
Submitted:
09 September 2023
Posted:
14 September 2023
You are already at the latest version
Abstract
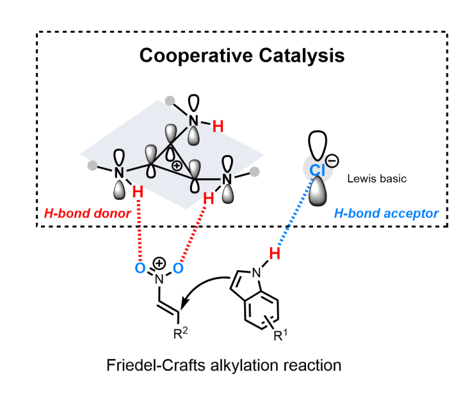
H-bonding, including H-bond donor (HBD) with electron-rich part of a substrate and H-bond acceptor (HBA) with electron-deficient part of a substrate, has achieved massive success. Faster transformation often correlates with more donicity of HBD. Besides the positive charge are employed to enhance the donicity of HBD, the electron withdrawing groups become a dissimilar approach for increasing the donicity of HBD. We describe newly designed H-bond donor catalysts, tris(monoalkylamino)cyclopropenium, implemented by vicinal positive charged on the cyclopropenium core. The counter anion became potential HBA to activate the electron-deficient part of a substrate. The tris(phenylamino)cyclopropenium chloride (TPAC·Cl) as a representative catalyst was applied in Friedel–Crafts alkylation of indoles with nitroalkenes. X-ray analyses of a single crystal of TPAC·Cl described the 3D architecture and the delocalized cationic charge in the solid state. Unit formal positive charge turned N–H moieties into H-bond donor (HBD) and the counter chloride anion exhibited potential H-bond acceptor (HBA). The HBD and HBA displayed cooperative organocatalysis in Friedel–Crafts alkylation of indoles with nitroalkenes. A new class of hydrogen bonding catalysis and working mechanism were proposed.
Keywords:
Cyclopropenium
; Friedel–Crafts alkylation
; Hydrogen bond
; Organocatalysis
1. Introduction
Hydrogen bonding catalysis has become a thriving and vital domain area in the past two decades [1,2,3,4,5,6,7,8,9]. Its behavioral mode is coordination of an H-bond donor (HBD) with electron-rich part of a substrate and/or coordination of an H-bond acceptor (HBA) with electron-deficient part of a substrate. Elaboration on control and selectivity is realized by HBD and HBA cocatalysis [2,10,11,12]. Fast reaction rate often correlates with high donicity of HBD [13,14,15]. One category of charged HBD, deriving from protonated super strong Brønsted base, shows high donicity due to pi-delocalization of the positive charge, such as in guanidiniums [16,17,18,19,20] (Scheme 1, a). Neutral HBD enhanced by the deliberate installation of electron withdrawing groups, such as in ureas and thioureas, has been developed as one mature direction [9,10,12,21,22,23,24] (Scheme 1, b). Another strategy for increasing the donicity of HBD is electron-withdrawing via sigma-bond to vicinal positive charged atom(s), such as in quaternary ammonium [25,26,27,28] (Scheme 1, c).
We suggested that the minimal Hϋckel aromatic ring cyclopropenium [29,30] in a pattern of tri-substitution with amino groups of NHR, i.e. tris(monoalkylamino)cyclopropenium, would behave as an H-bond donor on the N–H moieties (Scheme 1, d). The donicity of the N–H moieties in this new type of H-bond donor was implemented by vicinal positive charges on the cyclopropenium core. Herein, we demonstrated, for the first time, the tris(monoalkylamino)cyclopropenium as H-bonding catalyst to promote Friedel–Crafts alkylation of indoles with nitroalkenes.
2. Results and Discussion
Friedel–Crafts alkylation (F–C alkylation) of indoles is an important reaction for the formation of new carbon-carbon bonds and construction of versatile indole-containing scaffolds [35,36]. Organocatalysis in this area has received tremendous success under nearly all the major catalytic activation modes. Particular interest was focused on H-bonding catalytic F–C alkylation due to the mildness of reaction conditions and the broad tolerance of functional groups [1,36,37,38]. A valuable unprotected indole N–H moiety made the F–C adducts readily accessible to bioactive compounds [37]. Importantly, a free N–H readily accepts H-bond donor from a nucleophilic catalyst.
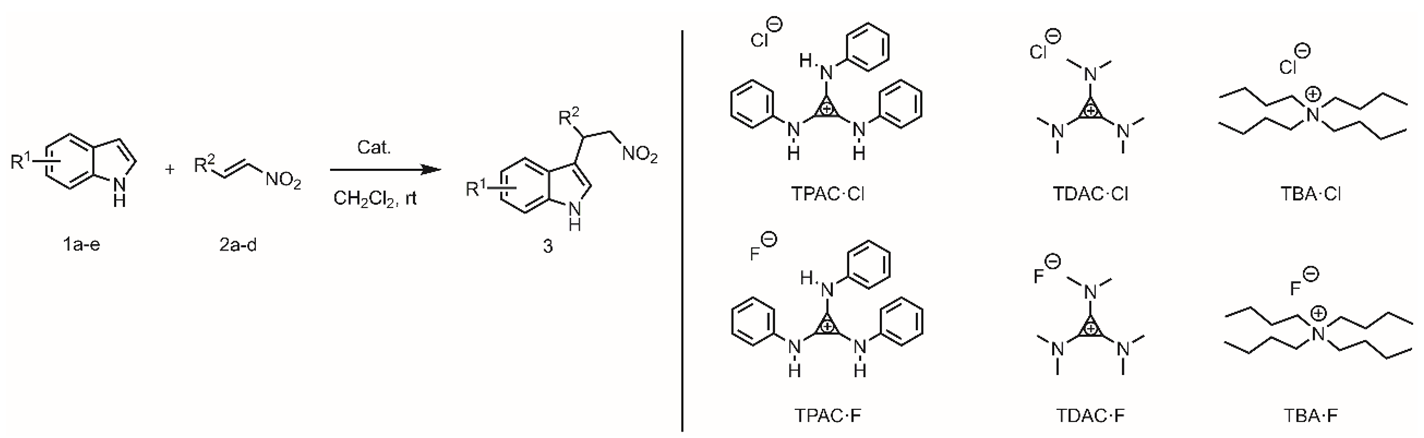
In the context of the ordinary H-bond [36,37,39] and the metal-enhanced H-bond catalysis,[40] we expanded exploration of the novel H-bond donor implemented by the vicinal positive on the cyclopropenium in F–C alkylation of indole 1 with trans-β-nitroalkene 2. Tri(phenylamino)cyclopropenium chloride (TPAC·Cl) was selected as a quintessential HBD catalyst (Table 1). The TPAC·Cl was able to promote the F–C alkylation of 1a with 2a at 25 °C in dichloromethane efficiently (Table 1, entry 2); in contrast, the noncatalyzed background reaction was negligible (entry 1). The TPAC·F ion pair, replacing the TPAC·Cl, performed no activity in the same reaction by changing the chloride to fluoride anion (entry 3). Poor catalytic performance implied that the chloride anion may be necessary for F–C alkylation. A possible reason was that the fluoride anion was easily coordinated with the HBD of the cationic TPAC, similar to the normal fluoride anion receptors with HBD structures [9,41,42]. Poor catalytic performance suggested that TPAC·F formed a tight ion pair [43] and the cationic TPAC preferably paired with the fluoride anion rather than to activate a substrate.
Dialkylamino-substituted cyclopropenium was tested on the F–C alkylation to verify the assumption that N–H moieties of the TPAC as an HBD were essential in the H-bonding catalysis (entries 4 and 5). The tris(dimethylamino)cyclopropenium chloride (TDAC·Cl) displayed no catalytic performance on the F–C alkylation (entry 4). The possible reason was the lack of N–H moiety on the cationic core TDAC and loss of the ability to work as an H-bond donor. Although fluoride anion, possessing strong nucleophilic, was supposed an excellent H-bond acceptor, the TDAC·F as catalysts was not workable on F–C alkylation (entry 5). These experimental results supported that the N–H moieties of TPAC as an HBD were essential.
The tetrabutylammonium chloride (TBA·Cl) along with tetrabutylammonium fluoride (TBA·F), counterparts to TPAC·Cl and TPAC·F, respectively, performed inactive in the benchmark F–C alkylations (entries 6 and 7). The discrepancy between TPAC·Cl and TBA·Cl suggested the necessity of the cationic structure of tris(monoalkylamino)cyclopropenium. We suggested that the H-bond donor realized by the vicinal cyclopropenium show excellent catalytic performance.
With primary success, we expanded substrates by using various indoles along with nitroalkenes (entries 8 to 15). Indoles 1a–e bearing different substituents on both the benzene ring and the pyrrole ring were conductive in reactions with nitroalkene 2a. Whereas the reactions of bare indole 1a and indoles with electron-donating groups (1b and 1c) afforded the corresponding products 3aa, 3ba, and 3ca in good yields (Table 1, entries 8–9), an electron-withdrawing chlorine on the 5-position caused 1d proceeded in a moderate yield of 3da (Table 1, entry 10). Steric hindrance at 7-position retarded the reaction (Table 1, entry 11), which could be accounted for by the unfavorable interference to chloride anion. The applicability of the catalyst was further supported by the variation of the nitroalkene partners. Nitroalkenes with substitutions on the benzene ring (2b–d), both electron-donating and -withdrawing, decreased the yields (Table 1, entries 12–14) as compared with a non-substituted one. Nevertheless, thienly nitroalkene 2d reacted stably with indole 1a to obtain the corresponding products 3ad in good yield (Table 1, entry 14).
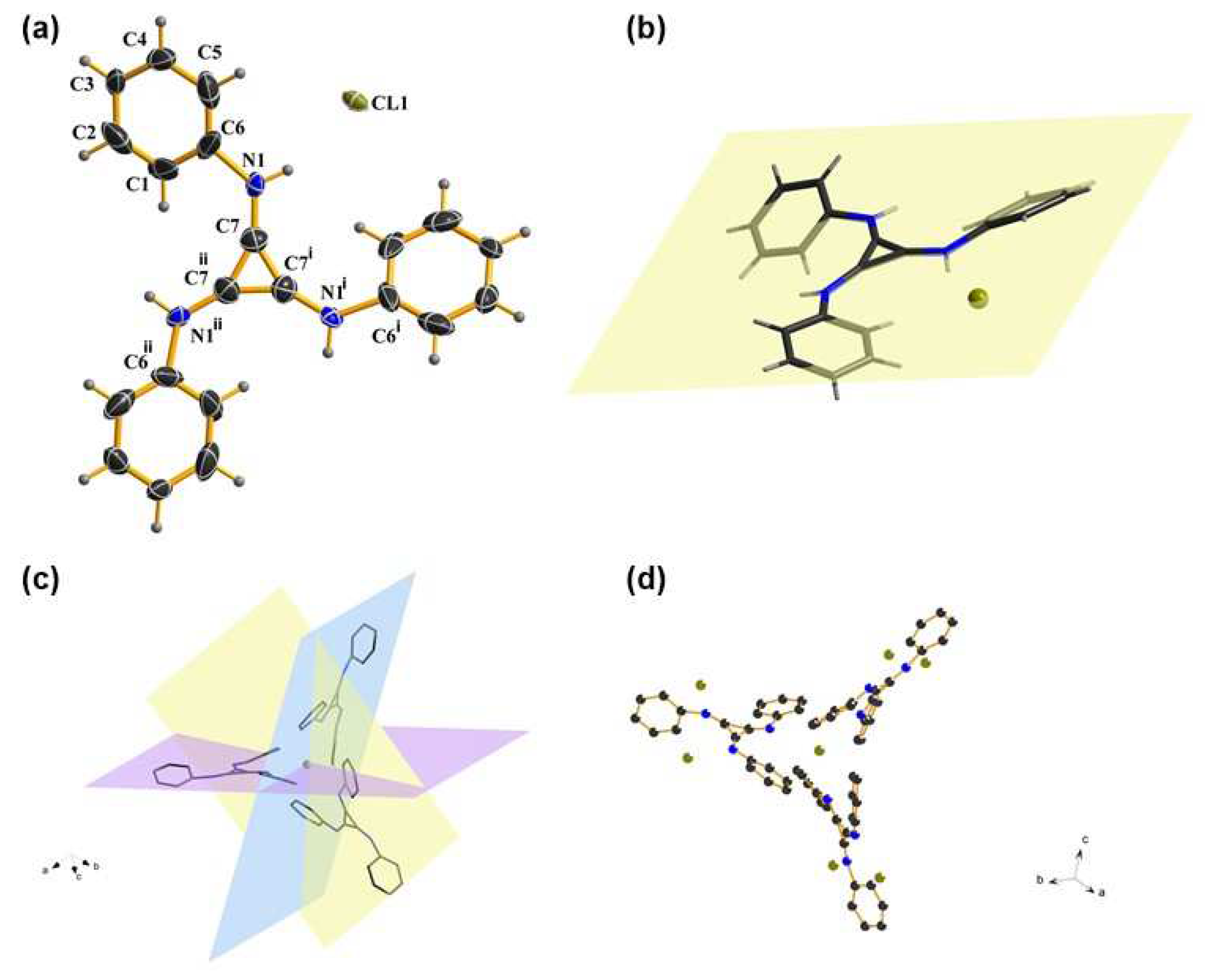
The single crystal of TPAC·Cl was prepared to show the visual view of the construction in the solid, including the interionic distances. A cubic system was confirmed by an X-ray diffraction analysis of the catalyst of TPAC·Cl. The chloride anion was closer to the benzene ring than the formally cationic (C3N3)+ core (Figure 1, a). The chloride anion is coplanar with cyclopropenium core (Figure 1, b), but each phenyl group is slightly skewed out of the plane. Three TPAC coordinate to one Cl (Figure 1, c), while each TPAC aligns in one of the three orthogonal planes of x, y, and z (Figure 1, d). The structure of TPAC·Cl is C3v symmetry. The distances, between the chloride anion and the positive core (C3)+ of the three carbons, showed distinctly larger distances than the normal ones, viz. 4.1086 Å, 4.8124 Å and 5.3485 Å, respectively [44]. These data described the 3D architecture of TPAC·Cl in the solid state.
Two plausible mechanisms were proposed (Scheme 2) based on the HBD implemented by cyclopropenium. One possible competitive interaction was the H-bonding attraction between the cationic TPAC and the counter anion, which decreased the activation of cationic HBD to the substrate, especially the fluoride anion (Scheme 2, A). Thus, the changes of the anion from fluoride to chloride anion promoting the catalytic performance may be the reason for the weak attraction between the TPAC and the chloride anion. The high-lying closed-shell HOMO of aminocyclopropenium cation contending against the closed-shell HOMO of chloride anion will counteract the ionic electrostatic attractions [45]. The phenomenon is called “ion pair strain” [46,47]. The cationic TPAC and the anionic Cl–, in this case, will keep away from each other due to the resistance, and reach obviously larger interionic distances under the readjusted dynamic equilibrium. The counter chloride anion is potential HBA to activate the substrate nucleophilically. The TPAC as an H-bond donor, implemented by vicinal positive charged on the cyclopropenium core, coordinated to the two oxygen of the triangle planar nitro group (Scheme 2 B). The counter chloride anion Cl–, cooperatively, possibly coordinating to hydrogen of the N–H on the indole ring, played the role of an H-bond acceptor (HBA). Different from the prevailing viewpoint of HBD and Lewis base as co-catalysis, we preferred to suggest tris(monoalkylamino)cyclopropenium as HBD and counter chloride anion as potential HBA in cooperative catalysis.
NMR titration experiments were performed to verify the H-bonding interaction between the N–Hs of TPAC·Cl and the substrate of nitroalkene 2a (Figure 2). The chemical shifts of the methine of 2a exhibited downfield shifts from 8.247 to 8.267 ppm by increasing the ratio of [TPAC·Cl]/[2a]0 from 0 to 2 (Figure 2). The two different methines were due to the geometric isomerism of the 2a by C=C. These shifts were important evidence that the catalyst cation of TPAC·Cl as HBD could activate the nitro compounds of 2a by H-bonding. To validate the chloride anion as a potential H-bond acceptor (HBA) with N–H of indole 1a, NMR titration experiments were performed (Figure 3). The chemical shifts of the H-bonding N–H of indole exhibited downfield shifts from 11.103 to 11.307 ppm by increasing the ratio of [TPAC·Cl]/[1a]0 from 0 to 2 (Figure 3). These were important evidence that the counter anion of catalyst TPAC·Cl as HBA could activate the indole 1a by H-bonding.
3. Materials and Methods
The organic solution was concentrated using Buchi rotary evaporator or IKA rotary evaporator. The machine of nuclear magnetic resonance was a type of Bruker-AV-400 (400 MHz). The detecting temperature was 25 °C and the protic solvent was CHCl3 or DMSO. The substrates of indoles and nitroalkenes were purchased from Sigma Aldrich without additional purification. All experiments were executed by standard Schlenk reaction techniques under an argon atmosphere. Dichloromethane was stirred with CaH2 for 10 h and distilled under an argon atmosphere. The purified dichloromethane was stored in 3 Å molecular sieve pellets. Toluene, sodium and diphenyl ketone were heated and stirred until dark purple color came flooding out. The purified toluene was deposited in 3 Å molecular sieve pellets.
Argon airflow was employed to protect all operations progress under standard Schlenk techniques. Freshly distilled 3.0 mL aniline mixed with 1.7 mL chlorotrimethylsilane in 20.0 mL dry benzene at reflux for 1 h. Aniline hydrochloride was separated out in this system and removed by filtration. The filtrate was dried by rotary evaporator to obtain N-trimethylsilylaniline as a yellow oil: 2.46 g, 76 % yield. 1H NMR (400 MHz, CDCl3) δ 7.17 (dd, J = 8.5, 7.5 Hz, 2H), 6.73 (t, J = 7.5 Hz, 1H), 6.68 (d, J = 8.5 Hz, 2H), 3.45 (brs, 1H), 0.30 (s, 9H).
- Preparation of the catalyst TPAC·Cl [34]
Argon airflow was employed to protect all operations progress under standard Schlenk techniques. Freshly prepared 2.10 g N-trimethylsilylaniline was placed to 0.5 mL tetrachlorocyclopropene in 50.0 mL dry dichloromethane and stirred for 6 h. The white precipitate was precipitated gradually. The dichloromethane was used to clean up the white solid. Finally, the white solid was recrystallized from methanol: 0.78 g, 52 % yield; m.p, 207.3 °C (decomp); 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s,3H), 7.41 (t, J = 7.8 Hz, 6H), 7.36 (d, J = 7.6 Hz, 6H), 7.16 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO) δ 138.78, 129.68, 123.94, 118.02, 112.83; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H18N3 312.1495; Found 312.1463.
- The general method for Friedel-Crafts Alkylation catalyzed by TPAC·Cl
Argon airflow was employed to protect all operations progress under standard Schlenk techniques. Thin-layer chromatography (TLC), combined with UV light, was used to monitor the reaction process. Purification was performed by flash column chromatography with silica gel 60 N (Kanto Chemical Co., Inc) or Isolera one with SNAP Ultra Column. In 10 mL reaction tube, 1.0 mmol nitroalkenes 2a–d along with the 0.0347 g TPAC·Cl were weighted in 10.0 mL dichloromethane, the 1.5 mmol indoles 1a–e were placed. The reaction tube was then placed at room temperature for 24 h, the product 3 was obtained by column chromatography (n-hexane/EtOAc mixtures).
4. Conclusions
In summary, we first observed the tris(monoalkylamino)cyclopropenium cation as an H-bond donor (HBD) and the counter anion as potential H-bond acceptor (HBA) by cooperative organocatalysis. The donicity of the N–H moieties in this new type of H-bond donor was implemented by vicinal positive charges on the cyclopropenium core. Tris(phenylamino)cyclopropenium chloride (TPAC·Cl) as a representative H-bonding catalyst was used in Friedel–Crafts alkylation of indoles with nitroalkenes. The X-ray analyses verify the 3D architecture of the TPAC·Cl in the solid state. The TPAC exhibited an H-bond donating ability to the nitroalkene by vicinal cyclopropenium. The counter chloride anion as a potential HBA activated the indole. The proposed mechanisms were certified by using NMR measurements and supported the cooperative activation mechanism. Taken together, an HBD/HBA as cooperative organocatalysis by TPAC·Cl displayed initial mode in organic transformations. Investigations on a new class of hydrogen bonding catalysis in the wider scope of catalysis and transformations are currently underway.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Zhenjiang Li; Funding acquisition, Zhenjiang Li; Investigation, Xuesuo Ma, Jiaxi Xu, Jingjing Liu, Jun He, Qingbiao Yang, Ning Li and Dong Qian; Methodology, Xuesuo Ma, Jiaxi Xu and Jingjing Liu; Project administration, Zhenjiang Li; Resources, Zhenjiang Li; Validation, Jun He; Writing – original draft, Xuesuo Ma, Jiaxi Xu and Tong Chang; Writing – review & editing, Xuesuo Ma, Tong Chang and Zhenjiang Li.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (22078150), the Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), the project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Jiangsu Synergetic Innovation Center for Advanced Bio-Manufacture (XTB2201), and the Top-Notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schreiner, P. R. , Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A. G.; Jacobsen, E. N. , Small-molecule H-bond donors in asymmetric catalysis. Chemical Reviews 2007, 107, 5713–5743. [Google Scholar] [CrossRef]
- Taylor, M. S.; Jacobsen, E. N. , Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Malerich, J. P.; Hagihara, K.; Rawal, V. H. , Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [PubMed]
- Phipps, R. J.; Hamilton, G. L.; Toste, F. D. , The progression of chiral anions from concepts to applications in asymmetric catalysis. Nat. Chem. 2012, 4, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Auvil, T. J.; Schafer, A. G.; Mattson, A. E. , Design Strategies for Enhanced Hydrogen- Bond Donor Catalysts. Eur. J. Org. Chem. 2014, 2014, 2633–2646. [Google Scholar] [CrossRef]
- Min, C.; Seidel, D. , Asymmetric Bronsted acid catalysis with chiral carboxylic acids. Chem. Soc. Rev. 2017, 46, 5889–5902. [Google Scholar] [CrossRef]
- Nishikawa, Y. , Recent topics in dual hydrogen bonding catalysis. Tetrahedron Lett. 2018, 59, 216–223. [Google Scholar] [CrossRef]
- Zhang, Z. G.; Schreiner, P. R. , (Thio)urea organocatalysis - What can be learnt from anion recognition? Chem. Soc. Rev. 2009, 38, 1187–1198. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Takemoto, Y. , Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef]
- Berkessel, A.; Cleemann, F.; Mukherjee, S.; Müller, T. N.; Lex, J. , Highly Efficient Dynamic Kinetic Resolution of Azlactones by Urea-Based Bifunctional Organocatalysts. Angew. Chem. Int. Ed. 2005, 44, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Connon, S. J. , Organocatalysis Mediated by (Thio)urea Derivatives. Chem. Eur. J. 2006, 12, 5418–5427. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K. H.; Sigman, M. S. , Systematically Probing the Effect of Catalyst Acidity in a Hydrogen-Bond-Catalyzed Enantioselective Reaction. Angew. Chem. Int. Ed. 2007, 46, 4748–4750. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K. H.; Sigman, M. S. , Evaluation of Catalyst Acidity and Substrate Electronic Effects in a Hydrogen Bond-Catalyzed Enantioselective Reaction. J. Org. Chem. 2010, 75, 7194–7201. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, H.; Zhang, B.; Li, J.; Zhang, L.; Luo, S.; Cheng, J. P. , Physical Organic Study of Structure–Activity–Enantioselectivity Relationships in Asymmetric Bifunctional Thiourea Catalysis: Hints for the Design of New Organocatalysts. Chem. Eur. J. 2010, 16, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Corey, E. J.; Grogan, M. J. , Enantioselective synthesis of alpha-amino nitriles from N-benzhydryl imines and HCN with a chiral bicyclic guanidine as catalyst. Org. Lett. 1999, 1, 157–160. [Google Scholar] [CrossRef]
- Terada, M.; Nakano, M.; Ube, H. , Axially chiral guanidine as highly active and enantioselective catalyst for electrophilic amination of unsymmetrically substituted 1,3-dicarbonyl compounds. J. Am. Chem. Soc. 2006, 128, 16044–16045. [Google Scholar] [CrossRef]
- Uyeda, C.; Jacobsen, E. N. , Enantioselective Claisen rearrangements with a hydrogen-bond donor catalyst. J. Am. Chem. Soc. 2008, 130, 9228. [Google Scholar] [CrossRef]
- Selig, P. , Guanidine Organocatalysis. Synthesis 2013, 45, 703–718. [Google Scholar] [CrossRef]
- Fu, X.; Tan, C.-H. , Mechanistic considerations of guanidine-catalyzed reactions. Chem. Commun. 2011, 47, 8210–8222. [Google Scholar] [CrossRef]
- Wittkopp, A.; Schreiner, P. R. , Metal-Free, Noncovalent Catalysis of Diels–Alder Reactions by Neutral Hydrogen Bond Donors in Organic Solvents and in Water. Chem. Eur. J. 2003, 9, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T. P.; Jacobsen, E. N. , Highly enantioselective thiourea-catalyzed nitro-Mannich reactions. Angew. Chem. Int. Ed. 2005, 44, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Malerich, J. P.; Hagihara, K.; Rawal, V. H. , Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [PubMed]
- Aleman, J.; Parra, A.; Jiang, H.; Jorgensen, K. A. , Squaramides: Bridging from Molecular Recognition to Bifunctional Organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899. [Google Scholar] [CrossRef]
- Reetz, M. T.; Huette, S.; Goddard, R. , Tetrabutylammonium salts of CH-acidic carbonyl compounds: real carbanions or supramolecules? J. Am. Chem. Soc. 1993, 115, 9339–9340. [Google Scholar] [CrossRef]
- Shirakawa, S.; Liu, S.; Kaneko, S.; Kumatabara, Y.; Fukuda, A.; Omagari, Y.; Maruoka, K. , Tetraalkylammonium Salts as Hydrogen-Bonding Catalysts. Angew. Chem. Int. Ed. 2015, 54, 15767–15770. [Google Scholar] [CrossRef]
- Kaneko, S.; Kumatabara, Y.; Shimizu, S.; Maruoka, K.; Shirakawa, S. , Hydrogen-bonding catalysis of sulfonium salts. Chem. Commun. 2017, 53, 119–122. [Google Scholar] [CrossRef]
- Kumatabara, Y.; Kaneko, S.; Nakata, S.; Shirakawa, S.; Maruoka, K. , Hydrogen-Bonding Catalysis of Tetraalkylammonium Salts in an Aza-Diels-Alder Reaction. Chem. Asian J. 2016, 11, 2126–2129. [Google Scholar] [CrossRef]
- Krebs, A. W. , Cyclopropenylium Compounds and Cyclopropenones. Angew. Chem. Int. Ed. 1965, 4, 10–22. [Google Scholar] [CrossRef]
- Breslow, R.; Groves, J. T.; Ryan, G. , Cyclopropenyl cation. J. Am. Chem. Soc. 1967, 89, 5048–5048. [Google Scholar] [CrossRef]
- Fuchter, M. J.; Smith, C. J.; Tsang, M. W. S.; Boyer, A.; Saubern, S.; Ryan, J. H.; Holmes, A. B. , Clean and efficient synthesis of O-silylcarbamates and ureas in supercritical carbon dioxide. Chem. Commun. 2008, 2152–2154. [Google Scholar] [CrossRef] [PubMed]
- Gaul, D. A.; Just, O.; Rees, W. S. , Synthesis and characterization of a series of zinc bis (alkyl)(trimethylsilyl)amide compounds. Inorg. Chem. 2000, 39, 5648–5654. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. P.; Cumming, W. D. , Conformational preferences of the N-trimethylsilyl and O-trimethylsilyl groups. J. Am. Chem. Soc. 1971, 93, 928–932. [Google Scholar] [CrossRef]
- Weiss, R.; Hertel, M. , NITROGEN ANALOG OF DELTIC ACID. J. Chem. Soc., Chem. Commun. 1980, 223–224. [Google Scholar] [CrossRef]
- Bartoli, G.; Bencivenni, G.; Dalpozzo, R. , Organocatalytic strategies for the asymmetric functionalization of indoles. Chem. Soc. Rev. 2010, 39, 4449–4465. [Google Scholar] [CrossRef]
- Herrera, R. P.; Sgarzani, V.; Bernardi, L.; Ricci, A. , Catalytic enantioselective Friedel-Crafts alkylation of indoles with nitroalkenes by using a simple thiourea organocatalyst. Angew. Chem. Int. Ed. 2005, 44, 6576–6579. [Google Scholar] [CrossRef]
- Lancianesi, S.; Palmieri, A.; Petrini, M. , Synthetic Approaches to 3-(2-Nitroalkyl) Indoles and Their Use to Access Tryptamines and Related Bioactive Compounds. Chem. Rev. 2014, 114, 7108–7149. [Google Scholar] [CrossRef]
- Narumi, T.; Tsuzuki, S.; Tamamura, H. , Imidazolium Salt-Catalyzed Friedel-Crafts-Type Conjugate Addition of Indoles: Analysis of Indole/Imidazolium Complex by High Level ab Initio Calculations. Asian J. Org. Chem. 2014, 3, 497–503. [Google Scholar] [CrossRef]
- Zhuang, W.; Hazell, R. G.; Jorgensen, K. A. , Enantioselective Friedel-Crafts type addition of indoles to nitro-olefins using a chiral hydrogen-bonding catalyst - synthesis of optically active tetrahydro-[small beta]-carbolines. Org. Biomol. Chem. 2005, 3, 2566–2571. [Google Scholar] [CrossRef]
- Nickerson, D. M.; Mattson, A. E. , Transition Metal and Hydrogen Bond Donor Hybrids: Catalysts for the Activation of Alkylidene Malonates. Chem. Eur. J. 2012, 18, 8310–8314. [Google Scholar] [CrossRef]
- Boiocchi, M.; Del Boca, L.; Gómez, D. E.; Fabbrizzi, L.; Licchelli, M.; Monzani, E. , Nature of Urea−Fluoride Interaction: Incipient and Definitive Proton Transfer. J. Am. Chem. Soc. 2004, 126, 16507–16514. [Google Scholar] [CrossRef] [PubMed]
- Cametti, M.; Rissanen, K. , Recognition and sensing of fluoride anion. Chem. Commun. 2009, 2809–2829. [Google Scholar] [CrossRef] [PubMed]
- Lacour, J.; Moraleda, D. , Chiral anion-mediated asymmetric ion pairing chemistry. Chem. Commun. 2009, 7073–7089. [Google Scholar] [CrossRef]
- Komatsu, K.; Kitagawa, T. , Cyclopropenylium Cations, Cyclopropenones, and Heteroanalogues Recent Advances. Chem. Rev. 2003, 103, 1371–1428. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Schwab, O.; Hampel, F. , Ion-Pair Strain as the Driving Force for Hypervalent Adduct Formation between Iodide Ions and Substituted Iodobenzenes: Structural Alternatives to Meisenheimer Complexes. Chem. Eur. J. 1999, 5(3), 968–974. [Google Scholar] [CrossRef]
- Weiss, R.; Brenner, T.; Hampel, F.; Wolski, A. , The Consequences of an Electrostatic “Forced Marriage” between Two Electron-Rich Particles: Strained Ion Pairs. Angew. Chem. Int. Ed. 1995, 34, 439–441. [Google Scholar] [CrossRef]
- Weiss, R.; Rechinger, M.; Hampel, F.; Wolski, A. , Stable 1 : 1 Adducts from Iodoacetylenes and Iodide Ions: Ion Pair Strain as an Additional Driving Force? Angew. Chem. Int. Ed. 1995, 34, 441–443. [Google Scholar] [CrossRef]
Scheme 1.
(a) H-bond donor is enhanced by pi-delocalization of the positive charge; (b) H-bond donor is enhanced by electron withdrawing groups; (c) H-bond donor is enhanced by vicinal positive charged atom; (d) H-bond donor was implemented by vicinal positive charged on the cyclopropenium core, such as tris(monoalkylamino)cyclopropenium.
Scheme 1.
(a) H-bond donor is enhanced by pi-delocalization of the positive charge; (b) H-bond donor is enhanced by electron withdrawing groups; (c) H-bond donor is enhanced by vicinal positive charged atom; (d) H-bond donor was implemented by vicinal positive charged on the cyclopropenium core, such as tris(monoalkylamino)cyclopropenium.
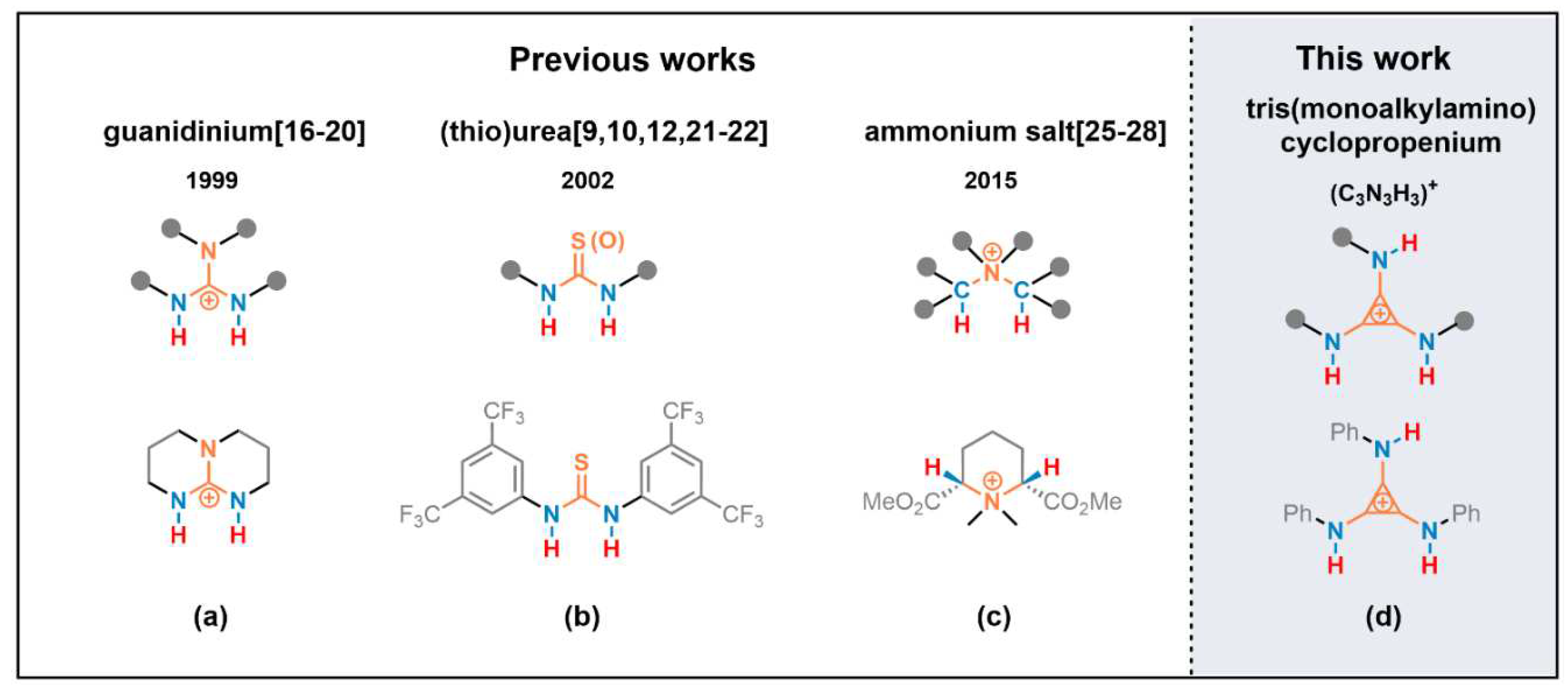
Figure 1.
(a) Crystal structure of TPAC·Cl. (b) Coplanar TPAC·Cl. (c) Side-on view of the three orthogonal planes that the cations aligned around one chloride anion. (d) Side-on view of the coordination between chloride anion and cation.
Figure 1.
(a) Crystal structure of TPAC·Cl. (b) Coplanar TPAC·Cl. (c) Side-on view of the three orthogonal planes that the cations aligned around one chloride anion. (d) Side-on view of the coordination between chloride anion and cation.
Scheme 2.
Two possible cooperative activations of the catalyst TPAC·Cl in Friedel–Crafts alkylation.
Scheme 2.
Two possible cooperative activations of the catalyst TPAC·Cl in Friedel–Crafts alkylation.
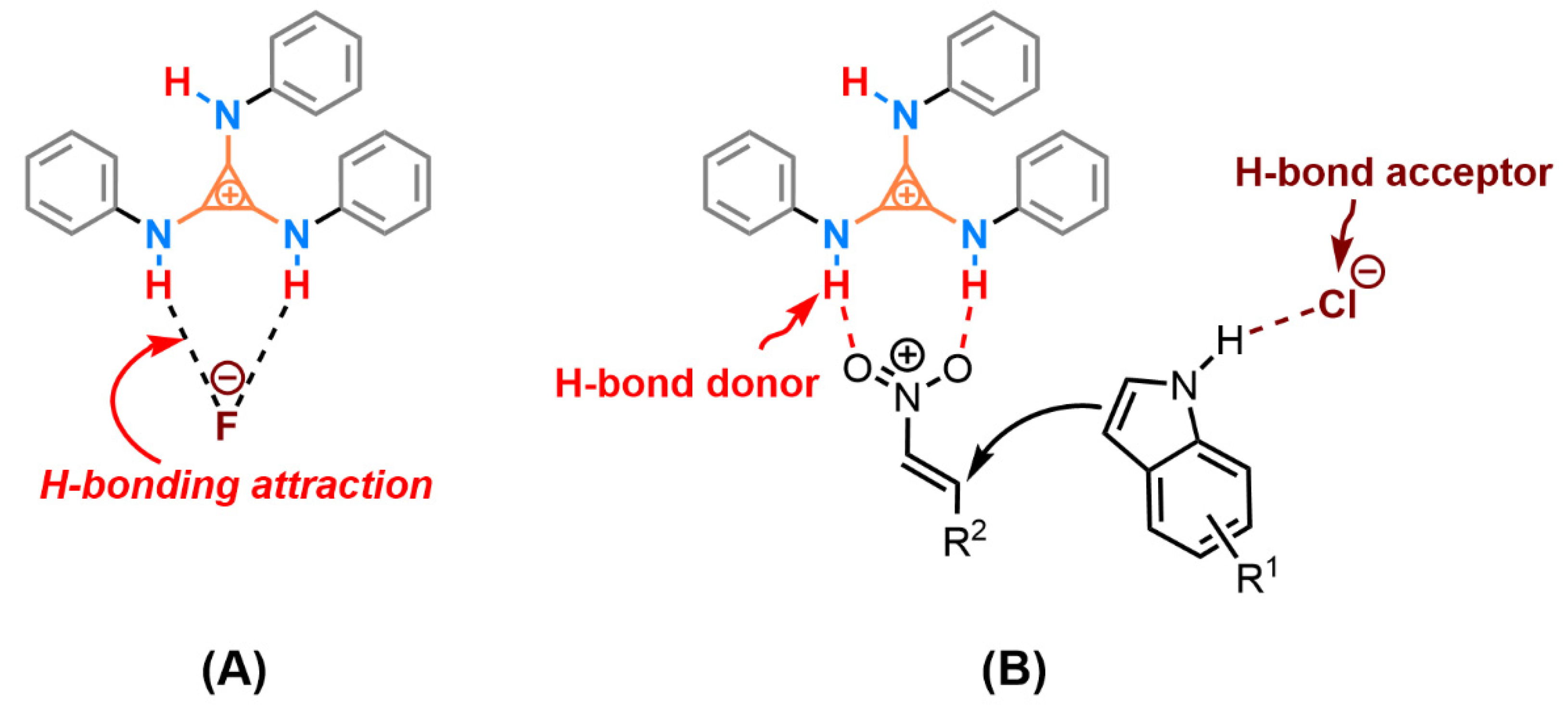
Figure 2.
The chemical shifts of the methine of 2a in the 1H NMR spectrum (DMSO-d6) observed by titration of TPAC·Cl with 2a: (1) free 2a, (2) TPAC·Cl/2a = 0.1/1, (3) TPAC·Cl/2a = 0.5/1, (4) TPAC·Cl/2a = 1/1, (5) TPAC·Cl/2a = 1.5/1, (6) TPAC·Cl/2a = 2/1.
Figure 2.
The chemical shifts of the methine of 2a in the 1H NMR spectrum (DMSO-d6) observed by titration of TPAC·Cl with 2a: (1) free 2a, (2) TPAC·Cl/2a = 0.1/1, (3) TPAC·Cl/2a = 0.5/1, (4) TPAC·Cl/2a = 1/1, (5) TPAC·Cl/2a = 1.5/1, (6) TPAC·Cl/2a = 2/1.
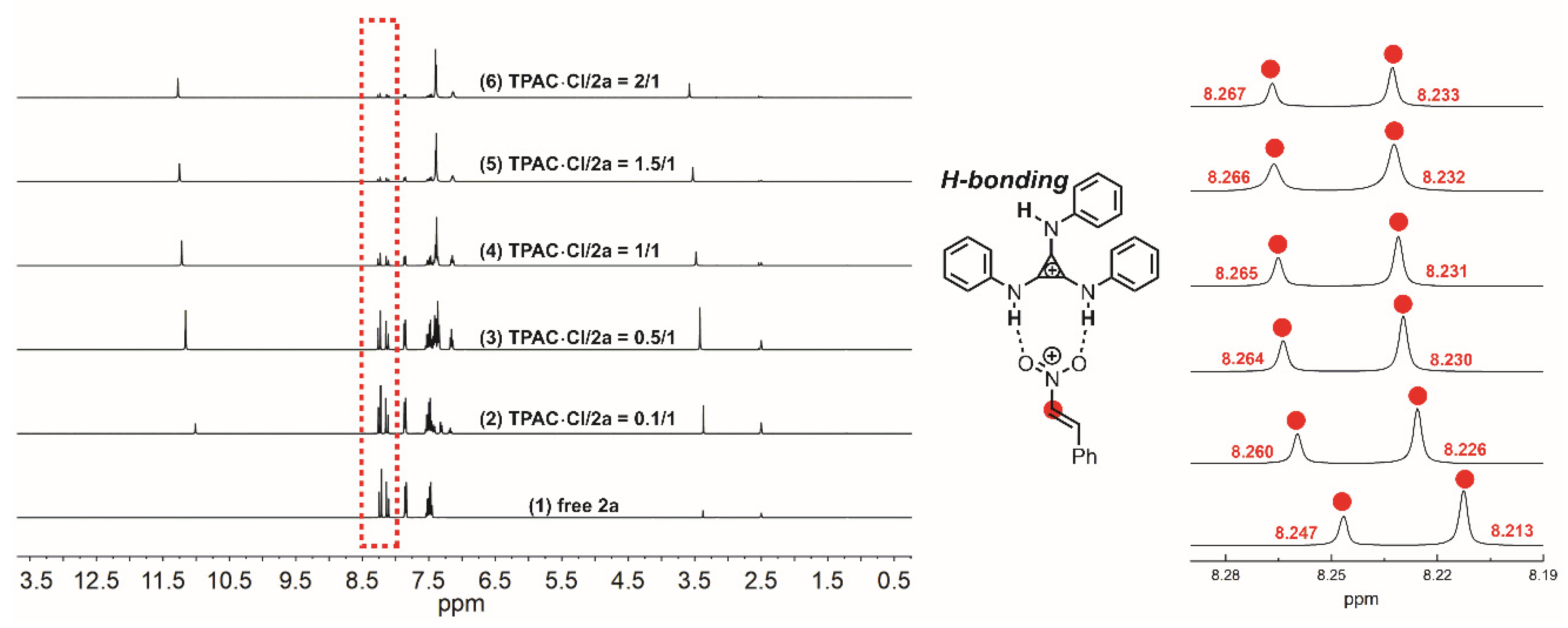
Figure 3.
The chemical shifts of the H-bonding N–Hs of 1a in the 1H NMR spectrum (DMSO-d6) observed by titration of TPAC·Cl with 1a: (1) free 1a, (2) TPAC·Cl/1a = 0.1/1, (3) TPAC·Cl/1a = 0.5/1, (4) TPAC·Cl/1a = 1/1, (5) TPAC·Cl/1a = 1.5/1, (6) TPAC·Cl/1a = 2/1.
Figure 3.
The chemical shifts of the H-bonding N–Hs of 1a in the 1H NMR spectrum (DMSO-d6) observed by titration of TPAC·Cl with 1a: (1) free 1a, (2) TPAC·Cl/1a = 0.1/1, (3) TPAC·Cl/1a = 0.5/1, (4) TPAC·Cl/1a = 1/1, (5) TPAC·Cl/1a = 1.5/1, (6) TPAC·Cl/1a = 2/1.
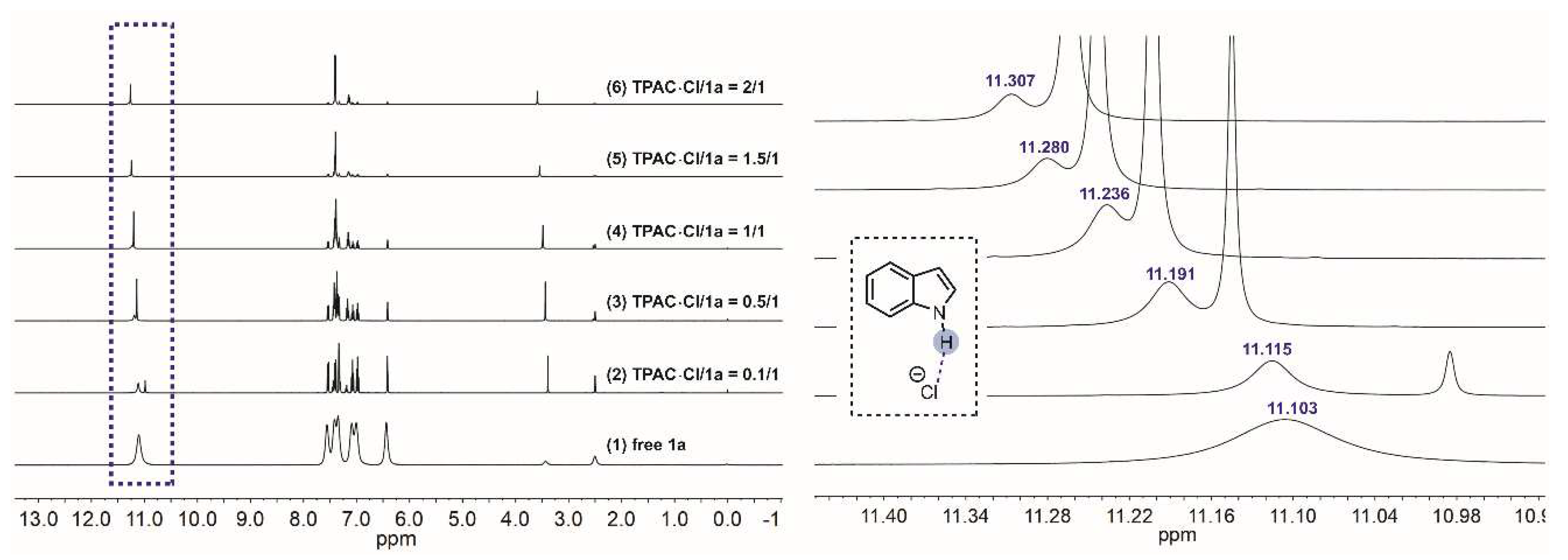

Table 1.
F–C alkylation of indoles 1a–e with nitroalkenes 2a–da.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Ion pair catalyst | Indole | R1 | R2 | Nitroalkene | product | Time / h | Yield / %b |
| 1 | - | 1a | H | Ph | 2a | 3aa | 24 | trace |
| 2 | TPAC·Cl | 1a | H | Ph | 2a | 3aa | 24 | 78 |
| 3 | TPAC·F | 1a | H | Ph | 2a | 3aa | 24 | trace |
| 4 | TDAC·Cl | 1a | H | Ph | 2a | 3aa | 24 | trace |
| 5 | TDAC·F | 1a | H | Ph | 2a | 3aa | 24 | trace |
| 6 | TBA·Cl | 1a | H | Ph | 2a | 3aa | 24 | trace |
| 7 | TBA·F | 1a | H | Ph | 2a | 3aa | 24 | trace |
| 8 | TPAC·Cl | 1b | 2-Me | Ph | 2a | 3ba | 24 | 86 |
| 9 | TPAC·Cl | 1c | 5-OMe | Ph | 2a | 3ca | 24 | 88 |
| 10 | TPAC·Cl | 1d | 5-Cl | Ph | 2a | 3da | 24(72)d | 16(52) |
| 11 | TPAC·Cl | 1e | 7-Me | Ph | 2a | 3ea | 24 | 57 |
| 12 | TPAC·Cl | 1a | H | 4-MeC6H4 | 2b | 3ab | 24 | 33 |
| 13 | TPAC·Cl | 1a | H | 4-MeOC6H4 | 2c | 3ac | 24 | 55 |
| 14 | TPAC·Cl | 1a | H | 2-thienyl | 2d | 3ad | 24 | 71 |
a 1.5 mmol indole 1 along with the 1 mmol nitroalkene 2 were added to 1 mL dichloromethane at 25 °C. b Yield of isolated product. c Without catalyst. d Reaction time was 72 h.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



