
Submitted:
14 September 2023
Posted:
14 September 2023
You are already at the latest version
Abstract
Several rheumatologic diseases are primarily distinguished by their involvement of bone tissue, where it not only serves as a mere target of the condition but often plays a pivotal role in its pathogenesis. This scenario is particularly prominent in chronic inflammatory arthritis such as rheumatoid arthritis (RA) and spondyloarthritis (SpA). Given the immunological and systemic nature of these diseases, in this review, we report an overview of the pathogenic mechanisms underlying the specific bone involvement, focusing on the complex interactions that occur between bone tissue's own cells and the molecular and cellular actors of the immune system, a recent and fascinating field of interest defined osteoimmunology. Specifically, we have comprehensively elaborated on the distinct pathogenic mechanisms of bone erosion seen in both rheumatoid arthritis and spondyloarthritis, as well as the characteristic process of aberrant bone formation observed in spondyloarthritis. Lastly, chronic inflammatory arthritis lead to systemic bone involvement, resulting in systemic bone loss and consequent osteoporosis, along with increased skeletal fragility.
Keywords:
osteoimmunology
; rheumatoid arthritis
; erosions
; spondyloarthritis
; bone tissue
; new bone formation
1. Introduction
Bone tissue is frequently involved in Rheumatologic diseases, particularly chronic inflammatory diseases, such as Rheumatoid Arthritis (RA) and Spondyloarthritis (SpA). In recent years, osteoimmunology has provided extensive support for the role of skeletal tissue in the pathogenesis of many rheumatological diseases. In RA the bone tissue is predominantly affected as the object of skeletal erosions, which constitute the permanent damage of the disease. In spondyloarthritis, appositional and erosive damage coexist, giving this group of diseases peculiar and only partially known characteristics.
In addition to local structural damage, chronic arthritis is also characterized by systemic involvement resulting in skeletal fragility secondary to systemic osteoporosis.
The aim of this review is to provide a comprehensive overview of the pathogenetic mechanisms underlying bone tissue involvement in chronic arthritis.
2. Bone remodelling and homeostasis in health
Under physiological conditions, bone tissue structure and mass are maintained through a timely balanced process called bone remodeling. Bone remodeling consists of the tightly coupled actions of bone removal and apposition operated by resident specialized cells: osteoclasts and osteoblasts, respectively [1].
Bone remodeling cellular events take place in a chronologically hierarchical fashion, with osteoclasts demineralizing and eroding the bone matrix in discrete anatomical areas called resorption lacunae (or Howship lacunae), followed by the apposition of new matrix components and subsequent matrix mineralization completed by osteoblasts [2].
Osteoclasts are fused, multinucleated cells of hematopoietic origin. Two molecules are essential for osteoclast differentiation. The first step of osteoclast precursors proliferation and maturation towards the monocytes/macrophages lineage is driven by the macrophage-colony stimulating factor (M-CSF) [3]. Subsequent osteoclastogenesis processes are determined by the binding of the tumor necrosis factor (TNF)-superfamily cytokine receptor activator of the nuclear factor kappa B (NF-κB) ligand (RANKL) to the receptor activator of NF-κB (RANK) expressed on pre-osteoclasts membranes. RANK-RANKL interaction allows downstream signaling pathways of NF-κB, activator protein 1 (AP-1), and Nuclear Factor Of Activated T Cells 1 (NFATc1), and ultimately leads to cellular fusion and final osteoclast maturation and activation [4,5]. RANKL is mainly produced by stromal cells such as osteoblasts, osteocytes and fibroblasts, as well as by immune cells [5,6]. To downwardly tune the osteoclast activation and bone removal in healthy conditions, a decoy receptor called osteoprotegerin (OPG) is produced by osteoblasts to disrupt the RANK-RANKL interaction [7].
Bone-forming cells osteoblasts originate from pluripotent mesenchymal stem cells (MSC), guided in their commitment by different signals such as the parathyroid hormone (PTH), vitamin D 1,25(OH)2 , transforming growth factor (TGF)- β, fibroblast growth factor (FGF), as well as mechanical loading [8,9,10,11]. These factors promote the cascade of the bone morphogenic protein (BMP) and the Wingless and int-1 (Wnt)/ β-catenin pathways, vital intracellular mechanisms that positively regulate the expression of runt-related factor 2 (Runx2) and osterix to drive osteoblasts maturation [12], and molecules like OPG and alkaline phosphatase (ALP) to exert their bone-forming properties [13]. Appropriate regulation of osteoblasts differentiation relies upon three main inhibitor proteins. Sclerostin, mainly produced by osteoclasts, and proteins member of the Dickoppf (Dkk) family act as antagonists of the low-density lipoproteins (LPR)-5/6, essential Wnt co-receptors [14], while the secreted frizzled-related protein family (sFRP) and Wnt inhibitory factor (WIF-1) inhibit the interaction between Wnt and its receptor [15].
A key director role in bone remodeling is played by osteocytes, former osteoblasts embedded throughout the bone matrix during the process of bone deposition. As they become entombed, osteocytes cellular bodies situated deep into lacunae change shape and grow dendritic processes that run capillary into canaliculi to reach periosteal and endosteal surfaces and other lacunae, to communicate with other bone cells (osteoblasts, osteoclasts, and osteocytes), blood vessels and with the bone marrow stroma [16]. This way, osteocytes can sense both mechanical stimuli and local and circulating factors to regulate the bone remodeling processes through the calibrated release of sclerostin, OPG and RANKL [17].
Lastly, bone cells and microenvironment are enriched by connections with and regulation by immune cells and their products, above all cytokines [18]. The bone-immunity interplay is capable of tuning the abovementioned bone resorption and formation mechanisms, laying the pathogenetic ground for bone disease observed in rheumatic disorders such as inflammatory arthritis [19].
3. Bone involvement in Rheumatoid Arthritis
3.1. Immune cells
Contrary to other resident cells in the bone, osteoclasts stem from progenitors of the monocytic/macrophage lineage, specifically from pre-osteoclastic progenitors within the bone or from circulating peripheral precursors such as monocytes. However, several in vitro studies in the presence of an inflammatory milieu [20,21] and in vivo data from animal models of aseptic bone inflammation [22] have demonstrated the ability of dendritic cells to transdifferentiate into cells with an osteoclast-like phenotype, fueling bone remodelling imbalance and subsequent net bone loss. Dendritic cells (DC) have the same embryological origin and share certain cellular markers with osteoclasts, such as αvβ3 integrin, an integral membrane glycoprotein that exerts cell fusion for DCs and adhesion to the bone surface at the site of resorption for osteoclasts. It is not yet known whether the differentiation of dendritic cells into osteoclast-like cells is solely mediated by Receptor Activator of NF-κB Ligand (RANK-L) and Macrophage Colony-Stimulating Factor (M-CSF), or if there are additional factors specific to the inflammatory microenvironment that actively contribute to this process [23]
In the last years, the newly emerging field of osteoimmunology has received increasing attention regarding the multiple and tangled interactions between the immune and skeletal systems in physiologic and pathological conditions. One of the key aspects of osteoimmunology is the interaction between T cells and bone homeostasis, for example through the RANK/RANK-L/OPG pathway. T cells can differentiate into different T helper (Th) cell subsets: Th1, Th2, Th17, and regulatory T cells (Tregs), each with distinct effects on bone metabolism. It was observed that the Th1 and Th2 subsets inhibit RANK-L and M-CSF-dependent differentiation of osteoclast progenitors [24] in an in vitro model of osteoclastogenesis. In the context of osteoclastogenesis, Interferon- γ (IFN-γ) and interleukin-4 (IL-4), mainly produced by Th1 and Th2 cells respectively, can inhibit the expression of the RANK receptor on osteoclast progenitors and reduce their sensitivity to the differentiation signals induced by RANK-L. Conversely, Th17 cells have been shown to promote osteoclastic activity through the secretion of IL-17. This cytokine does not directly act on osteoclast precursors, but induces the expression of RANK-L by osteoblasts, thereby promoting osteoclast differentiation and activity. Treg cells play a central role in maintaining homeostasis and immunological tolerance and exert a protective effect on bone tissue regardless of the presence of an inflammatory microenvironment [25], prompting a direct effect on the bone remodelling process. In patients with RA, Treg lymphocytes are functionally altered. However, it is not yet fully understood how this may impact bone loss [26].
3.2. Cytokines
The pro-inflammatory cytokine orchestra produced by macrophages, T lymphocytes, and synovial fibroblasts plays a crucial role in the bone involvement of RA, both in the genesis of focal bone damage and in systemic bone loss (Figure 1). The central inflammatory cytokines involved are Tumor Necrosis Factor-α (TNF-α), IL-1β, and IL-6, targeted by biotechnological drugs that have dramatically improved the prognosis of patients with RA unresponsive to conventional disease-modifying antirheumatic drugs (DMARDs) therapy.
3.2.1. TNF- α
TNF-α significantly alters bone remodeling with different mechanisms. This cytokine acts indirectly on the process of osteoclastogenesis by promoting the expression of RANKL, a member of the TNF superfamily, and by stimulating the autocrine/paracrine expression of IL-1 and Interleukin-1 Receptor 1 (IL-1R1) [27]. A significant portion of the osteoclast-inducing activity of TNF-α is mediated by IL-1, as evidenced by the approximately 50% reduction in TNF-induced osteoclastogenesis observed in IL-1R1-deficient mice [28]. This result highlights the reinforcing and synergistic interplay between these two inflammatory cytokines in promoting bone resorption. IL-1 is the pro-inflammatory cytokine with the highest dose-dependent osteoclastogenic effect in monocellular cultures of mouse bone marrow-derived macrophages, at permissive concentrations of RANKL, compared with TNF-α, IL-6, IL-17, and IL-23 [27]. Furthermore, TNF- α might exert a direct, RANKL-independent, effect in promoting the differentiation of mouse monocytic-macrophage lineage precursors towards an osteoclast phenotype. This effect has observed when TNF- α was added to the culture system of osteoclastogenesis at lower concentrations of RANKL. [27]
When the direct effect of TNF-α on an osteocyte population was investigated, It was demonstrated that TNF-α induces an increase in the expression of RANKL-positive osteocytes. This effect was associated with the up regulation of phosphorylation of extracellular signal-regulated Kinases (ERK) 1/2, p38 mitogen-activated protein kinases (p38), and c-Jun N-terminal kinases (JNKs) signaling pathway proteins [29].
The overexpression of TNF-α in RA also occurs in the osteogenic side of bone remodeling with the overexpression of Dickkopf-1 (DKK-1), one of the main negative regulators of the canonical Wnt-β-catenin pathway that is strongly implicated in the processes of proliferation, differentiation, and maturation of osteoblasts. An increased expression of DKK-1 in inflamed synovial tissue in patients with RA was reported [30], speculating on the active role of this mediator in the pathogenesis of bone loss. However, conflicting results have emerged regarding the serum concentrations of this soluble mediator, as a meta-analysis of 136 RA patients and 232 controls found no difference between the two populations [31]. A more recent second meta-analysis [32], which included 9 studies with a total of 1305 RA patients and 504 controls, yielded conclusive evidence of a statistically significant increase in serum concentrations of DKK-1 in patients with RA that contribute to the uncoupling between bone resorption and new bone formation at a systemic level.
The link between TNF-α and sclerostin (SOST), a protein encoded by the SOST gene and produced by osteocytes that impair osteoblastic activity by downregulating the Wnt-β-catenin pathway, is more controversial. Chronic exposure to TNF-α promotes the upregulation of sclerostin and in a rat adjuvant-induced arthritis (AIA) model SOST and DKK-1 were overexpressed in the early stages of arthritis onset [33]. Furthermore, a recent meta-analysis that grouped more than 1000 patients with RA and 561 controls highlighted significantly higher levels of sclerostin in patients with RA [34]. However, both in Sost−/− human TNF-α transgenic (hTNFtg) mice and in hTNFtg mice treated with neutralizing antibodies against murine and human SOST, an exacerbation of arthritic disease has been observed, with an increase in synovial membrane formation and progression in erosive rate [35]. This might suggest a local paracrine action of SOST that acts negatively on TNF-driven inflammation in an inflammatory microenvironment. This issue raised caution regarding the use of romosozumab, a monoclonal antibody that binds and inhibits sclerostin, due to a hypothetical worsening of RA and other autoimmune diseases that involve TNF-dependent inflammation. However, neither during the clinical development of the drug nor in subsequent meta-analyses, did such a side effect emerge [36,37]. Furthermore, in a study comparing the efficacy of denosumab, a monoclonal antibody targeting RANKL, and romosozumab in patients with RA undergoing chronic treatment with glucocorticoids, a similar improvement in disease activity between the two groups was observed [38].
3.2.2. IL-6
IL-6 is an inflammatory cytokine with a broad range of pleiotropic activities resulting in diverse biological effects on various types of cells. It is a member of a family of cytokines that also includes IL-11, IL-27, IL-31, oncostatin M (OSM), leukaemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF), cardiotrophin 1 (CT-1), and cardiotrophin-like cytokine factor 1 (CLCF1). IL-6 is a pivotal factor in the inflammation-related joint damage typical of RA; furthermore, this cytokine acts as an acute-phase protein that promotes and intensifies systemic inflammation and its associated comorbidities, such as anemia due to disturbances of iron metabolism or accelerated atherosclerosis. High levels of this inflammatory mediator also affect bone homeostasis favoring the expression of RANKL [39] resulting in systemic bone loss that leads to osteoporosis and an increased risk of fractures [40]. Additionally, the selective overexpression of IL-6 affects bone formation by hindering osteoblast functionality [41]. In an in vitro study IL-6 strongly increased Osteoblast apoptosis during late differentiation stages of murine preosteoblastic cells in culture [42].
3.2.3. OSCAR
Finally, evidence has been reported regarding the action of pro-inflammatory cytokines on the osteoclastic co-stimulation pathway osteoclast-associated receptor (OSCAR) / immunoreceptor tyrosine-based activation motif (ITAM). A secreted form of OSCAR (sOSCAR) has been identified in human blood serum [43]. In this study, the concentration of this soluble mediator was found to be reduced in the serum of patients with RA compared to healthy controls, and it was observed to increase in RA patients treated with TNF inhibitors (TNFi) therapy. This finding has led to scientific speculation on the role of sOSCAR resulting from cleavage of the extracellular portion of OSCAR, competitively binding to OSCAR ligands and modulating osteoclastogenic stimuli through a negative feedback mechanism. As a result, in chronic inflammatory contexts, TNF-α, IL-1, and IL-6 may inhibit the cleavage of OSCAR and perpetuate the osteoclastogenic signal, thus contributing to inflammatory osteoclastogenesis [44].
3.3. Autoantibodies
The role of RA autoimmunity in the pathogenesis of systemic bone loss has been extensively investigated over the years, specifically the action of anti-citrullinated protein (ACPA) antibodies, Immunoglobulin G (IgG) subclass of antibodies, which target citrullinated proteins such as vimentin, fibrinogen, and collagen. Citrullination is a post-translational modification by which the amino acid arginine is converted into citrulline through the action of specific enzymes called peptidyl arginine deiminases (PAD). ACPA are highly specific for RA, whereas Rheumatoid Factor (RF) can also be found among healthy individuals and patients with other autoimmune diseases or infection. The presence and titer of FR and ACPAs have a diagnostic and prognostic role, as these autoantibodies are associated with a more severe disease phenotype, as well as increased extra-articular involvement of organs and tissues [45].
ACPAs are postulated to affect bone remodelling independently, with a prominent role concerning bone mass loss in the pre-clinical phase of RA, meant as patients with the presence of these antibodies without synovial inflammation [46]. The in vivo increased osteoclastogenesis and systemic bone loss ACPAs-related were first demonstrated in 2012, following the injection of purified human ACPAs from the serum of patients with RA into lymphocyte-deficient Recombination activating gene 1 (Rag1) –/– mice compared with control IgG injections.Hence ACPAs can directly bind to citrullinated proteins, such as vimentin, present on the surface of pre-osteoclasts, resulting in an increase in osteoclastogenesis and differentiation into mature osteoclasts [47].
Furthermore, the interaction between ACPA immune complexes and the receptor for IgG Fc γ receptor (FcγR) present on osteoclasts results in the activation of the spleen tyrosine kinase (SYK), leading to an increase in intracellular calcium concentration and subsequent activation of MAP kinases (MAPK) [48]. This signal transduction cascade is very similar to the one engaged by OSCAR activation, which could reasonably explain the direct pro-osteoclastogenic activity of these immune complexes. Finally, the interaction between these ACPA immune complexes and macrophage cells via FcγR results in the release of TNF-α [49], which, as extensively reviewed earlier, strongly contributes to the enhancement of bone resorption and serves as a bridge among autoimmunity, inflammation and bone loss. Noteworthy ACPAs have been shown to have a significant clinical impact as an independent factor in systemic bone mass loss in various cohorts of early arthritis patients, comparing bone mineral density values between ACPA-positive and ACPA-negative individuals. [50,51]
Recently Anti-carbamylated protein (CarP) antibodies have been identified in the serum of patients with RA. These antibodies target epitopes that have undergone post-transcriptional modification of carbamylation, a chemical reaction between lysine residues and cyanate forming homocitrulline [52]. Nevertheless, the current lack of standardized methods for their detection limits the execution of clinical studies to assess their diagnostic and prognostic role in this subset of patients.
These autoantibodies can exert in vitro increased resorptive activity in osteoclast cultures in the presence of CarP-IgG immune complexes at a permissive concentration of RANKL [53], consistent with a direct impact of these autoantibodies on bone remodelling, similar to what has been observed with ACPA. However, additional studies are warranted to explore this hypothesis further.
3.4. Bone erosions in rheumatoid arthritis
Bone erosions represent central features of RA and are the primary manifestations occurring within the first 8 weeks from disease onset in as much as 10% of patients, with more than 50% of patients developing erosive RA within the first year from disease onset [54,55]. Bone erosions are typically recognized by plain radiographs as interruptions in the cortical bone with loss of adjacent trabecular bone in peculiar peri-articular locations where the synovium enters in direct contact with the bone, known as “bare areas’’ [56]. Similar to systemic bone loss, joint structural abnormalities observed in patients with RA result from an imbalance between upregulated mechanisms of bone resorption and downregulated mechanisms of bone formation.
The areas of periarticular bone loss are enriched in osteoclasts both in RA patients and animal models of arthritis [57,58,59] while showing a paucity in active osteoblasts [60]. Mechanisms of abnormal osteoclast activation in RA are multiple, closely mirroring RA pathogenic mechanisms [61], and involve interactions between autoantibodies, pro-inflammatory cytokines, immune cells, and synovium resident cells with local bone microenvironment. Interestingly, not all the events that lead to bone resorption take place inside the joint cavity – within the synovial membrane. In fact, an additional stimulus to focal bone loss appears to come from the bone marrow niche surrounding the erosions, where a higher degree of marrow aggregates characterized by an increased number of osteoclasts adherent to the subchondral bone have been observed [62]. Before dissecting the role of extracellular factors influencing osteoclasts’ abnormal activity, it is noteworthy that RA osteoclasts precursors per se demonstrate characteristics of enhanced cellular differentiation and activation [63]. In particular, both circulating pre-osteoclasts, synovial pre-osteoclasts and peri-resorption sites mature osteoclasts in RA show an abundance of OSCAR expression[43,64], somehow suggesting that RA bone damage may be partly due to OSCAR-mediated differentiation of imprinted osteoclasts precursors and excessive activation of pathologic mature osteoclasts [65]. In addition, the triggering receptor expressed on myeloid cells 2 (TREM2) and DAP12 were also found upregulated in inflammatory arthritis-derived osteoclasts ([44,66], explaining how priming of these cells is also partly mediated by an upregulation of calcium-mediated auto-amplification of nuclear factor of activated T cells 1 (NFATc1) [67].
Noteworthy, many of the mechanisms of action underlying ACPAs and pro-inflammatory cytokines in the pathogenesis of erosive damage are also involved in systemic bone loss, as reviewed in the previous section.
Approximately ten years ago, the first evidence of ACPAs binding to osteoclasts and the demonstration of bone loss induced by RA patients’ ACPAs transfer to mice was published [46]. It was demonstrated that the osteoclast binding ability was limited to IgG ACPAs, with polyclonal IgGs unable to exert the same osteoclastogenic capabilities. It was suggested that osteoclasts and osteoclasts precursors might represent preferential targets for ACPAs due to the physiologic display of PADs and citrullinated proteins by all osteoclast lineage cells[68], thus explaining features of ACPAs-induced osteoclastogenesis before disease onset, even in absence of inflammation [47]. Recently, erosion was also described in patients ACPA positive without clinical joint infammation, but with tenosynovitis and osteitis on RMI imaging supporting the role of biomarker of early RA [47].
Osteoclasts differentiation and activation promoted by autoantibodies occur by direct and indirect interactions. Mainly operating through the activation of the FcγR on osteoclasts lineage cells with subsequent differentiation of osteoclasts precursors [69], another Fc-independent mechanism of direct ACPA-induced osteoclasts activation is mediated by ACPA-originating Fab fragments, which induce osteoclastogenesis presumably through their binding to citrullinated molecules expressed on pre-osteoclasts surface [46].
RF does not seem to exert any direct effect on osteoclasts. However, the pentameric structure of its most common IgM isotype allows it to enhance the formation and stability of IgG-ACPAs-containing immune complexes, thus increasing the autoantibodies’ osteoclastogenic properties [70,71]. Coherently, a synergistic role for both autoantibodies was observed in a study in which a higher burden of erosive changes in patients with RA was associated with the concomitant presence of RF and ACPAs compared to ACPAs alone [72]. Another key factor influencing IgG ACPAs’ ability to induce osteoclast activation is Fc glycosylation. In particular, the lower levels of sialic acid residues (sialylation) on the terminal Fc glycan observed on RA ACPAs increase their affinity to FcγR and thus their erosive properties [73].
The indirect mechanisms of ACPA-induced osteoclastogenesis partially overlap with another mechanism of bone resorption, through the promotion of TNF-α by FcγR-expressing macrophages [73]. Moreover, macrophages are activated by ACPAs via binding to the citrullinated glucose-related protein 78 (cit-GRP78) [74]. Thus, the indirect ACPAs contribution to bone loss occurs through the release of inflammatory cytokines.
As already reported above, TNF-α is a key cytokine in RA pathogenesis and bone loss (71,72]. TNF-α exerts diverse effects on osteoclastogenesis. First, the cytokine increases the production of RANKL by osteocytes [29] and mesenchymal cells [56], triggering osteoclast activation. Furthermore, TNF-α and RANKL act in concert in the bone destruction process inducing downstream activating pathways such as NF-κB, activator protein (AP)-1 and NFATc-1, signalling mechanisms involved in osteoclastogenesis [77]. Lastly, another mechanism of TNF-induced bone loss is the increase in circulating osteoclast precursors through bone marrow haematopoietic stem cells’ fate commitment via the up-regulation of colony-stimulating factor-1 (c-CSF) expression [78]. Not only TNF-α induce osteoclast proliferation and activation, but it also disrupts osteoblasts pathways through the degradation of the pro-osteoclastogenic factor Runx2 [79] and the induction of Wnt inhibitors DKK-1 and SOST by synovium-resident cells [35,80].
Macrophages, T cells and synovial fibroblasts are the major producers of synovial IL-6 [81]. Like TNF-α, IL-6 contributes to osteoclastogenesis stimulating RANKL production by osteoblasts through signal transducer and activator of transcription 3 (STAT3) activation [82], although IL-6 osteoclastogenic properties is still debated [83]. IL-6 shares another feature with TNF-α, that is the ability to suppress osteoblast-mediated bone formation. Both cytokines promote the expression of the metallopeptidase ADAMTS4, which in turn cleaves the osteoblast-enhancing semaphorin Sema3A in favour of the inhibitor semaphorin Sema4D [84].
Even though it was initially postulated a lack in osteoclasts IL-1 signalling [85], a potential for IL-1 to replace RANKL during the late stages of osteoclast differentiation [86], while simultaneously reducing osteoprotegerin (OPG) production by osteoblasts [87]. The influence of IL-1 on bone loss mechanisms is paralleled by the observation of a reduction in radiographic scores with the use of IL-1β inhibitor Anakinra [88].
Last but not least, IL-17 plays a decisive role in RA synovitis and bone loss [89]. Whether a direct contribution of IL-17 on bone cells is exerted or not is still to be elucidated. The presence of RANKL and M-CSF was able to induce an IL-17-mediated osteoclast activation [90]. However, the same effect was not replicated by other observations [91]. Overall, IL-17 seems to play an indirect effect on osteoclastogenesis by increasing the production of pro-inflammatory cytokines by stromal and immune cells [92], by activating innate immune cells, osteoblasts and synovial fibroblasts to express RANKL [93]. IL-17 is produced by a specific subset of CD4+ T helper cells, called Th17. CD4+ T cells infiltrate RA synovium where they display multiple effector functions [94]. IL-17 expression is only one of the osteoclastogenic mechanisms offered by Th17 cells. These T helper cells are major actors in antibodies’ Fc desialylation [95], promoting immune complexes-mediated osteoclastogenesis. Th17 also reportedly increases pre-osteoclast recruitment through the production of chemokines by mesenchymal stromal cells (MSCs) located in the bone marrow [96]. Under normal conditions, immunological balance is established with the aid of FOXP3-expressing T regulatory (Treg) cells[97]. Other than suppressing inflammation, high amounts of IL-10 and Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) expressed by Treg act as inhibiting factors on osteoclasts and bone destruction [98]. In detail, CTLA4-expressing Treg cells bind to CD80/86 presented on osteoclast precursors’ surfaces, leading to intracellular activation of indoleamine-2,3-dioxygenase followed by the apoptosis of osteoclast precursor cells [99]. On the other hand, secreted IL-10 induces the upregulation of OPG and downregulates the production of RANKL [100]. Although committed, Treg cells show plastic properties and when exposed to an inflammatory environment, they lose their FOXP3 expression and transform into arthritogenic Th17 cells (called exFOXP3Th17 cells) [101]. These cells copiously produce cytokines and chemokines that can induce more profound osteoclastogenesis than naive CD4+T cells-derived TH17 cells [101,102]. Both Th17 and exFOXP3Th17 cells play a conspicuous role in RA bone loss by interacting with one of the major contributors to developing erosions, that is synovial fibroblasts.
Synovial fibroblasts (also renamed as type B fibroblast-like synoviocytes, FLS) are stromal tissue-resident MSCs that, under physiological conditions, cohabit the synovial lining layer with type A macrophages-like synoviocytes. In the lining layer, FLS and macrophage-like synoviocytes serve as a barrier function. Moreover, FLS more loosely populate the sub-lining layer among less densely packed tissue matrix [103] Synovial fibroblasts ensure the joint cavity with lubricating molecules and plasma-derived nutrients, as well as playing a landscaping function with the constantly fine-tuned regulation of the extracellular matrix (ECM) composition through the production of ECM components (such as collagens, fibronectin, vitronectin, and proteoglycans), ECM degrading matrix metalloproteinases (MMPs), and their inhibitors [104]. Moreover, FLS act as immune sentinels, in that they display features of innate immune cells such as Toll-like receptors (TLRs) 2, 3, and 4 through which FLS activate the classical NF-κB and AP-1 pathways to generate chemokines and MMPs [105]. Lastly, FLS serve as bridges between the innate and adaptive immune responses thanks to the expression of the molecule CD40, through which they induce the production of pro-inflammatory cytokines and autoantibodies by activated T-cells and CD40-expressing B lymphocytes, respectively [106].
The synovium in RA patients undergoes profound hyperplastic modifications, with the thickening of the lining layer growing from the two-to-three-cells deep physiologic layer to an excessively proliferated layer of ten to twenty cells (both FLS and macrophages-like synoviocytes) in depth [103]. Moreover, at the synovium border, the thickened tissue transforms into a mass of “pannus” rich in FLS and osteoclasts that invades the adjacent articular cartilage and subchondral bone [107].
Such aggressive phenotypic modifications seem to be closely related to immune cell recruitment as well as to FLS phenotypic changes. FLS shows a state of overproliferation coupled with a pro-inflammatory, invasive phenotype that is maintained even throughout several cellular generations and in the absence of activating triggers as a persistently imprinted make-up [108].
Different subsets of RA-related FLS distinguished by the differential expression of surface markers have been identified and their aggressive phenotype is seemingly obtained through interactions in the inflamed synovial microenvironmen[109]. Such FLS populations differentially localize in the lining and/or sub-lining and exert several pathological functions, although two main subsets exist: the sub-lining located inflammatory CD34-FAPα+THY1+ FLS, and the lining-resident tissue-destructive CD34-FAPα+THY1- FLS [110] such major subpopulations account for the two principal mechanisms through which FLS mediate bone loss: a direct, RANKL- and MMP- mediated bone loss, and an inflammatory cytokines-driven bone injury.
The aggressive front of the synovial pannus is mainly composed of macrophages and fibroblasts that secrete tissue-degrading enzymes causing cartilage and bone damage [111].Pathological, synovial lining-FLS shows a discrete pattern of surface protein expression. Under the influence of inflammatory cytokines these cells express high levels of membrane-bound podoplanin (PDPN) and CD55 that mediate features of migration, tissue aggression and invasiveness [109]. Moreover, the molecular analysis of the aggressive milieu in which lining-FLS operate demonstrates a niche in which pannus-resident FLS have protected from apoptosis thanks to the upregulation of the transcription factor p53 and the downregulation of the tumor suppressor PTEN[73]. Aggressive synovial fibroblasts produce a variety of MMPs to degrade ECM and articular cartilage tissue under the promotion of three main inducers: pro-inflammatory cytokines (among which IL-1 is probably the most potent inducer), growth factors like (FGF) and platelet-derived growth factor (PDGF), and matrix molecules such as collagen and fibronectin [112]. There is good evidence that transcription factors and promoter activators overlapping with immune functions (like various MAPK families like ERK, JNK, p38, as well as AP-1, NF-kB activators and STAT and ETS transcriptional factors) regulate the increased MMPs production by FLSs[108,113].
In addition, synovial fibroblasts proved to be the main source of articular, soluble RANKL when activated by the interaction with inflammatory cytokines [101]. In collagen-induced arthritis (CIA) models, mice lacking RANKL expression in FLS, but not those lacking RANKL expression in T- or B-cells, demonstrated protection against bone erosions [114]. These data indicate the FLS as the main producers of RANKL in inflammatory arthritis and support the concept of “tissue-destructive synovial fibroblasts’’ [115]. Another determinant contribution of FLS in bone damage is through the inhibition of osteoblasts’ bone-forming properties. It has been demonstrated that activated synovial fibroblasts produce DKK-1 from the earliest stages of disease [116], thus cushioning osteoblasts functions and enhancing the destructive profile of FLS. Moreover, it was observed that, in turn, DKK-1 exacerbated the inflammation and tissue-degrading enzyme production by FLS [117]. Other than DKK-1, FLS inhibit the Wnt pathway and osteoblast bone-forming properties by secreting a high amount of sclerostin [35], even though data regarding inhibition or deletion of sclerostin in TNF-α-dependent models of arthritis (but not in TNF-α-independent arthritis models) induced acceleration of the synovial pannus formation and subsequent local inflammation and bone injuries, suggesting a more sophisticated role of sclerostin in the TNF-α signalling pathways [118].
As orchestrating homeostatic cells, physiological and pathological FLS show intricate interplay with various cellular and humoral environments. In particular, FLS are particularly sensible to inflammatory cytokines. Macrophages- and TH17-derived TNF-α, IL-17 and IL-22 mediate the proliferation of FLS and promote the production of pro-inflammatory cytokines (such as IL-6, TNF-α and IL-1) and chemokines by these cells [119]. The inflammatory milieu of the synovium promotes the activation of inflammatory pathways by FLS by upregulating the expression of cadherin-11 on fibroblasts membranes, which in turn creates abundant homotypic interactions between cadherin-11 molecules, thus activating the NF-kB pathway to increase the production of pro-inflammatory cytokines, in particular IL-6 [120]. Another mechanism of FLS inflammatory phenotype activation is mediated by the interaction of FLS-expressed CD40 with CD40L molecules on activated T cells. CD40-CD40L binding enhances the expression of IL-6 as well as adhesion molecules like intracellular adhesion molecule 1 (ICAM1) and vascular adhesion molecule 1 (VCAM1) [121]. The secretion of pro-inflammatory cytokines and adhesion molecules helps maintain an inflammatory microenvironment through a vicious circle amplified by FLS. IL-6 produced by FLS is both a key promoter of Th17 differentiation and an important molecule in the conversion of FOXP3+ T cells in exFOXP3Th17 cells, while in turn IL-17 further stimulates IL-6 production by FLS in a positive feedback loop renamed “IL-6 amplifier” [122]. Other than VCAM1 and ICAM1 enhance the interactions between T cells and synovial fibroblasts [123], FLS participate in the recruitment of inflammatory infiltrates into the joint through the production of high levels of inflammatory chemokines [124]. Stimulated fibroblasts produce both neutrophil-attracting chemokines like chemokine (C-X-C motif) ligand (CXCL8), CXCL5 and CXCL1 [125], as well as CX3CL1 (fractalkine) recruiting CX3CR1-expressing T cells, CXCL10 recruiting CXCR3+ Th1 cells, and CCL20 recruiting Th17 cells to inflammatory synovium [126,127]. Furthermore, FLS promote the survival of B cells through the secretion of VCAM1 and CXCL12 [128], as well as through the B cells differentiation and production of the survival factors APRIL and BAFF induced by FLS-TLR3 ligands [129], thus enhancing the pathogenic roles of autoantibodies.
Notably, a particular macrophage subset expressing CX3CR1hiLy6CintF4/80+I-A/I-E+ termed arthritis-associated osteoclastogenic macrophages (AtoMs) have been identified as possible pathogenic osteoclast precursors in RA [130]. As such, FLS would further participate in synovial pathology and bone loss mechanisms via their enhanced production of CX3CL1, allowing for the migration of pathogenic osteoclast precursors to inflamed joints.
Lastly, a recently discovered mechanism of monocyte commitment towards the osteoclast lineage involves the participation of phagocytic, innate-immune cells neutrophils and their aberrant expression of neutrophils extracellular traps (NETs) [131]. Furthermore, in addition to the established role of NETs in perpetuating the exposure to post-translationally modified autoantigens and PADs in the synovial milieu [132], they promote tissue injury by releasing cartilage-degrading elastase [133] and by potentiating the IL-17-mediated inflammation [134].
3.5. Osteoporosis in rheumatoid arthritis
Osteoporosis is the most common skeletal disorder characterized by progressive decreases in bone mass and microarchitecture impairment, resulting in an increased risk of fractures. [135].
Several studies have established a significantly increased risk of osteoporosis in patients affected by RA compared to healthy age-sex-matched controls. Furthermore, a meta-analysis of 25 cohort studies and almost 250000 RA patients showed a 1.6-fold greater risk of fragility fractures [136,137]
The association between osteoporosis and RA is multifactorial and does not solely reflect the use of glucocorticoids, immobility, disability and the consequent increased risk of falls, but also the specific role of rheumatoid arthritis as a systemic autoimmune inflammatory process that can lead to extra-articular involvement, including bone tissue [138]. Pro-inflammatory cytokines overexpression along with immune cells hyperactivation and the presence of specific autoantibodies account for this detrimental interaction between bone remodelling and rheumatoid arthritis through several pathogenic mechanisms that can enhance osteoclastogenesis, osteoclast lifespan and activity, and conversely can impair bone apposition.
4. Bone involvement in spondyloarthritis
SpA represent a group of interconnected chronic inflammatory conditions characterized by both peripheral joints and axial involvement. As in other rheumatic diseases, bone resorption markers are elevated, leading to bone loss and an elevated risk of vertebral fractures. On the other hand, SpA is also associated with new bone formation in the peripheral and axial joints, in particular, the so-called syndesmophytes result in increased rigidity and ankylosing of the spine. SpA also have a negative effect on bone: in ankylosing spondylitis (AS) elevated bone resorption leads to increased bone loss and, as stated, an increased vertebral fracture risk.
4.1. HLA-B27
HLA (human leukocyte antigen) class I molecules (A, B and C), are expressed on the cellular membranes of nearly all nucleated cells. These molecules present endogenous antigens to cytotoxic T cells, facilitating the development of loss of tolerance to self-antigens [139]. It is widely recognized that SpA exhibits a genetic predisposition linked to the HLA-B27 allele [140]. This allele encompasses over 100 subtypes, which exhibit varying distribution patterns across diverse populations [141]. Prevalence and incidence of AS are positively correlated with the prevalence of the HLA-B27 allele in the population [142]. The highest prevalence of HLA-B27 is found in patients with ankylosing spondylitis (AS) and non-radiographic axial SpA (nr-axSpA) (75-90%) while patients with inflammatory bowel disease-related SpA (SpA-IBD) show the lowest prevalence of HLA-B27 (10-40%) [143].
Several research groups have explored the role of HLA-B27 in bone remodelling, both in normal physiological conditions and in the presence of inflammatory states. For instance, in HLA-B27+ transgenic mice, the administration of exogenous TNF-ɑ leads to a 2.5-fold increase in osteoclastogenesis compared to wild-type mice [144]. In cells expressing HLA-B27, exposure to TNF-α leads to an overstimulated production of both IL-1α and IFN-β, suggesting an intricate interplay between TNF-α and HLA-B27. However, despite the dual secretion of pro-osteoclastogenic (IL-1α) and anti-osteoclastogenic (IFN-β) molecules after TNF-α exposure, the former appears to dominate, fostering an overall pro-osteoclastogenic environment [144]. Additional studies have unveiled a heightened production of pro-inflammatory cytokines in HLA-B27+ mice, correlating with decreased bone mass and strength [145]. This result has primarily been attributed to increased bone resorption rather than decreased bone formation. Other studies have also indicated that HLA-B27+ mice exhibit a reduced OPG/RANKL ratio and a state of osteoporosis [146]. Collectively, the studies mentioned above converge to demonstrate that HLA-B27 organisms are predisposed to an imbalance in OPG/RANKL-related mechanisms of bone remodelling, heightened osteoclastogenesis, and elevated bone resorption. Furthermore, in transgenic mice it has been demonstrated a lower bone mass, trabecular bone density, and mineral-to-matrix ratio, but higher levels of c-terminal telopeptide (CTX), pyridinoline/divalent collagen cross-link ratio (a marker of mechanical competence), proteoglycans (which inhibit matrix mineralization), and alveolar bone loss [147]. Other in vitro data seem to report a direct positive influence of HLA-B27 on osteoclastogenesis [148].
Molecules inhibiting OPG activity result in lower bone mineral density (BMD): as written above for RA; recently autoantibodies against OPG have also been found in SpA, correlating to lower BMD and a history of fractures[149]. Moreover, it has been showcased that elevated soluble RANKL (sRANKL) levels and an increased soluble RANKL/OPG ratio among AS patients were associated with decreased BMD values [150]. Furthermore, these findings correlated with radiological signs of disease damage, higher clinimetrics scores, CRP and other pathogenetical cytokines, providing support to the presence of a direct link between joint and systemic inflammation, altered levels of important mediators of bone homeostasis and osteoclast activation, and overall BMD.
4.2. Cytokines
4.2.1. IL-23 and IL-17
Pro-inflammatory cytokines like IL-23, implicated in SpA pathogenesis, also contribute to bone loss [151]. Mainly produced by Paneth cells in the gut and joint macrophages, IL-23 triggers IL-17 and IL-22 production by Th17 cells, both involved in systemic inflammation. IL-17 relates to bone loss, while IL-22 is associated with osteoproliferation [152]. IL-23 drives T-helper cells to become IL17-producing Th17 in osteoblast-free cultures, fostering osteoclastogenesis via a RANKL-independent pathway [153]. Additionally, IL-23 prompts osteoclastogenesis and enhances RANK expression in osteoclast precursors [154]. However, several studies suggest that such a catabolic action of IL-23 is possible exclusively through the mediation of IL-17 since IL-23 alone demonstrates an inhibitory effect on osteoclast [155]. IL-17A is primarily generated by Th17 lymphocytes, which indeed have demonstrated osteoclastogenic activity [24]. IL-17A is released in response to TNF-β, IL1β, IL21, and IL23 stimulation, but suppressed by IFN-α [156]. Experimental studies indicate that exposure to very low or high IL-17A concentrations triggers increased RANK expression and JAK2-STAT3 signalling activation in circulating human MSCs (hMSCs), promoting their differentiation into mature osteoclasts. Interestingly, intermediate IL-17A levels yield the opposite effect on osteoclast precursors [157]. Synergistically with TNF-α, IL-17A increases the production of IL-1a, IL-1b and IL-6, which in turn stimulate osteoclast activity [158]. Finally, IL-17A induces the secretion of RANKL by osteocytes, with a consequential increase in the RANKL/OPG ratio and TNF-α concentration [157]. Interestingly, in transgenic mice in which the expression of IL17A was increased, no alterations in either the number or activity of osteoblast and osteoclast or in bone mass were found. This suggests that IL17A may only have a detrimental effect on bone in an inflammatory context, thus acting in concert with other pro-inflammatory cytokines, while on its own it is unable to alter bone metabolism. Research using mouse models of Psoriatic Arthritis (PsA) has revealed a correlation between elevated circulating IL-17A and bone loss [159]. In these models, IL-17A stimulates RANKL production by affected skin cells and hinders osteoblasts and osteocytes through the SOST/DKK-1 pathway [160]. Additional support for IL-17A’s role comes from anti-IL17 treatments like secukinumab and ixekizumab used in PsA management, which bring about bone stability, decreased bone erosion, and reduced bone damage progression [161].
4.2.2. TNF-α
TNF-α and IL-17A can mutually enhance each other’s effects, resulting in diverse outcomes in distinct settings. For instance, within synovial joints, the coexistence of osteoclasts (OCs), driven by elevated RANKL levels prompted by these cytokines, leads to bone erosions [162] but OCs differentiation could also be induced in a RANKL-indipendent pathway[163].
TNF-α amplifies the presence of circulating CD14+/CD11b+ osteoclast precursors both in transgenic mice featuring TNF-α persistent activation and in mice receiving TNF parenteral administration [164]. This correlation is further substantiated by evidence showing that the administration of TNF-α inhibitors (TNFi) to patients leads to a reduction in the count of circulating progenitors [165].
4.2.3. Other cytokines
Few data have been published on the role of growth differentiation factor (GDF)-15, also known as macrophage inhibitory cytokine 1 (MIC-1) in SpA, which appears to be elevated in the synovial fluid but not in sera of SpA patients [166]. This cytokine is a member of the TGF-β superfamily but its pro-inflammatory or anti-inflammatory activity is still debated. It rises after TNF-α stimulus, secreted by osteocytes after hypoxic damage, with consensual osteoclast activation [167]. This cytokine seems to be higher in SpA sera, with respect to healthy controls, and it is positively correlated with CRP levels, with the presence of erosion in the sacroiliac joint (SIJ), and with a high degree of X-ray scores [162].
Finally, in early SpA IL-31 seems to be associated with low BMD and lower syndesmophytes development, evaluated as modified Stoke Ankylosing Spondylitis Spinal Score (mSASS) <1. IL-31 is an IL-6 superfamily member and a Th1-Th2 border cytokine. It binds its receptor (heterodimer of IL-31RA and Oncostatin M) activating JAK/STAT, PhosphatidylInositol 3-Kinase/AKT (PI3K/AKT) and MAPK pathways. IL-31 is produced by memory Th2 lymphocytes, macrophages and dendritic cells. IL-31 activity on bone remodelling is still debated: it induces metalloproteinases and osteoclastogenic cytokines, inducing higher differentiation of osteoclast progenitors in mature cells [169], and it is correlated with pro-inflammatory cytokines in early SpA. However, some studies have demonstrated its anti-inflammatory action in different mucous membranes [170,171]. Its serum levels correlate with those of TNF-α and IL-6, but not of IL-17. A study demonstrated a correlation between IL-31 and DKK-1 levels, resulting in an inhibition of bone formation and activation of bone resorption by RANK-RANKL pathway [172].
As opposed to RA, skeletal abnormalities observed along the SpA spectrum appear multifaceted, with simultaneous evidence of bone loss and bone formation (Figure 2).
A fundamental pathway enhancing osteoblast differentiation and activity revolves around wingless (Wnt). Two important down-regulators of the WNT/beta-catenin pathway are DKK-1 and SOST, although data on their serum levels in patients with SpA are conflicting. Both of them are directly related to CRP and inflammation but only SOST is minimally reduced under TNFi treatment [173]. Increased levels of expression of DKK-1 are observed both in the synovium and serum of mice subjected to increased levels of TNF-α, RANKL and other inflammatory cytokines [30]. This suggests that DKK-1 is over-expressed under conditions of inflammation. The same study identified DKK-1 as a molecule capable of inducing osteoclastogenesis and, on the other hand, of down-regulating osteophyte formation, depending on the microenvironment. Indeed in enthesis, in mice transgenic for constitutive expression of TNF, the inhibition of DKK-1 resulted in osteophyte production, while the simultaneous expression of both molecules preserved bone from osteoproductive damage [30]. Moreover, these results are supported in the same context by the finding of increased local and serum levels of osteocalcin and OPG when mice were treated with anti-DKK-1 antibodies.This observation lends further support to the existence of a cross-talk between DKK-1 and the RANKL-OPG system, suggesting an inhibitory effect of DKK-1 on OPG. The resorptive effect of DKK-1, in fact, seems to be driven by its inhibition of OPG. In mice treated with anti-DKK-1, the concurrent suppression of OPG leads to the reactivation of osteoclastogenesis. Conversely, when OPG is administered subsequently, this reactivation is once again halted. Interestingly, it is worth noting that these processes do not appear to influence osteophyte formation [30].
Other important molecules involved in osteoblastogenesis, as well as in endochondral ossification, are the so-called bone morphogenetic proteins (BMPs), which belong to the TGF family. These proteins, as well as the BMPs/DKK-1 ratio, were higher in the serum of patients affected by AS than in healthy controls, and positively correlated with disease activity, structural damage, and disease duration, suggesting a role of BMPs in the formation of pathological new bone. Intriguingly, although in other studies DKK-1 levels were found to be lower in AS patients compared to healthy controls, a positive correlation between DKK-1 levels and the risk of vertebral fractures in AS patients was observed [174]. This underscores the complex interplay of these molecules in AS and suggests that the role of DKK-1 in bone metabolism is not straightforward and may involve intricate interactions with other factors. Because lower levels of DKK-1 and SOST were found in AS patients, the authors suggested that the main cause of osteoporosis in AS should be searched for in other alterations such as immobilization, systemic inflammation, and biomechanical factors.
While in RA the onset of a state of systemic osteoporosis seems to follow the “so-called” juxta-articular osteopenia, in SpA this has not been demonstrated [175]. An assay of the levels of OPG expressed by resident cell populations in the synovial joint membrane [176] confirmed such suggestion, demonstrating that OPG expression by endothelial cells and lining macrophages was absent in patients with active RA, contrarily to what is observed in patients with SpA and even inactive RA. At the same time, the study demonstrated an increased expression of RANKL in both RA and SpA patients.
4.3. Erosions in Spondyloarthritis
Early literature characterized SpA as non-erosive inflammatory conditions. However, subsequent research has changed this perspective, revealing that erosions can indeed occur even in SpA. It’s worth noting that the data showing comparatively less local bone damage in SpA might be explained by various factors such as the direct joint damage mediated by autoantibodies in RA.
A clear elucidation of how the microenvironment driven by T helper cell polarizations can either activate or inhibit osteoclastogenesis was provided by Sato and colleagues in a previously mentioned in vitro study [24]. Co-cultures of osteoclast precursors and osteoblasts were established, introducing Th1 and Th2 cells to examine their effects on osteoclastogenesis, measured by tartrate-resistant acid phosphatase (TRAP) levels. Interestingly, the presence of Th1 or Th2 cells alone did not trigger osteoclastogenesis. Conversely, the introduction of Th17 cells, particularly after exposure to IL-23, stimulated osteoclastogenesis. This effect of Th17 cells is linked to IL-17, and the significance of this cytokine in osteoclast activation was further confirmed by the fact that mice lacking the IL-17 gene did not exhibit increased osteoclastogenesis [24]. Moreover, IL-23 itself has been demonstrated to induce osteoclastogenesis [177] and this concords with the results of clinical trials in which the use of IL-23 inhibitors (IL-23i) or IL-17 inhibitors (IL-17i) antibodies prevent radiographic progression in PsA patients [177,178].
A clear illustration of the complex interplay between different pathways of innate immunity in the context of inflammatory arthritis emerged from a study in which adenoviral vectors containing genes for both IL-17 and TNF-α were introduced into the knee of a CIA mice model. The outcome demonstrated a more pronounced inflammation and increased bone destruction compared to the effects of each cytokine alone. This combined effect was also marked by a synergistic up-regulation of the NF-kB pathway, elevated MMP production, and more extensive cartilage damage. These findings were substantiated by their subsequent investigation involving the injection of IL-17R and TNF-binding protein (TNF-BP) to act as decoy receptors for these inflammatory cytokines. This intervention led to a substantial reduction in joint inflammation, further supporting the pivotal role of IL-17 and TNF-α in the inflammatory process. Importantly, the study revealed that only the concurrent presence of both cytokines resulted in irreversible bone and cartilage erosions, in contrast to TNF-α alone [179].
Psoriatic arthritis (PsA) is the more prevalent disease in the SpA family showing a tendency to periarticular bone erosions. Erosive disease seen during PsA is associated with higher levels of RANKL at the synovial lining layer, both absolute and relative to those of OPG, and with higher levels of circulating CD14+/CD11b+ pre-osteoclasts [165]. Some authors found in the serum of PsA patients, with and without erosive disease, higher levels of DKK-1 than in healthy controls [180]. In the same study, higher levels of M-CSF were found in the subgroup of PsA patients showing erosive disease. The number of circulating pre-OC and the levels of serum M-CSF and sRANKL were associated with radiographic damage scores, including erosions and osteolysis. A more recent paper confirmed the finding of higher serum DKK-1 in PsA patients and demonstrated that it represented a risk factor for bone erosions, with a significant OR of 4.4 [181]. Noteworthy, studies are conflicting because some groups confirmed the higher levels of DKK-1 in PsA but others described even lower levels with respect to healthy controls [182].
Other cytokines involved in PsA pathogenesis and related bone disease are IL-20 and IL-24. These pro inflammatory molecules seem to exert their effect on bone through the induction of MCP1, which is a recruiting cytokine for osteoclast precursor [183]. The IL-20 family of cytokines is very large including, other than IL-20 and IL-24, IL-19 (which exert principally anti-inflammatory effects), IL-26, and IL-22. The latter is produced by Th22 and Th17 under IL-1b, IL-6 and IL-23 stimulation [183]. In particular, IL-22 showed damaging properties, being associated with joint destruction causing erosions in arthritis animal models [184]. Nevertheless, a definite role for IL-22 as a biomarker or therapeutic target in axSpA has yet not been identified [185], and its effects on bone do not appear to be solely confined to erosive properties.
4.4. New Bone Formation in Spondyloarthritis
New bone formation is a key process in the pathophysiology of SpA. It could affect both the axial joints (i.e. the SIJ and the spine) and peripheral sites, as well as periarticular tissues such as the enthesis. Molecular mechanisms of new bone formation have been largely studied in axSpA, while fewer data are available on PsA. To the best of the actual knowledge, the most credited hypothesis is that the new bone formation process represents the final step in the continuum of SpA bone lesions. Indeed, bone overproduction generally occurs where bone marrow edema (inflammation) and, subsequently, fat metaplasia take place [186]. Nevertheless, two main controversial points persist. First, nearly one-third of the patients affected by axSpA will never develop structural changes during the disease course [187]. Second, in some patients, new bone formation happens even when the disease is well controlled with optimal treatment and it could be partially explained if new bone formation is finalistically intended as an effort of the spine to increase its stability [188]. On the other hand, histological analysis of the spine and the SIJ of patients with AS detects inflammatory infiltrates in the subchondral bone marrow even where inflammatory lesions have not been seen by imaging techniques, suggesting that sometimes MRI might miss mild inflammatory changes [189,190]. Therefore, despite the arrival of biological DMARDs (bDMARDs) having dramatically improved the quality of life and the physical performance of patients affected by SpA, the relationship between new bone formation and chronic inflammation remains not completely understood. Therefore, while some studies suggest the existence of a coupling between inflammation - and disease activity - and new bone formation in patients with AS [191,192,193], others demonstrate that ossification progresses independently from the suppression of inflammation involving adjacent but distinct sites [194,195]. Anyway, most findings from clinical trials show less bone formation in patients with axSpA treated with bDMARDs whose disease is well controlled by the therapy, suggesting that new bone formation in the context of a clinically-controlled disease could be explained by a subclinical, persistent non-detectable joint inflammation [196,197].
4.5. The enthesis milieu
The enthesis is the site of attachment of tendons, ligaments, fasciae, and joint capsules to the bone [198]. During the last decades, several studies have contributed to evolving the concept of the enthesis considered merely as an anatomical site to a biologically complex entity including the tendon, the entheseal fibrocartilage, and the subchondral bone marrow[199]. Enthesitis, which is considered a hallmark of SpA, is the result of the combination of the inflammatory process and mechanical load. The relevance of enthesis in SpA and the key role of IL-17 and TNF-α were also confirmed in a study, which demonstrated a population of tissue-resident memory CD4 + and CD8 + T cells with an immunomodulatory phenotype but still able to produce IL-17 and TNF at the level of enthesis [194]. Although bone formation could be considered the final step of the inflammation and a mechanism of repair of bone loss, an interesting study demonstrated that new bone formation in the Achilles tendon of patients with PsA occurs at distinct sites than erosions [201]. The mechanical load takes part actively in the pathogenesis of enthesitis, as reported in a study involving transgenic mice prone to develop inflammatory features similar to SpA. Indeed, new bone formation was strongly associated with mechanical load and correlated with inflammation and interestingly the unloading of the enthesis significantly reduces the inflammation compared to weight-bearing mice [202]. Overall, the process of bone spurs formation at entheseal sites resembles a response-to-injury process, mimicking the healing from a fracture through a sequence of endochondral bone formation, where a cartilage scaffold is then replaced by bone tissue [203].
The new bone formation at entheseal sites has been further deepened by a Chinese study in which ossifying spinal enthesis of 10 patients with AS and 10 controls were analyzed finding higher expression of BMP-2 and enhanced osteogenic differentiation of hMSCs in the enthesis of AS patients rather than controls [204]. In particular, an imbalance between BMP-2 and its antagonist Nogging has been demonstrated in patients with ankylosing spondylitis [205].
Osteogenic precursor cells (OPCs), the MSCs responsible for new bone formation in SpA, could be resident or recruited, but a yet unsolved question is how recruited OPCs reach the enthesis and few data have been published. To this purpose, CXCL12 has been found to be overexpressed in spinal ligaments sampled from patients with AS compared to healthy controls and OPCs were attracted to sites where CXCL12 is overexpressed [206].
4.6. New bone formation pathways
4.6.1. Wnt/ß-catenin
In both axial and peripheral SpA, the role of the pathways of Wnt-ßcatenin - and its regulator molecules - in the process of new bone formation has been extensively studied, most often with discordant results.
During the last 15 years, several studies tried to analyse the role of DKK-1 in SpA pathogenesis. Higher levels of DKK-1 in the serum of patients with AS have been reported compared to RA patients and healthy controls and the addition of serum of AS patients to lymphocytes cells culture promoted Wnt activity [207]. Despite this could seem paradoxical, a dysfunctional activity of DKK-1 could partially explain the results. However, a recent meta-analysis including 40 studies on the measurement of serum DKK-1 levels did not find any differences between AS patients and healthy controls and higher serum levels of DKK-1 do not increase the risk of developing AS [208]. Further, as demonstrated in other studies conducted in cohorts of axSpA patients as the DESIR cohort, DKK-1 levels are not related to structural damage and with levels of DKK-1 single nucleotide polymorphisms (SNPs), suggesting that DKK-1 production is not genetically determined in patients affected by axSpA [209,210].
DKK-1 has been studied also in patients with peripheral SpA such as psoriatic arthritis - although less extensively - with controversial results even in this case. An Italian study demonstrated lower serum levels of DKK-1 in patients with PsA compared to healthy controls and RA patients for the first time[182].This evidence differs from other previous and subsequent studies in which - similarly to SpA - higher levels of DKK-1 in PsA patients were reported [180,211]. Furthermore, in an observational case-control study including 50 patients with PsA and 50 healthy controls, DKK-1 serum levels were significantly higher in PsA patients and positively correlated with both inflammation, erosions, and calcification at entheseal sites, evaluated by ultrasound using the Madrid Sonographic Enthesitis Index (MASEI) [212]. However, conclusive data on DKK-1 levels in patients with SpA still lacking [173].
Data on SOST in patients with SpA are controversial as well. Interestingly, SOST seems to be overexpressed in HLA-B27 positive people, independently from a diagnosis of SpA [213], as well as in males and elderly SpA patients ([209]. However, several studies found discordant results, finding SOST serum levels not significantly different between patients with AS and healthy controls [173] or even lower in patients with early axSpA in the DESIR cohort compared to healthy controls [209]. In the absence of specific serological markers useful to detect subclinical inflammation in people with SpA under treatment, a study on 30 patients with AS treated with TNFi and 30 healthy subjects found that SOST serum levels were significantly lower in AS patients compared to controls and even lower in patients with elevated CRP, suggesting that lower levels of serum SOST could reflect the presence of underlying inflammation [214] In addition, in a work conducted on serum and tissue samples from zygoapophyseal joints of patients with AS, RA, OA, and healthy controls, a lower expression of SOST in both serum and chondrocytes of patients with AS compared with OA and healthy controls was found. Furthermore, SOST levels were reported to be inversely correlated with syndesmophyte formation [215]. Moreover, an Italian study reported lower levels of SOST and higher anti-SOST-IgG serum levels in patients with axial SpA-IBD compared to patients with only IBD and they negatively correlated with the duration of musculoskeletal symptoms [216].
As well as for AS, the role of SOST in the pathogenesis of new bone formation in patients with PsA is not univocally demonstrated. Most of the studies reported higher levels of serum SOST in PsA patients compared to healthy subjects, even if a correlation with disease activity or with structural damage has not been demonstrated [217,218]. In conclusion, although a reduced activity of DKK-1 and SOST in patients with SpA could be assumed to be a relevant mechanism of new bone formation, current evidence based on in vitro and on human biological samples are discordant and not conclusive.
Indian Hedgehog (IHH) is produced by chondrocytes and it is an important regulator of endochondral ossification [219], mechanistically similar to bone formation in SpA. However, the role of IHH in SpA has not been widely evaluated so far and only one study analyzed serum levels of IHH of patients with AS, RA, and healthy subjects and the effect of such serum on chondrocyte cell cultures, finding higher levels of IHH in patients with AS in comparison with AR patients, as well as in AS patients treated with TNFi compared to those not receiving therapy[220]. Furthermore, the molecule antagonist noggin inhibits the onset and progression of spontaneous arthritis and endochondral bone formation in a preventive and therapeutic way. The endogenous noggin seems to affect the progression of joint remodelling and slows the ossification process [221].
4.6.2. Cytokines
Many cytokines participate in the pathogenesis of SpA and are targeted by bDMARDs. As discussed above, the relationship between inflammation and new bone formation is still debated, therefore how pro inflammatory cytokines could affect the ossification in SpA is only partially known.
TNFi has been the first class of bDMARDs approved for treating SpA. TNF is a pro inflammatory cytokine which binds two receptors: TNFR1 and TNFR2. As TNFR1 delivers pro inflammatory and catabolic stimuli, on the other hand, TNFR2 has opposite functions. Therefore, while the TNFR2-mediated signalling increases new bone formation via the inactivation of osteoclasts, TNFR1 promotes bone loss [222]. As mentioned above, treatment with TNFi seems to increase bone mass in patients with SpA but as TNF plays an important role in the inflammatory process in patients with AS, its effect on bone formation and ankylosis are still not understood even if early initiation and long-term treatment with TNFi demonstrated to slow radiographic progression [223,224].
Despite the evidence of the direct and indirect pro-osteoclastogenetic properties of IL-17 and its role in systemic and focal bone loss in RA and SpA (discussed above), surprisingly, several studies report IL-17 as a promoter of osteoblastogenesis, leading to higher bone formation, probably via a JAK2/STAT3 pathway [225]. Anyway, the overall in vivo effect of cytokines and other biological regulators is hard to analyze because many other factors take part in the microenvironment of the joint and the enthesis of patients with SpA. A very interesting study conducted on bone marrow samples of healthy subjects demonstrated that IL-17A acts as an osteogenic factor, stimulating the osteogenic differentiation of human bone marrow-derived mesenchymal stem cells (MSCs) and blocking RANKL, assisted by TNF [162]. Overall, the biological net effect of IL-17 in patients with SpA could seem contradictory. However, a possible explanation for these conflicting results could reside in the stage of differentiation of bone-forming cells exposed to IL-17 and the timing of exposure to the cytokine, as IL-17 acts as a stimulator of MSCs osteoclastogenic differentiation [162,226], while it exerts anti-differentiating effects on osteoblast-committed cells[227].
Translating these findings to clinical practice, the advent of IL-17i and IL-12/23i created interest in the potential inhibition of both inflammation and new bone formation, allowing to delay radiographic progression. The relevance of IL-17 as an osteogenic stimulus has been confirmed first in vitro, demonstrating that IL-17i could reduce both the inflammation and the ossification process [228], then in clinical trials of secukinumab and ixekizumab, two IL-17i, which has been approved for the treatment of axSpA and PsA demonstrating to be effective in achievement of clinical remission and in reducing new bone formation in the spine [229]. However, the inhibition of IL-23 signalling in patients with axSpA did not achieve the expected results. Indeed, the ustekinumab (IL-12/23i) and risankizumab (IL-23i) trials on ankylosing spondylitis have failed and it is probably explained by the very early intervention of IL-23 in the pathogenesis of axSpA and by the evidence that IL-17 could be produced independently from IL-23 regulation [230,231,232].
IL-22 is another important cytokine in SpA. It is a pleiotropic cytokine with an important function in the regulation of adaptive immunity([233]. Serum levels of IL-22 were increased in patients with axSpA - particularly in patients with longstanding disease - compared to healthy subjects in a recent study. However, IL-22 serum levels were not associated with disease activity nor with radiographic progression [185]. These results do not differ from what was yet demonstrated. Indeed, the concept that IL-23-mediated synthesis of IL-22 is believed to contribute to new bone formation [234]. Interestingly, in an in vitro study IL-22 drove the migration and the osteogenic differentiation of hMSCs only when IFN-γ and TNF were added to the culture, demonstrating once again how biological processes are regulated by highly complex systems [235].
Apart from the classic regulators of bone metabolism, the potential role of many other molecules has been studied. The most important are represented by adipokines - such as leptin - and serotonin. Leptin is an adipokine produced by adipocytes that seems to promote osteoblast and inhibit osteoclast activity in vitro [236]. However, studies on leptin levels in patients with SpA are once again not conclusive. Data from the ENRADAS trial reported a protective role of leptin against radiographic progression [237]. Interestingly, considering the usually higher levels of leptin in women, it has been suggested that leptin could partially explain the lower rate of new bone formation in women with axSpA [237].
Serotonin is a polyhedral substance produced by both nerve cells in the central nervous system and the enterochromaffin cells of the pancreas. Among its functions, gastrointestinal serotonin acts as an inhibitor of osteoblasts proliferation in vitro and, consequently, of the bone formation process[238]. A study published a few years ago demonstrated lower levels of serotonin in patients with AS compared to RA and even lower levels in patients with AS under treatment with TNFi ([239]. Such findings gave way to the acknowledgement of the so-called gut-bone axis and a potential role of serotonin in the regulation of bone metabolism in patients with SpA has been hypothesized.
4.7. Osteoporosis in Spondyloarthritis
Various studies indicate that the prevalence of osteoporosis in SpA ranges from 12% to 34% with a four-fold increase in the risk of vertebral fractures compared to healthy individuals [240].Specifically, factors contributing to the occurrence of fractures include male gender, longer duration of the disease, concurrent inflammatory bowel disease, elevated disease activity scores, reduced femur BMD, and the presence of syndesmophytes [241]. The role of skin involvement as a risk factor for the development of osteoporosis is still debated. A recent study conducted on a large cohort of SpA patients found no disparity in the prevalence of osteoporosis based on phenotypes (axial or peripheral) in the presence of skin psoriasis [242]. Among spondyloarthritis related to inflammatory bowel disease (IBD), it can be suggested that a heightened risk of osteoporosis exists, most likely due to malabsorption issues stemming from these gastrointestinal disorders.
5. Conclusions
To conclude, while the underlying mechanisms of bone erosions have been extensively elucidated, giving some convincing mechanistic evidence especially about RA pathogenetic processes, the bone involvement in SpA is still far from being fully elucidated.
No bone markers have been identified to help the clinical management nor provide prognostic certainties on radiographic progression in both RA and SpA.
Further studies need to clarify the interplay between bone tissue and rheumatic chronic arthropathies.
Author Contributions
Conceptualization, F.O. and C.C.; writing—review and editing, F.O., G.C., R.D.T.,A.M., F.M.; supervision, M.V. and R.C.; All authors have read and agreed to the published version of the manuscript.”.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bolamperti, S.; Villa, I.; Rubinacci, A. Bone Remodeling: An Operational Process Ensuring Survival and Bone Mechanical Competence. Bone Res 2022, 10, 48. [CrossRef]
- Parfitt, A.M. Osteonal and Hemi-Osteonal Remodeling: The Spatial and Temporal Framework for Signal Traffic in Adult Human Bone. J Cell Biochem 1994, 55, 273–286. [CrossRef]
- Takayanagi, H. Osteoimmunology: Shared Mechanisms and Crosstalk between the Immune and Bone Systems. Nat Rev Immunol 2007, 7, 292–304. [CrossRef]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol Rev 2017, 97, 1295–1349. [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev Cell 2002, 3, 889–901. [CrossRef]
- Yeo, L.; Toellner, K.-M.; Salmon, M.; Filer, A.; Buckley, C.D.; Raza, K.; Scheel-Toellner, D. Cytokine MRNA Profiling Identifies B Cells as a Major Source of RANKL in Rheumatoid Arthritis. Ann Rheum Dis 2011, 70, 2022–2028. [CrossRef]
- Wehmeyer, C.; Pap, T.; Buckley, C.D.; Naylor, A.J. The Role of Stromal Cells in Inflammatory Bone Loss. Clin Exp Immunol 2017, 189, 1–11. [CrossRef]
- Silva, B.C.; Bilezikian, J.P. Parathyroid Hormone: Anabolic and Catabolic Actions on the Skeleton. Curr Opin Pharmacol 2015, 22, 41–50. [CrossRef]
- Nakashima, K.; de Crombrugghe, B. Transcriptional Mechanisms in Osteoblast Differentiation and Bone Formation. Trends Genet 2003, 19, 458–466. [CrossRef]
- Chen, G.; Deng, C.; Li, Y.-P. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int J Biol Sci 2012, 8, 272–288. [CrossRef]
- Titorencu, I.; Pruna, V.; Jinga, V.V.; Simionescu, M. Osteoblast Ontogeny and Implications for Bone Pathology: An Overview. Cell Tissue Res 2014, 355, 23–33. [CrossRef]
- Kawane, T.; Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, H.; Yoshida, C.A.; Matsuura-Kawata, V.K.D.S.; Sakane, C.; Matsuo, Y.; Nagai, K.; et al. Runx2 Is Required for the Proliferation of Osteoblast Progenitors and Induces Proliferation by Regulating Fgfr2 and Fgfr3. Sci Rep 2018, 8, 13551. [CrossRef]
- Cawley, K.M.; Bustamante-Gomez, N.C.; Guha, A.G.; MacLeod, R.S.; Xiong, J.; Gubrij, I.; Liu, Y.; Mulkey, R.; Palmieri, M.; Thostenson, J.D.; et al. Local Production of Osteoprotegerin by Osteoblasts Suppresses Bone Resorption. Cell Rep 2020, 32, 108052. [CrossRef]
- Westendorf, J.J.; Kahler, R.A.; Schroeder, T.M. Wnt Signaling in Osteoblasts and Bone Diseases. Gene 2004, 341, 19–39. [CrossRef]
- De Santis, M.; Di Matteo, B.; Chisari, E.; Cincinelli, G.; Angele, P.; Lattermann, C.; Filardo, G.; Vitale, N.D.; Selmi, C.; Kon, E. The Role of Wnt Pathway in the Pathogenesis of OA and Its Potential Therapeutic Implications in the Field of Regenerative Medicine. Biomed Res Int 2018, 2018, 7402947. [CrossRef]
- Bonewald, L.F. The Amazing Osteocyte. J Bone Miner Res 2011, 26, 229–238. [CrossRef]
- Bellido, T. Osteocyte-Driven Bone Remodeling. Calcif Tissue Int 2014, 94, 25–34. [CrossRef]
- Lorenzo, J.; Horowitz, M.; Choi, Y. Osteoimmunology: Interactions of the Bone and Immune System. Endocr Rev 2008, 29, 403–440. [CrossRef]
- Guder, C.; Gravius, S.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. Osteoimmunology: A Current Update of the Interplay Between Bone and the Immune System. Front Immunol 2020, 11, 58. [CrossRef]
- Rivollier, A.; Mazzorana, M.; Tebib, J.; Piperno, M.; Aitsiselmi, T.; Rabourdin-Combe, C.; Jurdic, P.; Servet-Delprat, C. Immature Dendritic Cell Transdifferentiation into Osteoclasts: A Novel Pathway Sustained by the Rheumatoid Arthritis Microenvironment. Blood 2004, 104, 4029–4037. [CrossRef]
- Speziani, C.; Rivollier, A.; Gallois, A.; Coury, F.; Mazzorana, M.; Azocar, O.; Flacher, M.; Bella, C.; Tebib, J.; Jurdic, P.; et al. Murine Dendritic Cell Transdifferentiation into Osteoclasts Is Differentially Regulated by Innate and Adaptive Cytokines. Eur J Immunol 2007, 37, 747–757. [CrossRef]
- Maitra, R.; Follenzi, A.; Yaghoobian, A.; Montagna, C.; Merlin, S.; Cannizzo, E.S.; Hardin, J.A.; Cobelli, N.; Stanley, E.R.; Santambrogio, L. Dendritic Cell-Mediated in Vivo Bone Resorption. J Immunol 2010, 185, 1485–1491. [CrossRef]
- Wu, L.; Luo, Z.; Liu, Y.; Jia, L.; Jiang, Y.; Du, J.; Guo, L.; Bai, Y.; Liu, Y. Aspirin Inhibits RANKL-Induced Osteoclast Differentiation in Dendritic Cells by Suppressing NF-ΚB and NFATc1 Activation. Stem Cell Res Ther 2019, 10, 375. [CrossRef]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 Functions as an Osteoclastogenic Helper T Cell Subset That Links T Cell Activation and Bone Destruction. J Exp Med 2006, 203, 2673–2682. [CrossRef]
- Zaiss, M.M.; Frey, B.; Hess, A.; Zwerina, J.; Luther, J.; Nimmerjahn, F.; Engelke, K.; Kollias, G.; Hünig, T.; Schett, G.; et al. Regulatory T Cells Protect from Local and Systemic Bone Destruction in Arthritis. J Immunol 2010, 184, 7238–7246. [CrossRef]
- Flores-Borja, F.; Jury, E.C.; Mauri, C.; Ehrenstein, M.R. Defects in CTLA-4 Are Associated with Abnormal Regulatory T Cell Function in Rheumatoid Arthritis. Proc Natl Acad Sci U S A 2008, 105, 19396–19401. [CrossRef]
- Moon, S.-J.; Ahn, I.E.; Jung, H.; Yi, H.; Kim, J.; Kim, Y.; Kwok, S.-K.; Park, K.-S.; Min, J.-K.; Park, S.-H.; et al. Temporal Differential Effects of Proinflammatory Cytokines on Osteoclastogenesis. Int J Mol Med 2013, 31, 769–777. [CrossRef]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 Mediates TNF-Induced Osteoclastogenesis. J Clin Invest 2005, 115, 282–290. [CrossRef]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.-R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-α Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Frontiers in Immunology 2019, 10.
- Diarra, D.; Stolina, M.; Polzer, K.; Zwerina, J.; Ominsky, M.S.; Dwyer, D.; Korb, A.; Smolen, J.; Hoffmann, M.; Scheinecker, C.; et al. Dickkopf-1 Is a Master Regulator of Joint Remodeling. Nature Medicine 2007 13:2 2007, 13, 156–163. [CrossRef]
- Zhang, L.; Ouyang, H.; Xie, Z.; Liang, Z.-H.; Wu, X.-W. Serum DKK-1 Level in the Development of Ankylosing Spondylitis and Rheumatic Arthritis: A Meta-Analysis. Exp Mol Med 2016, 48, e228. [CrossRef]
- Ma, Y.; Zhang, X.; Wang, M.; Xia, Q.; Yang, J.; Wu, M.; Han, R.; Chen, M.; Hu, X.; Yuan, Y.; et al. The Serum Level of Dickkopf-1 in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Int Immunopharmacol 2018, 59, 227–232. [CrossRef]
- Courbon, G.; Lamarque, R.; Gerbaix, M.; Caire, R.; Linossier, M.-T.; Laroche, N.; Thomas, M.; Thomas, T.; Vico, L.; Marotte, H. Early Sclerostin Expression Explains Bone Formation Inhibition before Arthritis Onset in the Rat Adjuvant-Induced Arthritis Model. Sci Rep 2018, 8, 3492. [CrossRef]
- Mao, Y.-M.; Liao, T.; Ye, Q.-L.; Wu, G.-C.; Zhang, Q.; Tao, S.-S.; Zhao, C.-N.; Wu, Q.; Dan, Y.-L.; Pan, H.-F.; et al. Increased Circulating Sclerostin Levels in Rheumatoid Arthritis Patients: An Updated Meta-Analysis. Z Rheumatol 2023, 82, 51–58. [CrossRef]
- Wehmeyer, C.; Frank, S.; Beckmann, D.; Böttcher, M.; Cromme, C.; König, U.; Fennen, M.; Held, A.; Paruzel, P.; Hartmann, C.; et al. Sclerostin Inhibition Promotes TNF-Dependent Inflammatory Joint Destruction. Science Translational Medicine 2016, 8, 330ra35-330ra35. [CrossRef]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N Engl J Med 2016, 375, 1532–1543. [CrossRef]
- Singh, S.; Dutta, S.; Khasbage, S.; Kumar, T.; Sachin, J.; Sharma, J.; Varthya, S.B. A Systematic Review and Meta-Analysis of Efficacy and Safety of Romosozumab in Postmenopausal Osteoporosis. Osteoporos Int 2022, 33, 1–12. [CrossRef]
- Kobayakawa, T.; Miyazaki, A.; Kanayama, Y.; Hirano, Y.; Takahashi, J.; Suzuki, T.; Nakamura, Y. Comparable Efficacy of Denosumab and Romosozumab in Patients with Rheumatoid Arthritis Receiving Glucocorticoid Administration. Mod Rheumatol 2023, 33, 96–103. [CrossRef]
- Sims, N.A. Influences of the IL-6 Cytokine Family on Bone Structure and Function. Cytokine 2021, 146, 155655. [CrossRef]
- Shi, X.; Jiang, J.; Hong, R.; Xu, F.; Dai, S. Circulating IGFBP-3 and Interleukin 6 as Predictors of Osteoporosis in Postmenopausal Women: A Cross-Sectional Study. Mediators Inflamm 2023, 2023, 2613766. [CrossRef]
- Franchimont, N.; Wertz, S.; Malaise, M. Interleukin-6: An Osteotropic Factor Influencing Bone Formation? Bone 2005, 37, 601–606. [CrossRef]
- Li, Y.; Bäckesjö, C.-M.; Haldosén, L.-A.; Lindgren, U. IL-6 Receptor Expression and IL-6 Effects Change during Osteoblast Differentiation. Cytokine 2008, 43, 165–173. [CrossRef]
- Herman, S.; Müller, R.B.; Krönke, G.; Zwerina, J.; Redlich, K.; Hueber, A.J.; Gelse, H.; Neumann, E.; Müller-Ladner, U.; Schett, G. Induction of Osteoclast-Associated Receptor, a Key Osteoclast Costimulation Molecule, in Rheumatoid Arthritis. Arthritis Rheum 2008, 58, 3041–3050. [CrossRef]
- Crotti, T.N.; Dharmapatni, A.A.; Alias, E.; Zannettino, A.C.; Smith, M.D.; Haynes, D.R. The Immunoreceptor Tyrosine-Based Activation Motif (ITAM) -Related Factors Are Increased in Synovial Tissue and Vasculature of Rheumatoid Arthritic Joints. Arthritis Research & Therapy 2012, 14, R245. [CrossRef]
- Hecht, C.; Schett, G.; Finzel, S. The Impact of Rheumatoid Factor and ACPA on Bone Erosion in Rheumatoid Arthritis. Ann Rheum Dis 2015, 74, e4. [CrossRef]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.-J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of Osteoclastogenesis and Bone Loss by Human Autoantibodies against Citrullinated Vimentin. J Clin Invest 2012, 122, 1791–1802. [CrossRef]
- Kleyer, A.; Finzel, S.; Rech, J.; Manger, B.; Krieter, M.; Faustini, F.; Araujo, E.; Hueber, A.J.; Harre, U.; Engelke, K.; et al. Bone Loss before the Clinical Onset of Rheumatoid Arthritis in Subjects with Anticitrullinated Protein Antibodies. Annals of the Rheumatic Diseases 2014, 73, 854–860. [CrossRef]
- Seeling, M.; Hillenhoff, U.; David, J.P.; Schett, G.; Tuckermann, J.; Lux, A.; Nimmerjahn, F. Inflammatory Monocytes and Fcγ Receptor IV on Osteoclasts Are Critical for Bone Destruction during Inflammatory Arthritis in Mice. Proc Natl Acad Sci U S A 2013, 110, 10729–10734. [CrossRef]
- Mathsson, L.; Lampa, J.; Mullazehi, M.; Rönnelid, J. Immune Complexes from Rheumatoid Arthritis Synovial Fluid Induce FcgammaRIIa Dependent and Rheumatoid Factor Correlated Production of Tumour Necrosis Factor-Alpha by Peripheral Blood Mononuclear Cells. Arthritis Res Ther 2006, 8, R64. [CrossRef]
- Bugatti, S.; Bogliolo, L.; Vitolo, B.; Manzo, A.; Montecucco, C.; Caporali, R. Anti-Citrullinated Protein Antibodies and High Levels of Rheumatoid Factor Are Associated with Systemic Bone Loss in Patients with Early Untreated Rheumatoid Arthritis. Arthritis Res Ther 2016, 18, 226. [CrossRef]
- Amkreutz, J.A.M.P.; de Moel, E.C.; Theander, L.; Willim, M.; Heimans, L.; Nilsson, J.-Å.; Karlsson, M.K.; Huizinga, T.W.J.; Åkesson, K.E.; Jacobsson, L.T.H.; et al. Association Between Bone Mineral Density and Autoantibodies in Patients With Rheumatoid Arthritis. Arthritis Rheumatol 2021, 73, 921–930. [CrossRef]
- Shi, J.; Knevel, R.; Suwannalai, P.; van der Linden, M.P.; Janssen, G.M.C.; van Veelen, P.A.; Levarht, N.E.W.; van der Helm-van Mil, A.H.M.; Cerami, A.; Huizinga, T.W.J.; et al. Autoantibodies Recognizing Carbamylated Proteins Are Present in Sera of Patients with Rheumatoid Arthritis and Predict Joint Damage. Proceedings of the National Academy of Sciences 2011, 108, 17372–17377. [CrossRef]
- O’Neil, L.J.; Oliveira, C.B.; Sandoval-Heglund, D.; Barrera-Vargas, A.; Merayo-Chalico, J.; Aguirre-Aguilar, E.; Kaplan, M.J.; Carmona-Rivera, C. Anti-Carbamylated LL37 Antibodies Promote Pathogenic Bone Resorption in Rheumatoid Arthritis. Front Immunol 2021, 12, 715997. [CrossRef]
- Machold, K.P.; Stamm, T.A.; Nell, V.P.K.; Pflugbeil, S.; Aletaha, D.; Steiner, G.; Uffmann, M.; Smolen, J.S. Very Recent Onset Rheumatoid Arthritis: Clinical and Serological Patient Characteristics Associated with Radiographic Progression over the First Years of Disease. Rheumatology 2007, 46, 342–349. [CrossRef]
- Svensson, B.; Andersson, M.L.E.; Gjertsson, I.; Hafström, I.; Ajeganova, S.; Forslind, K. Erosion-Free Rheumatoid Arthritis: Clinical and Conceptional Implications—a BARFOT Study. BMC Rheumatology 2022, 6, 88. [CrossRef]
- Schett, G.; Gravallese, E. Bone Erosion in Rheumatoid Arthritis: Mechanisms, Diagnosis and Treatment. Nat Rev Rheumatol 2012, 8, 656–664. [CrossRef]
- Bromley, M.; Woolley, D.E. Histopathology of the Rheumatoid Lesion. Arthritis & Rheumatism 1984, 27, 857–863. [CrossRef]
- Gravallese, E.M.; Harada, Y.; Wang, J.T.; Gorn, A.H.; Thornhill, T.S.; Goldring, S.R. Identification of Cell Types Responsible for Bone Resorption in Rheumatoid Arthritis and Juvenile Rheumatoid Arthritis. Am J Pathol 1998, 152, 943–951.
- Kong, Y.-Y.; Feige, U.; Sarosi, I.; Bolon, B.; Tafuri, A.; Morony, S.; Capparelli, C.; Li, J.; Elliott, R.; McCabe, S.; et al. Activated T Cells Regulate Bone Loss and Joint Destruction in Adjuvant Arthritis through Osteoprotegerin Ligand. Nature 1999, 402, 43–47. [CrossRef]
- Berardi, S.; Corrado, A.; Maruotti, N.; Cici, D.; Cantatore, F.P. Osteoblast Role in the Pathogenesis of Rheumatoid Arthritis. Mol Biol Rep 2021, 48, 2843–2852. [CrossRef]
- Harre, U.; Schett, G. Cellular and Molecular Pathways of Structural Damage in Rheumatoid Arthritis. Semin Immunopathol 2017, 39, 355–363. [CrossRef]
- Bugatti, S.; Caporali, R.; Manzo, A.; Vitolo, B.; Pitzalis, C.; Montecucco, C. Involvement of Subchondral Bone Marrow in Rheumatoid Arthritis: Lymphoid Neogenesis and in Situ Relationship to Subchondral Bone Marrow Osteoclast Recruitment. Arthritis Rheum 2005, 52, 3448–3459. [CrossRef]
- Allard-Chamard, H.; Carrier, N.; Dufort, P.; Durand, M.; de Brum-Fernandes, A.J.; Boire, G.; Komarova, S.V.; Dixon, S.J.; Harrison, R.E.; Manolson, M.F.; et al. Osteoclasts and Their Circulating Precursors in Rheumatoid Arthritis: Relationships with Disease Activity and Bone Erosions. Bone Reports 2020, 12, 100282. [CrossRef]
- Crotti, T.N.; Flannery, M.; Walsh, N.C.; Fleming, J.D.; Goldring, S.R.; McHugh, K.P. NFATc1 Regulation of the Human Β3 Integrin Promoter in Osteoclast Differentiation. Gene 2006, 372, 92–102. [CrossRef]
- Nedeva, I.R.; Vitale, M.; Elson, A.; Hoyland, J.A.; Bella, J. Role of OSCAR Signaling in Osteoclastogenesis and Bone Disease. Frontiers in Cell and Developmental Biology 2021, 9.
- Chen, D.-Y.; Yao, L.; Chen, Y.-M.; Lin, C.-C.; Huang, K.-C.; Chen, S.-T.; Lan, J.-L.; Hsieh, S.-L.E. A Potential Role of Myeloid DAP12-Associating Lectin (MDL)-1 in the Regulation of Inflammation in Rheumatoid Arthritis Patients. PLOS ONE 2014, 9, e86105. [CrossRef]
- Jung, Y.-K.; Kang, Y.-M.; Han, S. Osteoclasts in the Inflammatory Arthritis: Implications for Pathologic Osteolysis. Immune Netw 2019, 19, e2. [CrossRef]
- Krishnamurthy, A.; Joshua, V.; Hensvold, A.H.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.J.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a Novel Chemokine-Dependent Molecular Mechanism Underlying Rheumatoid Arthritis-Associated Autoantibody-Mediated Bone Loss. Annals of the Rheumatic Diseases 2016, 75, 721–729. [CrossRef]
- Negishi-Koga, T.; Gober, H.-J.; Sumiya, E.; Komatsu, N.; Okamoto, K.; Sawa, S.; Suematsu, A.; Suda, T.; Sato, K.; Takai, T.; et al. Immune Complexes Regulate Bone Metabolism through FcRγ Signalling. Nat Commun 2015, 6, 6637. [CrossRef]
- Edwards, J.C.; Cambridge, G. Rheumatoid Arthritis: The Predictable Effect of Small Immune Complexes in Which Antibody Is Also Antigen. Br J Rheumatol 1998, 37, 126–130. [CrossRef]
- Song, Y.W.; Kang, E.H. Autoantibodies in Rheumatoid Arthritis: Rheumatoid Factors and Anticitrullinated Protein Antibodies. QJM: An International Journal of Medicine 2010, 103, 139–146. [CrossRef]
- Hecht, C.; Englbrecht, M.; Rech, J.; Schmidt, S.; Araujo, E.; Engelke, K.; Finzel, S.; Schett, G. Additive Effect of Anti-Citrullinated Protein Antibodies and Rheumatoid Factor on Bone Erosions in Patients with RA. Annals of the Rheumatic Diseases 2015, 74, 2151–2156. [CrossRef]
- Buckley, C.D.; Ospelt, C.; Gay, S.; Midwood, K.S. Location, Location, Location: How the Tissue Microenvironment Affects Inflammation in RA. Nat Rev Rheumatol 2021, 17, 195–212. [CrossRef]
- Lu, M.-C.; Lai, N.-S.; Yu, H.-C.; Huang, H.-B.; Hsieh, S.-C.; Yu, C.-L. Anti-Citrullinated Protein Antibodies Bind Surface-Expressed Citrullinated Grp78 on Monocyte/Macrophages and Stimulate Tumor Necrosis Factor Alpha Production. Arthritis Rheum 2010, 62, 1213–1223. [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid Arthritis. Nat Rev Dis Primers 2018, 4, 1–23. [CrossRef]
- Weitzmann, M.N.; Ofotokun, I. Physiological and Pathophysiological Bone Turnover — Role of the Immune System. Nat Rev Endocrinol 2016, 12, 518–532. [CrossRef]
- Luo, G.; Li, F.; Li, X.; Wang, Z.-G.; Zhang, B. TNF-α and RANKL Promote Osteoclastogenesis by Upregulating RANK via the NF-κB Pathway. Molecular Medicine Reports 2018, 17, 6605–6611. [CrossRef]
- Yao, Z.; Li, P.; Zhang, Q.; Schwarz, E.M.; Keng, P.; Arbini, A.; Boyce, B.F.; Xing, L. Tumor Necrosis Factor-α Increases Circulating Osteoclast Precursor Numbers by Promoting Their Proliferation and Differentiation in the Bone Marrow through Up-Regulation of c-Fms Expression *. Journal of Biological Chemistry 2006, 281, 11846–11855. [CrossRef]
- Gilbert, L.; He, X.; Farmer, P.; Rubin, J.; Drissi, H.; Wijnen, A.J. van; Lian, J.B.; Stein, G.S.; Nanes, M.S. Expression of the Osteoblast Differentiation Factor RUNX2 (Cbfa1/AML3/Pebp2αA) Is Inhibited by Tumor Necrosis Factor-α *. Journal of Biological Chemistry 2002, 277, 2695–2701. [CrossRef]
- Yeremenko, N.; Zwerina, K.; Rigter, G.; Pots, D.; Fonseca, J.E.; Zwerina, J.; Schett, G.; Baeten, D. Tumor Necrosis Factor and Interleukin-6 Differentially Regulate Dkk-1 in the Inflamed Arthritic Joint. Arthritis Rheumatol 2015, 67, 2071–2075. [CrossRef]
- Srirangan, S.; Choy, E.H. The Role of Interleukin 6 in the Pathophysiology of Rheumatoid Arthritis. Therapeutic Advances in Musculoskeletal 2010, 2, 247–256. [CrossRef]
- Chang, P.-Y.; Wu, H.-K.; Chen, Y.-H.; Hsu, Y.-P.; Cheng, M.-T.; Yu, C.-H.; Chen, S.-K. Interleukin-6 Transiently Promotes Proliferation of Osteoclast Precursors and Stimulates the Production of Inflammatory Mediators. Mol Biol Rep 2022, 49, 3927–3937. [CrossRef]
- Yoshitake, F.; Itoh, S.; Narita, H.; Ishihara, K.; Ebisu, S. Interleukin-6 Directly Inhibits Osteoclast Differentiation by Suppressing Receptor Activator of NF-ΚB Signaling Pathways *. Journal of Biological Chemistry 2008, 283, 11535–11540. [CrossRef]
- Takagawa, S.; Nakamura, F.; Kumagai, K.; Nagashima, Y.; Goshima, Y.; Saito, T. Decreased Semaphorin3A Expression Correlates with Disease Activity and Histological Features of Rheumatoid Arthritis. BMC Musculoskeletal Disorders 2013, 14, 40. [CrossRef]
- Akatsu, T.; Takahashi, N.; Udagawa, N.; Imamura, K.; Yamaguchi, A.; Sato, K.; Nagata, N.; Suda, T. Role of Prostaglandins in Interleukin-1-Induced Bone Resorption in Mice in Vitro. Journal of Bone and Mineral Research 1991, 6, 183–190. [CrossRef]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The Mechanism of Osteoclast Differentiation Induced by IL-11. The Journal of Immunology 2009, 183, 1862–1870. [CrossRef]
- Tanabe, N.; Maeno, M.; Suzuki, N.; Fujisaki, K.; Tanaka, H.; Ogiso, B.; Ito, K. IL-1α Stimulates the Formation of Osteoclast-like Cells by Increasing M-CSF and PGE2 Production and Decreasing OPG Production by Osteoblasts. Life Sciences 2005, 77, 615–626. [CrossRef]
- Jiang, Y.; Genant, H.K.; Watt, I.; Cobby, M.; Bresnihan, B.; Aitchison, R.; McCabe, D. A Multicenter, Double-Blind, Dose-Ranging, Randomized, Placebo-Controlled Study of Recombinant Human Interleukin-1 Receptor Antagonist in Patients with Rheumatoid Arthritis: Radiologic Progression and Correlation of Genant and Larsen Scores. Arthritis & Rheumatism 2000, 43, 1001–1009. [CrossRef]
- Gremese, E.; Tolusso, B.; Bruno, D.; Perniola, S.; Ferraccioli, G.; Alivernini, S. The Forgotten Key Players in Rheumatoid Arthritis: IL-8 and IL-17 – Unmet Needs and Therapeutic Perspectives. Frontiers in Medicine 2023, 10.
- Adamopoulos, I.E.; Suzuki, E.; Chao, C.-C.; Gorman, D.; Adda, S.; Maverakis, E.; Zarbalis, K.; Geissler, R.; Asio, A.; Blumenschein, W.M.; et al. IL-17A Gene Transfer Induces Bone Loss and Epidermal Hyperplasia Associated with Psoriatic Arthritis. Annals of the Rheumatic Diseases 2015, 74, 1284–1292. [CrossRef]
- Yago, T.; Nanke, Y.; Ichikawa, N.; Kobashigawa, T.; Mogi, M.; Kamatani, N.; Kotake, S. IL-17 Induces Osteoclastogenesis from Human Monocytes Alone in the Absence of Osteoblasts, Which Is Potently Inhibited by Anti-TNF-α Antibody: A Novel Mechanism of Osteoclastogenesis by IL-17. Journal of Cellular Biochemistry 2009, 108, 947–955. [CrossRef]
- Mills, K.H.G. IL-17 and IL-17-Producing Cells in Protection versus Pathology. Nat Rev Immunol 2023, 23, 38–54. [CrossRef]
- Le Goff, B.; Bouvard, B.; Lequerre, T.; Lespessailles, E.; Marotte, H.; Pers, Y.-M.; Cortet, B. Implication of IL-17 in Bone Loss and Structural Damage in Inflammatory Rheumatic Diseases. Mediators Inflamm 2019, 2019, 8659302. [CrossRef]
- Chemin, K.; Gerstner, C.; Malmström, V. Effector Functions of CD4+ T Cells at the Site of Local Autoimmune Inflammation—Lessons From Rheumatoid Arthritis. Frontiers in Immunology 2019, 10.
- Pfeifle, R.; Rothe, T.; Ipseiz, N.; Scherer, H.U.; Culemann, S.; Harre, U.; Ackermann, J.A.; Seefried, M.; Kleyer, A.; Uderhardt, S.; et al. Regulation of Autoantibody Activity by the IL-23–TH17 Axis Determines the Onset of Autoimmune Disease. Nat Immunol 2017, 18, 104–113. [CrossRef]
- Ciucci, T.; Ibáñez, L.; Boucoiran, A.; Birgy-Barelli, E.; Pène, J.; Abou-Ezzi, G.; Arab, N.; Rouleau, M.; Hébuterne, X.; Yssel, H.; et al. Bone Marrow Th17 TNFα Cells Induce Osteoclast Differentiation, and Link Bone Destruction to IBD. Gut 2015, 64, 1072–1081. [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How Regulatory T Cells Work. Nat Rev Immunol 2008, 8, 523–532. [CrossRef]
- Zaiss, M.M.; Axmann, R.; Zwerina, J.; Polzer, K.; Gückel, E.; Skapenko, A.; Schulze-Koops, H.; Horwood, N.; Cope, A.; Schett, G. Treg Cells Suppress Osteoclast Formation: A New Link between the Immune System and Bone. Arthritis Rheum 2007, 56, 4104–4112. [CrossRef]
- Fischer, L.; Herkner, C.; Kitte, R.; Dohnke, S.; Riewaldt, J.; Kretschmer, K.; Garbe, A.I. Foxp3+ Regulatory T Cells in Bone and Hematopoietic Homeostasis. Front Endocrinol (Lausanne) 2019, 10, 578. [CrossRef]
- Zhu, L.; Hua, F.; Ding, W.; Ding, K.; Zhang, Y.; Xu, C. The Correlation between the Th17/Treg Cell Balance and Bone Health. Immun Ageing 2020, 17, 30. [CrossRef]
- Komatsu, N.; Takayanagi, H. Mechanisms of Joint Destruction in Rheumatoid Arthritis — Immune Cell–Fibroblast–Bone Interactions. Nat Rev Rheumatol 2022, 18, 415–429. [CrossRef]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving Concepts in Bone–Immune Interactions in Health and Disease. Nat Rev Immunol 2019, 19, 626–642. [CrossRef]
- Orr, C.; Vieira-Sousa, E.; Boyle, D.L.; Buch, M.H.; Buckley, C.D.; Cañete, J.D.; Catrina, A.I.; Choy, E.H.S.; Emery, P.; Fearon, U.; et al. Synovial Tissue Research: A State-of-the-Art Review. Nat Rev Rheumatol 2017, 13, 463–475. [CrossRef]
- Nygaard, G.; Firestein, G.S. Restoring Synovial Homeostasis in Rheumatoid Arthritis by Targeting Fibroblast-like Synoviocytes. Nat Rev Rheumatol 2020, 16, 316–333. [CrossRef]
- Brentano, F.; Schorr, O.; Ospelt, C.; Stanczyk, J.; Gay, R.E.; Gay, S.; Kyburz, D. Pre-B Cell Colony-Enhancing Factor/Visfatin, a New Marker of Inflammation in Rheumatoid Arthritis with Proinflammatory and Matrix-Degrading Activities. Arthritis Rheum 2007, 56, 2829–2839. [CrossRef]
- Tu, J.; Huang, W.; Zhang, W.; Mei, J.; Zhu, C. Two Main Cellular Components in Rheumatoid Arthritis: Communication Between T Cells and Fibroblast-Like Synoviocytes in the Joint Synovium. Frontiers in Immunology 2022, 13.
- Ouboussad, L.; Burska, A.N.; Melville, A.; Buch, M.H. Synovial Tissue Heterogeneity in Rheumatoid Arthritis and Changes With Biologic and Targeted Synthetic Therapies to Inform Stratified Therapy. Frontiers in Medicine 2019, 6.
- Buckley, C.; Filer, A. Fibroblasts and Fibroblastlike Synoviocytes. In Firestein and Kelley’s Textbook of Rheumatology; Elsevier, Inc.: Philadelphia, 2021; pp. 222–240.
- Wu, Z.; Ma, D.; Yang, H.; Gao, J.; Zhang, G.; Xu, K.; Zhang, L. Fibroblast-like Synoviocytes in Rheumatoid Arthritis: Surface Markers and Phenotypes. International Immunopharmacology 2021, 93, 107392. [CrossRef]
- Matsuda, K.; Shiba, N.; Hiraoka, K. New Insights into the Role of Synovial Fibroblasts Leading to Joint Destruction in Rheumatoid Arthritis. International Journal of Molecular Sciences 2023, 24, 5173. [CrossRef]
- Firestein, G.S. Evolving Concepts of Rheumatoid Arthritis. Nature 2003, 423, 356–361. [CrossRef]
- Grillet, B.; Pereira, R.V.S.; Van Damme, J.; Abu El-Asrar, A.; Proost, P.; Opdenakker, G. Matrix Metalloproteinases in Arthritis: Towards Precision Medicine. Nat Rev Rheumatol 2023, 19, 363–377. [CrossRef]
- Noss, E.H.; Chang, S.K.; Watts, G.F.M.; Brenner, M.B. Cadherin-11 Engagement Modulates Matrix Metalloproteinase Production by Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheum 2011, 63, 3768–3778. [CrossRef]
- Danks, L.; Komatsu, N.; Guerrini, M.M.; Sawa, S.; Armaka, M.; Kollias, G.; Nakashima, T.; Takayanagi, H. RANKL Expressed on Synovial Fibroblasts Is Primarily Responsible for Bone Erosions during Joint Inflammation. Annals of the Rheumatic Diseases 2016, 75, 1187–1195. [CrossRef]
- Komatsu, N.; Win, S.; Yan, M.; Huynh, N.C.-N.; Sawa, S.; Tsukasaki, M.; Terashima, A.; Pluemsakunthai, W.; Kollias, G.; Nakashima, T.; et al. Plasma Cells Promote Osteoclastogenesis and Periarticular Bone Loss in Autoimmune Arthritis. J Clin Invest 2021, 131, e143060, 143060. [CrossRef]
- Juarez, M.; Toellner, D.S.; Karouzakis, E.; Hardy, R.; Yeo, L.; Bayley, R.; De Paz, B.; Raza, K.; Cooper, M.; Gay, S.; et al. Early Rheumatoid Arthritis and Resolving Fibroblasts Segregate According to Dickkopf Related Protein 1 Expression. The Lancet 2013, 381, S57. [CrossRef]
- Zheng, L.; Hu, F.; Bian, W.; Li, Y.; Zhang, L.; Shi, L.; Ma, X.; Liu, Y.; Zhang, X.; Li, Z. Dickkopf-1 Perpetuated Synovial Fibroblast Activation and Synovial Angiogenesis in Rheumatoid Arthritis. Clin Rheumatol 2021, 40, 4279–4288. [CrossRef]
- Barranco, C. Sclerostin: A Novel Role in TNF Arthritis? Nat Rev Rheumatol 2016, 12, 251–251. [CrossRef]
- Komatsu, N.; Takayanagi, H. Inflammation and Bone Destruction in Arthritis: Synergistic Activity of Immune and Mesenchymal Cells in Joints. Frontiers in Immunology 2012, 3.
- Chang, S.K.; Noss, E.H.; Chen, M.; Gu, Z.; Townsend, K.; Grenha, R.; Leon, L.; Lee, S.Y.; Lee, D.M.; Brenner, M.B. Cadherin-11 Regulates Fibroblast Inflammation. Proceedings of the National Academy of Sciences 2011, 108, 8402–8407. [CrossRef]
- Yellin, M.J.; Winikoff, S.; Fortune, S.M.; Baum, D.; Crow, M.K.; Lederman, S.; Chess, L. Ligation of CD40 on Fibroblasts Induces CD54 (ICAM-1) and CD106 (VCAM-1) up-Regulation and IL-6 Production and Proliferation. J Leukoc Biol 1995, 58, 209–216. [CrossRef]
- Ogura, H.; Murakami, M.; Okuyama, Y.; Tsuruoka, M.; Kitabayashi, C.; Kanamoto, M.; Nishihara, M.; Iwakura, Y.; Hirano, T. Interleukin-17 Promotes Autoimmunity by Triggering a Positive-Feedback Loop via Interleukin-6 Induction. Immunity 2008, 29, 628–636. [CrossRef]
- Van Seventer, G.A.; Shimizu, Y.; Horgan, K.J.; Shaw, S. The LFA-1 Ligand ICAM-1 Provides an Important Costimulatory Signal for T Cell Receptor-Mediated Activation of Resting T Cells. J Immunol 1990, 144, 4579–4586.
- Sato, H.; Muraoka, S.; Kusunoki, N.; Masuoka, S.; Yamada, S.; Ogasawara, H.; Imai, T.; Akasaka, Y.; Tochigi, N.; Takahashi, H.; et al. Resistin Upregulates Chemokine Production by Fibroblast-like Synoviocytes from Patients with Rheumatoid Arthritis. Arthritis Research & Therapy 2017, 19, 263. [CrossRef]
- Cambier, S.; Gouwy, M.; Proost, P. The Chemokines CXCL8 and CXCL12: Molecular and Functional Properties, Role in Disease and Efforts towards Pharmacological Intervention. Cell Mol Immunol 2023, 20, 217–251. [CrossRef]
- Lee, J.-H.; Kim, B.; Jin, W.J.; Kim, H.-H.; Ha, H.; Lee, Z.H. Pathogenic Roles of CXCL10 Signaling through CXCR3 and TLR4 in Macrophages and T Cells: Relevance for Arthritis. Arthritis Research & Therapy 2017, 19, 163. [CrossRef]
- Hirota, K.; Hashimoto, M.; Yoshitomi, H.; Tanaka, S.; Nomura, T.; Yamaguchi, T.; Iwakura, Y.; Sakaguchi, N.; Sakaguchi, S. T Cell Self-Reactivity Forms a Cytokine Milieu for Spontaneous Development of IL-17+ Th Cells That Cause Autoimmune Arthritis. J Exp Med 2007, 204, 41–47. [CrossRef]
- Burger, J.A.; Zvaifler, N.J.; Tsukada, N.; Firestein, G.S.; Kipps, T.J. Fibroblast-like Synoviocytes Support B-Cell Pseudoemperipolesis via a Stromal Cell–Derived Factor-1– and CD106 (VCAM-1)–Dependent Mechanism. J Clin Invest 2001, 107, 305–315. [CrossRef]
- Bombardieri, M.; Kam, N.-W.; Brentano, F.; Choi, K.; Filer, A.; Kyburz, D.; McInnes, I.B.; Gay, S.; Buckley, C.; Pitzalis, C. A BAFF/APRIL-Dependent TLR3-Stimulated Pathway Enhances the Capacity of Rheumatoid Synovial Fibroblasts to Induce AID Expression and Ig Class-Switching in B Cells. Ann Rheum Dis 2011, 70, 1857–1865. [CrossRef]
- Hasegawa, T.; Kikuta, J.; Sudo, T.; Matsuura, Y.; Matsui, T.; Simmons, S.; Ebina, K.; Hirao, M.; Okuzaki, D.; Yoshida, Y.; et al. Identification of a Novel Arthritis-Associated Osteoclast Precursor Macrophage Regulated by FoxM1. Nat Immunol 2019, 20, 1631–1643. [CrossRef]
- O’Neil, L.J.; Oliveira, C.B.; Wang, X.; Navarrete, M.; Barrera-Vargas, A.; Merayo-Chalico, J.; Aljahdali, R.; Aguirre-Aguilar, E.; Carlucci, P.; Kaplan, M.J.; et al. Neutrophil Extracellular Trap-Associated Carbamylation and Histones Trigger Osteoclast Formation in Rheumatoid Arthritis. Annals of the Rheumatic Diseases 2023, 82, 630–638. [CrossRef]
- Wigerblad, G.; Kaplan, M.J. Neutrophil Extracellular Traps in Systemic Autoimmune and Autoinflammatory Diseases. Nat Rev Immunol 2023, 23, 274–288. [CrossRef]
- Carmona-Rivera, C.; Carlucci, P.M.; Goel, R.R.; James, E.; Brooks, S.R.; Rims, C.; Hoffmann, V.; Fox, D.A.; Buckner, J.H.; Kaplan, M.J. Neutrophil Extracellular Traps Mediate Articular Cartilage Damage and Enhance Cartilage Component Immunogenicity in Rheumatoid Arthritis. JCI Insight 2020, 5, e139388, 139388. [CrossRef]
- Kim, T.S.; Silva, L.M.; Theofilou, V.I.; Greenwell-Wild, T.; Li, L.; Williams, D.W.; Ikeuchi, T.; Brenchley, L.; NIDCD/NIDCR Genomics and Computational Biology Core; Bugge, T.H.; et al. Neutrophil Extracellular Traps and Extracellular Histones Potentiate IL-17 Inflammation in Periodontitis. Journal of Experimental Medicine 2023, 220, e20221751. [CrossRef]
- Glaser, D.L.; Kaplan, F.S. Osteoporosis. Definition and Clinical Presentation. Spine (Phila Pa 1976) 1997, 22, 12S-16S. [CrossRef]
- Tong, J.-J.; Xu, S.-Q.; Zong, H.-X.; Pan, M.-J.; Teng, Y.-Z.; Xu, J.-H. Prevalence and Risk Factors Associated with Vertebral Osteoporotic Fractures in Patients with Rheumatoid Arthritis. Clin Rheumatol 2020, 39, 357–364. [CrossRef]
- Jin, S.; Hsieh, E.; Peng, L.; Yu, C.; Wang, Y.; Wu, C.; Wang, Q.; Li, M.; Zeng, X. Incidence of Fractures among Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Osteoporos Int 2018, 29, 1263–1275. [CrossRef]
- Conforti, A.; Di Cola, I.; Pavlych, V.; Ruscitti, P.; Berardicurti, O.; Ursini, F.; Giacomelli, R.; Cipriani, P. Beyond the Joints, the Extra-Articular Manifestations in Rheumatoid Arthritis. Autoimmun Rev 2021, 20, 102735. [CrossRef]
- Hacquard-Bouder, C.; Ittah, M.; Breban, M. Animal Models of HLA-B27-Associated Diseases: New Outcomes. Joint Bone Spine 2006, 73, 132–138. [CrossRef]
- Bowness, P. HLA-B27. Annu. Rev. Immunol. 2015, 33, 29–48. [CrossRef]
- Gui, L.; Gu, J. The Study of the Effect of HLA-B27 on THP-1 Monocytic Cells Survival and Its Mechanism. Int J Rheum Dis 2023, 26, 1474–1484. [CrossRef]
- Khan, M.A.; Yong, S.-B.; Wei, J.C.-C. Ankylosing Spondylitis: History, Epidemiology, and HLA-B27. Int J Rheum Dis 2023, 26, 413–414. [CrossRef]
- Kavadichanda, C.G.; Geng, J.; Bulusu, S.N.; Negi, V.S.; Raghavan, M. Spondyloarthritis and the Human Leukocyte Antigen (HLA)-B*27 Connection. Front. Immunol. 2021, 12, 601518. [CrossRef]
- Layh-Schmitt, G.; Yang, E.Y.; Kwon, G.; Colbert, R.A. HLA-B27 Alters the Response to Tumor Necrosis Factor α and Promotes Osteoclastogenesis in Bone Marrow Monocytes From HLA-B27-Transgenic Rats: Cellular Effects of HLA-B27 Misfolding in Transgenic Rats. Arthritis & Rheumatism 2013, 65, 2123–2131. [CrossRef]
- Papet, I.; Yousfi, M.E.; Godin, J.-P.; Mermoud, A.-F.; Davicco, M.-J.; Coxam, V.; Breuillé, D.; Obled, C. HLA-B27 Rats Develop Osteopaenia through Increased Bone Resorption without Any Change in Bone Formation.
- Rauner, M.; Stupphann, D.; Haas, M.; Fert, I.; Glatigny, S.; Sipos, W.; Breban, M.; Pietschmann, P. The HLA-B27 Transgenic Rat, a Model of Spondyloarthritis, Has Decreased Bone Mineral Density and Increased RANKL to Osteoprotegerin MRNA Ratio. J Rheumatol 2009, 36, 120–126. [CrossRef]
- Gamsjaeger, S.; Srivastava, A.K.; Wergedal, J.E.; Zwerina, J.; Klaushofer, K.; Paschalis, E.P.; Tatakis, D.N. Altered Bone Material Properties in HLA-B27 Rats Include Reduced Mineral to Matrix Ratio and Altered Collagen Cross-Links: BONE QUALITY IN HLA-B27 RATS. J Bone Miner Res 2014, 29, 2382–2391. [CrossRef]
- Rauner, M.; Thiele, S.; Fert, I.; Araujo, L.M.; Layh-Schmitt, G.; Colbert, R.A.; Hofbauer, C.; Bernhardt, R.; Bürki, A.; Schwiedrzik, J.; et al. Loss of Bone Strength in HLA-B27 Transgenic Rats Is Characterized by a High Bone Turnover and Is Mainly Osteoclast-Driven. Bone 2015, 75, 183–191. [CrossRef]
- Hauser, B.; Zhao, S.; Visconti, M.R.; Riches, P.L.; Fraser, W.D.; Piec, I.; Goodson, N.J.; Ralston, S.H. Autoantibodies to Osteoprotegerin Are Associated with Low Hip Bone Mineral Density and History of Fractures in Axial Spondyloarthritis: A Cross-Sectional Observational Study. Calcif Tissue Int 2017, 101, 375–383. [CrossRef]
- Kim, H.-R.; Kim, H.-Y.; Lee, S.-H. Elevated Serum Levels of Soluble Receptor Activator of Nuclear Factors- B Ligand (SRANKL) and Reduced Bone Mineral Density in Patients with Ankylosing Spondylitis (AS). Rheumatology 2006, 45, 1197–1200. [CrossRef]
- Tan, Z.Y.; Bealgey, K.W.; Fang, Y.; Gong, Y.M.; Bao, S. Interleukin-23: Immunological Roles and Clinical Implications. The International Journal of Biochemistry & Cell Biology 2009, 41, 733–735. [CrossRef]
- Ciccia, F.; Rizzo, A.; Triolo, G. Subclinical Gut Inflammation in Ankylosing Spondylitis. Current Opinion in Rheumatology 2016, 28, 89–96. [CrossRef]
- Yago, T.; Nanke, Y.; Kawamoto, M.; Furuya, T.; Kobashigawa, T.; Kamatani, N.; Kotake, S. IL-23 Induces Human Osteoclastogenesis via IL-17 in Vitro, and Anti-IL-23 Antibody Attenuates Collagen-Induced Arthritis in Rats. Arthritis Res Ther 2007, 9, R96. [CrossRef]
- Li, X.; Kim, K.-W.; Cho, M.-L.; Ju, J.-H.; Kang, C.-M.; Oh, H.-J.; Min, J.-K.; Lee, S.-H.; Park, S.-H.; Kim, H.-Y. IL-23 Induces Receptor Activator of NF-ΚB Ligand Expression in Fibroblast-like Synoviocytes via STAT3 and NF-ΚB Signal Pathways. Immunology Letters 2010, 127, 100–107. [CrossRef]
- Chisălău, B.; Crînguș, L.-I.; Vreju, F.; Pârvănescu, C.; Firulescu, S.; Dinescu, Ștefan; Ciobanu, D.; Tica, A.; Sandu, R.; Siloși, I.; et al. New Insights into IL-17/IL-23 Signaling in Ankylosing Spondylitis (Review). Exp Ther Med 2020. [CrossRef]
- Chalise, J.; Narendra, S.; Paudyal, B.; Magnusson, M. Interferon Alpha Inhibits Antigen-Specific Production of Proinflammatory Cytokines and Enhances Antigen-Specific Transforming Growth Factor Beta Production in Antigen-Induced Arthritis. Arthritis Res Ther 2013, 15, R143. [CrossRef]
- Tang, M.; Lu, L.; Yu, X. Interleukin-17A Interweaves the Skeletal and Immune Systems. Front. Immunol. 2021, 11, 625034. [CrossRef]
- Van Bezooijen, R.L.; Farih-Sips, H.C.M.; Papapoulos, S.E.; Löwik, C.W.G.M. Interleukin-17: A New Bone Acting Cytokine In Vitro. J Bone Miner Res 1999, 14, 1513–1521. [CrossRef]
- Uluçkan, Ö.; Jimenez, M.; Karbach, S.; Jeschke, A.; Graña, O.; Keller, J.; Busse, B.; Croxford, A.L.; Finzel, S.; Koenders, M.; et al. Chronic Skin Inflammation Leads to Bone Loss by IL-17-Mediated Inhibition of Wnt Signaling in Osteoblasts. Sci Transl Med 2016, 8, 330ra37. [CrossRef]
- Raimondo, A.; Lembo, S.; Di Caprio, R.; Donnarumma, G.; Monfrecola, G.; Balato, N.; Ayala, F.; Balato, A. Psoriatic Cutaneous Inflammation Promotes Human Monocyte Differentiation into Active Osteoclasts, Facilitating Bone Damage. European Journal of Immunology 2017, 47, 1062–1074. [CrossRef]
- Wu, D.; Li, C.; Zhang, S.; Wong, P.; Cao, Y.; Griffith, J.F.; Zhang, X.; Gu, J.; Tam, L.-S. Effect of Biologics on Radiographic Progression of Peripheral Joint in Patients with Psoriatic Arthritis: Meta-Analysis. Rheumatology 2020, 59, 3172–3180. [CrossRef]
- Osta, B.; Lavocat, F.; Eljaafari, A.; Miossec, P. Effects of Interleukin-17A on Osteogenic Differentiation of Isolated Human Mesenchymal Stem Cells. Frontiers in Immunology 2014, 5. [CrossRef]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor Necrosis Factor Alpha Stimulates Osteoclast Differentiation by a Mechanism Independent of the ODF/RANKL-RANK Interaction. J Exp Med 2000, 191, 275–286. [CrossRef]
- Li, P.; Schwarz, E.M. The TNF-Alpha Transgenic Mouse Model of Inflammatory Arthritis. Springer Semin Immunopathol 2003, 25, 19–33. [CrossRef]
- Ritchlin, C.T.; Haas-Smith, S.A.; Li, P.; Hicks, D.G.; Schwarz, E.M. Mechanisms of TNF-α– and RANKL-Mediated Osteoclastogenesis and Bone Resorption in Psoriatic Arthritis. J Clin Invest 2003, 111, 821–831. [CrossRef]
- Lambrecht, S.; Coudenys, J.; De Keyser, F.; Verbruggen, G.; Deforce, D.; Elewaut, D. Reduced Levels of the TGFb Family Member GDF15 in Spondyloarthritis versus Other Rheumatic Diseases. Annals of the Rheumatic Diseases 2011, 70, A88–A88. [CrossRef]
- Hinoi, E.; Ochi, H.; Takarada, T.; Nakatani, E.; Iezaki, T.; Nakajima, H.; Fujita, H.; Takahata, Y.; Hidano, S.; Kobayashi, T.; et al. Positive Regulation of Osteoclastic Differentiation by Growth Differentiation Factor 15 Upregulated in Osteocytic Cells under Hypoxia. J Bone Miner Res 2012, 27, 938–949. [CrossRef]
- Song, Y.; Cui, Y.; Zhang, X.; Lin, H.; Zhang, G.; Zeng, H.; Zeng, Y. Increased Serum Levels of MIC1/GDF15 Correlated with Bone Erosion in Spondyloarthritis: A Pilot Study. Medicine (Baltimore) 2018, 97, e13733. [CrossRef]
- De Martinis, M.; Sirufo, M.M.; Suppa, M.; Ginaldi, L. IL-33/IL-31 Axis in Osteoporosis. IJMS 2020, 21, 1239. [CrossRef]
- Perrigoue, J.G.; Li, J.; Zaph, C.; Goldschmidt, M.; Scott, P.; De Sauvage, F.J.; Pearce, E.J.; Ghilardi, N.; Artis, D. IL-31–IL-31R Interactions Negatively Regulate Type 2 Inflammation in the Lung. The Journal of Experimental Medicine 2007, 204, 481–487. [CrossRef]
- Perrigoue, J.G.; Zaph, C.; Guild, K.; Du, Y.; Artis, D. IL-31-IL-31R Interactions Limit the Magnitude of Th2 Cytokine-Dependent Immunity and Inflammation Following Intestinal Helminth Infection. The Journal of Immunology 2009, 182, 6088–6094. [CrossRef]
- Rosine, N.; Etcheto, A.; Hendel-Chavez, H.; Seror, R.; Briot, K.; Molto, A.; Chanson, P.; Taoufik, Y.; Wendling, D.; Lories, R.; et al. Increase In Il-31 Serum Levels Is Associated With Reduced Structural Damage In Early Axial Spondyloarthritis. Sci Rep 2018, 8, 7731. [CrossRef]
- Lems, W.; Miceli-Richard, C.; Haschka, J.; Giusti, A.; Chistensen, G.L.; Kocijan, R.; Rosine, N.; Jørgensen, N.R.; Bianchi, G.; Roux, C. Bone Involvement in Patients with Spondyloarthropathies. Calcif Tissue Int 2022, 110, 393–420. [CrossRef]
- Rossini, M.; Viapiana, O.; Idolazzi, L.; Ghellere, F.; Fracassi, E.; Troplini, S.; Povino, M.R.; Kunnathully, V.; Adami, S.; Gatti, D. Higher Level of Dickkopf-1 Is Associated with Low Bone Mineral Density and Higher Prevalence of Vertebral Fractures in Patients with Ankylosing Spondylitis. Calcif Tissue Int 2016, 98, 438–445. [CrossRef]
- Szentpetery, A.; Heffernan, E.; Haroon, M.; Kilbane, M.; Gallagher, P.; McKenna, M.J.; FitzGerald, O. Striking Difference of Periarticular Bone Density Change in Early Psoriatic Arthritis and Rheumatoid Arthritis Following Anti-Rheumatic Treatment as Measured by Digital X-Ray Radiogrammetry. Rheumatology 2016, 55, 891–896. [CrossRef]
- Haynes, D.R. Osteoprotegerin Expression in Synovial Tissue from Patients with Rheumatoid Arthritis, Spondyloarthropathies and Osteoarthritis and Normal Controls. Rheumatology 2003, 42, 123–134. [CrossRef]
- Kavanaugh, A.; Puig, L.; Gottlieb, A.B.; Ritchlin, C.; Li, S.; Wang, Y.; Mendelsohn, A.M.; Song, M.; Zhu, Y.; Rahman, P.; et al. Maintenance of Clinical Efficacy and Radiographic Benefit Through Two Years of Ustekinumab Therapy in Patients With Active Psoriatic Arthritis: Results From a Randomized, Placebo-Controlled Phase III Trial. Arthritis Care & Research 2015, 67, 1739–1749. [CrossRef]
- Van Der Heijde, D.; Landewé, R.B.; Mease, P.J.; McInnes, I.B.; Conaghan, P.G.; Pricop, L.; Ligozio, G.; Richards, H.B.; Mpofu, S. Brief Report: Secukinumab Provides Significant and Sustained Inhibition of Joint Structural Damage in a Phase III Study of Active Psoriatic Arthritis: INHIBITION OF JOINT STRUCTURAL DAMAGE WITH SECUKINUMAB. Arthritis & Rheumatology 2016, 68, 1914–1921. [CrossRef]
- Koenders, M.I.; Marijnissen, R.J.; Devesa, I.; Lubberts, E.; Joosten, L.A.B.; Roth, J.; Van Lent, P.L.E.M.; Van De Loo, F.A.; Van Den Berg, W.B. Tumor Necrosis Factor-Interleukin-17 Interplay Induces S100A8, Interleukin-1β, and Matrix Metalloproteinases, and Drives Irreversible Cartilage Destruction in Murine Arthritis: Rationale for Combination Treatment during Arthritis. Arthritis & Rheumatism 2011, 63, 2329–2339. [CrossRef]
- Dalbeth, N.; Pool, B.; Smith, T.; Callon, K.E.; Lobo, M.; Taylor, W.J.; Jones, P.B.; Cornish, J.; McQueen, F.M. Circulating Mediators of Bone Remodeling in Psoriatic Arthritis: Implications for Disordered Osteoclastogenesis and Bone Erosion. Arthritis research & therapy 2010, 12. [CrossRef]
- Chung, Y.; Li, Z.-C.; Sun, X.-L.; Liu, Y.-Y.; Shao, M.; Gan, Y.-Z.; Li, Y.-M.; Li, Y.-H.; Zhang, X.-W. Elevated Serum Dickkopf-1 Is a Biomarker for Bone Erosion in Patients with Psoriatic Arthritis. Chinese Medical Journal 2021, 134, 2583–2588. [CrossRef]
- Fassio, A.; Idolazzi, L.; Viapiana, O.; Benini, C.; Vantaggiato, E.; Bertoldo, F.; Rossini, M.; Gatti, D. In Psoriatic Arthritis Dkk-1 and PTH Are Lower than in Rheumatoid Arthritis and Healthy Controls. Clinical Rheumatology 2017, 36, 2377–2381. [CrossRef]
- Kragstrup, T.W.; Andersen, T.; Heftdal, L.D.; Hvid, M.; Gerwien, J.; Sivakumar, P.; Taylor, P.C.; Senolt, L.; Deleuran, B. The IL-20 Cytokine Family in Rheumatoid Arthritis and Spondyloarthritis. Front. Immunol. 2018, 9, 2226. [CrossRef]
- Marijnissen, R.J.; Koenders, M.I.; Smeets, R.L.; Stappers, M.H.T.; Nickerson-Nutter, C.; Joosten, L.A.B.; Boots, A.M.H.; Van Den Berg, W.B. Increased Expression of Interleukin-22 by Synovial Th17 Cells during Late Stages of Murine Experimental Arthritis Is Controlled by Interleukin-1 and Enhances Bone Degradation: IL-1-Driven Regulation of IL-22 in Chronic Arthritis. Arthritis & Rheumatism 2011, 63, 2939–2948. [CrossRef]
- Sagiv, M.; Adawi, M.; Awisat, A.; Shouval, A.; Peri, R.; Sabbah, F.; Rosner, I.; Kessel, A.; Slobodin, G. The Association between Elevated Serum Interleukin-22 and the Clinical Diagnosis of Axial Spondyloarthritis: A Retrospective Study. International Journal of Rheumatic Diseases 2022, 25, 56–60. [CrossRef]
- Poddubnyy, D.; Sieper, J. Mechanism of New Bone Formation in Axial Spondyloarthritis. Curr Rheumatol Rep 2017, 19, 55. [CrossRef]
- Sieper, J.; van der Heijde, D. Review: Nonradiographic Axial Spondyloarthritis: New Definition of an Old Disease? Arthritis Rheum 2013, 65, 543–551. [CrossRef]
- Neerinckx, B.; Lories, R. Mechanisms, Impact and Prevention of Pathological Bone Regeneration in Spondyloarthritis. Curr Opin Rheumatol 2017, 29, 287–292. [CrossRef]
- Gong, Y.; Zheng, N.; Chen, S.-B.; Xiao, Z.-Y.; Wu, M.-Y.; Liu, Y.; Zeng, Q.-Y. Ten Years’ Experience with Needle Biopsy in the Early Diagnosis of Sacroiliitis. Arthritis Rheum 2012, 64, 1399–1406. [CrossRef]
- Appel, H.; Loddenkemper, C.; Grozdanovic, Z.; Ebhardt, H.; Dreimann, M.; Hempfing, A.; Stein, H.; Metz-Stavenhagen, P.; Rudwaleit, M.; Sieper, J. Correlation of Histopathological Findings and Magnetic Resonance Imaging in the Spine of Patients with Ankylosing Spondylitis. Arthritis Research & Therapy 2006, 8, R143. [CrossRef]
- Baraliakos, X.; Haibel, H.; Listing, J.; Sieper, J.; Braun, J. Continuous Long-Term Anti-TNF Therapy Does Not Lead to an Increase in the Rate of New Bone Formation over 8 Years in Patients with Ankylosing Spondylitis. Annals of the rheumatic diseases 2014, 73, 710–715. [CrossRef]
- Braun, J.; Baraliakos, X.; Deodhar, A.; Baeten, D.; Sieper, J.; Emery, P.; Readie, A.; Martin, R.; Mpofu, S.; Richards, H.B. Effect of Secukinumab on Clinical and Radiographic Outcomes in Ankylosing Spondylitis: 2-Year Results from the Randomised Phase III MEASURE 1 Study.. [CrossRef]
- Ramiro, S.; Heijde, D.V.D.; Tubergen, A.V.; Stolwijk, C.; Dougados, M.; Bosch, F.V.D.; Landewé, R. Higher Disease Activity Leads to More Structural Damage in the Spine in Ankylosing Spondylitis: 12-Year Longitudinal Data from the OASIS Cohort.. [CrossRef]
- Heijde, D.V.D.; Landewé, R.; Einstein, S.; Ory, P.; Vosse, D.; Ni, L.; Lin, S.L.; Tsuji, W.; Davis, J.C. Radiographic Progression of Ankylosing Spondylitis after up to Two Years of Treatment with Etanercept. Arthritis and Rheumatism 2008, 58, 1324–1331. [CrossRef]
- van Duivenvoorde, L.M.; Dorris, M.L.; Satumtira, N.; van Tok, M.N.; Redlich, K.; Tak, P.P.; Taurog, J.D.; Baeten, D.L. Relationship between Inflammation, Bone Destruction, and Osteoproliferation in the HLA-B27/Human Β2 -Microglobulin-Transgenic Rat Model of Spondylarthritis. Arthritis Rheum 2012, 64, 3210–3219. [CrossRef]
- Llop, M.; Moreno, M.; Navarro-Compán, V.; Juanola, X.; de Miguel, E.; Almodóvar, R.; Quintana, E.C.; Sanz, J.S.; Beltrán, E.; Montesinos, M.D.R.; et al. Sustained Low Disease Activity Measured by ASDAS Slow Radiographic Spinal Progression in Axial Spondyloarthritis Patients Treated with TNF-Inhibitors: Data from REGISPONSERBIO. Arthritis Research & Therapy 2022, 24, 30. [CrossRef]
- van der Heijde, D.; Østergaard, M.; Reveille, J.D.; Baraliakos, X.; Kronbergs, A.; Sandoval, D.M.; Li, X.; Carlier, H.; Adams, D.H.; Maksymowych, W.P. Spinal Radiographic Progression and Predictors of Progression in Patients With Radiographic Axial Spondyloarthritis Receiving Ixekizumab Over 2 Years. J Rheumatol 2022, 49, 265–273. [CrossRef]
- Apostolakos, J.; Durant, T.J.; Dwyer, C.R.; Russell, R.P.; Weinreb, J.H.; Alaee, F.; Beitzel, K.; McCarthy, M.B.; Cote, M.P.; Mazzocca, A.D. The Enthesis: A Review of the Tendon-to-Bone Insertion. Muscles Ligaments Tendons J 2014, 4, 333–342.
- Russell, T.; Bridgewood, C.; Rowe, H.; Altaie, A.; Jones, E.; McGonagle, D. Cytokine “Fine Tuning” of Enthesis Tissue Homeostasis as a Pointer to Spondyloarthritis Pathogenesis with a Focus on Relevant TNF and IL-17 Targeted Therapies. Semin Immunopathol 2021, 43, 193–206. [CrossRef]
- Watad, A.; Rowe, H.; Russell, T.; Zhou, Q.; Anderson, L.K.; Khan, A.; Dunsmuir, R.; Loughenbury, P.; Borse, V.; Rao, A.; et al. Normal Human Enthesis Harbours Conventional CD4+ and CD8+ T Cells with Regulatory Features and Inducible IL-17A and TNF Expression. Ann Rheum Dis 2020, 79, 1044–1054. [CrossRef]
- McGonagle, D.; Wakefield, R.J.; Tan, A.L.; D’Agostino, M.A.; Toumi, H.; Hayashi, K.; Emery, P.; Benjamin, M. Distinct Topography of Erosion and New Bone Formation in Achilles Tendon Enthesitis: Implications for Understanding the Link between Inflammation and Bone Formation in Spondylarthritis. Arthritis Rheum 2008, 58, 2694–2699. [CrossRef]
- Jacques, P.; Lambrecht, S.; Verheugen, E.; Pauwels, E.; Kollias, G.; Armaka, M.; Verhoye, M.; Van der Linden, A.; Achten, R.; Lories, R.J.; et al. Proof of Concept: Enthesitis and New Bone Formation in Spondyloarthritis Are Driven by Mechanical Strain and Stromal Cells. Ann Rheum Dis 2014, 73, 437–445. [CrossRef]
- Gravallese, E.M.; Schett, G. Effects of the IL-23–IL-17 Pathway on Bone in Spondyloarthritis. Nat Rev Rheumatol 2018, 14, 631–640. [CrossRef]
- Zheng, G.; Xie, Z.; Wang, P.; Li, J.; Li, M.; Cen, S.; Tang, S.; Liu, W.; Ye, G.; Li, Y.; et al. Enhanced Osteogenic Differentiation of Mesenchymal Stem Cells in Ankylosing Spondylitis: A Study Based on a Three-Dimensional Biomimetic Environment. Cell Death Dis 2019, 10, 1–11. [CrossRef]
- Xie, Z.; Wang, P.; Li, Y.; Deng, W.; Zhang, X.; Su, H.; Li, D.; Wu, Y.; Shen, H. Imbalance Between Bone Morphogenetic Protein 2 and Noggin Induces Abnormal Osteogenic Differentiation of Mesenchymal Stem Cells in Ankylosing Spondylitis. Arthritis Rheumatol 2016, 68, 430–440. [CrossRef]
- Cui, H.; Li, Z.; Chen, S.; Li, X.; Chen, D.; Wang, J.; Li, Z.; Hao, W.; Zhong, F.; Zhang, K.; et al. CXCL12/CXCR4-Rac1–Mediated Migration of Osteogenic Precursor Cells Contributes to Pathological New Bone Formation in Ankylosing Spondylitis. Science Advances 2022, 8, eabl8054. [CrossRef]
- Daoussis, D.; Liossis, S.N.C.; Solomou, E.E.; Tsanaktsi, A.; Bounia, K.; Karampetsou, M.; Yiannopoulos, G.; Andonopoulos, A.P. Evidence That Dkk-1 Is Dysfunctional in Ankylosing Spondylitis. Arthritis and Rheumatism 2010, 62, 150–158. [CrossRef]
- Fang, X.; Chen, C.; Wang, Z.-X.; Zhao, Y.; Jiang, L.-Q.; Fang, Y.; Zhang, R.-D.; Pan, H.-F.; Tao, S.-S. Serum DKK-1 Level in Ankylosing Spondylitis: Insights from Meta-Analysis and Mendelian Randomization. Frontiers in Immunology 2023, 14. [CrossRef]
- Nocturne, G.; Pavy, S.; Boudaoud, S.; Seror, R.; Goupille, P.; Chanson, P.; Heijde, D.V.D.; Gaalen, F.V.; Berenbaum, F.; Mariette, X.; et al. Increase in Dickkopf-1 Serum Level in Recent Spondyloarthritis. Data from the DESIR Cohort. 2015. [CrossRef]
- Cortes, A.; Maksymowych, W.P.; Wordsworth, B.P.; Inman, R.D.; Danoy, P.; Rahman, P.; Stone, M.; Corr, M.; Gensler, L.S.; Gladman, D.; et al. Association Study of Genes Related to Bone Formation and Resorption and the Extent of Radiographic Change in Ankylosing Spondylitis. Annals of the Rheumatic Diseases 2015, 74, 1387–1393. [CrossRef]
- Hamid, H.S.A. el; Ibrahim, N.H.; Morsi, M.H.; Al-Tabbakh, A.-S.M.; EL-Melouk, M.S. Elevated Serum Dickkopf-1 Levels as a Biomarker for Disease Activity and Severity in Psoriatic Arthritis Patients. The Egyptian Journal of Hospital Medicine 2022, 89, 6445.
- Wahba, M.A.W.A.; El-Gazzar, N.M.; Elsharaby, R.M.; Tabra, S.A. DKK-1 in Psoriatic Arthritis: Correlation with Disease Activity and Enthesopathy. Reumatología Clínica 2023. [CrossRef]
- Aschermann, S.; Englbrecht, M.; Bergua, A.; Spriewald, B.M.; Said-Nahal, R.; Breban, M.; Schett, G.; Rech, J. Presence of HLA-B27 Is Associated with Changes of Serum Levels of Mediators of the Wnt and Hedgehog Pathway. Joint Bone Spine 2016, 83, 43–46. [CrossRef]
- Saad, C.G.S.; Ribeiro, A.C.M.; Moraes, J.C.B.; Takayama, L.; Goncalves, C.R.; Rodrigues, M.B.; de Oliveira, R.M.; Silva, C.A.; Bonfa, E.; Pereira, R.M.R. Low Sclerostin Levels: A Predictive Marker of Persistent Inflammation in Ankylosing Spondylitis during Anti-Tumor Necrosis Factor Therapy? Arthritis Res Ther 2012, 14, R216. [CrossRef]
- Appel, H.; Ruiz-Heiland, G.; Listing, J.; Zwerina, J.; Herrmann, M.; Mueller, R.; Haibel, H.; Baraliakos, X.; Hempfing, A.; Rudwaleit, M.; et al. Altered Skeletal Expression of Sclerostin and Its Link to Radiographic Progression in Ankylosing Spondylitis. Arthritis and Rheumatism 2009, 60, 3257–3262. [CrossRef]
- Luchetti, M.M.; Ciccia, F.; Avellini, C.; Benfaremo, D.; Guggino, G.; Farinelli, A.; Ciferri, M.; Rossini, M.; Svegliati, S.; Spadoni, T.; et al. Sclerostin and Antisclerostin Antibody Serum Levels Predict the Presence of Axial Spondyloarthritis in Patients with Inflammatory Bowel Disease. The Journal of Rheumatology 2018, 45, 630–637. [CrossRef]
- Sorour, N.E.; Abdel Hafeez, N.A.; Akl, E.M.; Mowafy, M.A.; Mohamed, A.A. Evaluation of Serum Level of Sclerostin in Patients with Psoriatic Arthritis. Benha Journal of Applied Sciences 2021, 6, 11–15. [CrossRef]
- Fayed, A.; Elgohary, R.; Fawzy, M. Evaluating the Role of Serum Sclerostin as an Indicator of Activity and Damage in Egyptian Patients with Rheumatoid Arthritis: University Hospital Experience. Clin Rheumatol 2020, 39, 1121–1130. [CrossRef]
- Ohba, S. Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action. Int J Mol Sci 2020, 21, 6665. [CrossRef]
- Daoussis, D.; Filippopoulou, A.; Liossis, S.-N.; Sirinian, C.; Klavdianou, K.; Bouris, P.; Karamanos, N.K.; Andonopoulos, A.P. Anti-TNFα Treatment Decreases the Previously Increased Serum Indian Hedgehog Levels in Patients with Ankylosing Spondylitis and Affects the Expression of Functional Hedgehog Pathway Target Genes. Semin Arthritis Rheum 2015, 44, 646–651. [CrossRef]
- González-Chávez, S.A.; Quiñonez-Flores, C.M.; Pacheco-Tena, C. Molecular Mechanisms of Bone Formation in Spondyloarthritis. Joint Bone Spine 2016, 83, 394–400. [CrossRef]
- Lata, M.; Hettinghouse, A.S.; Liu, C. Targeting Tumor Necrosis Factor Receptors in Ankylosing Spondylitis. Ann N Y Acad Sci 2019, 1442, 5–16. [CrossRef]
- Haroon, N.; Inman, R.D.; Learch, T.J.; Weisman, M.H.; Lee, M.; Rahbar, M.H.; Ward, M.M.; Reveille, J.D.; Gensler, L.S. The Impact of TNF-Inhibitors on Radiographic Progression in Ankylosing Spondylitis. Arthritis Rheum 2013, 65, 2645–2654. [CrossRef]
- Koo, B.S.; Oh, J.S.; Park, S.Y.; Shin, J.H.; Ahn, G.Y.; Lee, S.; Joo, K.B.; Kim, T.-H. Tumour Necrosis Factor Inhibitors Slow Radiographic Progression in Patients with Ankylosing Spondylitis: 18-Year Real-World Evidence. Annals of the Rheumatic Diseases 2020, 79, 1327–1332. [CrossRef]
- Jo, S.; Wang, S.E.; Lee, Y.L.; Kang, S.; Lee, B.; Han, J.; Sung, I.-H.; Park, Y.-S.; Bae, S.-C.; Kim, T.-H. IL-17A Induces Osteoblast Differentiation by Activating JAK2/STAT3 in Ankylosing Spondylitis. Arthritis Research & Therapy 2018, 20, 115. [CrossRef]
- Ono, T.; Okamoto, K.; Nakashima, T.; Nitta, T.; Hori, S.; Iwakura, Y.; Takayanagi, H. IL-17-Producing Γδ T Cells Enhance Bone Regeneration. Nat Commun 2016, 7, 10928. [CrossRef]
- Kim, Y.-G.; Park, J.-W.; Lee, J.-M.; Suh, J.-Y.; Lee, J.-K.; Chang, B.-S.; Um, H.-S.; Kim, J.-Y.; Lee, Y. IL-17 Inhibits Osteoblast Differentiation and Bone Regeneration in Rat. Arch Oral Biol 2014, 59, 897–905. [CrossRef]
- Tok, M.N.; Duivenvoorde, L.M.; Kramer, I.; Ingold, P.; Pfister, S.; Roth, L.; Blijdorp, I.C.; Sande, M.G.H.; Taurog, J.D.; Kolbinger, F.; et al. Interleukin-17A Inhibition Diminishes Inflammation and New Bone Formation in Experimental Spondyloarthritis. Arthritis Rheumatol 2019, 71, 612–625. [CrossRef]
- Mease, P.J.; Landewé, R.; Rahman, P.; Tahir, H.; Singhal, A.; Boettcher, E.; Navarra, S.; Readie, A.; Mpofu, S.; Delicha, E.M.; et al. Secukinumab Provides Sustained Improvement in Signs and Symptoms and Low Radiographic Progression in Patients with Psoriatic Arthritis: 2-Year (End-of-Study) Results from the FUTURE 5 Study. RMD Open 2021, 7, e001600. [CrossRef]
- Deodhar, A.; Gensler, L.S.; Sieper, J.; Clark, M.; Calderon, C.; Wang, Y.; Zhou, Y.; Leu, J.H.; Campbell, K.; Sweet, K.; et al. Three Multicenter, Randomized, Double-Blind, Placebo-Controlled Studies Evaluating the Efficacy and Safety of Ustekinumab in Axial Spondyloarthritis. Arthritis Rheumatol 2019, 71, 258–270. [CrossRef]
- Baeten, D.; Østergaard, M.; Wei, J.C.-C.; Sieper, J.; Järvinen, P.; Tam, L.-S.; Salvarani, C.; Kim, T.-H.; Solinger, A.; Datsenko, Y.; et al. Risankizumab, an IL-23 Inhibitor, for Ankylosing Spondylitis: Results of a Randomised, Double-Blind, Placebo-Controlled, Proof-of-Concept, Dose-Finding Phase 2 Study. Ann Rheum Dis 2018, 77, 1295–1302. [CrossRef]
- McGonagle, D.; Watad, A.; Sharif, K.; Bridgewood, C. Why Inhibition of IL-23 Lacked Efficacy in Ankylosing Spondylitis. Frontiers in Immunology 2021, 12. [CrossRef]
- Dudakov, J.A.; Hanash, A.M.; van den Brink, M.R.M. Interleukin-22: Immunobiology and Pathology. Annu Rev Immunol 2015, 33, 747–785. [CrossRef]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.-C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 Induces Spondyloarthropathy by Acting on ROR-Γt+ CD3+CD4−CD8− Entheseal Resident T Cells. Nat Med 2012, 18, 1069–1076. [CrossRef]
- El-Zayadi, A.A.; Jones, E.A.; Churchman, S.M.; Baboolal, T.G.; Cuthbert, R.J.; El-Jawhari, J.J.; Badawy, A.M.; Alase, A.A.; El-Sherbiny, Y.M.; McGonagle, D. Interleukin-22 Drives the Proliferation, Migration and Osteogenic Differentiation of Mesenchymal Stem Cells: A Novel Cytokine That Could Contribute to New Bone Formation in Spondyloarthropathies. Rheumatology (Oxford) 2017, 56, 488–493. [CrossRef]
- Upadhyay, J.; Farr, O.M.; Mantzoros, C.S. The Role of Leptin in Regulating Bone Metabolism. Metabolism 2015, 64, 105–113. [CrossRef]
- Hartl, A.; Sieper, J.; Syrbe, U.; Listing, J.; Hermann, K.-G.; Rudwaleit, M.; Poddubnyy, D. Serum Levels of Leptin and High Molecular Weight Adiponectin Are Inversely Associated with Radiographic Spinal Progression in Patients with Ankylosing Spondylitis: Results from the ENRADAS Trial. Arthritis Res Ther 2017, 19, 140. [CrossRef]
- Karsenty, G.; Yadav, V.K. Regulation of Bone Mass by Serotonin: Molecular Biology and Therapeutic Implications. Annu Rev Med 2011, 62, 323–331. [CrossRef]
- Klavdianou, K.; Liossis, S.-N.; Papachristou, D.J.; Theocharis, G.; Sirinian, C.; Kottorou, A.; Filippopoulou, A.; Andonopoulos, A.P.; Daoussis, D. Decreased Serotonin Levels and Serotonin-Mediated Osteoblastic Inhibitory Signaling in Patients With Ankylosing Spondylitis. Journal of Bone and Mineral Research 2016, 31, 630–639. [CrossRef]
- Clunie, G.; Horwood, N. Loss and Gain of Bone in Spondyloarthritis: What Drives These Opposing Clinical Features? Ther Adv Musculoskelet Dis 2020, 12, 1759720X20969260. [CrossRef]
- Ghozlani, I.; Ghazi, M.; Nouijai, A.; Mounach, A.; Rezqi, A.; Achemlal, L.; Bezza, A.; El Maghraoui, A. Prevalence and Risk Factors of Osteoporosis and Vertebral Fractures in Patients with Ankylosing Spondylitis. Bone 2009, 44, 772–776. [CrossRef]
- Puche-Larrubia, M.Á.; Ladehesa-Pineda, L.; Font-Ugalde, P.; Escudero-Contreras, A.; Moltó, A.; López-Medina, C.; Collantes-Estévez, E. Distribution of Comorbidities in Spondyloarthritis with Regard to the Phenotype and Psoriasis: Data from the ASAS-COMOSPA Study. Therapeutic Advances in Musculoskeletal 2021, 13, 1759720X2110452. [CrossRef]
Figure 1.
Cellular and molecular interplay involved in the pathogenesis of bone damage in rheumatoid arthritis. ACPAs, anti-citrullinated protein antibodies; RANKL, receptor activator of nuclear factor kappa-Β ligand; OPG, osteoprotegerin; TNFα, tumor necrosis factor-alpha; FLS, fibroblast-like synoviocytes; NETs, neutrophil extracellular traps; MMPs, matrix metalloproteinases; FcγR, fc gamma receptor; IL-1, interleukin-1; IL-6, interleukin-6; IL-17, interleukin 17.
Figure 1.
Cellular and molecular interplay involved in the pathogenesis of bone damage in rheumatoid arthritis. ACPAs, anti-citrullinated protein antibodies; RANKL, receptor activator of nuclear factor kappa-Β ligand; OPG, osteoprotegerin; TNFα, tumor necrosis factor-alpha; FLS, fibroblast-like synoviocytes; NETs, neutrophil extracellular traps; MMPs, matrix metalloproteinases; FcγR, fc gamma receptor; IL-1, interleukin-1; IL-6, interleukin-6; IL-17, interleukin 17.
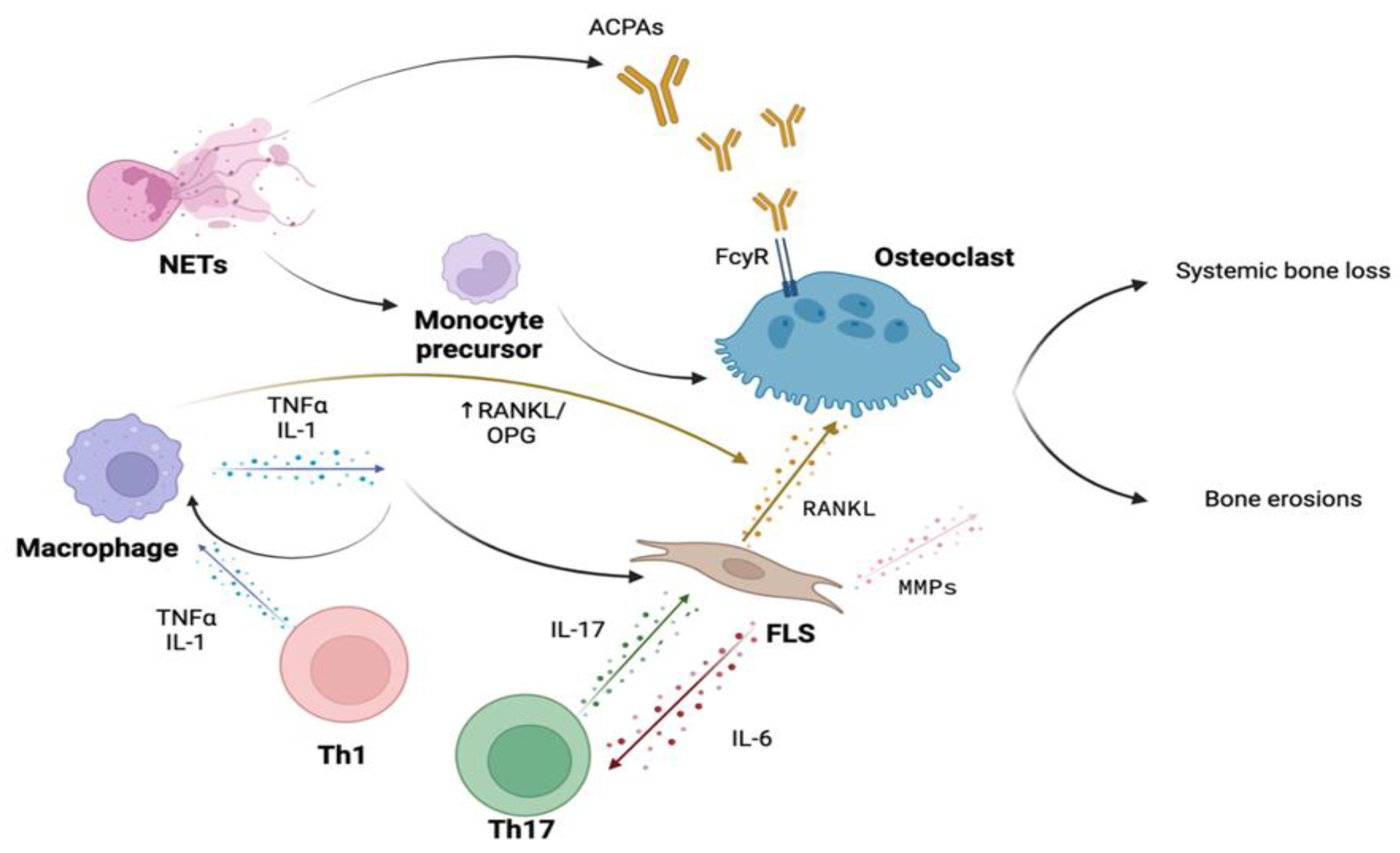
Figure 2.
Interaction between the Immune System, Osteoclasts, and Osteoblasts in the Pathogenesis of Bone Involvement in Spondyloarthritis. MSC, mesenchymal stromal cells; ILC, Innate lymphoid cells; hPBMCs, human peripheral blood mononuclear cells; HLA-B27, human leukocyte antigen - B27; RANKL, receptor activator of nuclear factor kappa-Β ligand; OPG, osteoprotegerin; DKK1, Dickkopf-related protein 1; TNFα, tumor necrosis factor-alpha; TNFR1-2, tumor necrosis factor receptor 1-2; IL-1, interleukin-1; IL-6, interleukin-6; IL-17A, interleukin-17A; IL-22, interleukin-22; IL-23, interleukin-23.
Figure 2.
Interaction between the Immune System, Osteoclasts, and Osteoblasts in the Pathogenesis of Bone Involvement in Spondyloarthritis. MSC, mesenchymal stromal cells; ILC, Innate lymphoid cells; hPBMCs, human peripheral blood mononuclear cells; HLA-B27, human leukocyte antigen - B27; RANKL, receptor activator of nuclear factor kappa-Β ligand; OPG, osteoprotegerin; DKK1, Dickkopf-related protein 1; TNFα, tumor necrosis factor-alpha; TNFR1-2, tumor necrosis factor receptor 1-2; IL-1, interleukin-1; IL-6, interleukin-6; IL-17A, interleukin-17A; IL-22, interleukin-22; IL-23, interleukin-23.
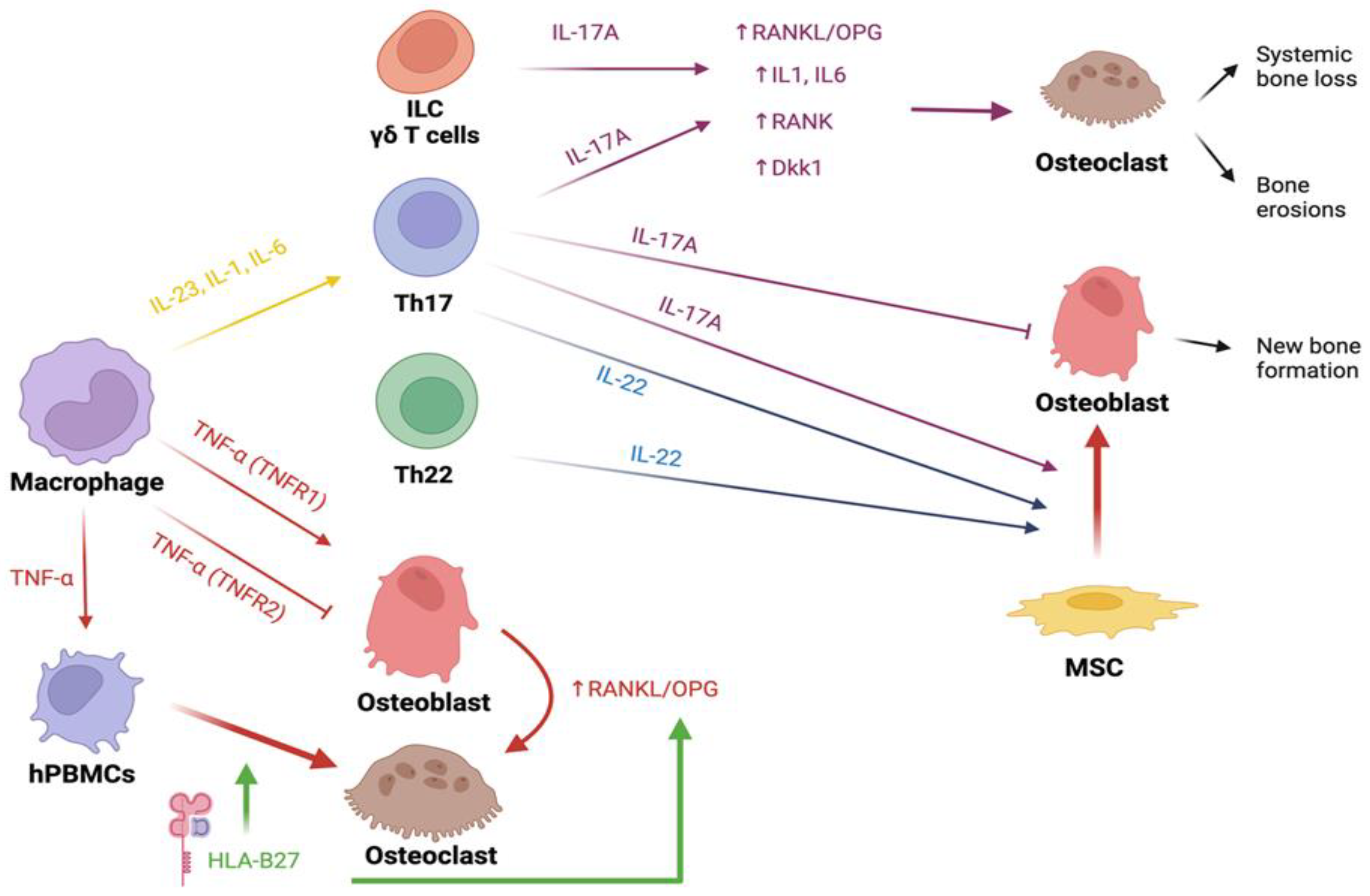
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



