
Submitted:
18 September 2023
Posted:
20 September 2023
You are already at the latest version
Abstract
Since decades, oxybutynin hydrochloride is prescribed to improve bladder control in cases of incontinence and excessive urination frequency. This review summarizes synthetic methods enabling preparation of the racemic drug and, in a detailed manner, preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid, a key intermediate in the synthesis of (S)-oxybutynin. Mode of action and metabolism are briefly commented in order to explain the main adverse effects associated with its use and to justify the evolution observed in the diverse commercial formulations. Repositioning opportunities are discussed in terms of clinical trials for the management of hyperhidrosis, hot flashes, and obstructive sleep apnea.
Keywords:
bladder
; breast cancer
; hot flashes
; hyperhidrosis
; obstructive sleep apnea
; oxybutynin
; prostate cancer
1. Introduction
Better knowledge of signaling pathways and therapeutic targets allows to find novel applications for marketed drugs or drug candidates failing in late stages of clinical trials. The so-called “drug repositioning” or “drug repurposing” concept was introduced in 2004 by Ashburn and Thor [1]. It enables pharmaceutical companies to save time and money because efficient procedures of preparation at laboratory and pilot scales have already been developed. In addition, results of most pre-clinical and clinical assays have already been accumulated. Drug repositioning also provides new tools to physicians, giving them the opportunity of proposing innovative, but sometimes experimental, medications to their patients. Examples of successfully repositioned drugs (Figure 1) include, in a non-exhaustive list, aspirin (1), sildenafil (2), and thalidomide (3). Aspirin, well-known for its antipyretic and analgesic effects, eases blood circulation [2] and could exhibit beneficial effects in some cancers [3]. Sildenafil was initially developed to treat cardiovascular problems but it failed in the corresponding clinical trials. The molecule is now prescribed in the case of erectile dysfunction [4] and extensively studied for its antitumoral activities [5]. Thalidomide [6], on the other hand, was commercialized to relieve nausea in pregnant women but was soon abandoned due to its teratogenic consequences. Thalidomide is now manufactured as an effective agent against erythema nodosum leprosum and multiple myeloma.
Many other approved drugs have been screened or are still screened in order to find them new applications for the treatment of, among others, Alzheimer’s disease [7], asthma [8], and more recently COVID-19 [9].
The racemic oxybutynin (4, Figure 2) is an antimuscarinic agent clinically used, under its hydrochloride form (4.HCl), in the therapy of overactive bladder for almost five decades. Interestingly, a series of reports indicated that it could emerge as a promising medication for managing hyperhidrosis, hot flashes, and, hopefully, obstructive sleep apnea. Therefore, we thought it useful to summarize, for the first time in one single paper, the knowledge acquired on syntheses, mode of action, metabolism, and formulations of this substance. Repositioning opportunities are highlighted.
2. Chemical Names
The Chemical Abstracts registry number of the racemic form of oxybutynin and its hydrochloride are 5633-20-5 and 1508-65-2, respectively. Its index name is benzeneacetic acid, α-cyclohexyl-α-hydroxy-, 4-(diethylamino)-2-butynyl ester.
Registry numbers of the (R)- and (S)-enantiomers (Figure 2) are 119618-21-2 and 119618-22-3 respectively. The corresponding registry numbers for the hydrochlorides are 1207344-05-5 and 230949-16-3. The denominations aroxybutynin [10] for the (R)-enantiomer and esoxybutynin [11] for the (S)-enantiomer can also be found in the literature.
The IUPAC name is 4-diethylaminobut-2-ynyl 2-cyclohexyl-2-hydroxy-2-phenylethanoate.
Sometimes, oxybutynin is considered as a derivative
- of glycolic acid: 4-diethylamino-2-butynyl phenylcyclohexylglycolate,
- or of phenylacetic acid: 4-diethylamino-2-butynyl 2-cyclohexyl-2-hydroxy-2-phenylacetate.
The name oxybutynin is often used, indifferently, to designate the free base as well as its hydrochloride. Therefore, in order to stay consistent and to lighten the text of the review, we shall distinguish both species by numbering them 4 and 4.HCl respectively.
3. Syntheses
3.1. Syntheses of the Racemic
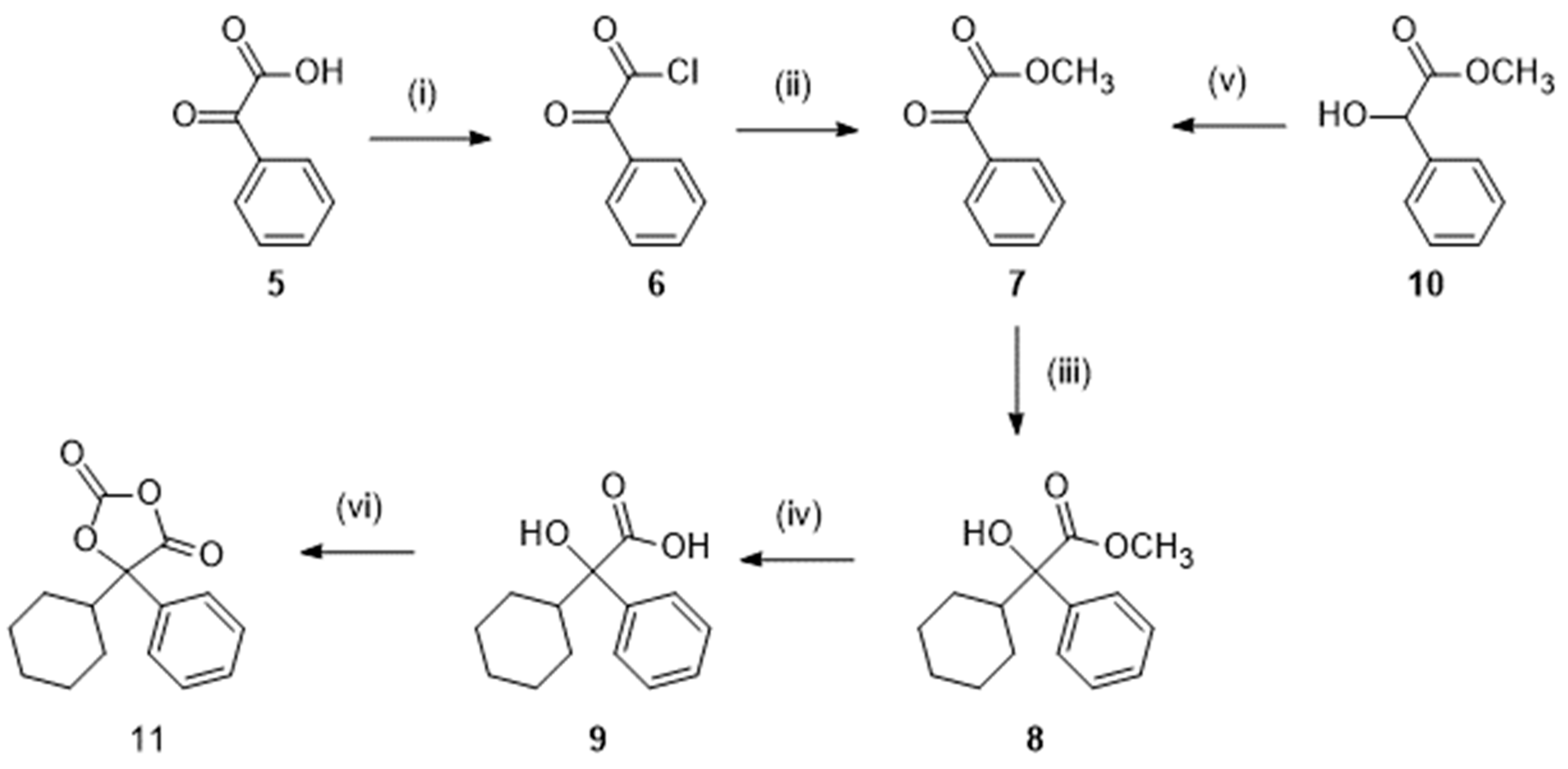
Practically, oxybutynin can be prepared by a convergent synthesis requiring two key reagents, namely methyl 2-cyclohexyl-2-hydroxy-2-phenylacetate (8, Scheme 1) or the corresponding acid 9 and 2-propyn-1-ol (propargyl alcohol; 12, Scheme 2) or a derivative, which are coupled in an esterification or transesterification reaction.
3.1.1. Preparation of methyl 2-cyclohexyl-2-hydroxy-2-phenylacetate (8) and the corresponding acid (9)
There are two routes (Scheme 1) to obtain those reagents 8 and 9. One route starts from 2-oxo-2-phenylacetic acid (phenylglyoxylic acid; 5), which is first converted into the acid chloride 6 by treatment with thionyl chloride. Further reaction with methanol yielded the ester 7. Action of bromocyclohexane under conditions of a Grignard reaction afforded methyl 2-cyclohexyl-2-hydroxy-2-phenylacetate 8, which could be hydrolyzed to the corresponding acid 9 [12,13].
In the alternative route, methyl 2-hydroxy-2-phenylacetate (methyl mandelate; 10) was the starting material that could readily be oxidized into 7 before the Grignard reaction [13].
Optionally, activation of the acid 9 under the form of 5-cyclohexyl-5-phenyl-1,3-dioxolane-2,4-dione 11 (Scheme 1) by reaction with trichloromethylchloroformate has been reported [14].
Scheme 1.
Preparation of the intermediates 8, 9, and 11. Reagent(s); catalyst; solvent(s); yield(s). (i) SOCl2; toluene; 92 % [12]. (ii) CH3OH; 87 % [12]. (iii) Bromocyclohexane, Mg; I2; tetrahydrofuran; 65 % [12], 57 % [13]. (iv) NaOH; H2O, CH3OH; 77 % [13]. (v) Pyridinium chlorochromate; CH2Cl2; 76 % [13]. (vi) N-methylpiperidine, trichloromethylchloroformate; tetrahydrofuran; 79 % [14].
Scheme 1.
Preparation of the intermediates 8, 9, and 11. Reagent(s); catalyst; solvent(s); yield(s). (i) SOCl2; toluene; 92 % [12]. (ii) CH3OH; 87 % [12]. (iii) Bromocyclohexane, Mg; I2; tetrahydrofuran; 65 % [12], 57 % [13]. (iv) NaOH; H2O, CH3OH; 77 % [13]. (v) Pyridinium chlorochromate; CH2Cl2; 76 % [13]. (vi) N-methylpiperidine, trichloromethylchloroformate; tetrahydrofuran; 79 % [14].
3.1.2. Preparation of derivatives of 2-propyn-1-ol
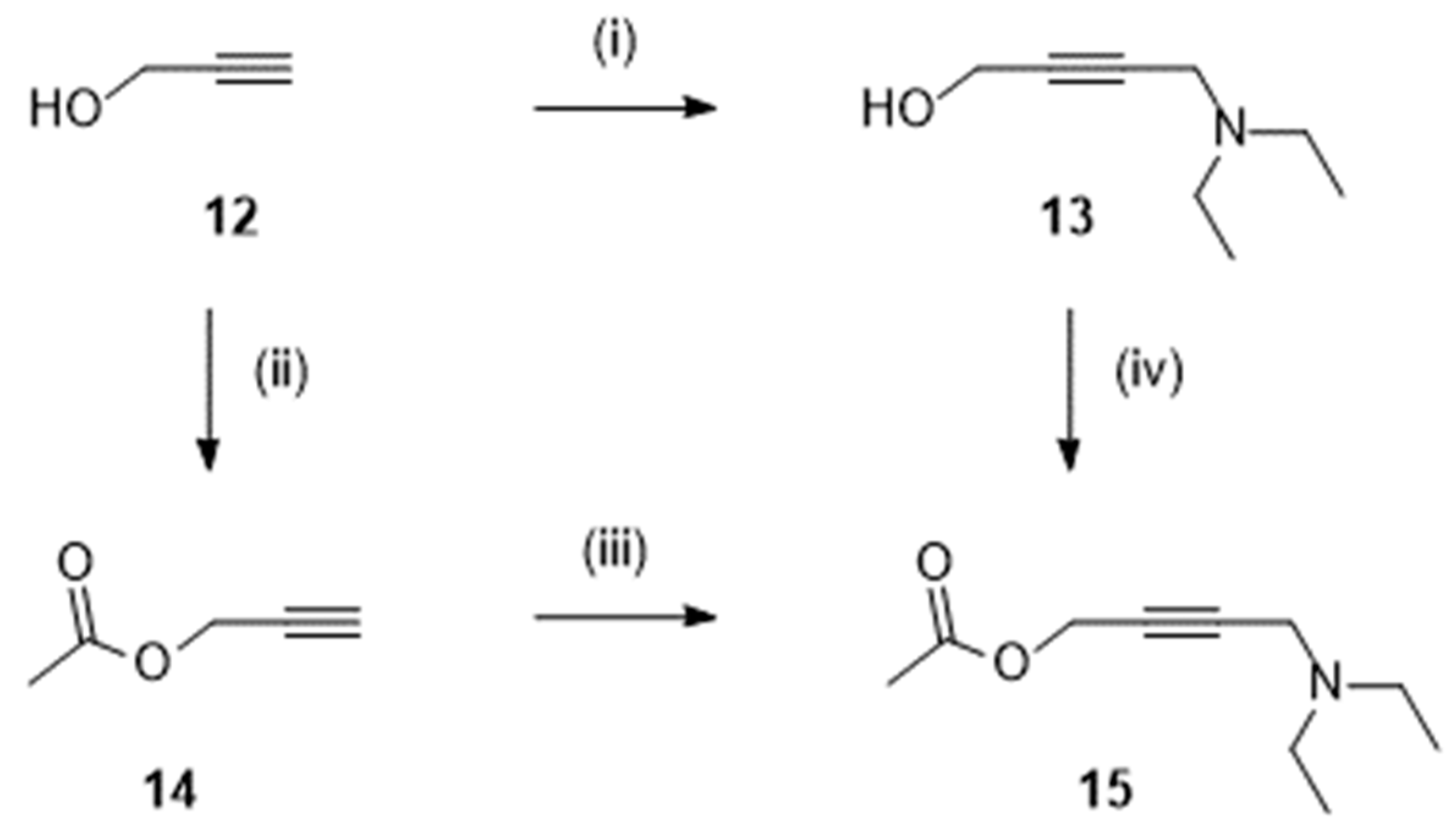
In most procedures, the butynyl alcohol 13 (Scheme 2) was prepared by a Mannich reaction involving 2-propyn-1-ol 12 (150 g; [13]), formadehyde and diethylamine. In some works, 12 (300 g; [12]) was esterified with acetyl chloride (to give 14) before the Mannich reaction to finally afford the acetate 15 [12,14]. The later could also be obtained from 13 and a mixture of acetic acid and acetic anhydride [15].
Scheme 2.
Preparation of derivatives 13-15. Reagent(s); catalyst; solvent(s); yield(s). (i) HCHO, (C2H5)2NH; CuSO4; H2O; 23 % [13]; (ii) CH3COCl, (C2H5)3N; CH2Cl2; 91 % [12]. (iii) HCHO, (C2H5)2NH; CuCl; 1,4-dioxane; 89 % [12]. (iii) HCHO, (C2H5)2NH; (CH3CO2)2Cu; 1,4-dioxane; 84 % [15]. (iv) (CH3CO)2O; H2SO4; CH3CO2H; 81 % [15].
Scheme 2.
Preparation of derivatives 13-15. Reagent(s); catalyst; solvent(s); yield(s). (i) HCHO, (C2H5)2NH; CuSO4; H2O; 23 % [13]; (ii) CH3COCl, (C2H5)3N; CH2Cl2; 91 % [12]. (iii) HCHO, (C2H5)2NH; CuCl; 1,4-dioxane; 89 % [12]. (iii) HCHO, (C2H5)2NH; (CH3CO2)2Cu; 1,4-dioxane; 84 % [15]. (iv) (CH3CO)2O; H2SO4; CH3CO2H; 81 % [15].
3.1.3. Final step
Ultimately, oxybutynin 4 and its hydrochloride 4.HCl were synthesized (Scheme 3) by coupling the ester 8 or the acid 9 and the alcohol 13 or the ester 15. Details on published sequences, experimental procedures, and yields can be found in Scheme 3.
Overall yields, calculated from commercially available precursors 2-oxo-2-phenylacetic acid 5 [12] or methyl 2-hydroxy-2-phenylacetate 10 [13] and 2-propyn-1-ol 12, ranged from a modest 23 % [12] to a poor 6 % [13].
Oxybutynin hydrochloride (4.HCl) [13]. M.p. 122-126 °C. 1H NMR (400MHz, DMSO-d6) δ: 1.02-1.78 (m, 16H), 2.16-2.22 (m, 1H), 2.96-3.07 (m, 4H), 4.11 (br s, 2H), 4.87 (br s, 2H), 5.73 (s, exchangeable with D2O, 1H), 7.26-7. 36 (m, 3H), 7.54-7.56 (m, 2H), 10.70 (br, exchangeable with D2O, 1H) ppm. IR (KBr) ν: 3511, 3444, 3323, 3098, 3040, 2930, 2857, 2774, 2617, 2564, 2476, 1742, 1624, 1534, 1457, 1393, 1347, 1249, 1209 cm-1.
Additionally, a recent Chinese patent [20] mentioned, as illustrated in Scheme 4, the possibility of preparing the ester 17 from 9 and 3-chloroprop-1-yne 16. The Mannich reaction was then performed in the last reaction of the sequence yielding 4.HCl. A sequence allowing to access the ester 17 from 9 and 2-propyn-1-ol 12 has also been adapted to synthesize some deuterated derivatives of oxybutynin [13,21,22]. However, experimental details were not clearly disclosed in any of those four references.
3.2. Enantioselective Syntheses
Enantioselective synthesis of the (S)-enantiomer of oxybutynin has attracted much interest and deserves a detailed attention, even if oxybutynin is commercially available for medical use as a racemic mixture only. Most efforts have been made during the first decade of this century and were dedicated to the obtention of the (S)-enantiomer of 2-cyclohexyl-2-hydroxy-2-phenylacetic acid 9. The simplest way to isolate it was to treat, as described in the patent of Bakale et al [23], the racemic (100 g) with L-tyrosine methyl ester in order to afford the expected (S)-oxybutynin in 42 % yield.
(S)-Oxybutynin hydrochloride ((S)-4.HCl) [14]. M.p. 117-118 °C. 1H NMR (400 MHz, DMSO-d6): δ 0.9 to 1.1 (m, 4H), 1.1 to 1.2 (m, 7H), 1.3 (m, 1H), 1.4 (m, 1H), 1.6 (m, 2H), 1.7 (m, 1H), 2.9 (d, 4H), 4.1 (s, 2H), 4.8 (s, 2H), 5.7 (s, 1H), 7.2 (m, 1H), 7.3 (m, 2H), 7.5 (m, 2H), 11.4 (s, 1H) ppm. 13C NMR (100 MHz, DMSO-d6): δ 8.9, 25.2, 25.1, 25.8, 25.9, 25.9, 40.5, 45.7, 46.8, 52.4, 75.4, 80.9, 83.8, 125.7, 127.2, 127.9,141.1, 173.5 ppm.
Besides separation of diastereoisomers, more sophisticated methods have been described to prepare (S)-9 and they are summarized hereafter. Evidently, those protocols can be adapted to afford the (R)-enantiomer of 9.
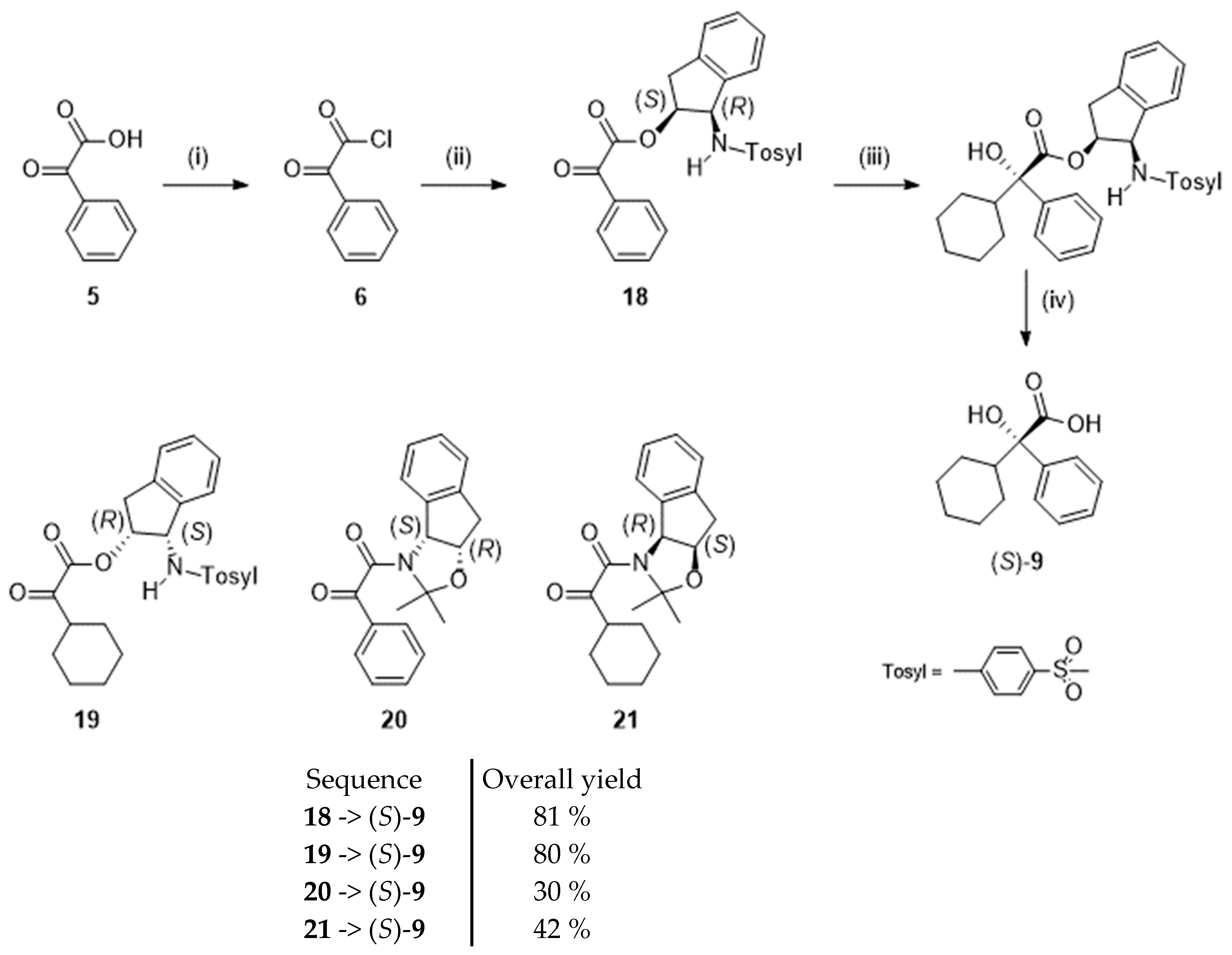
For example, Senanayake et al. [24] obtained (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid in enantiomeric excesses higher than 98 % by forcing a Grignard reagent to preferentially attack one face rather than the other face of 2-oxo-2-phenylacetic acid or 2-oxo-cyclohexylacetic acid derivatives. To achieve that goal, the authors used bulky chiral auxiliaries based on substituted cis-1-amino-2-indanol moieties (1-para-tolylsulfonyl group and acetonide). One representative case is depicted in scheme 5. Thus, 2-oxo-2-phenylacetic acid (5) was converted into its acyl chloride and then reacted with cis-(1S,2R)-2-para-tolylsulfonamidoindanol to afford the ester 18. Subsequent Grignard reaction and hydrolysis of the ester bond yielded the targeted (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9. Other structurally-related chiral intermediates (19-21) evaluated in the study are represented in Scheme 4. Best yields were obtained from the esters 18 and 19 bearing the N-para-tolylsulfonamidoindanyl group.
Scheme 5.
Preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [24]. Reagent(s); catalyst; solvent(s); yield(s). (i) (COCl)2; HCON(CH3)2; CH2Cl2; 95 %. (ii) Cis-(1S,2R)-2-para-tolylsulfonamidoindanol; (C2H5)3N; tetrahydrofuran; yield not mentioned. (iii) Bromocyclohexane, Mg; ZnCl2; tetrahydrofuran; 55%; (iv) NaOHaq; CH3OH; > 95 %.
Scheme 5.
Preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [24]. Reagent(s); catalyst; solvent(s); yield(s). (i) (COCl)2; HCON(CH3)2; CH2Cl2; 95 %. (ii) Cis-(1S,2R)-2-para-tolylsulfonamidoindanol; (C2H5)3N; tetrahydrofuran; yield not mentioned. (iii) Bromocyclohexane, Mg; ZnCl2; tetrahydrofuran; 55%; (iv) NaOHaq; CH3OH; > 95 %.
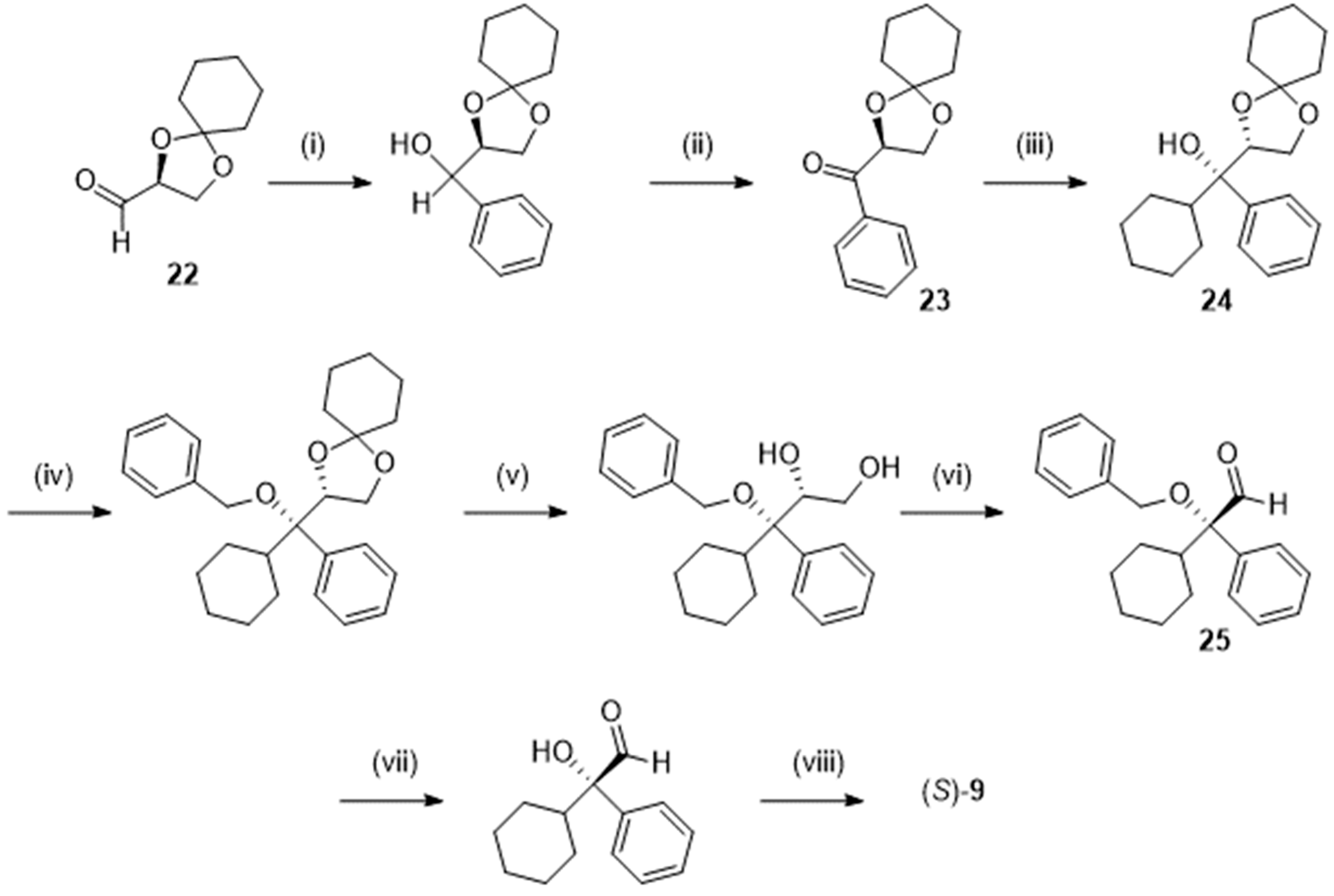
Later, Chattopadhyay et al. [25] described another stereocontrolled Grignard reaction (Scheme 6) by addition of cyclohexylmagnesium bromide on the phenyl ketone 23, obtained from (R)-2,3-O-cyclohexylydene-D-glyceraldehyde 22. Hydrolysis of the acetal 24 in fairly good yield required a preliminary benzylation of the alcohol function, thus lengthening the sequence giving access, via the aldehyde 25, to the expected S-9. An acceptable overall yield of 25 % was obtained at the end of that 8-step procedure.
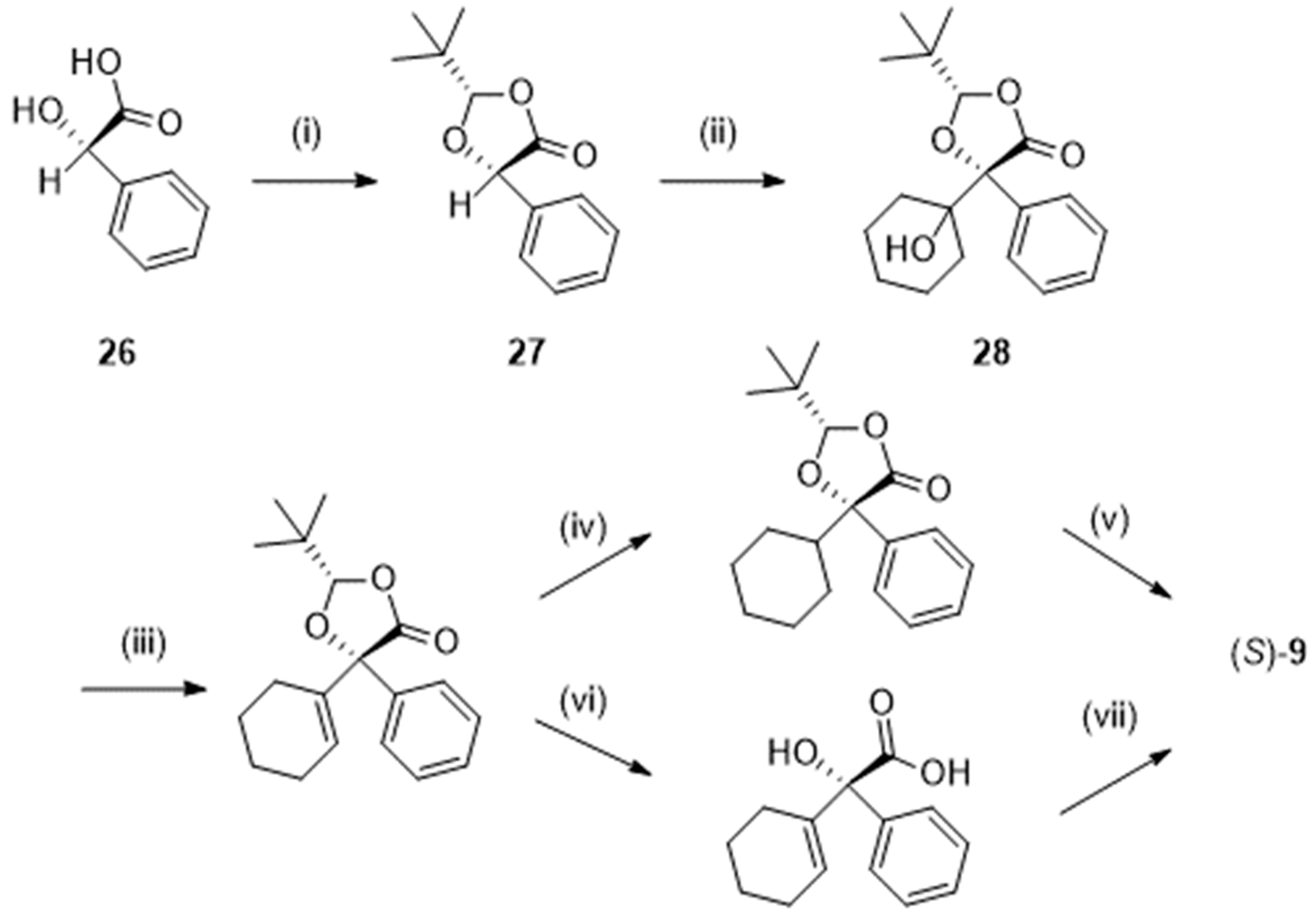
In another approach, that could be performed at a 100 g scale (Scheme 7), trimethylacetaldehyde (pivaldehyde) was acetalized with (S)-2-hydroxy-2-phenylacetic acid (26; (S)- mandelic acid). The resulting dioxolone (27) was deprotonated and stereoselectively coupled with cyclohexanone at – 78 °C. Dehydration of the so-formed alcohol (28) followed by hydrolysis and hydrogenation (or the inverse sequence) yielded (S)-9 in excellent enantiomeric excess (> 99.9 %) and an overall yield of 66% [26].
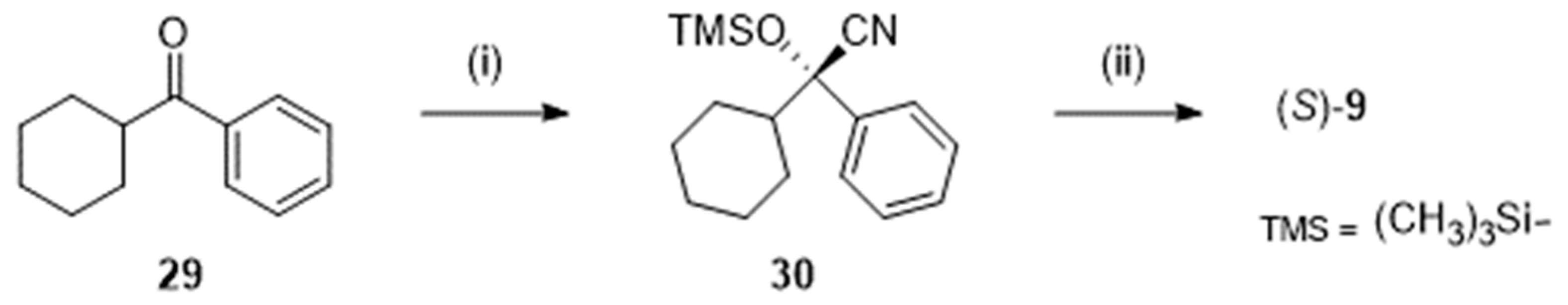
Catalytic enantioselective cyanosilylation of cyclohexyl phenyl ketone 29 (Scheme 8) with a chiral gadolinium complex was the first step of the procedure reported by Shibasaki et al. [27]. Subsequent reduction, desilylation, and oxidation afforded the expected (S)-9. Performed from 100 g of 29, (S)-9 was isolated in 80 % yield with an enantiomeric excess higher than 99.5 %. To be complete, let us mention that, at the mmol scale, enzymatic resolution of racemic cyanohydrins structurally related to 30 has been the subject of a study of Gotor et al. [28].
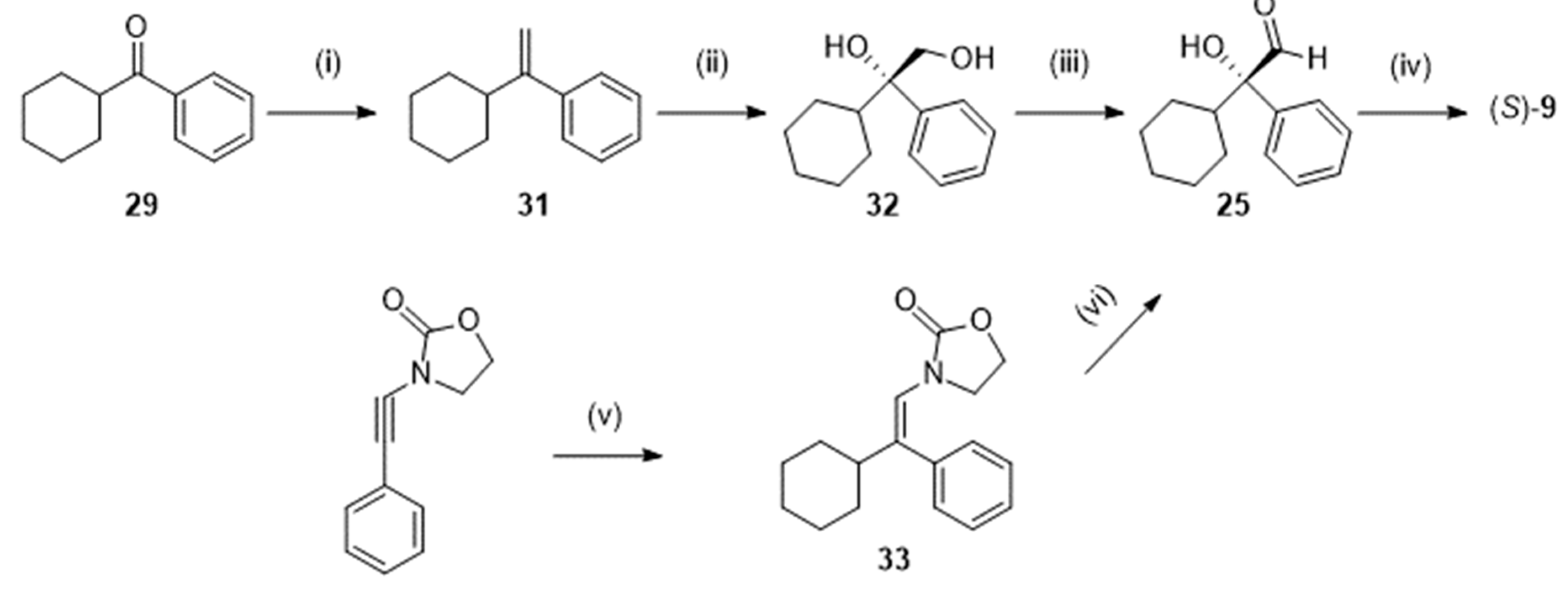
Alternatively (Scheme 9), the ketone 29 was introduced in a Wittig reaction with methylenetriphenylphosphorane yielding the alkene 31. Asymmetric dihydroxylation with osmium tetraoxide under Sharpless conditions gave 32. Then, a Swern reaction oxidized the diol into the corresponding aldehyde 25, which could be further oxidized to the acid (S)-9 with an overall yield of 45 % [29]. Notice that the aldehyde 25 has also been obtained from 3-[(Z)-2-phenyl-2-cyclohexylvinyl]oxazolidin-2-one 33 (scheme 9) as proposed by Gourdet and Lam [30].
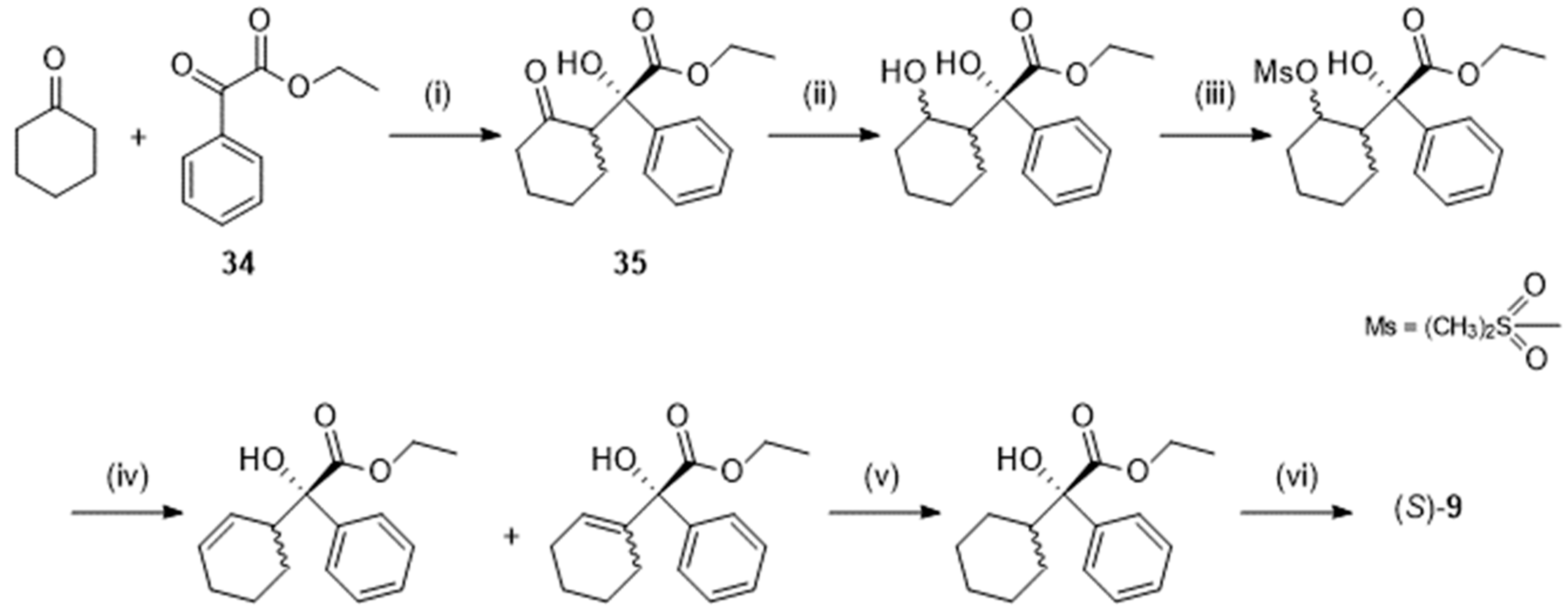
Following Maruoka et al. [31], (S)-9 could be prepared through a L-proline-catalyzed asymmetric aldol reaction between cyclohexanone (in 10-fold excess) and ethyl 2-oxo-2-phenylacetate (34, Scheme 10). That reaction yielded the ester 35 in good yield (79 %), good diastereoselectivity (dr = > 20:1) and good enantiomeric excess (96 %). However, obtention of the pure corresponding acid (S)-9 was not straightforward so that the authors had to design a tedious 5-step sequence starting from 32. The overall yield, calculated on 34, fell to 40 %.
In the work of Trost et al. [32], the initial precursor of (S)-9 was the commercially available cyclohex-2-en-1-ol 36 (Scheme 11), which was converted, in basic medium by treatment with carbon dioxide and then 2-bromoacetophenone, into the ketocarbonate 37. Deprotonation of 37 and protection of the enol by tert-butyldimethylsilyl chloride was accompanied by an intramolecular rearrangement affording 38. In the subsequent step, an internal allylic alkylation involving a chiral palladium catalyst gave the aldehyde 39. Reduction of the cyclohexenyl ring, oxidation and deprotection afforded the expected (S)-acid 9 with an enantiomeric excess higher than 99 % but an overall yield of 22 %.
Having in hands (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid 9 (or the (R) enantiomer), optically active oxybutynin (hydrochloride) could be synthesized using one of the procedures described in Scheme 3 and Scheme 4. The activated (S)-acid 11 has been involved, with the butynyl alcohol 13, in the multigram preparation (40 g) of (S)-oxybutynin hydrochloride [14].
3.3. Resolution of the Racemic
Although high-performance thin-layer chromatography has been cited [33], enantiomers of oxybutynin have been generally separated by high-performance liquid chromatography. The silica-based support of the columns was covalently bonded to ovomucoid [34] or, more often, coated with polysaccharides such as amylose-tris(3,5-dimethylphenylcarbamate) [35,36] or cellulose-tris(4-methylbenzoate) [37]. Also reported and noteworthy, the use of β-cyclodextrin derivatives, essentially hydroxypropyl-β-cyclodextrin, as chiral selectors for the separation by high-performance liquid chromatography [38] as well as by electrophoresis [39], liquid-liquid reactive extraction [40], and recycling high-speed counter-current chromatography [41].
Astonishingly, preparation of diastereoisomers was seldom described. One example could be found in the patents of Molnar and Johnston [18,19]. The inventors treated the racemic (86.1 g), under its free base form, with D-malic acid in 2-propanol and isolated the D-malate salt of (R)-oxybutynin in 41 % yield. In the most recent patent [19], the same inventors claimed that “eleven other chiral acids were tested for production of (R)-oxybutynin salt from racemic oxybutynin: L-tartric acid, D-tartric acid, L-(+)-lactic acid, D-glucuronic acid, D-gluconic acid, L-malic acid, (1R,3S)-(+)-camphoric acid, (S)-(+)-mandelic acid, (1R)-(-)-10-camphorsulfonic acid, L-pyroglutamic acid, and D-(-)-quinic acid. None of the other eleven chiral acids were successful in chiral resolution to produce an (R)-oxybutynin salt from racemic oxybutynin.”
4. Mode of action and metabolism
Micturition is mainly controlled by coordinated actions of the detrusor muscle and the bladder neck muscle. During bladder filling, the detrusor muscle is relaxed whereas the neck muscle is contracted. The situation is reversed during urine elimination. Too frequent urinary urgencies, which are caused by abnormal contractions of the detrusor muscle, are manifestations of the so-called overactive bladder condition. Those contractions occur, in brief, when acetylcholine, released from parasympathetic nerves, binds to M3 muscarinic receptors located on the detrusor muscle. Therefore, blockade of those M3 muscarinic receptors emerged as a strategy of choice for treating overactive bladder [42,43].
Oxybutynin is one of the most popular antagonists of muscarinic receptors and it is widely prescribed in the therapy of overactive bladder since decades. However, muscarinic receptors are present in many parts of the body, including salivary glands, gut smooth muscle, eyes, heart, and brain [42,44]. That explains the series of side-effects observed by administration of the drug, among which dry mouth, the most common and frequent side-effect, decrease sweating, constipation, blurred vision, tachycardia, and nausea. Impairments of cognitive function has also been reported and should be linked to the ability of the drug to cross the blood-brain barrier, due to its lipophilic character [43,45,46,47]. That adverse effect is observed in elderly patients especially, since permeability of the barrier increases with aging.
Interestingly, it was demonstrated that, in the gut and the liver, oxybutynin is rapidly metabolized by cytochrome P450 isozyme 3A4 into N-desethyloxybutynin (40; Figure 3) giving rise to concentrations of N-desethyloxybutynin in serum 4 to 10 times higher than those of the initial drug. However, that first metabolite also exhibits high affinity for the M3 muscarinic receptors and especially for those located in the parotid gland. N-desethyloxybutynin is thought to be the main responsible agent of the dry mouth side-effect associated with the oral administration of oxybutynin [42,48,49].
Mention should be made that oxybutynin also exerts spasmolytic and local anesthetic effects on the bladder smooth muscle. However, those effects are far much weaker than the antimuscarinic activity. Nevertheless, the efficacy of each enantiomer in both pathways has been studied. There were conflicting reports on the spasmolytic effects. The (S)-enantiomer being claimed to exhibit higher [42,50] or similar [51,52,53] spasmolytic action when compared with the (R)-enantiomer. On the other hand, all studies underlined the lower antimuscarinic activity of the (S)-enantiomer when compared with the (R)-enantiomer, thus explaining the better tolerability of that former [48,51,54]. However, up to now, those characteristics did not justify the marketing of any single enantiomer.
5. Formulations and Brand Names
Usually, physicians prescribe 5 to 20 mg of oxybutynin daily for the treatment of overactive bladder. Tablets of Ditropan® were approved by the U.S. Food and Drug Administration (FDA, Silver Spring, MD, USA) on July 16, 1975. Ditropan® syrup was FDA-approved on November 29, 1979.
As earlier mentioned, oxybutynin is rapidly converted into N-desethyloxybutynin upon gastric and hepatic metabolism and that first metabolite could be responsible for the well-known dry mouth side-effect. Therefore, it is not surprising that the original formulations of the racemic drug have successfully evolved to extended-release systems and transdermal administrations.
Ditropan XL® tablet (FDA-approved on December 16, 1998), an extended-release formulation of the drug, was commercialized by Janssen Pharmaceuticals Inc. (Raritan, NJ, USA). It consisted in a core containing a layer of the drug and excipients and a second layer made of osmotic agents. The drug layer was surrounded by a drilled semipermeable membrane enabling controlled entry of water and release of oxybutynin [55].
Ditropan® and Ditropan XL® have now given way to more than 200 generics [56].
The first transdermal formulations of oxybutynin were the Oxytrol® patches marketed by Allergan USA Inc. (Madison, NJ, USA) further to the FDA-approval published on February 26, 2003. Oxytrol® patches, which should be applied on the abdomen, hip, or buttock, are diffusion-controlled delivery systems (5.7 X 7.6 cm2) dispensing 3.9 mg of oxybutynin per day [57].
The product was approved on June 15, 2004 by the European Medicines Agency (EMA) under the brand name Kentera® (Teva B.V., Haarlem, The Netherlands) [58].
Gelnique® is a 10% oxybutynin gel available in sachets of 1 g. It was FDA-approved on January 27, 2009 and is manufactured by Allergan USA Inc. [59]. On December 8, 2011, FDA approved a 3% oxybutynin gel supplied in a metered-dose pump dispenser, sold by Antares Pharma Inc. (Ewing, NJ, USA) [60]. A gel preparation of a nanosuspension of oxybutynin is actually under study in order to minimize skin irritation and improve the permeation efficiency [61].
Advantages, efficacy, and safety of the transdermal administration of oxybutynin were the subject of several clinical trials discussed in the works of Cohn [62] and Vozmediano-Chicharro [49]. Original pharmacokinetic parameters for different oral and transdermal formulations can be found in the study of Kennelly [48]. Roughly, oxybutynin tablet (5 mg PO bid-qid) achieved a maximum concentration in serum of 12 ng/mL within 1 hour with a half-life time of 2-3 hours whereas the extended-release formulation (5-30 mg PO qd) enabled to reach a maximum concentration of 4 ng/mL within 5 hours and exhibited a half-live time of 13 hours. Little differences between the pharmacokinetic parameters of each enantiomer, when orally administered as the racemate, have been detected [63].
Other alternatives enabling to decrease the plasma concentration of N-desethyloxybutynin, and consequently to reduce side-effects, have been studied and include rectal, intravesical, as well as vaginal administrations.
Although still not commercially available, suppositories of oxybutynin were the subject of several clinical trials, the first report appearing in 1998 [64]. The daily dose was similar to that used in oral administration and varied between 5 and 20 mg. Suppositories administered in the study of Winkler [64] contained 5 mg of oxybutynin for 1.25 g of fat. It is noteworthy that the topic remains of actuality since suppositories loaded with oxybutynin microparticles were recently described and evaluated by Bedse et al. [65].
Intravesical solutions were initially obtained by dissolving oxybutynin tablets (5 mg) in distilled water (10 mL) and were instilled in the bladder by mean of a catheter twice a day [66]. Discomfort associated with such a daily treatment led to the development of a delivery system (UROS™, Situs Corp., San Diego, CA, USA) constituted by a reservoir allowing to release the drug over a period of a month and that can be removed by cystoscopy [67,68]. The device knew, however, little success. Improved systems of instillation still attract attention as evidenced by the recent patents literature [69,70].
Intravaginal gels containing oxybutynin [71,72] have been the subject of several in vivo studies on an animal model (rabbits) but, to the best of our knowledge, there was no clinical trial involving such gels. A preliminary determination of the efficacy of an insert releasing oxybutynin in the vagina of rabbits was performed by the group of Levin in 2000 [73]. Other vaginal rings have been designed and some were successfully evaluated in humans but still not commercialized [74,75,76].
6. Repositioning Opportunities
To date, 69 registered clinical trials can be retrieved in the database [79] of the U.S. National Library of Medicine, when using “oxybutynin” as search term. The majority of them (50 trials) are studies on the safety and/or efficacy of oxybutynin for the treatment of dysregulations of the bladder activity and, anecdotally (one trial; NCT03877289), for the treatment of cystitis in children. A Phase 2 trial analyzed the effects of a combination of oxybutynin and desloratadine (41; Figure 4) in cases of seasonal allergic rhinitis (NCT00816972; started in 2005) and another Phase 2 trial studied the potential of a combination of oxybutynin and clonidine (42; Figure 4) in reducing excessive salivation (sialorrhea) in patients with Parkinson’s disease (NCT01370811; started in 2011).
As detailed below, the remaining trials provided repositioning opportunities for the management of hyperhidrosis (9 trials but one, NCT02099695, has been withdrawn) and hot flashes (4 trials). More recently, there was an increase of interest for the use of oxybutynin in combination (see heading 6.3) as a pharmacotherapy for obstructive sleep apnea (4 trials).
6.1. Hyperhidrosis
Hyperhidrosis [80,81,82] is the term used to define an excessive sweating without any link with a normal response of the body to heat or effort. Armpits, soles of the feet, palms of the hands, and face are the body regions that are the most frequently touched by hyperhidrosis. Because stimulation of cholinergic receptors on eccrine sweat glands is responsible for perspiration, hyperfunctioning of those glands could be controlled by use of anticholinergic agents [83]. That approach has been evaluated in the absence of sympathetomy or after such a chirurgical intervention.
The first registered clinical trial dedicated to the efficacy of oxybutynin in the treatment of axillary hyperhidrosis was launched in Brazil in 2007 (NCT01118429). Positive results were published in 2011 [84] since the authors claimed that “more than 80% of the patients experienced an improvement in axillary hyperhidrosis. … The side effects were minor, dry mouth being the most frequent (73.5%).”.
Among the other clinical trials, let us mention one Phase 3 trial (NCT01855256), which started in 2013, and one Phase 4 trial (NCT01310712), which started in 2010 [85]). Today, oxybutynin may be prescribed by physicians for the treatment of hyperhidrosis [e.g. 86,87,88,89]
6.2. Hot Flashes (vasomotor symptoms)
Hot flashes are sudden episodes of sensation of excessive heat of the body, often accompanied by uncontrolled sweating and appearance of red blotches on the face, the chest, and the neck [91,92,93]. Those episodes can be associated, at least in part, with a decrease of the level of estrogen and they involve numerous neurotransmitters, among which norepinephrine, serotonin, and acetylcholine. Frequent in healthy peri- and postmenopausal women, vasomotor symptoms are also observed in patients under hormone-suppression therapies, among which women having been diagnosed some breast cancers and men under treatment for prostate cancer [94,95].
Amid various drugs prescribed to decrease the frequency and/or intensity of hot flashes [96], antimuscarinic agents, including oxybutynin, had been patented as earlier as in 2007 [97]. The inventor (K.D. LaGuardia) had been involved in the first registered clinical trial (Phase 2) on the subject and entitled “The effect of extended-release oxybutynin chloride on vasomotor symptoms in healthy post-menopausal women” (NCT00990886; started in 2004). Results were published in a Conference Paper in 2007 [98] and in a manuscript much later [99]. More recently, a Phase 3 trial (NCT02961790; started in 2016) successfully evaluated the efficacy of lower doses of oxybutynin in the management of hot flashes in women under hormonotherapy or not [100]. Further to those promising results, oxybutynin, even if not FDA-approved for that application, is suggested as an alternative medication by some oncologists and gynecologists [101,102,103]. Two more trials are still recruiting. One (Phase 2) for the study of the efficacy of oxybutynin in men treated for prostate cancer (NCT04600336; started in 2021). The second (Phase 3) will compare oxybutynin and paroxetine (43; Figure 5), an antidepressant, in women with hormone-dependent breast cancer (NCT05637671; started in 2022).
6.3. Obstructive Sleep Apnea
Reduction of the activity of the upper airway dilator muscles is a normal phenomenon during sleep. However, in the situation of obstructive sleep apnea, the reduction reaches such an extent that it obstructs the flow of air into the lungs and temporarily blocks breathing, perturbating oxygenation of blood.
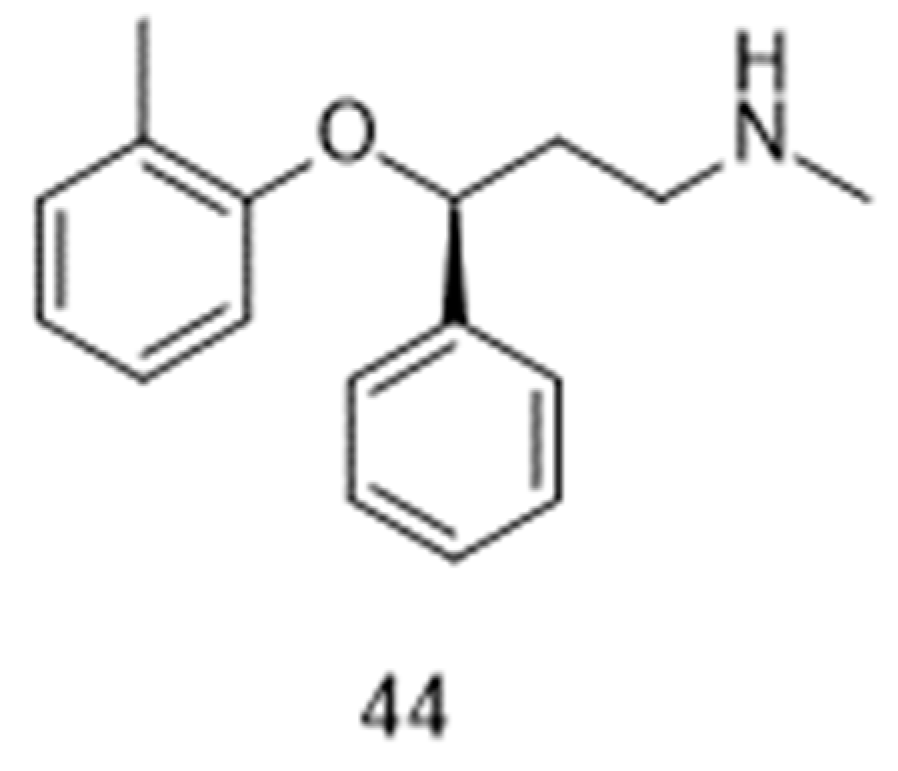
The possibility of restoring the activity of pharyngeal muscles under the influence of a noradrenergic agent [104] and an antimuscarinic agent [105] had been highlighted years ago. However, it is recently only that combinations of such drugs [106,107,108] emerged as a challenging opportunity for the treatment of obstructive sleep apnea. More specifically, the combination oxybutynin and atomoxetine (44, Figure 6) was the subject of two Phase 2 registered clinical trials, which started in 2016 (NCT02908529; [107]) and 2020 (NCT04115878). Two other Phase 2 trials (NCT05550246; NCT05944965) should start in the second half of 2023, as part of the Sleep Disorders Research Program of the Brigham and Women’s Hospital at Boston (MS, USA).
Two additional Phase 2 trials, sponsored by Apnimed (Cambridge, MA, USA), can be retrieved in the database [79] of the U.S. National Library of Medicine, but, surprisingly, they did not appear with the search term “oxybutynin”. In one trial (NCT 04445688), a combination of oxybutynin (5 mg) and atomoxetine (80 mg) was defined under the code name AD036. The conclusion of the evaluation was that “AD036 significantly improved obstructive sleep apnea severity in patients with moderate pharyngeal collapsibility. Atomoxetine may account for the majority of improvement in obstructive sleep apnea severity, while the addition of oxybutynin may mitigate the disruptive effect of atomoxetine on sleep and further improve ventilation” [109]. Soon after, the company sponsored another Phase 2 trial (NCT04631107) with the combination of the (R)-enantiomer of oxybutynin and atomoxetine, in not well-defined proportions and under the code name AD109. In the published results [10], it was intriguingly reported that “the current study is with aroxybutynin (AD109), a new enantiomerically pure form of oxybutynin…”. Anyway, the authors concluded that “this study provides additional support that a pharmacological intervention for obstructive sleep apnea, namely the combination of atomoxetine and aroxybutynin, offers promising results. Additional development of this compound and others is warranted.”
7. Conclusion
Oxybutynin improves bladder control in cases of incontinence and excessive urination frequency by blocking M3 muscarinic receptors on the detrusor muscle. The drug, also named 4-diethylamino-2-butynyl 2-cyclohexyl-2-hydroxy-2-phenylacetate, possesses a chiral center and is commercialized under the form of the racemate. Its synthesis requires three key reactions, namely (i) a Grignard reaction; (ii) a Mannich reaction; and (iii) an esterification or a transesterification in order to bound an acidic segment containing the chiral center and the chain containing the triple bond. Despite the fact that enantiomerically pure isomers are not in clinical use, there has been, in the years 2000-2010, a marked interest in the preparation of (S)-oxybutynin and more specifically in the design of protocols yielding essential intermediates required to afford it, namely (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid and analogs. Very recently, the (R)-enantiomer of oxybutynin attracted the attention because of a potential efficacy, in combination with atomoxetine, in the treatment of obstructive sleep apnea.
In the gut and the liver, oxybutynin is rapidly converted into the active N-desethyloxybutynin metabolite, which is thought to be responsible for the most frequent dry mouth side-effect observed when taking the drug. That observation gave rise to intense efforts in order to find means of by-passing the first metabolic step. That led to successful and extensively prescribed extended-release and transdermal formulations, among others.
A number of clinical trials also indicated that oxybutynin could help managing hyperhidrosis, hot flashes, and, in combination with atomoxetine, obstructive sleep apnea. This represents a new hope to ease daily life of many persons, and among them patients being under hormonotherapy. A turn in the story of oxybutynin?
Funding
This research received no external funding
Conflicts of Interest
The author declares no conflict of interest.
References
- Ashburn, T.T.; Thor, K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C. Aspirin as an antiplatelet drug N. Engl. J. Med. 1994, 330, 1287–1294. [Google Scholar] [CrossRef]
- Patrono, C.; Rocca, B. Aspirin at 120: Retiring, recombining, or repurposing? Res Pract Thromb Haemost. 2021, 5, e12516. [Google Scholar] [CrossRef]
- Goldstein, I.; Burnett, A.L.; Rosen, R.C.; Park, P.W.; Stecher, V.J. The serendipitous story of sildenafil: An unexpected oral therapy for erectile dysfunction Sex. Med. Rev. 2019, 7, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Burgos, M.; Losada-Garcia, A.; Cruz-Hernández, C.D.; Cortés-Ramı́rez, S.A.; Camacho-Arroyo, I.; Gonzalez-Covarrubias, V.; Morales-Pacheco, M.; Trujillo-Bornios, S.I.; Rodrı́guez-Dorantes, M. New approaches in oncology for repositioning drugs: The case of PDE5 inhibitor sildenafil Front. Oncol. 2021, 11, 627229. [Google Scholar] [CrossRef]
- Vargesson, N.; Stephens, T. Thalidomide: History, withdrawal, renaissance, and safety concerns Expert Opin. Drug Saf. 2021, 20, 1455–1457. [Google Scholar] [CrossRef]
- Bauzon, J.; Lee, G.; Cummings, J. Repurposed agents in the Alzheimer’s disease drug development pipeline Alzheimer's Res. Ther. 2020, 12, 98. [Google Scholar] [CrossRef]
- Anderson, S.D. Repurposing drugs as inhaled therapies in asthma Adv. Drug Deliv. Rev. 2018, 113, 19–33. [Google Scholar] [CrossRef]
- Vanden Eynde, J.J. COVID-19: Failure of the DisCoVeRy clinical trial, and now–New hopes? Pharmaceuticals 2021, 14, 664. [Google Scholar] [CrossRef]
- Rosenberg, R.; Abaluck, B.; Thein, S. Combination of atomoxetine with the novel antimuscarinic aroxybutynin improves mild to moderate OSA. J. Clin. Sleep Med. 2022, 18, 2837–2844. [Google Scholar] [CrossRef]
- Esoxybutynin. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Esoxybutynin (accessed on 17 September 2023).
- Pai, N.R.; Dubhashi, D.S. Design, synthesis and evaluation of substituted benzeneacetic acid ester derivatives as potential antiinflammatory and analgesic agents. Int. J. Pharmtech Res. 2010, 2, 443–452. [Google Scholar]
- Gant, T.G.; Sarshar, S. Substituted phenylcyclohexylglycolates. US patent 2009/0247628A1 (2009).
- Vandenbossche, C.P.; de Croos, P.; Singh, S.P.; Bakale, R.P.; Wagler, T.R. Formation of (S)-5-cyclohexyl-5-phenyl-1,3-dioxolane-2,4-dione: A key intermediate in the synthesis of (S)-oxybutynin hydrochloride. Org. Process Res. Dev. 2010, 14, 921–925. [Google Scholar] [CrossRef]
- Ray, P.C.; Sethi, M.; Mahajan, S.; Tyagi, O.D. Crystalline oxybutynin and process for preparing the same. PCT Int. Appl. WO 2009/122429 A2 (2009).
- Campbel, K.N.; Majewski, R.F. Substituted aminobutynyl acetates. US patent 3,76,019 (1965).
- Scigel, R.; Smrz, R. Process for preparing oxybutynin. Czech Republic patent CZ 296056 B6 ( 2005.
- Molnar, D; Johnston, S. Preparation of polymorphic forms of (R)-oxybutynin hydrochloride. PCT Int. Appl. WO 2021226020 A1 (2021).
- Johnston, S.; Molnar, D. Solid forms of (R)-oxybutynin d-malate. PCT Int. Appl. WO 2022235726 A1 ( 2022.
- Yang, P.; Yuan, H.; Zhou, M. Preparation method of oxybutynin hydrochloride from α-cyclohexyl-α-hydroxyphenylacetic acid via esterification, Mannich reaction and salifying. Chinese patent CN-114805100-A (2022).
- Czarnik, A.W. Deuterium-enriched oxybutynin. US patent 2008/02992.19 A1 (2008).
- Li, F.; Jiang, W.; Czarnik, A.W.; Li, W. Combinatorial synthesis of deuterium-enriched (S)-oxybutynin. Mol. Divers. 2016, 20, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Bakale, R.P.; Lopez, J.L.; McConville, F.X.; Vandenbossche, C.P.; Senanayake, C.H. Carbonate intermediates useful in the preparation of optically active cyclohexylphenylglycolate esters. US patent 5,973, 182 1999. [Google Scholar]
- Senanayake, C.H.; Fang, K.; Grover, P.; Bakale, R.P.; Vandenbossche, C.P.; Wald, S.A. Tetrahedron Lett.1999, 40, 819-822. [CrossRef]
- Roy, S.; Sharma, A.; Chattopadhyaya, N.; Chattopadhyay, S. An efficient asymmetric synthesis of (S)-2-cyclohexyl-2-phenylglycolic acid, the acid segment of oxybutynin. Tetrahedron Lett. 2006, 47, 7067–7069. [Google Scholar] [CrossRef]
- Grover, P.T.; Bhongle, N.N.; Wald, S.A.; Senanayake, C.H. Chiral mandelic acid template provides a highly practical solution for (S)-oxybutynin synthesis. J. Org. Chem. 2000, 65, 6283–6287. [Google Scholar] [CrossRef]
- Masumoto, S.; Suzuki, M.; Kanaia, M.; Shibasakia, M. A practical synthesis of (S)-oxybutynin. Tetrahedron Lett. 2002, 43, 8647–8651. [Google Scholar] [CrossRef]
- Recuero, V.; Ferrero, M.; Gotor-Fernandez, V.; Brieva, R.; Gotor, V. Enzymatic resolution of hindered cyanohydrins, key precursors of muscarinic receptor antagonists. Tetrahedron: Asymmetry 2007, 18, 994–1002. [Google Scholar] [CrossRef]
- Gupta, P.; Fernandes, R.A.; Kumar, P. An asymmetric dihydroxylation route to (S)-oxybutynin. Tetrahedron Lett. 2003, 44, 4231–4232. [Google Scholar] [CrossRef]
- Gourdet, B.; Lam, H.W. Catalytic asymmetric dihydroxylation of enamides and application to the total synthesis of (+)-tanikolide. Angew. Chem. Int. Ed. 2010, 49, 8733–8737. [Google Scholar] [CrossRef]
- Tokuda, O.; Kano, T.; Gao, W.-G.; Ikemoto, T.; Maruoka, K. A practical synthesis of (S)-2-cyclohexyl-2-phenylglycolic acid via organocatalytic asymmetric construction of a tetrasubstituted carbon center. Org. Lett. 2005, 7, 5103–5105. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Xu, J.; Reichle, M. Enantioselective synthesis of α-tertiary hydroxyaldehydes by palladium-catalyzed asymmetric allylic alkylation of enolates. J. Am. Chem. Soc. 2007, 129, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Sitadevi, P.; Rao, P.L.K.M. Development and validation of a method for the enantioseparation of oxybutynin hydrochloride by HPTLC. Anal. Chem.: Indian J. 2010, 9, 378–383. [Google Scholar]
- Walker, T.A. The chiral separation of oxybutynin enantiomers using an ovomucoid column. J. Liq. Chromatogr. Relat. Technol. 2000, 23, 841–853. [Google Scholar] [CrossRef]
- Miyamoto, E.; Demizu, Y.; Murata, Y.; Yamada, Y.; Kawashima, S.; Kontani, H.; Sakai, T. High-performance liquid chromatographic preparation of oxybutynin enantiomers on a chiral stationary phase. J. Chromatogr. A 1993, 653, 135–137. [Google Scholar] [CrossRef]
- Zin, L.C.; Silva, C.F.; Guimaraes, L.; Borges, K.B.; Nascimento, C.S. Jr. Enantioselective separation of oxybutynin: a theoretical and experimental investigation. Quim. Nova 2022, 45, 263–267. [Google Scholar] [CrossRef]
- Cai, L.; Xue, M.; Lun, J.; Li, S.; Yu, J.; Guo, X. Enantioseparation and molecular modeling study of eight psychoactive drugs on a coated polysaccharide-based chiral stationary phase. Electrophoresis 2020, 41, 2092–2101. [Google Scholar] [CrossRef]
- Lu, Y.; Sun, G. Hydroxypropyl-β-cyclodextrin encapsulated stationary phase based on silica monolith particles for enantioseparation in liquid chromatography. J. Sep. Sci. 2021, 44, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Song, P.; Wen, X.; Deng, M.; Wang, J.; Guo, X. Chiral separation of 12 pairs of enantiomers by capillary electrophoresis using heptakis-(2,3-diacetyl-6-sulfato)-β-cyclodextrin as the chiral selector and the elucidation of the chiral recognition mechanism by computational methods. J. Sep. Sci. 2017, 40, 2999–3007. [Google Scholar] [CrossRef]
- Tang, K.; Cai, J.; Yang, C.; Liu, Y.; Zhang, P.; Liu, Y. Kinetic study on reactive extraction for chiral separation of oxybutynin enantiomers. Sep. Purif. Technol. 2012, 92, 30–35. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, G.; Tang, K.; Yang, W.; Sui, G.; Zhou, C. Enantiomeric separation of oxybutynin by recycling high-speed counter-current chromatography with hydroxypropyl-β-cyclodextrin as chiral selector. J. Sep. Sci. 2014, 37, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Dmochowski, R. Improving the tolerability of anticholinergic agents in the treatment of overactive bladder. Drug Saf. 2005, 28, 583–600. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Ito, Y.; Nishijima, S.; Kadekawa, K.; Sugaya, K. Basic and clinical aspects of antimuscarinic agents used to treat overactive bladder. Pharmacol. Ther. 2018, 189, 130–148. [Google Scholar] [CrossRef] [PubMed]
- Abrams, P.; Andersson, K.-E.; Buccafusco, J.J.; Chapple, C.; de Groat, W.C.; Fryer, A.D.; Kay, G.; Laties, A.; Nathanson, N.M.; Pasricha, P.J.; Wein, A.J. Muscarinic receptors: Their distribution and function in body systems, and the implications for treating overactive bladder. Br. J. Pharmacol. 2006, 148, 565–578. [Google Scholar] [CrossRef]
- Welk, B.; Richardson, K.; Panicker, J.N. The cognitive effect of anticholinergics for patients with overactive bladder. Nat. Rev. Urol. 2021, 18, 686–700. [Google Scholar] [CrossRef]
- Welk, B.; Etaby, K.; McArthur, E.; Chou, Q. The risk of delirium and falls or fractures with the use of overactive bladder anticholinergic medications. Neuroulol. Urodyn. 2022, 41, 348–356. [Google Scholar] [CrossRef]
- Nishtala, P.S.; Chyou, T.-Y. Risk of delirium associated with antimuscarinics in older adults: A case-time-control study. Pharmacoepidemiol. Drug Saf. 2022, 31, 883–891. [Google Scholar] [CrossRef]
- Kennelly, M.J. A comparative review of oxybutynin chloride formulations: Pharmacokinetics and therapeutic efficacy in overactive bladder. Rev Urol. 2010, 12, 12–19. [Google Scholar] [CrossRef]
- Vozmediano-Chicharro, R.; Blasco Hernández, P.; Madurga-Patuel, B. Insights into the management of overactive bladder with transdermal oxybutynin: A practical review. Res. Rep. Urol. 2020, 12, 321–330. [Google Scholar] [CrossRef]
- Dmochowski, R.R. (S)-oxybutynin (Sepracor). Curr. Opin. Investig. Drugs, 2002, 3, 1508–11. [Google Scholar]
- Kachur, J.F.; Peterson, J.S.; Carter, J.P.; Rzeszotarski, W.J.; Hanson, R.C.; Noronha-Blob, L. R and S enantiomers of oxybutynin: Pharmacological effects in guinea pig bladder and intestine. J. Pharmacol. Exp. Ther. 1988, 247, 867–872. [Google Scholar] [PubMed]
- Noronha-Blob, L.; Kachur, J.F. Enantiomers of oxybutynin: In vitro pharmacological characterization at M1, M2 and M3 muscarinic receptors and in vivo effects on urinary bladder contraction, mydriasis and salivary secretion in guinea pigs. J. Pharmacol. Exp. Ther. 1991, 256, 562–567. [Google Scholar] [PubMed]
- Starkman, J.S.; Dmochowski, R. Management of overactive bladder with transdermal oxybutynin. Rev. Urol. 2006, 8, 93–103. [Google Scholar] [PubMed]
- Kretschmar, M.; Suleiman, A.A.; Krause, P.; Albrecht, U.; Stein, R.; Rubenwolf, P.; Fuhr, U.; Taubert, M. A population pharmacokinetic model of (R)- and (S-) oxybutynin and its active metabolites after oral and intravesical administration to healthy volunteers. J. Clin. Pharmacol. 2021, 21, 961–971. [Google Scholar] [CrossRef]
- Ditropan, XL. Available online: https://www.janssenlabels.com/package-insert/product-monograph/prescribing-information/DITROPAN+XL-pi.pdf (accessed on 17 September 2023).
- Oxybutynin. Available on line: https://go.drugbank.com/drugs/DB01062 (accessed on 17 September 2023).
- Oxytrol. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021351s005lbl.pdf (accessed on 17 September 2023).
- Kentera (previously Oxybutynin Nicobrand). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kentera-previously-oxybutynin-nicobrand (accessed on 17 September 2023).
- Gelnique. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/022204s010lbl.pdf (accessed on 17 September 2023).
- Anturol. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202513s000lbl.pdf (accessed on 17 September 2023).
- Sheng, Y.; Zhang, S.; Ling, J.; Hu, C.; Zhang, Z.; Lv, H. Oxybutynin nanosuspension gel for enhanced transdermal treatment for overactive bladder syndrome. Pharm. Dev. Technol. 2022, 27, 459–468. [Google Scholar] [CrossRef]
- Cohn, J.A.; Brown, E.T.; Reynolds, W.S.; Kaufman, M.R.; Milam, D.F.; Dmochowski, R.R. An update on the use of transdermal oxybutynin in the management of overactive bladder disorder. Ther. Adv. Urol. 2016, 8, 83–90. [Google Scholar] [CrossRef]
- Sathyan, G.; Chancellor, M.B.; Gupta, S.K. Effect of OROS® controlled-release delivery on the pharmacokinetics and pharmacodynamics of oxybutynin chloride. Br. J. Clin. Pharmacol., 2001, 52, 409–417. [Google Scholar] [CrossRef]
- Winkler, H.A.; Sand, P.K. Treatment of detrusor instability with oxybutynin rectal suppositories. Int. Urogynecol. J. Pelvic Floor Dysfunct. 1998, 9, 100–102. [Google Scholar] [CrossRef]
- Bedse, A. : Mahajan, H.; Dhamane, S. Formulation of oxybutynin chloride microparticle-loaded suppositories: in vitro characterization and in vivo pharmacokinetic study. Future J. Pharm. Sci. 2022, 8, 22. [Google Scholar] [CrossRef]
- Brendler, C.B.; Radebaugh, L.C.; Mohler, J.L. Topical oxybutynin chloride for relaxation of dysfunctional bladders. J. Urol., 1989, 141, 1350–1352. [Google Scholar] [CrossRef]
- Fraser, M.O.; Lavelle, J.P.; Sacks, M.S.; Chancellor, M.B. The future of bladder control—Intravesical drug delivery, a pinch of pepper, and gene therapy. Rev Urol. 2002, 4, 1–11. [Google Scholar] [PubMed]
- Moon, H.S. Research on novel intravesical drug delivery devices. Int. Neurourol. J. 2016, 20, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Kuehbacher, A.; Zeppek, C.; Thielicke, W. Syringe sterilization method. PCT Int. Appl. WO 2022223174 A1 ( 2022.
- Gordon, A.; Malchi, N.; Nasser, T.; Touitou, D. Compositions and methods for drug instillation into the urinary bladder. PCT Int. Appl. WO 2023062638 A1 ( 2023.
- Levin, R.M.; Whitbeck, C.; Borow, A.; Burden, O.; Leggett, R.E. Effectiveness of vaginally administered oxybutynin on rabbit bladder function. Urology, 2003, 61, 1273–1277. [Google Scholar] [CrossRef]
- Tugcu-Demiröz, F.; Acartürka, F.; Erdogan, D. Development of long-acting bioadhesive vaginal gels of oxybutynin: Formulation, in vitro and in vivo evaluations. Int. J. Pharm. 2013, 457, 25–39. [Google Scholar] [CrossRef]
- Schröder, A.; Levin, R.M.; Kogan, B.A.; Das, A.K.; Kay, F.; Mahashabde, A. Absorption of oxybutynin from vaginal inserts: Drug blood levels and the response of the rabbit bladder. Urology, 2000, 56, 1063–1067. [Google Scholar] [CrossRef]
- Woolfson, A.D.; Malcolm, R.K.; Gallagher, R.J. Design of a silicone reservoir intravaginal ring for the delivery of oxybutynin. J. Control. Release, 2003, 91, 465–476. [Google Scholar] [CrossRef]
- Gittelman, M.; Weiss, H.; Seidman, L. A phase 2, randomized, double-blind, efficacy and safety study of oxybutynin vaginal ring for alleviation of overactive bladder symptoms in women. J. Urol., 2014, 191, 1014–21. [Google Scholar] [CrossRef]
- de Laat, W.; Pagan, L.; Malcolm, R.K.; Wiegerinck, M.; Nickolson, V.; Huisman, B.; Stuurman, R.; van Esdonk, M.; Klarenbeek, N. First-in-human study to assess the pharmacokinetics, tolerability, and safety of single-dose oxybutynin hydrochloride administered via a microprocessor-controlled intravaginal ring. Drug Deliv. 2023, 30, 2180113. [Google Scholar] [CrossRef]
- Palugan, L.; Cerea, M.; Cirilli, M.; Moutaharrik, S.; Maroni, A.; Zema, L.; Melocchi, A.; Uboldi, M.; Filippin, I.; Foppoli, A.; Gazzaniga, A. Intravesical drug delivery approaches for improved therapy of urinary bladder diseases. Int. J. Pharm.: X 2021, 3, 100100. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, S.; Cardozo, L. The vagina as a route for drug delivery: A review. Int. Urogynecol. J., 2013, 24, 537–543. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine - Clinical Trials. Available online: https://clinicaltrials.gov/ct2/home (accessed on 17 September 2023).
- Nawrocki, S.; Cha, J. The etiology, diagnosis, and management of hyperhidrosis: A comprehensive review. J. Am. Acad. Dermatol. 2019, 81, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Gregoriou, S.; Sidiropoulou, P.; Kontochristopoulos, G.; Rigopoulos, D. Clin. Cosmet. Investig. Dermatol. 2019, 12, 733–738. [CrossRef] [PubMed]
- Wohlrab, J.; Bechara, F.G.; Schick, C.; Naumann, M. Hyperhidrosis: A central nervous dysfunction of sweat secretion. Dermatol. Ther. 2023; 13, 453–463. [Google Scholar]
- Wong. N.S.; Adlam, T.M.; Potts, G.E.; Farshchian, M. Hyperhidrosis: A review of recent advances in treatment with topical anticholinergics. Dermatol. Ther. 2022, 12, 2705–2714. [Google Scholar] [CrossRef]
- Wolosker, N.; Milanez de Campos, J.R.; Kauffman, P.; Neves, S.; Munia, M.A.; BiscegliJatene, F.; Puech-Leão, P. The use of oxybutynin for treating axillary hyperhidrosis. J. Vasc. Surg. 2011, 25, 1057–1062. [Google Scholar] [CrossRef]
- Wolosker, N.; Milanez de Campos, J.R.; Kauffman, P.; Puech-Leão, P. A randomized placebo-controlled trial of oxybutynin for the initial treatment of palmar and axillary hyperhidrosis. J. Vasc. Surg. 2012, 55, 1696–1700. [Google Scholar] [CrossRef]
- Wolosker, N.; Kauffman, P.; de Campos, J.R.M.; Faustino, C.B.; da Silva, M.F.A.; Teivelis, M.P.; Puech-Leão, P. Long-term results of the treatment of primary hyperhidrosis with oxybutynin: follow-up of 1,658 cases. Int. J. Dermatol. 2020, 59, 709–715 doiorg/101111/ijd14872. [Google Scholar] [CrossRef]
- El-Samahy, M.; Mouffokes, A.; Badawy, M.M.; Amro, S.; Fayad, T.; Abdelwahab, O.A. Safety and efficacy of oxybutynin in patients with hyperhidrosis: systematic review and meta-analysis of randomized controlled trials. Arch. Dermatol. Res. 2023; Ahead of Print. [Google Scholar] [CrossRef]
- Oxybutynin as treatment for hyperhidrosis. Available online: https://duradry.com/blogs/hyperhidrosis/oxybutynin-for-the-treatment-of-hyperhidrosis (accessed on 17 September 2023).
- Oxybutynine/Ditropan contre la transpiration/hyperhidrose. Available online: https://observatoire-hyperhidrose.fr/2020/05/11/effet-oxybutynine-hyperhidrose/#google_vignette (accessed on 17 September 2023).
- Hyperhidrosis. Available online: https://www.farmacotherapeutischkompas.nl/bladeren/indicatieteksten/hyperhidrosis (accessed on 17 September 2023).
- Ravnikar, V. Physiology and treatment of hot flushes. Obstet. Gynecol. 1990, 75, 3S–8S. [Google Scholar] [CrossRef]
- Kronenberg, F. Hot flashes: Phenomenology, quality of life, and search for treatment options. Exp. Gerontol. 1994, 29, 319–336. [Google Scholar] [CrossRef]
- Bansal, R.; Aggarwal, N. Menopausal hot flashes: A concise review. J. Mid-life Health 2019, 10, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Faubion, S.S.; Loprinzi, C.L.; Ruddy, K.J. Management of hormone deprivation symptoms after cancer. Mayo Clin. Proc. 2016, 91, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Loprinzi, C.L.; Deville, C. Oxybutynin for hot flashes due to androgen deprivation in men. N. Engl. J. Med. 2018, 378, 1745–1746. [Google Scholar] [CrossRef]
- Leon-Ferre, R.A.; Majithia, N.; Loprinzi, C.L. Management of hot flashes in women with breast cancer receiving ovarian function suppression. Cancer Treat. Rev. 2017, 52, 82–90. [Google Scholar] [CrossRef]
- LaGuardia, K.D. Treatment of hot flashes using muscarinic receptor antagonists. US patent US 2007/028 1998 A1.
- Simon, J.A.; LaGuardia, K.D. Extended-release oxybutynin relieves vasomotor symptoms in healthy postmenopausal women. 55th Annual Clinical Meeting of the American College of Obstetricians and Gynecologists, May 5-9, 2007, San Diego (CA, USA). [CrossRef]
- Simon, J.A.; Gaines, T.; LaGuardia, K.D. Extended-release oxybutynin therapy for vasomotor symptoms in women: a randomized clinical trial. Menopause 2016, 23, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Leon-Ferre, R.A.; Novotny, P.J.; Wolfe, E.G.; Faubion, S.S.; Ruddy, K.J.; Flora, D.; Dakhil, C.S.R.; Rowland, K.M.; Graham, M.L.; Le-Lindqwister, N.; Smith, T.J.; Loprinzi, C.L. Oxybutynin vs placebo for hot flashes in women with or without breast cancer: A randomized, double-blind clinical trial (ACCRU SC-1603). JNCI Cancer Spectr. 2020, 4, pkz088. [Google Scholar] [CrossRef] [PubMed]
- Le manuel MSD – Menopause. Available online: https://www.msdmanuals.com/fr/professional/gyn%C3%A9cologie-et-obst%C3%A9trique/m%C3%A9nopause/m%C3%A9nopause (accessed on 17 September 2023).
- Kaiser Permanente - Nonhormonal prescription medications for menopause symptoms. Available online: https://mydoctor.kaiserpermanente.org/ncal/article/nonhormonal-prescription-medications-for-menopause-symptoms-2209311 (accessed on 17 September 2023).
- HealthCentral - What to know about menopause medications. Available on line: https://www.healthcentral.com/condition/menopause/menopause-drugs-medications (accessed on 17 September 2023).
- Chan, E.; Steenland, H.W.; Liu, H.; Horner, R.L. Endogenous excitatory drive modulating respiratory muscle activity across sleep-wake states. Am. J. Respir. Crit. Care Med. 2006, 174, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Grace, K.P.; Hughes, S.W.; Horner, R.L. Identification of the mechanism mediating genioglossus muscle suppression in REM sleep. Am. J. Respir. Crit. Care Med. 2013, 187, 311–319. [Google Scholar] [CrossRef]
- Lim, R.; Carberry, J.C.; Wellman, A.; Grunstein, R.; Eckert, D.J. Reboxetine and hyoscine butylbromide improve upper airway function during nonrapid eye movement and suppress rapid eye movement sleep in healthy individuals. Sleep, 2019, 42, zsy261 doiorg/101093/sleep/zsy261. [Google Scholar] [CrossRef]
- Taranto-Montemurro, L.; Messineo, L.; Sands, S.A.; Azarbarzin, A.; Marques, M.; Edwards, B.A.; Eckert, D.J.; White, D.P.; Wellman, A. The Combination of Atomoxetine and Oxybutynin Greatly Reduces Obstructive Sleep Apnea Severity: A Randomized, Placebo-Controlled, Double-Blind Crossover Trial. Am. J. Respir. Crit. Care Med. 2019, 199, 1267–1276. [Google Scholar] [CrossRef]
- Taranto-Montemurro, L.; Pho, H.; White, D.P. Development of a combination of noradrenergic and antimuscarinic drugs for the treatment of obstructive sleep apnea: Challenges and progress. Front. Sleep, 2023, 2, 1148282. [Google Scholar] [CrossRef]
- Schweitzer, P.K.; Maynard, J.P.; Wylie, P.E.; Emsellem, H.A.; Sands, S.A. Efficacy of atomoxetine plus oxybutynin in the treatment of obstructive sleep apnea with moderate pharyngeal collapsibility. Sleep Breath. 2023, 27, 495–503. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure of aspirin (1), sildenafil (2), and thalidomide (3).
Figure 2.
Structure of oxybutynin (4) and its (S)-enantiomer.
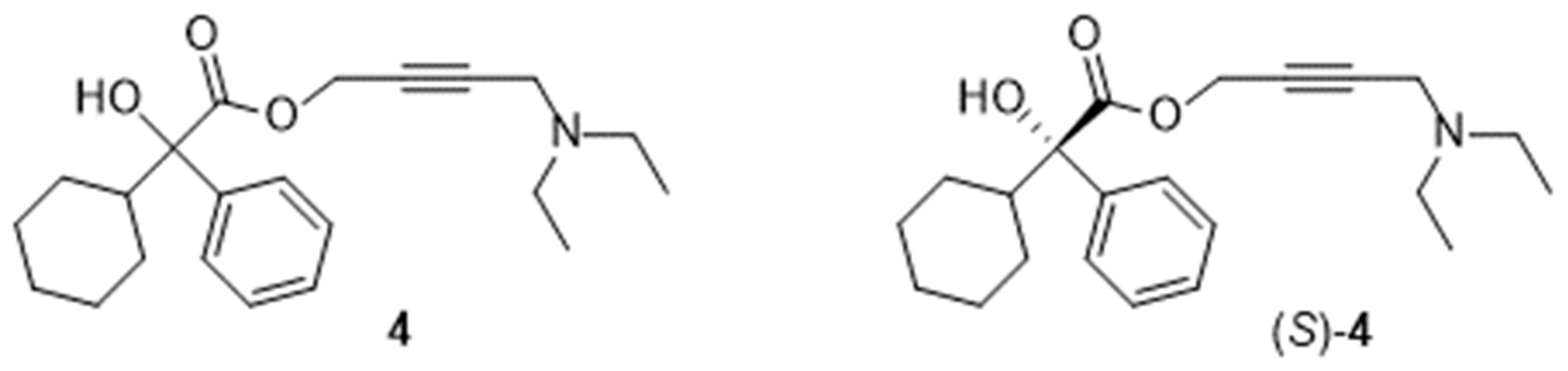
Scheme 3.
Final step in the preparation of oxybutynin 4. Sequence: reagent(s); catalyst; solvent(s); yield(s). 8 + 15 -> [4] -> 4.HCl: (i) CH3ONa; n-heptane then (ii) HClaq; H2O; 86 % [12], 51 % [16]. 8 + 15 -> 4: (i) CH3ONa; n-heptane; 73 % [16]. 8 + 13 -> 4: (i) NaOH; H2O; 91 % [17]. 9 +13 -> [4] -> 4.HCl: (i) 1-Hydroxybenzotriazole hydrate, N-methylmorpholine, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride; CH2Cl2 then (ii) HClg; CH3OH; 27 % [13]. 4.HCl -> 4: (iii) NaOH; H2O, n-heptane; 96 % [16]; (iii) NaOH; H2O, 95 % [18,19]
Scheme 3.
Final step in the preparation of oxybutynin 4. Sequence: reagent(s); catalyst; solvent(s); yield(s). 8 + 15 -> [4] -> 4.HCl: (i) CH3ONa; n-heptane then (ii) HClaq; H2O; 86 % [12], 51 % [16]. 8 + 15 -> 4: (i) CH3ONa; n-heptane; 73 % [16]. 8 + 13 -> 4: (i) NaOH; H2O; 91 % [17]. 9 +13 -> [4] -> 4.HCl: (i) 1-Hydroxybenzotriazole hydrate, N-methylmorpholine, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride; CH2Cl2 then (ii) HClg; CH3OH; 27 % [13]. 4.HCl -> 4: (iii) NaOH; H2O, n-heptane; 96 % [16]; (iii) NaOH; H2O, 95 % [18,19]
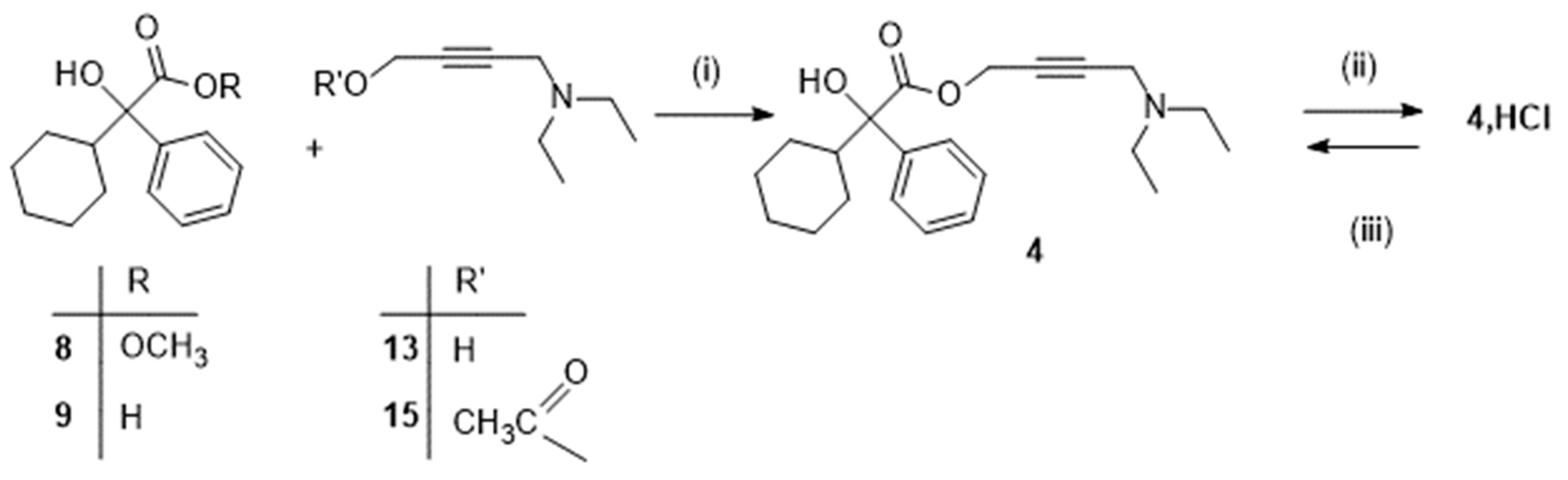
Scheme 4.
Alternative route yielding oxybutynin 4.
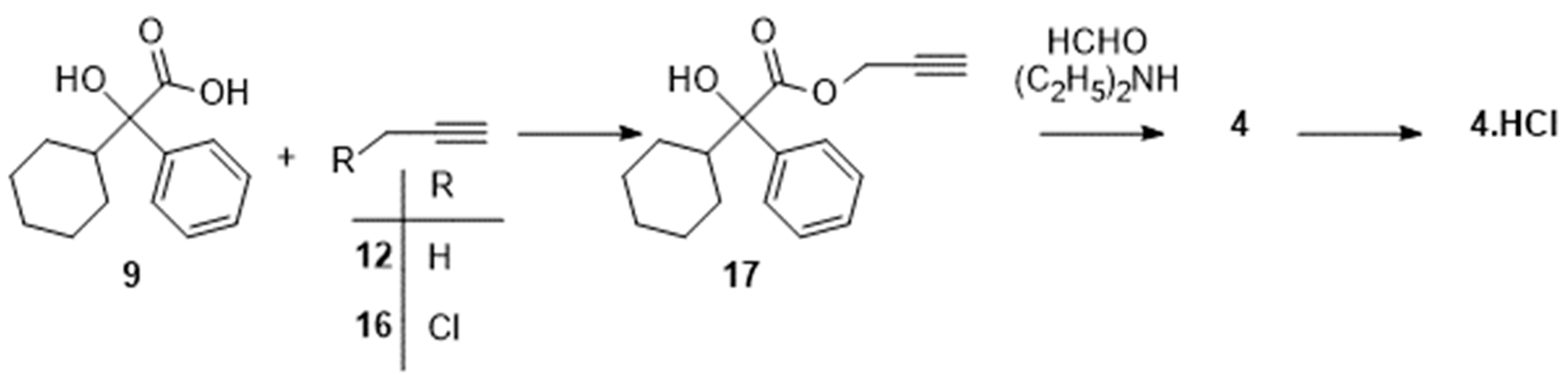
Scheme 6.
Preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [25]. Reagent(s); catalyst; solvent(s); yield(s). (i) Phenylmagnesium bromide; tetrahydrofuran; 88 %. (ii) Pyridinium chlorochromate, CH3CO2Na; CH2Cl2; 84 %. (iii) Cyclohexylmagnesium bromide; tetrahydrofuran; 87 %; (iv) NaH, C6H5CH2Br; tetrahydrofuran; 88 %; (v) HClaq; CH3OH; 92 %. (vi) NaIO4; CH3CN, H2O; 81 %. (vii) Dichlorodicyanobenzoquinone; CH2Cl2; 88 %. (vii) NaClO2; 68 %.
Scheme 6.
Preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [25]. Reagent(s); catalyst; solvent(s); yield(s). (i) Phenylmagnesium bromide; tetrahydrofuran; 88 %. (ii) Pyridinium chlorochromate, CH3CO2Na; CH2Cl2; 84 %. (iii) Cyclohexylmagnesium bromide; tetrahydrofuran; 87 %; (iv) NaH, C6H5CH2Br; tetrahydrofuran; 88 %; (v) HClaq; CH3OH; 92 %. (vi) NaIO4; CH3CN, H2O; 81 %. (vii) Dichlorodicyanobenzoquinone; CH2Cl2; 88 %. (vii) NaClO2; 68 %.
Scheme 7.
Preparation of S-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [26]. Reagent(s); catalyst; solvent(s); yield(s). (i)(CH3)3CHO; trifluoromethylmethanesulfonic acid; pentane; 96 %. (ii) Lithium bis(trimethylsilyl)amide; cyclohexanone, tetrahydrofuran; 76 %. (iii) SOCl2, pyridine; tetrahydrofuran; > 98 %. (iv) H2; Pd/C; CH3OH; 95 %. (vi) KOH; CH3OH; then HCl; 96 %. (vi) KOH; CH3OH; then HCl; 96 %. (vii) H2; Pd/C; CH3OH; 95 %.
Scheme 7.
Preparation of S-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [26]. Reagent(s); catalyst; solvent(s); yield(s). (i)(CH3)3CHO; trifluoromethylmethanesulfonic acid; pentane; 96 %. (ii) Lithium bis(trimethylsilyl)amide; cyclohexanone, tetrahydrofuran; 76 %. (iii) SOCl2, pyridine; tetrahydrofuran; > 98 %. (iv) H2; Pd/C; CH3OH; 95 %. (vi) KOH; CH3OH; then HCl; 96 %. (vi) KOH; CH3OH; then HCl; 96 %. (vii) H2; Pd/C; CH3OH; 95 %.
Scheme 8.
Preparation of S-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [27]. Reagent(s); catalyst; solvent(s); yield(s). (i) Trimethylsilyl cyanide; gadolinium (O-isopropyl)3; C2H5CN; 100 %. (ii) Diisobutylaluminium hydride; toluene then HClaq; tetrahydrofuran then NaClO2, NaH2PO4, 2-methyl-2-butene; H2O, tert-butanol; 80 %.
Scheme 8.
Preparation of S-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [27]. Reagent(s); catalyst; solvent(s); yield(s). (i) Trimethylsilyl cyanide; gadolinium (O-isopropyl)3; C2H5CN; 100 %. (ii) Diisobutylaluminium hydride; toluene then HClaq; tetrahydrofuran then NaClO2, NaH2PO4, 2-methyl-2-butene; H2O, tert-butanol; 80 %.
Scheme 9.
Preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [29,30]. Reagent(s); catalyst; solvent(s); yield(s). (i) Ph3P+CH3I-, n-C4H9Li; tetrahydrofuran; 92 %. (ii) OsO4, K3Fe(CN)6, hydroquinine 1,4-phthalazinediylether; tert-ButOH, H2O; 70 %. (iii) (COCl)2, (C2H5N)3; dimethylsulfoxide, CH2Cl2; not isolated. (iv) NaClO2, NaHPO4.2H2O, 2-methyl-2-butene; tert-ButOH; 70 %. (v) Cyclohexylmagnesium bromide, (CH3CO)2Cu; tetrahydrofuran; 74 %. (vi) K2OsO2(OH)2,K3Fe(CN)6, hydroquinine 1,4-phthalazinediylether; tert-butanol; 80 %.
Scheme 9.
Preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [29,30]. Reagent(s); catalyst; solvent(s); yield(s). (i) Ph3P+CH3I-, n-C4H9Li; tetrahydrofuran; 92 %. (ii) OsO4, K3Fe(CN)6, hydroquinine 1,4-phthalazinediylether; tert-ButOH, H2O; 70 %. (iii) (COCl)2, (C2H5N)3; dimethylsulfoxide, CH2Cl2; not isolated. (iv) NaClO2, NaHPO4.2H2O, 2-methyl-2-butene; tert-ButOH; 70 %. (v) Cyclohexylmagnesium bromide, (CH3CO)2Cu; tetrahydrofuran; 74 %. (vi) K2OsO2(OH)2,K3Fe(CN)6, hydroquinine 1,4-phthalazinediylether; tert-butanol; 80 %.
Scheme 10.
Preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [31]. Reagent(s); catalyst; solvent(s); yield(s). (i) L-proline; dimethylsulfoxide; 79 %; (ii) BH3-(CH3)2S; tetrahydrofuran, CH3OH; not purified. (iii) Methanesulfonyl chloride, (C2H5N)3; CH2Cl2; 80 % (iv) LiCl; hexamethylphosphoramide; 81 %. (v) H2; Pd/C; C2H5OH; 94 %. (vi) NaOH; CH3OH; 93 %.
Scheme 10.
Preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [31]. Reagent(s); catalyst; solvent(s); yield(s). (i) L-proline; dimethylsulfoxide; 79 %; (ii) BH3-(CH3)2S; tetrahydrofuran, CH3OH; not purified. (iii) Methanesulfonyl chloride, (C2H5N)3; CH2Cl2; 80 % (iv) LiCl; hexamethylphosphoramide; 81 %. (v) H2; Pd/C; C2H5OH; 94 %. (vi) NaOH; CH3OH; 93 %.
Scheme 11.
Preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [32]. Reagent(s); catalyst; solvent(s); yield(s). (i) NaH, CO2; then C6H5COCH2Br; HCON(CH3)2; 42 %. (ii) Sodium bis(trimethylsilyl)amide, tert-butyldimethylsilyl chloride; tetrahydrofuran; 83 %. (iii) Tris(dibenzylideneacetone)dipalladium(0)-chloroform adduct; ligand; 1,4-dioxane; 94 %. (iv) H2; Pd/C; C2H5OH; 94 %. (v) NaClO2, NaHPO4.2H2O, 2-methyl-2-butene; tert-butanol, H2O; 95 %.
Scheme 11.
Preparation of (S)-2-cyclohexyl-2-hydroxy-2-phenylacetic acid (S)-9 following [32]. Reagent(s); catalyst; solvent(s); yield(s). (i) NaH, CO2; then C6H5COCH2Br; HCON(CH3)2; 42 %. (ii) Sodium bis(trimethylsilyl)amide, tert-butyldimethylsilyl chloride; tetrahydrofuran; 83 %. (iii) Tris(dibenzylideneacetone)dipalladium(0)-chloroform adduct; ligand; 1,4-dioxane; 94 %. (iv) H2; Pd/C; C2H5OH; 94 %. (v) NaClO2, NaHPO4.2H2O, 2-methyl-2-butene; tert-butanol, H2O; 95 %.
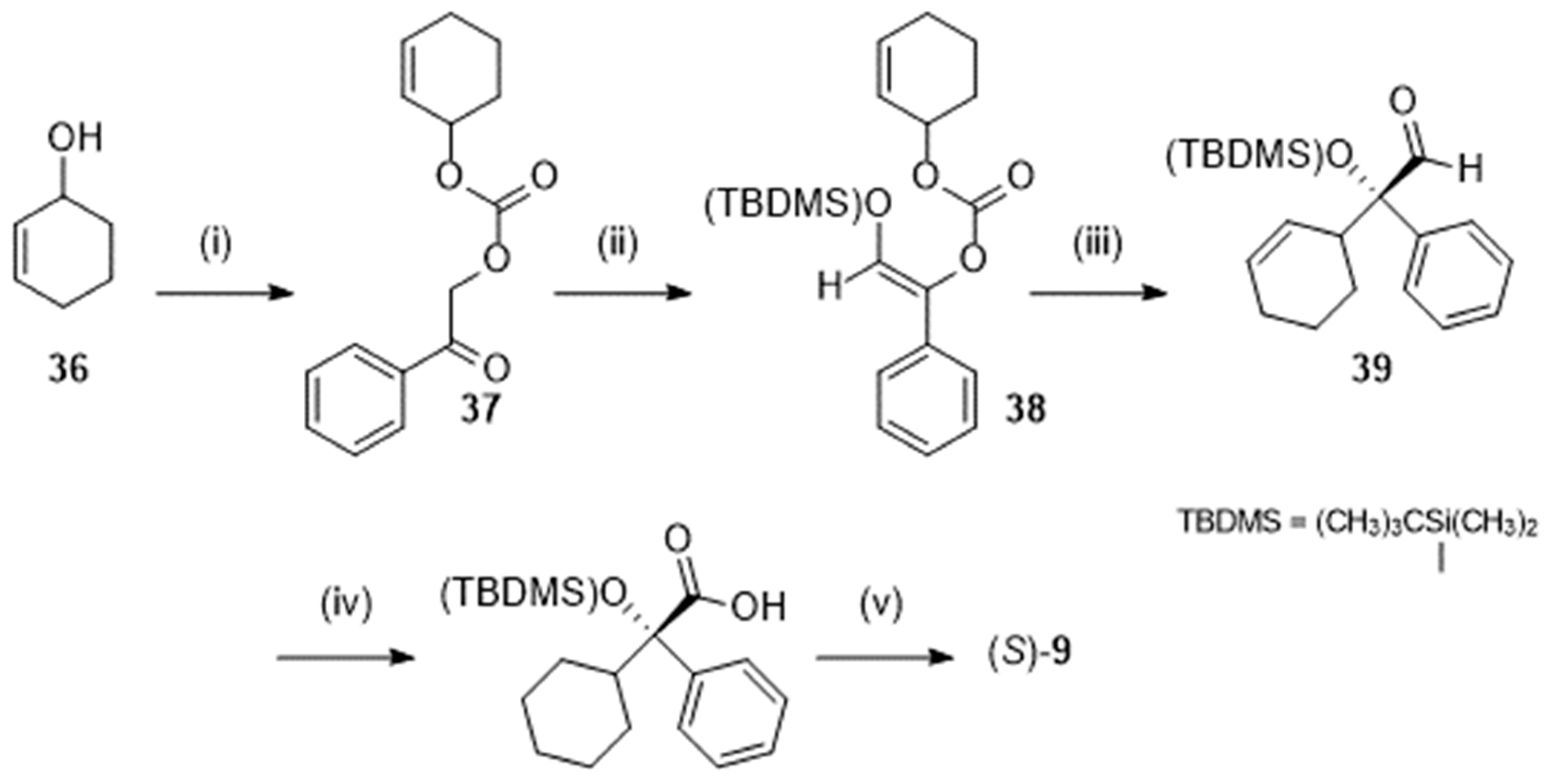
Figure 3.
Chemical structure of N-desethyloxybutynin 40.
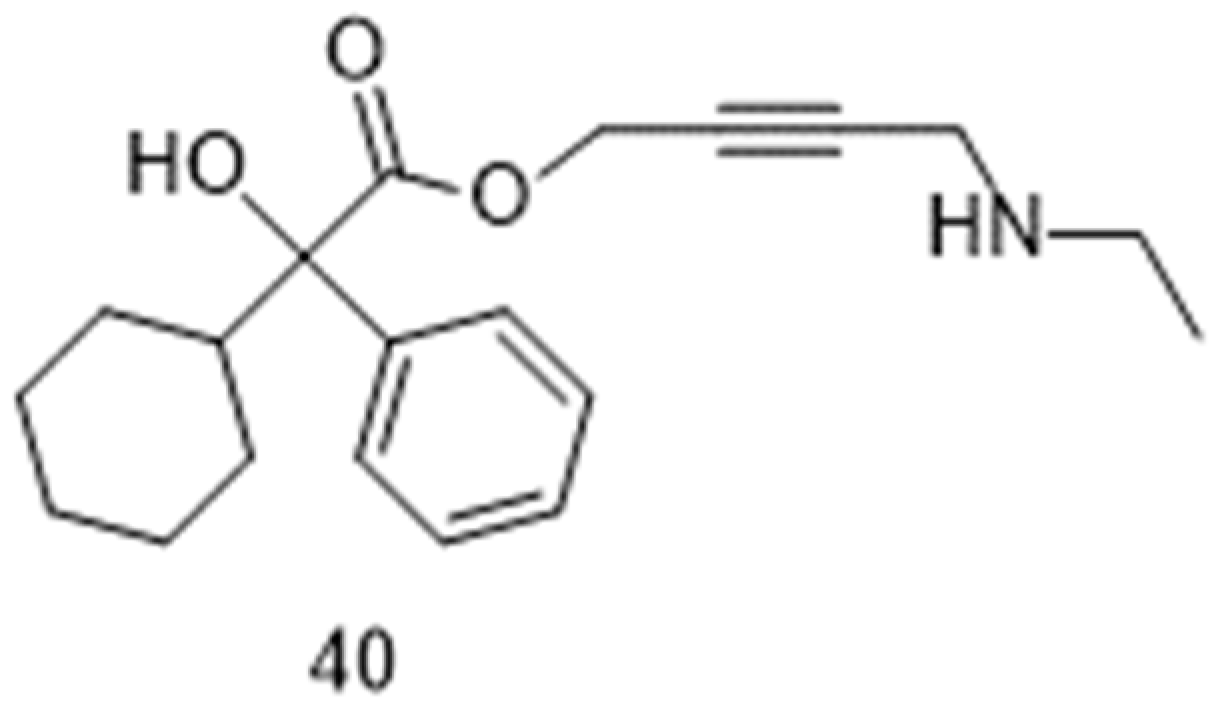
Figure 4.
Chemical structures of desloratadine 41 and clonidine 42.
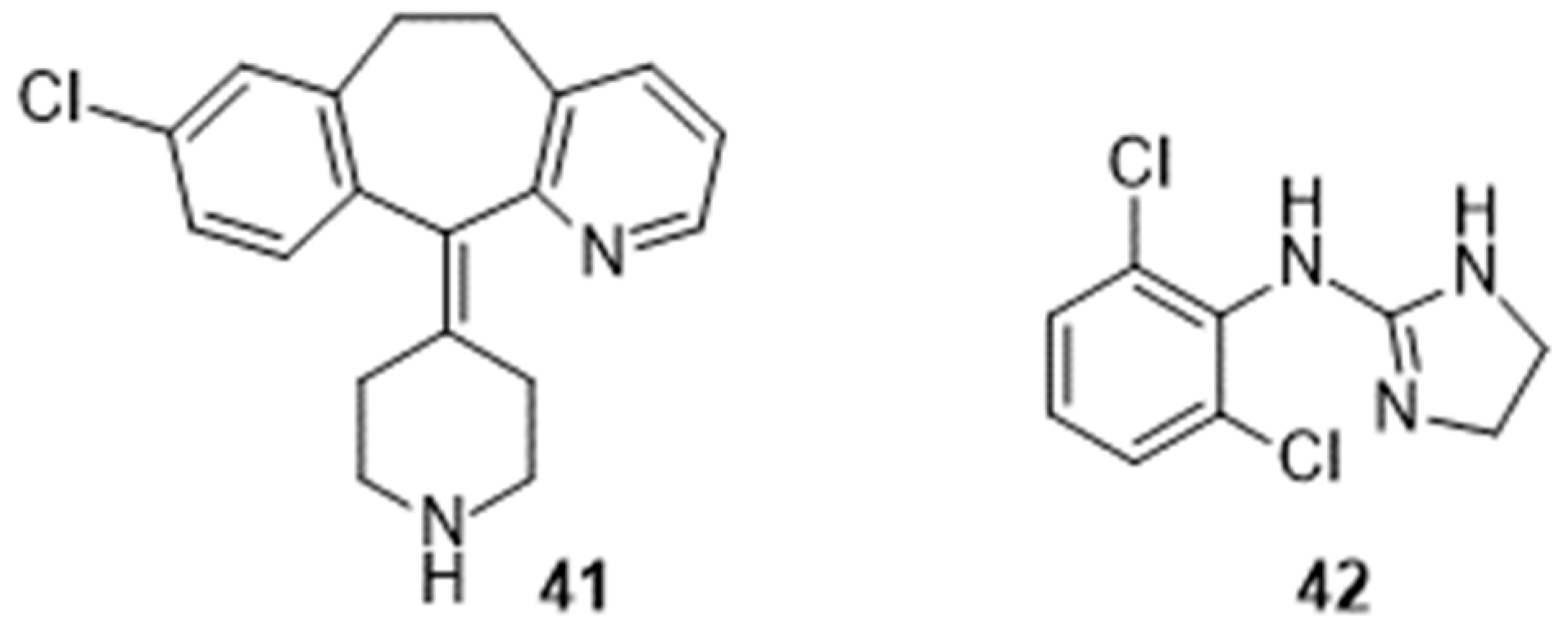
Figure 5.
Chemical structure of paroxetine 43.
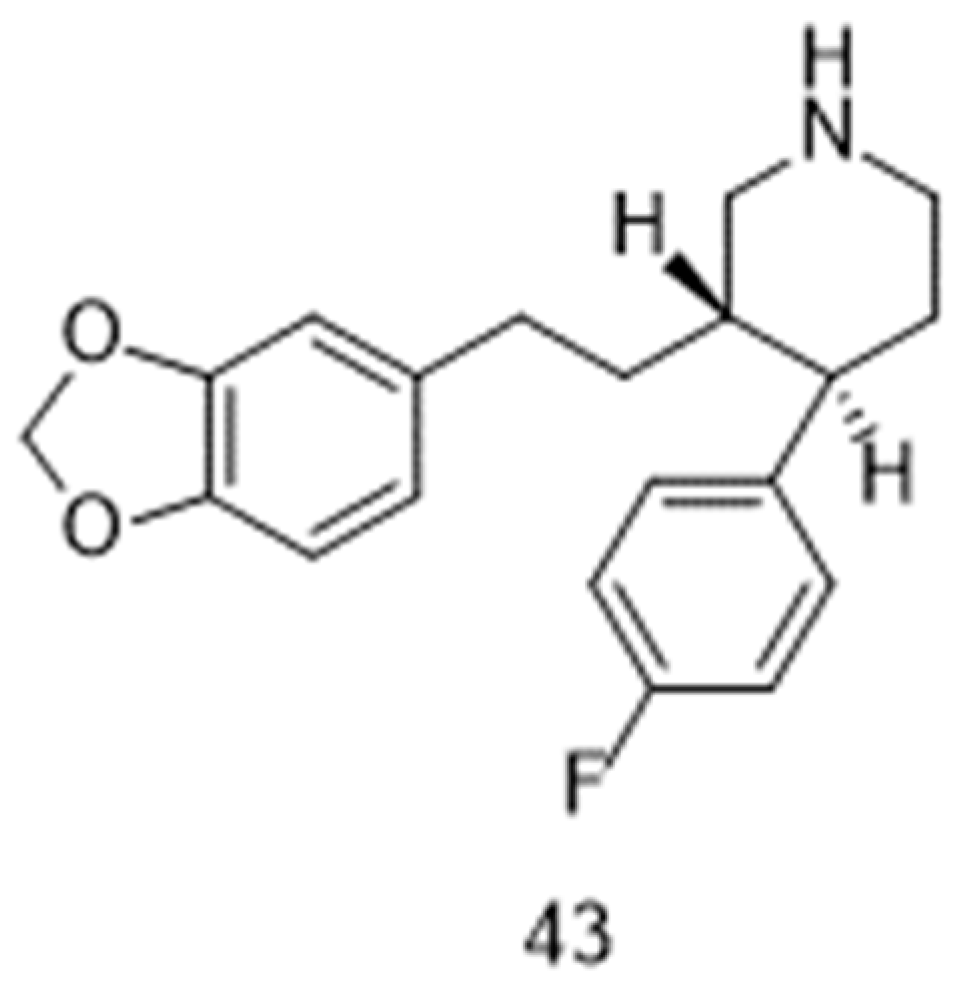
Figure 6.
Chemical structure of atomoxetine 44.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



