
Submitted:
22 September 2023
Posted:
26 September 2023
You are already at the latest version
Abstract
New complexes of manganese(II) hexafluoroacetylacetonate [Mn(hfac)2] with 2-(1-R-3-pyrazol-4-yl)-4,4,5,5-tetramethyl-2-imidazoline-3-oxide-1-oxyl (R = CHF2, CH2CH2F, CH2CHF2 or CH2CF3) were synthesised and characterised structurally and magnetically. All complexes were prepared under similar conditions. Nonetheless, their crystal structures were considerably different. Depending on the structure of fluorinated alkyl substituent R, the complexation reaction led to complexes of three types: chain-polymeric complexes with the head-to-head or head-to-tail motif and complexes of molecular structure. All complexes show strong antiferromagnetic behaviour in a high-temperature region (150−300 K) and weak ferro- or antiferromagnetic exchange interactions at low temperatures. The stronger antiferromagnetic exchange, –101.7 ± 1.5 or –136 ± 10 cm–1, –82.3 ± 1.3 cm–1 and –87.4 ± 1.3 cm–1, was attributed to the magnetic interaction in three- or two-spin clusters: {>N∸O–Mn2+–O∸N<} or {>N∸O–Mn2+}, respectively. The weaker antiferromagnetic interaction, –0.005, between three-spin clusters or ferromagnetic interactions, 0.18–0.81 cm–1, between two-spin clusters are realised through the pyrazole ring or intermolecular contacts.
Keywords:
nitronyl nitroxides
; metal-nitroxide complexes
; crystal structures
; magnetic properties
1. Introduction
Nitroxide radicals have been proved to be promising ligands for the construction of molecular magnetic materials [1,2,3,4]. The ease of modification of nitronyl nitroxide radicals gives versatility to the design of various interesting systems of the metal–nitroxide type. Such systems have a large variety of topological structures and manifest intriguing bulk and molecular magnetic properties [5,6,7,8,9,10,11,12]. The direct linking of nitronyl nitroxides to metal ions results in the strongest possible magnetic coupling, which reduces quantum tunnelling of the magnetisation and gives long relaxation time [13,14]. So far, a number of molecular magnets based on metal–nitroxide complexes have been created, for example, cobalt-radical coordination magnets with high coercivity blocking temperature [15,16,17]. In addition, discrete complexes with defined geometric structure of paramagnetic metal ions and nitronyl nitroxide radical ligands are good candidates for basic research on magneto-structural correlations [18,19,20].
In a preceding paper, we investigated an influence of step-by-step fluorination of different paramagnetic systems on their structure and magnetism [21,22,23,24,25,26,27,28]. The motivation was that the introduction of fluorine atom(s) into molecules alters physical and chemical properties of the compounds because of fluorine’s electronegativity, its low polarisability and high bond strength [29]. In addition, there are many examples of the influence of substitution with fluorine on inter-molecular interactions and the use of ‘organic fluorine’ in crystal engineering or in systematic design of functional materials [30,31,32,33,34]. Many research groups have performed comprehensive analyses of crystal structures of fluorine-containing compounds and published excellent reviews on the specific role of fluorine in crystal packing [35,36]. In some reviews, fluorine has been poetically described as the odd man out [37], the little atom that could [29], the atom with many faces [38], and the chameleon of noncovalent interactions [39]. Although there is ample research on inter-molecular interactions of the C−F group in various types of compounds, this kind of interactions has not been studied in a systematic series of complexes of paramagnetic metal ions with radicals. Intuitively, it is expected that the introduction of fluorine into metal–radical systems can have a significant influence on their packing in the crystals thereby changing inter-molecular exchange interaction channels and eventually magnetic characteristics of molecular magnetic materials.
Herein, we used a series pyrazolyl-substituted nitronyl nitroxides bearing different fluorinated alkyl groups in the pyrazole nucleus, namely 2-(1-R-3-pyrazol-4-yl)-4,4,5,5-tetramethyl-2-imidazoline-3-oxide-1-oxyl, where R = CHF2, CH2CH2F, CH2CHF2 or CH2CF3. The complexation reaction of [Mn(hfac)2] with these radicals under identical conditions led to the formation of complexes of dimer structure, head-to-head chain-polymeric structure or head-to-tail chain-polymeric structure depending on the structure of the fluorinated alkyl substituent. Magnetic analyses showed that there is a strong antiferromagnetic interaction between the Mn(II) ion and the directly coordinated N–O group in the high-temperature region and weak ferro- or antiferromagnetic exchange interactions below 100 K.
2. Materials and Methods
2.1. General procedures and materials
2-(1-R-3-Pyrazol-4-yl)-4,4,5,5-tetramethyl-2-imidazoline-3-oxide-1-oxyls (LR), where R = CHF2, CH2CH2F, CH2CHF2 or CH2CF3, were synthesised according to methods from the literature [25]. All other chemicals were of reagent grade and used without purification. Toluene was distilled under an argon stream and kept in an argon atmosphere. Other solvents were of reagent quality and used without additional purification. The reactions were monitored by thin-layer chromatography on silica gel 60 F254 aluminium sheets from Merck. The chromatography was carried out using silica gel (0.050–0.160 mm) for column chromatography. The yields are given for pure substances obtained after recrystallisation. IR spectra were obtained from KBr pellets by means of a Bruker VECTOR 22 infrared spectrometer. Elemental analyses for C, H and N were carried out using Perkin-Elmer elemental analyzer model 240.
2.2. Synthesis of [Mn(hfac)2(NN-Pz-CHF2)]n
A solution of 42 mg (0.08 mmol) of Mn(hfac)2⋅2H2O in 5 mL of dry toluene was heated under reflux for 40 min. After that, the solution was cooled to the room temperature, and a solution of NN-Pz-CHF2 (22 mg, 0.08 mmol) in 1 mL of toluene was added. The resulting mixture was allowed to stand at –15 °C for a week to obtain blue blocks of crystals (30.9 mg, 57.5%). Anal. calcd. for C21H17F14MnN4O6: C, 33.98; H, 2.31; N, 7.55. Found: C, 33.99; H, 2.20; N, 7.57%. IR (KBr, cm–1): 3149(m), 2997(m), 2947(w), 1645(s), 1611(m), 1562(m), 1536(m), 1501(s), 1488(s), 1466(m), 1407(m), 1395(m), 1381(m), 1348(s), 1328(s), 1260(s), 1223(s), 1142(s), 1099(s), 1020(m), 1002(w), 965(w), 946(w), 892(m), 869(m), 838(s), 807(s), 766(w), 742(m), 697(w), 666(s), 609(w), 585(m), 542(m), 528(w), 483(w),470(w), 419(w).
2.3. Synthesis of [Mn(hfac)2(NN-Pz-CH2CH2F)]n
A solution of 42 mg (0.08 mmol) of Mn(hfac)2⋅2H2O in 5 mL of dry toluene was heated under reflux for 40 min. Next, the solution was cooled to room temperature, and a solution of NN-Pz-CH2CH2F (22 mg, 0.08 mmol) in 1 mL of toluene was introduced. The resulting mixture was kept at –15 °C for several days to obtain blue blocks of crystals (32.2 mg, 52.4% based on Mn ion). Anal. calcd. for C22H20F13MnN4O6: C, 35.79; H, 2.73; N, 7.59. Found: C, 35.52; H, 2.71; N, 7.81%. IR (KBr, cm–1): 3153(m), 2996(w), 1649(s), 1610(w), 1556(w), 1530(m), 1500(m), 1350(m), 1301(w), 1257(s), 1206(s), 1146(s), 1097(w), 1039(w), 1019(w), 884(w), 863(w), 798(m), 758(w), 741(w), 665(m), 584(m), 544(w), 527(w), 507(w), 464(w), 435(w).
2.4. Synthesis of [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8 and [Mn(hfac)2(NN-Pz-CH2CHF2)]n
A solution of 42 mg (0.08 mmol) of Mn(hfac)2⋅2H2O in 5 mL of dry toluene was heated under reflux for 40 min. Then, the solution was cooled to room temperature, and a solution of NN-Pz-CH2CHF2 23 mg (0.08 mmol) in 1 mL of toluene was added. The resultant mixture was incubated at –15 °C for ~20 h to obtain blue blocks of crystals of solvate complex [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8. The crystals were filtered off and dried on air. Yield 40.9 mg (60.2%). The complex [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8 was stored for a month in vial at ambient condition that led to its desolvation. Anal. calcd. for C22H19F14MnN4O6: C, 34.94; H, 2.53; N, 7.41. Found: C, 34.78; H, 2.60; N, 8.18%. IR (KBr, cm–1): 3152(m), 2996(w), 2948(w), 1646(s), 1611(m), 1559(m), 1532(s), 1501(s), 1401(w), 1353(s), 1373 sh, 1320(w), 1303(w), 1259(s), 1214(s), 1192 sh, 1144(s), 1090(s), 1069(m), 1019(w), 1007(w), 946(w), 886(w), 867(w), 798(m), 742(w), 666(s), 642(w), 605(w), 585(m), 558(w), 544(w), 527(w), 484(w), 469(w), 436(w).
2.5. Synthesis of [Mn(hfac)2(NN-Pz-CH2CF3)]2⋅2C7H8
A solution of 34 mg (0.07 mmol) of Mn(hfac)2⋅2H2O in 5 mL of dry toluene was heated under reflux for 40 min. After that, the solution was cooled to room temperature, and a solution of NN-Pz-CH2CF3 22 mg (0.07 mmol) in 3 mL of toluene was added. This mixture was allowed to stand at –15 °C for ~24 h to obtain blue blocks of crystals (44.5 mg, 76.3%). Anal. calcd. for C44H36F30Mn2N8O12⋅2(C7H8): C, 40.20; H, 3.02; N, 6.47. Found: C, 40.12; H, 3.01; N, 6.31%. IR (KBr, cm–1): 3153(m), 3024(w), 2996(w), 2948(w), 1647(s), 1610(m), 1558(m), 1532(m), 1501(s), 1481 sh, 1438 sh, 1400(w), 1356(m), 1315(m), 1257(s), 1197(s), 1146(s), 1096 sh, 1020(m), 933(m), 889(w), 868(w), 841(w), 800(m), 766(w), 742(w), 664(m), 584(m), 542(w), 469(w), 433(w).
2.6. X-ray Crystallography Details
X-ray diffraction (XRD) data on monocrystals of [Mn(hfac)2(NN-Pz-CHF2)]n, [Mn(hfac)2(NN-Pz-CH2CH2F)]n, [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8 and [Mn(hfac)2(NN-Pz-CH2CF3)]2⋅2C7H8 were collected at 100 K on a four-circle Rigaku Synergy S diffractometer equipped with a HyPix6000HE area-detector (kappa geometry, shutterless ω-scan technique) using mono-chromatised Cu Kα-radiation. The intensity data were integrated and corrected for absorption and decay in the CrysAlisPro software [40]. The structures were solved by direct methods using SHELXT [41] and refined on F2 with the help of SHELXL-2018 [42] in OLEX2 [43]. All non-hydrogen atoms were refined with individual anisotropic displacement parameters. All hydrogen atoms were placed in ideal calculated positions and refined as riding atoms with relative isotropic displacement parameters. A rotating group model was applied to methyl groups.
XRD data on monocrystals of [Mn(hfac)2(NN-Pz-CH2CHF2)]n and [Mn(hfac)2(NN-Pz-CH2CHF2)]nn[Mn(hfac)2(NN-Pz-CH2CHF2)H2O]) were collected at 100 K on a Bruker D8 QUEST diffractometer. Single-crystal X-ray analyses were carried out in the APEX3 software [44]. The collected data were then integrated in SAINT. SADABS was used for scaling, empirical absorption corrections and for generating data files for structure solution and refinement.
CCDC 2272057 (for [Mn(hfac)2(NN-Pz-CHF2)]n), 2272056 (for [Mn(hfac)2(NN-Pz-CH2CH2F)]n), 2239233 (for [Mn(hfac)2(NN-Pz-CH2CHF2)]n), 2239234 for [Mn(hfac)2(NN-Pz-CH2CHF2)]nn[Mn(hfac)2(NN-Pz-CH2CHF2)H2O]), 2272055 [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8 and 2272058 (for [Mn(hfac)2(NN-Pz-CH2CF3)]2⋅2C7H8) contain the supplementary crystallographic data for the manganese–nitroxide complexes.
2.7. Magnetic measurements
The variable-temperature magnetic-susceptibility measurements were carried out in the temperature range 2–300 K with a Quantum Design PPMS-9 or Quantum Design MPMSXL SQUID magnetometer. None of the complexes showed any field dependence of molar magnetic susceptibility at low temperatures. The molar magnetic susceptibility was corrected for the sample holder and diamagnetic contributions of all constituent atoms by means of Pascal’s constants [45].
3. Results and discussion
3.1. Synthesis and crystal structures of manganese–nitroxide complexes
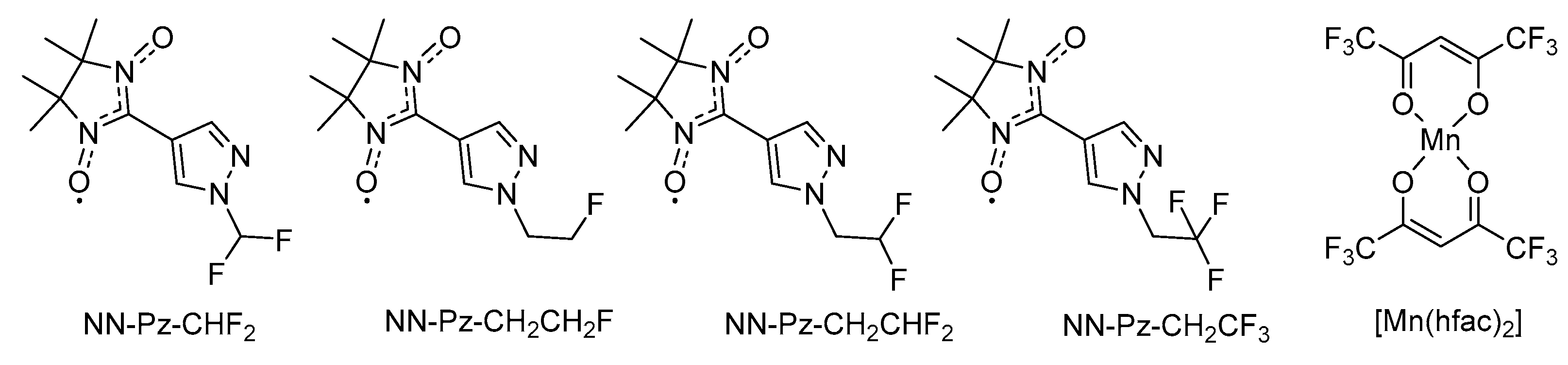
Complexes [Mn(hfac)2(NN-Pz-R)]n were prepared by interaction of manganese(II) hexafluoroacetylacetonate ([Mn(hfac)2]) with nitronyl nitroxides (NNs), namely 2-(1-R-3-pyrazol-4-yl)-4,4,5,5-tetramethyl-2-imidazoline-3-oxide-1-oxyls (R = CHF2, CH2CH2F, CH2CHF2 or CH2CF3), in dry toluene at –15 °C. The single-crystal X-ray analysis revealed that compound [Mn(hfac)2(NN-Pz-CHF2)]n crystallises in orthorhombic space group I2/a. The structure of [Mn(hfac)2(NN-Pz-CHF2)]n consists of 1D coordination chains shown in Figure 1a. One can see that the paramagnetic NN-Pz-CHF2 is a bridging bidentate ligand coordinated via the oxygen atom of one of the N–O groups and the nitrogen atom of the pyrazole ring. The coordination chains have a ‘head-to-head’ motif, and the nitroxide oxygen atoms and the nitrogen atom are coordinated to Mn in a trans-configuration. Each manganese ion is sixfold coordinated and has almost isometric octahedral geometry. For the MnO6 unit, the Mn–ONO bond lengths are 2.143 Å, whereas the average Mn–Ohfac bond length is 2.139 Å. These values are close to those observed for other complexes of [Mn(hfac)2] with nitronyl nitroxides [46,47]. For the MnO4N2 unit, the Mn–N bond lengths are 2.297 Å, whereas the Mn–Ohfac bond lengths are 2.132 and 2.138 Å. In the coordinated nitroxide group, the bond length [1.303(2) Å] is slightly elongated as compared with the non-coordinated one [1.273(2) Å]. The shortest inter-chain distances between O atoms of the nitroxide groups exceed 5 Å (the sum of van der Waals radii of O atoms is 3.04 Å); this arrangement allows one to expect only very weak exchange interactions between the chains.
Figure 1b shows a schematic diagram of possible magnetic interactions within the manganese–nitroxide chains; the direct coordination bonding of two O atoms to Mn(II) should give the largest exchange interaction, which is denoted as J1, whereas magnetic couplings between Mn(II) ions and the nitroxide group via the pyrazole ring (denoted as J2) should be weaker than J1. The sign and magnitude of these interactions are discussed in the Magnetic Properties section.
Complex [Mn(hfac)2(NN-Pz-CH2CH2F)]n was found to crystallise in monoclinic space group P21/c. The structure of [Mn(hfac)2(NN-Pz-CH2CH2F)]n also consists of 1D coordination chains (Figure 2) formed by bridging bidentate ligand coordination via the nitroxide O atom and the N atom of the pyrazole ring. As opposed to [Mn(hfac)2(NN-Pz-CHF2)]n, the coordination chains have a head-to-tail motif, and the nitroxide O atom and N atom are coordinated to Mn in a cis-configuration. The coordination polyhedron of manganese is nearly isometric. The Mn–ONO bond length is 2.145(2) Å, the Mn–N bond length is 2.236(2) Å, whereas the Mn–Ohfac bond lengths are 2.130(2), 2.144(2), 2.172(2) and 2.176(2) Å. In the coordinated nitroxide group, the bond length [1.305(3) Å] is slightly elongated as compared with the non-coordinated one [1.278(3) Å]. The shortest inter-chain distance between the oxygen atoms of nitroxide groups is 4.266 Å, which is also considerably greater than the sum of van der Waals radii of O atoms (3.04 Å).
The interaction of [Mn(hfac)2] with NN-Pz-CH2CHF2 in dry toluene at –15 °C gave rise to complex [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8 in the form of a solvate with toluene. Complex [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8 was found to crystallise in the same monoclinic space group, P21/n. The structure of [Mn(hfac)2(NN-Pz-CH2CHF2)]n also consists of 1D coordination chains (Figure 3) with the head-to-tail motif and cis-configuration of the Mn surroundings. The coordination of the manganese polyhedron is nearly isometric: the Mn–ONO bond length is 2.138(3) Å, the Mn–N bond length is 2.245(3) Å whereas the Mn–Ohfac bond lengths lie in the range 2.126(2)–2.169(2) Å. In the coordinated nitroxide group, the bond length [1.309(4) Å] is slightly elongated as compared with the non-coordinated one [1.274(4) Å]. The shortest inter-chain distances between the oxygen atoms of nitroxide groups exceed 5.0 Å. After storage under ambient conditions, the complex [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8 loses the solvate molecules while retaining the quality of the crystals. According to XRD data, crystal structure of desolated complex [Mn(hfac)2(NN-Pz-CH2CHF2)]n belongs to space group P21/c. In [Mn(hfac)2(NN-Pz-CH2CHF2)]n, geometric parameters are almost the same as in its solvate with toluene, for example, the Mn–ONO bond length is 2.147(2) Å, and the Mn–N bond length is 2.247(2) Å, whereas the Mn–Ohfac bond lengths lie in the range 2.126(2)–2.178(2) Å. N–O bond lengths are 1.310(3) and 1.277(3) Å for coordinated and non-coordinated nitroxide groups, respectively (Figure S1).
In accordance with the structure of complexes [Mn(hfac)2(NN-Pz-CH2CH2F)]n and [Mn(hfac)2(NN-Pz-CH2CHF2)]n, there are two kinds of magnetic interactions in the chains: Mn(II)–nitroxide direct magnetic interactions, J1, and Mn(II)–nitroxide interactions through the pyrazole ring: J2 (Figure 3b). It is known that nitroxide radicals directly bound to Mn(II) centres through the N–O group always exhibit much stronger magnetic coupling than that through a donor atom of the nitronyl nitroxide substituent [48,49,50]. Therefore, from a magnetic point of view, the coordination chain can be described as strongly coupled two-spin clusters with relatively weak exchange between them: J2. The quantification of these magnetic interactions is discussed in the Magnetic Properties section.
The reaction of Mn(hfac)2 with NN-Pz-CH2CF3 led to the formation solvate complex [Mn(hfac)2(NN-Pz-CH2CF3)]2⋅2C7H8 having structure of a cyclic dimer (Figure 4). Crystal structure of the complex belongs to the triclinic P-1 space group. The structure of the complex results from the bridging bidentate ligand coordination via the nitroxide O atom and the N atom of the pyrazole ring. The Mn–ONO bond length is 2.110(2) Å, and the Mn–N bond length is 2.278(2) Å, which are comparable to those observed in previously reported cyclic metal–nitroxide dimers [51,52,53]. In the coordinated nitroxide group, the bond length is also slightly elongated [1.303(2) Å] as compared with the non-coordinated one [1.272(3) Å]. The Mn···Mn distance in the intra-dimer is 6.248 Å, which is shorter than the shortest inter-dimer Mn···Mn distance of 8.984 Å. The shortest distances between the uncoordinated NO groups is 4.886 Å. The packing diagram for [Mn(hfac)2(NN-Pz-CH2CF3)]2⋅2C7H8 is shown in Figure S2. The cyclic dimers are arranged parallel to each other along the a-axis thereby forming stacks, between which the shortest contacts that are established by oxygen atoms (O1) of the uncoordinated nitroxide groups and hydrogen atoms (H6C) of methyl groups are 2.532 Å. It is also noteworthy that unlike complex [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8, solvate complex [Mn(hfac)2(NN-Pz-CH2CF3)]2⋅2C7H8 is stable under ambient conditions and does not lose the solvent molecules.
In [Mn(hfac)2(NN-Pz-CH2CF3)]2, there are mainly two kinds of magnetic interactions for the present four-spin magnetic system, i.e. the magnetic interaction between the Mn(II) ion and the directly coordinated nitroxide group (J1) and the magnetic coupling between the Mn(II) ion and nitroxide group through the pyrazole rings (J2) (Figure 4b). The second kind of magnetic coupling will be shown below to be weak and ferromagnetic (see the Magnetic Properties section).
The experimental powder XRD patterns of all complexes matched well the simulated XRD patterns based on the structures refined by single-crystal XRD analysis. In addition, elemental analyses yielded satisfactory results for all manganese–nitroxide complexes. The preparation of the complexes and their characterisation were repeated at least three times; the results (yields, crystal structures) were reproducible.
Notably, the reaction of [Mn(hfac)2] with NN-Pz-RF under the same conditions produced complexes of different types: a chain-polymeric complex with a head-to-head motif or head-to-tail motif as well as molecular complex. The reason is a fine influence of fluorinated alkyl substituents R in NN-Pz on a set of equilibrium constants predetermining concentrations of different species and their solubility (Scheme 1). In NN-Pz-CH2CF3, the fluoroalkyl substituent somewhat reduces electron density on the donor nitrogen atom, and this effect obviously should favour the formation of a soluble Pz-NN−{Mn} complex with a coordinated NO group. If we assume that the acceptor ability of the manganese atom in Pz-NN−{Mn} is not sufficient to bind one more ligand, then the formation of a cyclic dimer with pairwise coordination bonds becomes preferable and therefore leads to precipitation of [Mn(hfac)2(NN-Pz-CH2CF3)]2. Incidentally, the reasons may be the same for the frequent formation of such dimers during the interaction of [Mn(hfac)2] with hetaryl-substituted nitronyl nitroxides [54,55,56,57,58,59].
On the contrary, only one case is known when the reaction of [Mn(hfac)2] with a hetaryl-substituted nitronyl nitroxide produces a chain-polymeric complex, moreover, having a head-to-head motif [60]. In our case, the [Mn(hfac)2(NN-Pz-CHF2)]n complex has similar structure, and its formation can be explained by the influence of the CHF2 substituent, which reduces electron density on the donor nitrogen atom to such an extent that the formation of the corresponding dimer becomes thermodynamically unfavourable. Therefore, the process goes further along the path of coordination of another radical by the manganese ion through the NO group with the formation of a three-spin molecule: Pz-NN−{Mn}−NN-Pz. The interaction of the latter with the [Mn(hfac)2] acceptor matrix eventually gives a poorly soluble chain-polymeric complex with a head-to-head motif (Scheme 1). Incidentally, one can notice an interesting detail: only in this case does the complex precipitate into a solid phase very slowly (for approximately a week), whereas solid phases of the other complexes described here form much faster, within several hours. This finding indirectly indicates that the concentration of the Pz-NN−{Mn}−NN-Pz form that is necessary for the assembly of the solid phase of the [Mn(hfac)2(NN-Pz-CHF2)]n complex is too low in the solution.
The structure of the two remaining complexes, [Mn(hfac)2(NN-Pz-CH2CH2F)]n and [Mn(hfac)2(NN-Pz-CH2CHF2)]n, is unprecedented. These are the first examples of manganese–nitroxide chain-polymeric complexes with a head-to-tail motif in which hetaryl-substituted nitronyl nitroxides serve as bidentate-bridging ligands. Their formation can be explained as follows: a decrease in electron acceptor properties of substituent R possibly enhances the donor ability of the nitrogen atom of the paramagnetic ligand to such an extent that the NN-Pz−{Mn} complex becomes preferable in solution, thereby causing crystallisation of the chain head-to-tail complexes (Scheme 1).
The effect of RF substituents on the course of the complexation reactions can also be observed visually. As mentioned above, the [Mn(hfac)2(NN-Pz-CHF2)]n complex precipitates very slowly, and after 24 h, one can see the emergence of a small amount of the product (Figure 5). In case of [Mn(hfac)2(NN-Pz-CH2CH2F)]n, after 24 h under the same conditions, a gel-like product initially arises, retaining the entire mother liquor. This product, upon further incubation of the reaction mixture for several days, transitions to a crystalline [Mn(hfac)2(NN-Pz-CH2CH2F)]n complex. The two latter complexes, [Mn(hfac)2(NN-Pz-CH2CH2F)]n and [Mn(hfac)2(NN-Pz-CH2CHF2)]n, form faster and after 24 h almost completely precipitate with the emergence of crystalline phases.
Thus, the fluorinated substituents in NN-Pz have a substantial effect on the structure of heterospin manganese–nitroxide complexes. It is noteworthy that their solid phases have different sets of exchange channels, and therefore a difference in magnetic behaviour is expected.
3.2. Magnetic properties
The temperature dependence of effective magnetic moment μeff for [Mn(hfac)2(NN-Pz-CHF2)]n is shown in Figure 6. The μeff value at 300 K is 5.17 μB and slightly decreases to reach a plateau of 4.99 μB in the temperature range 150–10 K. The observed μeff values in the temperature range 10–300 K are considerably less than the theoretical spin-only value of 6.16 μB for the non-interacting spin system of Mn2+ and nitroxide based on the {Mn(hfac)2(NN-Pz-CHF2)} moiety. It is reasonable to explain the observed magnetic behaviour of [Mn(hfac)2(NN-Pz-CHF2)]n by the strong antiferromagnetic interactions in {>N∸O–Mn2+–O∸N<} three-spin exchange clusters, in which the spins of the coordinated N∸O groups partially compensate the spin of the Mn2+ ion (SMn = 5/2). In the range from 150 to 10 K, the μeff values are close to theoretical value μteor = (0.5⋅15 + 0.5⋅35)1/2 = 5 μB, taking into account that the magnetic susceptibility contains contributions only from residual moments of the three-spin exchange clusters having a quartet ground state (S = 3/2) and from the moments of the Mn2+ ions located in MnO4N2 coordination units (SMn = 5/2). The decline of μeff below 10 K down to 4.69 μB at 2 K is explained by the weak inter-chain antiferromagnetic interactions (Figure 1b). Analysis of the experimental μeff(T) dependence, using a trimer model for the {>N∸O–Mn2+–O∸N<} exchange cluster (spin-Hamiltonian H = –2J(SR1SMn + SMnSR2)) while taking into account magnetic susceptibility of the Mn2+ ions located in MnO4N2 coordination units according to Curie law, allows to estimate exchange interaction energy. The best-fit values of g-factors and exchange interaction parameter J are 2.01 ± 0.01 and –101.7 ± 1.5 cm–1, respectively. The weaker exchange interactions, Jn, are approximated as mean-field parameterθ thus giving –0.005 ± 0.001 cm–1.
For complexes [Mn(hfac)2(NN-Pz-CH2CH2F)]n and [Mn(hfac)2(NN-Pz-CH2CHF2)]n with a head-to-tail motif of chains, the μeff(T) dependences are similar (Figure 7). The μeff values are 5.34 and 5.37 μB and slightly decrease with diminishing temperature. The μeff values in the temperature range 300–50 K are close to the theoretical spin-only value of 4.90 μB for one paramagnetic centre with S = 2, indicating realisation of strong antiferromagnetic exchange interactions in {Mn2+–O∸N<} exchange clusters. Below 50 K, μeff values drop rapidly, which is caused by the weak inter-cluster interactions within the chains. Analysis of the experimental μeff(T) dependences using two-spin model (spin-Hamiltonian H = –2JSMnSR) as reported in ref. [61] enabled us to estimate exchange interaction energy in {Mn2+–O∸N<} exchange clusters. The best-fit values of g-factors and exchange interaction parameter J1 (see Figure 3b) are 2.029 ± 0.003 and –136 ± 10 cm–1 for [Mn(hfac)2(NN-Pz-CH2CH2F)]n, and 2.041 ± 0.002 and –82.3 ± 1.3 cm–1, respectively, for [Mn(hfac)2(NN-Pz-CH2CHF2)]n. The weak exchange interactions between paramagnetic centers are approximated as mean-field parameterθ thus giving 0.78 ± 0.01 cm–1 and 0.18 ± 0.01 cm–1, respectively. The small positive value of θ may be attributed with FM exchange between the Mn(II) ion and the nitronyl nitroxide moiety through the pyrazole ring.
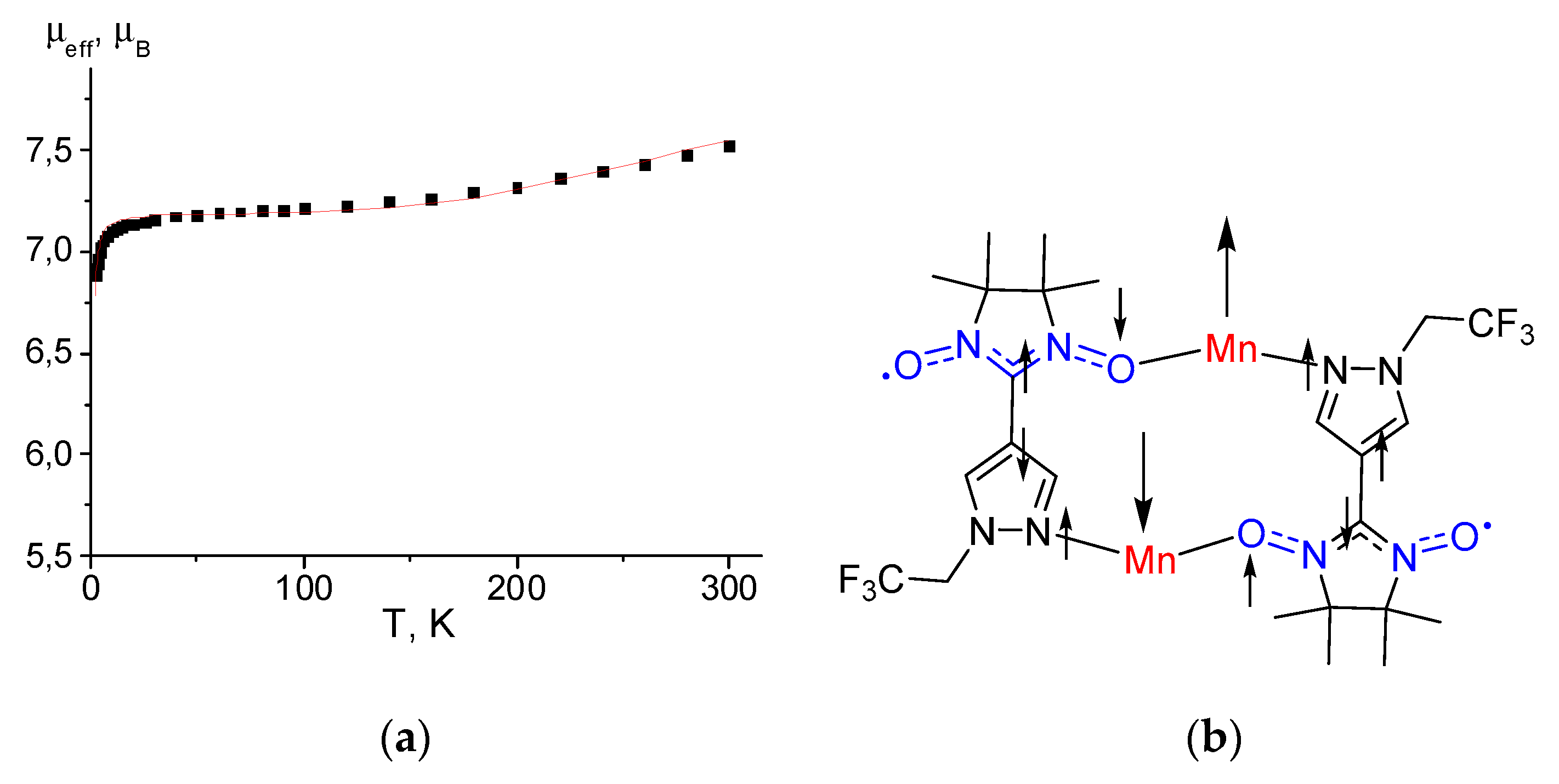
For cyclic dimer [Mn(hfac)2(NN-Pz-CH2CF3)]2, the temperature dependence of μeff is presented in Figure 8a. The room temperature μeff value is approximately 7.52 μB. As the temperature is lowered, μeff slightly decreases, reaching a plateau of 7.20 μB at 100 K and then drops rapidly at temperatures below 15 K. In the temperature range of plateau 30–100 K, the observed μeff values are considerably less than the theoretical spin-only value (8.72 μB) for the non-interacting spin system of two Mn2+ ions and two nitroxide ligands based on the unit with the [Mn(hfac)2(NN-Pz-CH2CF3)]2 formula. As in the previous complexes, the observed magnetic behaviour of [Mn(hfac)2(NN-Pz-CH2CF3)]2 can be explained by the strong antiferromagnetic interactions in {Mn2+–O∸N<} exchange clusters, in which the spins of the coordinated N∸O groups are completely coupled to the two spins of Mn2+. Therefore, the magnetic susceptibility has contributions only from the residual moments of two exchange clusters (S = 2) (the theoretical spin-only magnetic moment of μteor = 6.93 μB). The further decrease in μeff at T < 10 can be attributed to the weak interactions between two-spin {Mn2+–O∸N<} exchange clusters. Analysis of the experimental μeff(T) dependences with a tetramer model using spin-Hamiltonian H = –2J1(SMn1SR1 + SMn2SR2) – 2J2(SMn1SR2 + SMn2SR1) as reported in refs. [58,62] gives the best fit values of g-factors and exchange interaction parameters J1 and J2: 2.0036 ± 0.002, –87.4 ± 1.3 cm–1 and 0.81 ± 0.01 cm–1, respectively. Parameter J1 corresponds to strong antiferromagnetic exchange in the {Mn2+–O∸N<} moieties, and J2 corresponds to weak interactions between spins of the Mn2+ ion and the nitroxide coordinated via the N atom.
The strong antiferromagnetic interaction can be attributed to the effective overlap between the π-SOMO orbital containing the unpaired electrons of the nitronyl nitroxide moiety and d orbitals of the Mn(II) ion [63]. The experimental J1 value in [Mn(hfac)2(NN-Pz-CH2CF3)]2 has the same order of magnitude as that observed in different dimeric complexes (Table S2). The observed difference in antiferromagnetic coupling (from –74 to –208 cm–1) may be ascribed to the different coordination geometry of the manganese–nitroxide spin cluster affecting the overlap of the magnetic orbitals. The small positive J2 value shows that a weak ferromagnetic coupling exists between the two {Mn–O-N} moieties and is mediated by the pyrazole rings (Figure 8b) thus giving rise to the non-magnetic ground spin state. This weak ferromagnetic interaction can be explained by the spin polarization mechanism.
4. Conclusions
In summary, the complexation reaction of [Mn(hfac)2] with pyrazolyl-substituted nitronyl nitroxides bearing various fluorinated alkyl groups (–CHF2, –CH2CH2F, –CH2CHF2 or –CH2CF3) in the pyrazole core was investigated. The most interesting result of this investigation is that depending on the structure of the fluorinated alkyl substituent, the complexation reaction of [Mn(hfac)2] with these radicals under identical conditions affords complexes of head-to-head chain-polymeric structure, head-to-tail chain-polymeric structure or molecular structure. In all complexes, nitronyl nitroxide ligands act as a bridging ligand linking Mn(II) ions through the O atom of the nitroxide group and the N atom of the pyrazole ring, thereby creating three-spin {>N∸O–Mn2+–O∸N<} or two-spin {Mn2+–O∸N<} exchange clusters. According to magnetic measurements, in all the clusters, the Mn2+ ion strongly interacts antiferromagnetically with the coordinated nitroxide group. Our investigation shows that the structure of the metal–nitroxide complexes can be modulated by the step-by-step fluorination of the radical ligand thus making it possible to obtain coordination compounds with a previously unknown motif. Further studies on magnetostructural correlations of related organic and inorganic composite systems are in progress.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: ORTEP drawings of a fragment of structure in co–crystal [Mn(hfac)2(NN–Pz–CH2CHF2)]n⋅n[Mn(hfac)2(NN–Pz–CH2CHF2)H2O]; Figure S2: The packing diagram for [Mn(hfac)2(NN–Pz–CH2CF3)]2⋅2C7H8; Figure S3: Experimental and calculated powder X-ray diffraction pattern of [Mn(hfac)2(NN-Pz-CHF2)]n; Figure S4: Experimental and calculated powder X-ray diffraction pattern of [Mn(hfac)2(NN-Pz-CH2CH2F)]n; Figure S5: Experimental and calculated powder X-ray diffraction pattern of [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8; Figure S6: Experimental and calculated powder X-ray diffraction pattern of [Mn(hfac)2(NN-Pz-CH2CHF2)]n; Figure S7: Experimental and calculated powder X-ray diffraction pattern of [Mn(hfac)2(NN-Pz-CH2CF3)]2⋅2C7H8. Table S1: Selected geometric parameters and experimental J1 values in the [Mn(hfac)2(NN–Pz–CH2CF3)]2 dimer and cousin dimeric complexes of Mn(hfac)2 with nitronyl nitroxides; Table S2: Selected bond distances in [Mn(hfac)2(NN–Pz–CHF2)]n; Table S3: Selected bond distances in [Mn(hfac)2(NN–Pz–CH2CH2F)]n; Table S4: Selected bond distances in [Mn(hfac)2(NN–Pz–CH2CHF2)]n; Table S5: Selected bond distances in [Mn(hfac)2(NN–Pz–CH2CHF2)]n⋅nC7H8; Table S6: Selected bond distances in [Mn(hfac)2(NN–Pz–CH2CF3)]2⋅2C7H8; Tables S7 and S8: XRD data on the manganese–nitroxide complexes. References [64,65,66] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, E.T.; methodology, B.U. and N.E.; validation, N.E. and A.B.; formal analysis, A.K. and A.B.; investigation, E.K., A.S., B.U, T.D., D.N., A.K., M.Z., N.E. and A.B.; writing—original draft preparation, E.T.; writing—review and editing, E.T.; supervision, N.E. and E.T.; project administration, E.T. All authors have read and agreed to the published version of the manuscript.
Funding
The authors thank the Russian Science Foundation for funding (project No. 21-73-20079).
Data Availability Statement
The data that support the findings of this study are available upon reasonable request from the authors.
Acknowledgments
The crystal structures of [Mn(hfac)2(NN-Pz-CHF2)]n, [Mn(hfac)2(NN-Pz-CH2CH2F)]n, [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8 and [Mn(hfac)2(NN-Pz-CH2CF3)]2⋅C7H8 were solved in the Department of Structural Studies at N. D. Zelinsky Institute of Organic Chemistry, Moscow. The crystal structure of the [Mn(hfac)2(NN-Pz-CH2CHF2)]n complex was solved at the Center for Molecular Composition Studies at INEOS RAS (contract No. 075-03-2023-642 with the Ministry of Science and Higher Education of the Russian Federation).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Benelli, C.; Gatteschi, D. Magnetism of Lanthanides in Molecular Materials with Transition-Metal Ions and Organic Radicals. Chem. Rev. 2002, 102, 2369–2388. [Google Scholar] [CrossRef] [PubMed]
- Tretyakov, E.V.; Ovcharenko, V.I.; Terent'ev, A.O.; Krylov, I.B.; Magdesieva, T.V.; Mazhukin, D.G.; Gritsan, N.P. Conjugated nitroxides. Russ. Chem. Rev. 2022, 91, RCR5025. [Google Scholar] [CrossRef]
- Ovcharenko, V. Metal-Nitroxide Complexes: Synthesis and Magnetostructural Correlations. In Stable Radicals; Hicks, R.G., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2010; pp. 461–506. [Google Scholar] [CrossRef]
- Ferrando-Soria, J.; Vallejo, J.; Castellano, M.; Martínez-Lillo, J.; Pardo, E.; Cano, J.; Castro, I.; Lloret, F.; Ruiz-García, R.; Julve, M. Molecular magnetism, quo vadis? A historical perspective from a coordination chemist viewpoint. Coord. Chem. Rev. 2017, 339, 17–103. [Google Scholar] [CrossRef]
- Bernot, K.; Pointillart, F.; Rosa, P.; Etienne, M.; Sessoli, R.; Gattesch, D. Single molecule magnet behaviour in robust dysprosium–biradical complexes. Chem. Commun. 2010, 35, 6458–6460. [Google Scholar] [CrossRef]
- Wang, J.; Miao, H.; Xiao, Z.X.; Zhou, Y.; Deng, L.D.; Zhang, Y.Q.; Wang, X.Y. Syntheses, structures and magnetic properties of the lanthanide complexes of the pyrimidyl-substituted nitronyl nitroxide radical. Dalton Trans. 2017, 31, 10452–10461. [Google Scholar] [CrossRef]
- Kadilenko, E.M.; Gritsan, N.P.; Tretyakov, E.V.; Fokin, S.V.; Romanenko, G.V.; Bogomyakov, A.S.; Gorbunov, D.E.; Schollmeyer, D.; Baumgarten, M.; Ovcharenko, V.I. A black-box approach to the construction of metal-radical multispin systems and analysis of their magnetic properties. Dalton Trans. 2020, 49, 16916–16927. [Google Scholar] [CrossRef]
- Sun, J.; Xi, L.; Xie, J.; Wang, K.; Li, L.C.; Sutter, J.P. A loop chain and a three-dimensional network assembled from a multi-dentate nitronyl nitroxide radical and M(hfac)2 (M = CoII, CuII). Dalton Trans. 2018, 47, 14630–14635. [Google Scholar] [CrossRef]
- Li, H.D.; Sun, J.; Yang, M.; Sun, Z.; Xie, J.; Ma, Y.; Li, L.C. Functionalized nitronyl nitroxide biradical bridged one-dimensional lanthanide chains: slow magnetic relaxation in the Tb and Dy analogues. New J. Chem. 2017, 41, 10181–10188. [Google Scholar] [CrossRef]
- Haraguchi, M.; Tretyakov, E.; Gritsan, N.; Romanenko, G.; Gorbunov, D.; Bogomyakov, A.; Maryunina, K.; Suzuki, S.; Kozaki, M.; Shiomi, D.; et al. (Azulene-1,3-diyl)-bis(nitronyl nitroxide) and (Azulene-1,3-diyl)-bis(iminonitroxide) and Their Copper Complexes. Chem Asian J. 2017, 12, 2929–2941. [Google Scholar] [CrossRef]
- Barskaya, I.Y.; Veber, S.L.; Suturina, E.A.; Sherin, P.S.; Maryunina, K.Y.; Artiukhova, N.A.; Tretyakov, E.V.; Sagdeev, R.Z.; Ovcharenko, V.I.; Gritsan, N.P.; et al. Spin-state-correlated optical properties of copper(II)–nitroxide based molecular magnets. Dalton Trans. 2017, 46, 13108–13117. [Google Scholar] [CrossRef]
- Kaszub, W.; Marino, A.; Lorenc, M.; Collet, E.; Bagryanskaya, E.G.; Tretyakov, E.V.; Ovcharenko, V.I.; Fedin, M.V. Ultrafast Photoswitching in a Copper-Nitroxide-Based Molecular Magnet. Angew. Chem., Intern. Ed. 2014, 53, 10636–10640. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.D.; Fang, M.; Evans, W.J.; Long, J.R. A N23– Radical-Bridged Terbium Complex Exhibiting Magnetic Hysteresis at 14 K. J. Am. Chem. Soc. 2011, 133, 14236–14239. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.D.; Fang, M.; Evans, W.J.; Long, J.R. Strong exchange and magnetic blocking in N23−-radical-bridged lanthanide complexes. Nat. Chem. 2011, 3, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Novitchi, G.; Shova, S.; Train, C. Investigation by Chemical Substitution within 2p-3d-4f Clusters of the Cobalt(II) Role in the Magnetic Behavior of [vdCoLn]2 (vd = Verdazyl Radical). Inorg. Chem. 2022, 61, 17037–17048. [Google Scholar] [CrossRef]
- Ishii, N.; Okamura, Y.; Chiba, S.; Nogami, T.; Ishida, T. Giant coercivity in a one-dimensional cobalt-radical coordination magnet. J. Am. Chem. Soc. 2008, 130, 24–25. [Google Scholar] [CrossRef]
- Maria, G.F.V.; Cassaro, R.A.A.; Akpinar, H.; Schlueter, J.A.; Lahti, P.M.; Novak, M.A. A Cobalt Pyrenylnitronylnitroxide Single-Chain Magnet with High Coercivity and Record Blocking Temperature. Chem. Eur. J. 2014, 20, 5460–5467. [Google Scholar] [CrossRef]
- Barskaya, I. Yu.; Veber, S.L.; Fokin, S.V.; Tretyakov, E.V.; Bagryanskaya, E.G.; Ovcharenko, V.I.; Fedin, M.V. Structural specifics of light-induced metastable states in copper(II)–nitroxide molecular magnets. Dalton Trans. 2015, 44, 20883–20888. [Google Scholar] [CrossRef]
- Tretyakov, E.V.; Romanenko, G.V.; Veber, S.L.; Fedin, M.V.; Polushkin, A.V.; Tkacheva, A.O.; Ovcharenko, V.I. Cu(hfac)2 Complexes with Nitronyl Ketones Structurally Mimicking Nitronyl Nitroxides in Breathing Crystals. Aust. J. Chem. 2015, 68, 970–980. [Google Scholar] [CrossRef]
- Furui, T.; Suzuki, S.; Kozaki, M.; Shiomi, D.; Sato, K.; Takui, T.; Okada, K.; Tretyakov, E.V.; Tolstikov, S.E.; Romanenko, G.V.; et al. Preparation and Magnetic Properties of Metal-Complexes from N-t-Butyl-N-oxidanyl-2-amino-(nitronyl nitroxide). Inorg. Chem. 2014, 53, 802–809. [Google Scholar] [CrossRef]
- Tretyakov, E.V.; Fedyushin, P.A.; Panteleeva, E.V.; Stass, D.V.; Bagryanskaya, I. Yu.; Beregovaya, I.V.; Bogomyakov, A.S. Substitution of a Fluorine Atom in Perfluorobenzonitrile by a Lithiated Nitronyl Nitroxide. J. Org. Chem. 2017, 82, 4179–4185. [Google Scholar] [CrossRef]
- Tretyakov, E.; Fedyushin, P.; Panteleeva, E.; Gurskaya, L.; Rybalova, T.; Bogomyakov, A.; Zaytseva, E.; Kazantsev, M.; Shundrina, I.; Ovcharenko, V. Aromatic SNF-Approach to Fluorinated Phenyl tert-Butyl Nitroxides. Molecules 2019, 24, 4493. [Google Scholar] [CrossRef]
- Politanskaya, L.V.; Fedyushin, P.A.; Rybalova, T.V.; Bogomyakov, A.S.; Asanbaeva, N.B.; Tretyakov, E.V. Fluorinated Organic Paramagnetic Building Blocks for Cross-Coupling Reactions. Molecules 2020, 25, 5427. [Google Scholar] [CrossRef] [PubMed]
- Fedyushin, P.; Rybalova, T.; Asanbaeva, N.; Bagryanskaya, E.; Dmitriev, A.; Gritsan, N.; Kazantsev, M.; Tretyakov, E. Synthesis of Nitroxide Diradical Using a New Approach. Molecules 2020, 25, 2701. [Google Scholar] [CrossRef] [PubMed]
- Serykh, A.; Tretyakov, E.; Fedyushin, P.; Ugrak, B.; Dutova, T.; Lalov, A.; Korlyukov, A.; Akyeva, A.; Syroeshkin, M.; Bogomyakov, A.; et al. N-Fluoroalkylpyrazolyl-substituted Nitronyl Nitroxides. J. Mol. Struct. 2022, 1269, 133739. [Google Scholar] [CrossRef]
- Tretyakov, E.V.; Fedyushin, P.A. Polyfluorinated organic paramagnets. Russ. Chem. Bull. 2021, 70, 2298–2314. [Google Scholar] [CrossRef]
- Zayakin, I.A.; Korlyukov, A.A.; Gorbunov, D.E.; Gritsan, N.P.; Akyeva, A. Ya.; Syroeshkin, M.A.; Stass, D.V.; Tretyakov, E.V.; Egorov, M.P. Au–Au Chemical Bonding in Nitronyl Nitroxide Gold(I) Derivatives. Organometallics 2022, 41, 1710–1720. [Google Scholar] [CrossRef]
- Tretyakov, E.; Fedyushin, P.; Bakuleva, N.; Korlyukov, A.; Dorovatovskii, P.; Gritsan, N.; Dmitriev, A.; Akyeva, A.; Syroeshkin, M.; Stass, D.; et al. Series of Fluorinated Benzimidazole-Substituted Nitronyl Nitroxides: Synthesis, Structure, Acidity, Redox Properties, and Magnetostructural Correlations. J. Org. Chem. 2023, 88, 10355–10370. [Google Scholar] [CrossRef]
- Reichenbächer, K.; Süss, H.I.; Hulliger, J. Fluorine in crystal engineering—“the little atom that could”. Chem. Soc. Rev. 2005, 34, 22–30. [Google Scholar] [CrossRef]
- Schwarzer, A.; Weber, E. Influence of Fluorine Substitution on the Crystal Packing of N-Phenylmaleimides and Corresponding Phthalimides. Cryst. Growth Des. 2008, 8, 2862–2874. [Google Scholar] [CrossRef]
- Singla, L.; Yadav, H.R.; Choudhury, A.R. Structural and Computational Analysis of Organic Fluorine-Mediated Interactions in Controlling the Crystal Packing of Tetrafluorinated Secondary Amides in the Presence of Weak C–H···O=C Hydrogen Bonds. Cryst. Growth Des. 2022, 22, 1604–1622. [Google Scholar] [CrossRef]
- Levina, E.O.; Chernyshov, I.Y.; Voronin, A.P.; Alekseiko, L.N.; Stash, A.I.; Vener, M.V. Solving the enigma of weak fluorine contacts in the solid state: a periodic DFT study of fluorinated organic crystals. RSC Adv. 2019, 9, 12520–12537. [Google Scholar] [CrossRef] [PubMed]
- Hebeisen, P.; Weiss, U.; Alker, A.; Kuhn, B.; Müller, K.; Wang, F.; Prakash, G.K.S. Molecular Structure and Crystal Packing of Monofluoromethoxyarenes. Eur. J. Org. Chem. 2018, 3724–3734. [Google Scholar] [CrossRef]
- Berger, R.; Resnati, G.; Metrangolo, P.; Weberd, E.; Hulliger, J. Organic fluorine compounds: a great opportunity for enhanced materials properties. Chem. Soc. Rev. 2011, 40, 3496–3508. [Google Scholar] [CrossRef]
- Chopra, D.; Guru Row, T.N. Role of organic fluorine in crystal engineering. CrystEngComm 2011, 13, 2175–2186. [Google Scholar] [CrossRef]
- Cole, J.C.; Taylor, R. Intermolecular Interactions of Organic Fluorine Seen in Perspective. Cryst. Growth Des. 2022, 22, 1352–1364. [Google Scholar] [CrossRef]
- Dunitz, J.D. Organic Fluorine: Odd Man Out. ChemBioChem. 2004, 5, 614–621. [Google Scholar] [CrossRef]
- Murray, J.S.; Seybold, P.G.; Politzer, P. The Many Faces of Fluorine: Some Noncovalent Interactions of Fluorine Compounds. J. Chem. Thermodyn. 2021, 156, 106382. [Google Scholar] [CrossRef]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Nature of Halogen-Centered Intermolecular Interactions in Crystal Growth and Design: Fluorine-Centered Interactions in Dimers in Crystalline Hexafluoropropylene as a Prototype. J. Comput. Chem. 2019, 40, 1836–1860. [Google Scholar] [CrossRef]
- CrysAlisPro. Version 1.171.41.106a. Rigaku Oxford Diffraction, 2021.
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 229–341. [Google Scholar] [CrossRef]
- Bruker APEX3, RLATT, CELL_NOW, TWINABS, SAINT-Plus and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA, 2016.
- Carlin, R.L. Magnetochemistry; Springer: Berlin, Germany, 1986. [Google Scholar]
- Romanenko, G.V.; Fokin, S.V.; Vasilevskii, S.F.; Tretyakov, E.V.; Shvedenkov, Yu. G.; Ovcharenko, V.I. Dimeric Complexes of Manganese(II) and Nickel(II) Hexafluoroacetylacetonates with Pyrazole-Containing Nitronylnitroxyl Radicals. Russ. J. Coord. Chem. 2001, 27, 360–367. [Google Scholar] [CrossRef]
- Maryunina, K.; Fokin, S.; Ovcharenko, V.; Romanenko, G.; Ikorskii, V. Solid solutions: An efficient way to control the temperature of spin transition in heterospin crystals MxCu1 − x(hfac)2L (M = Mn, Ni, Co; L = nitronyl nitroxide). Polyhedron 2005, 24, 2094–2101. [Google Scholar] [CrossRef]
- Caneschi, A.; Gatteschi, D.; Sessoli, R. Magnetic properties of a layered molecular material comprising manganese hexafluoroacetylacetonate and nitronyl nitroxide radicals. Inorg. Chem. 1993, 32, 4612–4616. [Google Scholar] [CrossRef]
- Okada, K.; Nagao, O.; Mori, H.; Kozaki, M.; Shiomi, D.; Sato, K.; Takui, T.; Kitagawa, Y.; Yamaguchi, K. Preparation and Magnetic Properties of Mn(hfac)2-Complexes of 2-(5-Pyrimidinyl)- and 2-(3-Pyridyl)-Substituted Nitronyl Nitroxides. Inorg. Chem. 2003, 42, 3221–3228. [Google Scholar] [CrossRef]
- Zhu, M.; Lou, D.; Deng, X.; Zhang, L.; Zhang, W.; Lü, Y. A functional nitroxide ligand builds up two 2p–3d complexes with different spin ground states and a 2p–3d–4f chain of rings. CrystEngComm 2018, 20, 2583–2592. [Google Scholar] [CrossRef]
- Okada, K.; Beppu, S.; Tanaka, K.; Kuratsu, M.; Furuichi, K.; Kozaki, M.; Suzuki, S.; Shiomi, D.; Sato, K.; Takui, T.; et al. Preparation, structure, and magnetic interaction of a Mn(hfac)2-bridged [2-(3-pyridyl)(nitronyl nitroxide)–Mn(hfac)2]2 chain complex. Chem. Commun. 2007, 2485–2487. [Google Scholar] [CrossRef]
- Field, L.M.; Lahti, P.M.; Palacio, F. 1∶1 Complexes of 5-(4-[N-tert-butyl-N-aminoxyl]phenyl)pyrimidine with manganese(II) and copper(II) hexafluoroacetonylacetonate. Chem. Commun. 2002, 636–637. [Google Scholar] [CrossRef]
- Wang, H.-M.; Liu, Z.-L.; Liu, C.-M.; Zhang, D.-Q.; Lu, Z.-L.; Geng, H.; Shuai, Z.-G.; Zhu, D.-B. Coordination Complexes of 2-(4-Quinolyl)nitronyl Nitroxide with M(hfac)2 [M = Mn(II), Co(II), and Cu(II)]: Syntheses, Crystal Structures, and Magnetic Characterization. Inorg. Chem. 2004, 43, 4091–4098. [Google Scholar] [CrossRef]
- Zhou, N.; Wang, Y.-L.; Wang, H.-L.; Li, W.-J.; Guo, Z.; Ma, Y.; Li, L.-C.; Wang, Q.-L.; Cheng, P.; Liao, D.-Z. Structural design and magnetic properties study on two nitronyl nitroxide radicals–MnII complexes with hetero chain or mononuclear tri-spin structures. Polyhedron 2015, 89, 96–100. [Google Scholar] [CrossRef]
- Liu, R.-N.; Li, L.-C.; Xing, X.-Y.; Liao, D.-Z. Cyclic metal–radical complexes based on 2-[4-(1-imidazole)phenyl]-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide: Syntheses, crystal structures and magnetic properties. Inorganica Chim. Acta 2009, 362, 2253–2258. [Google Scholar] [CrossRef]
- Fokin, S.V.; Tolstikov, S.E.; Tret’yakov, E.V.; Romanenko, G.V.; Bogomyakov, A.S.; Veber, S.L.; Sagdeev, R.Z.; Ovcharenko, V.I. Molecular magnets based on chain polymer complexes of copper(II) bis(hexafluoroacetylacetonate) with isoxazolylsubstituted nitronyl nitroxides. Russ. Chem. Bull. 2011, 60, 2470–2484. [Google Scholar] [CrossRef]
- Yang, M.; Xie, S.; Liang, X.; Zhang, Y.; Dong, W. A novel functional nitronyl nitroxide and its manganese and cobalt complexes: Synthesis, structures and magnetic properties. Polyhedron 2019, 161, 132–136. [Google Scholar] [CrossRef]
- Wang, C.; Ma, Y.; Wang, Y.; Wang, Q.; Li, L.; Cheng, P.; Liao, D. A New Quinoxalinyl-Substituted Nitronyl Nitroxide Radical and its Five-Spin CuII and Four-Spin MnII Complexes: Syntheses, Crystal Structures, and Magnetic Properties. Aust. J. Chem. 2012, 65, 672–679. [Google Scholar] [CrossRef]
- Mei, Z.; Dingshuo, L.; Xiaochun, D.; Li, Z.; Wei, Z.; Yaohong, L. A functional nitroxide ligand builds up two 2p–3d complexes with different spin ground states and a 2p–3d–4f chain of rings. CrystEngComm 2018, 20, 2583–2592. [Google Scholar] [CrossRef]
- Maryunina, K.; Fokin, S.; Ovcharenko, V.; Romanenko, G.; Ikorskii, V. Solid solutions: An efficient way to control the temperature of spin transition in heterospin crystals MxCu1−x(hfac)2L (M = Mn, Ni, Co; L = nitronyl nitroxide). Polyhedron 2005, 24, 2094–2101. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Liu, C.-M.; Zhang, D.-Q.; Gao, S.; Zhu, D.-B. Syntheses, crystal structures, and magnetic properties of two cyclic dimer M2L2 complexes constructed from a new nitronyl nitroxide ligand and M(hfac)2 (M = Cu2+, Mn2+). Inorganica Chim. Acta 2007, 360, 3553–3559. [Google Scholar] [CrossRef]
- Borra´s-Almenar, J.J.; Clemente-Juan, J.M.; Coronado, E.; Tsukerblat, B.S. MAGPACK1 A package to calculate the energy levels, bulk magnetic properties, and inelastic neutron scattering spectra of high nuclearity spin clusters. J. Comput. Chem. 2001, 22, 985–991. [Google Scholar] [CrossRef]
- Caneschi, A.; Gatteschi, D.; Sessoli, R.; Rey, P. Toward molecular magnets: the metal-radical approach. Acc. Chem. Res. 1989, 22, 392–398. [Google Scholar] [CrossRef]
- Caneschi, A.; Gatteschi, D.; Sessoli, R.; Rey, P. Structure and magnetic properties of a ring of four spins formed by manganese(II) and a pyridine substituted nitronyl nitroxide. Inorganica Chim. Acta 1991, 184, 67–71. [Google Scholar] [CrossRef]
- Guo, J.-N.; Wang, J.-J.; Sun, G.-F.; Li, H.-D.; Li, L.-C. A novel nitronyl nitroxide radical containing thiophene and pyridine rings and its manganese(II) complex: synthesis, structure, and magnetic properties. J. Coord. Chem. 2017, 70, 1926–1935. [Google Scholar] [CrossRef]
- Rancurel, C.; Leznoff, D.B.; Sutter, J.-P.; Golhen, S.; Ouahab, L.; Kliava, J.; Kahn, O. Synthesis, Structure, and Magnetism of Mono- and Binuclear Manganese(II) Compounds of Nitronyl Nitroxide Substituted Phosphine Oxides. Inorg. Chem. 1999, 38, 4753–47538. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) A fragment of chain structure in [Mn(hfac)2(NN-Pz-CHF2)]n (colour code: Mn: violet, N: blue, O: red, F: yellow, C: gray; H atoms are not shown for the sake of clarity). (b) Magnetic exchange pathways within and between the repeating units (the red and blue balls denote the SMn = 5/2 and SR = 1/2 spins, respectively; the arrows indicate the spin alignment in the ground state).
Figure 1.
(a) A fragment of chain structure in [Mn(hfac)2(NN-Pz-CHF2)]n (colour code: Mn: violet, N: blue, O: red, F: yellow, C: gray; H atoms are not shown for the sake of clarity). (b) Magnetic exchange pathways within and between the repeating units (the red and blue balls denote the SMn = 5/2 and SR = 1/2 spins, respectively; the arrows indicate the spin alignment in the ground state).
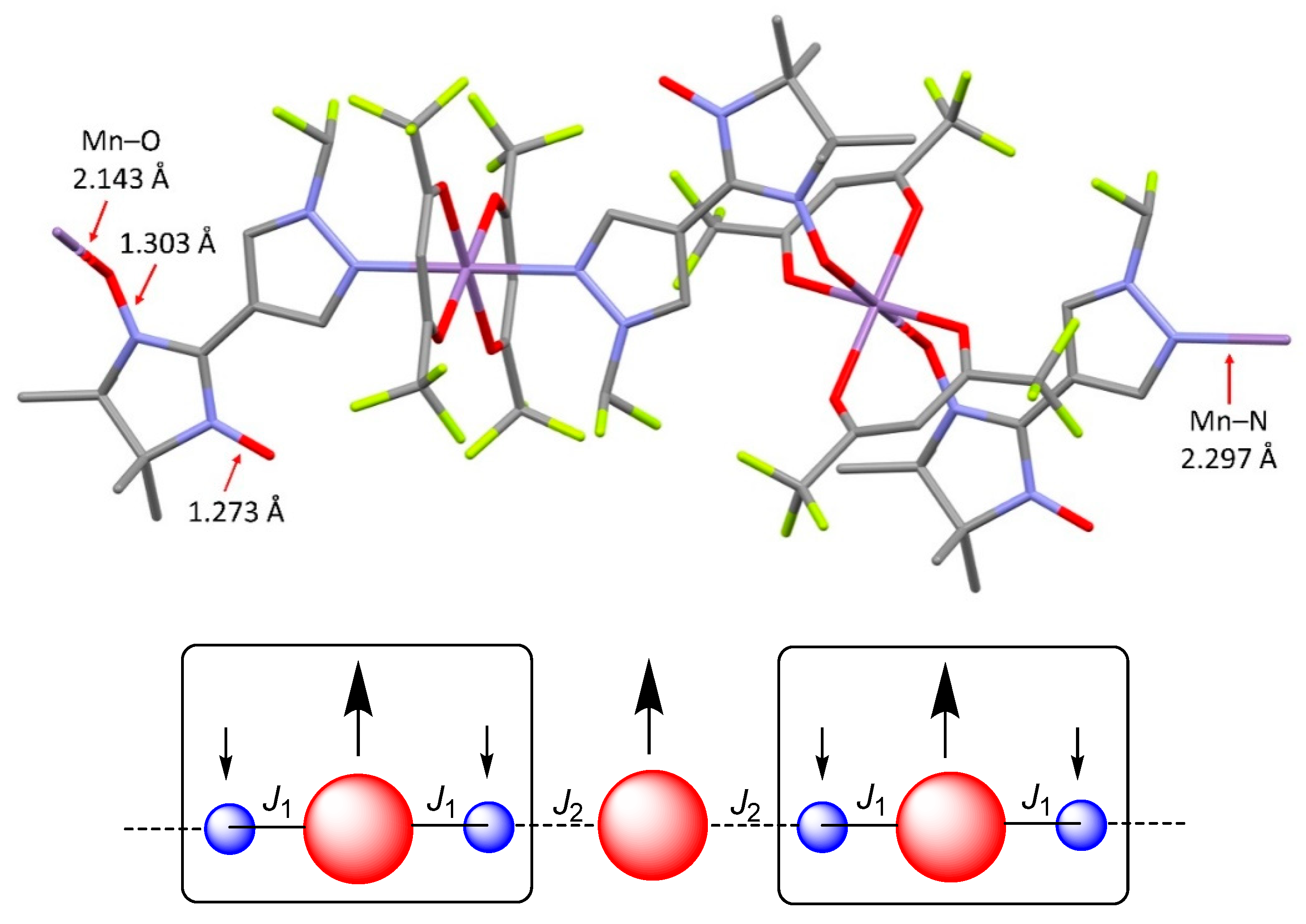
Figure 2.
A fragment of chain structure in [Mn(hfac)2(NN-Pz-CH2CH2F)]n (colour code: Mn: violet, N: blue, O: red, F: yellow, C: gray; H atoms are not shown for the sake of clarity).
Figure 2.
A fragment of chain structure in [Mn(hfac)2(NN-Pz-CH2CH2F)]n (colour code: Mn: violet, N: blue, O: red, F: yellow, C: gray; H atoms are not shown for the sake of clarity).
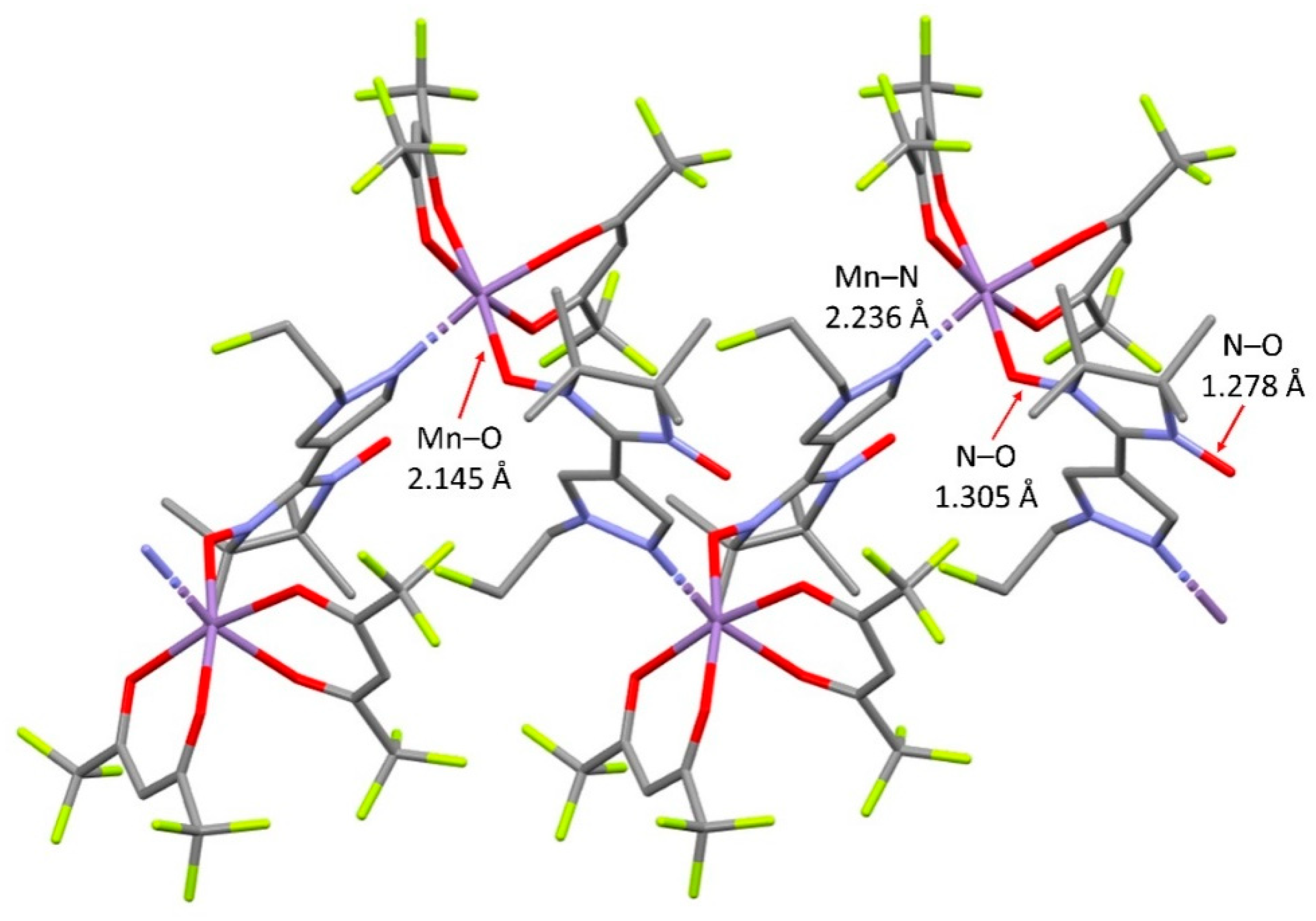
Figure 3.
(a) A fragment of chain structure in [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8 (colour code: Mn: violet, N: blue, O: red, F: yellow, C: gray; the solvate molecules and H atoms are not shown). (b) Magnetic exchange pathways within and between the two spin clusters (the red and blue balls denote the SMn = 5/2 and SR = 1/2 spins, respectively; the arrows indicate the spin alignment in the ground state).
Figure 3.
(a) A fragment of chain structure in [Mn(hfac)2(NN-Pz-CH2CHF2)]n⋅nC7H8 (colour code: Mn: violet, N: blue, O: red, F: yellow, C: gray; the solvate molecules and H atoms are not shown). (b) Magnetic exchange pathways within and between the two spin clusters (the red and blue balls denote the SMn = 5/2 and SR = 1/2 spins, respectively; the arrows indicate the spin alignment in the ground state).
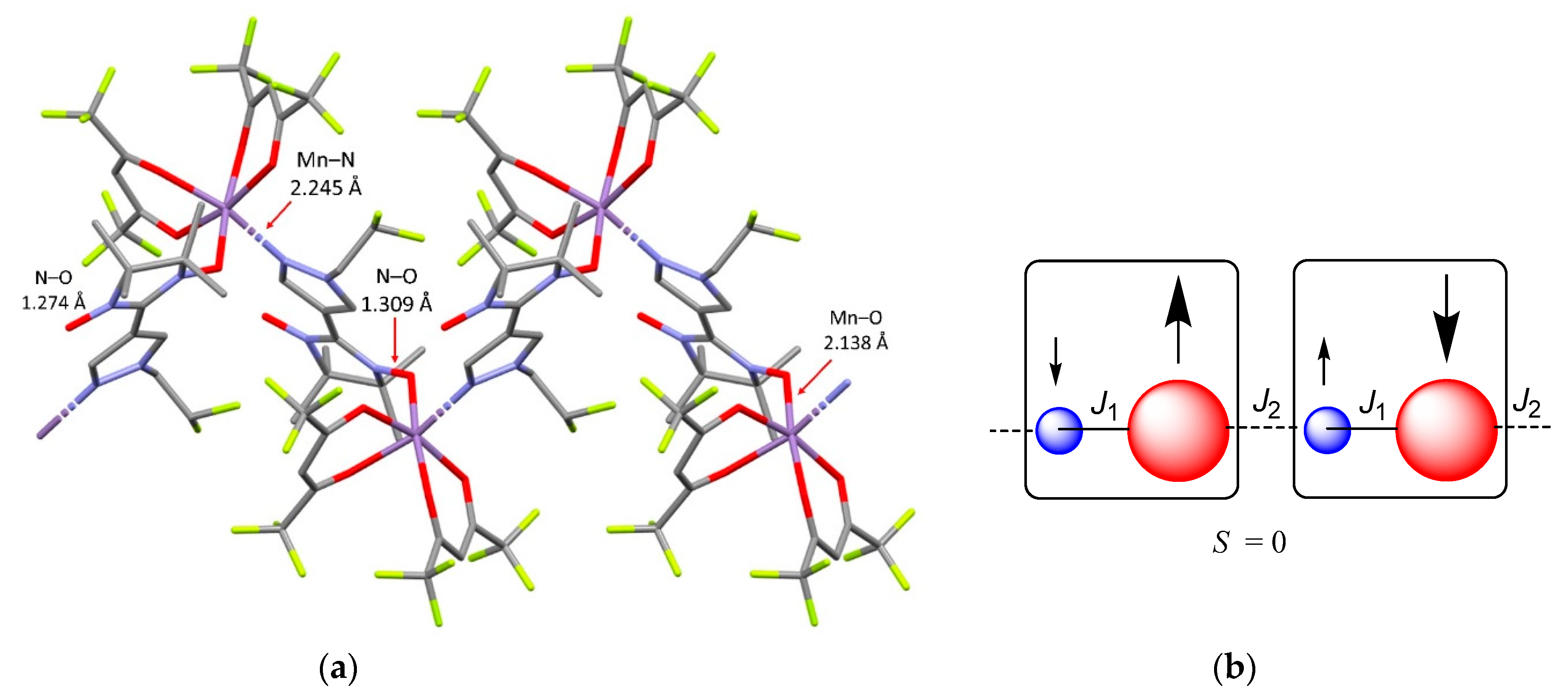
Figure 4.
(a) The cyclic dimer in the structure of [Mn(hfac)2(NN-Pz-CH2CF3)]2⋅2C7H8 (colour code: Mn: violet, N: blue, O: red, F: yellow, C: gray; the solvate molecules and H atoms are not shown). (b) Magnetic exchange pathways within the dimer (the red and blue balls represent the SMn = 5/2 and SR = 1/2 spins, respectively; the arrows indicate the spin alignment in the ground state).
Figure 4.
(a) The cyclic dimer in the structure of [Mn(hfac)2(NN-Pz-CH2CF3)]2⋅2C7H8 (colour code: Mn: violet, N: blue, O: red, F: yellow, C: gray; the solvate molecules and H atoms are not shown). (b) Magnetic exchange pathways within the dimer (the red and blue balls represent the SMn = 5/2 and SR = 1/2 spins, respectively; the arrows indicate the spin alignment in the ground state).
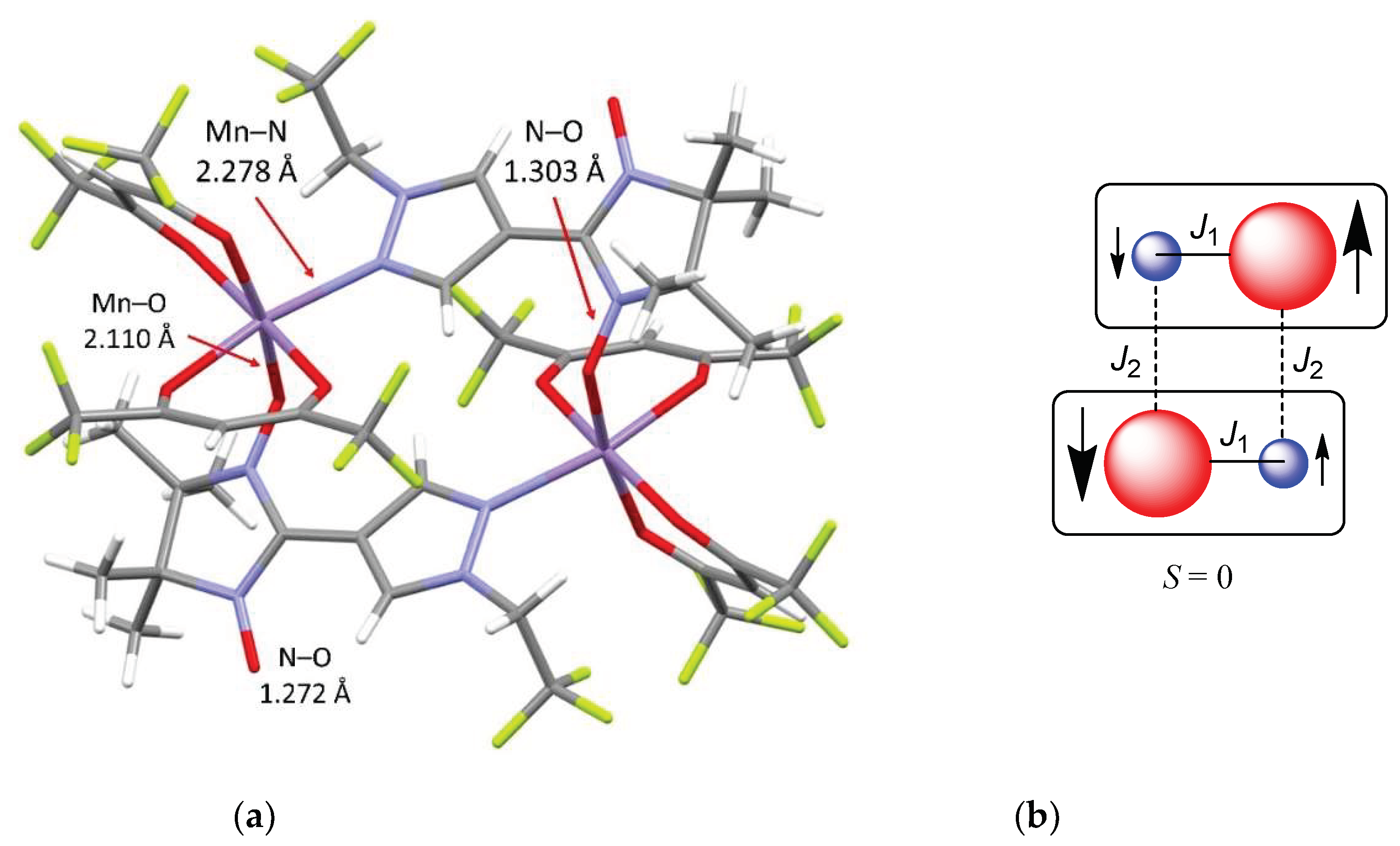
Scheme 1.
Plausible pathways to different Mn–nitroxide complexes.
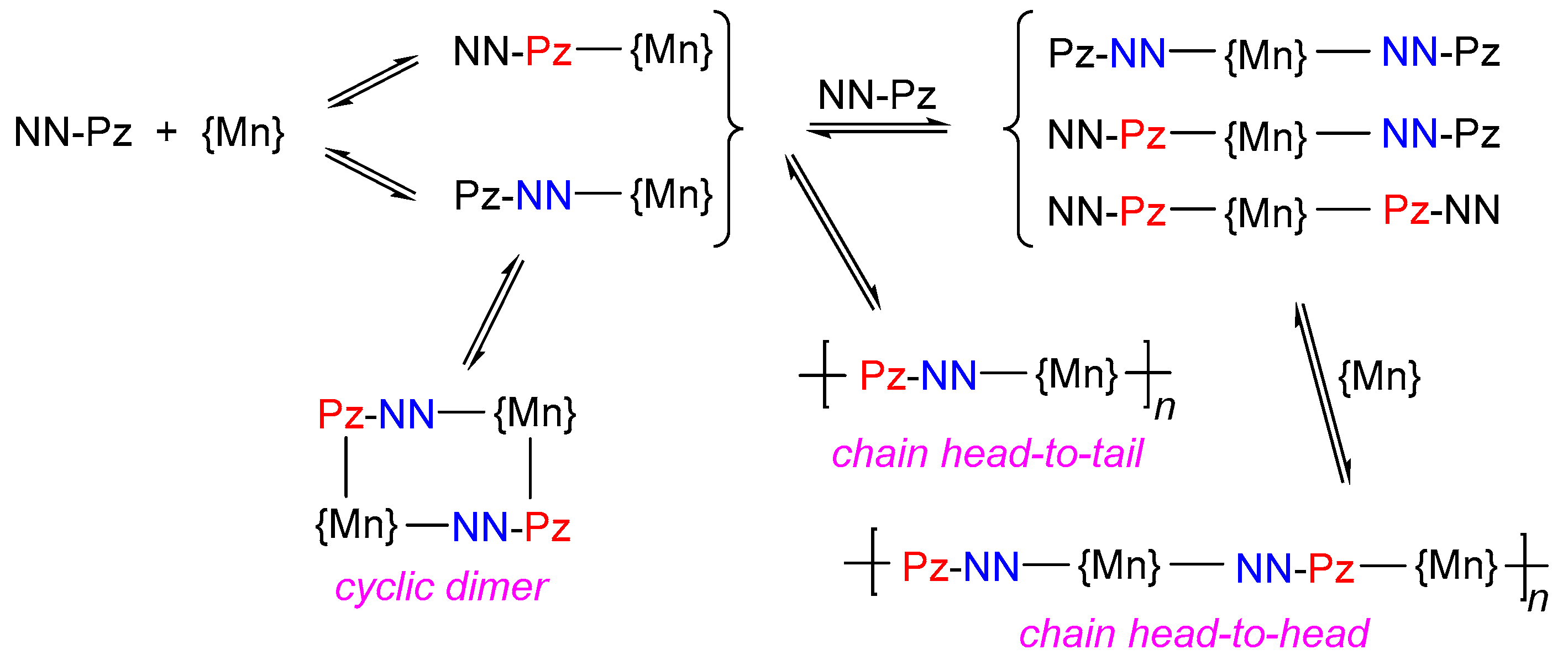
Figure 5.
Images of inverted reaction vessels with reaction mixtures [Mn(hfac)2] + NN-Pz-CHF2 (a) and [Mn(hfac)2] + NN-Pz-CH2CH2F (b) after incubation at –15 °C for 24 h.
Figure 5.
Images of inverted reaction vessels with reaction mixtures [Mn(hfac)2] + NN-Pz-CHF2 (a) and [Mn(hfac)2] + NN-Pz-CH2CH2F (b) after incubation at –15 °C for 24 h.

Figure 6.
The temperature dependence of μeff for complex [Mn(hfac)2(NN-Pz-CHF2)]n. The solid curve corresponds to the best theoretical fit.
Figure 6.
The temperature dependence of μeff for complex [Mn(hfac)2(NN-Pz-CHF2)]n. The solid curve corresponds to the best theoretical fit.
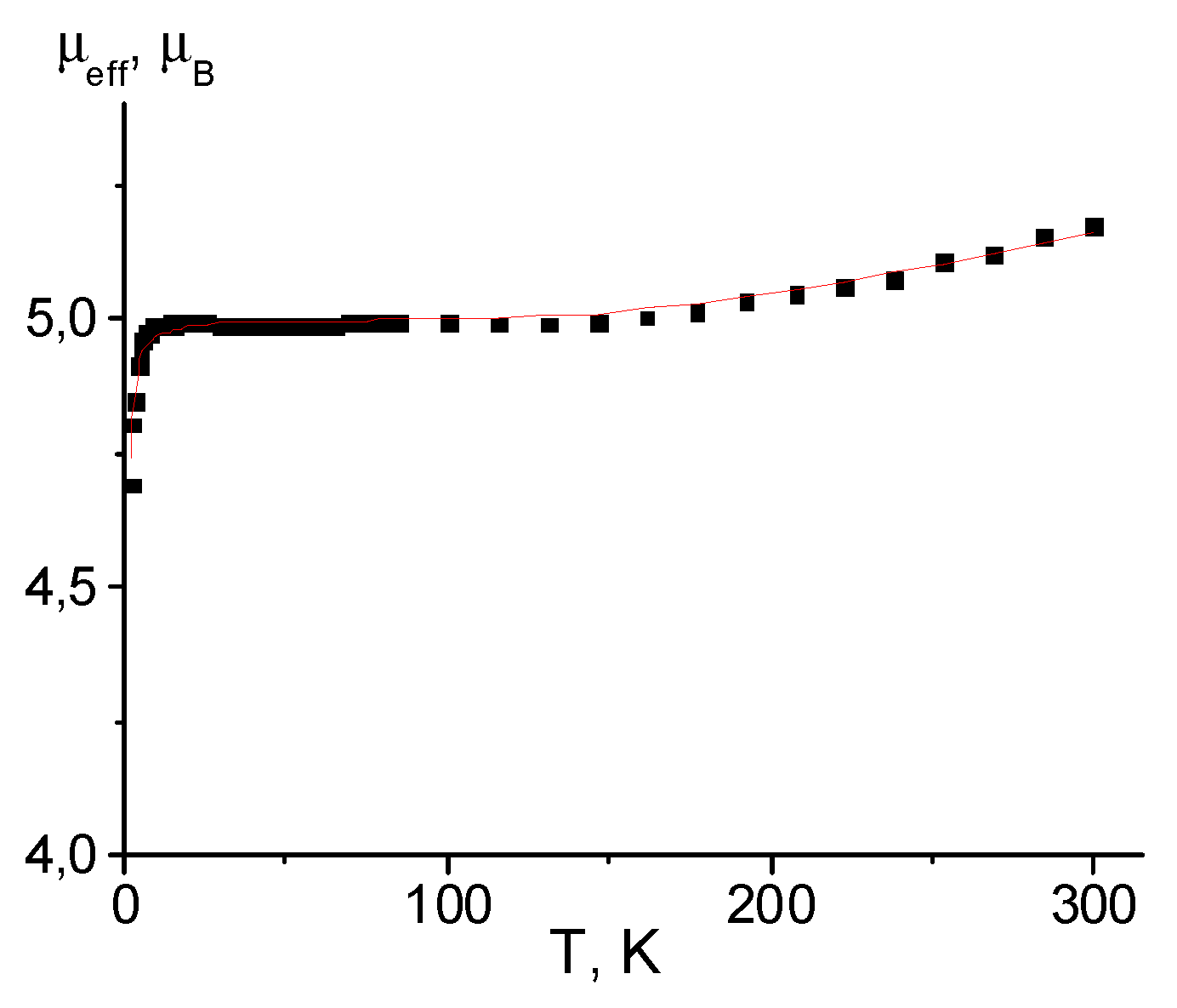
Figure 7.
The μeff(T) dependencies for complexes [Mn(hfac)2(NN-Pz-CH2CH2F)]n (a) and [Mn(hfac)2(NN-Pz-CH2CHF2)]n (b). Solid curves are theoretical ones.
Figure 7.
The μeff(T) dependencies for complexes [Mn(hfac)2(NN-Pz-CH2CH2F)]n (a) and [Mn(hfac)2(NN-Pz-CH2CHF2)]n (b). Solid curves are theoretical ones.
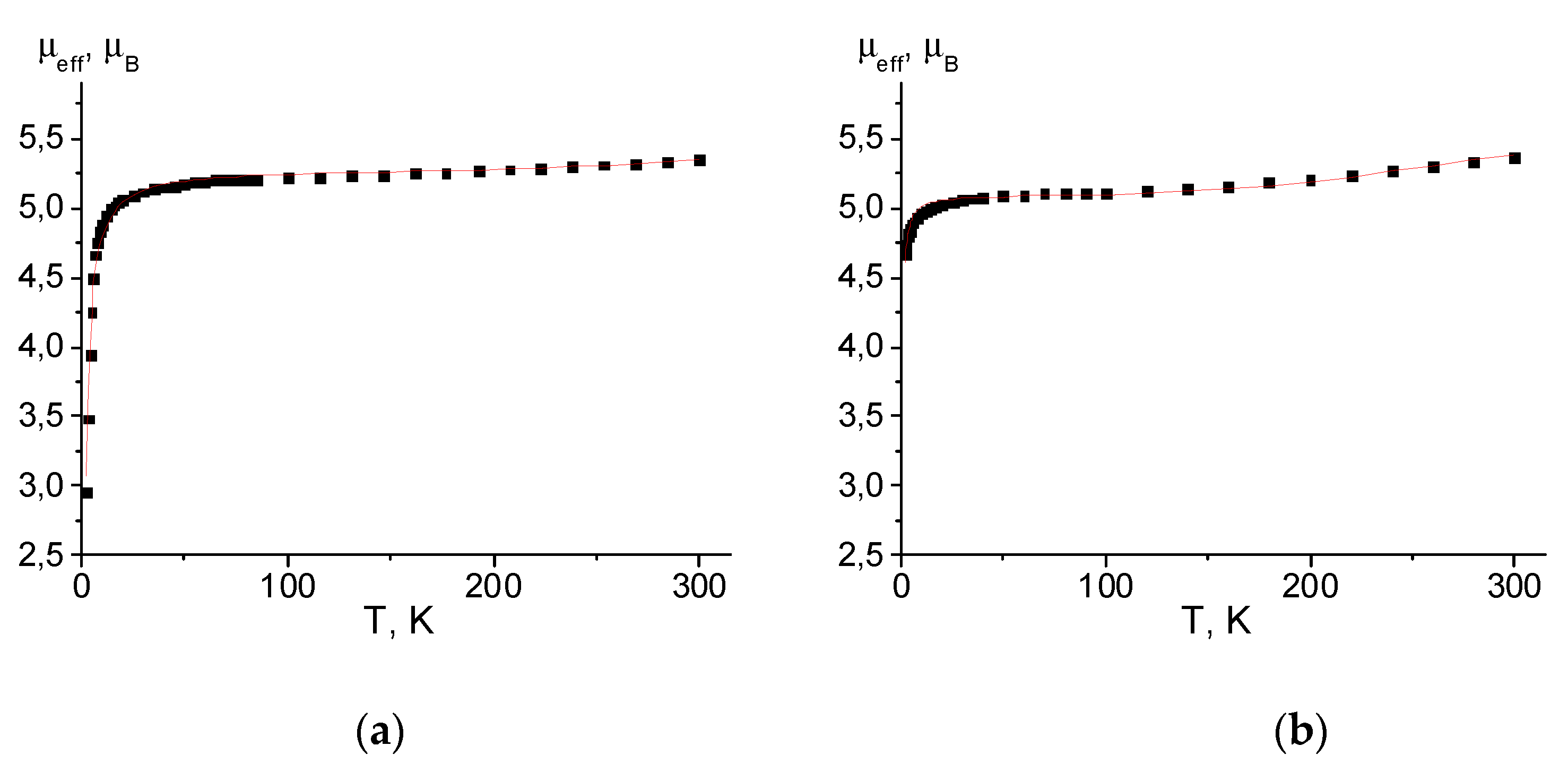
Figure 8.
(a) The μeff(T) dependencies for complex [Mn(hfac)2(NN-Pz-CH2CF3)]2. The solid curve corresponds to the best theoretical fit. (b) The spin polarisation mechanism for the intra-dimer magnetic coupling.
Figure 8.
(a) The μeff(T) dependencies for complex [Mn(hfac)2(NN-Pz-CH2CF3)]2. The solid curve corresponds to the best theoretical fit. (b) The spin polarisation mechanism for the intra-dimer magnetic coupling.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



