
Submitted:
21 September 2023
Posted:
22 September 2023
You are already at the latest version
Abstract
Chronic myelomonocytic leukemia (CMML) is a hematological neoplasm characterized by monocytosis, splenomegaly, thrombocytopenia and anemia. Moreover, it is associ-ated with SRSF2 mutations and rarely with CSF3R variants. The presented case is an 84-year-old patient with persistent anemia and monocytosis. Due to the presence of dysmorphic granulocytes, monocyte atypia and myeloid precursors in peripheral blood, the patient was submitted to bone marrow examination. The diagnosis was consistent with CMML type 2. Hemocoagulative test showed an increase of fibrinolysis markers. Next generation targeted sequencing showed TET2 and SRSF2 mutations, along with an unexpected CSF3R germline missense variant, rarely encountered in CMML. The pa-tient started Azacitidine treatment reaching normal hemostatic process values. In con-clusion, we identified a heterozygous germline mutation that, cooperating with TET2 and SRSF2 variants, have been responsible for the hemorrhagic manifestation.
Keywords:
chronic myelomonocytic leukemia
; CSF3R
; bleeding disorders
; predisposing genes
; NGS
1. Introduction
Chronic myelomonocytic leukemia (CMML) is a myeloid malignancy with overlapping features of myelodysplastic syndrome (MDS) and myeloproliferative neoplasms (MPN), typically characterized by peripheral monocytosis along with dysplastic features in the bone marrow (BM). Symptoms are usually related to splenomegaly and cytopenias, thrombocytopenia and anemia [1]. Bleeding is an unusual event in CMML, and when present, it is generally related to thrombocytopenia. Molecular aberrations often involve epigenetic, splicing, and signaling genes. The co-occurrence of mutations in TET2, SRSF2, ASXL1 and genes of the RAS signaling pathway is a common feature, with a specific high frequency of the combination of SRSF2 and TET2 alterations [2]. Somatic mutations in the gene encoding the colony-stimulating factor 3 receptor (CSF3R) have been identified in a minority of CMML patients [3], while they have been described in more than 80% of patients affected by chronic neutrophilic leukemia (CNL), a rare BCR-ABL1-negative myeloid malignancy characterized by mature granulocytosis and a high incidence of hemorrhage [4]. Moreover, biallelic germline CSF3R nonsense variants are responsible of severe congenital neutropenia (SCN) [5] and have been described in cases refractory to G-CSF treatment [6]. Heterozygous CSF3R mutations have been also reported as a predisposing condition to the development of lymphoid and myeloid malignancies, as multiple myeloma and acute lymphoblastic leukemia [7]. Here we report the case of a CMML patient presenting with atypical bleeding tendency and a germline CSF3R variant.
2. Case presentation
An 84-year-old patient, suffering from autoimmune thyroiditis and atrophic gastritis, reported the appearance of symptoms including asthenia and hyporexia (overall loss of 10% of body weight), swelling of the proximal joints of the hands and ankles. She was therefore admitted to the internal medicine department of a private institution on suspicion of a flare-up of autoimmune manifestations. The thyroid-stimulating hormone (TSH) was elevated, and the esophagogastroduodenoscopy (EGDS) confirmed the associated atrophic gastritis. Blood tests, initially interpretable as an immune-mediated condition, showed persistent normochromic normocytic anemia in the absence of absolute iron or vitamin B12/folic acid deficiency, mild and fluctuating thrombocytopenia (96-111 x 10^9/L) and marked monocytosis (the latter progressively worsening from 1.69 to 4.02 x 10^9/L).
Radiological examinations - including a computerized tomography (CT) scan of the thorax - showed a 9-mm nodule in the middle-lower third of the right lung field, which was not considered suspicious. Therapy with low-dose corticosteroids and a change in levothyroxine dosage resulted in a clinical benefit, an improvement in platelet count (149 x 10^9/L) and a partial reduction in monocyte count (1.4 x 10^9/L). However, a few weeks after discharge, numerous bruises of the lower limbs appeared, and peripheral blood smears showed dysmorphic granulocytes, monocyte atypia and myeloid precursors (myelocytes, 2%). The patient was then referred to our Haematology Unit. BM examination showed monocytosis associated with multilineage dysplasia and increased blastic cells, leading to a diagnosis of CMML type 2 (Figure 1A). Cytogenetic analysis reported a normal female karyotype (46, XX on 20 metaphases). At the end of the procedure, she showed significant bleeding from the biopsy site, thus requiring hospitalization and antifibrinolytic therapy with intravenous and local tranexamic acid. Hemostasis secured, immediately after discharge the patient reported domestic trauma, and the clinical picture was further complicated by the appearance of hemarthrosis in the right knee. Hemocoagulative tests showed prothrombin time-international normalized ratio (PT-INR) and activated partial thromboplastin time (aPTT) in the normal range, but a marked increase in fibrinolysis markers (D-dimer above detectability ranges) and initial fibrinogen consumption (73 mg/dL). The International Society on Thrombosis and Haemostasis (ISTH) Criteria for Disseminated Intravascular Coagulation (DIC) were not compatible with overt DIC, but the patient was still admitted to our Hematology Department given the clinical risks.
We opted for a better characterization of the disease at its onset from a molecular and coagulative point of view. Details on samples collection and analyses are reported in the Additional File 1.
We performed mutational analysis by next generation sequencing (NGS, Sophia Myeloid Solution, a 30 genes panel by SOPHia GENETICS-), and we evaluated markers of platelet activation by flow cytometry (in particular fibrinogen receptor expression, that was determined by procaspase-activating compound-1 (PAC-1) antibody and global coagulation assays (rotational thromboelastometry, ROTEM).
NGS analysis identified the following variants: a P95L missense mutation in the SRSF2 gene, a nonsense and a frameshift TET2 variant (G1825* and L615Afs*23) and the E808K missense mutation in the CSF3R gene.
The PAC-1 binding capacity was severely impaired (CD61+PAC-1+ events = 1%). Accordingly, hemostatic analysis revealed impaired fibrin polymerization after exposure to tissue factor (TF), with deleterious effects on clot formation in both the intrinsic (INTEM) and extrinsic (EXTEM) pathways. In particular, FIBTEM clotting time (CT) was extremely prolonged (3593 seconds (s). EXTEM data showed a rise in CT (148 s versus a normal range of 38-79 s). In EXTEM, INTEM and APTEM analysis clotting formation time (CFT) was increased (318 s, 260 s and 328 s, respectively), underlying an impairment in the initial rate of fibrin polymerization and a decrease in maximum clot firmness (MCF), with a consequent reduced viscoelastic strength of the clot (Figure 1 B).
Azacitidine (AZA) treatment was started at the standard dosage of 75 mg/mq for 7 days every 28. Treatment was well tolerated and led to an improvement of the hematologic parameters as well as of the coagulative tests, consistent with the resolution of bleeding complications. PAC-1 binding capacity was slightly increased after AZA exposure (51.4% of CD61+PAC-1+ cells at diagnosis versus 62.1% after the first AZA cycle, data not shown).
In the course of treatment all values, including global coagulation tests, showed a progressive shift towards a normal haemostatic process (Figure 1 C).
In order to characterize the mutational landscape of the patient, targeted NGS resequencing were performed at diagnosis and after AZA cycles 1 and 9, on BM and/or peripheral blood (PB) samples (Table 1).
The allelic ratio (AR) of TET2 and SRSF2 mutations was significantly reduced both in BM and in PB samples after 9 AZA cycles, while the variant allele frequency (VAF) of CSF3R E808K persisted at 50% at all the time points. To validate CSF3R mutation and to test the hypothesis of a germline origin of this variant, we performed Sanger sequencing of CSF3R exon 17 on DNA isolated from all the samples and from both saliva and blood CD3+ and CD3− cells as control, confirming the presence of a heterozygous germline variant (supplementary figure 1). The main clinical and laboratory information are summarized in Figure 1D.
3. Discussion
CMML is a clonal hematopoietic disease usually associated with the presence of mutations in genes involved in splicing regulation, epigenetic and proliferation control [8]. Somatic mutations of the CSF3R gene are rarely found in CMML, with a frequency of only 4-7% [9], while they have been largely reported in CNL [4],[10] and have been included in the CNL diagnostic criteria according to the WHO classification 1. Specifically, somatic T618I mutation is a hallmark of CNL and atypical chronic myeloid leukemia (aCML) [11], while to date, only 10 cases of CMML carrying T618I have been reported [12,13,14].
CSF3R encodes for the receptor of granulocyte colony stimulating factor (G-CSF), a cytokine essential for granulocyte proliferation and differentiation [15]. Acquired CSF3R mutations are responsible for SCN, which is considered a preleukemic bone marrow failure syndrome presenting around 20% of risk to transformation into acute myeloid leukemia (AML) or MDS, especially when they occur in association with mutations targeting other genes as RUNX1. These mutations usually develop early in the disease history and drive leukemogenesis [16].
Our patient harbored a well-known mutation, the E808K, that has been previously described both as somatic and germline in patients affected by myeloid malignancies, particularly MDS or MDS/MPN. The mutation lies within the cytoplasmic domain of the CSF3R protein and is described as a predisposing leukemia variant since the majority of patients evolve into AML [17]. Functional studies confirmed that this variant has a leukemogenic potential, as it is able to transform Ba/F3 cells and to induce a sustained STAT5 activation [18].
Therefore, the germline heterozygous CSF3R mutation detected in our patient could have predisposed to CMML development, in combination with the acquisition of somatic mutations within the TET2 and SRSF2 genes, which are commonly detected in this myeloid malignancy [8]. The presence of the germline CSF3R variant was not pathogenetic per se, since the patient did not present a personal history of hematological abnormalities, nor neutrophilia or neutropenia.
From the clinical point of view, our patient showed a normal bleeding time, and his platelet number was only slightly decreased. However, her clinical behavior was characterized by a severe bleeding tendency at diagnosis, which is pretty unusual in CMML and generally related to thrombocytopenia. Conversely, hemorrhagic diathesis is frequent in CNL, presenting a considerable number of cases with cerebral hemorrhage, despite the platelet count. Bleeding in CNL has been attributed to fibrinogen consumption likely because of a paraneoplastic process or a defect of platelet function [19] or vascular leukocytic infiltration [20]. Coagulation parameters analysis in our patient demonstrated that the hemorrhages are the consequences of a fibrinogen impairment, with prolonged CT and CFT, underlying an impairment in the initial rate of fibrin polymerization. Moreover, there was a clear reduction of the viscoelastic strength of the clot. The hemorrhagic tendency in CSF3R mutated CNL led us to speculate that the E808K germline CSF3R mutation could have a role in the hemorrhagic phenotype observed in our CMML patient, who did not present a critical value of thrombocytopenia to justify her bleeding manifestations.
4. Conclusions
In conclusion, we identified a heterozygous germline mutation that could have predisposed to the development of CMML, likely after the acquisition of TET2 and SRSF2 mutations as definitive leukemia drivers, and to a paraneoplastic hyperfibrinolysis that could have been responsible for the severe hemorrhagic clinical manifestations. Further efforts are needed to better understand the role of CSF3R mutations in inducing bleeding.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Additional file 1 and Supplemetary figure 1.
Author Contributions
MTB: molecular analysis, manuscript drafting; SA: anatomical pathologist analisys; MG: sample collection, ROTEM and flow cytometry assays; GMic: patient management; GS: molecular analysis; AL: manuscript revision, patient management, supervision. All authors were involved in interpreting the results and approved the final version to be published. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was approved by Comitato Etico della Romagna (protocol 5805/2019, #NCT04298892) and was carried out in accordance with the principles laid down in the 1964 Declaration of Helsinki. Written informed consent was received from the patient prior to inclusion in the study.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
The data supporting the findings of this study are available within the article [and in its supplementary files].
Acknowledgments
This work was partly supported thanks to the contribution of Ricerca Corrente by the Italian Ministry of Health within the research line "Precision, gender and ethnicity-based medicine and geroscience: genetic-molecular mechanisms in the development, characterization and treatment of tumors". The authors acknowledge Matteo Paganelli, Lorenzo Ledda and Giovanni Marconi from IRCCS Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) “Dino Amadori”, Meldola (FC), Italy and Andrea Ghelli Luserna di Rorà from Fondazione Pisana per la Scienza ONLUS, San Giuliano Terme, Italy for their contribution to the research and for the useful discussion on clinician and molecular data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. Epub ahead of print 2016. [CrossRef]
- Malcovati L, Papaemmanuil E, Ambaglio I, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood; 124. Epub ahead of print 2014. [CrossRef]
- Ouyang Y, Qiao C, Chen Y, et al. Clinical significance of CSF3R, SRSF2 and SETBP1 mutations in chronic neutrophilic leukemia and chronic myelomonocytic leukemia. Oncotarget; 8. Epub ahead of print 2017. [CrossRef]
- Li YP, Chen N, Ye XM, et al. Eighty-year-old man with rare chronic neutrophilic leukemia caused by CSF3R T618I mutation: A case report and review of literature. World J Clin Cases; 8. Epub ahead of print 2020. [CrossRef]
- Touw IP, Beekman R. Severe congenital neutropenia and chronic neutrophilic leukemia: An intriguing molecular connection unveiled by oncogenic mutations in CSF3R. Haematologica; 98. Epub ahead of print 2013. [CrossRef]
- Triot A, Järvinen PM, Arostegui JI, et al. Inherited biallelic CSF3R mutations in severe congenital neutropenia. Blood; 123. Epub ahead of print 2014. [CrossRef]
- Trottier AM, Druhan LJ, Kraft IL, et al. Heterozygous germ line CSF3R variants as risk alleles for development of hematologic malignancies. Blood Adv. Epub ahead of print 2020. [CrossRef]
- Patnaik MM, Tefferi A. Chronic myelomonocytic leukemia: 2018 update on diagnosis, risk stratification and management. Am J Hematol; 93. Epub ahead of print 2018. [CrossRef]
- Kosmider O, Itzykson R, Chesnais V, et al. Mutation of the colony-stimulating factor-3 receptor gene is a rare event with poor prognosis in chronic myelomonocytic leukemia. Leukemia. Epub ahead of print 2013. [CrossRef]
- Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R Mutations in Chronic Neutrophilic Leukemia and Atypical CML . N Engl J Med. Epub ahead of print 2013. [CrossRef]
- Pardanani A, Lasho TL, Laborde RR, et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia. Epub ahead of print 2013. [CrossRef]
- Zhang X, Ghiuzeli C, Jou E, et al. CSF3R T618I mutant myelodysplastic/myeloproliferative neoplasm in the elderly: An age-related disease with unfavorable prognosis. Leuk Res Reports. Epub ahead of print 2022. [CrossRef]
- Bezerra ED, Lasho TL, Finke CM, et al. CSF3R T618I mutant chronic myelomonocytic leukemia (CMML) defines a proliferative CMML subtype enriched in ASXL1 mutations with adverse outcomes. Blood Cancer Journal. Epub ahead of print 2021. [CrossRef]
- Kwon A, Ibrahim I, Le T, et al. CSF3R T618I mutated chronic myelomonocytic leukemia: A proliferative subtype with a distinct mutational profile. Leuk Res Reports. Epub ahead of print 2022. [CrossRef]
- Dwivedi P, Greis KD. Granulocyte colony-stimulating factor receptor signaling in severe congenital neutropenia, chronic neutrophilic leukemia, and related malignancies. Experimental Hematology. Epub ahead of print 2017. [CrossRef]
- Skokowa J, Steinemann D, Katsman-Kuipers JE, et al. Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: A unique pathway in myeloid leukemogenesis. Blood. Epub ahead of print 2014. [CrossRef]
- Adema V, Hirsch CM, Przychodzen B, et al. Molecular Spectrum of CSF3R variants Correlate with Specific Myeloid Malignancies and Secondary Mutations. Blood. Epub ahead of print 2018. [CrossRef]
- Zhang H, Reister Schultz A, Luty S, et al. Characterization of the leukemogenic potential of distal cytoplasmic CSF3R truncation and missense mutations. Leukemia. Epub ahead of print 2017. [CrossRef]
- Hossfeld DK, Lokhorst HW, Garbrecht M. Neutrophilic leukemia accompanied by hemorrhagic diathesis: Report of two cases. Blut; 54. Epub ahead of print 1987. [CrossRef]
- Noguchi T, Ikeda K, Yamamoto K, et al. Severe bleeding tendency caused by leukemic infiltration and destruction of vascular walls in chronic neutrophilic leukemia. Int J Hematol; 74. Epub ahead of print 2001. [CrossRef]
Figure 1.
A) Morphological characterization of BM biopsy. Hematoxylin and eosin (H&E, 20x and 40x respectively) and Immunohistochemistry (IHC) stains for CD68 (CD68/PGM1, 20x) showing hypercellular bone marrow with prominent granulopoiesis, atypical megakaryocytes and increased CD68/PGM1 positive monocytes. Analysis of coagulation parameters by ROTEM at diagnosis (B) and after cycle 1 of Azacitidine treatment (C). Units and reference values are displayed in brackets (Clotting Time: CT, Clot Formation Time: CFT, Amplitude 5 min after CT: A5, Amplitude 10 min after CT: A10, Amplitude 20 min after CT: A20, Maximum Clot Firmness: MCF, Maximum Lysis: ML, Normality Range: NR). D) Timeline displaying therapy schedule on the upper part and the clinical history and molecular data on the bottom part.
Figure 1.
A) Morphological characterization of BM biopsy. Hematoxylin and eosin (H&E, 20x and 40x respectively) and Immunohistochemistry (IHC) stains for CD68 (CD68/PGM1, 20x) showing hypercellular bone marrow with prominent granulopoiesis, atypical megakaryocytes and increased CD68/PGM1 positive monocytes. Analysis of coagulation parameters by ROTEM at diagnosis (B) and after cycle 1 of Azacitidine treatment (C). Units and reference values are displayed in brackets (Clotting Time: CT, Clot Formation Time: CFT, Amplitude 5 min after CT: A5, Amplitude 10 min after CT: A10, Amplitude 20 min after CT: A20, Maximum Clot Firmness: MCF, Maximum Lysis: ML, Normality Range: NR). D) Timeline displaying therapy schedule on the upper part and the clinical history and molecular data on the bottom part.
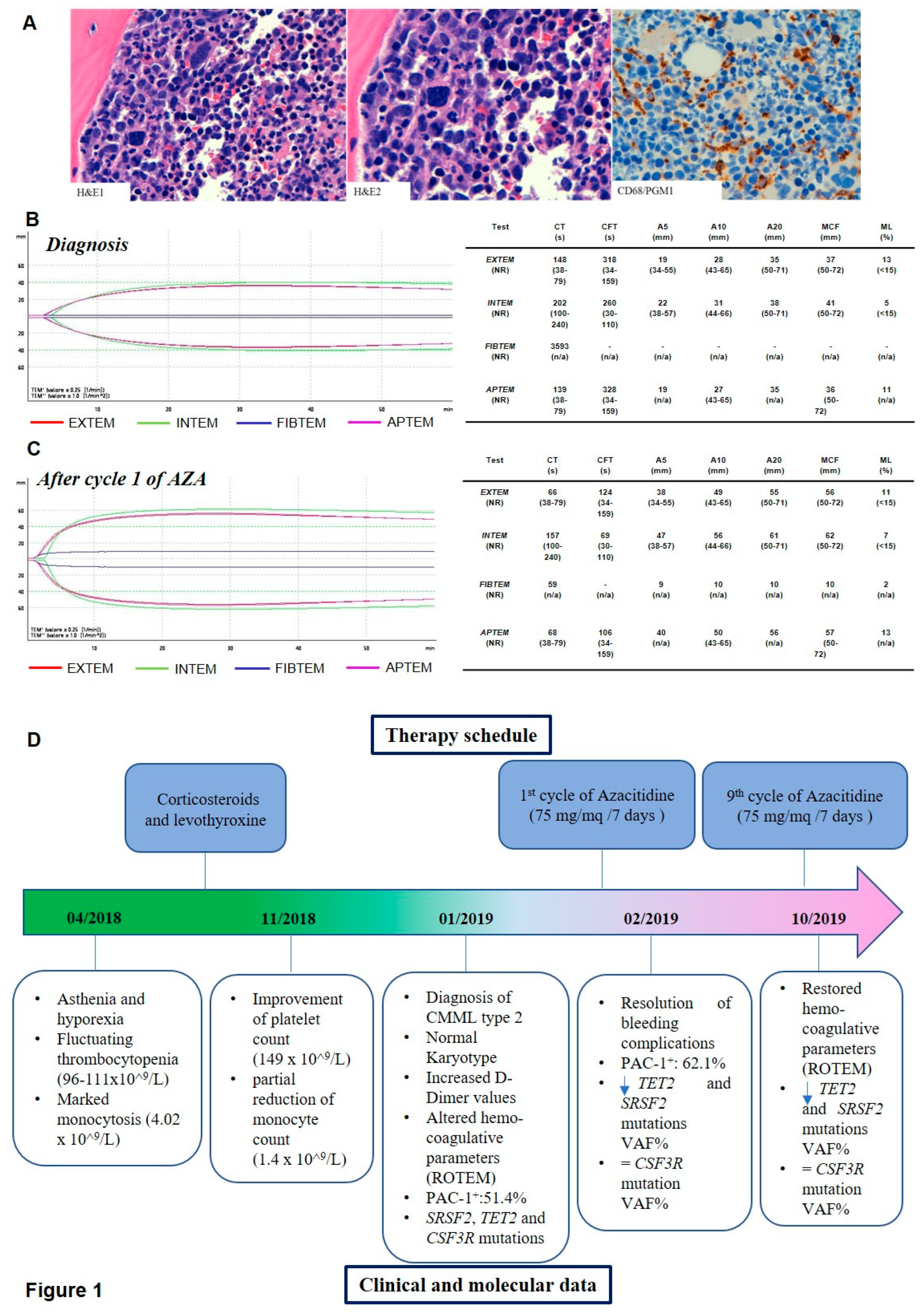

Table 1.
Mutational profile of the bone marrow and peripheral blood samples at diagnosis and after 1 and 9 Azacitidine cycles.
Table 1.
Mutational profile of the bone marrow and peripheral blood samples at diagnosis and after 1 and 9 Azacitidine cycles.
| Tissue | Gene | Locus | NM_ID | Exon | Type | Coding | Amino Acid Change | VAF (%) | Coding Consequence | Depth | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DIAGNOSIS | BONE MARROW | TET2 | chr4:106197140 | NM_001127208 | 11 | SNP | c.5473C>T | p.(Gln1825*) | 41,7 | nonsense | 5854 |
| TET2 | chr4:106156935 | NM_017628 | 3 | INDEL | c.1842dupG | p.(Leu615Alafs*23) | 41,5 | frameshift | 6031 | ||
| CSF3R | chr1:36932047 | NM_000760 | 17 | SNP | c.2422G>A | p.(Glu808Ly) | 49,5 | missense | 5329 | ||
| SRSF2 | chr17:74732959 | NM_003016 | 1 | SNP | c.284C>T | p.(Pro95Leu) | 43 | missense | 4443 | ||
| PERIPHERAL BLOOD | TET2 | chr4:106156935 | NM_017628 | 3 | INDEL | c.1842dupG | p.(Leu615Alafs*23) | 37,7 | frameshift | 6111 | |
| TET2 | chr4::106197140 | NM_001127208 | 11 | SNP | c.5473C>T | p.(Gln1825*) | 36,8 | nonsense | 5889 | ||
| CSF3R | chr1:36932047 | NM_000760 | 17 | SNP | c.2422G>A | p.(Glu808Ly) | 47,7 | missense | 5389 | ||
| SRSF2 | chr17:74732959 | NM_003016 | 1 | SNP | c.284C>T | p.(Pro95Leu) | 35,9 | missense | 4803 | ||
| POST 1 AZA CYCLE | PERIPHERAL BLOOD | TET2 | chr4:106156935 | NM_017628 | 3 | INDEL | c.1842dupG | p.(Leu615Alafs*23) | 37,7 | frameshift | 4642 |
| TET2 | chr4::106197140 | NM_001127208 | 11 | SNP | c.5473C>T | p.(Gln1825*) | 41,6 | nonsense | 4590 | ||
| CSF3R | chr1:36932047 | NM_000760 | 17 | SNP | c.2422G>A | p.(Glu808Ly) | 49,8 | missense | 4012 | ||
| SRSF2 | chr17:74732959 | NM_003016 | 1 | SNP | c.284C>T | p.(Pro95Leu) | 39,3 | missense | 3628 | ||
| POST 9 AZA CYCLES | BONE MARROW | TET2 | chr4:106197140 | NM_001127208 | 11 | SNP | c.5473C>T | p.(Gln1825*) | 18,4 | nonsense | 7019 |
| TET2 | chr4:106156935 | NM_017628 | 3 | INDEL | c.1842dupG | p.(Leu615Alafs*23) | 20,2 | frameshift | 7406 | ||
| CSF3R | chr1:36932047 | NM_000760 | 17 | SNP | c.2422G>A | p.(Glu808Lys) | 49,9 | missense | 6845 | ||
| SRSF2 | chr17:74732959 | NM_003016 | 1 | SNP | c.284C>T | p.(Pro95Leu) | 20,5 | missense | 5869 | ||
| PERIPHERAL BLOOD | TET2 | chr4:106156935 | NM_017628 | 3 | INDEL | c.1842dupG | p.(Leu615Alafs*23) | 28,6 | frameshift | 8666 | |
| TET2 | chr4:106197140 | NM_001127208 | 11 | SNP | c.5473C>T | p.(Gln1825*) | 29,9 | nonsense | 8235 | ||
| CSF3R | chr1:36932047 | NM_000760 | 17 | SNP | c.2422G>A | p.(Glu808Lys) | 49,5 | missense | 7662 | ||
| SRSF2 | chr17:74732959 | NM_003016 | 1 | SNP | c.284C>T | p.(Pro95Leu) | 27,3 | missense | 7175 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



