
Submitted:
26 September 2023
Posted:
27 September 2023
You are already at the latest version
Abstract
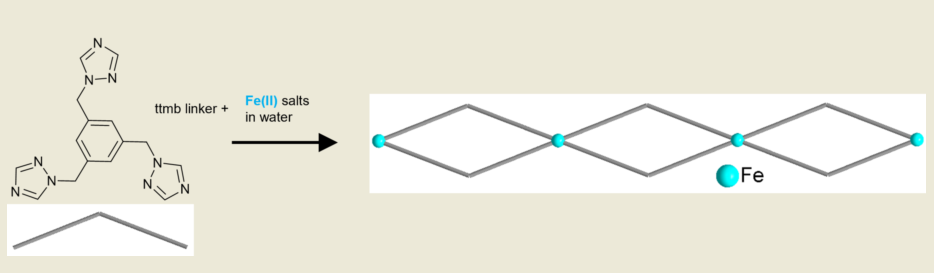
Three novel iron(II) coordination polymers, [Fe(H2O)2(ttmb)2](ClO4)2·4H2O (1), [Fe(H2O)2(ttmb)2](BF4)2·4H2O (2), [Fe(ttmb)2(NCS)2] (3), were synthesized with the linker 1,3,5-tris(1H-1,2,4-triazol-1-yl)methyl)benzene (ttmb). The single-crystal structures show that all three compounds form a double chain structure with the adjacent iron atoms being bridged by two ttmb linkers. The iron(II) ions are octahedrally surrounded by four N4 donor atom from the 1,2,4-triazol-1-yl groups of four different ttmb linkers which form an equatorial plane and two trans-coordinated aqua ligands in 1 and 2 or isothiocyanato ligands in 3 in the axial positions. In view of the neutral bridging ttmb linker, there is a counter-anion in all structures; uncoordinated ClO4– and BF4–in 1 and 2 or coordinated NCS–in 3. Compounds 1 and 2 are isostructural. Interestingly, the ttmb linker only utilizes two of its three potentially coordinating triazole groups. All iron(II) coordination networks are colorless or have a light-yellow color, being indicative of the high-spin state.
Keywords:
coordination polymers
; coordination networks
; triazole ligand
; iron(II)
; self-assembly
; hydrogen bonds
1. Introduction
According to the definition of the International Union of Pure and Applied Chemistry (IUPAC), a coordination polymer is a one-, two- or three-dimensional (1D, 2D or 3D) coordination compound consisting of repeating units [1,2,3,4]. Coordination polymers are often formed by self-assembly processes and are constructed from metal ions as connectors and organic ligands as linkers [5,6,7]. They can provide a wide range of topologies and structures [8,9,10,11]. Self-assembly describes a process in which the molecules arrange themselves into an ordered pattern based on interactions without external forces [6,12,13,14,15,16,17]. For the construction of 2D or 3D networks, tripodal ligands with three coordinating groups are attractive linkers to connect between three metal ions as a basis for higher dimensionality [18,19,20,21,22,23,24]. Triazole based ligands are appropriate as linkers in coordination polymers since they can be deprotonated to the corresponding azolate anions in which the nitrogen atoms are strong donor atoms for d-metal ions [25,26,27,28,29,30,31,32,33,34]. Not only the dimensionality but also the stability of networks increases with the number of donor atoms and coordination sites [35]. The geometry of coordination polymers not only depends on the structure of the linker, it also depends on the presence of non-coordinating anions [36]. Particularly non-coordinating anions not only balance the charges of the complex, but they also function as templates [37,38,39,40,41]. A template provides an organized arrangement of atoms to achieve a specific linkage of atoms [39,40]. In addition to the anions, solvent molecules can act as templates and control the arrangement of a network [31,42,43,44,45,46,47,48]. Depending on the size and properties of the anion the effect on the geometry can vary [49,50]. Even if the anion is uncoordinated to the metal ion, then there are intermolecular interaction with the surrounding network [51], such as anion-π, hydrogen bonding or Lewis base-acid interactions [52,53]. Also, hydrogen bonds within the coordination network have an influence on the structure [7,54].
2. Materials and Methods
The used chemicals were all commercially obtained and no further purification was done (see Suppl. Mater., Section S1). The used water was deionized.
FT-IR measurements were carried out on a Bruker TENSOR 37 IR spectrometer (Bruker, Billerica, MA, USA) in ATR mode (Platinum ATR-QL, Diamond) in the range 4000-500 cm–1. NMR spectra were collected with a Bruker Avance III – 300 (Bruker, Billerica, MA, USA) (1H: 300 MHz); 13C{1H}: 75 MHz). Elemental analyses were measured on a PerkinElmer 2400 series II elemental analyzer (PerkinElmer, Waltham, MA, USA) (accuracy of 0.5%). Thermogravimetric analyses were carried out using a Netzsch TG209 F3 Tarsus (Netzsch, Selb, Germany) under nitrogen atmosphere with a ramp of 5 K min–1 up to 1000°C. X-ray powder diffraction measurements were performed on a Rigaku MiniFlex600 (Rigaku, Tokyo, Japan) (600 W, 40 kV, 15 mA) at room temperature with Cu-Kα radiation (λ = 1.54184 Å). The low-background silicon holder in the PXRD device is the cause of the rising baseline below 5° 2Theta. The highest reflex was normalized to 1. The simulated powder patterns were derived from the single crystal data using the MERCURY 2020.3.0 software [55].
Under a polarized-light Leica M80 microscope (Leica, Wetzlar, Germany) suitable single crystals were carefully selected and covered with oil on a cryo-loop. The single crystal diffraction measurement for compounds 1 and 3 were carried out on a Rigaku XtaLAB Synergy S diffractometer (Rigaku, Tokyo, Japan) with a hybrid pixel array detector and a micro-focus sealed X-ray tube, PhotonJet copper X-ray source (λ = 1.54184 Å). For cell refinement, data reduction and absorption correction CRYSALISPRO was used [56]. For compound 2 the measurement was performed on a Bruker Kappa APEX2 CCD X-ray diffractometer (Bruker, Billerica, MA, USA) with a microfocus sealed tube molybdenum X-ray source (λ = 0.71073 Å) and a multi-layer mirror monochromator. Cell refinement was performed with APEX2, data reduction with SAINT and adsorption correction with SADABS [57,58,59]. The crystal structures for compounds 1-3 were solved using OLEX2 with SHELXT and the refinement was done with SHELXL [60,61,62,63]. The graphics were drawn with the DIAMOND 4.0 software [64].
2.1. Synthesis
2.1.1 Synthesis of 1,3,5-tris(1H-1,2,4-triazol-1-yl)methyl)benzene (ttmb)
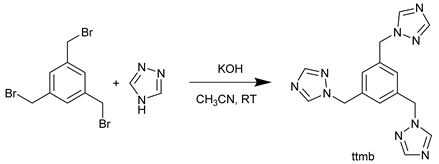
The linker ttmb was synthesized with a modified synthesis procedure of Shang et al. [65] as shown in Equation (1). 0.85 g (12.3 mmol) of 1,2,4-triazole and 1.32 g(23.5 mmol) of potassium hydroxide were stirred in 30 mL of acetonitrile for 30 min at room temperature. Afterwards, a solution of 1.0 g (2.8 mmol) of 1,3,5-tris(bromo methyl) benzene in 20 mL of acetonitrile was added. The resulting solution was stirred for an additional 30 min at room temperature. After filtration the solvent was removed in vacuo. Next, the resulting oil was dissolved in 20 mL of deionized water and extracted with chloroform (5 x 50 mL). Once the organic phase was dried with MgSO4, the solvent was again removed using rotatory evaporation. The product crystallized over night and was then dried in vacuum at 60 °C. Yield: 0.45 g (46%). C15H15N9: calc. C 56.1, H 4.7, N 39.2; exp. C 55.5, H 4.6, N 38.5 %. IR: ṽ [cm–1]: 3114, 3096, 3034, 2994, 2955, 3034, 2993, 2956, 2849, 1798, 1757, 1711, 1609, 1503, 1466, 1445, 1430, 1373, 1338, 1298, 1270, 1207, 1170, 1137, 1096, 1018, 987, 960, 917, 895, 880, 858, 800, 742, 680, 648, 601, 570. 1H-NMR (300 MHz, DMSO-d6): δ [ppm]: 8.63 (s, 3H), 7.97 (s, 3H), 7.12 (s, 3H), 5.39 (s, 6H). 13C{1H}-NMR (75 MHz, DMSO-d6: δ [ppm]: 151.81, 144.31, 137.26, 51.62.
2.1.2. Synthesis of [Fe(H2O)2(ttmb)2](ClO4)2·4H2O (1)
Please note, that perchlorates are potentially explosive and should be handled with care! TGA shows an explosion at around 200 °C after the sample was dried at 60 °C in vacuo beforehand. The amount of 49 mg (0.19 mmol) of Fe(ClO4)2·xH2O and 84 mg (0.26 mmol) of ttmb were dissolved in 3 mL of H2O and stored in an pre-heated oven at 60 °C for 20 h. After cooling down to room temperature over a period of 4 h, yellow crystals were obtained. The crystals were washed with water (3 x 3 mL) and stored in H2O. Yield: 73 mg (38%). C30H42Cl2FeN18O14: calc. C 40.2, H 3.4, N 28.0; exp. C 39.5, H 3.6, N 27.8 %. IR: ṽ [cm–1]: 3509, 3455, 3357, 3269, 3126, 3036, 2357, 1767, 1679, 1632, 1612, 1517, 1466, 1438, 1373, 1359, 1340, 1302, 1283, 1218, 1179, 1163, 1134, 1078, 1026, 989, 977, 962, 915, 886, 853, 778, 765, 747, 693, 677, 654, 621, 577.
2.1.3. Synthesis of [Fe(H2O)2(ttmb)2](BF4)2·4H2O (2)
For the synthesis of 2 two solutions were prepared. The first solution contained 122.1 mg (0.36 mmol) of Fe(BF4)2ˑ6H2O and 62.9 mg (0.36 mmol) of ascorbic acid in 1.5 mL of H2O. The second solution was composed of 57.9 mg (0.18 mmol) of ttmb dissolved in 1.5 mL of EtOH. Both solutions were heated up to 80 °C and then combined. The mixture was stored in the preheated oven at 60 °C for 24 h and cooled down for 4 h. The resulting colorless crystals were washed with a 1:1 (v:v) mixture of H2O:EtOH (3 x 3 mL) and later stored in that mixture as well. Yield: 68.5 mg (56 %). C30H42B2F8FeN18O14: calc. C 36.8, H 4.3, N 25.8; exp. C: 36.9, H 4.2, N 25.4 %. IR: ṽ [cm–1]: 3537, 3460, 3264, 3129, 3033, 2360, 1766, 1634, 1612, 1518, 1469, 1439, 1374, 1360, 1341, 1284, 1218, 1180, 1163, 1144, 1133, 1047, 1023, 990, 977, 913, 884, 852, 795, 778, 765, 747, 693, 677, 654, 577.
2.1.4. Synthesis of [Fe(ttmb)2(NCS)2] (3)
A modified procedure of Garcia et al. [66] was used. Three solutions were prepared. Solution 1 contained 19.6 mg (0.05 mmol) of (NH4)2Fe(SO4)2·6H2O and 32 mg (0.18 mmol) of ascorbic acid in 1 mL of H2O. The second solution embodied 7 mg (0.09 mmol) of NH4SCN in 1 mL H2O. Solution 3 was composed of 29 mg (0.09 mmol) of ttmb in 1 mL of deionized water. After heating each solution near its boiling point, solution 2 was added to solution 1. Solution 3 was then added dropwise to this combined solution. After 48 h at 60 °C colorless crystals were obtained. The crystals were washed with water (3 x 3 mL) and stored in H2O. Yield: 26 mg (65%). C32H30FeN20S2: calc.: C = 47.2, H = 3.7, N = 34.4; exp.: C = 46.5, H = 3.8, N = 33.9. IR: ṽ [cm–1]: 3580, 3436, 3140, 3126, 3110, 2961, 2845, 2338, 2162, 2046, 1779, 1757, 1611, 1522, 1502, 1466, 1430, 1359, 1340, 1311, 1298, 1275, 1202, 1178, 1159, 1131, 1019, 988, 974, 956, 923, 884, 847, 789, 759, 747, 682, 673, 652, 633, 583, 569.
3. Results and Discussion
The linker 1,3,5-tris(1H-1,2,4-triazol-1-yl)methyl)benzene (ttmb) was synthesized by a nucleophilic substitution reaction between 1H-1,2,4-triazole and 1,3,5-tris(bromomethyl)benzene in acetonitrile (see Equation (1)). The authentification was done by 1H, 13C NMR and IR spectroscopy. The results of thermographic analysis show a thermal stability up to 310 °C (see Supplementary Materials, Section S2).
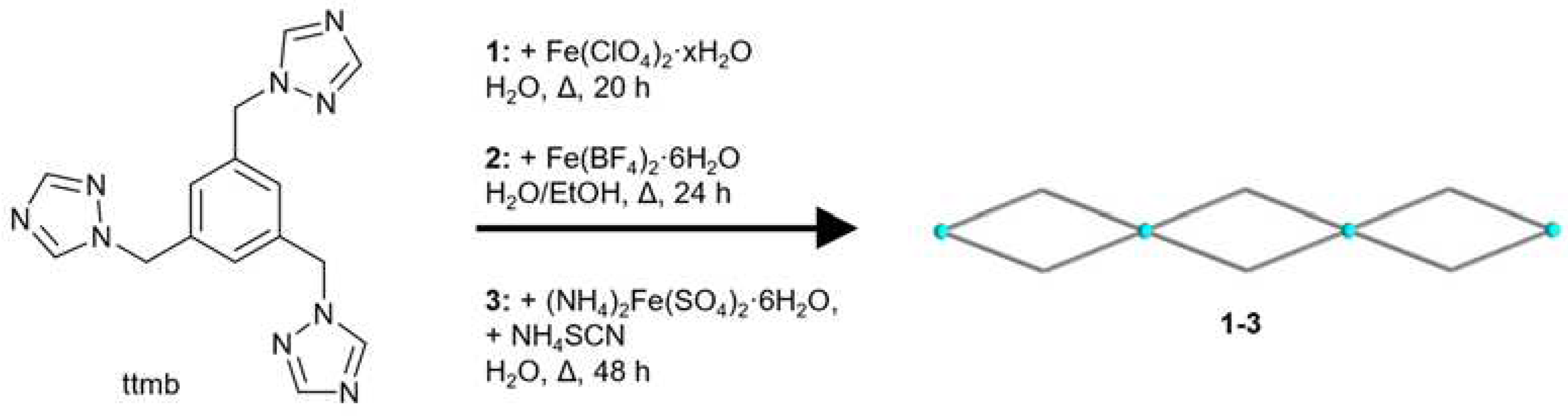
A schematic presentation of the synthesis of the complexes 1-3 is given in Scheme 1. The compounds had to be prepared with different molar metal:ligand ratios, which were optimized beforehand, in order to obtain crystals of sufficient quality for single crystal X-ray analysis. The molar M:L ratio for 1 was 2:3, for compound 2 it was 2:1 and for 3 it was 1:2. The synthesis for the crystals of 1 was carried out by combining the metal salt and the linker in water. For compound 2 the preparation was different: Two separate warm solutions were assembled and later combined. The first solution contained the metal salt with ascorbic acid and the second one contained the linker. This approach was chosen to avoid rapid precipitation as powders. A similar synthesis was done for 3: Additionally, a third warm solution with NH4NCS was prepared and in order to form the intermediate product, ferrous nitrate, the solution of the metal salt and ascorbic acid were combined with this solution first. After that the solution of the linker was added [67]. The crystals have a colorless or a light-yellow color and the microscopic images of the crystals can be found in the Supplementary Materials, Section S5.
Scheme 1.
Schematic presentation of the synthesis of the coordination polymers 1-3 with their double chain structures. The blue spheres represent Fe(II) atoms.
Scheme 1.
Schematic presentation of the synthesis of the coordination polymers 1-3 with their double chain structures. The blue spheres represent Fe(II) atoms.
The IR spectra of the coordination polymers 1-3 (see Supplementary Materials, section S3) show their indicative bands for the ttmb linker at around 977 cm–1 for ν(C=C) and 1519 cm–1 for ν(C=N). Additionally, the characteristic bands for the anions of the compounds can be detected for the ClO4– of 1 at ν(Cl–O) = 621 cm–1, 1078 cm–1. The band of the anion BF4– of 2 can be observed at ν(B–F) = 1047 cm–1 and the anion of 3 at ν(NCS) = 2046 cm–1 [68].
Thermogravimetric analysis (at a heating rate of 5 K min–1, see Supplementary Materials, Section S4) revealed that the decomposition for compounds 1 and 2 already starts at around 70 °C due to the loss of crystal water. The main decomposition starts for compound 1 at 190 °C and for 2 at 250 °C. Compound 3, which does not contain solvent of crystallization, is thermally stable up to 300 °C where the decomposition of the ligand starts.
The experimental powder X-ray diffraction patterns of 1-3 could be positively matched to the simulated pattern from the single-crystal X-ray analysis which indicated a high the phase purity for the crystalline part of each compound (see Supplementary Materials, Section S8). At the same time, light microscopy images of the batches of compound 1-3 showed almost exclusively crystalline matter (see Supplementary Materials, Section S5).
3.1. Crystal structure of [Fe(H2O)2(ttmb)2](X)2·4H2O (1: X = ClO4; 2: X = BF4)
Single crystal X-ray analysis reveals that the isostructural complexes 1 and 2 crystallize in the monoclinic space group P21/n. The asymmetric unit consists of one half of an Fe(II) atom (on an inversion center), one ttmb ligand, one coordinated and two uncoordinated water molecules, and one uncoordinated ClO4– or BF4– anion (Figure 1). Due to the steric hindrance of the ttmb ligand and the interactions with the anions, the metal center possesses a somewhat distorted octahedral geometry with a FeO2N4 coordination environment (see Supplementary Materials, Section S7).
The equatorial positions are occupied by four nitrogen atoms which are provided by four pyrazole groups of four ttmb units. The oxygen atoms of the water molecules are trans-positioned around the Fe(II) atom (Figure 1).
For compound 1 the distances of the Fe—N bonds range from 2.14 and 2.22 Å. The Fe—N bond lengths for comound 2 are close to the ones in 1 with distances from 2.08 to 2.22 Å. The Fe–O length is about 2.07 Å for 1 and 2.08 Å for 2.
Every ttmb ligand functions as a twofold bridge and connects two Fe(II) atoms with an anti-conformation of the coordinating pyrazole groups. The spanned distance between the Fe atoms is 13.46 Å. Every Fe(II) atom is connected to its neighbor by two ttmb linkers in a double chain structure (Figure 2). Since the perchlorate and tetrafluoroborate anion have similar sizes, similar geometries and also similar chemical hardness, it was expected that both compounds 1 and 2 have the same structure [69]. The ionic radius of ClO4– is 2.37 Å the one of BF4– is 2.29 Å [36].
The ClO4– and BF4– anions have C–H···O and C–H···F, respectively, hydrogen bond interactions from the triazole groups of the ttmb linker [69]. For compound 1 Figure 3 shows that a ClO4– anion interacts with one C-H of each triazole ring of the ttmb linker. For the C–H···O interactions in 1 the hydrogen bond lengths range from 2.44 to 2.65 Å (Supplementary Materials, Table S4). The C–H···F contacts in 2 are in the range of 2.45–2.55 Å (Supplementary Materials, Table S4).
3.2. Crystal Structure of [Fe(ttmb)2(NCS)2] (3)
The X-ray crystallographic analysis reveals that compound 3 crystallizes in the triclinic space group . The asymmetric unit is composed one half of an Fe(II) atom (on an inversion center), one ttmb ligand and one coordinated NCS– anion, as shown in Figure 4. The Fe(II) metal is octahedrally coordinated by six nitrogen donor atoms from four triazole rings in the equatorial plane and two trans-coordinated NCS– in the axial positions.
The distances of the Fe–N bonds lie between 2.07 and 2.23 Å. Similar to the structures of 1 and 2, two Fe(II) atoms are linked by two ttmb linkers in an anti-conformation to form a double chain structure (Figure 5). The distance between two Fe(II) atoms is 13.79 Å.
4. Conclusions
The new iron(II) coordination polymers [Fe(H2O)2(ttmb)2](ClO4)2·4H2O (1), [Fe(H2O)2(ttmb)2](BF4)2·4H2O (2), [Fe(ttmb)2(NCS)2] (3) with the linker 1,3,5-tris(1H-1,2,4-triazol-1-yl)methyl)benzene (ttmb) were structurally characterized by single-crystal X-ray diffraction. It was hoped for that the potentially tritopic ttmb linker would utilize its three triazole N-donor groups for metal coordination to form at least 2D if not even 3D coordination networks. Somewhat to a surprise, only two of the triazole groups coordinated to iron. This could have still given a 2D framework if each of the four ttmb linkers around an iron atom would connect to a different iron. Yet, two ttmb linkers each did connect to the same iron atom through a double bridge. Subsequently, 1D coordination polymers with a double chain structure and a 1:2 Fe:ttmb stoichiometry resulted. We note that the metal ions were even offered in excess during the optimized synthesis with a molar M:L ratio of 2:3 for 1 and 2:1 for 2. We hypothesize that it is not so much the counter anions which exert a templating effect in the structures of 1-3 but the water solvent (or water/ethanol for 2) from which the compounds were crystallized. Future work using different solvents and also other metal ions for the crystallization should try to invoke the coordination of all triazole groups of the ttmb linker.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Section S1: Used Chemicals; Section S2: Ligand analyses; Section S3: Infrared spectra of 1-3; Section S4: Thermogravimetric analyses of 1-3; Section S5: Crystal images of 1-3; Section S6: Crystal data of 1-3; Section S7: Distortion of the coordination polyhedron of 1-3; Section S8: Powder X-ray diffraction patterns of 1-3; Section S9: References.
Author Contributions
Conceptualization, A.L., D.N.J., L.P.C., Y.G. and C.J.; methodology, A.L., D.N.J. and L.P.C.; validation, A.L., D.N.J. and L.P.C.; formal analysis, A.L, D.N.J., T.S. and L.P.C.; investigation, A.L, D.N.J., T.S. and L.P.C.; resources, C.J.; data curation, A.L., D.N.J. and T.S.; writing—original draft preparation, A.L.; writing—review and editing, C.J.; visualization, A.L. and C.J.; supervision, C.J.; project administration, C.J.; funding acquisition, C.J. and Y.G. All authors have read and agreed to the published version of the manuscript.
Funding
Fonds De La Recherche Scientifique—FNRS (PDR T.0095.21, CDR J.0064.23) and by the Deutsche Forschungsgemeinschaft (DFG) under grant 440366605 (for the Rigaku diffractometer), grant INST 208/589-1 (for the Bruker diffractometer) and within the Priority Program SPP 1928/2 COORNETs (C.J. grant Ja466/43-1).
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The CCDC numbers 2297073-2297075 for 1–3, respectively, contain the supplementary crystallographic data reported in this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif (accessed on 25 September 2023).
Acknowledgments
The authors thank Birgit Tommes for IR measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Batten, S.R.; Neville, S.M.; Turner, D.R. Coordination polymers. Design, analysis and application; Royal Society of Chemistry: Cambridge, UK, 2009; ISBN 978-0-85404-837-3. [Google Scholar]
- Batten, S.R.; Champness, N.R.; Chen, X.-M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O'Keeffe, M.; Suh, M.P.; Reedijk, J. Coordination polymers, metal–organic frameworks and the need for terminology guidelines. CrystEngComm 2012, 14, 3001. [Google Scholar] [CrossRef]
- Batten, S.R.; Champness, N.R.; Chen, X.-M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’Keeffe, M.; Paik Suh, M.; Reedijk, J. Terminology of metal–organic frameworks and coordination polymers (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1715–1724. [Google Scholar] [CrossRef]
- Kuznetsova, A.; Matveevskaya, V.; Pavlov, D.; Yakunenkov, A.; Potapov, A. Coordination Polymers Based on Highly Emissive Ligands: Synthesis and Functional Properties. Materials 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Leong, W.L.; Vittal, J.J. One-dimensional coordination polymers: complexity and diversity in structures, properties, and applications. Chem. Rev. 2011, 111, 688–764. [Google Scholar] [CrossRef]
- Kitagawa, S.; Kitaura, R.; Noro, S. Functional porous coordination polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef]
- Robin, A.Y.; Fromm, K.M. Coordination polymer networks with O- and N-donors: What they are, why and how they are made. Coord. Chem. Rev. 2006, 250, 2127–2157. [Google Scholar] [CrossRef]
- Naik, A.D.; Dîrtu, M.M.; Railliet, A.P.; Marchand-Brynaert, J.; Garcia, Y. Coordination Polymers and Metal Organic Frameworks Derived from 1,2,4-Triazole Amino Acid Linkers. Polymers 2011, 3, 1750–1775. [Google Scholar] [CrossRef]
- Hoskins, B.F.; Robson, R. Infinite polymeric frameworks consisting of three dimensionally linked rod-like segments. J. Am. Chem. Soc. 1989, 111, 5962–5964. [Google Scholar] [CrossRef]
- Horike, S.; Shimomura, S.; Kitagawa, S. Soft porous crystals. Nat. Chem. 2009, 1, 695–704. [Google Scholar] [CrossRef]
- Kitagawa, S.; Noro, S.; Nakamura, T. Pore surface engineering of microporous coordination polymers. Chem. Commun. 2006, 701–707. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular chemistry: Concepts and perspectives : a personal account built upon the George Fisher Baker lectures in chemistry at Cornell University [and] Lezioni Lincee, Accademia nazionale dei Lincei, Roma; VCH: Weinheim, New York, 1995; ISBN 3527293124. [Google Scholar]
- Mu, Y.; Han, G.; Ji, S.; Hou, H.; Fan, Y. Coordination polymers based on a flexible bis(triazole) ligand and aromatic polycarboxylate anions: syntheses, topological structures and photoluminescent properties. CrystEngComm 2011, 13, 5943. [Google Scholar] [CrossRef]
- Du, M.; Bu, X.-H.; Huang, Z.; Chen, S.-T.; Guo, Y.-M.; Diaz, C.; Ribas, J. From metallacyclophanes to 1-D coordination polymers: role of anions in self-assembly processes of copper(II) and 2,5-bis(3-pyridyl)-1,3,4-oxadiazole. Inorg. Chem. 2003, 42, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Chand, D.K.; Biradha, K.; Fujita, M.; Sakamoto, S.; Yamaguchi, K. A molecular sphere of octahedral symmetry. Chem. Commun. 2002, 2486–2487. [Google Scholar] [CrossRef]
- Friese, V.A.; Kurth, D.G. From coordination complexes to coordination polymers through self-assembly. Current Opinion in Colloid & Interface Science 2009, 14, 81–93. [Google Scholar] [CrossRef]
- Li, W.; Kim, Y.; Li, J.; Lee, M. Dynamic self-assembly of coordination polymers in aqueous solution. Soft Matter 2014, 10, 5231–5242. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-L.; Sun, W.-Y.; Zhu, H.-L.; Yu, K.-B.; Tang, W.-X. A two-dimensional network constructed via 72-membered heart-shaped macrocycles, a copper(II) complex with 1,3,5-tris(imidazol-1-ylmethyl)-2,4,6-trimethylbenzene and diethylenetriamine ligands. Inorg. Chim. Acta 1999, 295, 129–135. [Google Scholar] [CrossRef]
- Shi, X.; Tan, D.; Liu, Y.; Liang, G.; Zhang, X. Syntheses, structures and fluorescence properties of two novel polymers based on a flexible tripodal ligand 1,3,5-tris((1H-1,2,4-triazol -1-yl)methyl)benzene. J. Mol. Struct. 2014, 1074, 134–139. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, X.; Li, X.; Hou, H.; Fan, Y. Structure analysis and catalytic property of a microporous framework based on a flexible tripodal ligand with novel conformations. J. Mol. Struct. 2011, 996, 110–114. [Google Scholar] [CrossRef]
- Yin, X.-J.; Zhou, X.-H.; Gu, Z.-G.; Zuo, J.-L.; You, X.-Z. Syntheses and physical properties of three-dimensional coordination polymers with the flexible tripodal ligand 1,3,5-tris(1,2,4-triazol-1-ylmethyl)benzene. Inorg. Chem. Commun. 2009, 12, 548–551. [Google Scholar] [CrossRef]
- Shi, Z.; Pan, Z.; Zhang, C.; Zheng, H. Syntheses, structures, and properties of six cobalt(II) complexes based on a tripodal tris(4-(1H-1,2,4-triazol-1-yl)phenyl)amine ligand. Dalton Trans. 2015, 44, 16854–16864. [Google Scholar] [CrossRef]
- Li, B.Z.; Liu, X.G.; Wang, Z.H.; Li, B.L.; Zhang, Y. A novel two-dimensional network cadmium(II) coordination polymer containing one 1,4-bis(1,2,4-triazol-1-yl)butane and double dicyanamide bridges. Acta Crystallogr. C 2006, 62, m10–2. [Google Scholar] [CrossRef] [PubMed]
- Woschko, D.; Millan, S.; Ceyran, M.-A.; Oestreich, R.; Janiak, C. Synthesis of a Chiral 3,6T22-Zn-MOF with a T-Shaped Bifunctional Pyrazole-Isophthalate Ligand Following the Principles of the Supramolecular Building Layer Approach. Molecules 2022, 27, 5374. [Google Scholar] [CrossRef]
- Aromí, G.; Barrios, L.A.; Roubeau, O.; Gamez, P. Triazoles and tetrazoles: Prime ligands to generate remarkable coordination materials. Coordination Chemistry Reviews 2011, 255, 485–546. [Google Scholar] [CrossRef]
- Haasnoot, J.G. Mononuclear, oligonuclear and polynuclear metal coordination compounds with 1,2,4-triazole derivates as ligands. Coord. Chem. Rev. 2000, 200-202, 131–185. [Google Scholar] [CrossRef]
- Garcia, Y.; Adarsh, N.N.; Naik, A.D. Crystal engineering of Fe(II) spin crossover coordination polymers derived from triazole or tetrazole ligands. Chimia (Aarau) 2013, 67, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Yi, L.; Cheng, P.; Liao, D.-Z.; Yan, S.-P. Synthesis and characterization of a 3D coordination polymer based on trinuclear triangular CuII as secondary building units. Inorg. Chem. 2006, 45, 5799–5803. [Google Scholar] [CrossRef]
- Ouellette, W.; Liu, H.; O'Connor, C.J.; Zubieta, J. Solid-state coordination chemistry of copper(II) tetrazolates: anion control of frameworks constructed from trinuclear copper(II) building blocks. Inorg. Chem. 2009, 48, 4655–4657. [Google Scholar] [CrossRef]
- Ouellette, W.; Hudson, B.S.; Zubieta, J. Hydrothermal and structural chemistry of the zinc(II)- and cadmium(II)-1,2,4-triazolate systems. Inorg. Chem. 2007, 46, 4887–4904. [Google Scholar] [CrossRef]
- Zhang, J.-P.; Chen, X.-M. Crystal engineering of binary metal imidazolate and triazolate frameworks. Chem. Commun. 2006, 1689–16990. [Google Scholar] [CrossRef]
- Wang, H.-P.; Wang, H.-L.; Li. B.-L. Synthesis, Structure, Lumiescence and Thermal Stability Properties of a New (3,4)-Connected 2D Zn Coordination Polymer. J. Struct. Chem. 2020, 10, 1835–1840. [Google Scholar] [CrossRef]
- Li, H.; Liu, G.; Liu, T.T.; Zhang, H.Y.; Yue, F.; Wang, J.D. Syntheses of triazole-bridged cadmium coordination polymer with luminescence properties. Russ J Coord Chem 2011, 37, 8–11. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Luan, J.; Wang, X.-L.; Zhang, J.-W.; Liu, G.-C.; Tian, A.-X. Construction and properties of cobalt(ii )/copper(ii ) coordination polymers based on N-donor ligands and polycarboxylates mixed ligands. RSC Adv 2014, 4, 62430–62445. [Google Scholar] [CrossRef]
- Schweifer, J.; Weinberger, P.; Mereiter, K.; Boca, M.; Reichl, C.; Wiesinger, G.; Hilscher, G.; van Koningsbruggen, P. J.; Koojiman, H.; Grunert, M.; Linert, W. catena-[µ-Tris(1,2-bis(tetrazol-1-yl)ethane-N4,N4´)iron(II)] bis(tetrafluoroborate): synthesis, structure, spectroscopic and magnetic characterization of a chain-type coordination polymer spin-crossover compound. Inorg. Chim. Acta 2002, 339, 297–306. [Google Scholar] [CrossRef]
- Absmeier, A.; Bartel, M.; Carbonera, C.; Jameson, G.N.L.; Werner, F.; Reissner, M.; Caneschi, A.; Létard, J.-F.; Linert, W. Mutual Influence of Spacer Length and Noncoordinating Anions on Thermal and Light-Induced Spin-Crossover Properties of Iron(II)–α,ω-Bis(tetrazol-1-yl)alkane Coordination Polymers. Eur J Inorg Chem 2007, 2007, 3047–3054. [Google Scholar] [CrossRef]
- Li, D.-P.; Zhou, X.-H.; Liang, X.-Q.; Li, C.-H.; Chen, C.; Liu, J.; You, X.-Z. Novel Structural Diversity of Triazolate-Based Coordination Polymers Generated Solvothermally with Anions. Crystal Growth & Design 2010, 10, 2136–2145. [Google Scholar] [CrossRef]
- Schottel, B.L.; Chifotides, H.T.; Shatruk, M.; Chouai, A.; Pérez, L.M.; Bacsa, J.; Dunbar, K.R. Anion-pi interactions as controlling elements in self-assembly reactions of Ag(I) complexes with pi-acidic aromatic rings. J. Am. Chem. Soc. 2006, 128, 5895–5912. [Google Scholar] [CrossRef] [PubMed]
- Busch, D.H. Structural Definition of Chemical Templates and the Prediction of New and Unusual Materials. Am. Chem. Soc. 1992, 12, 389–395. [Google Scholar] [CrossRef]
- Anderson, S.; Anderson, H.L.; Sanders, J.K.M. Expanding roles for templates in synthesis. Acc. Chem. Res. 1993, 26, 469–475. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, L.-Y.; Liu, X.-G.; Wang, J.; Li, B.-L.; Li, H.-Y. Structural versatility of seven copper(ii) coordination polymers constructed with the long flexible ligand 1,4-bis(1,2,4-triazol-1-yl)butane. CrystEngComm 2011, 13, 6090. [Google Scholar] [CrossRef]
- Tanaka, D.; Kitagawa, S. Template Effects in Porous Coordination Polymers. Chem. Mater. 2008, 20, 922–931. [Google Scholar] [CrossRef]
- Halper, S.R.; Do, L.; Stork, J.R.; Cohen, S.M. Topological Control in Heterometallic Metal-Organic Frameworks by Anion Templating and Metalloligand Design. J. Am. Chem. Soc. 2006, 128, 15255–15268. [Google Scholar] [CrossRef] [PubMed]
- Beer, P.D.; Gale, P.A. Anion Recognition and Sensing: The State of the Art and Future Perspectives. Angew. Chem. Int. Ed. 2001, 40, 486–516. [Google Scholar] [CrossRef]
- Huang, X.-C.; Zhang, J.-P.; Chen, X.-M. A new route to supramolecular isomers via molecular templating: nanosized molecular polygons of copper(I) 2-methylimidazolates. J. Am. Chem. Soc. 2004, 126, 13218–13219. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-C.; Zhang, J.-P.; Lin, Y.-Y.; Chen, X.-M. Triple-stranded helices and zigzag chains of copper(I) 2-ethylimidazolate: solvent polarity-induced supramolecular isomerism. Chem. Commun. 2005, 2232–2234. [Google Scholar] [CrossRef]
- Vilar, R. Anion-templated synthesis. Angew. Chem. Int. Ed Engl. 2003, 42, 1460–1477. [Google Scholar] [CrossRef]
- Wang, H.; Feng, Y.; Liang, N.; Li, B.; Zhang, Y. Two nickel coordination polymers with flexible ligand 1,3,5-tri(1,2,4-triazol-1-ylmethyl)-2,4,6-trimethylbenzene. Inorg. Chem. Commun. 2009, 12, 1161–1163. [Google Scholar] [CrossRef]
- Gimeno, N.; Vilar, R. Anions as templates in coordination and supramolecular chemistry. Coordination Chemistry Reviews 2006, 250, 3161–3189. [Google Scholar] [CrossRef]
- Kim, H.-J.; Zin, W.-C.; Lee, M. Anion-directed self-assembly of coordination polymer into tunable secondary structure. J. Am. Chem. Soc. 2004, 126, 7009–7014. [Google Scholar] [CrossRef]
- Vilar, R. Anion-templated synthesis. Angew. Chem. Int. Ed Engl. 2003, 42, 1460–1477. [Google Scholar] [CrossRef]
- Wang, H.; Feng, Y.; Liang, N.; Li, B.; Zhang, Y. Two nickel coordination polymers with flexible ligand 1,3,5-tri(1,2,4-triazol-1-ylmethyl)-2,4,6-trimethylbenzene. Inorganic Chemistry Communications 2009, 12, 1161–1163. [Google Scholar] [CrossRef]
- Lankshear, M.D.; Beer, P.D. Strategic anion templation. Coordination Chemistry Reviews 2006, 250, 3142–3160. [Google Scholar] [CrossRef]
- Shi, Q.; Cao, R.; Sun, D.-F.; Hong, M.-C.; Liang, Y.-C. Solvothermal syntheses and crystal structures of two metal coordination polymers with double-chain structures. Polyhedron 2001, 20, 3287–3293. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; Wood, P.A. Mercury 4.0: from visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Agilent; Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014.
- APEX2, data collection program for the CCD area-detector system, Version 2.1-0, Bruker Analytical X-ray Systems, Madison (WI), USA,1997–2014.
- SAINT, data reduction and frame integration program for the CCD area-detector system, Bruker Analytical X-ray Systems, Madison(WI), USA, 1997–2014.
- Sheldrick, G.M. SADABS: Area-Detector Absorption Correction, University of Göttingen, Göttingen, Germany, 1996.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2 : a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. , Diamond (Version 4.5), Crystal and Molecular Structure Visualization, Crystal Impact – K. Brandenburg & H. Putz Gbr, Bonn, Germany, 2009–2022.
- Shang, Q.; Zeng, T.; Gao, K.; Liu, N.; Cheng, Q.; Liao, G.; Pan, Z.; Zhou, H. A novel nitrogen heterocyclic ligand-based MOF: synthesis, characterization and photocatalytic properties. New J. Chem. 2019, 43, 16595–16603. [Google Scholar] [CrossRef]
- Garcia, Y.; Bravic, G.; Gieck, C.; Chasseau, D.; Tremel, W.; Gütlich, P. Crystal structure, magnetic properties, and 57Fe Mössbauer spectroscopy of the two-dimensional coordination polymers M(1,2-bis(1,2,4-triazol-4-yl)ethane)2(NCS)2 (MII = Fe, Co). Inorg. Chem. 2005, 44, 9723–9730. [Google Scholar] [CrossRef]
- Lavrenova, L.G.; Yudina, N.G.; Ikorskii, V.N.; Varnek, V.A.; Oglezneva, I.M.; Larionov, S.V. Spin-crossover and thermochromism in complexes of iron(II) iodide and thiocyanate with 4-amino-1,2,4-triazole. Polyhedron 1995, 14, 1333–1337. [Google Scholar] [CrossRef]
- Hesse, M.; Meier, H.; Zeeh, B.; Bienz, S.; Bigler, L.; Fox, T. Spektroskopische Methoden in der Organischen Chemie; 8 Auflage; Georg Thieme Verlag: Stuttgart, Germany, 2012; ISBN 9783135761084. [Google Scholar]
- Jordan, D.N.; Straßburg, P.G.; Woschko, D.; Carrella, L.M.; Cuignet, L.P.; Eickmeier, K.; Dronskowski, R.; Garcia, Y.; Rentschler, E.; Janiak, C. Interpenetration Phenomena via Anion Template Effects in Fe(II) and Co(II) Coordination Networks with a Bis-(1,2,4-triazole) Ligand. Polymers 2023, 15, 3286. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Extended asymmetric unit of (a) 1 and (b) 2 (50 % thermal ellipsoids and H atoms with arbitrary radii). The distorted F atoms of the BF4– anion of compound 2 are presented as transparent atoms. Symmetry transformation: (a): i=-x+1, -y+1, -z; ii=-x+1, -y+1, -z+1; iii=x, y, z-1; iv=x, y, z+1; v=-x+2, -y+1, -z+1; vi=x-1/2, -y+3/2, z-1/2; vii=-x+3/2, y-1/2, -z+3/2, viii=x+1, y, z+1. (b): i = −x+2, −y+1, −z+2; ii = −x+2, −y+1, −z+1; iii = x, y, z+1; iv = x, y, z−1; v = −x+1, −y+1, −z+1; vi = −x+3/2, y+1/2, −z+3/2.
Figure 1.
Extended asymmetric unit of (a) 1 and (b) 2 (50 % thermal ellipsoids and H atoms with arbitrary radii). The distorted F atoms of the BF4– anion of compound 2 are presented as transparent atoms. Symmetry transformation: (a): i=-x+1, -y+1, -z; ii=-x+1, -y+1, -z+1; iii=x, y, z-1; iv=x, y, z+1; v=-x+2, -y+1, -z+1; vi=x-1/2, -y+3/2, z-1/2; vii=-x+3/2, y-1/2, -z+3/2, viii=x+1, y, z+1. (b): i = −x+2, −y+1, −z+2; ii = −x+2, −y+1, −z+1; iii = x, y, z+1; iv = x, y, z−1; v = −x+1, −y+1, −z+1; vi = −x+3/2, y+1/2, −z+3/2.
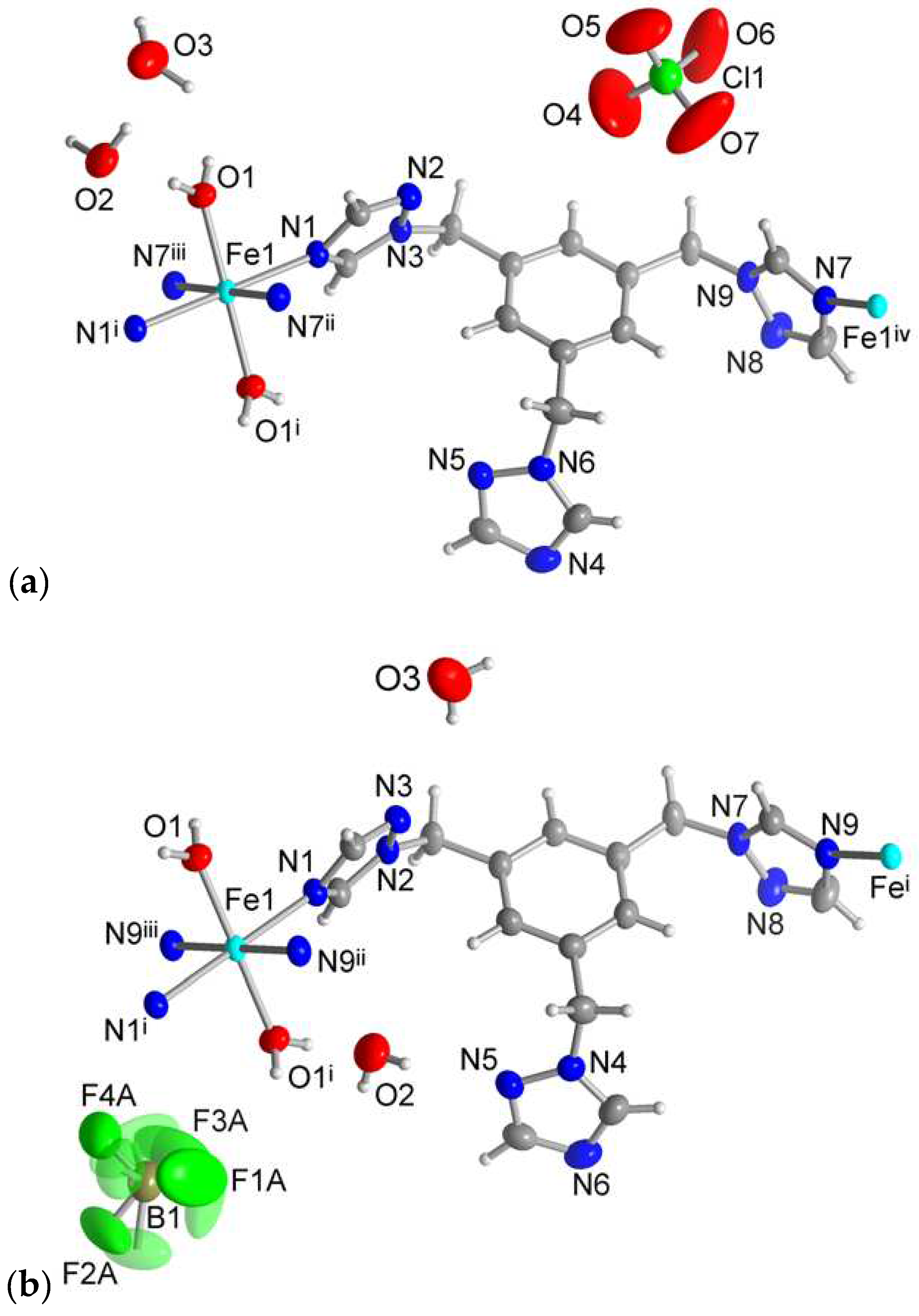
Figure 2.
View of the double chain of (a) 1 and (b) 2 with the surrounding crystal water and anions. For clarity the H atoms have been omitted on the ttmb linkers. (c) Presentation of the octahedral iron(II) environment in 1 (identical to 2).
Figure 2.
View of the double chain of (a) 1 and (b) 2 with the surrounding crystal water and anions. For clarity the H atoms have been omitted on the ttmb linkers. (c) Presentation of the octahedral iron(II) environment in 1 (identical to 2).
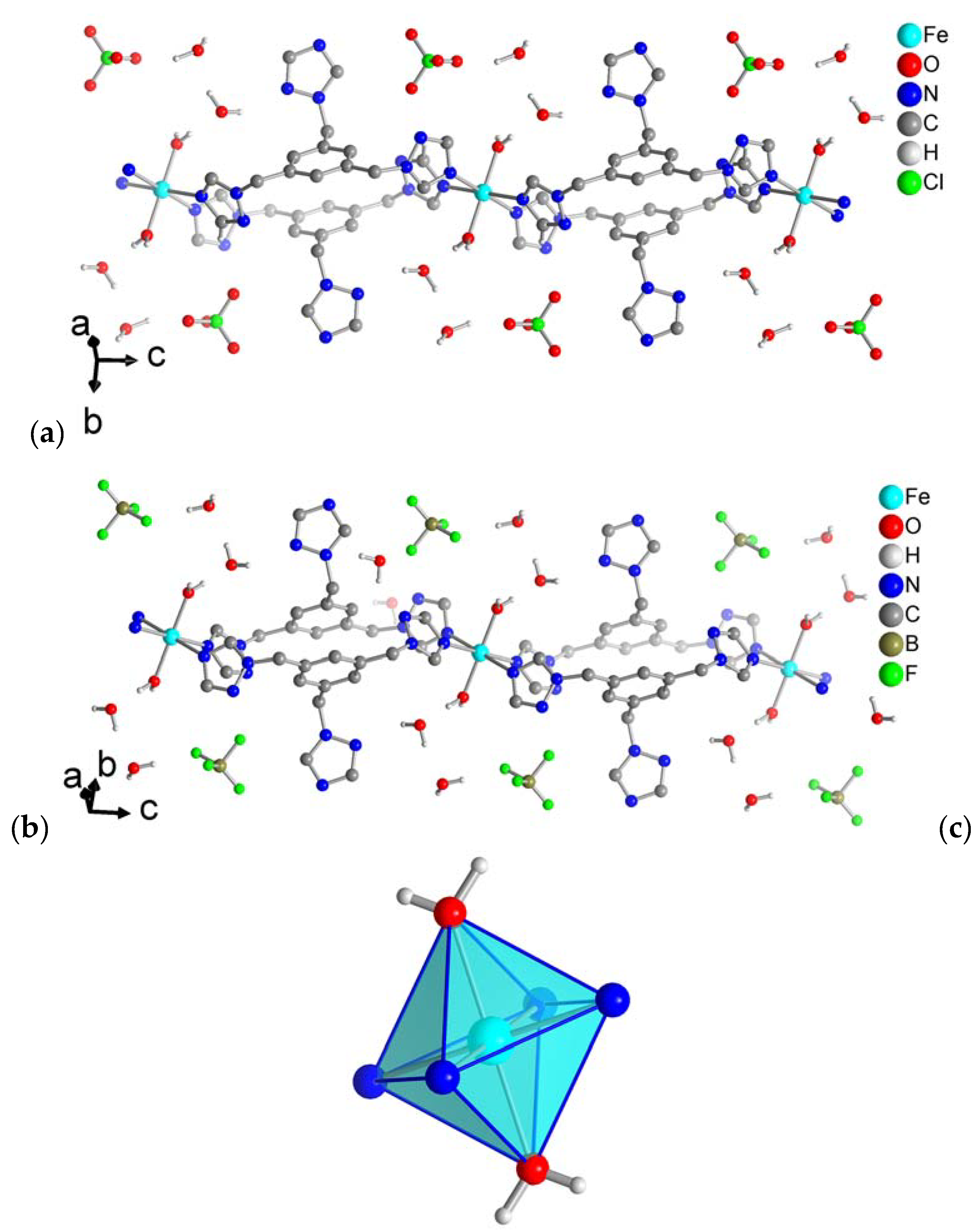
Figure 3.
Hydrogen-bonding interactons in (a) 1 and (b) 2 as orange dashed lines. See Supplementary Materials, Table S4 for distances and angles.
Figure 3.
Hydrogen-bonding interactons in (a) 1 and (b) 2 as orange dashed lines. See Supplementary Materials, Table S4 for distances and angles.
Figure 4.
Extended asymmetric unit of 3 (50 % thermal ellipsoids and H atoms with arbitrary radii). Symmetry transformations: i = -x+1, -y+2, -z; ii = x, y+1, z-1; iii = -x+1, -y+1, -z+1; iv = x, y-1, z+1; v = -x, -y+1, -z+1; vi = x, y-1, z.
Figure 4.
Extended asymmetric unit of 3 (50 % thermal ellipsoids and H atoms with arbitrary radii). Symmetry transformations: i = -x+1, -y+2, -z; ii = x, y+1, z-1; iii = -x+1, -y+1, -z+1; iv = x, y-1, z+1; v = -x, -y+1, -z+1; vi = x, y-1, z.
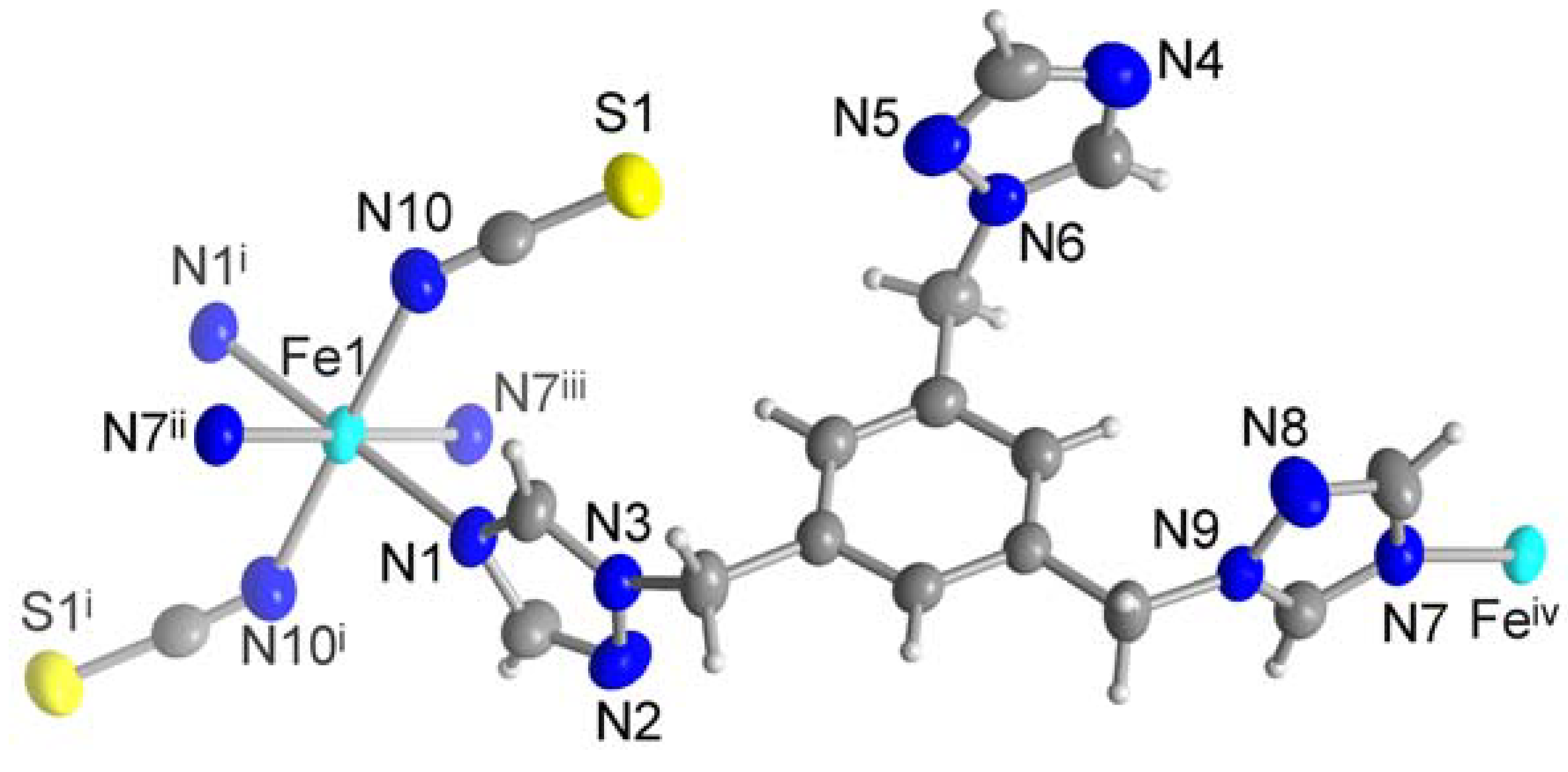
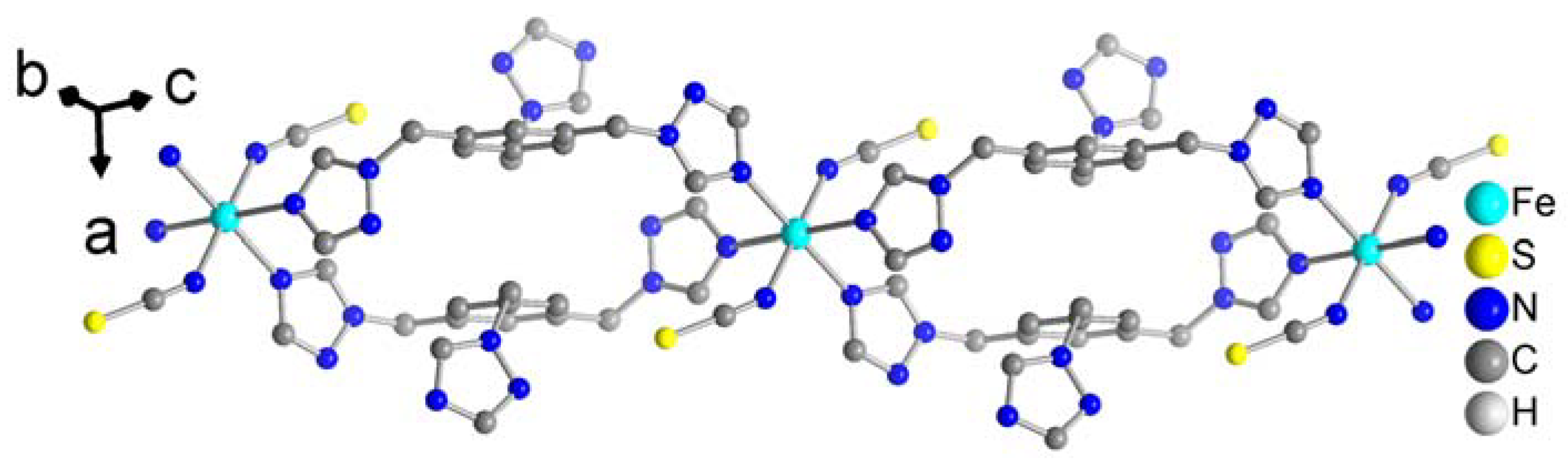
Figure 5.
The double chain of 3. For the sake of clarity the H atoms have been omitted.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



