
Submitted:
19 September 2023
Posted:
21 September 2023
Read the latest preprint version here
Abstract
Alzheimer’s disease (AD) and vascular dementia (VaD) together contribute to almost 90% of all dementia cases leading to major health challenges of our time with a substantial global socioeconomic burden. While in Alzheimer’s disease, the improved understanding of Amyloid beta (Aß) mismetabolism and tau hyperphosphorylation as pathophysiological hallmarks has led to significant clinical breakthroughs, similar advances in VaD are lacking. We thus highlight here the importance of shared pathomechanisms found in AD and VaD: Endothelial damage, blood brain barrier (BBB) breakdown and hypoperfusion inducing oxidative stress, inflammation and demyelination and thus trophic uncoupling in the neurovascular unit. A dysfunctional endothelium and BBB lead to the accumulation of neurotoxic molecules and Aß through impaired clearance, which in turn leads to neurodegeneration. In this context we discuss possible neuropathological parameters, which might serve as biomarkers and thus improve diagnostic accuracy or reveal targets for novel therapeutic strategies for both forms of dementia.
Keywords:
Alzheimer`s
; vascular dementia
; neurovascular unit
; neurovascular coupling
; impaired blood brain barrier
; neurotoxic metabolites
; endothelial damage
1. Introduction
Dementia has emerged as one of the top public health challenges of our time. Due to the aging of the world population and the lack of available causative therapies, the number of affected individuals, estimated at 55 million worldwide by the WHO in 2021 [1], is anticipated to reach approximately 78 million by 2030 and 139 million by 2050. The global financial burden has already outpaced cancer and heart disease [2] with costs of US$ 1.3 trillion in 2019 and may rise to US$ 2.8 trillion by 2030. Although the anticipated “dementia epidemic” has forced world leaders to develop national plans to deal with the tremendous socioeconomic impact and enhance schemes of research funding, the development of novel treatment strategies, which span the translational gap from promising preclinical results to sustained improvement or even prevention of cognitive decline in patients, remains challenging.
Initially, the discovery of the amyloid precursor protein and its metabolism leading to different variants of Aß peptides as the core features of the amyloid cascade hypothesis [3] and the parallel identification of the microtubules-associated tau protein originating and accumulating in transentorhinal and hippocampal neurons [4] as dominated the pathophysiological research on AD. However, it has recently become accepted that by the time when neurodegeneration in AD has resulted in first clinical symptoms, additional pathologies in the brain, which interact with Aß and tau pathology and can be traced back to distinct molecular and cellular processes, contribute to the clinical manifestation, mainly via synaptic dysfunction and degeneration:
A.) Among those are microglia and astrocytes, which support the functioning of neurons under physiological conditions as components of the innate immune system of the brain. However, in the case of AD misfolded and aggregated proteins bind to microglia and astroglia receptors and trigger an innate immune response with release of pro-inflammatory cytokines and other immunological modulators promoting AD progression [5,6,7,8,9]. In this respect, several candidate genes, which have been identified as risk factors for AD, encode for proteins that regulate microglial and astroglial clearance of misfolded proteins and other inflammatory responses, e.g., the ABC transporter A7 which is involved in clearance of Amyloid-β from the cerebrospinal fuid (CSF), Apolipoprotein E which regulates lipid metabolism, and TREM2 which is involved in microglia-mediated clearance of tau. Thus, these pathophysiological pathways may deliver promising treatment targets in AD [10,11].
B.) Vascular dysfunction and cerebrovascular pathologies may contribute to neurodegeneration in AD [12,13,14], Vascular dysfunction may have a major impact in the pathogenesis of AD [15,16], although the mechanisms of interaction and cause – effects sequences are not fully understood [15]. Vascular pathology is common in elders and increases with advanced age [16]. For instance, 79.9 % of the 4,629 patients diagnosed with AD based on neuropathological criteria from the NACC database showed vascular pathology [17,18]. Furthermore, 32.2 % from those patients had cerebrovascular disease [17]. In another study, cerebrovascular lesions coexisted with AD lesions in up to 50 % of cases. In these cases, other vascular lesions such as lacunes and microinfarcts were also observed [15,17]. Thus, vascular pathology may simply be co-existing with AD [15] and pure AD may be rare [19]. This is reflected by the Mayo Clinic Brain Bank from 2007 to 2016, in which the majority of AD cases showed other pathologies and comorbidities with advanced age [20].
In fact, AD and VaD depict many similarities on different levels: (1) Epidemiological studies show that almost all risk factors for AD reported so far have a vascular component which reduces cerebral perfusion [12]. (2) All approved medication to symptomatically treat AD also improve cerebral perfusion [21]. (3) Cerebral capillary degeneration has been shown to be present in practically all brains of AD patients examined postmortem and in cortical biopsy material from pathologically confirmed AD [22,23,24]. (4) Finally, there is evidence that the largest proportion of dementia cases have mixed pathology, comprising features of AD (amyloid plaques and neurofibrillary tangles) as well as ischemic lesions [15,25,26].
Targeted therapies beginning to enter clinical practice make a deeper understanding of biomarker classification, thorough diagnosis, identification of co-pathologies and biologically based staging of AD necessary. We thus discuss in this review relevant common and distinct neuropathological mechanisms in both AD and VD. We first highlight the clinical presentation, including risk factors, disease patterns, course of diseases and further diagnostic parameters. We then focus on the known pathology of both diseases and highlight neuropathological parameters, which might serve as (new) biomarkers, helping to improve diagnoses, or which may be even new anchor points for the development of novel therapeutic strategies.
2. Clinical features
2.1. Prevalence and risk factors
Age is the most important risk factor for developing dementia. The risk of developing VaD doubles every 5.3 years, an exponential rise which is slightly less pronounced than in AD, where rates double every 4.5 years [27]. Furthermore, around 15-30% of stroke survivors develop dementia within 3 months after stroke [28]. Having a stroke, also increases the risk of developing dementia in the long-term, with about 20-25% of stroke survivors revealing a delayed dementia [28]. However, discussion have been risen if post-stroke dementia is a pathology on its own, as the development of this disorder is very heterogenous in its nature. E.g. it remains sometimes unclear, if the stroke has just unmasked an already present cognitive impairment, whether the vascular pathology is responsible on its own for the cognitive decline or wether the vascular pathology fortified a neurodegenerative picture as seen in AD. A long-term autopsy follow-up study in stroke survivors aged over 75 years [29] claimed that vascular but not degenerative dementia was the cause of the dementia in 75% of the cases. In addition, in regions where stroke is very common e.g., in East Asia, the incidence of vascular dementia exceeds that of AD [30].
Both subtypes of dementia have most risk factors in common (see Figure 1 and Figure 2) [27]. Female sex is an unmodifiable risk factor for Alzheimer`s dementia [31,32,33], while the relationship between female sex and vascular dementia is less clear with studies showing no association at least post-stroke [28]. Only few genetic risk factors are known for vascular dementia [34]. Also, in most cases AD does not have a single genetic cause. However, more than 70 genetic regions have been identified to be associated with Alzheimer`s [35]. One well-known gene, which is a high risk factor in particular in women, is the apolipoprotein E (APOE) gene. APOE is involved in cholesterol transportation and the metabolism of other fat types in the bloodstream. Problems in this process are thought to contribute to the development of Alzheimer`s, while the involvement of APOE in the development of VaD is less obvious [34]. Three rare single-gene variants are known to cause Alzheimer`s disease, including Amyloid precursor protein (APP) on chromosome 21, Presenilin 1 (PSEN1) on chromosome 14 and Presenilin 2 (PEN2) on chromosome 1.
For both disease entities vascular risk factors are important (Figure 2) hinting at a common vascular pathomechanism: Several studies revealed that for a similar burden of Alzheimer`s pathology, clinical symptom manifestation was greater when there was a comorbid vascular disease [27]. There is also strong evidence linking midlife hypertension and diabetes to both, Alzheimer`s and vascular dementia [36]. Midlife cholesterol levels and obesity were associated with later-life dementia [36]. Smoking was major risk factor for cognitive decline both, in AD and VaD [36,37,38]. Furthermore, late life depression increased the risk for cognitive decline in both diseases [39] and associations were found providing a plausible mechanistic link between late life depression and several vascular abnormalities. In contrast, a higher cognitive reserve, higher education, social networks, cognitive and physical activity are protective factors preventing cognitive decline [31,36].
2.2. Clinical presentation
It is often difficult to distinguish patients diagnosed with AD from those with VaD, based on pure clinical presentation (Table 1) and cognitive performance (Table 2). In AD, symptom progression is somehow reasonably well defined with a relentlessly progressive memory impairment- in particular episodic memory- which converts over months and years to disorientation, personality and judgment dysfunction, speech abnormalities, and apraxias, among other signs of cortical dysfunction [40]. In contrast to AD a well-defined single neuropsychological profile has been challenging to establish for VaD [41]: Clinical symptoms and signs vary depending on the location and size of the stroke lesion(s) and their distribution [42], but generally, deficits in executive functions are most pronounced.
Abrupt deterioration of cognitive functioning (e.g., when a stroke directly hits important cognitive areas) is described as well as a course of fluctuating intensity of cognitive symptoms or stepwise deterioration [43,44]. Patients with a vascular cause of cognitive decline tend to reveal a more impaired semantic memory, deficits in attentional functioning, visuospatial and perceptual skills and in particular executive dysfunction as the most prominent symptom [43,45,46]. Patients with VaD also tend to show more symptoms of depression, apathy and loss of drive than in AD: In emotional recognition tests AD patients outperformed patients with vascular dementia in identifying emotions depicted in the photographs [44]. Table 2 lists differences and similarities when applying neuropsychological tests: E.g. while the Mini Mental State Exam (MMSE) provides good results in detecting cognitive decline in Alzheimer`s, the test is quite indifferent for delivering reasonable results for vascular cognitive decline. In contrast, the Montreal Cognitive Assessment (MoCA) has been validated in particular in settings after stroke and suspicion of vascular cognitive impair. However, if these test are to be used in clinical settings, they should be applied cautiously and in conjunction with other information, such as medical history, behavioral observations, imaging, and information from relatives, when contributing to a diagnosis [44]. Importantly, patients with VaD may also present with other neurological symptoms such as locomotor abnormalities with gait disturbances and autonomous dysregulation such as bladder dysfunction [47].
2.3. Typical clinical diagnosis parameters
While in the 1980s the Aß peptide was identified and shortafter mutations in the amyloid precursor protein (APP) were detected in cases of familiar forms of AD, biomarkers for the diagnosis of AD have been steadily established forming the basis for relatively clear consensus statements for diagnosing AD such as the currently revised National Institute of Aging and the Alzheimer`s Association (NIA-AA) Criteria for the Diagnosis of Alzheimer`s Disease (https://aaic.alz.org/nia-aa.asp). These criteria have been consistently applied throughout the last years with the three classification systems A (Aß), T (tau) and N (neurodegeneration in MRI or hypometabolism in PET) for the diagnosis of AD [48]. Eight different AT(N) profiles were identified, and individuals were staged basedon integrating biomarker profile and the severity of the clinical impairment. Three new additional categories have been recently added – including I (Inflammation), V (vascular) and S (alpha-synuclein)- to open up biomarkers for AD towards a more differentiated descriptive approach and to include highly relevant co-pathologies.
For VaD the picture is less obvious. Even the term vascular dementia has been controversially discussed for many years as VaD can be caused by a reduced cerebral blood flow supplying the brain which may or may not be associated with a stroke. It can also be caused by a single major stroke strategically destroying important areas for cognitive processing such as the hippocampus or by multiple microstrokes. Subsequently, the term vascular dementia did not fully reflect the entire spectrum of cognitive alterations caused by vascular factors. Thus, the term vascular cognitive impairment (VCI) was proposed [49] and widely adopted. Furthermore, as no clear consensus exists on pathological criteria for VaD, different classification systems such as the National Institute of Neurological Disorders and Stroke and the Association Internationale pour la Recherche et l’Enseignement en Neurosciences (NINDS-AIREN) criteria or the Alzheimer's Disease Diagnostic and Treatment Centers (ADDTC) criteria have been developed over time. While the NINDS-AIREN criteria have been proven to be most specific but less sensitive and have been used in most relevant studies [27], the opposite was found for the ADDTC criteria. The latest published Vascular Impairment of Cognition Classification Consensus Study (VICCCS) guidelines [50] require for the diagnosis of vascular cognitive impairment at least “clinically significant deficits in one cognitive domain which are sufficient to cause a severe disruption of activities of daily living and imaging evidence for a cerebrovascular disease”. For imaging, a magnetic resonance imaging (MRI) scan is required with identified vascular lesions. According to the VICCS criteria (Iadecola et al., 2019), VaD is classified into four main subtypes (Figure 3): 1) post-stroke dementia, which is defined as dementia disclosing within 6 months after stroke; 2) subcortical ischemic vascular dementia; 3) multi-infarct dementia and 4) mixed dementia. For mixed dementia the predominant case - either Alzheimer`s or vascular dementia- is further specified.
Typical assessment features in MRI for AD include the assessment of general brain atrophy with a focus on medial temporal lobe atrophy and hippocampal atrophy [51,52]. Neuroimaging of VaD should examine also the following [31,50]: 1) General brain atrophy, ventricular size and medial temporal lobe atrophy as well as 2) white matter hyperintensities, 3) infarcts (number, size - stratified for large (>1 cm) or small (3 to 10mm) lesions, location) and 4) haemorrhage (number, size - stratified for large (>1 cm) or small (< 1cm) lesions and location). For the diagnosis of AD further instruments such as Amyloid PET (positron emission tomography) and functional MRI, hold promise to assess structural and functional losses and are on the way in academic centers to become part of the diagnostic routine, in particular in case of difficult diagnosis finding. Furthermore, blood and cerebrospinal biomarkers exists for AD: In addition to decreased Amyloid-beta 1-42 (Aß 1-42) or increased phospho-Tau [53,54], there are new, emerging plasma biomarkers that will make biological AD diagnosis generally feasible in clinical practice and which perform well to discriminate e.g., AD from other forms of dementia such as frontotemporal dementia and Lewy body dementia [55]. Other markers now mentioned in the revised NIA-AA Criteria include neurogranin as a marker of post-synaptic injury and degeneration while SNAP-25 and GAP-43 are markers of pre-synaptic degeneration and dysfunction. NfL is a marker of large caliber axonal injury that can be measured in CSF or plasma and was used already in various disorders in clinics [56,57]. However, specific molecular biomarkers are missing supporting the diagnosis of vascular cognitive impairment.
3. Pathology
3.1. Similar and distinct vascular features of Alzheimer`s versus vascular dementia
Artherosclerosis, arterioloscleroisis, macro- and micro-infarcts and cerebral amyloid angiopathy (CAA) increase the likelihood that a person will exhibit dementia symptoms [58]. The different vascular features (Table 3) and their impact for the development of Alzheimer`s or vascular dementia are reviewed (Figure 4) in the following paragraphs.
3.1.1. Atherosclerosis
Atherosclerosis is a chronic inflammatory condition that affects large and medium-sized arteries [59,60]. Different risk factors such as dyslipidemia, arterial hypertension and diabetes mellitus are associated with the emergence of atherosclerosis [59,61]. Formation of atherosclerotic plaques and their calcification aggravates fibrosis and degenerative processes of vessel walls, inducing infarcts due to thrombosis or embolism. Beach, Wilson [62] reported that in particular the atherosclerosis of the Circle of Willis was more severe in subjects with AD and VaD than in control subjects, while it was equivalent between control subjects and subjects with non-AD dementias. Severe circle of Willis atherosclerosis was even more common in VaD. With an increasing severity of atherosclerosis, the odds ratios (OR) for the diagnoses of both, AD and VaD, are pronounced, also for neuritic plaque density and higher Braak neurofibrillary tangle stages in the case of AD. A similar result was found in a populated-based cohort study in which 678 patients developed dementia (476 were diagnosed with AD, 52 with mixed pathology, and 78 with VaD). Atherosclerosis- this time predominantly at the carotid artery- was associated with an increased risk for dementia [63].
Atherosclerosis very often coexists with small vessel disease (SVD), an umbrella term that includes various pathological findings, including small infarcts, microscopic infarcts, CAA and arteriolosclerosis [31]. Together, atherosclerosis and SVD are among the most frequently associated vessel pathologies found in VaD: Changes of intracranial small vessels lead to Binswanger’s disease or subcortical ischemic vascular dementia [27,30,47,64]. In addition, atherosclerosis starting already in midlife was rather associated with the development of VaD and SVD [65] than with the development of AD.
3.1.2. Mechanisms of endothelial dysfunction
Although it is somewhat common sense that endothelial dysfunction may lead to the insufficient supply of neurons with oxygen and glucose – the mechanisms behind are not well understood and may be multifaceted.
Cerebral endothelial cells are next to astrocytes and pericytes an essential part for the blood-brain barrier (BBB) formation and function. They are connected via tight junction and adherens junctions, creating a barrier [15,18]. While an intact blood brain barrier is crucial for the homeostasis of the central nervous system (CNS) and protects from toxins and pathogens, endothelial dysfunction and alterations in BBB permeability probably precede several pathological alterations, thus playing a major role in the pathology of different neurovascular diseases [67].
BBB dysfunction was found both, in patients with AD and VaD [68]. Moreover, BBB breakdown seems to take place early in the course of both diseases, in particular affecting the hippocampus in Alzheimer`s disease [69]. It was thus even suggested that BBB dysfunction precedes hippocampal atrophy seen later in AD. A recently published review on dysfunctional BBB in AD [70], showed, that BBB breakdown was detectable early in the course of AD on MRI. In addition to the hippocampus, disruption of BBB in early AD was detectable on dynamic contrast-enhanced MRI (DCE-MRI) also in other grey and white matter regions associated with cognitive decline and dementia [71,72].
A common explanation for endothelial dysfunction may be endothelial damage – induced by a figurative vicious cycle of hypoperfusion and oxidative stress- which in turn results in reduced resting cerebral blood flow (CBF) in the marginally perfused white matter and in an altered BBB [15]. As a result, hypoxia and additional oxidative stress induce inflammatory pathways and altogether lead to injured oligodendrocytes, demyelination and trophic uncoupling in the neurovascular unit (a unit of neurons, astrocytes, oligodendrocytes and vascular cells involved in CBF and BBB function) [15,73,74]. Damage to the neurovascular unit- of any of the components - vice versa- contributes to impaired vascular cells (Figure 4). Supporting this, CBF reduction in patients with early AD correlated with an increased BBB leakage rate [75,76,77].
In addition, APOE4, a major risk factor for AD but also detected in VaD [62], leads to a reduced cerebral blood flow but also activates a pro-inflammatory pathway resulting in a dysfunctional BBB and neuronal uptake of blood-derived neurotoxic proteins [76,78,79,80]. Carriers of APOE4 allele were distinguishable from non-carriers by BBB breakdown in the hippocampus and medial temporal lobe. These findings were even more severe in APOE4 carriers with cognitive impairment [80] and support the two-hit hypothesis which is discussed for the development of AD [71,81]: According to the hypothesis, cerebrovascular damage is the initial insult, the so-called “first hit”. Damage to brain microcirculation leads to dysfunctional BBB and diminished brain perfusion, which in turn induces hypoperfusion and accumulation of neurotoxic molecules. This mediates neuronal dysfunction and disturbed Aß-clearance and thus Aß accumulation in the brain (second hit). Increased levels of Aß amplify neuronal dysfunction and accelerate neurodegeneration [18,81,82].
Extensive literature also discusses other molecular mechanisms underlying a dysfunctional BBB: P-glycoprotein, for example, is an efflux pump, which is expressed at endothelial cells of the BBB and is involved in the transportation of Aß [83,84,85]. It was shown, that vessels with high P-glykoprotein expression revealed no Aß-accumulation in their walls, suggesting, that impaired clearance of P-glycoprotein leads to Aß accumulation, which increases the risk for cerebral amyloid angiopathy (CAA) and AD [83]. Activity of P-glycoprotein could be assessed in vivo and was found to be reduced in frontal, parietal, temporal, and occipital cortices as well as in anterior and posterior cingulate cortex in patients with AD [84], while only the parietotemporal, frontal, and posterior cingulate cortices and hippocampus were affected in patients with mild AD [86]. Altered P-glykoprotein levels were also identified in several animal models targeting neurovascular risk factors such as diabetes mellitus and hypertension: Maeng, Kim [87] investigated the clearance of cyclosporin A via P-glykoprotein in streptozotocin-induced diabetic rats in vivo. The clearance of cyclosporin A was reduced and was associated with an increased mRNA and protein level of P-glycoprotein [87]. Another study detected an increased expression of P-glycoprotein in the BBB-damaged hippocampal vessels of stroke-prone spontaneously hypertensive rats (SHRSP) [88]. The increased levels of P-glykoprotein could be a compensatory strategy in vascular dementia or may even reflect a different molecular mechanism at the BBB level in the pathology of AD and VaD, as in AD disappearing P-glycoprotein levels were measured in smooth muscle cell layers of arterioles were Aß was deposited. However, at the same time P-glycoprotein levels were increased in capillaries. Thus, P-glycoprotein seems to serve as a gatekeeper to the BBB with even a suspected neuroprotective role in Alzheimer`s [85] while the same was not reported for vascular dementia.
3.1.3. Cerebral blood flow
Alterations in cerebral blood flow have been linked to age-related cognitive impairment since the time of Alois Alzheimer, best known for identifying the condition now called Alzheimer`s disease, who proposed that stiffening of arteries would impair the ability of cerebral blood vessels to relax and adjust the delivery of blood to the metabolic needs of the brain, causing hypoperfusion, neuronal death and dementia [31]. This concept prevailed for many years, till measurements of cerebral blood flow demonstrated that cerebral blood vessels were able to increase cerebral blood flow also in cognitively impaired individuals [90]. However, resting cerebral blood flow (rCBF) was shown to be reduced in patients with hypertension in prefrontal, anterior cingulate and occipital areas over time [91]. Thus, reduced CBF might represent an early pathological process in both diseases (see Korte, Nortley [92], Cohen [93] and Bracko, Cruz Hernández [76]).
In particular the deep white matter is vulnerable to decreased CBF [15], alongside other susceptible brain regions such as the basal ganglia and the hippocampus [94]. Patients with AD and with subcortical ischemic vascular dementia (SIVD) showed reductions of the CBF in the same regions- the frontal, parietal and the temporal cortex [95,96,97,98].
However, reductions of the CBF tend to be more prominent in the posterior brain areas in AD [99]. Specifically, CBF was reduced in posterior cingulate and precuneus cortex in mild cognitive impairment (MCI), whereas AD was associated with more global and severe CBF reduction [98,100] and the involvement of other areas such as the bilateral parietotemporal, frontal and occipital cortex, parahippocampal gyrus, hippocampus and entorhinal cortex [98].
In contrast, reduction of CBF tends to be anterior-dominant in VaD [99], preferentially involving the frontal lobe [101,102]. Compared to normal controls a reduced CBF has also been reported in the right thalamus, left caudate nucleus and in the cingulate, bilateral superior temporal and left ventral subcallosal gyri in subcortical VaD [103]. In line with the described anterior-dominant lower CBF perfusion, resting CBF (rCBF) was reduced in the frontal and temporal white matter in patients with VaD [97].
Furthermore, Aβ, the major pathogenetic marker for Alzheimer`s disease, is discussed of having a vascular effect in reducing cerebral blood flow alongside endothelial dysfunction [15]. Also APOE4 is associated with BBB disruption and reduced CBF [76,78,79,80,98]. Several studies showed greater reduction of CBF in carriers of APOE4 compared to non-carriers [104-107]. Differences were in particular observed in the frontal, parietal and temporal area [104] and in mid-life individuals (aged 40 – 59) carrying the APOE4 allele [107]. Other authors [16] also discussed a dysfunction of the cholinergic system, which is reported for both, AD as well as VaD [108,109,110], causing a reduction of the CBF.
3.1.4. Cerebral amyloid angiopathy (CAA)
Cerebral amyloid angiopathy (CAA) is common in older individuals and the most common cause of intracerebral hemorrhage, contributing to cognitive decline [40,111]. The pathology is characterized by the deposition of amyloid, mainly Aβ, in the media and adventitia of small and mid-sized cerebral and leptomeningeal vessels, accompanied by degeneration of smooth muscle cells (SMC) [112,113,114]. Subsequently, CAA leads to a disrupted vascular architecture and BBB [18,64,112]. Furthermore, CAA causes not only hemorrhages, including microbleeds, but also capillary occlusion and blood flow disturbances as well as infarcts and white matter lesions by hypoperfusion and dysfunctional vascular autoregulation [64,112,113,115].
Clinically, CAA manifests in 5 - 20 % of all cases with lobar intracerebral hemorrhages [113], indicating the strong connection between CAA and cerebrovascular lesions. CAA is found in both, patients with AD and VaD. While the prevalence of CAA in VaD is still unclear [47], the cerebrovasculature of AD patients is very often affected by CAA, occurring in over 90 % of the cases [15,40,112,115]. However, recent findings [81] indicate, that pathological vascular alterations are not only a consequence of Aβ deposition, but that vice versa, impaired cerebrovasculature may contribute to the accumulation of Aβ plaques: In mouse models of Aβ pathology, experimental manipulation inducing deficient endothelial cells or pericytes and impaired blood brain barrier augmented Aβ plaque load. Therefore, disrupted Aβ clearance through the cerebrovascular system may be a pivotal process in the development of AD.
CAA is most frequently and severely affecting the occipital lobe, which might be due to a higher number of tortuous occipital vessels, leading to decreased perivascular clearance [113,116,117]. CAA was also frequently located in the frontal, temporal, or parietal lobes [116,117,118,119,120]. In all regions, the leptomeningeal vessels seem to be in particular susceptible for CAA [116]. A study by Haglund, Sjöbeck [121] described in this context more amyloid-positive vessels in the leptomeninges and in the cortex in patients with mixed dementia pathology than in patients with a Alzheimer`s diagnosis only. For VaD, a preferentially affected region could not be identified in the literature so far.
APOE e4, APOE e2, mutations in CR1 and APP gene polymorphism are connected to AD and vascular lesions. APOE e4 is not only an established susceptibility gene for AD, but is also associated with an increased risk of CAA, which emphasizes the link between AD and CAA [15,27,112,113,122,123]. For VaD, there are inconsistent reports of an association between APOE e4 and an increased risk for the disease [124]. APOE e2 is described to be a risk factor for CAA-related hemorrhage [112,115,125] and thus might intercept with vascular dementia. However, Rannikmäe, Samarasekera [122] found no association between APOE e2 and the presence of CAA. Furthermore, CR1 gene polymorphism is known to increase the risk for AD and was also associated with the severity of CAA and with CAA-associated intracerebral hemorrhage [126].
Mutations in presenilin 1 or presenilin 2 genes are associated with familial AD and severe CAA [115]. According to Pandey, Pradhan [127] Presenilin 1 allele 1 is susceptible to degenerative dementia, but not to vascular dementia. In addition, mutations in the APP gene were identified with familial forms of AD and with sporadic CAA, as well as familial (or hereditary forms of) Aß and non-Aß-CAA [15,109,117,128]. Hereditary forms of CAA with mutations in the APP gene do also contribute, although not consistent, to cerebral vasculopathies [117,128]. In general, hereditary forms of CAA are characterized by multiple hemorrhages and infarcts in addition to severe amyloid depositions in the walls of vessels and can lead to vascular cognitive impairment or VaD [47]. For instance, lobar intracerebral hemorrhage and infarcts were observed in the Dutch mutation (NFT and plaques were rarely observed), while stroke, cognitive decline, and seizures were associated with the Italian mutation. Cortical hemorrhages and subcortical infarcts were in particular observed in the Iowa mutation of hereditary Aß-CAA [117]. Lobar intracerebral hemorrhage in non-Aß-CAA was rarely observed, except for the Iceland type [128].
3.1.5. Hemorrhagic lesions
Damaged blood vessels and a dysfunctional blood brain barrier lead to blood leakage and extravasation into the perivascular space, which manifests as microbleeds [15,18,98]. Microbleeds are visualized as small, dot-like hypotense abnormalities in the MRI and are associated with hypertension and white matter disease [15,129].
Microbleeds occur with cerebrovascular disease, dementia, and normal aging [130] and are thus observed in both, AD and VaD, although with different frequencies. The prevalence of microbleeds in VaD ranges from 35% to 85% [47,129]. Microbleeds are also one of the three features referred to as surrogate markers in small vessel disease (SVD), which is a substrate of subtype subcortical ischemic vascular dementia. In AD, microbleeds are usually detected at lower percentages using MR imaging. For example, 18 % of patients with AD exhibited microbleeds compared to 65 % of patients with VaD [131]. Interestingly, in another study, patients with early AD or mild cognitive impairment showed a relatively high prevalence of microbleeds using ultra-high field strength 7Tesla MRI [132].
The location of microbleeds is related to their etiology: a hypertensive vasculopathy presumably leads to deep and infratentorial microbleeds, while CAA most likely plays a role in lobar microbleeds [129,130]. According to Yates, Villemagne [133], the majority of microbleeds in AD might be caused by CAA and thus tend to appear in posterior cortical regions, especially in the occipital lobe [18,130,133,134,135]. Pettersen, Sathiyamoorthy [136] for example reported a lobar predominance of microbleeds in 92 % of patients with AD on T2-weighted and proton density-weighted scans. 57 % of these microbleeds were detected in the occipital lobe. However, another study observed an anterior-posterior decreasing gradient of the cortical microbleeds in AD patients, who showed more cortical microbleeds in the superior frontal, inferior temporal, the rectus and the cinguli gyrus, and in the insular cortex compared to controls [134].
Depending on the underlying pathology, microbleeds in VaD can be located in deep or lobar regions. In this context a hypertensive vasculopathy plays a dominating role in VaD [129]. As already mentioned, hypertensive-associated microbleeds are particularly found in deep and infratentorial regions such as the basal ganglia, thalamus, brainstem and cerebellum [130]. In hereditary VaD (CADASIL), microbleeds are scattered throughout the brain with a preference for subcortical lesions, white matter, thalamus and brainstem – but also lobar regions [129,137]. Furthermore, a study comparing cortical microbleeds in VaD patients versus patients with mixed dementia, found microbleeds preferentially located in the frontal lobe and the cerebellum in VaD patients [138].
Looking for genetic risk factors for microbleeds, the APOE gene again appears as a major player associated with sporadic microbleeds. Both alleles, APOE e2 and APOE e4, have each been reported to be related to lobar microbleeds [133]. Maxwell, Jackson [139] also observed an association of APOE e4 with deep microbleeds, while another study found only an association of APOE e4 with lobar microbleeds, but not with deep and infratentorial ones [140]. As CADASIL presents with microbleeds and is subject to the mutation of NOTCH3, mutation of NOTCH3 can be also considered as a genetic risk factor for microbleeds.
3.1.6. Ischemic lesions
3.1.6.1. Lacunar infarcts
Lacunar infarcts are small (1-15 mm), discrete and often multiple and bilateral irregular lesions [47,141], which are located in deep parts of the brain, but not in the cerebral or cerebellar cortices [141]. These kinds of lesions are thought to be an age-related neurovascular disorder [142], originating either from vessel occlusion related to SVD or from embolic events [64].
Lacunar infarcts are part of the vascular pathology detectable in AD [143], having been associated with the same risk factors as in AD, among which advancing age is particularly noteworthy [142]. In the study of Snowdon, Greiner [144] 39 % (n=24) of the 61 participants with AD showed at least one infarct. However, lacunar infarcts are in particular a characteristic pathological feature as well as one of the three surrogate markers for VaD and SVD [47]. Around 68 % of the cerebrovascular lesions seen in pure VaD (VaD without AD pathology) are subcortical lacunar infarcts [143]. Moreover, it is reported, that lacunar infarcts are identified in 25 % of all ischemic strokes [142]. Lacunar infarcts are also the most common feature in more than 50 % of elderly patients with ischemic VaD [47].
Considering the localization of lacunar infarcts, for both disease entities, AD and VaD lacunar infarcts typically occur in the basal ganglia, thalamus, and white matter (see table 10 in Jellinger [112], Tikka et al., 2014). However, for patients with AD and severe cerebrovascular disease (mixed dementia) larger and more lobar infarcts are found than small subcortical lacunar infarcts (Attems and Jellinger [143]. In VaD, lacunar infarcts predominate in the deep white matter [145] and/or subcortical brain areas (in particular the putamen) and are associated with intracerebral hemorrhage [47]. Especially in CADASIL, lacunar infarcts are described as typical features and of diagnostic importance [15,27,146]. T2-weighted changes and lacunar infarcts in the MRI located in the external capsule and the anterior part of the temporal lobe are highly suggestive of CADASIL.
Genetically, a linkage between AD and VaD may exist for ischemic lesions. In a genome-wide association study [147] evidence for a shared genetic contribution between AD and small vessel stroke was found. Furthermore, another study reports an association of APOE4 with acute ischemic stroke [148]. For VaD three hereditary small vessel diseases (SVD), typically leading to lacunar infarcts, are known: CADASIL, CARASIL [149,150] and the COL4 disorder. As already described in the above chapter, CADASIL is caused by the defective gene NOTCH3. On the other hand, CARASIL (cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy) is a rare and recessively inherited disease [150], in which mutation of the gene HTRA1 (high-temperature requirement A serine peptidase 1) was identified [15,150,151]. The COL4 disorder is another rare condition that presents with multiple lacunar infarcts in the white matter and the pons and is caused by mutations in the COL4A1 gene, encoding the type IV collagen alpha 1 chain [15,47].
3.1.6.2. Microinfarcts
Microinfarcts are small cystic or gliotic, pale lesions, which occur throughout the brain. They induce neuronal loss and show in the acute stage an inflammatory response [15,47,152]. The size of the microinfarcts is often described to be not visible to the eye or only upon microscopy indicating the limited use of the MRI for their detection. Only ultra-high field strength clinical MR scanners might detect microinfarcts in patients [152]. Microinfarcts are in particular found in small vessel disease (SVD). But they can also originate from embolic events due to atherosclerotic plaques [152,153].
Microinfarcts (especially multiple cortical microinfarcts) come along with an increased risk of developing dementia [154]. Microinfarcts are common, their number usually increases with age [155] and they are detected in particular in VaD (62 %), but also in AD (43 %) or mixed pathology (AD with cerebrovascular disease; 33%) [152].
Microinfarcts can be localized cortically and subcortically [156]. In particular, for AD patients, microinfarcts are found cortically, in close proximity to CAA [157], indicating that cortical microinfarcts are probably caused by amyloid angiopathy. Moreover, these cortical microinfarcts were predominantly located in the occipital lobe in AD [157]. Some studies also reported cortical microinfarcts in AD patients in the motor cortex with existing motor impairment [156].
Although some cortical microinfarcts are also described to be present in SVD [158] and are found e.g., in the frontal lobe and the cerebellum, with cerebral arteriolosclerosis thought to be the main cause [138], subcortical infarcts are the determining pathological feature of SVD [159], and thus also for VaD [30].
3.1.6.3. Leukoaraiosis
Leukoaraiosis represents abnormal modifications of the white matter, detectable as hyperintensities in the MR and include demyelination, axon loss, astrogliosis and microglia activation [64]. Leukoaraiosis is associated with vascular risk factors [160,161] and was found to be related to CAA and small vessel disease [160]. Although the exact neuropathological mechanisms for the development of leukaoraiosis remain obscure, ischemia, a dysfunctional blood brain barrier (BBB) and endothelial dysfunction are being discussed.
Leukaoraiosis in the form of white matter lesions is found both in AD and VaD. Englund [162] could detect white matter lesions in 33 of 60 (55 %) patients with AD. In addition, the review of van Gijn [161] reported an occurrence of white matter lesions in 25 to 60% of patients with AD. Moreover, white matter lesions were more frequently found in late-onset AD and in AD without parietal predominance [163]. White matter lesions are a hallmark feature of VaD [47,164]. In an imaging study, comparing white matter lesions in VaD and AD, all patients with VaD had white matter changes, while in AD only a small proportion revealed these features, leading to the author’s assumption, that white matter lesions might be a useful diagnostic tool for differentiation [164].
White matter lesions can be diffusely distributed in the brain or very focal in both disease entities, AD as well as VaD. In AD patients with microbleeds but also in individuals with lobar intracerebral hemorrhage, white matter lesions were predominantly distributed posterior [133]. Others [162] however, also reported white matter lesions in the frontal deep white matter. White matter degeneration was situated next to advanced cortical degeneration in the temporal lobes. Thus, it is suggested that white matter lesions in AD are a possible consequence of wallerian degeneration of nerve fibers caused by neuronal loss [165]. Similar mechanisms can be true for VaD, however on a much higher level [166]: In the hereditary forms of VaD- CARASIL and CADASIL -white matter lesions are typical. Characteristic for CADASIL are white matter hyperintensities in the temporo-polar region and capsula externa as well as a periventricular spotty leukoaraiosis [149,150]. MRI findings in CARASIL are mostly similar to those in CADASIL. However, white matter lesions in CARASIL appeared to develop more homogenously than in CADASIL [150].
As before for the other ischemic lesions, APOE2 and APOE4 have been described to give rise to white matter lesions and may lead to increased vulnerability and chronic hypoperfusion of the white matter [167]. Furthermore, studies could find an association of APOE4 with white matter lesions in the deep white matter in AD [168] and of APOE2/3 with microangiopathy-related cerebral damage [169]. Polymorphism of homozygous MTHFR 677TT and of ACE D/D seemed to contribute to white matter lesions and may also be of interest for VaD [170]: Elevated levels of homocysteine, which cause endothelial dysfunction and are a known risk factor for VaD, can be associated with the MTHFR 677TT variant, whereas ACE D/D polymorphism is presumably interfering with the vaso-regulatory system [167]. Both genetic defects can therefore lead to chronic hypoperfusion of the white matter, which in turn is most likely causing white matter lesions [167]. Specific genetic factors for VaD are the above-mentioned hereditary leukoencephalopathies CADASIL and CARASIL, which were described as type 7 of VaD in Konno, Meyer [30] or can be classified as a subclass of subcortical vascular dementia [15,171].
3.2. Similar and diverse neuronal features of Alzheimer`s versus vascular dementia
3.2.1. Brain areas differently affected in Alzheimer`s and vascular dementia
3.2.1.1. Macroscopical changes
Dementia is related to neuronal loss that leads to brain atrophy years before the manifestation of clinical symptoms [172].
For AD a stereotypical and predictable progression of brain atrophy was described and a classification of different disease stages (Figure 3) was introduced by Braak and Braak [4]: Stage I and II include the findings of neurofibrillary tangles in the transentorhinal region of the brain. Stages III and IV are used, if neurofibrillary tangle involvement is also confined to the limbic regions such as the hippocampus and stages V and VI when there is extensive neocortical involvement. In accordance with Braak’s stages, the earliest pathological lesions are preferentially located in the medial temporal lobe, especially affecting the amygdala and hippocampal formation and there, in particular the subiculum and CA1 [40]. The cortical thinning, which is significantly affecting the hippocampus, leads to a dilatation of the adjacent temporal horn of the lateral ventricle and together with medial temporal lobe atrophy it can be interpreted as a possible early pathomorphological sign of AD on MRI [173,174]. But compared to the typical medial temporal lobe atrophy, atrophy of the entorhinal cortex was found to have a higher diagnostic accuracy [175]. Other important regions showing cortical atrophy are the parahippocampal and the superior frontal gyrus. Very little cortical atrophy occurs in the occipital lobe and cingulate gyrus [176,177].
Brain atrophy is also evident in VaD. An investigation of brain atrophy on serial MRI in patients with Lewy Body dementia, AD and VaD, revealed higher rates of brain atrophy in subjects with dementia compared to control subjects, without any significant differences between the three dementia groups [178]. Atrophy in VaD is generalized and similar to AD also located in the medial temporal lobe [27,47]. Moreover, the hippocampus is also affected in VaD, in which van de Pol, Gertz [179] could detect a similar reduction in the volume (11.6 %) compared to AD (16.6 %). Also, the pattern of hippocampal atrophy was similar in both disease entities [180]. Another study indicated that both, hippocampal and cortical atrophy, correlated best with subcortical vascular dementia and AD. The characteristic feature of CADASIL leukoaraiosis, but also the disease itself, were associated with focal cortical thinning [181,182]. In the study of Seo, Lee [182] deep white matter hyperintensities were associated with cortical thinning in the frontal and lingual gyrus. In addition, brain atrophy in CADASIL was associated with the volume of lacunar lesions and progressed three times more rapidly compared to normal aging [149]. Cortical thinning in subcortical vascular dementia was most evident in the frontal regions (bilateral inferior frontal, superior temporal gyri and right medial frontal and orbitofrontal lobes) and more prominent than in AD patients, where the right medial temporal region was significantly more affected [183].
3.2.1.2. Changes on cellular and subcellular structures
Both disease entities, AD and VaD, present with neuronal and synaptic loss [174], including dysfunctional neurotransmitter systems [40] (Table 4). Areas in particular affected by neuronal and synaptic loss for both forms of dementia are as already mentioned above, the hippocampus, the dorsolateral prefrontal cortex and the cholinergic nucleus basalis Meynert [180,184,185,186]. The hippocampus is known for its high degree of neuroplasticity. But this feature might also be the reason for the pronounced vulnerability of the hippocampus to ischemia and chronic stress [187]. Damage to the hippocampus is most evident in CA1 hippocampal neurons, in which a brief episode of cerebral ischemia already results in cell death [187]. Furthermore, regional differences in antioxidants and inflammatory reactions may contribute to the vulnerability of the CA1 region [187]. The pyramidal cell volumes in layer III and layer V of the dorsolateral prefrontal cortex were also reduced by 30 – 40 % in post-stroke and vascular dementia, and in mixed dementia and AD patients compared to subjects without dementia symptoms and controls [186]. Furthermore, neuron numbers and nucleolar volumes in the cholinergic nucleus basalis Meynert were decreased in pure AD and in mixed dementia (AD and VaD), although pure multi-infarct dementia showed, on average, no significant changes [185,188].
For Alzheimer`s disease, region-specific neuronal degeneration was also reported in other subcortical areas such as the locus coeruleus, substantia nigra pars compacta, and the dorsal raphe nucleus [173,189,190]. The suggestion arose that of these subcortical areas, the locus coaeruleus is the site where the first pathological alterations in AD commence [190]. It was shown that intraneuronal lesions associated with AD occur early, affecting the locus coaeruleus and thus the noradrenergic system [191]. Studies reported a neuronal loss of 60% in the locus coaeruleus in AD compared to controls [192]. In the serotoninergic system, alterations have been initially described particularly in the temporal and frontal cortex. Further studies observed a substantial loss of serotonin type 1 and 2 receptors in the amygdala, neocortex and hippocampus [193].
In contrast to AD, microinfarcts with neuronal loss can be found in essentially all brain areas in VaD [16]. However, for CADASIL e.g., neuronal apoptosis was specifically observed in layers III and V of the neocortex. Neuronal loss correlated with the extent of ischemic lesions and axonal damage in the underlying white matter [194].
On the subcellular level, synaptic loss is a characteristic pathomorphological feature that precedes neuronal loss, as in particular shown for AD. (Sheppard & Coleman, 2020). The pattern of this neuronal and synaptic degeneration in AD matches with that of the appearance of neurofibrillary tangles and plaques [174]. Neurodegeneration starts in layer II of the entorhinal cortex and spreads into the hippocampus, temporal cortex, frontoparietal cortex and subcortical nuclei [195,196]. Furthermore, microglia activation has been found to play a considerable role in neuroinflammation spreading fastest along highly connected synaptic pathways. This trans-synapic propagation was in many ways similar as the trans-synpatic progragation of tau occurring through anatomically connected synapsis [197]. These results could be further confirmed in humans using translocator protein (TSPO) PET imaging [198,199].
Considering dysfunctional neurotransmitter systems, in AD (and for some cases also in VaD) in particular the glutamatergic [200,201], the serotoninergic [110,202] and the cholinergic systems are impaired [16,109,110]. For VaD neurochemical studies found abnormalities in the cholinergic transmitter system in the basal forebrain [16]. Dysfunction of the cholinergic system was also found in the temporal cortex, in which choline acetyltransferase activity revealed a more decreased activity in patients with AD and mixed dementia (AD and VaD) than in controls and patients with “pure” VaD [203]. Furthermore, CADASIL exhibits especially severe cholinergic deficits in the frontal and temporal cortices [204]. And even for the serotoninergic system a decreased serotonin metabolism was identified in VaD patients in the hypothalamus and caudate nucleus [110].
3.2.2. Genetic and cell-cycle related changes involved in neuronal loss and brain atrophy
Some genetic risk factors for AD (APP, APOE4, presenilin 1 and 2) cause accumulation of Aß which subsequently leads to neuronal and glial pathology in brain regions important for memory and cognition [205]. Genetic risk factors for VaD with associated neuronal and/or synaptic dysfunction are genes involved in the genesis of CADASIL, but also ERBB3. ERBB3 plays a role in neuroprotection and was found to be downregulated in the hippocampus of a vascular dementia rat model with modified two-vessel occlusion (2-VO) [206]. In this model, the ERBB3 downregulation was associated with vascular cognitive impairment and loss of CA1 pyramidal cells [207].
Furthermore, it was suggested that cell cycle-related phenomena may play a key role in the formation of AD pathology and neuronal cell death in AD, as well as in cerebrovascular diseases [208]. Smith, Nagy [208] observed elevated levels of Cyclin B1 expression in the CA1 region of patients with cerebrovascular disease, while cyclin E expression was elevated in the CA4 region in patients with mixed dementia (AD and cerebrovascular disease). The authors assumed that cell cycle arrest may lead to pathological changes related either to AD or VaD.
3.2.3. Amyloid-ß and tau pathology
Amyloid plaques and the formation of neurofibrillary tangles are known as the “hallmark” lesions of AD [20,40,43,174]. Dysfunctional processing of the amyloid precursor protein and imbalances in the production and clearance pathways cause amyloid plaques, which are extracellular deposits formed by the accumulation of Aß peptides (Aß40 and Aß42) [20]. In contrast, neurofibrillary tangles are part of the tau pathology, where insoluble misfolded deposits of hyperphosphorylated tau are aggregated in neurons.
As characteristic neuropathological features for AD the neuritic plaque density and Braak stage for neurofibrillary tangles were greater in AD (2.57 and 4.71 respectively) compared to VaD (1.87 and 3.67 respectively) [62]. According to Mukaetova-Ladinska, Abdel-All [53] tangle density was 9-fold higher in AD subjects than in patients diagnosed VaD. However, also in neurovascular diseases, Aß is present (Table 4). According to Kalaria and Ballard [209] about one-third of patients with VaD would have AD-type pathology at autopsy. Furthermore, 30 % of individuals with post-stroke dementia or subcortical VaD displayed concurrent Aß pathology in in-vivo Aß PET studies [210,211]. Aß-42 accumulation was also evident in patients with multi-infarct dementia and was suggested to be similar to the Aß-42 accumulation observed in older patients (<70 years) with AD [110]. In fact, Lewis, Beher [212] demonstrated that especially Aß-42 accumulation was present in AD as well as in VaD, although the mean amount of total Aß-42 in VaD was approximately 50 % of that in AD. Aging might contribute to the severity of Aß-42 accumulation in patients with VaD, as patients older than 80 years had comparable Aß-42 concentration with those in AD in the temporal cortex [212]. Furthermore, Bibl, Mollenhauer [54] evaluated the patterns of Aß peptides, total tau and phosphor-tau in the cerebrospinal fluid (CSF) and found a similar pattern of these neurochemical components in AD and AD with cerebrovascular disease. Tau-levels were increased in patients with probable AD diagnosis, definite AD with cerebrovascular disease and probable VaD diagnosis. Aß1-42 levels measured by ELISA were diminished in all dementia groups. Patients with suspected vascular dementia displayed AD-like changes only for Aß1-40ox and Aß1-42%.
The spreading pattern of Aß in AD-related cognitive impairment differs from patients with subcortical vascular cognitive impairment (SVCI) [211]. In patients with AD-related cognitive impairment, Aß accumulated first in the parietal and fronto-temporal regions before the occipital region. However, in VaD Aß is first excessively deposited in the parietal and occipital region, which may be connected with the predominant appearance of CAA in the occipital lobe [211] or an increased vulnerability for dysfunctional microvasculature in the posterior circulation [213]. Moreover, protein analysis by indirect ELISA revealed a distinct pattern of t-tau (total-tau) and p-tau (phosphorylated tau) distribution in both forms of dementia: Compared to VaD and controls, t-tau protein levels were higher in the frontal lobe and p-tau (for antibodies Ser202/Thr105 and Ser262) was increased in the temporal and frontal lobes of AD patients. Moreover, p-tau was increase, while Aß1-42 levels were decreased in the CSF of subjects with probable AD. In contrast p-tau and Aß1-42 levels in the CSF were not altered in patients with VaD [54]. Therefore, the authors conclude that p-tau and Aß1-42 levels in the CSF sufficiently discriminate between probable AD and VaD. For VaD, the study of Mukaetova-Ladinska, Abdel-All [53] did also reveal a selective loss of t-tau protein compared with controls and AD patients, which are supposed to be rather related to dysfunctional microvasculature architecture than neurofibrillary or amyloid pathology. There is also a link between age of AD symptom onset and the extent of tau pathology in certain mainly affected regions and accelerated tau spreading has been associated with faster cognitive decline. Brain Hubs were identified where tau aggregation was mainly present and these regions were essential parts of controlling and maintaining the cognitive performance in AD. A stronger tau signal in those brain regions in the PET predicted faster tau accumulation and suggested tau spreading through highly connected brain regions [214].
There is mounting evidence of Aß having powerful vascular effects (vascular Aß is a potent vasoconstrictor) [15,215]. Aß may induce ischemia and hypoperfusion. But also vascular insufficiency, resulting in hypoperfusion and hypoxia, may promote tau and tau phosphorylation as well as Aß production [15,215]. Indications for a potential link between hypoxic events and Aß production were provided by the study of Zhang, Zhou [216] in which hypoxia-inducible factor-1 (HIF-1), a marker of hypoxia, supposedly mediated Aß production and the hypoxic effect of BACE1 (ß-secretase). Eventually, this leads to a vicious circle in which vascular insufficiency and Aß progressively affect each other negatively. In this sense, it was also suggested that cerebral small vessel disease impairs the clearance of Aß, explaining the possible mechanistic link between AD and cerebral small vessel disease. In addition, tau formation was associated with increased cerebrovascular pathology in animal studies [211].
3.2.4. Brain metabolism
Glucose is the main substrate for brain energy metabolism to support all cellular functions and can be measured by positron emission tomography (PET) [217,218,219]. Thus, regional cerebral metabolic rates for glucose reflect regional brain function.
For both forms of dementia, AD and VaD, glucose hypometabolism was reported (Table 5) [218]. Reduced glucose metabolism was found in the posterior parietal, precuneus, posterior cingulate, prefrontal and anterior hippocampal regions for both, AD and VaD [220]. Of these areas, glucose metabolism was in particular low for patients with AD in the precuneus, prefrontal area and the parahippocampal region [177]. Others also report a pronounced glucose hypometabolism in the association areas for AD patients [218]. However, comparing the pattern of glucose metabolism among both forms of dementia, patients with VaD showed a more widespread pattern of hypometabolism with the deep gray nuclei, cerebellum, primary cortices, middle temporal gyrus, and anterior cingulate gyrus mainly affected [218,220]. Similar results were obtained when comparing the preclinical stages of AD (amnestic mild cognitive impairment) and VaD (subcortical vascular cognitive impairment): In patients with preclinical AD hypometabolic regions were detected bilaterally in the parahippocampal and posterior cingulate gyri, the left superior temporal gyri, the left inferior parietal lobule, and right inferior frontal gyrus, while in patients with preclinical VaD a decreased glucose metabolism mainly affected the thalamus, the insula, the superior temporal gyrus, the anterior cingulate gyrus, cingulum, right basal ganglia, cerebellum, and brainstem [221].
A strong link between an altered glucose metabolism and a genetic risk factor for AD is the APOE allele [222]. It was demonstrated that glucose hypometabolism in APOE4 carriers was evident before cognitive symptoms were present [222,223]. A list of AD genetic risk factors associated with glucose metabolism is provided byCho, Lee [222].
4. Conclusion
As elaborated in this review, both- the diagnosis and the development of novel therapeutic strategies- are challenging for both, AD and VaD. We thus focused here to review similarities and differences between the two disease entities to identify factors which might improve diagnostic accuracy and conceptual strategies for new treatment approaches.
For improved diagnostic accuracy for the early detection of neurodegenerative signs but in particular the differentiation between AD and VaD we propose the following features:
- 1)
- MR Imaging can reveal medial temporal lobe atrophy or entorhinal cortex atrophy as early neuromorphological markers for AD [173,174,175]. While medial temporal lobe atrophy can also be detected in VaD, brain atrophy in VaD is more globally distributed [27,47]. Other imaging biomarkers are microbleeds, lacunar infarcts, microinfarcts and leukoaraiosis. Their location can provide additional diagnostic clues: E.g. Microbleeds are located preferentially lobar, mainly in the occipital lobe in AD [136]. In contrast, a scattered distribution throughout the brain (with described involvement of frontal lobe and cerebellum) would be typical for VaD [129,137].
- 2)
- CBF reduction and BBB breakdown are early pathological alterations in both diseases. Posterior-dominant reduction of CBF in AD and anterior-dominant reduction of CBF in VaD [99], as well as molecular factors related to the BBB, such as P-glycoprotein and plasma cyclophilin A levels can serve as early makers when distinguishing VaD and AD.
- 3)
- 4)
- Glucose metabolism measured by positron emission topography (PET) show a different pattern of hypometabolism in both forms of dementia.
- 5)
- Alzheimer’s presents with a typical disease progression with the hippocampal region being affected first, whereas in VaD the course of the disease depends on the subtype with an abrupt, stepwise or gradual development of cognitive decline (Figure 3).
Therapeutic treatment have been challenging for VaD, due to the heterogeneity of the underlying pathology [15], but also for Alzheimer’s disease drug development, especially targeting Aß accumulation and tau pathology has been demanding [225,226,227]. The here reviewed neuropathological features highlight the considerable overlap of pathological alterations (Table 6) and shared basic pathomechanisms found in AD and VaD (Figure 4). Endothelial damage, BBB breakdown and hypoperfusion induce oxidative stress, inflammation and demyelination and thus trophic uncoupling in the neurovascular unit [15], aggravating damage to the endothelium and BBB. Leakage of the BBB and embolic events result in microbleeds and lacunar infarcts, microinfarcts and leukoaraiosis. Furthermore, a dysfunctional endothelium and BBB lead to the accumulation of neurotoxic molecules and Aß through impaired clearance, which in turn leads to neurodegeneration [18,81,82]. Deposition of Aß in the media and adventitia of small and mid-sized cerebral and leptomeningeal vessels aggrevates vascular dysfunction, reinforcing a vicious cycle for the development of hemorrhagic and ischemic lesions with consecutive undersupply of essential metabolites to neurons resulting in neuronal dysfunction and ultimately neuronal death.
With regard to the vascular contribution in Alzheimer’s, it is thus essential to target the vascular pathology in the treatment of Alzheimer’s instead of solely focusing on the pure reduction of the Aß or tau pathology. The interaction of Alzheimer’s pathology with vascular pathology is still obscure. Further studies are necessary to evaluate the origin and impact of the vascular pathology and the destroyed homeostasis between neurons, glia and endothelial cells in AD and VaD to provide insight into novel therapeutic strategies. Improving the endothelial integrity and CBF might be a reasonable focus in novel treatment studies for AD, as well as VaD, as BBB dysfunction and reduced CBF are early pathomechanisms in both forms of dementia.
Author Contributions
A.V., A.C.S, L.F. and A.S.W. collected relevant literature. A.V. designed tables and figures with input from A.S.W. A.V., L.F. and A.S.W wrote the manuscript with input from all authors.
Funding
This work was supported by the Dementia Research Switzerland/ Synpasis Foundation Switzerland and the UZH Foundation, the Hurka Foundation as well as the Branco Weiss Fellowship and the Wrangell Habilitation Fellowship to A.S.W.
Acknowledgements
Some figure panels were created with BioRender.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- World Health Organization. Dementia. 2023.
- Hurd, M.D.; Martorell, P.; Delavande, A.; Mullen, K.J.; Langa, K.M. Monetary costs of dementia in the United States. N Engl J Med. 2013, 368, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992, 256, 184–185. [Google Scholar] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [PubMed]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer's disease. Nat Immunol. 2015, 16, 229–236. [Google Scholar]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate Immunity and Neurodegeneration. Annu Rev Med. 2018, 69, 437–449. [Google Scholar] [CrossRef]
- Le Page A, Dupuis G, Frost EH, Larbi A, Pawelec G, Witkowski JM, et al. Role of the peripheral innate immune system in the development of Alzheimer's disease. Exp Gerontol. 2018, 107, 59–66. [Google Scholar]
- Tejera, D.; Heneka, M.T. Microglia in Alzheimer's disease: the good, the bad and the ugly. Curr Alzheimer Res. 2016, 13, 370–380. [Google Scholar] [CrossRef]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019, 51, 404–413. [Google Scholar]
- McQuade, A.; Blurton-Jones, M. Microglia in Alzheimer's Disease: Exploring How Genetics and Phenotype Influence Risk. J Mol Biol. 2019, 431, 1805–1817. [Google Scholar] [PubMed]
- de la Torre, JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke. 2002, 33, 1152–1162. [Google Scholar] [PubMed]
- Pąchalska, M.; Bidzan, L.; Bidzan, M.; Góral-Półrola, J. Vascular Factors and Cognitive Dysfunction in Alzheimer Disease. Med Sci Monit. 2015, 21, 3483–3489. [Google Scholar] [PubMed]
- Nucera A, Hachinski V. Cerebrovascular and Alzheimer disease: fellow travelers or partners in crime? J Neurochem. 2018, 144, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron. 2013, 80, 844–866. [Google Scholar]
- Jellinger, K.A. Pathology and pathogenesis of vascular cognitive impairment-a critical update. Front Aging Neurosci. 2013, 5, 17. [Google Scholar] [CrossRef]
- Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, et al. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain. 2013, 136, 2697–2706. [Google Scholar]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018, 14, 133–150. [Google Scholar]
- Pugazhenthi, S. Metabolic Syndrome and the Cellular Phase of Alzheimer's Disease. Prog Mol Biol Transl Sci. 2017, 146, 243–258. [Google Scholar]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019, 14, 32. [Google Scholar]
- de la Torre, JC. Alzheimer's disease is a vasocognopathy: a new term to describe its nature. Neurol Res. 2004, 26, 517–524. [Google Scholar] [PubMed]
- Claudio, L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer's disease patients. Acta Neuropathol. 1996, 91, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Miyakawa, T.; Katsuragi, S. Vascular changes in the brains with Alzheimer's disease. Jpn J Psychiatry Neurol. 1991, 45, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Hedera, P. Differential degeneration of the cerebral microvasculature in Alzheimer's disease. Neuroreport. 1995, 6, 477–480. [Google Scholar] [CrossRef]
- Launer LJ, Petrovitch H, Ross GW, Markesbery W, White LR. AD brain pathology: vascular origins? Results from the HAAS autopsy study. Neurobiol Aging. 2008, 29, 1587–1590. [Google Scholar] [CrossRef]
- Schneider, J.A. High blood pressure and microinfarcts: a link between vascular risk factors, dementia, and clinical Alzheimer's disease. J Am Geriatr Soc. 2009, 57, 2146–2147. [Google Scholar] [CrossRef]
- O'Brien, J.T.; Thomas, A. Vascular dementia. Lancet. 2015, 386, 1698–1706. [Google Scholar] [CrossRef]
- Pendlebury, S.T.; Rothwell, P.M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 2009, 8, 1006–1018. [Google Scholar]
- Allan LM, Rowan EN, Firbank MJ, Thomas AJ, Parry SW, Polvikoski TM, et al. Long term incidence of dementia, predictors of mortality and pathological diagnosis in older stroke survivors. Brain. 2011, 134, 3716–3727. [Google Scholar] [CrossRef]
- Konno, S.; Meyer, J.S.; Terayama, Y.; Margishvili, G.M.; Mortel, K.F. Classification, diagnosis and treatment of vascular dementia. Drugs Aging. 1997, 11, 361–373. [Google Scholar] [CrossRef]
- Iadecola C, Duering M, Hachinski V, Joutel A, Pendlebury ST, Schneider JA, et al. Vascular Cognitive Impairment and Dementia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2019, 73, 3326–3344. [Google Scholar]
- Scheyer O, Rahman A, Hristov H, Berkowitz C, Isaacson RS, Diaz Brinton R, et al. Female Sex and Alzheimer's Risk: The Menopause Connection. J Prev Alzheimers Dis. 2018, 5, 225–230. [Google Scholar]
- Podcasy, J.L.; Epperson, C.N. Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin Neurosci. 2016, 18, 437–446. [Google Scholar] [PubMed]
- Markus, H.S.; Schmidt, R. Genetics of Vascular Cognitive Impairment. Stroke. 2019, 50, 765–772. [Google Scholar]
- 35. National Institute on Aging. Alzheimer's Disease Genetics Fact Sheet.
- Dichgans, M.; Zietemann, V. Prevention of vascular cognitive impairment. Stroke. 2012, 43, 3137–3146. [Google Scholar]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer's disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar]
- Román, G.C. Vascular dementia prevention: a risk factor analysis. Cerebrovasc Dis. 2005, 20 Suppl. 2, 91–100. [Google Scholar]
- Diniz BS, Butters MA, Albert SM, Dew MA, Reynolds CF, 3rd. Late-life depression and risk of vascular dementia and Alzheimer's disease: systematic review and meta-analysis of community-based cohort studies. Br J Psychiatry. 2013, 202, 329–335. [Google Scholar]
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer disease. Dis Mon. 2010, 56, 484–546. [Google Scholar]
- Lee, A.Y. Vascular dementia. Chonnam Med J. 2011, 47, 66–71. [Google Scholar]
- Braaten, A.J.; Parsons, T.D.; McCue, R.; Sellers, A.; Burns, W.J. Neurocognitive differential diagnosis of dementing diseases: Alzheimer's Dementia, Vascular Dementia, Frontotemporal Dementia, and Major Depressive Disorder. Int J Neurosci. 2006, 116, 1271–1293. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, S.; Wicklund, A.H.; Salmon, D.P. The neuropsychological profile of Alzheimer disease. Cold Spring Harb Perspect Med. 2012, 2, a006171. [Google Scholar] [CrossRef] [PubMed]
- Mathias, J.L.; Burke, J. Cognitive functioning in Alzheimer's and vascular dementia: a meta-analysis. Neuropsychology. 2009, 23, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.L.; Emery, T.; Hodges, J.R. Distinctive cognitive profiles in Alzheimer's disease and subcortical vascular dementia. J Neurol Neurosurg Psychiatry. 2004, 75, 61–71. [Google Scholar]
- Murray, M.E.; Knopman, D.S.; Dickson, D.W. Vascular dementia: clinical, neuroradiologic and neuropathologic aspects. Panminerva Med. 2007, 49, 197–207. [Google Scholar]
- Kalaria, R.N. The pathology and pathophysiology of vascular dementia. Neuropharmacology. 2018, 134, 226–239. [Google Scholar]
- Jack CR, Jr. , Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar]
- Hachinski, V.C.; Bowler, J.V. Vascular dementia. Neurology. 1993, 43, 2159–2160. [Google Scholar] [CrossRef]
- Skrobot OA, Black SE, Chen C, DeCarli C, Erkinjuntti T, Ford GA, et al. Progress toward standardized diagnosis of vascular cognitive impairment: Guidelines from the Vascular Impairment of Cognition Classification Consensus Study. Alzheimers Dement. 2018, 14, 280–292. [Google Scholar]
- Scheltens P, Leys D, Barkhof F, Huglo D, Weinstein HC, Vermersch P, et al. Atrophy of medial temporal lobes on MRI in "probable" Alzheimer's disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry. 1992, 55, 967–972. [Google Scholar]
- Rau, A.; Urbach, H. The MTA score-simple and reliable, the best for now? Eur Radiol. 2021, 31, 9057–9059. [Google Scholar] [CrossRef] [PubMed]
- Mukaetova-Ladinska EB, Abdel-All Z, Mugica ES, Li M, Craggs LJ, Oakley AE, et al. Tau proteins in the temporal and frontal cortices in patients with vascular dementia. J Neuropathol Exp Neurol. 2015, 74, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Bibl M, Mollenhauer B, Esselmann H, Schneider M, Lewczuk P, Welge V, et al. Cerebrospinal fluid neurochemical phenotypes in vascular dementias: original data and mini-review. Dement Geriatr Cogn Disord. 2008, 25, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Thijssen EH, Verberk IMW, Kindermans J, Abramian A, Vanbrabant J, Ball AJ, et al. Differential diagnostic performance of a panel of plasma biomarkers for different types of dementia. Alzheimers Dement (Amst). 2022, 14, e12285. [Google Scholar]
- Leuzy, A.; Mattsson-Carlgren, N.; Palmqvist, S.; Janelidze, S.; Dage, J.L.; Hansson, O. Blood-based biomarkers for Alzheimer's disease. EMBO Mol Med. 2022, 14, e14408. [Google Scholar]
- Teunissen CE, Verberk IMW, Thijssen EH, Vermunt L, Hansson O, Zetterberg H, et al. Blood-based biomarkers for Alzheimer's disease: towards clinical implementation. Lancet Neurol. 2022, 21, 66–77. [Google Scholar]
- Arvanitakis, Z.; Capuano, A.W.; Leurgans, S.E.; Bennett, D.A.; Schneider, J.A. Relation of cerebral vessel disease to Alzheimer's disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 2016, 15, 934–943. [Google Scholar]
- Lusis, AJ. Atherosclerosis. Nature. 2000, 407, 233–241. [Google Scholar]
- Shabir, O.; Berwick, J.; Francis, S.E. Neurovascular dysfunction in vascular dementia, Alzheimer's and atherosclerosis. BMC Neurosci. 2018, 19, 62. [Google Scholar]
- Fava, C.; Montagnana, M. Atherosclerosis Is an Inflammatory Disease which Lacks a Common Anti-inflammatory Therapy: How Human Genetics Can Help to This Issue. A Narrative Review. Front Pharmacol. 2018, 9, 55. [Google Scholar] [CrossRef]
- Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R, et al. Circle of Willis atherosclerosis: association with Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 2007, 113, 13–21. [Google Scholar]
- van Oijen M, de Jong FJ, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Atherosclerosis and risk for dementia. Ann Neurol. 2007, 61, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Grinberg, L.T.; Attems, J. Vascular dementia: different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp Gerontol. 2012, 47, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, A.M.; van Westen, D.; Stomrud, E.; Engström, G.; Nägga, K.; Hansson, O. Midlife Atherosclerosis and Development of Alzheimer or Vascular Dementia. Ann Neurol. 2020, 87, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Genetics of atherosclerosis. Trends Genet. 2012, 28, 267–275. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Skoog I, Wallin A, Fredman P, Hesse C, Aevarsson O, Karlsson I, et al. A population study on blood-brain barrier function in 85-year-olds: relation to Alzheimer's disease and vascular dementia. Neurology. 1998, 50, 966–971. [Google Scholar] [CrossRef]
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Kurz, C.; Walker, L.; Rauchmann, B.S.; Perneczky, R. Dysfunction of the blood-brain barrier in Alzheimer's disease: Evidence from human studies. Neuropathol Appl Neurobiol. 2022, 48, e12782. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Pa, J.; Sweeney, M.D.; Toga, A.W.; Zlokovic, B.V. Brain imaging of neurovascular dysfunction in Alzheimer's disease. Acta Neuropathol. 2016, 131, 687–707. [Google Scholar]
- van de Haar HJ, Burgmans S, Jansen JF, van Osch MJ, van Buchem MA, Muller M, et al. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology. 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The neurovascular unit - concept review. Acta Physiol (Oxf). 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Bell, A.H.; Miller, S.L.; Castillo-Melendez, M.; Malhotra, A. The Neurovascular Unit: Effects of Brain Insults During the Perinatal Period. Front Neurosci. 2019, 13, 1452. [Google Scholar] [CrossRef]
- van de Haar HJ, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, Wong SM, et al. Neurovascular unit impairment in early Alzheimer's disease measured with magnetic resonance imaging. Neurobiol Aging. 2016, 45, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Bracko, O.; Cruz Hernández, J.C.; Park, L.; Nishimura, N.; Schaffer, C.B. Causes and consequences of baseline cerebral blood flow reductions in Alzheimer's disease. J Cereb Blood Flow Metab. 2021, 41, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Ali M, Falkenhain K, Njiru BN, Murtaza-Ali M, Ruiz-Uribe NE, Haft-Javaherian M, et al. VEGF signalling causes stalls in brain capillaries and reduces cerebral blood flow in Alzheimer's mice. Brain. 2022, 145, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular defects and faulty amyloid-β vascular clearance in Alzheimer's disease. J Alzheimers Dis. 2013, 33 Suppl. 1, S87–S100. [Google Scholar] [CrossRef]
- Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer's disease. Biochim Biophys Acta. 2016, 1862, 887–900. [Google Scholar] [PubMed]
- Vogelgesang S, Warzok RW, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, et al. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2004, 1, 121–125. [Google Scholar] [CrossRef] [PubMed]
- van Assema DM, Lubberink M, Bauer M, van der Flier WM, Schuit RC, Windhorst AD, et al. Blood-brain barrier P-glycoprotein function in Alzheimer's disease. Brain. 2012, 135, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Pathan, N.; Shende, P. Tailoring of P-glycoprotein for effective transportation of actives across blood-brain-barrier. J Control Release. 2021, 335, 398–407. [Google Scholar] [CrossRef]
- Deo AK, Borson S, Link JM, Domino K, Eary JF, Ke B, et al. Activity of P-Glycoprotein, a β-Amyloid Transporter at the Blood-Brain Barrier, Is Compromised in Patients with Mild Alzheimer Disease. J Nucl Med. 2014, 55, 1106–1111. [Google Scholar] [CrossRef]
- Maeng HJ, Kim MH, Jin HE, Shin SM, Tsuruo T, Kim SG, et al. Functional induction of P-glycoprotein in the blood-brain barrier of streptozotocin-induced diabetic rats: evidence for the involvement of nuclear factor-kappaB, a nitrosative stress-sensitive transcription factor, in the regulation. Drug Metab Dispos. 2007, 35, 1996–2005. [Google Scholar] [CrossRef]
- Ueno M, Nakagawa T, Huang CL, Ueki M, Kusaka T, Hosomi N, et al. The expression of P-glycoprotein is increased in vessels with blood-brain barrier impairment in a stroke-prone hypertensive model. Neuropathol Appl Neurobiol. 2009, 35, 147–155. [Google Scholar] [CrossRef]
- Gong, M.; Jia, J. Contribution of blood-brain barrier-related blood-borne factors for Alzheimer's disease vs. vascular dementia diagnosis: A pilot study. Front Neurosci. 2022, 16, 949129. [Google Scholar]
- Hachinski VC, Iliff LD, Zilhka E, Du Boulay GH, McAllister VL, Marshall J, et al. Cerebral blood flow in dementia. Arch Neurol. 1975, 32, 632–637. [Google Scholar] [CrossRef]
- Beason-Held, L.L.; Moghekar, A.; Zonderman, A.B.; Kraut, M.A.; Resnick, S.M. Longitudinal changes in cerebral blood flow in the older hypertensive brain. Stroke. 2007, 38, 1766–1773. [Google Scholar] [CrossRef]
- Korte, N.; Nortley, R.; Attwell, D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer's disease. Acta Neuropathol. 2020, 140, 793–810. [Google Scholar]
- Cohen, R.A. Hypertension and cerebral blood flow: implications for the development of vascular cognitive impairment in the elderly. Stroke. 2007, 38, 1715–1717. [Google Scholar] [PubMed]
- Román, G.C. Brain hypoperfusion: a critical factor in vascular dementia. Neurol Res. 2004, 26, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Schuff N, Matsumoto S, Kmiecik J, Studholme C, Du A, Ezekiel F, et al. Cerebral blood flow in ischemic vascular dementia and Alzheimer's disease, measured by arterial spin-labeling magnetic resonance imaging. Alzheimers Dement. 2009, 5, 454–462. [Google Scholar]
- Yao, H.; Sadoshima, S.; Kuwabara, Y.; Ichiya, Y.; Fujishima, M. Cerebral blood flow and oxygen metabolism in patients with vascular dementia of the Binswanger type. Stroke. 1990, 21, 1694–1699. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.Z.; Zhang, J.J.; Liu, H.; Wu, G.Y.; Xiong, L.; Shu, M. Regional cerebral blood flow and cerebrovascular reactivity in Alzheimer's disease and vascular dementia assessed by arterial spinlabeling magnetic resonance imaging. Curr Neurovasc Res. 2013, 10, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci. 2018, 21, 1318–1331. [Google Scholar]
- Yoshikawa T, Murase K, Oku N, Imaizumi M, Takasawa M, Rishu P, et al. Heterogeneity of cerebral blood flow in Alzheimer disease and vascular dementia. AJNR Am J Neuroradiol. 2003, 24, 1341–1347. [Google Scholar]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017, 18, 419–434. [Google Scholar]
- Enciu, A.M.; Constantinescu, S.N.; Popescu, L.M.; Mureşanu, D.F.; Popescu, B.O. Neurobiology of vascular dementia. J Aging Res. 2011, 2011, 401604. [Google Scholar] [CrossRef]
- Starkstein SE, Sabe L, Vazquez S, Teson A, Petracca G, Chemerinski E, et al. Neuropsychological, psychiatric, and cerebral blood flow findings in vascular dementia and Alzheimer's disease. Stroke. 1996, 27, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.W.; Kim, B.S.; Park, J.K.; Kim, S.Y.; Kim, E.N.; Sohn, H.S. Analysis of cerebral blood flow of subcortical vascular dementia with single photon emission computed tomography: adaptation of statistical parametric mapping. J Neurol Sci. 2002, 203-204, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty M, Beason-Held L, An Y, Kraut MA, Resnick SM. APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol. 2010, 67, 93–98. [Google Scholar]
- Filippini N, Ebmeier KP, MacIntosh BJ, Trachtenberg AJ, Frisoni GB, Wilcock GK, et al. Differential effects of the APOE genotype on brain function across the lifespan. Neuroimage. 2011, 54, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Michels L, Warnock G, Buck A, Macauda G, Leh SE, Kaelin AM, et al. Arterial spin labeling imaging reveals widespread and Aβ-independent reductions in cerebral blood flow in elderly apolipoprotein epsilon-4 carriers. J Cereb Blood Flow Metab. 2016, 36, 581–595. [Google Scholar] [CrossRef]
- McKiernan EF, Mak E, Dounavi ME, Wells K, Ritchie C, Williams G, et al. Regional hyperperfusion in cognitively normal APOE ε4 allele carriers in mid-life: analysis of ASL pilot data from the PREVENT-Dementia cohort. J Neurol Neurosurg Psychiatry. 2020, 91, 861–866. [Google Scholar] [CrossRef]
- Tomimoto, H.; Ohtani, R.; Shibata, M.; Nakamura, N.; Ihara, M. Loss of cholinergic pathways in vascular dementia of the Binswanger type. Dement Geriatr Cogn Disord. 2005, 19, 282–288. [Google Scholar] [CrossRef]
- Santos, C.Y.; Snyder, P.J.; Wu, W.C.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic relationship between Alzheimer's disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis. Alzheimers Dement (Amst). 2017, 7, 69–87. [Google Scholar]
- Kalaria, R. Similarities between Alzheimer's disease and vascular dementia. J Neurol Sci. 2002, 203-204, 29–34. [Google Scholar] [CrossRef]
- Kozberg, M.G.; Perosa, V.; Gurol, M.E.; van Veluw, S.J. A practical approach to the management of cerebral amyloid angiopathy. Int J Stroke. 2021, 16, 356–369. [Google Scholar] [CrossRef]
- Jellinger, K.A. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm (Vienna). 2002, 109, 813–836. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Jellinger, K.; Thal, D.R.; Van Nostrand, W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2011, 37, 75–93. [Google Scholar] [PubMed]
- Hecht, M.; Krämer, L.M.; von Arnim, C.A.F.; Otto, M.; Thal, D.R. Capillary cerebral amyloid angiopathy in Alzheimer's disease: association with allocortical/hippocampal microinfarcts and cognitive decline. Acta Neuropathol. 2018, 135, 681–694. [Google Scholar] [PubMed]
- Yamada, M.; Naiki, H. Cerebral amyloid angiopathy. Prog Mol Biol Transl Sci. 2012, 107, 41–78. [Google Scholar]
- Attems, J.; Jellinger, K.A.; Lintner, F. Alzheimer's disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol. 2005, 110, 222–231. [Google Scholar] [PubMed]
- Pezzini, A.; Del Zotto, E.; Volonghi, I.; Giossi, A.; Costa, P.; Padovani, A. Cerebral amyloid angiopathy: a common cause of cerebral hemorrhage. Curr Med Chem. 2009, 16, 2498–2513. [Google Scholar] [CrossRef]
- Tian, J.; Shi, J.; Bailey, K.; Mann, D.M. Negative association between amyloid plaques and cerebral amyloid angiopathy in Alzheimer's disease. Neurosci Lett. 2003, 352, 137–140. [Google Scholar] [CrossRef]
- Masuda, J.; Tanaka, K.; Ueda, K.; Omae, T. Autopsy study of incidence and distribution of cerebral amyloid angiopathy in Hisayama, Japan. Stroke. 1988, 19, 205–210. [Google Scholar]
- Xu, D.; Yang, C.; Wang, L. Cerebral amyloid angiopathy in aged Chinese: a clinico-neuropathological study. Acta Neuropathol. 2003, 106, 89–91. [Google Scholar] [CrossRef]
- Haglund, M.; Sjöbeck, M.; Englund, E. Severe cerebral amyloid angiopathy characterizes an underestimated variant of vascular dementia. Dement Geriatr Cogn Disord. 2004, 18, 132–137. [Google Scholar] [CrossRef]
- Rannikmäe, K.; Samarasekera, N.; Martînez-Gonzâlez, N.A.; Al-Shahi Salman, R.; Sudlow, C.L. Genetics of cerebral amyloid angiopathy: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2013, 84, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M. Cerebral amyloid angiopathy: emerging concepts. J Stroke. 2015, 17, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Verghese, P.B.; Castellano, J.M.; Holtzman, D.M. Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 2011, 10, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Nicoll JA, Burnett C, Love S, Graham DI, Dewar D, Ironside JW, et al. High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann Neurol. 1997, 41, 716–721. [Google Scholar] [CrossRef]
- Biffi A, Shulman JM, Jagiella JM, Cortellini L, Ayres AM, Schwab K, et al. Genetic variation at CR1 increases risk of cerebral amyloid angiopathy. Neurology. 2012, 78, 334–341. [Google Scholar] [CrossRef]
- Pandey, P.; Pradhan, S.; Mittal, B. Presenilin gene predisposes to late-onset degenerative but not vascular dementia: a comparative study of PS1 and ApoE genes in a North Indian Cohort. Dement Geriatr Cogn Disord. 2007, 24, 151–161. [Google Scholar] [CrossRef]
- Biffi, A.; Greenberg, S.M. Cerebral amyloid angiopathy: a systematic review. J Clin Neurol. 2011, 7, 1–9. [Google Scholar] [CrossRef]
- Van der Flier, W.M.; Cordonnier, C. Microbleeds in vascular dementia: clinical aspects. Exp Gerontol. 2012, 47, 853–857. [Google Scholar] [CrossRef]
- Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009, 8, 165–174. [Google Scholar] [CrossRef]
- Cordonnier, C.; van der Flier, W.M.; Sluimer, J.D.; Leys, D.; Barkhof, F.; Scheltens, P. Prevalence and severity of microbleeds in a memory clinic setting. Neurology. 2006, 66, 1356–1360. [Google Scholar] [CrossRef]
- Brundel M, Heringa SM, de Bresser J, Koek HL, Zwanenburg JJ, Jaap Kappelle L, et al. High prevalence of cerebral microbleeds at 7Tesla MRI in patients with early Alzheimer's disease. J Alzheimers Dis. 2012, 31, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Yates, P.A.; Villemagne, V.L.; Ellis, K.A.; Desmond, P.M.; Masters, C.L.; Rowe, C.C. Cerebral microbleeds: a review of clinical, genetic, and neuroimaging associations. Front Neurol. 2014, 4, 205. [Google Scholar] [CrossRef] [PubMed]
- De Reuck J, Auger F, Durieux N, Deramecourt V, Cordonnier C, Pasquier F, et al. Topography of Cortical Microbleeds in Alzheimer's Disease with and without Cerebral Amyloid Angiopathy: A Post-Mortem 7.0-Tesla MRI Study. Aging Dis. 2015, 6, 437–443. [Google Scholar] [CrossRef]
- Shams S, Martola J, Granberg T, Li X, Shams M, Fereshtehnejad SM, et al. Cerebral microbleeds: different prevalence, topography, and risk factors depending on dementia diagnosis—the Karolinska Imaging Dementia Study. AJNR Am J Neuroradiol. 2015, 36, 661–666. [Google Scholar] [CrossRef]
- Pettersen JA, Sathiyamoorthy G, Gao FQ, Szilagyi G, Nadkarni NK, St George-Hyslop P, et al. Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch Neurol. 2008, 65, 790–795. [Google Scholar]
- Dichgans, M.; Holtmannspötter, M.; Herzog, J.; Peters, N.; Bergmann, M.; Yousry, T.A. Cerebral microbleeds in CADASIL: a gradient-echo magnetic resonance imaging and autopsy study. Stroke. 2002, 33, 67–71. [Google Scholar] [CrossRef]
- De Reuck J, Auger F, Durieux N, Deramecourt V, Maurage CA, Cordonnier C, et al. Frequency and topography of small cerebrovascular lesions in vascular and in mixed dementia: a post-mortem 7-tesla magnetic resonance imaging study with neuropathological correlates. Folia Neuropathol. 2017, 55, 31–37. [Google Scholar]
- Maxwell SS, Jackson CA, Paternoster L, Cordonnier C, Thijs V, Al-Shahi Salman R, et al. Genetic associations with brain microbleeds: Systematic review and meta-analyses. Neurology. 2011, 77, 158–167. [Google Scholar] [CrossRef]
- Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Niessen WJ, Hofman A, et al. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology. 2008, 70, 1208–1214. [Google Scholar] [CrossRef]
- Caplan, L.R. Lacunar infarction and small vessel disease: pathology and pathophysiology. J Stroke. 2015, 17, 2–6. [Google Scholar] [CrossRef]
- Cai Z, Wang C, He W, Tu H, Tang Z, Xiao M, et al. Cerebral small vessel disease and Alzheimer's disease. Clin Interv Aging. 2015, 10, 1695–1704. [Google Scholar]
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer's disease--lessons from pathology. BMC Med. 2014, 12, 206. [Google Scholar] [CrossRef]
- Snowdon, D.A.; Greiner, L.H.; Mortimer, J.A.; Riley, K.P.; Greiner, P.A.; Markesbery, W.R. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. Jama. 1997, 277, 813–817. [Google Scholar] [CrossRef] [PubMed]
- De Reuck J, Auger F, Durieux N, Cordonnier C, Deramecourt V, Pasquier F, et al. Topographic distribution of white matter changes and lacunar infarcts in neurodegenerative and vascular dementia syndromes: A post-mortem 7.0-tesla magnetic resonance imaging study. Eur Stroke J. 2016, 1, 122–129. [Google Scholar] [CrossRef]
- Uchino, M. [The pathomechanism and treatment of CADASIL]. Rinsho Shinkeigaku. 2011, 51, 945–948. [Google Scholar] [CrossRef]
- Traylor M, Adib-Samii P, Harold D, Dichgans M, Williams J, Lewis CM, et al. Shared genetic contribution to Ischaemic Stroke and Alzheimer's Disease. Ann Neurol. 2016, 79, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Liu Y, Laakso MP, Karonen JO, Vanninen RL, Nuutinen J, Soimakallio S, et al. Apolipoprotein E polymorphism and acute ischemic stroke: a diffusion- and perfusion-weighted magnetic resonance imaging study. J Cereb Blood Flow Metab. 2002, 22, 1336–1342. [Google Scholar] [CrossRef]
- Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil. Lancet Neurol. 2009, 8, 643–653.
- Tikka S, Baumann M, Siitonen M, Pasanen P, Pöyhönen M, Myllykangas L, et al. CADASIL and CARASIL. Brain Pathol. 2014, 24, 525–544. [Google Scholar] [CrossRef]
- Khandelwal D, Mathur V, Vyas A, Ghunawat J, Bagaria AK. CARASIL - A Review of Patients from India. Neurol India. 2021, 69, 1359–1362. [Google Scholar]
- Brundel, M.; de Bresser, J.; van Dillen, J.J.; Kappelle, L.J.; Biessels, G.J. Cerebral microinfarcts: a systematic review of neuropathological studies. J Cereb Blood Flow Metab. 2012, 32, 425–436. [Google Scholar] [CrossRef]
- Hartmann DA, Hyacinth HI, Liao FF, Shih AY. Does pathology of small venules contribute to cerebral microinfarcts and dementia? J Neurochem. 2018, 144, 517–526. [CrossRef]
- Arvanitakis, Z.; Leurgans, S.E.; Barnes, L.L.; Bennett, D.A.; Schneider, J.A. Microinfarct pathology, dementia, and cognitive systems. Stroke. 2011, 42, 722–727. [Google Scholar] [CrossRef]
- Corrada, M.M.; Sonnen, J.A.; Kim, R.C.; Kawas, C.H. Microinfarcts are common and strongly related to dementia in the oldest-old: The 90+ study. Alzheimers Dement. 2016, 12, 900–908. [Google Scholar] [CrossRef]
- Smith, E.E.; Schneider, J.A.; Wardlaw, J.M.; Greenberg, S.M. Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 2012, 11, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Ihara, M.; Fujita, Y.; Ito, H.; Takahashi, R.; Tomimoto, H. Cortical microinfarcts in Alzheimer's disease and subcortical vascular dementia. Neuroreport. 2009, 20, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Rost, N.S.; Etherton, M. Cerebral Small Vessel Disease. Continuum (Minneap Minn). 2020, 26, 332–352. [Google Scholar] [CrossRef] [PubMed]
- Vinke EJ, Yilmaz P, van der Toorn JE, Fakhry R, Frenzen K, Dubost F, et al. Intracranial arteriosclerosis is related to cerebral small vessel disease: a prospective cohort study. Neurobiol Aging. 2021, 105, 16–24. [Google Scholar] [CrossRef]
- O'Sullivan, M. Leukoaraiosis. Pract Neurol. 2008, 8, 26–38. [Google Scholar] [CrossRef] [PubMed]
- van Gijn, J. Leukoaraiosis and vascular dementia. Neurology. 1998, 51, S3–S8. [Google Scholar] [CrossRef]
- Englund, E. Neuropathology of white matter changes in Alzheimer's disease and vascular dementia. Dement Geriatr Cogn Disord. 1998, 9 Suppl. 1, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Wallin, A.; Uhlemann, C.; Gottfries, C.G. White-matter lesions on CT in Alzheimer patients: relation to clinical symptomatology and vascular factors. Acta Neurol Scand. 1991, 83, 187–193. [Google Scholar] [CrossRef]
- Erkinjuntti T, Ketonen L, Sulkava R, Sipponen J, Vuorialho M, Iivanainen M. Do white matter changes on MRI and CT differentiate vascular dementia from Alzheimer's disease? J Neurol Neurosurg Psychiatry. 1987, 50, 37–42.
- Bozzali M, Falini A, Franceschi M, Cercignani M, Zuffi M, Scotti G, et al. White matter damage in Alzheimer's disease assessed in vivo using diffusion tensor magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2002, 72, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Barber R, Scheltens P, Gholkar A, Ballard C, McKeith I, Ince P, et al. White matter lesions on magnetic resonance imaging in dementia with Lewy bodies, Alzheimer's disease, vascular dementia, and normal aging. J Neurol Neurosurg Psychiatry. 1999, 67, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Szolnoki, Z. Pathomechanism of leukoaraiosis: a molecular bridge between the genetic, biochemical, and clinical processes (a mitochondrial hypothesis). Neuromolecular Med. 2007, 9, 21–33. [Google Scholar] [CrossRef]
- Bronge L, Fernaeus SE, Blomberg M, Ingelson M, Lannfelt L, Isberg B, et al. White matter lesions in Alzheimer patients are influenced by apolipoprotein E genotype. Dement Geriatr Cogn Disord. 1999, 10, 89–96. [Google Scholar] [CrossRef]
- Schmidt R, Schmidt H, Fazekas F, Schumacher M, Niederkorn K, Kapeller P, et al. Apolipoprotein E polymorphism and silent microangiopathy-related cerebral damage. Results of the Austrian Stroke Prevention Study. Stroke. 1997, 28, 951–956. [Google Scholar] [CrossRef]
- Szolnoki, Z.; Somogyvári, F.; Kondacs, A.; Szabó, M.; Fodor, L. Evaluation of the roles of common genetic mutations in leukoaraiosis. Acta Neurol Scand. 2001, 104, 281–287. [Google Scholar] [CrossRef]
- Dichgans, M. CADASIL: a monogenic condition causing stroke and subcortical vascular dementia. Cerebrovasc Dis. 2002, 13 Suppl 2, 37–41. [Google Scholar] [CrossRef]
- Tariq, S.; Barber, P.A. Dementia risk and prevention by targeting modifiable vascular risk factors. J Neurochem. 2018, 144, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Perl, D.P. Neuropathology of Alzheimer's disease. Mt Sinai J Med. 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Enkirch SJ, Traschütz A, Müller A, Widmann CN, Gielen GH, Heneka MT, et al. The ERICA Score: An MR Imaging-based Visual Scoring System for the Assessment of Entorhinal Cortex Atrophy in Alzheimer Disease. Radiology. 2018, 288, 226–333. [Google Scholar] [CrossRef] [PubMed]
- Koval I, Schiratti JB, Routier A, Bacci M, Colliot O, Allassonnière S, et al. Spatiotemporal Propagation of the Cortical Atrophy: Population and Individual Patterns. Front Neurol. 2018, 9, 235. [Google Scholar] [CrossRef]
- Koval I, Bône A, Louis M, Lartigue T, Bottani S, Marcoux A, et al. AD Course Map charts Alzheimer's disease progression. Sci Rep. 2021, 11, 8020. [Google Scholar] [CrossRef]
- O'Brien JT, Paling S, Barber R, Williams ED, Ballard C, McKeith IG, et al. Progressive brain atrophy on serial MRI in dementia with Lewy bodies, AD, and vascular dementia. Neurology. 2001, 56, 1386–1388. [Google Scholar] [CrossRef]
- van de Pol L, Gertz HJ, Scheltens P, Wolf H. Hippocampal atrophy in subcortical vascular dementia. Neurodegener Dis. 2011, 8, 465–469. [Google Scholar] [CrossRef]
- Kril, J.J.; Patel, S.; Harding, A.J.; Halliday, G.M. Patients with vascular dementia due to microvascular pathology have significant hippocampal neuronal loss. J Neurol Neurosurg Psychiatry. 2002, 72, 747–751. [Google Scholar] [CrossRef]
- Duering M, Righart R, Csanadi E, Jouvent E, Hervé D, Chabriat H, et al. Incident subcortical infarcts induce focal thinning in connected cortical regions. Neurology. 2012, 79, 2025–2028. [Google Scholar] [CrossRef]
- Seo SW, Lee JM, Im K, Park JS, Kim SH, Kim ST, et al. Cortical thinning related to periventricular and deep white matter hyperintensities. Neurobiol Aging. 2012, 33, 1156–1167. [Google Scholar] [CrossRef]
- Kim CH, Seo SW, Kim GH, Shin JS, Cho H, Noh Y, et al. Cortical thinning in subcortical vascular dementia with negative 11C-PiB PET. J Alzheimers Dis. 2012, 31, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Vinters HV, Ellis WG, Zarow C, Zaias BW, Jagust WJ, Mack WJ, et al. Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol. 2000, 59, 931–945. [Google Scholar] [CrossRef]
- Mann, D.M.; Yates, P.O.; Marcyniuk, B. The nucleus basalis of Meynert in multi-infarct (vascular) dementia. Acta Neuropathol. 1986, 71, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Foster V, Oakley AE, Slade JY, Hall R, Polvikoski TM, Burke M, et al. Pyramidal neurons of the prefrontal cortex in post-stroke, vascular and other ageing-related dementias. Brain. 2014, 137, 2509–2521. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, T.; Wulff, P. The hippocampus in aging and disease: From plasticity to vulnerability. Neuroscience. 2015, 309, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.K.; Chang, R.C.; Pearce, R.K.; Gentleman, S.M. Nucleus basalis of Meynert revisited: anatomy, history and differential involvement in Alzheimer's and Parkinson's disease. Acta Neuropathol. 2015, 129, 527–540. [Google Scholar] [CrossRef]
- Lyness, S.A.; Zarow, C.; Chui, H.C. Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta-analysis. Neurobiol Aging. 2003, 24, 1–23. [Google Scholar] [CrossRef]
- Gannon, M.; Che, P.; Chen, Y.; Jiao, K.; Roberson, E.D.; Wang, Q. Noradrenergic dysfunction in Alzheimer's disease. Front Neurosci. 2015, 9, 220. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Slater, C.; Wang, Q. Alzheimer's disease: An evolving understanding of noradrenergic involvement and the promising future of electroceutical therapies. Clin Transl Med. 2021, 11, e397. [Google Scholar] [CrossRef]
- Aaldijk, E.; Vermeiren, Y. The role of serotonin within the microbiota-gut-brain axis in the development of Alzheimer's disease: A narrative review. Ageing Res Rev. 2022, 75, 101556. [Google Scholar] [CrossRef] [PubMed]
- Gray, F.; Polivka, M.; Viswanathan, A.; Baudrimont, M.; Bousser, M.G.; Chabriat, H. Apoptosis in cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy. J Neuropathol Exp Neurol. 2007, 66, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, O.; Coleman, M. Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis. In: Huang X, editor. Alzheimer’s Disease: Drug Discovery. Brisbane (AU): Exon Publications Copyright: The Authors.; 2020.
- Rao, Y.L.; Ganaraja, B.; Murlimanju, B.V.; Joy, T.; Krishnamurthy, A.; Agrawal, A. Hippocampus and its involvement in Alzheimer's disease: a review. 3 Biotech. 2022, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Rauchmann BS, Brendel M, Franzmeier N, Trappmann L, Zaganjori M, Ersoezlue E, et al. Microglial Activation and Connectivity in Alzheimer Disease and Aging. Ann Neurol. 2022, 92, 768–781. [Google Scholar] [CrossRef]
- Xiang X, Wind K, Wiedemann T, Blume T, Shi Y, Briel N, et al. Microglial activation states drive glucose uptake and FDG-PET alterations in neurodegenerative diseases. Sci Transl Med. 2021, 13, eabe5640. [CrossRef]
- Kirvell, S.L.; Elliott, M.S.; Kalaria, R.N.; Hortobágyi, T.; Ballard, C.G.; Francis, P.T. Vesicular glutamate transporter and cognition in stroke: a case-control autopsy study. Neurology. 2010, 75, 1803–1809. [Google Scholar] [CrossRef]
- Benarroch, E.E. Glutamatergic synaptic plasticity and dysfunction in Alzheimer disease: Emerging mechanisms. Neurology. 2018, 91, 125–132. [Google Scholar] [CrossRef]
- Prokšelj, T.; Jerin, A.; Muck-Seler, D.; Kogoj, A. Decreased platelet serotonin concentration in Alzheimer's disease with involuntary emotional expression disorder. Neurosci Lett. 2014, 578, 71–74. [Google Scholar] [CrossRef]
- Perry, E.; Ziabreva, I.; Perry, R.; Aarsland, D.; Ballard, C. Absence of cholinergic deficits in "pure" vascular dementia. Neurology. 2005, 64, 132–133. [Google Scholar] [CrossRef] [PubMed]
- Keverne, J.S.; Low, W.C.; Ziabreva, I.; Court, J.A.; Oakley, A.E.; Kalaria, R.N. Cholinergic neuronal deficits in CADASIL. Stroke. 2007, 38, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Podlisny, M.B. Deciphering the genetic basis of Alzheimer's disease. Annu Rev Genomics Hum Genet. 2002, 3, 67–99. [Google Scholar] [CrossRef] [PubMed]
- Hu Z, Hu K, Wang R, Gu Y, Ouyang W, Zhou J, et al. Differentially expressed genes accompanying neurobehavioral deficits in a modified rat model of vascular dementia. Neurosci Lett. 2021, 750, 135774. [Google Scholar] [CrossRef] [PubMed]
- Gooch, J.; Wilcock, D.M. Animal Models of Vascular Cognitive Impairment and Dementia (VCID). Cell Mol Neurobiol. 2016, 36, 233–239. [Google Scholar] [CrossRef]
- Smith, M.Z.; Nagy, Z.; Esiri, M.M. Cell cycle-related protein expression in vascular dementia and Alzheimer's disease. Neurosci Lett. 1999, 271, 45–48. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Ballard, C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord. 1999, 13 Suppl 3, S115–S123. [Google Scholar] [CrossRef]
- van der Flier WM, Skoog I, Schneider JA, Pantoni L, Mok V, Chen CLH, et al. Vascular cognitive impairment. Nature Reviews Disease Primers. 2018, 4, 18003. [Google Scholar] [CrossRef]
- Kim SE, Kim HJ, Jang H, Weiner MW, DeCarli C, Na DL, et al. Interaction between Alzheimer's Disease and Cerebral Small Vessel Disease: A Review Focused on Neuroimaging Markers. Int J Mol Sci. 2022, 2022.
- Lewis H, Beher D, Cookson N, Oakley A, Piggott M, Morris CM, et al. Quantification of Alzheimer pathology in ageing and dementia: age-related accumulation of amyloid-beta(42) peptide in vascular dementia. Neuropathol Appl Neurobiol. 2006, 32, 103–118. [Google Scholar] [CrossRef]
- Noh Y, Seo SW, Jeon S, Lee JM, Kim JH, Kim GH, et al. White matter hyperintensities are associated with amyloid burden in APOE4 non-carriers. J Alzheimers Dis. 2014, 40, 877–886. [Google Scholar] [CrossRef]
- Frontzkowski L, Ewers M, Brendel M, Biel D, Ossenkoppele R, Hager P, et al. Earlier Alzheimer's disease onset is associated with tau pathology in brain hub regions and facilitated tau spreading. Nat Commun. 2022, 13, 4899. [Google Scholar] [CrossRef]
- van Norden AG, van Dijk EJ, de Laat KF, Scheltens P, Olderikkert MG, de Leeuw FE. Dementia: Alzheimer pathology and vascular factors: from mutually exclusive to interaction. Biochim Biophys Acta. 2012, 1822, 340–349. [Google Scholar] [CrossRef]
- Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF, et al. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef]
- Pellerin, L. Food for thought: the importance of glucose and other energy substrates for sustaining brain function under varying levels of activity. Diabetes Metab. 2010, 2010 Suppl 3, S59–S63. [Google Scholar] [CrossRef]
- Heiss WD, Zimmermann-Meinzingen S. PET imaging in the differential diagnosis of vascular dementia. J Neurol Sci. 2012, 322, 268–273. [Google Scholar] [CrossRef]
- McKenna, M.C.; Schuck, P.F.; Ferreira, G.C. Fundamentals of CNS energy metabolism and alterations in lysosomal storage diseases. J Neurochem. 2019, 148, 590–599. [Google Scholar] [CrossRef]
- Kerrouche N, Herholz K, Mielke R, Holthoff V, Baron JC. 18FDG PET in vascular dementia: differentiation from Alzheimer's disease using voxel-based multivariate analysis. J Cereb Blood Flow Metab. 2006, 26, 1213–1221. [Google Scholar] [CrossRef]
- Seo, S.W.; Cho, S.S.; Park, A.; Chin, J.; Na, D.L. Subcortical vascular versus amnestic mild cognitive impairment: comparison of cerebral glucose metabolism. J Neuroimaging. 2009, 19, 213–219. [Google Scholar] [CrossRef]
- Cho, S.; Lee, H.; Seo, J. Impact of Genetic Risk Factors for Alzheimer's Disease on Brain Glucose Metabolism. Mol Neurobiol. 2021, 58, 2608–2619. [Google Scholar] [CrossRef]
- Jagust, W.J.; Landau, S.M. Apolipoprotein E, not fibrillar β-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci. 2012, 32, 18227–18233. [Google Scholar] [CrossRef]
- Stefani A, Bernardini S, Panella M, Pierantozzi M, Nuccetelli M, Koch G, et al. AD with subcortical white matter lesions and vascular dementia: CSF markers for differential diagnosis. J Neurol Sci. 2005, 237, 83–88. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Weller, J.; Budson, A. Current understanding of Alzheimer's disease diagnosis and treatment. F1000Res. 2018, 7. [Google Scholar] [CrossRef]
- Pinheiro, L.; Faustino, C. Therapeutic Strategies Targeting Amyloid-β in Alzheimer's Disease. Curr Alzheimer Res. 2019, 16, 418–452. [Google Scholar] [CrossRef]
- Yarchoan M, Xie SX, Kling MA, Toledo JB, Wolk DA, Lee EB, et al. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain. 2012, 135, 3749–3756. [Google Scholar] [CrossRef]
- Pasi M, Boulouis G, Fotiadis P, Auriel E, Charidimou A, Haley K, et al. Distribution of lacunes in cerebral amyloid angiopathy and hypertensive small vessel disease. Neurology. 2017, 88, 2162–2168. [Google Scholar] [CrossRef]
- Yip AG, McKee AC, Green RC, Wells J, Young H, Cupples LA, et al. APOE, vascular pathology, and the AD brain. Neurology. 2005, 65, 259–265. [Google Scholar] [CrossRef]
- Zarow, C.; Zaias, B.; Lyness, S.A.; Chui, H. Cerebral amyloid angiopathy in Alzheimer disease is associated with apolipoprotein E4 and cortical neuron loss. Alzheimer Dis Assoc Disord. 1999, 13, 1–8. [Google Scholar] [CrossRef]
Figure 1.
Depicted are genetic risk factors for Alzheimer’s disease and vascular dementia (inner grey ring). These genetic risk factors indicated in different colors are associated with one or more pathological features (as indicated in the surrounding red ring).
Figure 1.
Depicted are genetic risk factors for Alzheimer’s disease and vascular dementia (inner grey ring). These genetic risk factors indicated in different colors are associated with one or more pathological features (as indicated in the surrounding red ring).
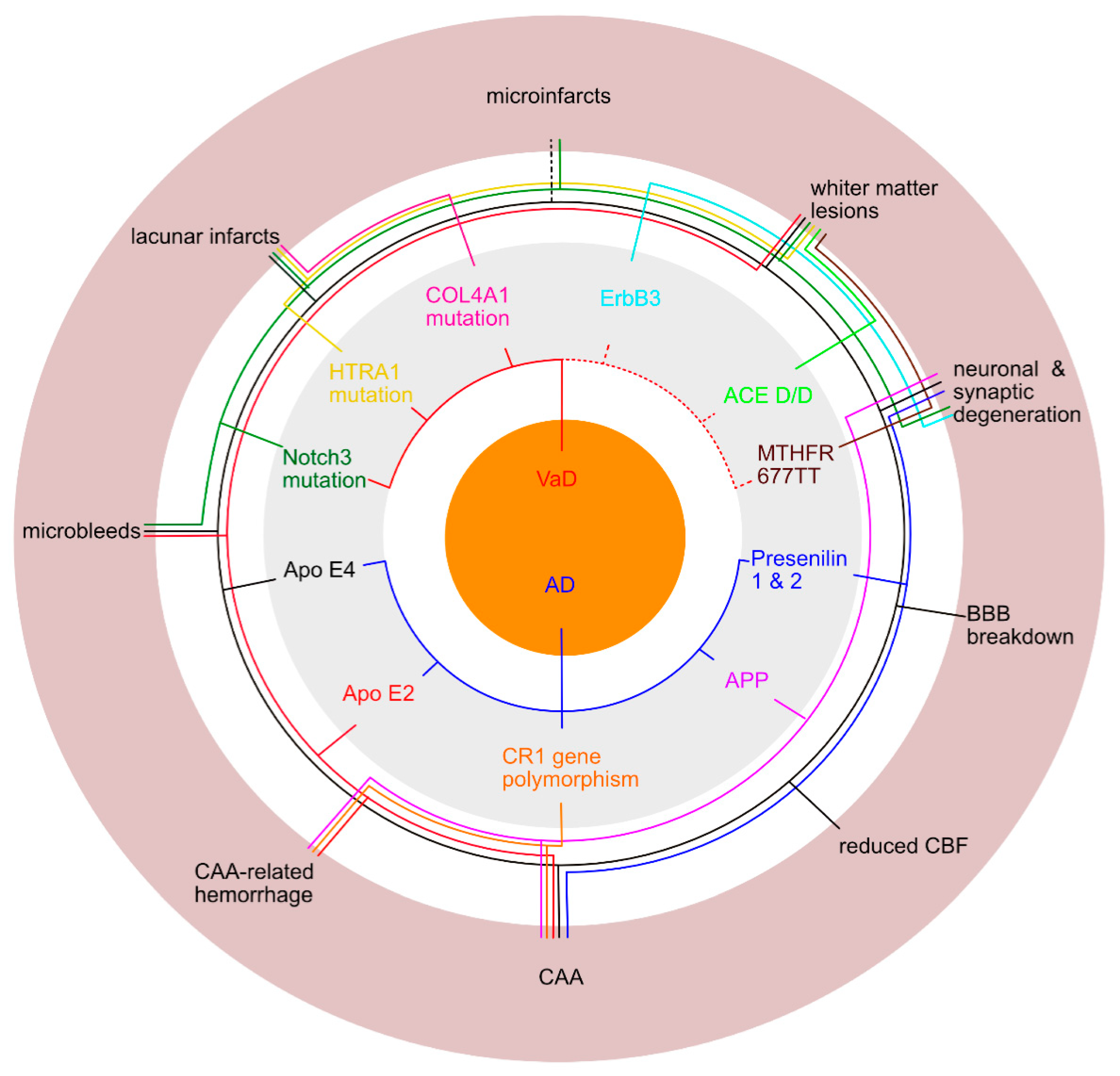
Figure 2.
Vascular risk factors. Alzheimer’s disease and vascular dementia (VaD) have the most risk factors in common, whereby stroke is especially associated with VaD. Those risk factors are related to one or more pathological mechanisms, such as blood-brain barrier breakdown or reduced cerebral perfusion, leading to diverse microscopic changes.
Figure 2.
Vascular risk factors. Alzheimer’s disease and vascular dementia (VaD) have the most risk factors in common, whereby stroke is especially associated with VaD. Those risk factors are related to one or more pathological mechanisms, such as blood-brain barrier breakdown or reduced cerebral perfusion, leading to diverse microscopic changes.
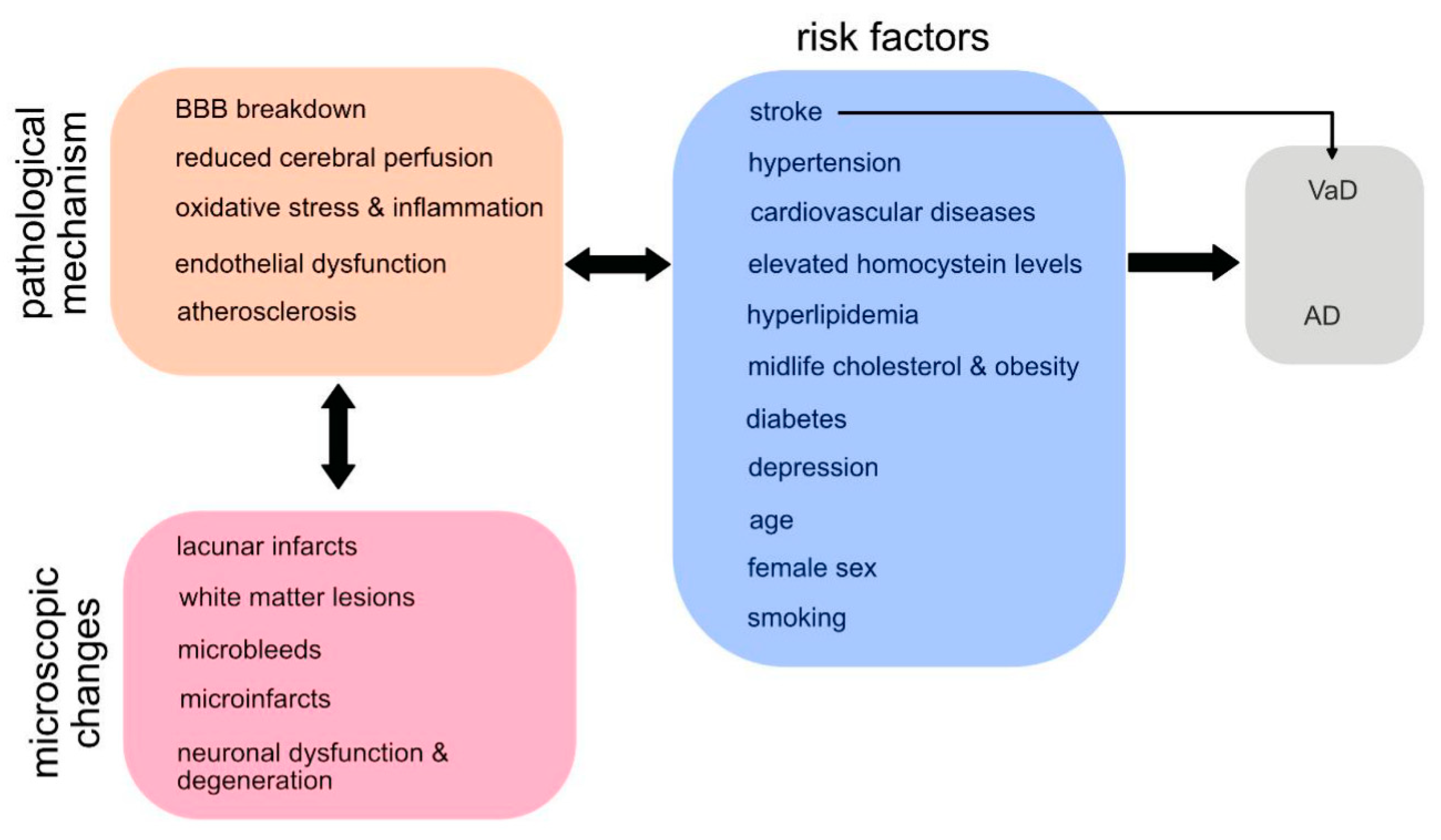
Figure 3.
Disease course in Alzheimer’s disease versus vascular dementia Alzheimer’s disease has a well-defined and predictable disease progression with different disease stages introduced by Braak and Braak [4]. Neurofibrillary tangles appear first in the transentorhinal and entorhinal region of the brain in (stage I and II) and gradually extend to limbic structures (stages III and IV) and association cortices (stages V and VI). In mild cognitive impairment, the first signs of cognitive decline are apparent. Cognition progressively worsens as the diseases progress. In comparison, vascular dementia can be classified into four subtypes (post-stroke dementia (PSD), subcortical ischemic vascular dementia, multi-infarct dementia and mixed dementia) with different lesion types. Cognitive decline depends on the lesion type and site of lesion and can therefore be abrupt, slow and gradual or stepwise.
Figure 3.
Disease course in Alzheimer’s disease versus vascular dementia Alzheimer’s disease has a well-defined and predictable disease progression with different disease stages introduced by Braak and Braak [4]. Neurofibrillary tangles appear first in the transentorhinal and entorhinal region of the brain in (stage I and II) and gradually extend to limbic structures (stages III and IV) and association cortices (stages V and VI). In mild cognitive impairment, the first signs of cognitive decline are apparent. Cognition progressively worsens as the diseases progress. In comparison, vascular dementia can be classified into four subtypes (post-stroke dementia (PSD), subcortical ischemic vascular dementia, multi-infarct dementia and mixed dementia) with different lesion types. Cognitive decline depends on the lesion type and site of lesion and can therefore be abrupt, slow and gradual or stepwise.
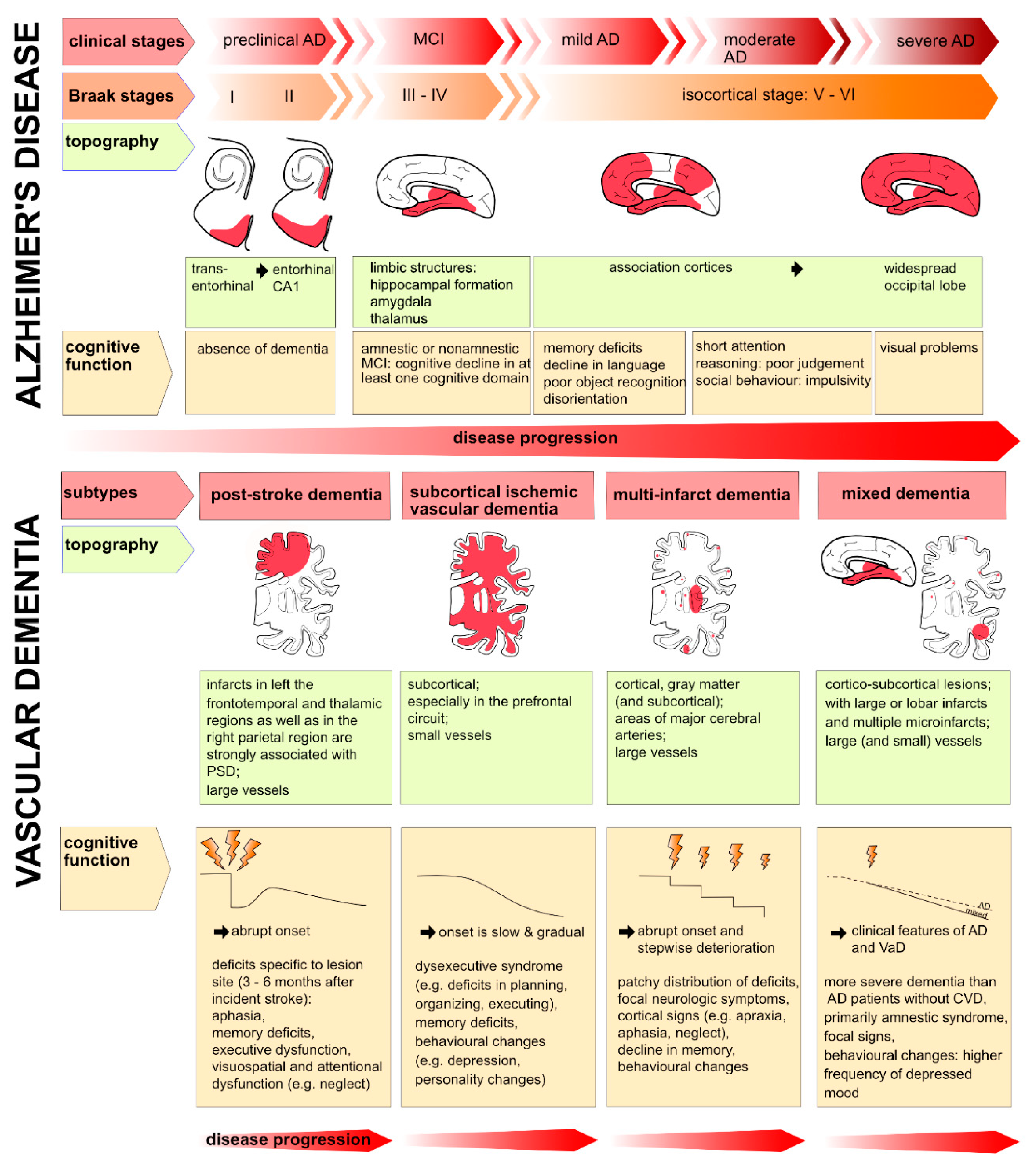
Figure 4.
Common pathomechanism in Alzheimer`s and vascular dementia: Cerebral endothelium, astrocytes and pericytes are an essential part of the formation and function of the blood-brain barrier (BBB). Endothelial damage leads to BBB breakdown and reduced cerebral blood flow (CBF), which induce on the one hand oxidative stress, inflammation and demyelination and thus trophic uncoupling in the neurovascular unit and on the other hand accumulation of neurotoxic molecules and Aß through impaired clearance, leading to neurodegeneration. Trophic uncoupling in the neurovascular units as well as deposition of Aß in the media and adventitia of small and mid-sized cerebral and leptomeningeal vessels aggravates vascular dysfunction. Hemorrhagic and ischemic lesions (red boxes) originate from these basic pathomechanisms.
Figure 4.
Common pathomechanism in Alzheimer`s and vascular dementia: Cerebral endothelium, astrocytes and pericytes are an essential part of the formation and function of the blood-brain barrier (BBB). Endothelial damage leads to BBB breakdown and reduced cerebral blood flow (CBF), which induce on the one hand oxidative stress, inflammation and demyelination and thus trophic uncoupling in the neurovascular unit and on the other hand accumulation of neurotoxic molecules and Aß through impaired clearance, leading to neurodegeneration. Trophic uncoupling in the neurovascular units as well as deposition of Aß in the media and adventitia of small and mid-sized cerebral and leptomeningeal vessels aggravates vascular dysfunction. Hemorrhagic and ischemic lesions (red boxes) originate from these basic pathomechanisms.
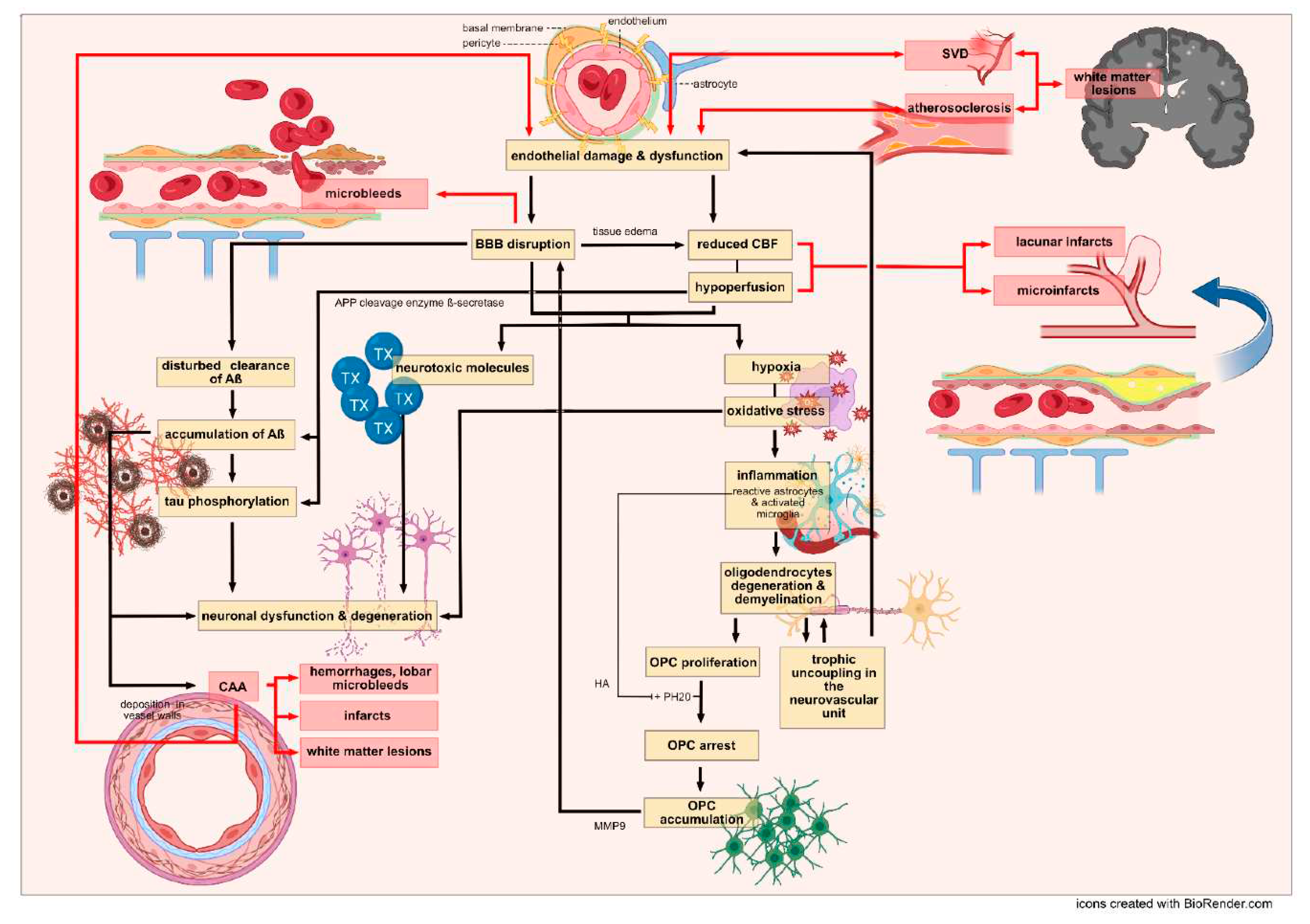

Table 1.
Clinical picture of Alzheimer’s disease and vascular dementia.
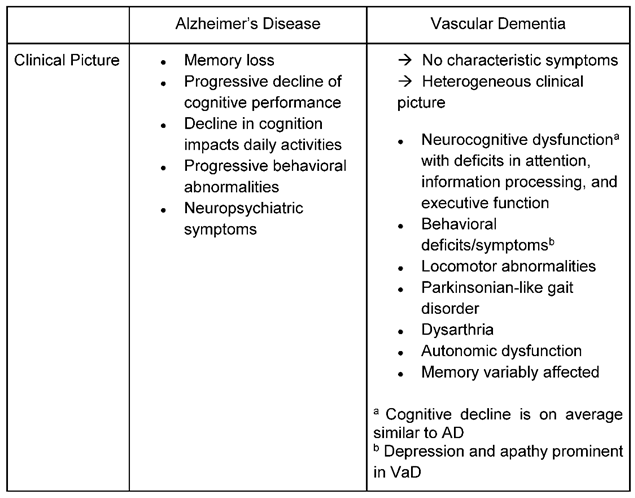 |
Table 1 depicts typical clinical features for both, Alzheimer`s and vascular dementia.
Table 2.
Neuropsychological profile of Alzheimer’s disease and vascular dementia.
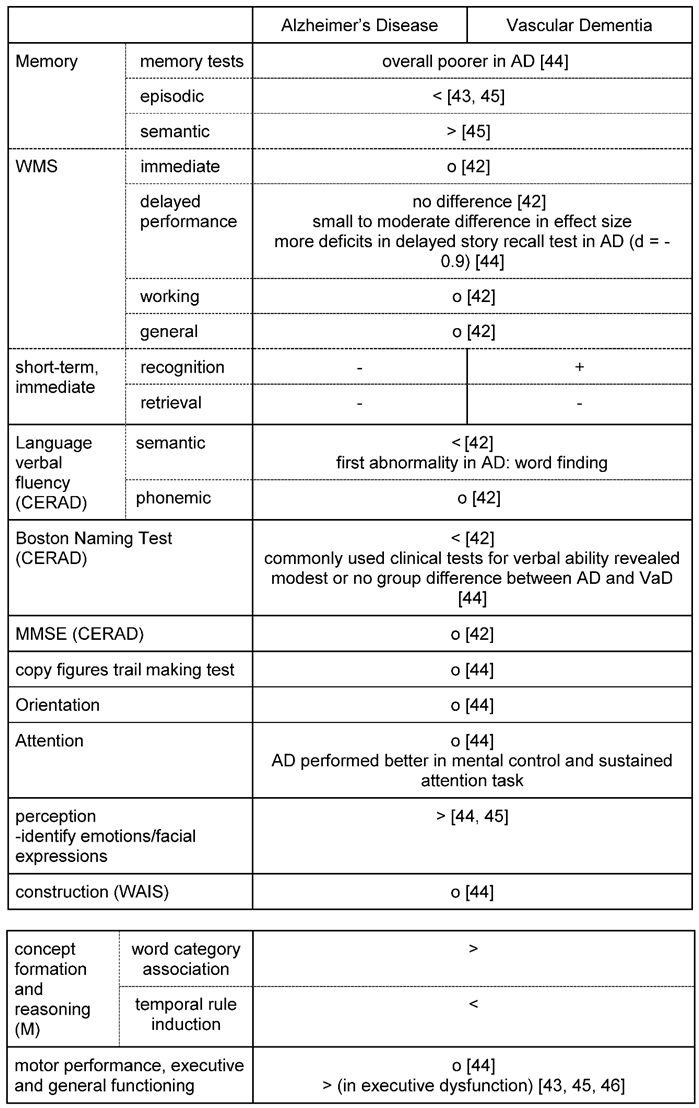 |
Table 2 reveals differences and similarities of neuropsychological test results in Alzheimer patients or subjects diagnosed with vascular dementia. The neuropsychological tests compared in the table are usually part of a general neuropsychological assessment provided in memory clinics during diagnostics. Abbreviations: “-:” not intact; “+”: intact; “o”: no difference;” <,>”: worse or better than; AD: Alzheimer’s disease; VaD: vascular dementia; CERAD: Consortium to Establish a Registry for Alzheimer’s disease; WAIS: Wechsler Adult Intelligence Scales.
Table 3.
Vascular pathology.
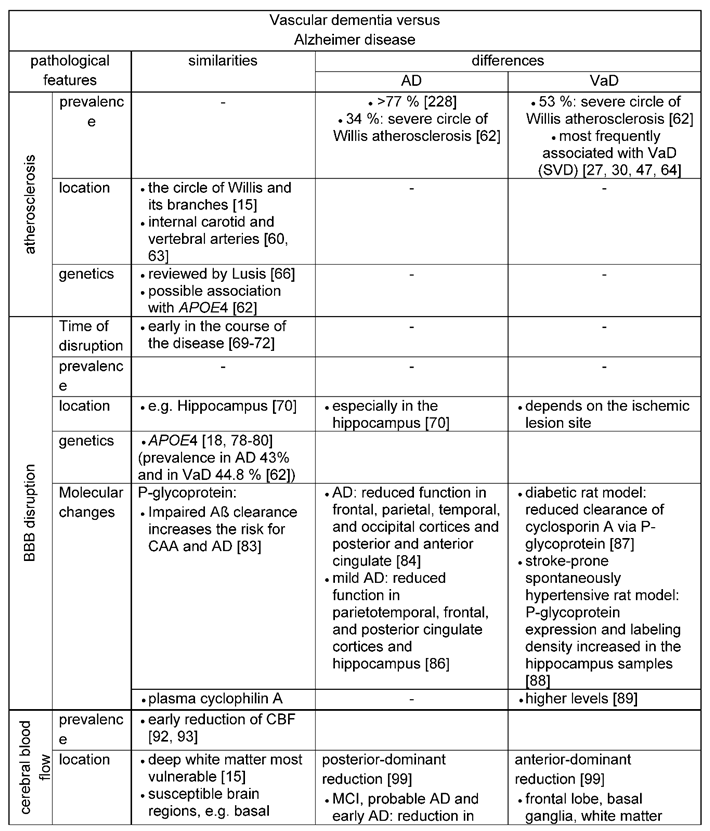 |
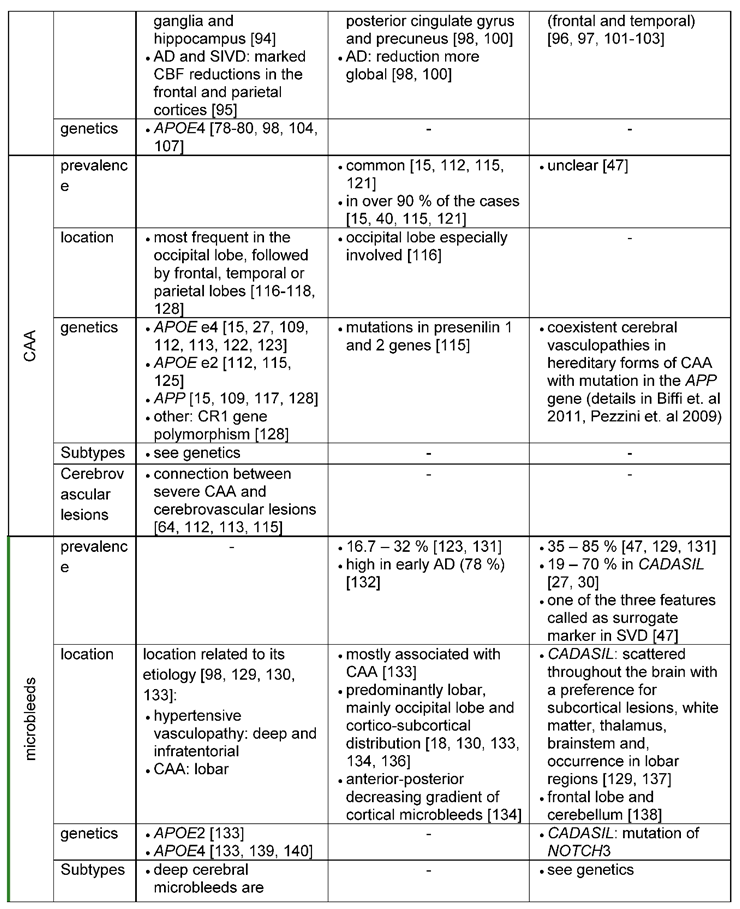 |
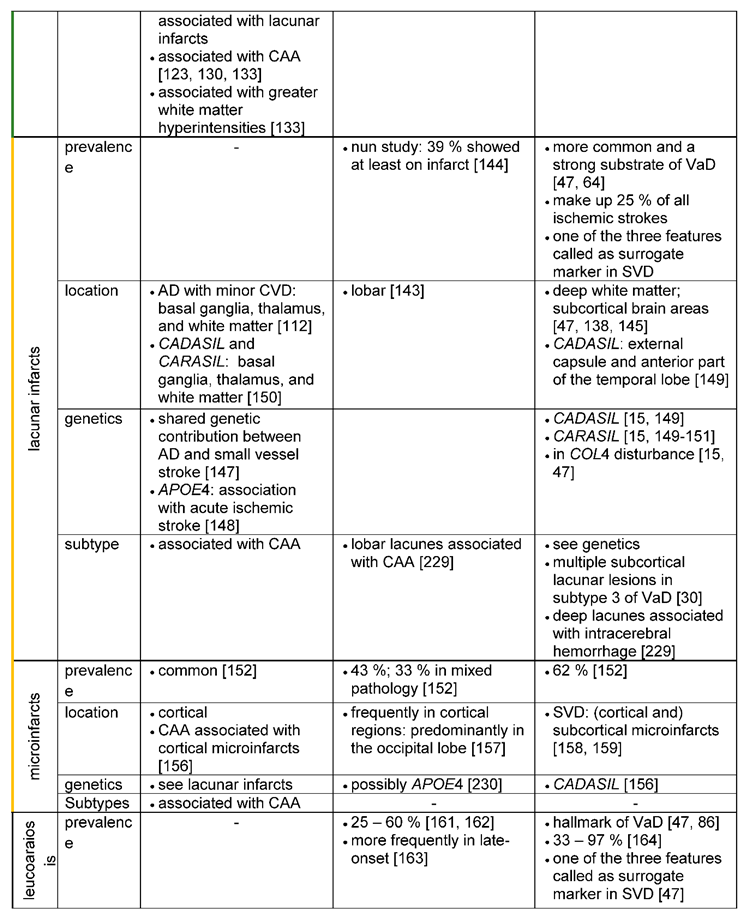 |
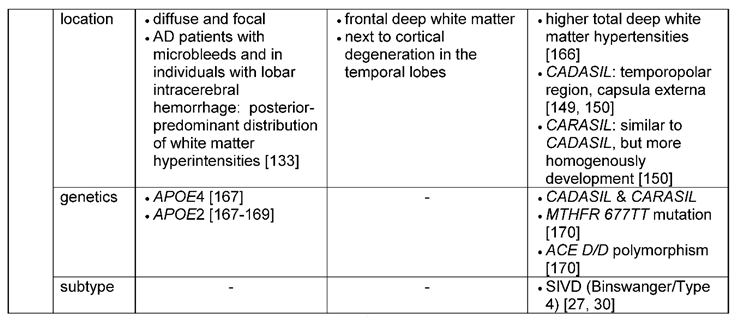 |
Table 3 summarizes typical vascular pathological features found in Alzheimer`s and vascular dementia and their similar or divers peculiarity. Abbreviations: AD : Alzheimer’s disease; VaD: vascular dementia; SVD: small vessel disease; CAA: cerebral amyloid angiopathy; SIVD: subcortical ischemic vascular dementia; CBF: cerebral blood flow; MCI: mild cognitive impairment; CVD: cerebrovascular disease; orange edge: hemorrhagic lesions; yellow edge: ischemic lesions.
Table 4.
AD-like pathology.
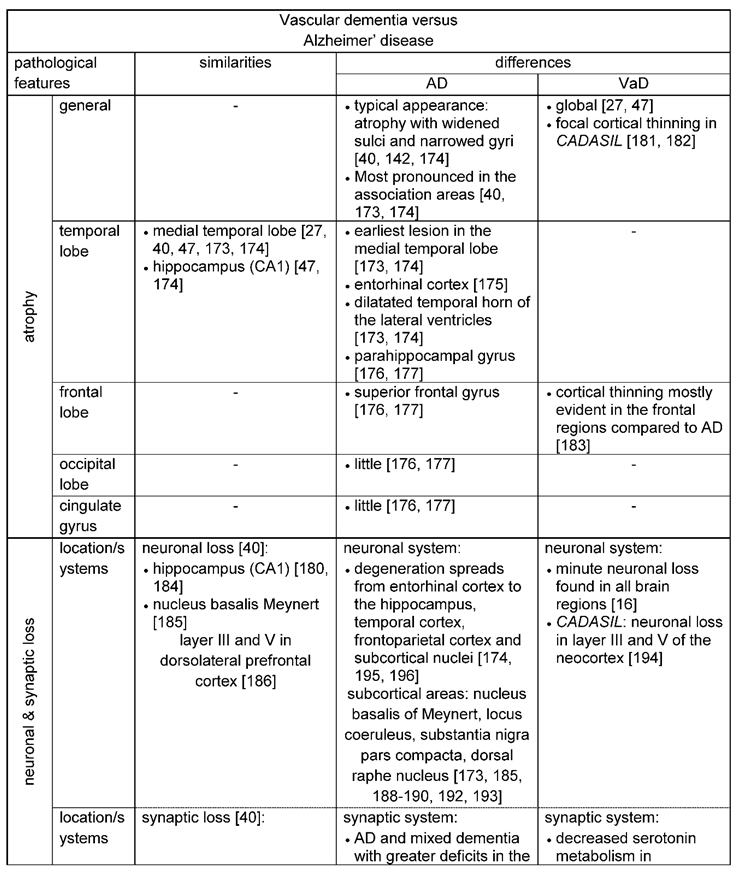 |
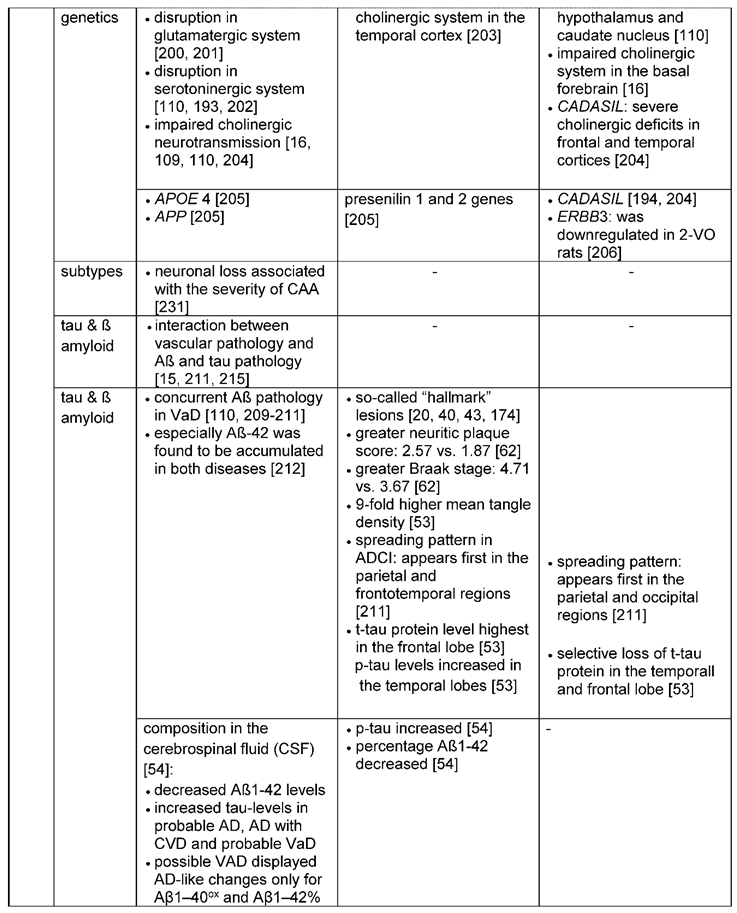 |
Table 4 summarizes similar and diverse neuronal pathological features in Alzheimer`s versus vascular dementia. Abbreviations: AD: Alzheimer’s disease; VaD: vascular dementia; ADCI: Alzheimer’s disease related cognitive impairment; CVD: cerebrovascular disease.
Table 5.
metabolism in vascular dementia and Alzheimer’s disease.
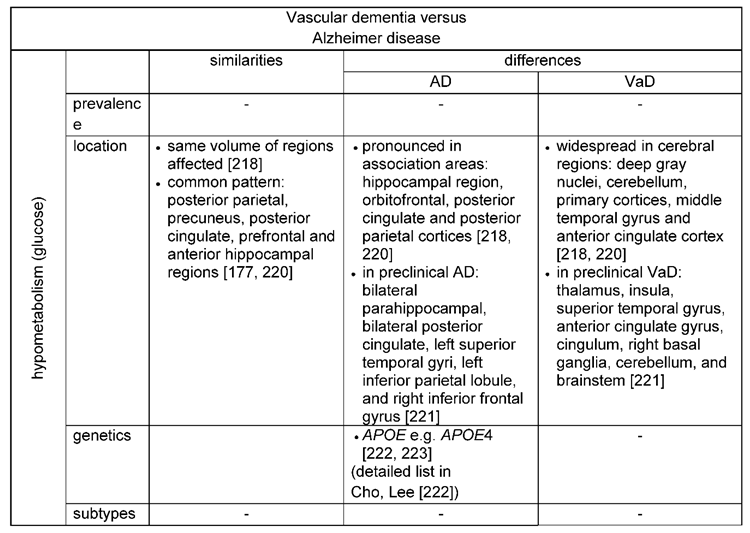 |
Table 5 compares features of brain metabolism in Alzheimer`s versus vascular dementia. Abbreviations: AD: Alzheimer’s disease; VaD: vascular dementia.
Table 6.
Overview of the compared pathological features.
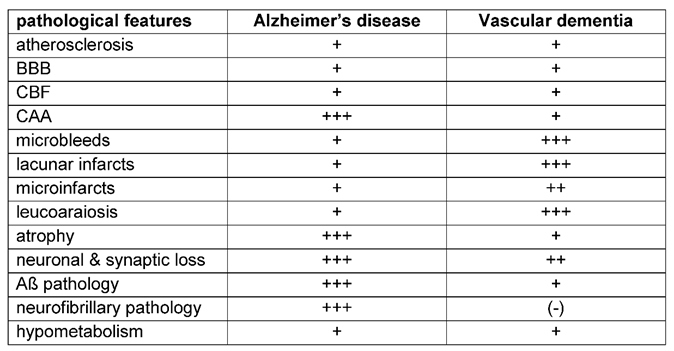 |
Table 6 compares the degree of different vascular and neuronal pathological features between Alzheimer`s and vascular dementia. Abbreviations: BBB: blood-brain barrier; CBF: cerebral blood flow; CAA: cerebral amyloid angiopathy.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



