
Submitted:
11 September 2023
Posted:
13 September 2023
You are already at the latest version
Abstract
Staphylococcus aureus is a common pathogen in human. Methicillin resistant Staphylococcus aureus (MRSA) infection poses a big and perplexing difficulty in terms of therapy. The acquisition of the non-native gene PBP2a, which has a decreased tolerance for β-lactam antibiotics, frequently confers resistance. PBP2a has a less attraction for methicillin and it helps bacteria to continue peptidoglycan biosynthesis, and it is cell wall’s core component in bacteria. So, even in the presence of methicillin or any other antibiotic, bacteria develop resistance. Due to resistance-causing genes, S. aureus becomes MRSA. The main premise of the resistance mechanism is well understood. The current demand for novel antibiotics is legitimate in the face of therapeutic concerns posed by resistant micro-organisms. The emphasis of this review is on PBP2a scaffolds and the different screening approaches used to find PBP2a inhibitors. Penicillin, Cephalosporins, Pyrazole-Benzimidazole based derivatives, Oxadiazole containing derivatives, non-β-lactam allosteric inhibitors, 4-(3H)-Quinazolinones, Pyrrolylated chalcone, Bis-2-Oxoazetidinyl macrocycle (β-lactam antibiotics with 1, 3-Bridges), Macrocycle-embedded β-lactams as novel inhibitors, Pyridine-Coupled Pyrimidinones, novel Naphthalimide corbelled aminothiazoximes, non-covalent inhibitors, Investigational-β-lactam antibiotics Carbapenem, novel Benzoxazole derivatives, Pyrazolylpyridine analogues, and other miscellaneous classes of scaffolds for PBP2a are also represented as well as with their biological activity is discussed. The penicillin-binding protein is also discussed, which is the crucial target for the cell wall of MRSA. Various aspects of PBP2a, the cell wall of bacteria, peptidoglycans, different crystal structures of PBP2a, synthetic routes for PBP2a inhibitors, and future perspectives of MRSA inhibitors are also enumerated.
Keywords:
Penicillin-binding protein 2a (PBP2a)
; Mur enzymes
; heterocycles
; High-throughput screenings
; transpeptidation
1. Introduction:
Methicillin resistant Staphylococcus aureus (MRSA) is called a Superbug1, when we see the word MRSA, we immediately think of S.aureus. For the better understanding of MRSA, we need to know about S. aureus and its disease-causing capacity as well as its resistance mechanism. MRSA is produced when S. aureus that is methicillin-susceptible S. aureus (MSSA) produces the mecA gene, which is a mutated one. This means methicillin-resistant depends on the production of the mecA gene2. S. aureus is a common human pathogen as well as commensal bacterium. This causes a variety of clinical infections, such as bacteraemia, some device-related infections (implantable cardiac device infections, intravascular catheter infections, and other prostheses), pleuropulmonary infections, osteoarticular infections (osteomyelitis, native joint septic and prosthetic joints), skin and soft tissue infections, and infective endocarditis. However, it rarely causes urinary tract infections, which have been reported3. Osteomyelitis, Ritter's disease, endocarditis, and bacteraemia in the developed world4 also septicaemias are caused by S. aureus5. Although, commonly, S. aureus is responsible for infections in the community and amongst healthcare professionals. The infection is of different types, from superficial skin and soft tissue infections to invasive infections. It can also lead to sepsis, and death. This pathogen can adapt to many environmental situations. Therefore, S. aureus is likely the pathogen of greatest concern6,7. Due to the wide variety of infection causing capacities of S.aureus,it is known as a “superbug”8. S. aureus (MRSA) appears to be the most commonly diagnosed bacteria in many parts of the world. S. aureus pathogen is causing resistance to all β-lactam antibiotics andvarious other classes of antibiotics that could inhibit staphylococcus aureus9. Various antibiotics are used to treat such infections such as β-lactam antibiotics. The capacity of β-lactam antibiotics to access and enact penicillin-binding proteins is crucial due to their efficiency against bacteria10. In the family Staphylococcus, there are now 29 species and 8 subspecies that have been identified. Due to the variety of species, understanding and finding the reason for resistance is quite a difficult task11. Millions of deaths occur each year because of antibiotics causing resistance, and this resistance then causes infection12.S. aureus is the cause of Nosocomial and community-acquired bacterial infections all over the world13. To combat penicillin-resistant Staphylococcus aureus14, a penicillinase-resistant penicillin called methicillin was introduced in the year 1959. However, within a year, the late Professor Patricia Jevons reported the first methicillin-resistant human S. aureus strains in a hospital in UK15. During the early 1960s, the introduction of semisynthetic β-lactam antibiotics like methicillin and oxacillin caused a general frequency decrease of multidrug-resistant bacteria16. As antibiotics develop resistance to different antibiotics, it results in various infection formations such as VRSA, MSSA, and MRSA. MRSA has been found to be the most common gram-positive pathogen in nosocomial infections. According to (NNIS) National Nosocomial Infections Surveillance, it is a type of infection that occurs mainly in the intensive care unit17. The nosocomial wound infection occurs due to S. aureus and all over the world, it is responsible for hospital infection18. Three different discoveries in the 1950s, during the century when methicillin-resistant S. aureus (MRSA) had been documented, led to an acknowledgment of the potential public-health consequences that S aureus could have19.The first major nosocomial epidemic of MRSA was described in 1963. In the 1970s, the USA reported a nosocomial infection caused by MRSA. Themethicillin resistances are of two types: homogeneous resistance and heterogeneous resistance. Heterogeneous strains may be made up of two types of populations of relatively susceptible and highly resistant cells. Unlike homogeneous strains, which contain a single cell population, all of these strains tend to be highly resistant to S. aureus.Acquisition of multiple nosocomial infections due to some strains may even produce toxins that may trigger primary and secondary bacteraemia and various infections20. When methicillin-resistant S. aureus was first described to the medical community, it was a major publication, published in a British medical magazine fifty years ago21. Outside of the therapeutic setting, MRSA has become a common pathogen. The starting cases of MRSA were first reported in the community in the late 1980s; in those patients who had not been exposed to hospitals or nursing homes22.
1.2. History and Emergence of MRSA:
MRSA is a multi-drug resistant bacteria that causes serious infections in communities and hospitalized patients worldwide, as per report23. Methicillin-resistant Staphylococcus aureus (MRSA) is the cause for outbreaks of nosocomial infections and community-associated infections that have been reported worldwide24. Accordingly, the multilocus sequencing (MLST) identified five different clonal complexes, namely they are, CC45, CC30, CC22, CC8, and CC5, and these six strain types constituted 88.2% of all infections of MRSA25,26. The first MRSA strains resistant to various antibiotics emerged in the second half of the 1960s, and a multidrug-resistant MRSA has been identified across several centrally located European nations27, including England, Australia, and India. MRSA strains with simultaneous susceptibility topenicillin, streptomycin, and tetracycline, but rarely erythromycin, were accounted as 15% of all S. aureus strains reported in Denmark in 197128. Infections acquired in intensive care unit accounts for nearly 60% of nosocomial S. aureus infections in the USA29. Many MRSA strains have been accounted for many nosocomial S. aureus surgical site and bloodstream infections that have spread in the USA, UK, Japan, Finland, Turkey, Netherlands, and so many countries30.These are especially the Americas, Europe, the Middle East, North Africa and East Asia, where MRSA is a genotype which has become a serious pathogen in nosocomial and community-based settings over the last decade31. MRSA is considered a contagion of multi-resistant nosocomial infections including bloodstream infection and nosocomial pneumonia, and S. aureus is a gram-positive bacterium that is the cause of skin and respiratory infections32. In India, infections due to S. aureus are rising significantly, putting a growing strain on healthcare resources. MRSA infections in hospitals are especially widespread33. Patients in ICUs, particularly surgical wounds, drains, and intrusive surveillance systems, that cause skin breaches and increase the risk of infection. Methicillin-resistant S. aureus has been discovered in 13–47% of India. Because of growing resistance to many kinds of antibiotics, the therapeutic options for combating MRSA are dwindling34. A teaching hospital, which was located in North-West India, reported infection due to MRSA35. Methicillin-resistant S. aureus is among the most prevalent bacteria that causes complicated skin and skin structure infections (cSSSIs) and other hospital-acquired illnesses, such as bloodstream infections (BSIs), including ventilator-associated pneumonia (VAP)36.According to the data from the Japanese Ministry of Agriculture, Forestry, and Fisheries animal quarantine services, MRSA ST398 has been transmitted through the swine production system. To see how common MRSA is in Japanese pigs,some surveys were conducted in Asian countries like China, Malaysia, and Singapore to check the prevalence of MRSA. It seems that isolated patients remain resistant to antibiotics. Despite using an enrichment culture method with a 7.5 percent NaCl broth medium in one study, the isolation rate of MRSA from pigs in Japan (0.9 percent) appears to be lower than in Europe and North America37 (for more refer to http://www.maff.go.jp/aqs/tokei/toukeinen.html). In the special care baby unit (2011), MRSA outbreak was noted in the United Kingdom. This incident occurred at Cambridge University Hospital National Health Service Foundation Trust (CUH)38. In the past decade, a considerable increase in MRSA transmission and disease within healthy subjects has been documented in Taiwan. In up to 9.5 percent of normal Taiwanese children's nasal passages, MRSA infections accounted for more than half of all paediatric CA pathogens. The total population was also impacted, but to a lesser extent39. In the United States alone and other countries like Japan, the US, and many more, MRSA caused more than 80,000 invasive infections in 2011. MRSA brings the total to 53 million people globally infected by the infection40. The nosocomial infection spreads in various countries; namely the Netherlands and the whole Scandinavian area, Greece, France, Europe, Italy, Spain, and Germany41. MRSA prevalence was observed in various other countries like Australia (1965), the United States (1968), the UK (1970’s), Japan (2003) and China (2011). In these countries, outbreaks of MRSA were observed and a report was made42. Because of multi-resistant to many antibiotics, sepsis-induced MRSA infection had a worse outcome, leading to death from septic shock43. In the United Kingdom (England and Wales), it is critical to focus on actions to prevent healthcare-associated infections. Such actions include better and easier selection of antibiotic, infected patients isolation, using gloves to handle them, enhanced handwashing hygiene,etc. Those efforts lowered MRSA infections from April 2010 to March 2011 throughout the National Health Service, causing a 50% reduction from the 2008 and 2009 reported cases. In 2012, 12 patients in the Neurosurgery Department at Jiangsu University, affiliated with the hospital, faced a suspected MRSA epidemic44. It's still up for debate if MRSA is more virulent than MSSA45. According to this research and reported data, MRSA has also surpassed MSSA as the most common S aureus in community-acquired illnesses. Incision and drainage with antibiotic treatment were used in 60% of cases, while incision and evacuation alone were used in 19%. In 64 percent of cases where antibiotic therapy was administered, a beta-lactam antibiotic was employed, including 57 percent of MRSA patients having treated with antibiotics. It resolved 96.1% of the cases, with no link with antimicrobial medication. The need for drainage of a cutaneous abscess is a universally recognized problem. But the reasons for using antibiotics to support drainage are still unclear46. In many aspects, CA-MRSA infections differ from HA-MRSA infections. The epidemiologic differences between different strains of the same clonal CA-MRSA complex are also unclear. The first case of CA-MRSA was reported in 1993, in Western Australia, and infection with MRSA was initially identified in Australia in 1965, and the first case of hospital MRSA was reported in Boston in the year 196842.
2. PBPs, Types of PBP, its location and contribution in transpeptidation reaction:
The cell wall-producing enzymes with penicillin sensitivity are focused on the β-lactam antibiotics. Their propensity to bind radio-labelled penicillin makes them easily detectable. Penicillin-binding proteins are named after this (for domains of PBPs refer Figure 10)47. PBPs are serine acyltransferases that, along with facilitating the production of cross-linked peptidoglycan, are also target for β-lactam antibiotics due to their transpeptidase-related catalytic activity. Because β-lactam antibiotics become connected covalently with the active site serine after adhering to the PBP catalytic cleavage, the resultant acyl-enzyme is only hydrolysed slowly, decreasing the quantity of peptidoglycan cross-linking48. β-lactam antibiotics and penicillin-binding proteins (PBPs) as their primary targets and are present in the periplasmic membrane of the bacterial cell wall49. PBPs are membrane-bound transpeptidase50. Allowing the medicine to reach the active site without having to pass through the cytoplasmic membrane51. The penicillin-binding proteins (PBPs) are historically divided into molecular mass or molecular weight penicillin-binding proteins (PBPs). High molecular weight penicillin-binding proteins (HMW-PBPs) are divided into two categories. One is class A, and the other one is class B. Low-molecular-weight PBPs (LMW-PBPs) are also divided into four subclasses, and they have a tertiary structure. Penicillin-binding proteins with high molecular weight are required for mobile survival, which is the actual target of β-lactam antibiotics. When the Class-A penicillin-binding proteins (PBPs) catalyse the biosynthesis of a specific linkage of monosaccharides called the O-glycosidic linkage of monosaccharides, the reaction we also called trans-glycosylation. Each category of penicillin-binding proteins, namely category A and category B of penicillin-binding proteins (PBPs), causes the cross-coupling reaction to catalyse during the peptidoglycan biosynthesis52. PBPs have transpeptidases or carboxypeptidases in their penicillin-binding domains, which are involved in peptidoglycan metabolism. Those penicillin-binding proteins, which have a low molecular weight, are not necessary for laboratory stipulations, so it signifies very few antibiotic goals that consist of β-lactam antibiotics53. The penicillin-binding protein contains a donor strand. The D-Ala-D-Ala residues of the 'Donor' peptidoglycan strand are hydrolysed at the ends of amino acid chains as an acyl intermediate is generated between the carbonyl of the penultimate D-Ala and the active site serine of the penicillin-binding proteins. The 'Donor' peptidoglycan binds to PBP so that the D-Ala-D-Ala moiety is vulnerable to assault by the PBP active site's nucleophilic serine hydroxyl group54. All bacteria contain PBPs. However, the size, number, quantity, and affinity for β-lactam antibiotics differ from one species to the next55. When we are talking about Staphylococcus aureus, we are talking about gram-positive bacteria. According to scientists, the basis of differentiation between gram-positive and gram-negative cells is the cell's core component. Peptidoglycan is a significant factor in determining whether bacteria are gram-positive or gram-negative. When we talk about core components, we know that peptidoglycan is the core and central part of the bacterial cell wall. When we think about the mechanochemical properties of the bacterial cells and the pressure potentials, the highly complex multicomponent structure in the cell wall is known as peptidoglycan. It is required for movement (mechanical) strength, which counteracts the intracellular pressure of bacterial cells. As the peptidoglycan is a core component of the cell wall, it is responsible for forming a continuous, mesh-like structure around the bacterial cell membrane, which protects cells from destruction. Thus, peptidoglycan plays a vital role in the cell wall’s biosynthesis ofS. aureus bacteria and many other gram-positive bacterial cell walls. Peptidoglycan is also called murein, or glycopeptide, or mucopeptide. Peptidoglycan is a polymer made up of long glycan chains connected by a flexible peptide bridge56. A cell wall is present around the cytoplasmic membrane to maintain the pressure inside the bacterial cell and prevent leakage of essential components from being leaked through a semipermeable membrane.Protection of the cellular content from the external environment is due to the pressure inside the cells, and thus it is helpful to balance the shape of the bacterial cell. The bacterial cell layer gives the desired auxiliary astuteness for bacteria or microbes, which then helps to resist the osmotic pressure differences between the cell divider and the exterior of the cell50. We conclude that the bacterial cell wall is a crucial component for differentiating distinct strains of bacteria. It is reported in various papers that gram-positive bacteria contain folds of the cell wall; approximately 20-fold thicker cell walls of about 15-80 nm. The cell wall is responsible for maintaining bacterial strength by simply managing microbes' shape, size, and integrity. The core component of the bacterium is peptidoglycan, which is a macromolecule composed of O-glycosidic linkages of main monosaccharides (glycan strands). This is then cross-linked through movable and species-specific peptide bridges required to produce a mesh-like structure surrounding the bacterial cell. Glycan strands are repeating units of N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) amino sugars are then attached with β-(1,4) glycosidic bonds in an alternating manner. Each N-acetylmuramic acid (MurNAc) residue is joined to the short amino acid chain; D-glutamine, L-alanine,D-alanine and L-lysine which are in contact with pentaglycine bridges, and these cause cross-bridge formation. MurNAc and GlcNAc pentapeptides are used for peptidoglycan biosynthesis57. To give better mechanical strength and flexibility throughout entire cell cycles of bacterial life cycles, bacterial growth occurs when bacteria get what they need for development. Crosslinking and glycan polymerization are two reactions. They are two enzymatic activities. Both of them are required for cell wall biosynthesis. Transpeptidase enzymes subsequently cross-link the polymerized glycans by forming amide bonds between bound peptides58. β-lactam antibiotics act like suicide inhibitors and they bind to PBPs. Their mechanism is explained as; the active site serine cleaves the β-lactam ring's carbonyl, causing the ring to open, which results in the formation of covalent acyl-enzyme complex. This product is eventually hydrolyzed, thereby inhibiting additional reactions59. The response in which core components are formed leads to the generation of a backbone called stem peptides. In this phase, amino acids are cross-coupled and create peptidoglycan, and this phase occurs in the outer cytoplasmic membrane; therefore, we call it a transpeptidation reaction60. Below is the scheme for penicillin-binding protein.
Figure 1.
Penicillin-binding proteins are susceptible to β-lactams61.
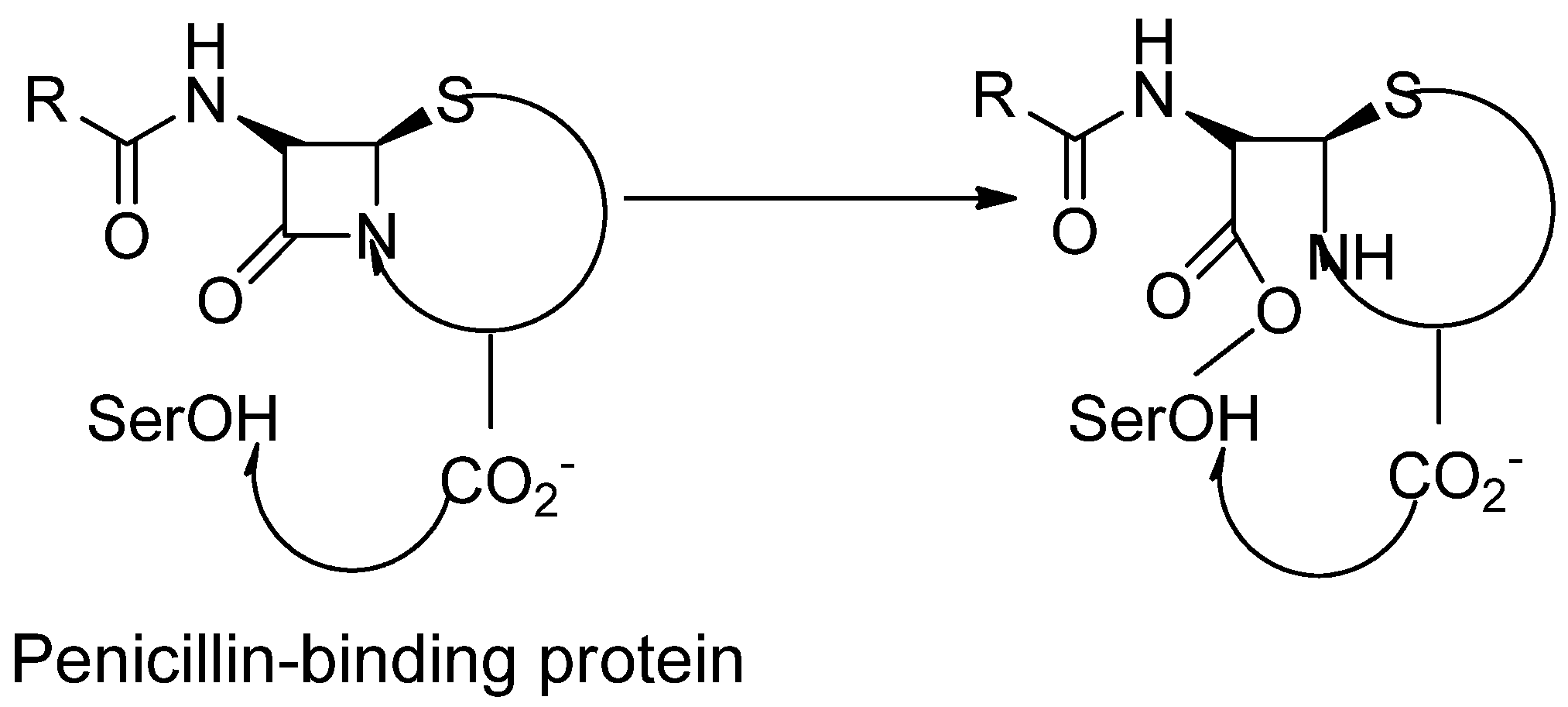
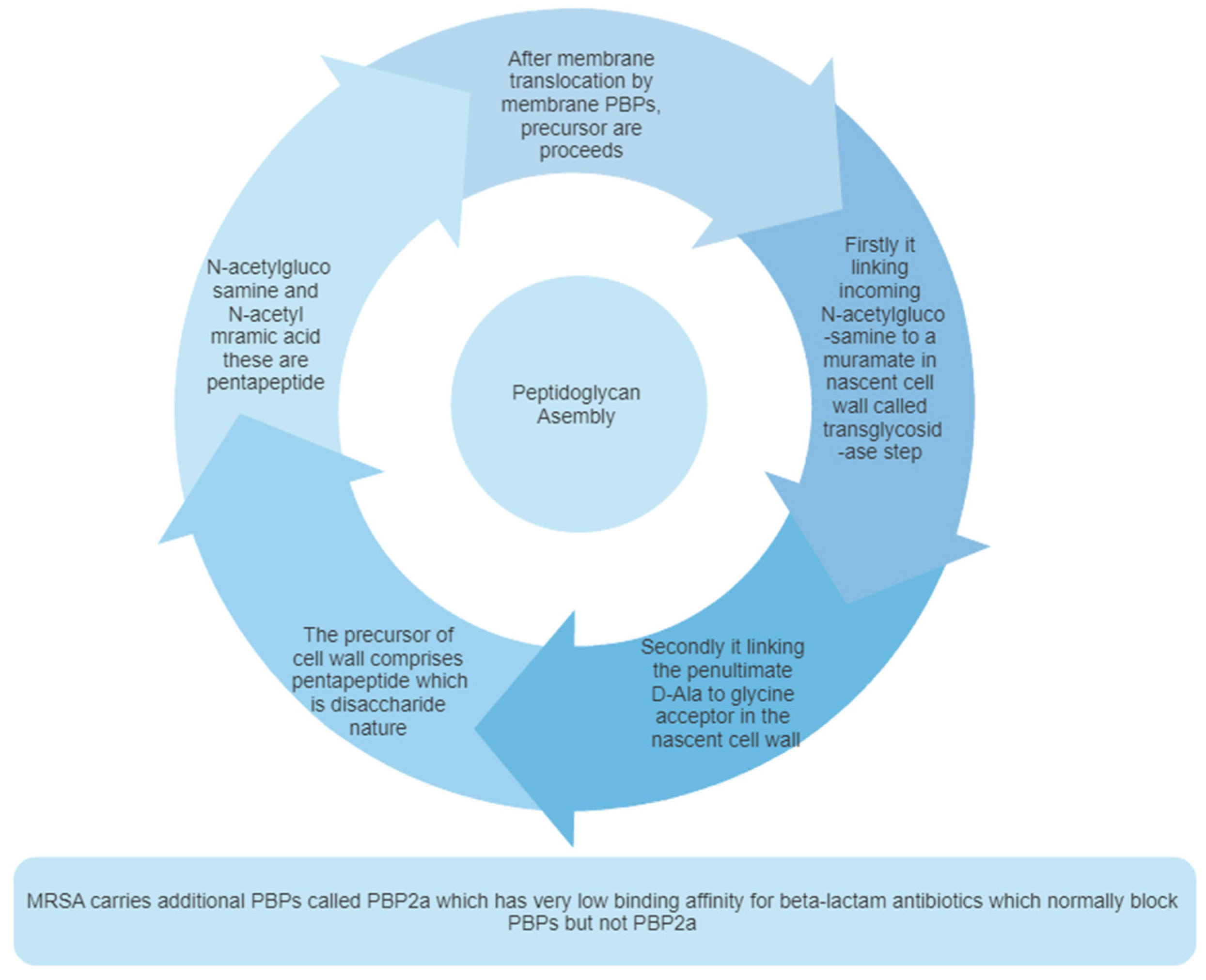
Figure 2.
Cell Wall assembly.
2.1. PBP2a:
The penicillin-binding proteins (PBP2) of S. aureus is an enzyme that is involved in the final step of peptidoglycan assembly. It is a crucial factor in the pathogen’s virulence factor. PBP2, a class A enzyme in Staphylococcus aureus, needs its ligand to localize to the septum as well as engage in the division of cell62. Antibiotic use causes a distinct systemic reform within the mecA analog's production, transforming it into an antibiotic resistance determinant. This determinant also allowed S. aureus to express the precursor in its genetic history. S. aureus bacteria bear the S. sciuri mecA homolog and are responsible for the production of PBP2A-like protein, which is produced by species of Staphylococcussciuri. As we know, that mecA is not a native, but a mutated gene in S. aureus, the way of acquisition of the mecA gene mechanism is unkown63. Along with the four PBPs seen in all the staphylococcal strains, all clinical isolates of MRSA have an additional PBP2a. Although the presence of β-lactam antibiotics is necessary for continuous cell wall production as well as bacterial proliferation64. "The genetic architecture of the mec element downstream of the mecA gene in 34 methicillin-resistant S. aureus (MRSA) clinical isolates contains 13 of the most common mecA mutations and reflects the major epidemic clones of MRSA are discussed in the article"65. The mecA gene product PBP2a is generally considered to act as a surrogate enzyme. From the normal complement of S. aureus PBPs, this enzyme takes over the task of cell wall synthesis 66. PBPs are involved in peptidoglycan biosynthesis67. Transpeptidase activity is present in PBP2a. All β-lactam antibiotics have a poor affinity for PBP2a68. PBP2a shows resistance to β-lactam antibiotic changes and remains a capable catalyst for physiological processes69. For β-lactam antibiotics, PBP2 is a target to bind to. PBP2a provides resistance to all β-lactam medicines that are currently available70. During interactions with β-lactam antibiotics, the enzymes experience a significant conformational shift, which could have ramifications for PBP2a’s catalytic activities in bacterial cell wall cross-linking71. PBP2a is a resistance-causing gene, but it is not the sole cause of resistance. It depends on PBP2 to show resistance. MecI and MecR1 proteins control the production of PBP2a. The blaZ system’s signalling proteins play a role here as well. From the study, three main things that are required for the resistance are mentioned below:
1) PBP2a is reliant on PBP2's transglycosylase activity.
2) The length of the stem peptides must be correct and the normal sequence of peptides for PBP2a must be included to function well.
3) The pentaglycine cross-bridge must be present for PBP2a to work72.
2.2. Peptidoglycan biosynthesis:
Bacterial cells are embedded in a macromolecular mesh-like structure known as peptidoglycan, which is required to survive the bacteria73. But the presence of peptidoglycan causes resistance to not only cell division but also osmotic pressure74. Peptidoglycan is composed of 30-70% of the total cell wall. It also consists of teichoic acid and lipoteichoic acid75. The peptidoglycan of bacteria is composed of two polysaccharides pentapeptide units; GlcNAc and MurNAc. Peptidoglycan of bacterial cell wall contains alternating N- acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) and they are cross-linked by this short peptide. The bacterial cell wall stability depends on peptidoglycan's strength and helps keep the bacteria's rod shape. Peptidoglycan is necessary for the development of cells and also viability. Inhibition of the synthesis of PBP2a leads to cell wall lysis and the bacteria’s death. PBPs have a catalytic site on the outside portion of the bacterial cell wall, which allows to access the ligand without crossing the lipid bilayer76.It provides a rigid mechanism for stability due to its highly cross-linked lattice structure. Two sugar units generate the lattice structure, one is N-acetyl glucosamine, and the another one is N-acetyl muramic acid. These sugar units are also called pentapeptides. The glycan chain is a linear strand formed by the alteration of these two amino sugar units, which are responsible for the biosynthesis of highly complex structures77.

Table 1.
In the biosynthesis of the peptidoglycan, Mur enzymes play an essential role. Below is a list of Mur enzymes that are implicated in peptidoglycan production78,79,80.
Table 1.
In the biosynthesis of the peptidoglycan, Mur enzymes play an essential role. Below is a list of Mur enzymes that are implicated in peptidoglycan production78,79,80.
| Sr. no | Name of Mur enzymes | Role of enzyme in the biosynthesis of peptidoglycan |
|---|---|---|
| 1 | MurA | The enzyme MurA is responsible for catalysing the intracellular stage of peptidoglycan biosynthesis, which is the first stage in peptidoglycan biosynthesis. MurA enzyme brings such a reaction simply by transferring enol pyruvate. This enol pyruvate is produced by conversion from phosphoenolpyruvate to UDP-N-acetylglucosamine. |
| 2 | MurB | Reduce UDP-GlcNAc-enol pyruvate to UDP-acetylmuramic acid. |
| 3 | The ATP-dependent enzyme, Mur ligases enzymes, Enzymes from MurC to MurF | These enzymes are responsible for linking five amino acid residues. These enzymes relate to the UDP-MurNAc and thus help to form the UDP-MurNAc-pentapeptide. |
| 4 | MraY | Produce lipid I. |
| 5 | MurG | MurG adds GlcNAc moiety to lipid I resulting in the formation of lipid II. |
| 6 | MurJ | To export lipid II to the outer leaflet, MurJ switches between inward and outward conformations. Lipid II reversal is involved by MurJ81. |
There are three steps in the biosynthesis of the peptidoglycan, which are discussed as follows:
Peptidoglycan Synthesis is done in a Nutshell and the Role of Mur enzymes is very important in peptidoglycan biosynthesis (refer ref. 82 and 83)82, 83. Undecaprenyl N-acetyl muramic acid (UDP-MurNAc) is a pentapeptide, which is then shifted to Undecaprenyl phosphate (UNDP) via the Mur Y enzyme, which works as a membrane-associated carrier. Mur G is a peripherally membrane-associated protein to which GlcNAc is coupled and is responsible for producing Lipid II84.
Step 1: The nucleotide sugar-linked precursor is produced. They are, namely, UDP-N-Acetylmuramyl-pentapeptide and UDP-N-acetylglucosamine pentapeptide. Synthesis of uridyl diphosphate (UDP) acetyl muramyl pentapeptide involves acetyltransferase and uridyl transfer using acetyl Co-enzyme A (AcCoA) and UTP, respectively. From glucosamine-1-phosphate, uridyl diphosphate N-acetylglucosamine is formed where Glmu catalyzes the reaction. Here Glmu is called (N-acetyl-1-phosphate glucosamine uridyltransferase). Peptidoglycan is a highly complex polysaccharide made up of long glycan chains that are then cross-linked by short peptide chains85.
Step 2: It is also known as a lipid-linked step involving the lipid carrier undecaprenyl phosphate. Phosphor-N-acetylmuramyl pentapeptide and N-acetylglucosamine of them are transferred directly to the lipophilic carrier. Undecaprenyl phosphate and various disaccharides (pentapeptide)-pyrophosphate-undecaprenol are then formed. It is a reduction reaction. (Refer Figure 3 B)
Step 3: In this step, polymerization and cross-linking of the cell wall on the cell surface occur, and the complete portion is transferred to the peptidoglycan, which is in the growing stage. Cross-bridge formation occurs simultaneously during this stage and leads to secondary modification of newly-synthesized peptidoglycan86.
2.3. Pentaglycine bridge and transpeptidation reaction in cell wall:
The composition of peptidoglycan may be varied between different species. FemXAB protein family synthesizes cross bridges that contain five glycines. Staphylococcus aureus, a gram-positive pathogen, commands a highly complicated cross-coupled peptidoglycan due to the cross-bridge for transpeptidation (for the transpeptidation refer Figure 4 A). The factor essential for methicillin-resistant is known as the fem factor. The FemA contributes significantly to the second and third glycine, and FemB contributes to the fourth and fifth glycine. The pentaglycine bridge is required to cross-link bacterial peptidoglycans, so by transpeptidation reaction, cross-linking of adjacent strands in the peptidoglycan assembly is carried out as shown in (refer Figure 4 C), forming fibrous-like segments that wrap around the bacterial cell wall. Structural properties of the peptidoglycan give flexibility and robustness to the envelope of the cell, which is essential to oppose or fight pressure, which is not typical for the bacterial cell derived from intracellular tension and its normal state of turbidity87. Bacterial trans-glycosylation is a vital process responsible for polymerizing intermediates into cell wall peptidoglycan88. Membrane-embedded glycosyltransferase enzymes polymerize the glycan strand by using disaccharides to which lipid is linked, and it contains the pentapeptide precursor lipid II. The pentaglycine side-chain is formed by adding glycine residues to the membrane-bound lipid precursor (Lipid II), which is localized in the cytoplasm (see Figure 4 D). The glycine residues are added in a very progressive way. The pentaglycine interpeptide generation is assisted by FemA and FemB factors.
2.4. Mechanism of Methicillin Resistances in S. Aureus:
Information from the Emerging Infection Program (EIP) is obtained from which it is concluded that MRSA is an infectious disease-causing bacterium. The overall population of MRSA-affecting patients is continuously monitored from obtained data (2005-2016) and Premier and Cerner Electronic Health Record Databases (2012-2017), which describes the global trends in community-onset and hospital-onset MRSA and MSSA. According to the study, S. aureus bloodstream infections are a significant source of mortality and morbidity in the United States. S. aureus is a common cause of infections in the community and among healthcare workers which range from superficial skin infections to invasive infections, sepsis, and even death also93.
MRSA infection can be gained either through the horizontal gene transmission of mobile genetic elements (MGEs) or from transposons, which are bacteria genetic mobile elements, and the staphylococcal cassette chromosome element (SCCmec), which are mutated genes that produce encoded mecA genes, or from chromosomal mutations that may alter the binding sites of the drugs on molecular targets; or by increase in the expression of endogenous efflux pumps or by alteration of the enzymatic activities of genes (refer Figure 5)94.The overall effect of which leads to decreased availability of drugs for the target, hence less amount of drug is present for action, which is the main reason for mutation because bacteria then bio-synthesized peptidoglycan as the drug is not available for inhibition. When we talk about efflux proteins, it means that they are responsible for lowering the bioavailability of the drug, which is then responsible for resistance.
MRSA acquires resistances to all β-lactam antibiotics as well as other antibiotics, probably through two different pathways:
- 1)
- By formation of PBPs (PBP2a): It is a transpeptidase that mainly causes inhibition resistance by all β-lactam antibiotics and other antibiotics. It is also assisted in maintaining the cross-linking of peptidoglycan. Hence, it allows various MRSA strains for survival, even in the presence of β-lactam antibiotics and other antibiotics.
- 2)
- Production of β-lactamase: It hydrolyses the amide bond, which is essentially present in the four-membered β-lactam ring and causes inactivation of β-lactam antibiotics penicilloic acid before they reach the PBP target95.
β-lactam inhibits the transpeptidation reaction during cell wall biosynthesis. These antibiotics act as substrate analogues of the D-Ala-D-Ala peptidoglycan side chains. A long-lasting or longish covalent acyl-enzyme layer is formed between two functional groups. One is serine nucleophile, and another is β-lactam antibiotics that act on the PBP of the specific molecular substrate and then cause inhibition of the transpeptidation reaction within the cell wall. Modification of PBP to PBP2a is the most common and essential target for cell wall inhibition. β-lactam antibiotics and other antibiotics are acting on PBP2a, and it seems that the PBP2a is one of the most common causes of resistance in Staphylococcus aureus96. PBP2a causes catalysis of the DD-transpeptidation reaction of peptidoglycan. Because PBP2a has a low attraction for β-lactam antibiotics, DD- transpeptidation can functionally cooperate with the PBP2a and trans-glycosylase to bring biosynthesis peptidoglycans97. β-lactamase enzyme expression is induced when bacterialagents engage with antibiotics in the identical patch, and there is ametabolic cost related to β-lactamase production98.
Figure 6.
(A) The domains of penicillin-binding protein and penicillin-binding protein 2a of the target strain and their locations are depicted in a diagram99. (B)Schematic representation of resistance in Staphylococcus aureus. β-lactamase is a bacterial enzyme. This enzyme breaks down the β-lactam ring, which makes antibiotics useless before they contact the PBP target100.
Figure 6.
(A) The domains of penicillin-binding protein and penicillin-binding protein 2a of the target strain and their locations are depicted in a diagram99. (B)Schematic representation of resistance in Staphylococcus aureus. β-lactamase is a bacterial enzyme. This enzyme breaks down the β-lactam ring, which makes antibiotics useless before they contact the PBP target100.
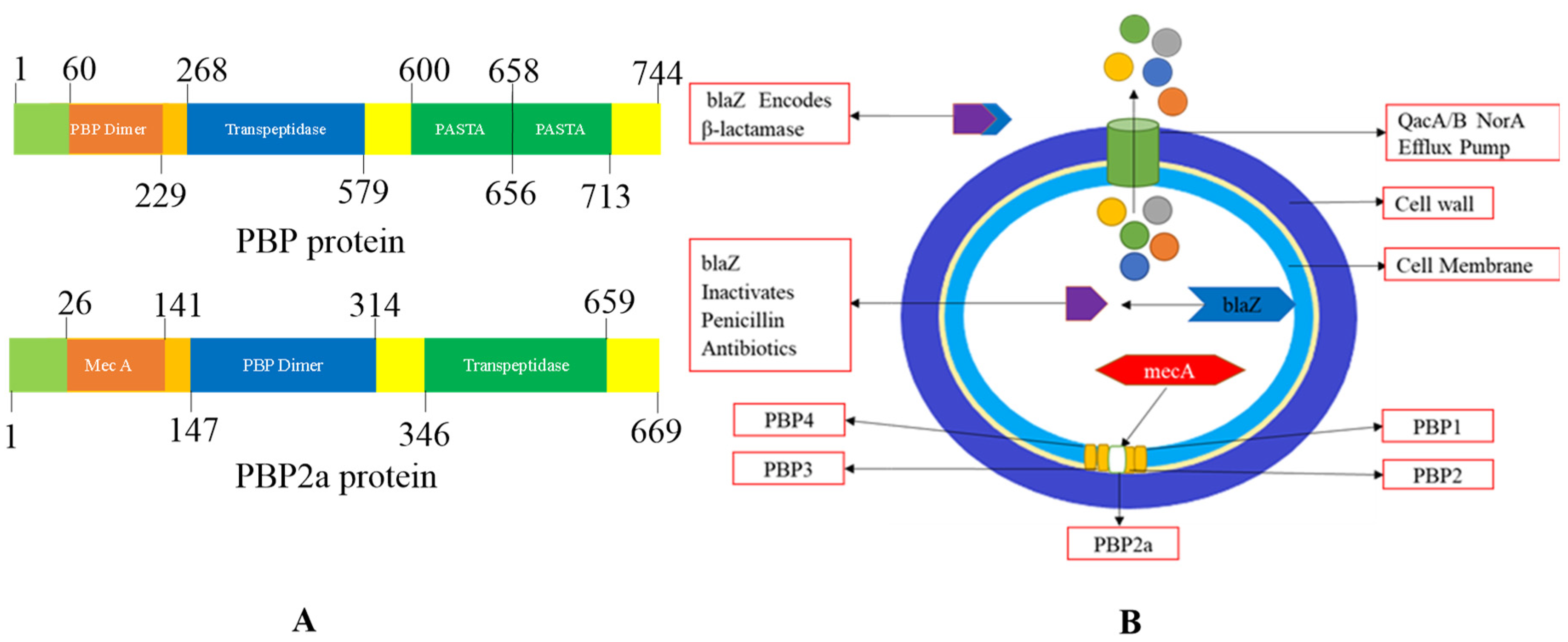
Antibiotics, mainly β-lactam antibiotics, cause resistance simply by acquiring the mecA gene, which is mutated once, then this ‘mecA encoded PBP2a (transpeptidase)’101. The multi-drug efflux proteins like; QacA/B, Nora, and smr are present on the surface of the S. aureus cell membrane. MRSA is shielded by efflux proteins of Qac, qacA, and qacB, and ene is linked with resistance increase to non-β lactam antibiotics (refer Figure 5 A). In both MSSA and MRSA, the resistant genes to identification of various antibiotics. In penicillin, the mutated gene is blaZ. In erythromycin, the mutated gene is ermC and Erma likewise in clindamycin mainly ermC encoded inducible resistance. In tetracyclines, tetK and tetL and trimethoprim, dfrA and dfrK mutated genes are found. In Staphylococcus aureus, the adoption of the Blaz gene generated more β-lactamase enzymes, which are responsible for the inactivation of β-lactam antibiotics, which helps them from reaching their targets95.
2.5. Staphylococcus Cassette chromosome:
When methicillin-susceptible S. aureus (MSSA) receives a methicillin resistance gene called mecA from a mobile genetic element called staphylococcal cassette chromosome mec (SCCmec), MRSA is produced, which is thought to be highly infectious among staphylococcal species (SCCmec). On the other hand, the source of the mecA gene for methicillin-resistant is still unknown. Understanding the history of MRSA, on the other hand, necessitates locating the significance of the mecA gene. In one of the animal-related Staphylococcus species, S. fleurettii, the mecA gene is most likely to have originated from the mecA gene of S. fleurettii, which was discovered on the chromosome connected to critical genes for staphylococci formation but not to SCCmec102,103. There are some staphylococci with various peptidoglycan types. Staphylococcus sciuri was named after a group of staphylococci with a distinctive peptidoglycan type104. The staphylococcal cassette chromosome mec gene is a chromosomal genetic component, also known as a mobile genetic element, which carries the mecA gene and encodes PBP2a in methicillin-resistant staphylococci105. (SCCmec) Staphylococcus cassette chromosome mec gene is a mutated gene and has been considered a larger heterologous mobile genetic element, which includes the central element of MRSA. SCCmec is incorporated further into the chromosome at the exact 3' end of the orfx locus, closely located at the origin of S. aureus replication106. This plays a vital role in the immediate transcription of imported antibiotics during the initial stage, and imported antibiotics are immediately transcribed, which are then recognized as resistance genes. Ccr gene (cassette chromosome recombinase) consists of one or two site-specific recombinase genes, vital for Staphylococcal cassette chromosome mec gene (SCCmec) movement107. It contains three J regions. By the combination of two mutated genes, one is the mec gene, and the other one is the ccr gene, a complex is produced, which is then strongly mutated; one complex is formed and the presence of the location of insertion of sequences, five main gene complex types are recognized largely (for types and subtypes of SCC)108.
The mec gene complex encodes two genes. One is the ccr gene complex, which encodes the recombinases, and another one is the mec gene complex. This ccr is essential for its movement and makes up the Staphylococcal cassette chromosome mec (SCCmec). The mec gene complex is divided into four categories, while the ccr gene group is divided into three allotypes109. PBP2a is produced by the staphylococcal cassette chromosome mec (SCCmec) motif, which encodes a viable mecA gene. Hence, for PBP2a formation, SCC is mainly responsible110. The nomenclature of SCCmec is given by an international working group (IWG-SCC) that established this type of nomenclature to help people. They provided consensus guidelines on SCCmec nomenclature; more details on the SCCmec gene are given on-site111. SCCmec comes in a variety of kinds and subclasses, and they all have two key elements that are responsible for resistance. These two main subgroups are mentioned below:
- a)
- The mecA gene is a complex which contains mecA genes coding for a penicillin-binding protein (PBP2) with low affinity for beta-lactam antibiotics.
- b)
- Site-specific recombinase is encoded by the cassette chromosome recombinase (ccr) gene complex112.
The SCCmec nomenclature guidelines should be observed, and it seems that the IWG-SCC should be constantly updated. The best strategy for identifying SCCmec elements, particularly for strains apart from S. aureus or MRSA, acquired from animals or the environment, is still unclear113.
3. Structure basis for PBP2A:
Lim and Strynadka published the first crystal composition of PBP2a. It consists of an acyl-PBP complex of PBP2a with nitrocefin, penicillin G, and methicillin, which allows for comparing apo and acylated forms. This protein structure is present in apo and acylated forms. This crystal structure of PBP2a is a soluble one and gives resolution at 1.8 Å; This is a transpeptidase enzyme and it seemed to be an extended protein. It contains a transpeptidase loop and a non-penicillin binding region. It also holds an N-terminal domain that is elongated and its substructure, which makes up the whole PBP2a. All these domains seem to be according to their structural arrangement. For characterization purpose, we can generate a soluble protein simply by deleting the transmembrane anchor region from the full-length protein. The architecture of structures like apo-PBP2a and nitrocefin-acylated PBP2a demonstrated a closed interference pattern that an unmodified β-lactam could never approach. As a result, an architectural shift at a binding center is required to support antibacterial activity. Then the resistance in MRSA can be explained by a closed active site but not by PBP2a physiological reaction. As a result, a structural change in the cell surface is required to meet the needs of antibiotics. PBP2a can be operated as a single transpeptidase in cell wall synthesis. β-lactam antibiotics have avoided PBP2a development. The active site of PBP2a from S. aureus was found in an extended groove at the N-terminus with Ser 403, which is nucleophilic, according to the researcher. The conserved oxyanion hole is formed by the backbone nitrogen of Ser 403 and Thr 600. The sensitivity of PBP2a can be explained by the closed active site of PBP2a and not by the closed active site of PBP2. This enzyme is capable of successfully performing the peptidoglycan binding reaction. PBP2a necessitates the functional site's ability to accept the two layers of the cell membrane, which have a volume of over 1000 Å. PBP2a is a high-molecular-weight class B PBP found only in resistant bacteria. PBP2a is a high molecular-weight peptide included in the class B penicillin-binding protein. PBP2a resistance is only found in a few other organisms, including Staphylococcus aureus. The active site consists of a serine (S403) nucleophilic amino acid located at the N-terminus. It consists of the α2 helices in the sequence motif of SXXK inside the transpeptidase catalytic site of PBP2a 115. (PDB ID: 1VQQ) at 1.80Å resolution, 1VQQ is the structure of PBP2a from methicillin-resistant strain 27r, which also contains two chains, namely, chain A and chain B, each having 646 sequence lengths (refer Figure 7). Table 2 includes 17 crystal structures consisting of bounded ligands to protein structures.
3.1. β-lactam as substrate analogues:
The β-lactam antibiotics target the transpeptidase enzyme (PBP2a), which is involved in bacterial cell wall synthesis, and the cell wall provides mechanical strength to the bacteria. The transpeptidase enzyme catalyses the cross-linkage between peptidoglycans, and it also recognizes the acyl-D-Ala-D-Ala moiety as its native substrate118. β-lactam ring containing antibiotics are particularly useful conjugates. This is because of the D-Ala-D-Ala groups in the cell wall. This then consists of chain termini under which a PBP is placed. PBP operates and generates a long-lasting suppressive covalent acyl-enzyme complex. This complex is embedded with a nucleophilic serine group, which is present inside the enzyme activation site. The location of the active site that accommodates the deacylation acceptor moiety of an adjacent peptidoglycan strand or an attainable hydrolysing water molecule typically stays occupied by the fused ring system of the β-lactam; hence, the antibiotic-bound PBP can't undergo its common subsequent deacylation. With the enzyme now inactivated, the consequent loss of cell wall cross-linking leads to cell lysis119. Ligand binding to the antibiotics and the peptidoglycan or either antibiotics or peptidoglycan depends on substrate accessibility. At the allosteric region, proteins adopt a structural state system that exposes the active site to substrate analogues. The universality of this allosteric principle was demonstrated by using multiple-lactam antibiotics as a substrate protein of the cell membrane peptide core120. As we know β-lactam ring is active ring in the beta lactam antibiotics, which bind to the PBPs. The β-lactam antibiotics that binds to the serine hydroxyl groups in the active site of bacterial enzymes which brings irreversible inactivation by forming inactive o-acyl enzymes. Thereaction shows that β-lactam antibiotics containing carbonyl groups are targeted by serine residues present inside the active site during acylation121.
3.2. About Crystal structures for PBP2a:
The crystal structure of PBP2A, PDB ID, and publication date are arranged by chronological sequencein the PROTEIN DATA BANK (PDB). The PDB ID, resolution at which the crystal structure was resolved, species of S. aureus , a ligand which was bound, and synthetic routes for a few ligands which are attached and referenced are as follows. It is to be noted that ligand-bound complexes are only presented here:
Table 3.
Crystal structure of PBP2a, their PDB ID, ligand bound to protein structure coupled with their date of publication in the Protein Data Bank (PDB) arranged in chronological sequence, and crystal structure resolutions and species of Staphylococcus aureus, with references.
Table 3.
Crystal structure of PBP2a, their PDB ID, ligand bound to protein structure coupled with their date of publication in the Protein Data Bank (PDB) arranged in chronological sequence, and crystal structure resolutions and species of Staphylococcus aureus, with references.
| Sr. no | Release Year | PDB ID | Resolution | Bound ligand | Species | Ref. |
|---|---|---|---|---|---|---|
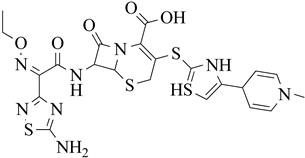
| 1 | 2019 | 6Q9N | 2.5 |
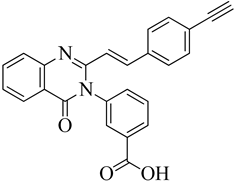 
|
S. aureus subsp. Mu50 | 122 |
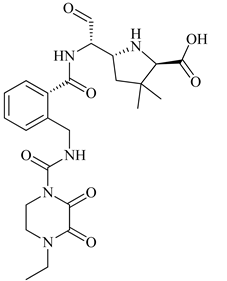
| 2 | 2019 | 6H5O | 2.82 | 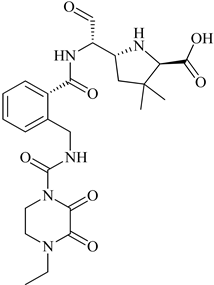 |
S. aureus Subsp.Mu50 | 123 |
| 3 | 2015 | 4CJN | 1.947 | 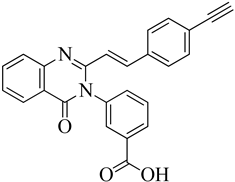 |
S. aureus | 124 |
| 4 | 2013 | 3ZG0 | 2.6 |
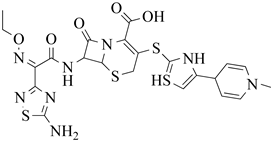 
|
S. aureus Subsp.Mu50 | 125 |
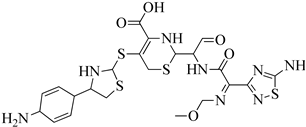
| 5 | 2013 | 3ZFZ | 2.25 |
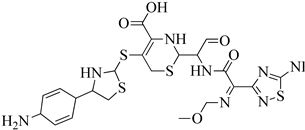 
|
S. aureus subsp. Mu50 | 125 |
| 6 | 2002 | 1MWT | 2.45 | 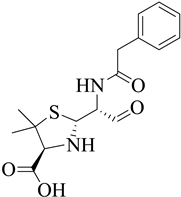 |
S. aureus | 116 |
| 7 | 2002 | 1MWU | 2.60 | 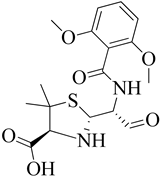 |
S. aureus | 116 |
4. Development of PBP2a inhibitors:
PBPs are membrane proteins that cause catalysis of the trans-peptide reaction during the building of the bacterial cell wall and hence play a crucial role in the bacterial cell cycle and turnover of the cell wall. When we talk about cell wall biosynthesis, PBPs are needed. Bacteria require their normal life to preserve their regular shape and function. By interacting with PBPs, penicillin and other antibiotics block the manufacturing of bacterial cell walls, resulting in bacterial cell death and serving as bactericides. In the case of four penicillin-binding proteins, MRSA contains a fifth PBP, expressed as penicillin-binding protein 2a (PBP 1, PBP 2, PBP 3, and PBP 4). Penicillin-binding protein 2a (PBP2a) is an essential protein involved in methicillin-resistant S. aureus (MRSA) acquiring resistance to beta-lactam drugs and is a potential antibacterial target. We know that PBP2a and β-lactam antibiotics have a poor affinity for each other, and many antibiotics cannot bind to PBP2a126. PBP2a is a one-of-a-kind transpeptidase enzyme that causes resistance to β-lactam antibiotics. As a result, even when other PBPs have transpeptidase activity, they are blocked. It can maintain peptidoglycan cross-linking in the presence of these antibiotics.
The following are the criteria used to choose compounds having antibacterial activity:
1) If Pa>Pi, then components seem to have a greater chance of being antibacterial.
2) If Pa<Pi, then compounds show less antibacterial activity127.
4.1. Penicillin:
Methicillin-resistant S. aureus has produced substantial issues with the use of two fundamental types of antibiotics, namely cephalosporins and penicillin. Others are (ceftaroline, ceftobiprole, ceftazidime, cefepime), penicillin (methicillin and oxacillin), carbapenems, quinolones, and aminoglycosides, but the bacteria developed resistances. As a result, this bacterium is multi-drug resistant (MDR)128. PBP2a has a decreased susceptibility to adhering to β-lactam antibiotics (penicillin, cephalosporins, and carbapenems). MRSA is susceptible to all β-lactam drugs and also to many other antibiotic agents129. Penicillin are chemically 4-thia-1-azabicyclo-(3.2.0) heptane’s (Figure 8 A)130.
The β-lactamase inhibitor tazobactam protects piperacillin from being hydrolyzed by penicillinase. Piperacillin can then bind to PBP2a to prevent transpeptidation, resulting in cell-wall biosynthesis inhibition. Meropenem, a new β-lactam antibiotic that belongs to the carbapenem class of antibiotics, interacts with PBP1 and PBP2a at allosteric sites; when bound to PBP1, it inhibits transpeptidation, and when attached to PBP2a, it causes opening of the active site. However, both in-vivo and in-vitro studies were performed to check the synergistic activity of three β-lactam antibiotics. Still, additional studies are required to validate these observations before considering the triple β-lactam combination used therapeutically (refer Figure 8 B)131.
4.2. Cephalosporins:
The US-FDA has approved ceftaroline, which is the active metabolite of an N-phosphono prodrug called ceftaroline fosamil, which is used for the management of acute bacterial skin and skin structure diseases as well as community-acquired pneumococcal disease. This was recently approved in European countries133,134. This antibiotic inhibits cell wall formation by attaching to penicillin-binding proteins (PBP). It seems to have a greater affinity for PBP2a, which is linked to methicillin resistance135. Ceftaroline interacts with PBP2a, a Methicillin-resistant S. aureus PBP with limited specificity for all other β-lactam antibiotics136. Ceftaroline affects MRSA by causing a structural shift in penicillin-binding protein2a. It mainly affects an allosteric site on (PBP2a). Cephalosporins like ceftaroline and ceftobiprole show novel antibiotic-resistant mechanisms acting on allosteric sites. This can lead to the opening of an active region of the substrate and cause inhibition of penicillin-binding protein 2a (PBP2a)137.Ceftaroline has the same antibacterial effect as other β-lactams; it binds to penicillin-binding proteins and interferes with cell wall synthesis. Ceftaroline binds to multiple PBPs, including 1 to 4, and has a high PBP2a (mecA) affinity associated with methicillin resistance. This attraction for PBP2a differentiates it from other cephalosporins135. Ceftaroline is considered a strong, safe, and newly globalized drug. Cephalosporin is used for the treatment of cSSSIs, including those affected by community-associated-MRSA as well as hospital-associated-MRSA. Due to its broad inhibitory activity, bactericidal activity, safety profile, and fewer adverse effects it is a practical and valuable activity. Ceftaroline is used against MRSA by targeting its allosteric region. Ceftaroline is indeed a first-line curative choice for both the therapies of cSSSIs, including bacterial infection with speculation of MRSA. tolerances138. Ghana has reported a high incidence of ceftaroline resistant139. The majority of ceftobiprole trials have been completed, and phase III development is still carried on. Most trials' outcomes will be closely followed140. New anti-MRSA agents need to be added to the available anti-infective drugs. Despite the world view that MRSA pathogens are uniformly resistant to all β-lactam antimicrobials, Ceftobiprole is a possible new therapy option for infections caused by the pathogens141. Cephalosporin antibiotics contain a bicyclic ring structure comprising a four-membered lactam group. It is reported that there is a dihydrothiazine ring, which is a six-membered ring. This ring is also known as cephem, and it contains a carbon double bond between positions 3 and 4. Changing its 7-acylamino side chain by replacing the cephem ring created cephalosporin drugs with varied activity patterns, reported in various papers (ceftobiprole interaction with the PBP2a for this refer Figure 9)142. CB-181963 is a new cephalosporin. CB-181963, the azomethine substituent, is in position 3 of the cephem core, with antimicrobial efficacy against MRSA. It is one of the essential properties of this cephalosporin. This is one of cephalosporin's most critical qualities. There is no data based on CB-181963's affinity for S. aureus PBP2a. CB-181963's regulatory registration will ultimately be contingent on good clinical effectiveness and safety profiles based on the reported data. There is a need to do more antimicrobial studies and clinical trials to know the safety profile. (See Figure 8 C)143.
4.3. Novel pyrazole-benzimidazole based derivatives:
A novel quinazolinone-based small molecule was effective as a non-β-lactam antibiotic against MRSA pathogen by targeting the penicillin-binding protein 2a (PBP2a), thus inhibiting cell wall biosynthesis. New scaffolds have the same pharmacophoric features through bioisosteric modifications of the lead compound novel pyrazole and benzimidazole-based small molecules quinazolinone. Three compounds exhibited moderate antibacterial activity against vancomycin and linezolid resistant MRSA. Providing potential leads for further optimization as novel antimicrobials against MRSA infection. Preliminary modelling studies were carried out for pyrazole-benzimidazole to ensure the design strategy's compliance with the lead compound's reported binding mode (refer Figure 10 A).
In drug discovery, benzimidazoles are heterocyclic ring systems that fuse and have enticed numerous researchers throughout the world to investigate their therapeutic potential. Benzimidazole nucleus is essential in lead discovery144. Quinazolinone and benzimidazole show antibacterial activity145. The benzimidazole ring represents an essential heterocyclic pharmacophore146. The lead compound (E)-3-(3-carboxyphenyl)-2- (4-cyanostyryl) quinazoline-4(3H) replaced the quinazolinone core with phenyl pyrazole and benzimidazole scaffolds. (E)-methyl (3-(2styryl-1H-benzo[d] imidazole-1yl) phenyl sulfamate 8th structure is a moderately active compound that showed a considerable MIC of 16 μg/mL (refer Figure 10 B). The synthetic scheme for structure 8th one shows moderate activity, and it is a good start for optimization to introduce a newer class of PBP2a inhibitors. It is maybe considered a potent anti-MRSA agent147.
4.4. Novel 1,2,4-oxadiazole-containing derivatives:
Oxadiazole was found via in-silico screening, and it is a newer derivative of non-beta lactam antibiotics. Antibiotics with excellent oral bioavailability and Gram-positive activity were discovered via lead optimization and in-vivo and in-vitro testing148; by fixing the 4-indole ring as common at the C-5 position and varying the aromatic substitution in the C-3 position, a new 1,2,4-oxadiazole containing derivatives were designed (refer Figure 11 A). This compound is then synthesized based on amidoxime and carboxylic acid heterocyclization and shows synergistic activity with Oxacillin against MRSA149,150.
4.5. Non-β-lactam allosteric inhibitors:
Firstly, acquisition of the mecA gene occurs, then encodes the PBP2a gene, which is responsible for resistance in S. aureus due to mutation. Preliminary modeling studies demonstrated that the reported molecules like eMol26314565 and eMol26313223 inhibit PBP2a in-vivo and in-vitro studies (refer Figure 12). They then passed in-vivo and in-vitro inhibition tests with antibiotic-resistant strains. According to the ADMET investigation, these chemicals are non-toxic and have a high oral absorption rate. Allosteric site inhibition against doubly mutant PBP2a is investigated using quinazolinone (QNZ) pharmacophore screening, but it shows less binding affinity than the reported molecules below.
4.6. 4-(3H)-Quinazolinones:
Several quinazolinone analogues have been suggested as potential MRSA antibacterial agents. Molecular docking studies aided the development of new quinazolinone analogues also, and in-silico research was followed by MM-GBSA studies. The medicinal chemistry approach synthesized newly designed compounds, which IR and NMR then characterized. It also uses HR-MS techniques. Some drugs like streptomycin, kanamycin, and linezolid were used as standard drugs in the biological evaluation study. This study was performed on various compounds to study resistance caused by MRSA bacterial strains. The MIC study of these compounds was carried out by using the microbroth dilution method. They found that compound 9, which was synthesized by using the synthetic route discussed below. Then this compound was subjected to in-vitro evaluation, is active, and has a MIC value of 31.25 μg/mL for MRSA. This MIC value was obtained by inserting a methylene linker between the amide group and the 4-methoxy phenyl substituents at the 3rd position of the aromatic ring of quinazolinone. While the 4-chloro styryl substitution gives good activity for MRSA with MIC of ≥ 15.625 μg/mL. This research shows how non β-lactam can block cell walls in the same way that β-lactam can. In the future, if someone wants to optimize a lead molecule, this work will be useful in identifying prospective lead compounds using virtual screening. The in-vitro assessment results showed that the compounds were effective against the resistant MRSA bacterial strains153(see Figure 13 A structure 1 and 2) and for synthesis, scheme refer (scheme C). 4-(3H)-Quinazolinone antibiotics namely known as (E)-3-(3-carboxyphenyl)-2-(4-cyanostyryl) quinazoline-4(3H)-one. Compound 3 has a MIC value of 2 µg/mL for S. aureus ATCC 29213. Compound 4 had an IC50 of 63 ± 1. This antibiotic impairs cell-wall biosynthesis. This does so by binding to DD-transpeptidases, which are involved in the cross-linking process of the cell wall. And it is documented by functional assays, which are reported (see Figure 13 A structure 3 and 4)154. For the synthesis of the compound, refer to (scheme B).
4.7. Pyrrolylated-chalcones:
According to the docking results, the reported ligand (pyrrolylated-chalcone) showed similar bonding interactions in the PBP2a active site to the standard medication, chlorhexidine (CHX). Their docking scores are -7.0 kcal/mol and 8.2 kcal/mol respectively. However, they did not conduct a MIC study for PBP2a against MRSA. In-vivo zebrafish model was used to study blood vessel development, apoptosis, and normal embryonic development during 24-hour. Scanning electron microscopy analysis verifications are just being carried out even further. Reported compound bactericidal properties are highlighted by the uniformly shaped S. aureus cells. Post-treatment, they were dissolved and turned into irregular shapes.Furthermore, no adverse effects on embryonic development were observed. The chemical 9 (meta-hydroxy) shows promise as an MRSA inhibitor. The zebrafish model was used to profile this chemical for in-vivo toxicity. In addition, they used all collected information as a starting point for hit-to-lead compound155(see Figure 14).
4.8. Bis-2-oxoazetidinyl macrocycle (β-lactams with 1,3-bridges):
β-lactams with 1,3-bridges incorporated into macrocycles as possible penicillin-binding inhibitors (refer Figure 15 A). A dimerization occurred when a bis-2-oxazetidinyl macrocycle was unexpectedly generated under ring-closing metathesis. Some bis-2-oxazetidinyl macrocycles demonstrated anti-PBP2a activity. Unsaturated compounds frequently occupy the same local minima as saturated compounds. Their close affinities are significantly altered. The structure of our new inhibitors is opposite to that of penicillins or cephalosporins. It disproves the β-lactam antibiotics' old paradigm. The old belief about β-lactam antibiotics is debunked. All these so-called non-symmetrical dimers mentioned below, which resemble like screws due to intramolecular H-bonds that impose geometrical limitations. Their screw pitch varies depending on macrocycle size but is best for 28-membered cycles. That, paired with their great conformational adaptability, could explain, for example, how the compounds can slip into and adapt to the PBP2a active site's closed conformation. In molecular docking, reactivity happens by ring opening of β-lactam, which occurs by nucleophilic attack at the serine active site. According to research, all of these compounds were tested for two high molecular weight transpeptidases, which is responsible for bacterial resistance to β-lactam antibiotics156.
4.9. Macrocycle-embedded β-lactams as novel inhibitors:
Traditional antibiotics have a more challenging time forming acyl-enzyme complexes with PBP2a. These are the bicyclic β-lactam antibiotics with considerable flexibility in design. Potential inhibitors are a family of bis-2-oxo-azetidinyl macrocycles. The molecule is made up of “head-to-head" cyclodimers of 1-(ω-alkenoyl)-3-(S) -(ω'-alkenoylamino)-2-azetidinones via varied alkene chain lengths obtained through two metatheses is reaction using the Grubbs catalyst. With the reaction, except for one β-lactam, all compounds serve as acylating inhibitors of PBP2a. The compounds embedded in the larger ring consist of (32 atoms), and it exhibits activity similar to that of ceftobiprole. A novel pharmacophore has been proposed based on conformational analysis, theoretical reactivity models, and docking investigation in the PBP2a cavity. Reported compounds are given below, which show the best inhibitor within a series of said macrocycle-embedded β-lactam antibiotics. In the three conserved motifs of R39, the calculation of the compound involves 228 atoms and 660 basis set functions. The compound count includes 228 atoms plus 660 base sets. Functions are documented in the R39 with three protected diagrams. This molecular structure was formed by superimposing the methanol solvent component of the SER403 model on a matching X-ray piece during the docking process in PBP2a. This study proposes a new method for rationally designing active β-lactam antibiotics. More studies are needed to determine its toxicity and ADMET profile (refer Figure 15 B)156.
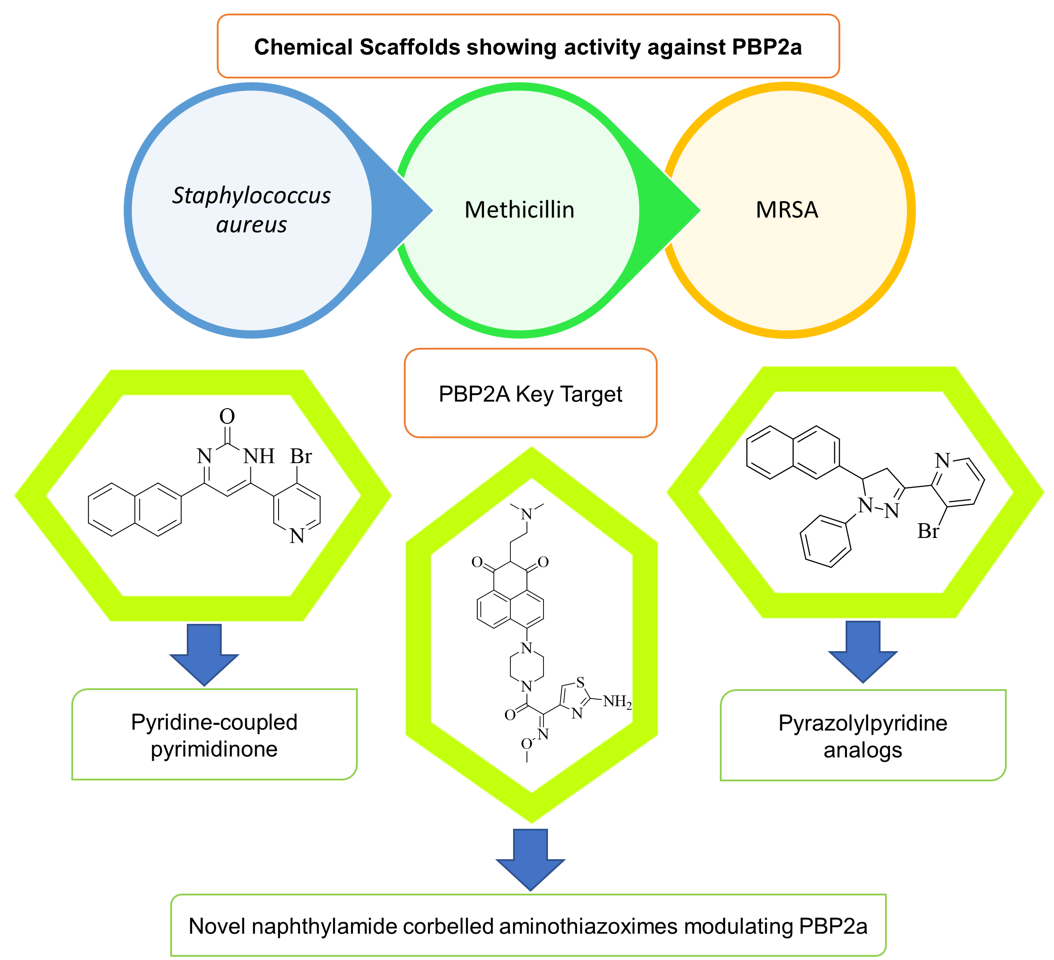
4.10. Pyridine-coupled pyrimidinones/pyrimidinthiones:
In silico studies, ADMET, molecular docking, and MD simulation are performed for these derivatives. This study is performed to check the in-vitro method. Two potent compounds show better activity, but when subjected to molecular docking experiments andsimulations of molecular dynamics which are used to check the stability of matter and the targeted PBP2a protein, only one derivative is there which shows better activity. The reported compound shows binding activity towards PBP2a with stability. The antimicrobial activity was confirmed by molecular dynamic modelling. Different spectrum technologies were used to design, manufacture, and characterize new pyridine-coupled pyrimidinones and pyrimidinthiones. The synthesized substances were tested for minimum inhibitory concentration (MIC) and has a MIC value of 8 µg. This compound shows good activity for MRSA (see Figure 15 D). There are three parts to the molecules,
1) Naphthyl ring – Lyophilic unit
2) Pyridine ring – Hydrophilic unit
3) Pyrimidinone ring – (Hydrogen-Donor-Acceptor) (See Figure 15 C).
It is designed so that one end contains lyophilic unit and the other unit contains hydrophilic unit. The Hydrogen-Acceptor-Donor (HAD) unit is linked with these two ends. Among the pyrimidinone and pyrimidinthiones series, the pyrimidinone structure (see below structure) had better anti-MRSA activity compared to pyrimidinthiones. For the antibacterial activity, the substituent of a pyrimidine for a pyridine ring is very important. Thus, alteration of the substituent at the different positions was done to investigate the anti-MRSA activity's impact. There was an enhancement in the anti-MRSA activity in the designed molecule, due to bromo and chloro moiety as they are halogens and produce stearic hindrance. Among the bromo and chloro compounds, compared to chloro compounds, bromo showed good activity due to the stability of the bromo group.
4.11. Novel naphthalimide corbelled aminothiazoximes:
Some additional drugs, such as oxadiazole and flavonoids, work the same way as QNZ, binding to allosteric sites and causing changes in conformation that lead to the active site opening. Naphthalimide is DNA targeting chemotherapeutic moiety. Against the MRSA, all of the compounds were found to be effective. At the 4th position of the naphthalimide core, the aminothiazoxime group and the piperazine bridges were incorporated. N-position was substituted with various substituents for the exploitation the naphthalimide core to produce a novel class of antibiotics affecting PBP2a. This compound has an MIC value 0.5 µg/mL (see Figure 16 A). The method used to test naphthalimide-corbeled aminothiazoximes paved the way for developing their future derivatives. Compounds with a high affinity for lipase can more easily attach to cell membranes. Membrane depolarization and turbulence can destroy the cytoplasmic material, while cell membrane degradation can damage the cell wall. Allosteric interactions of naphthalimide-corbeled aminothiazoximes with PBP2a, the active site might be exposed, resulting in older drug regeneration. The multistep synthesis process was used to absorb the unique naphthalimide-corbeled aminothiazoxime as a new antibacterial.
To synthesize the said compounds 4a from the preliminary procedure for naphthalimide corbelled aminothiazoximes, starting material was 6-bromobenzo[de]isochromene-1,3-dione used. To produce naphthalimide intermediate, coupling agent lipophilic alkylamines or acyclic amines were used. Further, reaction with piperazine and S-(benzothiazol-2-yl) (Z)-2-(2-aminothiazol-4-yl)-2-(methoxyimino) ethanethioate forming various groups of naphthalimide corbelled aminothiazoximes. In scheme B, 6-bromobenzo[de]isochromene-1,3-dione was used as starting material. To produce naphthalimide intermediate (See Figure 16C), coupling agent lipophilic alkylamines or acyclic amines were used. Further, react with piperazine and S-(benzothiazole-2-yl) (Z)-2-(2-aminothiazol-4-yl)-2-(methoxyimino) ethanethioate to form various groups of naphthalimide corbelled aminothiazoximes.
4.12. Non-covalent inhibitors:
Anthranilic acid derivatives: Screening and development of anthranilic acid derivatives show PBP2a inhibition action, hypothesized reported compounds (see Figure 17 A)159.
4.13. Investigational β-lactam antibiotics:
Several carbapenems were prepared with 7-acylated imidazo [5,1-b] thiazole-2-yl groups, and their bactericidal inhibitory substance properties were examined. ME1036, originally CP5609, is a carbapenem skeleton in the C-2 condition 7- (1-carbamoyl-methyl pyridinium-3-yl) carbonyl imidazo [5,1-b] thiazole-2-yl group. Clinical trials of this drug are now underway. ME1036 demonstrated a stronger affinity for PBP 2a and MF535 mec for PBP 1, PBP 2, PBP 3, and PBP 4. ME1036 has a wide antibacterial action spectrum and strong in vitro activity against methicillin-resistant staphylococci (refer Figure 17 B). These findings reveal an affinity for PBP2a, the new carbapenem being further evaluated. By comparing two assays, one is a kinetic assay and another one is SDS/PAGE assay, it is found that out of reported soluble PBP2a inhibitors, L-695,256 was more potent (see Figure 17 C). The filter binding assay described in the literature and the electrophoretic assay using methicillin-resistant S. aureus membrane for PBP2a. The binding relationships of numerous carbapenems and β-lactams are well correlated161.
Tomopenem has a half-life approximately twice that of other carbapenems such as imipenem and meropenem. Tomopenem has been shown to have bactericidal action against MRSA clinical isolates, and it has been reported. Tomopenem is in the second stage of development (see Figure 17 D). Meropenem and imipenem/cilastatin are commonly used to treat various nosocomial infections; however, they are ineffective against MRSA162.
4.14. Carbapenem:
In MRSA, tomopenem has a greater affinity for PBP2a than imipenem or meropenem, members of the beta-methylcarbapenem family. Since 2006, the US Food and Drug Administration approved to treat SSSIs. By delocalizing PBP2 and thereby stimulating cell autolysis and limiting biofilm production, epicatechin gallate can inhibit PBP2a-mediated methicillin resistance. Some alkaline boronic acids are effective against MRSA because they target PBP1 and PBP2a163. Corilagin belongs to the polyphenolic compound family. Corilagin has a high antibiotic effect against MRSA, interfering with the formation of bacterial cell walls, mainly affecting PBP2a activity (see Figure 17 E)164.
4.15. Novel benzoxazole derivatives:
Adenine has elemental structural isotopes and guanine bases, which are the most important heterocyclic compounds in the shape of nucleic acids. This form benzoxazole rings, and it is believed that derivatives with this ring device may also have the ability to inhibit microbial contamination inhibiting nucleic acid synthesis. 2-(p-test-butyl phenyl)-5-(3-substituted propanamido). Antibiotic properties of benzoxazole derivatives were studied, developed, manufactured, and tested (see Figure 18 A). The substance was more efficiently evaluated against reference tablets with a MIC value of 8 µg / ml against MRSA and VREF. The PBP2a allosteric site and the PBP4 active site revealed a 2D / 3D connection. They are showing potential antibacterial activity.
4.16. Pyrazolylpyridine analogues:
The bacterial efficacy of the compound was confirmed by monitoring the destruction of MRSA cellular membranes. MRSA cell membrane damage was determined by monitoring potassium flow and cellular content such as DNA leakage. Based on the data, it was found that as the dose of the drug increases, the rate of DNA leakage also increases, which is due to damage to the MRSA cell membrane. Research has shown that compounds 1 and 2 bind the amino acid residues HIS293, VAL277, TYR446, HIS583, and THR582 to the active and allosteric sites of PBP2a. This suggests from spectroscopic techniques such as FTIR, NMR, mass spectroscopy, and molecular docking results that compound 2 is effective against PBP2a. Biofilm protects against MRSA, but compound 2 is responsible for the inhibition of biofilm and acts on the active site and the allosteric site of PBP2a, and helps combat MRSA. This antibacterial study is validated using in-silico studies. Then, combination therapy is showing promising action. It can also be used to prevent MRSA bacteria from becoming resistant. MIC value of compound 2 was found to be 30 ±0.4 μg/mL (see Figure 19 A).
4.17. Miscellaneous PBP2a inhibitors:
4.17.1. Flavonoids:
Flavonoids belong to the plant's kingdom, also known as secondary metabolites. The major constituents of plants are potential classes of chemicals against MRSA, especially non-glycosylated types. A combination of both glycosylated derivatives, flavonolignan silibinin A, proved to restore the inhibitory action of ampicillin against MRSA. To examine the mechanism of action of this class, they were presented in a comprehensive inverse virtual screening (VS), in which penicillin-binding protein 2a (PBP2a) was a mediator in the antibacterial antibiotic-synergistic activity of flavonoids. Most of them are phenolic compounds such as hesperidin and its aglycon hesperetin, Rutin, and aglycon quercetin. Polyhydroxylated flavonoids are showing promising synergistic activity against PBP2a (see Figure 34) among the various flavonoids undergoing virtual screening. Hesperetin ((S) -5,7-dihydroxy-2- (3-hydroxy-4-methoxyphenyl) chroman-4-one) can be detected as a flavonoid that can be used for continued in vitro and in-vivo evaluation. Methicillin is used for MRSA therapeutic testing (refer Figure 20 A)167.
4.17.2. Demethoxycurcumin:
The goal of this trial was to see if DMC could prevent MRSA from causing a pathogenicity.A set ofdrug susceptibility assays to determine DMC's inhibitory effect on various MRSA strain. Synergistic interactions with β-lactam antibiotics were discovered, as were several essential resistance genes. Demethoxycurcumin (see Figure 20 B) shows a sound effect for treating MRSA in holistic treatment-resistant bacteraemia. Research on dimethoxy curcumin expects to continue in the future. In the future, demethoxycurcumin will be used to combat MRSA. The checkerboard method and time-kill experiment have been used to detect the action of antibiotics and the synergistic action of dimethoxy curcumin. And from reported data MIC of the dimethoxy curcumin was 62.5 g/mL169. At a 250 g / mL dose, curcumin in MRSA was found to reduce PBP2a levels. When they were combined with oxacillin, such protein deficiency was confirmed. This decrease in PBP2a suggests that curcumin may act by interfering with translation or transcription activities. They were combining its synergistic effect with oxacillin. Which completely reduced PBP2a levels compared to the synergistic effect of curcumin170.
4.17.3. Quercetin 3-o-Rutinoside:
The allosteric and active sites of PBP2a were better bound by Quercetin 3-o-Rutinoside and Ceftaroline and are also helpful for combination therapy. When cephalosporins such as cefixime and ceftriaxone, and quercetin 3-o-rutinoside cause reduction. When used alone, they are less effective than when used together. On the surface of the bacteria, the mixture undergoes morphological changes. The effectiveness of combination of rutinoside and cefixime is same as that of cefixime and rut on a single bacterial surface and cell wall. Overall, the findings suggest that rutinoside has promising potential for MRSA combination therapy. But more research is needed to confirm the effectiveness of rutinoside using synergistic strategies. More research is required to understand quercetin 3-o-role rutinoside as PBP2a modulators against MRSA infection. Further research is needed to confirm quercetin 3-o-role Rutinoside as PBP2a inhibitors171,172.
4.17.4. Ursolic acid 3-O-α-L-arabinopyranoside with oxacillin:
Oxacillin is a β-lactam antibiotic that reduces cell wall peptidoglycans biosynthesis. Simply by attaching them and restricting them competitively with PBPs. The synthesis of PBP2a creates resistance in S. aureus. A Protein inherited by mecA that has a low attraction for β-lactam antibiotics. PBP2a affected the regular functioning of PBPs by blocking their effects. As a result, restricting PBP2a expression to restore MRSA antibiotic sensitivity is still a sound approach (see Figure 20 C) 173.
4.17.5. Thioridazine:
Thioridazine is used to treat various diseases which are caused primarily due to the antibiotic-resistant but gram-positive microorganisms like MRSA. Compared to traditional synthesis methods, thioridazine reproduces itself, and photoproducts produced by laser radiation show rapid and low-cost development. MRSA membrane proteins show a more potential adverse effect of radiation thioridazine solution which then blocks PBP2a expression by sulforidazine and mesoridazine (see Figure 20 D)174.
4.17.6. Metronidazole-Triazole Hybrids:
MRSA resistance was found in all the drugs produced. The largest protective molecule, oxacillin, co-ordinated well with the reference drug. Compounds up to 50 M concentrations appear to be non-toxic to cell line THP-1. In addition, the molecules had appropriate pharmacokinetic parameters, and they adhered to lipinski's five rules. As a result, researchers have speculated that these compounds could be used as a starting point for developing new anti-MRSA drugs. 1,4-dissociated 1,2,3-triazole, which are bioisosteres of amide links, are formed by copper catalyst azide-alkyne cycloaddition synthesis. This compound was effective at a MIC value of 4µg/mL for nine MRSA strains (refer Figure 20 E, F and G)175.
4.17.7. Aspermerodione:
Small agents that attach to the allosteric protein surface of PBP2a here may help to make MRSA more susceptible to conventional β-lactam antibiotics. By using small molecules attached to the allosteric cell surface of PBP2a. The inhibition of PBP2a helps overcome MRSA. Aspermerodione is a new complex terpene-polyketide hybrid of meroterpenoid with polycyclic and a unique carbocyclic core. It damages the bacterial cell wall or cell membrane. It acts as a possible PBP2a inhibitor, which works in tandem with oxacillin and piperacillin, and antibacterial action against MRSA will be provided by β-lactam drugs. Aspermerodione is the spiro [bicyclo [3.2.2] nonane-2,1′-cyclohexane] ring system a 2,6-dioxabicyclo [2.2.1] heptane group (see Figure 20 H)176.
4.17.8. Chitosan-ferulic acid conjugates:
Chitosan is a mucopolysaccharide that occurs naturally. It is biodegradable and biocompatible and seems to have low toxicity. The in-vitro bactericidal efficacy of the chitosan-ferulic acid compound in combination with other antibiotics against MRSA was reported, but more study is required to get more data about safety profiles and then use for inhibition of the mecA gene and PBP2A synthesis. Chitosan-ferulic acid compounds reduced the intensity of the mecA gene, which depends on the dose (See Figure 20 I)129.
4.17.9. Indole-nitroimidazole conjugates:
The penicillin-binding protein 2a (PBP2a) found in MRSA is a protein that can complete cell structure cross-linking and reduce resistance to β-lactam inactivation. According to the bioassay, it seems that this compound shows satisfactory inhibition of MRSA with a MIC value of 01 µg/mL (see Figure 20 J, K and L). It is bound to PBP2a and decreased resistant related gene expression in MRSA.
4.17.10. Polyphenol:
Tellimagrandin II is a polyphenol which is extracted from a plant called trapabispinosa shell of this plant used for the extraction led to inhibition to does regulation of MRSA de novo mutations. It also reduces the penicillin-binding capacity of mecA transcription and PBP2a. Tellimagrandin II inhibit MRSA causes β-lactam resistance by reshaping bacteria and inhibiting PBP2a-mediated resistance. Due to its significant efficacy in preventing the development of MRSA and MSSA strain which has minimum inhibitory concentration (MIC) of 128 g / mL (refer Figure 20 M), it has been described as a new anti-Staphylococcus agent. The antibacterial effect of tellimagrandin II against MRSA reduces the dose of current antibiotics and helps overcome drug-resistant of S. aureus.
5. Screening technologies:
There are several screening methods available like computer-aided screening, High Throughput Screening (HTS), pharmacophore-based screening, inverse virtual screening (VS), reverse virtual screening (RVs), and Surface Plasmon Resonance (SPR) assay for the discovery of novel PBP2a inhibitors. The following methods and in-silico techniques have already been used to identify different types of PBP2a inhibitors:
5.1. High throughput screening:
High throughput screening is the exposure of large to identify hit compounds; a vast number of chemical compositions are compared to the defined target. The advantages of HTS over other screening techniques are that it is a very effective method within a short period. This method brings good results, simple techniques to handle, cost-effective, excellent effectiveness, and the ability to generate high-quality data on ligand-target interactions. Virtual high throughput screening (VHTS) is performed for carbapenem derivatives. In the next century, carbapenem and penicillin-binding proteins were used as blockers for PBP2a. The first natural carbapenem derivative is thienamycin. Thousands of thienamycin derivatives have been formulated, and 273 non-toxic chemicals have been selected for future research. Based on the molecular dynamic simulation used after docking to ensure the docking is stable for pharmacophores, only a few shows penicillin-binding protein 2a inhibition activity. Five compounds, imipenem, meropenem, doripenem, ertapenem, and thienamycin, are selected as lead depending on the docking analysis. The results showed that practically all molecules interact with the reference ligands, and thus the active site of PBP2a was discovered by proteomics research. Compared to known inhibitors, imipenem and meropenem have similar action against PBP2a (refer Figure 21).
5.2. Computer-aided screening:
Virtual screening is widely used to identify new inhibitors for target180. Computer-assisted drug design seems to be a starting point for finding new compounds, and this data is used in drug discovery. ADMET and docking experiments have been used to evaluate drug-similarity in the effectiveness of various herbal substances. Computer-aided drug design techniques are a primary way of examining potential therapeutic agents as they facilitate drug development approaches to overcome many difficulties. Finding lead molecules with strong pharmacological characteristics and drug similarities in the drug discovery process is time-consuming. Computer-aided technology makes it easy to find chemically active molecules and friendly ADMETs. For best results, data from in-silico research are then used in in-vitro tests because it selects chemical components with strong pharmacological and druggist properties. The computer-aided technique provides a quick and effective screening process. Of the 74 compounds derived from medicinal plants, only a handful was determined to be superior for MRSA. Lead molecules can be more effective than vancomycin in meliantriol and related compounds. According to computer-aided research, plant-derived inhibitors have a high therapeutic value and should be tested in vitro. Computer-assisted ADME testing and toxicity estimation were preferred to screen the best ligands with strong pharmacological activity. Herbal substances with meliantriol, β-Sitosterol, and other related compounds have been found to have potent inhibitory effects against MRSA PBP2a181.
5.3. In-silico docking screening:
In-silico screening was performed to detect new compounds that attack specific PBP2a allosteric sites in acidic media examined chemical databases such as eMolecules, ChEMBL, and ChEBI for possible inhibitors resembling quinazolinone. By Screening obtained, the PBP2a binding mode, this was calculated using molecular docking using a crystallized ligand with quinazolinone as a reference. A total of 35 compounds are subjected to virtual screening, but only two compounds show promising inhibitor activity depending on their binding affinity. Two compounds, the inhibitory effect of eMol26313223 and eMol26314565 against mutant PBP2a, seem promising for in vitro and in vivo suppression. Moreover, it is confirmed using tests with antibiotic-resistant bacterial strains. According to post-dynamics assessments, two detectable blockers with mutant PBP2a have high stability and ligand binding, according to post-dynamics assessments152. PBP2a and its ligands were represented according to the data in Figure 22 A. X-ray composition of N-acetylmuramic acid (NAM) -pentapeptide (medium, coloured according to molecular type; yellow carbon) was found at one end of the allosteric site cavity. Due to its dynamics, Ligand's NAG moiety was also absent in the electron density. When the peptidoglycan attaches to the allosteric site, the active site opens, allowing the two strands of peptidoglycan to be accommodated for the cross-linking process. The cross-linked peptidoglycan (coloured by atom kinds; carbon in dark grey) is shown at 1 o'clock coupled to the active site as the reaction's result, reported by the researcher. After the enzyme rotates 90 degrees on the y-axis in the viewer's direction, the extension provides a stereo picture of the cross-linked peptidoglycan attached to the chemical structure beneath it. Quantum mechanical/molecular mechanical (QM / MM) calculations were used to calculate the structure of bound cross-linked peptidoglycan.
In-silico docking result followed by the discovery of (E)-3-(3-carboxyphenyl)-2-(4-cyanostyryl) quinazoline-4(3H)-one as an antibacterial efficacious against methicillin-resistant S. aureus proceeded by screening and chemical synthesis of a collection of quinazolinones (MRSA). This family of antibiotics is effective in treating MRSA infections. The compound has an IC50 of 63 ± 1 μg/mL154. Antibiotics have promising effects in fighting MRSA infections. The compound has an IC50 of 63 ± 1 μg/mL154. Many computer tests were performed on 87 herbs and 2000 chemicals, which found suitable drugs to inhibit the essential MRSA proteins called PBP2a, also called transpeptidase. It is reported that three compounds have the highest binding, namely berbamine, hypericin and galangin, with the binding affinity (kCal/mol) -11.5, -8.3, and -8.3, respectively183.
5.4. Multiple virtual screening techniques:
Inhibitors of the penicillin-binding protein 2a of S. aureus have been discovered. PBP2a (penicillin-binding protein 2a) is a major protein involved in methicillin-resistant S. aureus gaining beta-lactam drug resistance and could be used as a target for many antibiotics. In the present study, a virtual screening was done with a biological assessment to uncover novel medications. PBP2a inhibitors are a relatively recent class of drugs. We used a drug-likeness-based hybrid virtual screening technique. Docking can be LibDock or CDOCKER docking, and various docking-based testing is also used. The assessment must comply with Lipinski's Rule Five and ADMET. New PBP2a inhibitors were discovered in commercially available chemical compounds using virtual screens. Eleven chemicals were picked from the final hits and are now being studied in the lab. The anti-MRSA ATCC 33591 activity of Hit 1, Hit 2, and Hit 3 was excellent with low toxicity (see Figure 23).
The three medicines' affinity for PBP2a was Surface plasmon resonance (SPR) measurements with molecular dynamics (MD) simulations were used to reach this conclusion. According to the study of inter-complex interactions. All hit compounds effectively optimized all the allosteric locations of the PBP2a protein. Hit two as well (with the best binding). The affinity for PBP2a (KD = 1.29 x 107 M) inhibits MRSA clinical isolates substantially. Both of them get along swimmingly. Further studies will be conducted on three of the hit compounds, notably Hit2, which have the potential to be effective non-lactam antibiotics against MRSA. In the future, this research could lead to the development of specific drugs that inhibit the interaction with the target of PBP2a126.
5.5. X-ray crystallography:
Brainstorm discovery based on structure, including fragment-based drug discovery X-ray crystallography, is beneficial. This is the only process that provides comprehensive results. It provides information about the three-dimensional structure and the interaction of a small molecule with its macromolecular target. It is the only method for screen conduction of a fragment library, or it can also be employed as the final technique in a screening campaign to confirm putative "hit" compounds which is identified by a variety of biochemical and/or biophysical screening techniques against target macromolecule. The successive result of X-ray crystallography-based ligand screening depends on the nature of the crystal structure employed in the study184. The cephalosporin ceftobiprole is the first β-lactam antibiotic that shows Anti-MRSA activity by targeting PBP2a, a key determinant of β-lactam resistance to MRSA. It is active against MRSA and Enterococcus faecalis, and Streptococcus pneumonia. Ceftobiprole is a pyrrolidinone cephalosporin containing the oxyimino aminothiadiazolyl substitute (R1 group) attached to the 7-amino group of the cephalosporin nucleus, which provides hydrolysis stability to many β-lactamases. Vinyl-pyrrolidinone moat (R2 group) at location 3 enhances contact with the narrow groove of the PBP2a active site and promotes PBP2a oscillation or movement119.Ceftaroline is a recently approved β-lactam antibiotic that can inhibit PBP2a by triggering an allosteric conformation that opens an active site185. β-lactam antibiotics cause irreversible acylation of the active site serine, and transpeptidase activity of PBP in susceptible bacteria is lost125.
As mentioned earlier, PBP2a is a crucial target for MRSA drug development. For broad-spectrum β-lactam antibiotics that cause MRSA strain resistance, a key determinant is PBP2a which has a low binding affinity to β-lactam antibiotics. PBP2a allows bacteria cell wall production even in the presence of β-lactam antibiotics. β-lactam antibiotics usually block PBPs, but not PBP2a. The crystal structure of (PDB ID- 1VQQ, 1MWS, 1MWT, 1MWR, and 1MWU) PBP2a apo and acetylated complexes with nitrocefin, penicillin G, and methicillin are compared. The active site of PBP2a in this form is analysed, revealing the structural basis for its resistance and the features of newly discovered lactams that are critical for high binding116.
5.6. Miscellaneous method of screening:
5.6.1. Inverse virtual screening:
Flavonoids, a plant-derived secondary metabolite, are a potential class of drugs against MRSA, especially non-flavonoids. In combination with flavonolignan silibinin A, glycosylated derivatives were able to restore the inhibitory action of ampicillin against MRSA. They were presented in a comprehensive inverse virtual screening (IVS) to examine the mechanism of action of this class, which then revealed the penicillin-binding protein 2a (PBP2a) as a target candidate mediating antibiotic-synergistic activities in this family. In addition to supporting virtual screening, in-vitro investigations of molecular docking, molecular dynamics, and PBP2a binding modes are performed168.
5.6.2. Reverse virtual screening:
To evaluate the antibacterial mechanism of icariin derivative (Figure 24 A and B), reverse virtual screening was performed. The surface plasmon resonance (SPR) assay works to confirm the binding affinity of PBP2a; this study found that this compound has a high binding affinity toward PBP2a186.The icariin derivative in which sulfonyl in C-3 substituted position binds to (PBP2a) penicillin-binding protein 2a, then causes inhibition of cell wall synthesis186.
5.6.3. Microtiter plate-based assay:
A high level of β-lactam resistance in methicillin-resistant S. aureus is caused due to PBP2a mutated PBP. There are various methods for screening PBP2a inhibitors. One is the microtiter plate-based assay, an effective and convenient method for developing and characterizing new inhibitors designed for targeting PBP2a. PBP2a inhibitor binding has been described in this method based on radio-labelled β-lactam binding. Various penicillins, cephalosporins, and carbapenem and their derivatives were used for this study to determine the binding of molecules to target187.
5.6.4. ab initio methods of screening:
Development of large ring 1,3-bridged 2-azetidinones, carbapenem parent structures (2-hydroxyethyl side chains on C3 and thienamycin stereochemistry), cephalosporin family (2-hydroxyethyl side chains on C3) and thiamine. Stereochemistry), as well as the cephalosporin family (2-hydroxyethyl side chains on C3 and thienamycin stereochemistry) (C3 and acylamino side chains on penicillin stereochemistry) ab initio screening techniques, is carried out to determine the binding affinity of PBP2a inhibitors and new hits for MRSA infection. Out of 12 compounds, only one showed good to excellent inhibition activity, and two showed modest activity against PBP2a188.
6. Conclusion:
As we search for MRSA literature, we see that MRSA is a pandemic disease, and most people are suffering from this. As mentioned above, it is a multi-resistant pathogen, and to overcome this problem, many studies are in the process of discovering new Anti-MRSA. This compound is needed to minimize the drug-resistance. The need for anti-MRSA compounds to reduce the risk of treatment resistance is now a significant concern. PBP2a is an important enzyme in the formation of bacterial cell walls. Because a range of scaffolds blocks it, PBP2a is a unique target for preventing cell wall production. As a result, PBP2a can now be targeted for anti-MRSA drug production. Both in-vivo and in-vitro studies have shown anti-MRSA action in seventeen different scaffolds. Several PBP2a inhibitors have been found using screening techniques including high throughput screening, virtual screening, and other methods including computer-aided design, molecular docking screening, X-ray diffraction screening, and some miscellaneous screening like ab initio method, reverse-virtual screening, inverse-virtual screening, and microtiter-plate based assay are used to screen PBP2a inhibitors. PBP2a activity has been studied using a variety of scaffolding. It is namely novel pyrazole-benzimidazole based derivatives, novel 1,2,4-oxadiazole containing derivatives, non-β-lactam allosteric inhibitors, 4-(3H)-quinazolinone, pyrrolylated chalcones, β-lactam with 1,3-bridges, macrocycle-embedded β-lactams as novel inhibitors, pyridine-coupled pyrimidinones or pyrimidinthiones, novel naphthalimide corbelled aminothiazoximes, non-covalent inhibitors, some investigational β-lactam antibiotics, carbapenem, novel benzoxazole derivatives and miscellaneous PBP2a inhibitors like flavonoids, demethoxycurcumin, Quercetin-3-O-Rutinoside, thioridazine, metronidazole-triazole hybrid, aspermerodione, chitosan-ferulic acid conjugates, indole-nitrobenzmidazole conjugates were found against PBP2a.
The enzymes involved in the S. aureus cell wall biosynthesis are an attractive alternative to MRSA drug detection and target-based drug or structured-based drug discovery. According to the material review, they are inhibiting PBP2a. This increases the chances of finding and identifying new drug candidates to combat antibiotic resistance. Some efforts can be made to improve the in-vivo properties of drugs by focusing on both pharmacokinetics and pharmacodynamic aspects to find PBP2a inhibitors. For that, indeed, pharmacokinetics and pharmacodynamic properties are taken into consideration. PBP2a is a promising option for MRSA and the development of Anti-MRSA compounds. To obtain a good result, novel agents are to be combined for future treatment.
7. The Outlook:
1. In methicillin-resistant Staphylococcus aureus, peptidoglycan precursor generation is critical.
2. It is better and more convenient to discover novel medicines against MRSA using a ligand-based method.
3. The following are the main problems in creating novel anti-MRSA agents:
a) How to use the knowledge you have gained to create anti-virulence medications.
b) How to incorporate those findings into the still-developing field of bacterial interaction in the human microbiome189.
4) Widespread knowledge from various articles should be used to discover new antibiotics as they are needed to combat MRSA infections (Figure 25). As antibiotic resistance poses a hazard to public health, present antibiotics classes must be improved or altogether new classes discovered.
Author Contributions
SA: VG, RB, PB and RC planned the review, collected data, wrote the draft.
Funding resources
None to declare.
Conflicts of Interest
None to declare.
References
- Tansey, E. M. Superbugs And Superdrugs: A History Of Mrsa The Transcript of a Witness Seminar Held by the Wellcome Trust Centre. 2008, 32 (July 2006). 20 July.
- Campbell, J.; Singh, A. K.; Santa, J. P.; Jr, M.; Kim, Y.; Swoboda, J. G.; Mylonakis, E.; Wilkinson, B. J.; Walker, S. NIH Public Access. 2012, 6 (1), 106–116. [CrossRef]
- Tong, S. Y. C.; Davis, J. S.; Eichenberger, E.; Holland, T. L.; Fowler, V. G. Staphylococcus Aureus Infections : Epidemiology , Pathophysiology , Clinical Manifestations , and Management. 2015, 623. [CrossRef]
- Guo, Y.; Song, G.; Sun, M.; Wang, J.; Wang, Y.; Wang, Y. Prevalence and Therapies of Antibiotic-Resistance in Staphylococcus Aureus. 2020, 10 (March), 1–11. [CrossRef]
- I, J. F. D. C.; I, Í. M. D. A.; Borges, K.; Rocha, F. Oxacillin Magnetically Targeted for the Treatment Of. 32 (1), 46–55.
- Lowy, F. D.; Lowy, F. D. Antimicrobial Resistance : The Example of Staphylococcus Aureus Find the Latest Version : Antimicrobial Resistance : The Example of Staphylococcus Aureus. 2003, 111 (9), 1265–1273. [CrossRef]
- Aureus, T. The Chromosome, as Well as the Extrachromosomal El- Ements. 6 These Genes Are Transferred between Staphy- Lococcal Strains, Species, or Other Gram-Positive Bacte- Rial Species through the Extrachromosomal Elements. 7. 1998.
- Foster, T. J.; Foster, T. J. The Staphylococcus Aureus “ Superbug ” The Staphylococcus Aureus “ Superbug .” 2004, 114 (12), 1693–1696.
- Saraiva, F. B.; de Araújo, A. C. C.; de Araújo, A. É. V.; Senna, J. P. M. Monoclonal Antibody Anti-PBP2a Protects Mice against MRSA (Methicillin-Resistant Staphylococcus Aureus) Infections. PLoS ONE2019, 14 (11), 1–13. [CrossRef]
- Deng, J. Z. Methicillin/per-6-(4-Methoxylbenzyl)-Amino-6-Deoxy-β-Cyclodextrin 1:1 Complex and Its Potentiation in Vitro against Methicillin-Resistant Staphylococcus Aureus. Journal of Antibiotics2013, 66 (9), 517–521. [CrossRef]
- Vernozy-rozand, C.; Mazuy, C.; Lasne, Y.; Fiedler, F.; Etienne, J. Staphylococcus Fleurettii Sp . Nov ., Isolated from Goat ’ s Milk Cheeses. 2000, 1521–1527.
- Berndsen, R.; Cunningham, T.; Kaelin, L.; Callender, M.; Boldog, W. D.; Viering, B.; King, A.; Labban, N.; Pollock, J. A.; Miller, H. B.; Blackledge, M. S. Identi Fi Cation and Evaluation of Brominated Carbazoles as a Novel Antibiotic Adjuvant Sca Ff Old in MRSA. 2022. [CrossRef]
- Hao, H.; Dai, M.; Wang, Y.; Huang, L.; Yuan, Z. Key Genetic Elements and Regulation Systems in Methicillin-Resistant Staphylococcus Aureus. Future Microbiology2012, 7 (11), 1315–1329. [CrossRef]
- Barber, M.; Rozwadowska-dowzenko, M. Patients 2. 1948, 641–644.
- Sciences, V. Review Article. 2015.
- Lyont, B. R.; Skurray, R. O. N. Antimicrobial Resistance of Staphylococcus Aureus : Genetic Basis. 1987, 51 (1), 88–134.
- Kumosani, T. Nosocomial Infections in Saudi Arabia Caused by Methicillin Resistance Staphylococcus Aureus (MRSA). Clinical Microbiology: Open Access2014, 03 (03). [CrossRef]
- Z; vizdi, AmraHuki, M. STAPHYLOCOCCUS AUREUS ( MRSA ) AS A CAUSE OF NOSOCOMIAL WOUND. 10 (1), 32–37.
- Grundmann, H.; Aires-de-sousa, M.; Boyce, J.; Tiemersma, E. Emergence and Resurgence of Meticillin-Resistant Staphylococcus Aureus as a Public-Health Threat. 2006, 874–885. [CrossRef]
- Liscano, Y.; Amú, A.; González, A.; Oñate-Garzón, J.; Salamanca, C. H. In Silico Characterization of the Interaction between the Pbp2a “Decoy” Protein of Resistant Staphylococcus Aureus and the Monomeric Units of Eudragit e-100 and Poly(Maleic Acid-Alt-Octadecene) Polymers. Polymers (Basel)2021, 13 (14). [CrossRef]
- Jr, R. C. M. MRSA: The First Half Century. 2012, No. October 2011, 4–11. 20 October. [CrossRef]
- Oehler, R. L. MRSA: Historical Perspective Richard L. Oehler, MD. 2000, 1961 (Table 1).
- Andrade, M. M.; Luiz, W. B.; Oliveira, S.; Amorim, J. H. The History of Methicillin-Resistant Staphylococcus Aureus in Brazil. 2020, 2020.
- Haamann, F.; Dulon, M.; Nienhaus, A. MRSA as an Occupational Disease : A Case Series. 2011, 259–266. [CrossRef]
- Diep, B. A.; Carleton, H. A.; Chang, R. F.; Sensabaugh, G. F. Roles of 34 Virulence Genes in the Evolution of Hospital- and Community-Associated Strains of Methicillin-Resistant Staphylococcus Aureus. 2006, 94110, 1495–1503.
- Enright, M. C.; Robinson, D. A.; Randle, G.; Feil, E. J.; Grundmann, H.; Spratt, B. G. The Evolutionary History of Methicillin-Resistant Staphylococcus Aureus ( MRSA ). 2002, 99 (11).
- Stefani, S.; Varaldo, P. E. Epidemiology of Methicillin-Resistant Staphylococci in Europe. 2003.
- Janout, V. CURRENT KNOWLEDGE OF METHICILLIN-RESISTANT STAPHYLOCOCCUS AUREUS AND COMMUNITY-ASSOCIATED METHICILLIN-RESISTANT STAPHYLOCOCCUS AUREUS Ivanka Matouskova , Vladimir Janout. 2008, 152 (2), 191–202.
- Majed, H.; Johnston, T.; Kelso, C.; Monachino, E.; Jergic, S.; Dixon, N. E.; Mylonakis, E.; Kelso, M. J. Structure-Activity Relationships of Pyrazole-4-Carbodithioates as Antibacterials against Methicillin−Resistant Staphylococcus Aureus. Bioorganic & Medicinal Chemistry Letters2018. [CrossRef]
- Since January 2020 Elsevier Has Created a COVID-19 Resource Centre with Free Information in English and Mandarin on the Novel Coronavirus COVID- Research That Is Available on the COVID-19 Resource Centre - Including This for Unrestricted Research Re-Use and Analyses in Any Form or by Any Means with Acknowledgement of the Original Source . These Permissions Are Meticillin-Resistant Staphylococcus Aureus. 2020, No. January.
- Ippolito, G.; Leone, S.; Lauria, F. N.; Nicastri, E.; Wenzel, R. P. International Journal of Infectious Diseases Methicillin-Resistant Staphylococcus Aureus : The Superbug. 2010, 4, 7–11. [CrossRef]
- Pantosti, A.; Venditti, M. What Is MRSA? 2009, 34 (5), 1190–1196. [CrossRef]
- Wong, S. C. Y.; Tse, H.; Chen, J. H. K. Multidrug-Resistant Staphylococcus Aureus , Colistin-Resistant Enterobacteriaceae Carrying the Mcr-1 Gene among Patients in Hong Kong. 2016, 22 (9), 2015–2016.
- Mehta, Y.; Hegde, A.; Pande, R.; Zirpe, K. G.; Gupta, V.; Ahdal, J.; Qamra, A.; Motlekar, S. Methicillin-Resistant Staphylococcus Aureus in Intensive Care Unit Setting of India : A Review of Clinical Burden , Patterns of Prevalence , Preventive Measures , and Future Strategies. 2020, 0–7.
- Correspondence; 2016; Vol. 132, pp 371–372. [CrossRef]
- Ld, I. M.; David, M. G.; Esposito, S.; Garau, J.; Lina, G.; Mazzei, T.; Peters, G. International Journal of Antimicrobial Agents New Insights into Meticillin-Resistant Staphylococcus Aureus ( MRSA ) Pathogenesis , Treatment and Resistance ଝ. 2012, 39, 96–104. [CrossRef]
- Baba, K.; Ishihara, K.; Ozawa, M.; Tamura, Y.; Asai, T. International Journal of Antimicrobial Agents Isolation of Meticillin-Resistant Staphylococcus Aureus ( MRSA ) from Swine in Japan. International Journal of Antimicrobial Agents2010, 36 (4), 352–354. [CrossRef]
- Outbreak, H. W.; Toleman, M. S.; Reuter, S.; Coll, F.; Harrison, E. M.; Peacock, S. J. Local Persistence of Novel MRSA Lineage after Community-Acquired Clostridium Difficile Infection,. 2016, 22 (9), 2011–2012.
- Huang, Y.; Chen, C. International Journal of Antimicrobial Agents Community-Associated Meticillin-Resistant Staphylococcus Aureus in Children In. International Journal of Antimicrobial Agents2011, 38 (1), 2–8. [CrossRef]
- Peacock, S. J.; Paterson, G. K. Mechanisms of Methicillin Resistance in Staphylococcus Aureus. Annual Review of Biochemistry2015, 84, 577–601. [CrossRef]
- Id, C. B.; Schwab, F.; Id, R. V. Infection Control Measures in Nosocomial MRSA Outbreaks — Results of a Systematic Analysis. 2021, 1–10. [CrossRef]
- Kumar, P. A Review on Quinoline Derivatives as Anti-Methicillin Resistant Staphylococcus Aureus (MRSA) Agents. BMC Chemistry2020, 14 (1), 1–14. [CrossRef]
- Jiang, W.; Li, B.; Zheng, X.; Liu, X.; Cen, Y.; Li, J.; Pan, X.; Cao, H.; Zheng, J.; Zhou, H. International Immunopharmacology Artesunate in Combination with Oxacillin Protect Sepsis Model Mice Challenged with Lethal Live Methicillin-Resistant Staphylococcus Aureus ( MRSA ) via Its Inhibition on Proin Fl Ammatory Cytokines Release and Enhancement on Antibacterial Activity of Oxacillin. International Immunopharmacology2011, 11 (8), 1065–1073. [CrossRef]
- Kong, Z.; Zhao, P.; Liu, H.; Yu, X.; Qin, Y.; Su, Z. Whole-Genome Sequencing for the Investigation of a Hospital Outbreak of MRSA in China. 2016, 1–12. [CrossRef]
- Stryjewski, M. E.; Corey, G. R. Methicillin-Resistant Staphylococcus Aureus : An Evolving Pathogen. 2014, 58 (Suppl 1), 10–19. [CrossRef]
- Mrsa, C. S. Methicillin-Resistant Staphylococcus Aureus Infections. 2008, 16 (5).
- Sahare, P.; Moon, A. Penicillin Binding Proteins: An Insight Into Novel Antibacterial Drug Target. 2014, 5 (8), 13–23.
- Goffin, C.; Ghuysen, J. Biochemistry and Comparative Genomics of SxxK Superfamily Acyltransferases Offer a Clue to the Mycobacterial Paradox : Presence of Penicillin-Susceptible Target Proteins versus Lack of Efficiency of Penicillin as Therapeutic Agent. 2002, 66 (4), 702–738. [CrossRef]
- Gordon, E.; Mouz, N.; Due, E.; Dideberg, O.; Horowitz, J. The Crystal Structure of the Penicillin-Binding Protein 2x from Streptococcus Pneumoniae and Its Acyl-Enzyme Form : Implication in Drug Resistance. 2000, 477–485. [CrossRef]
- Frere_Joris_1985_CRC_299.Pdf.
- Mouz, N.; Gordon, E.; Hopkins, J.; Dideberg, O. Crystal Structure of PBP2x from a Highly Penicillin-Resistant Streptococcus Pneumoniae Clinical Isolate. 2001, 276 (48), 45106–45112. [CrossRef]
- Sauvage, E.; Terrak, M.; Ayala, J. A.; Charlier, P. The Penicillin-Binding Proteins : Structure and Role in Peptidoglycan Biosynthesis. 2008, 32, 234–258. [CrossRef]
- EZervosen, A.; Sauvage, E.; Frère, J.; Charlier, P.; Luxen, A. Development of New Drugs for an Old Target — The Penicillin Binding Proteins. 2012, 12478–12505. [CrossRef]
- Green, D. W. The Bacterial Cell Wall as a Source of Antibacterial Targets. Expert Opinion on Therapeutic Targets2002, 6 (1), 1–20. [CrossRef]
- Resistance, B. MINIREVIEW Penicillin-Binding Proteins and Bacterial Resistance to , 3-Lactams. 1993, 37 (10), 2045–2053.
- Scheffers, D.; Pinho, M. G. Bacterial Cell Wall Synthesis : New Insights from Localization Studies. 2005, 69 (4), 585–607. [CrossRef]
- Anderson, J. S.; Matsuhashi, M.; Haskin, M. A.; Strominger, J. L. Biosythesis of the Peptidoglycan of Bacterial Cell Walls. II. Phospholipid Carriers in the Reaction Sequence. Journal of Biological Chemistry1967, 242 (13), 3180–3190. [CrossRef]
- Id, P. D. A. R.; Buss, J.; Id, S. I. S.; Id, G. R. S.; Srisuknimit, V.; Id, M. S.; Cho, H.; Sjodt, M.; Id, A. C. K.; Id, E. C. G.; Walker, S.; Kahne, D. E.; Id, T. G. B. A Central Role for PBP2 in the Activation of Peptidoglycan Polymerization by the Bacterial Cell Elongation Machinery. 2018, 1–25.
- Contreras-martel, C.; Vernet, T. Penicillin-Binding Proteins and b -Lactam Resistance. 2008. [CrossRef]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J. A.; Charlier, P. The Penicillin-Binding Proteins: Structure and Role in Peptidoglycan Biosynthesis. FEMS Microbiology Reviews2008, 32 (2), 234–258. [CrossRef]
- Dave, K.; Palzkill, T.; Pratt, R. F. Neutral β-Lactams Inactivate High Molecular Mass Penicillin-Binding Proteins of Class B1, Including PBP2a of MRSA. ACS Medicinal Chemistry Letters2014, 5 (2), 154–157. [CrossRef]
- Pinho, M. G.; Errington, J. Recruitment of Penicillin-Binding Protein PBP2 to the Division Site of Staphylococcus Aureus Is Dependent on Its Transpeptidation Substrates. 2005, 55, 799–807. [CrossRef]
- hang Wei Wu; De Lencastre, H.; Tomasz, SA. Recruitment of the MecA Gene Homologue of Staphylococcus Sciuri into a Resistance Determinant and Expression of the Resistant Phenotype in Staphylococcus Aureus. Journal of Bacteriology2001, 183 (8), 2417–2424. [CrossRef]
- De Jonge, B. L. M.; Chang, Y. S.; Gage, D.; Tomasz, A. Peptidoglycan Composition of a Highly Methicillin-Resistant Staphylococcus Aureus Strain. The Role of Penicillin Binding Protein 2A. Journal of Biological Chemistry1992, 267 (16), 11248–11254. [CrossRef]
- Oliveira, D. C.; Wu, S. W. Genetic Organization of the Downstream Region of the MecA Element in Methicillin-Resistant Staphylococcus Aureus Isolates Carrying Different Polymorphisms of This Region. 2000, 44 (7), 1906–1910. [CrossRef]
- Lencastre, H. De; Jonge, B. L. M. De; Matthews, P. R.; Tomasz, A. Review Molecular Aspects of Methicillin Resistance in Staphylococcus Aureus. 1994, 7–24.
- Murakami, K. Nucleotide Sequence of the Structural Gene for the Penicillin-Binding Protein 2 of Staphylococcus Aureus and the Presence of a Homologous Gene in Other Staphylococci. 1994, 117, 131–136. [CrossRef]
- Filipe, R. R.; Pinho, M. G.; Tomasz, A. Complementation of the Essential Peptidoglycan Transpeptidase Function of Penicillin-Binding Protein 2 ( PBP2 ) by the Drug Resistance Protein PBP2A in Staphylococcus Aureus. 2001, 183 (22), 6525–6531. [CrossRef]
- Fuda, C.; Hesek, D.; Lee, M.; Morio, K. I.; Nowak, T.; Mobashery, S. Activation for Catalysis of Penicillin-Binding Protein 2a from Methicillin-Resistant Staphylococcus Aureus by Bacterial Cell Wall. J Am Chem Soc2005, 127 (7), 2056–2057. [CrossRef]
- Hakenbeck, R.; Coyette, J. Resistant Penicillin-Binding Proteins. 1998, 54, 332–340. [CrossRef]
- Fuda, C.; Suvorov, M.; Vakulenko, S. B.; Mobashery, S. The Basis for Resistance to β-Lactam Antibiotics by Penicillin-Binding Protein 2a of Methicillin-Resistant Staphylococcus Aureus. Journal of Biological Chemistry2004, 279 (39), 40802–40806. [CrossRef]
- Stapleton, P. D.; Taylor, P. W. Europe PMC Funders Group Methicillin Resistance in Staphylococcus Aureus : Methicillin Resistance. 2007, 85 (Pt 1), 1–14.
- Srisuknimit, V.; Qiao, Y.; Schaefer, K.; Kahne, D.; Walker, S. Peptidoglycan Cross-Linking Preferences of Staphylococcus Aureus Penicillin-Binding Proteins Have Implications for Treating MRSA Infections. J Am Chem Soc2017, 139 (29), 9791–9794. [CrossRef]
- Contreras-martel, C.; Martins, A.; Ecobichon, C.; Trindade, D. M.; Matteï, P.; Hicham, S.; Hardouin, P.; Ghachi, M. El; Boneca, I. G.; Dessen, A. Molecular Architecture of the PBP2–MreC Core Bacterial Cell Wall Synthesis Complex. Nature Communications 1–10. [CrossRef]
- Schleifer, K. H.; Kandler, O. Peptidoglycan Types of Bacterial Cell Walls and Their Taxonomic Implications. Bacteriol Rev1972, 36 (4), 407–477. [CrossRef]
- Contreras-martel, C.; Amoroso, A.; Woon, E. C. Y.; Zervosen, A.; Inglis, S.; Martins, A.; Verlaine, O.; Rydzik, A. M.; Job, V. Structure-Guided Design of Cell Wall Biosynthesis Inhibitors That Overcome β -Lactam Resistance in Staphylococcus Aureus (MRSA). 2011, 943–951. [CrossRef]
- Tahir99-VRG & Vip.Persianss.Ir.
- Hrast, M.; Rožman, K.; Jukič, M.; Patin, D.; Gobec, S.; Sova, M. Synthesis and Structure–Activity Relationship Study of Novel Quinazolinone-Based Inhibitors of MurA. Bioorganic and Medicinal Chemistry Letters2017, 27 (15), 3529–3533. [CrossRef]
- Nikolaidis, I.; Dessen, A. Resistance to Antibiotics Targeted to the Bacterial Cell Wall. 2014, 23, 243–259. [CrossRef]
- Lewis, R. J. Structural Basis for the Coordination of Cell Division with the Synthesis of the Bacterial Cell Envelope. 2019, No. August, 2042–2054. [CrossRef]
- Caveney, N. A.; Li, F. K.; Strynadka, N. C. Enzyme Structures of the Bacterial Peptidoglycan and Wall Teichoic Acid Biogenesis Pathways. Current Opinion in Structural Biology2018, 53, 45–58. [CrossRef]
- Macheboeuf, P.; Contreras-martel, C.; Job, V.; Dideberg, O. Penicillin Binding Proteins : Key Players in Bacterial Cell Cycle and Drug Resistance Processes. 2006, 30, 673–691. [CrossRef]
- Gautam, A.; Vyas, R.; Tewari, R. Peptidoglycan Biosynthesis Machinery : A Rich Source of Drug Targets. 2011, 31 (April 2010), 295–336. 20 April. [CrossRef]
- Naclerio, G. A.; Sintim, H. O. Multiple Ways to Kill Bacteria via Inhibiting Novel Cell Wall or Membrane Targets. 2020.
- Kovac, A. Cytoplasmic Steps of Peptidoglycan Biosynthesis. 2008, 32, 168–207. [CrossRef]
- Rogers, H. J.; Perkins, H. R.; Ward, J. B. Biosynthesis of Peptidoglycan. Microbial Cell Walls and Membranes1980, 239–297. [CrossRef]
- Monteiro, J. M.; Covas, G.; Rausch, D.; Filipe, S. R.; Schneider, T.; Sahl, H. G.; Pinho, M. G. The Pentaglycine Bridges of Staphylococcus Aureus Peptidoglycan Are Essential for Cell Integrity. Scientific Reports2019, 9 (1), 1–10. [CrossRef]
- Goldman, R. C.; Gange, D. Inhibition of Transglycosylation Peptidoglycan Synthesis Involved in Bacterial. 2000, No. 609, 801–820.
- Gondokesumo, M. E.; Kurniawan, I. M. Molecular Docking Study of Sappan Wood Extract to Inhibit PBP2A Enzyme on Methicillin-Resistant Staphylococcus Aureus (MRSA). Journal of Basic and Clinical Physiology and Pharmacology2019, 30 (6), 1–9. [CrossRef]
- Young, K. D. Peptidoglycan. 2001, 1–11.
- Walsh, C. T. Enzymes in the D-Alanine Branch of Bacterial Cell Wall Peptidoglycan Assembly. Journal of Biological Chemistry1989, 264 (5), 2393–2396. [CrossRef]
- Berger-Bächi, B.; Tschierske, M. Role of Fem Factors in Methicillin Resistance. Drug Resistance Updates1998, 1 (5), 325–335. [CrossRef]
- Kourtis, A. P.; Hatfield, K.; Baggs, J.; Mu, Y.; See, I.; Epson, E.; Nadle, J.; Kainer, M. A.; Dumyati, G. Vital Signs : Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus Aureus Bloodstream Infections — United States. 2020.
- Cedraro, N.; Cannalire, R.; Astolfi, A.; Mangiaterra, G.; Felicetti, T.; Vaiasicca, S.; Cernicchi, G.; Massari, S.; Biavasco, F.; Sabatini, S. From Quinoline to Quinazoline-Based S . Aureus NorA Efflux Pump Inhibitors by Coupling a Focused Scaffold Hopping Approach and a Pharmacophore Search. 2021, 3044–3059. [CrossRef]
- Lade, H.; Kim, J. S. Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus Aureus. Antibiotics2021, 10 (4). [CrossRef]
- Kaczor, A.; Witek, K.; Podlewska, S.; Czekajewska, J.; Lubelska, A.; Zesławska, E.; Nitek, W.; Latacz, G.; Alibert, S.; Pagès, J. M.; Karczewska, E.; Kieć-Kononowicz, K.; Handzlik, J. 5-Arylideneimidazolones with Amine at Position 3 as Potential Antibiotic Adjuvants against Multidrug Resistant Bacteria. Molecules2019, 24 (3). [CrossRef]
- Jousselin, A.; Manzano, C.; Biette, A.; Reed, P.; Pinho, M. G.; Rosato, A. E.; Kelley, W. L.; Renzoni, A. The Staphylococcus Aureus Chaperone PrsA Is a New Auxiliary Factor of Oxacillin Resistance Affecting Penicillin-Binding Protein 2A. Antimicrobial Agents and Chemotherapy2016, 60 (3), 1656–1666. [CrossRef]
- Murphy, J. T.; Walshe, R.; Devocelle, M. A Computational Model of Antibiotic-Resistance Mechanisms in Methicillin-Resistant Staphylococcus Aureus (MRSA). Journal of Theoretical Biology2008, 254 (2), 284–293. [CrossRef]
- Mohamed, S. B.; Adlan, T. A.; Khalafalla, N. A.; Abdalla, N. I.; Ali, Z. S. A.; Munir KA, A.; Hassan, M. M.; Elnour, M. A. B. Proteomics and Docking Study Targeting Penicillin-Binding Protein and Penicillin-Binding Protein2a of Methicillin-Resistant Staphylococcus Aureus Strain SO-1977 Isolated from Sudan. Evolutionary Bioinformatics2019, 15. 2019; 15. [CrossRef]
- Babic, M.; Hujer, A. M.; Bonomo, R. A.; Stokes, L.; Medical, A. What ’ s New in Antibiotic Resistance ? Focus on Beta-Lactamases. 2006, 9, 142–156. [CrossRef]
- Wielders, C. L. C.; Fluit, A. C.; Brisse, S.; Verhoef, J.; Schmitz, F. J. MecA Gene Is Widely Disseminated in Staphylococcus Aureus Population. 2002, 40 (11), 3970–3975. [CrossRef]
- Tsubakishita, S.; Kuwahara-arai, K.; Sasaki, T.; Hiramatsu, K. Origin and Molecular Evolution of the Determinant of Methicillin Resistance in Staphylococci. 2010, 54 (10), 4352–4359. [CrossRef]
- Webster, J. A.; Bannerman, T. L.; Hubner, R. J.; Ballard, D. N.; Cole, E. M.; Bruce, J. L.; Fiedler, F.; Schubert, K.; Kloos, W. E. Identification of the Staphylococcus Sciuri Species Group with EcoRI Fragments Containing RRNA Sequences and Description of Staphylococcus Vitulus Sp . Nov . 1994, 460 (July), 454–460.
- Schleifer, K. H.; Geyer, U.; Kilpper-balz, R.; Devriese, L. A. Elevation of Staphylo Coccus Sciuri Subsp . Lentus ( Kloos et al .) to Species Status : Staphylococcus Lentus ( Kloos et al .) Comb . Nov . 1983, 387, 382–387. [CrossRef]
- Ouchenane, Z.; Agabou, A.; Smati, F.; Rolain, J. M.; Raoult, D. Staphylococcal Cassette Chromosome Mec Characterization of Methicillin-Resistant Staphylococcus Aureus Strains Isolated at the Military Hospital of Constantine/Algeria. Pathologie Biologie2013, 61 (6), 280–281. [CrossRef]
- Chromosome, S. C. Crossm Evolutionary Origin of the Staphylococcal Cassette Chromosome. 2017, 61 (6), 1–16.
- Katayama, Y.; Ito, T.; Hiramatsu, K. A New Class of Genetic Element , Staphylococcus Cassette Chromosome Mec , Encodes Methicillin Resistance in Staphylococcus Aureus. 2000, 44 (6), 1549–1555.
- Liu, J.; Chen, D.; Peters, B. M.; Li, L.; Li, B.; Xu, Z.; Shirliff, M. E. Staphylococcal Chromosomal Cassettes Mec (SCCmec): A Mobile Genetic Element in Methicillin-Resistant Staphylococcus Aureus. Microbial Pathogenesis2016, 101, 56–67. [CrossRef]
- Ito, T.; Ma, X. X.; Takeuchi, F.; Okuma, K.; Yuzawa, H.; Hiramatsu, K. Novel Type V Staphylococcal Cassette Chromosome Mec Driven by a Novel Cassette Chromosome Recombinase , CcrC. 2004, 48 (7), 2637–2651. [CrossRef]
- Ender, M.; Berger-Bächi, B.; McCallum, N. Variability in SCCmecN1 Spreading among Injection Drug Users in Zurich, Switzerland. BMC Microbiology2007, 7, 1–10. [CrossRef]
- hongtrakool, P.; Ito, T.; Ma, X. X.; Trakulsomboon, S.; Tiensasitorn, C.; Jamklang, M.; Chavalit, T.; Song, J.; Chongtrakool, P.; Ito, T.; Ma, X. X.; Kondo, Y.; Trakulsomboon, S. Staphylococcal Cassette Chromosome Mec ( SCC Mec ) Typing of Methicillin-Resistant Staphylococcus Aureus Strains Isolated in 11 Asian Countries : A Proposal for a New Nomenclature for SCC Mec Elements Staphylococcal Cassette Chromosome Mec ( SCC Mec ) Typing of Methicillin-Resistant Staphylococcus Aureus Strains Isolated in 11 Asian Countries : A Proposal for a New Nomenclature for SCC Mec Elements. 2006. [CrossRef]
- Tsubakishita, S.; Kuwahara-arai, K.; Baba, T.; Hiramatsu, K. Staphylococcal Cassette Chromosome Mec -Like Element in Macrococcus Caseolyticus. 2010, 54 (4), 1469–1475. [CrossRef]
- Uehara, Y. Current Status of Staphylococcal Cassette. 2022, 1–12. [CrossRef]
- Berger-Bächi, B.; Rohrer, S. Factors Influencing Methicillin Resistance in Staphylococci. Archives of Microbiology2002, 178 (3), 165–171. [CrossRef]
- Fishovitz, J.; Hermoso, J. A.; Chang, M.; Mobashery, S. Penicillin-Binding Protein 2a of Methicillin-Resistant Staphylococcus Aureus. IUBMB Life2014, 66 (8), 572–577. [CrossRef]
- Lim, D.; Strynadka, N. C. J. Structural Basis for the β-Lactam Resistance of PBP2a from Methicillin-Resistant Staphylococcus Aureus. Nature Structural Biology2002, 9 (11), 870–876. [CrossRef]
- Kumar, K. M.; Anitha, P.; Sivasakthi, V.; Bag, S.; Lavanya, P.; Anbarasu, A.; Ramaiah, S. In Silico Study on Penicillin Derivatives and Cephalosporins for Upper Respiratory Tract Bacterial Pathogens. 2014, 241–251. [CrossRef]
- Chiang, Y. C.; Wong, M. T. Y.; Essex, J. W. Molecular Dynamics Simulations of Antibiotic Ceftaroline at the Allosteric Site of Penicillin-Binding Protein 2a (PBP2a). Israel Journal of Chemistry2020, 60 (7), 754–763. [CrossRef]
- Lovering, A. L.; Gretes, M. C.; Safadi, S. S.; Danel, F.; Castro, L. De; Page, M. G. P.; Strynadka, N. C. J. Structural Insights into the Anti-Methicillin-Resistant Staphylococcus Aureus ( MRSA ) Activity of Ceftobiprole *. 2012, 287 (38), 32096–32102. [CrossRef]
- Hermoso, J. A.; Chang, M.; Mobashery, S. HHS Public Access. 2018, 139 (5), 2102–2110. [CrossRef]
- My, N. H.; Hirao, H.; Van, D. U.; Morokuma, K. Computational Studies of Bacterial Resistance to β-Lactam Antibiotics: Mechanism of Covalent Inhibition of the Penicillin-Binding Protein 2a (PBP2a). Journal of Chemical Information and Modeling2011, 51 (12), 3226–3234. [CrossRef]
- Janardhanan, J.; Bouley, R.; Peng, Z.; Batuecas-mordillo, M.; Meisel, J. E.; Ding, D.; Schroeder, V. A.; Wolter, W. R.; Mahasenan, K. V; Hermoso, J. A.; Mobashery, S.; Chang, M. Crossm The Quinazolinone Allosteric Inhibitor of PBP 2a Synergizes. 2019, 63 (5), 1–12.
- Janardhanan, J.; Bouley, R.; Peng, Z.; Batuecas-mordillo, M.; Meisel, J. E.; Ding, D.; Schroeder, V. A.; Wolter, W. R.; Mahasenan, K. V; Hermoso, J. A.; Mobashery, S.; Chang, M. Crossm The Quinazolinone Allosteric Inhibitor of PBP 2a Synergizes. 2019, 63 (5), 1–12.
- Bouley, R.; Kumarasiri, M.; Peng, Z.; Otero, L. H.; Song, W.; Mark, A.; Schroeder, V. A.; Wolter, W. R.; Lastochkin, E.; Antunes, N. T.; Pi, H.; Vakulenko, S.; Hermoso, J. A.; Chang, M.; Mobashery, S. Discovery of Antibiotic ( E ) -3- ( 3-Carboxyphenyl ) -2- ( 4- Cyanostyryl ) Quinazolin-4 ( 3 H ) -One. 2015, 4. [CrossRef]
- Otero, L. H.; Rojas-Altuve, A.; Llarrull, L. I.; Carrasco-López, C.; Kumarasiri, M.; Lastochkin, E.; Fishovitz, J.; Dawley, M.; Hesek, D.; Lee, M.; Johnson, J. W.; Fisher, J. F.; Chang, M.; Mobashery, S.; Hermoso, J. A. How Allosteric Control of Staphylococcus Aureus Penicillin Binding Protein 2a Enables Methicillin Resistance and Physiological Function. Proc Natl Acad Sci U S A2013, 110 (42), 16808–16813. [CrossRef]
- Lv, N.; Kong, Q.; Zhang, H.; Li, J. Discovery of Novel Staphylococcus Aureus Penicillin Binding Protein 2a Inhibitors by Multistep Virtual Screening and Biological Evaluation. Bioorganic and Medicinal Chemistry Letters2021, 41 (April), 2–9. [CrossRef]
- Lavanya, P.; Ramaiah, S.; Anbarasu, A. A Molecular Docking and Dynamics Study to Screen Potent Anti-Staphylococcal Compounds Against Ceftaroline Resistant MRSA. Journal of Cellular Biochemistry2016, 117 (2), 542–548. [CrossRef]
- Matys, A.; Podlewska, S.; Witek, K.; Witek, J.; Bojarski, A. J.; Schabikowski, J.; Otrębska-Machaj, E.; Latacz, G.; Szymańska, E.; Kieć-Kononowicz, K.; Molnar, J.; Amaral, L.; Handzlik, J. Imidazolidine-4-One Derivatives in the Search for Novel Chemosensitizers of Staphylococcus Aureus MRSA: Synthesis, Biological Evaluation and Molecular Modeling Studies. European Journal of Medicinal Chemistry2015, 101, 313–325. [CrossRef]
- Eom, S.; Kang, S.; Lee, D.; Myeong, J.; Lee, J.; Kim, H.; Kim, K.; Je, J.; Jung, W.; Kim, Y. Synergistic Antibacterial Effect and Antibacterial Action Mode of Chitosan - Ferulic Acid Conjugate against Methicillin-Resistant Staphylococcus Aureus. 2016, 26 (0), 784–789. [CrossRef]
- Kumar, M.; Kumar, D.; Raj, V. Studies on Imidazole and Its Derivatives with Particular Emphasis on Their Chemical/Biological Applications as Bioactive Molecules/Intermediated to Bioactive Molecule. Current Synthetic and Systems Biology2017, 05 (01), 1–10. [CrossRef]
- Bush, K. Synergistic MRSA Combinations. Nature Publishing Group2015, 11 (11), 832–833. [CrossRef]
- Gonzales, P. R.; Pesesky, M. W.; Bouley, R.; Ballard, A.; Biddy, B. A.; Suckow, M. A.; Wolter, W. R.; Schroeder, V. A.; Burnham, C. D.; Mobashery, S.; Chang, M.; Dantas, G. Combinations Suppress Resistance in MRSA. 2015, No. september. [CrossRef]
- Stryjewski, M. E.; Jones, R. N.; Corey, G. R. SC. Diagnostic Microbiology and Infectious Disease2014. [CrossRef]
- Pii, I. PT SC. Integrative Medicine Research2018. [CrossRef]
- Saravolatz, L. D.; Stein, G. E.; Johnson, L. B. Ceftaroline: A Novel Cephalosporin with Activity against Methicillin-Resistant Staphylococcus Aureus. Clinical Infectious Diseases2011, 52 (9), 1156–1163. [CrossRef]
- Soriano, A. Ceftaroline. Revista Espanola de Quimioterapia2021, 34 (7), 29–31. [CrossRef]
- Otero, L. H.; Rojas-Altuve, A.; Llarrull, L. I.; Carrasco-López, C.; Kumarasiri, M.; Lastochkin, E.; Fishovitz, J.; Dawley, M.; Hesek, D.; Lee, M.; Johnson, J. W.; Fisher, J. F.; Chang, M.; Mobashery, S.; Hermoso, J. A. How Allosteric Control of Staphylococcus Aureus Penicillin Binding Protein 2a Enables Methicillin Resistance and Physiological Function. Proc Natl Acad Sci U S A2013, 110 (42), 16808–16813. [CrossRef]
- Hernandez, P. O.; Lema, S.; Tyring, S. K.; Mendoza, N. Ceftaroline in Complicated Skin and Skin-Structure Infections. Infection and Drug Resistance2012, 5 (1), 23–35. [CrossRef]
- Schaumburg, F.; Peters, G.; Alabi, A.; Becker, K.; Idelevich, E. A. Missense Mutations of PBP2a Are Associated with Reduced Susceptibility to Ceftaroline and Ceftobiprole in African MRSA. 2016, No. October 2015, 41–44. [CrossRef]
- Livermore, D. M. Can β-Lactams Be Re-Engineered to Beat MRSA? Clinical Microbiology and Infection2006, 12 (SUPPL. 2), 11–16. (SUPPL. 2). [CrossRef]
- Vidaillac, C.; Rybak, M. J. Ceftobiprole: First Cephalosporin with Activity against Methicillin-Resistant Staphylococcus Aureus. Pharmacotherapy2009, 29 (5), 511–525. [CrossRef]
- Nigo, M.; Lorena Diaz, Lina P. Carvajal, Truc T. Tran, R. R.; Panesso, D.; Garavito, J. D.; Miller, W. R.; Wanger, A.; George Weinstock; Munita, J. M.; Arias, C. A.; (Commentator), H. F. C. Ceftaroline-Resistant, Daptomycin- Tolerant, and Heterogeneous Vancomycin-Intermediate Methicillin- Resistant Staphylococcus Aureus Causing Infective Endocarditis. Antimicrobial Agents and Chemotherapy2017, 61 (3), 1–7.
- Miller, K.; Storey, C.; Stubbings, W. J.; Hoyle, A. M.; Hobbs, J. K.; Chopra, I. Antistaphylococcal Activity of the Novel Cephalosporin. 2005, No. February, 579–582. [CrossRef]
- Yadav, G.; Ganguly, S. Structure Activity Relationship (SAR) Study of Benzimidazole Scaffold for Different Biological Activities: A Mini-Review. European Journal of Medicinal Chemistry2015, 97 (1), 419–443. [CrossRef]
- Malasala, S.; Ahmad, N.; Akunuri, R.; Shukla, M. Jo Ur Na l P Re Of. European Journal of Medicinal Chemistry2020, 112996. [CrossRef]
- Tunçbilek, M.; Kiper, T.; Altanlar, N. Synthesis and in Vitro Antimicrobial Activity of Some Novel Substituted Benzimidazole Derivatives Having Potent Activity against MRSA. European Journal of Medicinal Chemistry2009, 44 (3), 1024–1033. [CrossRef]
- Shalaby, M.-A.; Dokla, E.; Serya, R.; Abouzid, K. Identification of Novel Pyrazole and Benzimidazole Based Derivatives as PBP2a Inhibitors: Design, Synthesis, and Biological Evaluation. Archives of Pharmaceutical Sciences Ain Shams University2019, 3 (2), 228–245. [CrossRef]
- O’Daniel, P. I.; Peng, Z.; Pi, H.; Testero, S. A.; Ding, D.; Spink, E.; Leemans, E.; Boudreau, M. A.; Yamaguchi, T.; Schroeder, V. A.; Wolter, W. R.; Llarrull, L. I.; Song, W.; Lastochkin, E.; Kumarasiri, M.; Antunes, N. T.; Espahbodi, M.; Lichtenwalter, K.; Suckow, M. A.; Vakulenko, S.; Mobashery, S.; Chang, M. Discovery of a New Class of Non-β-Lactam Inhibitors of Penicillin-Binding Proteins with Gram-Positive Antibacterial Activity. J Am Chem Soc2014, 136 (9), 3664–3672. [CrossRef]
- Buommino, E.; Marino, S. De; Sciarretta, M.; Auria, M. V. D.; Festa, C. With Oxacillin against Methicillin-Resistant Staphylococcus Aureus. 2021, 1–16.
- Verma, S. K.; Verma, R.; Kumar, K. S. S.; Banjare, L.; Shaik, A. B.; Bhandare, R. R.; Rakesh, K. P.; Rangappa, K. S. A Key Review on Oxadiazole Analogs as Potential Methicillin-Resistant Staphylococcus Aureus (MRSA) Activity: Structure-Activity Relationship Studies. European Journal of Medicinal Chemistry2021, 219, 113442. [CrossRef]
- Shalaby, M. A. W.; Dokla, E. M. E.; Serya, R. A. T.; Abouzid, K. A. M. Penicillin Binding Protein 2a: An Overview and a Medicinal Chemistry Perspective. European Journal of Medicinal Chemistry2020, 199, 112312. [CrossRef]
- Ibrahim, M. A. A.; Abdeljawaad, K. A. A.; Abdelrahman, A. H. M.; Alzahrani, O. R.; Alshabrmi, F. M.; Khalaf, E.; Moustafa, M. F.; Alrumaihi, F.; Allemailem, K. S.; Soliman, M. E. S.; Paré, P. W.; Hegazy, M. E. F.; Atia, M. A. M. Non-β-Lactam Allosteric Inhibitors Target Methicillin-Resistant Staphylococcus Aureus: An in Silico Drug Discovery Study. Antibiotics2021, 10 (8). [CrossRef]
- Qureshi, S. I.; Chaudhari, H. K. Design, Synthesis, in-Silico Studies and Biological Screening of Quinazolinone Analogues as Potential Antibacterial Agents against MRSA. Bioorganic and Medicinal Chemistry2019, 27 (12), 2676–2688. [CrossRef]
- Soc, J. A. C. HHS Public Access. 2015, 137 (5), 1738–1741. [CrossRef]
- Gunasekharan, M.; Choi, T. I.; Rukayadi, Y.; Latif, M. A. M.; Karunakaran, T.; Faudzi, S. M. M.; Kim, C. H. Preliminary Insight of Pyrrolylated-Chalcones as New Anti-Methicillin-Resistant Staphylococcus Aureus (Anti-Mrsa) Agents. Molecules2021, 26 (17). [CrossRef]
- Chem, M.; Marchand-brynaert, J. MedChemComm. 2012, No. 3, 344–351. [CrossRef]
- Nanjundaswamy, S.; Bindhu, S.; Renganathan, R. R. A.; Nagashree, S.; Mallu, P.; Rai, V. R.; Bindhu, S.; Renganathan, R. R. A.; Nagashree, S. Design , Synthesis of Pyridine Coupled Pyrimidinone / Pyrimidinthione as Anti-MRSA Agent : Validation by Molecular Docking and Dynamics Simulation. Journal of Biomolecular Structure and Dynamics2021, 0 (0), 1–12. [CrossRef]
- Zhang, P.; Gopala, L.; Zhang, S.; Cai, G. European Journal of Medicinal Chemistry An Unanticipated Discovery towards Novel Naphthalimide Corbelled Aminothiazoximes as Potential Anti-MRSA Agents and Allosteric Modulators for PBP2a. European Journal of Medicinal Chemistry2022, 229, 114050. [CrossRef]
- Shalaby, M. A. W.; Dokla, E. M. E.; Serya, R. A. T.; Abouzid, K. A. M. Penicillin Binding Protein 2a: An Overview and a Medicinal Chemistry Perspective. European Journal of Medicinal Chemistry2020, 199, 112312. [CrossRef]
- Kurazono, M.; Ida, T.; Yamada, K.; Hirai, Y.; Maruyama, T.; Shitara, E.; Yonezawa, M. In Vitro Activities of ME1036 ( CP5609 ), a Novel Parenteral Carbapenem , against Methicillin-Resistant Staphylococci. 2004, 48 (8), 2831–2837. [CrossRef]
- Toney, J. H.; Hammond, G. G.; Leiting, B.; Pryor, K. A. D.; Wu, J. K.; Cuca, G. C.; Pompliano, D. L. Soluble Penicillin-Binding Protein 2a: β-Lactam Binding and Inhibition by Non-β-Lactams Using a 96-Well Format. Analytical Biochemistry1998, 255 (1), 113–119. [CrossRef]
- Manuscript, A. NIH Public Access. 2013, 8 (9), 1095–1116. [CrossRef]
- Hao, H.; Cheng, G.; Dai, M.; Wu, Q.; Yuan, Z. Inhibitors Targeting on Cell Wall Biosynthesis Pathway of MRSA. Molecular BioSystems2012, 8 (11), 2828–2838. [CrossRef]
- Shiota, S.; Shimizu, M.; Sugiyama, J.; Morita, Y.; Mizushima, T.; Tsuchiya, T. Mechanisms of Action of Corilagin and Tellimagrandin I That Remarkably Potentiate the Activity of  -Lactams against Methicillin-Resistant Staphylococcus Aureus. 2004, 48 (1), 67–73. [CrossRef]
- Erol, M.; Celik, I.; Temiz-Arpaci, O.; Kaynak-Onurdag, F.; Okten, S. Design, Synthesis, Molecular Docking, Density Functional Theory and Antimicrobial Studies of Some Novel Benzoxazole Derivatives as Structural Bioisosteres of Nucleotides. Journal of Biomolecular Structure and Dynamics2021, 39 (9), 3080–3091. [CrossRef]
- Karthik, C. S.; Ananda, A. P.; Nagashree, S.; Mallu, P.; Rai, V. R. Design , Synthesis , and in-Silico Studies of Pyrazolylpyridine Analogues : A Futuristic Antibacterial Contender against Coagulase Positive. Journal of Molecular Structure2022, 1255, 132400. [CrossRef]
- Sangeetha, M.; Saranya, T. S.; Sathianarayanan, S.; Hima Vyshnavi, A. M.; Krishnan Namboori, P. K. Design and Development of Potential Flavonoid Moiety for Pbp2a Inhibition for Mrsa Therapy-A Computational Technique. Biomedical and Pharmacology Journal2020, 13 (2), 687–692. 2020; 13, 687–692. [CrossRef]
- Alhadrami, H. A.; Hamed, A. A.; Hassan, H. M.; Belbahri, L.; Rateb, M. E.; Sayed, A. M. Flavonoids as Potential Anti-MRSA Agents through Modulation of PBP2A: A Computational and Experimental Study. Antibiotics2020, 9 (9), 1–16. [CrossRef]
- Li, Q. Q.; Kang, O. H.; Kwon, D. Y. Study on Demethoxycurcumin as a Promising Approach to Reverse Methicillin-Resistance of Staphylococcus Aureus. International Journal of Molecular Sciences2021, 22 (7). [CrossRef]
- Neto, D. A.; Ramos, A.; Max, W.; Silva, J.; Silva, E.; Coelho, B.; Odorico, M.; Moraes, D. Microbial Pathogenesis Anti-MRSA Activity of Curcumin in Planktonic Cells and Biofilms and Determination of Possible Action Mechanisms. 2021, 155 (March). [CrossRef]
- Rani, N.; Saravanan, V.; Lakshmi, P. T. V. Allosteric Site Mediated Active Site Inhibition of PBP2a Using Quercetin 3-O-Rutinoside and Its Combination. 2015, 1102 (September). [CrossRef]
- Rani, N.; Vijayakumar, S.; Thanga Velan, L. P.; Arunachalam, A. Quercetin 3-O-Rutinoside Mediated Inhibition of PBP2a: Computational and Experimental Evidence to Its Anti-MRSA Activity. Molecular BioSystems2014, 10 (12), 3229–3237. [CrossRef]
- Zhou, T.; Li, Z. H. I.; Kang, O. K. H. W. A.; Mun, S. U. H.; Seo, Y. U. N. S. O. O.; Kong, R. Antimicrobial Activity and Synergism of Ursolic Acid 3-O- α -L-Arabinopyranoside with Oxacillin against Methicillin-Resistant Staphylococcus Aureus. 2017, 1285–1293. [CrossRef]
- Tozar, T.; Costa, S. S.; Udrea, A. M.; Nastasa, V.; Couto, I.; Viveiros, M.; Pascu, M. L.; Romanitan, M. O. Anti - Staphylococcal Activity and Mode of Action of Thioridazine Photoproducts. Scientific Reports2020, 1–12. [CrossRef]
- Negi, B.; Kumar, D.; Kumbukgolla, W.; Jayaweera, S.; Ponnan, P.; Singh, R.; Agarwal, S.; Rawat, D. S. Anti-Methicillin Resistant Staphylococcus Aureus Activity, Synergism with Oxacillin and Molecular Docking Studies of Metronidazole-Triazole Hybrids. European Journal of Medicinal Chemistry2016, 115, 426e–4437. [CrossRef]
- Qiao, Y.; Zhang, X.; He, Y.; Sun, W.; Feng, W.; Liu, J.; Hu, Z.; Xu, Q.; Zhu, H.; Zhang, J.; Luo, Z.; Wang, J.; Xue, Y.; Zhang, Y. Aspermerodione, a Novel Fungal Metabolite with an Unusual 2,6-Dioxabicyclo[2.2.1]Heptane Skeleton, as an Inhibitor of Penicillin-Binding Protein 2a. Scientific Reports2018, 8 (1), 2–12. [CrossRef]
- Li, Z. Z.; Tangadanchu, V. K. R.; Battini, N.; Bheemanaboina, R. R. Y.; Zang, Z. L.; Zhang, S. L.; Zhou, C. H. Indole-Nitroimidazole Conjugates as Efficient Manipulators to Decrease the Genes Expression of Methicillin-Resistant Staphylococcus Aureus. European Journal of Medicinal Chemistry2019, 179, 723–735. [CrossRef]
- Chang, Y. W.; Huang, W. C.; Lin, C. Y.; Wang, W. H.; Hung, L. C.; Chen, Y. H. Tellimagrandin Ii, a Type of Plant Polyphenol Extracted from Trapa Bispinosa Inhibits Antibiotic Resistance of Drug-Resistant Staphylococcus Aureus. International Journal of Molecular Sciences2019, 20 (22), 1–17. [CrossRef]
- Chowdhury, A.; Paul, P.; Bhattacharjee, A.; Talukdar, A. Das; Choudhury, M. D. Virtual High Throughput Screening of Carbapenem Derivatives as New Generation Carbapenemase and Penicillin Binding Protein Inhibitors : A Hunt to Save Drug of Last Resort. 2015, 18–23. [CrossRef]
- Lian, X.; Xia, Z.; Li, X.; Karpov, P.; Jin, H.; Tetko, I. V; Xia, J.; Wu, S. Bioorganic Chemistry Anti-MRSA Drug Discovery by Ligand-Based Virtual Screening and Biological Evaluation. Bioorganic Chemistry2021, 114 (2), 105042. 2021; 114. [CrossRef]
- Skariyachan, S.; Krishnan, R. S.; Siddapa, S. B.; Salian, C.; Bora, P.; Sebastian, D. Computer Aided Screening and Evaluation of Herbal Therapeutics against MRSA Infections. Bioinformation2011, 7 (5), 222–233. [CrossRef]
- Chang, M.; Mahasenan, K. V; Hermoso, J. A.; Mobashery, S. Unconventional Antibacterials and Adjuvants. 2021. [CrossRef]
- Abedi, F.; Balmeh, N.; Sanjari, S.; Plantago-, A. Informatics in Medicine Unlocked In-Silico Investigation of Antibacterial Herbal Compounds in Order to Find New Antibiotic against Staphylococcus Aureus and Its Resistant Subtypes ☆. Informatics in Medicine Unlocked2022, 28, 100843. [CrossRef]
- Davies, D. R. Chapter 23 Screening Ligands by X-Ray Crystallography. 2014, 1140, 315–323. [CrossRef]
- Fishovitz, J.; Rojas-altuve, A.; Otero, L. H.; Dawley, M.; Carrasco-lo, C.; Chang, M.; Hermoso, J. A.; Mobashery, S. Of Resistance to Antibiotics. 2014, 1–4.
- Articles, O. Synthesis and Antibacterial Activity of Novel Icariin Derivatives. 2019, 74 (ii), 73–78. [CrossRef]
- Mart, A.; Pascual, M.; Sheth, C. C.; Veses, V. Icariin in Combination with Amoxycillin-Clavulanate and Ampicillin , but Not Vancomycin , Increases Antibiotic Sensitivity and Growth Inhibition against Methicillin-Resistant Staphylococcus Aureus. 2022.
- Marchand-brynaert, J.; Urbach, A.; Dive, G.; Tinant, B. European Journal of Medicinal Chemistry Large Ring 1 , 3-Bridged 2-Azetidinones : Experimental and Theoretical Studies. 2009, 44, 2071–2080. [CrossRef]
- Cheung, G. Y. C.; Bae, J. S.; Otto, M. Pathogenicity and Virulence of Staphylococcus Aureus ABSTRACT. Virulence2021, 12 (1), 547–569. [CrossRef]
Figure 3.
Steps in the biosynthesis of peptidoglycan.
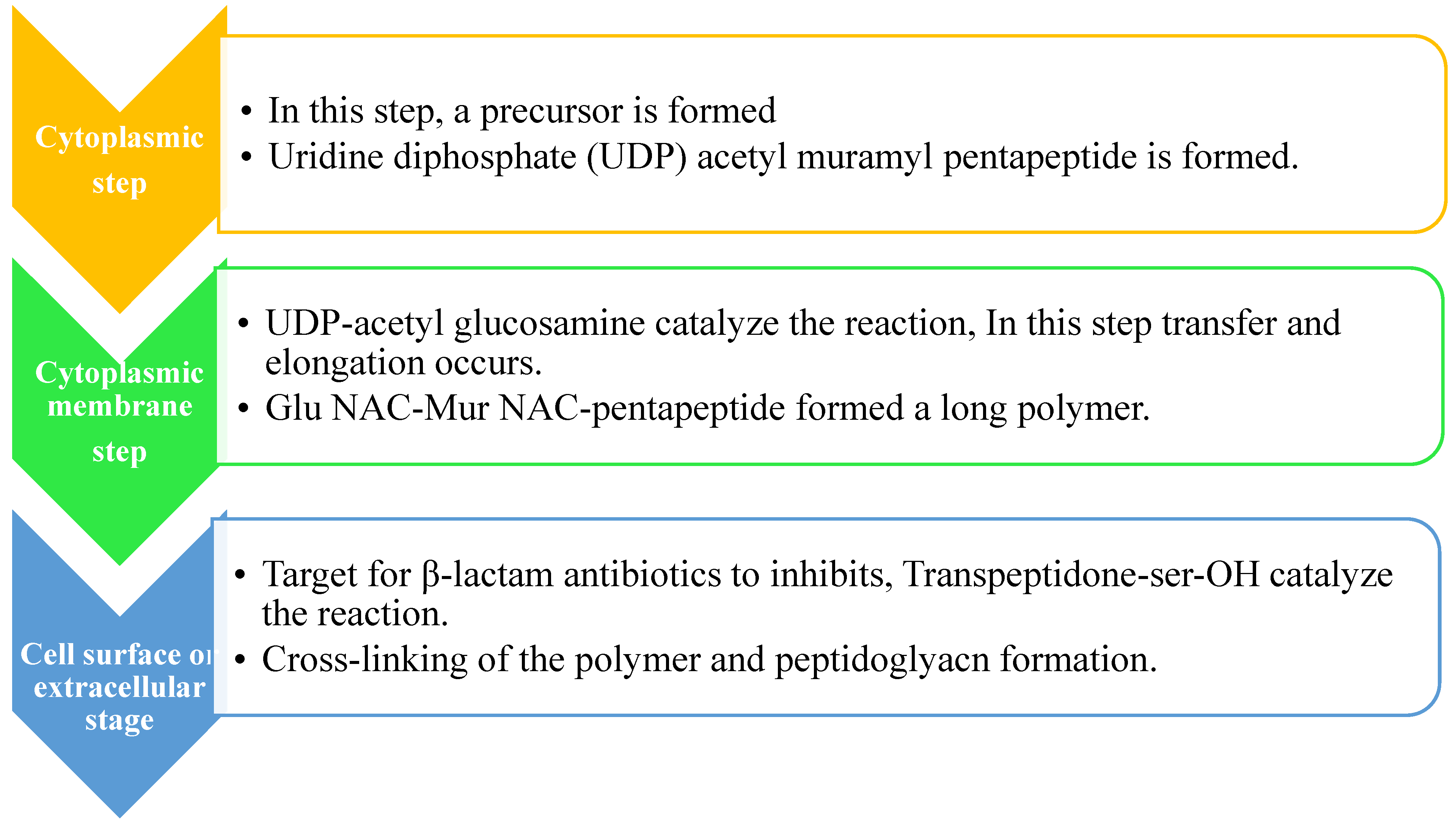
Figure 4.
(A)Depiction of the transpeptidation reaction shows cross-linking within the bacterial cell wall. Ceftobiprole led to inhibition of PBP2a and this is responsible for stopping the growth of cell wall89. (B)Illustration shows the development of the small molecule groups and disaccharide constituents of peptidoglycan in the cytoplasmic membrane.At this stage, enzymes encoded by genes successively add amino acids to the lactyl group of N-acetylmuramicacid (NAM). Just before the complete subunit is transported across the membrane. Two amino acid residues, namely two D-alanine (D-Ala) residues, are covalently attached to produce a dipeptide and are linked as a single entity at the end of the side-chain N-acetylglucosamine (NAG)90. (C)Adjacent strands are cross-linked in peptidoglycan assembly by transpeptidation91. (D)Synthesis of pentaglycine interpeptide, and this occurs on lipid II92.
Figure 4.
(A)Depiction of the transpeptidation reaction shows cross-linking within the bacterial cell wall. Ceftobiprole led to inhibition of PBP2a and this is responsible for stopping the growth of cell wall89. (B)Illustration shows the development of the small molecule groups and disaccharide constituents of peptidoglycan in the cytoplasmic membrane.At this stage, enzymes encoded by genes successively add amino acids to the lactyl group of N-acetylmuramicacid (NAM). Just before the complete subunit is transported across the membrane. Two amino acid residues, namely two D-alanine (D-Ala) residues, are covalently attached to produce a dipeptide and are linked as a single entity at the end of the side-chain N-acetylglucosamine (NAG)90. (C)Adjacent strands are cross-linked in peptidoglycan assembly by transpeptidation91. (D)Synthesis of pentaglycine interpeptide, and this occurs on lipid II92.
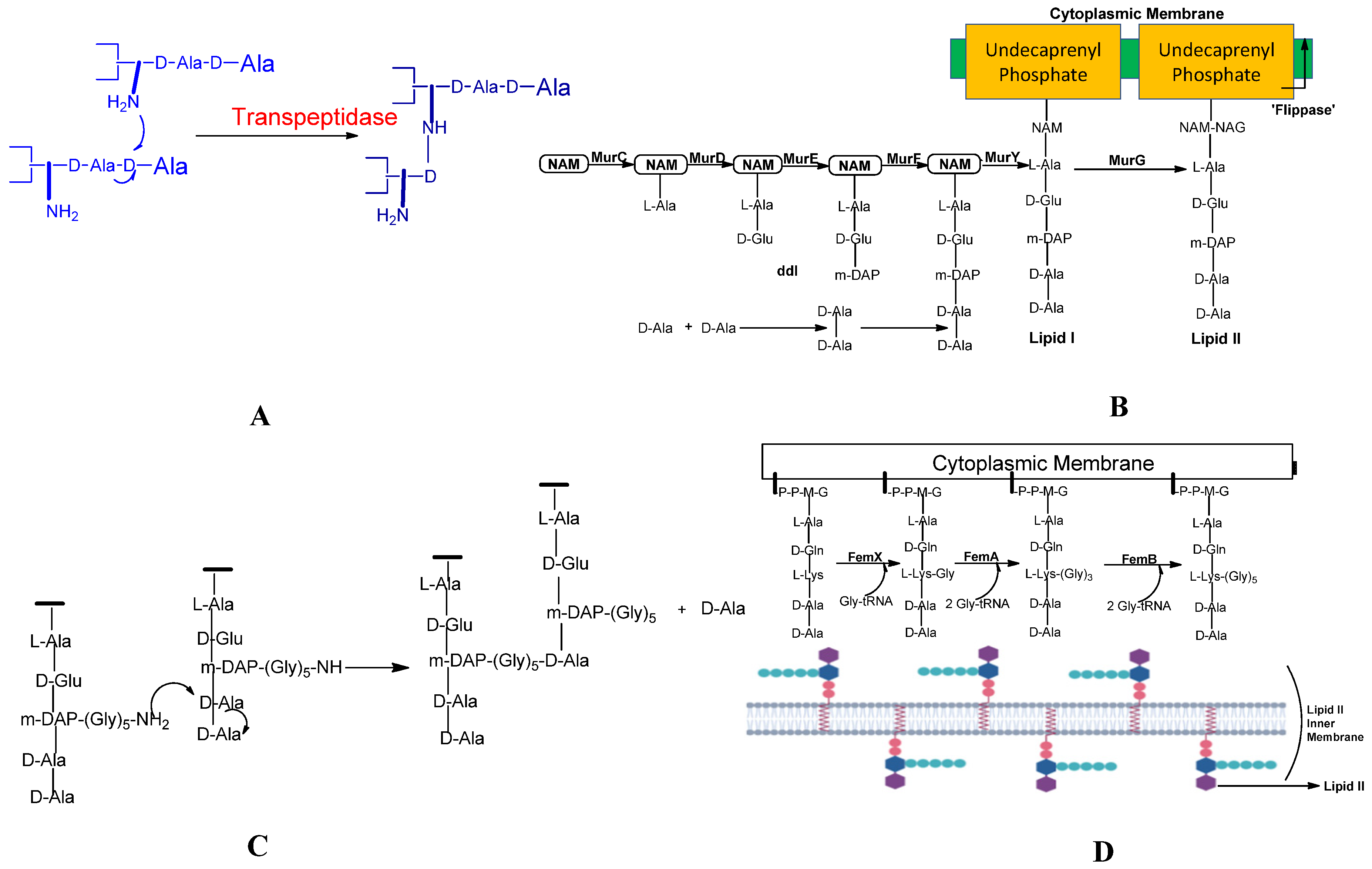
Figure 5.
A Detailed illustration of MRSA resistance.
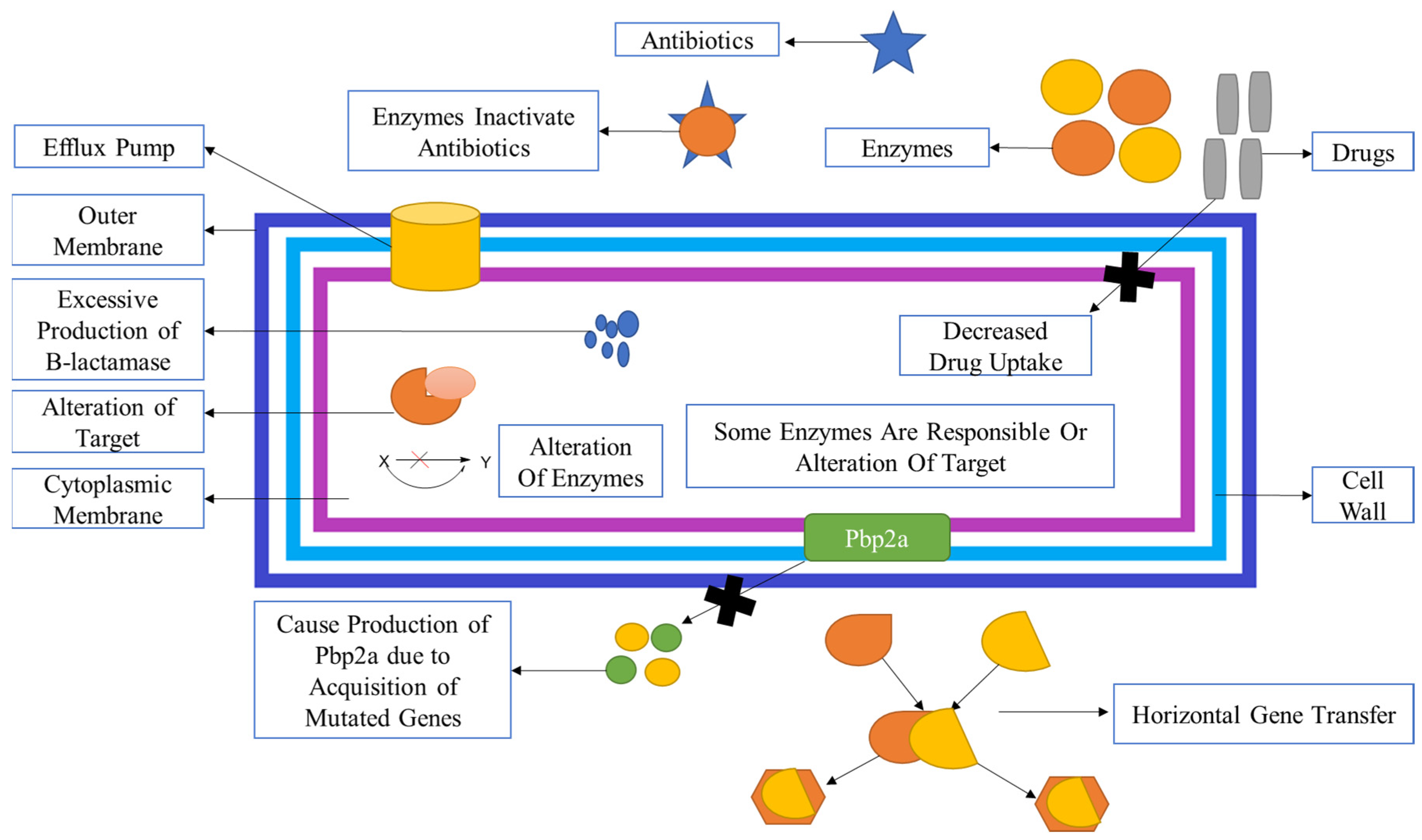
Figure 7.
The surface interface of PBP2a (PDB ID. 1VQQ), it is a soluble structure of MRSA strain 27r, which is determined at 1.80Å resolution. This provides the highest resolution for high molecular mass PBP116. The active site residues of penicillin-binding protein 2a (PBP2a) are Ser403, Lys406, Arg445, Tyr446, Glu447, Ile459, Glu460, Ser403, Ser462, Asp463, Asn464117.
Figure 7.
The surface interface of PBP2a (PDB ID. 1VQQ), it is a soluble structure of MRSA strain 27r, which is determined at 1.80Å resolution. This provides the highest resolution for high molecular mass PBP116. The active site residues of penicillin-binding protein 2a (PBP2a) are Ser403, Lys406, Arg445, Tyr446, Glu447, Ile459, Glu460, Ser403, Ser462, Asp463, Asn464117.
Figure 8.
(A) Penicillin’s (methicillin and oxacillin) targeting PBPs, (B) β-lactam antibiotics (piperacillin, tazobactam, meropenem) showing PBP2a inhibition when given in combination132. (C) Cephalosporin’s targeting PBPs (ceftobiprole, ceftaroline, nitrocefin, CB-181963).
Figure 8.
(A) Penicillin’s (methicillin and oxacillin) targeting PBPs, (B) β-lactam antibiotics (piperacillin, tazobactam, meropenem) showing PBP2a inhibition when given in combination132. (C) Cephalosporin’s targeting PBPs (ceftobiprole, ceftaroline, nitrocefin, CB-181963).
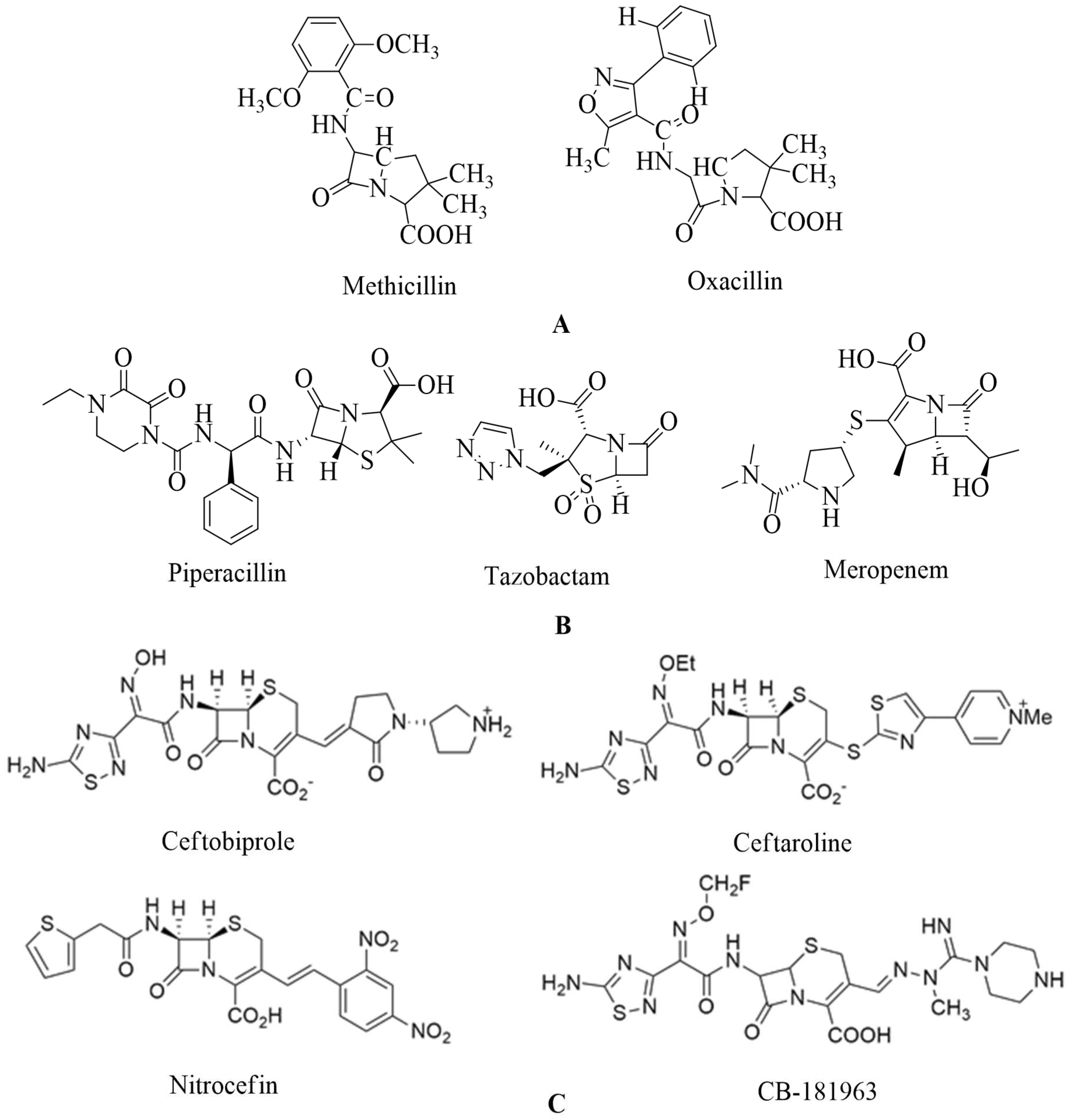
Figure 9.
Depiction of Ceftobiprole interaction with PBP2a (PDB ID. 4DKI); (A) Structure shows ceftobiprole bound with PBP2a and neighbouring amino acids which interact with ligand, and (B) showing 2D interaction of same119.
Figure 9.
Depiction of Ceftobiprole interaction with PBP2a (PDB ID. 4DKI); (A) Structure shows ceftobiprole bound with PBP2a and neighbouring amino acids which interact with ligand, and (B) showing 2D interaction of same119.
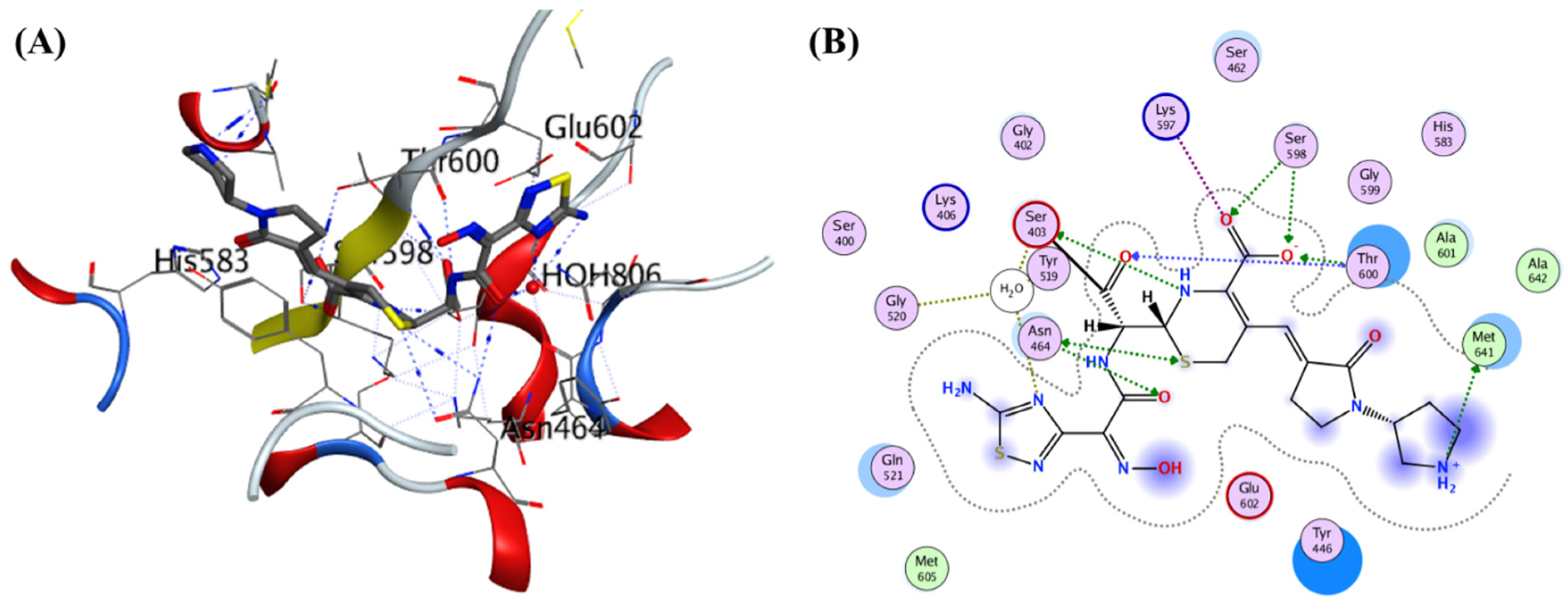
Figure 10.
(A) Structures of novel pyrazole-based derivatives, (B) Structure of novel benzimidazole-based derivatives, (C) Synthesis route for benzimidazole-based derivates, Reaction details: Chemical agent name and reagent used are (1) 1-fluoro-2-nitrobenzene added to (2) 2-phenylethanamine reagent (a) K2CO3, DMF added and reflux for overnight (3) 2-nitro-N-phenethylaniline obtained to which (b) aldehyde, Sodium dithionite, DMSO added and kept overnight gives (4) 2-(3-nitrophenyl)-1-phenethyl-1H-benzo[d] imidazole procured by adding reducing agent (c) SnCl2.2H2O, EtOAc final N-(3-(1-phenethyl-1H-benzo[d] imidazole -2yl) phenyl) methanesulfonamide obtained147.
Figure 10.
(A) Structures of novel pyrazole-based derivatives, (B) Structure of novel benzimidazole-based derivatives, (C) Synthesis route for benzimidazole-based derivates, Reaction details: Chemical agent name and reagent used are (1) 1-fluoro-2-nitrobenzene added to (2) 2-phenylethanamine reagent (a) K2CO3, DMF added and reflux for overnight (3) 2-nitro-N-phenethylaniline obtained to which (b) aldehyde, Sodium dithionite, DMSO added and kept overnight gives (4) 2-(3-nitrophenyl)-1-phenethyl-1H-benzo[d] imidazole procured by adding reducing agent (c) SnCl2.2H2O, EtOAc final N-(3-(1-phenethyl-1H-benzo[d] imidazole -2yl) phenyl) methanesulfonamide obtained147.
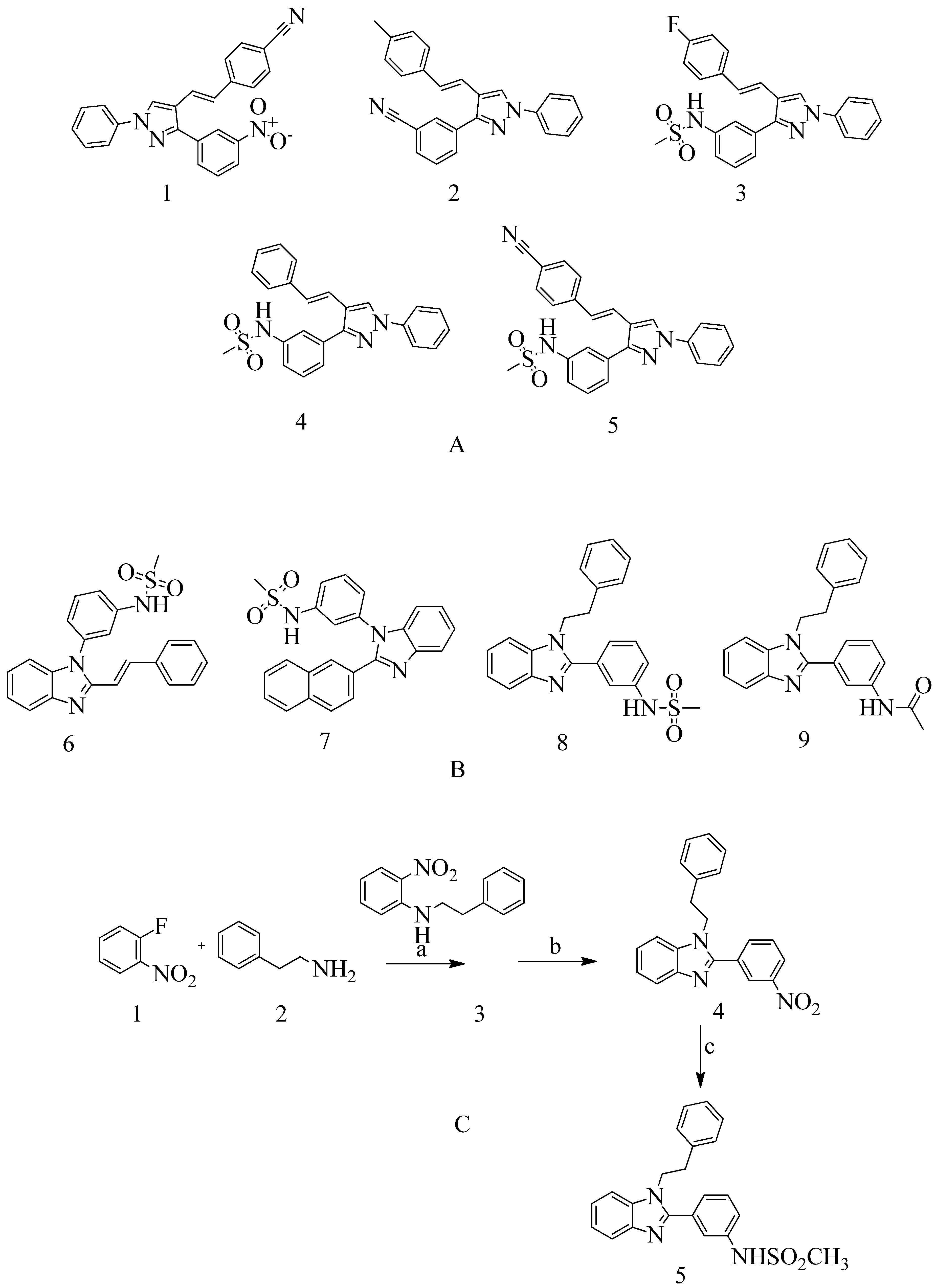
Figure 11.
(A) Novel structure of 1,2,4-Oxadiazole-containing derivatives151, (B) Synthesis route for 1,2,4-oxadiazole-containing derivatives; Reagent used for reaction are: a) CuI, CsCO3, N, N-dimethyl glycine HCl, 1,4-dioxane b) Hydroxylamine ethanol c) Net(i-Pr)2, CH2Cl2 d) THF, (Bu)4NF e) Toluene f) 1 eq. THF, TBAF g) NO2BzCl, toluene h) Fe, H2O, HCl, EtOH148.
Figure 11.
(A) Novel structure of 1,2,4-Oxadiazole-containing derivatives151, (B) Synthesis route for 1,2,4-oxadiazole-containing derivatives; Reagent used for reaction are: a) CuI, CsCO3, N, N-dimethyl glycine HCl, 1,4-dioxane b) Hydroxylamine ethanol c) Net(i-Pr)2, CH2Cl2 d) THF, (Bu)4NF e) Toluene f) 1 eq. THF, TBAF g) NO2BzCl, toluene h) Fe, H2O, HCl, EtOH148.
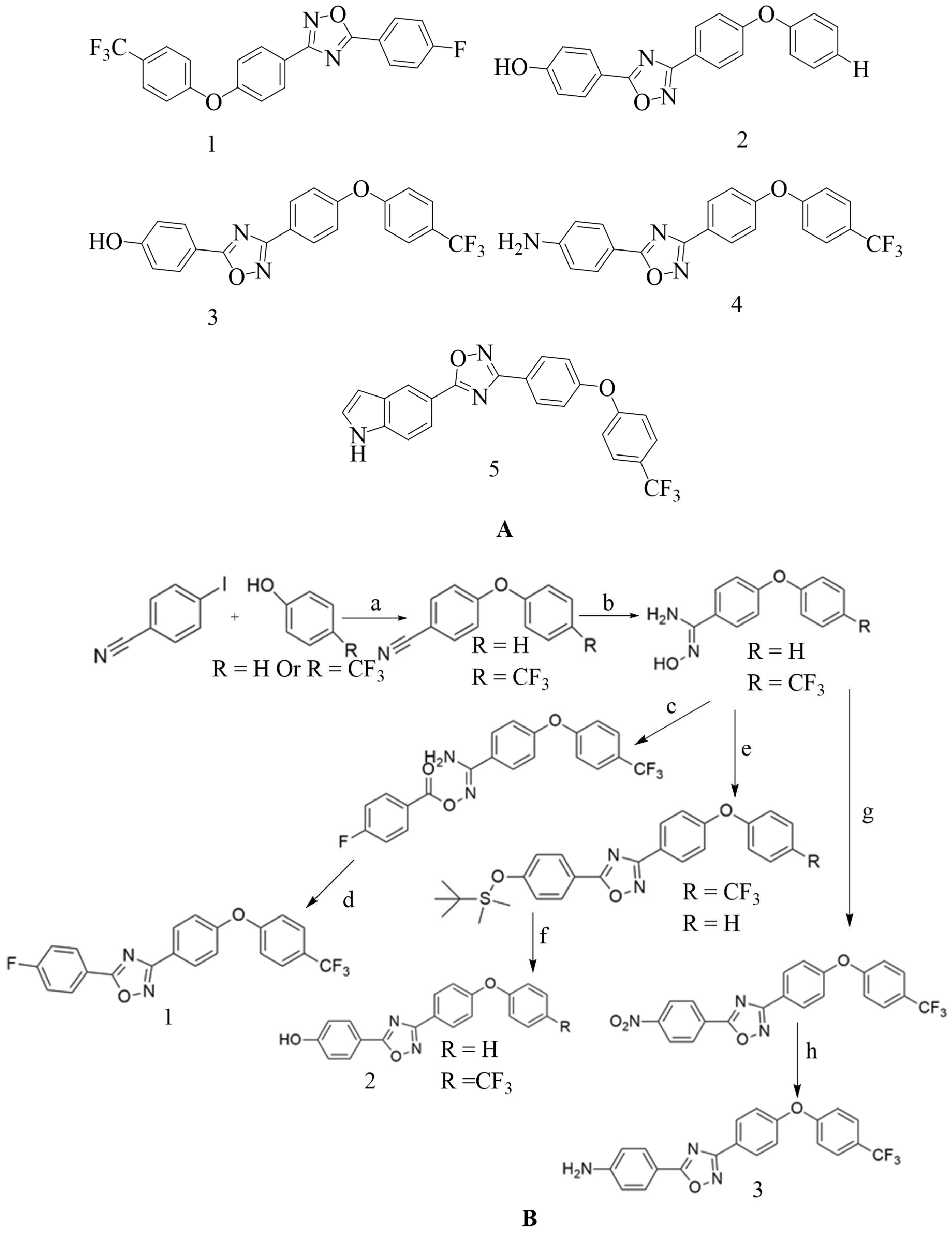
Figure 12.
Non β-lactam allosteric inhibitors152.
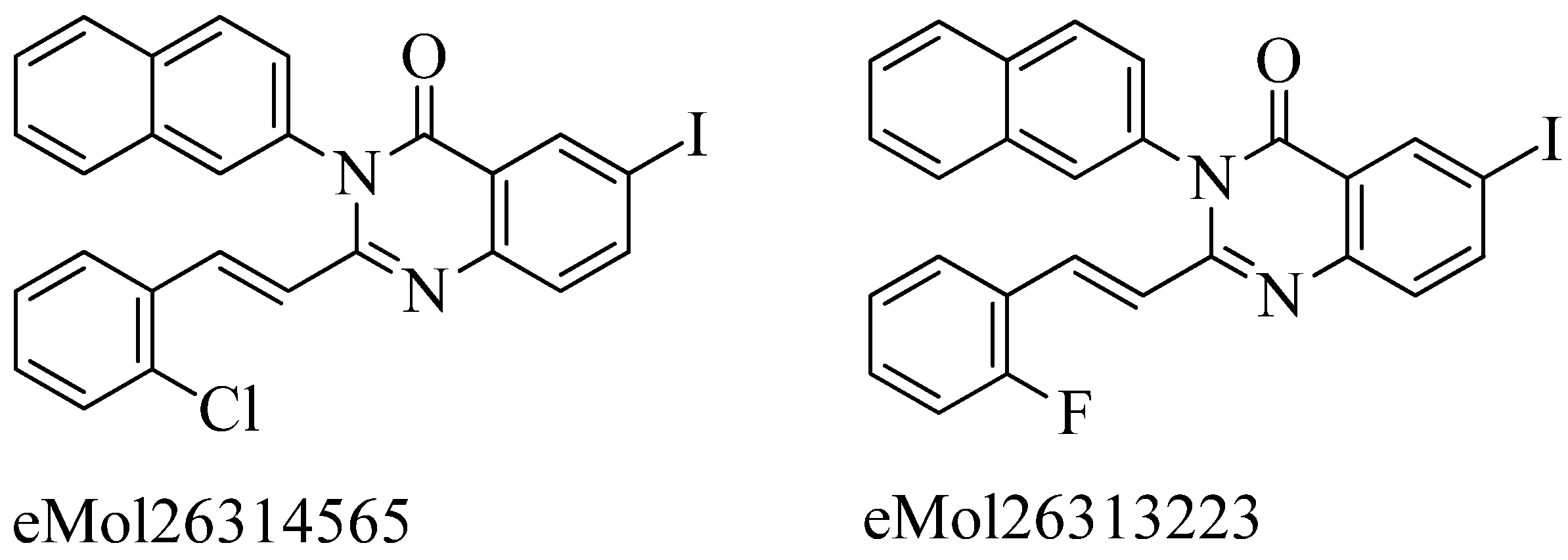
Figure 13.
(A)Structure of 4-(3H)-quinazolinone, (B) Synthesis route of 4-(3H)-quinazolinone (compound 4 (see Figure 12 A)); a) quinazolinone, anthranilic acid, b) acetic acid, c)aldol-type condensation reaction, aromatic aldehyde154.(C) Synthesis route of 4-(3H)-quinazolinone (compounds 1 and 2(see Figure 12 A)); Reagents and conditions used for synthesis: SOCl2, triethylamine (TEA), DCM, put overnight; b) Fe, CaCl2, Ethanol: H2O, reflux for 5 to 6 hours) AcOH, reflux for 1 to 2 hours, d) AcOH, reflux for 1 to 2 hours, e) AcOH, reflux for 24 to 48 hours.
Figure 13.
(A)Structure of 4-(3H)-quinazolinone, (B) Synthesis route of 4-(3H)-quinazolinone (compound 4 (see Figure 12 A)); a) quinazolinone, anthranilic acid, b) acetic acid, c)aldol-type condensation reaction, aromatic aldehyde154.(C) Synthesis route of 4-(3H)-quinazolinone (compounds 1 and 2(see Figure 12 A)); Reagents and conditions used for synthesis: SOCl2, triethylamine (TEA), DCM, put overnight; b) Fe, CaCl2, Ethanol: H2O, reflux for 5 to 6 hours) AcOH, reflux for 1 to 2 hours, d) AcOH, reflux for 1 to 2 hours, e) AcOH, reflux for 24 to 48 hours.
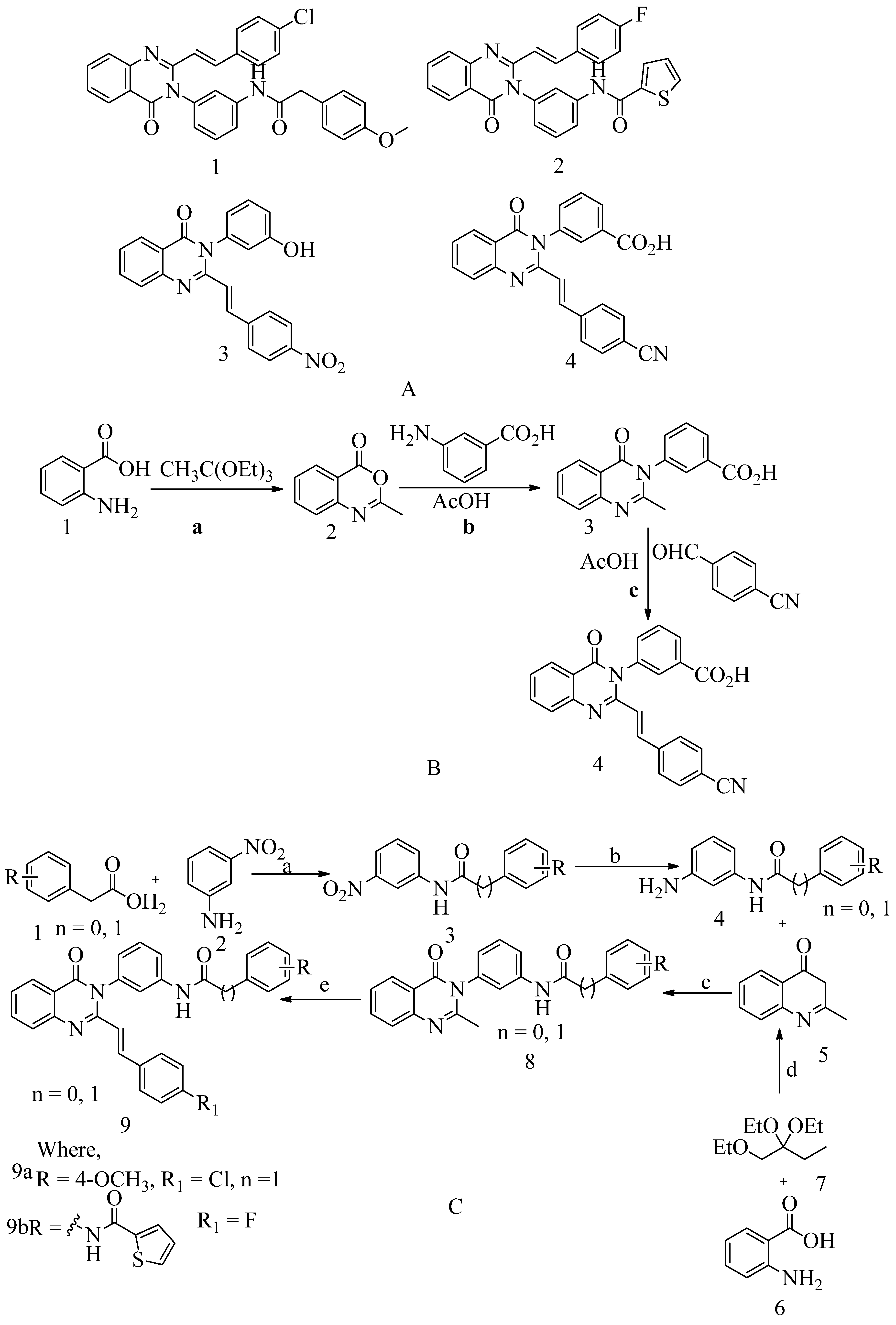
Figure 14.
A)Pyrrolylated-chalcone, (B)Synthetic route for pyrrolylated-chalcone. It is a Claisen-Schmidt condensation reaction. 2-Acetopyrrole reacting with substituted benzaldehyde in ethanolic sodium hydroxide for 24hrs at room temperature155.
Figure 14.
A)Pyrrolylated-chalcone, (B)Synthetic route for pyrrolylated-chalcone. It is a Claisen-Schmidt condensation reaction. 2-Acetopyrrole reacting with substituted benzaldehyde in ethanolic sodium hydroxide for 24hrs at room temperature155.
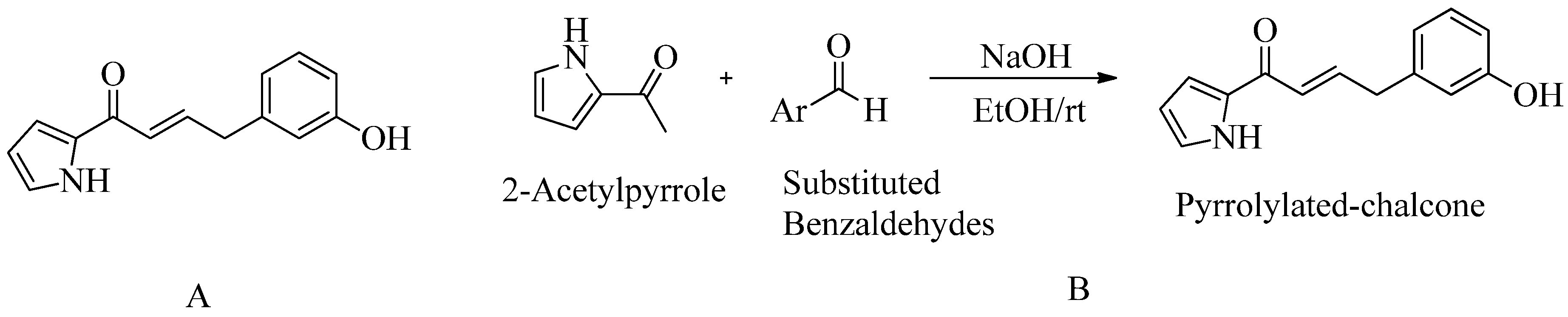
Figure 15.
(A) β-lactams with 1,3-bridges inhibiting PBP2a, (B) Macrocycle-embedded β-lactam antibiotics, (C) Pharmacophore model for proposed compound, (D) Pyridine-coupled pyrimidinone157.
Figure 15.
(A) β-lactams with 1,3-bridges inhibiting PBP2a, (B) Macrocycle-embedded β-lactam antibiotics, (C) Pharmacophore model for proposed compound, (D) Pyridine-coupled pyrimidinone157.
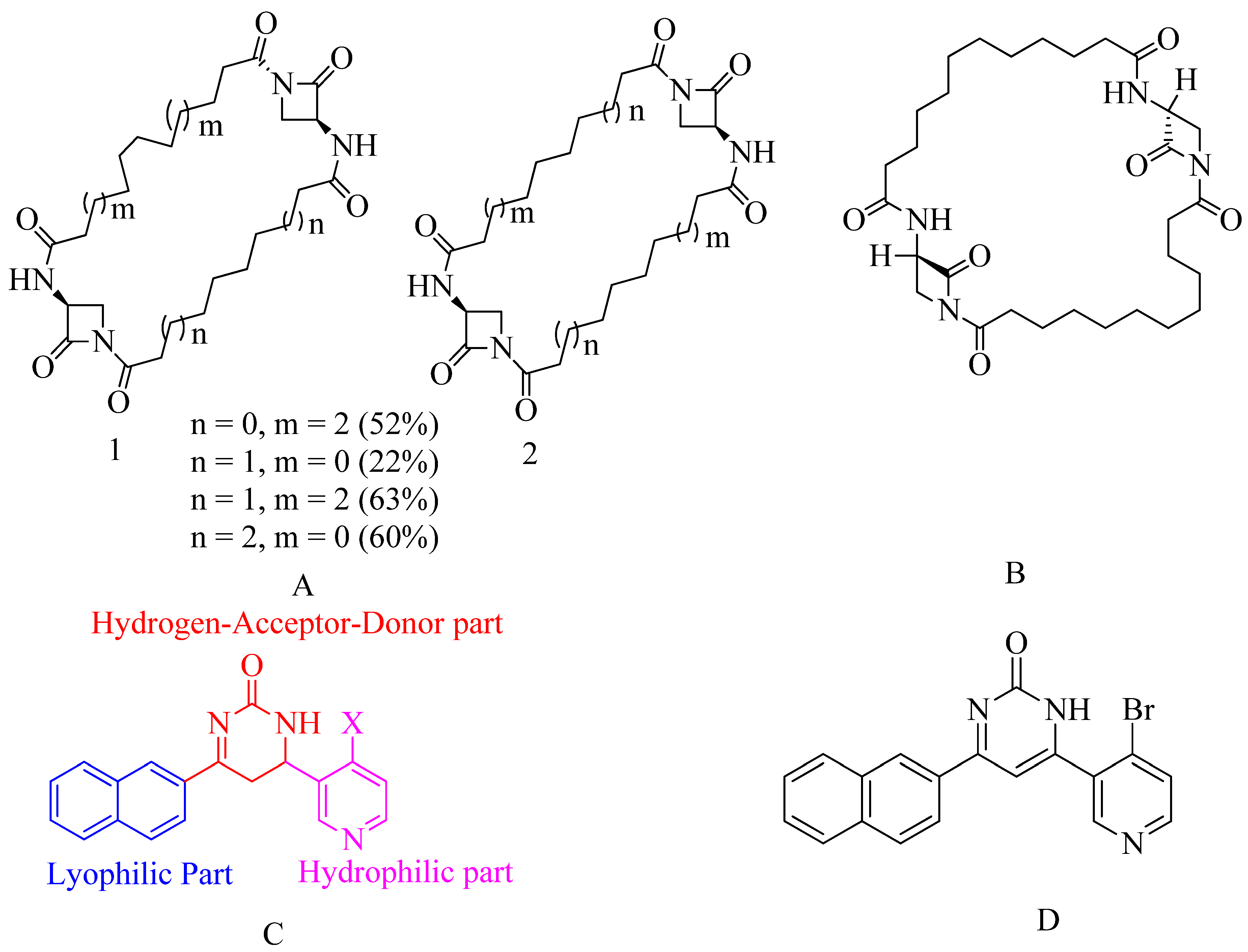
Figure 16.
(A) A Novel naphthalimide corbelled aminothiazoximes modulating PBP2a, (B)Preliminary procedure for naphthalimide corbelled aminothiazoximes, (C) Synthesis route for novel naphthalimide corbelled aminothiazoximes PBP2a modulator; a) 2-(2-amino-4-thiazolyl)-2-methoxyiminoacetic (361 mg, 1.03 mmol), b) 10 mL of ethanol, c) 104 mg, 1.03 mmol of triethylamine at 25 C. 4a was used as starting material to synthesized this one. This containing ethyl unit-connected to the hydrophilic group. However, by incorporation of the dimethylenediamine group reduce the MIC value to 0.05 µg/mL, effectively suppressing MRSA development to twice of tetracycline, 4 times of vancomycin, 8 times of ciprofloxacin, and 32 times of norfloxacin from reported data158.
Figure 16.
(A) A Novel naphthalimide corbelled aminothiazoximes modulating PBP2a, (B)Preliminary procedure for naphthalimide corbelled aminothiazoximes, (C) Synthesis route for novel naphthalimide corbelled aminothiazoximes PBP2a modulator; a) 2-(2-amino-4-thiazolyl)-2-methoxyiminoacetic (361 mg, 1.03 mmol), b) 10 mL of ethanol, c) 104 mg, 1.03 mmol of triethylamine at 25 C. 4a was used as starting material to synthesized this one. This containing ethyl unit-connected to the hydrophilic group. However, by incorporation of the dimethylenediamine group reduce the MIC value to 0.05 µg/mL, effectively suppressing MRSA development to twice of tetracycline, 4 times of vancomycin, 8 times of ciprofloxacin, and 32 times of norfloxacin from reported data158.
Figure 17.
(A)Non-covalent inhibitors, (B) Novel carbapenem ME1036160, (C) Novel carbapenem L-695,256, (D) Tomopenem, (E) Structure of carbapenem, flavan-3-ol, and alkyl boronic acid shows inhibitory action against PBP2a...
Figure 17.
(A)Non-covalent inhibitors, (B) Novel carbapenem ME1036160, (C) Novel carbapenem L-695,256, (D) Tomopenem, (E) Structure of carbapenem, flavan-3-ol, and alkyl boronic acid shows inhibitory action against PBP2a...
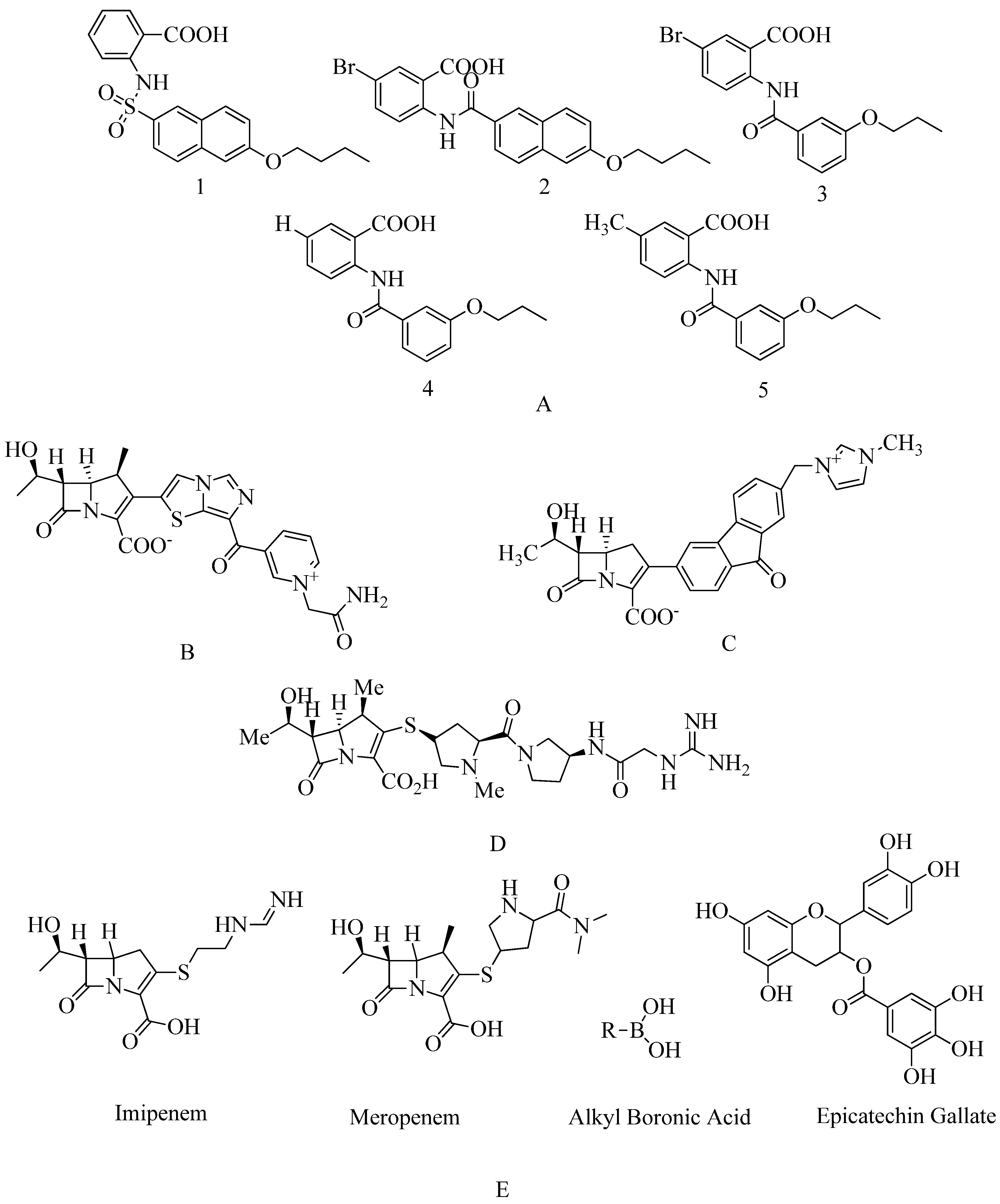
Figure 18.
(A) Novel Benzoxazole Derivatives, (B) Synthetic route for novel Benzoxazole Derivatives165.
Figure 18.
(A) Novel Benzoxazole Derivatives, (B) Synthetic route for novel Benzoxazole Derivatives165.
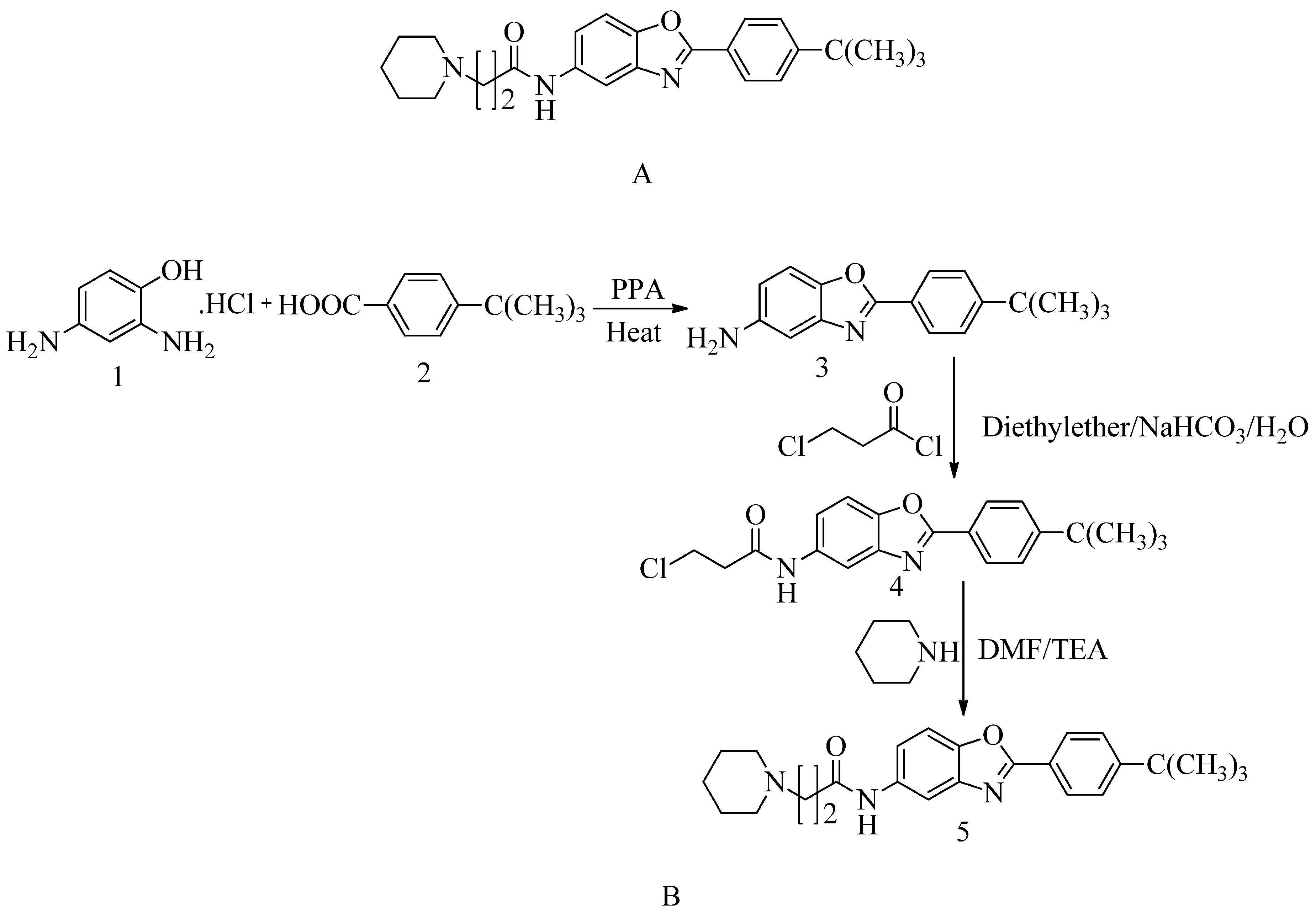
Figure 19.
A)Pyrazolylpyridine analogues, (B)Synthetic route for pyrazolylpyridine analogues; 1) phenylhydrazine, 2) 1,3 dicetone, and 3) pyridine carboxaldehyde, refluxed for 3-4 hrs using basic medium (sodium ethoxide)166.
Figure 19.
A)Pyrazolylpyridine analogues, (B)Synthetic route for pyrazolylpyridine analogues; 1) phenylhydrazine, 2) 1,3 dicetone, and 3) pyridine carboxaldehyde, refluxed for 3-4 hrs using basic medium (sodium ethoxide)166.
Figure 20.
(A)Structure of polyhydroxylated flavonoids is showing promising synergistic activity against PBP2a168, (B) Structure of demethoxycurcumin169, (C) Ursolic acid 3-O-α-L-arabinopyranoside structure, (D) Structure of Thioridazine, (E) Formation of hybrid, (F)Structure of Metronidazole-Triazole Hybrids, (G) Synthetic route for Metronidazole-Triazole Hybrids; reagent used in synthesis and procedure: Reagent (a) p-TsCl, Pyridine, (b) NaN3, DMF (c) t-BuOH: H2O (1:1), Sodium ascorbate; CuSO4.5H2On used for the, synthesis of metronidazole-triazole hybrids175, (H) Structure of Aspermerodione, (I) Synthesis route of chitosan-ferulic acid conjugate, (J) Indole-Nitroimidazole conjugates177, (K) Synthesis route of Indole-Nitroimidazole conjugates; (a) potassium carbonate, (b) dimethylformamide, and a phosphorus oxychloride catalyst, promoters, piperidine, and glacial acetic acid177, (L) Indole-Nitroimidazole conjugates intermediates, (M) Structure of Tellimagrandin II178.
Figure 20.
(A)Structure of polyhydroxylated flavonoids is showing promising synergistic activity against PBP2a168, (B) Structure of demethoxycurcumin169, (C) Ursolic acid 3-O-α-L-arabinopyranoside structure, (D) Structure of Thioridazine, (E) Formation of hybrid, (F)Structure of Metronidazole-Triazole Hybrids, (G) Synthetic route for Metronidazole-Triazole Hybrids; reagent used in synthesis and procedure: Reagent (a) p-TsCl, Pyridine, (b) NaN3, DMF (c) t-BuOH: H2O (1:1), Sodium ascorbate; CuSO4.5H2On used for the, synthesis of metronidazole-triazole hybrids175, (H) Structure of Aspermerodione, (I) Synthesis route of chitosan-ferulic acid conjugate, (J) Indole-Nitroimidazole conjugates177, (K) Synthesis route of Indole-Nitroimidazole conjugates; (a) potassium carbonate, (b) dimethylformamide, and a phosphorus oxychloride catalyst, promoters, piperidine, and glacial acetic acid177, (L) Indole-Nitroimidazole conjugates intermediates, (M) Structure of Tellimagrandin II178.
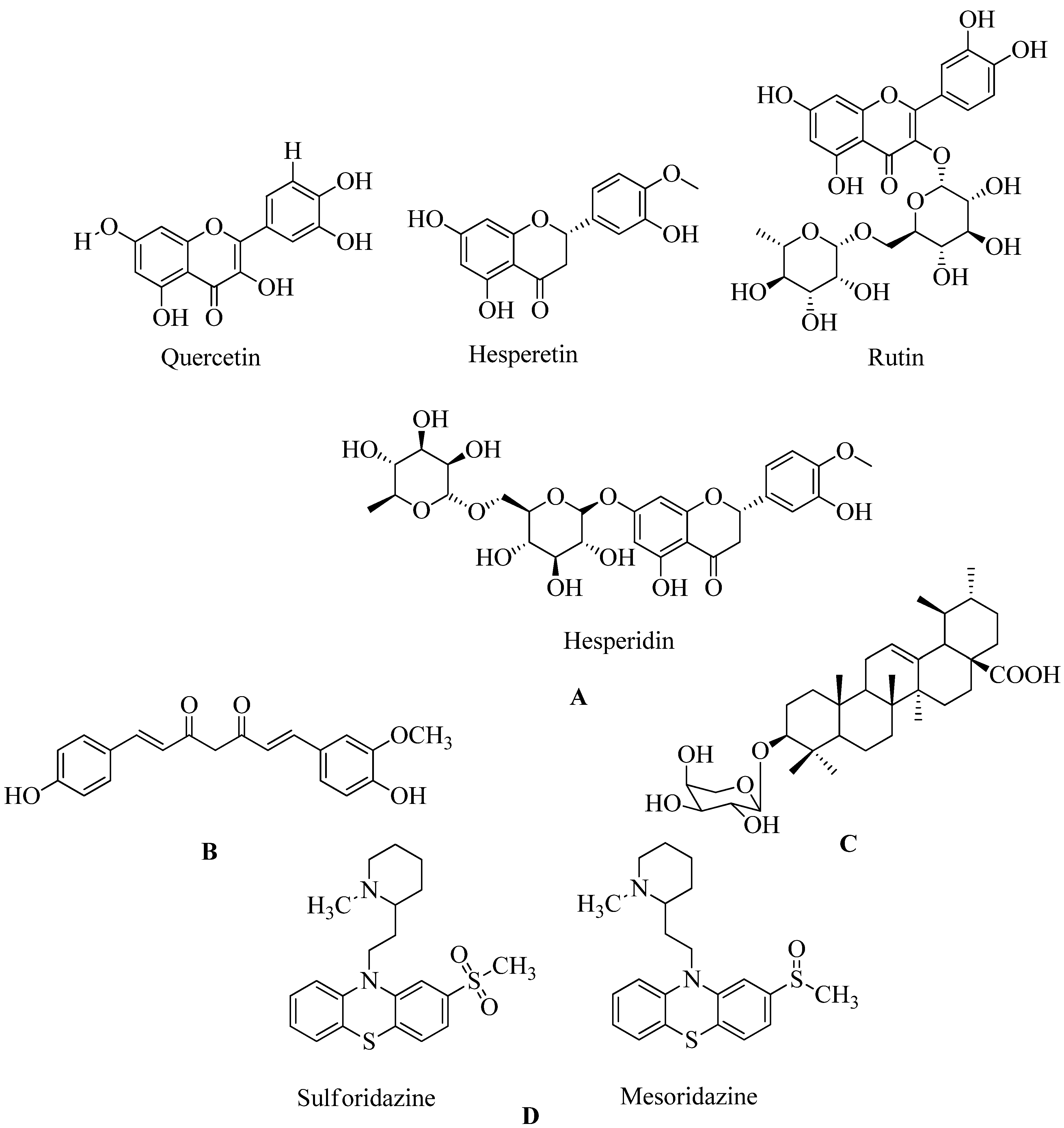
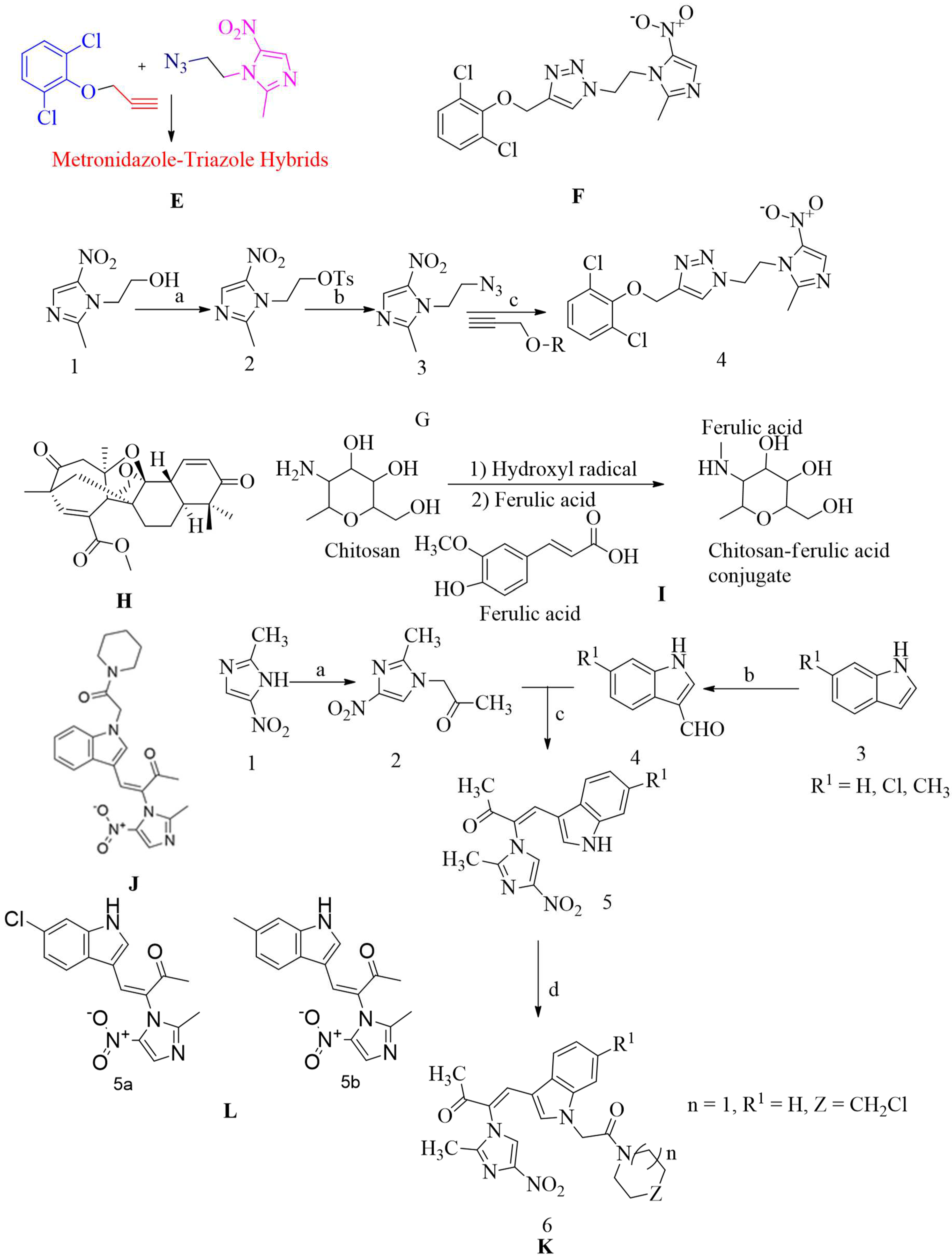
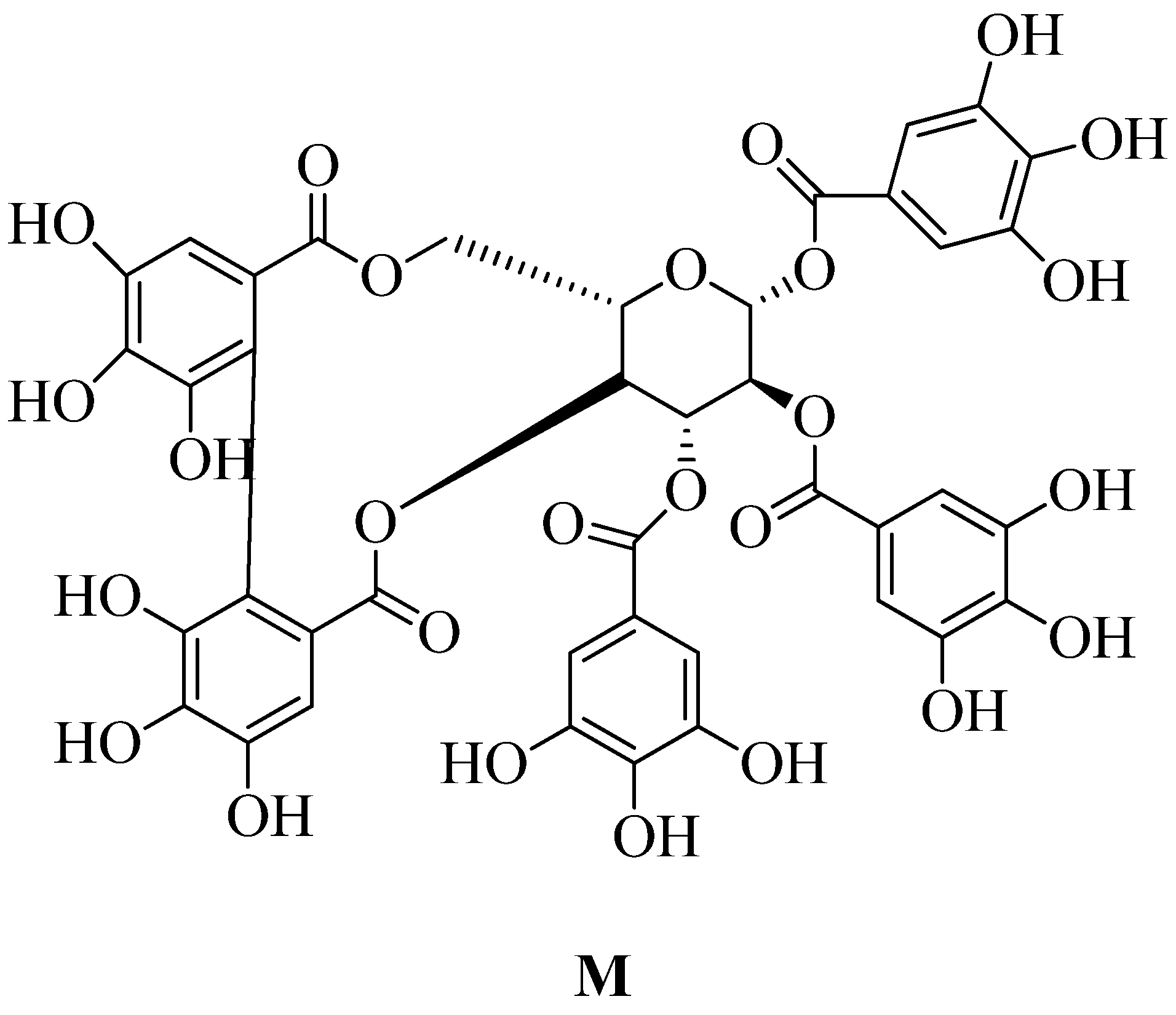
Figure 21.
Binding pattern of imipenem and meropenem from 1VQQ crystal structure179.
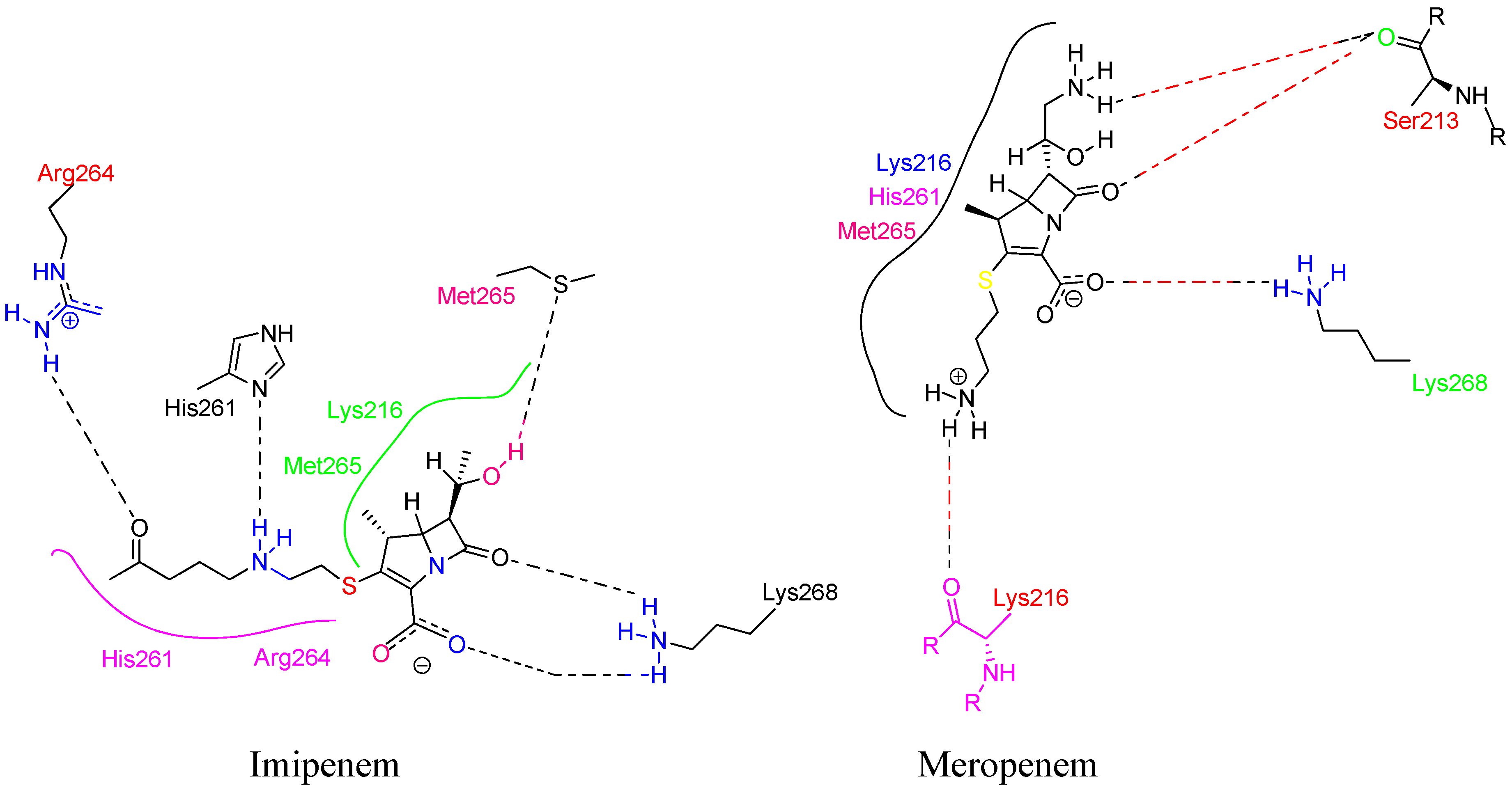
Figure 22.
(A) The catalyst is enabled at the active site by binding peptidoglycan to the allosteric site182 (Copyright permission @ACS), (B) PBP2a ligands detected by computer search. X-ray composition of S. aureus PBP2a is shown as a grey surface that the solvent can penetrate. At 6 o'clock, the protein attaches to the plasma membrane’s surface. Oxadiazole 1 is shown as a CPK model in a docked position at the active site at 1 o'clock, coded by atomic colour type (carbon in dark Gray, nitrogen in blue, oxygen in red, fluorine in green). After the enzyme rotates 90 degrees in the viewer's direction along the Y-axis, the extension provides a stereo picture of the attached oxadiazole; all is reported data182 (Copyright permission @ACS), (C)Chemicals most closely related to PBP2a. Interactions involving PBP2a and even berberine molecules are summarized in A1 and A2. The composition of PBP2a is shown in purple, while the composition of the berbamine compound is shown in red. The H bond between PBP2a and the berbamine molecule appears in A3. (Readers are directed to the web version of this article to clarify the colour references in the narrative of this figure.)183(Copyright permission @Elsevier).
Figure 22.
(A) The catalyst is enabled at the active site by binding peptidoglycan to the allosteric site182 (Copyright permission @ACS), (B) PBP2a ligands detected by computer search. X-ray composition of S. aureus PBP2a is shown as a grey surface that the solvent can penetrate. At 6 o'clock, the protein attaches to the plasma membrane’s surface. Oxadiazole 1 is shown as a CPK model in a docked position at the active site at 1 o'clock, coded by atomic colour type (carbon in dark Gray, nitrogen in blue, oxygen in red, fluorine in green). After the enzyme rotates 90 degrees in the viewer's direction along the Y-axis, the extension provides a stereo picture of the attached oxadiazole; all is reported data182 (Copyright permission @ACS), (C)Chemicals most closely related to PBP2a. Interactions involving PBP2a and even berberine molecules are summarized in A1 and A2. The composition of PBP2a is shown in purple, while the composition of the berbamine compound is shown in red. The H bond between PBP2a and the berbamine molecule appears in A3. (Readers are directed to the web version of this article to clarify the colour references in the narrative of this figure.)183(Copyright permission @Elsevier).
Figure 23.
Virtual screening and biological evaluation of some of the novel PBP2a inhibitors.
Figure 24.
[A] (A) 3D model of the interaction between compound with PBP2a (PDB 4CJN). (B) 2D binding mode within the receptor PBP2a allosteric site pocket. These figures were produced using the Discovery Studio 2017 R2 software186 (Copyright permission @Avoxa), [B] Structure of icariin derivative186.
Figure 24.
[A] (A) 3D model of the interaction between compound with PBP2a (PDB 4CJN). (B) 2D binding mode within the receptor PBP2a allosteric site pocket. These figures were produced using the Discovery Studio 2017 R2 software186 (Copyright permission @Avoxa), [B] Structure of icariin derivative186.
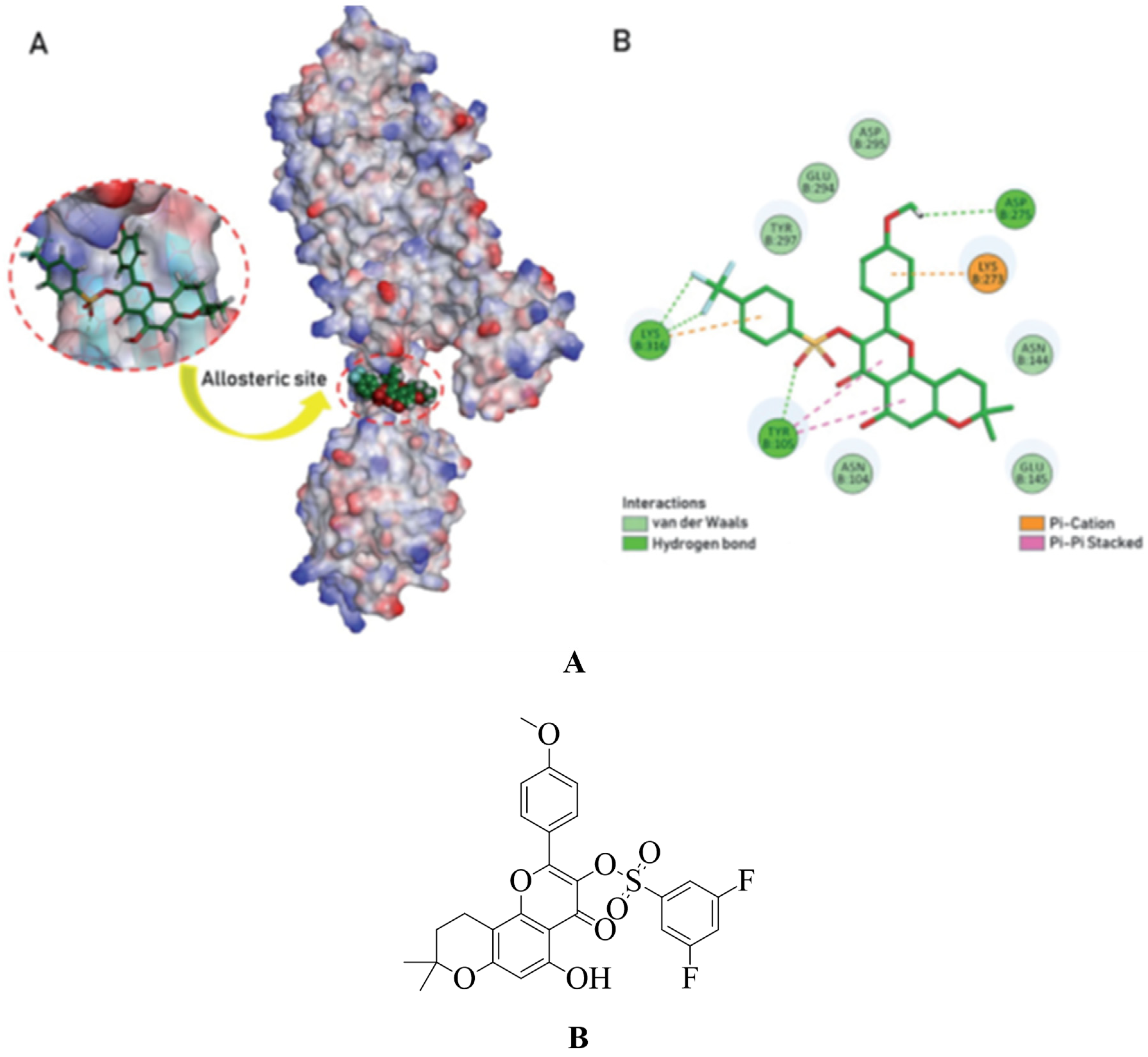
Figure 25.
Schematic representation showing a target and different scaffold targeting PBP2a especially the five membered and six membered heterocyclic scaffolds including miscellaneous drugs.
Figure 25.
Schematic representation showing a target and different scaffold targeting PBP2a especially the five membered and six membered heterocyclic scaffolds including miscellaneous drugs.
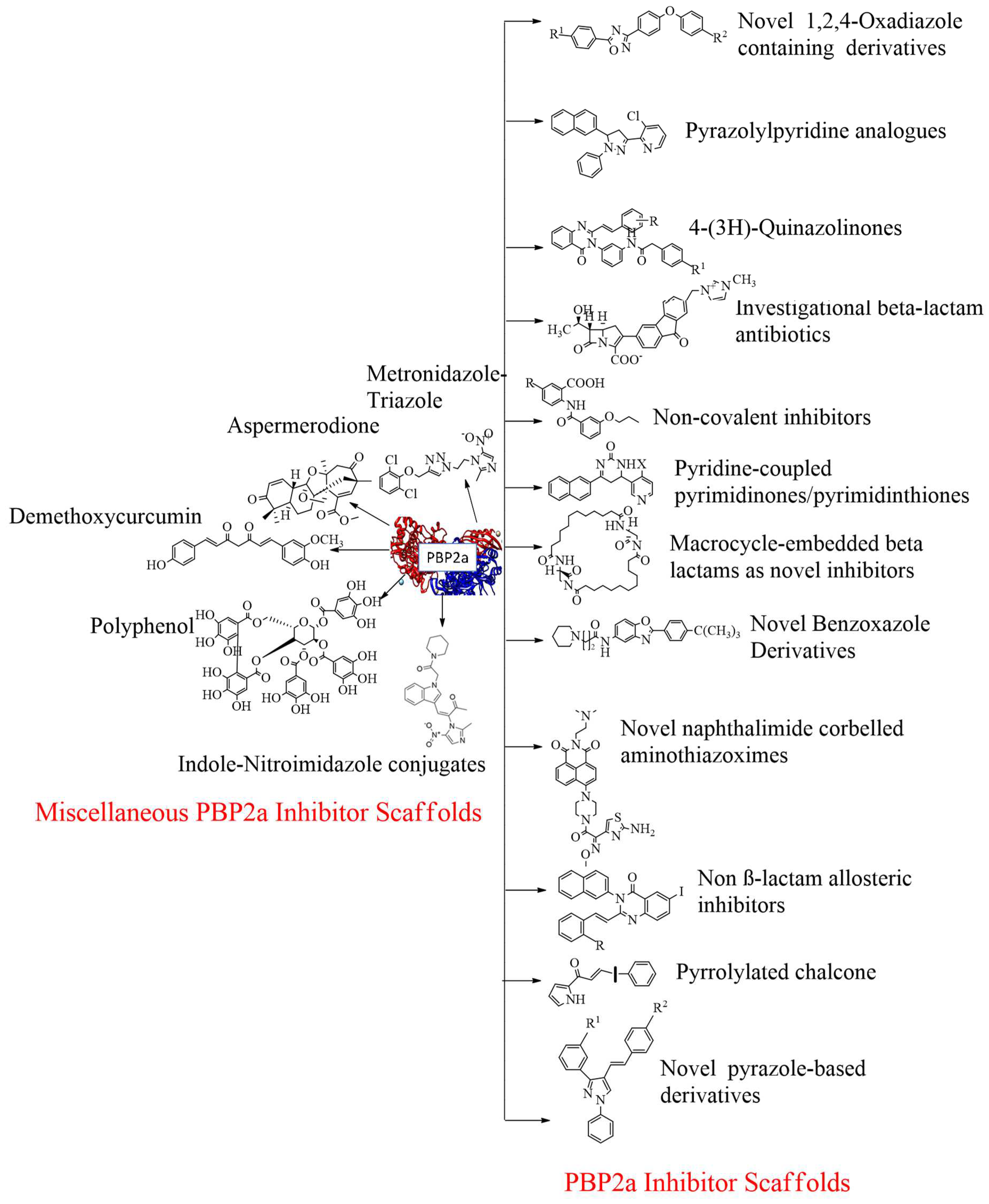
Table 2.
Some crucial genes are listed here, which are responsible for resistance in S. aureus 92,114.
Table 2.
Some crucial genes are listed here, which are responsible for resistance in S. aureus 92,114.
| Sr. No | Name of genes | Role in MRSA |
|---|---|---|
| s1 | Mec (mecA, mecR1-mecIs) | The close relationship between two regulatory elements, one is mec and the second one is bla. BlaR2 or a close relative may be participating in mecA activation here too. |
| 2 | Chromosomal gene | FEM means factors essential for methicillin resistance. Its inactivation reduces methicillin-resistant and the aux (auxiliary) factors usually present in staphylococcal aureus. The majority of elements are engaged in cell wall biosynthesis and some turnover of bacteria. |
| 3 | The PBP2a operon in normal S. aureus contributes to resistance | The mecA is responsible for the production of PBP2a, which helps in the biosynthesis of bacteria's cell walls. However, its role in the resistance of S. aureus is still unclear. |
| 4 | Fmt | Reduce methicillin resistance. |
| 5 | femX, femAB | When reducing the length of the glycine side chain occurs, it is because of the inactivation of the femAB factor. This inactivation and reduced length lead to impairment in the growth of peptidoglycan and also the turnover of cell-wall being reduced. All these reactions are responsible for lowering peptidoglycan cross-linking and hypersusceptibility to all β-lactam antibiotics and other antibiotics, which leads to the decrease in methicillin resistance. |
| 6 |
glnRA (femC) (Mutated gene) |
The precursors of muropeptide use an amino acid which is a non-amidated D-glutamate amino acid, and it takes part in the stem peptide of the transpeptidation reactions less readily, which leads to a reduction of methicillin resistance |
| 7 | glmM (femD, femR) | When the rate of precursor formation for peptidoglycan is reduced, it reduces methicillin resistance. It increases teicoplanin susceptibility and thus decreases methicillin resistance in MRSA. |
| 8 | Lytic enzymes (Hydrolysis Enzyme) |
Murein, which is present in Staphylococcus aureus, causes hydrolysis. It is then needed for the growth of peptidoglycan, which is a precious cell wall content. This play one of the leading roles in peptidoglycan growth. |
| 9 | murE (femF) | In the formation of peptidoglycan precursors, MurE necessitates the presence of this factor. Because the abnormal precursors of peptidoglycan are present, which causes a decline in methicillin resistance, it is also possible that there may be abnormal shortening precursors involved in the peptidoglycan biosynthesis. It is still unclear. |
| 10 | Llm gene | The llm gene encodes lipophilic membrane proteins, affecting methicillin resistance levels and necessarily causing bacterial cell lysis rate reduction. Its functions, however, are still unclear. |
| 11 | Global regulators: sar, agr, sigB | The global regulators like sar and agr control cell density-dependent synthesis of cell wall factors that are cell wall-associated and extracellular virulence factors. In heterogeneous MRSA, this global regulator appears to have just a slight effect on methicillin resistance in MRSA. |
| 12 | ctaA | Reduced the resistance to methicillin in MRSA |
| 13 | Blaz | The blaZ gene has been mutated in Staphylococcus aureus, and it is responsible for the breakdown of the β-lactam ring of mainly penicillin antibiotics. |
| 14 | blaI | blaI is a repressor protein that is responsible for preventing the transcription of mutated genes like blaZ or mecA. |
| 15 | blaR1 | blaR1 is a transmembrane protein that signals blaZor mecA transcription. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



