
Submitted:
21 September 2023
Posted:
22 September 2023
You are already at the latest version
Abstract
The 1,2,3-triazole derivative 2-(4-chlorophenyl)-5-(pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylic acid with potential anticancer activity was used as a ligand in complex formation with the lan-thanum(III) ion. The molecular structure and vibrational spectra of the complex were optimized at three DFT levels and the scaled IR and Raman spectra were compared to the experimental ones. Several scaling procedures were used. Through a detailed analysis, the structure predicted for the newly synthetized La(III) complex was confirmed by the good accordance of the calculat-ed-experimental IR and Raman spectra. The best DFT method appears to be M06-2X with the Lanl2mb basis set, followed closely by Lanl2dz. The effect of the lanthanide atom on the molecular structure and atomic charge distribution of the triazole ring was evaluated. Potential free radical scavenging activity of both, ligand and complex, was investigated in several radical-generating model systems. Potential mechanisms of antioxidant action (hydrogen atom transfer – HAT and single electron transfer - SET) were elucidated.

Keywords:
1
; 2
; 3-triazoles
; antioxidant
; lanthanum
; vibrational analysis
; scaling
1. Introduction
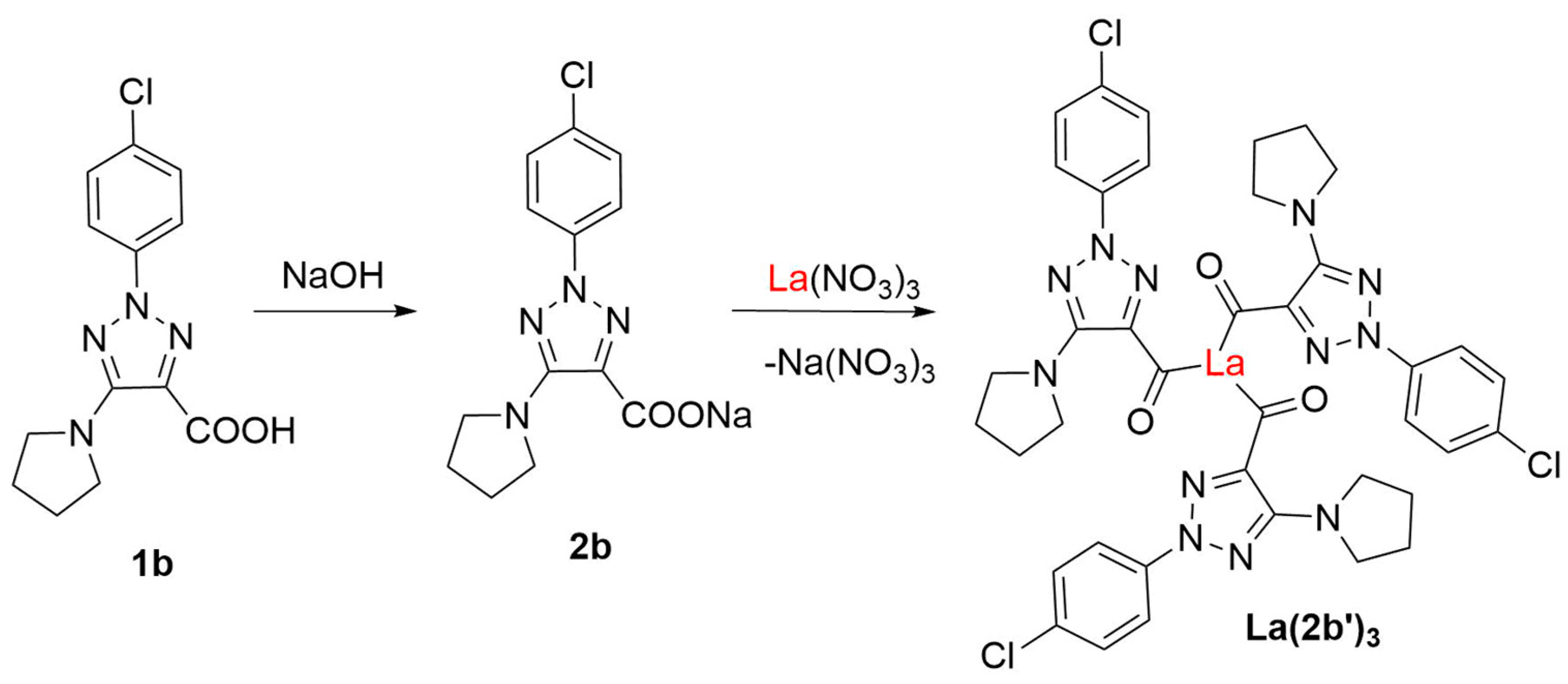
Transition metals and their various organometallic and coordination compounds are widely studied and extensively used in medical practice [1,2]. Among them, rare earth elements have quickly gained prominence in cancer diagnosis and therapy, owing to their versatile chemical and magnetic properties due to their 4f electronic configuration and their low toxicity. Design and synthesis of lanthanide complexes combining with organic ligands attract great attention owing to numerous fields of their practical applications in material chemistry and in biosciences [3,4,5,6]. It was found that complexation with Lanthanums could enhance antibacterial [7,8], antifungal [7,9], antioxidant [10] or anticancer activity [11] of free organic ligands. As the potential utility of lanthanides in these areas continues to increase, a study of a lanthanum(III) complex with a 1,2,3-triazole derivative as bioligand is shown in the present manuscript. The molecule selected as bioligand for the synthesis of La(III) complex was the sodium salt of 2-(4-chlorophenyl)-5-(pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylate, which is called molecule 2b according to notation of ref.[12].
Numerous lanthanide(III) complexes have been reported to have exhibited pronounced cytotoxic activity on a wide range of tumor cell populations of different origins, with these complexes always showing a significantly higher inhibition potential than the initial ligands and inorganic salts. This class of tumor-inhibiting metal complexes has been shown to be effective against tumors, resistant to classical platinum complexes. Their influence on the reactive species (RS) levels generated by several reactive oxygen species (ROS) model systems has also been studied [13] by us, confirming the antioxidant capacity of the lanthanide(III) complexes [14].
1,2,3-triazoles have been extensively studied for their antimicrobial, anticancer, antioxidant, etc. activities [15,16,17,18,19]. This wide range of pharmacological effects is due to their good accessibility, ease of specific structural modifications with a great variety of substitution patterns, and their photochemical and physicochemical properties, such as a high dipole moment, molecular rigidity, intermolecular interactions (dipole–dipole, hydrogen bonds, and π-stacking) with biotargets. Therefore, these biologically active compounds are the focus of consideration in several scientific areas including chemistry, biology, medicine, physics, etc.
The ligand 2-(4-chlorophenyl)-5-(pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylic acid (1b) in its anionic form (2b) was selected for several reasons: i) It is a triazole derivative, belonging to a class of molecules widely synthesized and extensively tested nowadays for their anticancer activities [4,11]. ii) It includes a 1,2,3-triazole ring, which has a remarkable biological potential through different modes of action [13,20]. iii) It is a novel compound, synthetized by us, bearing a strong-accepting group (the carboxylate) and in addition - aryl and pyrrolidine groups, providing the structure with liposolubility to facilitate cell membrane permeation. In the complex, La+3 ions are coordinated through the carboxylate group (COO−) in a three-dimensional system, labelled as La(2b’)3 as seen in Figure 1. This arrangement finds support in several previously reported lanthanide(III) coordination complexes with carboxylic acid derivatives [14,21].
In the present paper, the identification, structural and vibrational characterization of the newly synthetized lanthanum(III) coordination complex through theoretical and spectroscopic results was carried out particularly by means of spectral (FT-IR, FT-Raman) and elemental analysis. Since crystal structure data were not available, theoretical approaches at high level for determination of geometrical parameters, vibrational frequencies, and the binding mode of the model system are very useful for extracting reliable structural information.
Theoretical and experimental FTIR and Raman spectra of related triazole molecules have been reported [15], although not in such a large complex as presented here, nor in detailed form using accurate scaling procedures. Significant differences were observed in the bands of the IR and Raman spectra of the complex, compared to those of the free ligand. The comparative vibrational analysis allowed a good assignment of the functional group vibrations involved in the coordination and helped explain the ligand vibrational modes, which are sensitive to interaction with metal ions being in agreement with literature data and with theoretical predictions.
The impact of the ligand and its complex with La(III) on a variety of RS-generating model systems was assessed – hydroxyl radicals (OH●), superoxide (O2●−), hypochlorite (OCl−) with some interesting effects having been observed. Potential mechanisms of scavenging activity were additionally elucidated with the help of the 2,2-diphenyl-1-picrylhydrazyl (DPPH●) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS●+) stable radical model systems.
2. Materials and Methods
2.1. Experimental Details
The composition of the newly obtained metal complex was characterized by elemental analysis. The nature of La(III) complex was confirmed by IR and Raman spectroscopy. The compounds used for synthesis of the complexes were from Merck company, p.a.grade: La(NO3)3·6H2O. The sodium salt of 1b [12] was used as ligand. The complex was obtained via an interaction of lanthanum(III) inorganic salt and the ligand in amount equal to a metal:ligand molar ratio of 1:3. The synthesis was carried out by adding aqueous solution of La(III) nitrate to the aqueous solution of ligand. The reaction mixture was stirred with an electromagnetic stirrer at room temperature for one hour. As result of solutions mixing, the precipitate of the complex was obtained, which was filtered, washed with water, and dried in a desiccator until constant weight. The obtained complex was very slightly soluble in water, methanol and ethanol, and highly soluble in dimethyl sulfoxide (DMSO).
The carbon, hydrogen, and nitrogen contents of the obtained complex were determined by elemental analysis. Solid-state infrared spectra of the complex was recorded in KBr in the 4000–400 cm−1 frequency range by an FTIR IFS25 Bruker spectrometer. Raman spectra of the acidic form (1b) and the sodium salt (2b) of the ligand and its new La(III) complex were recorded with a Dilor microspectrometer (Horiba-Jobin-Yvon, model LabRam) equipped with 1800 grooves/mm holographic grating. The 514.5 nm line of an argon ion laser (Spectra Physics, model 2016) was used for the probes’ excitation. The spectra were collected in backscattering geometry with a confocal Raman microscope equipped with an OlympusLMPlanFL 50× objective. The detection of Raman signal was carried out with a Peltier-cooled CCD camera. Laser power of 100 mW was used in our measurements.
All UV-VIS measurements were carried out on a Shimadzu UV1601 spectrophotometer. LDCL measurements were performed LKB 1251 luminometer (Bioorbit, Turku, Finland) set at 37°C and connected with IT-type computer via serial interface. Three replicate measurements were carried out for each concentration tested. Each measurement represents an individual datapoint. Averages and standard deviations were calculated. Relative changes within the limits of experimental error were not discussed. One-way ANOVA, followed by Bonferroni post-test were used for statistical verification of the impact of the varying concentrations on the results obtained.
Radical scavenging assays: Preliminary research on the ligand showed its large potential antioxidant properties [39], which also appear in its lanthanum(III) complex that is now analyzed. To do that, additional radical-generating model systems (Fenton reaction, potassium superoxide, sodium hypochlorite) were applied to both ligand and complex, yielding somewhat unexpected and hence, interesting results.
2.2. Materials
Pro analysis grade materials SIGMA-ALDRICH (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) were used. Solutions were prepared with bi-distilled water and 95% ethanol. The Deoxyribose Degradation Assay involved the following solutions and reagents: 50mM K-Na-phosphate saline solution (PBS) with pH= 7.45; 6.0 mM water solution of deoxyribose; 3% water solution of trichloroacetic acid (TCA); 1% water solution of thiobarbituric acid (TBA). The Fenton reaction MTT assay involved: 3 mg/mL water solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT); Fenton reagent, containing FeCl2, H2O2 and Na2-EDTA; 4 mg/mL water solution of ascorbic acid. The ability to participate in HAT reactions was measured according to well-established protocols [47,48], using a 0,6 mM stock ethanol solution of 2,2-diphenyl-1-picrylhydrazyl radical (DPPH●). The ability of the substances to participate in SET was established using the 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic free radical (ABTS) assay, as described by Erel et al [49,50]. The following solutions were prepared: 0.4 mM acetate buffer, pH=5.8; 0.03 mmol acetate buffer solution, pH =3.6 with ABTS and H2O2 in order to produce the stable radical-ion ABTS●+. Scavenging of superoxide radical-ions (O2∙-), derived from potassium superoxide (KO2) and hypochlorite (ClO−), derived from sodium hypochlorite (NaClO) was assessed with luminol-dependent chemiluminescence (LDCL). 5-Amino-2,3-dihydro-1,4-phthalazinedione (luminol) was dissolved in a 0.1 M NaOH, diluted to 1·10-3 M in PBS of pH 7.45, and pH was adjusted again to 7.45. 1 mM KO2 solution in dehydrated dimethyl sulfoxide (DMSO) and 4·10-4 M aqueous solution of NaOCl were prepared prior measurements.
2.3. Methods
2.3.1. Deoxyribose Degradation Assay
A modified protocol [51] was applied to generate hydroxyl radicals (OH●) via water radiolysis [52]. 2-Deoxyribose degradation to MDA-like products is elucidated with the aid of the TBA assay. Every sample contains the following solutions: 0.5 mL 2-deoxyribose, the compound under investigation and PBS up to 5.0 mL. Controls did not contain the investigated compounds. After 30 minutes’ irradiation with an UV lamp (220-400 nm), 1.0 mL from each 5.0 mL volume was withdrawn. To each of these 1.0 mL volumes was added 0.6 mL TCA and 0.6 mL TBA, followed by 30 minutes’ cultivation in water bath at 100 ̊С. Absorbance was measured at λ=532 nm. The degree of 2-deoxyribose degradation is presented as Spectrophotometric Scavenging Index (SPh-SI):
2.3.2. Fenton reaction MTT assay
Fenton-generated OH● reduce yellow MTT to purple formazan. Ascorbic acid increases RS production in the model system. Formazan production is registered as absorbance increase at λ= 578 nm, using the kinetic function of the apparatus: 10 seconds lag time, 600 seconds measuring time. Sample composition (2.0 mL total volume) is as follows (Table 1):
Results are presented as Radicals-scavenging activity (RSA), calculated with the formula:
where: ΔАcontrol is the relative change in absorbance at 578 nm in presence of the Fenton reagent, but in absence of the investigated compound. ΔАsample is the relative change in absorbance at 578 nm in presence of the Fenton reagent together with the investigated compound. ΔАblank is the relative change in absorbance at 578 nm in absence of the Fenton reagent, but in presence of the investigated compound.
2.3.3. DPPH assay
The characteristic signal of DPPH● at λ=517 nm was measured in three types of samples – “blank”, “control” and “sample” (Table 2).
Total volume of each sample is 2.0 mL. Absorbance decrease was measured, using the kinetic function of the apparatus: 10 seconds lag time, 300 seconds measuring time. Results are presented as RSA:
2.3.4. ABTS assay
Two solutions are prepared, as previously described [49] – Reagent 1 (R1) and Reagent 2 (R2). R1 is Na-acetate buffer with pH=5,8. R2 contains ABTS, in Na-acetate buffer with pH=3,8 and H2O2 to produce the stable ABTS●+ radical-ion. Total volume of each sample is 1.0 mL. The characteristic signal for ABTS●+ was measured at 660 nm. Absorbance decrease was measured, using the kinetic function of the apparatus: 10 seconds lag time, 300 seconds measuring time. Results are presented as RSA – same as with the DPPH assay. Sample composition is as follows (Table 3).
2.3.5. LDCL in presence of KO2
1.0 mL “control” volume was included 0.05 mL luminol, 0.05 mL KO2 and PBS. 1.0 mL “sample” volume included 0.05 mL luminol, 0.05 mL KO2, the investigated compound at the desired concentration and PBS. Kinetics were measured with 2 seconds delay for a duration of 10 seconds. Integral intensities were used in calculations. Results were calculated as chemiluminometric scavenging index (CL-SI):
Background signal was measured in absence of KO2 and was subtracted from both “control” and “sample”.
2.3.6. LDCL in presence of NaClO
1,0 mL “control” volume included 0.15 mL NaClO, 0.05 mL luminol and PBS. 1,0 mL “control” volume included 0.15 mL NaClO, 0.05 mL luminol, the investigated compound at the desired concentration and PBS. Background signal was measured in absence of NaClO and was subtracted from both “control” and “sample”. Results were presented as CL-SI, as with the KO2 assay.
2.4. Computational Details
Density Functional methods (DFT) [53] were only used for the calculations due to the large size of the La(2b’)3 complex. DFT computations have also provided results in biomolecules which are quantitatively in good accordance to those obtained at MP2 level [54,55]. The Minnesota functional M06-2X DFT method was select because it is the best choice among other meta-generalized gradient functionals to examine dispersion–bound systems[56,57]. Since, non-covalent weak interactions are expected in our large system, it was the preferred method. In addition, it also shows broad applicability in chemistry [58]. The B3LYP DFT method was also used because it leads to excellent results in the IR and Raman vibrational wavenumber calculations [59,60]. All these methods appear implemented in the GAUSSIAN-16 program package [61]. The UNIX version with standard parameters of this package was running in the Brigit super computer of the University Complutense at Madrid.
For lanthanide atom, few basis sets appear available. The Lanl2dz basis set was chosen because it gave good results in a La(III) complex with a carboxylic acid derivative [14]. Similar to that, the Lanl2mb basis set appears that was also used. Another type is the Cep-4g. They are very small basis set that are available, but should be only used for compounds with heavy metal atoms, as the lanthanide atom.
Harmonic wavenumber calculations were carried out at the same level of the corresponding optimization process. All optimized structures show only positive harmonic vibrations, which indicates a local minima energy. The calculated Raman scattering activities (Si) were converted into relative Raman intensities (Ii) using relationship derived from the basic theory of Raman scattering [62],
2.4.1. Scaling the Wavenumbers
To correct the overestimation in the frequency calculation by the theoretical methods used, it is necessary to use different scaling procedures specially selected for each level [59,60]. Therefore, a remarkable improvement in the results will be obtained. It is the regular procedure followed to get an accurate assignment of the experimental bands. The linear scaling equation procedure (LSE) and the polynomic scaling equation procedure (PSE) were mainly used, which are as follows:
νscal = 56.8 + 0.9147 νcal at M06-2X/Lanl2dz level
νscal = 44.8 + 0.9579 νcal − 0.000039· (νcal )2 at M06-2X/Lanl2mb level
νscal = −82.9 + 1,0917 νcal − 0.0000579· (νcal )2 at B3LYP/Cep-4g level
3. Results
One of the goals of the present manuscript was the identification and characterization of the synthetized La(2b’)3 coordination complex through a detailed study of the spectroscopic results. In addition, it would be interesting to observe the effect of the La(III) ion on the molecular structure and atomic charge distribution of 2b ligand, particularly in the triazole ring. Different basis sets were tested and the best one was selected.
3.1. Synthesis
N-2-aryl-triazoles 1b and 2b were prepared according to the literature procedures. The complex La(2b’)3 was obtained via treating triazole salt 2b with La(NO3)3 in the ratio of 3:1 in a water solution (Scheme 1) in excellent yield (80%) and characterized by elemental analysis data and IR spectroscopy.
Scheme 1.
Synthesis of the complex La(2b’)3.
3.2. Molecular structure of the complex
Since lanthanide metals coordinate more preferably to oxygen atoms than to nitrogen atoms [14], the starting structure optimized was that with the lanthanum(III) ion coordinated with three 2b ligands through the carboxylate group (COO−), Figure 1.
Figure 1.
Labelling of the atoms and plot of the optimized La(2b’)3 structure with 2b: sodium 2-(4-chlorophenyl)-5-(pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylate. Front and lateral view forms were included in the optimized structures at the: (a) M06-2X/Lanl2dz level, and (b) M06-2X/Lanl2mb level. Several bond length values of interest calculated are included in the figure, together with the energy values of the system. 1 AU = 2625.5 kJ/mol.
Figure 1.
Labelling of the atoms and plot of the optimized La(2b’)3 structure with 2b: sodium 2-(4-chlorophenyl)-5-(pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylate. Front and lateral view forms were included in the optimized structures at the: (a) M06-2X/Lanl2dz level, and (b) M06-2X/Lanl2mb level. Several bond length values of interest calculated are included in the figure, together with the energy values of the system. 1 AU = 2625.5 kJ/mol.
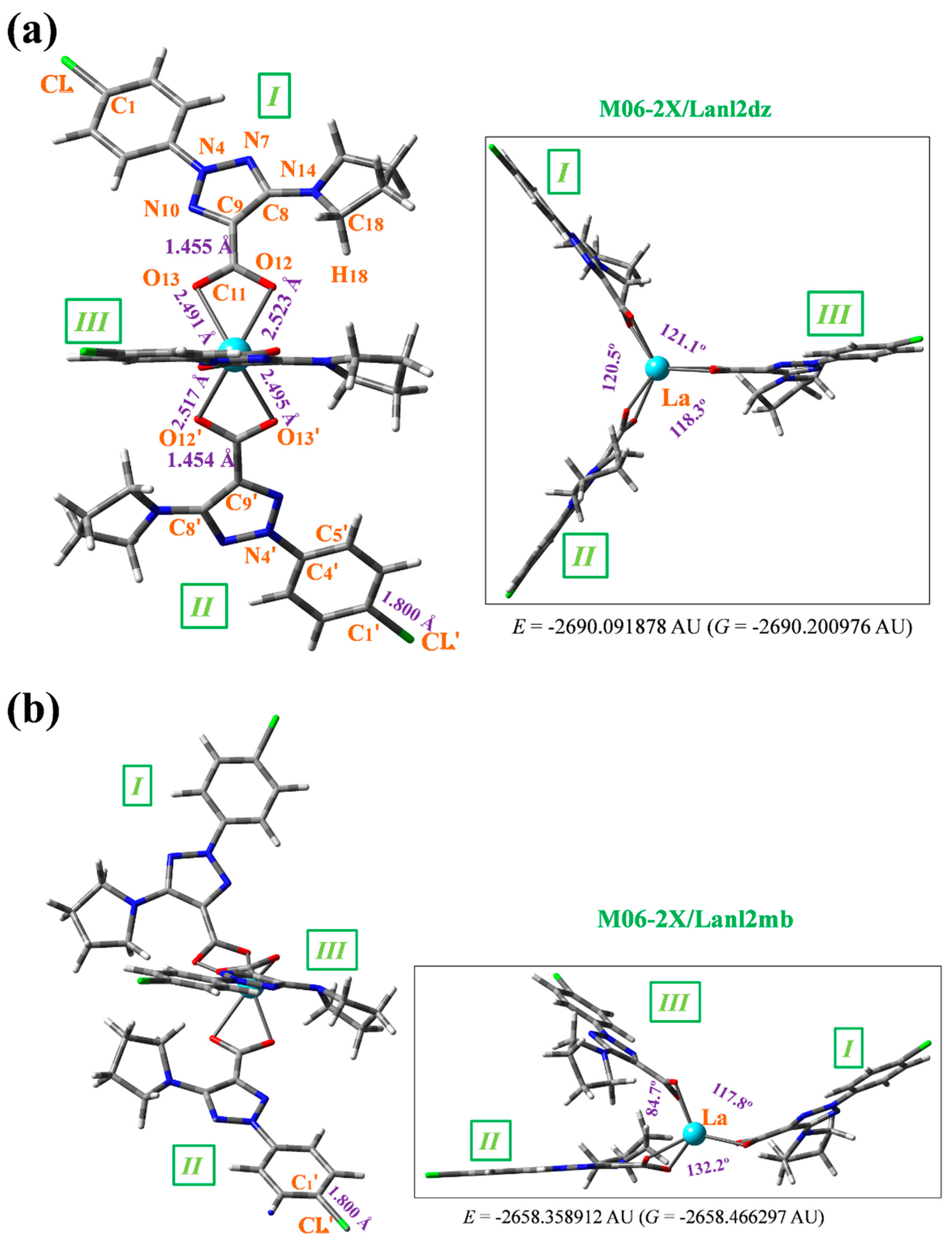
Two views of the optimized structure at the M06-2X/lanl2dz level are shown in Figure 1a, with an almost symmetrical arrangement. By rotation of the carboxylate group around the C9-C11 bond, another conformer can be obtained, but is slightly less stable than that plotted in this figure. The notation used for labelling the atoms is in accordance with the one previously reported for this very same 2b ligand [12]. Few bond length values are also included in this figure. In the bottom of it, the total energy (E) value is shown, which includes the ZPE (zero-point vibrational energy) correction, and the Gibbs energy (G). Several selected optimized geometrical parameters, namely bond lengths, bond angles and torsional angles of interest in one of the ligands (labelled as I) are collected in Table 4. For comparison purposes, the MP2 values obtained in the isolated 2b structure [22] are listed in the last column of the table.
The intramolecular H-bond between the carbonyl oxygen O12 and the pyrrolidine hydrogen H18 is very weak by M06-2X (2.443 Å) in the La-(2b’)3 complex due to La-O12 bond formation, but it is present in the isolated ligand by MP2 (2.034 Å) [22].
The average calculated value of the La-O bond length (2.5 Å) agrees well with the experimental value of the odd diacids and it is specific for the monometallic complexes of lanthanides [14,23]. An increase in this value has been related to a closing of the OCO angle [24]. The calculated bond angle C=O12-La (94.0º) is slightly different to the C=O13-La (95.7º) angle, Table 4, which indicates that the C-O-La bond is not fully linear due to the slightly higher negative charge of O13 (−1.266e), compared to O12 (−1.250e), Table 5. A higher negative charge on O13 leads to a shortened of the O13-La bond length (2.491 Å) vs. O12-La (2.523 Å), and therefore an opening of the C=O13-La angle. This very slight asymmetry is also observed in the O12-La-O′13 angle (103.0º), which is slightly more open than O12-La-O′13 (102.0º). These features also lead to rotation of the ligands, and thus the torsional angle C11-O12···O′12-C′11 (13.2º) is quite different from the C9-C11···C′11-C′9 angle (−3.2º), as well as the (C11···La·· C′11) angle (121.1º), compared to (C′11···La···C′′11) (118.3º).
In this optimized structure, at the M06-2X/lanl2dz level, the aryl ring is fully planar and almost coplanar with the triazole ring, with a C5-C4-N4-N10 torsional angle of 0.8º, although it appears slightly rotated (−2.3º) by MP2. The triazole ring is also planar with values of the torsional angles lower than 1º. The pyrrolidine ring, however, is noticeably out-of-plane with a value of the torsional angle N14-C15-C16-C17 of −32.2º, in accordance to that found in a cyclopyrrol ring. This pyrrolidine ring is also non-coplanar with the triazole ring plane with a value of the torsional angle C9-C8-N14-C18 of −18.3º, although it is 32.0º by MP2 indicating a failure in this angle calculation by M06-2X method with lower values.
The carbon C11 atom of the carboxylate group appears slightly titled out of the triazole ring plane and remarkably rotated, but this rotation is larger with O13, than with O12, with a value of the torsional angle N10-C9-C=O13 of −11.2º vs. C8-C9-C=O12 of −7.5º. For the binding to the lanthanide ion, the OCO angle closes up to 117.4º vs. 131.3º by MP2 in the free ligand, and the CO bond lengths are noticeably lengthened. This large flexibility of the carboxylic oxygens facilitates bonding to the lanthanum(III) ion. The lengthening of the CO bonds leads to a shortening of the C9-C11 bond length, which noticeably reduces the double bond character of N7=C8 and C9=N10 bonds of the triazole ring and, consequently, the N4-N10 and C8-N14 bonds are shortened. This feature slightly modifies the bond angles of the triazole ring with the lanthanide binding.
The optimized structure at the M06-2X/lanl2mb level appears deformed, Figure 1b, perhaps due to its smaller basis set. However, this more planar arrangement can be more probably expected to be present for the solid state sample due to the crystal packing forces compressing the structure. The selected optimized geometric parameters at this level are also included in Table 4. It is noted that the intramolecular O12···H18 H-bond only appears in ligand I, and it is very weak in ligands II and III. The C-N and N-N bond lengths are noticeably larger and the La-O bonds - shorter than by M06-2X/lanl2dz. Because of the structural deformation, the C=O-La and O12-La-O′13 angles are closer and the O12-La-O′13 angle larger than with the lanl2dz basis set. The pyrrolidine and carboxylate groups are also more rotated related to triazole ring, and the arrangement of the ligands differ remarkably, with a (C′11···La···C′′11) angle of 84.7º vs. 118.3º with lanl2mb basis set, and torsional angle (C11-O12···O′12-C′11) of −169.5º vs. 13.2º with lanl2mb basis set. However, the value of the OCO angle remains almost the same in both levels.
Due to the optimized structure at the B3LYP/Cep-4g appearing largely deformed, it was included in Figure S1 (Supplementary Material). To this feature contributes the low basis set used, as well as that the B3LYP method does not consider long range interactions that stabilize the system to a symmetric arrangement as in Figure 1a. The optimized values at this level appear with noticeably longer bond lengths than by M06-2X and MP2. With longer bond lengths, the triazole substituents are more twisted, especially the pyrrolidine ring, which can interact with other ligands.
3.3. Atomic charges and molecular properties calculation
The lanthanide ion has the highest positive charge, which is 3.164e by M06-2X/Lanl2dz and 2.884e by B3LYP/Cep-4G, Table 5. Uncommonly, it is rather small (2.066e) by M06-2X/Lanl2mb. As expected, the highest negative charge corresponds to the oxygen atoms, with the charge on O13 slightly higher in negative value than on O12, and in contrast to that calculated in the free 2b molecule by MP2. These oxygens appear as the most reactive and therefore they are bonded to the lanthanide ion. They are expected to participate in the biological activity of these molecules. The negative charge on the nitrogen atoms N4 and N14 is lower but it is expected that they can also contribute to the biological activity of this compound. By contrast, a high positive charge appears in the carbon atoms C8 and C11 because they are bonded to large negative atoms.

Table 5.
APT (Atomic Polar Tensor) charges calculated at different DFT levels in La-(2b’)3 complex. The values of ligand I are only shown.
Table 5.
APT (Atomic Polar Tensor) charges calculated at different DFT levels in La-(2b’)3 complex. The values of ligand I are only shown.
| M06-2X | B3LYP | MP2 | ||
| atom | Lanl2dz | Lanl2MB | Cep-4G | 6-31G(d,p)a |
| La CL C1 C4 N4 N7 C8 C9 N10 C11 O12 O13 N14 |
3.164 −0.423 0.452 0.518 −0.566 −0.293 0.714 −0.835 0.340 1.821 −1.250 −1.266 −0.924 |
2.066 −0.496 0.476 0.723 −0.832 −0.069 0.465 −0.716 0.398 1.506 −0.925 −1.033 −0.771 |
2.884 −0.254 0.267 0.620 −0.742 −0.066 0.373 −0.844 0.401 1.381 −1.014 −1.089 −0.713 |
- −0.054 −0.080 0.210 −0.073 −0.402 0.419 0.031 −0.240 0.955 −0.887 −0.839 −0.557 |
aCalculated in isolated 2b molecule.
Several thermodynamic parameters, rotational constants and dipole moments were also calculated and included in Table 6. Because of the symmetry of the complex at M06-2X/Lanl2dz level, the rotational constant values in the three directions (A, B, C) have almost the same value. They differ, however, by the other DFT methods. One value is larger (A axis) - corresponding to the ligand noted as I. Two values are lower (B and C axes) - corresponding to ligands II and III. The Cv and entropy (S) values were close to M06-2X and lower than those calculated by B3LYP. This feature may be due to the lower symmetry of the complex obtained by B3LYP. The calculated dipole moment value indicates that this complex presents a slight water solubility, which could facilitate its biomedical use.
With the HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) values the global chemical reactivity descriptors were calculated [25,26] to better understand the reactivity and stability of the La(2b’)3 complex under study. The energy gap (Eg) between HOMO and LUMO frontier orbitals is a significant characteristic of molecules and helps characterize their chemical reactivity and kinetic stability. A high Eg in a molecule indicates that it is less polarizable. It is generally associated with low chemical reactivity and high kinetic stability. Therefore, our low calculated Eg values in the La(2b’)3 complex by all three DFT methods indicates large chemical reactivity and small excitation energies to the manifold of excited states.
The following formulae were used to calculate the global chemical reactivity descriptors:
The calculated value for the ionization potential (IP) is somewhat low in accordance with the large reactivity of the complex. The electron affinity (EA) is remarkably lower than IP and positive, with the exception of the M06-2X/Lanl2mb calculation. The electronegativity (χ) is low in accordance with a neutral system. Chemical hardness (η) and global softness (S) express the resistance of a system to a change in its number of electrons. When η is weak the molecule is called soft and it has a small HOMO–LUMO gap and when it is high, the molecule is called hard. Our low values correspond to a soft molecule, with a small gap, and with an electron density that can change easily.
3.4. Vibrational Analysis
For the identification and characterization of the synthesized La(2b’)3 coordination complex, a detailed analysis of the calculated and experimental spectra was carried out. The wavenumbers with high IR or Raman intensity only were included in Table 7, while in Table S1 to Table S3 (Supplementary Material) were collected all calculated values corresponding to M06-2X/Lanl2dz, M06-2X/Lanl2mb and B3LYP/Cep-4g levels, respectively. All data correspond to the most stable conformer, with the ligands orientation such as it is plotted in Figure 1.
The first column lists the calculated wavenumbers at the M06-2X/Lanl2mb or M06-2X/Lanl2dz levels. Three values appear for each vibrational mode corresponding to the three ligands in the complex. Of these three values, the one with higher calculated IR intensity is shown in bold type, while that with higher Raman intensity is shown in italic type. This notation is omitted if these three values are the same, or with almost null IR or Raman intensity. The second and third columns collect the relative IR and Raman intensities (%) calculated in the wavenumber shown in bold type and in italic type, respectively. The relative intensities are obtained by normalizing each calculated value to the intensity of the strongest one in the spectrum. The scaled values by the LSE or PSE procedures are included in another column. The notation used for the values was the same as in the first column. In the next two columns the experimental IR and Raman values observed in the spectra, are listed. The last column corresponds to the main characterization of the vibrations at the M06-2X/Lanl2mb or M06-2X/Lanl2dz levels. In few cases, the % contribution of the different modes to a computed value (PEDs) is included. The calculated values at the B3LYP/Cep-4g level were only included in Table S3 due to their large differences.
The whole IR spectra in the 3750-400 cm-1 range obtained with the scaled values at the three DFT levels used is shown in Figure S2. It can be easily compared to the experimental one plotted at the same scale. In a general comparison of these IR spectra the following is observed:
- A very broad band at ca. 3500 cm-1 is observed in the experimental spectrum, which can only correspond to the O-H stretching ν(O-H) mode of hydration water strongly H-bonded to the ligands in the La-(2b’)3 complex system. Because the spatial arrangement of the ligands in the complex, many holes appear in the structure that can be occupied by water molecules associated with the synthesis of the complex. Due to the large negative charge around the three carboxylate groups, water molecules can be H-bonded through their hydrogen atoms. Moreover, this band does not appear in the Raman spectrum, as it is expected.
- A broad and very strong band centered at 1577.8 cm-1 in the experimental spectrum, in which the in-plane bending δ(O-H) mode of these hydrated water molecules contributes to its broadness.
- A large similarity between the scaled spectra at the M06-2X/Lanl2dz and M06-2X/Lanl2mb levels with the experimental one appears, while the B3LYP/Cep-4g level differs remarkably. This feature is in accordance with a more symmetric and better optimized structure by M06-2X method, compared to B3LYP. The best accordance appears at the M06-2X/Lanl2mb level, although the characterization obtained at the M06-2X/Lanl2dz level was also used for the assignment of experimental spectrum.
- The coordination of the 2b ligands to the lanthanide ion noticeably changes the IR and Raman spectra. They seem different of those obtained with the 2b ligand molecule alone [22].
The same comparison but in the whole Raman spectra is included in Figure S3. Unfortunately, the experimental spectrum shows large background noise that impedes the detection of all weak bands. In this case, the three theoretical scaled spectra appear to have a large similarity to the experimental one. The main difference appears in the experimental Raman line observed at 72 cm-1, which was not reproduced in the theoretical scaled ones.
For a detailed and better analysis of the different experimental and scaled vibrational wavenumbers of these figures, the spectra are divided in three ranges: from 3750-2600 cm-1 (Figure 2), from 1800-1000 cm-1 (Figure 3) and from 1000-400 cm-1 (Figure 4). Due to a lower quality of the experimental Raman spectrum, Figure 5 shows only the spectra comparison in the 1800-800 cm-1 range. The theoretical comparison in other spectral ranges is included as Supplementary Material. Therefore, the region from 3400-2600 cm-1 is shown in Figure S4, from 1800-1000 cm-1 in Figure S5, and from 1000-50 cm-1 in Figure S6. The assignment of the strong and characteristic Raman lines is included in these figures.
In a general comparison of the scaled spectra, it is noted that most of the calculated normal modes appear in their expected ranges. This feature, together with the fact that most of the scaled strongest vibrations by M06-2X method appear close in its wavenumber to the experimental ones, confirm both the scaling carried out on the calculated wavenumbers, as well as the M06-2X method used. Therefore, the assignments in general could be considered correct.
Because the main objective was the identification and characterization of the synthetized La(2b’)3 coordination complex, we focus the main attention in the most characteristics and stronger IR and Raman bands to confirm the structure proposed and optimized in Figure 1. Because the spectra at the B3LYP/Cep-4g level were the worst, their values were omitted in the discussion. For this purpose, the assignment and further discussed was done under the following sections: (i) The COO group modes, (ii) the triazole ring modes, and (iii) the aryl ring modes.
3.4.1. The carboxylate COO group modes
In the isolated state of 2b ligand, the νas(COO) stretching mode was predicted (scaled) with very strong IR intensity at 1710 cm-1 [22]. However, in the La(2b’)3 complex it is expected to be significantly red shifted to lower wavenumbers because of the noticeable lengthening of the CO bonds to form the six O-La bonds. At the M06-2X/Lanl2mb level, this stretching mode was clearly identified in the calculated wavenumber with the strongest IR intensity at 1595 cm-1 (scaled at 1472 cm-1) in very good accordance to the experimental very strong IR band at 1484.5 cm-1. However, at the M06-2X/Lanl2dz level, the symmetric νs(COO) mode was clearly characterized first at higher wavenumbers, compared to the asymmetric mode. It was at 1596 cm-1 (scaled at 1516 cm-1), with strong IR intensity, and at 1607 cm-1 (scaled at 1527 cm-1) with very strong Raman intensity. Both scaled values were in good concordance with the experimental strong IR band at 1500.2 cm-1 and the strong Raman line at 1504 cm-1. However, this assignment differs from that found at the M06-2X/Lanl2mb level.The scaled spectrum at M06-2X/Lanl2mb level appears to be a slightly better fit with the experimental ones (Figure 3). The asymmetric stretching vibrations for the carboxylate group (COO−) are reported to appear as strong IR absorption near 1600-1560 cm-1 for solid state samples [28,29] and at higher wavenumbers than the symmetric ones. For these reasons we assign the experimental IR spectrum mainly to that calculated at the M06-2X/Lanl2mb level.
The symmetric νs(COO) stretching mode appears scaled at the M06-2X/Lanl2mb level at 1368 cm-1 (IR) and at 1372 cm-1 (Raman) with strong intensity, in excellent agreement with the experimental, very strong IR band at 1372.3 cm-1 and the Raman line at 1375 cm-1. These values are also in accordance the ones reported in solid state samples of related compounds [28,29] - near the 1420-1400 cm-1 range for this symmetric stretching mode. At the M06-2X/Lanl2dz level, the asymmetric (instead of the symmetric) mode was the one characterized in calculated wavenumber at 1440 cm-1 (scaled at 1375 cm-1), also in good accordance with the experimental spectra at 1372.3 cm-1 (IR) and 1375 cm-1 (Raman).
3.4.2. The triazole ring modes
The characteristic normal modes of the 1,2,3-triazole ring have been reported [30,31,32], which are in accordance with our calculations. For simplicity, the discussion was only focused on the assignment of the strongest bands.
NNN modes: The νs(NNN) stretching appears strongly coupled with the νs(COO) mode, as well as with other ring modes. At the M06-2X/Lanl2mb level, the highest contribution of this mode was identified in the scaled wavenumber at 1375 cm-1 with strong Raman intensity, in excellent fit with the very strong Raman line at the same value, 1375 cm-1. A large contribution of this mode was also observed in the scaled wavenumber at 1368 cm-1, whose major contribution corresponds to the νs(COO) stretching mode. At the M06-2X/Lanl2dz level this mode was identified at the same scaled wavenumber, but strongly coupled with the νas(COO) mode. In the isolated state of 2b ligand, it was scaled at 1360 cm-1 and related to the experimental IR band at 1340.5 cm-1 [22]. Another triazole ring stretching mode with symmetric character and strongly coupled with an aryl ring mode appears identified in the scaled wavenumber at 1097 cm-1 and in good relation to the very strong experimental IR band at 1091.2 cm-1 and to the Raman line at 1091 cm-1.
The νas(NNN) stretching mode appears characterized at the M06-2X/Lanl2mb level in the scaled wavenumber with very strong IR intensity at 1304 cm-1, also in excellent agreement with the very strong experimental IR band at 1302.0 cm-1.
C8-N14 modes: The stretching mode is predicted at the M06-2X/Lanl2mb level with very high IR intensity and medium Raman activity at 1574 cm-1 in excellent agreement with the very strong and broad band observed in the IR spectrum and centered at 1577.8 cm-1, in which this mode contributes in addition to δ(O-H) of the hydrated water molecules. A similar result was obtained at the M06-2X/Lanl2dz level, but it was predicted at 1581 cm-1 with the highest IR intensity of the spectrum. In the 2b molecule alone [22], it was scaled at 1556 cm-1 in accordance to the experimental IR band with medium intensity at 1543.9 cm-1 and to the Raman line at 1550.6 cm-1.
3.4.3. The aryl ring modes
The assignments for several aryl ring modes are obvious and require no further discussion, therefore the attention was focused only on the strongest vibrations to confirm the structure of the synthesized complex. For the assignments of the ring modes was followed the Varsanyi notation [27] for a 1,4-disubstituted benzene.
The aromatic C-H stretching vibrations are generally observed in the 3200-2950 cm-1 range and they are predicted theoretically as almost pure modes (100% PED) with weak and very weak IR intensity and weak Raman activity. Therefore, the only 2b mode with the scaled wavenumber at 2956 cm-1 was related to the experimental IR band at 2968.1 cm-1.
The aromatic C-C stretching vibration, modes 8a and 8b, appears characterized as almost pure modes with %PED higher than 90%, and they are observed in the experimental spectra at similar wavenumbers. However, mode 8a is predicted with almost null IR intensity and with the highest Raman intensity. It is in good agreement with the very strong Raman line observed at 1597 cm-1. Mode 8b is predicted with almost null IR and Raman intensity and thus it was not observed in the experimental spectra.
Mode 19a is predicted at the M06-2X/Lanl2mb level with strong IR and Raman intensity at 1511 cm-1 in good accordance to the experimental strong IR band at 1500.2 cm-1 and to the Raman line 1504 cm-1. The C-C stretching, mode 14, appears predicted at 1234 cm-1 with medium IR intensity and null Raman intensity in accordance with the experimental IR band with medium intensity observed at 1218.0 cm-1 and to its non-detection in the Raman spectrum.
3.4.4. Low-frequency vibrations
Low-lying molecular vibrations have been studied by different authors [33,34,35,36] and their presence indicates a high flexibility in the molecular structure that may be caused by various factors. Moreover, in our La(2b’)3 complex the normal vibrational modes that include the lanthanide ion mainly appear below 200 cm-1. Thus, this kind of vibrations may play an important role in some biological functions, such as transcription and replication of the double DNA helix, specific interactions with proteins and drugs, and gene expression [36]. Our ligand molecule 2b possesses high conformational flexibility, since it was characterized by ten vibrational modes below 200 cm-1. These modes may be also important for local recognition in the protein cavity. Moreover, the pattern of individual vibrations for every ligand also reflects the details of its conformation. Taking into account the high deformability of our molecule 2b, these low frequencies should be very sensitive to changes in the local environment, such as hydration and interactions with other biomolecules.
Additionally, the spectra in the frequency region under 200 cm−1 are quite interesting, since they offer information about metal–ligand vibrations. Thus, in the present work a theoretical analysis of the low-lying vibrations of our La(2b’)3 complex with frequencies below 200 cm-1 was carried out. Because the large number of vibrational modes below 200 cm-1 in our complex, 42 in total, only the six most characteristic ones were considered and plotted in schematic form in Figure 6. For simplicity and clarity, only the carboxylic and triazole ring vibrations of two of the three ligands were included in this figure. The larger number of these vibrations and its shape, compared to the 2b ligand, agree well with a noticeable increase in the La(2b’)3 complex flexibility.
The first detail observed in these vibrations is that the lanthanide ion motions do not follow the bond length lines with the carboxylic oxygen atoms. It appears that its motion depends on the modulus and direction of the displacement vectors of the surrounding oxygen atoms. The second detail observed is that most of the vibrational modes follow the displacement vectors calculated in the dimer form of benzoic acid (BA) [37] and in agreement with those found by far-infrared [38]. The lanthanide ion does not appear to affect the displacement vectors of the carboxylic group.
As it is expected in the low frequency range, in some atoms the in-plane and out-of-plane character of the displacement vectors is mixed, which makes it difficult to assign it. Figure 6 shows only the most characteristic modes that can be well related to those corresponding to BA molecule and in which the lanthanide ion has a noticeable large displacement vector. In the remaining vibrational modes, the motions of other groups remarkably prevail over those of the carboxylic group and the lanthanide ion and therefore, they were not included in this Figure 6. There are also modes that are difficult to be characterized. An analysis of the shape of the selected vibrations reveals that most of the modes involves a strong deformation of complex, and the motion of the whole structure seems, for example as a γ (butterfly) and as a γ (tilting “slanting”).
3.5. Radical-Scavenging assays
It has been reported[39] that the sodium salt of the ligand (2b), together with its non-ionic, conjugate acid (1b) suppress in vitro 2-deoxyribose degradation at a concentration of 3·10-5 M, or higher. Scavenging of DPPH● was not observed with 2b, mild effect was observed with 1·10-4 M 1b (RSA=10±2%), decreasing in a concentration-dependent manner to practically zero at 1·10-5 M. Both compounds scavenge ABTS●+ at 1·10-4 M (RSA= 17±1% with 2b and RSA= 13±2% with 1b), again – the effect drops to zero at 1·10-5 M. Those results will be presented together with the new data, derived from the equivalent experiments on the complex La(2b’)3.
3.5.1. Impact of 1b, 2b and La(2b’)3 on 2-Deoxyribose degradation
The effect of 2b, and La(2b’)3 compounds on the UV-induced degradation of 2-deoxyribose are presented in Figure 7. They were investigated at concentrations between 1·10-6 M and 1·10-4 M. A statistically significant concentration-dependent effect with respect to scavenging of OH● is observed with these three compounds. Unlike 2b and 1b, the complex manifests significant scavenging activity at a lower concentration (SPh-SI= 72±2% at 1·10-5 M). At concentrations above 3·10-6 М the scavenging activity of the complex is not different than the scavenging activity of 2b at three times higher concentration. Each La(III) ion coordinates three 2b ligands. This feature may be a clue that all three ligands in the complex participate equally in the scavenging process.
3.5.2. Impact of 1b, 2b, and La(2b’)3 on a model system containing the stable radical DPPH●
The ability of the tested compounds to exchange hydrogen with is presented in Figure 8. Previous research showed that 2b practically does not interact with DPPH●, while its conjugate acid 1b interacts only weakly at 1·10-4 M (RSA= 10±2%), the effect decreasing in a concentration-dependent manner to zero at 1·10-5 M. For this reason, lower concentrations were not studied. The La(III) complex was investigated at three times lower concentrations, manifesting very mild activity within the tested concentration range. An additional concentration of 1·10-6 M yielded a result of RSA= 0,5±0,4%. The low activity toward DPPH● means all three compounds tend to scavenge it via HAT only weakly. One has to consider that DPPH● has a large molecule and steric hindrances may play a part in the observed low activity. The model system with 2-deoxyribose produces OH● that are known to be scavenged via HAT reactions [40]. Unlike DPPH●, OH● is small in size and very mobile. La(2b’)3 and 2b demonstrate clear scavenging effect toward OH●, generated by water radiolysis at concentrations higher than 1·10-6 M. The mild, concentration-dependent DPPH-scavenging activity of 1b may be due to the presence of an active carboxyl hydrogen atom in its molecule. La(2b’)3 is more active than concentrations three times higher of 2b. That may be due to the impact of the La(III) ion on electron density distribution within the coordinated ligands, thus yielding active hydrogen atoms, increasing interaction with DPPH●.
3.5.3. Impact of 2b, 1b and La(2b’)3 on a model system containing the stable radical ABTS●+
The ability of the tested compounds to participate in SET reactions with ABTS●+ is presented in Figure 9. At the highest tested concentration of 1·10-4 M 2b and 1b have a mild effect on this model system, decreasing to practically statistically zero at 1·10-5 M. For that reason, lower concentrations were not tested. La(2b’)3 was investigated at three times lower concentrations. Its activity at 3·10-5 M (RSA= 9±1%) was greater than that of 2b at the same concentration (RSA= 5±1%). On the other hand, 1·10-4 M 2b (three times greater concentration) is more active (RSA= 17.3±0,8%) than 3·10-5 M La(2b’)3. A similar, statistically significant observation is noted when comparing 1·10-5 M La(2b’)3 (RSA= 2.1±1.6%) with 3·10-5 M 2b (RSA= 5±1%). At concentrations of 1·10-5 M the ligand and the complex do not demonstrate significant electron exchange with ABTS●+. La(2b’)3 tends to be less active than three times more concentrated 2b. That may be due to the fact that the active sites for electron exchange in the ligand might be sterically hindered after complexation with La(III) occurs. Such complexation may also cause electron density redistribution due to the presence of the metal coordination center.
3.5.4. Impact of 2b and La(2b’)3 on MTT-formazan transformation via Fenton reaction derived hydroxyl radicals
The Fenton reaction is a well-known chemical process with clear clinical significance [41]. “Free” transition metal ions within living organisms create the conditions for its fast progress that yields the highly reactive hydroxyl radicals. OH● tend to attack molecular sites bearing conjugated double bonds, causing a free radical chain reaction, molecular fragmentation, lipid peroxidation and MDA formation. Antioxidants can stop the aforementioned processes, thus preventing damage to biomolecules and possible pathogenesis.
The water radiolysis model system can serve as a good demonstration of the interaction between OH● and a potential antioxidant. On the other hand, the Fenton reaction is a significant biochemical process that takes place at physiological conditions in living organisms. Therefore, what would be expected is similarity between the results of these model systems. That is what makes the results presented in Figure 10 even more interesting.
The complex was tested at concentrations between 3·10-6 M and 3·10-5 M. The ligand 2b was tested at three times greater molarities. In presence of both compounds, the behavior of this model system was extremely unstable. Experiments were repeated twice, on different days to lower the probability of human error or equipment failure as much as possible. Based on observations made, both compounds increase formazan production, i.e. increase RS formation, acting as prooxidants. At concentrations up to 3·10-5 M the ligand 2b manifests no statistically significant impact on absorbance at 578 nm, hence on RSA. At 1·10-4 M the effect is strong and statistically significant (RSA= 115±72%). At three times lower molarity, La(2b’)3 also seems to acts as a prooxidant, though to a smaller degree (RSA= 68±16%). At lower concentrations the effect statistically is zero. These results are unexpected for the authors – in the water radiolysis system both ligand and complex act as potent hydroxyl radical scavengers while at the same time MTT-formazan transformation, caused by Fenton-generated OH● seems to be potentiated by these same substances.
3.5.5. Impact of 2b and La(2b’)3 on LDCL in presence of KO2
The superoxide radical-ion is a type of RS that normally occurs in the human body, produced by one-electron reduction of oxygen. A by-product of oxygen metabolism, and various dedicated enzymes it also plays a role in defence against pathogens and in a variety of cell signalling pathways [42,43]. The ability of the ligand 2b and Ln(2b’)3 to scavenge KO2 derived superoxide is presented in Figure 11.
At concentrations 3·10-3 M and below both compounds are inactive. At 1·10-4 M the ligand increases LDCL slightly (CL-SI= 108±7%). The effect becomes more evident at 3·10-4 M (CL-SI= 121±2.%) – evidence for slight prooxidant action. At 1·10-4 M La(2b’)3 has CL-SI= 118±6.%. The limited solubility of both compounds does not allow testing higher molarities. In terms of in vitro KO2 superoxide scavenging both ligand and its complex could be viewed as relatively inert.
3.5.6. Impact of 2b and La(2b’)3 on LDCL in presence of NaClO
Hypochlorous acid is naturally produced in the human body as an important component of immune defence [44]. It is produced by neutrophils during the respiratory burst - the enzyme myeloperoxidase uses H2O2 and chloride ions as substrates to synthesize this highly toxic specie, a strong oxidant that damages a variety of molecular targets, acting as a bactericide. Its high chemical reactivity and non-specific action are the reason HClO is associated with variety of human pathologies [45]. The ability of the ligand 2b and Ln(2b’)3 to scavenge in vitro NaClO derived superoxide is presented in Figure 12.
When it comes to in vitro interaction with hypochlorite, the divergent trends of behaviour are quite stark. At the lowest tested concentration, the ligand 2b increases LDCL (CL-SI= 139±12%) quite significantly, compared to the control. As molarity rises, CL-SI decreases in a concentration-dependent manner down to 64±40% at 1·10-4 M. The opposite is observed with Ln(2b’)3. At 1·10-6 M it does not significantly impact LDCL (CL-SI=104±4%). At the next, higher concentration (3·10-6 M) CL-SI increases and is statistically the same as that of 2b. That increase continues right up to 201±10% at 1·10-4 M. A previous study [46], using the same model system with another La(III) complex has demonstrated clear correlation between the behavior of both ligand and complex. In this case, the coordination of 2b with La(III) seems to show some kind of drastic change in behavior.
4. Discussion and Conclusions
A new lanthanum(III) complex of 2-aryl-1,2,3-triazole was synthesized and characterized by elemental analysis, IR and Raman spectroscopy. It was analyzed in detail to confirm the molecular arrangement of the ligands in the newly synthetized complex. The most important findings were the following:
- A structural study of the lanthanum(III) complex was carried out at three DFT levels. The starting structure optimized was that with the La(III) ion coordinated with three 2b ligands through the carboxylate group. The optimized structure at the M06-2X/lanl2dz level shows an almost symmetric arrangement. By rotation around the C9-C11 bond length another conformer can be obtained but it is less stable.
- Global chemical reactivity descriptors were calculated in the La(2b’)3 complex. The low energy gap calculated indicates a large chemical reactivity and small excitation energies to the manifold of excited states.
- The coordination of 2b ligand to La(III) ion noticeable changes its IR and Raman spectra, which seem different of those obtained with 2b alone.
- Several new scaling equations were used to improve the calculated spectra. The scaled spectra at M06-2X/Lanl2dz and M06-2X/Lanl2mb levels appear close to the experimental one, while those at the B3LYP/Cep-4g level differ remarkably. The best accordance corresponds to M06-2X/Lanl2mb level.
- The scaled wavenumbers of the most intense IR and Raman vibrations appear in good accordance by both, frequency and intensity, to the experimental most intense IR and Raman bands. Therefore, the spatial arrangement of the ligands in the synthetized La(III) complex was confirmed.
- The main low-lying molecular vibrations in the La(2b’)3 complex were characterized. Its number indicates a high flexibility of its molecular structure.
- The complex shows moderate HAT activity with the stable DPPH● radical, but the activity of ligand 2b in this model system is much lower, and almost zero. This lack of activity may be due to steric factors that hinder hydrogen transfer between the bulky DPPH● and the possible hydrogen-donating active sites in the 2b and La(2b’)3 molecules. When interacting with the small and mobile OH●, generated by UV-induced water radiolysis, both ligand and complex behave as scavengers (3·10−6 M or higher), the latter being the more potent at equimolar concentrations.
- The ligand and the complex participate in SET with the ABTS●+ radical-ion only moderately, in a concentration-dependent manner. Steric factors also seem to be at play here, as the complex appears to be less active than the ligand at three times the concentration.
- Both compounds appear to have little impact on KO2-derived superoxide. Only at the highest tested concentrations, 3·10-4 M for 2b and 1·10-4 M for La(2b’)3, do they seem to show a slight significant pro-oxidant effect.
- Though 2b and La(2b’)3 scavenge OH●, a different model system (Fenton reaction), generating the same RS yields completely opposite results. At low concentrations both compounds seem to be inactive. Increasing the molarity to 3·10-5 M and 1·10-4 M for La(2b’)3 and 2b, respectively. Yields results demonstrating that these compounds behave as pro-oxidants in this model system. As possible causes of this behavior, it is proposed that the ligand interacts with one or more components of the system, rather than the OH● generated by it.
- The La(III) ion dramatically changes the behavior of 2b toward hypochlorite once coordination has taken place. The higher the concentration, the better hypochlorite scavenger 2b seems to be. The behavior of its La(III) complex is the opposite, manifesting itself as a potent pro-oxidant at the highest concentration tested.
The structural and spectroscopic characterization of this new triazole derivative La(III) complex with possible anticancer and antioxidant activity could facilitate the characterization of new complexes with other lanthanides. Its in vitro behavior toward a wide range of RS-generating model systems certainly reveals some interesting possibilities for potential therapeutic applications: compounds that act as hydroxyl scavengers in certain conditions, but as potential prooxidants in others. The La(III) ion is known to be able to compete with iron, displacing it from binding sites in transport proteins and iron-dependent enzymes. Thus both enzyme inhibition and “free” iron release take place simultaneously. Free iron produces RS through the Fenton reaction. A La(III)-bearing complex that can impair iron-dependent physiological processes, release “free” iron, allow the Fenton reaction to take place and in addition enhances that reaction may very well be a promising avenue for further investigation in the oncology field. LDCL demonstrates that while 2b may act as a hypochlorite scavenger, La(2b’)3 may well be a hypochlorite “enhancer”, bearing a coordination center with intrinsic antibacterial activity – another possible application antimicrobial that may well be worth investigating.
Supplementary Materials
The following supporting information has been submitted together in addition to the main body.
Author Contributions
Conceptualization, I.K. and M.P.; methodology, I.K., N.B., M.P. and L.T.; software, M.P.; validation, I.K., N.B., M.P. and L.T.; formal analysis, M.P. and L.T.; investigation, M.P. and L.T.; resources, I.K., N.B., M.P. and L.T.; data curation, M.P. and L.T.; writing—original draft preparation, I.K., N.B., M.P. and L.T.; writing—review and editing, I.K., N.B., M.P. and L.T.; visualization, M.P., N.B. and L.T.; supervision, I.K. and M.P.; project administration, I.K., M.P. and L.T.; funding acquisition, M.P. and I.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study is financed by the European Union-NextGenerationEU, through the National Recovery and Resilience Plan of the Republic of Bulgaria, project № BG-RRP-2.004-0004-C01.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kostova, I.; Soni, R. Bioinorganic Chemistry. 2011.
- Goswami, A.K.; Kostova, I. Medicinal and Biological Inorganic Chemistry; Walter de Gruyter GmbH & Co KG, 2022. [Google Scholar]
- Patyal, M.; Kaur, K.; Bala, N.; Gupta, N.; Malik, A.K. Innovative Lanthanide Complexes: Shaping the future of cancer/tumor Chemotherapy. Journal of Trace Elements in Medicine and Biology 2023, 127277. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, S. Applications of rare earth elements in cancer: Evidence mapping and scientometric analysis. Frontiers in Medicine 2022, 9, 946100. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bettinelli, M.; Boffi, A.; Botta, M.; De Simone, G.; Luchinat, C.; Marengo, E.; Mei, H.; Aime, S. Rare earth elements (REE) in biology and medicine. Rendiconti Lincei. Scienze Fisiche e Naturali 2020, 31, 821–833. [Google Scholar] [CrossRef]
- Bao, G. Lanthanide complexes for drug delivery and therapeutics. Journal of Luminescence 2020, 228, 117622. [Google Scholar] [CrossRef]
- Fouad, R. Synthesis and characterization of lanthanide complexes as potential therapeutic agents. Journal of Coordination Chemistry 2020, 73, 2015–2028. [Google Scholar] [CrossRef]
- Paswan, S.; Anjum, A.; Yadav, N.; Jaiswal, N.; Singh, R.K.P. Synthesis, thermal, photo-physical, and biological properties of mononuclear Yb3+, Nd3+, and Dy3+ complexes derived from Schiff base ligands. Journal of Coordination Chemistry 2020, 73, 686–701. [Google Scholar] [CrossRef]
- Taha, Z.A.; Hijazi, A.K.; Al Momani, W.M. Lanthanide complexes of the tridentate Schiff base ligand salicylaldehyde-2-picolinoylhydrazone: Synthesis, characterization, photophysical properties, biological activities and catalytic oxidation of aniline. Journal of Molecular Structure 2020, 1220, 128712. [Google Scholar] [CrossRef]
- Wu, H.; Pan, G.; Bai, Y.; Zhang, Y.; Wang, H.; Shi, F.; Wang, X.; Kong, J. Study on synthesis, crystal structure, antioxidant and DNA-binding of mono-, di-and poly-nuclear lanthanides complexes with bis (N-salicylidene)-3-oxapentane-1, 5-diamine. Journal of Photochemistry and Photobiology B: Biology 2014, 135, 33–43. [Google Scholar] [CrossRef]
- Zou, H.-H.; Meng, T.; Chen, Q.; Zhang, Y.-Q.; Wang, H.-L.; Li, B.; Wang, K.; Chen, Z.-L.; Liang, F. Bifunctional mononuclear dysprosium complexes: single-ion magnet behaviors and antitumor activities. Inorganic Chemistry 2019, 58, 2286–2298. [Google Scholar] [CrossRef]
- Safronov, N.E.; Kostova, I.P.; Palafox, M.A.; Belskaya, N.P. Combined NMR Spectroscopy and Quantum-Chemical Calculations in Fluorescent 1, 2, 3-Triazole-4-carboxylic Acids Fine Structures Analysis. International Journal of Molecular Sciences 2023, 24, 8947. [Google Scholar] [CrossRef]
- Alam, M.M. 1, 2, 3-Triazole hybrids as anticancer agents: A review. Archiv der Pharmazie 2022, 355, 2100158. [Google Scholar] [CrossRef] [PubMed]
- Peica, N.; Kostova, I.; Kiefer, W. Theoretical and experimental studies on binding mode of 3, 5-pyrazoledicarboxylic acid in its new La (III) complex. Chemical physics 2006, 325, 411–421. [Google Scholar] [CrossRef]
- Hrimla, M.; Oubella, A.; Laamari, M.R.; Bahsis, L.; Ghaleb, A.; Auhmani, A.; Morjani, H.; Julve Olcina, M.; Stiriba, S.E.; Itto, M.Y.A. Click synthesis, anticancer activity, and molecular docking investigation of some functional 1, 2, 3-triazole derivatives. Biointerface Research in Applied Chemistry, 2022, vol. 12, num. 6, p. 7633-7667, 2022. [Google Scholar]
- Poonia, N.; Kumar, A.; Kumar, V.; Yadav, M.; Lal, K. Recent progress in 1H-1, 2, 3-triazoles as potential antifungal agents. Current Topics in Medicinal Chemistry 2021, 21, 2109–2133. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Sun, X.; Li, W.; Hou, G.; Gao, F. 1, 2, 3-Triazole-containing compounds as anti–lung cancer agents: Current developments, mechanisms of action, and structure–activity relationship. Frontiers in pharmacology 2021, 12, 661173. [Google Scholar] [CrossRef]
- Dong, G.; Jiang, Y.; Zhang, F.; Zhu, F.; Liu, J.; Xu, Z. Recent updates on 1, 2, 3-, 1, 2, 4-, and 1, 3, 5-triazine hybrids (2017–present): The anticancer activity, structure–activity relationships, and mechanisms of action. Archiv der Pharmazie 2023, 356, 2200479. [Google Scholar] [CrossRef]
- Wu, X.; Wang, J.; Xia, S.; Cheng, S.; Shi, Y. 1, 2, 3-Triazole Derivatives with Anti-breast Cancer Potential. Current Topics in Medicinal Chemistry 2022, 22, 1406–1425. [Google Scholar] [CrossRef]
- Slavova, K.I.; Todorov, L.T.; Belskaya, N.P.; Palafox, M.A.; Kostova, I.P. Developments in the application of 1, 2, 3-triazoles in cancer treatment. Recent Patents on Anti-Cancer Drug Discovery 2020, 15, 92–112. [Google Scholar] [CrossRef]
- Kostova, I.; Valcheva-Traykova, M. New samarium (III) complex of 5-aminoorotic acid with antioxidant activity. Applied Organometallic Chemistry 2015, 29, 815–824. [Google Scholar] [CrossRef]
- Palafox, M.A.; Belskaya, N.P.; Kostova, I.P. Study of the Molecular Architectures of 2-(4-Chlorophenyl)-5-(Pyrrolidin-1-Yl)-2H-1, 2, 3-Triazole-4-Carboxylic Acid as the Potential Anticancer Drug by Their Vibrational Spectra and Quantum Chemical Calculations. 2023.
- Rogachev, A.; Kuzmina, N.; Nemukhin, A. Theoretical modeling of the heterobimetallic complex [La (pta) 3Cu (salen)] and its precursors. Journal of alloys and compounds 2004, 374, 335–338. [Google Scholar] [CrossRef]
- Mehring, M.; Mansfeld, D.; Schürmann, M. The Stereochemical Activity of the Lone Pair in [Bi (NO3) 3 {(iPrO) 2 (O) PCH2P (O)(OiPr) 2} 2]—Comparison of Bismuth, Lanthanum and Praseodymium Nitrate Complexes. Zeitschrift für anorganische und allgemeine Chemie 2004, 630, 452–461. [Google Scholar] [CrossRef]
- Mishra, V.R.; Sekar, N. Photostability of coumarin laser dyes-a mechanistic study using global and local reactivity descriptors. Journal of fluorescence 2017, 27, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Chemical hardness and density functional theory. Journal of Chemical Sciences 2005, 117, 369–377. [Google Scholar] [CrossRef]
- Varsányi, G.; Láng, L.; Kovner, M.A.e.; Lempert, K. Assignment for vibrational spectra of seven hundred benzene derivatives. (No Title), 1974. [Google Scholar]
- Tammer, M. G. Sokrates: Infrared and Raman characteristic group frequencies: tables and charts: Wiley, Chichester, 2004. ISBN 0-470-09307-2, 347 pages, paperback; US $60. 2004.
- George, S. Infrared and Raman characteristic group frequencies: tables and charts. Wiley, Chichester 2001, 82, 85–87. [Google Scholar]
- Aziz, S.G.; Elroby, S.A.; Alyoubi, A.; Osman, O.I.; Hilal, R. Experimental and theoretical assignment of the vibrational spectra of triazoles and benzotriazoles. Identification of IR marker bands and electric response properties. Journal of molecular modeling 2014, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Törnkvist, C.; Bergman, J.; Liedberg, B. Geometry and vibrations of the 1, 2, 3-triazole anion. A theoretical and experimental study. The Journal of Physical Chemistry 1991, 95, 3119–3123. [Google Scholar] [CrossRef]
- El-Azhary, A.; Suter, H.; Kubelka, J. Experimental and theoretical investigation of the geometry and vibrational frequencies of 1, 2, 3-triazole, 1, 2, 4-triazole, and tetrazole anions. The Journal of Physical Chemistry A 1998, 102, 620–629. [Google Scholar] [CrossRef]
- Shishkin, O.V.; Pelmenschikov, A.; Hovorun, D.M.; Leszczynski, J. Theoretical analysis of low-lying vibrational modes of free canonical 2-deoxyribonucleosides. Chemical Physics 2000, 260, 317–325. [Google Scholar] [CrossRef]
- Hovorun, D.; Mishchuk, Y.R.; Yurenko, Y.P. Low-frequency Raman spectra of polycrystalline ribonucleosides. Biopolymers and Cell 2002, 18, 219–226. [Google Scholar] [CrossRef]
- Martel, P.; Hennion, B.; Durand, D.; Calmettes, P. Low-frequency vibrations of a nucleoside analog. Journal of Biomolecular Structure and Dynamics 1994, 12, 401–411. [Google Scholar] [CrossRef]
- Pelmenschikov, A.; Hovorun, D.M.; Shishkin, O.V.; Leszczynski, J. A density functional theory study of vibrational coupling between ribose and base rings of nucleic acids with ribosyl guanosine as a model system. The Journal of Chemical Physics 2000, 113, 5986–5990. [Google Scholar] [CrossRef]
- Palafox, M.A.; Nunez, J.; Gil, M. Theoretical quantum chemical study of benzoic acid: Geometrical parameters and vibrational wavenumbers. International journal of quantum chemistry 2002, 89, 1–24. [Google Scholar] [CrossRef]
- Zelsmann, H.; Mielke, Z. Far-infrared spectra of benzoic acid. Chemical physics letters 1991, 186, 501–508. [Google Scholar] [CrossRef]
- Todorov, L.; Hristova, N.; Belskaya, N.; Kostova, I. Antioxidant properties of a novel triazole ligand. Macedonian Pharmaceutical Bulletin 2022, 68, 2. [Google Scholar] [CrossRef]
- Galano, A. Free radicals induced oxidative stress at a molecular level: The current status, challenges and perspectives of computational chemistry based protocols. Journal of the Mexican Chemical Society 2015, 59, 231–262. [Google Scholar] [CrossRef]
- Kell, D.B. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC medical genomics 2009, 2, 1–79. [Google Scholar]
- Che, M.; Wang, R.; Li, X.; Wang, H.-Y.; Zheng, X.S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug discovery today 2016, 21, 143–149. [Google Scholar] [CrossRef]
- Trist, B.G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide dismutase 1 in health and disease: how a frontline antioxidant becomes neurotoxic. Angewandte Chemie International Edition 2021, 60, 9215–9246. [Google Scholar] [CrossRef]
- Pattison, D.; Davies, M. Reactions of myeloperoxidase-derived oxidants with biological substrates: gaining chemical insight into human inflammatory diseases. Current medicinal chemistry 2006, 13, 3271–3290. [Google Scholar] [CrossRef]
- Strzepa, A.; Pritchard, K.A.; Dittel, B.N. Myeloperoxidase: A new player in autoimmunity. Cellular immunology 2017, 317, 1–8. [Google Scholar] [CrossRef]
- Todorov, L.T.; Traykova, M.L.; Kostova, I.P. In Vitro Interaction of 5-aminoorotic Acid and Its Lanthanum (III) Complex With Superoxide and Hypochlorite Radicals. Der Pharma Chemica 2020, 12, 10. [Google Scholar]
- Kedare, S.B.; Singh, R. Genesis and development of DPPH method of antioxidant assay. Journal of food science and technology 2011, 48, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, P. The use of the stable free radical diphenylpicrylhydrazyl (DPPH) for estimating antioxidant activity. Songklanakarin J. sci. technol 2004, 26, 211–219. [Google Scholar]
- Erel, O. A novel automated direct measurement method for total antioxidant capacity using a new generation, more stable ABTS radical cation. Clinical biochemistry 2004, 37, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Erel, O. A novel automated method to measure total antioxidant response against potent free radical reactions. Clinical biochemistry 2004, 37, 112–119. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.; Aruoma, O.I. The deoxyribose method: a simple “test-tube” assay for determination of rate constants for reactions of hydroxyl radicals. Analytical biochemistry 1987, 165, 215–219. [Google Scholar] [CrossRef]
- Burns, W.G.; Sims, H.E. Effect of radiation type in water radiolysis. Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases 1981, 77, 2803–2813. [Google Scholar] [CrossRef]
- Seminario, J.M. Modern density functional theory: a tool for chemistry; Elsevier, 1995. [Google Scholar]
- Palafox, M.A.; Iza, N. Tautomerism of the natural thymidine nucleoside and the antiviral analogue D4T. Structure and influence of an aqueous environment using MP2 and DFT methods. Physical Chemistry Chemical Physics 2010, 12, 881–893. [Google Scholar] [CrossRef]
- Brovarets’, O.h.O.; Hovorun, D.M. Prototropic tautomerism and basic molecular principles of hypoxanthine mutagenicity: An exhaustive quantum-chemical analysis. Journal of Biomolecular Structure and Dynamics 2013, 31, 913–936. [Google Scholar] [CrossRef]
- Riley, K.E.; Hobza, P. Noncovalent interactions in biochemistry. Wiley Interdisciplinary Reviews: Computational Molecular Science 2011, 1, 3–17. [Google Scholar] [CrossRef]
- Riley, K.E.; Pitonák, M.; Jurecka, P.; Hobza, P. Stabilization and structure calculations for noncovalent interactions in extended molecular systems based on wave function and density functional theories. Chemical Reviews 2010, 110, 5023–5063. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Applications and validations of the Minnesota density functionals. Chemical Physics Letters 2011, 502, 1–13. [Google Scholar] [CrossRef]
- Alcolea Palafox, M. Scaling factors for the prediction of vibrational spectra. I. Benzene molecule. International Journal of Quantum Chemistry 2000, 77, 661–684. [Google Scholar] [CrossRef]
- Palafox, M.A. DFT computations on vibrational spectra: Scaling procedures to improve the wavenumbers. Physical Sciences Reviews 2018, 3, 20170184. [Google Scholar] [CrossRef]
- Frisch, M.e.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16, revision C. 01. 2016. [Google Scholar]
- Srivastav, G.; Yadav, B.; Yadav, R.K.; Yadav, R. DFT studies of molecular structures conformers and vibrational characteristics of sulfanilamide. Computational and Theoretical Chemistry 2019, 1167, 112588. [Google Scholar] [CrossRef]
Figure 2.
Comparison of the scaled IR spectra at the M06-2X/Lanl2dz, M06-2X/Lanl2mb and B3LYP/Cep-4G levels by different procedures with the experimental ones in the 3750-2600 cm-1 range.
Figure 2.
Comparison of the scaled IR spectra at the M06-2X/Lanl2dz, M06-2X/Lanl2mb and B3LYP/Cep-4G levels by different procedures with the experimental ones in the 3750-2600 cm-1 range.
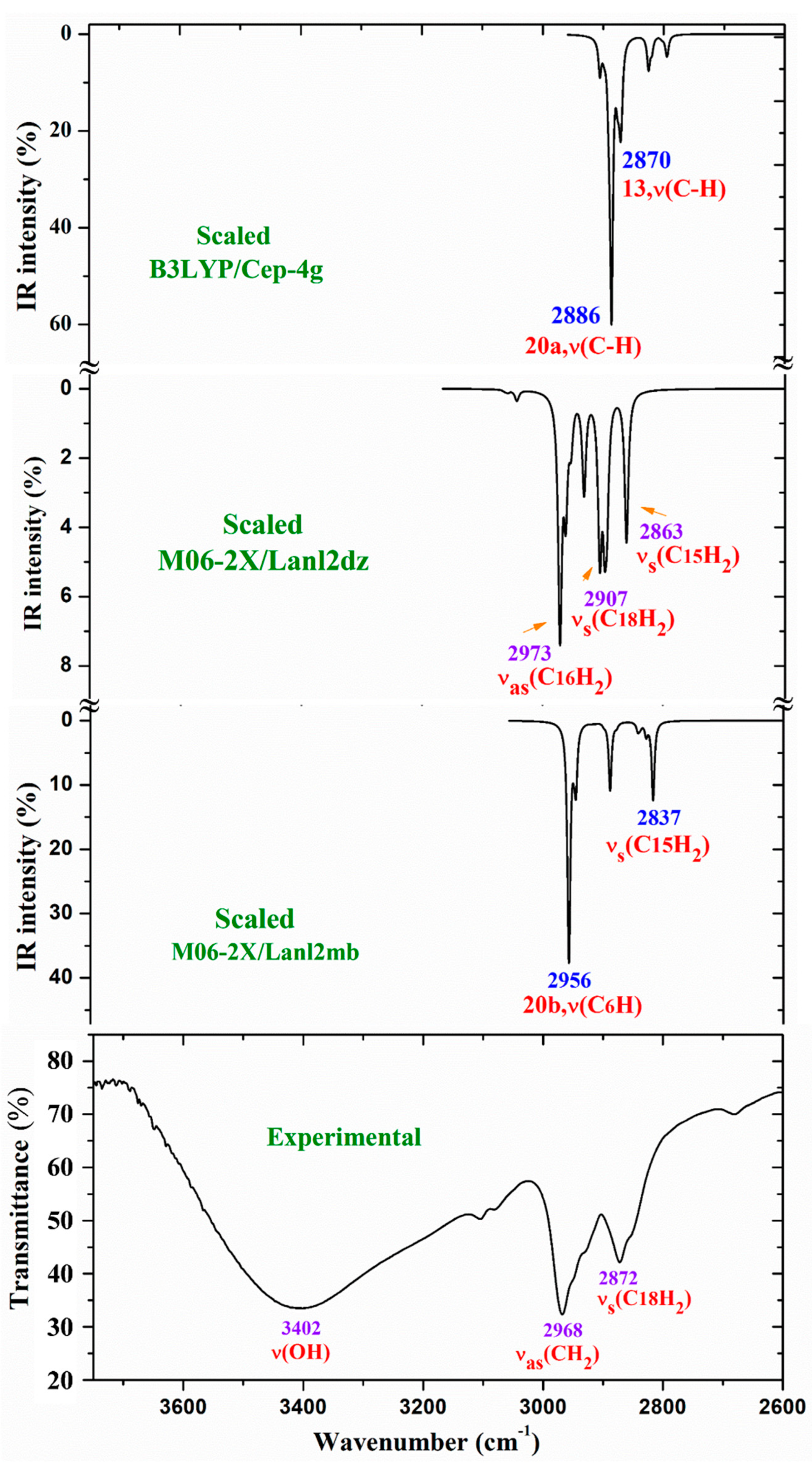
Figure 3.
Comparison of the scaled IR spectra at different levels and procedures with the experimental ones in the 1800-1000 cm-1 range.
Figure 3.
Comparison of the scaled IR spectra at different levels and procedures with the experimental ones in the 1800-1000 cm-1 range.
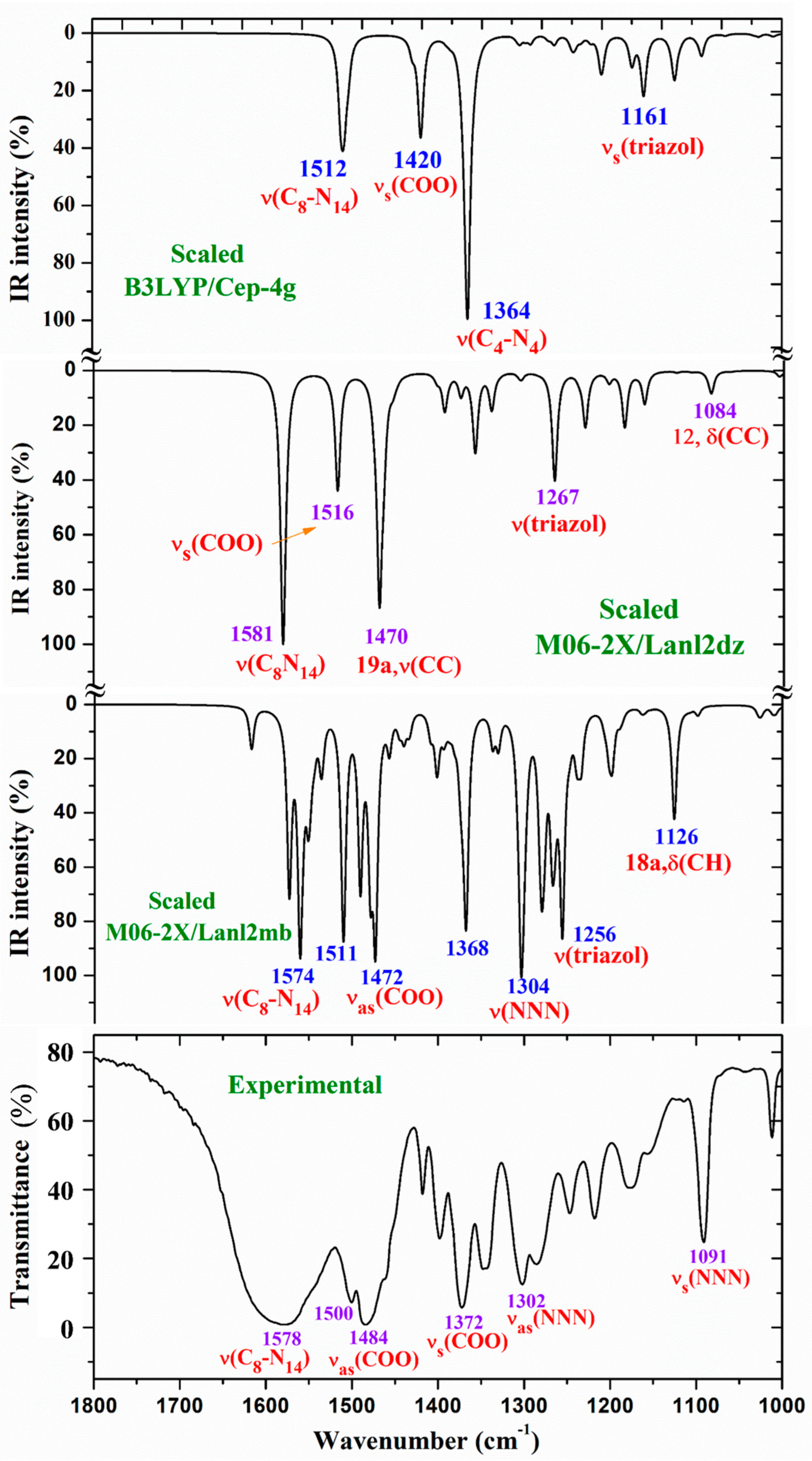
Figure 4.
Comparison of the scaled IR spectra at different levels and procedures with the experimental ones in the 1000-400 cm-1 range.
Figure 4.
Comparison of the scaled IR spectra at different levels and procedures with the experimental ones in the 1000-400 cm-1 range.

Figure 5.
Comparison of the scaled Raman spectrum at the M06-2X/Lanl2dz level and scaled following the LSE procedure with the experimental one in the 1800-800 cm-1 range.
Figure 5.
Comparison of the scaled Raman spectrum at the M06-2X/Lanl2dz level and scaled following the LSE procedure with the experimental one in the 1800-800 cm-1 range.
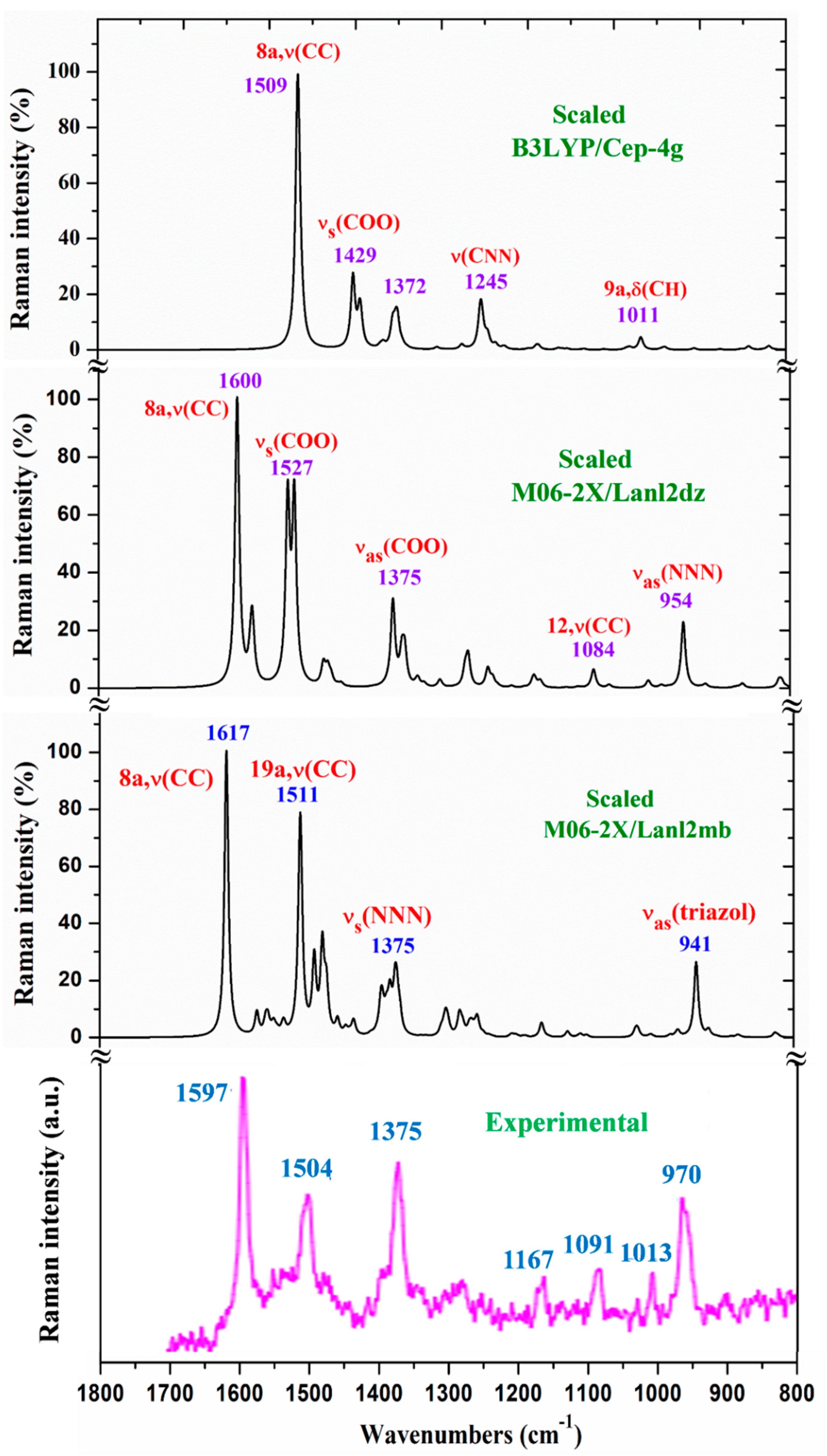
Figure 6.
Low frequency vibrational modes characterized in the calculated spectrum at the M06-2X/Lanl2dz level. The ⊕ and ⊝ symbols correspond to out-of-plane motions, in one direction or in its opposite way.
Figure 6.
Low frequency vibrational modes characterized in the calculated spectrum at the M06-2X/Lanl2dz level. The ⊕ and ⊝ symbols correspond to out-of-plane motions, in one direction or in its opposite way.
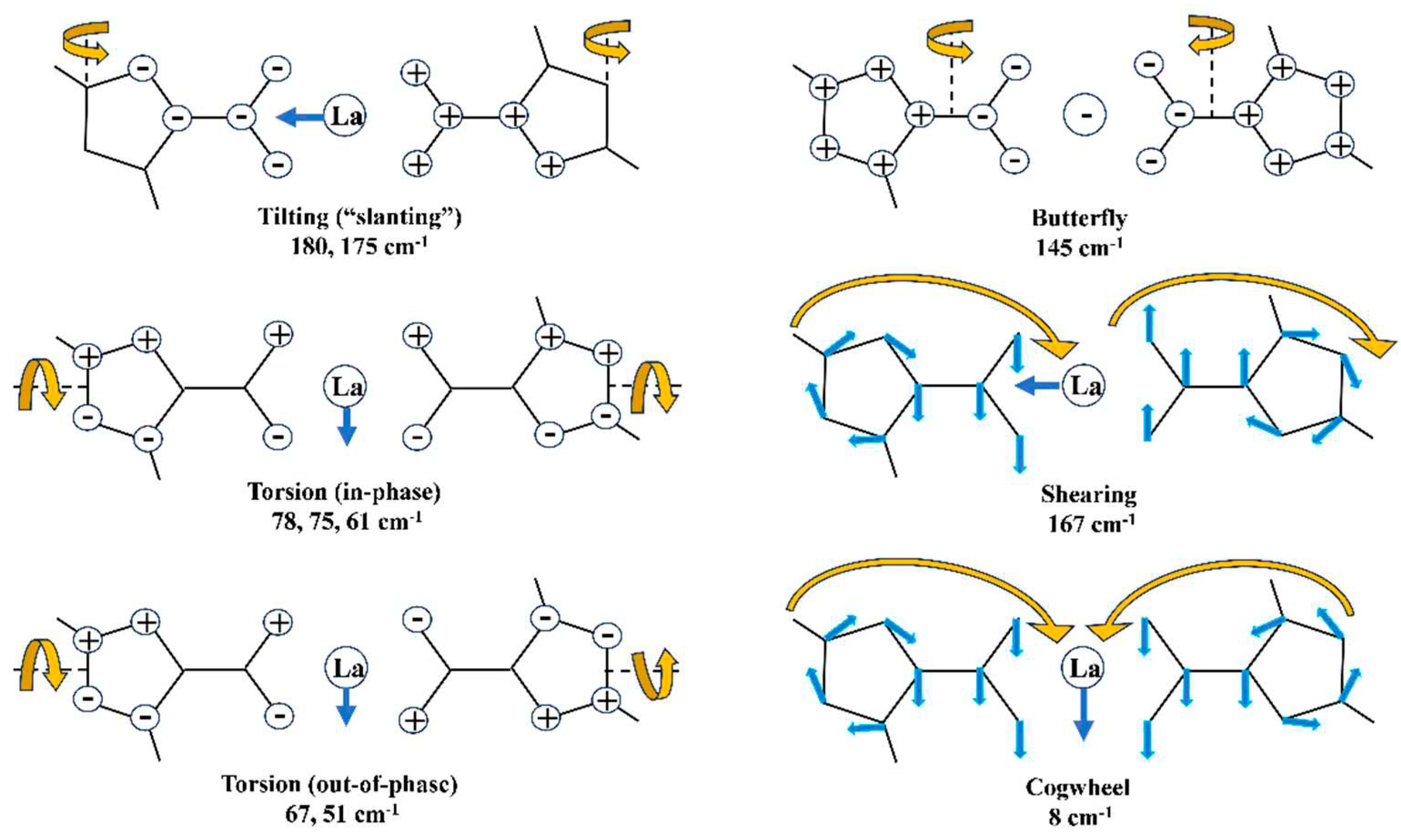
Figure 7.
Impact of 1b, 2b and La(2b’)3 on 2-deoxyribose degradation, resulting from UV-induced water radiolysis. Data= Mean±StDev, p<0.05, N=3. Lower results mean higher scavenging activity.
Figure 7.
Impact of 1b, 2b and La(2b’)3 on 2-deoxyribose degradation, resulting from UV-induced water radiolysis. Data= Mean±StDev, p<0.05, N=3. Lower results mean higher scavenging activity.
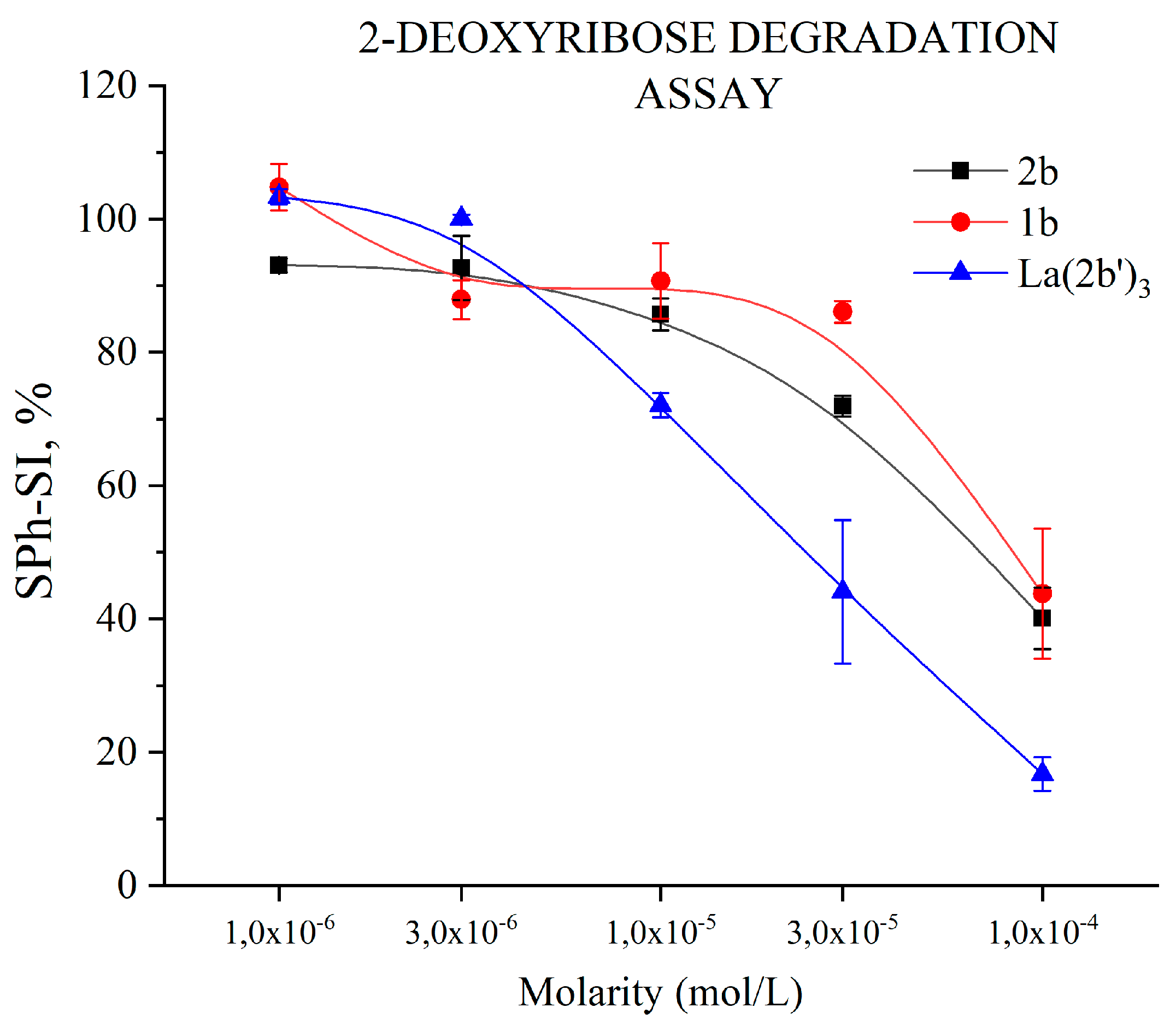
Figure 8.
Impact of 2b, 1b and La(2b’)3 on DPPH●. Data= Mean±StDev, p<0.05, N=3. Higher result means higher scavenging activity.
Figure 8.
Impact of 2b, 1b and La(2b’)3 on DPPH●. Data= Mean±StDev, p<0.05, N=3. Higher result means higher scavenging activity.
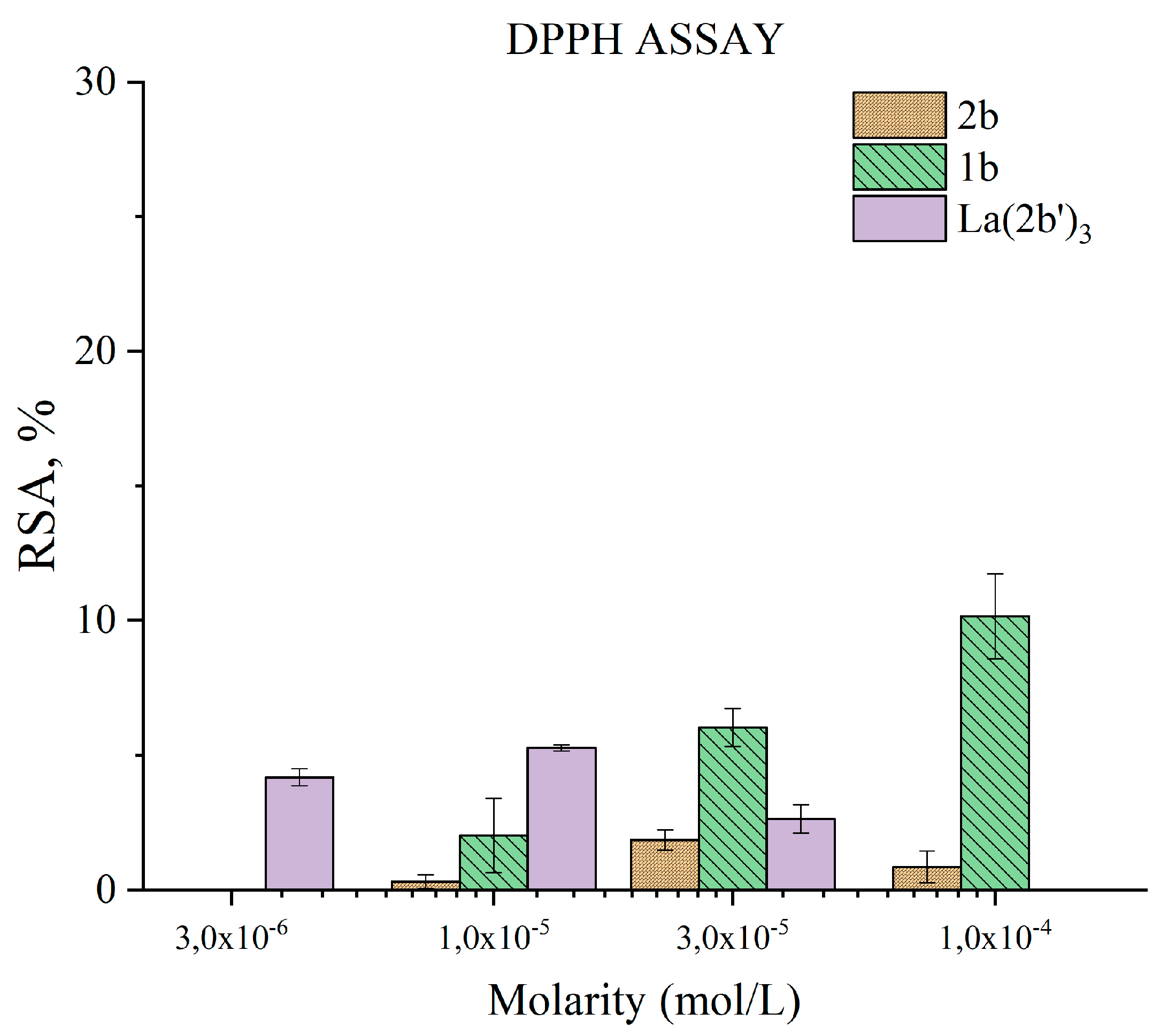
Figure 9.
Impact of 2b, 1b and La(2b’)3 on ABTS●+. Data= Mean±StDev, p<0.05, N=3. Higher result means higher scavenging activity.
Figure 9.
Impact of 2b, 1b and La(2b’)3 on ABTS●+. Data= Mean±StDev, p<0.05, N=3. Higher result means higher scavenging activity.
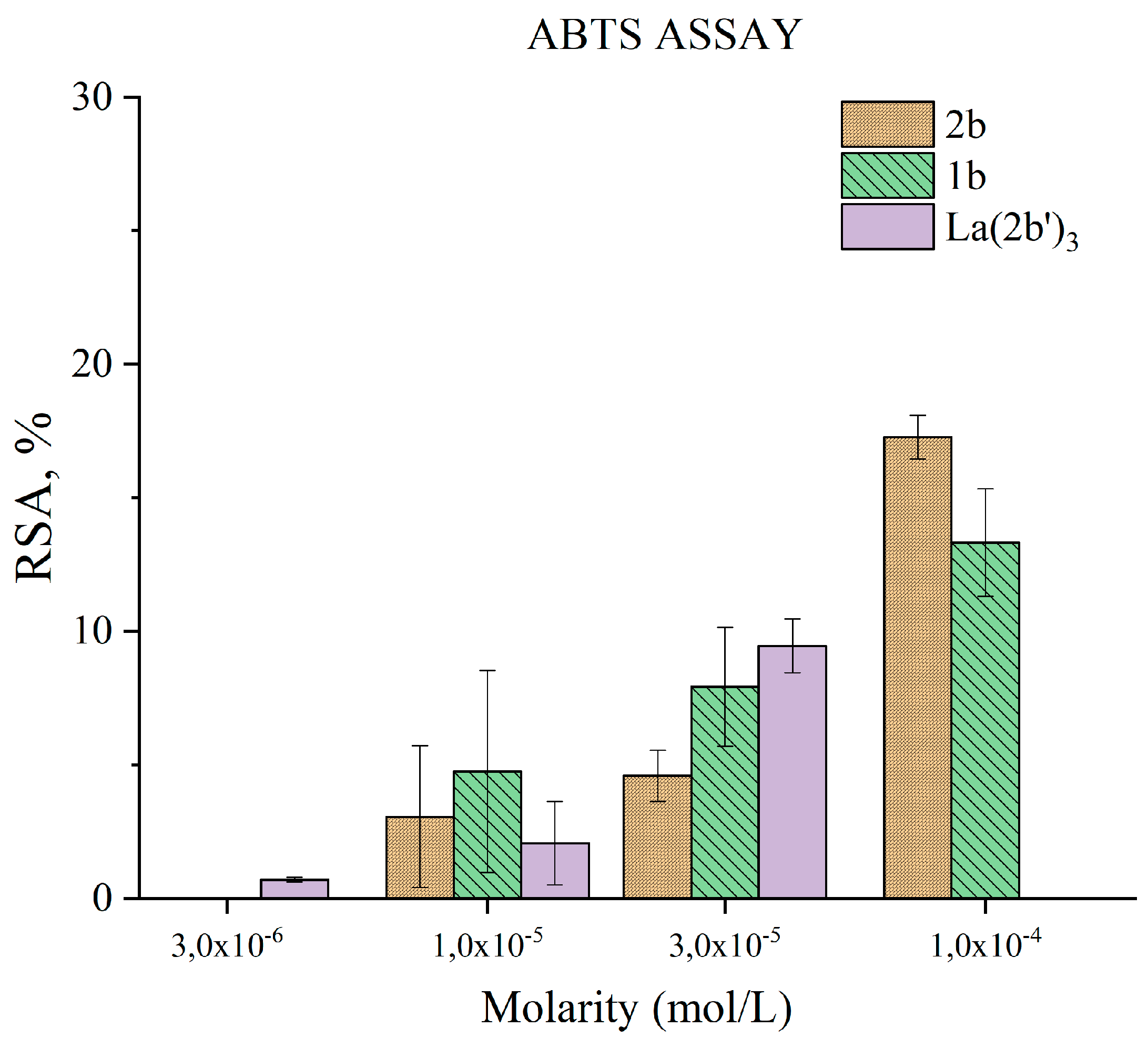
Figure 10.
Impact of 2b and La(2b’)3 on MTT-Formazan transformation by Fenton-generated OH●. Data= Mean±StDev, p< 0.05, N=3. Higher result means higher scavenging activity.
Figure 10.
Impact of 2b and La(2b’)3 on MTT-Formazan transformation by Fenton-generated OH●. Data= Mean±StDev, p< 0.05, N=3. Higher result means higher scavenging activity.
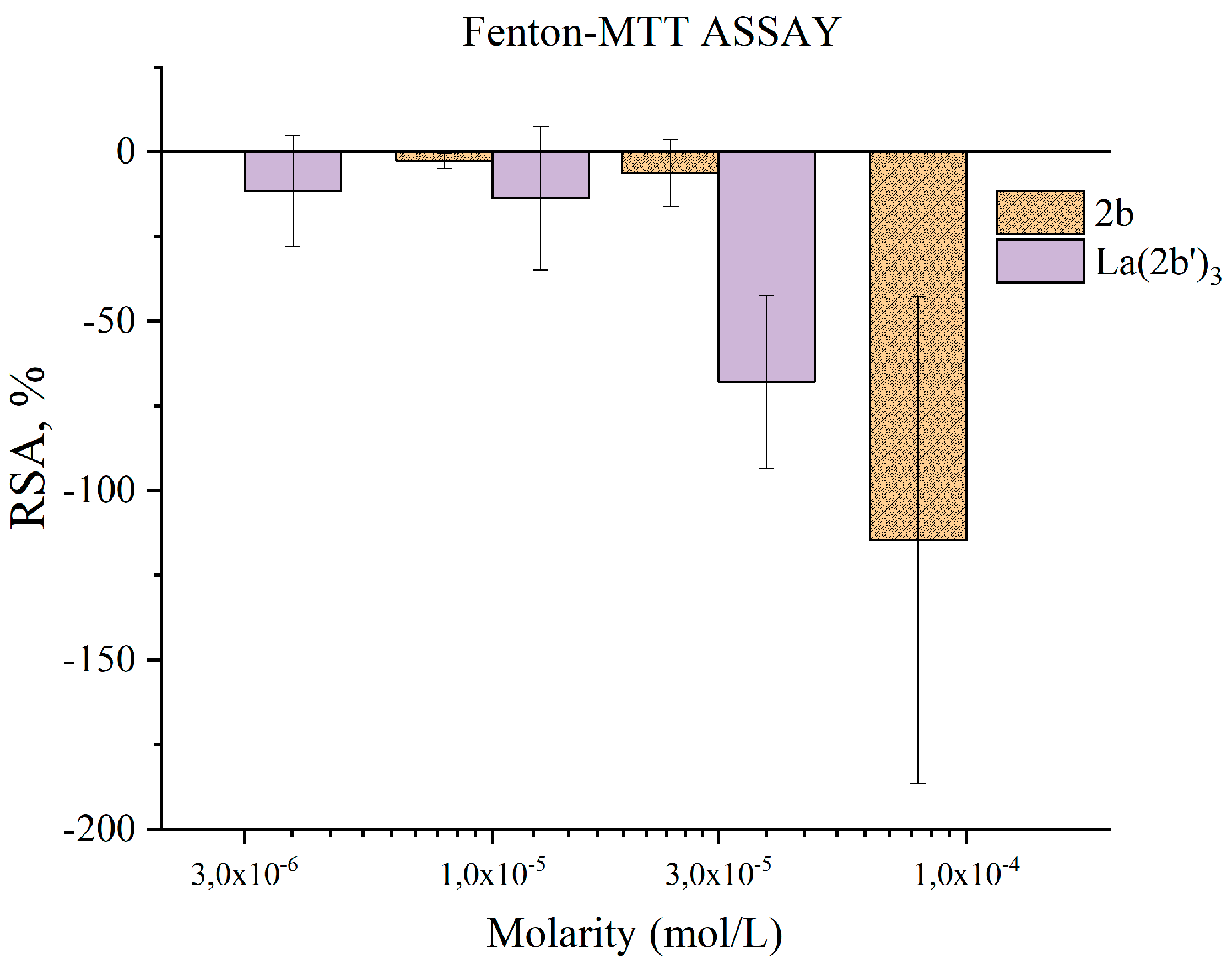
Figure 11.
Impact of 2b and La(2b’)3 on LDCL from KO2. Data= Mean±StDev, p< 0.05, N=3. Lower result means higher scavenging activity.
Figure 11.
Impact of 2b and La(2b’)3 on LDCL from KO2. Data= Mean±StDev, p< 0.05, N=3. Lower result means higher scavenging activity.
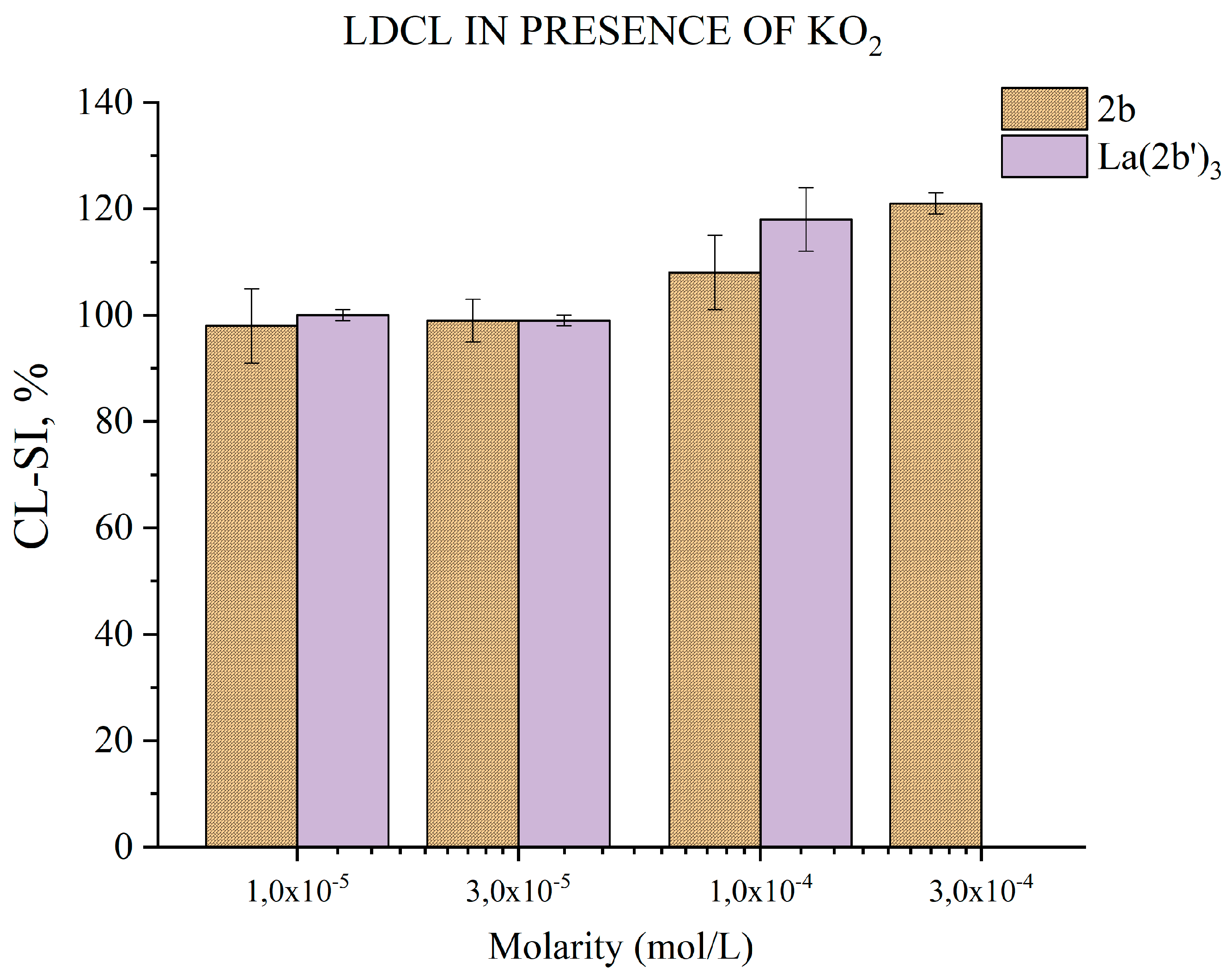
Figure 12.
Impact of 2b and La(2b’)3 on LDCL from hypochlorite. Data= Mean±StDev, p< 0.05, N=3. Lower result means higher scavenging activity.
Figure 12.
Impact of 2b and La(2b’)3 on LDCL from hypochlorite. Data= Mean±StDev, p< 0.05, N=3. Lower result means higher scavenging activity.
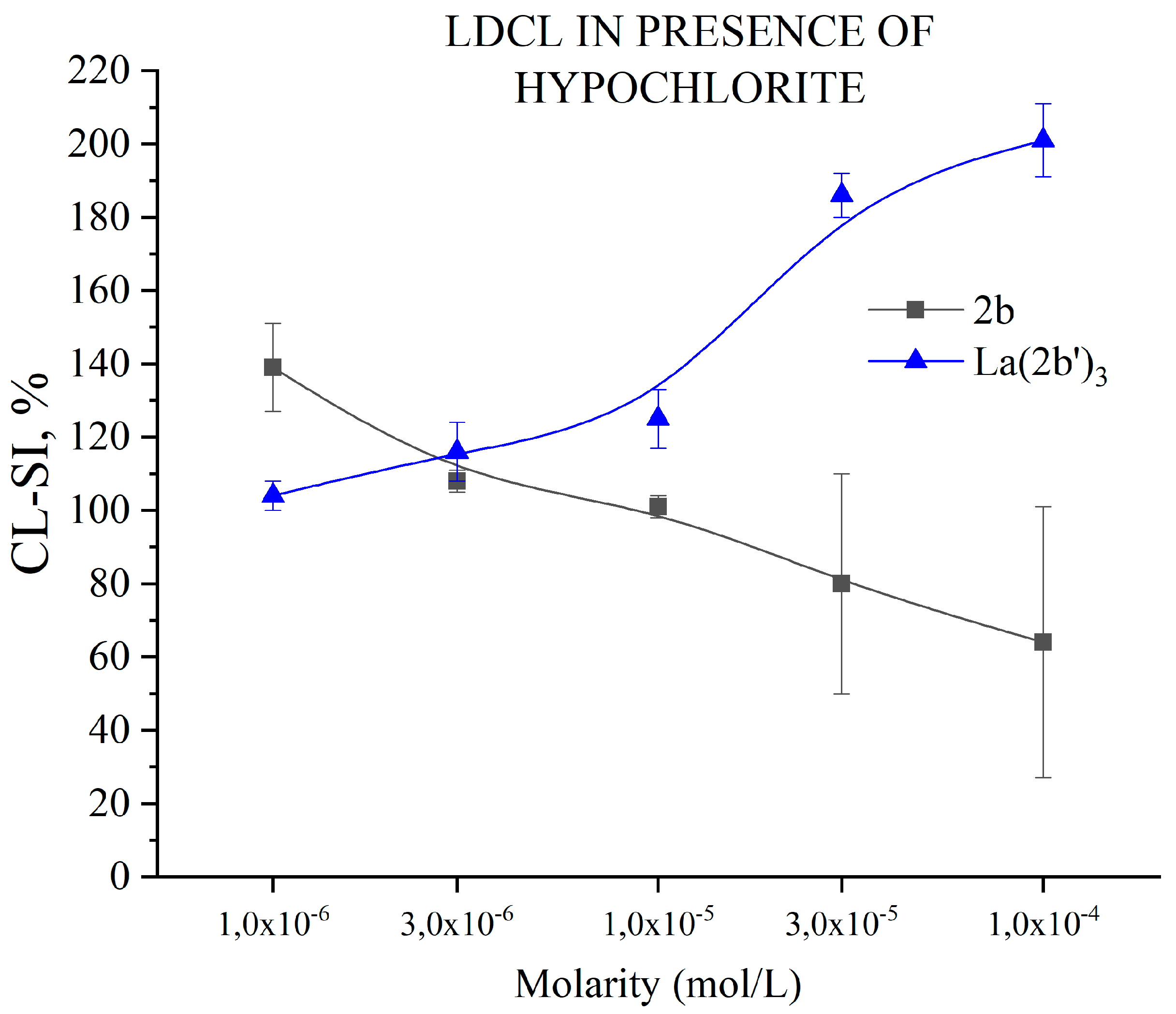
Table 1.
Fenton reaction-MTT assay sample composition.
| Control | Sample | Blank | |
|---|---|---|---|
| Tested compound | no | 200 L | 200 L |
| MTT | 200 L | 200 L | 200 L |
| Fe2+/H2O2/Na2-EDTA | 100 L | 100 L | no |
| Ascorbic acid | 100 L | 100 L | no |
| Bi-destilled water | up to 2.0 mL | дo 2.0 mL | дo 2.0 mL |
Table 2.
DPPH assay sample composition.
| Blank | Control | Sample | |
|---|---|---|---|
| Tested compound | 200 L | no | 200 L |
| DPPH | no | 1800 L | 1800 L |
| Ethanol | 1800 L | no | no |
| Bi-distilled water | no | 200 L | no |
Table 3.
ABTS assay sample composition.
| Blank | Control | Sample | |
|---|---|---|---|
| Tested compound | 100 L | no | 100 L |
| R1 | 860 L | 860 L | 860 L |
| R2 | no | 40 L | 40 L |
| Bi-destilled water | 40 L | 100 L | no |
Table 4.
Several selected optimized geometrical parameters calculated at different DFT levels in the La-(2b’)3 system. Bond lengths (r) in Å, bond angles and dihedral angles (∠) in degrees. The data were from ligand I, Figure 1.
Table 4.
Several selected optimized geometrical parameters calculated at different DFT levels in the La-(2b’)3 system. Bond lengths (r) in Å, bond angles and dihedral angles (∠) in degrees. The data were from ligand I, Figure 1.
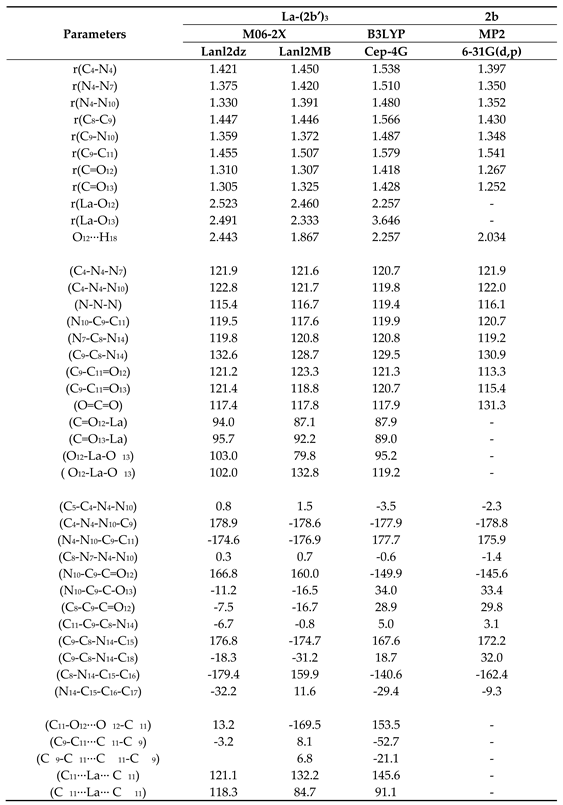 |
Table 6.
Molecular properties and global chemical descriptors (eV) calculated at three DFT levels in the La-(2b’)3 complex.
Table 6.
Molecular properties and global chemical descriptors (eV) calculated at three DFT levels in the La-(2b’)3 complex.
| Molecular properties | M06-2X | B3LYP | |
|---|---|---|---|
| Lanl2dz | Lanl2MB | Cep-4G | |
| Rotational constants: A (GHz) B C |
0.019 0.017 0.012 |
0.040 0.015 0.014 |
0.035 0.013 0.012 |
| Cv (cal/mol·K) S (cal/mol·K) |
202.97 348.72 |
200.6 345.3 |
229.86 376.53 |
| Dipole moment (Debye) | 4.085 | 3.323 | 4.377 |
| HOMO LUMO Eg IP EA S |
-0.262 -0.052 0.210 0.262 0.052 0.157 0.105 0.052 |
-0.189 0.009 0.198 0.189 -0.009 0.090 0.099 0.049 |
-0.253 -0.146 0.107 0.253 0.146 0.199 0.054 0.027 |
Table 7.
Calculated, scaled and experimental wavenumbers (ν, cm-1) in the La(2b’)3 complex. Relative infrared intensity (A) in %, relative Raman intensity (S) in %, and Raman depolarization ratios for plane (DP) and unpolarized incident light (DU). For each vibration of the trimer, the wavenumber with the highest IR intensity is indicated in bold type and that with the highest Raman intensity is indicated in italic type. The relative IR and Raman intensities were shown only for these wavenumbers. DP and DU values were from most intense Raman line. The number of the ring mode corresponds to Wilson’s notation [27].
Table 7.
Calculated, scaled and experimental wavenumbers (ν, cm-1) in the La(2b’)3 complex. Relative infrared intensity (A) in %, relative Raman intensity (S) in %, and Raman depolarization ratios for plane (DP) and unpolarized incident light (DU). For each vibration of the trimer, the wavenumber with the highest IR intensity is indicated in bold type and that with the highest Raman intensity is indicated in italic type. The relative IR and Raman intensities were shown only for these wavenumbers. DP and DU values were from most intense Raman line. The number of the ring mode corresponds to Wilson’s notation [27].
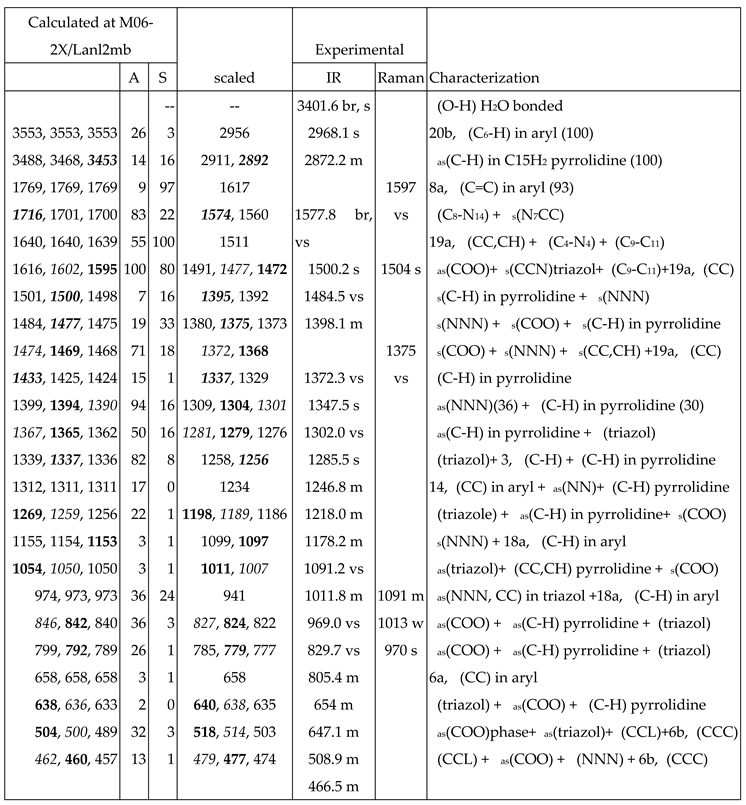 |
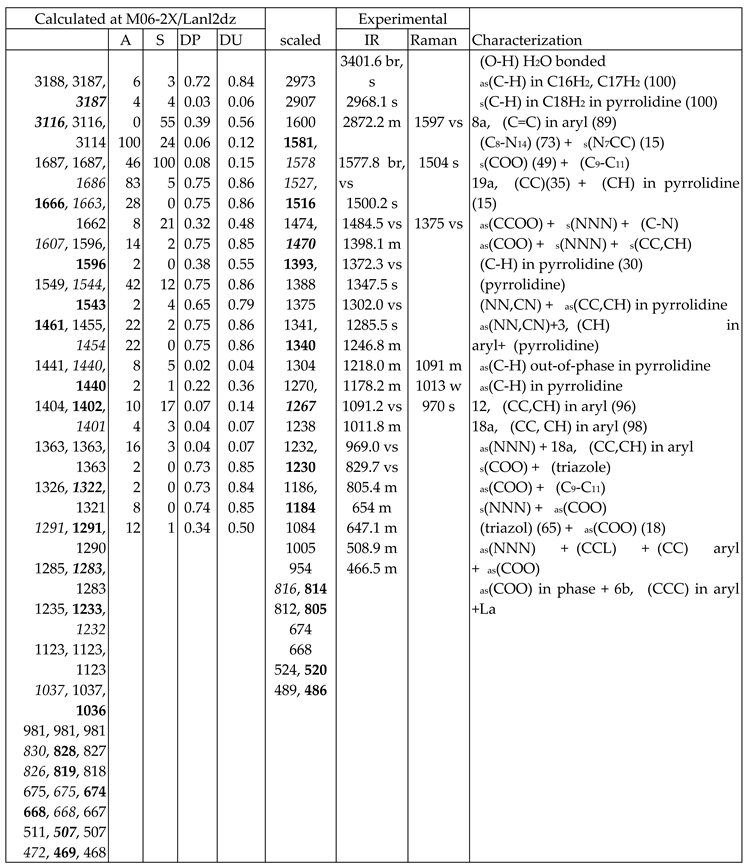 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



