
Submitted:
12 September 2023
Posted:
13 September 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
We tested the possibility of using “adverse events count” (AEC) as a drug-risk indicator and side-effect severity indicator. Data from 3938 adverse event (AE) reports for COVID-19 vaccines (Comirnaty, Moderna, Vaxzevria, and Janssen) and 6869 AE reports for other medicines were collected from the Bulgarian Drug Agency database (01.01.2018–31.03.2022). AEC was modeled with zero-adjusted negative binomial (ZANBI) and zero-adjusted Poisson inverse Gaussian (ZAPIG) regression models, which account for zero absence and overdispersion. The models’ fit was checked with residual diagnostic plots and parametric correspondence to normality. Explanatory variables were: age, sex, sequence number (order of submission), severity of AE, and vaccine type. Average AEC was higher in severe vs. non-severe AE, and in females vs. males; it decreased with age and was lower in Comirnaty than other vaccines. Variability of AEC for COVID-19 data decreased with sequence number in severe AE and increased in non-severe AE. Full ZANBI models had greater sensitivity than parametric ZANBI or ZAPIG models. Results showed correlation between AEC and AE severity, suggesting AEC as a simple and reliable measure of drug-risk and side-effect severity for clinicians and regulators, especially for COVID-19 vaccines.
Keywords:
COVID-19 vaccines
; SARS-CoV-2
; vaccine safety
; pharmacovigilance
; vaccination campaigns
; adverse event
; negative binomial distribution
; Poisson-inverse Gaussian distribution
; model diagnostics
; real-world data
1. Introduction
The coronavirus disease 2019 (COVID-19) pandemic was followed by a global vaccination campaign on an unprecedented scale that turned the public attention towards the benefit-risk balance of the vaccines used for combating the disease.
Analysis of adverse events (AE), or in the case of vaccines, adverse events following immunization (AEFI), is an important component of the assessment of the benefit-risk balance. During the controlled conditions in the pre- and post-authorization clinical trials, there is enough information for each participant (doses taken, AE after each dose including no AE, as well as other variables like sex, age, severity, time after dose, etc.) to enable a proper comparison between control and treatment arms and unbiased conclusions for vaccines short term side-effects. Conversely, in spontaneous, continuous, post-marketing AE reporting much of the needed information is lacking and, moreover, additional hard-to-assess variables appear, like reporting rate and its variability over such factors as age, sex, education, location, etc. Although statistical approaches to the analysis of such data exist, with some methods more promising than others, these data are so limited that it is recommended they should be considered only for specific purposes like detecting very rare AE. Problems associated with the use of spontaneous reports in drug safety include (1) confounding by indication (i.e., patients taking a particular drug may have a disease that is itself associated with a higher incidence of the AE (e.g., anti-COVID-19 immunization after SARS-CoV-2 or another viral infection), (2) systematic underreporting, (3) questionable representativeness of patients, (4) effects of publicity in the media on numbers of reports, (5) extreme duplication of reports, (6) attribution of the event to a single drug when patients may be exposed to multiple drugs, and (7) extensive amounts of missing data. These limitations degrade the capacity for optimal data mining and analysis of AEs [1,2].
Time-independent risk prediction models for AEs belong mainly to two types: logistic regression and Poisson regression. Response variables for binary logistic regression are most often AE occurrence (no AE, AE) or severity (non-severe, severe). Severity might be divided to more than two levels and assessed with multinomial or ordinal logistic regression. These models require data from a clinical trial or at least from a clinical setting in which AE occurrence is followed up and severity of the adverse effect is assessed by one or more physicians as objectively as possible. Response variables for Poisson regression are usually the number of AE or number of individuals with AE. The data can also come from a population trial in which one has the information mentioned above about each individual.
Such models applied to data from spontaneous AE reporting would be misleading for the reasons outlined above. For example, severity of the AE is for the most part self-assessed by the reporting person and thus is subjective. The number of AE or number of reports depends on the total number of vaccine / drug doses taken, the reporting rate, its variation in different subgroups, media publicity, etc. On the other hand, spontaneous reporting has an important advantage over clinical trials, which is their greater coverage: whereas the typical sample size of experimental studies is <1000, spontaneous reports number several thousands to several tens (or even hundreds) of thousands depending on the drug(s) / vaccine(s) in consideration.
The pharmacovigilance database of the Bulgarian pharmaceutical regulator, the Bulgarian Drug Agency (BDA), records up to 20–25 AEs for each spontaneous report. The number of AEs in an individual report which we abbreviate as “adverse events count” (AEC) is here used as an indicator of drug risk that does not vary with the total number of reports or doses in different subgroups. It also obviates the need for a control group. In our opinion, this variable has the potential to alleviate many of the problems associated with spontaneously reported AE: (1) an eventual disease underlying the reported AEs may increase AEC but it can be discovered on follow-up and the report taken out from the analysis, (2) underreporting is not a problem since AEC does not depend on the number of reports, (3) unequal representation of some subgroups does not change the AEC reported from them, (4) AEC are not expected to be influenced much by media publicity at least not to such extent as number of reports, (5) report duplication would randomly increase the weights of some AEC values resulting in no systematic bias, (6) exposition to multiple drugs can increase AEC and, therefore, remains a problem; however, most AE reporting forms have an item for additional drugs and data can be culled for suspiciously high AEC with untypical AEs in presence of multiple drugs; another approach is to include the number of additional drugs as an independent variable in the model and assess its effect on AEC, (7) the amount of missing data depend on the number of independent variables included in the model; the bulk result from not filling an item in the AE reporting form. They are of the type missing-completely-at-random (MCAR) and do not affect the AEC distribution.
1.1. AER distributions
Looked at from a theoretical point of view, AEC provides a sufficient statistic as defined by Fisher [3]. More specifically, the AEC random variable has a definite statistical distribution from which a likelihood function can be calculated. From the latter, when applied to the sample, maximal likelihood estimates for the parameters of the distribution are found and on these, a statistical model is built. The distribution parameters are a sufficient statistic because they contain all information extracted from the data and no other information is needed to build the model.
AEC are integer values resulting from counts. If they were distributed randomly and independently, they would follow the Poisson distribution:
where y is the AEC count (), e is the Euler number () and ! is the factorial function. The positive real number is the single distribution parameter that is equal to the mean and to the variance (equidispersion).
It can be seen, however, that AEC violate both the conditions of randomness (they occur in clusters as several AE per report and not one at a time) and independence (the AE within the cluster (report) are correlated, i. e., they depend on each other). The result of such within-report correlation is that the variance of AEC would be larger than the mean, a phenomenon known as overdispersion. The overdispersion may be modeled as follows [4,5]: first we assume that conditional on the cluster (report) effect, denoted by we have
Second, we assume that the random cluster effect has = 1, and
. Therefore
regardless of the distribution assigned to . Here is a random variable that summarizes the effect of our inability to model all pertinent independent variables and other sources of variation. In the above formula, indicates the presence of overdispersion, underdispersion, and equidispersion. Under equidispersion, the Poisson model is the most commonly used model to analyze count data. In the other cases, it is assumed that the distribution of the response variable Y has a discrete probability function , conditional on a continuous random variable u whose distribution is . Then the marginal (or unconditional) probability function of Y is given by
where is the range of u. The resulting distribution of Y is called a continuous mixture of discrete distributions. For the clustered AEC data, the conditional distribution is the Poisson-like
where instead of , there is . Substituting (5) in (4), one obtains
To complete the model, one must specify , the distribution of u. The usual assumption is that u follows a Gamma distribution
Substituting (7) in (6), solving the integral and rearranging, one obtains a negative binomial distribution as the final marginal distribution for AEC [6]
where and are the parameters. is the Gamma function, which is a special function replacing factorial expressions when the arguments are not integers. Equation (8) is the negative binomial type I, NBI(,), probability distribution. Such a genesis of the negative binomial distribution as a mixture of Poisson and Gamma distributions is quite distinct from its classical development as the distribution of the number of failures until the success in independent Bernoulli trials, and was originally developed by Greenwood and Yule [7].
NBI(,) is quite flexible with many different parametrizations and has been used to model a variety of data with mild to moderate overdispersion from various fields of study: CD4 counts in HIV-infected women [8], microbiome counts in mouse gut [9], rainfall counts [10], postfire conifer regeneration counts to examine seedling distributions [11], data from single-cell RNA sequencing [12] and dental caries count indices [13], prediction of micronuclei frequency as a biomarker for genotoxic exposure and cancer risk [14], a clinical trial to evaluate the effectiveness of a prehabilitation program in preventing functional disease among physically frail, community-living older persons [15], investigating the social and demographic factors associated with public awareness of health warnings on the harmful effects of environmental tobacco smoke [16], assessing highway crash frequency by injury severity [17], improving planning and management of urban trail traffic [18], modeling overdispersion in ecological count data, e. g., bird migration [19], and other applications of NB in ecology and biodiversity reviewed in [20], male satellites counts in the popular horseshoe crab data [21], modeling the dependence of tropical storm counts in the North Atlantic basin on climate indices [22], estimation of the reproduction number for SARS 2003 coronavirus by the number of secondary cases [23], predicting length of stay from an electronic patient record for patients with knee replacement [24], or for elderly patients [25], serial clustering of extratropical cyclones [26], or of intense European storms [27], digital gene expression counts [28].
The variance of NBI(,) is , hence, the overdispersion is a quadratic in as provisioned in formula (3). Reparametrization of to in NBI(,) gives NBII(,) in which the variance is linear in : . For data that have a high initial peak, clumped heavily at 1 and 2, and that may be skewed to the far right as well as data that are highly Poisson overdispersed, another distribution for the random effect u may provide a better fit. For such data, the Inverse Gaussian distribution is commonly used [29]:
Substituting (9) in (6) and solving the integral for u, one obtains the Poisson-inverse Gaussian distribution denoted by PIG(,) [5]
for , where , , and . is the modified Bessel function of the second kind given by
The Bessel function in general presents challenges in computation; however, when its order is a half-integer, as it is in (10), computations are simplified considerably as
and use is made of the recurrence relation
It can be shown with certain parametrizations of PIG that its overdispersion is cubic in . Compared with NBI, with greater mean values of PIG come increasingly greater values of the variance. With a dispersion parameter >1, larger values of the mean of the response variable in a PIG regression provide for adjustment of larger amounts of overdispersion than does the NBI model. Simply put, PIG models can better deal with highly overdispersed data than can NBI regression [29] (p. 163).
The PIG models are not as common as the NB models for several reasons: (1) in many cases, the NB models are adequate to handle the amount of overdispersion present in the data, (2) authors are not aware of the availability of PIG, and that a PIG model can fit their data better than a Poisson or NB model, (3) no commercial statistical software offers the PIG model as a component of its official offerings; the only software that supports PIG at present is the R package GAMLSS [30].
Still, PIG models are not completely lacking and there is a variety of fields that these are used: PIG and NB distributions have been fitted to the popular horseshoe crab data and it was found that PIG provides a better fit [21], identification of the relationship between farm management practices and risk of cow mastitis [5], comparing PIG and NB for the analysis of automobile claim frequency data [31], modeling overdispersed dengue data in the city of Campo Grande, MS, Brazil [32], application of NBI, PIG, and Negative Binomial-Inverse Gaussian (NBIG) models to car insurance claim frequency [33,34], application of generalized PIG for analysis of bibliometric data sets [35], modeling species abundance data [36], application to the bonus-malus system in car insurance claims [37], modeling infectious disease count data [38], application to health services: number of hospital stays [39], assessing microdata disclosure risk [40], modeling the number of HIV and AIDS cases in two districts in Indonesia [41], analysing long-term survival data in patients with skin cancer [42], modeling the numbers of pauci-baciliary and multi-baciliary leprosy cases with geographical conditions factors in West Sumatera, Indonesia [43], application to accident data for men in a soap factory and for women working with highly explosive shells [44], number of falls in Parkinson’s disease patients over a 10-week period, analysing both the mean () and variability () parameters [45], analysis of data from a Phase III cutaneous melanoma clinical trial (E1690 data) [46], transportation origin–destination analysis by introducing predictory variables-based models which incorporate different transport modeling phases and also allow for direct probabilistic inference on link traffic based on Bayesian predictions [47], analysis of motor vehicle crash data [48].
AEC data structurally exclude zero counts as each report includes at least one AE. Conversely, Poisson, NB, and PIG distributions have a non-zero probability for a zero count and if one tries to fit them to the AEC data there would be some misfit just because these distributions do not accommodate the data property of zero exclusion. This situation is solved by “hurdle models”, so called because the deviating value (in this case, the zero count) is seen as a hurdle which is passed with some probability , and the remaining probability is reserved for the other values which are distributed according to the corresponding distribution. In the case of AEC, , so that values higher than 0 occur almost surely. In this case, more appropriate are the so-called zero-altered or zero-adjusted (ZA) distributions, in which the 0 has a point mass distribution, known in physics as Dirac delta function. In other words, if the count is 0 then otherwise or, more precisely
The coefficient c is calculated from the parameters , , and is different for the different distributions but in any case, if then . Please note that the ZA distributions are already three-parametric with the additional parameter .
In this paper, we propose a model applied to AE data in the Bulgarian Drug Agency (BDA) pharmacovigilance database, whose dependent variable is the AEC. Two data sets: AEC due to vaccines against COVID-19 and AEC due to the administration of drugs other than COVID-19 vaccines were studied separately. To our knowledge, this is the first attempt to determine the distribution of AEC and use it in a model.
We also aim, using the increased sensitivity of the model to infer differences between the COVID-19 vaccines and the other drugs in terms of AEC also taking into account additional explanatory variables such as sex, age, vaccine, declared severity and sequence number of the report.
2. Results
The data with COVID-19 vaccines (“COVID vaccines”) includes 3938 spontaneous AEFI reports for the four vaccines used for immunization in Bulgaria: two mRNA vaccines (Comirnaty [Pfizer-BioNTech] deployed on 27.12.2020 and Spikevax [Moderna] deployed on 14.01.2021), and two adenovirus vaccines (Vaxzevria [AstraZeneca] deployed on 07.02.2021 and Jcovden [Janssen] deployed on 28.04.2021).
The data with medicines other than COVID vaccines (“other drugs”) includes 6869 spontaneous AE reports for 1557 medicines for the 51 months in the period 01.01.2018 – 03.31.2022.
The AEC for the COVID vaccines data contains no outliers and no extreme points (see Materials and Methods) while the AEC for the other drugs data contains 329 outliers from which 124 are extreme points. A perusal of these outliers showed that 7–9 AEC are considered an outlier and ≥10 AEC are considered an extreme point. We did not remove these outliers from the data because under the hypothesis that high AEC count might be correlated with severity and also taking into account the high asymmetry of the distribution, they might contain the signal. While 45.28% of the total other drugs data are reported as severe, this percentage rises to 75.08% in the outliers and to 84.68% in the extreme points lending support to the hypothesized correlation between AEC and severity.
2.1. Parametric GAMLSS models
The top five distributions with the smallest information criteria out of 32 discrete distributions in the GAMLSS package that were examined for goodness of fit, are shown in Table 1.
ZANBI and ZAPIG fit both datasets with the smallest AIC and BIC criteria values. These two distributions differ slightly between each other, while their differences with the other distributions are significantly larger. The difference between ZANBI and ZAPIG criteria, for COVID vaccines is about 11 units, while for the other drugs the difference is about 32 units. The differences with the third distribution are much larger, about 400 units for the COVID vaccines and about 300 units for the other drugs, respectively. The significant difference between the two datasets, however, is that ZANBI fits better than ZAPIG for COVID vaccines, while the opposite is true for other drugs. The other drugs AEC data have slightly higher overdispersion than the AEC from COVID vaccines data and the fact that ZAPIG fits better these data is in unison with the theoretical conclusion that ZAPIG is a distribution that accommodates data with high overdispersion.
Table 1 shows also that the zero-adjusted distributions fit the data better than the other distributions. This is because there is no report with a zero AEC count present in the data, while both NBI and PIG allow for zero value data. Therefore, these distributions need to be adjusted by removal of the zero value.
Formulae for ZANBI and ZAPIG distributions as well as the NBI and PIG distributions from which they are formed, were given in the Introduction, respectively (14), (8), and (10). Parametric models with ZANBI and ZAPIG have been fitted to both COVID vaccines and other drugs data in order to define if the slight difference between the two distributions can be detected with the diagnostic methods.
The parameter is significant and is non-significant in all four models while is significant in the first three models and non-significant in the last model (ZAPIG on the other drugs data). The parameter is close to zero, i.e. negligibly small as expected(Table 2).
Comparing the two distributions on the same data set, means are larger, and variance, skewness, and kurtosis are smaller in ZANBI, which is true for both data sets. Comparing the data sets, AEC for COVID vaccines have higher means and variances and lower skewness and kurtosis than AEC for “other drugs” valid for both distributions. Because there are several ways for comparing skewness and kurtosis, a more precise statement would be that other drugs is more moment skew to the right [49](p. 292) and more moment kurtotic [49](p. 300) than COVID vaccines.
Graphical comparison helps to visualize the striking difference in distributions between the two data sets (Figure 1). The large difference between COVID vaccines (a and b) and other drugs (c and d) is due to the above mentioned smaller mean (peak shifted to the left) and bigger skewness (longer tail) and kurtosis (sharper peak). Differences between ZANBI and ZAPIG (a,c vs. b,d) are smaller but observable. For example, comparing a and b, differences are in 1, 3, 4, 6, 7 and comparing b and d, differences are in 1, 2 and 4.
The diagnostic plots in Figure 2 confirm the main findings from the comparison in Figure 1: the differences between the two data sets are easily observable in all diagnostic plots while the differences between the two distributions that were fitted on the same data are more subtle. More specifically, COVID vaccines show large positive autocorrelation with both distributions as more than half of the vertical lines cross the upper limit (Figure 2a,e). The autocorrelation in other drugs is much smaller (Figure 2i,n). A possible reason is the small number of COVID vaccines compared to other drugs (4 vs. 1557). Supposedly, reports with the same vaccine would correlate more than reports with different vaccines. If this is the case, it is expected that a model in which vaccine is an explanatory variable would display noticeably lower autocorrelation than the parametric model.
Density plots of COVID vaccines (Figure 2b,f) have a front shoulder more apparent in the ZAPIG fit (Figure 2f) which is a deviation from the Gaussian curve. Density plots for other drugs (Figure 2k,o) have a shape that is close to Gaussian except the peaks which are concave or flat. The ZAPIG curve (Figure 2o) is more symmetric than the ZANBI curve (Figure 2k). All Q-Q plots (Figure 2c, g, l, p) look similar with the residuals lying on the diagonal line for most of its length except for its upper end where they form a variably deviating loop above the line. This loop is smaller in the ZAPIG fits (Figure 2g, p) than in the respective ZANBI fits (Figure 2c, l). Also, in all fits but ZAPIG on other drugs (Figure 2p) some smaller deviations are seen at the lower end of the Q-Q plots.
The deviations observed in the Q-Q plots are emphasized in the worm plots that are, in fact, series of detrended Q-Q plots [50]. Figure 2d, h, m, q present worm plots averaged from 40 bootstrapped curves resulting from permutations of the respective residuals order. In the worm plots for COVID vaccines (Figure 2d, h), there are no problems with variance and skewness but the mean is slightly problematic especially with the ZAPIG fit (Figure 2h) where the worm goes beyond the confidence limits at two places. The raised tail on the right shows that the residuals distributions are leptokurtic (with sharper peak than normal) which means that the fitted distributions (ZANBI and ZAPIG) are platykurtic (flatter than normal). Leptokurtosis is higher in the ZANBI fit (Figure 2d) where the tail goes beyond the upper confidence limit. The mean is less problematic with the other drugs data (Figure 2m, q) as the worm goes closer to the central line and deviations do not occur much beyond the confidence limits (compare the grey bootstrapped points). With these data, the residuals distributions are also leptokurtic with the excess kurtosis lower in the ZAPIG fit (Figure 2q). The lower leptokurtosis in the two ZAPIG fits compared with the ZANBI fits corresponds with the fact that the ZAPIG distributions have higher excess kurtosis than the ZANBI distributions for both data sets (cf. Table 2).
The goodness of fit through the residuals distribution can be assessed more accurately from the actual values of the moments of this distribution and from the Filliben correlation coefficient, which are shown in Table 3. In general, the moments are reasonably close to those of the standard normal distribution and the Filliben correlation coefficient approximates 1 with an accuracy to the third digit. The range of deviations from standard normal are: mean – 0.14% to 0.49%; variance – 0.94% to 1.57%; skewness – 2.19% to 7.86%; kurtosis – 2.52% to 9.36%. Judging by variances, COVID vaccines residuals pdf’s are wider, and other drugs ones are narrower than standard normal pdf. The skewness is also in different direction: COVID vaccines residuals pdf’s are skewed to the left (negative skewness) while other drugs ones are skewed to the right (positive skewness). This can be indistinctly seen in the density plots (Figure 2b,f,k,o) where COVID vaccines have a shoulder to the left and other drugs have slightly longer tails to the right which is more noticeable in the ZANBI fit (Figure 2k). The kurtosis values show that ZAPIG can model better kurtosis in both data sets and that the kurtosis in COVID vaccines is modeled better than in other drugs by both ZANBI and ZAPIG.
The goodness of fit measured by the Filliben correlation coefficient is best in other drugs modeled with ZAPIG and worst in COVID vaccines modeled with ZANBI. Other drugs data are modeled better than COVID vaccines with both ZANBI and ZAPIG whereas ZAPIG models are better in both data sets. The latter is in disagreement with the fit assessment by AIC and BIC criteria (Table 1) which shows a better ZANBI than ZAPIG fit on the COVID vaccines data. We have no immediate explanation for this discrepancy rather than point out that the differences between ZANBI and ZAPIG both by AIC/BIC criteria and Filliben coefficient are very small and that effects of the explanatory variables which may affect AIC/BIC and Filliben coefficient to a different extent are not taken into account in the parametric models.
2.2. Full GAMLSS models
In this study, we tested how the values of the response variable (AEC) change as a result of changes in the values of some explanatory variables. We explored the effects of two quantitative explanatory variables (covariates) and three categorical explanatory variables (factors):
- (1)
- Sequential number (covariate, integers);
- (2)
- Age (covariate, integers);
- (3)
- Sex (factor with two levels – female and male);
- (4)
- Severity (factor with two levels – non-severe and severe);
- (5)
- Vaccine type (factor with four levels – Comirnaty, Spikevax, Vaxzevria, Jcovden) only for the COVID vaccine models.
The sequential steps of the selection are presented in Table 4. The models were calculated with the function gamlss() starting from single explanatory variables and gradually complicating models depending on BIC values. It was found that interactions did not improve the models and that ZAPIG generally performed worse that ZANBI in terms of BIC and convergence. Smoothers like pb(), cs(), and others (pbm(), pbz(), lo(), nn(), results not shown) did not improve models; although some of the smoothers led to lower global deviance than the respective non-smoothed model, they resulted in bigger number of occupied degrees of freedom and bigger BIC. Without smoothers, the model remains linear.
Models for were done on the basis of the best (lowest BIC) models for by adding terms to the right hand side of the equation for in (19). For other drugs, it was found that including Age and Sequence_number instead of Age and Severity after modeling with Severity improved the model with 1.6 BIC.
A large part of the fixed effects in the model for other drugs may come from the drugs themselves. However, they cannot be included directly in the model because of the large number of different drugs (too many levels of the categorical variable). One way to bypass this problem is to treat the drugs effect as random, rather than fixed. A model based on Model 29.4.1 with additional term random(Suspect_drug) gave a small global variance (15952.25) but too large BIC (19803.88). In addition, diagnostic plots and goodness of fit were much worse than those of the corresponding parametric fit. Another way to deal with the many levels of Suspect_drug is to use the pcat() smoother that merges the different levels of a factor into a set in which similar levels are combined. The attempt to add pcat(Suspect_drug) failed, however, because of lack of memory with the program announcing the error “cannot allocate vector of size 14.6 Gb”.
Inclusion of an explanatory variable is justified if it results in a better-fitted model. Indeed, the parametric ZANBI model for COVID vaccines differs from Model 15.6 with 2753.83 BIC units (17254.8 vs. 14500.97) and the one for other drugs differs from Model 29.4.1 with 5038.27 BIC units (22898.13 vs. 17859.86). The improvement of fit is also evident in the diagnostics of the best full models (Figure 3) compared with the diagnostics of the respective parametric models (Figure 2).
For COVID vaccines, the biggest improvement is seen in the autocorrelation plots (compare Figure 2a and Figure 3a): most of the autocorrelations are within limits and many are close to zero. Such reduction of autocorrelations shows that they are for the most part due to the explanatory variables especially Vaccines. The left shoulder in the density curve is less apparent and the curve is more symmetric (compare Figure 2b and Figure 3b). The Q-Q plot is straighter and closer to the diagonal line, the upper loop is smaller and there are no deviating points in the lower left (compare Figure 2c and Figure 3c). A smaller part of the worm plot comes outside the limits especially in the right hand tail which is lower (compare Figure 2d and Figure 3d).
For other drugs, the improvement in autocorrelation is not distinct (5 vs. 4 out-of-limit autocorrelations) (Figure 2i and Figure 3i); density plot is less inclined and more Gaussian-shaped and symmetric (Figure 2k and Figure 3k); the loop in the Q-Q plot is smaller (Figure 2l and Figure 3l); less of the black points in the worm plot are out of the confidence limits especially in the right tail (Figure 2m and Figure 3m).
Although most of the moments for the residuals of the best full models (Model 15.6 for COVID vaccines and Model 29.4.1 for other drugs) are farther from the standard normal distribution than the respective ZANBI parametric distributions diagnostics, their Filliben coefficients show a better fit to the standard normal (compare Table 3 and Table 5).
2.3. Effects of the explanatory variables (fixed effects)
Because of model-dependent row removal, the data for Model 15.6 have 3308 rows (reports) and those for Model 29.4.1 have 5226 rows. The results of the full models are given in Table 6. For the COVID vaccines model, reference levels for the factors are: Vaccine: Vaxzevria; Sex: female; Severity: non-severe. These levels are equated to 0 and the other levels are compared to the reference (not seen in Table 6). For the parameter , the following factor levels are significant: the vaccine Comirnaty causes significantly less AEC than Vaxzevria; men have significantly less AEC than women; self-reported severe AEs correlate with more AEC than reported non-severe AEs. Of the covariates, decreases significantly with increasing age. The vaccines Jcovden and Spikevax do not cause significantly less AEC than Vaxzevria. For the parameter , responsible for overdispersion (), reports with severe AEs have significantly larger overdispersion than reports with non-severe AEs; and overdispersion significantly increases with increasing report sequence number. However, there is a significant interaction Sequence_number × Severity that is expected to cause the lines for Severe and Non-severe levels to intersect (see below).
For the other drugs model, of AEC decreases significantly with increasing age and decreases, but not significantly, with increasing sequence report number. Reports with severe AEs have significantly larger parameter than reports with non-severe AE.
Table 7 presents a parametric comparison (mean and standard deviation) of raw AEC data and the values of the parameter derived from Model 15.6. The means of in the subgroups are on average 6.3% (4.0–10.0%) smaller than those of the raw AEC data. The standard deviations are practically constant for the raw data () and for the modeled values (); however, the standard deviation for the raw data is times bigger than the one for the modeled values. The main conclusions from both raw data and modeled values coincide: mean AEC for Vaxzevria, Jcovden, and Spikevax are above the mean and AEC for Comirnaty is below it. In the other two factors, Female and Severe are above the mean while Male and Non-severe are below it.
However, the above way of comparison by parametric statistics presumes a normal or at least a symmetric distribution while, as seen above, the actual distribution is highly asymmetric. Thus, a more appropriate comparison is calculating non-parametric statistics from the raw data and representing the modeled values with the coefficients and the standard errors output from the model (Table 6). This information is extracted and plotted with the GAMLSS function term.plot() while the non-parametric statistics for raw data are plotted with boxplot() (Figure 4).
The term plots (Figure 4a, b, c) show the estimates for each level which are the respective coefficients of the terms in the model of formula (19) together with the 95% confidence intervals calculated on the basis of the standard errors for each coefficient as given in Table 6. These standard errors are bigger than those calculated from the standard deviations for modeled values in Table 7; for example, the standard error for Comirnaty in the model is 0.024956 and the one calculated from standard deviation is 0.01834, for Jcovden – 0.07376 vs. 0.04827, for Spikevax – 0.06029 vs. 0.03054. The standard error in the model is obtained from the variance-covariance matrix created during the process for finding the maximal likelihood, which accounts for the actual distribution. Thus, the standard errors output from the model are more realistic than those calculated from the standard deviations since the latter imply an assumed symmetric distribution that is not observed. The model means are also different – they are coefficients that contribute to the logarithm of rather than being a result of averaging. Despite the differences, ZANBI and non-parametric models come to similar conclusions: Vaxzevria, Jcovden, and Spikevax have similar effects on AEC while Comirnaty has lower effect (significant in the model); women and severe AE have more AEC than for men and non-severe AE, respectively (significant in the model). Box plots show the asymmetry of the distribution by the longer upper whiskers and the “outliers” on the upper side.
Model 15.6 includes a single covariate (Age) for . Rather than modeling it separately (a single descending line in the term plot), it would be more informative to model the whole right hand side of the equation for in (19). The latter model includes a single covariate (Age) and three factors (Vaccine, Sex, Severity). Such type of a multiple regression model that does not include interactions is of the subtype additive model. The additive model consists of a family of straight lines with a common slope (because there are no interactions). The number of these lines is a product of factor levels [30] (p. 227–231). In our model, we have Vaccine type (4 levels), Sex (2 levels), and Severity of AE (2 levels) so we expect theoretically lines.
Figure 5 presents two models for (linear and non-linear) compared with raw COVID vaccines data. The lines in the linear model (Figure 5b) are exponential rather than straight because the function fitted() for obtaining exponentiates the right term side in (19): . A non-linear model, in which Age is modeled with penalized B-splines (Figure 5a) is included since it is difficult to decide if it is better than the linear Model 15.6. The AIC for the non-linear model (Figure 5a) is 14416.5 vs. 14427.27 for Model 15.6; therefore, the non-linear model is fitting better. On the other hand, BIC for the non-linear model is 14500.97 while for Model 15.6 it is 14500.52; therefore, Model 15.6 is more parsimonious. In principle, parsimony is preferred before good fit. The two models are almost equally parsimonious because the difference in BIC values is very small. The linear regressions fitted on the models in Figure 5a and Figure 5b (red lines) are equal (equal intercepts and slopes) but the loess fits (blue lines) are different with the model on Figure 5a having a distinct convexity between 25 and 50 years. This makes the non-linear model better corresponding to the raw data whose loess fit has a less discernable convexity at this age range.
The two models are similar in the relative positions of points and the number and distances between regression lines. The number of distinct lines are 9–12 for the different ages. As concerns points, one has to bear in mind that each point is a different combination of age on one side, and sex, type of vaccine, and AE severity levels on the other; therefore, there may be multiple reports for one point. To understand what combinations of factor levels the lines represent, we made vertical sections through the data for some age values (Table 8).
Of note is that (i) each point in Figure 5a and Figure 5b may include multiple reports (repeats) with the same combination of Age-Vaccine-Sex-Severity level, and (ii) a single age does not contain all level combinations resulting in discontinuities in some lines. The numbering of lines is from top to bottom. It is evident that in general reports designated with severe AE have higher ’s, women have higher ’s for the same combinations of other factor levels, and Comirnaty has lower ’s for the same combinations of other factor levels.
The model for in Model 15.6 ( in formula (19)) is a multiple regression with a covariate Sequence_number and factor Severity with two levels; thus two lines on the graph are seen (Figure 6). The presence of a significant interaction Sequence_number × Severity, however, complicates the model (interaction model). The pure effect of level Severe is highly positive and significant. Exponentiation gives the initial positions of Non-severe at 0.076 and Severe at 0.482 (practically, taking into account the standard errors, these are, respectively, 0.074 and 0.473). Therefore, the top line in Figure 6 represents the Severe level. The interaction of this level with Sequence_number has a negative sign, which is represented by the slight downward slope of the line with severe. Conversely, interaction of Non-severe with Sequence_number has a positive sign giving an upward slope of the bottom line which, because of the preponderance of Non-severe points () becomes steeper with increasing sequence numbers. The intersection of the two lines occurs around sequence number 5168 and from then on the increase in is mainly because of the Non-severe points (blue loess line on Figure 6).
The best model for in the other drugs data (Model 29.4.1) is not a multiple regression because it contains two covariates (Age and Sequence_number) instead of one (Figure 7). In order to understand it, it would be helpful to compare Model 29.4.1 with the second best model - Model 29.4 that is a multiple regression containing the covariate Age and the factor Severity (Figure 7b). Model 29.4 is two descending lines (decreasing with increasing age) corresponding to Severe (top line) and Non-severe (bottom line). The slope of Age in Model 29.4 is comparable to that in Model 15.4 (–0.0039 vs. –0.0049) but the factor Severity for is not significant in Model 29.4 although it is essential for the model fit as its inclusion decreases AIC and BIC.
Changing Severity to Sequence_number generates Model 29.4.1 (Figure 7a) that differs from Model 29.4 (Figure 7b) only for the inclusion of the variability in factor Severity. More specifically, the points in the two lines of Figure 7b span the whole gap between the lines in Figure 7a with more Severe points being closer to the top and more Non-severe closer to the bottom. This distribution is stochastic meaning that the two levels are mixed in different proportions along a vertical.
Parameter in Model 29.4.1 is modeled only by the factor Severity which is highly significant with Severe level having a large positive effect (Table Figure 6). Thus, the bigger part of is contributed by reports designated as severe.
2.4. Overdispersion
Dispersion measured by the method of Cameron-Trivedi does not depend on the particular model, distribution, or explanatory variables used since it builds its own initial Poisson model with all explanatory variables and then using from the initial model checks for the constancy through a t-test in an ordinary least squares model. All that is required for the Cameron-Trivedi test is the data frame and indication of the response and explanatory variables.
Overdispersion is significant in both data sets with p-values ∼ 0. The Cameron-Trivedi test gives -statistic 10.303 for the COVID vaccines data and 10.734 for the other drugs data. This test shows that the other drugs data have slightly higher overdispersion.
Unlike the Cameron-Trivedi method, the method using the Pearson dispersion statistic [51] depends on the model and the distribution. The overdispersion parameters for the different models and distributions used here are shown in Table 9.
Judging from the Pearson dispersion statistic, COVID vaccines data have higher overdispersion than other drugs data with most distributions, a result opposite to the one obtained by the Cameron-Trivedi method. Zero-adjusted Poisson (ZAP) results in even higher overdispersion than the pure Poisson showing that the zero-adjustment of this particular distribution fails to alleviate the overdispersion. On the other hand, pure NBI and PIG eliminate too much of the overdispersion resulting in severe underdispersion, PIG more so than NBI. In contrast, ZANBI and ZAPIG on the parametric models and ZANBI on the full models eliminate just enough of the overdispersion to make the models equidispersed.
Full models largely mirror the parametric models having slightly smaller overdispersion than the respective parametric models. An exception is ZAPIG on full models, which does not converge with the concomitant large increase in overdispersion, especially with the other drugs data.
As a whole, the Pearson dispersion statistic does not correlate with model fit in that lower statistic which does not mean a better fit. The best models are those in which the statistic is close to 1, that is, equidispersed model. In our case, these are the parametric models of other drugs with ZAPIG and ZANBI and the full model of other drugs with ZANBI all of which have p-values close to 0.5.
It is also evident that the Pearson dispersion is not a good statistic in the sense that the interval is very short resulting in abrupt changes from significant to non-significant close to the value 1 of the statistic. For this reason, we consider the -statistic more reliable since the t-distribution changes the significance values in a wider interval.
3. Discussion
To our knowledge, this is the first study attempting to determine the distribution of AEC and use it in a model. By systematically fitting different discrete distributions to AEC from COVID vaccine and other drugs data, it was found that they were best fitted by ZANBI and ZAPIG, respectively, as predicted on the theoretical basis presented in the Introduction. Furthermore, the AEC for the other drugs data, which have bigger overdispersion according to the Cameron-Trivedi test, is better fitted by ZAPIG while the AEC for COVID vaccines data are better fitted by ZANBI according to the theoretical conclusion that ZAPIG fits overdispersed data better than ZANBI. Nevertheless, in both data sets the difference in fit between the two distributions is small compared with the all other available in the GAMLSS package discrete distributions that were tested.
When fitted on the two data sets (COVID vaccines and other drugs), ZANBI and ZAPIG have only minor visible differences between each other. Judging by their parameters and moments, ZAPIG, indeed, handles better a larger overdispersion than ZANBI. For example, ZAPIG has smaller means but larger all other three moments (variance, skewness, and especially kurtosis). Fit deficiencies, in general, are most evident in kurtosis and skewness because they are the highest moments (powers of the distribution generating function), fourth and third, respectively, and any deviation would be magnified when multiplied several times.
Differences between the two distributions are observed in the diagnostic residual plots with ZAPIG handling better data with higher overdispersion, as theoretically predicted. Thus, ZAPIG residuals have smaller excess kurtosis as evidenced by the smaller loops in the Q-Q plots, and, respectively, the lower right tails in the worm plots. The moments of the ZAPIG residual distributions are generally closer to the standard normal distribution than those of ZANBI.
Kurtosis and skewness show the biggest deviations from the standard normal in line with what was said above. Filliben coefficients, which reflect the degree of fit for the residuals, are almost the same for the ZANBI and ZAPIG distributions on the COVID vaccine data (difference in the sixth digit after the decimal point) and slightly different for the other drugs data (advantage for ZAPIG in the fourth digit after the decimal point). This confirms the results from the analysis of AIC and BIC criteria. As a whole, differences in diagnostic plots between the two distributions are minor compared to the differences between the two sets of data.
Part of the misfit of a distribution can be due to the effects of explanatory variables (fixed effects). Correlation within the same level of an explanatory variable is bigger than correlation among levels. For example, data obtained from subjects immunized with Comirnaty are stronger correlated than data from Comirnaty and those from other COVID-19 vaccines because of the differences of action and consequences of immunization with different vaccines. When explanatory variables are included into the model, their fixed effects are accounted for as separate coefficients, which reduces or eliminates the correlation within levels (autocorrelation). This is seen in our residual autocorrelation plots for the parametric models of the COVID vaccine data, which are highly autocorrelated. Low autocorrelation means more independence and randomness which are the implied conditions in the first equation in the systems of equations (15) and (19) in Materials and Methods. Because the distributions used to model the data, in our case ZANBI and ZAPIG, fit theoretically perfect random data, the more random are the real data, the better is the fit, that is, the higher is the likelihood that the distribution fits the data. Including the explanatory variables in the model makes data more random, simultaneously increasing likelihood and decreasing variance. Since the increased likelihood results in better fit, the full model has lower residual variance and AIC value than the parametric model.
These assertions are confirmed by our results. The high autocorrelation in the parametric model of the COVID vaccines is drastically reduced in the full model, which is supposedly due primarily to the inclusion of the vaccines fixed effects in the full model. Simultaneously, AIC and BIC decrease with inclusion of each new term; which is the primary criterion (AIC and BIC decrease) by which model selection is done. In principle, the AIC decrease and, correspondingly, likelihood increase, could continue for as many terms as we have with no guarantee that the more complex models are better than the simpler ones. BIC is preferred here since its second term is a penalty for model complexity that takes into account the balance between free and occupied degrees of freedom. The BIC drops from parametric to full model are considerable, 2754 and 5038 BIC units for the COVID vaccines and other drugs, respectively, which shows a high response to fixed effects and, therefore, a high sensitivity of the model.
During the model selection, ZANBI performed better than ZAPIG in that the former converged on more term combinations and had lower BIC even at the combinations where both distributions converged. Convergence depends on the model, distribution and its algorithmic implementation as well as the data. Increase in the sample size usually results in better convergence. It has been shown that dispersion is related to convergence though this relationship is still distribution-, model-, and data-dependent. For example, Fernandez and Vatcheva [52] in a methodological paper examining data for hospital length of stay as well as simulated data with various dispersion measured through Pearson dispersion statistics, found that NB and zero-inflated NB (ZINB), but not Poisson and zero-inflated Poisson (ZIP), had less than 100% convergence rate occurring only in data with very low overdispersion. In our parametric models, all distributions had 100% convergence on both data sets. In the full models, NBI converged everywhere, and PIG did not converge only in the most complicated model – with all terms up to the 5-way interactions for both and parameters.
We found that both data sets have significant overdispersion according to Pearson dispersion statistic and Cameron-Trivedi tests, but they differ in the degree and direction of overdispersion. The Cameron-Trivedi test suggests that the other drugs data have slightly higher overdispersion than the COVID vaccines data, while the Pearson dispersion statistic suggests the opposite for most distributions. This discrepancy may be due to different assumptions and sensitivities of the two tests, or to model misspecification.
The results also show that different models and distributions have different effects on overdispersion. Zero-adjusted Poisson (ZAP) increases overdispersion, while pure negative binomial (NBI) and Poisson inverse Gaussian (PIG) reduce it too much, resulting in underdispersion. Zero-adjusted negative binomial (ZANBI) and zero-adjusted Poisson inverse Gaussian (ZAPIG) on the parametric models and ZANBI on the full models eliminate just enough of the overdispersion to make the models equidispersed. These results suggest that zero-inflation and excess dispersion should be accounted for separately and appropriately in count data models.
The results also indicate that the Pearson dispersion statistic is not a good indicator of model fit, as lower values do not necessarily imply better fit. The best models are those that have values close to one, indicating equidispersion. In this case, these are the parametric models of other drugs with ZAPIG and ZANBI and the full model of other drugs with ZANBI. The Pearson dispersion statistic also has a very short interval of non-significance around one, making it sensitive to small changes in values. The Cameron-Trivedi test may be more robust as it uses a wider interval based on the t-distribution.
Zero-adjustment was found to have a negative effect on convergence for both distributions though ZANBI converged on more full models than ZAPIG. This means that ZANBI can fit more complex models with multiple explanatory variables, while ZAPIG may struggle with complex models due to convergence issues. This can be explained with the fact that ZAPIG is more difficult to calculate and the necessity for the algorithms to use the recurrence relation (13). This is supported by the observation that the ZAPIG algorithm is several times slower than the ZANBI one especially with the more complex models. Another reason for the worse performance of ZAPIG could be the implementation of the zero alteration to the algorithm. Using zero-truncated PIG (ZTPIG) rather than zero-altered PIG (ZAPIG) would likely perform better than ZAPIG or ZANBI in both parametric and full models even on highly overdispersed data and large samples.
Because of the ZAPIG non-convergence on full models, the best of them use ZANBI. Nevertheless, these models still have better fit than the respective parametric ZAPIG models as evident by their BIC criteria, diagnostic plots, and Filliben coefficients. Since one of the most important aims of modeling is to estimate the effects of the explaining variables, i.e., the fixed effects, it is interesting to explore the extent to which the replacement of ZAPIG by ZANBI would change the fixed effect estimates. We could not observe in practice the change of the fixed effects on replacement of distribution in the best models because ZAPIG did not converge with these term combinations. With our data, however, which have very close fits of ZAPIG and ZANBI and similar overdispersion, it is expected that change of distribution would minimally influence the fixed effects.
Such an expectation is additionally supported by theory. From formula (6) which represents the two distributions before mixing, it is seen that only the first, Poisson-like, distribution of formula (5) contains the parameter. The second distribution, , which could be Gamma (for NB) or inverse Gaussian (for PIG) is a distribution for u, which is more connected to than to . True, the distinction between the two initial distributions is to a large extent obliterated after mixing but nevertheless the possibility to have alternative distributions for suggests that the choice of is not critical for the fixed effects.
This assumption is rigorously proven by the theoretical study of Weems and Smith [53] assessing the robustness of the fixed-effect estimators when fitting Poisson mixed models. The nominal distribution F (e.g., PIG) when contaminated with a proportion of the contaminating distribution G (e.g., NB) becomes a contaminated distribution . Then the Gâteaux derivative in the direction of G of the functional form connecting the explanatory and response variables of the model, becomes . The Gâteaux derivative can be viewed as a measure of the sensitivity of T to small perturbations of F and by definition it follows that T is robust against distribution misspecification if is bounded for all G. For PIG, this definition can be restated by replacing with the integral of the conditional expected score function , where and . Weems and Smith [53] proved that the integral of the conditional expected score matrix is uniformly bounded and found the bounds by using properties of Bessel functions and by relating the Bessel functions to the PIG probabilities. Numerical integration in R showed that the upper and lower bounds for and are similar and the distance between the upper and lower bounds for increases at a faster rate than the distance between the bounds corresponding to the regression parameters which suggests that may be more sensitive to PIG misspecification than . The authors investigated further with simulation study done with the gamlss package the performance of maximum likelihood estimates (MLEs) when the assumed PIG distribution is misspecified in different ways ( and ). The simulations generated 1000 iid covariates, random effects, and sample responses. Monte Carlo estimates were obtained for , and , and their statistical behavior is examined. The bias of and is very small, regardless of the true value of , both when there is a small amount of contamination and when there is complete PIG misspecification. Their standard errors also increase gradually as increases, but their behavior is not affected by the PIG misspecification. Conversely, the bias of increases rapidly as increases, and there is a considerable bias of for under complete PIG misspecification. The Q-Q plots suggest normality of the estimates, but there is some deviation from a straight line in the tails of the distribution for . Overall, Weems and Smith [53] conclude that is not affected in both parametric () and full () models, while both bias and standard error of are significantly affected upon misspecification of the mixing distribution which in the present case involves the replacement of PIG by NBI in the full models.
While the differences between ZANBI and ZAPIG on the same data set are minimal, the differences between the two data sets are significant and easily observable in the histograms of the experimental data (Figure 1). In particular, the peak in the theoretical distributions of COVID vaccines is shifted to the right with a clearly delineated left shoulder while in the other drugs data, the peak is at the very beginning and there is no left shoulder. This is reflected in differences between the parameters and moments of the distributions between the two data sets. The most important and of greatest practical implication is the difference between locations (means) of the two data sets: 1.60 and 2.19 times larger mean for COVID vaccines compared to other drugs for ZANBI and ZAPIG, respectively. Such large difference in means, taken directly, shows that COVID vaccines, on average, cause much more AECs than the other drugs combined. This can be explained by the reactogenicity of vaccines, which is connected to a large-scale systemic immunological response. The vaccines against diseases other that COVID in the other drugs data probably have AECs comparable to those in COVID vaccines and this could be a subject for a further study.
Variance is larger and skewness is smaller in COVID vaccines approximately in the same ratio interval as the mean while excess kurtosis is 3.63 and 4.06 bigger in the other drugs for ZANBI and ZAPIG, respectively. These differences are carried over in the diagnostic residuals plots, especially in the autocorrelation (discussed above), density, and worm plots.
Fixed effects (i.e., the effects of explanatory variables on the response variable AEC that are established in the full models) are practically important and the theoretical conclusion outlined above that they are robust in relation to contaminating distributions makes their estimates stable and accurate. We chose to estimate the effects of five explanatory variables: three factors (Severity, Vaccine, Sex) and two covariates (Age, Sequence_number) because these are more complete and easily processed from the data. Age and Sex are the most frequently used demographic characteristics; the Vaccine factor with levels Comirnaty, Spikevax, Vaxzevria and Jcovden is estimated only in COVID vaccine data; and the Sequence_number used in combination with one or more factors gives a time-dependent multiple regression.
The factor Severity with two levels (Severe, Non-Severe) in the data refers for the whole record and not for each AE contained in the record. Since patients submit about 90% of the reports, this explanatory variable suffers from high subjectivity. Despite this limitation, the presence of the Severity estimates for each report makes it possible to conclude about the correlation between event severity and AEC number. The following results in our study point unambiguously to a positive correlation between AEC and severity:
- (1)
- In the total other drug reports, 45.28% were reported as severe, but this percentage rose to 75.08% in the AEC outliers (7–9 AEC) and 84.68% in the extreme points ( AEC).
- (2)
- Severity has a significant positive effect on and for COVID vaccine data, as well as on in other drugs data. The latter effect interacts with report Sequence: severe AEs contribute most to in the beginning with a small linear decline while non-severe increase to become dominant factor for toward the end of the period.
- (3)
- In the age-dependent multiple regression plot for in COVID vaccines, severity was found to be the most important factor determining the high AEC number.
While further research is needed to fully understand the relationship between AEC and AE severity, these findings have important implications for pharmacovigilance. They give another meaning for AEC, which can be looked at as a proxy for AE severity in addition to its primary meaning as a measure for the multiplicity of symptoms.
The effect of Age on is negative and significant in both data sets suggesting that younger individuals may be more likely to experience numerous adverse events compared to older individuals, which pertains not only to COVID vaccines but to all medicinal products. Lack of model improvement with smoothers on Age shows that decrease of AEC with age is approximately linear. The significance of Age is seen from the result that on average it decreases AEC with about 1.3 (4.4–3.1) and 0.51 (1.72–1.21) in COVID vaccines and other drugs, respectively (red line on Figure 5b and Figure 7b). The multiple regressions on these figures show also that the Age effect is strongly modified by the different levels of the factors Severity, Sex, and Type of Vaccine in COVID vaccines and less affected by Severity in other drugs. The slopes of all lines in these multiple regressions are approximately equal so they do not intersect which means that there are no interactions among factors and between any of the factors and the Age covariate. This is confirmed in the full model table (Table 6), in which there are no significant interactions for the parameter.
Regarding the effect of Sex, males have significantly less AEC than females with COVID vaccines while this effect is not significant with the other drugs data. The higher number of reports from women supports the finding that females have more AEC (last column of Table 7).
The type of COVID vaccine also has an effect on for COVID vaccine data. Taking Vaxzevria as a reference level, only Comirnaty causes significantly lower number of AEC. The other two vaccines, Spikevax and Jcovden, are not significantly different from Vaxzevria. No significant interactions of Vaccines’ type with other factors were observed which can be seen from the non-intersecting lines in Figure 5.
When using parametric statistics assuming normality, the raw AEC data and the modeled values of the parameter have different means and standard deviations across the subgroups of vaccine type, sex, and AE severity. The raw data have higher means and larger variability than the modeled values, which may reflect the overdispersion and skewness of the data. The modeled values are based on the ZANBI distribution, which accounts for zero-inflation and excess dispersion in count data. The results also indicate that using parametric statistics to compare the raw data and the modeled values may not be appropriate, as the last assume a normal or symmetric distribution, which is not the case for the raw data.
With our data, both modeling and non-parametric methods lead to similar conclusions about the effects of vaccine type, sex, and severity on AEC. Vaxzevria, Jcovden, and Spikevax have higher AEC than Comirnaty; women and severe case reports have higher AEC than men and non-severe cases. However, these effects are more significant in the model than in the box plots, as shown by the confidence intervals and p-values. This suggests that the model is more sensitive and precise than the non-parametric statistics in detecting differences between subgroups.
We have modeled in addition to in the NB- and PIG-based models. This approach provides a more flexible model than one assuming a constant dispersion parameter . It allows for the possibility that the dispersion of the dependent variable may vary according to the values of one or more covariates. For example, it may be that the variance of the dependent variable increases or decreases as the value of an explanatory variable increases. Modeling can also improve the fit and efficiency of the model, as well as provide more accurate standard errors and confidence intervals for the coefficients. Heller et al. [45] in a simulation study confirmed that estimates in the model could be severely biased if the model is misspecified. However, using the () parametrization, model estimates are robust to misspecification of the model. This potentially has implications not only for PIG regression, but also for regression models for any response distribution, in which the scale parameter is not orthogonal to the mean.
3.1. Limitations
We prefer referring to the independent variables as “explanatory variables” rather than “predictory variables” because our modeled data have a limited predictive power. On one hand, the sample size is insufficient to establish permanent trends and differences in levels of explanatory variables, and, on the other, data are local pertaining to only one country (Bulgaria) with its specifics in health system, hospital care, vaccination coverage and profile of vaccinated population, ethnic and sex specifics, etc.
Some groups are too small to infer conclusions and their modeled values are imbalanced such as persons immunized with Jcovden and Spikevax and their subgroups. Furthermore, even trends obtained on groups with moderate size, such as the gradual decrease of AEC with age for all groups, may not occur in other countries or in summary AE databases, such as EudraVigilance or VAERS because of the above mentioned specifics of our modeled data.
The choice of groups (explanatory variables) was limited by their availability in the data. The set of explanatory variables used in the present study are clearly insufficient to encompass all the dependencies and interactions existing in the data. These may possibly be explained or modified by including additional explanatory variables such as dose sequence (first vs. second vs. booster doses), reporter (patient, medical personnel), medical office visits, hospitalized (yes, no), time after immunization, immunization venue, etc.
The causal link between AEC and AE severity remains unclear due to the lack of research on the topic. The present paper found a tentative positive correlation between self-reported AE severity and AEC number. However, due to the above data limitations and the uncertainty in the severity designation, these results are to be considered as preliminary. When speaking about this correlation, we are fully aware that it holds in statistical sense, i.e., only if averaged on a large number of reports. This certainly does not exclude cases of a single AE that is more severe than multiple AEs in different organs and systems. Although severity in the other drugs data correlated with higher AEC, this result was not significant presumably because of the high variability generated by the great variety of drugs used. Mixed-effects models in which the drugs are modeled as a random effect can be used for a further study. Better conclusions could be made from clinical studies where the severity of each AE can be assessed objectively by one or more skilled physicians.
These different limitations and the circumstance that this is the first model using AEC number as a dependent variable makes this study more a proof-of-concept than a systematic investigation of all factors and covariates affecting the AEC number.
4. Materials and Methods
4.1. Data source and processing
The BDA pharmacovigilance (“Yellow card”) database started functioning in 2012 and fully complies with European regulation. The data base with AE was divided into two parts: 1) reports of AEs for the four vaccines against COVID administered in Bulgaria for the period 01.01.2021 – 03.31.2022 and 2) all AEs except those for vaccines against COVID for the period 01.01.2018 – 03.31.2022. Data processing and statistical analysis were done in the R version 4.1.1 programming environment.
The following data columns (vectors) were extracted from the pharmacovigilance database for further processing: Sequence number of the report, Sex and Age of the patient, Suspected drug, self-reported Severity of adverse event, AE1 to AElast. It should be noted that the last AE in the data of 2018 and 2019 is AE25 while in the data from 2020, 2021, and 2022 the last AE is AE20.
Data were shuffled, partitioned, merged and cleaned using the data processing packages contained in the tidyverse superpackage. After processing, the data were collected into tibbles (type of data frames) for easier processing in the models. Further processing involved counting the AEs in each report: the data were formatted in “long” format using the gather() function, specifying the columns with AEs, which were converted into rows: one row for each column. The filter() function removed the rows that contained a blank value in the AE column, and then, using group_by() the rows that contained the same sequence number were merged and AEs for each sequence number were counted with summarize(). The data containing AEC were included in an additional column “Counts”.
4.2. Outliers identification
Identification of count outliers was done by non-parametric (boxplot) method involving calculation of quartiles (Q1 to Q4) and the interquartile range (IQR). Computation was done with the function identify_outliers() from the package rstatix. Values above Q3 + 1.5 × IQR or below Q1 – 1.5 × IQR are considered as outliers. Values above Q3 + 3 × IQR or below Q1 – 3 × IQR are considered as extreme points (or extreme outliers).
4.3. Determination of overdispersion
Overdispersion was determined by two ways: according to the Pearson dispersion statistic and according to the method of Cameron and Trivedi [54].
The Pearson dispersion statistic is calculated by dividing the model’s Pearson statistic by the corresponding degrees of freedom which was calculated with the bespoke function overdisp_fun() (see the R code in the Supplementary Materials).
The Cameron-Trivedi method, implemented in the overdisp function and package, tests for overdispersion in count data models. It considers both the dependent variable and the explanatory variables in the hypothesis test. The method assumes that the variance of the dependent variable Y follows a quadratic function of its mean , that is, . The null hypothesis is that , which implies no overdispersion, and the alternative hypothesis is that , which implies overdispersion. The test statistic is based on the following procedure:
- (1)
- Estimate a Poisson regression model for each observation ;
- (2)
- Obtain the fitted mean values from the Poisson model;
- (3)
- Compute the auxiliary values , which measure the deviation of each observation from its fitted mean;
- (4)
- Estimate an auxiliary linear model , where is a random error, and obtain the coefficient .
According to Cameron and Trivedi [55], the t-statistic for testing is asymptotically normal under the null hypothesis. The null hypothesis is rejected if the p-value of the test statistic is less than a pre-specified significance level , where .
4.4. Statistical modeling
Model building and analysis were performed primarily using the GAMLSS package, which is specifically tailored to the modeling of distributions, including their selection, analysis, and parametrization [30]. In fact, GAMLSS is an upgrade of GAM models [56] for nonlinear regression, allowing modeling of all parameters of distributions (location, scale and shape).
The model of the parameters of the distribution only, ignoring the fixed effects caused by the inclusion of explanatory variables, is as follows (parametric model)
where is the response variable distribution; , are design matrices that include the additive linear terms in the model; are the linear coefficients for the parameters; , , , and are the linearized predictors for , , , and , respectively.
The functions are the link functions, or simply links, because they link with . Because the discrete models are usually exponential, i.e., they have the form where is some parameter, they use a logarithmic link (log link) to utilize the property that the log link transforms the relationship from exponential to linear. In other words, taking the natural logarithm from both sides of the equation one obtains avoiding the exponent.
The above GAMLSS parametric model is fitted to experimental data by assessment of maximal likelihood. For parametric models, it is preferable to use the gamlssML() function instead of gamlss(). gamlssML() uses numerical optimization methods that are faster than the algorithm used by gamlss(). The function histDist() from the gamlss package makes a histogram from the observed AEC values (counts on the X axes), uses gamlssML() to fit the parametric distributions, calculates the theoretical value for each count and overlays the latter over the observed values. The ChooseDist() function with the “counts” argument fits each of the 32 discrete distributions (applicable to count data) to the data by computing the maximal log-likelihood ℓ and tabulating the Akaike and Bayes information criteria. The getOrder() function sorts the rows in this table in ascending order.
The first equation in (15) implies a likelihood function defined as the probability of observing the sample (the respective data) in the form
On the right-hand side is the product of likelihoods for each observation given that the data are distributed according to the chosen distribution.
Hence the logarithm of the likelihood function (log-likelihood) is
The parametric models give the estimates for (eta.mu), (eta.sigma), (eta.nu), i.e. the link functions for the respective parameters. For and the link functions are logarithms, e.g. and to obtain the parameter these have to be exponentiated: . For , the link function is logit and the parameter is obtained by “anti-logit” operation: .
In the present context, significance of a parameter means how much it contributes to log-likelihood ℓ: if the logarithm of the parameter is not significantly different from 0, the parameter does not contribute to ℓ. The moments were calculated from the parameters by the formulae given in [49] (p. 512 for ZANBI, and p. 513 for ZAPIG).
The log-likelihood function is used in the Akaike and Bayes information criteria AIC and BIC as follows
where L is the maximal likelihood, k is the number of free degrees of freedom, and n is the number of observations. Unlike AIC, BIC applies a larger penalty for adding additional parameters, i.e. for increased model complexity, resulting in a more parsimonious model.
The full models require that there is no missing data in any of the columns with explanatory variables so before modeling any row with missing data is removed. Hence, the data for the respective models contains fewer sample points than the parametric models. Inclusion of explanatory variables complicates the GAMLSS model because the coefficients for the respective explanatory variables are added to the parametric terms in the right hand side. The model acquires the following general form (full model)
where with are the coefficients for the respective explanatory variables, or predictors and are various smoothing functions that make the model non-linear like splines of various kinds, loess curve fitting, polynomials, random effects, ridge regression, and non-linear parametric fits. The smoothing functions can be applied or not depending on whether they contribute to decreasing AIC and BIC. For the full models, the GAMLSS model with the gamlss() function was used as it has more flexible and suitable for the relatively large data.
Most smoothing functions used by GAMLSS can be written as
where is a basis matrix depending on values of , and is a set of coefficients subject to the quadratic penalty where is a vector or scalar of hyperparameter(s). It is shown [57] that the algorithm used for fitting the GAMLSS model for fixed values of the hyperparameters maximizes a penalized likelihood function given by
where ℓ is the log-likelihood function in (17).
The general GAMLSS model with explanatory variables is shown in formula (19) where a term for each explanatory variable is added to the parametric terms in the right hand side. Addition of explanatory variables is done not only to the parameter forming the mean () but also to the other parameters (in our case, ). Thus, the effect of explanatory variables is modeled on all parameters of the distribution.
Selection of explanatory variables for inclusion in the model can in principle be done in three ways: backward selection starts with the complete set of explanatory variables and drops them one at a time; forward selection, conversely, starts with the simplest model and adds explanatory variables one at a time; stepwise regression alternatively adds one explanatory variable and deletes another. The GAMLSS package, like other modeling packages, has functions that automate the model selection process. Here, we do the selection manually (forward) in order to have more control on each step of the selection. The only criterion for model adequacy that we use is BIC, because it selects models not only by the goodness of fit but also by parsimony.
4.5. Model diagnostics
The goodness of fit for parametric and full models is determined from the residuals. These are the differences between observed and theoretical value for each experimental point (report). In an ideal fit, residuals are distributed exactly in a standard normal distribution, i.e., a normal distribution with mean = 0, variance = 1, skewness = 0, and kurtosis = 3.
The following diagnostic plots are used:
- (1)
- Autocorrelation function (ACF) is a measure of independence between observations. Independent observations should have the vertical lines close to zero and between the two limits (hashed lines);
- (2)
- Density (pdf) should be as close to the symmetric bell-shaped normal curve as possible;
- (3)
- Normal quantile-quantile (Q-Q) plot should have the residuals lying on the red diagonal line on all its length;
- (4)
- Worm plot [58] has an information on all moments of the residuals distribution as follows: mean (above central line – too high; below central line – too low), variance (positive slope – too high; negative slope – too low), skewness (U-shape – positive skewness; inverted U-shape – negative skewness), kurtosis (S-shape with right bent up – leptokurtosis; S-shape with right bent down – platykurtosis) [30](p. 428). The gamlss package has two functions for worm plots: wp() for plots without bootstrap and rqres.plot() for plots with bootstrap.
In addition, we use the capability of the histDist() function to calculate and display the moments of the residuals distribution. The closer the moments are to those of the standard normal distribution mentioned above, the better is the fit. This information also includes the Filliben correlation coefficient [59] which is a test statistic for the composite hypothesis of normality defined as the product moment correlation coefficient between the ordered observations and the order statistic medians from a standard normal distribution.
5. Conclusions
Analysis of parametric models by BIC and residual diagnostics show that ZANBI and ZAPIG are the distributions that fit best AEC data for COVID-19 vaccines, and for other drugs, respectively. These results confirm the theoretical predictions. Distribution parameters but not distribution fits differ substantially between the two data sets. The full models reveal significant effects of age, sex, and self-assessed severity on the AEC means in COVID-19 vaccines data, and of severity on the AEC variance in other drugs data. Results in our study point unambiguously to a positive correlation between AEC and AE severity. It is worthwhile to test the hypothesis for AEC as a risk indicator on clinical trials data.
Author Contributions
Conceptualization, L.A. and B.K.; methodology, L.A.; software, L.A.; validation, K.D.; formal analysis, L.A.; investigation, L.A.; resources, B.K.; data curation, L.A.; writing—original draft preparation, L.A.; writing—review and editing, K.D. and B.K.; visualization, L.A.; supervision, K.D; project administration, B.K.; funding acquisition, B.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The processed data containing the explanatory and response variables (counts) are openly available in ResearchGate at DOI:10.13140/RG.2.2.31116.28806 [COVID Vaccine Data], and DOI:10.13140/RG.2.2.26083.12329 [Other Drugs Data].
Acknowledgments
The authors thank Dr. Maria Popova-Kiradjieva for providing us with the BDA pharmacovigilance data.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
References
- Hauben, M.; Horn, S.; Reich, L. Potential use of data-mining algorithms for the detection of surprise adverse drug reactions. Drug Safety 2007, 30, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, R.D.; Amatya, A.K. Statistical Methods for Drug Safety; CRC Press, 2016. [CrossRef]
- Fisher, R.A. On the mathematical foundations of theoretical statistics. Philosophical Transactions of the Royal Society of London. Series A, Containing Papers of a Mathematical or Physical Character 1922, 222, 309–368. [Google Scholar] [CrossRef]
- Cox, D.R. Some remarks on over-dispersion. Biometrika 1983, 70, 269–274. [Google Scholar] [CrossRef]
- Shoukri, M.M.; Asyali, M.H.; vanDorp, R.; Kelton, D.F. The Poisson inverse Gaussian regression model in the analysis of clustered counts data. Journal of Data Science 2004, 2, 17–32. [Google Scholar] [CrossRef]
- Hilbe, J.M. Negative Binomial Regression, ed., 2nd ed.; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar] [CrossRef]
- Greenwood, M.; Yule, G. An inquiry into the nature of frequency distributions representative of multiple happenings with particular reference to the occurrence of multiple attacks of disease or of repeated accidents. Journal of Royal Statistical Society 1920, 83, 255–279. [Google Scholar] [CrossRef]
- Yirga, A.A.; Melesse, S.F.; Mwambi, H.G.; Ayele, D.G. Negative binomial mixed models for analyzing longitudinal CD4 count data. Scientific Reports 2020, 10, 16742. [Google Scholar] [CrossRef]
- Zhang, X.; Mallick, H.; Tang, Z.; Zhang, L.; Cui, X.; Benson, A.K.; Yi, N. Negative binomial mixed models for analyzing microbiome count data. BMC Bioinformatics 2017, 18, 4. [Google Scholar] [CrossRef]
- Hashim, L.; Dreeb, N.; Hashim, K.; Shiker, M. An application comparison of two negative binomial models on rainfall count data. Journal of Physics: Conference Series 2021, 1818, 012100. [Google Scholar] [CrossRef]
- Crotteau, J.S.; Ritchie, M.W.; Varner, J.M. A mixed-effects heterogeneous negative binomial model for postfire conifer regeneration in Northeastern California, USA. Forest Science 2014, 60, 275–287. [Google Scholar] [CrossRef]
- Ren, X.; Kuan, P.F. Negative binomial additive model for RNA-Seq data analysis. BMC Bioinformatics 2020, 21, 171. [Google Scholar] [CrossRef]
- Cho, H.; Liu, C.; Preisser, J.S.; Wu, D. A bivariate zero-inflated negative binomial model and its applications to biomedical settings. Statistical Methods in Medical Research 2023, 0, 1–18. [Google Scholar] [CrossRef]
- Lehman, R.R.; Archer, K.J.; Rapallo, F. Penalized negative binomial models for modeling an overdispersed count outcome with a high-dimensional predictor space: Application predicting micronuclei frequency. PLoS One 2019, 14, e0209923. [Google Scholar] [CrossRef]
- Byers, A.L.; Allore, H.; Gill, T.M.; Peduzzi, P.N. Application of negative binomial modeling for discrete outcomes: A case study in aging research. Journal of Clinical Epidemiology 2003, 56, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Zewotir, T.; Ramroop, S. Application of negative binomial regression for assessing public awareness of the health effects of nicotine and cigarettes. African Safety Promotion: A Journal of Injury and Violence Prevention 2009, 7. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, C.; Chen, F.; Cui, S.; Cheng, J.; Wu, B. A random-parameter negative binomial model for assessing freeway crash frequency by injury severity: daytime versus nighttime. Sustainability 2022, 14, 9061. [Google Scholar] [CrossRef]
- Wang, X.; Lindsey, G.; Hankey, S.; Hoff, K. Estimating mixed-mode urban trail traffic using negative binomial regression models. Journal of Urban Planning and Development 2014, 140. [Google Scholar] [CrossRef]
- Lindén, A.; Mäntyniemi, S. Using the negative binomial distribution to model overdispersion in ecological count data. Ecology 2011, 92, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Stoklosa, J.; Blakey, R.V.; Hui, F.K. An overview of modern applications of negative binomial modeling in ecology and biodiversity. Diversity 2022, 14, 320. [Google Scholar] [CrossRef]
- Putri, G.; Nurrohmah, S.; Fithriani, I. Comparing Poisson-Inverse Gaussian Model and Negative Binomial Model on case study: horseshoe crabs data. Journal of Physics: Conference Series 2020, 1442, 012028. [Google Scholar] [CrossRef]
- Villarini, G.; Vecchi, G.; Smith, J. Modeling the dependence of tropical storm counts in the North Atlantic Basin on climate indices. Monthly Weather Review 2010, 138, 2681–2705. [Google Scholar] [CrossRef]
- Lloyd-Smith, J.; Schreiber, S.; Kopp, P.; Getz, W. Superspreading and the effect of individual variation on disease emergence. Nature 2005, 438, 355–359. [Google Scholar] [CrossRef]
- Carter, E.; Potts, H. Predicting length of stay from an electronic patient record system: a primary total knee replacement example. BMC Medical Informatics and Decision Making 2014, 14, 26. [Google Scholar] [CrossRef]
- Orooji, A.; Nazar, E.; Sadeghi, M.; Moradi, A.; Jafari, Z.; Esmaili, H. Factors associated with length of stay in hospital among the elderly patients using count regression models. Medical Journal of The Islamic Republic of Iran 2021, 35, 38–42. [Google Scholar] [CrossRef]
- Mailier, P.; Stephenson, D.; Ferro, C.; Hodges, K. Serial clustering of extratropical cyclones. Monthly Weather Review 2006, 134, 2224–2240. [Google Scholar] [CrossRef]
- Vitolo, R.; Stephenson, D.; Cook, I.; Mitchell-Wallace, K. Serial clustering of intense European storms. Meteorologische Zeitschrift 2009, 18, 411–424. [Google Scholar] [CrossRef]
- Robinson, M.; Smith, G. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 2007, 23, 2881–2887. [Google Scholar] [CrossRef] [PubMed]
- Hilbe, J.M. Modeling Count Data; Cambridge University Press New York USA, 2014. [CrossRef]
- Stasinopoulos, M.; Rigby, R.; Heller, G.; Voudouris, V.; De Bastiani, F. Flexible Regression and Smoothing Using GAMLSS in R; CRC Press Boca Raton FL USA, 2017. [CrossRef]
- Willmot, G.E. The Poisson-Inverse Gaussian distribution as an alternative to the negative binomial. Scandinavian Actuarial Journal 1987, 1987, 113–127. [Google Scholar] [CrossRef]
- Saraiva, E.; Vigas, V.; Flesch, M.; Gannon, M.; de Bragança Pereira, C. Modeling overdispersed dengue data via Poisson inverse Gaussian regression model: a case study in the city of Campo Grande, MS, Brazil. Entropy 2022, 24, 1256. [Google Scholar] [CrossRef]
- Tzougas, G.; Hoon, W.L.; Lim, J.M. The negative binomial-inverse Gaussian regression model with an application to insurance ratemaking. European Actuarial Journal 2019, 9, 323–344. [Google Scholar] [CrossRef]
- Dean, C.; Lawless, J.F.; Willmot, G.E. A mixed Poisson-inverse Gaussian regression model. Canadian Journal of Statistics 1989, 17, 171–181. [Google Scholar] [CrossRef]
- Sichel, H. Anatomy of the generalized inverse Gaussian-Poisson distribution with special applications to bibliometric studies. Information Processing and Management 1992, 28, 5–17. [Google Scholar] [CrossRef]
- Ord, J.; Whitmore, G. The Poisson-inverse Gaussian distribution as a model for species abundance. Communications in Statistics - Theory and Methods 1986, 15, 853–871. [Google Scholar] [CrossRef]
- Tremblay, L. Using the Poisson inverse Gaussian in bonus-malus systems. ASTIN Bulletin: The Journal of the IAA 1992, 22, 97–106. [Google Scholar] [CrossRef]
- Ouma, V.; Mwalili, S.; Kiberia, A. Poisson Inverse Gaussian (PIG) model for infectious disease count data. American Journal of Theoretical and Applied Statistics 2016, 5, 326–333. [Google Scholar] [CrossRef]
- Gómez-Déniz, E.; Calderín-Ojeda, E. Properties and applications of the Poisson-reciprocal inverse Gaussian distribution. Journal of Statistical Computation and Simulation 2018, 88, 269–289. [Google Scholar] [CrossRef]
- Carlson, M.A. Assessing microdata disclosure risk using the Poisson-inverse Gaussian distribution. Statistics in Transition 2002, 5, 901–925. [Google Scholar]
- Purhadi, E.; Noviyana, R.; Sutikno. Bivariate zero-inflated Poisson inverse Gaussian regression model and its application. International Journal on Advanced Science, Engineering and Information Technology 2021, 11, 2407–2415. [Google Scholar] [CrossRef]
- Rahimzadeh, M.; Kavehie, B. Promotion time cure model with generalized Poisson-Inverse Gaussian Distribution. Journal of Biostatistics and Epidemiology 2016, 2, 68–75. [Google Scholar]
- Amalia, J.; Purhadi.; Otok, B. Application of geographically weighted bivariate Poisson inverse Gaussian regression. In Proceedings of the AIP Conference Proceedings. AIP Publishing LLC Melville NY USA, 2020, Vol. 2268, p. 020001. [CrossRef]
- Shaban, S. On the discrete Poisson-inverse Gaussian distribution. Biometrical Journal 1981, 23, 297–303. [Google Scholar] [CrossRef]
- Heller, G.Z.; Couturier, D.L.; Heritier, S.R. Beyond mean modelling: Bias due to misspecification of dispersion in Poisson-inverse Gaussian regression. Biometrical Journal 2019, 61, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.K.; Cancho, V.G.; Louzada, F. The Poisson–Inverse-Gaussian regression model with cure rate: a Bayesian approach and its case influence diagnostics. Statistical Papers 2016, 57, 133–159. [Google Scholar] [CrossRef]
- Perrakis, K.; Karlis, D.; Cools, M.; Janssens, D. Bayesian inference for transportation origin-destination matrices: the Poisson-inverse Gaussian and other Poisson mixtures. Journal of the Royal Statistical Society: Series A (Statistics in Society) 2015, 178, 271–296. [Google Scholar] [CrossRef]
- Zha, L.; Lord, D.; Zou, Y. The Poisson Inverse Gaussian (PIG) generalized linear regression model for analyzing motor vehicle crash data. Journal of Transportation Safety and Security 2016, 8, 18–35. [Google Scholar] [CrossRef]
- Rigby, R.A.; Stasinopoulos, M.D.; Heller, G.Z.; De Bastiani, F. Distributions for Modeling Location, Scale, and Shape using GAMLSS in R; CRC Press Boca Raton FL USA, 2020. [CrossRef]
- van Buuren, S. Worm plot to diagnose fit in quantile regression. Statistical Modelling 2007, 7, 363–376. [Google Scholar] [CrossRef]
- Hilbe, J.M.; Robinson, A.P. Methods of Statistical Model Estimation; CRC Press Boca Raton FL USA, 2013. [CrossRef]
- Fernandez, G.; Vatcheva, K. A comparison of statistical methods for modeling count data with an application to hospital length of stay. BMC Medical Research Methodology 2022, 22, 211. [Google Scholar] [CrossRef]
- Weems, K.; Smith, P. Assessing the robustness of estimators when fitting Poisson inverse Gaussian models. Metrika 2018, 81, 985–1004. [Google Scholar] [CrossRef]
- Cameron, A.C.; Trivedi, P.K. Regression-based tests for overdispersion in the Poisson model. Journal of Econometrics 1990, 46, 347–364. [Google Scholar] [CrossRef]
- Cameron, A.C.; Trivedi, P.K. Regression Analysis of Count Data, 2nd ed. ed.; Cambridge University Press Cambridge UK, 2013. https://faculty.econ.ucdavis.edu/faculty/cameron/racd2/.
- Hastie, T.J.; Tibshirani, R.J. Generalized Additive Models; Chapman and Hall/CRC Boca Raton FL USA, 1990. [CrossRef]
- Rigby, R.; Stasinopoulos, D. Generalized additive models for location, scale and shape, (with discussion). Applied Statistics-Journal of the Royal Statistical Society Series C 2005, 54, 507–554. [Google Scholar] [CrossRef]
- van Buuren, S.; Fredriks, M. Worm plot: A simple diagnostic device for modelling growth reference curves. Statistics in Medicine 2001, 20, 1259–1277. [Google Scholar] [CrossRef]
- Filliben, J.J. The probability plot correlation coefficient test for normality. Technometrics 1975, 17, 111–117. [Google Scholar] [CrossRef]
Figure 1.
Observed and theoretical distributions from the parametric models. The panels are: (a) ZANBI on COVID vaccines data; (b) ZAPIG on COVID vaccines data; (c) ZANBI on other drugs data; (d) ZAPIG on other drugs data.
Figure 1.
Observed and theoretical distributions from the parametric models. The panels are: (a) ZANBI on COVID vaccines data; (b) ZAPIG on COVID vaccines data; (c) ZANBI on other drugs data; (d) ZAPIG on other drugs data.
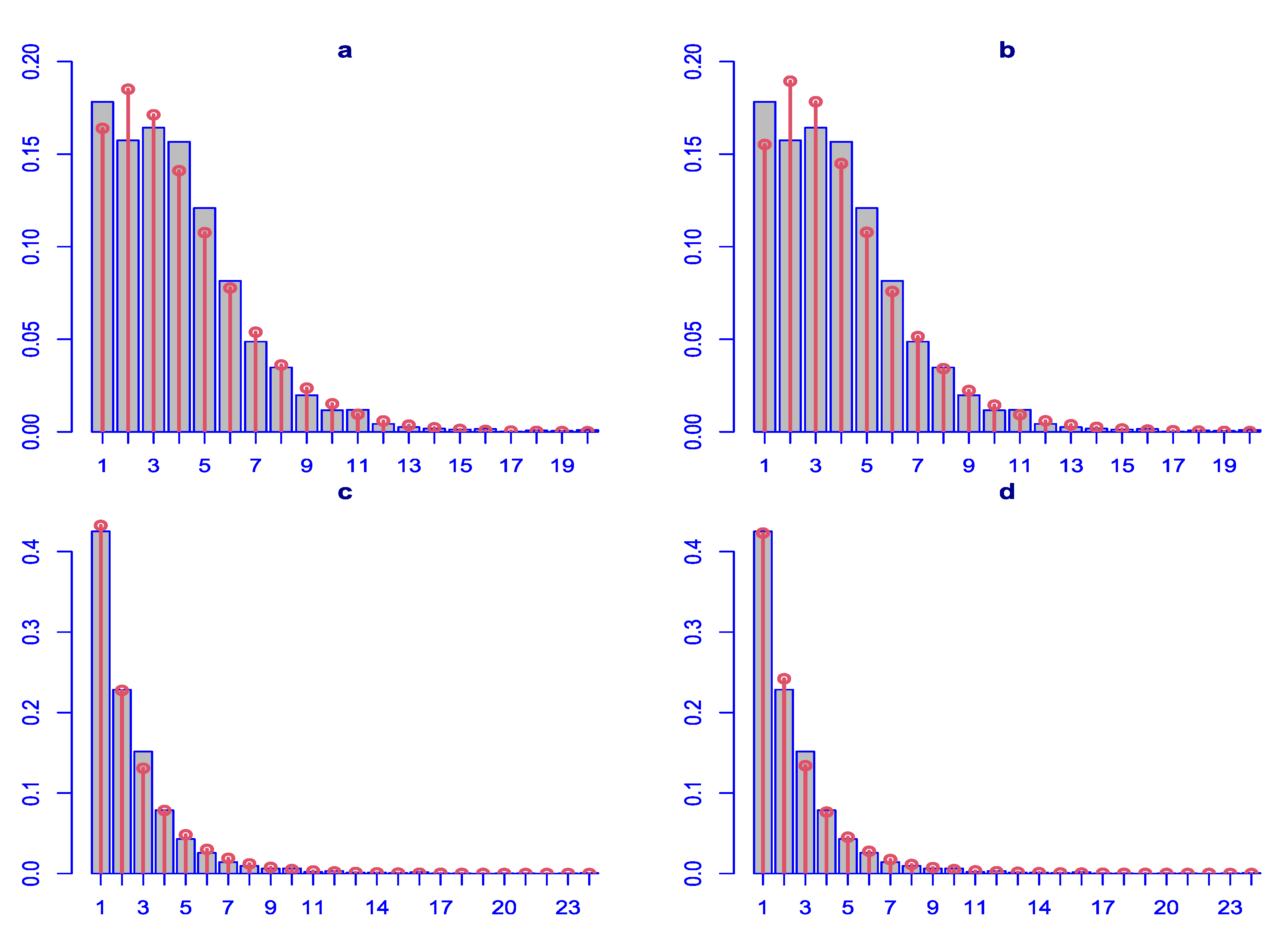
Figure 2.
Diagnostic plots for the residuals of the parametric distributions. First row (a, b, c, d): ZANBI on COVID vaccines data; second row (e, f, g, h): ZAPIG on COVID vaccines data; third row (i, k, l, m): ZANBI on other drugs data; fourth row (n, o, p, q): ZAPIG on other drugs data. Plots are the following types: first column (a, e, i, n): autocorrelation function (ACF); second column (b, f, k, o): probability density function (pdf); third column (c, g, l, p): Q-Q plots; fourth column (d, h, m, q): worm plots (black points) with bootstrap (gray points).
Figure 2.
Diagnostic plots for the residuals of the parametric distributions. First row (a, b, c, d): ZANBI on COVID vaccines data; second row (e, f, g, h): ZAPIG on COVID vaccines data; third row (i, k, l, m): ZANBI on other drugs data; fourth row (n, o, p, q): ZAPIG on other drugs data. Plots are the following types: first column (a, e, i, n): autocorrelation function (ACF); second column (b, f, k, o): probability density function (pdf); third column (c, g, l, p): Q-Q plots; fourth column (d, h, m, q): worm plots (black points) with bootstrap (gray points).
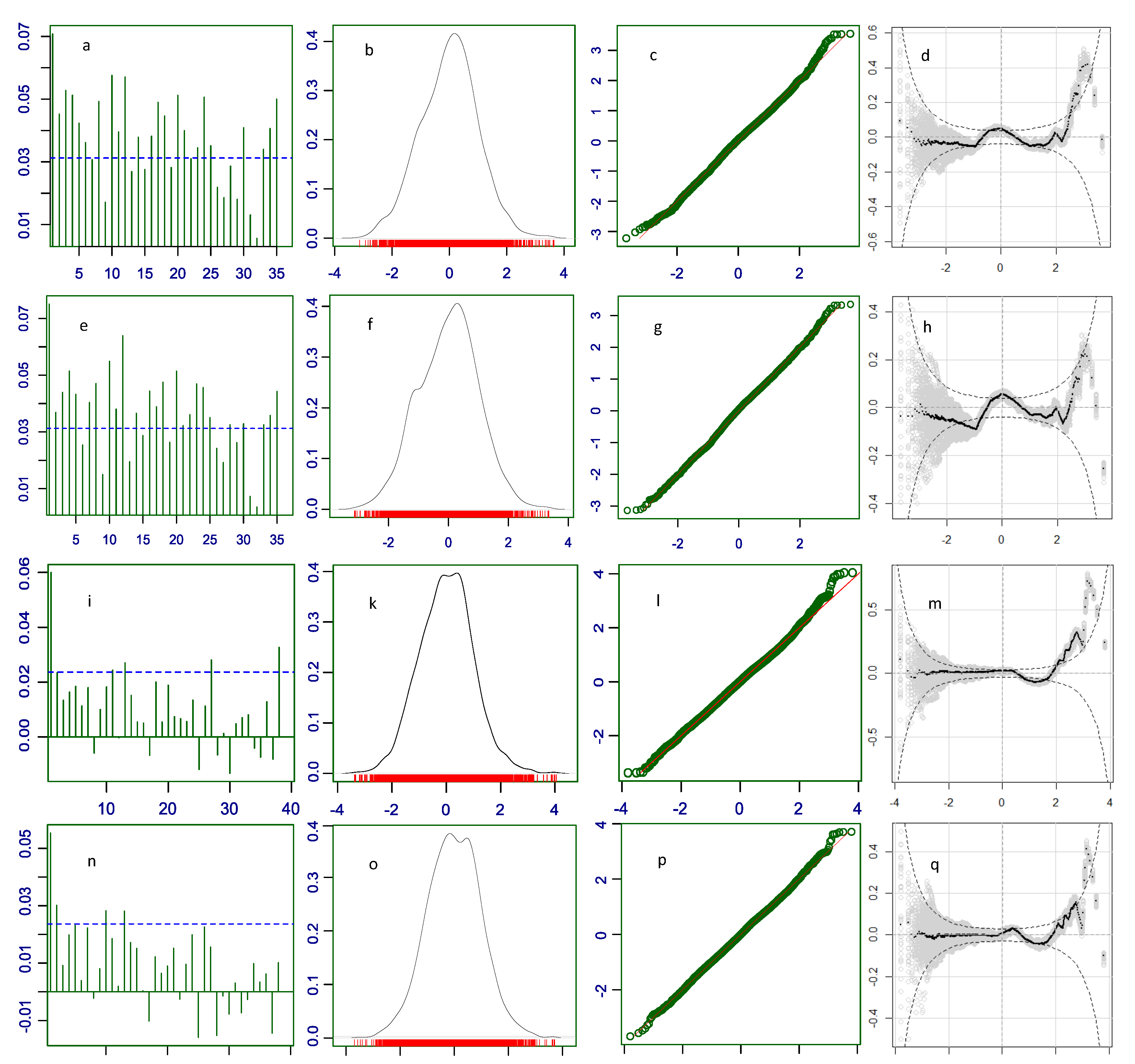
Figure 3.
Diagnostic plots for the residuals of the GAMLSS models with explanatory variables. First row (a, b, c, d): ZANBI on COVID vaccines data; second row (i, k, l, m): ZANBI on other drugs data. Plots are the following types: first column (a, i): autocorrelation function (ACF); second column (b, k): probability density function (pdf); third column (c, l): Q-Q plots; fourth column (d, m): worm plots (black points) with bootstrap (gray points). Letters for plots in this figure correspond to those in Figure 2.
Figure 3.
Diagnostic plots for the residuals of the GAMLSS models with explanatory variables. First row (a, b, c, d): ZANBI on COVID vaccines data; second row (i, k, l, m): ZANBI on other drugs data. Plots are the following types: first column (a, i): autocorrelation function (ACF); second column (b, k): probability density function (pdf); third column (c, l): Q-Q plots; fourth column (d, m): worm plots (black points) with bootstrap (gray points). Letters for plots in this figure correspond to those in Figure 2.
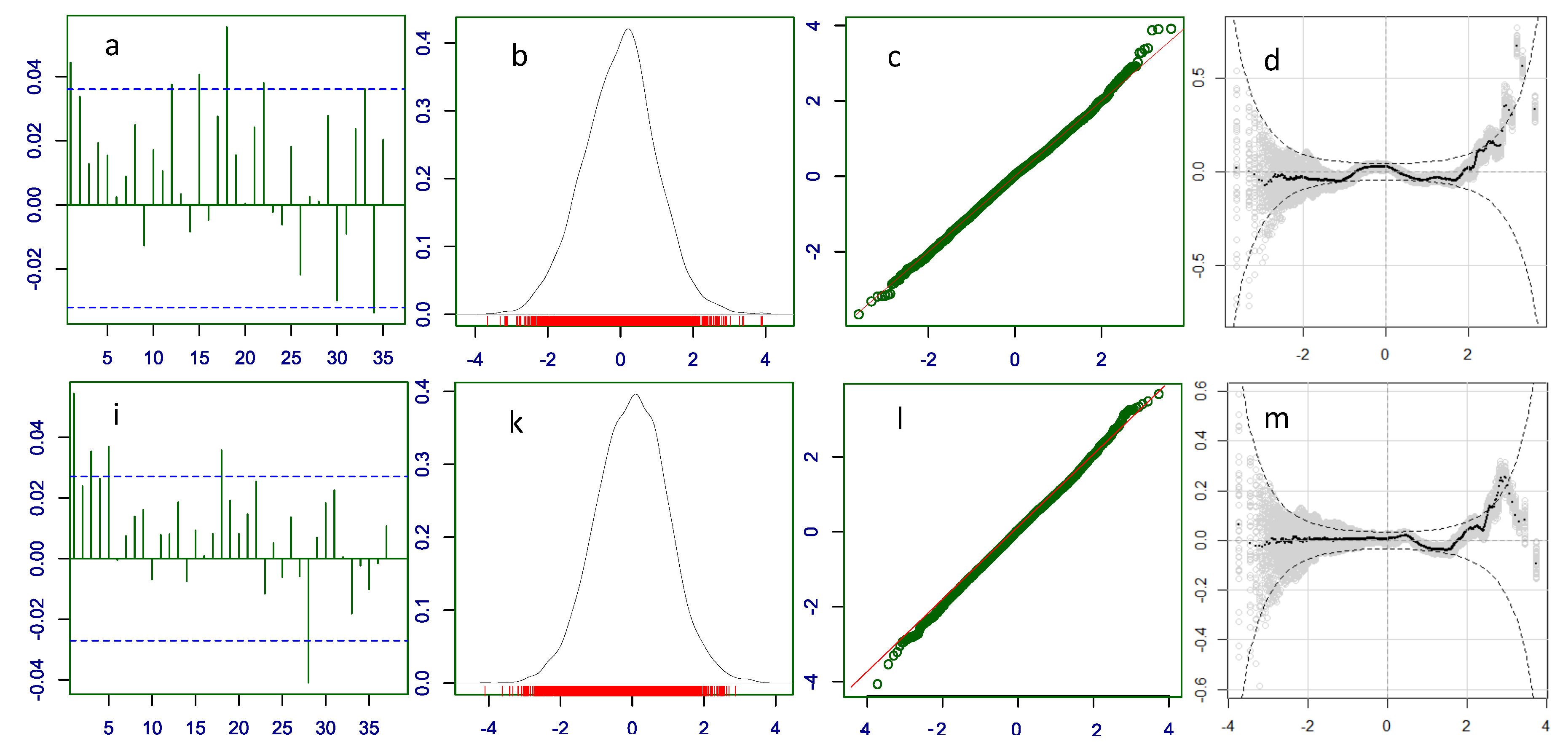
Figure 4.
Comparison of the factors Vaccine, Sex, and Severity by non-parametric statistics and from estimates for in Model 15.6. Variables are: Vaccine (a and d), Sex (b and e), Severity (c and f); term plots are top row (a, b, c), box plots are bottom row (d, e, f).
Figure 4.
Comparison of the factors Vaccine, Sex, and Severity by non-parametric statistics and from estimates for in Model 15.6. Variables are: Vaccine (a and d), Sex (b and e), Severity (c and f); term plots are top row (a, b, c), box plots are bottom row (d, e, f).
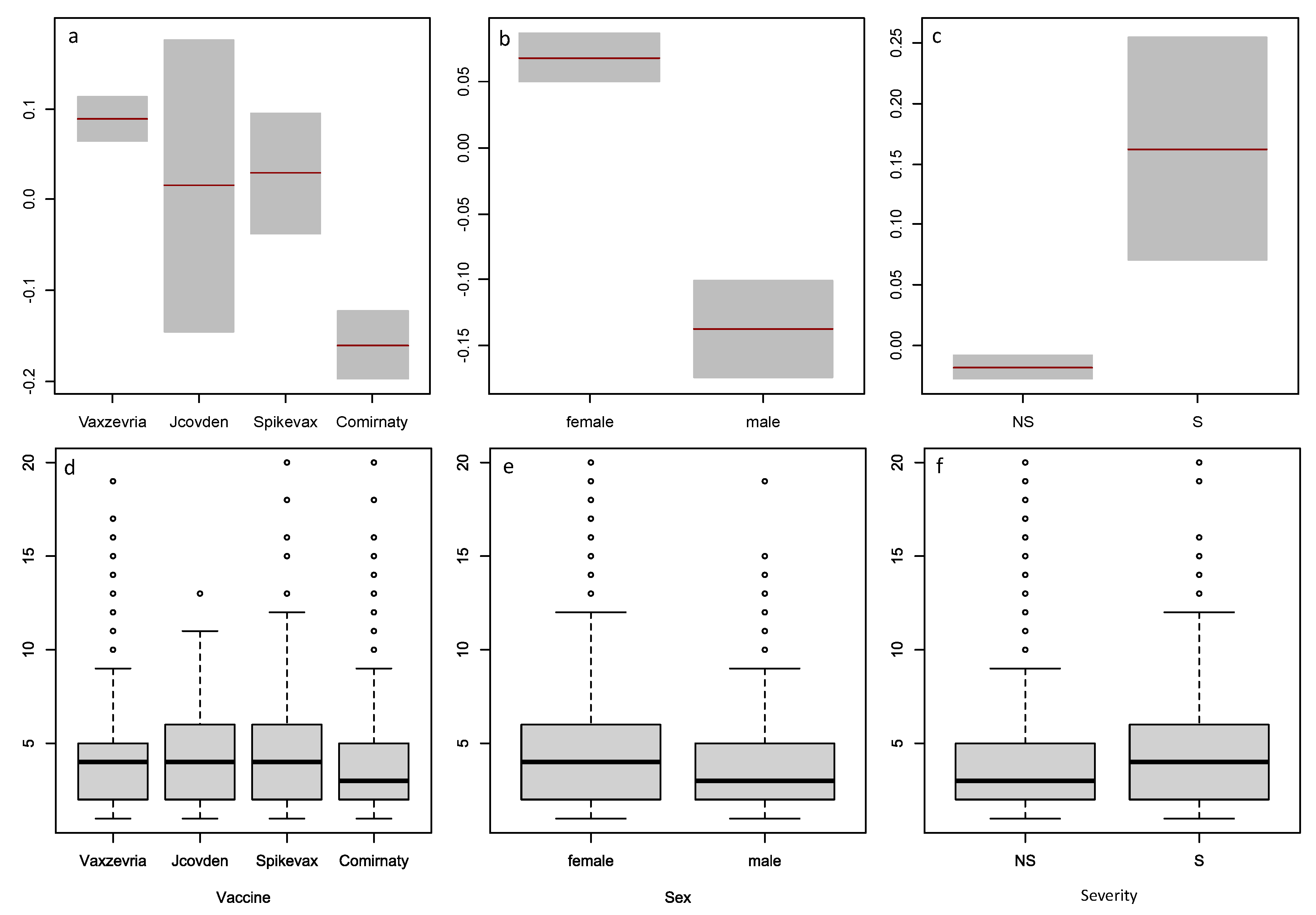
Figure 5.
Models of the parameter for the COVID vaccine data. a, model with penalized B-splines on age (pb(Age)); b, model linear in age (Model 15.6); c, raw data (observed AER counts). Red line: linear least squares regression; blue line: loess (a non-linear regression smoother) applied on raw or modeled data at the moment of plotting.
Figure 5.
Models of the parameter for the COVID vaccine data. a, model with penalized B-splines on age (pb(Age)); b, model linear in age (Model 15.6); c, raw data (observed AER counts). Red line: linear least squares regression; blue line: loess (a non-linear regression smoother) applied on raw or modeled data at the moment of plotting.
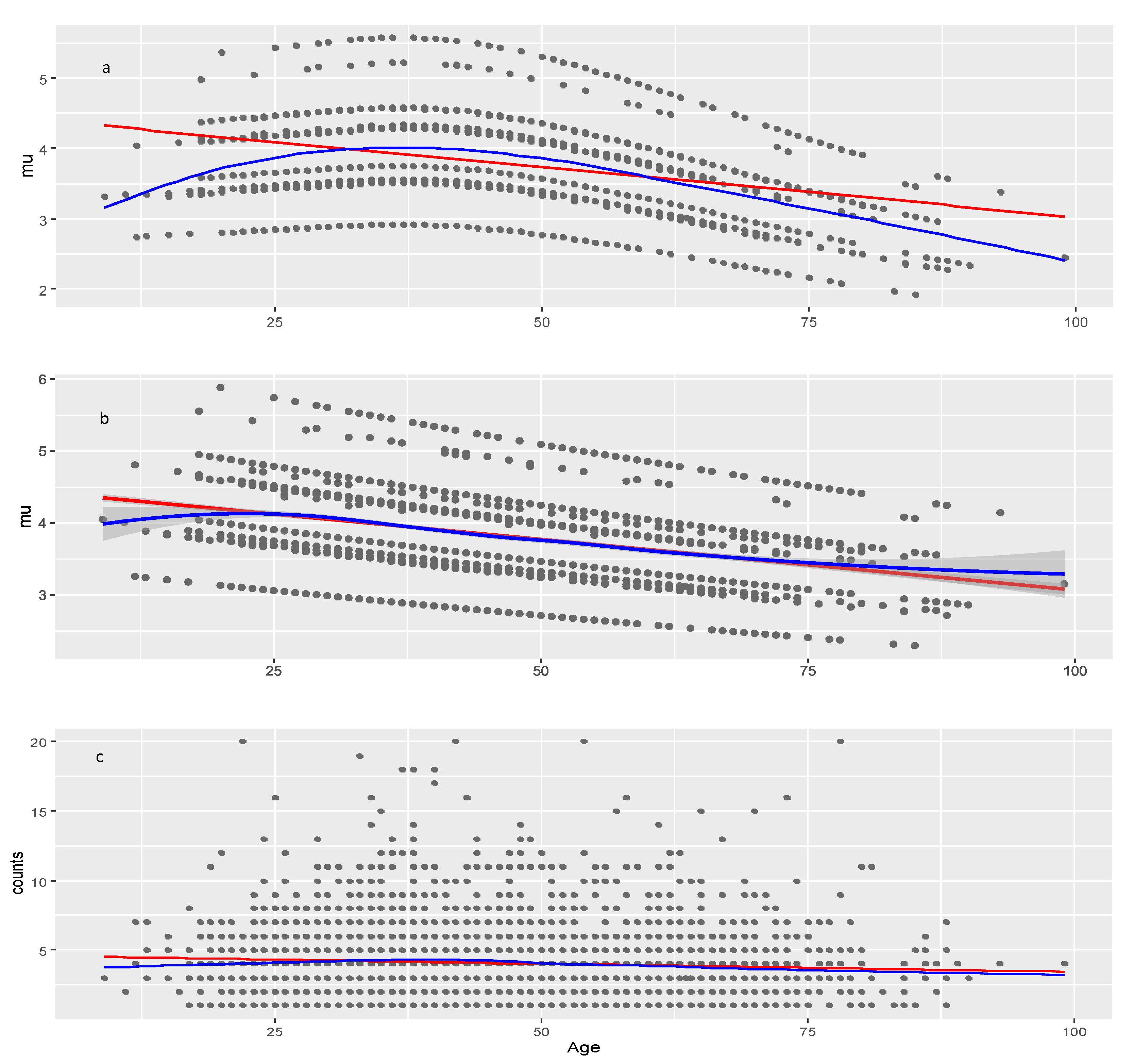
Figure 6.
Model of the parameter for the COVID vaccine data (Model 15.6). Red line: linear least squares regression; blue line: loess (a non-linear regression smoother) applied on raw or modeled data at the moment of plotting.
Figure 6.
Model of the parameter for the COVID vaccine data (Model 15.6). Red line: linear least squares regression; blue line: loess (a non-linear regression smoother) applied on raw or modeled data at the moment of plotting.
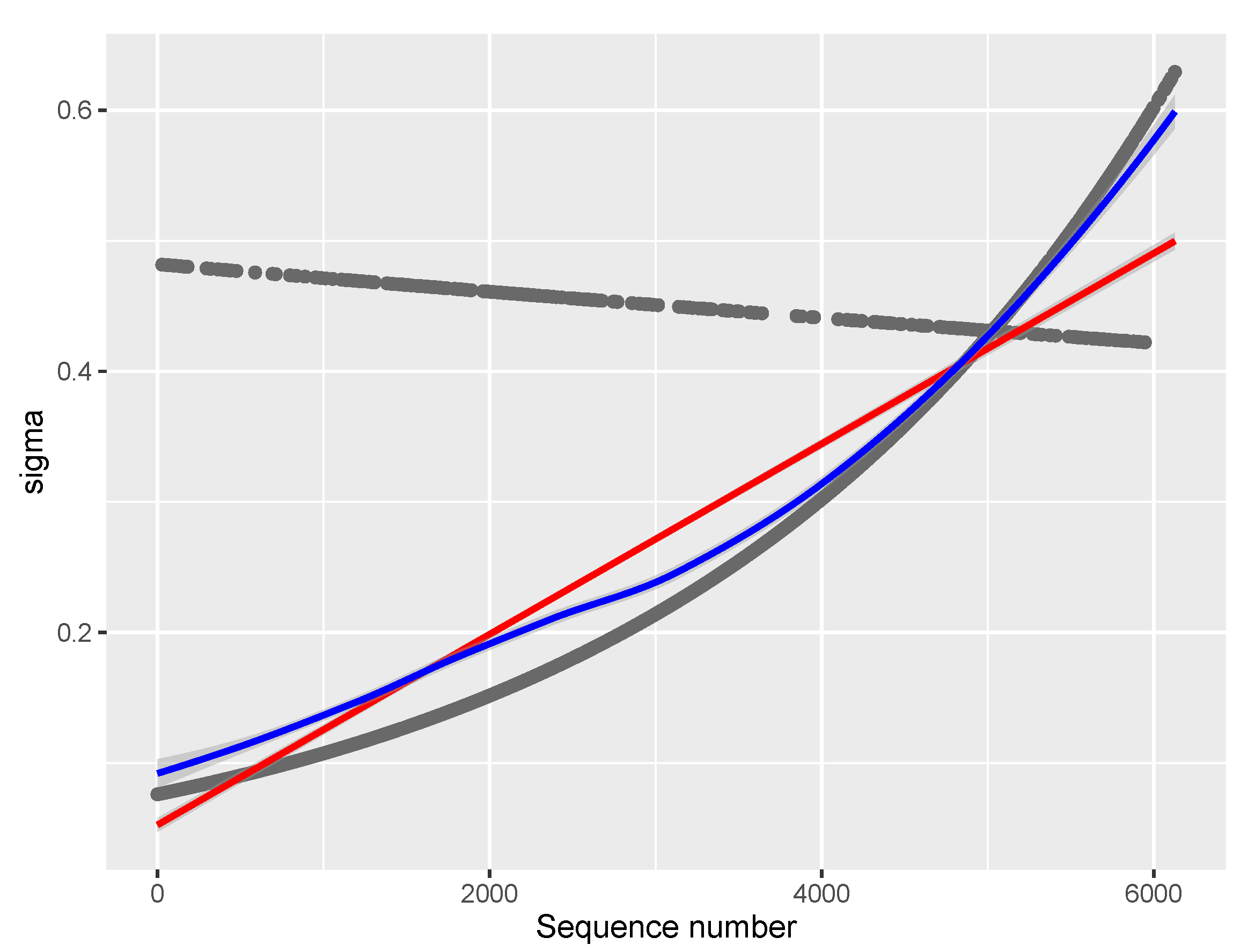
Figure 7.
Models of the parameter for the other drugs data. a, model with two covariates: Age and Sequence_number (29.4.1); b, multiple regression model (29.4) with covariate Age and factor Seriousness; c, raw data (observed AER counts). Red line: linear least squares regression; blue line: loess (a non-linear regression smoother) applied on raw or modeled data at the moment of plotting.
Figure 7.
Models of the parameter for the other drugs data. a, model with two covariates: Age and Sequence_number (29.4.1); b, multiple regression model (29.4) with covariate Age and factor Seriousness; c, raw data (observed AER counts). Red line: linear least squares regression; blue line: loess (a non-linear regression smoother) applied on raw or modeled data at the moment of plotting.
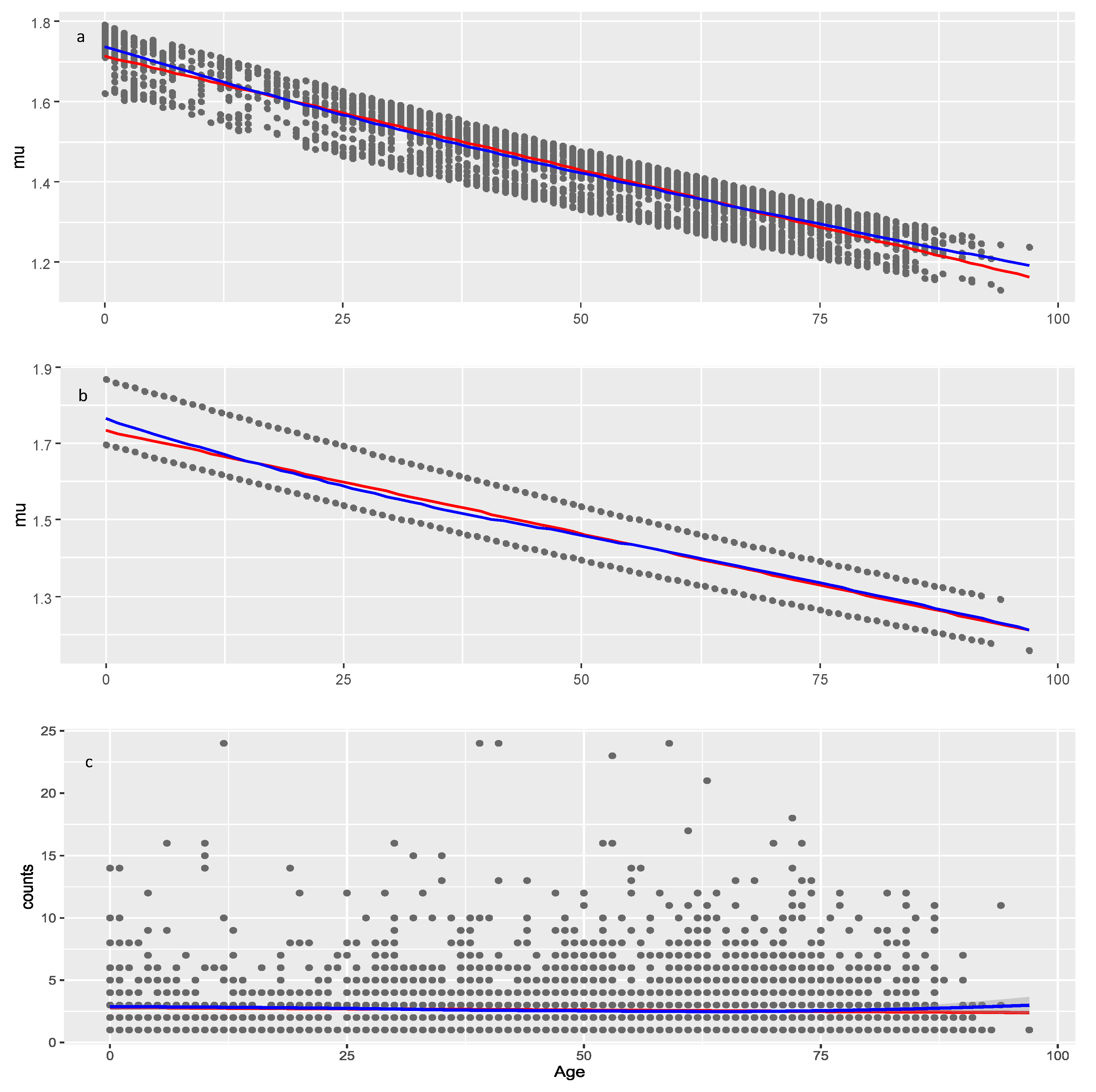

Table 1.
Distributions with the smallest information criteria
| COVID vaccines | |||||
|---|---|---|---|---|---|
| Information criteria | ZANBI * | ZAPIG | DEL | SI | SICHEL |
| Akaike ( AIC) | 17235.9 | 17246.74 | 17635.49 | 17658.71 | 17658.71 |
| Bayes ( BIC) | 17254.8 | 17265.58 | 17657.04 | 17677.55 | 17685.83 |
| other drugs | |||||
| Information criteria | ZAPIG | ZANBI | LG | ZALG | ZIPF |
| Akaike ( AIC) | 22877.62 | 22909.63 | 23127.03 | 23129.03 | 24771.36 |
| Bayes ( BIC) | 22898.13 | 22930.14 | 23133.86 | 23142.70 | 24778.19 |
* Abbreviations are the following distributions [49]: ZANBI – zero-adjusted negative binomial distribution type I; ZAPIG – zero-adjusted Poisson-inverse Gaussian distribution; DEL – Delaporte distribution; SI – Sichel distribution; SICHEL – Sichel distribution with as mean; LG – logarithmic distribution; ZALG – zero-adjusted logarithmic distribution; ZIPF – Zipf distribution
Table 2.
Parameters and moments from the parametric models
| Parameter/moment | COVID vaccines | Other drugs | ||
|---|---|---|---|---|
| ZANBI | ZAPIG | ZANBI | ZAPIG | |
| 3.61265 | 3.68168 | 1.15929 | 1.58990 | |
| 0.29823 | 0.27991 | 1.76746 | 1.07132 | |
| 1.57e-10 | 3.87e-09 | 1.60e-10 | 1.60e-10 | |
| c coefficient | 1.09429 | 1.00000 | 2.13765 | 1.05786 |
| mean | 3.95328 | 3.68168 | 2.47816 | 1.68190 |
| variance | 6.86600 | 7.47576 | 4.28757 | 4.39195 |
| skewness | 1.29504 | 1.31084 | 2.36928 | 2.60040 |
| excess kurtosis | 2.32031 | 2.88184 | 8.42981 | 11.69541 |
Table 3.
Moments and goodness of fit for the diagnostic residuals distributions
| Moment | COVID vaccines | Other drugs | ||
|---|---|---|---|---|
| ZANBI | ZAPIG | ZANBI | ZAPIG | |
| mean | – 0.00313 | 0.00145 | 0.00214 | 0.00489 |
| variance | 1.01015 | 1.01015 | 0.98431 | 0.99056 |
| skewness | – 0.02186 | – 0.03680 | 0.07862 | 0.02204 |
| kurtosis | 3.10751 | 2.92447 | 3.28073 | 3.13219 |
| Filliben coefficient | 0.9989902 | 0.9989915 | 0.9990338 | 0.9996147 |
Table 4.
Steps in the selection process of GAMLSS models with explanatory variables
| Model * | Terms ** | BIC | Distribution | Parameter | Converge |
|---|---|---|---|---|---|
| COVID vaccines | |||||
| Model 1 | 1 | 14651.28 | ZANBI | Yes | |
| Model 2 | 1 | 14659.57 | ZAPIG | Yes | |
| Model 3 | 2 | 14642.24 | ZANBI | Yes | |
| Model 4 | 2 | 14642.67 | ZAPIG | Yes | |
| Model 5 | 1, 2 | 14644.42 | ZANBI | Yes | |
| Model 6 | 1, 2 | 14644.59 | ZAPIG | Yes | |
| Model 7 | 1, 2, 1×2 | 14652.17 | ZANBI | Yes | |
| Model 8 | 1, 2, 1×2 | 14650.66 | ZAPIG | Yes | |
| Model 9 | 3 | 14611.18 | ZANBI | Yes | |
| Model 10 | 3 | 14647.72 | ZAPIG | Yes | |
| Model 11 | 2, 3 | 14602.40 | ZANBI | Yes | |
| Model 12 | 2, 3 | 14682.75 | ZAPIG | No | |
| Model 13 | 2, 3, 2×3 | 14609.92 | ZANBI | Yes | |
| Model 14 | 2, 3, 4 | 14583.51 | ZANBI | Yes | |
| Model 15 | 2, 3, 4, 5 | 14553.20 | ZANBI | Yes | |
| Model 16 | 2, 3, 4, 5, 2×4 | 14560.68 | ZANBI | Yes | |
| Model 17 | 2, 3, 4, 5, 4×5 | 14576.83 | ZANBI | Yes | |
| Model 18 | pb(2), 3, 4, 5 | 14556.41 | ZANBI | Yes | |
| Model 19 | cs(2,df=2), 3, 4, 5 | 14557.85 | ZANBI | Yes | |
| Model 15.1 | 1 | 14509.69 | ZANBI | Yes | |
| Model 15.2 | 1, 2 | 14510.32 | ZANBI | Yes | |
| Model 15.3 | 1, 3 | 14516.84 | ZANBI | Yes | |
| Model 15.4 | 1, 4 | 14504.85 | ZANBI | Yes | |
| Model 15.5 | 1, 4, 5 | 14508.38 | ZANBI | Yes | |
| Model 15.6 | 1+4+1×4 | 14500.97 | ZANBI | Yes | |
| Other drugs | |||||
| Model 20 | 1 | 18036.65 | ZANBI | No | |
| Model 21 | 1 | 19610.37 | ZAPIG | No | |
| Model 22 | 2 | 18032.81 | ZANBI | No | |
| Model 23 | 2 | 18114.44 | ZAPIG | Yes | |
| Model 24 | 3 | 18039.84 | ZANBI | No | |
| Model 25 | 3 | 18042.80 | ZAPIG | No | |
| Model 26 | 4 | 17899.01 | ZANBI | No | |
| Model 27 | 4 | 20016.47 | ZAPIG | No | |
| Model 28 | 1, 2 | 18036.91 | ZANBI | No | |
| Model 29 | 2, 4 | 17885.05 | ZANBI | Yes | |
| Model 30 | 2, 3, 4 | 17893.51 | ZANBI | Yes | |
| Model 31 | 1, 2, 4 | 17890.33 | ZANBI | Yes | |
| Model 32 | pb(2), 4 | 17894.02 | ZANBI | Yes | |
| Model 33 | cs(2,df=2), 4 | 17893.08 | ZANBI | No | |
| Model 29.1 | 1 | 17888.98 | ZANBI | Yes | |
| Model 29.2 | 2 | 17882.25 | ZANBI | No | |
| Model 29.3 | 3 | 17891.85 | ZANBI | Yes | |
| Model 29.4 | 4 | 17861.46 | ZANBI | Yes | |
| Model 29.5 | 2, 4 | 17865.46 | ZANBI | No | |
| Model 29.4.1 | 1, 2 | 17859.86 | ZANBI | Yes | |
* Models for are designated with consecutive numbers. The best model (with lowest BIC) is chosen to model (designated by versioning); ** Explanatory variables are designated by their numbers in the above list; the multiplication sign × designates interaction
terms; pb() is P-splines smoother, cs() is cubic spline smoother, df is degrees of freedom for the cs() smoother.
Table 5.
Moments and goodness of fit for the diagnostics of the best full models
| Moment | COVID vaccines | Other drugs |
|---|---|---|
| (Model 15.4) | (Model 29.4.1) | |
| mean | – 0.00574 | 0.00187 |
| variance | 1.01269 | 1.01149 |
| skewness | – 0.00122 | – 0.01752 |
| kurtosis | 3.16622 | 3.15641 |
| Filliben coefficient | 0.9990017 | 0.9995885 |
Table 6.
Statistics of the best full models for COVID vaccines and other drugs
| Estimate | Std. Error | t value | p value | Significance | |
|---|---|---|---|---|---|
| Model 15.6 (COVID vaccines) | |||||
| parameter | |||||
| Intercept | 1.68278 | 0.04475 | 37.608 | <2e–16 | *** |
| Age | – 0.00487 | 0.00093 | – 5.243 | 1.68e–07 | *** |
| Sex: male | – 0.20626 | 0.02737 | – 7.535 | 6.28e–14 | *** |
| Severity: Severe | 0.18047 | 0.05422 | 3.329 | 0.000883 | *** |
| Vaccine: Jcovden | – 0.07376 | 0.07277 | – 1.014 | 0.310837 | |
| Vaccine: Spikevax | – 0.06029 | 0.04124 | – 1.462 | 0.143916 | |
| Vaccine: Comirnaty | – 0.24956 | 0.02962 | – 8.426 | <2e–16 | *** |
| parameter | |||||
| Intercept | – 2.575e+00 | 1.021e–01 | – 25.214 | <2e–16 | *** |
| Sequence_number | 3.447e–04 | 5.753e–05 | 5.992 | 2.29e–09 | *** |
| Seriousness: Serious | 1.846e–00 | 2.276e–01 | 8.109 | 7.11e–16 | *** |
| Sequence_number×Serious | – 3.670e–04 | 1.487e–04 | – 2.468 | 0.0136 | * |
| Model 29.4.1 (other drugs) | |||||
| parameter | |||||
| Intercept | 6.936e–01 | 1.046e–01 | 6.630 | 3.71e–11 | *** |
| Age | – 3.806e–03 | 8.279e–04 | – 4.598 | 4.38e–06 | *** |
| Sequence_number | – 9.303e–06 | 5.834e–06 | – 1.595 | 0.111 | |
| parameter | |||||
| Intercept | – 0.39184 | 0.06498 | – 6.03 | 1.75e–09 | *** |
| Severity: Severe | 1.15604 | 0.08258 | 14.00 | <2e–16 | *** |
Table 7.
Parametric statistics of the factors that significantly affect
| Levels | Raw data | Modeled values | N | ||
|---|---|---|---|---|---|
| Mean | Std. deviation | Mean | Std. deviation | ||
| Vaxzevria | 4.25460 | 2.66433 | 4.08621 | 0.58845 | 1740 |
| Jcovden | 4.37323 | 2.76218 | 4.01120 | 0.57521 | 142 |
| Spikevax | 4.31649 | 2.64401 | 3.98038 | 0.59216 | 376 |
| Comirnaty | 3.65524 | 2.65239 | 3.29001 | 0.59439 | 1050 |
| Female | 4.29031 | 2.65346 | 4.03942 | 0.59382 | 2208 |
| Male | 3.64727 | 2.65225 | 3.37426 | 0.59445 | 1100 |
| Non-severe | 4.00973 | 2.65287 | 3.76593 | 0.59432 | 2979 |
| Severe | 4.68085 | 2.64759 | 4.29183 | 0.59326 | 1540 |
| Total | 4.07648 | 2.65287 | 3.81823 | 0.59432 | 3308 |
Table 8.
Factor level combinations for the lines in the parameter in Figure 5 for the non-linear model of COVID vaccine data
Table 8.
Factor level combinations for the lines in the parameter in Figure 5 for the non-linear model of COVID vaccine data
| Line No | Age | Vaccine | Sex | Seriousness | Repeats | |
|---|---|---|---|---|---|---|
| 1 | 50 | Vaxzevria | female | S | 5.37038 | 2 |
| 2 | 49 | Jcovden | female | S | 5.08064 | 1 |
| 3 | 62 | Spikevax | female | S | 4.55633 | 1 |
| 4 | 48 | Vaxzevria | male | S | 4.46271 | 3 |
| 5 | 50 | Vaxzevria | female | NS | 4.34896 | 19 |
| 6 | 32 | Jcovden | male | S | 4.32715 | 1 |
| 7 | 47 | Spikevax | male | S | 4.21830 | 2 |
| 8 | 50 | Comirnaty | female | S | 4.18823 | 1 |
| 9 | 50 | Jcovden | female | NS | 4.09347 | 1 |
| 10 | 50 | Spikevax | female | NS | 4.08612 | 3 |
| 11 | 50 | Vaxzevria | male | NS | 3.56603 | 10 |
| 12 | 51 | Comirnaty | male | S | 3.40940 | 1 |
| 13 | 50 | Comirnaty | female | NS | 3.39165 | 13 |
| 14 | 50 | Jcovden | male | NS | 3.35653 | 1 |
| 15 | 50 | Spikevax | male | NS | 3.35050 | 1 |
| 16 | 50 | Comirnaty | male | NS | 2.78106 | 4 |
Table 9.
Overdispersion in the parametric and full models with different distributions
| Model | Data | Distribution | Pearson disp. statistic | p-value |
|---|---|---|---|---|
| Parametric | COVID vaccines | Poisson | 1.512 | 0.0000 |
| Parametric | other drugs | Poisson | 1.368 | 0.0000 |
| Parametric | COVID vaccines | ZAP | 1.704 | 0.0000 |
| Parametric | other drugs | ZAP | 1.852 | 0.0000 |
| Parametric | COVID vaccines | NBI | 0.937 | 0.9955 |
| Parametric | other drugs | NBI | 0.842 | 1.0000 |
| Parametric | COVID vaccines | PIG | 0.920 | 0.9996 |
| Parametric | other drugs | PIG | 0.766 | 1.0000 |
| Parametric | COVID vaccines | ZANBI | 1.022 | 0.1803 |
| Parametric | other drugs | ZANBI | 0.997 | 0.5606 |
| Parametric | COVID vaccines | ZAPIG | 1.063 | 0.0062 |
| Parametric | other drugs | ZAPIG | 0.997 | 0.5529 |
| Model 15.6 | COVID vaccines | Poisson | 1.437 | 0.0000 |
| Model 29.4.1 | other drugs | Poisson | 1.360 | 0.0000 |
| Model 15.6 | COVID vaccines | ZAP | 1.641 | 0.0000 |
| Model 29.4.1 | other drugs | ZAP | 1.843 | 0.0000 |
| Model 15.6 | COVID vaccines | NBI | 0.937 | 0.9954 |
| Model 29.4.1 | other drugs | NBI | 0.862 | 1.0000 |
| Model 15.6 | COVID vaccines | PIG | 0.914 | 0.9998 |
| Model 29.4.1 | other drugs | PIG | 0.818 | 1.0000 |
| Model 15.6 | COVID vaccines | ZANBI | 1.019 | 0.2243 |
| Model 29.4.1 | other drugs | ZANBI | 1.001 | 0.4706 |
| Model 15.6 | COVID vaccines | ZAPIG * | 1.961 | 0.0000 |
| Model 29.4.1 | other drugs | ZAPIG * | 4.154 | 0.0000 |
* Did not converge
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



