. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Tang, J.; Li, P.; Si, S.; Yu, H.; Yang, X.; Tao, J.; Lv, Q.; Gu, M.; Yang, H.; et al. Chloroquine Enhances the Radiosensitivity of Bladder Cancer Cells by Inhibiting Autophagy and Activating Apoptosis. Cell. Physiol. Biochem. 2018, 45, 54–66. [Google Scholar] [CrossRef]
- Domagala, A.; Stachura, J.; Gabrysiak, M.; Muchowicz, A.; Zagozdzon, R.; Golab, J.; Firczuk, M. Inhibition of autophagy sensitizes cancer cells to Photofrin-based photodynamic therapy. BMC Cancer 2018, 18, 1–10. [Google Scholar] [CrossRef]
- Hao, C.; Liu, G.; Tian, G. Autophagy inhibition of cancer stem cells promotes the efficacy of cisplatin against non-small cell lung carcinoma. Ther. Adv. Respir. Dis. 2019, 13. [Google Scholar] [CrossRef]
- Liu, F.; Liu, D.; Yang, Y.; Zhao, S. Effect of autophagy inhibition on chemotherapy-induced apoptosis in A549 lung cancer cells. Oncol. Lett. 2013, 5, 1261–1265. [Google Scholar] [CrossRef]
- Ren, J.-H.; He, W.-S.; Nong, L.; Zhu, Q.-Y.; Hu, K.; Zhang, R.-G.; Huang, L.-L.; Zhu, F.; Wu, G.; Luo, S.; et al. Acquired Cisplatin Resistance in Human Lung Adenocarcinoma Cells Is Associated with Enhanced Autophagy. Cancer Biotherapy Radiopharm. 2010, 25, 75–80. [Google Scholar] [CrossRef]
- D’agostino, S.; Tombolan, L.; Saggioro, M.; Frasson, C.; Rampazzo, E.; Pellegrini, S.; Favaretto, F.; Biz, C.; Ruggieri, P.; Gamba, P.; et al. Rhabdomyosarcoma Cells Produce Their Own Extracellular Matrix With Minimal Involvement of Cancer-Associated Fibroblasts: A Preliminary Study. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Liu, P.; Fan, J.; Wang, Z.; Zai, W.; Song, P.; Li, Y.; Ju, D. The role of autophagy in the cytotoxicity induced by trastuzumab emtansine (T-DM1) in HER2-positive breast cancer cells. AMB Express 2020, 10, 1–10. [Google Scholar] [CrossRef]
- McCreery, K.P.; Xu, X.; Scott, A.K.; Fajrial, A.K.; Calve, S.; Ding, X.; Neu, C.P. Nuclear Stiffness Decreases with Disruption of the Extracellular Matrix in Living Tissues. Small 2021, 17, 2006699–e2006699. [Google Scholar] [CrossRef] [PubMed]
- Ignatius, M.S.; Chen, E.; Elpek, N.M.; Fuller, A.Z.; Tenente, I.M.; Clagg, R.; et al. In vivo imaging of tumor-propagating cells, regional tumor heterogeneity, and dynamic cell movements in embryonal rhabdomyosarcoma. Cancer Cell 2012, 21, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, E.R.; Anderson, J.R.; Hawkins, D.S.; Skapek, S.X.; Parham, D.M.; Teot, L.A. The World Health Organization classification of skeletal muscle tumors in pediatric rhabdomyosarcoma: A report from the Children's Oncology Group. Arch Pathol Lab Med 2015, 139, 1281–7. [Google Scholar] [CrossRef] [PubMed]
- Sleijfer, S.; Ray-Coquard, I.; Papai, Z.; Le Cesne, A.; Scurr, M.; Schöffski, P.; Blay, J.Y.; et al. Faculty Opinions recommendation of Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043). Journal of clinical oncology 2009, 27, 3126–3132. [Google Scholar] [CrossRef]
- van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Santoro, A.; Comandone, A.; Basso, U.; Parra, H.S.; De Sanctis, R.; Stroppa, E.; Marcon, I.; Giordano, L.; Lutman, F.R.; Boglione, A.; et al. Phase II prospective study with sorafenib in advanced soft tissue sarcomas after anthracycline-based therapy. Ann. Oncol. 2013, 24, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Wozniak, A.; Leahy, M.G.; Aamdal, S.; Rutkowski, P.; Bauer, S.; Richter, S.; Grünwald, V.; Debiec-Rychter, M.; Sciot, R.; et al. The tyrosine kinase inhibitor crizotinib does not have clinically meaningful activity in heavily pre-treated patients with advanced alveolar rhabdomyosarcoma with FOXO rearrangement: European Organisation for Research and Treatment of Cancer phase 2 trial 90101 ‘CREATE’. Eur. J. Cancer 2018, 94, 156–167. [Google Scholar] [CrossRef]
- Kim, A.; Widemann, B.C.; Krailo, M.; Jayaprakash, N.; Fox, E.; Weigel, B.; et al. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: a report from the Children's Oncology Group. Pediatric blood & cancer 2015, 62, 1562–1566. [Google Scholar]
- Santoro, A.; Comandone, A.; Basso, U.; Parra, H.S.; De Sanctis, R.; Stroppa, E.; Marcon, I.; Giordano, L.; Lutman, F.R.; Boglione, A.; et al. Phase II prospective study with sorafenib in advanced soft tissue sarcomas after anthracycline-based therapy. Ann. Oncol. 2013, 24, 1093–1098. [Google Scholar] [CrossRef]
- Weigel, B.; Malempati, S.; Reid, J.M.; Voss, S.D.; Cho, S.Y.; Chen, H.X.; et al. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children's Oncology Group. Pediatric blood & cancer 2014, 61, 452–456. [Google Scholar]
- Bisogno, G.; De Salvo, G.L.; Bergeron, C.; Melcón, S.G.; Merks, J.H.; Kelsey, A.; Martelli, H.; Minard-Colin, V.; Orbach, D.; Glosli, H.; et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, G.; Jenney, M.; Bergeron, C.; Melcón, S.G.; Ferrari, A.; Oberlin, O.; Carli, M.; Stevens, M.; Kelsey, A.; De Paoli, A.; et al. Addition of dose-intensified doxorubicin to standard chemotherapy for rhabdomyosarcoma (EpSSG RMS 2005): a multicentre, open-label, randomised controlled, phase 3 trial. Lancet Oncol. 2018, 19, 1061–1071. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Chi, Y.-Y.; Anderson, J.R.; Tian, J.; Arndt, C.A.; Bomgaars, L.; et al. Addition of vincristine and irinotecan to vincristine, dactinomycin, and cyclophosphamide does not improve outcome for intermediate-risk rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol 2018, 36, 2770–2777. [Google Scholar] [CrossRef] [PubMed]
- Weigel, B.J.; Lyden, E.; Anderson, J.R.; Meyer, W.H.; Parham, D.M.; Rodeberg, D.A.; et al. Intensive multiagent therapy, including dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high-risk rhabdomyosarcoma: a report from the Children's Oncology Group. Journal of Clinical Oncology 2016, 34, 117. [Google Scholar] [CrossRef]
- Setty, B.A.; Stanek, J.R.; Mascarenhas, L.; Miller, A.; Bagatell, R.; Okcu, F.; et al. VIncristine, irinotecan, and temozolomide in children and adolescents with relapsed rhabdomyosarcoma. Pediatric blood & cancer 2018, 65, e26728. [Google Scholar]
- Igarashi, K.; Kawaguchi, K.; Kiyuna, T.; Murakami, T.; Miwa, S.; Nelson, S.D.; Dry, S.M.; Li, Y.; Singh, A.S.; Kimura, H.; et al. Temozolomide combined with irinotecan caused regression in an adult pleomorphic rhabdomyosarcoma patient-derived orthotopic xenograft (PDOX) nude-mouse model. Oncotarget 2017, 8, 75874–75880. [Google Scholar] [CrossRef]
- Hugle, M.; Belz, K.; Fulda, S. Identification of synthetic lethality of PLK1 inhibition and microtubule-destabilizing drugs. Cell Death Differ. 2015, 22, 1946–1956. [Google Scholar] [CrossRef] [PubMed]
- Stehle, A.; Hugle, M.; Fulda, S. Eribulin synergizes with Polo-like kinase 1 inhibitors to induce apoptosis in rhabdomyosarcoma. Cancer Lett. 2015, 365, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Abbou, S.; Lanvers-Kaminsky, C.; Daudigeos-Dubus, E.; Le Dret, L.; Laplace-Builhe, C.; Molenaar, J.; Vassal, G.; Geoerger, B. Polo-like Kinase Inhibitor Volasertib Exhibits Antitumor Activity and Synergy with Vincristine in Pediatric Malignancies. Anticancer Res. 2016, 36, 599–609. [Google Scholar]
- Ignatius, M.S.; Hayes, M.N.; E Moore, F.; Tang, Q.; Garcia, S.P.; Blackburn, P.R.; Baxi, K.; Wang, L.; Jin, A.; Ramakrishnan, A.; et al. Author response: tp53 deficiency causes a wide tumor spectrum and increases embryonal rhabdomyosarcoma metastasis in zebrafish. Elife 2018, 7, e–37202. [Google Scholar] [CrossRef]
- Hayes, M.N.; McCarthy, K.; Jin, A.; Oliveira, M.L.; Iyer, S.; Garcia, S.P.; et al. Vangl2/RhoA signaling pathway regulates stem cell self-renewal programs and growth in rhabdomyosarcoma. Cell Stem Cell 2018, 22, 414–427. [Google Scholar] [CrossRef]
- Tenente, I.M.; Hayes, M.N.; Ignatius, M.S.; McCarthy, K.; Yohe, M.; Sindiri, S.; et al. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. Elife 2017, 6, e19214. [Google Scholar] [CrossRef]
Figure 1.
Chemical structures of frequently used chemotrophic agents for the treatment of RMS.
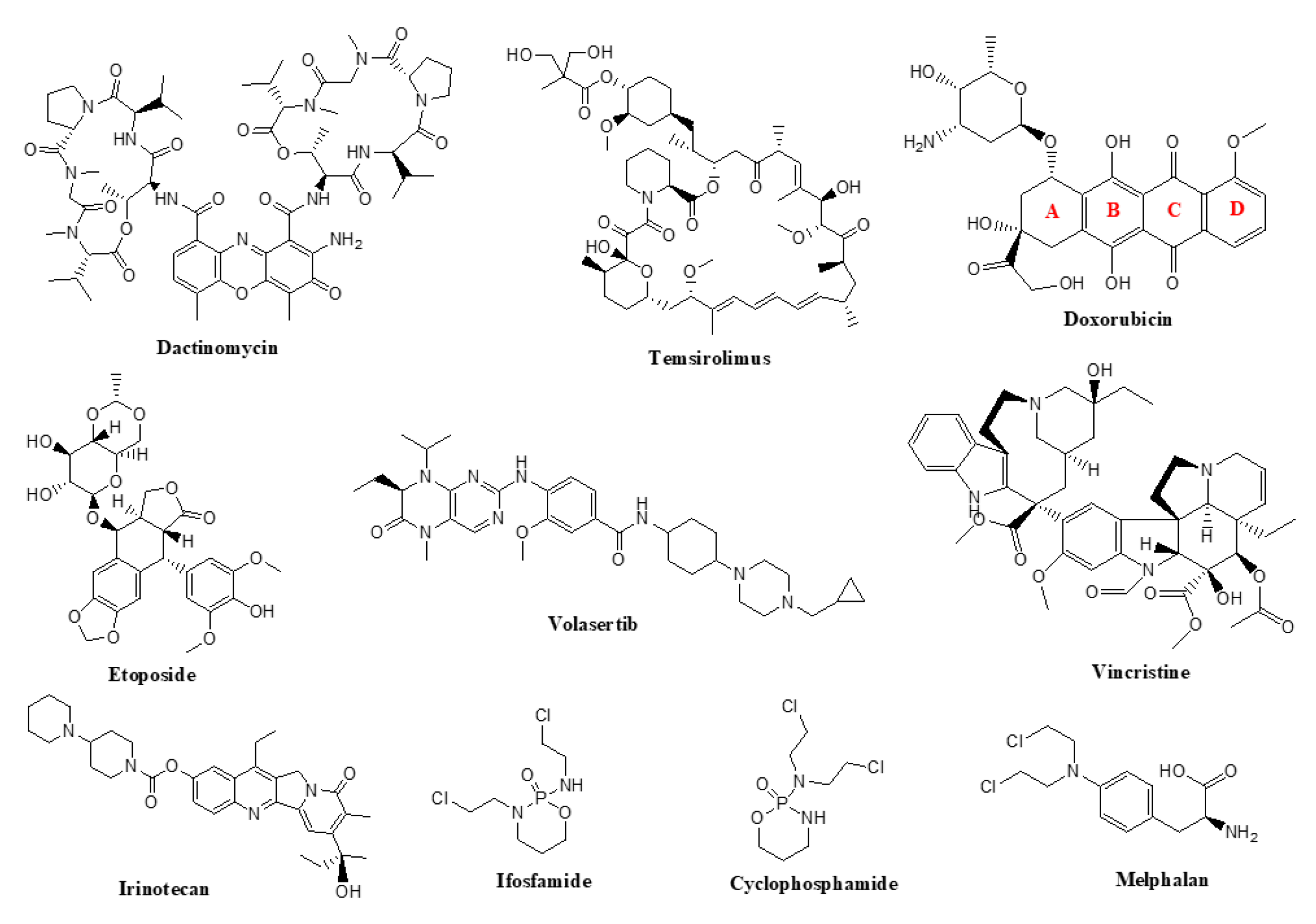
Figure 2.
A schematic overview of the molecular mechanism of apoptosis. Apoptotic cell death can be triggered by various stimuli, both intracellular (DNA damage or endoplasmic reticulum stress) and extracellular (TNF-α, TRAIL). The extracellular pathway is initiated by the binding of ligands to the plasma membrane-located death receptors. The stimulation of death receptors induces caspase 8 and caspase 10 activation with subsequent downstream cleavage of effector caspases, caspase 3 and caspase 7. The intrinsic pathway is regulated by the Bcl-2 family. It consists of pro-survival and pro-apoptotic members to the latter of which belong BH-3 only proteins. Overexpression of BH-3 only, as well as other pro-apoptotic members of the Bcl-2 family initiates programmed cell death. Bax and Bak are the main effectors of the Bcl-2 regulated pathway. When activated, they increase mitochondrial membrane permeability and allow for the release of apoptogenic cytochrome c into the cytosol. Cytochrome c release prompts the formation of protein complex called apoptosome, which turns pro-caspase 9 into caspase 9. Then, caspase 9 activates effector caspases, leading to apoptosis. Dotted arrows represent the interactions between chemotherapeutic agents and respective proteins involved in the process of apoptosis. Abbreviations: alkylating agents – cyclophosphamide and ifosfamide, BH3-only – Bcl-2 homology 3 only, BID – BH3-interacting domain death agonist, ER – endoplasmic reticulum, FADD – Fas-associated protein with death domain, tBID – truncated BID, TRAILR – TNF-related apoptosis-inducing ligand receptor.
Figure 2.
A schematic overview of the molecular mechanism of apoptosis. Apoptotic cell death can be triggered by various stimuli, both intracellular (DNA damage or endoplasmic reticulum stress) and extracellular (TNF-α, TRAIL). The extracellular pathway is initiated by the binding of ligands to the plasma membrane-located death receptors. The stimulation of death receptors induces caspase 8 and caspase 10 activation with subsequent downstream cleavage of effector caspases, caspase 3 and caspase 7. The intrinsic pathway is regulated by the Bcl-2 family. It consists of pro-survival and pro-apoptotic members to the latter of which belong BH-3 only proteins. Overexpression of BH-3 only, as well as other pro-apoptotic members of the Bcl-2 family initiates programmed cell death. Bax and Bak are the main effectors of the Bcl-2 regulated pathway. When activated, they increase mitochondrial membrane permeability and allow for the release of apoptogenic cytochrome c into the cytosol. Cytochrome c release prompts the formation of protein complex called apoptosome, which turns pro-caspase 9 into caspase 9. Then, caspase 9 activates effector caspases, leading to apoptosis. Dotted arrows represent the interactions between chemotherapeutic agents and respective proteins involved in the process of apoptosis. Abbreviations: alkylating agents – cyclophosphamide and ifosfamide, BH3-only – Bcl-2 homology 3 only, BID – BH3-interacting domain death agonist, ER – endoplasmic reticulum, FADD – Fas-associated protein with death domain, tBID – truncated BID, TRAILR – TNF-related apoptosis-inducing ligand receptor.
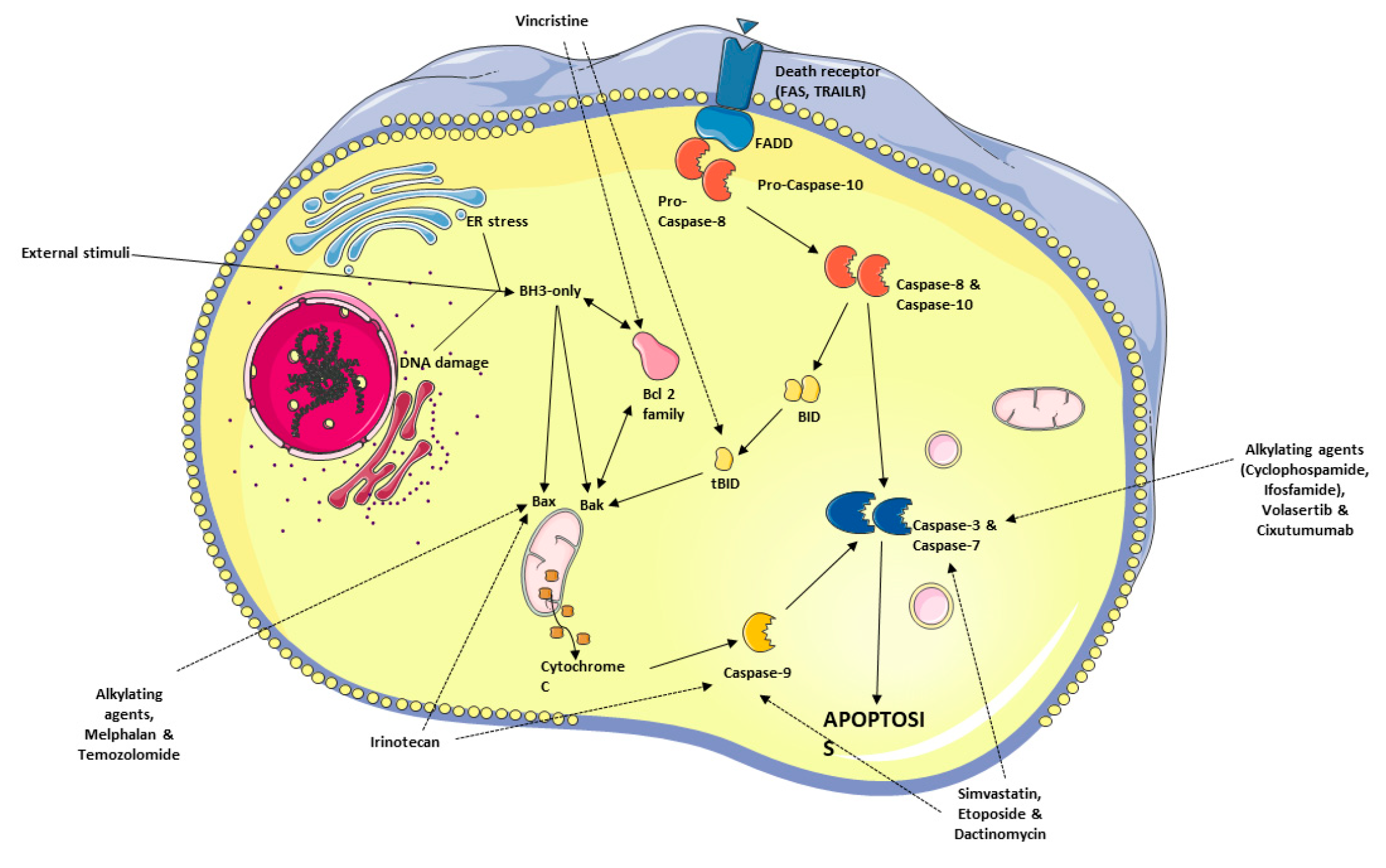
Figure 3.
Autophagy is divided into three major types. A) Macroautophagy, the lysosome is fused into the autophagosome to digest substrates. B) Chaperone-mediated autophagy (CMP), HSPA8 complex detects KFERQ motif on the substrate proteins and transports them to the lysosome. C) Microautophagy, the substrates are directly transported to the lysosome.
Figure 3.
Autophagy is divided into three major types. A) Macroautophagy, the lysosome is fused into the autophagosome to digest substrates. B) Chaperone-mediated autophagy (CMP), HSPA8 complex detects KFERQ motif on the substrate proteins and transports them to the lysosome. C) Microautophagy, the substrates are directly transported to the lysosome.
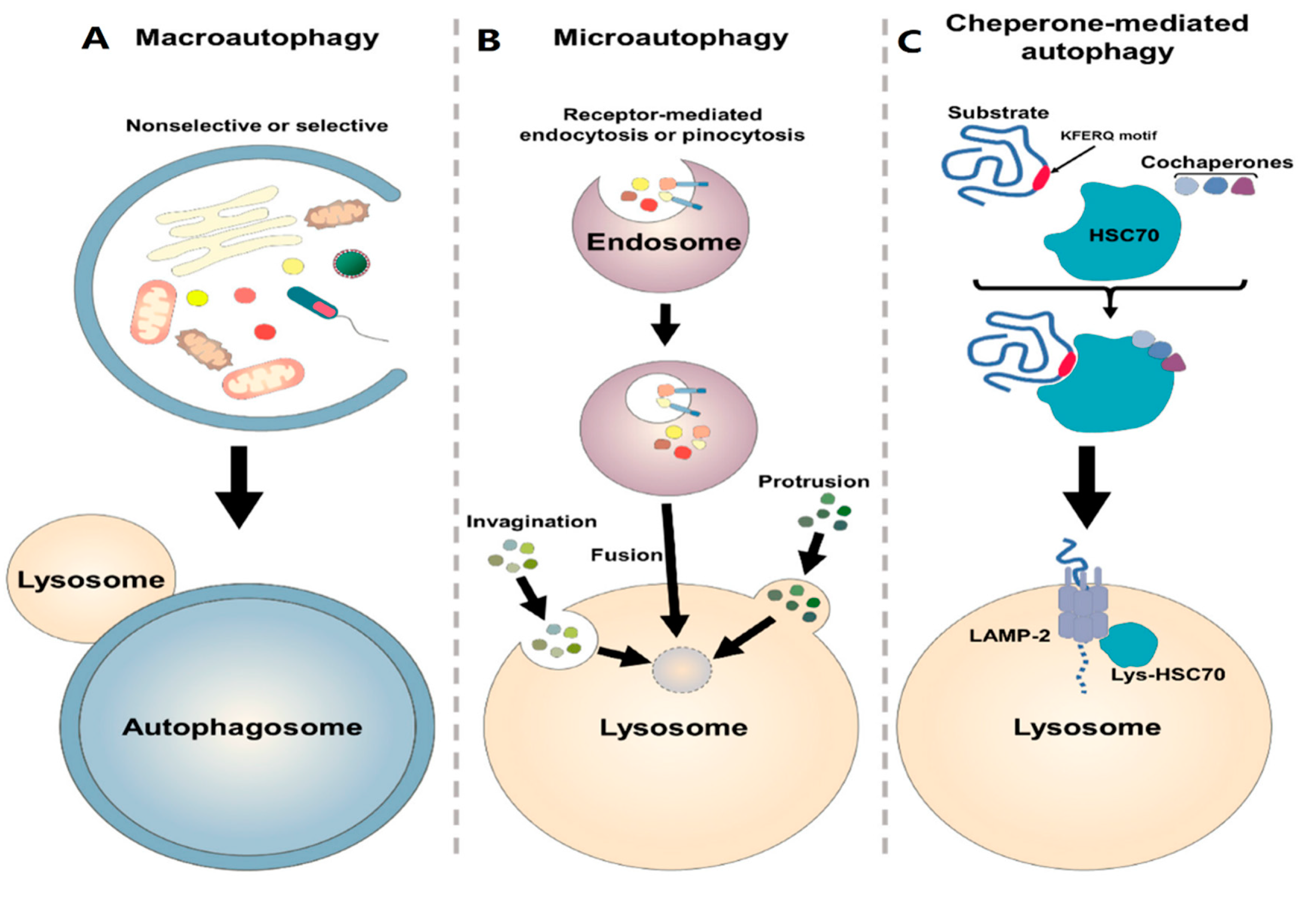
Figure 4.
A schematic overview of autophagy targeting in relation to RMS. Different therapeutic strategies produce synergistic or additive effects to treat RMS cancer cells and enhance their response to anticancer compounds. The treatment regimens can affect the autophagy flux. For example, TMZ, AF/AD, ATG7, Ros-mediated JNK pathway, lysosome acidification, SIRT, and ABCC subfamily (P-gp/MDR1/ABCB1) have increasing effect on autophagy flux; but inhibitory treatments like doxorubicin, simvastatin, vincristine, omeprazole, bortezomib, tenovin, and tesirolimus revert the autophagy process by affecting the targets mentioned above.
Figure 4.
A schematic overview of autophagy targeting in relation to RMS. Different therapeutic strategies produce synergistic or additive effects to treat RMS cancer cells and enhance their response to anticancer compounds. The treatment regimens can affect the autophagy flux. For example, TMZ, AF/AD, ATG7, Ros-mediated JNK pathway, lysosome acidification, SIRT, and ABCC subfamily (P-gp/MDR1/ABCB1) have increasing effect on autophagy flux; but inhibitory treatments like doxorubicin, simvastatin, vincristine, omeprazole, bortezomib, tenovin, and tesirolimus revert the autophagy process by affecting the targets mentioned above.
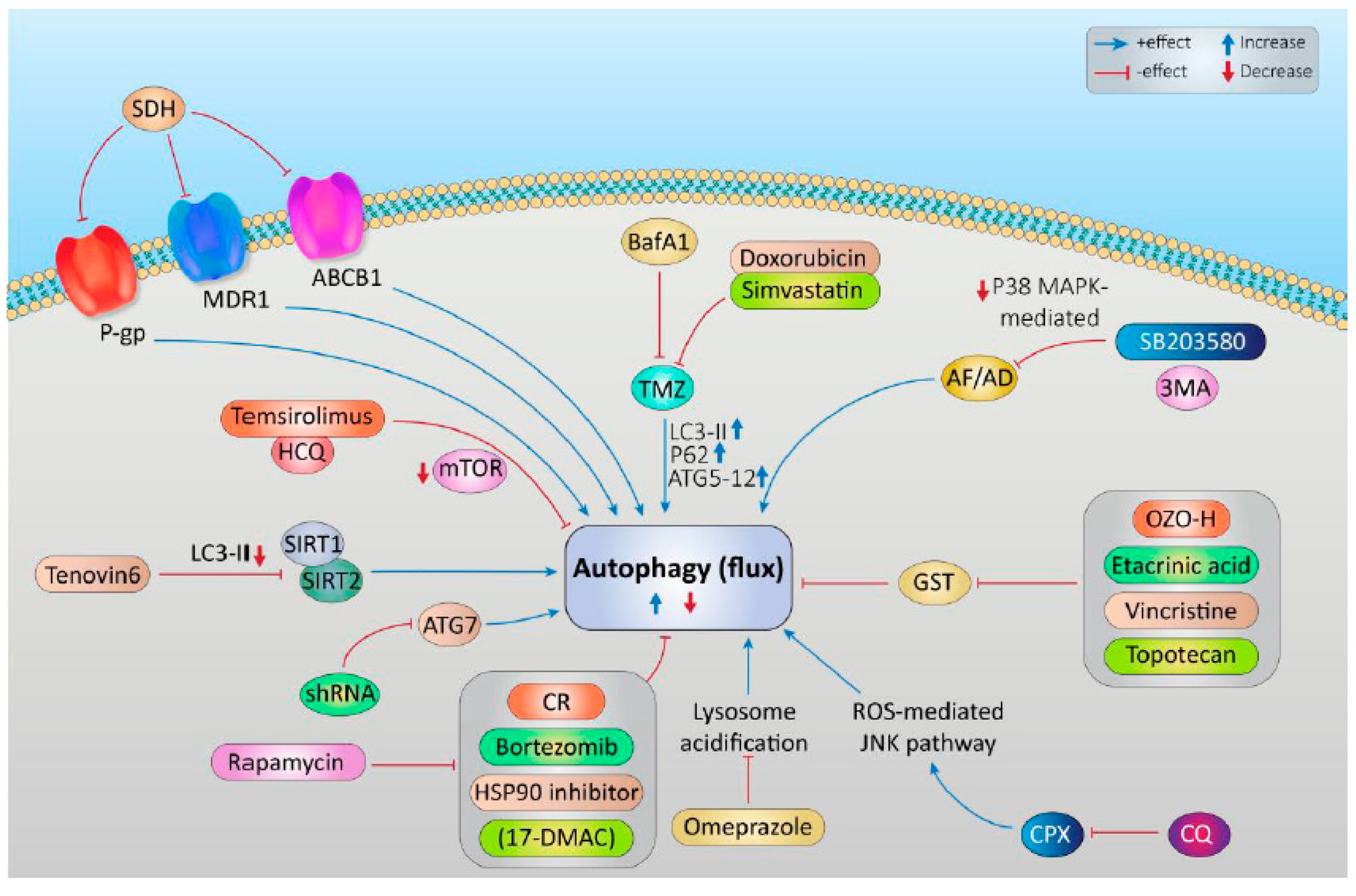
Figure 5.
Descriptive RMS animal model chart. A. Genetically-engineered mouse models (GEMM) demonstrated as one modeling approach used in RMS and the production of the model indicated as the most common technique named syngeneic allograft model (SAM) to monitor how tumor cells behave in the presence of immune response. B. Stem-cell humanized patient-derived xenograft mouse models (PDX) are commonly used as combinatorial approach with cell line-derived xenograft models (CDX) as complementary study. C. Cellular barcoding is demonstrated as one of the most important steps in producing targeted mouse models especially in GEMM and environmentally induced mouse models (EIMM). D. Human-mouse chimera is a type of EIMM-dependent mouse model, which could be used for RMS studies. E. Transplantation of tumor tissue into the models by inserting gene regulation vectors for producing PDX models. F. Inducible mouse model generated via gene regulation by iCas9 and shRNA. G. Different types of divergent methods used for producing metastatic and progressive mouse models. Reprinted from [357] with permission from MDPI.
Figure 5.
Descriptive RMS animal model chart. A. Genetically-engineered mouse models (GEMM) demonstrated as one modeling approach used in RMS and the production of the model indicated as the most common technique named syngeneic allograft model (SAM) to monitor how tumor cells behave in the presence of immune response. B. Stem-cell humanized patient-derived xenograft mouse models (PDX) are commonly used as combinatorial approach with cell line-derived xenograft models (CDX) as complementary study. C. Cellular barcoding is demonstrated as one of the most important steps in producing targeted mouse models especially in GEMM and environmentally induced mouse models (EIMM). D. Human-mouse chimera is a type of EIMM-dependent mouse model, which could be used for RMS studies. E. Transplantation of tumor tissue into the models by inserting gene regulation vectors for producing PDX models. F. Inducible mouse model generated via gene regulation by iCas9 and shRNA. G. Different types of divergent methods used for producing metastatic and progressive mouse models. Reprinted from [357] with permission from MDPI.
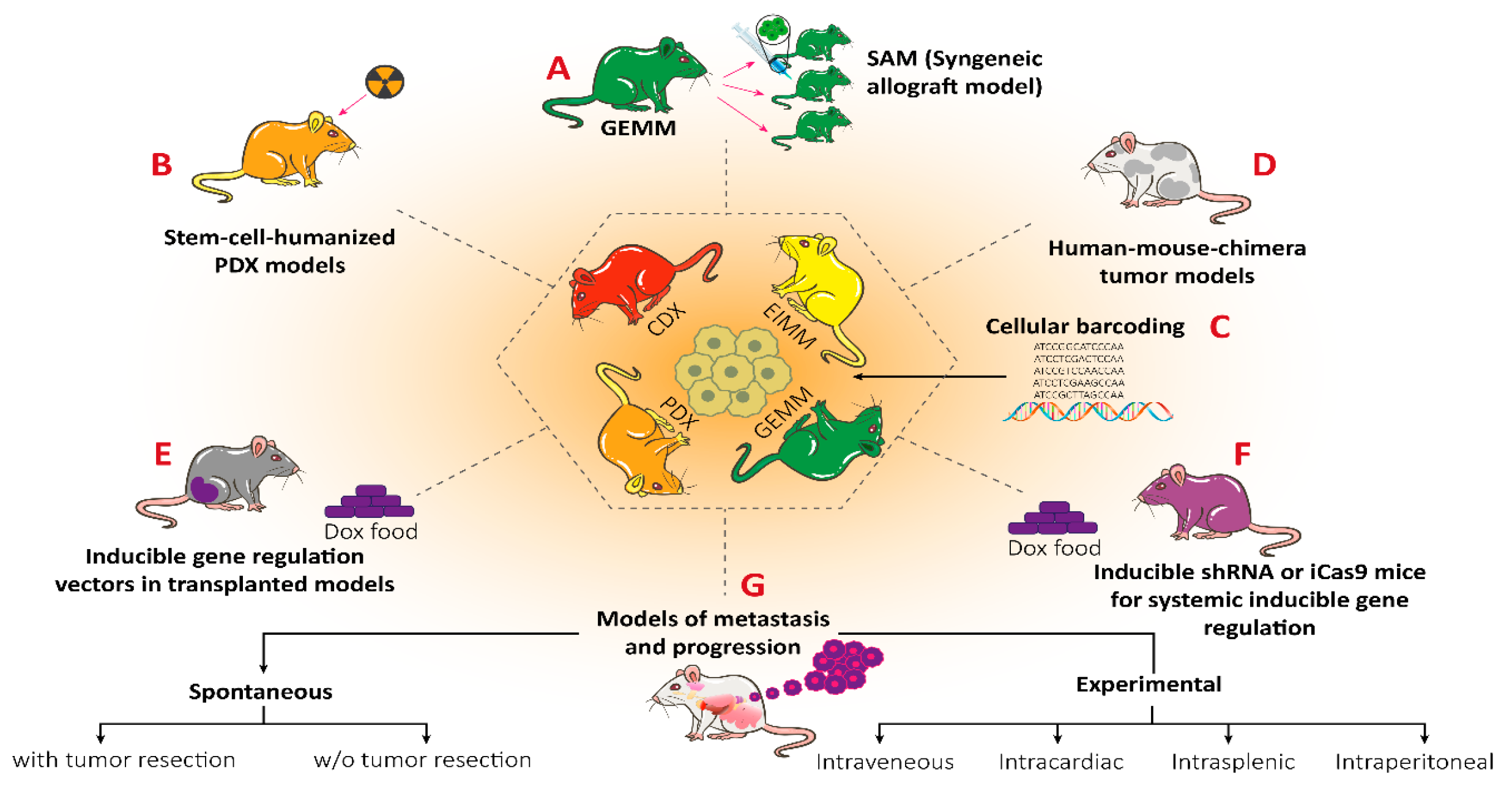
Figure 6.
Different applications for sarcoma mouse models. All colors represent the most proper animal modeling approach. GEMM is presented as green, PDX is presented as orange, CDX is shown as red, and EIMM is shown as yellow. EIMM cannot be used by itself, which needs some additional mouse models to be named the evaluation as precise. All colored letters in green present the certain application field of GEMM while yellow (EIMM), red (CDX), and orange (PDX) colored letters present the distinct application fields, which are specific to the mouse model. Reprinted from [357] with permission from MDPI.
Figure 6.
Different applications for sarcoma mouse models. All colors represent the most proper animal modeling approach. GEMM is presented as green, PDX is presented as orange, CDX is shown as red, and EIMM is shown as yellow. EIMM cannot be used by itself, which needs some additional mouse models to be named the evaluation as precise. All colored letters in green present the certain application field of GEMM while yellow (EIMM), red (CDX), and orange (PDX) colored letters present the distinct application fields, which are specific to the mouse model. Reprinted from [357] with permission from MDPI.
Figure 7.
Descriptive chart for GEMM modeling approaches. A. Spontaneous mutations are applied to generate randomized mutant colonies by targeting the gene. B. Externally induced mutation model generated by chemicals or radiations. C. Retroviral transduction is the technique used to breed the transgene via retrovirus. D. Microinjection of DNA constructs to donors. In vitro culture is the way of growing the egg and implanted female generates offspring with transgene. Reprinted from [360] with permission from MDPI.
Figure 7.
Descriptive chart for GEMM modeling approaches. A. Spontaneous mutations are applied to generate randomized mutant colonies by targeting the gene. B. Externally induced mutation model generated by chemicals or radiations. C. Retroviral transduction is the technique used to breed the transgene via retrovirus. D. Microinjection of DNA constructs to donors. In vitro culture is the way of growing the egg and implanted female generates offspring with transgene. Reprinted from [360] with permission from MDPI.
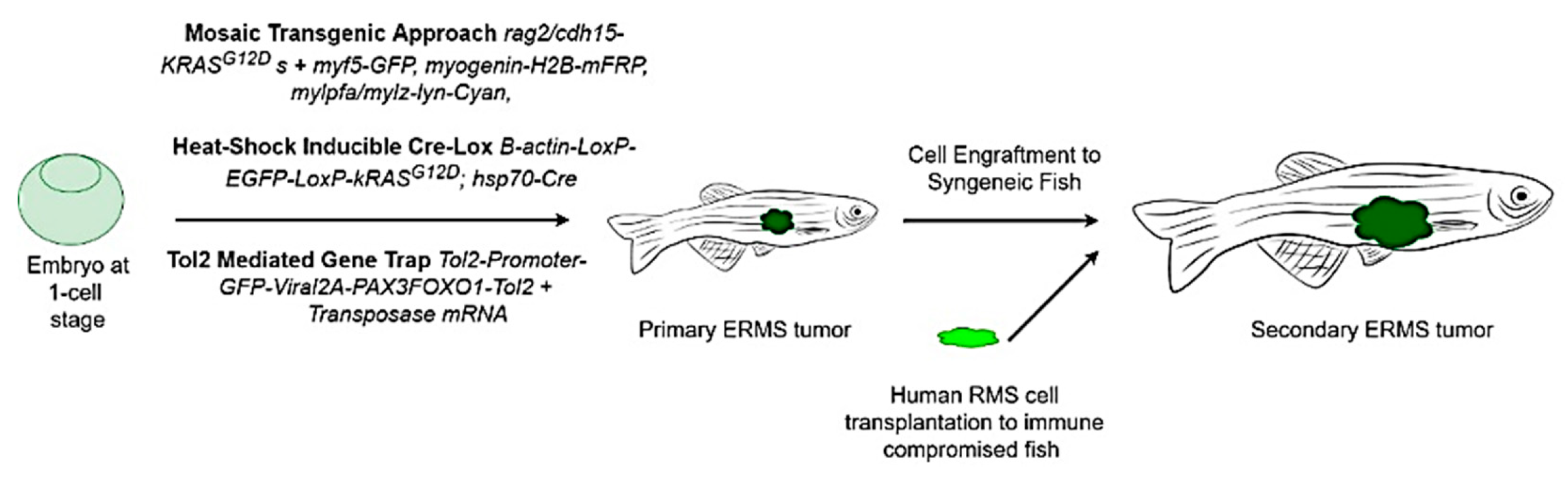
Figure 9.
KRAS-induced ERMS in zebrafish. (A) Co-injection of rag2-KRASG12D, myogenin-H2B-RFP and mylz2-lyn-cyan into myf5-GFP stable transgenic embryos at the one-cell stage. (B-C) A triple-labeled ERMS at 16 days of life. (D) Serial transplantation of myf5-GFP positive ERMS propagating cells. (E-G) A primary ERMS arising in syngeneic myf5-GFP; mylz2-mcherry transgenic zebrafish at 35 days post-fertilization. (E) Fluorescent and bright-field images of transgenic fish, (F) tumor histology, (G) FACS result of labeled ERMS cells, (H) Fluorescent and bright-field images of engrafted fish, (I) histology of tumor and (J) FACS of isolated ERMS cells. Adapted from [386] with get permission from Elsevier.
Figure 9.
KRAS-induced ERMS in zebrafish. (A) Co-injection of rag2-KRASG12D, myogenin-H2B-RFP and mylz2-lyn-cyan into myf5-GFP stable transgenic embryos at the one-cell stage. (B-C) A triple-labeled ERMS at 16 days of life. (D) Serial transplantation of myf5-GFP positive ERMS propagating cells. (E-G) A primary ERMS arising in syngeneic myf5-GFP; mylz2-mcherry transgenic zebrafish at 35 days post-fertilization. (E) Fluorescent and bright-field images of transgenic fish, (F) tumor histology, (G) FACS result of labeled ERMS cells, (H) Fluorescent and bright-field images of engrafted fish, (I) histology of tumor and (J) FACS of isolated ERMS cells. Adapted from [386] with get permission from Elsevier.
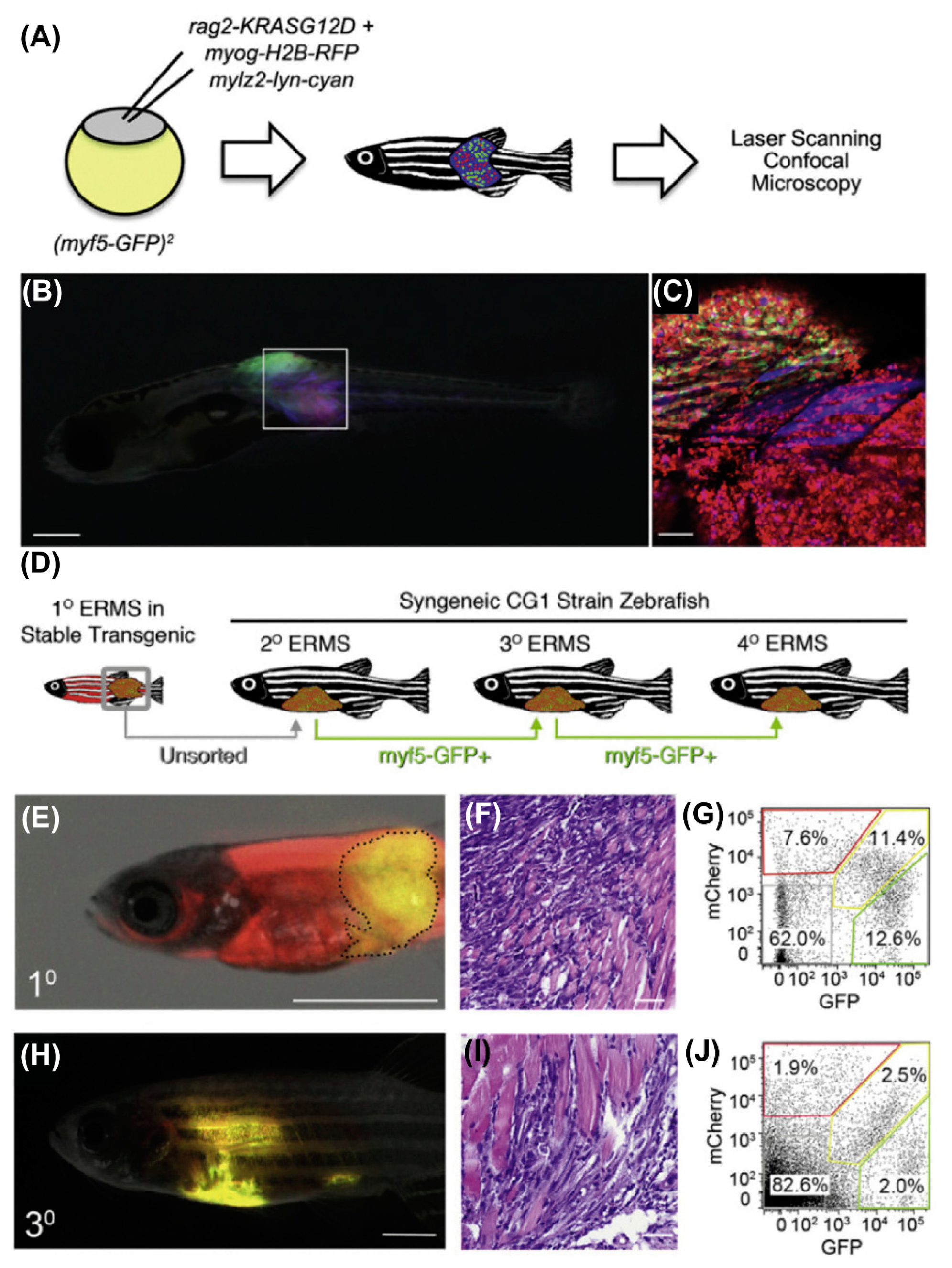
Figure 10.
Cre-mediated KRASG12D transgene expression in zebrafish. (A) β-actin-LoxP-EGFP-LoxP-kRASG12D transgene illustration. Transgenic zebrafish at 24 hr post-fertilization (B and C), and 44 days post-fertilization (D and E). (F) Heat-shock was performed for KRASG12D in situ hybridization of the transgenic embryos (double) at24 hr post-fertilization, and (F) without heat-shock. KRASG12D expressing cells were annotated with arrows. (H) The number of cells with KRASG12D -expression in single embryos with heat treatment from 4 to 5 hr post-fertilization and analyzed at 8, 12, 16, 20, and 24 hr post-fertilization (* is related to the P<0.001). Abbreviations: +HS= heat shock, NoHS= non-heat shocked. Reprinted from [389] with permission of PNAS.
Figure 10.
Cre-mediated KRASG12D transgene expression in zebrafish. (A) β-actin-LoxP-EGFP-LoxP-kRASG12D transgene illustration. Transgenic zebrafish at 24 hr post-fertilization (B and C), and 44 days post-fertilization (D and E). (F) Heat-shock was performed for KRASG12D in situ hybridization of the transgenic embryos (double) at24 hr post-fertilization, and (F) without heat-shock. KRASG12D expressing cells were annotated with arrows. (H) The number of cells with KRASG12D -expression in single embryos with heat treatment from 4 to 5 hr post-fertilization and analyzed at 8, 12, 16, 20, and 24 hr post-fertilization (* is related to the P<0.001). Abbreviations: +HS= heat shock, NoHS= non-heat shocked. Reprinted from [389] with permission of PNAS.
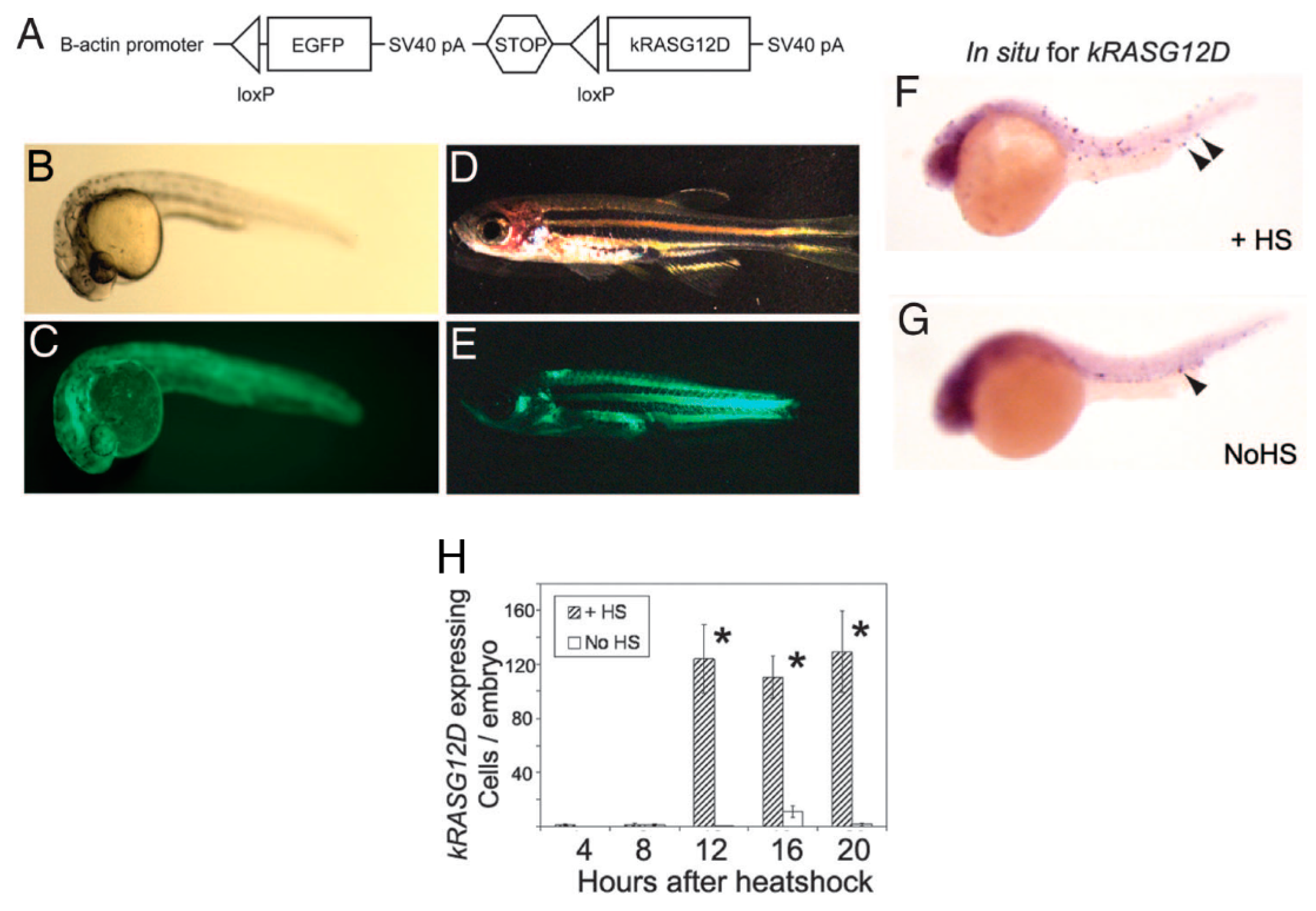
Figure 11.
Human PAX3-FOXO1 expression in zebrafish with different promoters using Tol2 trap gene system. (A) The Tol2-promoter-GFP-2A-PAX3-FOXO1-Tol2 was injected to the zebrafish at their single-cell stage and after evaluating the expression of GFP in 1-day embryos they were grown up to 19 months. (B) Production of PNET in the wild-type genetic zebrafish by PAX3-FOXO1 expressed by β-actin. (C) Creation of RMS in tp53M214K/M214K-sensitized genetic background via expression of PAX3-FOXO1 by CMV promoter. (D) PAX3-FOXO1 via ubiquitin promoter generated a non-differentiated sarcoma in the wild-type genetic zebrafish. Reprinted from [391].
Figure 11.
Human PAX3-FOXO1 expression in zebrafish with different promoters using Tol2 trap gene system. (A) The Tol2-promoter-GFP-2A-PAX3-FOXO1-Tol2 was injected to the zebrafish at their single-cell stage and after evaluating the expression of GFP in 1-day embryos they were grown up to 19 months. (B) Production of PNET in the wild-type genetic zebrafish by PAX3-FOXO1 expressed by β-actin. (C) Creation of RMS in tp53M214K/M214K-sensitized genetic background via expression of PAX3-FOXO1 by CMV promoter. (D) PAX3-FOXO1 via ubiquitin promoter generated a non-differentiated sarcoma in the wild-type genetic zebrafish. Reprinted from [391].
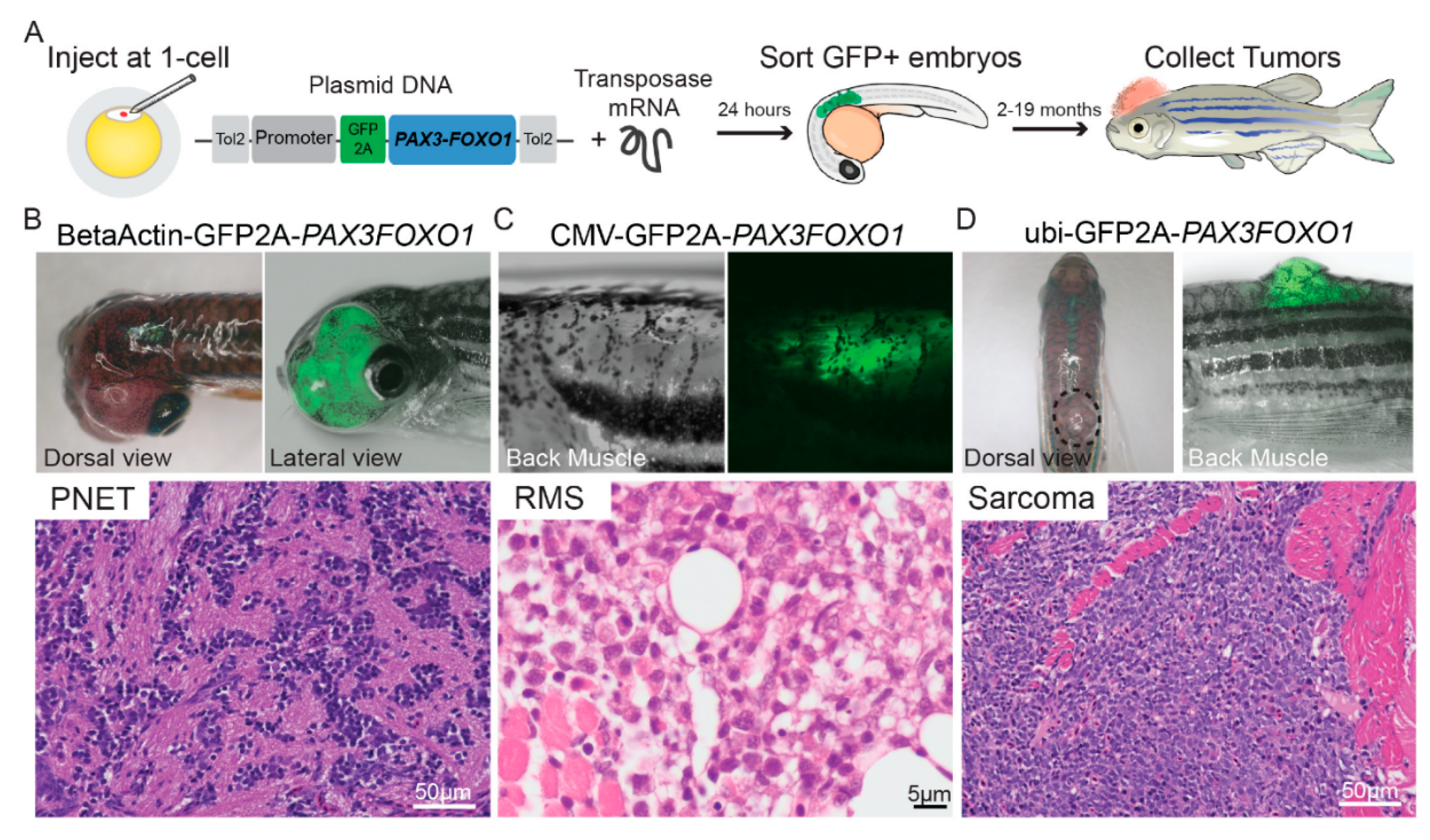
Figure 12.
Multichromatic imaging of ERMS tumor heterogeneity at single cell resolution. Florescent images of (A) ERMS tumor in different types of zebrafish (myf5-GFP, myogenin-H2B-mRFP, and mylpfa-lyn-cyan) and (B) flk1-mCherry, rag2E450fs Casper fish intra-muscularly engrafted with fluorescently labeled ERMS after 4 weeks of transplantation (n ¼ 4 animals). (C) Confocal images of mCherry-labelled vasculature indicated via dashed lines illustrated with X100 magnification (left) and X400 magnification (right). The number of differentiated cells (Diff) was less than the TPCs. Myosin-expressing differentiated cells (Diff.). Reprinted from [385].
Figure 12.
Multichromatic imaging of ERMS tumor heterogeneity at single cell resolution. Florescent images of (A) ERMS tumor in different types of zebrafish (myf5-GFP, myogenin-H2B-mRFP, and mylpfa-lyn-cyan) and (B) flk1-mCherry, rag2E450fs Casper fish intra-muscularly engrafted with fluorescently labeled ERMS after 4 weeks of transplantation (n ¼ 4 animals). (C) Confocal images of mCherry-labelled vasculature indicated via dashed lines illustrated with X100 magnification (left) and X400 magnification (right). The number of differentiated cells (Diff) was less than the TPCs. Myosin-expressing differentiated cells (Diff.). Reprinted from [385].
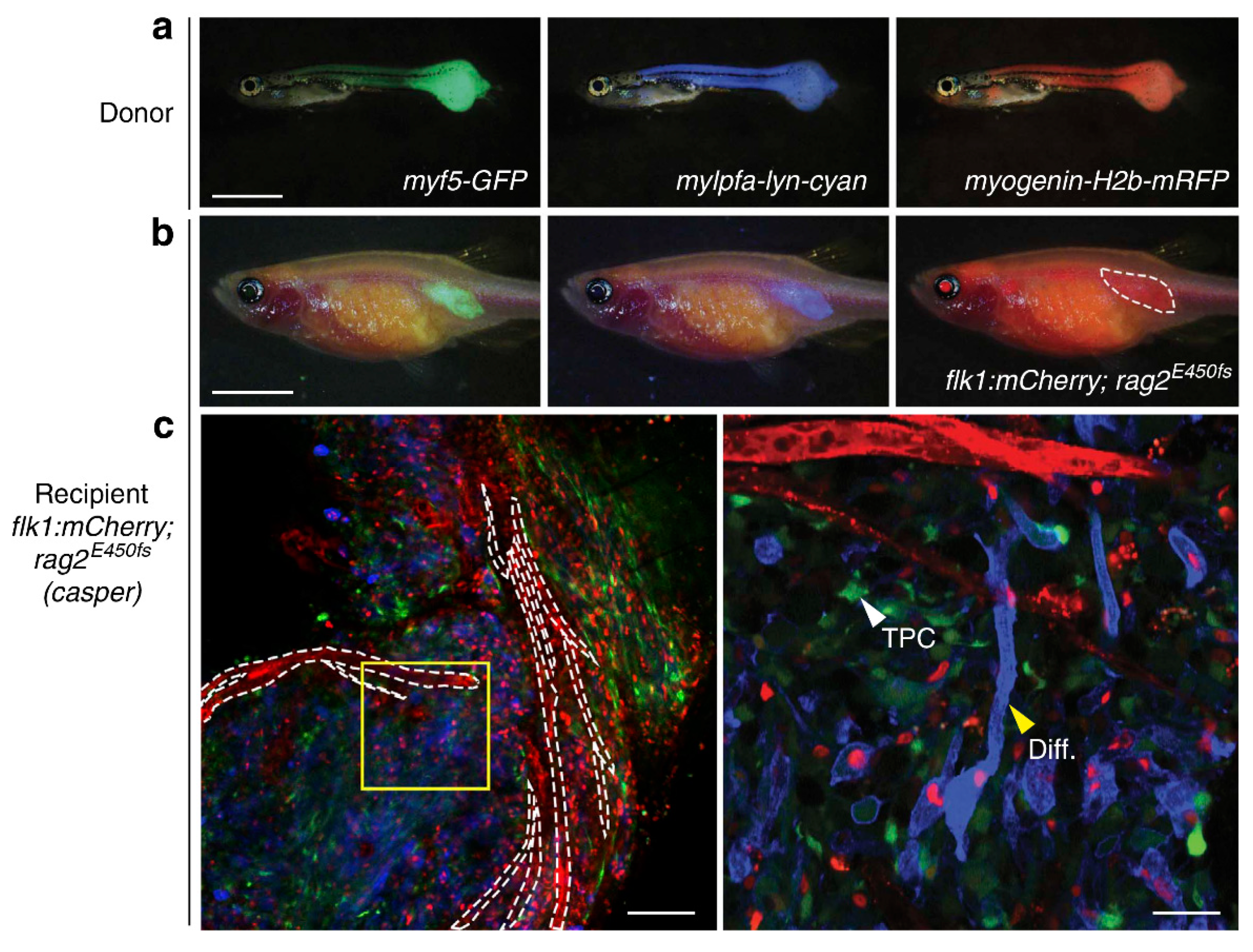
Figure 13.
TMZ and olaparib reduce the human RMS tumor growth in immunodeficient zebrafish. (A) Experimental design for growing GFP-expressing RMS cells in fish. Fish were selected at 7 days of post-transplantation (dpt), dosed with the drugs at 7, 14 and 21 dpt, sacrificed at 28 dpt. (B) Fluorescent images of tumor growth in engrafted animal after 7 days (before drug administration (left)) and 28 days (after 3 times of drug administration (right)) of post-transplantation. Histopathological analysis of RD engrafted sections stained by (C) Hematoxylin and eosin, (D) Ki67, and (E) TUNEL. (F) Relative growth of ERMS and ARMS cell lines after drug administration. Reprinted from [380] with the permission of Cell Press.
Figure 13.
TMZ and olaparib reduce the human RMS tumor growth in immunodeficient zebrafish. (A) Experimental design for growing GFP-expressing RMS cells in fish. Fish were selected at 7 days of post-transplantation (dpt), dosed with the drugs at 7, 14 and 21 dpt, sacrificed at 28 dpt. (B) Fluorescent images of tumor growth in engrafted animal after 7 days (before drug administration (left)) and 28 days (after 3 times of drug administration (right)) of post-transplantation. Histopathological analysis of RD engrafted sections stained by (C) Hematoxylin and eosin, (D) Ki67, and (E) TUNEL. (F) Relative growth of ERMS and ARMS cell lines after drug administration. Reprinted from [380] with the permission of Cell Press.
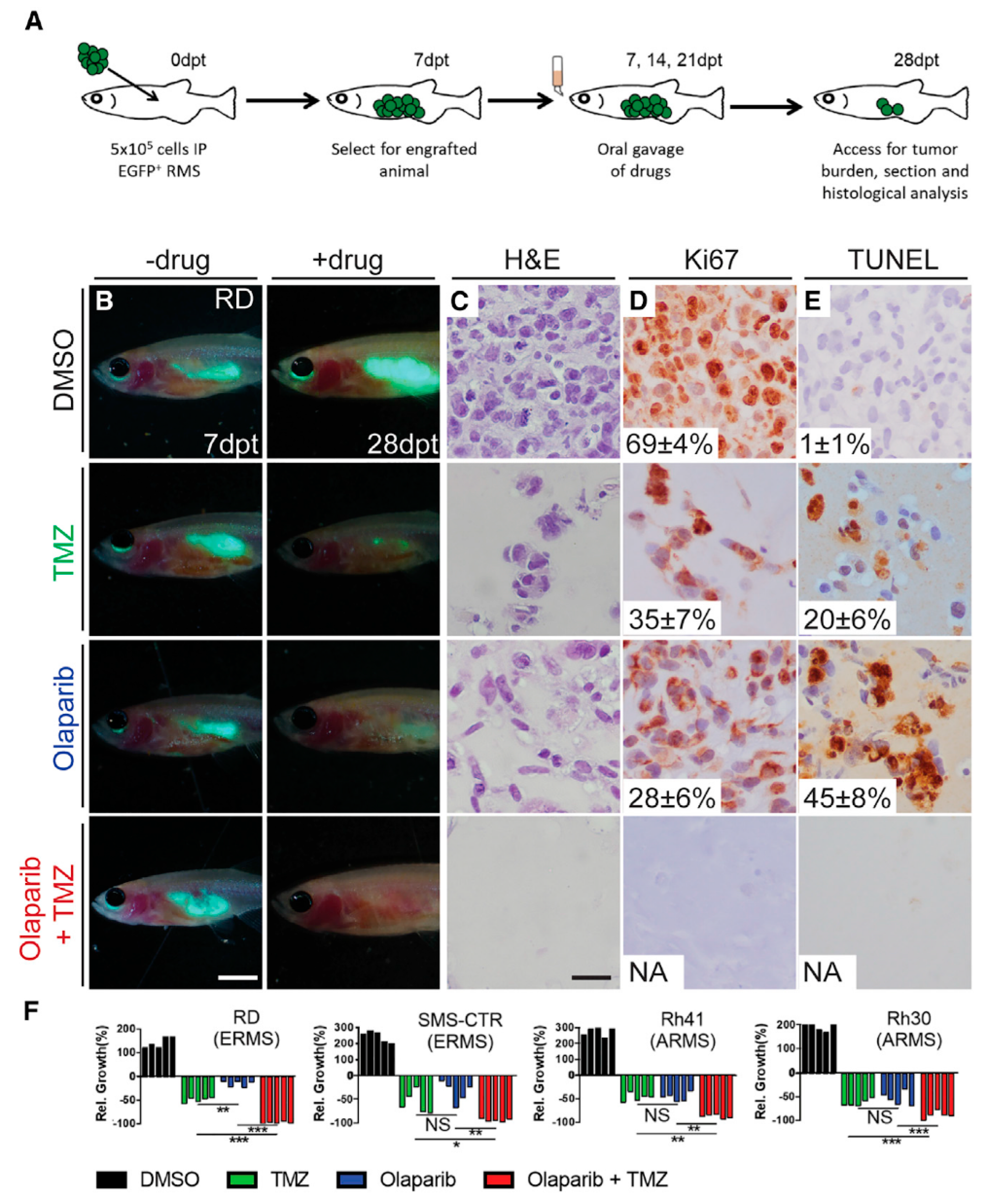
Figure 14.
(A) Confocal images of cell seeded scaffolds after 7 days using Phalloidin (Red)/DAPI (Blue) staining. The results of cell viability test (Alamar Blue) of (B) E0 and E1 groups without/with electrical stimulation (1.5 V), respectively and (C) cells treated with different formulations. Reprinted from [413].
Figure 14.
(A) Confocal images of cell seeded scaffolds after 7 days using Phalloidin (Red)/DAPI (Blue) staining. The results of cell viability test (Alamar Blue) of (B) E0 and E1 groups without/with electrical stimulation (1.5 V), respectively and (C) cells treated with different formulations. Reprinted from [413].
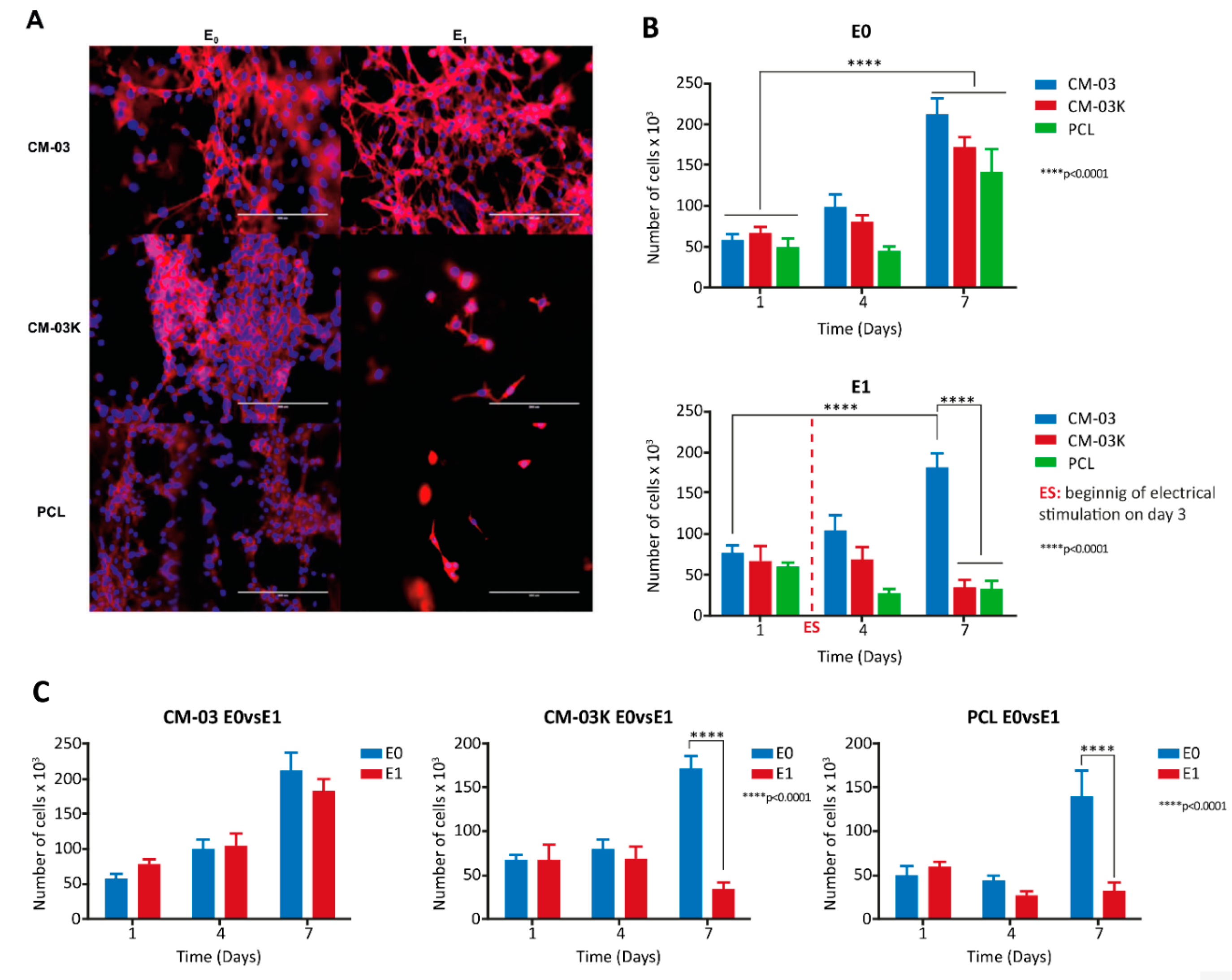
Figure 15.
In situ printing of scaffolds using a handheld bioprinter. (A) Bioprinting of cell-laden hydrogels for the VML injuries treatment. (B) A handheld bioprinter, which is able to crosslink the printed hydrogel scaffolds in situ using the provided UV light source. (C) Scaffold printed on a non-flat porcine skeletal muscle. (D) Printing an N-shaped scaffold on a glass slide. Reprinted from [414] with permission from ACS Publications.
Figure 15.
In situ printing of scaffolds using a handheld bioprinter. (A) Bioprinting of cell-laden hydrogels for the VML injuries treatment. (B) A handheld bioprinter, which is able to crosslink the printed hydrogel scaffolds in situ using the provided UV light source. (C) Scaffold printed on a non-flat porcine skeletal muscle. (D) Printing an N-shaped scaffold on a glass slide. Reprinted from [414] with permission from ACS Publications.
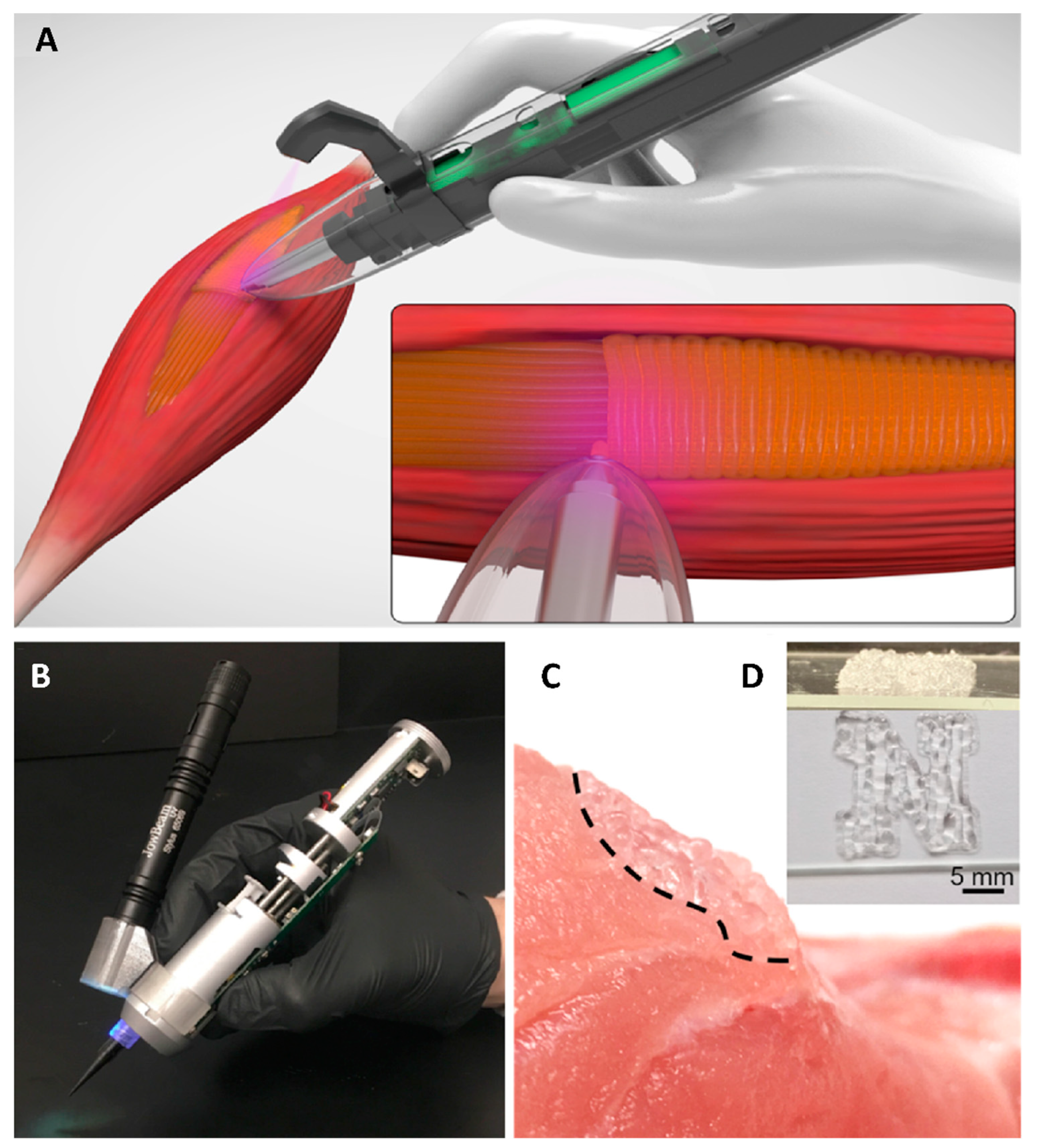
Figure 16.
Application of in situ printing in murine model with VML injuries. (A) Implanting the GelMA hydrogels into murine VML injury through in situ printing method. (B) Before and after VML surgery, and after printing the GelMA hydrogel in the injured site. (C) Histopathological analysis of the interface of the printed scaffold interface and the skeletal muscle tissue 4 weeks after surgery. Reprinted from [414] with permission from ACS Publications.
Figure 16.
Application of in situ printing in murine model with VML injuries. (A) Implanting the GelMA hydrogels into murine VML injury through in situ printing method. (B) Before and after VML surgery, and after printing the GelMA hydrogel in the injured site. (C) Histopathological analysis of the interface of the printed scaffold interface and the skeletal muscle tissue 4 weeks after surgery. Reprinted from [414] with permission from ACS Publications.
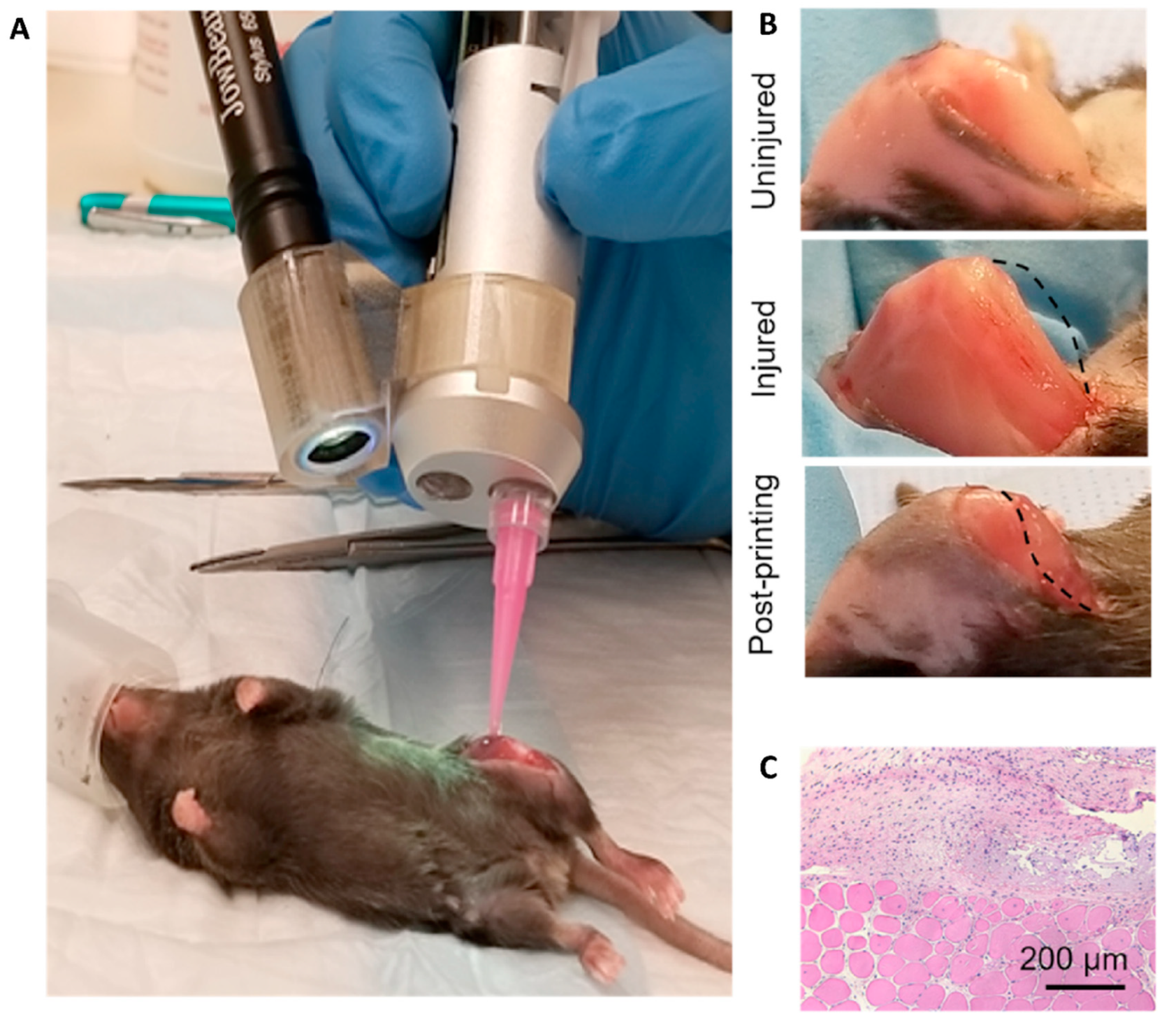
Figure 17.
(A) Coronal (Left) and Axial plane (Right) of liver tissue CT image; blue color is related to liver, and red and brown colors are related to the portal system and intrahepatic biliary tract –tumor, respectively. Green was also used for determining the rest of the tumor. (B) Model design procedure. (C) After production of all parts, inner parts were located inside of the printed molds and then 3D manufactured surgical planning prototype was produced by casting method using 1%wt agarose. Reprinted from [406] with permission from ACS Publications.
Figure 17.
(A) Coronal (Left) and Axial plane (Right) of liver tissue CT image; blue color is related to liver, and red and brown colors are related to the portal system and intrahepatic biliary tract –tumor, respectively. Green was also used for determining the rest of the tumor. (B) Model design procedure. (C) After production of all parts, inner parts were located inside of the printed molds and then 3D manufactured surgical planning prototype was produced by casting method using 1%wt agarose. Reprinted from [406] with permission from ACS Publications.
Figure 18.
Demonstration of the sheet structure distortion with alterations in the RD ratio: (A) Sheet morphology time course (t = 0 ~ 96 hr and scale bar=2 mm), (B) The RDs (green) and HSMMs (red) localization in heterogeneous sheets (t=96 hr) and the time course of HSMM cell in multilayered cell sheet, and (C) The HSMM sheet (both green and red cells are HSMMs) as the control, and (D) The mixture of 10% of RDs (green) and HSMM (red) sheet. Reprinted from [445] with permission from Elsevier.
Figure 18.
Demonstration of the sheet structure distortion with alterations in the RD ratio: (A) Sheet morphology time course (t = 0 ~ 96 hr and scale bar=2 mm), (B) The RDs (green) and HSMMs (red) localization in heterogeneous sheets (t=96 hr) and the time course of HSMM cell in multilayered cell sheet, and (C) The HSMM sheet (both green and red cells are HSMMs) as the control, and (D) The mixture of 10% of RDs (green) and HSMM (red) sheet. Reprinted from [445] with permission from Elsevier.
Figure 19.
The 3D TEM fabrication workflow for RMS in vitro studies: (A) PDMS mold fabrication using biopsy punch, (B) Thermal crosslinking of collagen hydrogel mixture with cells are conducted in 12-well plates for 45 min at 37°C before being cell culturing, (C) After 3D culturing of constructs, the drugs are added and their cytotoxic effect is analyzed using bright-field microscopy, immunocytochemistry, or live/dead viability assays after 48 or 96 hr cell exposure to the cytotoxic drugs. Reprinted from [446].
Figure 19.
The 3D TEM fabrication workflow for RMS in vitro studies: (A) PDMS mold fabrication using biopsy punch, (B) Thermal crosslinking of collagen hydrogel mixture with cells are conducted in 12-well plates for 45 min at 37°C before being cell culturing, (C) After 3D culturing of constructs, the drugs are added and their cytotoxic effect is analyzed using bright-field microscopy, immunocytochemistry, or live/dead viability assays after 48 or 96 hr cell exposure to the cytotoxic drugs. Reprinted from [446].
Table 1.
Characteristics of Malignant Rhabdomyosarcoma Subtypes.
| Embryonal | Alveolar | Pleomorphic | Spindle Cell/Sclerosing++ | |
|---|---|---|---|---|
| Prevalence | 2.6 (most common) [2]* | 1.0 (common) [2]* | Rare[2,20] | Rare [2,20] |
| Age | Bimodal distribution: peak incidence ages 0-4 > 14-18 [2,22] | Late childhood/adolescents [3,23] | 40-70yrs of age, peak during 6th decade of life [34] | Children [4,472] |
| Gender predominance | Male [4] | None | Male [3] | NA |
| Subtypes | Spindle cell and Botryoid subtypes2+ | NA+ | Classic, Round cell and Spindle cell subtypes4 | NA |
| Primary Tumour Location | Head/Neck, Superior nasal quadrants, eye socket, bladder and prostate [23] | Trunk and extremities, Inferior orbit [23] | Lower extremities [3,4] | Head /Neck region, paratesticular region [4,23] |
| Genetics | 80% have loss of heterozygosity at 11p15 (IGF-2 gene) [3] Associated with familial cancer syndromes eg LFS, NF1 |
60% are t(2:13)(q35:114): PAX3-FOXO1 positive [3] 20% are t(1;13)(p36;q14): PAX7-FOXO1 positive [1] 20% are, F.N.; resemble ERMS characteristics/prognosis [3] |
NA | NA |
| Histology | Immature rhabdomyoblast, less dense stromal rich background vs ARMS, lacks alveolar pattern [24] | Densely packed, small, round cells lining septations that resembles fetal alveoli [4] | Differentiated from high-grade soft tissue sarcomas by the presence of skeletal muscle proteins on immunohistochemistry [3,4] | NA |
IGF-2: Insulin Growth factor-II, LFS: Li-Fraumeni Syndrome, NF1: neurofibromatosis type 1, FFS: failure free survival, FOXO1: forkhead box protein O1, t(2:13): translocation between chromosomes 2 and 13, t(1:13): translocation between chromosomes 1 and 13, FN: fusion negative, ERMS: embryonal rhabdomyosarcoma. *Per 1, 000, 000 population in US, **Based on the current Children’s Oncology Group Soft Tissue Sarcoma Risk Stratification. +different sources divide botryoid as subtypes of ERMS [23] vs subtypes of ERMS and ARMS [4], ++Relatively new subtype, thus not much information is available.
Table 2.
RMS Targeted Therapies and their clinical trial status.
| Treatment | Clinical Trial Phase | Reference |
|---|---|---|
| Pazopanib | II | [242] |
| Pazopanib or Placebo | III | [241] |
| Sorafenib | II | [477] |
| Sorafenib | II | [478] |
| Crizotinib | II | [237] |
| Temsirolimus | II | [221] |
| Cixutumumab | II | [479] |
| Cixutumumab | II | [227] |
Table 4.
Recent clinical trials on novel therapeutic agents against RMS.
| Therapeutic agents | Clinical Trial ID | Number of participants | Study phase | Comments |
|---|---|---|---|---|
| Abemaciclib | NCT04238819 | 60 | I | Study recruiting |
| Temsirolimus or Bevacizumab | NCT01222715 | 87 | II | Study completed, has results |
| Cixutumumab | NCT00668148 | 113 | II | Study completed, has results |
| Cixutumumab | NCT00831844 | 116 | II | Study completed, has results |
| Cixutumumab or Temozolomide | NCT01055314 | 175 | II | Study completed, has results |
| Cixutumumab and Temsirolimus | NCT01614795 | 46 | II | Study completed, has results |
| Crizotinib | NCT01524926 | 582 | II | Study active, not recruiting |
| Onivyde and Talazoparib or Temozolomide | NCT04901702 | 160 | I/II | Study not yet recruiting |
| Palbociclib | NCT03709680 | 133 | I | Study recruiting |
| Pazopanib | NCT01532687 | 54 | II | Study completed, has results |
| Regorafenib | NCT02048371 | 150 | II | Study recruiting |
| Regorafenib | NCT02085148 | 62 | I | Study completed, has results |
| Sorafenib | NCT01502410 | 20 | II | Study completed, has results |
| Sorafenib | NCT02050919 | 20 | II | Study completed, has results |
| Temozolomide | NCT01355445 | 120 | II | Study completed, has results |
| Temsirolimus | NCT02567435 | 397 | III | Study recruiting |
| Temsirolimus | NCT00106353 | 71 | I & II | Study completed, has results |
| Temsirolimus | NCT00949325 | 24 | I & II | Study completed, has results |
| Trabectedin | NCT00070109 | 50 | II | Study completed, has results |
| Vinorelbine | NCT04994132 | 100 | III | Study not yet recruiting |
| Vinorelbine | NCT00003234 | 50 | II | Study completed, has results |
| Vinorelbine | NCT04994132 | 100 | III | Study not yet recruiting |
Table 5.
Genetic and pharmacological inhibition of autophagy synergize with therapeutic agents in RMS.
Table 5.
Genetic and pharmacological inhibition of autophagy synergize with therapeutic agents in RMS.
| Model | Therapeutic agent | Autophagy inhibitor | Act | Outcomes/Effects | Ref. | |
|---|---|---|---|---|---|---|
| Pharmacologic | Genetic | |||||
|
ARMS cell lines (RH30 & RH4) |
Temozolomide | Bafilomycin A1 | - | Inhibition of V-ATPase / ATG7 | Promoted chemotherapy efficacy | [289] |
| Human RMS cell line (hRD) | Doxorubicin | Simvastatin | - | Activation of mitochondrial apoptotic pathway (BAX) | Improved the sensitivity of cancer cells towards Dox and improved antitumor activity | [296] |
| ERMS CSC cell lines | Doxorubicin | Omeprazole | V0c siRNA |
Inhibition of V-ATPase / Lysosomal pH | Enhanced cytotoxic effect of chemotherapy and reduced the invasive potential of ERMS CSCs | [304] |
|
Human RMS cell lines (RH30 & hRD) |
Ciclopirox Olamine | Chloroquine | - | Inhibition of Lysosomal pH | Improved antitumor activity | [305] |
|
Human RMS cell lines (RH30 & hRD) |
Bortezomib & 17-DMAG |
Chloroquine | - | Inhibition of Lysosomal pH / UPS & HSR systems | Enhanced drug-induced apoptosis | [306] |
|
ERMS (RD) & ARMS (RMS13) cell lines |
Bortezomib | Bafilomycin A1 / ST80 | BAG3 siRNA | Inhibition of V-ATPase /ATG7 | Impaired cancer cell growth and increased cell death | [307] |
|
ERMS cell lines) )(RD, RH30 & RMS) |
Tenovin-6 | - | SIRT1 & SIRT2 siRNA | Inhibition of Sirtuins | Impaired cancer cell growth and increased apoptosis | [315] |
| Human RMS cell line (hRD) | Methotrexate & SDH | - | - | Inhibition of P-gp | Enhanced methotrexate mediated cytotoxicity | [320] |
| ARMS RH30 (FG+) | Vincristine | Etoposide | - | Inhibition of PLK1 / Activation of mitochondrial apoptotic pathway (BAX/BAK) | Improved antitumor activity and increased apoptosis | [486] |
| ARMS RH30 &ERMS RD, TE381.T (FG+) | Vincristine | Volasertib | - | |||
| ARMS RH30 (FG+) | Doxorubicin | Etoposide | - | |||
| ARMS RH30 (FG+) | Eribulin | Etoposide | - | [487] | ||
| ARMS RMS1 (FG+) | Etoposide | Volasertib | - | [488] | ||
ARMS: Alveolar Rhabdomyosarcoma, V-ATPase: Vacuolar H+ ATPase, ERMS: Embryonal Rhabdomyosarcoma, siRNA: small interfering RNA, V0c: V0c ATPase, Dox: Doxorubicin, CSC: Cancer Stem Cells, 17-DMAG: 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin, UPS: The ubiquitin-proteasome system, HSR: the heat shock response, ST80: The cytoplasmic histone deacetylase 6 inhibitor ST80, BAG3: Bcl-2-associated athanogene 3, PQC: Protein quality control system, SDH: Silibinin di-hemisuccinate, P-gp: P-glycoprotein, FG +: PAX3-FOXO1 fusion genes positive, PLK1: Polo-like kinase 1.
Table 6.
Studies for KRAS-induced RMS in Zebrafish.
| Parameter | Method | Tumor Onset | Outcomes | Ref |
|---|---|---|---|---|
| HDAC6 | CRISPR/Cas9 method for deletion of HDAC6*, constructs containing rag2-KRASG12D-U6-hdac6 guide RNA, rag2-Cas9 and myogenin-H2B-RFP injected in 1st-cell stage | 15-20 days of post-fertilization | HDAC6 was found to has significant role in ERMS tumorigenesis, promoting tumor growth, metastasis and self-renewal. | [388] |
| RAC1 | Engraftment of KRAS-driven zebrafish ERMS tumors co-expressing GFP and mutant RAC1** (RAC1V12), dorsal subcutaneous way | Tumor harvest after 3 weeks | Zebrafish expressing RAC1V12 exhibited more aggressive tumor growth and invasiveness compared to the control group (empty vector). | [388] |
| tp53 | KRAS-induced ERMS generated in tp53del/del zebrafish. | Tumors were tracked 90 days | Deletion of tp53 increased metastasis and invasion of ERMS cells, but not the total frequency of tumor cells. | [489] |
| Van Gogh-like 2 (Vangl2) | KRAS-induced ERMS is generated in fish with additional Vangl2 gene. | 15 days of post-fertilization, 90 days after transplantation | Expression of Vangl2 supports TPCs and has positive effect for their self-renewal. No effect of Vangl2 was found on the size, penetrability and latency of the ERMS tumors. | [490] |
| Intracellular NOTCH1 (ICN1) | KRAS-induced ERMS (KRASG12D and KRASG12D-ICN1) was generated in transgenic zebrafish expressing myf5-GFP and mylz2-mCherry | Tumors imaged over 100 days after transplantation to the recipient fish. | ICN1 enhanced the number of tumor propagating cells in zebrafish ERMS, by blocking the differentiation of zebrafish ERMS cells into self-renewing myf5 positive TPCs. | [383] |
| myf5 | KRAS-induced ERMS was generated in zebrafish with rag2-KRASG12D, with additional mylpfa-mCherry, myf5-GFP injection | Animals were imaged after 35 days post-fertilization | Re-expression of myf5 enhanced tumor formation and penetration, thus had a role in reprogramming of ERMS cells into TPCs. | [491] |
| GSK3 inhibitors screening | KRAS-induced ERMS in myf5-GFP and/or mylz2-mCherry transgenic fish | Tumor engraftment was monitored from 10 to 120 days after drug treatment | GSK3*** inhibitors suppressed ERMS growth, depleted TPCs and blocked self-renewal while activated the WNT/β-catenin pathway. | [382] |
| Screening of PD98059 and TPCK drugs | rag2-KRASG12D and rag2-DsRed transgenic zebrafish | Tumors were observed after 7-10 days of post-fertilization | Tumor growth was reduced with the drugs treatments, showing anticancer potential. | [390] |
HDAC6*: Histone deacetylase 6, RAC1**: Ras-related C3 botulinum toxin substrate 1, GSK3***: Glycogen synthase kinase 3.
Table 7.
Available animal model approaches for RMS and comparative evaluation.
| Animal Model | Injection Types | Pros | Cons |
|---|---|---|---|
| CDX |
Heterotopic (subcutaneous) engraftment – Easy to apply and used to monitor tumor growth. In therapeutic applications, drug response may differ from the orthotopic engraftment. Orthotopic engraftment – The most preferable injection type for clinical applications due to high prediction value. Technically, this injection technique is challenging and difficult to monitor the tumor growth. |
- Easy to scale - Low cost and high availability - Easy to manipulate |
- Low yield in tumor tissue observation - Therapeutic applications are limited. - Low clinical relevance |
| PDX | - High feasibility and good tumor reflection - Strong clinical relevance - High therapeutic prediction |
- High cost and prolonged time are required. - Therapeutic applications are limited - Low availability |
|
| EIMM | - Suitable for tumor initiation and progression observation - Cost is moderate. - Feasibility is moderate. |
- Low clinical relevance depending on the sarcoma type. | |
| GEMM | - Easy-to-manipulate the expression of genes - Wide variety of applications (i.e., tumorigenesis, tumor progression, and maintenance) - High therapeutic prediction |
- High cost - Difficult feasibility - Low availability for the rare type of sarcomas |
Table 8.
The specifications of phantom production methods.
| Phantom production method | Advantages | Disadvantages |
|---|---|---|
| FFF |
|
|
| SLA |
|
|
| Material Jetting |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



