Submitted:
12 September 2023
Posted:
13 September 2023
You are already at the latest version
Abstract
Myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) originate from preleukemic hematopoietic conditions, such as clonal hematopoiesis of indeterminate potential (CHIP) or clonal cytopenia of undetermined significance (CCUS) and have desolate outcomes. The prognosis is worse in patients with TP53 mutations which are often linked to complex karyotypes and contribute to worse responses to induction therapy, demethylating agents or venetoclax-based treatments. Survival of patients with TP53 gene mutations is often less than one year. Therefore, TP53-mutated MDS and AML are now classified separately in the unfavorable risk category. In the clinical setting, the wild-type p53 is reactivated pharmacologically by targeting p53/MDM2/MDM4 interactions and mutant p53 reactivation is achieved by refolding the DNA binding domain to wild-type-like conformation or via targeted degradation of the mutated protein. This review discusses our current understanding of p53 biology in MDS and AML and the promises and failures of p53 reactivation in the clinical trial setting.
Keywords:
MDS
; AML
; p53
; MDM2
; MDM4
; p73
; improved therapy
FACTS
- Management of TP53-mutated myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) remains a therapeutic challenge despite significant recent advancements.
- Clinical studies on p53 reactivation have yielded inconsistent results in both MDS and AML.
- Further research is necessary to explore the biology of p53 in the pre-leukemic population of hematopoietic stem cells (HSCs).
OPEN QUESTIONS
- Can the effectiveness of mutant p53 structure correctors in TP53-mutated MDS and AML be determined through predictive biomarker stratification?
- Is there a possibility of high-affinity MDM2 inhibitors being approved as standalone treatments for MDS and AML?
- Could the utilization of p53 reactivating compounds in combination therapies to impede key drivers potentially result in enhanced outcomes in MDS and AML?
- Is it possible to develop a biomarker discovery test that uses conformation-specific antibodies to stratify patients for mutant p53 reactivating drugs?
1. Introduction
Myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) are cognate, clonal hematological neoplasms and originate from pre-malignant, mutated hematopoietic stem cells (HSCs) that undergo clonal expansion after selection pressure, in a process called clonal hematopoiesis (CH) [1]. Extensive studies showed that HSCs are constricted to the lineage−CD34+CD38−CD90+CD45RA− compartment and bear driver mutations in CH [2]. CH results in an accumulation of large numbers of abnormal, immature myeloid cells in the bone marrow and peripheral blood called leukemic stem cells (Figure 1a). Clonal hematopoiesis is often occurring because of physiological aging and is associated with a higher risk of hematological cancers. The rate of CH progression to hematologic neoplasm is 0.5% - 1% per year [3,4].
In more detail. During the lifespan, HSCs undergo functional decline because of accumulated mutations resulting from the increased DNA damage or epigenetic reprogramming and reside bone marrow as a genetically heterogenous cell population [5]. Some HSCs that acquire somatic mutations in genetic modulators (DNMT3A, TET2, ASXL1) and in signaling molecules (JAK2V617F) gain a competitive fitness advantage in the presence of selective pressure and expand resulting in clonal hematopoiesis and the production of the clonal cells that exhibit maturation defects [6,7].
MDS and AML originate during CH from clonal hematopoiesis of indeterminate potential (CHIP) or clonal cytopenia of undetermined significance (CCUS), which falls into the category of clonal cytopenia (Figure 1a). CCUS was discovered thanks to the advancements in NGS techniques which allow to distinguish it from CHIP. It represents a continuum with MDS to which it progresses faster than CHIP after the acquisition of additional mutations and dysplasia [8,9]. In both, CHIP and CCUS the increased risk of progression to MDS and de novo AML occurs upon > 1 additional driver mutation, VAF >10% and acquisition of additional mutations [10]. For patients with CCUS the risk of progression to MDS/AML was reported to be 18% within 16 months and 95% in 10 years [3]. Other premalignant conditions can also progress to MDS or AML and include aplastic anemia, paroxysmal nocturnal hemoglobinuria and vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome [6].
Many MDS and AML subtypes share common driver alterations which might occur at different frequencies but target the same pathways; for example DNA methylation (TET2, DNMT3A, and IDH1/IDH2), chromatin/histone modification (MLL2, EZH2 and ASXL1), RNA splicing (SF3B1, SRSF2, U2AF1, U2AF2, and SF3A1) or p53, RAS and other signaling pathways [11,12]. MDS and AML are thus, heterogenous bone marrow disorders with common characteristics like expansion of clonal hematopoietic stem cells, cytopenia and marrow dysplasia.
Myelodysplastic syndrome (MDS)
MDS, especially high-grade MDS, has a high risk of transformation to a secondary AML [13]. MDS is the most common adult myeloid malignancy, has a blast count < 20%, and in about 30% transforms to AML, termed “secondary AML to MDS” or to bone marrow failure. According to International Consensus Classification (ICC) and the World Health Organization classification of lympho-hemopoietic neoplasms (WHO-HAEM5), the novel intermediate state, MDS/AML, is characterized by the presence of ≥10% but <20% of blasts which is applied to show a continuum of MDS to AML [6]. The new, 2022, classification system of MDS and AML, though of accurate in genetic markers classification and in regression of blast threshold, is troublesome for healthcare providers due to existing discrepancies in subclassification and diagnostic criteria which might affect the choice of standardized treatment or clinical trial eligibility [14]. Yet, it is outside the scope of this review to discuss the clinical weight and the shortcomings of the current classification system. Generally, MDS patients have a poor prognosis, with a median overall survival of only 5 years. MDS patients who progressed to AML, have high-grade MDS, with myeloblast count ≥ 20% and acquired/expanded abnormalities in TP53, RUNX1, or RAS genes [15]. Phenotypically, high-grade MDS has lower cell death rates when compared to lower-risk patients [16], and typically has inferior rates of complete remission, relapse-free survival, and overall survival compared with patients with de novo AML [15].
Acute myeloid leukemia (AML)
Chromosomal abnormalities, copy number variations and translocations and inversions are common genetic events in both, MDS and AML. AML is on average diagnosed in older patients, 68 years old or older, and has poor outcomes with the five-year overall survival of less than 30% and up to 50% in younger patients [17]. Regardless of the age of diagnosis, patients who have not responded to induction therapy, have dismaying outcomes [9]. The criterion for AML diagnosis might differ depending on the driver mutation, yet in the majority, AML patients have ≥ 10% or 20%, if misdiagnosis with chronic myeloid leukemia (CML) might occur. Secondary AML accounts for up to 25% to 35% of total AML cases [15] with most (60-80%) arising from MDS. Due to clinical impact, in the new ICC system a new subgroup was generated which constitutes a separate entity within the group of myeloid neoplasms with mutated TP53 which includes MDS, MDS/AML and AML with mutated TP53 [6,18,19]. TP53 mutations underly the aggressiveness of AML and, even though in MDS multihit TP53 mutations are required for diagnosis of MDS with mutated TP53, in AML and MDS/AML with mutated TP53, any pathogenic TP53 mutation VAF of ≥10% is sufficient for diagnosis [20]. This group of patients usually does not respond to standard induction treatment. In 2017 and later, eleven, new drugs or combinations were approved for AML by the Food and Drug Administration [21]. Among the new approvals, five drugs target known AML vulnerabilities; FLT3 (midostaurin, gilteritinib), IDH1 (ivosidenib), IDH2 (enasidenib) and BCL2 (venetoclax). Yet, the targeted treatments are not effective in high-risk TP53-mutated AML patients, who and have dismaying outcomes as assessed for the frontline treated patient group [22].
p53 tumor suppressor
p53 is a tumor suppressor and is encoded by the TP53 gene located at 17p13.1, a site undergoing chromosomal aberrations resulting in cytogenetic deletion at 17p13.1; loss of heterozygosity (LOH) at the 17p TP53 locus or mutations largely of missense type [18]. TP53 gene is often mutated in human cancers, in the majority in the DNA binding domain. Cancers with a high incidence of TP53 mutations are high-grade serous ovarian cancer, lung, colon, brain or pancreatic cancer [23]. The TP53 gene mutations of missense, nonsense, frameshift or in/dels types, may result in loss-of-function, gain-of-function or in the dominant negative effect [24] propensities of the mutated protein (reviewed in [25]). Yet, our understanding of the biology behind the multiple pathogenic variants is limited.
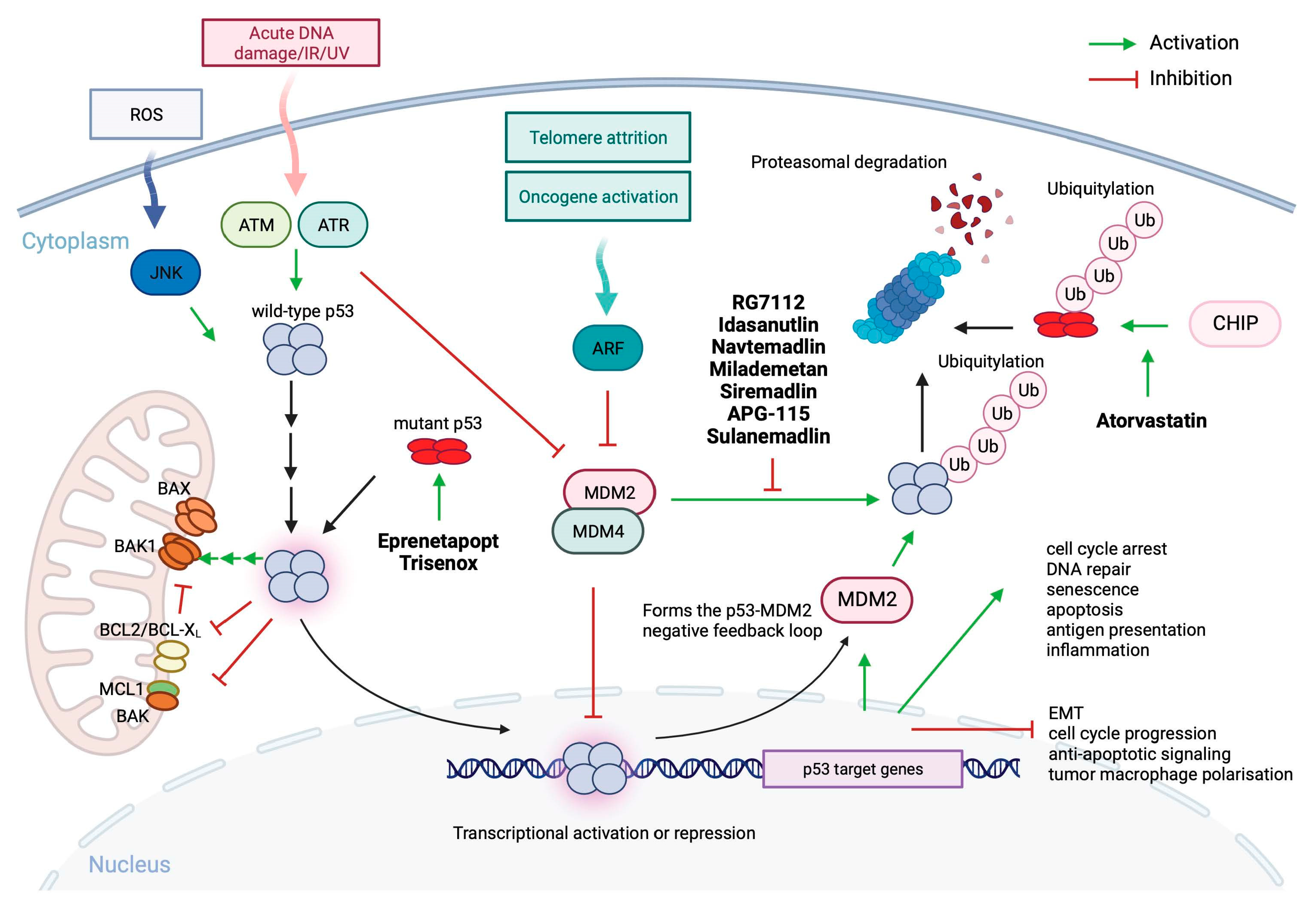
In cancers with intact TP53 gene, the functional protein is inactivated by overexpressed mouse double minute 2 (MDM2) and/or MDM4 which bind to the N-terminal domain and inhibit the transcriptional function of p53, or promote p53 mono- and/or polyubiquitination and nuclear export and proteasomal degradation [23]. p53 is a transcription factor which directly or indirectly activates or represses a range of target genes involved in a multitude of cellular processes like; cell cycle regulation, DNA repair, senescence, pro- and anti-oxidant response, apoptosis, ferroptosis, pyroptosis, cuproptosis, autophagy, immune response, inflammation, metabolism or fertility and stem cells renewal. The decision by which p53 drives the cell response to stress stimuli is complex and depends on the post-translational modifications and tissue/cell context (reviewed in [26]). In addition to regulating gene expression, p53 has also transcription-independent functions and can, upon cellular stress, localize to mitochondria and neutralizes BCL2 or BCL-XL oncogenes to release apoptosis associated, caspase-cleaved BH3 interacting domain death agonist, tBID or BCL2 associated X (BAX) from the inhibitory complex via direct binding to protein heterocomplex or bind to BCL2 antagonist/killer 1 (BAK1) pro-apoptotic protein and promote its dimerization through release form the anti-apoptotic MCL1 (Figure 2) (reviewed in [27,28]).
In MDS and AML, p53 inactivation and TP53 gene mutations represent an important resistance mechanism to DNA damaging chemotherapy [29] or to venetoclax [30] and are thus, underlying adverse prognosis. This review will highlight the contemporary status of the p53 re-activating drugs and emerging new, investigational therapies targeting the p53 pathway in MDS and AML.
2. p53 in MDS and AML
MDS and MDS/AML develop from mutated clones present in the hematopoietic compartment [31]. The presence of a TP53-mutated clone alone is yet not sufficient for the development of effective leukemogenesis (Figure 1b).
Within European Leukemia Net (ELN) 2017, TP53 mutations are associated with unfavorable risk category and decreased overall survival (OS < 2 years). Patients with AML with TP53 mutations and complex karyotype (CK) have inferior OS of 161 days vs 374 days compared with wild-type TP53 [32]. Even though, patients with mutant TP53 AML after complete remission receive allogeneic hematopoietic stem cell transplant, yet are among the group with high relapse rates [33]. Patients with MDS with excess blasts-2 (MDS-EB2) and TP53 mutations, share similar characteristics and clinical outcomes with mutant TP53, de novo AML patients. According to recent reports both, TP53 mutated AML and MDS-EB2, have practically undistinguishable biology; have blast count 15%-20% (20% cutoff is not considered specific any longer), in the majority (50% - 70%), have no co-existing driver mutations or rareness of NPM1 or FLT3 alterations [20,34], and possess high incidence of complex/monosomal karyotypes (80%–90%), which include abnormalities in chromosomes 5, 7, and 17 [20]. In therapy-related myeloid malignancies, TP53 mutations are not induced by the treatment itself but by existing progenitor clones with mutant TP53, that are resistant to DNA-damaging therapy, and expand in clonal hematopoiesis to give raise to TP53-mutated MDS/AML (Figure 1b) [1].
Congenital cancer predisposition syndromes predisposing to myeloid neoplasm are a separate entity according to WHO [35]. Hereditary cancer predisposition syndrome, Li Fraumeni (LFS), is an autosomal dominant condition connected with a high risk of a broad range of childhood- and adult-onset cancers. Patients with LFS develop multiple tumors during their lifespan; predominantly soft tissue sarcomas, osteosarcomas, pre-menopausal breast cancers, brain tumors, adrenocortical tumors and less frequently, pancreatic, ovarian or gastrointestinal cancers and other [36,37]. The incidence of leukemias is < 5% [38,39]. LFS is described by the heterozygous germline mutations in the TP53 gene [40]. Family history of inherited mutant (pathogenic variant) TP53 is a key criterion for the consideration of LFS yet, de novo mutations occur in ∼10%–20% of LFS cases [41]. Recent whole-genome sequence analysis combined with clock-like mutational signatures and MutationTimeR algorithm revealed that in LFS patients TP53 LOH occurs many years before tumor diagnosis, likely already in utero. It has been concluded that the copy number gains of mutant TP53 occur spontaneously in LFS patient cells and can readily outcompete diploid clones in a small number of generations [42]. In LFS patients all hematopoietic progenitor stem cells (HSPCs) carry TP53 mutations. Yet, the patients may mainly develop treatment-related myeloid neoplasm (t-NM) later in life (Figure 1b) and prognosticate a poor prognosis with standard therapies and even allogeneic stem cell transplant [38]. Thus, the risk of the development of t-MN in LFS patients should be taken into consideration when administering radiation treatment or myelosuppressive therapy.
Schwachman syndrome (SDS) with congenital mutations in Shwachman–Bodian–Diamond syndrome (SBDS) gene is a condition of high risk of developing myeloid neoplasms (MN) early in life [43]. In SDS, the SBDS protein which promotes the formation of the mature, translationally active 80S ribosome, is mutated, resulting in decreased ribosomal subunit joining and reduced translation efficiency and ribosomal stress (Figure 1b) [44]. Survival is poor in SDS patients who develop MDS or AML originating from CH [45,46]. It has been reported that the presence, number, persistence, and allele abundance of somatic TP53 mutations were not predictive of leukemia risk in SDS patients with CH, yet, the progression of TP53-mutated clones was found to be driven by the development of bi-allelic alterations of the TP53 locus via deletion, copy number (CN)-LOH, or point mutation (Figure 1b) [46]. It is hypothesized that continued ribosome stress in SDS HSPCs carrying a heterozygous TP53 mutation selects for clones that inactivate the second TP53 allele and lead to the development of TP53-mutated CH, but also to its progression to myeloid malignancy (Figure 1b) [1].
Because of the adverse outcomes for patients with TP53-altered AML/MDS as per norm, it should be strongly encouraged to enrol the patients into clinical trials. Such a strategy would let patients access promising treatments or new combinations with the potential to improve outcomes since the current gloom scenario shows dismal median survival of up to 10 months, irrespective of therapies used [47].
3. Targeting mutant p53 for improved therapy in MDS and AML
In TP53-mutated MDS, the “multihit” involvement with other genomic or chromosomal alterations is observed [48]. TP53 copy-number loss is prevalent in 70% of AML cases with a concomitant TP53 gene abnormality [22]. The recently investigated cohort of five hundred de novo and refractory AML patients shows that around 80% of patients harbor missense substitutions in the TP53 gene. Nonsense or in/del mutations are less common. In frontline patients, the predominant missense variations were R248, R273, R175 and Y220. CN loss with concomitant hot-spot TP53 variants is more deleterious in comparison with those with normal CN.
The most common missense mutations in TP53 can be classified into two groups; structural mutants which have altered conformation of the DNA binding domain and DNA-contact mutants; which have alterations in amino acids responsible for direct interactions with DNA. Contact mutants like p53-R273H or p53-R248Q display aberrant interactions with DNA, yet, unlike conformational mutants (e.g. p53-R175H), have a structure similar to wild-type protein [49].
Wild-type p53 is involved in multiple processes enabling tumor suppression and efficiently drives cell death in a transcription-dependent and independent manner under stress conditions (Figure 2). Cancer cells are predominantly loaded with mutant p53 protein due to disruption of the p53-MDM2 negative feedback loop (Figure 2). In addition, mutant p53 is stabilized in cells by interacting with heat-shock proteins (HSP) which halt degradation by MDM2 and other E3 ubiquitin ligases [50]. Thus, mutant p53 seems a plausible target for the development of targeted therapeutics (reviewed in [51]). Mutant p53 has many oncogenic properties (gain-of-function, GOF) which depend on the context and include, cooperation with other oncogenes like HIF1a to withstand the hostile hypoxic environment, promoting cytokine secretion, angiogenesis and persistent cell cycling [52,53], escaping cell death, immune evasion and enabling DNA damage repair and fueling nutrients [54].
The documented prevalence of TP53 missense variants in MDS and AML urges the development of a therapeutic approach that aims at the reactivation of mutant p53 to reinstate the tumor suppression function in malignant cells. The evidence from pre-clinical studies supports the feasibility of mutant p53 reactivation in cancer cells. The approaches which advanced to clinical trials include; refolding mutant p53 to wild-type-like conformation with small molecules, stabilizing the DNA core domain with Zn2+ chelators, mutant p53 degradation and gene therapies [55].
Mutant p53 conformation correctors
PRIMA-1MET/APR-246/Eprenetapopt
The first compound reported to act as a mutant p53 conformation corrector discovered in the protein-based screen, is CP-31398. It belongs to the Michael acceptors group of compounds [55,56]. In 2002 Wiman and colleagues discovered a small molecule PRIMA-1 which was killing tumor cells dependent on mutant p53 [57]. PRIMA-1 is a quinuclidinone and a soft electrophile, which was used as a scaffold to generate APR-246 (eprenetapopt), a methylated analog of PRIMA-1 (PRIMA-1MET) [58]. Both PRIMA-1 and APR-246 are converted to methylene quinuclidinone (MQ) [59]. MQ binds to cysteines in the p53 core domain via Michael addition and corrects the conformation to wild-type as detected using conformation specific-antibodies (Figure 2) [60]. PRIMA-1/PRIMA-1 MET reactivates p53 activity and induces cell death in multiple cancer cell lines with different contact and structural p53 mutants, including R110L, V157F, R175H, L194F, R213Q/Y234H, G245V, R248Q, R273C, R273H/P309S, R280K, and R282W and also targets redox balance (reviewed in [55]).
The firs-in-human clinical trial, phase 1b in hematological and prostate cancers (NCT00900614), allowed to estimate the maximal tolerated dose and clinical response was observed in several patients and one patient with TP53-mutated AML showed a reduction of blast percentage from 46% to 26% in the bone marrow [61] (Table 1).
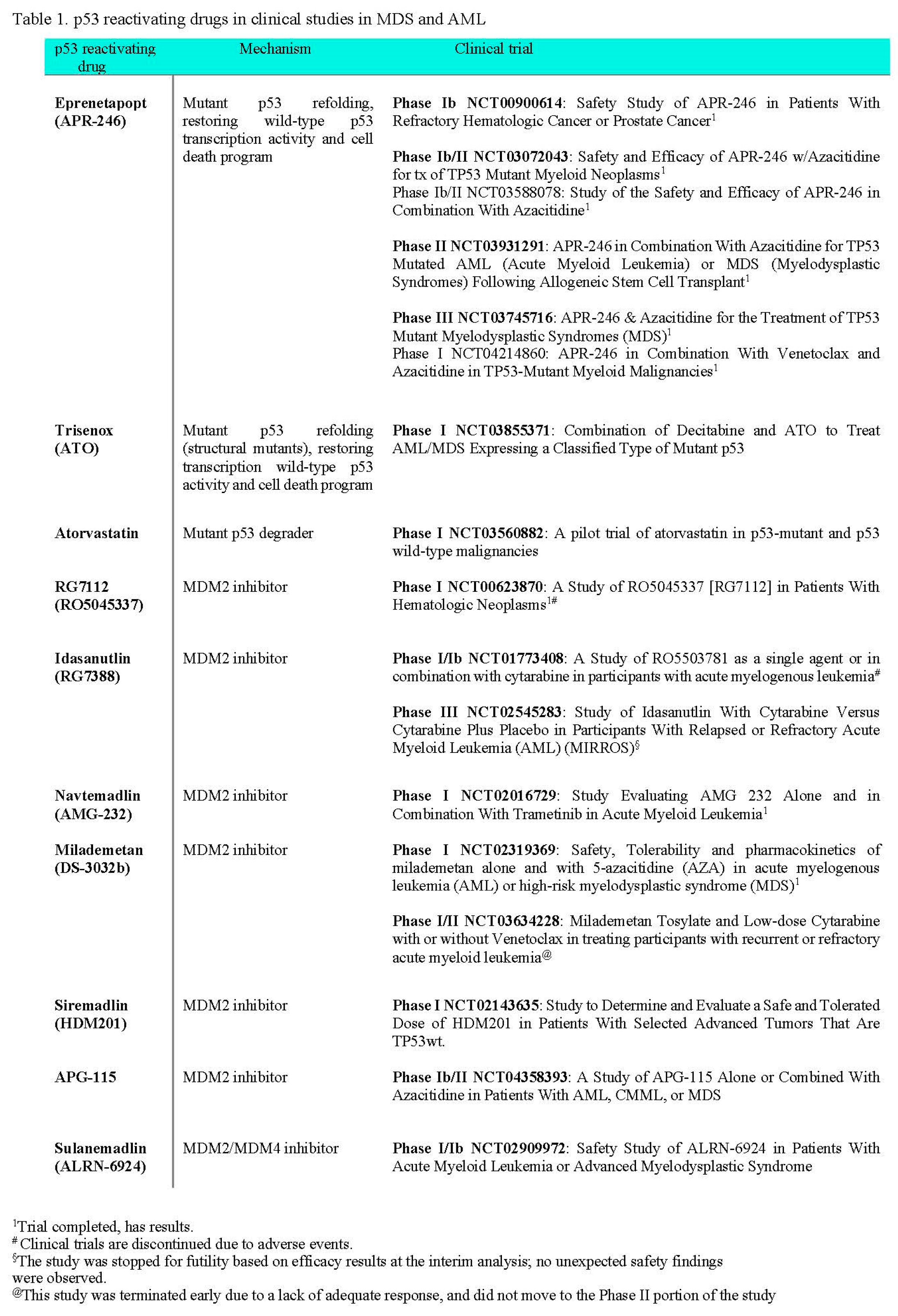
Two phase Ib/II clinical trials with APR-246 were concluded so far. One for the safety and efficacy of APR-246 in combination with azacitidine and to assess complete remission (CR) of the patients with TP53-mutated myeloid neoplasm alone and in combination with azacitidine (AZA, vidaza) (NCT03588078) [62] (Table 1), and one to determine the safety and recommended dose of APR-246 in combination with azacitidine as well as to see if this combination of therapy improves overall survival (OS) (NCT03072043) [63]. In the NCT03588078 trial, fifty-two TP53-mutated patients (34 MDS, 18 AML) were recruited. 80% of the patients had complex karyotype and median baseline mutant TP53 VAF was 20%. In MDS patients an overall response rate (ORR) was 62%, including 47% CR, with a median duration of response at 10.4 months. In AML patients the ORR was 33% including 17% CR. Of the patients who responded, 73% achieved mutant TP53 VAF < 5% determined by negativity of next-generation sequencing (NGS). The median follow-up was 9.7 months, median OS was 12.1 months in MDS patients, and 13.9. The combination was well tolerated and showed potentially higher ORR and CR rates, and longer OS than reported with AZA alone [62]. In the NCT03072043 trial, fifty-five patients with at least one TP53 mutation were treated. 89% of patients had a complex karyotype and/or multihit, e.g. > 1 TP53 mutation or deletion 17p/-17. The mutant TP53 median VAF in peripheral blood was 21%. 96% of patients had at least one mutation in the DNA binding domain. Azacitidine and eprenetapopt resulted in a 71% ORR and 44% CRR in the intention-to-treat population (50% for patients with MDS) with a median OS of 10.8 months, comparing favourably with single-agent azacytidine [63].
In the follow-up, a phase II study was performed (NCT03931291) to investigate the efficacy and safety of APR-246 in combination with azacitidine for TP53-mutated MDS or AML patients as post-hematopoietic stem-cell transplantation (HSCT) maintenance therapy [64]. Patients were screened pre-HSCT and from fifty-five patients screened post-HCT, thirty-three were enrolled and were treated with eprenetapopt in combination with azacitidine. In total, thirty patients had mutant TP53 detectable in the pre-HSCT sample. Among ten patients who completed all 12 treatment cycles and did not relapse, pre-HSCT mutant TP53 was detected in four patients and VAFs remained low during the treatment. At a median follow-up of 14.5 months, the median relapse-free survival (RFS) was 12.5 months. With a median follow-up of 17.0 months, the median OS was 20.6 months. It has been concluded that post-HSCT maintenance with eprenetapopt plus azacitidine was well tolerated with acceptable safety and may improve outcomes in mutant TP53 MDS or AML [64].
Phase III clinical trial (NCT03745716) was conducted to compare the rate of CR and duration of CR, in patients with TP53-mutated MDS who will receive APR-246 and azacitidine or azacitidine alone. In total 154 patients were recruited. At a median follow-up of 12 months, Aprea Therapeutics, the study sponsor, reported that the CR rate was 53% higher in eprenetapopt with AZA arm compared to AZA alone, but did not reach statistical significance and the study failed to meet the primary endpoint [65]. The findings of the study remain to be published.
Phase I trial (NCT04214860) was designed for dose-finding and cohort expansion study to determine the safety and preliminary efficacy of APR-246 in combination with venetoclax and azacitidine in patients with TP53-mutated myeloid malignancies. In total forty-nine patients were enrolled on the trial. Of the 49 patients who received study treatment, 20 (41%) had therapy-related acute myeloid leukaemia or had therapy-related secondary acute myeloid leukaemia, 24% of patients had more than one mutation of TP53 and 80% of patients had VAF > 50%. The overall response rate among patients receiving eprenetapopt and venetoclax with azacitidine was 64%, and the CRR was 38%. In NGS-tested patients the clearance of TP53 VAF < 5% was achieved in 26% of patients. The study showed that the combination of eprenetapopt and venetoclax with azacitidine had an acceptable safety profile in patients with previously untreated TP53-mutated acute myeloid leukemia [66].
Arsenic trioxide /ATO/Trisenox
Arsenic trioxide (ATO) is a standard of care in acute promyelocytic leukemia. ATO refolds p53 structural mutants to wild-type-like conformation and induces p53 transcription activity and cell death. The crystal structure of ATO-bound mutant p53 proteins showed, that alike APR-246, ATO binds covalently cysteine residues in the DNA binding domain, yet specifically targets residues in the allosteric cryptic site composed of three cysteines C124, C135 and C141 [67]. Phase I clinical study (NCT03855371), a combination of decitabine and ATO to treat AML/MDS expressing a classified type of mutant p53, evaluates the side effect and treatment potential of DAC+ATO in TP53 mutated high-risk MDS patients. According to the trial description, about two hundred AML/MDS patients will be recruited for TP53 sequencing. The mutant p53-positive AML/MDS patients will be treated with the combination.
Phase II study (NCT03381781) decitabine, cytarabine (Ara-C) and arsenic trioxide (ATO) in the treatment of acute myeloid leukemia with p53 mutations is designed to sequence one thousand five hundred MDS/AML patients and randomize around one hundred patients with TP53 mutations for the treatment. The outcomes of the studies remain to be reported.
Mutant p53 degraders
The most investigated clinically, so-called mutant p53 degraders are HSP90 inhibitor, ganetespib and the FDA-approved inhibitor of histone deacetylases, vorinostat. Yet, neither of the drugs is studied in patients based on stratification dependent on mutant TP53. Only one trial reported outcomes with vorinostat in combination with decitabine in patients with acute myeloid leukaemia or myelodysplastic syndrome, yet the status of TP53 was not reported [68].
Atorvastatin, is a statin approved by the FDA to prevent cardiovascular disease in patients with abnormal levels of lipids. Statins block the key enzyme in the mevalonate pathway (sterol synthesis pathway) and lower cholesterol levels. Blocking the 3-hydroxy-3-methylglutaryl-CoA reductase (HMG-CR) prevents the synthesis of mevalonate, a cholesterol precursor, which when inhibited prevents protein prenylation, G proteins signalling and EMT [69]. Atorvastatin, and other statins, were shown to promote degradation of misfolded mutant p53 by releasing it from the complex with HSPs and consequent degradation by E3 ligase CHIP (Figure 2) [70]. Phase I trial (NCT03560882), a pilot trial of atorvastatin in p53-mutant and p53 wild-type malignancies, will determine if atorvastatin will decrease the levels of conformational mutant p53 in solid tumors and in relapsed AML. The trial is currently ongoing.
Other p53 structure correctors which reached the clinical trials testing include COTI-2, or PEITC, yet they are not being evaluated in MDS or AML patients. APR-246/eprenetapopt has been tested or is tested in thirteen clinical trials in cancer and is, so far, the most clinically advanced and promising drug reactivating mutant p53 in myeloid malignancies (clinicaltrials.org).
Targeting p53/MDM2/MDMX with small molecules
In tumors which retain wild-type TP53, p53 protein is inactivated through two major routs: through binding of MDM2/MDM4 oncoproteins to the N-terminal domain and inhibition of p53 transcription function, and MDM2-, ubiquitin-mediated proteasomal degradation (Figure 2) [71].
In hematological malignancies, amplification of MDM2 was reported for AML, CML ALL with no concomitant mutations in exons 4-10 of the TP53 gene [72] and overexpression of MDM2 is associated with poor prognosis in AML [73]. Targeting the interactions between p53-MDMD2 has become a feasible strategy after the identification of the key p53 residues fitting into the MDM2 hydrophobic pocket in the crystal structure analysis which showed three sub-pockets within the MDM2 hydrophobic cleft that are occupied by the Leu26, Trp23, and Phe19 amino acid side chains of p53 [74]. The first drug ever developed to target p53-MDM2 protein complex was nutlin, a cis-imidazoline [75].
RG7112 and Idasanutlin/RG7388/RO5503781
RG7112 (RO5045337), is a cis-imidazoline, a derivative of nutlin and the first MDM2 antagonist tested in clinical trials. It showed clinical activity in AML patients in the Phase I trial (NCT00623870) (Table 1). A total of 116 patients were enrolled and at least 16 patients with wild-type p53 AML were treated. TP53 mutational analysis showed TP53 mutations in 19 of 96 patients tested and most mutant TP53 patients failed to show evidence of response. Ten genes, all p53 targets, were induced in wild-type p53 patients after treatment [76]. Yet, gastrointestinal toxicity, myelosuppression, and related complications resulted in the discontinuation of RG7112 clinical trials (reviewed in [77]).
Idasanutlin (RG7388, a selective MDM2 inhibitor) is widely studied in clinical trials. Idasanutlin is a pyrrolidine with enhanced potency, selectivity, and bioavailability compared to RG7112. It has been tested in Phase I/Ib trial (NCT01773408), a study of RO5503781 as a single agent or in combination with cytarabine in participants with acute myelogenous leukemia, yet, the final outcomes of the study were not published apart from the abstract [78]. Marker analysis of the patients enrolled in the study, using flow cytometry data for sixty-three evaluable patients showed that MDM2 expression in leukemic blasts was significantly associated with patients exhibiting a composite complete remission, and CR with incomplete hematologic recovery vs. no response. MDM2 per cent cell positivity in CD45dim/CD34+/CD117+ leukemic blasts also showed an association with clinical outcomes. Overall, the analysis supports improved MDM2 antagonist clinical outcomes in AML patients with higher levels of MDM2 protein expression and thus, MDM2 protein expression from blasts may serve as a stratification biomarker for AML patients likely to benefit from idasanutlin-based therapy [79].
Phase Ib/II study (NCT03850535), evaluating the safety and efficacy of idasanutlin in combination with cytarabine and daunorubicin in patients newly diagnosed with acute myeloid leukemia (AML) and the safety and efficacy of idasanutlin in the maintenance of first AML complete remission, is designed, to evaluate the safety, efficacy, and the sponsor decided not to continue the study based on the overall Company strategy in AML and a too-small group of patients enrolled.
In Phase III study (NCT02545283), study of idasanutlin with cytarabine versus cytarabine plus placebo in participants with relapsed or refractory acute myeloid leukemia (AML) (MIRROS), evaluated efficacy and safety of the treatment. A total of 447 patients were enrolled, 81% of patients had wild-type TP53. At the median duration of follow-up 6.7 months in both arms (drugs vs placebo), no subgroup showed a different outcome for OS. The median duration of CR was 13.9 months in the group with idasanutlin and 29.4 months in the placebo group. The myelosuppressive effect of idasanutlin was observed, yet prolonged neutropenia affected the response rates. The study did not meet the primary endpoint [80].
Milademetan/DS-3032b
Milademetan (DS-3032) tosylate hydrate is a specific and orally active MDM2 inhibitor [83]. Phase I study (NCT02319369) evaluated the safety, tolerability and pharmacokinetics of milademetan alone and with 5-azacitidine (aza) in acute myelogenous leukemia (AML) or high-risk myelodysplastic syndrome (MDS). Among 38 patients, two with AML and one with myelodysplastic syndrome had complete remission [77,84].
In the phase I study (NCT03552029), patients were enrolled to milademetan plus quizartinib combination study in FLT3-ITD mutant acute myeloid leukemia (AML). Ten patients were only recruited, and the study was terminated based on a business decision by the Sponsor.
Phase I/II study (NCT03634228), milademetan tosylate and low-dose cytarabine with or without venetoclax in treating participants with recurrent or refractory acute myeloid leukemia enrolled a total of 21 patients. Combination of MDM2 inhibition with BCL2 potentiated apoptotic response however, no meaningful clinical responses were reported. Noticeable gastrointestinal toxicities were reported, and the study was terminated early due to futility [85].
Siremadlin/ HDM201/CGM097
Siremadlin is a dihydroisoquinolinone derivative, the next-generation MDM2 inhibitor, evaluated in clinical studies [86]. Phase I (NCT02143635), a first-in-human dose-escalation study to determine and evaluate a safe and tolerated dose of HDM201 in patients with selected advanced tumors that are TP53wt, enrolled 115 patients with solid tumors and 93 patients with hematologic tumors (99% AML) [87]. A clear trend was observed for increases in serum GDF-15 (growth/differentiation factor-15), a biomarker for p53 transcription activity. Thirty-three per cent of evaluated patients had MDM2 amplification and fifty-three per cent of MDM2amp patients achieved either partial response or stable disease. The drug showed an acceptable safety profile [87].
Two trials with siremadlin are currently recruiting participants with AML; one, phase I/II (NCT05447663) to evaluate siremadlin alone and in combination with donor lymphocyte infusion in acute myeloid leukemia post-allogeneic stem cell transplant, second, phase I/II (NCT05155709) a study of siremadlin in combination with venetoclax plus azacitidine in adult participants with acute myeloid leukemia (AML) who are ineligible for chemotherapy. The results remain to be published.
APG-115
APG-115 belongs to spirooxindoles, class of potent MDM2 inhibitors of Ki < 1nM [88]. Phase Ib study (NCT04275518), of APG-115 single agent or in combination with azacitidine or cytarabine in patients with AML and MDS is recruiting one hundred two patients with relapse/refractory AML and relapsed/progressed high/very high risk MDS. Phase Ib/II study of APG-115 alone or in combination with azacitidine in patients with relapsed/refractory AML, CMML or MDS will enroll sixty-nine patients. The outcomes of the studies remain to be published.
Sulanemadlin/ALRN-6924
Sulanemadlin (ALRN-6924), the first cell-permeating, stabilized α-helical peptide which mimics the N-terminal domain of the p53 and binds with high affinity to both MDM2 and MDM4 to activate p53 signaling in cancer cells [89]. Phase I/Ib study (NCT02909972) safety study of ALRN-6924 in patients with acute myeloid leukemia or advanced myelodysplastic syndrome has recruited fifty-five patients and evaluates anti-tumor effects of ALRN-6924 alone or in combination with cytarabine. The outcome of the study remains to be published.
Currently other MDM2 inhibitors are under investigation in clinical trials, like BI-907828 (brigimadlin) [90] but are not evaluated in MDS or AML. Yet, the list of clinical trials discussed above may urge us to conclude that targeting MDM2 with high-affinity inhibitors has not so far delivered the expected clinical benefit in patients with AML and MDS. Likely, other strategies are needed to overcome the persevering problem of the insufficient response of patients due to persistent neutropenia and adverse events related to the gastrointestinal tract.
Beyond MDM2/MDM4 inhibitors
Another promising strategy to target MDM2 is to promote its degradation using protein degrader, PROTAC. A recent pre-clinical study in breast cancer demonstrated the feasibility of reconstituting the p53 tumor suppressor pathway in the presence of mutant p53, through activation of p73 [91]. p73 belongs to p53 protein family and together with p63 are ancestors of p53 in multicellular organisms. TP73 gene is rarely mutated in cancers and due to high structure and function homology, recognizes a plethora of p53 target genes involved in tumor suppression. It is therefore a promising target for improved cancer therapy in cancers with TP53 mutations [92]. We have shown that both, p53 and p73 proteins are reactivated by a repurposed drug, protoporphyrin IX, through targeting p53/MDMD2/MDM4 and p73/MDM2 interactions [93,94]. Drug repurposing emerges as a promising therapeutic approach in oncology, since the drugs studied have already been approved by the FDA for another indication and the safety profiles are known. Multiple clinical studies with purposed drugs in cancer are pending and promising outcomes have been reported.
The complexity of p53 protein biology in AML
Tuval et al., recently reported that the pre-leukemic clones with DNMT3A mutations have a selective advantage and an intrinsic chemoresistance as they pre-dominantly express pseudo-mutant p53[95]. The pseudo-mutant p53 protein is a misfolded wild-type p53 protein and has a limited transcriptional activity [96]. The protein exists in the equilibrium state in pre-leukemic blasts and was predominantly found in DNMT3A— mutated (wild-type TP53) AML enabling the clones’ enhanced self-renewal.
Refolding of pseudo-mutant with a structure-correcting peptide, pCAP-250, resulted in conformation refolding and restoration of p53 transcription activity in vitro and in vivo [95]. This implies that some sub-group of AML patients harbouring pseudo-mutant might profit from the therapy with p53 structure correctors rather than from the treatment with MDM2 inhibitors. Yet, due to the limited reports, the stratification strategy allowing to distinguish between p53 wild-type-like conformation and unfolded conformation is not applied in the clinical study design.
The emerging importance of pseudo-mutant p53 in CH, requires modifications to the current model of TP53-mutated CH (Figure 1b). Further studies are needed to evaluate the biology of pseudo-mutant in the pre-leukemic niche and to comprehend the co-existing factors contributing to clone evolution and fitness advantage in CH.
In conclusion, the role of p53 alterations in clonal hematopoiesis is still not fully depicted. The most advanced clinically drug, targeting p53 in hematological malignancies, is the mutant p53 reactivating compound, eprenetapopt (APR-246). So far, variable outcomes have been reported for MDM2 inhibitors and rational combination strategies may be crucial to enhanced efficacy with these compounds.
Author Contributions
Conceptualization, J.E. Z.; writing—original draft preparation, J.E. Z; visualization, J.E. Z
Funding
The work was supported by Cathrine Everts forsknigsstiftelse (to JZ-P) and Polish National Science Center (Narodowe Centrum Nauki), grant OPUS 20 no. 2020/39/B/NZ7/00757 (to JZ-P).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgements
The author apologizes other colleagues whose works were not cited due to space restrictions.
Conflicts of Interest
The author declares no conflict of interest.
References
- Warren, J.T.; Link, D.C. Clonal hematopoiesis and risk for hematologic malignancy. Blood 2020, 136, 1599–1605. [Google Scholar] [CrossRef] [PubMed]
- Arends, C.M.; Galan-Sousa, J.; Hoyer, K.; Chan, W.; Jäger, M.; Yoshida, K.; Seemann, R.; Noerenberg, D.; Waldhueter, N.; Fleischer-Notter, H.; et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia 2018, 32, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Vobugari, N.; Heuston, C.; Lai, C. Clonal cytopenias of undetermined significance: potential predictor of myeloid malignancies? Clin Adv Hematol Oncol 2022, 20, 375–383. [Google Scholar] [PubMed]
- Bullinger, L.; Döhner, K.; Döhner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef]
- Jajosky, A.N.; Sadri, N.; Meyerson, H.J.; Oduro, K.A.; Kelkar, A.; Fitzgerald, B.; Tomlinson, B.; Moore, E.M.; Beck, R.C. Clonal cytopenia of undetermined significance (CCUS) with dysplasia is enriched for MDS-type molecular findings compared to CCUS without dysplasia. Eur. J. Haematol. 2021, 106, 500–507. [Google Scholar] [CrossRef]
- Falini, B.; Martelli, M.P. Comparison of the International Consensus and 5th WHO edition classifications of adult myelodysplastic syndromes and acute myeloid leukemia. Am. J. Hematol. 2023, 98, 481–492. [Google Scholar] [CrossRef]
- Steensma, D.P. Clinical consequences of clonal hematopoiesis of indeterminate potential. Hematology Am Soc Hematol Educ Program 2018, 2018, 264–269. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Woll, P.S.; Yoshizato, T.; Hellström-Lindberg, E.; Fioretos, T.; Ebert, B.L.; Jacobsen, S.E.W. Targeting stem cells in myelodysplastic syndromes and acute myeloid leukemia. J. Intern. Med. 2022, 292, 262–277. [Google Scholar] [CrossRef]
- Shallis, R.M.; Daver, N.; Altman, J.K.; Komrokji, R.S.; A Pollyea, D.; Badar, T.; Bewersdorf, J.P.; Bhatt, V.R.; de Botton, S.; Burguera, A.d.l.F.; et al. Standardising acute myeloid leukaemia classification systems: a perspective from a panel of international experts. Lancet Haematol. 2023, 10, e767–e776. [Google Scholar] [CrossRef] [PubMed]
- Menssen, A.J.; Walter, M.J. Genetics of progression from MDS to secondary leukemia. Blood 2020, 136, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Allampallam, K.; Shetty, V.; Mundle, S.; Dutt, D.; Kravitz, H.; Reddy, P.L.; Alvi, S.; Galili, N.; Saberwal, G.S.; Anthwal, S.; et al. Biological Significance of Proliferation, Apoptosis, Cytokines, and Monocyte/Macrophage Cells in Bone Marrow Biopsies of 145 Patients With Myelodysplastic Syndrome. Int. J. Hematol. 2002, 75, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Ravandi, F.; Kadia, T.M.; DiNardo, C.D.; Short, N.J.; Borthakur, G.; Jabbour, E.; Kantarjian, H.M. De novo acute myeloid leukemia: A population-based study of outcome in the United States based on the Surveillance, Epidemiology, and End Results (SEER) database, 1980 to 2017. Cancer 2021, 127, 2049–2061. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, O.K.; Siddon, A.J.; Madanat, Y.F.; Gagan, J.; Arber, D.A.; Cin, P.D.; Narayanan, D.; Ouseph, M.M.; Kurzer, J.H.; Hasserjian, R.P. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood Adv. 2022, 6, 2847–2853. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Grob, T.; Al Hinai, A.S.A.; Sanders, M.A.; Kavelaars, F.G.; Rijken, M.; Gradowska, P.L.; Biemond, B.J.; Breems, D.A.; Maertens, J.; Kooy, M.v.M.; et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood 2022, 139, 2347–2354. [Google Scholar] [CrossRef]
- Shimony, S.; Stahl, M.; Stone, R.M. Acute myeloid leukemia: 2023 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2023, 98, 502–526. [Google Scholar] [CrossRef] [PubMed]
- Tashakori, M.; Kadia, T.M.; Loghavi, S.; Daver, N.G.; Kanagal-Shamanna, R.; Pierce, S.R.; Sui, D.; Wei, P.; Khodakarami, F.; Tang, Z.; et al. TP53 copy number and protein expression inform mutation status across risk categories in acute myeloid leukemia. Blood 2022, 140, 58–72. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, K.C.; Lee, E.E.; Tookmanian, E.M.; Kesserwan, C.A.; Manfredi, J.J.; Hatton, J.N.; Loukissas, J.K.; Zavadil, J.; Zhou, L.; Olivier, M.; et al. The TP53 Database: transition from the International Agency for Research on Cancer to the US National Cancer Institute. Cell Death Differ. 2022, 29, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Strasser, A.; Kelly, G. L. Should mutant TP53 be targeted for cancer therapy? Cell Death Differ. 2022, 29, 911–920. [Google Scholar] [CrossRef]
- Peuget, S.; Selivanova, G. p53-Dependent Repression: DREAM or Reality? Cancers (Basel) 2021, 13, 4850. [Google Scholar] [CrossRef]
- Marchenko, N.D.; Moll, U.M. Mitochondrial death functions of p53. Mol. Cell. Oncol. 2014, 1, e955995. [Google Scholar] [CrossRef]
- Vitale, I.; Pietrocola, F.; Guilbaud, E.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostini, M.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; et al. Apoptotic cell death in disease—Current understanding of the NCCD 2023. Cell Death Differ. 2023, 30, 1097–1154. [Google Scholar] [CrossRef]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D'Alessandro, A.; Culp-Hill, R.; D'Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Rose, D.; Haferlach, T.; Schnittger, S.; Perglerová, K.; Kern, W.; Haferlach, C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia 2017, 31, 11–17. [Google Scholar] [CrossRef] [PubMed]
- A Sallman, D.; Komrokji, R.; Vaupel, C.; Cluzeau, T.; Geyer, S.M.; McGraw, K.L.; Al Ali, N.H.; Lancet, J.; McGinniss, M.J.; Nahas, S.; et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia 2016, 30, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Ciurea, S.O.; Chilkulwar, A.; Saliba, R.M.; Chen, J.; Rondon, G.; Patel, K.P.; Khogeer, H.; Shah, A.R.; Randolph, B.V.; Perez, J.M.R.; et al. Prognostic factors influencing survival after allogeneic transplantation for AML/MDS patients with TP53 mutations. Blood 2018, 131, 2989–2992. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Othus, M.; Wood, B.L.; Walter, R.B.; Becker, P.S.; Percival, M.-E.; Abkowitz, J.L.; Appelbaum, F.R.; Estey, E.H. Comparison of myeloid blast counts and variant allele frequencies of gene mutations in myelodysplastic syndrome with excess blasts and secondary acute myeloid leukemia. Leuk. Lymphoma 2020, 62, 1226–1233. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li Fraumeni Syndrome: Clinical Characteristics of Families With p53 Germline Mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef]
- McBride, K.A.; Ballinger, M.L.; Killick, E.; Kirk, J.; Tattersall, M.H.N.; Eeles, R.A.; Thomas, D.M.; Mitchell, G. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 2014, 11, 260–271. [Google Scholar] [CrossRef]
- Swaminathan, M.; Bannon, S.A.; Routbort, M.; Naqvi, K.; Kadia, T.M.; Takahashi, K.; Alvarado, Y.; Ravandi-Kashani, F.; Patel, K.P.; Champlin, R.; et al. Hematologic malignancies and Li–Fraumeni syndrome. Cold Spring Harb Mol. Case Stud. 2019, 5, a003210. [Google Scholar] [CrossRef]
- Guha, T.; Malkin, D. Inherited TP53 Mutations and the Li-Fraumeni Syndrome. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- E Nichols, K.; Malkin, D.; E Garber, J.; Fraumeni, J.F.; Li, F.P. Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiology Biomarkers Prev. 2001, 10, 83–87. [Google Scholar]
- Correa, H. Li-Fraumeni Syndrome. J. Pediatr. Genet. 2016, 5, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Light, N.; Layeghifard, M.; Attery, A.; Subasri, V.; Zatzman, M.; Anderson, N.D.; Hatkar, R.; Blay, S.; Chen, D.; Novokmet, A.; et al. Germline TP53 mutations undergo copy number gain years prior to tumor diagnosis. Nat. Commun. 2023, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.S.; Myers, K.C. Diagnosis, Treatment, and Molecular Pathology of Shwachman-Diamond Syndrome. Hematol. Clin. North Am. 2018, 32, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.J. Molecular basis of the human ribosomopathy Shwachman-Diamond syndrome. Adv. Biol. Regul. 2018, 67, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.C.; Furutani, E.; Weller, E.; Siegele, B.; Galvin, A.; Arsenault, V.; Alter, B.P.; Boulad, F.; Bueso-Ramos, C.; Burroughs, L.; et al. Clinical features and outcomes of patients with Shwachman-Diamond syndrome and myelodysplastic syndrome or acute myeloid leukaemia: a multicentre, retrospective, cohort study. Lancet Haematol. 2020, 7, e238–e246. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Myers, K.C.; Bowman, J.; Gibson, C.J.; Camarda, N.D.; Furutani, E.; Muscato, G.M.; Klein, R.H.; Ballotti, K.; Liu, S.; et al. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Shallis, R.M.; Daver, N.G.; Altman, J.K.; Hasserjian, R.P.; Kantarjian, H.M.; Platzbecker, U.; Santini, V.; Wei, A.H.; Sallman, D.A.; Zeidan, A.M. TP53-altered acute myeloid leukemia and myelodysplastic syndrome with excess blasts should be approached as a single entity. Cancer 2023, 129, 175–180. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Moll, U.M. Depleting stabilized GOF mutant p53 proteins by inhibiting molecular folding chaperones: a new promise in cancer therapy. Cell Death Differ. 2017, 24, 3–5. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Fontemaggi, G.; Dell'Orso, S.; Trisciuoglio, D.; Shay, T.; Melucci, E.; Fazi, F.; Terrenato, I.; Mottolese, M.; Muti, P.; Domany, E.; et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol. 2009, 16, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Melino, G. Context is everything: extrinsic signalling and gain-of-function p53 mutants. Cell Death Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 77. [Google Scholar] [CrossRef]
- Nishikawa, S.; Iwakuma, T. Drugs Targeting p53 Mutations with FDA Approval and in Clinical Trials. Cancers 2023, 15, 429. [Google Scholar] [CrossRef]
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological Rescue of Mutant p53 Conformation and Function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 39. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in Vivo: A First-in-Human Study With p53-Targeting Compound APR-246 in Refractory Hematologic Malignancies and Prostate Cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.-B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al. Eprenetapopt Plus Azacitidine After Allogeneic Hematopoietic Stem-Cell Transplantation for TP53-Mutant Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Clin. Oncol. 2022, 40, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- Therapeutics, A. Aprea Therapeutics Announces Results of Primary Endpoint from Phase 3 Trial of Eprenetapopt in TP53 Mutant Myelodysplastic Syndromes (MDS). 2020.
- Garcia, G. Eprenetapopt combined with venetoclax and azacitidine in TP53-mutated acute myeloid leukaemia: a phase 1, dose-finding and expansion study. Manero.
- Chen, S.; Wu, J.-L.; Liang, Y.; Tang, Y.-G.; Song, H.-X.; Wu, L.-L.; Xing, Y.-F.; Yan, N.; Li, Y.-T.; Wang, Z.-Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239. [Google Scholar] [CrossRef]
- Kirschbaum, M.; Gojo, I.; Goldberg, S.L.; Bredeson, C.; Kujawski, L.A.; Yang, A.; Marks, P.; Frankel, P.; Sun, X.; Tosolini, A.; et al. A phase 1 clinical trial of vorinostat in combination with decitabine in patients with acute myeloid leukaemia or myelodysplastic syndrome. Br. J. Haematol. 2014, 167, 185–193. [Google Scholar] [CrossRef]
- Juarez, D.; Fruman, D.A. Targeting the Mevalonate Pathway in Cancer. Trends Cancer 2021, 7, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef]
- Oren, M. Regulation of the p53 Tumor Suppressor Protein. J. Biol. Chem. 1999, 274, 36031–36034. [Google Scholar] [CrossRef]
- Quesnel, B.; Preudhomme, C.; Oscier, D.; Lepelley, P.; Collyn-d’Hooghe, M.; Facon, T.; Zandecki, M.; Fenaux, P. Over-expression of the MDM2 gene is found in some cases of haematological malignancies. Br. J. Haematol. 1994, 88, 415–418. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Kornblau, S.M. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 2017, 31, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation Domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: an important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]
- Yee, K.; Martinelli, G.; Vey, N.; Dickinson, M.J.; Seiter, K.; Assouline, S.; Drummond, M.; Yoon, S.-S.; Kasner, M.; Lee, J.-H.; et al. Phase 1/1b Study of RG7388, a Potent MDM2 Antagonist, in Acute Myelogenous Leukemia (AML) Patients (Pts). Blood 2014, 124, 116. [Google Scholar] [CrossRef]
- Reis, B.; Jukofsky, L.; Chen, G.; Martinelli, G.; Zhong, H.; So, W.V.; Dickinson, M.J.; Drummond, M.; Assouline, S.; Hashemyan, M.; et al. Acute myeloid leukemia patients’ clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blasts. Haematologica 2016, 101, e185–e188. [Google Scholar] [CrossRef]
- Konopleva, M.Y.; Röllig, C.; Cavenagh, J.; Deeren, D.; Girshova, L.; Krauter, J.; Martinelli, G.; Montesinos, P.; Schäfer, J.A.; Ottmann, O.G.; et al. Idasanutlin Plus Cytarabine in Relapsed or Refractory Acute Myeloid Leukemia: Results of the MIRROS Trial. Blood Adv. 2022, 6, 4147–4156. [Google Scholar] [CrossRef]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef]
- Erba, H.P.; Becker, P.S.; Shami, P.J.; Grunwald, M.R.; Flesher, D.L.; Zhu, M.; Rasmussen, E.; Henary, H.A.; Anderson, A.A.; Wang, E.S. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019, 3, 1939–1949. [Google Scholar] [CrossRef]
- Arnhold, V.; Schmelz, K.; Proba, J.; Winkler, A.; Wünschel, J.; Toedling, J.; Deubzer, H.E.; Künkele, A.; Eggert, A.; Schulte, J.H.; et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 2018, 9, 2304–2319. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Rosenthal, J.; Andreeff, M.; Zernovak, O.; Kumar, P.; Gajee, R.; Chen, S.; Rosen, M.; Song, S.; Kochan, J.; et al. Phase 1 Dose Escalation Study of MDM2 Inhibitor DS-3032b in Patients with Hematological Malignancies - Preliminary Results. Blood 2016, 128, 593. [Google Scholar] [CrossRef]
- Senapati, J.; Muftuoglu, M.; Ishizawa, J.; Abbas, H.A.; Loghavi, S.; Borthakur, G.; Yilmaz, M.; Issa, G.C.; Dara, S.I.; Basyal, M.; et al. A Phase I study of Milademetan (DS3032b) in combination with low dose cytarabine with or without venetoclax in acute myeloid leukemia: Clinical safety, efficacy, and correlative analysis. Blood Cancer J. 2023, 13, 101. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, S.; Rebmann, R.; Berger, M.; Santacroce, F.; Albrecht, G.; Pollehn, K.; Sterker, D.; Wartmann, M.; Hueber, A.; Wiesmann, M.; Jensen, M. R.; Hofmann, F.; Sellers, W. R.; Holzer, P.; Jeay, S. Abstract 1224: Insights into the mechanism of action of NVP-HDM201, a differentiated and versatile Next-Generation small-molecule inhibitor of Mdm2, under evaluation in phase I clinical trials. In Experimental and Molecular Therapeutics; American Association for Cancer Research, 2016; pp. 1224–1224.
- Stein, E.M.; DeAngelo, D.J.; Chromik, J.; Chatterjee, M.; Bauer, S.; Lin, C.-C.; Suarez, C.; de Vos, F.; Steeghs, N.; Cassier, P.A.; et al. Results from a First-in-Human Phase I Study of Siremadlin (HDM201) in Patients with Advanced Wild-Type TP53 Solid Tumors and Acute Leukemia. Clin. Cancer Res. 2022, 28, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, A.; Lu, J.; Liu, L.; Du, D.; Bernard, D. Discovery of 4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-ethyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5 …. Journal of medicinal 2017.
- Guerlavais, V.; Sawyer, T.K.; Carvajal, L.; Chang, Y.S.; Graves, B.; Ren, J.-G.; Sutton, D.; Olson, K.A.; Packman, K.; Darlak, K.; et al. Discovery of Sulanemadlin (ALRN-6924), the First Cell-Permeating, Stabilized α-Helical Peptide in Clinical Development. J. Med. Chem. 2023, 66, 9401–9417. [Google Scholar] [CrossRef]
- LoRusso, P.; Yamamoto, N.; Patel, M.R.; Laurie, S.A.; Bauer, T.M.; Geng, J.; Davenport, T.; Teufel, M.; Li, J.; Lahmar, M.; et al. The MDM2-p53 Antagonist Brigimadlin (BI 907828) in Patients with Advanced or Metastatic Solid Tumors: Results of a Phase Ia, First-in-Human, Dose-Escalation Study. Cancer Discov. 2023, 13, 1802–1813. [Google Scholar] [CrossRef]
- Adams, C.M.; Mitra, R.; Xiao, Y.; Michener, P.; Palazzo, J.; Chao, A.; Gour, J.; Cassel, J.; Salvino, J.M.; Eischen, C.M. Targeted MDM2 Degradation Reveals a New Vulnerability for p53-Inactivated Triple-Negative Breast Cancer. Cancer Discov. 2023, 13, 1210–1229. [Google Scholar] [CrossRef]
- Zawacka-Pankau, J.E. The Undervalued Avenue to Reinstate Tumor Suppressor Functionality of the p53 Protein Family for Improved Cancer Therapy-Drug Repurposing. Cancers 2020, 12, 2717. [Google Scholar] [CrossRef]
- Jiang, L.; Malik, N.; Acedo, P.; Zawacka-Pankau, J. Protoporphyrin IX is a dual inhibitor of p53/MDM2 and p53/MDM4 interactions and induces apoptosis in B-cell chronic lymphocytic leukemia cells. Cell Death Discov. 2019, 5, 77. [Google Scholar] [CrossRef]
- Jiang, L.; Zawacka-Pankau, J. The p53/MDM2/MDMX-targeted therapies-a clinical synopsis. Cell Death Dis. 2020, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Tuval, A.; Brilon, Y.; Azogy, H.; Moskovitz, Y.; Leshkowitz, D.; Salame, T.M.; Minden, M.D.; Tal, P.; Rotter, V.; Oren, M.; et al. Pseudo-mutant P53 is a unique phenotype of DNMT3A-mutated pre-leukemia. Haematologica 2022, 107, 2548–2561. [Google Scholar] [CrossRef] [PubMed]
- Trinidad, A.G.; Muller, P.A.; Cuellar, J.; Klejnot, M.; Nobis, M.; Valpuesta, J.M.; Vousden, K.H. Interaction of p53 with the CCT Complex Promotes Protein Folding and Wild-Type p53 Activity. Mol. Cell 2013, 50, 805–817. [Google Scholar] [CrossRef]
- Shih, A.H.; Chung, S.S.; Dolezal, E.K.; Zhang, S.-J.; Abdel-Wahab, O.I.; Park, C.Y.; Nimer, S.D.; Levine, R.L.; Klimek, V.M. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica 2013, 98, 908–912. [Google Scholar] [CrossRef]
- Gaulin, C.; Kelemen, K.; Yi, C.A. Molecular Pathways in Clonal Hematopoiesis: From the Acquisition of Somatic Mutations to Transformation into Hematologic Neoplasm. Life 2022, 12, 1135. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Maiti, A.; Kadia, T.M.; Vyas, P.; Majeti, R.; Wei, A.H.; Garcia-Manero, G.; Craddock, C.; Sallman, D.A.; Kantarjian, H.M. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022, 12, 2516–2529. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging p53 in cancer: one protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef]
- Borrero, L.J.H.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. et Biophys. Acta (BBA) - Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2009, 1787, 414–420. [Google Scholar] [CrossRef]
Figure 1.
Origin of hematological neoplasms from clonal hematopoiesis (a) and TP53-mutated clonal hematopoiesis (b). (a) With age, hematopoietic stem cells (HSCs) acquire somatic mutations and the mutated clones are expanded during HSCs renewal. Early genetic events lead to clonal hematopoiesis (CH) and the origin of clonal hematopoiesis of indeterminate potential (CHIP) or clonal cytopenias of undetermined significance (CCUS), which have different genetic backgrounds, differ in cytopenia status but possess same variant allele frequencies of VAF ≥ 2%. Among these, CCUS have a higher potential of transformation to myelodysplastic syndrome (MDS) with a range of incidence 18-95%. Fitness variants occurring later, confer a growth advantage in the presence of selective pressures such as inflammation, cytotoxic treatment, radiotherapy, bone marrow microenvironment or aging. The consequent selection of high-risk variants, accumulation of the co-occurring mutations, increase in the VAF > 10% and the co-existing cytopenia are predictive of hematopoietic malignancy development and thus, accelerate the progression to hematological neoplasm; myelodysplastic syndrome (MDS) and MDS/AML. (b) TP53-mutated CH occurs in about 2-6% of cancer patients. TP53-mutated MDS and AML account for up to 13% of all de novo cases. In therapy-related myeloid neoplasm, TP53 gene mutations are present in 20-40% of cases [97]. The model shows the pathogenesis of MDS and AML in hereditary cancer syndromes; Li-Fraumeni syndrome (LFS) with congenital TP53 mutations and Schwachman syndrome (SDS) with congenital mutations in Shwachman–Bodian–Diamond syndrome (SBDS) gene and in therapy-related myeloid neoplasm (t-MN); diseases in which TP53 monoallelic mutations play a role in the progression from CH to myeloid malignancy. The incidence of malignant transformation is higher in the presence of selective pressure as delineated for SDS and t-MN. →→→ tandem of arrows indicates a multi-step process. Modified from [1,9,98]. Created with BioRender.com.
Figure 1.
Origin of hematological neoplasms from clonal hematopoiesis (a) and TP53-mutated clonal hematopoiesis (b). (a) With age, hematopoietic stem cells (HSCs) acquire somatic mutations and the mutated clones are expanded during HSCs renewal. Early genetic events lead to clonal hematopoiesis (CH) and the origin of clonal hematopoiesis of indeterminate potential (CHIP) or clonal cytopenias of undetermined significance (CCUS), which have different genetic backgrounds, differ in cytopenia status but possess same variant allele frequencies of VAF ≥ 2%. Among these, CCUS have a higher potential of transformation to myelodysplastic syndrome (MDS) with a range of incidence 18-95%. Fitness variants occurring later, confer a growth advantage in the presence of selective pressures such as inflammation, cytotoxic treatment, radiotherapy, bone marrow microenvironment or aging. The consequent selection of high-risk variants, accumulation of the co-occurring mutations, increase in the VAF > 10% and the co-existing cytopenia are predictive of hematopoietic malignancy development and thus, accelerate the progression to hematological neoplasm; myelodysplastic syndrome (MDS) and MDS/AML. (b) TP53-mutated CH occurs in about 2-6% of cancer patients. TP53-mutated MDS and AML account for up to 13% of all de novo cases. In therapy-related myeloid neoplasm, TP53 gene mutations are present in 20-40% of cases [97]. The model shows the pathogenesis of MDS and AML in hereditary cancer syndromes; Li-Fraumeni syndrome (LFS) with congenital TP53 mutations and Schwachman syndrome (SDS) with congenital mutations in Shwachman–Bodian–Diamond syndrome (SBDS) gene and in therapy-related myeloid neoplasm (t-MN); diseases in which TP53 monoallelic mutations play a role in the progression from CH to myeloid malignancy. The incidence of malignant transformation is higher in the presence of selective pressure as delineated for SDS and t-MN. →→→ tandem of arrows indicates a multi-step process. Modified from [1,9,98]. Created with BioRender.com.
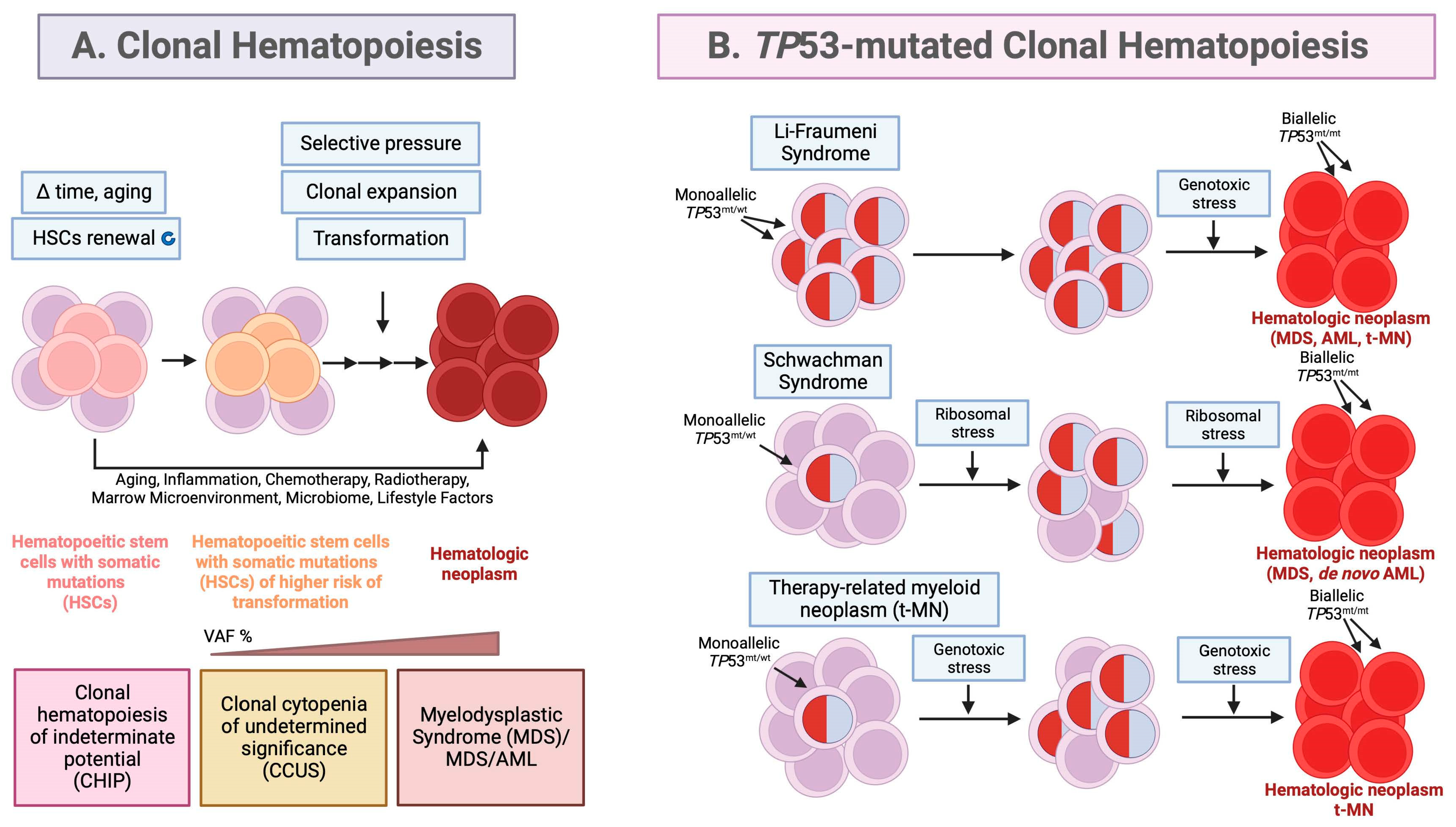
Figure 2.
A simplified scheme of p53 function and regulation and drugs currently evaluated in clinical trials for p53 reactivation for improved therapy in MDS and AML. In normal cells, p53 is activated by numerous stress conditions like, oncogene activation, telomere shortening, replication stress, DNA damage, or elevated levels of reactive oxygen species (ROS). Wild-type p53 is released from MDM2 and MDM4 by phosphorylation by ataxia telangiectasia mutated (ATM) or ataxia telangiectasia and Rad3-related protein (ATR) which releases p53 from the complex with MDM2 and MDM4. Consequent p53 acetylation and protein accumulation allows activation of p53 transcriptional function. ARF binds directly to MDM2 upon oncogenic stress and shifts the conformation so that p53 is released from the complex and becomes activated. In normal cells the negative feedback-loop; p53-MDM2 is responsible for p53 protein turnover. In cancer cells amplified MDM2 prevents p53 accumulation and activation. Mutant p53 accumulates in large quantities in cancer cells and escapes the regulation by the p53-MDM2 feedback loop. Drugs targeting wild-type and mutant p53 appraised in clinical studies for MDS and AML are marked in bold. Modified from [51,53,99,100,101,102]. Eprenetapopt (APR-246); Trisenox (ATO); Atorvastain; Idasanutlin (RG7388); Navtemadlin (AMG-232 oral, KRT-232); Milademetan (DS-3032b); Sulanemadlin (ALNR-6924) →→→ tandem of arrows indicates a multi-step process. Created with BioRender.com.
Figure 2.
A simplified scheme of p53 function and regulation and drugs currently evaluated in clinical trials for p53 reactivation for improved therapy in MDS and AML. In normal cells, p53 is activated by numerous stress conditions like, oncogene activation, telomere shortening, replication stress, DNA damage, or elevated levels of reactive oxygen species (ROS). Wild-type p53 is released from MDM2 and MDM4 by phosphorylation by ataxia telangiectasia mutated (ATM) or ataxia telangiectasia and Rad3-related protein (ATR) which releases p53 from the complex with MDM2 and MDM4. Consequent p53 acetylation and protein accumulation allows activation of p53 transcriptional function. ARF binds directly to MDM2 upon oncogenic stress and shifts the conformation so that p53 is released from the complex and becomes activated. In normal cells the negative feedback-loop; p53-MDM2 is responsible for p53 protein turnover. In cancer cells amplified MDM2 prevents p53 accumulation and activation. Mutant p53 accumulates in large quantities in cancer cells and escapes the regulation by the p53-MDM2 feedback loop. Drugs targeting wild-type and mutant p53 appraised in clinical studies for MDS and AML are marked in bold. Modified from [51,53,99,100,101,102]. Eprenetapopt (APR-246); Trisenox (ATO); Atorvastain; Idasanutlin (RG7388); Navtemadlin (AMG-232 oral, KRT-232); Milademetan (DS-3032b); Sulanemadlin (ALNR-6924) →→→ tandem of arrows indicates a multi-step process. Created with BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



