
Submitted:
19 September 2023
Posted:
21 September 2023
You are already at the latest version
Abstract
Aim: Complementary effects of dabigatran etexilate (DE), exercise training (ET) and combination (DE+ET) on the development and composition of the atherosclerotic lesions in diabetic apoE knockout (apoE-/-) mice. Methods: In 48 male apoE-/- diabetic mice, streptozotocin (STZ) was induced for 5 consecutive days. Mice received figh-fat diet (HFD) for 8 weeks and were randomized into 4 groups (1.Control/CG, 2.DEG: HFD with DE, 3.ETG: ET on treadmill, 4.DE+ETG: Combination DE and ET treatment). At the end of 8th week, all mice were euthanatized and morphometry of the aortic lesions at the level of aortic valve was obtained. Collagen, elastin, TNF-a, MCP-1, matrix-metalloproteinases (MMP-2,-3,-9), and TIMP-1 concentration within plaques at aortic valve were determined. Results: All active groups had significantly smaller aorta stenosis (DEG:7.9±2.2%, ETG:17.3±5.3%, DE+ETG:7.1±2.7%) compared to CG (23.3±5.5% p<0.05), reduced the relative intra-plaque concentrations of macrophages, MCP-1, MMP-3 and MMP-9, and considerably increased collagen, elastin and TIMP-1 (p<0.05). Group 4 showed the most pronounced results (p<0.05). Both DEG and DE+ETG significantly reduced MMP-2 and TNF-a concentrations compared to ETG and CG (p<0.010). Conclusion: DE and ET treatment in diabetic apoE-/- mice showed complementary amelioration of atherosclerotic lesions development and stability, mediated by the anti-inflammatory modulation of both DE and ET.
Keywords:
dabigatran etexilate
; exercise training
; atherosclerosis
; plaque stability
; matrix metalloproteinases
; inflammation
1. Introduction
Atherosclerosis-related complications are by far the leading cause of morbidity and mortality worldwide [1]. Atherosclerotic vulnerable plaques are prone to rupture and contain highly predictive information for future cardiovascular events. They are characterized by a large lipid core, thin fibrous cap, abundant inflammatory cell infiltration, inadequate elastin and collagen proteins and elevated proteolytic enzyme activity, such as matrix metalloproteinases (MMPs) [2]. The formation of atherothrombotic lesions after plaque rupture leads to devastating lumen obstruction. A plethora of studies have been published examining the impact of several pharmaceutical molecules, as potential candidates, of atherosclerosis inhibition [3,4]. Most importantly, the current preclinical research has shifted to pharmaceutical agents which may not reduce the atherosclerotic burden, but they alter the atherosclerotic plaques texture in order to make them less vulnerable and so less prone to rupture [5]. Notably, the co-existence of diabetes precipitates the atherosclerosis progression and magnifies the risk of cardiovascular events [6,7]. Therefore, therapeutic modalities targeting to prevent plaque destabilization and lumen thrombosis are of clinical importance among diabetic patients [5].
Thrombin is a pleiotropic enzyme that regulates hemostasis and nonhemostatic functions, including an array of vascular actions [8]. Physiologically, thrombin generation serves mainly to arrest bleeding, and exerts anti-coagulant properties depending on the local condition [9]. In contrast, vascular dysfunction shifts thrombin activities towards adverse thrombogenesis and inflammation. This shift may typically occur in the process of atherosclerosis [10,11]. Thus, the long-term inhibition of thrombin may create new ways of reducing atherosclerosis burden and its complications. Dabigatran etexilate (DE) is an oral, reversible, direct competitive thrombin (factor IIa) inhibitor [12]. DE has been shown as effective as warfarin in secondary venous thromboembolism (VTE) prevention and acute VTE treatment [13], with reduced major bleeding, and superior (DE 150mg b.i.d.) to warfarin in non-valvular atrial fibrillation [14]. However the impact of DE on atherosclerosis-related complications is undetermined based on current data from clinical trials [15,16]. Experimental data in transgenic, atherosclerosis-prone mice have recently supported the beneficial effects of DE on atherosclerosis development and texture [17,18]. The favorable, “pleiotropic” modification of inflammatory and oxidative mechanisms has been proposed as the underlying mechanisms, but more data are required.
Exercise training (ET) constitutes the cornerstone of primary and secondary prevention of atherosclerosis-induced complications [19]. The cardiovascular benefits derived from systemic ET are partly explained by the modulation of traditional cardiovascular risk factors (e.g. dyslipidemia, diabetes etc) [20], while additional “pleiotropic” actions may also predominantly contribute [21,22]. Multiple anti-atherosclerotic mechanisms including anti-inflammatory, anti-oxidative and other pathophysiological pathways have been proposed as the mediators for atherosclerotic lesions suppression or stabilization has been extensively proposed [23,24]. In clinical setting, atherosclerosis regression is not supported in exercise-trained participants, however, pleiotropic actions of systemic exercise can be beneficial for secondary prevention of atherosclerotic complications [25,26]. Notably, the exercise training exerts “pleiotropic” actions in diabetic animal models which attenuate plaque vulnerability [27]. Similarly, but to a lesser extent, there is indirect evidence of the atheroprotective, “pleiotropic” action of systemic ET on multiple cardiovascular risk factors in the diabetic population [28]. Among exercise-related atheroprotective mechanisms, the attenuation of coagulability has emerged as a promising one in cardiovascular disease-free adults [29]. However, data concerning the impact of exercise training on plaque stability through thrombin modification are absent.
In the present study, we hypothesized that the combination of the aforementioned therapeutic modalities (DE plus ET) would confer additive results on plaque stability compared to each intervention alone and thus their “pleiotropic” anti-inflammatory properties may advocate such a combined treatment.
2. Results
Results are listed in Table 1 and Table 2 and Figure 1, Figure 2 and Figure 3. Four mice died before the end of study (2 mice in DE+ETG, 1 mouse in DEG, 1 mouse in CG) for unexpected reasons. Those mice were replaced in order to maintain equivalent groups in statistical analysis. At the end of the study there were no significant differences in body-weight, total cholesterol, triglycerides and glucose levels between all groups (p>0.05). Exercise training conferred slight decline in the latter variables. As for fasting plasma glucose (FPG) levels, they highly increased from baseline (p<0.001). Furthermore, the magnitude of increment in FPG was marginally lower in ETG than in DEG (p=0.086) and CG (p=0.092) groups (Table 1).
2.1. Mean plaque area and plaque stability
Results are depicted in Table 2 and Figure 1. Compared to CG, all intervention groups had significantly smaller mean atherosclerotic plaque area at the aortic valve with lower percentage of lumen stenosis (p<0.001 and p<0.001, respectively). Notably, both DEG and DE+ETG groups had a significantly further reduction in atherosclerotic plaque burden compared to ETG (p<0.001), but they did not differ between them (Table 2 and Figure 1).
All intervention groups exhibited a more stable phenotype of the aortic atherosclerotic plaques characterized by a significantly higher collagen and elastin content, a thicker fibrous cap and less IEL breaks compared to CG (p<0.001). Notably, those effects were most pronounced and significant after combined treatment (DE+ET) rather than each intervention alone (p<0.05). In immunohistochemical analysis both DEG (p=0.040) and DE+ETG (p=0.023) showed higher concentrations of a-actin antibodies within plaques than CG, while the impact of ET on VSMCs was negligible (Table 2).

Table 2.
Atherosclerotic plaque area, lumen area and percentage of lumen stenosis (H&E staining), intra-plaque contents of elastin (orcein staining), collagen (sirius red staining), vascular smmoth muscle cells (VSMCs – a-actin) and macrophages (Mac-3).
Table 2.
Atherosclerotic plaque area, lumen area and percentage of lumen stenosis (H&E staining), intra-plaque contents of elastin (orcein staining), collagen (sirius red staining), vascular smmoth muscle cells (VSMCs – a-actin) and macrophages (Mac-3).
| CG (n=12) |
DEG (n=12) |
ETG (n=12) |
DE+ETG (n=12) |
p | |
|---|---|---|---|---|---|
| Plaque Area (x103 μm²) | 287.9±54.12 | 105.12±31.11a,c | 201.65±65.12a,b,d | 72.23±15.51 a,c | <0.001 |
| Lumen Area (x103 μm²) | 1323.56±265.3 | 1355.32±288.54 | 1222.1±243.51 | 1274.76±289.23 | 0.885 |
| Lumen Stenosis (%) | 23.3±5.5 | 7.9±2.2 a,c | 17.3±5.3a,b,d | 7.1±2.7a,c | <0.001 |
| Elastin (%) plaque | 9.79±2.92 | 21.62±6.52a,d | 18.91±5.07a,d | 30.24±7.72a,b,c | 0.002 |
| Collagen (%) plaque | 16.45±8.08 | 26.83±4.79 a,d | 21.44±3.1 a,d | 31.41±4.88 a,b,c | 0.001 |
| Fibrous cap thickness (μm) | 12.52±2.18 | 22.32±3.46 a,d | 19.63±3.02 a,d | 31.41±4.12 a,b,c | <0.001 |
| a-actin (VSMCs) (%) plaque | 15.91±4.98 | 23.79±5.54a | 18.51±4.31a,d | 25.22±6.18a,c | 0.039 |
| Mac-3 (macrophages) (%) plaque | 28.85±9.52 | 19.46±5.58a,d | 21.02±4.93a,d | 12.33±2.87a,b,c | <0.001 |
CG, control group; DEG, dabigatran etexilate group; ETG, exercise training group; DE+ETG, dabigatran etexilate + exercise training group. P, one-way ANOVA p value. Significant differences of each intervention group (p<0.05) compared to other groups based on post-hoc one-way ANOVA analysis: aCG, bDEG, cETG, dDE+ETG.
Figure 1.
a. All active groups (dabigatran etexilate, exercise training, combined treatment) significantly reduced plaque formation compared to controls in ApoE-/- mice. Representative images and quantifications of aortic valve sections stained with hematoxylin/eosin, across all groups.
Figure 1.
a. All active groups (dabigatran etexilate, exercise training, combined treatment) significantly reduced plaque formation compared to controls in ApoE-/- mice. Representative images and quantifications of aortic valve sections stained with hematoxylin/eosin, across all groups.
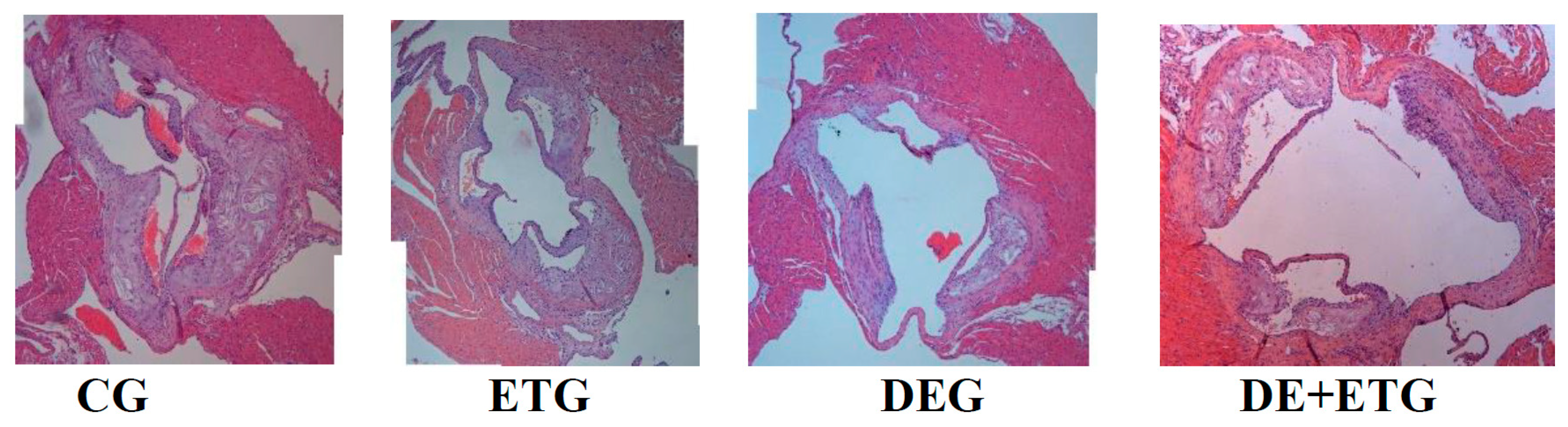
Figure 1.
b. All active groups (dabigatran etexilate, exercise training, combined treatment) significantly enhanced collagen and elastin concentrations compared to controls in ApoE-/- mice. Representative images of aortic valve sections stained with sirius-red (collagen) across all groups.
Figure 1.
b. All active groups (dabigatran etexilate, exercise training, combined treatment) significantly enhanced collagen and elastin concentrations compared to controls in ApoE-/- mice. Representative images of aortic valve sections stained with sirius-red (collagen) across all groups.

Section thickness was set at 5μm and original magnification at 100x. We analyzed 2-3 sections of each mouse (6 mice per group). CG, control group; DEG, dabigatran etexilate group; ETG, exercise training group; DE+ETG, dabigatran etexilate + exercise training group
2.2. Inflammatory mediators
In comparison to CG, all three intervention groups appeared with significantly reduced positive-stained atherosclerotic areas at the aortic valve for: macrophages (Mac-3 immunostaining, p<0.001), MMP-3 (p<0.05), MMP-9 (p<0.001), and MCP-1 (p<0.001). Notably, the most pronounced reduction in the above parameters was demonstrated in the DE+ETG rather than DEG and ETG (p<0.05). Both MMP-2 and TNF-a concentrations were reduced to a similar degree in DEG and DE+ETG compared to ETG and CG (p<0.010), implicating that DE administration predominantly drive the MMP-2 and TNF-a reduction. Inversely, in all intervention groups we observed increased intra-plaque levels of TIMP-1 compared to CG (p<0.05), while the amount of increment was greatest with combined interventions (DE+ET) compared to ET (p=0.018) or DE (p=0.025) therapy alone. The calculated MMP-9/TIMP-1 ratio was considerably decreased across all intervention groups (p<0.05). The aforementioned results are depicted in Figure 2 and Figure 3.
Figure 2.
Quantification of immunohistochemical staining with antibodies against MMP-2, MMP-3, MMP-9, TIMP-1, TNF-a, MCP-1. *p<0.05 vs CG, #p<0.05 vs DEG, ||p<0.05 vs ETG.
Figure 2.
Quantification of immunohistochemical staining with antibodies against MMP-2, MMP-3, MMP-9, TIMP-1, TNF-a, MCP-1. *p<0.05 vs CG, #p<0.05 vs DEG, ||p<0.05 vs ETG.
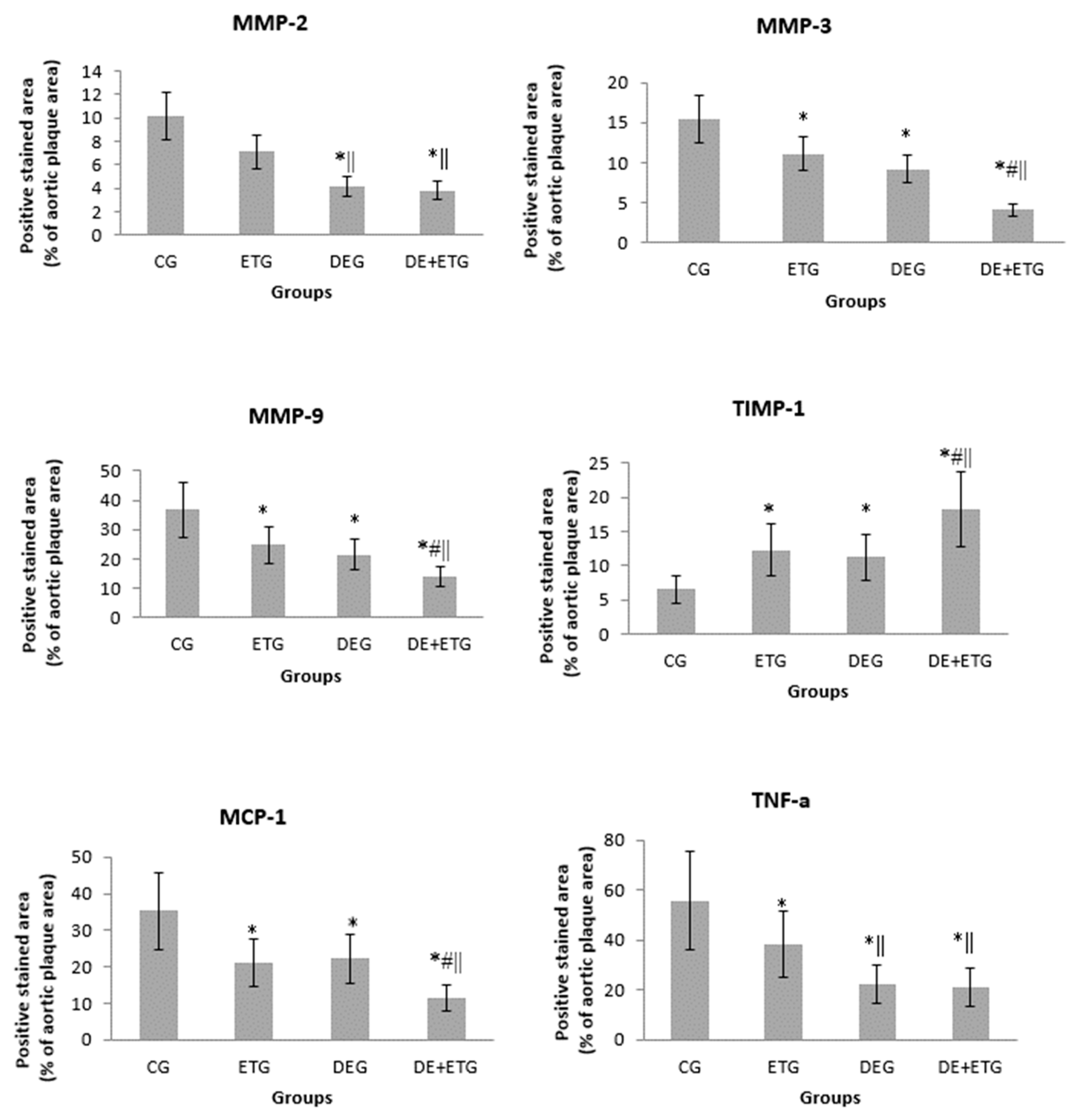
Figure 3.
Representative images of plaques at aortic valve sections stained with MMP-9 and MCP-1 across all groups.
Figure 3.
Representative images of plaques at aortic valve sections stained with MMP-9 and MCP-1 across all groups.
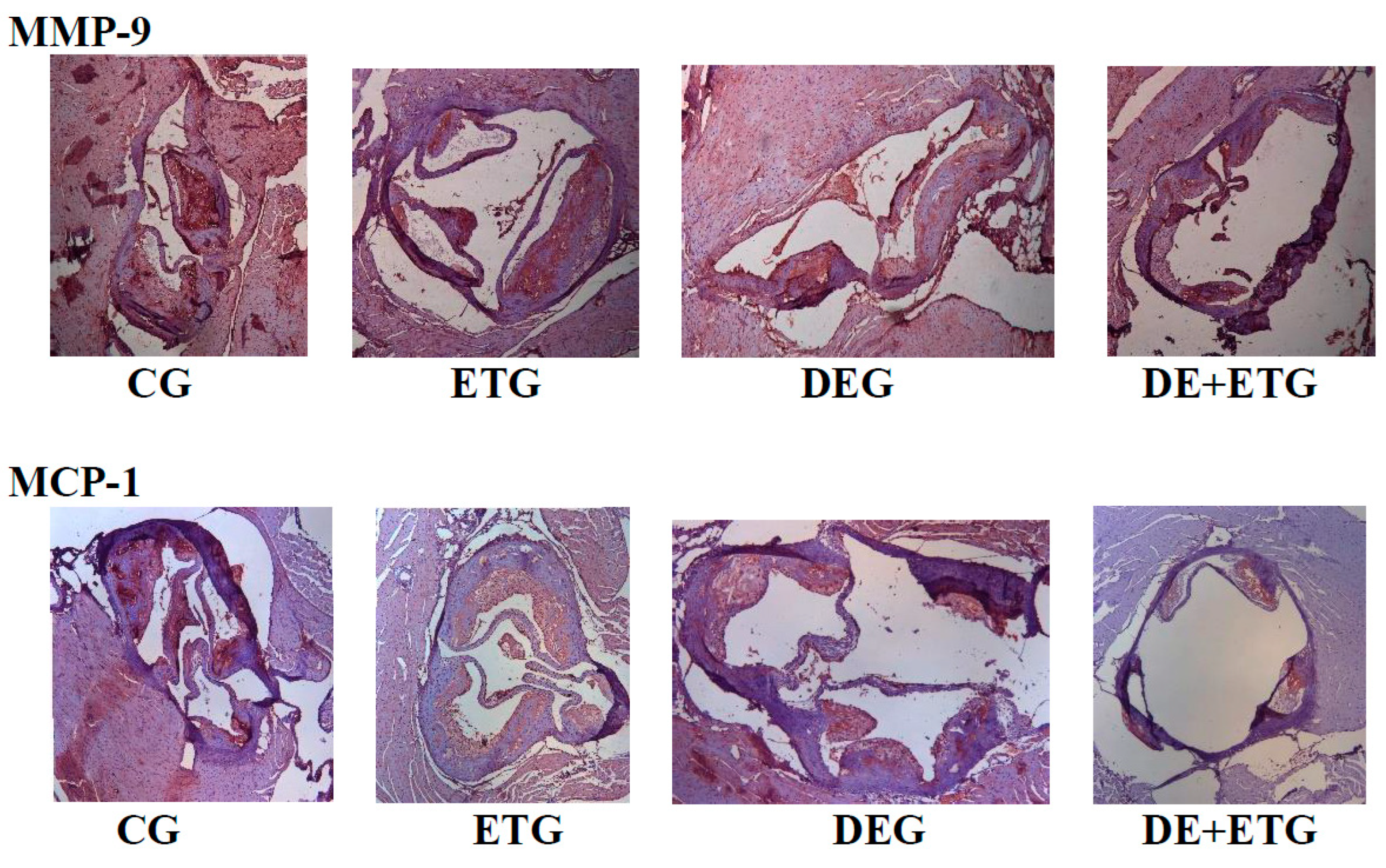
Section thickness was set at 5μm and original magnification at 100x. We analyzed 2-3 sections of each mouse (6 mice per group). CG, control group; DEG, dabigatran etexilate group; ETG, exercise training group; DE+ETG, dabigatran etexilate + exercise training group
3. Discussion
In our study, all three interventions (DE, ET, DE+ET) considerably suppressed atherosclerotic lesions development compared to untreated diabetic mice after 8-week therapy. Importantly, the stability of the atherosclerotic lesions was improved by all interventions, with the combined treatment (DE+ET) conferring the most pronounced results. The latter was attributed to the additive anti-inflammatory effects of ET and DE, while alterations in body weight and lipid profile were negligible across all groups.
As expected, we confirmed our previous data about the suppressive effects of either ET or direct thrombin inhibition on atherosclerosis progression [18,30]. The combination of DE and ET did not further decrease atherosclerosis burden, presumably due to the exaggerated atherosclerosis regression already achieved after DE administration. Plaque regression after ET was significant, but modest in agreement to our previous studies [17,27]. It is worth mentioning that the aforementioned results were observed despite the absence of any significant change in lipid levels and body weight. Three previously published studies have demonstrated a complex interplay between thrombin and atherosclerosis, using the same animal model of hypercholesterolemic mice [18,19,31]. In those studies, the direct inhibition of thrombin counteracted its pro-inflammatory, oxidative and pro-atherogenic actions, independent of lipid alterations. In the present study, DE and ET therapy inhibited not only plaque progression but also destabilization, by enhancing collagen and elastin contents, fibrous cap thickness and suppressing IEL ruptures. It is well-known that MMPs regulate extracellular matrix (ECM) remodelling and inflammatory cells infiltration [32], the mainstays of atherosclerosis development [33,34]. Thus, anti-inflammatory therapies targeting the inhibition of MMPs and inflammation have the potentiality of anti-atherosclerotic benefits [35]. Atherosclerosis regression after thrombin inhibition has been demonstrated only in animal models [36,37]. Clinical trials assessing atherosclerosis progression have significant disadvantages, like slow atherosclerosis progression, repeated expensive imaging modalities to detect any change in atherosclerosis burden, multifactorial pharmaceutical therapy confounding results [38,39,40]. The experimental atherosclerosis regression is resounding, but its extrapolation to the clinical setting is not feasible.
Atherosclerotic plaque texture predominantly determines the constant interaction between blood and plaque and the consequent atherothrombotic events [41]. In our study, all the examined parameters culminated in plaque stability, like elastin, collagen, inflammatory agents (macrophages TNF-a, MCP-1), and MMPs/TIMP-1 [42], were positively affected by each therapeutic modality, producing a more stable phenotype. We and other investigators have demonstrated the strong contribution of inflammation to atherosclerosis vulnerability [43,44]. The balance of proteolytic and anti-proteolytic enzymes (MMPs/TIMPs) has been long studied as a surrogate marker of plaque vulnerability. To our knowledge, this is the first study demonstrating the complementary, favorable changes in most of the aforementioned parameters of plaque stability after combined DE+ET therapy. In particular, the combination of DE and ET increased the intra-plaque concentration of collagen and elastin contents in an additive pattern. That effect was associated with a significant reduction in proteolysis expressed by downregulated MMPs (MMP-2,-3,-9) and increased TIMP-1 levels in all active groups. In addition to this, DE administration rather than ET stimulated the plaque infiltration by VSMCs, the cellular sources of the ECM [45]. Overall, the combined treatment magnified the derived benefits of each intervention (DE and ET) by shifting the balance between production and degradation of ECM in established atherosclerotic lesions towards increased intra-plaque collagen and elastin [46]. The observed complementary effects provide a clinical message of combining pharmaceutical inhibition of thrombin with life-style changes like exercise training.
We also noticed a complementary pattern after DE and ET in the modulation of inflammatory mediators, with the exception of MMP-2 and TNF-a. Experimental and clinical data have supported the relationship of inflammation with either low physical activity or thrombin action [47,48]. Previous studies have reported reduction of two multifunctional inflammatory agents, TNF-a [49] and MCP-1 [50] after exercise intervention. The impact of thrombin inhibitors on them is still unknown. In our study, single therapy with either DE or ET inhibited macrophages infiltration and decreased macrophage-derived atherogenic cytokines, like MCP-1 and TNF-a, within plaques [51]. The combination therapy seemed to precipitated further the suppression of MCP-1 and TNF-a and the consequent plaque stabilization with the highest, elastin, collagen and VSMCs contents and the lowest macrophages concentration. Plaque stabilization has been targeted by many pharmaceutical interventions. The clinical relevance of our preliminary data remains to be proved.
Macrophages are widely-recognized as the cornerstone of atherogenesis and the predominant cellular sources of MMPs [36]. Thus, the observed decrease in macrophages number provides a plausible explanation for the proportional reduction in protein concentration of MMP-2, MMP-3 and MMP-9 across our groups. The involvement of the latter MMP members in atherosclerotic plaque vulnerability is well-documented [52,53]. On the other hand, the wide-spectrum MMP inhibitor, TIMP-1, has been extensively reported to contribute to atherosclerotic plaque stability [54]. Therefore, the alteration in MMPs/TIMP-1 ratio to less proteolytic activity strongly supports a predominant atheroprotective mechanism. Any intervention which alters this balance in favor of less MMPs and higher TIMPs activity has been associated with more stable plaque phenotype and less adverse cardiovascular events [55,56]. Although we have previously demonstrated the beneficial effect of each intervention alone (ET or DE) on MMPs/TIMP-1 equilibrium [12,19], this is the first study where combined therapy confirmed their additive effects on it [18,24]. Those pleiotropic changes after combined intervention are quite promising for optimal ECM homeostasis, but they require unambiguously further validation.
The present study had several drawbacks. First of all, the histochemistry-based measurements provide a relative quantification of molecular concentration, but do not directly express the absolute protein activity and they do not reflect gene expression. Secondly, the estimation of plaque vulnerability is commonly indirect in apoE-/- mice fed HFD. A number of parameters associated with plaque texture are mostly used in this animal model, since plaque rupture is rarely observed in their atherosclerotic lesions. Nevertheless, apoE-/- mouse plaque morphology shows great similarities with that in humans. Third, we observed some unexpected deaths of DE-treated mice but we were unable to clarify the underlying reason. The detrimental or toxic effects of thrombin inhibition was not the object of the present study.
4. Conclusions
In conclusion, combined DE and ET treatment in diabetic atherosclerotic apoE-/- mice showed additive beneficial effects on most parameters of atherosclerotic lesion stability. Combined treatment yielded to a more stable plaque phenotype, which theoretically would have reduced the risk of rupture. Those effects paralleled a favourable modification of inflammatory mediators implicating a strong impact of both DE and ET on those crucial parameters of atherosclerosis progression. Our findings outline the clinical importance of pharmaceutical intervention accompanied with lifestyle alteration in the management of the atherosclerotic complications.
5. Materials and Methods
5.1. Study design
Forty-eight male mice with homozygous deficiency in apolipoprotein-E (apo-E-/-, C57BL/6J background), aged 8 weeks, were fed a western-type high-fat diet (HFD - Harlan, Teklad; 88137) for 8 weeks, in order to develop atherosclerotic lesions. Diabetes was induced at the beginning of the study by peritoneal injections of streptozotocin (STZ) for 5 consecutive days (0.05 mg/g body weight in 0.05 mol/L citrate buffer, pH 4.5). Mice maintaining fasting glucose levels >200 mg/dL throughout the course of the study were considered diabetic and were included in analyses. This is a valid animal model for diabetic atherosclerosis development widely used in pharmaceutical studies. Before beginning the study, mice were randomized to the following equivalent groups (n=12): 1) CG: No intervention was performed and served as controls. 2) DEG: HFD was supplemented with DE (10mg DE/g chow) for the whole study period. 3) ETG: Mice underwent ET on treadmill (Exer-6M Open Treadmill, Columbus Instruments, USA). Mice were gradually accustomed to the training program (5 times/week, 60 min/session, velocity:12m/min, slope: 5o). Some mechanical and electrical shock-plate stimuli were used during the first session on the treadmill, this was required to different degrees in individual mice[12]. 4) DE+ETG: Combination of DE and ET treatment, as described previously. All mice were euthanized at the end of the study (8th week) under isoflurane deep anesthesia. Diets were provided by Boehringer Ingelheim, Biberach, Germany. As we have previously reported, the daily absorbance of active substance was calculated to be ~2.3 mg of DE [18]. Just before euthanasia, body weights were measured and blood samples were obtained.
All the experiments were performed with prior approval by the Ethics Committee for Animal Experimentation of the Biomedical Research Foundation, Academy of Athens and the competent Veterinary Service, according to the European Directive 2010/63 (Permit number:43029201). All surgeries were performed under isoflurane anesthesia to minimize suffering. The animals were kept in a 12hr light/12 hr dark environment under constant temperature of 21οC, and were handled in accordance to the relevant international guidelines for the proper care and use of laboratory animals.
5.2. Glucose tolerance test
One week before euthanasia, 6-7 mice per group underwent an intraperitoneal glucose tolerance test (GTT). After an overnight fasting, anesthetized mice were intra-peritoneally injected glucose (D-Glucose 50% wt/vol solution) with a 27-gauge needle at a dose of 2 g/kg body weight. Blood samples were taken before glucose injection and after 30, 60, 90, and 120 min. Blood glucose was measured at each time point by Accu-Chek advantage glucose monitors (Roche Diagnostics, Indianapolis, IN, USA) and the area under the curve (AUC) was then determined using the trapezoid rule.
5.3. Histology
After euthanasia, the heart along with the aortic root was perfused with normal saline via heart puncture. the aortic root was excised and fixed in 10% buffered formalin overnight and then embedded in paraffin blocks. The procedure of histomorphometric analysis has been previously described [57]. In a microtome, serial sectioning (5μm thickness) started when aortic valve leaflets become apparent. For each staining per mouse, 3 nonconsecutive aortic slices (at equal intervals of 20μm intervals) were mounted on poly-D-lysine-coated slides following a standardized protocol which allowed the colocalization for the various measured variables. For quantitative morphometric analysis, we stained cross-sections with hematoxylin/eosin (H&E), sirius red and orcein, according to a standardized protocol [28]. Finally, by means of immunohistochemistry, we used antibodies for the staining of the corresponding antigens: monocytes chemoattractant protein-1 (MCP-1), tumor necrosis factor-a (TNF-a), MMP-2, MMP-3, MMP-9 (MBL, International Corporation, Woburn, MA, USA), Mac-3 antigen of murine macrophages (BD Pharmigen, Franklin Lakes, NJ, USA), alpha-smooth muscle isoform of actin (Biocare Medical, LLC, Concord, CA, USA) and TIMP-1 (Acris Antibodies GmbH, Herford, Germany).
5.4. Digital processing -Histomorphometry
All sections from the aortic valves were observed under x10 magnification in a bright-field microscope (Leica DM LS2, Wetzlar, Germany) and digital pictures were acquired and stored in a lossless format using a Leica DC 500 microscope camera (Heerbrugg, Switzerland) and “Altra 20 Soft Imaging System” computer software. In H&E-stained sections, we measured the extent of the atherosclerotic plaques (in μm2) and the total lumen area (in μm2) circumscribed by the internal elastic lamina (IEL). The proportion of the total lumen area occupied by the atherosclerotic plaques expressed the percentage of luminal stenosis, in each section. For the quantification of lesions in aortic sections per animal, we averaged plaque area and the luminal stenosis. In sirius-red and orcein-stained sections, we measured the relative concentrations of collagen and elastin, respectively, the fibrous cap thickness of each atherosclerotic plaque and the number of ruptures (i.e. discontinuities or fractures) of the IEL. We then averaged the values of all plaques per animal. For the measurement of the relative concentrations of the stained molecules by immunohistochemistry, the segmental stained plaque area was expressed as the percentage of the whole atherosclerotic plaque area [18]. We then averaged the values of all plaques per animal and then for each group.
5.5. Blood analyses
We drew all blood samples after an overnight fasting under isoflurane anaesthesia, via gastrocnemius muscle puncture. Fasting glucose, triglycerides and total cholesterol plasma levels were immediately assayed in an automatic enzymatic analyzer (Olympus AU560, Hamburg, Germany).
5.6. Statistical analysis
The results of the study are presented as mean values ± standard deviation. Normality of distribution was assessed using the Shapiro-Wilk test. For continuous variables, the comparisons within and between groups was made using paired samples t-test and one way ANOVA and post-hoc Tukey test, respectively. A two-tailed p value <0.05 was considered statistically significant. All statistical analyses were based on SPSS v26.0 (IBM, Armonk, NY, USA).
Author Contributions
N.K.: conceptualization, methodology, project administrator, funding acquisition, experiments; M.S.: Experiments, resources, data curation; E.G.: investigation, writing review and editing; E.C.: Experiments, resources, data curation; N.K.: technical support, resources; G.V.: conceptualization, supervision.
Funding
Nikolaos P.E. Kadoglou was awarded a grant by the Greek State Scholarship’s Foundation: EL BIO 003. This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Prefecture of Attika, Veterinary Service (K/3726, 1/7/2010).
Informed Consent Statement
Not applicable.
Acknowledgments
For Michail Katsimpoulas and Petros Moustardas for their technical support. We thank Boehringer Ingelheim for providing the substance of dabigatran etexilate and facilitating lab testing for our research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Moreno, P.R. Vulnerable plaque: Definition, diagnosis, and treatment. Cardiol Clin. 2010, 28, 1–30. [Google Scholar] [CrossRef]
- Christodoulou, E.; Kadoglou, N.P.E.; Stasinopoulou, M.; Konstandi, O.A.; Kenoutis, C.; Kakazanis, Z.I.; Rizakou, A.; Kostomitsopoulos, N.; Valsami, G. Crocus sativus L. aqueous extract reduces atherogenesis, increases atherosclerotic plaque stability and improves glucose control in diabetic atherosclerotic animals. Atherosclerosis. 2018, 268, 207–214. [Google Scholar] [CrossRef]
- Kadoglou, N.P.E.; Stasinopoulou, M. How to Use Statins in Secondary Prevention of Atherosclerotic Diseases: From the Beneficial Early Initiation to the Potentially Unfavorable Discontinuation. Cardiovasc Drugs Ther. 2023, 37, 353–362. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Khattab, E.; Velidakis, N.; Patsourakos, N.; Lambadiari, V. A new approach of statin therapy in carotid atherosclerosis: Targeting indices of plaque vulnerability on the top of lipid-lowering. A narrative review. Kardiol Pol. 2022, 80, 880–890. [Google Scholar] [CrossRef]
- Kadoglou, N.P.E.; Korakas, E.; Lampropoulos, S.; Maratou, E.; Kassimis, G.; Patsourakos, N.; Plotas, P.; Moutsatsou, P.; Lambadiari, V. Plasma nesfatin-1 and DDP-4 levels in patients with coronary artery disease: Kozani study. Cardiovasc Diabetol. 2021, 20, 166. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.E.; Kapetanios, D.; Korakas, E.; Valsami, G.; Tentolouris, N.; Papanas, N.; Lambadiari, V.; Karkos, C. Association of serum levels of osteopontin and osteoprotegerin with adverse outcomes after endovascular revascularisation in peripheral artery disease. Cardiovasc Diabetol. 2022, 21, 171. [Google Scholar] [CrossRef] [PubMed]
- Smyth, S.S.; McEver, R.P.; Weyrich, A.S.; Morrell, C.N.; Hoffman, M.R.; et al. Platelet Colloquium Participants. Platelet functions beyond hemostasis. J Thromb Haemost. 2009, 7, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.A.; Philippou, H.; Huntington, J.A. Directing thrombin. Blood. 2005, 106, 2605–2612. [Google Scholar] [CrossRef]
- Seehaus, S.; Shahzad, K.; Kashif, M.; Vinnikov, I.A.; Schiller, M.; Wang, H.; et al. Hypercoagulability inhibits monocyte transendothelial migration through protease-activated receptor-1-, phospholipase-Cbeta-, phosphoinositide 3-kinase-, and nitric oxide-dependent signaling in monocytes and promotes plaque stability. Circulation. 2009, 120, 774–784. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Spronk, H.M.; Heeneman, S.; ten Cate, H. Is thrombin a key player in the 'coagulation-atherogenesis' maze? Cardiovasc Res. 2009, 82, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Wienen, W.; Stassen, J.M.; Priepke, H.; Ries, U.J.; Hauel, N. In-vitro profile and ex-vivo anticoagulant activity of the direct thrombin inhibitor dabigatran and its orally active prodrug, dabigatran etexilate. Thromb Haemost. 2007, 98, 155–162. [Google Scholar]
- Schulman, S.; Kearon, C.; Kakkar, A.K.; Mismetti, P.; Schellong, S.; RE-COVER Study Group; et al. (2009) Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009, 361, 2342–2352. [Google Scholar] [CrossRef]
- Schirmer, S.H.; Baumhäkel, M.; Neuberger, H.R.; Hohnloser, S.H.; van Gelder, I.C.; et al. Novel anticoagulants for stroke prevention in atrial fibrillation: Current clinical evidence and future developments. J Am Coll Cardiol. 2010, 56, 2067–2076. [Google Scholar] [CrossRef]
- Oldgren, J.; Budaj, A.; Granger, C.B.; Khder, Y.; Roberts, J.; RE-DEEM Investigators; et al. Dabigatran vs. placebo in patients with acute coronary syndromes on dual antiplatelet therapy: A randomized, double-blind, phase II trial. Eur Heart J. 2011, 32, 2781–2789. [Google Scholar] [CrossRef]
- Uchino, K.; Hernandez, A.V. Dabigatran association with higher risk of acute coronary events: Meta-analysis of noninferiority randomized Controlled Trials. Arch Intern Med. 2012, 172, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.; Moustardas, P.; Katsimpoulas, M.; Kapelouzou, A.; Kostomitsopoulos, N.; et al. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice: Dabigatran etexilate and atherosclerosis. Cardiovasc Drugs Ther. 2012, 26, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.O.; Kratz, M.T.; Schirmer, S.H.; Baumhäkel, M.; Böhm, M. The effects of direct thrombin inhibition with dabigatran on plaque formation and endothelial function in apolipoprotein E-deficient mice. J Pharmacol Exp Ther. 2012, 343, 253–257. [Google Scholar] [CrossRef]
- Swift, D.L.; Lavie, C.J.; Johannsen, N.M.; Arena, R.; Earnest, C.P.; et al. Physical activity, cardiorespiratory fitness, and exercise training in primary and secondary coronary prevention. Circ J. 2013, 77, 281–292. [Google Scholar] [CrossRef]
- Teixeira-Lemos, E.; Nunes, S.; Teixeira, F.; Reis, F. Regular physical exercise training assists in preventing type 2 diabetes development: Focus on its antioxidant and anti-inflammatory properties. Cardiovasc Diabetol. 2011, 10, 12. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Iliadis, F.; Liapis, C.D. Exercise and carotid atherosclerosis. Eur J Vasc Endovasc Surg. 2008, 35, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.; Vrabas, I.S.; Kapelouzou, A.; Angelopoulou, N. The association of physical activity with novel adipokines in patients with type 2 diabetes. Eur J Intern Med. 2012, 23, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.E.; Stasinopoulou, M.; Christodoulou, E.; Valsami, G.; Kostomitsopoulos, N. Exercise training inhibits atherosclerosis progression and reduces VE-cadherin levels within atherosclerotic plaques in hypercholesterolemic mice. Biochem Biophys Res Commun. 2022, 623, 39–43. [Google Scholar] [CrossRef]
- Nakos, I.; Kadoglou, N.P.E.; Gkeka, P.; Tzallas, A.T.; Giannakeas, N.; Tsalikakis, D.G.; Katsimpoulas, M.; Mantziaras, G.; Kostomitsopoulos, N.; Liapis, C.D.; Kakisis, J. Exercise Training Attenuates the Development of Cardiac Autonomic Dysfunction in Diabetic Rats. In Vivo. 2018, 32, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Papagianni, G.; Panayiotou, C.; Vardas, M.; Balaskas, N.; Antonopoulos, C.; Tachmatzidis, D.; Didangelos, T.; Lambadiari, V.; Kadoglou, N.P.E. The anti-inflammatory effects of aerobic exercise training in patients with type 2 diabetes: A systematic review and meta-analysis. Cytokine. 2023, 164, 156157. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Fotiadis, G.; Kapelouzou, A.; Kostakis, A.; Liapis, C.D.; Vrabas, I.S. The differential anti-inflammatory effects of exercise modalities and their association with early carotid atherosclerosis progression in patients with type 2 diabetes. Diabet Med. 2013, 30, e41–e50. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Moustardas, P.; Kapelouzou, A.; Katsimpoulas, M.; Giagini, A.; Dede, E.; Kostomitsopoulos, N.; Karayannacos, P.E.; Kostakis, A.; Liapis, C.D. The anti-inflammatory effects of exercise training promote atherosclerotic plaque stabilization in apolipoprotein E knockout mice with diabetic atherosclerosis. Eur J Histochem. 2013, 57, e3. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Vrabas, I.S.; Kapelouzou, A.; Angelopoulou, N. The association of physical activity with novel adipokines in patients with type 2 diabetes. Eur J Intern Med. 2012, 23, 137–142. [Google Scholar] [CrossRef]
- Lockard, M.M.; Gopinathannair, R.; Paton, C.M.; Phares, D.A.; Hagberg, J.M. Exercise training-induced changes in coagulation factors in older adults. Med Sci Sports Exerc. 2007, 39, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.; Kostomitsopoulos, N.; Kapelouzou, A.; Moustardas, P.; Katsimpoulas, M.; et al. Effects of exercise training on the severity and composition of atherosclerotic plaque in apoE-deficient mice. J Vasc Res. 2011, 48, 347–356. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Otten, J.J.; Heeneman, S.; Leenders, P.; van Oerle, R.; Soehnlein, O.; et al. Genetic and pharmacological modifications of thrombin formation in apolipoprotein e-deficient mice determine atherosclerosis severity and atherothrombosis onset in a neutrophil-dependent manner. PLoS ONE. 2013, 8, e55784. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N. Vascular extracellular matrix in atherosclerosis. Cardiol Rev. 2013, 21, 270–288. [Google Scholar] [CrossRef] [PubMed]
- Siasos, G.; Tousoulis, D.; Kioufis, S.; Oikonomou, E.; Siasou, Z.; Limperi, M.; et al. Inflammatory mechanisms in atherosclerosis: The impact of matrix metalloproteinases. Curr Top Med Chem. 2012, 12, 1132–1148. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Daskalopoulou, S.S.; Perrea, D.; Liapis, C.D. Matrix metalloproteinases and diabetic vascular complications. Angiology 2005, 56, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Ogita, M.; Miyauchi, K.; Morimoto, T.; Daida, H.; Kimura, T.; Hiro, T.; et al. Association between circulating matrix metalloproteinase levels and coronary plaque regression after acute coronary syndrome--subanalysis of the JAPAN-ACS study. Atherosclerosis. 2013, 226, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Li, K.; Festenstein, S.; Karegli, J.; Wilkinson, H.; Leonard, H.; Wei, L.L.; Ma, N.; Xia, M.; Tam, H.; Wang, J.A.; Xu, Q.; McVey, J.H.; Smith, R.A.G.; Dorling, A. Regression of Atherosclerosis in ApoE-/- Mice Via Modulation of Monocyte Recruitment and Phenotype, Induced by Weekly Dosing of a Novel "Cytotopic" Anti-Thrombin Without Prolonged Anticoagulation. J Am Heart Assoc. 2020, 9, e014811. [Google Scholar] [CrossRef] [PubMed]
- Posthuma, J.J.; Posma, J.J.N.; van Oerle, R.; Leenders, P.; van Gorp, R.H.; Jaminon, A.M.G.; Mackman, N.; Heitmeier, S.; Schurgers, L.J.; Ten Cate, H.; Spronk, H.M.H. Targeting Coagulation Factor Xa Promotes Regression of Advanced Atherosclerosis in Apolipoprotein-E Deficient Mice. Sci Rep. 2019, 9, 3909. [Google Scholar] [CrossRef]
- Ishikawa, H.; Otsuka, K.; Kono, Y.; Hojo, K.; Yamaura, H.; Hirata, K.; Kasayuki, N.; Izumiya, Y.; Fukuda, D. Extent of coronary atherosclerosis is associated with deterioration of left ventricular global longitudinal strain in patients with preserved ejection fraction undergoing coronary computed tomography angiography. Int J Cardiol Heart Vasc. 2023, 44, 101176. [Google Scholar] [CrossRef]
- Huang, Q.; Liu, Z.; Wei, M.; Huang, Q.; Feng, J.; Liu, Z.; Xia, J. The atherogenic index of plasma and carotid atherosclerosis in a community population: A population-based cohort study in China. Cardiovasc Diabetol. 2023, 22, 125. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Pavlidis, G.; Kadoglou, N.; Makavos, G.; Katogiannis, K.; Kountouri, A.; Thymis, J.; Kostelli, G.; Kapniari, I.; Theodoropoulos, K.; Parissis, J.; Katsimbri, P.; Papadavid, E.; Lambadiari, V. Apremilast Improves Endothelial Glycocalyx Integrity, Vascular and Left Ventricular Myocardial Function in Psoriasis. Pharmaceuticals (Basel). 2022, 15, 172. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Sailer, N.; Moumtzouoglou, A.; Kapelouzou, A.; Gerasimidis, T.; Kostakis, A.; Liapis, C.D. Adipokines: A novel link between adiposity and carotid plaque vulnerability. Eur J Clin Invest. 2012, 42, 1278–1286. [Google Scholar] [CrossRef]
- Shah, P.K. Inflammation and plaque vulnerability. Cardiovasc Drugs Ther 2009, 23, 31–40. [Google Scholar] [CrossRef]
- Mitsis, A.; Kadoglou, N.P.E.; Lambadiari, V.; Alexiou, S.; Theodoropoulos, K.C.; Avraamides, P.; Kassimis, G. Prognostic role of inflammatory cytokines and novel adipokines in acute myocardial infarction: An updated and comprehensive review. Cytokine. 2022, 153, 155848. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.E.; Korakas, E.; Karkos, C.; Maratou, E.; Kanonidis, I.; Plotas, P.; Papanas, N.; Moutsatsou, P.; Ikonomidis, I.; Lambadiari, V. The prognostic role of RBP-4 and adiponectin in patients with peripheral arterial disease undergoing lower limb endovascular revascularization. Cardiovasc Diabetol. 2021, 20, 221. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, I.; Sciangula, A.; Perrotta, E.; Donato, G.; Cassese, M. Ultrastructural analysis and electron microscopic localization of Nox4 in healthy and atherosclerotic human aorta. Ultrastruct Pathol. 2011, 35, 1–6. [Google Scholar] [CrossRef]
- Wang, Y.; Johnson, J.A.; Fulp, A.; Sutton, M.A.; Lessner, S.M. Adhesive strength of atherosclerotic plaque in a mouse model depends on local collagen content and elastin fragmentation. J Biomech. 2013, 46, 716–722. [Google Scholar] [CrossRef]
- Kalz, J.; Ten Cate, H.; Spronk, H.M. Thrombin generation and atherosclerosis. J Thromb Thrombolysis. 2014, 37, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J.; Martinez, J.; Grammas, P. Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front Aging Neurosci. 2013, 5, 19. [Google Scholar] [CrossRef]
- Pina-Canseco Mdel, S.; Páez-Arenas, A.; Massó, F.; Pérez-Campos, E.; Martínez-Cruz, R.; Hernández-Cruz, P.; Majluf-Cruz, A.; et al. Protein C activation peptide inhibits the expression of ICAM-1, VCAM-1, and interleukin-8 induced by TNF-α in human dermal microvascular endothelial cells. Folia Histochem Cytobiol. 2012, 50, 407–413. [Google Scholar] [CrossRef]
- Wasinski, F.; Bacurau, R.F.; Moraes, M.R.; Haro, A.S.; Moraes-Vieira, P.M.; Estrela, G.R.; et al. Exercise and caloric restriction alter the immune system of mice submitted to a high-fat diet. Mediators Inflamm. 2013, 395672. [Google Scholar] [CrossRef]
- Bhaskar, V.; Yin, J.; Mirza, A.M.; Phan, D.; Vanegas, S.; Issafras, H.; et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis. 2011, 216, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.H.; Cho, C.H.; Kim, H.O.; Jo, Y.H.; Yoon, K.S.; Lee, J.H.; et al. Plaque rupture is a determinant of vascular events in carotid artery atherosclerotic disease: Involvement of matrix metalloproteinases 2 and 9. J Clin Neurol. 2011, 7, 69–76. [Google Scholar] [CrossRef]
- Kadoglou, N.P.E.; Stasinopoulou, M.; Christodoulou, E.; Valsami, G.; Kostomitsopoulos, N. Exercise training inhibits atherosclerosis progression and reduces VE-cadherin levels within atherosclerotic plaques in hypercholesterolemic mice. Biochem Biophys Res Commun. 2022, 623, 39–43. [Google Scholar] [CrossRef] [PubMed]
- de Vries, M.R.; Niessen, H.W.; Löwik, C.W.; Hamming, J.F.; Jukema, J.W.; Quax, P.H. (2012) Plaque rupture complications in murine atherosclerotic vein grafts can be prevented by TIMP-1 overexpression. PLoS ONE. 2012, 7, e47134. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Sailer, N.; Fotiadis, G.; Kapelouzou, A.; Liapis, C.D. The impact of type 2 diabetes and atorvastatin treatment on serum levels of MMP-7 and MMP-8. Exp Clin Endocrinol Diabetes. 2014, 122, 44–49. [Google Scholar] [CrossRef]
- Giagtzidis, I.T.; Kadoglou, N.P.; Mantas, G.; Spathis, A.; Papazoglou, K.O.; Karakitsos, P.; Liapis, C.D.; Karkos, C.D. The Profile of Circulating Matrix Metalloproteinases in Patients Undergoing Lower Limb Endovascular Interventions for Peripheral Arterial Disease. Ann Vasc Surg. 2017, 43, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Stasinopoulou, M.; Kadoglou, N.P.E.; Christodoulou, E.; Paronis, E.; Kostomitsopoulos, N.G.; Valsami, G.; Liapis, C.D.; Kakisis, J. Statins' Withdrawal Induces Atherosclerotic Plaque Destabilization in Animal Model-A "Rebound" Stimulation of Inflammation. J Cardiovasc Pharmacol Ther. 2019, 24, 377–386. [Google Scholar] [CrossRef]
Table 1.
Body weight, fasting plasma glucose (FPG), total cholesterol and triglycerides levels at baseline and at the end (8 weeks) of the study.
Table 1.
Body weight, fasting plasma glucose (FPG), total cholesterol and triglycerides levels at baseline and at the end (8 weeks) of the study.
| CG (n=12) |
DEG (n=12) |
ETG (n=12) |
DE+ETG (n=12) |
p | |
|---|---|---|---|---|---|
| Weight (g) | |||||
| Baseline | 27.8±4.6 | 28.5±7 | 28.3±5.1 | 27.2±4 | 0.881 |
| End | 31.1±4.1 | 32.23±6.3 | 29.8±2.7 | 29.7±5 | 0.321 |
| FPG (mg/dl) | |||||
| Baseline | 113±28 | 108±31 | 103±21 | 120±41 | 0.773 |
| End | 165±44* | 189±45* | 165±52* | 182±49* | 0.654 |
| TC (mg/dl) | |||||
| Baseline | 358±145 | 398±103 | 404±198 | 387±134 | 0.298 |
| End | 684±169* | 612±223* | 556±110* | 579±311* | 0.112 |
| TG (mg/dl) | |||||
| Baseline | 82±13 | 84±21 | 89±28 | 94±31 | 0.498 |
| End | 125±29* | 115±24* | 126±31* | 116±32* | 0.687 |
CG, control group; DEG, dabigatran etexilate group; ETG, exercise training group; DE+ETG, dabigatran etexilate + exercise training group. TC, total cholesterol; TG, triglycerides; P, one-way ANOVA. *p<0.05, within groups.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



