
Submitted:
24 September 2023
Posted:
26 September 2023
You are already at the latest version
Abstract
Temporal lobe epilepsy is a common, chronic disorder with spontaneous seizures that is often refractory to drug therapy. A potential cause of temporal lobe epilepsy is primary brain injury, making prevention of epileptogenesis after the initial event an optimal method of treatment. Despite this, no preventive therapy for epilepsy is currently available. The purpose of this study was to evaluate the effects of anakinra, lamotrigine, and their combination on epileptogenesis using the rat lithium-pilocarpine model of temporal lobe epilepsy. The study showed that the treated and untreated animals showed no significant difference in the number and duration of seizures. However, the severity of seizures was significantly reduced after treatment. Anakinra and lamotrigine, alone or in combination, significantly reduced neuronal loss in the CA1 hippocampus compared to the control group. However, the drugs administered alone were found to be more effective for CA3 than their combination. The treatment alleviated the impairments in activity level, exploratory behavior and anxiety, but had a relatively weak effect on TLE-induced impairments in social behavior and memory. The efficacy of the combination treatment did not differ from that of anakinra and lamotrigine monotherapy. These findings suggest that anakinra and lamotrigine, alone or in combination, may have clinical utility in preventing epileptogenesis.
Keywords:
temporal lobe epilepsy
; anakinra
; lithium–pilocarpine model
; behavior
; epileptogenesis
; hippocampus
; spontaneous recurrent seizures
; neuronal loss
1. Introduction
Epilepsy is a chronic neurological disorder characterized by recurrent, spontaneous seizures resulting from abnormal bioelectrical activity in the brain [1]. This prevalent neural pathology affects approximately 50-65 million individuals worldwide [2,3]. One of the most common forms of epilepsy is temporal lobe epilepsy (TLE), in which the epileptogenic focus is located in the temporal lobe [4]. The hallmark of TLE is the degeneration of hippocampal neurons, particularly in the areas of CA1 and CA3 [5].
TLE also results in the manifestation of associated cognitive and psychoemotional disturbances that adversely affect patients' quality of life [6]. Memory impairments are a common cognitive issue in patients with epilepsy [7], with patients suffering from TLE particularly affected due to epileptogenic foci affecting memory consolidation structures, including the hippocampus [8]. Additionally, epilepsy patients have a higher risk of developing anxiety disorders, personality disorders, psychosis, and attention deficit hyperactivity disorder [9]. Patients with TLE experience challenges in social interactions, specifically in comprehending the mental state of others and identifying emotions [10,11].
Despite continuous research and the availability of anti-seizure medications, approximately 30% of patients with epilepsy do not achieve complete remission [12]. Existing medications can prevent seizures but do not address the underlying mechanisms of epileptogenesis or prevent the development of epilepsy [13,14]. Additionally, many available anti-seizure medications have adverse psychological effects on patients [15,16]. Therefore, it is crucial to discover new treatment methods for epilepsy.
By focusing on the underlying disease mechanisms and primary signaling pathways, it may be possible to prevent or halt the progression of epileptogenesis and provide more effective treatments for epilepsy [14]. Unfortunately, the precise mechanisms of epileptogenesis are still not completely understood, and potential targets for therapy development remain hypothetical [17,18].
Several mechanisms contribute to the development of epilepsy, and neuroinflammation is one of them [19,20,21,22]. Neuroinflammation has been described as a common pathogenic mechanism promoting seizures in animal models of acquired epilepsy and drug-resistant epilepsy in humans. Neuroinflammation involves structural and functional changes in glial and immune cells in the central nervous system, along with dysfunction of the blood-brain barrier (BBB). This imbalance results in heightened production of inflammatory mediators such as IL-1β, IL-6, TNF-α, IFN-γ [20].
Previous studies, including our own, have demonstrated that anakinra, an interleukin-1 receptor antagonist, reduces epileptogenesis and improves seizure-related outcomes [23,24,25]. Anakinra blocks IL-1 receptor-mediated signaling, which might modulate the inflammatory response and decrease neuronal hyperexcitability. Administering anakinra during the latent phase of the lithium-pilocarpine model significantly reduced the duration and frequency of spontaneous recurrent seizures (SRS) in rats during the chronic phase. Additionally, anakinra prevented certain behavioral impairments, such as motor hyperactivity and disturbances in social interactions, during both the latent and chronic phases. Histological analysis showed that anakinra also reduced neuronal loss in the CA1 and CA3 regions of the hippocampus, but did not prevent astro- and microgliosis [23].
Lamotrigine, an anticonvulsant drug, is commonly prescribed for managing epilepsy. Its efficacy is linked to its ability to block voltage-gated sodium channels, which reduces abnormal electrical activity in the brain [26]. More recently, evidence indicates that lamotrigine has antiepileptogenic properties. Lamotrigine pretreatment, administered at doses of 10 and 20 mg/kg, resulted in a significant decrease in seizure stages and generalized seizure durations in the rat pentylenetetrazole kindling model [27]. Additionally, electrophysiological studies illustrated that lamotrigine pretreatment eliminated the heightened population spike amplitude in the hippocampus [27]. The study by Stratton et al. (2003) demonstrated the antiepileptogenic-like effects of lamotrigine in a rat amygdala kindling model [28]. However, a subsequent study by Nissinen et al. (2004) did not show whether lamotrigine had disease modifying or antiepileptogenic effects [29]. In a rat lithium-pilocarpine model of TLE, Wang et al. (2019) found that lamotrigine decreased the frequency of SRS in a dose-dependent manner, and limited neuronal loss as well as astrogliosis in the hippocampus [30].
Since both anakinra and lamotrigine have antiepileptogenic properties but work through differing mechanisms, we compared their individual and combined effects. We utilized the lithium-pilocarpine model to reproduce the major phases of epileptogenesis specific to temporal lobe epilepsy. Our assessment involved measuring the occurrence of SRS with different treatment options, as well as neuronal death and behavioral characteristics of the animals.
2. Results
2.1. The effect of treatment with anakinra, lamotrigine and their combination effect on neurological parameters
The study evaluated body weight changes and survival of rats treated with anakinra and/or lamotrigine for 10 days after pilocarpine administration. The TLE+A+L group receiving combination treatment showed no mortality after two days of treatment. Mortality was observed within the first 5 days in the TLE+A and TLE+L groups, and within the first 7 days in the untreated TLE group. However, due to the relatively low mortality across all groups, these differences did not reach statistical significance (Figure 1a; Long Rank test; χ2 = 4.27; p = 0.23).
After administration of pilocarpine, the body weight of all experimental animals decreased by approximately 15-20% and slowly recovered afterwards (Figure 1b). The body weight dynamics of TLE rats differed significantly from controls, according to two-way ANOVA analysis (F40, 620 = 6.45, p < 0.001). However, when excluding control animals, no significant differences were found between treated and untreated TLE animals (F30, 420 = 0.48, p = 0.99).
The severity of spontaneous recurrent seizures (SRS) was evaluated 3.5 months after pilocarpine administration. This was during the chronic phase of the model. Over a peri-od of 40 hours, the free behavior of rats was recorded. SRS occurred in 9 out of 15 (60%) untreated TLE animals, 6 out of 11 (54,5%) treated with anakinra, 2 out of 9 (22,2%) rats treated with lamotrigine, and 6 out of 12 (50%) rats receiving the combination treatment (Figure 1c).
The number and duration of seizures did not differ significantly between treated and untreated animals (Figure 1d, e). Analysis of seizure severity based on the Racine scale was only performed in animals with SRS. The study revealed a significant effect of treatment (H4, 22 = 7.90; p = 0.048), but intergroup differences were not statistically significant (Dunn's post hoc test, p > 0.05).
2.2. Anakinra, lamotrigine, or their combination can prevent neuronal death in the hippocampus following pilocarpine-induced status epilepticus.
A histological study was conducted to assess the effectiveness of administering anakinra, lamotrigine and their combinations in the lithium-pilocarpine model. Neuron counts in the pyramidal layers of the hippocampal CA1 and CA3 areas were obtained at 120 days post-SE using Nissl-stained brain sections (Figure 2). The counts were taken for the TLE(n = 9), matched control (n = 6), and after the administration of anakinra (TLE+A, n = 6), lamotrigine (TLE+L, n = 6), and the combination of these drugs (TLE+A+L, n = 9).
In the CA1 region, the variances displayed a significant difference (Levene's test, p = 0.001). Consequently, we applied Welch's one-way ANOVA, which indicated a substantial impact of status epilepticus induced by pilocarpine on neuronal death in the CA1 hippocampal regions (W4, 13.1 = 4.59, p < 0.001). The Games-Howell test demonstrated notable distinctions in the number of neurons in the hippocampal CA1 field between the control group and the group of animals after pilocarpine-induced seizures (Figure 3). Both administering lamotrigine and anakinra separately and in combination prevented neuronal death. The ANOVA contrast method indicated a significant difference between treated and untreated TLE rats (t = 2.93; p = 0.02), but no significant difference was detected between treating with anakinra and lamotrigine separately versus using them in combination (t = 0.4; p = 0.70).
As there were no significant differences in the variances of cell numbers in the CA3 field between groups (Levene's test, p = 0.75), we utilized one-way ANOVA. This analysis demonstrated significant differences in the number of neurons in the CA3 field of the hippocampus between the control group and the group of animals after pilocorpine-induced SE (F4, 31 = 6.28, p < 0.001). The ANOVA contrast method illustrated a notable difference between the untreated and treated TLE rats (t = 2.5; p = 0.02). However, in contrast to the CA1 region, a significant reduction in the number of neurons was observed in the CA3 region of the hippocampus, not only in the group of animals that experienced pilocarpine-induced status epilepticus without treatment, but also in the group that received a combination of lamotrigine and anakinra after status epilepticus. Tukey's post-hoc test confirmed this finding (Figure 3).
Thus, the use of lamotrigine, anakinra, or their combination was found to be generally effective in preventing neuronal death in the hippocampus after pilocarpine-induced status epilepticus. However, drugs administered separately were found more effective than their combination for CA3.
2.3. Behavioral disturbances may be improved with anakinra, lamotrigine, or their combination.
We previously demonstrated that epileptogenesis in the lithium-pilocarpine model of temporal lobe epilepsy results in motor activity disorders, altered levels of anxiety, communicative behavior changes, and memory impairments in experimental rats [31]. Therefore, this study aimed to investigate whether the therapy utilized - particularly combination therapy with anakinra and lamotrigine - could prevent TLE rats from developing behavioral disorders.
2.3.1. Treatment effects on activity levels, exploratory behavior, and anxiety in experimental and control animals in the Open field test.
The locomotor activity of rats in the Open field was analyzed in terms of locomotion time and traveled distance (Figure 4a, b, c). The TLE animals that did not receive treatment exhibited significantly higher locomotor activity than the control group. Specifically, the total distance increased by approximately 1.5-fold (F4, 67 = 13.1; p < 0.001) and the total locomotion time increased by 2-fold (F4, 67 =9.5; p < 0.001). None of the treatment variants were able to prevent these impairments (Table A1).
To evaluate the rats' overall activity level, we counted the number of distinct behavioral acts (Figure 4d). This measure significantly increased in both untreated and treated TLE rats (F4, 67 =10.0; p < 0.0001; Tukey's post-hoc test – untreated: p < 0.001 and treated p < 0.05). The impact of the treatment using anakinra and lamotrigine, alone or combined, was confirmed by the ANOVA contrast analysis (untreated vs. treated t = 2.7; p = 0.01). No difference was found between combination therapy and monotherapy (t = 1.15; p = 0.88) (Table A1).
We examined climbing as an indicator of exploratory behavior (Figure 4e). The number of climbs increased in TLE animals (F4, 65 = 4.49; p < 0.01), but the combination treatment prevented this change (Tukey's post-hoc test, p < 0.05). ANOVA contrast indicated efficacy of the treatment (t = 2.6; p = 0.013), but the combined treatment did not show a significant difference from monotherapy use (ANOVA contrast; t = 1.15; p = 0.25) (Table A1). The time of hole explorations, another indicator of exploratory behavior, did not differ significantly between groups (Figure 4f).
To evaluate the impact of the treatment on animal anxiety, we analyzed the duration of grooming (Figure 4g) and the time spent in the center area of the Open field (Figure 4h). The untreated TLE rats demonstrated an increase in grooming, indicating a higher level of anxiety (H5, 63 =14; p < 0.01; Dunn's post hoc test p < 0.01). This effect was not observed in any of the treated groups. Therefore, treatment decreased the anxiety increase caused by TLE. In addition, when anakinra was administered, a significant increase in time spent in the center of the open field was observed (W4, 28.5 = 6.0; p = 0.001), indicating reduced anxiety level compared to the untreated animals. The anakinra-treated group showed better results than the combination-treated group (Games-Howell post hoc test p < 0.05), while the combination-treated group showed no significant difference from the control group, which was confirmed by ANOVA contrast (t = 3.86; p = 0.001).
Thus, the treatment alleviated the impairments in activity levels, exploratory behavior, and anxiety. There was no difference in effectiveness between anakinra and lamotrigine monotherapy and the combined treatment for most parameters.
2.3.2. Social behavior
Social behavioral disturbances are one of the most prominent psychiatric disorders in humans with epilepsy [32] and in experimental animals in the lithium-pilocarpine model of this disease [33]. Using the Social test, we showed that the duration of communicative behavior in animals with TLE was reduced about 4-5 times compared to controls (Figure 5a; F4, 64 = 16.9; p < 0.001). These data indicate a severe impairment of social behavior in experimental rats. Although a slight increase in communication time was observed in all treated groups, none of the treatments was able to restore communicative behavior duration to the level of the control group.
In addition to reducing communication time, TLE also altered the structure of social behavior (Figure 5b). In control animals, genital sniffing accounted for 38% of total communication time during contact with a stranger, indicating a confident behavior of the resident. At the same time, sniffing of a stranger's tail is practically not observed in control rats, which, on the contrary, indicates an insecure behavior of the resident (0.5% of the total communication time). Genital sniffing disappeared almost completely in untreated TLE rats (4%), while tail sniffing increased (up to 8%). In lamotrigine-treated animals, genital sniffing reappeared but did not fully recover (TLE+L group - 10.1%; TLE+L+A - 13.1%).
Indicators of aggressive behavior - time and number of episodes of aggressive acts were group dependent (Figure 5c, d; H5, 64 = 13.1; p = 0.01 and H5,66 = 18.6; p = 0.001). However, these changes were not associated with an increase in aggression in untreated TLE rats, but with a decrease in aggressive behavior (compared to controls) in the group of rats receiving the combined treatment (Dunn's post hoc test p < 0.01). Thus, the treatment applied had a weak effect on the TLE-induced impairment of social behavior.
2.3.3. The memory impairments
Fear conditioning test
To evaluate fear-related conditioned memory, we utilized the Fear conditioning test (see Figure 6). On the test day, rats were initially placed in cage A, which was associated with shocks and where the conditioning occurred the previous day. Evaluating the total freezing time during this phase enabled us to assess contextual memory. Our findings indicate a significant decrease in freezing time in TLE rats, indicating impaired contextual memory (H5, 70 = 25.3; p < 0.001). The most significant impairments were found in untreated TLE and in the TLE+A groups (Dunn's post hoc test p < 0.01 vs. control). TLE rats treated with lamotrigine exhibited improvement in contextual memory and did not differ significantly from the control group (Figure 6a).
The rats were then transferred to an unfamiliar cage B, where no stimuli were presented for the first 3 min at this stage, the duration of freezing was short, and no significant differences were observed between groups (Figure 6b). This result indicates that there were no baseline differences between groups in the degree of freezing and that the observed differences were related to memory impairment.
To assess conditioned memory, rats in cage B were presented with a sound cue similar to that used in the learning phase (Figure 6c). Importantly, untreated TLE rats exhibited a significant (about 4-fold) decrease in their freezing response compared to controls. This finding suggests a significant impairment of memory for pain-associated stimuli (H5,68 = 45, p < 0.001). None of the treatments tested improved these abnormalities.
A high level of freezing was observed in control but not in TLE rats within 1 min after the end of the conditioned stimulus (Figure 6d; H5, 65 = 39.8, p < 0.001), probably indicating a high stress responsiveness in control animals. The duration of freezing did not differ between the treated and untreated TLE rats in this subtest.
Morris water maze
Long-term spatial memory was evaluated in the Morris water maze (Figure 7). Rats underwent four-day training sessions (with four trials daily) to locate a submerged platform. Improvement in skills was determined by a decrease in the distance traveled before reaching the platform. The training progress of the rats in the Morris water maze is depicted in Figure 7a. As training progressed, all groups showed an improvement in their ability to find the platform. However, the efficiency of the training varied between groups (mixed ANOVA: trials - F15, 975 = 24.8, p < 0.001; groups - F4, 65 = 5.34, p = 0.001; interaction - F60, 975 = 1.21, p = 0.14).
Control rats learned better than TLE rats. Both short-term and long-term memory impairments were observed in TLE rats. For example, in the third trial of the first day of training (short-term memory), treated TLE rats swam a longer distance to the platform compared to controls (Figure 7b, one-way ANOVA; W4, 27.3 = 5.3; p < 0.01), a similar result was observed in untreated TLE rats. In the first test on the second day, the worst results were found in untreated TLE animals (Figure 7c), but treated rats had similar results (one-way ANOVA; F4, 65 = 2.8; p = 0.03). Animals from different groups differed in the total distance traveled in all sixteen trials (Figure 7d; one-way ANOVA; W4, 24.8 = 8.6; p < 0.001), with the distance traveled being maximal in the TLE and TLE+A groups.
Overall, the analysis of learning showed that TLE leads to impaired learning in the Morris water maze, with no significant differences between treated and untreated rats (ANOVA contrast results are given in Table A1).
To test long-term spatial memory (Figure 7e, f, g), the platform was removed on the fifth experimental day and the animals were placed in the pool for 90 s. Staying in the target area where the platform was previously located for a long period of time was considered a high spatial memory performance. Because most control rats would first search for the platform in a given area and then, if they did not find it, swim away to search elsewhere, we examined the time spent in the target area not only during the entire test (90 s, Figure 7f), but also during the initial period (30 s, Figure 7e). In both cases, the groups differed significantly in the amount of time spent in the target area: 30 s - W4, 26.9 = 4.8; p < 0.01; 90 s - F4, 64 = 5.36; p < 0.001. Maximum memory impairment was observed in untreated TLE rats, but treated rats were also significantly different from controls (Tukey's post hoc test, p<0.05). Lamotrigine monotherapy reversed these abnormalities when the first 30 seconds of search were analyzed (Figure 7e, differences from control p>0.05). However, the ANOVA contrast method revealed no significant differences between animals receiving different treatments (Table A1).
Thus, TLE rats showed a decrease in spatial memory. Treatment with lamotrigine reduced memory abnormalities, but did not completely prevent them.
3. Discussion
In the present study, the rat lithium-pilocarpine TLE model [34] was utilized to examine the effects of anakinra and lamotrigine alone and in combination on epileptogenesis. Administration of these drugs during the initial 10-day period following pilocarpine-induced status epilepticus lessened the severity of SRS. In addition, anakinra and lamotrigine alone and in combination significantly ameliorated a number of behavioral deficits and reduced, but did not completely abolish, hippocampal neuronal loss. The effectiveness of the combined treatment did not significantly vary from that of anakinra and lamotrigine monotherapy. Thus, it can be inferred that both anakinra and lamotrigine have a disease-modifying effect in this TLE model.
Since acquired epilepsy often appears to be pharmacoresistant, prevention of epileptogenesis is an important goal [35]. Epileptogenesis is the complex process by which a healthy brain becomes epileptic. Several animal models are used to study epileptogenesis and provide valuable insights into the underlying mechanisms of epilepsy [36]. These models allow researchers to study various aspects of epileptogenesis, including seizure development, neuronal loss, synaptic reorganization, neuroinflammation, and metabolic changes in the brain. Some commonly used animal models to study epileptogenesis include: (1) kindling models, in which epileptic activity is induced by repeated application of low-dose convulsant drugs or electrical stimulation of specific brain regions, such as the amygdala [37]. (2) Post-status epilepticus (post-SE) models in which high doses of a convulsant agent such as kainate or pilocarpine are injected systemically [37]; (3) brain injury models, in which epilepsy develops after brain damage or stroke [38,39]. Compared to kindling models, post-SE lithium-pilocarpine model is deemed more reliable in identifying drugs with antiepileptogenic properties. For instance, in models of kindling, drugs are usually given before the daily kindling sessions [27,28], making it impossible to rule out the possibility that the anticonvulsant impact of the drugs mitigates kindling-dependent epileptogenesis [30].
Evidence of antiepileptogenic efficacy is increasing for numerous compounds. In a recent review, the authors listed 156 compounds with published reports of antiepileptogenic efficacy [40]. In this study, we tested antiepileptogenic efficacy of lamotrigine and anakinra. Lamotrigine is a conventional antiepileptic medication utilized to treat both focal and generalized epilepsy [41]. The pharmacological effect of lamotrigine involves the blockage of potential-dependent sodium [42] and N- and P/Q-type calcium channels on presynaptic nerve terminals [43,44]. Lamotrigine prevents excessive release of glutamate, protecting nerve cells from glutamate-induced neurotoxicity [45,46]. After status epilepticus induced by pilocarpine, neuronal networks become more excitable with pathologically high background activation of the glutamatergic system [47,48]. To minimize this effect, we employed lamotrigine in our study. Previously, various experimental models of epilepsy, including the lithium-pilocarpine model of temporal lobe epilepsy, have shown the neuroprotective and anti-epileptogenic effects of lamotrigine [30,49].
Another factor in favor of choosing lamotrigine was its ability to positively impact the mental state of patients, setting it apart from numerous other anti-epileptic medications that can trigger psycho-emotional and cognitive dysfunctions. Research has demonstrated that lamotrigine displays a strong antidepressant impact, making it suitable for mood stabilization in individuals with bipolar disorder [50]. It should be also noted that lamotrigine has demonstrated the potential to inhibit the release of proinflammatory cytokines in models of neuroinflammation [51].
Anakinra, a recombinant interleukin-1 receptor antagonist, has shown potential in the treatment of some types of epilepsy, particularly in febrile infection-related epilepsy syndrome (FIRES)[24,52] and as antiepileptogenic drug [23]. The mechanisms by which anakinra exerts its effects in epilepsy are not fully understood. However, it is believed to change expression of various genes, modulate the immune response and reduce inflammation [23,53,54]. Neuroinflammation is associated with increased production of proinflammatory cytokines such as IL- β, IL-6, TNF-α, and others. Increased expression of pro-inflammatory genes, particularly IL-1b and TNF-α, was found in the hippocampus and anterior temporal cortex of patients with TLE and hippocampal sclerosis [55,56]. High levels of IL-1β enhance excitation in the CNS by increasing the release of excitatory transmitters such as glutamate or ATP [57]. IL-1β enhances NMDA-mediated Ca2+ influx into the cell [58], which may induce the characteristic hippocampal neuronal death seen in epilepsy [59]. In addition, IL-1β may increase neuronal excitability by down-modulating the astrocytic glutamate transporter (GLT-1) [60], which results in impaired glutamate clearance that has been identified as one of the causative factors in drug-resistant epilepsy [61]. Increased levels of proinflammatory cytokines, including IL-1, may not only be associated with increased excitability, but may also be responsible for the development of behavioral abnormalities characteristic of epilepsy [62]. We and other researchers have previously demonstrated that IL-1 receptor blockade therapy decreases seizure development and neurodegenerative changes in the brain, as well as reducing the severity of comorbid behavioral disorders. However, it does not completely prevent them [23,63,64].
One of the key elements in epileptogenesis is neuronal death, which has been widely studied in the context of acquired epileptogenesis [65]. Traditionally, it has been proposed that neuronal death is necessary for epileptogenesis, as the loss of synaptic input from dying neurons is considered a critical signal to induce axonal sprouting and rewiring [65]. However, recent studies have challenged this concept and suggested that neuronal death may not be essential for epileptogenesis, particularly in the immature brain [65]. Our study demonstrates that both lamotrigine and anakinra provide neuroprotective effects and reduce neuronal death in the hippocampus, but they do not fully prevent spontaneous recurrent seizures. Thus, our data are consistent with the results of previous studies, which also showed that preventing neuronal loss is an important but not always sufficient factor to prevent epileptogenesis [66,67,68].
In our study, we also aimed to investigate the effect of drugs on behavioral disturbances in rats. This was motivated by the observation that a comorbid diagnosis of epilepsy and psychiatric disorders was found to predict pharmacoresistance [69]. Analogous findings have also been reported in a rat model of epilepsy [70]. Another point for this study is that epilepsy patients also have a higher risk of developing anxiety disorders, psychosis, attention deficit hyperactivity disorder, and various personality disorders [9]. Patients with TLE have difficulties in social interaction, especially in modeling the mental state of others and recognizing emotions [10,11]. In addition, patients with TLE and hippocampal sclerosis show impairments in several types of memory [8,71] and social behavior [72], which may also be related to hippocampal dysfunction [73].
Similar behavioral abnormalities are observed in animal models of temporal lobe epilepsy. Pilocarpine and lithium-pilocarpine rodent models of TLE are characterized by hyperactivity, impaired memory, social behavior, and anxiety [33,74,75], which was confirmed in the present study. Behavioral abnormalities often manifest in the latent period of the model when SRS is barely observed [76].
We have shown that treatment with anakinra, lamotrigine, or their combination in the early stages of epileptogenesis ameliorated impairments in motor activity, exploratory behavior, and anxiety in the open field, reduced aggression in the social test, but only slightly improved TLE-induced impairments in communicative behavior and memory. At the same time, the efficacy of the combined treatment was almost identical to that of anakinra and lamotrigine monotherapy.
We have previously shown that administration of anakinra for 10 days after pilocarpine-induced status epilepticus attenuates, but does not completely prevent, some behavioral deficits that develop during the latent and chronic phases of the lithium-pilocarpine model of TLE [23]. Mazarati et al. [62] found that intrahippocampal administration of interleukin-1 receptor antagonist, an analog of anakinra, ameliorated psychoemotional disturbances in the lithium-pilocarpine model of TLE in rats. The present study mainly confirms the previously obtained data.
Our data obtained during treatment with lamotrigine are consistent with clinical observations showing that lamotrigine also has a positive effect on the mental state of patients with epilepsy. For example, Miller et al. found that lamotrigine reduced depressive symptoms in patients with epilepsy [77], Kato et al. showed a decrease in aggression in patients with TLE after treatment with lamotrigine [78]. In a study performed in rats in the lithium-pilocarpine model of TLE, Mahfoz et al. found that administration of lamotrigine attenuated pilocarpine-induced spatial memory impairments in the Morris water maze [79]. However, in that study, lamotrigine was administered prior to the pilocarpine injection, which may account for a more pronounced effect than we found.
Overall, our study suggests that treatment with anakinra and lamotrigine has a protective effect on the development of some neurodegenerative and behavioral abnormalities during epileptogenesis. Although our study did not confirm the hypothesis that combined therapy is more effective than monotherapy with these drugs, the results suggest that both anakinra and lamotrigine, either alone or in combination, may have clinical value in preventing epileptogenesis.
4. Materials and Methods
4.1. Animals
This study used male Wistar rats that were eight-weeks-old and housed in standard cages with 5-6 rats per cage. The rats had unlimited access to water and food and were exposed to a 12-hour cycle (8 p.m. to 8 a.m. light and 8 a.m. to 8 p.m. dark). Rats from various litters were mixed to prevent potential genetic influence, and then allocated to either the control or experimental groups. All animal experiments were conducted in accordance with the regulations and standards set forth by the Animal Care and Use Committee of the Sechenov Institute of Evolutionary Physiology and Biochemistry of the RAS. These guidelines adhere fully to the EU Directive 2010/63/EU for animal experiments.
4.2. The lithium-pilocarpine model and treatment
The experimental plan is shown in Figure 8. Rats received an intraperitoneal (i.p.) injection of 127 mg/kg LiCl. After 24 hours, they were given the muscarinic receptor agonist pilocarpine (i.p.). To avoid any peripheral effects of pilocarpine, (-)-scopolamine methyl bromide (1 mg/kg, i.p.) was given 1 hour before the pilocarpine injection. Rats were injected with 10 mg/kg of pilocarpine every 30 minutes until they experienced seizures with a score of 4 on the Racine scale [80]. Rats that did not have seizures after the fourth injection (totaling 40 mg/kg of pilocarpine) were excluded from the study. 75 minutes after reaching stage 4 seizures, diazepam (10 mg/kg, i.p.) was given to stop the convulsions.
A total of 94 rats were included in the study. The control group of rats (Ctrl; n = 24) received LiCl and saline without pilocarpine injection. The experimental rats, injected with pilocarpine, were randomly allocated into one of four groups based on the type of treatment. One group remained untreated (TLE; n = 19), while the remaining three groups received the following treatments: 1) TLE+A group (n = 16), which was treated with anakinra on the following schedule: 5 injections of 100 mg/kg, i.p. once a day, then 5 injections of 50 mg/kg once a day; 2) TLE+L group (n = 20) was treated with lamotrigine dissolved in DMSO, 20 mg/kg, i.p. once a day; 3) TLE+A+L group (n = 15) received a combination of lamotrigine and anakinra in the doses described above. The first injection of all drugs was given one hour after the cessation of seizures.
4.3. Survival rate and Body Weight
Survival rate and body weight were monitored for ten days following pilocarpine-induced status epilepticus. The animals were initially given moist food and received subcutaneous injections of 5% glucose (2-3 ml) to increase their survival.
4.4. Spontaneous Recurrent Seizures (SRS)
110-112 days after receiving pilocarpine injections, the presence of SRS was assessed. Each rat was placed in a clear cage with unrestricted access to food and water, and their free behavior was videotaped for 40 hours. The collected data were analyzed to determine the total and average duration and frequency of SRS.
4.5. Behavioral Testing
Behavioral assessment was conducted in the chronic phase of the model. If seizures occurred in the rats during testing, testing was halted, and the results of that test were not taken into account. Experiments were performed between 6 pm and 11 pm. Video recordings of all tests were analyzed with Field 4W and Pole-Krest software programs (Institute of Experimental Medicine, St. Petersburg, Russia). The behavioral tests were conducted by an experienced researcher who was blinded to a group of animals.
4.5.1. Open field test
The Open field test was used to assess motor and exploratory activity. Each rat was placed in the center of a circular arena 1 m in diameter with 30 cm high walls. The arena had 4 cm diameter holes in the floor and was illuminated with a luminance of 8 Lx. The movements of each rat were monitored and recorded for 5 min. Several parameters were calculated to assess locomotor activity, including total distance traveled, time spent moving, and immobility. To assess the anxiety level of the rats, the time spent in the center of the arena (which is one third of the arena diameter) was measured. In addition, the frequency and duration of specific behaviors such as climbing, rearing, sniffing, and exploring holes (an indicator of exploratory activity), as well as autogrooming and freezing (indicators of anxiety) were determined. The arena was cleaned between trials to minimize the influence of olfactory cues.
4.5.2. Fear conditioning test
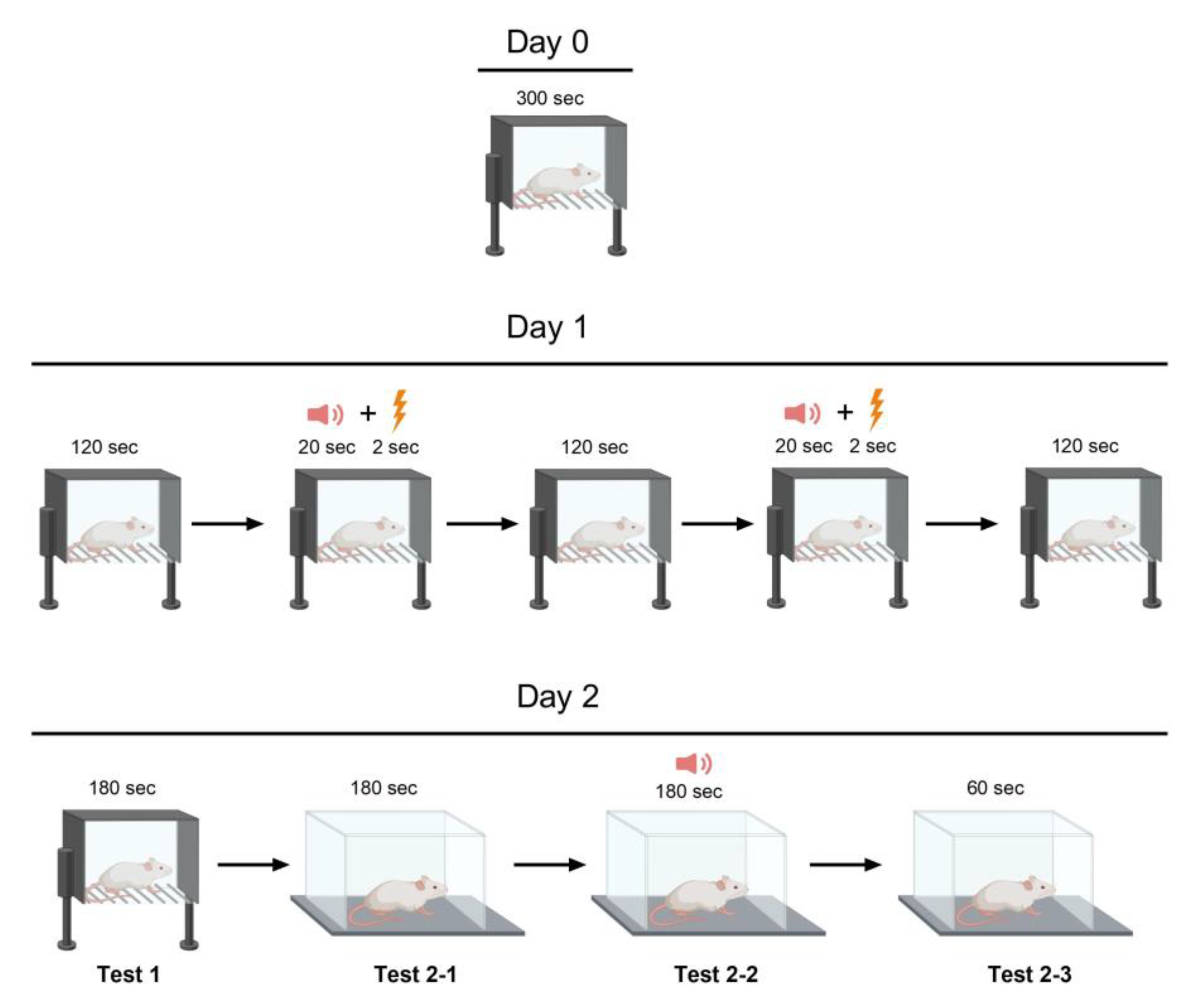
Two Plexiglas cages were used in this experiment (Figure 9). Cage A measured 45 × 30 cm with a height of 20 cm and had a conductive floor. Cage B was larger at 60 × 30 cm with a height of 40 cm and did not have a conductive floor. On Day 0, the rats were habituated to Cage A for five minutes. On day one, each rat was placed in cage A to undergo the conditioning stages. The habituation period lasted for two minutes, followed by the presentation of an 80-dB sound for 20 s as an auditory cue. Immediately following the auditory cue, a light foot shock of 0.6 mA lasting 2 s was administered as an aversive stimulus. After a two-minute break, the previous step was repeated. During the final two min of the test on Day 1, no sound was produced. On the second day, the rat was returned to Cage A for a 3-minute period without any cues or stimulation to evaluate its contextual fear memory (contextual conditioning test, test 1). The animal was then shifted to a new environment (Cage B) featuring dissimilar olfactory and visual cues (vanillin drops on the floor and geometrical figures on the walls). After 3 min of habituation (test 2-1), an 80-dB sound was emitted for 3 min to measure the rat's fear response to a current-associated sound stimulus (cued conditioning testing, test 2-2). During the final minute of the experiment, no stimuli were presented to the rat while its response to the conditioned stimulus or the cage was recorded as the time spent freezing (test 2-3). Fear-related memory was analyzed via the percentage of time spent freezing in relation to the duration of each step, which varied in length. Day 1's immediate post-foot-shock freezing response reflected short-term fear-associated memory, while Day 2's freezing response reflected long-term fear-associated memory.
4.5.3. Social interaction test
To assess social behavior, a social interaction test was conducted. First, a resident rat was placed in a 60 × 30 cm Plexiglas cage with a height of 40 cm for 30 minutes to acclimate to the new environment prior to the test. Then, an adult male intruder weighing at least 10 g less than the resident rat was placed in the same cage for 5 min. Behaviors exhibited during this interaction were quantified, including aggression, communication (such as grooming the intruder's body and sniffing the intruder's tail and genitals), and defense. The total and average time spent performing each behavior was analyzed.
4.5.4. Morris water maze
A Morris water maze was used to assess spatial memory and learning ability. A circular pool 150 cm in diameter with walls 70 cm high was filled with water laced with milk (to reduce visibility) and maintained at a temperature of 23 ± 1 °C. Four cues were placed on the walls to serve as spatial reference points: a circle, a square, a triangle, and a cross. The rats had to find a hidden platform (diameter 10 cm) located 1 cm below the water surface and 15 cm from the pool wall. Training was performed for four consecutive days, four trials per day for each rat, with a 5-min break between trials. The starting point for each trial was randomly rotated among the four locations (north, east, south, or west) so that all 4 locations were used during each training day. The platform location remained unchanged. The rat was left on the platform for 30 s after detection and then removed from the pool. An attempt was considered unsuccessful if the rat did not find the platform within 90 s. On test day 5, the platform was removed and the rat was placed in the pool for 90 s. Time spent in the target area (four times the diameter of the platform) where the platform was previously located was scored.
4.6. Histology
Rats were anesthetized with isoflurane, then decapitated and the brain removed. The brain was then fixed in 4% paraformaldehyde solution for 3-7 days at 4°C, cryoprotected in 30% sucrose, and stored at -80°C. Frontal serial sections of 20 µm thickness (-2.76 to -3.6 mm to bregma) were cut on a Bright OTF5000 cryostat (Bright Instrument Co Ltd., Huntingdon, UK).
Nissl staining was performed with 0.05% thionine solution as previously described [81]. Sections were analyzed on a Leica AF 7000 microscope (Leica Microsystems, Wetzlar, Germany) at ×400 magnification. For morphological analysis, neurons were counted on every 10th slice (a total of 8-10 slices from one rat hippocampus). The distance between analyzed slices was 200 μm. The number of neurons in the digital micrographs was counted in ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA) using the multi-point tool at 100 μm for the cell layer in two hippocampal regions: CA1 and CA3.
4.7. Statistical analysis
Statistical analysis was conducted using IBM SPSS Statistics 20 (IBM, Armonk, NY, USA) and Graph Pad Prism 8 (Graph Pad Software, San Diego, CA, USA) software. Outliers were identified using the Dixon's Q-test. The Kolmogorov-Smirnov test was used to test for normality of distribution. The Kruskal-Wallis H-test with Dunn's post hoc test was used to evaluate treatment effects for non-normally distributed data in pairwise comparison of groups. In the case of a normal distribution, we performed the Levene test to check for equal variances between groups. If the variances were not significantly different, we used one-way ANOVA and Tukey's post-hoc test. If the variances were significantly different, we used a variant of one-way ANOVA, Welch's test, and the Games-Howell test for post hoc analysis. The hypothesis of a difference between treated and untreated rats on the studied parameters was tested using the ANOVA contrast method [82,83]. To do this, four contrasts were examined: 1) untreated TLE (-3) vs. all treated animals (TLE+A; +1; TLE+L; +1; TLE+A+L; +1); 2) Monotherapy with anakinra (+1) and lamotrigine (+1) vs. combined therapy (-2). 3) Untreated TLE (-2) and treated with anakinra (TLE+A; +1; TLE+A+L; +1); 4) Untreated TLE (-2) and treated with lamotrigine (TLE+L; +1; TLE+A+L; +1). Contrasts were calculated for normally distributed data with significant values from a one-factor ANOVA.
Weight dynamics after pilocarpine administration, as well as the dynamics of successful rat learning in the Morris water maze, were analyzed by two-way mixed ANOVA. Survival analysis was performed using the log-rank Mantel-Cox test. Fisher's exact test was utilized to compare the proportions of rats with and without SRS in different groups. Differences were considered significant at p ≤ 0.05. Data is presented as mean ± standard error (for normal distribution) or median and interquartile range (for non-normal distribution).
Author Contributions
Conceptualization, O.E.Z. and A.V.Z.; Methodology, A.V.Z., O.E.Z., T.Y.P.; formal analysis, A.D.K., D.S.S., A.V.G., G.P.D.; investigation, A.D.K., D.S.S., A.V.G., G.P.D.; data curation, O.E.Z., A.D.K., G.P.D., T.Y.P.; writing—original draft preparation, O.E.Z., A.V.Z., T.Y.P.; writing—review and editing, A.V.Z.; visualization, O.E.Z.; D.S.S.; supervision, A.V.Z.; O.E.Z.; funding acquisition, A.V.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Russian Science Foundation, Project 21-15-00430.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Ethics Committee of the Institute of Evolutionary Physiology and Biochemistry of the Russian Academy of Sciences (ethical approval number: 13-k-a, dated 15 February 2018).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
Histology experiments were performed using the facilities of the Research Resource Centre for physiological, biochemical, and molecular–biological research of the Sechenov Institute of Evolutionary Physiology and Biochemistry of the Russian Academy of Sciences.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A.

Table A1.
Results of ANOVA contrasts.*.
| The indicators | Untreated TLE rats (-3) vs treated with anakinra (TLE+A; +1); lamotrigine (TLE+L; +1) and combined treatment (TLE+A+L; +1) | Untreated TLE rats (-2) vs treated with anakinra (TLE+A; +1) and combined treatment (TLE+A+L; +1) | Untreated TLE rats (-2) vs treated with lamotrigine (TLE+L; +1) and combined treatment (TLE+A+L; +1) | Monotherapy with anakinra (-1) and lamotrigine (-1) vs combined therapy (TLE+A+L; +2) |
| Histological studies | ||||
| Neuronal death in CA1 hippocampal regions | t = 2.93; t = 0.02 | t = 2.89; p = 0.02 | t = 2.85; p = 0.022 | t = 0.4; p = 0.70 |
| Neuronal death in CA3 hippocampal regions | t = 2.50; p = 0.022 | t = 2.08; p>0.05 | t = 1.56; p >0.05 | t = 1.56; p = 0.14 |
| The Open Field test | ||||
| Time of locomotion | t = 1.30; p = 0.20 | t = 1.10; p = 0.28 | t = 1.24; p = 0.22 | t = 0.30; p = 0.76 |
| Number of climbing | t = 2.55; p = 0.013 | t = 2.50; p = 0.015 | t = 2.68; p = 0.009 | t = 1.15; p = 0.25 |
| Number of acts | t = 2.66; p = 0.01 | t = 2.31; p = 0.02 | t = 2.74; p = 0.98 | t = 1.15; p = 0.88 |
| Time in the center | t = 1.40; p = 0.17 | t = 1.20; p = 0.24 | t = 0.24; p = 0.01 | t = 3.86; p = 0.001 |
| Social test | ||||
| Time of communication | t = 0.67; p = 0.50 | t = 0.37; p = 0.71 | t = 1.09; p = 0.28 | t = 0.60; p = 0.55 |
| Morris water maze | ||||
| Total distance during training | t = 0.25; p = 0.80 | t = 0.22; p = 0.83 | t = 0,56; p = 0.58 | t = 0.26; p = 0.80 |
| Time in the target area (during 90 s) | t = 0.085; p = 0.93 | t = 0.17; p = 0.87 | t = 0,19; p = 0.87 | t = 0.19; p = 0.85 |
| Time in the target area (during 30 s) | t = 0.33; p = 0.74 | t = 0.18; p = 0.86 | t = 0,39; p = 0.70 | t = 0.78; p = 0.45 |
* Contrasts were calculated for normally distributed data with significant values of one-factor ANOVA.
References
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in Adults. The Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, K.A.; Lopez Ramos, C.; Buch, V.P.; Mekary, R.A.; Amundson, J.R.; Shah, M.; Rattani, A.; Dewan, M.C.; Park, K.B. An Estimation of Global Volume of Surgically Treatable Epilepsy Based on a Systematic Review and Meta-Analysis of Epilepsy. J Neurosurg 2019, 130, 1127–1141. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Liu, Y.; Yu, X.; Wu, J.; Poon, A.N.; Demaio, A.; Wang, W.; Rudan, I.; Chan, K.Y. Prevalence of Epilepsy in China between 1990 and 2015: A Systematic Review and Meta–Analysis. J Glob Health 2017, 7. [Google Scholar] [CrossRef]
- Querol Pascual, M.R. Temporal Lobe Epilepsy: Clinical Semiology and Neurophysiological Studies. Seminars in Ultrasound, CT and MRI 2007, 28, 416–423. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.O. Cellular and Molecular Basis of Epilepsy. Journal of Neuroscience 1994, 14, 3413–3425. [Google Scholar] [CrossRef]
- Téllez-Zenteno, J.F.; Hernández-Ronquillo, L. A Review of the Epidemiology of Temporal Lobe Epilepsy. Epilepsy Res Treat 2012, 2012, 1–5. [Google Scholar] [CrossRef]
- Hendriks, M.P.H.; Aldenkamp, A.P.; van der Vlugt, H.; Alpherts, W.C.J.; Vermeulen, J. Memory Complaints in Medically Refractory Epilepsy: Relationship to Epilepsy-Related Factors. Epilepsy & Behavior 2002, 3, 165–172. [Google Scholar] [CrossRef]
- Tramoni-Negre, E.; Lambert, I.; Bartolomei, F.; Felician, O. Long-Term Memory Deficits in Temporal Lobe Epilepsy. Rev Neurol (Paris) 2017, 173, 490–497. [Google Scholar] [CrossRef]
- Swinkels, W.A.M.; Kuyk, J.; Dyck, R. van; Spinhoven, Ph. Psychiatric Comorbidity in Epilepsy. Epilepsy & Behavior 2005, 7, 37–50. [Google Scholar] [CrossRef]
- Bonora, A.; Benuzzi, F.; Monti, G.; Mirandola, L.; Pugnaghi, M.; Nichelli, P.; Meletti, S. Recognition of Emotions from Faces and Voices in Medial Temporal Lobe Epilepsy. Epilepsy & Behavior 2011, 20, 648–654. [Google Scholar] [CrossRef]
- Giovagnoli, A.R.; Franceschetti, S.; Reati, F.; Parente, A.; Maccagnano, C.; Villani, F.; Spreafico, R. Theory of Mind in Frontal and Temporal Lobe Epilepsy: Cognitive and Neural Aspects. Epilepsia 2011, 52, 1995–2002. [Google Scholar] [CrossRef] [PubMed]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The Consequences of Refractory Epilepsy and Its Treatment. Epilepsy & Behavior 2014, 37, 59–70. [Google Scholar] [CrossRef]
- Schmidt, D.; Schachter, S.C. Drug Treatment of Epilepsy in Adults. BMJ (Online) 2014, 348. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Schmidt, D. Modern Antiepileptic Drug Development Has Failed to Deliver: Ways out of the Current Dilemma. Epilepsia 2011, 52, 657–678. [Google Scholar] [CrossRef] [PubMed]
- Walia, K.S.; Khan, E.A.; Ko, D.H.; Raza, S.S.; Khan, Y.N. Side Effects of Antiepileptics- A Review. Pain Practice 2004, 4, 194–203. [Google Scholar] [CrossRef]
- Mula, M.; Kanner, A.M.; Schmitz, B.; Schachter, S. Antiepileptic Drugs and Suicidality: An Expert Consensus Statement from the Task Force on Therapeutic Strategies of the ILAE Commission on Neuropsychobiology. Epilepsia 2013, 54, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D. Is Antiepileptogenesis a Realistic Goal in Clinical Trials? Concerns and New Horizons. Epileptic Disorders 2012, 14, 105–113. [Google Scholar] [CrossRef]
- Galanopoulou, A.S.; Buckmaster, P.S.; Staley, K.J.; Moshé, S.L.; Perucca, E.; Engel Jr., J.; Löscher, W.; Noebels, J.L.; Pitkänen, A.; Stables, J.; et al. Identification of New Epilepsy Treatments: Issues in Preclinical Methodology. Epilepsia 2012, 53, 571–582. [Google Scholar] [CrossRef]
- Rana, A.; Musto, A.E. The Role of Inflammation in the Development of Epilepsy. J Neuroinflammation 2018, 15, 144. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The Role of Inflammation in Epilepsy. Nat Rev Neurol 2011, 7, 31–40. [Google Scholar] [CrossRef]
- Shimada, T.; Takemiya, T.; Sugiura, H.; Yamagata, K. Role of Inflammatory Mediators in the Pathogenesis of Epilepsy. Mediators Inflamm 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Ravizza, T.; Bedner, P.; Aronica, E.; Steinhäuser, C.; Boison, D. Astrocytes in the Initiation and Progression of Epilepsy. Nat Rev Neurol 2022, 18, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Dyomina, A.V.; Zubareva, O.E.; Smolensky, I.V.; Vasilev, D.S.; Zakharova, M.V.; Kovalenko, A.A.; Schwarz, A.P.; Ischenko, A.M.; Zaitsev, A.V. Anakinra Reduces Epileptogenesis, Provides Neuroprotection, and Attenuates Behavioral Impairments in Rats in the Lithium–Pilocarpine Model of Epilepsy. Pharmaceuticals 2020, 13, 340. [Google Scholar] [CrossRef] [PubMed]
- Dilena, R.; Mauri, E.; Aronica, E.; Bernasconi, P.; Bana, C.; Cappelletti, C.; Carrabba, G.; Ferrero, S.; Giorda, R.; Guez, S.; et al. Therapeutic Effect of Anakinra in the Relapsing Chronic Phase of Febrile Infection–Related Epilepsy Syndrome. Epilepsia Open 2019, 4, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Taraschenko, O.; Fox, H.S.; Zekeridou, A.; Pittock, S.J.; Eldridge, E.; Farukhuddin, F.; Al-Saleem, F.; Devi Kattala, C.; Dessain, S.K.; Casale, G.; et al. Seizures and Memory Impairment Induced by Patient-Derived Anti-N-Methyl-D-Aspartate Receptor Antibodies in Mice Are Attenuated by Anakinra, an Interleukin-1 Receptor Antagonist. Epilepsia 2021, 62, 671–682. [Google Scholar] [CrossRef]
- Costa, B.; Vale, N. Understanding Lamotrigine’s Role in the CNS and Possible Future Evolution. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Chen, Y.; He, X.; Sun, Q.; Fang, Z.; Zhou, L. Effect of Lamotrigine on Seizure Development in a Rat Pentylenetetrazole Kindling Model. Brain Behav 2017, 7. [Google Scholar] [CrossRef]
- Stratton, S.C.; Large, C.H.; Cox, B.; Davies, G.; Hagan, R.M. Effects of Lamotrigine and Levetiracetam on Seizure Development in a Rat Amygdala Kindling Model. Epilepsy Res 2003, 53, 95–106. [Google Scholar] [CrossRef]
- Nissinen, J.; Large, C.H.; Stratton, S.C.; Pitkänen, A. Effect of Lamotrigine Treatment on Epileptogenesis: An Experimental Study in Rat. Epilepsy Res 2004, 58, 119–132. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Y.; Wang, Q.; van Luijtelaar, G.; Sun, M. The Effects of Lamotrigine and Ethosuximide on Seizure Frequency, Neuronal Loss, and Astrogliosis in a Model of Temporal-Lobe Epilepsy. Brain Res 2019, 1712, 1–6. [Google Scholar] [CrossRef]
- Zubareva, O.E.; Dyomina, A.V.; Kovalenko, A.A.; Roginskaya, A.I.; Melik-Kasumov, T.B.; Korneeva, M.A.; Chuprina, A.V.; Zhabinskaya, A.A.; Kolyhan, S.A.; Zakharova, M.V.; et al. Beneficial Effects of Probiotic Bifidobacterium Longum in a Lithium–Pilocarpine Model of Temporal Lobe Epilepsy in Rats. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Mirabel, H.; Guinet, V.; Voltzenlogel, V.; Pradier, S.; Hennion, S. Social Cognition in Epilepsy: State of the Art and Perspectives. Rev Neurol (Paris) 2020, 176, 468–479. [Google Scholar] [CrossRef]
- Smolensky, I.V.; Zubareva, O.E.; Kalemenev, S.V.; Lavrentyeva, V.V.; Dyomina, A.V.; Karepanov, A.A.; Zaitsev, A.V. Impairments in Cognitive Functions and Emotional and Social Behaviors in a Rat Lithium-Pilocarpine Model of Temporal Lobe Epilepsy. Behavioural Brain Research 2019, 372, 112044. [Google Scholar] [CrossRef]
- Cavalheiro, E.A. The Pilocarpine Model of Epilepsy. The Italian Journal of Neurological Sciences 1995, 16, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H. Role of Multidrug Transporters in Pharmacoresistance to Antiepileptic Drugs. Journal of Pharmacology and Experimental Therapeutics 2002, 301, 7–14. [Google Scholar] [CrossRef]
- Löscher, W. Critical Review of Current Animal Models of Seizures and Epilepsy Used in the Discovery and Development of New Antiepileptic Drugs. Seizure 2011, 20, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Fahnestock, M.; Racine, R.J. Kindling and Status Epilepticus Models of Epilepsy: Rewiring the Brain. Prog Neurobiol 2004, 73, 1–60. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Bhimani, A.; Kuruba, R.; Park, M.J.; Sohrabji, F. Prospects of Modeling Poststroke Epileptogenesis. J Neurosci Res 2017, 95, 1000–1016. [Google Scholar] [CrossRef]
- Pitkänen, A.; Lukasiuk, K.; Dudek, F.E.; Staley, K.J. Epileptogenesis. Cold Spring Harb Perspect Med 2015, 5, a022822. [Google Scholar] [CrossRef]
- Hufthy, Y.; Bharadwaj, M.; Gupta, S.; Hussain, D.; Joseph, P.J.S.; Khan, A.; King, J.; Lahorgue, P.; Jayawardena, O.; Rostami-Hochaghan, D.; et al. Statins as Antiepileptogenic Drugs: Analyzing the Evidence and Identifying the Most Promising Statin. Epilepsia 2022, 63, 1889–1898. [Google Scholar] [CrossRef]
- Sills, G.J.; Rogawski, M.A. Mechanisms of Action of Currently Used Antiseizure Drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-C.; Lu, L. Characterization of Lamotrigine Inhibition of Na + Channels in Rat Hippocampal Neurones. Br J Pharmacol 1997, 121, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-J.; Huang, C.-C.; Hsu, K.-S.; Tsai, J.-J.; Gean, P.-W. Inhibition of N-Type Calcium Currents by Lamotrigine in Rat Amygdalar Neurones. Neuroreport 1996, 7, 3037–3040. [Google Scholar] [CrossRef] [PubMed]
- Stefani, A.; Spadoni, F.; Siniscalchi, A.; Bernardi, G. Lamotrigine Inhibits Ca2+ Currents in Cortical Neurons: Functional Implications. Eur J Pharmacol 1996, 307, 113–116. [Google Scholar] [CrossRef]
- Bacher, A.; Zornow, M.H. Lamotrigine Inhibits Extracellular Glutamate Accumulation during Transient Global Cerebral Ischemia in Rabbits. Anesthesiology 1997, 86, 459–463. [Google Scholar] [CrossRef]
- Farber, N.B.; Jiang, X.-P.; Heinkel, C.; Nemmers, B. Antiepileptic Drugs and Agents That Inhibit Voltage-Gated Sodium Channels Prevent NMDA Antagonist Neurotoxicity. Mol Psychiatry 2002, 7, 726–733. [Google Scholar] [CrossRef]
- Amakhin, D.V.; Malkin, S.L.; Ergina, J.L.; Kryukov, K.A.; Veniaminova, E.A.; Zubareva, O.E.; Zaitsev, A.V. Alterations in Properties of Glutamatergic Transmission in the Temporal Cortex and Hippocampus Following Pilocarpine-Induced Acute Seizures in Wistar Rats. Front Cell Neurosci 2017, 11, 264. [Google Scholar] [CrossRef]
- Diespirov, G.P.; Postnikova, T.Y.; Griflyuk, A.V.; Kovalenko, A.A.; Zaitsev, A.V. Alterations in the Properties of the Rat Hippocampus Glutamatergic System in the Lithium-Pilocarpine Model of Temporal Lobe Epilepsy. Biochemistry (Moscow) 2023, 88, 353–363. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, M.; Wang, W.; Ma, C.; He, N. Comparison and Effects of Acute Lamotrigine Treatment on Extracellular Excitatory Amino Acids in the Hippocampus of PTZ-Kindled Epileptic and PTZ-Induced Status Epilepticus Rats. Neurochem Res 2013, 38, 504–511. [Google Scholar] [CrossRef]
- Herman, E. Lamotrigine: A Depression Mood Stabiliser. European Neuropsychopharmacology 2004, 14, S89–S93. [Google Scholar] [CrossRef]
- Abu-rish, E.Y.; Dahabiyeh, L.A.; Bustanji, Y.; Mohamed, Y.S.; Browning, M.J. Effect of Lamotrigine on in Vivo and in Vitro Cytokine Secretion in Murine Model of Inflammation. J Neuroimmunol 2018, 322, 36–45. [Google Scholar] [CrossRef]
- Lai, Y.; Muscal, E.; Wells, E.; Shukla, N.; Eschbach, K.; Hyeong Lee, K.; Kaliakatsos, M.; Desai, N.; Wickström, R.; Viri, M.; et al. Anakinra Usage in Febrile Infection Related Epilepsy Syndrome: An International Cohort. Ann Clin Transl Neurol 2020, 7, 2467–2474. [Google Scholar] [CrossRef]
- Du, C.; Zheng, F.; Wang, X. Exploring Novel AEDs from Drugs Used for Treatment of Non-Epileptic Disorders. Expert Rev Neurother 2016, 16, 449–461. [Google Scholar] [CrossRef]
- Roginskaya, A.I.; Dyomina, A.V.; Kovalenko, A.A.; Zakharova, M.V.; Schwarz, A.P.; Melik-Kasumov, T.B.; Zubareva, O.E. Effect of Anakinra on the Gene Expression of Receptors Activated by the Peroxisome Proliferator in the Rat Brain in the Lithium Pilocarpine Model of Epilepsy. J Evol Biochem Physiol 2022, 58, 598–609. [Google Scholar] [CrossRef]
- Leal, B.; Chaves, J.; Carvalho, C.; Rangel, R.; Santos, A.; Bettencourt, A.; Lopes, J.; Ramalheira, J.; Silva, B.M.; da Silva, A.M.; et al. Brain Expression of Inflammatory Mediators in Mesial Temporal Lobe Epilepsy Patients. J Neuroimmunol 2017, 313, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Litovchenko, A.V.; Zabrodskaya, Yu.M.; Sitovskaya, D.A.; Khuzhakhmetova, L.K.; Nezdorovina, V.G.; Bazhanova, E.D. Markers of Neuroinflammation and Apoptosis in the Temporal Lobe of Patients with Drug-Resistant Epilepsy. J Evol Biochem Physiol 2021, 57, 1040–1049. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and Epilepsy: Excitability and Inflammation. Trends Neurosci 2013, 36, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Viviani, B.; Bartesaghi, S.; Gardoni, F.; Vezzani, A.; Behrens, M.M.; Bartfai, T.; Binaglia, M.; Corsini, E.; Di Luca, M.; Galli, C.L.; et al. Interleukin-1β Enhances NMDA Receptor-Mediated Intracellular Calcium Increase through Activation of the Src Family of Kinases. Journal of Neuroscience 2003, 23, 8692–8700. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Onufriev, M.V.; Moiseeva, J.V.; Novikova, M.R.; Gulyaeva, N.V. Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats. Int J Mol Sci 2021, 22, 5883. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Ehrlich, L.C.; Peterson, P.K.; Chao, C.C. Cytokine Effects on Glutamate Uptake by Human Astrocytes. Neuroimmunomodulation 2000, 7, 153–159. [Google Scholar] [CrossRef]
- Zaitsev, A.V.; Smolensky, I.V.; Jorratt, P.; Ovsepian, S.V. Neurobiology, Functions, and Relevance of Excitatory Amino Acid Transporters (EAATs) to Treatment of Refractory Epilepsy. CNS Drugs 2020, 34, 1089–1103. [Google Scholar] [CrossRef] [PubMed]
- Mazarati, A.M.; Pineda, E.; Shin, D.; Tio, D.; Taylor, A.N.; Sankar, R. Comorbidity between Epilepsy and Depression: Role of Hippocampal Interleukin-1β. Neurobiol Dis 2010, 37, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Suleymanova, E.M. Behavioral Comorbidities of Epilepsy and Neuroinflammation: Evidence from Experimental and Clinical Studies. Epilepsy & Behavior 2021, 117, 107869. [Google Scholar] [CrossRef]
- Noe, F.M.; Polascheck, N.; Frigerio, F.; Bankstahl, M.; Ravizza, T.; Marchini, S.; Beltrame, L.; Banderó, C.R.; Löscher, W.; Vezzani, A. Pharmacological Blockade of IL-1β/IL-1 Receptor Type 1 Axis during Epileptogenesis Provides Neuroprotection in Two Rat Models of Temporal Lobe Epilepsy. Neurobiol Dis 2013, 59, 183–193. [Google Scholar] [CrossRef]
- Dingledine, R.; Varvel, N.H.; Dudek, F.E. When and How Do Seizures Kill Neurons, and Is Cell Death Relevant to Epileptogenesis? In Advances in Experimental Medicine and Biology; 2014; Vol. 813, pp. 109–122. [Google Scholar] [CrossRef]
- Dyomina, A. V.; Kovalenko, A.A.; Zakharova, M. V.; Postnikova, T.Y.; Griflyuk, A. V.; Smolensky, I. V.; Antonova, I. V.; Zaitsev, A. V MTEP, a Selective MGluR5 Antagonist, Had a Neuroprotective Effect but Did Not Prevent the Development of Spontaneous Recurrent Seizures and Behavioral Comorbidities in the Rat Lithium–Pilocarpine Model of Epilepsy. Int J Mol Sci 2022, 23, 497. [Google Scholar] [CrossRef]
- Beesley, S.; Sullenberger, T.; Crotty, K.; Ailani, R.; D’Orio, C.; Evans, K.; Ogunkunle, E.O.; Roper, M.G.; Kumar, S.S. D-Serine Mitigates Cell Loss Associated with Temporal Lobe Epilepsy. Nat Commun 2020, 11, 4966. [Google Scholar] [CrossRef]
- Citraro, R.; Iannone, M.; Leo, A.; De Caro, C.; Nesci, V.; Tallarico, M.; Abdalla, K.; Palma, E.; Arturi, F.; De Sarro, G.; et al. Evaluation of the Effects of Liraglutide on the Development of Epilepsy and Behavioural Alterations in Two Animal Models of Epileptogenesis. Brain Res Bull 2019, 153, 133–142. [Google Scholar] [CrossRef]
- Hitiris, N.; Mohanraj, R.; Norrie, J.; Sills, G.J.; Brodie, M.J. Predictors of Pharmacoresistant Epilepsy. Epilepsy Res 2007, 75, 192–196. [Google Scholar] [CrossRef]
- Gastens, A.M.; Brandt, C.; Bankstahl, J.P.; Löscher, W. Predictors of Pharmacoresistant Epilepsy: Pharmacoresistant Rats Differ from Pharmacoresponsive Rats in Behavioral and Cognitive Abnormalities Associated with Experimentally Induced Epilepsy. Epilepsia 2008, 49, 1759–1776. [Google Scholar] [CrossRef]
- Rzezak, P.; Lima, E.M.; Gargaro, A.C.; Coimbra, E.; de Vincentiis, S.; Velasco, T.R.; Leite, J.P.; Busatto, G.F.; Valente, K.D. Everyday Memory Impairment in Patients with Temporal Lobe Epilepsy Caused by Hippocampal Sclerosis. Epilepsy & Behavior 2017, 69, 31–36. [Google Scholar] [CrossRef]
- Stewart, E.; Catroppa, C.; Gonzalez, L.; Gill, D.; Webster, R.; Lawson, J.; Sabaz, M.; Mandalis, A.; Barton, B.; McLean, S.; et al. Theory of Mind and Social Competence in Children and Adolescents with Temporal Lobe Epilepsy. Neuropsychology 2019, 33, 986–995. [Google Scholar] [CrossRef]
- Montagrin, A.; Saiote, C.; Schiller, D. The Social Hippocampus. Hippocampus 2018, 28, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.C.M.; Moura, P.J.; Cysneiros, R.M.; Colugnati, D.B.; Cavalheiro, E.A.; Scorza, F.A.; Xavier, G.F.; Zilbovicius, M.; Mercadante, M.T. Temporal Lobe Epilepsy and Social Behavior: An Animal Model for Autism? Epilepsy & Behavior 2008, 13, 43–46. [Google Scholar] [CrossRef]
- Jiang, Y.; Han, C.; Liu, H.; Wang, X.; Zhang, X.; Meng, F.; Zhang, J. Abnormal Hippocampal Functional Network and Related Memory Impairment in Pilocarpine-treated Rats. Epilepsia 2018, 59, 1785–1795. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Jung, S.; Lee, S.-Y.; Yang, H.; Kim, B.S.; Choi, J.; Bang, M.; Shin, H.-S.; Jeon, D. Early Deficits in Social Behavior and Cortical Rhythms in Pilocarpine-Induced Mouse Model of Temporal Lobe Epilepsy. Exp Neurol 2013, 241, 38–44. [Google Scholar] [CrossRef]
- Miller, J.M.; Kustra, R.P.; Vuong, A.; Hammer, A.E.; Messenheimer, J.A. Depressive Symptoms in Epilepsy. Drugs 2008, 68, 1493–1509. [Google Scholar] [CrossRef]
- Kato, H.; Fukatsu, N.; Noguchi, T.; Oshima, T.; Tadokoro, Y.; Kanemoto, K. Lamotrigine Improves Aggression in Patients with Temporal Lobe Epilepsy. Epilepsy & Behavior 2011, 21, 173–176. [Google Scholar] [CrossRef]
- Mahfoz, A.M.; Abdel-Wahab, A.F.; Afify, M.A.; Shahzad, N.; Ibrahim, I.A.A.; ElSawy, N.A.; Bamagous, G.A.; Al Ghamdi, S.S. Neuroprotective Effects of Vitamin D Alone or in Combination with Lamotrigine against Lithium-Pilocarpine Model of Status Epilepticus in Rats. Naunyn Schmiedebergs Arch Pharmacol 2017, 390, 977–985. [Google Scholar] [CrossRef]
- Racine, R.J. Modification of Seizure Activity by Electrical Stimulation. II. Motor Seizure. Electroencephalogr Clin Neurophysiol 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Postnikova, T.Y.; Diespirov, G.P.; Amakhin, D.V.; Vylekzhanina, E.N.; Soboleva, E.B.; Zaitsev, A.V. Impairments of Long-Term Synaptic Plasticity in the Hippocampus of Young Rats during the Latent Phase of the Lithium-Pilocarpine Model of Temporal Lobe Epilepsy. Int J Mol Sci 2021, 22, 13355. [Google Scholar] [CrossRef]
- Furr, R.M. A Contrast Analysis Approach to Change. Educational Research and Evaluation 2008, 14, 335–362. [Google Scholar] [CrossRef]
- Konietschke, F.; Bösiger, S.; Brunner, E.; Hothorn, L.A. Are Multiple Contrast Tests Superior to the ANOVA? International Journal of Biostatistics 2013, 9. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Effect of treatment with anakinra, lamotrigine and their combination on neurological parameters. (a) Kaplan-Meier survival curves. (b) Body weight dynamics. (c) Percentage of animals showing SRS during the 40 h observation period. (d) Number of SRS in the groups during 40 h of observation. (e) Duration of SRS episodes in the groups during 40 h of observation. (f) Seizure severity according to the Racine scale. Cntr – control rats; TLE – post-SE lithium-pilocarpine untreated group of rats; TLE+A, TLE+L, TLE+A+L – rats treated with anakinra, lamotrigine, and their combination. Each point represents one animal. Data are presented as mean ± standard error of the mean (b) or median and interquartile range (d-f).
Figure 1.
Effect of treatment with anakinra, lamotrigine and their combination on neurological parameters. (a) Kaplan-Meier survival curves. (b) Body weight dynamics. (c) Percentage of animals showing SRS during the 40 h observation period. (d) Number of SRS in the groups during 40 h of observation. (e) Duration of SRS episodes in the groups during 40 h of observation. (f) Seizure severity according to the Racine scale. Cntr – control rats; TLE – post-SE lithium-pilocarpine untreated group of rats; TLE+A, TLE+L, TLE+A+L – rats treated with anakinra, lamotrigine, and their combination. Each point represents one animal. Data are presented as mean ± standard error of the mean (b) or median and interquartile range (d-f).
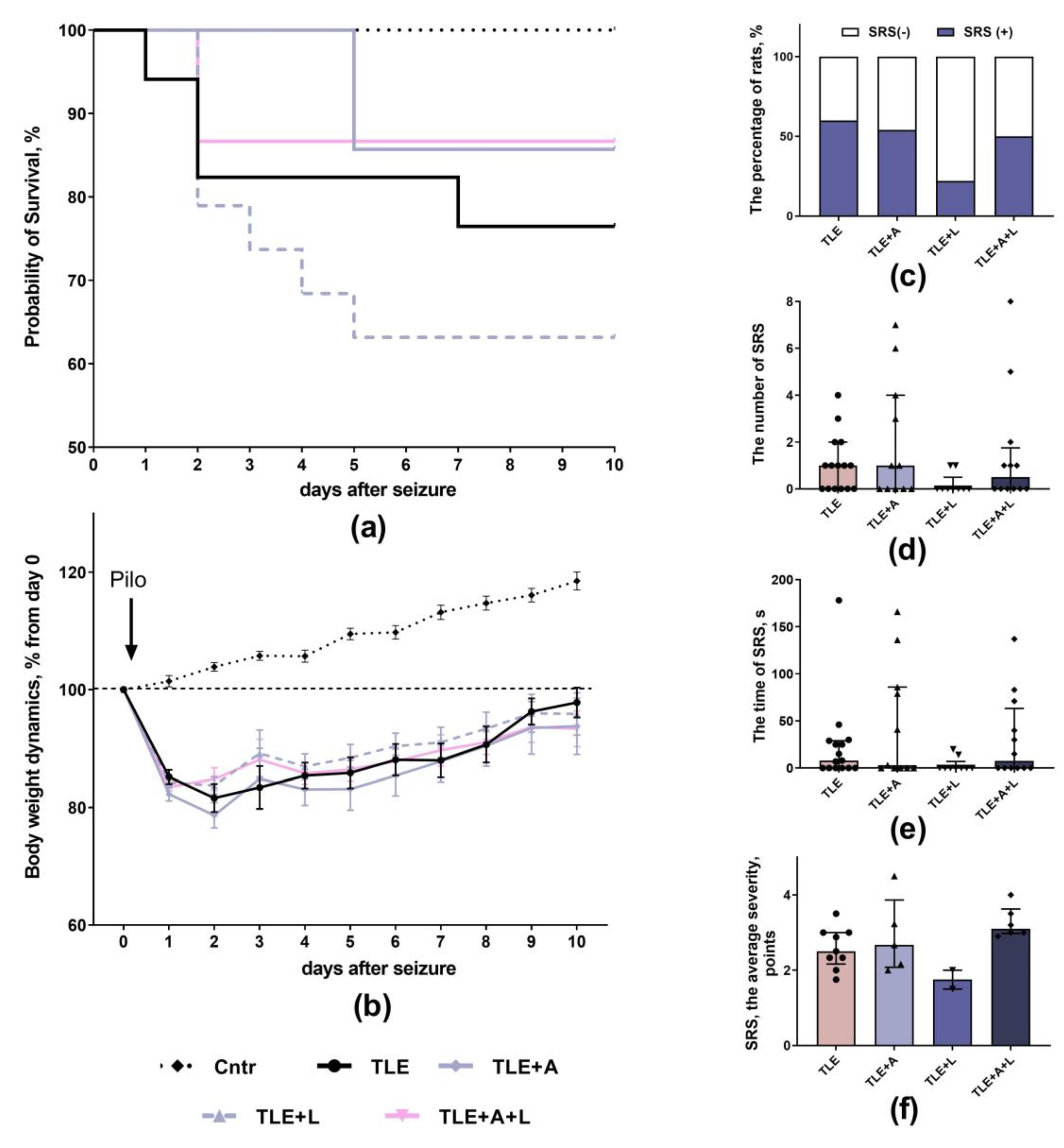
Figure 2.
Representative Nissl-stained sections of the hippocampus of control rat (Cntr), post-SE lithium-pilocarpine untreated rat (TLE) and post-SE rats treated with anakinra (TLE+A), lamotrigine (TLE+L), or their combination (TLE+A+L).
Figure 2.
Representative Nissl-stained sections of the hippocampus of control rat (Cntr), post-SE lithium-pilocarpine untreated rat (TLE) and post-SE rats treated with anakinra (TLE+A), lamotrigine (TLE+L), or their combination (TLE+A+L).
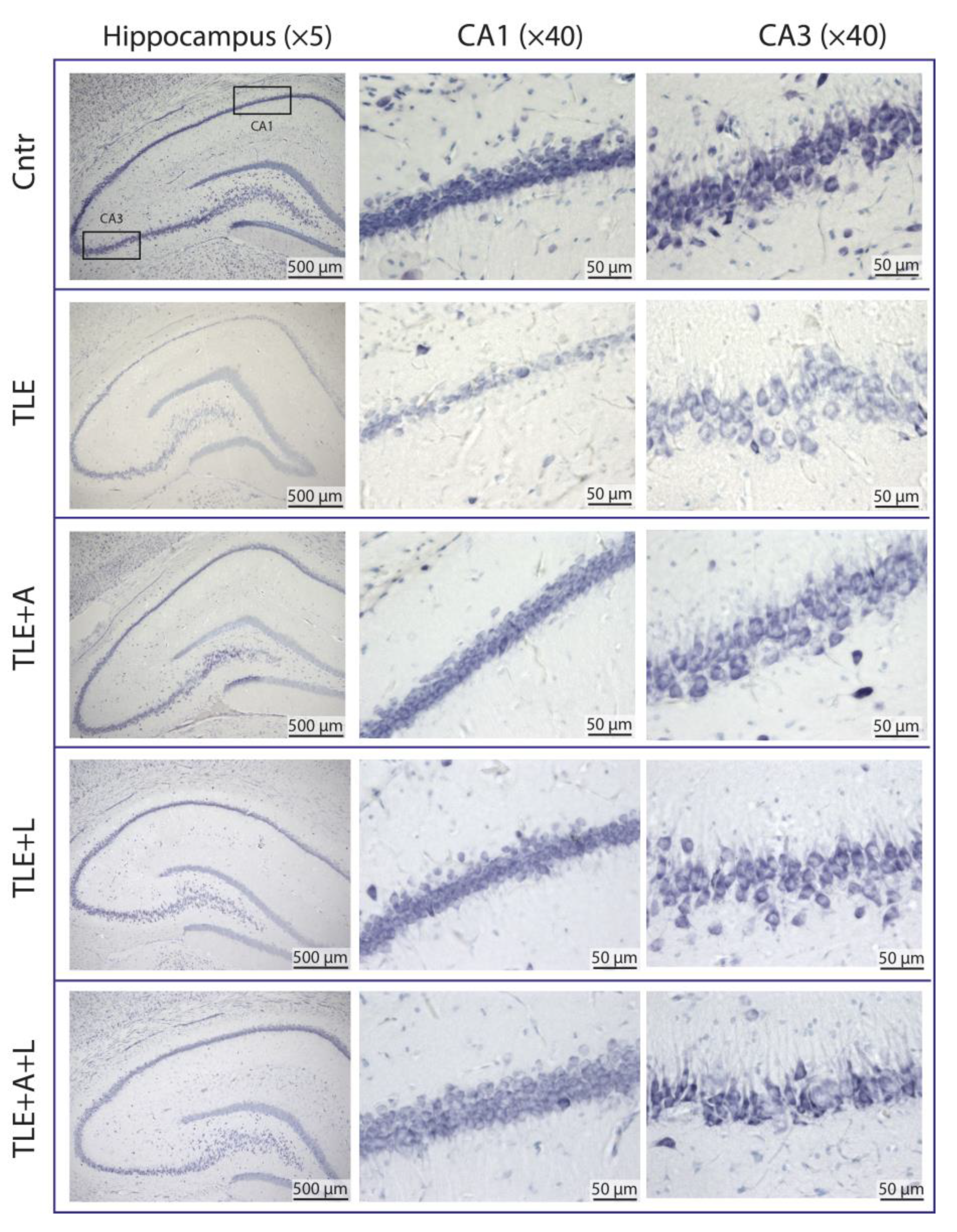
Figure 3.
Statistical data on the number of neurons per 100 µm length of cell layer in hippocampal areas CA1 and CA3 of control rats (Cntr), post-SE lithium-pilocarpine untreated rats (TLE), and post-SE rats treated with anakinra (TLE+A), lamotrigine (TLE+L), or their combination (TLE+A+L). Markers indicate individual values per rat. Columns show mean values and error bars show standard error of the mean. * p < 0.05, ** p < 0.01, *** p < 0.001 (post-hoc Games-Howell test for CA1; post-hoc Tukey's test for CA3).
Figure 3.
Statistical data on the number of neurons per 100 µm length of cell layer in hippocampal areas CA1 and CA3 of control rats (Cntr), post-SE lithium-pilocarpine untreated rats (TLE), and post-SE rats treated with anakinra (TLE+A), lamotrigine (TLE+L), or their combination (TLE+A+L). Markers indicate individual values per rat. Columns show mean values and error bars show standard error of the mean. * p < 0.05, ** p < 0.01, *** p < 0.001 (post-hoc Games-Howell test for CA1; post-hoc Tukey's test for CA3).
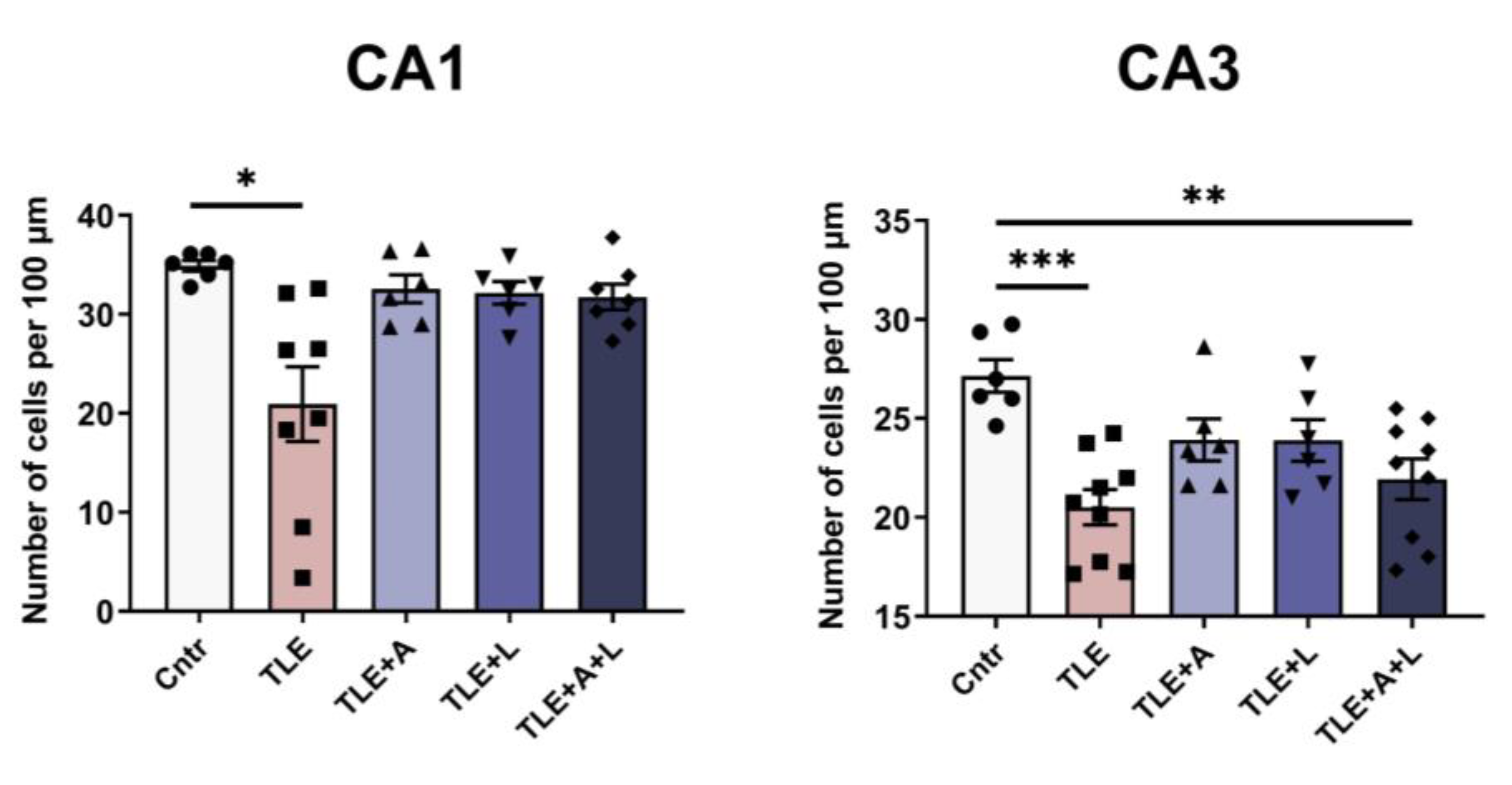
Figure 4.
Behavior of rats in the Open field test. (a) Representative examples of tracks in the open field of control rat (Cntr), post-SE lithium-pilocarpine untreated rat (TLE), and post-SE rats treated with anakinra (TLE+A), lamotrigine (TLE+L), or their combination (TLE+A+L). Statistical data on total distance traveled by rats in the open field (b), time of locomotion (c); number of acts (d); number of climbs (e); time of hole exploration (f); time of grooming (g); time in the center of the field (h). Data are presented as mean and standard error of the mean (b-f, h) or median and interquartile range (g). * p < 0.05, ** p < 0.01; *** p < 0.001,**** p < 0.0001, Tukey's post hoc test (normally distributed data) or Dunn's multiple comparison test (non-normally distributed data). Each point represents the value of a different animal.
Figure 4.
Behavior of rats in the Open field test. (a) Representative examples of tracks in the open field of control rat (Cntr), post-SE lithium-pilocarpine untreated rat (TLE), and post-SE rats treated with anakinra (TLE+A), lamotrigine (TLE+L), or their combination (TLE+A+L). Statistical data on total distance traveled by rats in the open field (b), time of locomotion (c); number of acts (d); number of climbs (e); time of hole exploration (f); time of grooming (g); time in the center of the field (h). Data are presented as mean and standard error of the mean (b-f, h) or median and interquartile range (g). * p < 0.05, ** p < 0.01; *** p < 0.001,**** p < 0.0001, Tukey's post hoc test (normally distributed data) or Dunn's multiple comparison test (non-normally distributed data). Each point represents the value of a different animal.
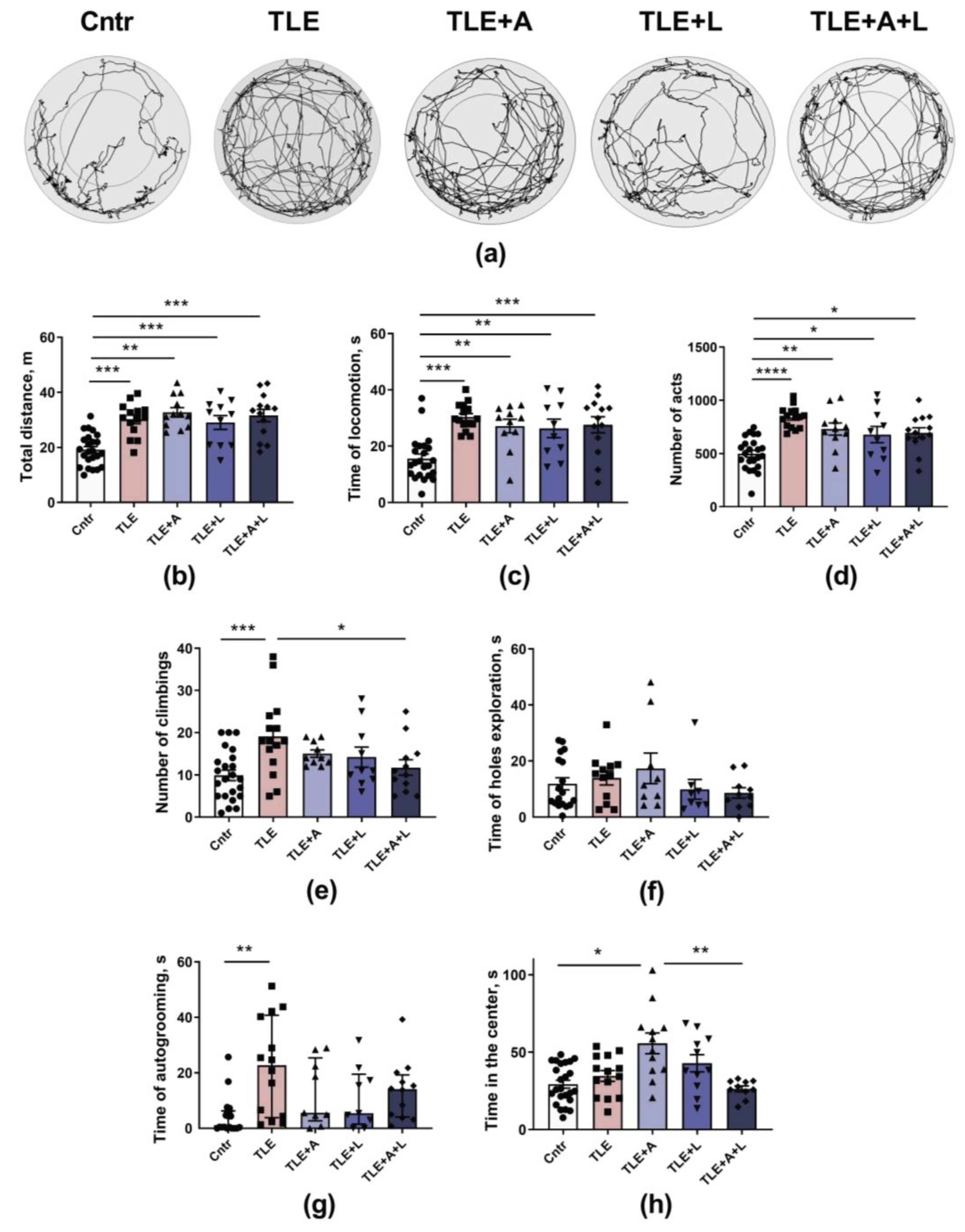
Figure 5.
The behavior of the rats in the Social test. (a) Time of communication. (b) Percentage of time of different types of communicative behaviors. (c) Time of aggressive behavior. (d) Number of acts of aggressive behavior. * p < 0.05, ** p < 0.01; *** p < 0.001,**** p < 0.0001; Tukey's post hoc test (normally distributed data) or Dunn's multiple comparison test (non-normally distributed data). Data are presented as mean and standard error of the mean (a) or median and interquartile range (c, d). Each point represents the value of a different animal.
Figure 5.
The behavior of the rats in the Social test. (a) Time of communication. (b) Percentage of time of different types of communicative behaviors. (c) Time of aggressive behavior. (d) Number of acts of aggressive behavior. * p < 0.05, ** p < 0.01; *** p < 0.001,**** p < 0.0001; Tukey's post hoc test (normally distributed data) or Dunn's multiple comparison test (non-normally distributed data). Data are presented as mean and standard error of the mean (a) or median and interquartile range (c, d). Each point represents the value of a different animal.
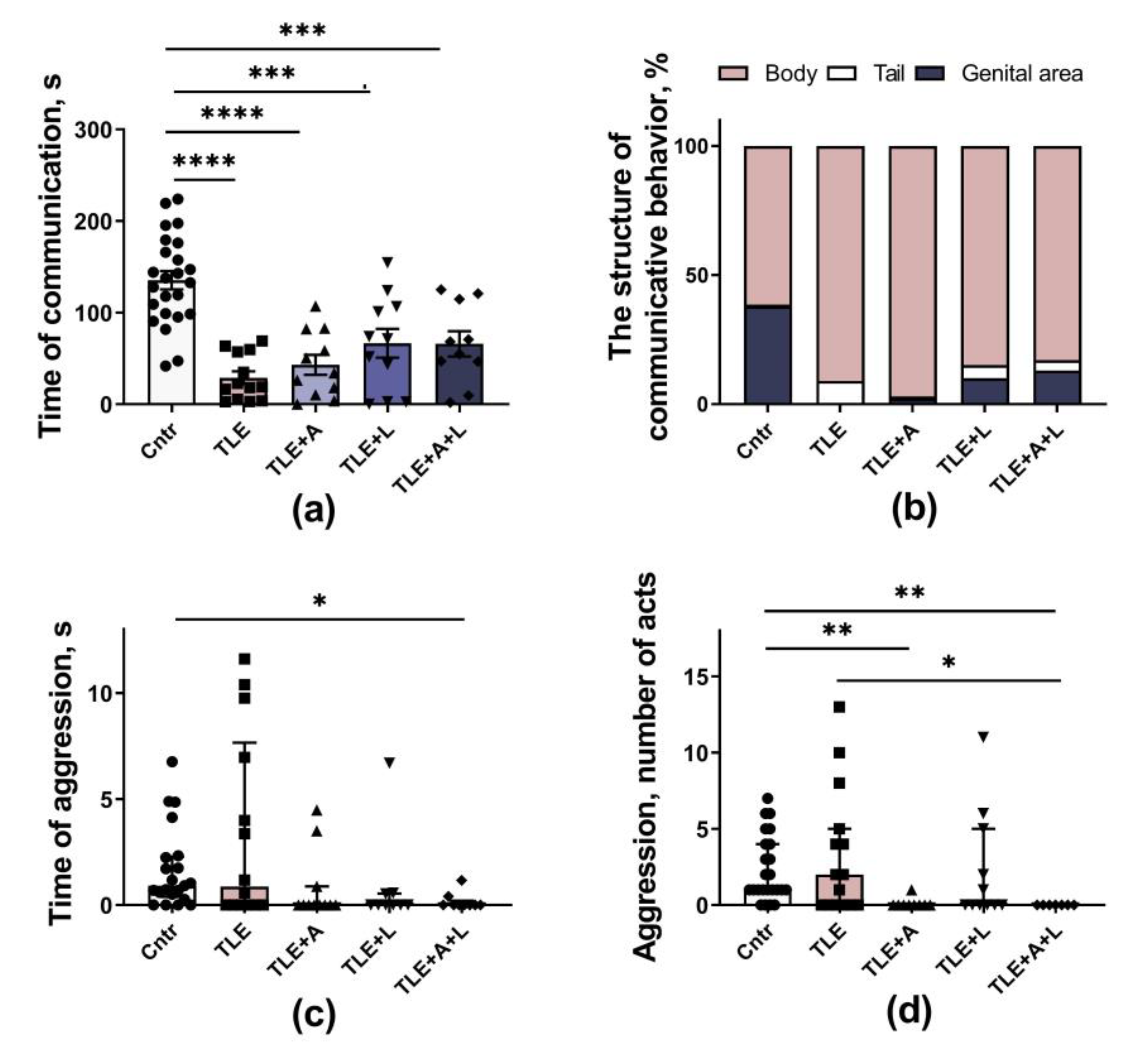
Figure 6.
Behavior of rats in the Fear conditioning test. (a) Time of freezing in cage A. (b) Time of freezing during adaptation to an unfamiliar cage B. (c) Time of freezing in the new cage B in response to a tone, a pain-associated stimulus. (d) Time of freezing in the novel cage B after cessation of the pain-associated stimulus. * p < 0.05, *** p < 0.001, **** p < 0.0001; Dunn's multiple comparisons test. The data are presented as median and interquartile range (a-b) or as mean and standard error of the mean (c-d).
Figure 6.
Behavior of rats in the Fear conditioning test. (a) Time of freezing in cage A. (b) Time of freezing during adaptation to an unfamiliar cage B. (c) Time of freezing in the new cage B in response to a tone, a pain-associated stimulus. (d) Time of freezing in the novel cage B after cessation of the pain-associated stimulus. * p < 0.05, *** p < 0.001, **** p < 0.0001; Dunn's multiple comparisons test. The data are presented as median and interquartile range (a-b) or as mean and standard error of the mean (c-d).
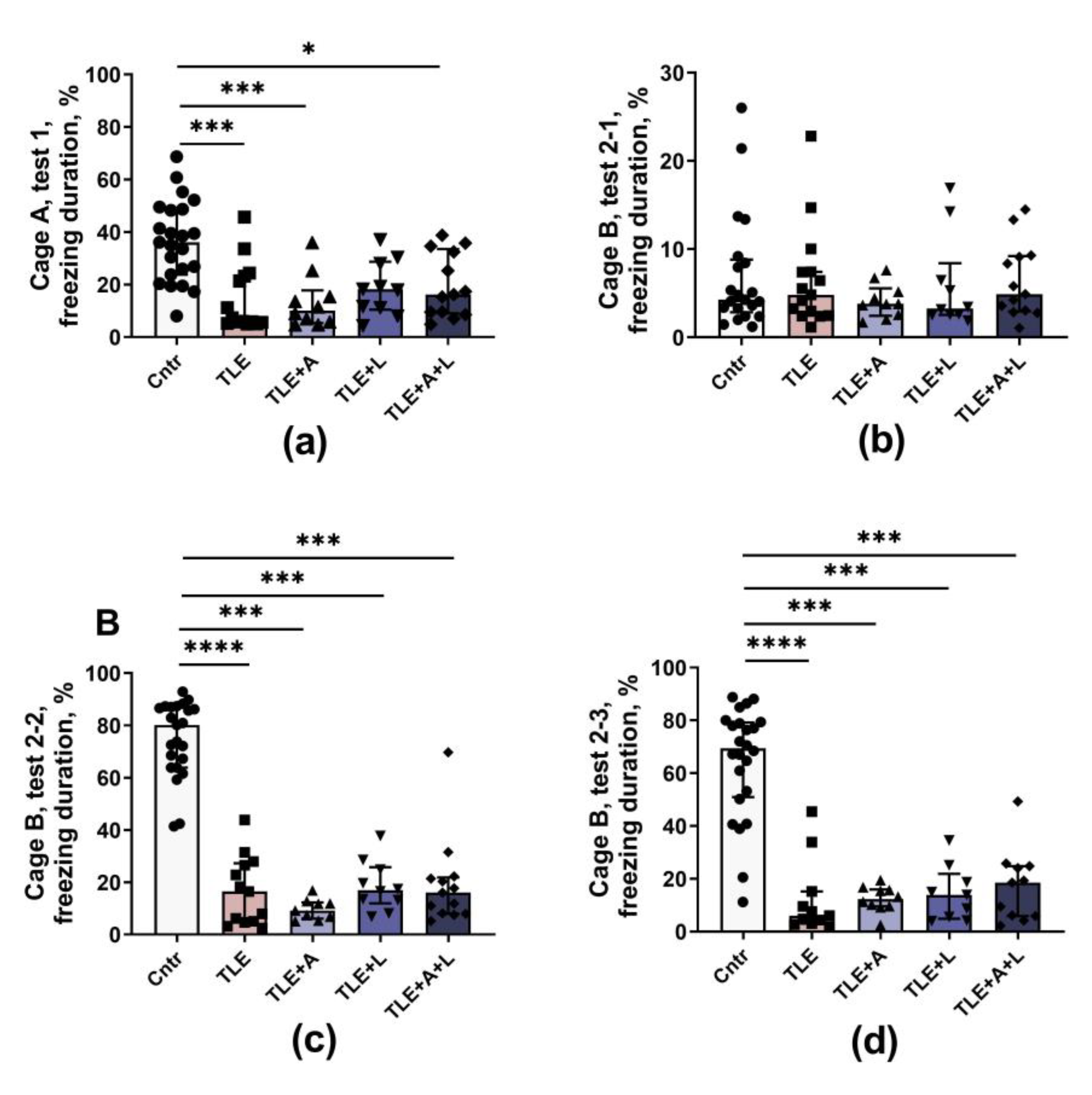
Figure 7.
Spatial learning and memory of rats in the Morris water maze. (a) Training dynamics (distance traveled to find the platform) over four training days. (b) Distance traveled before finding the platform in the third trial of the first training day. (c) Distance traveled before finding the platform on the first attempt of the second training day. (d) Total distance traveled before finding the platform on all trials for 4 training days. (e-g) Long-term spatial memory test on the fifth experimental day, time spent in the target area where the platform was previously located: (e) first 30 seconds; (f) for the entire test (90 seconds). (g) Examples of tracks during platform retrieval on test day. Data are shown for the entire test (90 seconds). The area where the platform was previously located is highlighted. *-p<0.05; **p<0.01, Tukey's or Games-Howell's post hoc test. Data are presented as mean and standard error of the mean. Each point represents the value of a different animal.
Figure 7.
Spatial learning and memory of rats in the Morris water maze. (a) Training dynamics (distance traveled to find the platform) over four training days. (b) Distance traveled before finding the platform in the third trial of the first training day. (c) Distance traveled before finding the platform on the first attempt of the second training day. (d) Total distance traveled before finding the platform on all trials for 4 training days. (e-g) Long-term spatial memory test on the fifth experimental day, time spent in the target area where the platform was previously located: (e) first 30 seconds; (f) for the entire test (90 seconds). (g) Examples of tracks during platform retrieval on test day. Data are shown for the entire test (90 seconds). The area where the platform was previously located is highlighted. *-p<0.05; **p<0.01, Tukey's or Games-Howell's post hoc test. Data are presented as mean and standard error of the mean. Each point represents the value of a different animal.
Figure 8.
The experimental design.
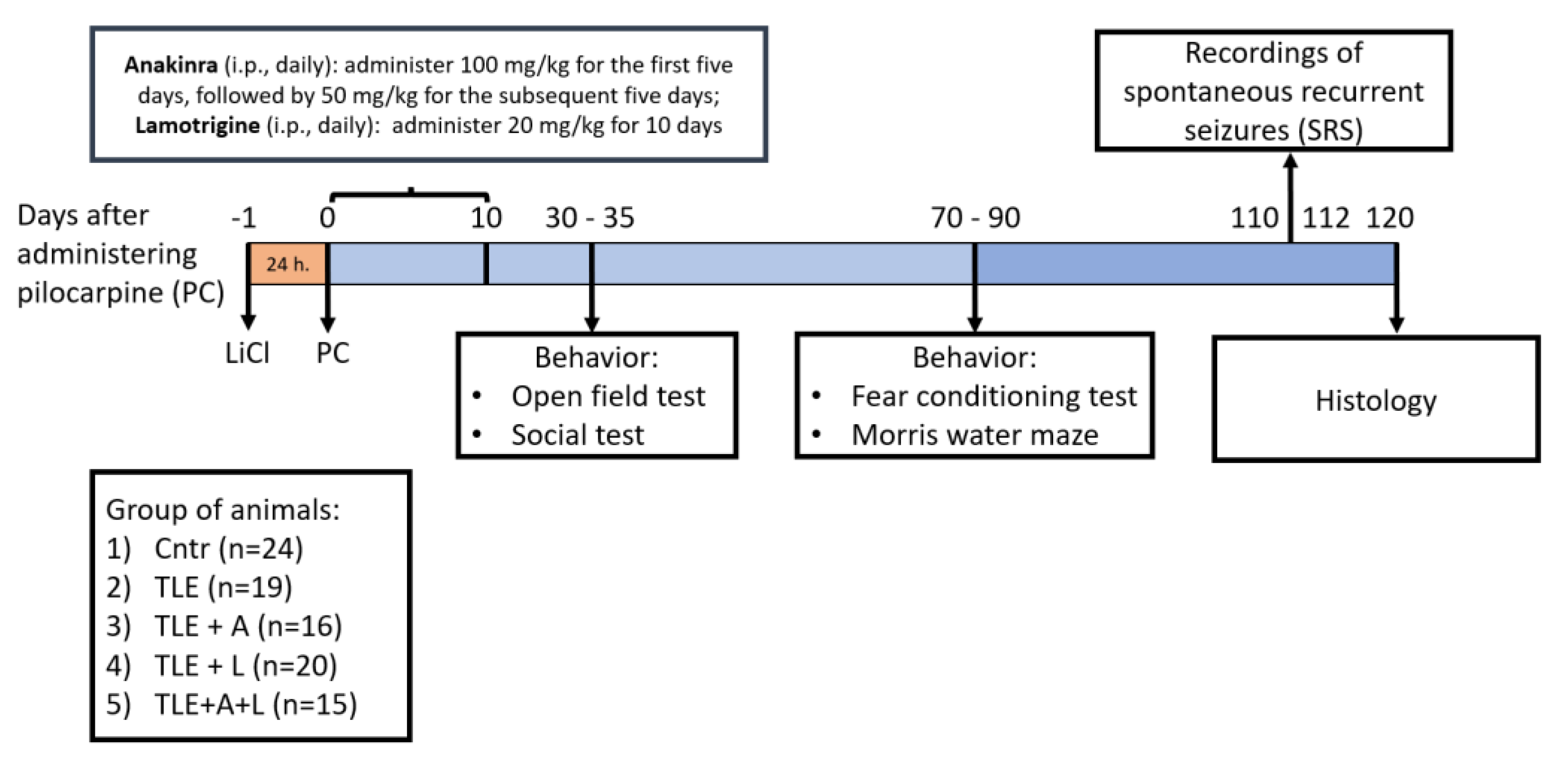
Figure 9.
Fear Conditioning Test Scheme.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



