
Submitted:
27 September 2023
Posted:
28 September 2023
You are already at the latest version
Abstract
Dysfunction and selective loss of retinal ganglion cells (RGCs) is a known cause of vision loss in glaucoma and other neuropathies where ocular hypertension (OHT) is the major risk factor. We investigated the impact of transient non-ischemic OHT spikes (spOHT) on RGC function and viability in vivo to identify cellular pathways linking low-grade repetitive mechanical stress to RGC pathology. We found that repetitive spOHT had an unexpectedly high impact on intraocular homeostasis and RGC viability, while exposure to steady OHT (stOHT) of similar intensity and duration failed to induce pathology. The repetitive spOHT induced a rapid activation of the inflammasome, marked by the upregulation of NLRP1, NLRP3, AIM2, caspases -1, -3/7, -8, gasdermin D (GsdmD) and the release of interleukin-1β (IL-1β) and other cytokines into the vitreous. Similar effects were also detected after 5 weeks of exposure to chronic OHT in an induced glaucoma model. The onset of these immune responses in both spOHT and glaucoma models preceded a 50% deficit in pattern electroretinograms (PERG) amplitude, and significant loss of RGCs at 7 days post-injury. Inactivation of inflammasome complexes in NLRP1-/- , Casp1-/- and GsdmD-/- knockout animals, significantly suppressed the spOHT induced inflammatory response and protected RGCs. Our results demonstrate that mechanical stress produced by acute repetitive spOHT or chronic OHT is mechanistically linked to inflammasome activation, which leads to RGC dysfunction and death.
Keywords:
ocular hypertension
; intraocular pressure spikes
; retinal ganglion cells
; inflammasome
; caspase-1
; interleukin-1β
; neuroinflammation
1. Introduction
The eye challenged with chronic OHT, like in the ocular hypertension type of glaucoma, is exposed to multiple mechanical, ischemic and metabolic stresses that affect essential RGC functions, including axonal transport, energy homeostasis, electrophysiological outputs and glial activation[1,2,3,4,5]. Repetitive short-term OHT episodes, caused by intraocular pressure (IOP) “spikes”, are also recognized as important risk factors [6,7], though these are much less studied than chronic OHT. Mounting clinical and experimental evidence from animal studies indicate that transient IOP spikes are more damaging to vision when they are recurrent and/or reach the ischemic level, such as with eye rubbing [8,9]. Recurrent OHT spikes are typically induced by medications, eye surgeries, intraocular drug injections, head-down postures during post-surgical recovery, or some lifestyle activities, including playing wind instruments, weightlifting or head-down positions in yoga [10,11,12,13]. Repeated incidence of OHT spikes have been linked to nerve fiber layer thinning and vision loss in human eyes [8,14]. For example, repetitive intraocular drug injections were associated with vascular hypoperfusion [15,16,17], nerve fiber layer thinning [17,18] and it has also been suggested to play a role in normal tension glaucoma [9,13,19]. Although spOHT and chronic OHT modalities differ dramatically by the type of the initial injury, both could produce similar RGC and axon injuries clinically presented as glaucomatous [8,14]. To the best of our knowledge, the present work is the first to compare neuroinflammatory responses and their link to RGC pathology following spOHT and chronic OHT.
Neuroinflammation and accumulation of pro-inflammatory cytokines, the two common end-products of active inflammasomes[20,21,22,23], have been detected in animal[24] and human[25,26,27,28] eyes with glaucoma along with an increase in extracellular ATP, a common inducer of the inflammasome[29]. At the cellular level, neuroinflammatory responses mediated by the release of IL-1b and tumor necrosis factor (TNF-a) have been confirmed in astrocytes, Muller glia, microglia, and in RGCs in glaucomatous eyes [25,30,31,32]. Recent reports from us and others have shown very early activation of neuronal NLRP1/3 inflammasome, caspase-1, and the release of IL-1β in a rodent model of ischemic OHT injury [33,34,35], suggesting that inflammasome signaling may serve as the key trigger of glial responses. The key role of inflammation and inflammatory caspases, like Casp8, in the pathogenesis of glaucoma induced by non-ischemic OHT injury has been recently shown [36,37].
An active role of inflammasome activation in the pathogenesis of retinal OHT damage [38,39] could shift the current paradigm where inflammation is considered secondary to optic nerve damage and RGC pathology [40]. To gain insight into how the short-term repetitive non-ischemic OHT spikes lead to RGC injury, we investigated the impact of activation of retinal inflammasomes on RGC function and viability using a mouse model of IOP spike-induced (spOHT) injury. Here we report that repetitive episodes of non-ischemic IOP spikes lead to impaired RGC electrophysiological function and cell death. Our results strongly suggest that the effects of spiking OHT are mediated predominantly by acute innate immune responses triggered by the activation of the endogenous neuronal NLRP1 inflammasome. Our results also identify key components of the inflammasome, along with its upstream regulator the mechanosensitive Panx1 channels, as therapeutic targets for OHT-mediated RGC degeneration.
The present work is an important step towards a better understanding of the underlying molecular mechanism through spiking OHT-induced degenerative stress in RGCs. Furthermore, our results from multiple inflammasome knockout mice indicate that acute activation of inflammasomes and their downstream products are mechanistically involved in the OHT-induced RGC dysfunction and loss, while their inactivation protected RGCs.
2. Materials and Methods
Animals. Animal handling, anesthetic procedures, experiments and post-surgical care were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals and according to the University of Miami, Institutional Animal Care and Use Committee approved protocols #18-025 and #21-036. Wildtype (WT, C57BL/6J) and transgenic mice, including Casp1-/- (Casp1/Casp4(11)del strain B6N.129S2-Casp1tm1Flv/J, Jax # 016621); NLRP1b-/- (B6.129S6-Nlrp1btm1Bhk/J, Jax # 021301), and GsdmD-/- (C57BL/6N-Gsdmdem4Fcw/J, Jax # 032410), were obtained from Laboratories Depository (Bar Harbor, ME). Mice expressing ASC-citrine for ASC speck visualization were provided by Dr. D. Golenbock [41], University of Massachusetts Med. School. All animals were bred in the University of Miami animal facility and housed under standard conditions of temperature and humidity with a 12-h light/dark cycle and free access to food and water.
Reagents. Antibodies were purchased from commercial sources: anti-GFAP (Dako, cat#z0334), anti-AIF1/Iba1 (Wako, cat#019-19741), anti-Casp1 (Novus Biologicals, cat#IMG5028); caspase-1, p-20 (Adipogen, cat# AG-20B-0042); anti-IL-1β (Cell Signaling, cat#8689S), anti-NLRP1 (Novus cat# NB100-561148SS); anti-NLRP3 (R&D, cat#AF7010); anti-RBPMS (GeneTex, cat#118619); anti-Brn3a (SantaCruz, cat# sc31984); anti- Class III β-Tubulin (clone TUJ1, Covance); anti-GSDMDC1 (A7, Santa Cruz, cat# sc-271054); anti-Casp8 monoclonal antibody (1G12) (Enzo, cat# ALX-804-447); anti-ASC-1 (F-9, Santa Cruz, cat# sc-271054); anti-CD11b (Santa Cruz, cat# sc-271050); anti-CD45 (Santa Cruz, cat# sc-271024).
The Spiking OHT injury model. The mean arterial pressure in murine eyes, is 112 mm Hg [42] and transient non-ischemic IOP elevations to 30-40 mm Hg were shown to be not injurious [43,44,45]. The spiking OHT model, however, was administered by seven consecutive IOP elevations of 40 mmHg. During the procedure animals were under isoflurane (3%) gas anesthesia and topical analgesia was induced with 0.5% proparacaine HCl (Bausch & Lomb Pharmaceuticals, Rochester, NY). Pupils were dilated with 1% tropicamide and 2.5% phenylephrine hydrochloride (NutraMax Products, Inc., Gloucester, MA) to aid in placement of the pressure input needle. IOP was elevated by cannulation of the anterior chamber with a 29G needle connected to a reservoir of normal saline (0.9% NaCl) that was placed 54 cm above the eye level to achieve an H2O pressure equivalent to 40 mm Hg, as described [33,43]. A digital in-line digital mini-pressure gauge (Centurion Compass CUHG; precision ±1.5 mm Hg) was used to monitor IOP elevation and potential loss of pressure from leaks. The spOHT experimental paradigm, shown in Figure 1A, consisted of seven consecutive 1-minute IOP spikes (spOHT) achieved by a rapid rise and consecutive lowering of the reservoir, with a 1-minute interval of normotension between spikes and quick transition between normal and elevated IOP. In the steady OHT group, IOP was elevated by a gradual 1-minute increase to 40 mmHg, which was maintained for 7 minutes, then returned to baseline gradually over 1 minute. Thus, both models produced the same maximum IOP exposure to 40 mm Hg for a total of 7 min and were induced by a single needle insertion through the peripheral cornea. Sham control procedure was performed by a single cannulation of the anterior chamber with no elevation of the reservoir (i.e. no IOP elevation) under anesthesia for the same duration as the experimental groups.
The Y437H-Myoc-induced OHT model. In this model, IOP was elevated by over expressing the pathogenic Y437H variant of human Myocilin, as described earlier by Grotegut and Kuehn [46,47]. To over express Y437H human Myocilin, the anterior chamber of each eye of an animal was injected with 1.4 µl of Ad5-Myoc suspension (5X107 pfu/eye) using 33G needle. A gradual IOP increase to 25-35mm was observed at 6-8 weeks in all mouse lines tested. Eyes were harvested and inner retinas collected at 3, 5 and 8 weeks after vector injection.
The retinal ischemia-reperfusion model. Retinal ischemia was achieved by increasing IOP above systolic blood pressure to 110 mm Hg for 45 min by direct cannulation of the anterior chamber with a 29 G needle connected to a normal saline (0.9% NaCl)-filled reservoir placed at 150 cm above the eye to create a pressure of 150 cm H2O (equivalent to 110 mm Hg), as previously described[33]. The pressure changes in the tube connected to the needle were calibrated using an in-line Centurion Compass CUHG1 digital pressure transducer (Centurion Medical Products Inc) prior to experiments. The contralateral eyes, cannulated at normal IOP, served as normotensive controls. Complete retinal ischemia was confirmed as the whitening of the anterior segment and blanching of the retinal arteries. Eyes were harvested 24h post-injury, mice were euthanized by CO2 overdose and eyes were collected. The vitreous body was harvested for cytokine analysis by ELISA, and retinas were dissected out, fixed and processed.
Intravitreal cytokine activity assay. To collect the vitreous, mice were perfused with phosphate-buffered saline (PBS), eyes were collected, placed on ice, and immediately dissected. Vitreous fluid was collected with three consecutive flushes of the vitreous cavity with 20 µl sterile PBS containing a protease inhibitor cocktail. All flush samples were combined, spun for 5 min in a refrigerated centrifuge, and stored at -80ºC. ELISA kits for mouse IL-1b (R&D ID# MBL00C) or Ella SimplePlex for IL1b, TNFa, and MCP1/CCL2 (Protein Simple) as described in [48] were used to measure cytokine released into the vitreous. Sample aliquots were processed for protein analyses in parallel with standards and controls following the manufacturer’s instructions. A colorimetric assay was done using a FLUOstar Omega plate reader (BMG Labtech) and analyzed using MARS data analysis software (BMG Labtech). Values from the wells containing blank samples were subtracted from the background. To validate the significance of measurements at the lowest reading, the limit of detection (LOD) and limit of quantification (LOQ) ratios were calculated from empirical data obtained in the ‘‘zero’’ wells of each plate, as described[49]. A minimum of three (N= 3) biological repeats were used for each data point. Significance was calculated using one-way analyses of variance (ANOVA) followed by Tukey’s test for multiple comparisons. To measure the co-release of IL-1b, TNFa and CCL2 cytokines we used the Ella Simple Plex microfluidic technology (Protein Simple) with internal calibration as described in [48].
In vivo retinal electrophysiology and data analysis. An optimized protocol for PERG (pattern electroretinogram) recording in mice was previously described [50,51]. Briefly, animals were anesthetized with ketamine/xylazine (80/10 mg/kg) and gently restrained in an animal holder. PERG signals were recorded simultaneously from both eyes from subdermal electrodes in the snout in response to horizontal bars that maximized PERG amplitude and minimized the noise (spatial frequency 0.05 cycles/deg, temporal frequency 1 Hz, contrast 100%, robust averaging of 2232 sweeps). The PERG signal-to-noise ratio was of the order of 10, and the test–retest variability was of the order of 30% [52]. Balanced salt solution (BSS) drops were applied to maintain cornea hydration.
RGC loss assessment. To assess RGC loss, retinas were collected at 7 days post-injury 7 dpi, fixed in 4% paraformaldehyde, and flat-mounted. RGCs were identified by RBPMS (RNA binding protein with multiple splicing) immunolabeling, visualized by confocal microscopy. RBPMS-positive cells were counted with ImageJ plugin open-source software after thresholding and manual exclusion of artifacts. Each retina was sampled from 16 fields in 4 retinal quadrants in 3 regions of the same eccentricities (0.5 mm, 1.0 mm, 1.5 mm from the optic disk) as previously described [33]. RGC loss was calculated as a percentage of RBPMS-positive cells in experimental eyes relative to sham-operated contralateral control eyes. The cell density data (n ≥5) were averaged for each group/genotype; statistical analyses data were analyzed with one-way ANOVA followed by the Tukey test for multiple comparisons; P values ≤0.05 were considered statistically significant.
Inflammasome detection using the citrine-labeled ASC-speck complex in vivo has been described earlier [34]. ASC-citrine was previously shown to incorporate into oligomerizing inflammasome complexes, thus providing a surrogate inflammasome activation in mouse tissues [53]. The bioindicator mice expressing ASC fusion protein with a C-terminal citrine protein (fluorescent GFP isoform) that brightly labels the filamentous ASC specks in vivo and allows for visualization in vivo were provided by Dr. D. Golenbock (University of Massachusetts, MA, USA).
Real-time PCR. Gene expression was assessed by real-time PCR using gene-specific primer pairs (primer pairs were validated to span an intron and to amplify only one product (see Supplementary data Table S1 for details). Total RNA from 2-4 pooled retinas was extracted using Trizol and quality controlled by Nanodrop. cDNA was synthesized with the Reverse Transcription System (Promega, Fitchburg, WI, USA). Real-time PCR was performed in the Rotor-Gene 6000 Cycler (Corbett Research, Mortlake, Australia) using the SYBR GREEN PCR MasterMix (Qiagen, Valencia, CA). Relative expression was calculated by comparison with a standard curve following normalization to the β-actin or Gapdh genes.
Immunohistochemistry. Eyes were enucleated, fixed in 4% paraformaldehyde for 1 h, and cryoprotected with 30% sucrose. Retinas were embedded into the OCT media and frozen-sectioned to a thickness of 10 µm on a microtome (Leica). Slides were washed in PBS, permeabilized in PBS with 0,2% Tween20, and incubated with a primary antibody for 4–16 h. Retinal flat-mounts were incubated with primary antibodies for 3–5 days at 4ºC to ensure even staining. To identify target proteins, specific antibodies were diluted and incubated 4-16h. After washes with PBS-Tween 20, secondary antibodies were applied for 2-4h for frozen sections and for 16h for whole mounts. Secondary AlexaFluor dye-labeled antibodies (Thermo Fisher Scientific, Waltham, MA) were applied for imaging with the Leica TSL AOBS SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany); controls with primary antibodies omitted were used for specificity tests.
Statistical Analysis. Statistical comparisons of PERG data were made using non-parametric Mann–Whitney test and Kruskal-Wallis test followed by post hoc Dunn’s Multiple Comparisons. Protein assay (ELISA, SimplePlex) data were presented as the mean ± standard deviation (SD) or standard error (SEM) for RGC survival data. Real-time PCR data were analyzed with one-way ANOVA followed by the Tukey test for multiple comparisons. For single comparisons, Student’s t-test was applied; one-way ANOVA was used for between-group comparisons. GraphPad Prism software (version 6.07; GraphPad Software, La Jolla, CA, USA) was used for statistical analysis. A minimum of three biological repeats per treatment was used for in vivo IL-1β release assessment and for gene expression analysis by quantitative RT-PCR. Groups of data were compared using ANOVA or two-tailed unpaired Student’s t-tests. Cell density data were analyzed with one-way ANOVA followed by Tukey’s test for multiple comparisons. For two group comparisons, Student’s t-test were carried out. P values < 0.05 were considered statistically significant for all analyses.
Description of common methods, namely immunohistochemistry and Western blot analysis is provided in the Supplementary information.
3. Results
3.1. Acute inflammasome induction after spOHT.
To characterize the effects of spOHT challenge on the inflammasome pathway, we measured changes in transcript levels of the genes encoding for IL-1β, TNFα, caspases -1, -3 and -8, Gasdermin-D (GsdmD) and the NLRP1b and NLRP3 inflammasome sensors in sham-operated vs experimental retinas. Significant increases in mRNA for il1b, tnfa, casp1, casp8, and nlrp3 genes (n=3, p<0.05) were detected in WT retinas after spOHT (red bars, Figure 1B), while expression of the nlrp1, casp3 and GsdmD genes remained unchanged. In contrast, no significant changes in expression of these genes, were detected in retinas challenged with stOHT (blue and gray bars, Figure 1B).
Inflammasome activation in the retina was previously shown to be induced within hours after ischemic-level IOP elevation [27,33,34,35]. To determine the time course of inflammasome activation in the spOHT model, we measured IL-1β levels in the vitreous body and IL1β gene expression in the retina of the C57BL6/J (WT) eyes after spHOT. A rapid increase in IL-1β was detected as early as 6 h post-spOHT, peaked at 12h and remained elevated at 24h (Figure 1C). Relative to the baseline, the levels of IL-1β in the vitreous increased by 3.8-, 16.8- and 18.7-fold at 6, 12 and 24h post-spOHT, respectively. IL-1β levels in eyes after stOHT were not significantly elevated, showing changes of 1.15-, 0.83- and 1.0- fold relative to corresponding baselines at 6, 12, and 24 h post-stOHT (Figure 1C).
To confirm the absence of ischemic conditions in spOHT retinas, we measured the expression of the ischemia marker protein HIF1α which was not elevated after spOHT. However, it was significantly upregulated in retinas that underwent IR (110mmHg IOP for 45 min) (Figure 1D), indicating that spOHT is a non-ischemic event.
3.2. SpOHT induces functional and structural damage to RGCs.
OHT-induced inflammasome activation has been previously shown to inversely correlate with the electrophysiological responsiveness of RGCs [54]. We measured PERG to assess RGC function after spOHT and stOHT (Figure 2A,B). SpOHT induced a persistent, but not progressive decrease in mean PERG amplitude, (35.57±8.13%, 31.07±7.83%, and 37.28±9.83%) as compared to the baseline at 7, 14, and 21 days after spOHT, respectively (Figure 2C). A progressive increase in PERG latency was also observed, averaged 6.43+/-4.34%, 8.37+/-2.92%, and 16.20+/-6.97% at 7, 14, and 21 days post spOHT, respectively (Supplement Figure S1). These data suggest that the impairment of RCG electrophysiological function is long-lasting and not reversible. SpOHT treatment also resulted in a significant RGC loss, averaging 20.41±4.83% (p<0.01) on day 7 post spOHT, 20.4±4.83% on day 14, and 10.52±11.73% on day 21. RGC loss in stOHT-treated eyes, on the other hand, was minimal (Figure 2D). These data indicate that IOP elevation to 40 mm Hg has dramatically different impacts on RGC function and survival when applied as seven repetitive 1-minute spikes vs. a single 7 min-long spike.
3.3. Inflammasome activity coordinates retinal neuroinflammation.
We used Casp1-/- mice with genetic inactivation of canonical inflammasomes, NLRP1b-/- mice with targeted inactivation of the neuron-specific inflammasome[55,56] and GsdmD-/- mice with the ablation of the gasderminD pore-forming protein, the IL-1β release conduit in many cell types, including CNS neurons and glia [34,57,58]. ELISA data showed that Casp1-/- and GsdmD-/- mice had a complete blockade of intravitreal IL-1β release levels (a non-significant 1.35-fold and 1.6-fold increase vs 12.1-fold increase in WT, relative to corresponding pre-injury baselines, respectively) at 24h post-spOHT. Importantly, the spOHT-induced levels in these knockout strains were not significantly different from those in the WT sham-treated eyes and in the naïve WT control eyes challenged with stOHT. The NLRP1-/- eyes, however, did not show any suppression of IL-1β release (14.4-fold increase) relative to its baseline, but the mean IL-1b release level in the vitreous of NLRP1-/- eyes was 2.7-fold lower than that in WT eyes after spOHT (Figure 3).
The measurements of concurrent release of IL-1β, TNFα and CCL2/MCP-1 cytokines in the same samples performed as SimplePlex assays showed strong co-regulation between the release of CCL2/MCP-1 and IL-1β in Casp1-/- and GsdmD-/- mice but not in Panx1-/- or NLRP1-/-. Thus, the release of TNFα was significantly suppressed in Casp1-/-, GsdmD-/- and Panx1-/-, while NLRP1-/- showed only partial suppression. These data indicate that the global increase in IL-1β is largely controlled by the activities of the canonical Casp1 inflammasomes and GsdmD cytokine release pores.
3.4. Panx1 is the mechanosensitive regulator of the inflammasome.
Panx1 is a cell surface mechanosensory Ca2+-permeable channel protein implicated in the pathophysiology of RGCs in retinal ischemia-reperfusion injury and glaucoma [33,34,39,54]. It is known to become activated by membrane stretch [59] like in the spOHT model used in this study. In the retina, it is expressed in many cell types but is particularly enriched in RGCs [33]. To investigate the potential role of Panx1 in the spOHT-induced injury to RGCs, we used Panx1-/- Casp11+/+ mouse strain with global genetic ablation of Panx1 and normal activity of Casp11 [60]. Consistent with relative suppression of genes encoding for IL-1β, TNFα, and CCL2 cytokines in the retinas of Panx1-/- mice, we found no significant increase in the release of IL-1β, TNFα, and CCL2 in the vitreous samples from the Panx1-/- mice 24 hrs after sp-OHT (Figure 3 and Figure 4), which contrasted with a significant upregulation in response spOHT challenge in WT eyes. Among the inflammasome structural genes, Panx1-/- retinas showed significant suppression of the casp1 and nlrp3 genes and partial suppression of nlrp1 transcripts (Figure 4); no suppression of the gsdmD gene was observed in these retinas.
Using electropysiological PERG recordings, we showed that inactivation of either Casp1, Nlrp1 or GsdmD genes significantly suppressed the rate of OHT-induced decline in PERG amplitude relative to pre-injury baselines in WT spOHT-challenged mice (Figure 5A). The between-group comparison showed no statistically significant decrease in PERG amplitude at 7dpi in experimental eyes of Casp1-/- (1.77±9.69% N=12 p=0.99), NLRP1-/- (10.33±8.43% N=14; p=0.89), GsdmD-/- (-24.38 ±9.96% N=11 p=0.13) and Panx1-/-(18.73±3.28% N=7 p=0.55). In addition, the PERG latency at 7dpi increased, although insignificantly, in both Casp1-/- (2.83±3.78% p=0.45), NLRP1-/- (4.49±2.56% p=0.1), GsdmD-/- mice (11.06±5.72% N=8 p=0.13) and Panx1-/- (3.70±4.22% N=8, p=0.28) eyes following the spOHT challenge. These results demonstrate that RGC dysfunction after the spOHT challenge requires the activities of Casp1, NLRP1, and the substrate GsdmD, which is known to be highly induced by these neurons after acute OHT challenge[34].
3.5. Inactivation of the inflammasome protects RGCs
To test the hypothesis that inflammasome activity plays a key role in RGC pathology after spiking IOP elevation, we first analyzed changes in PERG amplitude and RGC density. We compared changes in Casp1-/-, NLRP1b-/-, GsdmD-/-, and Panx1-/- Casp11+/+ mice with that in the WT at 7 days in spOHT and stOHT injury models. The rate of the spOHT-induced changes in PERG amplitude was presented as relative to their pre-spOHT baselines (Figure 5A). The between-group comparison showed statistically non-significant changes in PERG amplitude: 1.77±9.69% in Casp1-/- mice (N=12 p=0.99), 10.33±8.43% in NLRP1-/- mice (N=14; p=0.89), 24.38 ±9.96% in GsdmD-/- mice (N=11 p=0.13), and 18.73±3.28% in Panx1-/-mice (N=7 p=0.55). In addition, changes in PERG latency were insignificant in Casp1-/- (2.83±3.78% p=0.45), NLRP1-/- (4.49±2.56% p=0.1), GsdmD-/- mice (11.06±5.72% N=8 p=0.13) and Panx1-/- mice (3.70±4.22% N=8, p=0.28). These results demonstrate that RGC dysfunction after the spOHT challenge requires the activities of NLRP1, Casp1, and its substrate GsdmD, which has been shown to be highly induced by these neurons in the acute OHT challenge [34].
SpOHT induced RGC loss was studied in Casp1-/-, NLRP1b-/-, and GsdmD-/- mice. RBPMS-positive RGCs were counted in retinas harvested 7 days after spOHT or stOHT challenges. The rate of RBPMS+ RGC loss in experimental vs. naive age-matched retinas was significantly higher following spOHT (20.41±4.83% at 7dpi, 23.57±4.8% at 14 dpi, and 10.52±11.7 % at 21 dpi, p<0.01) (Figure 5A). No significant loss was observed after stOHT (6.95±4.58% p=0.21 change in RGC density at 7dpi, -0.05±7.96% at 14 dpi and 0.01±11.23% at 21 dpi) (Figure 5B). We then assessed RGC loss in knockout mice with inactivation of the inflammasome. The change in RGC density relative to that in naïve sham-operated eyes of the same genetic background at 7 dpi averaged 7.71 ±3.89% in Casp1-/- mice, 5.89 ±4.1% in the NLRP1-/- mice, and 4.65±2.46% in animals with inactivation of GsdmD. Similar to NLRP1-/- retinas, GsdmD-/- retinas were significantly protected from dysfunction and fully protected from structural loss (Figure 5A,B). In comparison, the rate of RGC loss in WT mice following spOHT was 20.41±4.83% (p<0.01) (Figure 5B), whereas no significant RGC loss was observed in the eyes after stOHT (6.95±4.58% p=0.21) (Figure 5B).
Protection by inhibition of Casp1 or other inflammasome signaling proteins could suggest that RGCs death in this model occurred via pyroptosis, like in the retinal ischemia-reperfusion model [34]. However, inflammasome can facilitate casp3-mediated apoptotic death via the combination of Casp3-mediated disruption of GSDMS pore formation, resulting in apoptotic death, or gasdermin-mediated release of mitochondrial cytochrome C that activates Casp3 [61,62]. We, therefore, tested whether Casp3 activity is essential for spOHT induced cell death using caspase-3 specific inhibitor DEVD. When intravitreally injected 1 hr prior to spOHT in WT mice, DEVD completely protected RGCs (Figure 5B).
3.6. Inner retina cell types that activate inflammasome.
Two complementary approaches were used to determine the retinal cell types that activate inflammasomes in the spOHT injury by detection of 1) proteolytically activated caspase-1, using intravitreally injected FLICA-Casp1-FM660 substrate at 1h prior to end points, or 2) in vivo formation of citrin-labelled ASC-speck, using the bioindicator ASC-citrine mice expressing the ASC core inflammasome scaffold protein fused to the green fluorescent marker citrine [34,41]. The analysis of retinal wholemounts for cell types that became fluorescently-labelled due to activity of caspase-1 FLICA-660 substrate showed Casp1+ cells in the ganglion cell layer (GCL) of spOHT-challenged retinas, which were identified as RGCs with RBPMS+ co-staining (Figure 6C). Next, we used the same approach to detect the activities of pro-apoptotic caspase-8 and caspases-3/7 using intravitreal co-delivery of FLICA Casp8-660 and FLICA Casp 3/7 Alexa 488 dyes. In the GCL, active Casp8-labelled cells co-localized with Casp1+ RBPMS+ RGCs in (Supplemental Figure S2). Casp3/7-specific activity among inner retinal cell types was detected in 1) RBPMS+ RGCs, 2) GFAP+ astrocytes and 3) blood vessel endothelial cells (elongated nuclei aligned with blood vessels). However, neither astrocytes nor blood endothelial cells were positive for Casp1 activity in the spOHT-challenged retinas at 6h post-injury (Figure 6A). These data show that co-activation of pro-inflammatory Casp1 with pro-apoptotic Casp3/7 and Casp8 occurs exclusively in RGCs at the early post-injury stage. In the control stOHT-challenged retinas Casp1+ and Casp8+ RGCs were not detected (Figure 6B). Some Casp3/7+-positive cells were detected among GFAP+ astrocytes and, likely, blood vessel endothelial cells (characteristic elongated nuclei aligned along blood capillaries) but Casp1+ cells were absent in these samples (Figure 6B).
Next, we used co-localization analysis of retinal multiplex immunostaining to determine cell types that activate the major pro-inflammatory cytokines and the major retinal inflammasome complexes NLRP1, NLRP3, and AIM2, that we previously characterized in the inner retina [34]. In the spOHT samples, specific labeling for IL-1β co-localized to neurons in the INL and was particularly strong in the GCL layers of the inner retina (Figure 7). The TNFα-specific labeling was confined to distinct cell types, including GFAP+ astroglia (arrows), CD11b+/Iba1+ microglia and infiltrating CD45+.CD11b+ monocytes/macrophages (white arrowheads, Figure 7) in the retina 48h after the spOHT challenge. Importantly, the citrine-positive ASC-specks, representing the most active inflammasomes in the retina, co-localized with the GFAP+/TNFα+ astrocytes at the inner surface of the retina (yellow arrows, Figure 7). In contrast, retinas from sham and from steady OHT controls had IL-1β- and TNFα-specific labeling significantly reduced along with lack of infiltrating monocytes (Figure 7 bottom panel). These data indicate that, in contrast to a steady non-ischemic OHT stress, the repetitive spOHT robustly induce acute inflammasome activation that is restricted to inner retinal neurons, particularly RGCs in the inner retina.
Previous research identified three canonical inflammasomes in the retina: NLRP1, NLRP3 and AIM2, with few reports showing activation of NLRC4- and NLRP12- containing complexes [24,34,35,63]. We analyzed cellular localization of the canonical complexes by immunostaining in thin slices of retinas 48h after spOHT and stOHT challenges. These data showed an increase in NLRP1 immunoreactivity in in the GCL, co-localizing with TUJ1+ RGCs and their axons after spOHT, which was significantly stronger than in sham-operated controls (Figure 8). The NLRP3-specific labeling co-localized with some RGCs and glial cells in sham controls, but showed little change after the spOHT challenge (Figure 8). Both NLRP1 and NLRP3 sensors were also expressed by infiltrating monocytes (yellow arrowheads, Figure 8). The AIM2 sensor-specific labeling co-localized exclusively with GlutSyn+ Muller glia and also showed an incresad immunoreactivity in spOHT challenged retinasrelative to stOHT controls. The infiltration of monocytes/macrophages co-expressing CD45+ and CD11b+ markers was detected in the retina and optic nerve of the spOHT-challenged eyes, but not in the stOHT or sham-treated controls (Figure 9). These cells were numerous at the inner retina surface, in the GCL and IPL layers and in the optic nerve after repetitive spOHT-induced injury (marked by yellow arrowheads in Figure 9), indicating a contribution of the adaptive immune system to the pathogenesis.
3.7. Inflammasome activation in glaucoma.
To answer the question whether inflammasomes play a pivotal role in chronic OHT/glaucoma as in the spOHT stress, we utilized the Ad5Myoc-induced chronic OHT glaucoma model, developed by Grotegut and Kuehn [46,47] and characterized by a reproducible pattern of early onset of mild IOP elevation. We correlated IOP dynamics in this model with changes in PERG amplitude and IL-1β activity in the vitreous body at 3, 5 and 8 weeks after OHT induction. As expected, PERG amplitude declined significantly starting at 2 weeks post Ad5-Myoc injection in WT eyes (Figure 10A) in strong correlation with IOP increase (Figure 10B). In WT eyes, we observed a significant increase in the levels of vitreous IL-1β at these time points, peaking at 5 weeks at 2.5 ± 0.5 pg/ml (Figure 10C). When compared to the maximum IL-1β elevation in the positive control eyes, injected with 3mM ATP- or challenged with 1h ischemia-reperfusion, vitreous IL-1β levels in Ad5-Myoc-injected eyes were elevated approximately 38% and 44% at 3 weeks, 67% and 59% at 5 weeks, 18 and 16% at 8 weeks post-injected, respectively. This persistent cytokine “tide” in this glaucoma model correlated with 29.8 ± 3.5% % loss of RGCs at 8 weeks post-OHT induction (Figure 10D). The analysis of the ASC speck formation to detect retinal cell types that activate inflammasomes in the chronic OHT-induced glaucoma showed that the RBPMS+ RGCs accounted for the majority of ASC citrine-positive cells (≥60%) in retinal wholemounts, whereas, the second most common type were GFAP+ astrocytes (~25%) and only a minor (<10%) proportion of AS specks co-localized with retinal blood vessels (Figure 10E-G). Combined, these data indicated that RGCs and astrocytes are the two key types of inner retinal cells contributing to IL-1β release into the vitreous during progression of the OHT-induced glaucoma.
4. Discussion
In this study, we investigated the impact of immune responses induced by repetitive (spiking) non-ischemic IOP elevations on function and viability of RGCs in mouse eyes. We demonstrated that, in contrast to the stOHT of similar magnitude and duration, the spOHT challenge caused rapid activation of the inflammasome in the retina, resulting in the release of pro-inflammatory cytokines. These events preceded an irreversible dysfunction and caspase-3 dependent loss of RGCs. A lower grade activation of the inflammasome was also observed in 3 weeks post-induction in retinas challenged with chronic OHT in the Ad5-MYOC-induced glaucoma model. Despite major differences in the types of OHT insult in these models, both spOHT and chronic OHT challenges induced RGC dysfunction and death, whereas its inactivation prevented RGC deficits in both models. Our results suggest mechanistic involvement of Casp1, Casp8, NLRP1, NLRP3, GsdmD proteins and inflammatory cytokines expressed by neuronal, glial and infiltrating immune cells in the spOHT-induced RGC pathology.
Acute and progressive inflammasome activation in the spOHT model. In the spOHT model, the production of pro-inflammatory cytokines TNFα and monocyte chemoattractant protein (MCP-1 or CCL2) was dependent on the activity of the Panx1-inflammasome signaling axis. We obtained evidence of mechanistic involvement of the endogenous NLRP1 inflammasome, and its downstream targets GsdmD and Casp1 in the spOHT-induced pathophysiology of RGCs by using gene knockout strains and drug inhibitors. In addition, we also confirmed an essential role of the upstream mechanosensitive regulator of NLRP1 and NLRP3 and the Panx1 channel. RGC dysfunction and loss strongly correlated with activation of NLRP1, NLRP3, and their products, Casp1, IL-1β cytokine, and GsdmD. Importantly, ablation of the nlrp1b gene was profoundly neuroprotective in the chronic model (Figure 10D). The alternative IL-1β convertase, Casp8, which is both pro-inflammatory and pro-apoptotic via activation of Casp3, was also synergistically co-activated with Casp1 and in RGC cells (Supplement Figure S2). Finally, AIM2, known to drive pyroptotic death and sensitized by intracellular self-DNA [64,65], was specifically active in Muller glia, the cell type also implicated in the glaucomatous loss of RGCs [66,67]. Significantly, in control stOHT-challenged eyes, we did not detect any significant increase in inflammasome activity (release of cytokines), induction of Casp1, Casp8, and RGC damage, thus allowing us to conclude that the spiking pattern of OHT is a key trigger of retinal inflammation and injury. To summarize, our model provided new mechanistic insights into 1) very early post-spOHT pro-inflammatory events, and 2) mechanisms driving excessive neurotoxicity after repetitive vs. single, non-repetitive IOP spikes. Finally, mechanistic similarities between the acute spOHT and chronic OHT-induced glaucoma, including the inflammasome/Casp1-dependent RGC dysfunction and death, suggest that spOHT can be instrumental in studies of the earliest pathophysiological events in the OHT-challenged retina.
Mechanosensor signaling drives inflammasome activation. Most inflammasomes are induced by a combination of internal (loss of ionic and energetic homeostasis) and external signals via surface receptors. These are integrated into Signal 1 that facilitates transcriptional induction of inflammasome components via TLR, TNF, IL-1, C5a receptors, and alarmins, and Signal 2, which facilitates complex assembly in response to lysosomal damage and extracellular ATP and K+ elevations via the Panx1-P2X4/7 activation [68,69,70,71,72]. Abundant literature evidence supports a model, where spOHT-induced stimulation of mechanosensitive Panx1 [33,73,74], Piezo-1[75] and transient receptor potential (TRP) vanilloid TRPV1 and TRPV4 [3,76,77] channels in neural cell plasma membrane initiates pro-inflammatory signaling [66,78,79,80]. A recent evidence that a highly mechanosensitive Piezo1 is an upstream interaction partner of Panx1 [39,81,82] makes it feasible to suggest that a serial activation of the Piezo1–Panx1 by repetitive IOP spikes produces progressively higher levels of ATP release and inflammasome induction[34]. In support of the key role of mechanosensory Panx1-inflammasome signaling in the retina, recent reports showed its pivotal role in both acute ischemia-induced and glaucomatous degeneration, where its activity strongly correlated with the induction of inflammasome, production of IL-1β [33,54]. The activity of TRP channels were linked to neuroinflammation via release of glial TNFα and infiltration of immune cells [66,78,79,80]. Consistently, our results showed that blockade of Panx1 provided suppression of inflammation and protection of RGC similar to the blockade of inflammasome activity by ablation of caspase-1 and GsdmD.
The neurotoxic pathways downstream of the inflammasome include pyroptotic and non-pyroptotic extracellular paracrine pathways, such as the GsdmD-NT pore-mediated release of IL-1β, several types of alarmins, miRNAs and ASC speck complexes [83,84,85], as well as cleavage-activation of Casp1 and Casp8 substrates. Most of these pathways facilitate strong pro-inflammatory responses via local glial activation, blood neutrophil attraction, tissue migration, [86,87,88,89,90,91], and spreading inflammation [92]. In particular, activation of GsdmD-NT pores has previously been shown to mediates cytokine release that sustains and propagates proinflammatory signaling in the retina in a way similar to a “cytokine storm” during sepsis or viral infection[93,94].
Furthermore, similar to other gasdermins, GsdmD activation by the inflammasome was reported to facilitate mitochondrial damage [95,96] and pro-apoptotic signaling via the release of mitochondrial DNA and cytochrome C [61,95,97]. Since mitochondrial dysfunction and subsequent loss of RGC functionality and viability are the key pathological events in cerebral ischemia [98] and glaucoma[99,100,101], we obtained direct evidence of GsdmD involvement in dysfunction and death of RGCs using GsdmD-/- retinas. Overall, since functional and structural damage to RGCs was prevented by the ablation of the nlrp1, panx1 or gsdmD genes, we concluded that the NLRP1 and NLRP3 inflammasomes, expressed by RGCs and macrophages, are key mediators of neurotoxicity in the spOHT injury model.
Finally, the high level of neuroinflammation and cytokine release strongly correlated with infiltration of blood monocytes. The concurrent release of IL-1β, TNFα, and particularly MCP-1/CCL2 cytokines, observed in the vitreous and retina of the spOHT-challenged eyes, is also known to weaken the blood-retina-barrier and promote infiltration of blood-borne monocytes and leukocytes elsewhere [45,102,103]. Such infiltration was evident at 48h post-OHT induction, as confirmed by abundant CD45+ CD11b+ cells in the retina and optic nerve (Figure 9), and strongly correlated with unexpectedly high for such a mild injury, levels of cytokines. Importantly, monocyte and leukocyte infiltration is implicated in various retinal degenerations, including OHT-induced ischemia-reperfusion[104] , AMD[105] and glaucoma[86,106].
Pyroptosis vs. apoptosis.The activation of the inflammasome end-products caspase-1 and GsdmD in response to spOHT challenge suggested cell death via pyroptosis ([107,108]; however, we used Casp1/3/8-FLICA labeling and DEVD blockade of Casp3 activation to obtain evidence that the spOHT challenge triggered RGCs loss via apoptosis. This conclusion was made despite our data indicating the key roles of Casp1 and GsdmD in RGC death, which are typically associated with pyroptotic death, as we reported earlier in the retinal ischemia model [34]. The molecular mechanism for Casp1- and Casp8-dependent activation of Casp3 is well-characterized [61,62,109]. Moreover, the most recent insights in non-canonical functions of GsdmD and Casp8 showed a non-pyroptotic GSDMD-ASC-Casp8 interaction as well as alternative cleavage of GsdmD by activated Casp3 into a non-pore forming fragment [59]. Both of these events prevent pyroptosis and facilitate apoptosis in a heightened inflammatory environment. In support of this mechanism, we observed the co-activation of Casp1 and Casp3/7 specifically in RGCs in spOHT-challenged retinas (Figure 6), which was blocked in either Casp1-/- retinas or in WT retinas treated with the Casp1 inhibitor VX-765 both of which protected RGCs functionally and structurally (Figure 5). It is reasonable to conclude that the neurotoxic activity of Casp1 is directly essential for endogenous OHT-induced RGC death since it is present in RGCs directly following injury, and its ablation caused a significant inhibition of IL-1β release into the vitreous, resulting in RGC protection [34,54]. At the same time, the activity of retinal Casp1 may also affect the viability of RGCs indirectly, since the bulk of Casp1-containing inflammasomes are not RGC-specific and are activated by astrocytes, microglia, and retina-infiltrating immune cells.
5. Conclusions
In conclusion, repetitive IOP spikes in the spOHT model induce an acute dysfunction and degeneration of about 1/5 of the RGC population via activity of the canonical inflammasomes and GsdmD. The extent of degeneration and RGC death were similar to that observed in most models of OHT- induced glaucoma [110,111,112,113]. Our results provide evidence of mechanistic involvement of inflammasomes from both endogenous and blood-borne cells in the pathophysiology of RGC loss after repetitive spiking IOP. Our results suggest that repetitive IOP spikes can both initiate de novo or accelerate an ongoing retinal pathology. As suggested in this and other studies [103,114], post spOHT inflammatory damage is unexpectedly high and can serve as facilitating risk factor in predisposed individuals, including those with lowered arterial blood pressure [115], systemic diseases, advanced age, pre-existing glaucoma and NTG patients [116,117,118], a certain proportion of which suffer from IOP spike-induced damage [119]. Current attempts to control IOP spikes in clinic using conventional IOP-lowering drugs failed [120], indicating a need for neuroprotective strategies targeting inflammasome-mediated mechanisms of injury.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Real-time PCR gene-specific primer pairs. Figure S1: Co-activation of Casp1 and Casp8 in RGCs.Figure S2. Representative micrographs of wholemount retinas stained for RBPMS-positive cells. Figure S3. A diagram depicting experimental setup for a PERG recording experiment. Figure S4. Meta-analysis of publicly available murine transcriptomic data. Figure S5. (full version of Figure 9) Monocyte/macrophages infiltration into the inner retina and optic nerves after spOHT challenge. Figure S6. Meta-analysis of RGC subtype-specific scRGAseq data for inflammasome gene expression. Figure S7. Meta-analysis of scRGAseq data from photoreceptors, bypolar horizontal and non-neuronal cells. Figure S8. Meta-analysis of scRGAseq data from photoreceptors, bipolar horizontal and non-neuronal cells for inflammasome gene expression. Supplementary Methods. Immunohistochemistry & Western Blot.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, V.S., and V.P.; methodology, V.S., M.S.; V.P., T-SC, and J.P.deRV data collection and validation, WA, GR, DPh, RW, HW, MB, G.S, SK, OJG, formal analysis, V.S.,, T-SC, PJdRV, V.P., S.K.; data curation, S.K., resources, PJdRV, S.K., writing—original draft preparation, V.S., M.S., and VP; writing—review and editing, V.S., M.S., PdeRV., RW, V.P., S.K.; funding acquisition, V.S., V.P. All authors have read and agreed to the published version of the manuscript.”
Funding
This work was supported by National Institute of Health grants R01EY021517(VS), R21 EY032261 (VS), DOD VRP grant VR2000079 (VS) F31 FP00001673 (MS), NIH R24EY028785 (VP) , NIH R01EY031492 (RW), National Institute of Health P30 center grant EY014801, an unrestricted Research to Prevent Blindness and Department of Defense grant W81XWH-13-1-0048 to the Department of Ophthalmology.
Institutional Review Board Statement
The animal study protocol # 21-036 LF was approved by the Institutional Review Board (IACUC) of the University of Miami for studies involving animals.
Informed Consent Statement
Notapplicable.
Data Availability Statement
All Data are contained within this article or supplementary material. Histology data used for cell counting are available upon request.
Acknowledgments
We acknowledge Dr. R. Brambilla for a gift of Cd45 antibodies and consultation on immunostaining infiltrating cells.
Conflicts of Interest
MS, WA, GR, DPh, RW, HW, MB, G.S, SK, OJG, VP, and VSH declare no conflicts of interest. JPdRV is a co-founder and managing member of InflamaCORE, LLC and has licensed patents on inflammasome proteins as biomarkers of injury and disease as well as on targeting inflammasome proteins for therapeutic purposes. JPdRV is a Scientific Advisory Board Member of ZyVersa Therapeutics. T-H Chou is currently an employee of AbbVie Inc., Irvine, CA.
References
- Osborne, N.N.; Ugarte, M.; Chao, M.; Chidlow, G.; Bae, J.H.; Wood, J.P.; Nash, M.S. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv Ophthalmol 1999, 43 Suppl 1, S102-128. [CrossRef]
- Osborne, N.N. Mitochondria: Their role in ganglion cell death and survival in primary open angle glaucoma. Exp Eye Res 2010, 90, 750-757, doi:S0014-4835(10)00083-7 [pii]. [CrossRef]
- 10.1016/j.exer.2010.03.008. [CrossRef]
- Sappington, R.M.; Sidorova, T.; Long, D.J.; Calkins, D.J. TRPV1: Contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Invest Ophthalmol Vis Sci 2009, 50, 717-728, doi:iovs.08-2321 [pii]. [CrossRef]
- 10.1167/iovs.08-2321. [CrossRef]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci 2000, 41, 764-774.
- Calkins, D.J. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retin Eye Res 2012, 31, 702-719. [CrossRef]
- Caprioli, J.; Coleman, A.L. Intraocular pressure fluctuation a risk factor for visual field progression at low intraocular pressures in the advanced glaucoma intervention study. Ophthalmology 2008, 115, 1123-1129 e1123. [CrossRef]
- Asrani, S.; Zeimer, R.; Wilensky, J.; Gieser, D.; Vitale, S.; Lindenmuth, K. Large diurnal fluctuations in intraocular pressure are an independent risk factor in patients with glaucoma. J Glaucoma 2000, 9, 134-142. [CrossRef]
- McMonnies, C.W. Intraocular pressure spikes in keratectasia, axial myopia, and glaucoma. Optom Vis Sci 2008, 85, 1018-1026. [CrossRef]
- Savastano, A.; Savastano, M.C.; Carlomusto, L.; Savastano, S. Bilateral Glaucomatous Optic Neuropathy Caused by Eye Rubbing. Case Rep Ophthalmol 2015, 6, 279-283. [CrossRef]
- Baskaran, M.; Raman, K.; Ramani, K.K.; Roy, J.; Vijaya, L.; Badrinath, S.S. Intraocular pressure changes and ocular biometry during Sirsasana (headstand posture) in yoga practitioners. Ophthalmology 2006, 113, 1327-1332. [CrossRef]
- Jasien, J.V.; Jonas, J.B.; de Moraes, C.G.; Ritch, R. Intraocular Pressure Rise in Subjects with and without Glaucoma during Four Common Yoga Positions. PLoS ONE 2015, 10, e0144505. [CrossRef]
- Schuman, J.S.; Massicotte, E.C.; Connolly, S.; Hertzmark, E.; Mukherji, B.; Kunen, M.Z. Increased intraocular pressure and visual field defects in high resistance wind instrument players. Ophthalmology 2000, 107, 127-133. [CrossRef]
- Krist, D.; Cursiefen, C.; Junemann, A. [Transitory intrathoracic and -abdominal pressure elevation in the history of 64 patients with normal pressure glaucoma]. Klin Monbl Augenheilkd 2001, 218, 209-213. [CrossRef]
- Ren, R.; Zhang, X.; Wang, N.; Li, B.; Tian, G.; Jonas, J.B. Cerebrospinal fluid pressure in ocular hypertension. Acta Ophthalmol 2011, 89, e142-148. [CrossRef]
- Barash, A.; Chui, T.Y.P.; Garcia, P.; Rosen, R.B. Acute Macular and Peripapillary Angiographic Changes with Intravitreal Injections. Retina 2020, 40, 648-656. [CrossRef]
- Song, J.; Huang, B.B.; Ong, J.X.; Konopek, N.; Fawzi, A.A. Hemodynamic Effects of Anti-Vascular Endothelial Growth Factor Injections on Optical Coherence Tomography Angiography in Diabetic Macular Edema Eyes. Transl Vis Sci Technol 2022, 11, 5. [CrossRef]
- Soheilian, M.; Karimi, S.; Montahae, T.; Nikkhah, H.; Mosavi, S.A. Effects of intravitreal injection of bevacizumab with or without anterior chamber paracentesis on intraocular pressure and peripapillary retinal nerve fiber layer thickness: A prospective study. Graefes Arch Clin Exp Ophthalmol 2017, 255, 1705-1712. [CrossRef]
- Wang, L.; Swaminathan, S.S.; Yang, J.; Barikian, A.; Shi, Y.; Shen, M.; Jiang, X.; Feuer, W.; Gregori, G.; Rosenfeld, P.J. Dose-Response Relationship between Intravitreal Injections and Retinal Nerve Fiber Layer Thinning in Age-Related Macular Degeneration. Ophthalmol Retina 2021, 5, 648-654. [CrossRef]
- Pecora, L.; Sibony, P.; Fourman, S. Eye-rubbing optic neuropathy. Am J Ophthalmol 2002, 134, 460-461. [CrossRef]
- Lu, W.; Albalawi, F.; Beckel, J.M.; Lim, J.C.; Laties, A.M.; Mitchell, C.H. The P2X7 receptor links mechanical strain to cytokine IL-6 up-regulation and release in neurons and astrocytes. J Neurochem 2017, 141, 436-448. [CrossRef]
- Li, A.; Zhang, X.; Zheng, D.; Ge, J.; Laties, A.M.; Mitchell, C.H. Sustained elevation of extracellular ATP in aqueous humor from humans with primary chronic angle-closure glaucoma. Exp Eye Res 2011, 93, 528-533. [CrossRef]
- Xia, J.; Lim, J.C.; Lu, W.; Beckel, J.M.; Macarak, E.J.; Laties, A.M.; Mitchell, C.H. Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J Physiol 2012, 590, 2285-2304. [CrossRef]
- Zhang, X.; Li, A.; Ge, J.; Reigada, D.; Laties, A.M.; Mitchell, C.H. Acute increase of intraocular pressure releases ATP into the anterior chamber. Exp Eye Res 2007, 85, 637-643. [CrossRef]
- 10.1016/j.exer.2007.07.016. [CrossRef]
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappaB pathway in acute glaucoma. J Neuroinflammation 2015, 12, 137. [CrossRef]
- Tezel, G.; Yang, X.; Luo, C.; Cai, J.; Powell, D.W. An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Invest Ophthalmol Vis Sci 2012, 53, 4220-4233. [CrossRef]
- Markiewicz, L.; Pytel, D.; Mucha, B.; Szymanek, K.; Szaflik, J.; Szaflik, J.P.; Majsterek, I. Altered Expression Levels of MMP1, MMP9, MMP12, TIMP1, and IL-1beta as a Risk Factor for the Elevated IOP and Optic Nerve Head Damage in the Primary Open-Angle Glaucoma Patients. Biomed Res Int 2015, 2015, 812503. [CrossRef]
- Lin, C.; Wu, F.; Zheng, T.; Wang, X.; Chen, Y.; Wu, X. Kaempferol attenuates retinal ganglion cell death by suppressing NLRP1/NLRP3 inflammasomes and caspase-8 via JNK and NF-kappaB pathways in acute glaucoma. Eye (Lond) 2019, 33, 777-784. [CrossRef]
- Zhang, Y.; Xu, Y.; Sun, Q.; Xue, S.; Guan, H.; Ji, M. Activation of P2X7R- NLRP3 pathway in Retinal microglia contribute to Retinal Ganglion Cells death in chronic ocular hypertension (COH). Exp Eye Res 2019, 188, 107771. [CrossRef]
- Lu, W.; Hu, H.; Sevigny, J.; Gabelt, B.T.; Kaufman, P.L.; Johnson, E.C.; Morrison, J.C.; Zode, G.S.; Sheffield, V.C.; Zhang, X.; et al. Rat, mouse, and primate models of chronic glaucoma show sustained elevation of extracellular ATP and altered purinergic signaling in the posterior eye. Invest Ophthalmol Vis Sci 2015, 56, 3075-3083. [CrossRef]
- Howell, G.R.; Macalinao, D.G.; Sousa, G.L.; Walden, M.; Soto, I.; Kneeland, S.C.; Barbay, J.M.; King, B.L.; Marchant, J.K.; Hibbs, M.; et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest 2011, 121, 1429-1444. [CrossRef]
- Yang, X.; Luo, C.; Cai, J.; Powell, D.W.; Yu, D.; Kuehn, M.H.; Tezel, G. Neurodegenerative and inflammatory pathway components linked to TNF-alpha/TNFR1 signaling in the glaucomatous human retina. Invest Ophthalmol Vis Sci 2011, 52, 8442-8454. [CrossRef]
- Soto, I.; Howell, G.R. The complex role of neuroinflammation in glaucoma. Cold Spring Harb Perspect Med 2014, 4. [CrossRef]
- Dvoriantchikova, G.; Ivanov, D.; Barakat, D.; Grinberg, A.; Wen, R.; Slepak, V.Z.; Shestopalov, V.I. Genetic ablation of Pannexin1 protects retinal neurons from ischemic injury. PLoS ONE 2012, 7, e31991. [CrossRef]
- Pronin, A.; Pham, D.; An, W.; Dvoriantchikova, G.; Reshetnikova, G.; Qiao, J.; Kozhekbaeva, Z.; Reiser, A.E.; Slepak, V.Z.; Shestopalov, V.I. Inflammasome Activation Induces Pyroptosis in the Retina Exposed to Ocular Hypertension Injury. Front Mol Neurosci 2019, 12, 36. [CrossRef]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc Natl Acad Sci U S A 2014, 111, 11181-11186. [CrossRef]
- Krishnan, A.; Kocab, A.J.; Zacks, D.N.; Marshak-Rothstein, A.; Gregory-Ksander, M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J Neuroinflammation 2019, 16, 184. [CrossRef]
- Yang, X.; Zeng, Q.; Tezel, G. Regulation of distinct caspase-8 functions in retinal ganglion cells and astroglia in experimental glaucoma. Neurobiol Dis 2021, 150, 105258. [CrossRef]
- Zhao, M.; Li, S.; Matsubara, J.A. Targeting Pyroptotic Cell Death Pathways in Retinal Disease. Front Med (Lausanne) 2021, 8, 802063. [CrossRef]
- Krizaj, D.; Ryskamp, D.A.; Tian, N.; Tezel, G.; Mitchell, C.H.; Slepak, V.Z.; Shestopalov, V.I. From mechanosensitivity to inflammatory responses: New players in the pathology of glaucoma. Curr Eye Res 2014, 39, 105-119. [CrossRef]
- Evangelho, K.; Mogilevskaya, M.; Losada-Barragan, M.; Vargas-Sanchez, J.K. Pathophysiology of primary open-angle glaucoma from a neuroinflammatory and neurotoxicity perspective: A review of the literature. Int Ophthalmol 2019, 39, 259-271. [CrossRef]
- Tzeng, T.C.; Schattgen, S.; Monks, B.; Wang, D.; Cerny, A.; Latz, E.; Fitzgerald, K.; Golenbock, D.T. A Fluorescent Reporter Mouse for Inflammasome Assembly Demonstrates an Important Role for Cell-Bound and Free ASC Specks during In Vivo Infection. Cell Rep 2016, 16, 571-582. [CrossRef]
- Mattson, D.L. Comparison of arterial blood pressure in different strains of mice. Am J Hypertens 2001, 14, 405-408. [CrossRef]
- Morrison, J.C.; Cepurna, W.O.; Tehrani, S.; Choe, T.E.; Jayaram, H.; Lozano, D.C.; Fortune, B.; Johnson, E.C. A Period of Controlled Elevation of IOP (CEI) Produces the Specific Gene Expression Responses and Focal Injury Pattern of Experimental Rat Glaucoma. Invest Ophthalmol Vis Sci 2016, 57, 6700-6711. [CrossRef]
- Bui, B.V.; Edmunds, B.; Cioffi, G.A.; Fortune, B. The gradient of retinal functional changes during acute intraocular pressure elevation. Invest Ophthalmol Vis Sci 2005, 46, 202-213. [CrossRef]
- Xu, Q.; Rydz, C.; Nguyen Huu, V.A.; Rocha, L.; Palomino La Torre, C.; Lee, I.; Cho, W.; Jabari, M.; Donello, J.; Lyon, D.C.; et al. Stress induced aging in mouse eye. Aging Cell 2022, e13737. [CrossRef]
- Grotegut, P.; Kuehn, S.; Meissner, W.; Dick, H.B.; Joachim, S.C. Intravitreal S100B Injection Triggers a Time-Dependent Microglia Response in a Pro-Inflammatory Manner in Retina and Optic Nerve. Mol Neurobiol 2020, 57, 1186-1202. [CrossRef]
- Zeng, H.; Dumitrescu, A.V.; Wadkins, D.; Elwood, B.W.; Gramlich, O.W.; Kuehn, M.H. Systemic Treatment with Pioglitazone Reverses Vision Loss in Preclinical Glaucoma Models. Biomolecules 2022, 12. [CrossRef]
- Weaver, C.; Cyr, B.; de Rivero Vaccari, J.C.; de Rivero Vaccari, J.P. Inflammasome Proteins as Inflammatory Biomarkers of Age-Related Macular Degeneration. Transl Vis Sci Technol 2020, 9, 27. [CrossRef]
- Shrivastava, A.; Saxena, P.; Gupta, V.B. Spectrophotometric estimation of tamsulosin hydrochloride by acid-dye method. Pharm Methods 2011, 2, 53-60. [CrossRef]
- Porciatti, V. The mouse pattern electroretinogram. Doc Ophthalmol 2007, 115, 145-153. [CrossRef]
- Chou, T.H.; Romano, G.L.; Amato, R.; Porciatti, V. Nicotinamide-Rich Diet in DBA/2J Mice Preserves Retinal Ganglion Cell Metabolic Function as Assessed by PERG Adaptation to Flicker. Nutrients 2020, 12. [CrossRef]
- Porciatti, V.; Saleh, M.; Nagaraju, M. The pattern electroretinogram as a tool to monitor progressive retinal ganglion cell dysfunction in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci 2007, 48, 745-751. [CrossRef]
- Scheiblich, H.; Schlutter, A.; Golenbock, D.T.; Latz, E.; Martinez-Martinez, P.; Heneka, M.T. Activation of the NLRP3 inflammasome in microglia: The role of ceramide. J Neurochem 2017, 143, 534-550. [CrossRef]
- Dvoriantchikova, G.; Pronin, A.; Kurtenbach, S.; Toychiev, A.; Chou, T.H.; Yee, C.W.; Prindeville, B.; Tayou, J.; Porciatti, V.; Sagdullaev, B.T.; et al. Pannexin 1 sustains the electrophysiological responsiveness of retinal ganglion cells. Sci Rep 2018, 8, 5797. [CrossRef]
- de Rivero Vaccari, J.P.; Lotocki, G.; Marcillo, A.E.; Dietrich, W.D.; Keane, R.W. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci 2008, 28, 3404-3414. [CrossRef]
- de Rivero Vaccari, J.P.; Lotocki, G.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D.; Keane, R.W. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab 2009, 29, 1251-1261. [CrossRef]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol 2018, 48, 584-592. [CrossRef]
- Li, J.; Hao, J.H.; Yao, D.; Li, R.; Li, X.F.; Yu, Z.Y.; Luo, X.; Liu, X.H.; Wang, M.H.; Wang, W. Caspase-1 inhibition prevents neuronal death by targeting the canonical inflammasome pathway of pyroptosis in a murine model of cerebral ischemia. CNS Neurosci Ther 2020, 26, 925-939. [CrossRef]
- Bao, L.; Locovei, S.; Dahl, G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett 2004, 572, 65-68. [CrossRef]
- S001457930400866X [pii].
- Qu, Y.; Misaghi, S.; Newton, K.; Gilmour, L.L.; Louie, S.; Cupp, J.E.; Dubyak, G.R.; Hackos, D.; Dixit, V.M. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol 2011, 186, 6553-6561. [CrossRef]
- 10.4049/jimmunol.1100478. [CrossRef]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 2019, 10, 1689. [CrossRef]
- Rogers, C.; Alnemri, E.S. Gasdermins: Novel mitochondrial pore-forming proteins. Mol Cell Oncol 2019, 6, e1621501. [CrossRef]
- Chen, H.; Deng, Y.; Gan, X.; Li, Y.; Huang, W.; Lu, L.; Wei, L.; Su, L.; Luo, J.; Zou, B.; et al. NLRP12 collaborates with NLRP3 and NLRC4 to promote pyroptosis inducing ganglion cell death of acute glaucoma. Mol Neurodegener 2020, 15, 26. [CrossRef]
- Adamczak, S.E.; de Rivero Vaccari, J.P.; Dale, G.; Brand, F.J., 3rd; Nonner, D.; Bullock, M.R.; Dahl, G.P.; Dietrich, W.D.; Keane, R.W. Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J Cereb Blood Flow Metab 2014, 34, 621-629. [CrossRef]
- Jakobs, C.; Perner, S.; Hornung, V. AIM2 Drives Joint Inflammation in a Self-DNA Triggered Model of Chronic Polyarthritis. PLoS ONE 2015, 10, e0131702. [CrossRef]
- Li, Q.; Cheng, Y.; Zhang, S.; Sun, X.; Wu, J. TRPV4-induced Muller cell gliosis and TNF-alpha elevation-mediated retinal ganglion cell apoptosis in glaucomatous rats via JAK2/STAT3/NF-kappaB pathway. J Neuroinflammation 2021, 18, 271. [CrossRef]
- Hu, X.; Xu, M.X.; Zhou, H.; Cheng, S.; Li, F.; Miao, Y.; Wang, Z. Tumor necrosis factor-alpha aggravates gliosis and inflammation of activated retinal Muller cells. Biochem Biophys Res Commun 2020, 531, 383-389. [CrossRef]
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923-932. [CrossRef]
- Parzych, K.; Zetterqvist, A.V.; Wright, W.R.; Kirkby, N.S.; Mitchell, J.A.; Paul-Clark, M.J. Differential role of pannexin-1/ATP/P2X(7) axis in IL-1beta release by human monocytes. FASEB J 2017, 31, 2439-2445. [CrossRef]
- Hung, S.C.; Choi, C.H.; Said-Sadier, N.; Johnson, L.; Atanasova, K.R.; Sellami, H.; Yilmaz, O.; Ojcius, D.M. P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PLoS ONE 2013, 8, e70210. [CrossRef]
- PONE-D-13-15721 [pii].
- El-Maadawy, W.H.; Hassan, M.; Badawy, M.H.; AbuSeada, A.; Hafiz, E. Probenecid induces the recovery of renal ischemia/reperfusion injury via the blockade of Pannexin 1/P2X7 receptor axis. Life Sci 2022, 308, 120933. [CrossRef]
- Rusiecka, O.M.; Tournier, M.; Molica, F.; Kwak, B.R. Pannexin1 channels-a potential therapeutic target in inflammation. Front Cell Dev Biol 2022, 10, 1020826. [CrossRef]
- Bruzzone, R.; Hormuzdi, S.G.; Barbe, M.T.; Herb, A.; Monyer, H. Pannexins, a family of gap junction proteins expressed in brain. Proc Natl Acad Sci U S A 2003, 100, 13644-13649. [CrossRef]
- Bargiotas, P.; Krenz, A.; Hormuzdi, S.G.; Ridder, D.A.; Herb, A.; Barakat, W.; Penuela, S.; von Engelhardt, J.; Monyer, H.; Schwaninger, M. Pannexins in ischemia-induced neurodegeneration. Proc Natl Acad Sci U S A 2011, 108, 20772-20777. [CrossRef]
- Morozumi, W.; Inagaki, S.; Iwata, Y.; Nakamura, S.; Hara, H.; Shimazawa, M. Piezo channel plays a part in retinal ganglion cell damage. Exp Eye Res 2020, 191, 107900. [CrossRef]
- Ryskamp, D.A.; Witkovsky, P.; Barabas, P.; Huang, W.; Koehler, C.; Akimov, N.P.; Lee, S.H.; Chauhan, S.; Xing, W.; Renteria, R.C.; et al. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J Neurosci 2011, 31, 7089-7101. [CrossRef]
- Lakk, M.; Young, D.; Baumann, J.M.; Jo, A.O.; Hu, H.; Krizaj, D. Polymodal TRPV1 and TRPV4 Sensors Colocalize but Do Not Functionally Interact in a Subpopulation of Mouse Retinal Ganglion Cells. Front Cell Neurosci 2018, 12, 353. [CrossRef]
- Wang, S.; He, H.; Long, J.; Sui, X.; Yang, J.; Lin, G.; Wang, Q.; Wang, Y.; Luo, Y. TRPV4 Regulates Soman-Induced Status Epilepticus and Secondary Brain Injury via NMDA Receptor and NLRP3 Inflammasome. Neurosci Bull 2021, 37, 905-920. [CrossRef]
- Liu, Y.; Fan, H.; Li, X.; Liu, J.; Qu, X.; Wu, X.; Liu, M.; Liu, Z.; Yao, R. Trpv4 regulates Nlrp3 inflammasome via SIRT1/PGC-1alpha pathway in a cuprizone-induced mouse model of demyelination. Exp Neurol 2021, 337, 113593. [CrossRef]
- Nishinaka, A.; Tanaka, M.; Ohara, K.; Sugaru, E.; Shishido, Y.; Sugiura, A.; Moriguchi, Y.; Toui, A.; Nakamura, S.; Shimada, K.; et al. TRPV4 channels promote vascular permeability in retinal vascular disease. Exp Eye Res 2023, 228, 109405. [CrossRef]
- Wang, A.Y.; Lee, P.Y.; Bui, B.V.; Jobling, A.I.; Greferath, U.; Brandli, A.; Dixon, M.A.; Findlay, Q.; Fletcher, E.L.; Vessey, K.A. Potential mechanisms of retinal ganglion cell type-specific vulnerability in glaucoma. Clin Exp Optom 2020, 103, 562-571. [CrossRef]
- Desplat, A.; Penalba, V.; Gros, E.; Parpaite, T.; Coste, B.; Delmas, P. Piezo1-Pannexin1 complex couples force detection to ATP secretion in cholangiocytes. J Gen Physiol 2021, 153. [CrossRef]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35-44 e36. [CrossRef]
- Volchuk, A.; Ye, A.; Chi, L.; Steinberg, B.E.; Goldenberg, N.M. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun 2020, 11, 4561. [CrossRef]
- Broz, P. Unconventional protein secretion by gasdermin pores. Semin Immunol 2023, 69, 101811. [CrossRef]
- Chen, H.; Cho, K.S.; Vu, T.H.K.; Shen, C.H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat Commun 2018, 9, 3209. [CrossRef]
- Williams, P.A.; Braine, C.E.; Kizhatil, K.; Foxworth, N.E.; Tolman, N.G.; Harder, J.M.; Scott, R.A.; Sousa, G.L.; Panitch, A.; Howell, G.R.; et al. Inhibition of monocyte-like cell extravasation protects from neurodegeneration in DBA/2J glaucoma. Mol Neurodegener 2019, 14, 6. [CrossRef]
- Gramlich, O.W.; Godwin, C.R.; Heuss, N.D.; Gregerson, D.S.; Kuehn, M.H. T and B Lymphocyte Deficiency in Rag1-/- Mice Reduces Retinal Ganglion Cell Loss in Experimental Glaucoma. Invest Ophthalmol Vis Sci 2020, 61, 18. [CrossRef]
- Tang, J.; Tang, Y.; Yi, I.; Chen, D.F. The role of commensal microflora-induced T cell responses in glaucoma neurodegeneration. Prog Brain Res 2020, 256, 79-97. [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355-361. [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med 2017, 214, 1351-1370. [CrossRef]
- Hulse, J.; Bhaskar, K. Crosstalk Between the NLRP3 Inflammasome/ASC Speck and Amyloid Protein Aggregates Drives Disease Progression in Alzheimer’s and Parkinson’s Disease. Front Mol Neurosci 2022, 15, 805169. [CrossRef]
- Shestopalov, V.I.; Spurlock, M.; Gramlich, O.W.; Kuehn, M.H. Immune Responses in the Glaucomatous Retina: Regulation and Dynamics. Cells 2021, 10. [CrossRef]
- Gupta, K.K.; Khan, M.A.; Singh, S.K. Constitutive Inflammatory Cytokine Storm: A Major Threat to Human Health. J Interferon Cytokine Res 2020, 40, 19-23. [CrossRef]
- de Torre-Minguela, C.; Gomez, A.I.; Couillin, I.; Pelegrin, P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J 2021, 35, e21757. [CrossRef]
- de Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ 2019, 26, 146-161. [CrossRef]
- Kondolf, H.C.; D’Orlando, D.A.; Dubyak, G.R.; Abbott, D.W. Protein engineering reveals that gasdermin A preferentially targets mitochondrial membranes over the plasma membrane during pyroptosis. J Biol Chem 2023, 299, 102908. [CrossRef]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J Neuroinflammation 2018, 15, 242. [CrossRef]
- Williams, P.A.; Harder, J.M.; Cardozo, B.H.; Foxworth, N.E.; John, S.W.M. Nicotinamide treatment robustly protects from inherited mouse glaucoma. Commun Integr Biol 2018, 11, e1356956. [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756-760. [CrossRef]
- Williams, P.A.; Harder, J.M.; John, S.W.M. Glaucoma as a Metabolic Optic Neuropathy: Making the Case for Nicotinamide Treatment in Glaucoma. J Glaucoma 2017, 26, 1161-1168. [CrossRef]
- Gramlich, O.W.; Teister, J.; Neumann, M.; Tao, X.; Beck, S.; von Pein, H.D.; Pfeiffer, N.; Grus, F.H. Immune response after intermittent minimally invasive intraocular pressure elevations in an experimental animal model of glaucoma. J Neuroinflammation 2016, 13, 82. [CrossRef]
- Resta, V.; Novelli, E.; Vozzi, G.; Scarpa, C.; Caleo, M.; Ahluwalia, A.; Solini, A.; Santini, E.; Parisi, V.; Di Virgilio, F.; et al. Acute retinal ganglion cell injury caused by intraocular pressure spikes is mediated by endogenous extracellular ATP. Eur J Neurosci 2007, 25, 2741-2754, doi:EJN5528 [pii]. [CrossRef]
- 10.1111/j.1460-9568.2007.05528.x. [CrossRef]
- Khanh Vu, T.H.; Chen, H.; Pan, L.; Cho, K.S.; Doesburg, D.; Thee, E.F.; Wu, N.; Arlotti, E.; Jager, M.J.; Chen, D.F. CD4(+) T-Cell Responses Mediate Progressive Neurodegeneration in Experimental Ischemic Retinopathy. Am J Pathol 2020, 190, 1723-1734. [CrossRef]
- Daftarian, N.; Zandi, S.; Piryaie, G.; Nikougoftar Zarif, M.; Ranaei Pirmardan, E.; Yamaguchi, M.; Behzadian Nejad, Q.; Hasanpour, H.; Samiei, S.; Pfister, I.B.; et al. Peripheral blood CD163(+) monocytes and soluble CD163 in dry and neovascular age-related macular degeneration. FASEB J 2020, 34, 8001-8011. [CrossRef]
- He, C.; Xiu, W.; Chen, Q.; Peng, K.; Zhu, X.; Wang, Z.; Xu, X.; Chen, Y.; Zhang, G.; Fu, J.; et al. Gut-licensed beta7(+) CD4(+) T cells contribute to progressive retinal ganglion cell damage in glaucoma. Sci Transl Med 2023, 15, eadg1656. [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660-665. [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci 2017, 42, 245-254. [CrossRef]
- Heilig, R.; Dilucca, M.; Boucher, D.; Chen, K.W.; Hancz, D.; Demarco, B.; Shkarina, K.; Broz, P. Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci Alliance 2020, 3. [CrossRef]
- Morrison, J.C.; Cepurna, W.O.; Johnson, E.C. Modeling glaucoma in rats by sclerosing aqueous outflow pathways to elevate intraocular pressure. Exp Eye Res 2015, 141, 23-32. [CrossRef]
- Gramlich, O.W.; Godwin, C.R.; Wadkins, D.; Elwood, B.W.; Kuehn, M.H. Early Functional Impairment in Experimental Glaucoma Is Accompanied by Disruption of the GABAergic System and Inceptive Neuroinflammation. Int J Mol Sci 2021, 22. [CrossRef]
- Sappington, R.M.; Carlson, B.J.; Crish, S.D.; Calkins, D.J. The microbead occlusion model: A paradigm for induced ocular hypertension in rats and mice. Invest Ophthalmol Vis Sci 2010, 51, 207-216. [CrossRef]
- Ruiz-Ederra, J.; Verkman, A.S. Mouse model of sustained elevation in intraocular pressure produced by episcleral vein occlusion. Exp Eye Res 2006, 82, 879-884. [CrossRef]
- Xu, Q.; Rydz, C.; Nguyen Huu, V.A.; Rocha, L.; Palomino La Torre, C.; Lee, I.; Cho, W.; Jabari, M.; Donello, J.; Lyon, D.C.; et al. Stress induced aging in mouse eye. Aging Cell 2022, 21, e13737. [CrossRef]
- Hu, J.; Bui, K.M.; Patel, K.H.; Kim, H.; Arruda, J.A.; Wilensky, J.T.; Vajaranant, T.S. Effect of hemodialysis on intraocular pressure and ocular perfusion pressure. JAMA Ophthalmol 2013, 131, 1525-1531. [CrossRef]
- Esen, F.; Eraslan, M.; Cerman, E.; Celiker, H.; Kazokoglu, H. Diurnal Spikes of Intraocular Pressure in Uveitic Glaucoma: A 24-hour Intraocular Pressure Monitoring Study. Semin Ophthalmol 2020, 35, 246-251. [CrossRef]
- Demirel, S.; Yanik, O.; Batioglu, F.; Ozmert, E. Intraocular pressure changes related to intravitreal injections of ranibizumab: Analysis of pseudophakia and glaucoma subgroup. Int Ophthalmol 2015, 35, 541-547. [CrossRef]
- Pollmann, A.S.; Zhang, A.; Shuba, L.M. Intraocular pressure fluctuations in a professional woodwind musician with advanced glaucoma. Can J Ophthalmol 2022, 57, e184-e185. [CrossRef]
- Koz, O.G.; Turkcu, M.F.; Yarangumeli, A.; Koz, C.; Kural, G. Normotensive glaucoma and risk factors in normotensive eyes with pseudoexfoliation syndrome. J Glaucoma 2009, 18, 684-688. [CrossRef]
- Kandarakis, A.; Soumplis, V.; Karampelas, M.; Panos, C.; Kyriakos, N.; Baxevanakis, A.; Karagiannis, D. Efficacy of brimonidine in preventing intraocular pressure spikes following phacoemulsification in glaucoma patients. Eur J Ophthalmol 2010, 20, 994-999. [CrossRef]
Figure 1.
Repetitive IOP spikes induce activation of inflammatory biomarkers, RGC dysfunction and loss. A. Experimental paradigm of the spiking OHT (spOHS) model and steady OHT (stOHT) control; B. qRT-PCR data on relative changes in transcript abundances of inflammasome pathway genes in retinas. Mean fold change vs. naïve ±SE, * P, 0.05, n=3-5. C. intravitreal IL1-β measured at 6, 12 and 24 hours after spOHT (red) and stOHT eyes (OD, blue). Mean ± SE, *P <0.05, n=5. Negative control: Casp1-/- eyes (Cs1KO). D. Quantification for HIF1a fluorescent labeling in retina wholemounts from eyes exposed to sham, spOHT expression sha or spOHT. Mean percentile change vs. sham SE, ** P< 0.05, n=5.
Figure 1.
Repetitive IOP spikes induce activation of inflammatory biomarkers, RGC dysfunction and loss. A. Experimental paradigm of the spiking OHT (spOHS) model and steady OHT (stOHT) control; B. qRT-PCR data on relative changes in transcript abundances of inflammasome pathway genes in retinas. Mean fold change vs. naïve ±SE, * P, 0.05, n=3-5. C. intravitreal IL1-β measured at 6, 12 and 24 hours after spOHT (red) and stOHT eyes (OD, blue). Mean ± SE, *P <0.05, n=5. Negative control: Casp1-/- eyes (Cs1KO). D. Quantification for HIF1a fluorescent labeling in retina wholemounts from eyes exposed to sham, spOHT expression sha or spOHT. Mean percentile change vs. sham SE, ** P< 0.05, n=5.
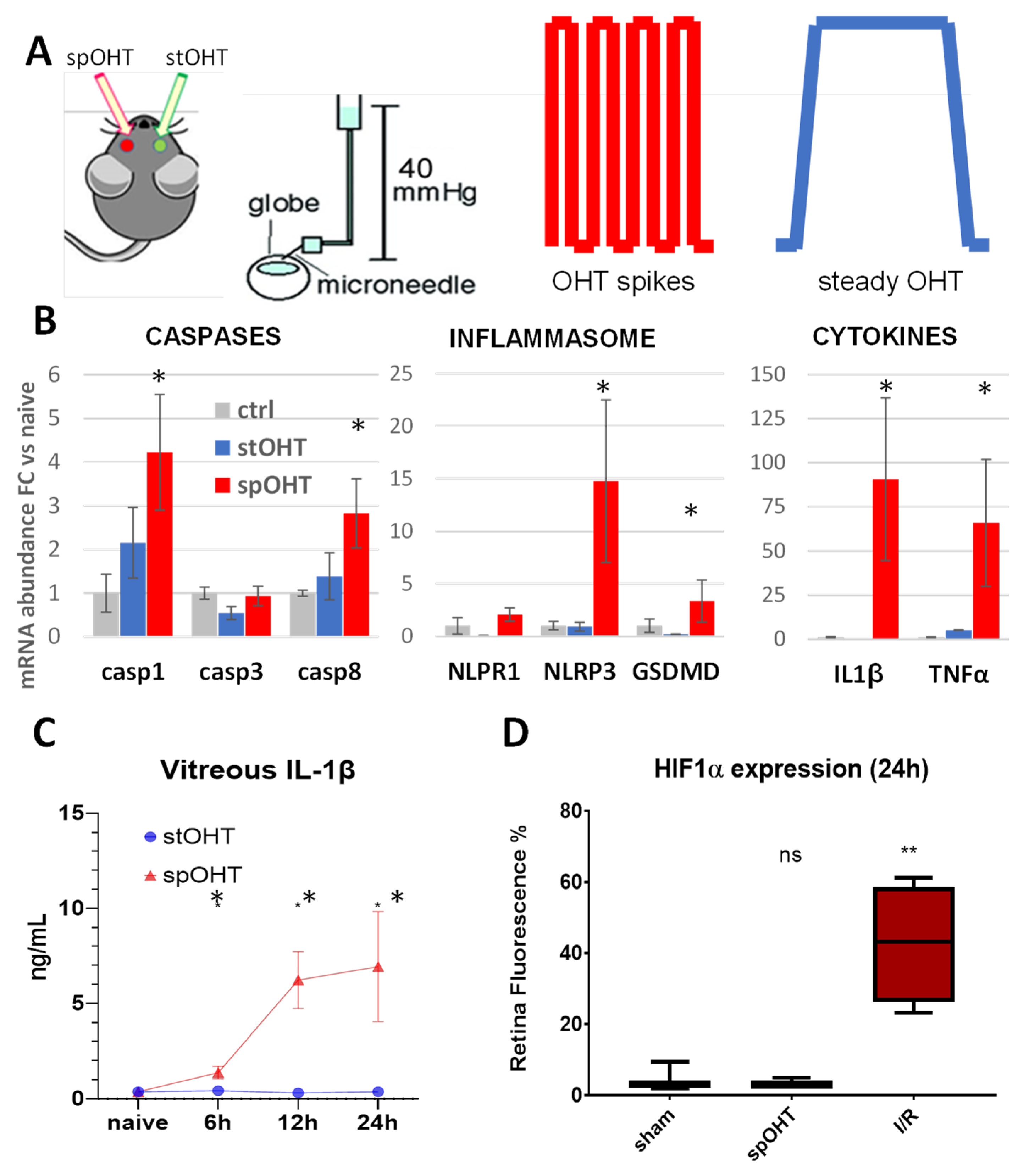
Figure 2.
PERG changes after single spOHT challenge are stable over time. Electro-physiological recordings of changes in PERG amplitude in the retinas exposed to either spOHT stress or stOHT (green bars) elevation for 7, 14 and 21 dpi. *P<0.05 . A. Representative PERG grand-average waveforms recorded in WT eyes prior to (baseline, blue) or 7d (red) after the challenge. B. Representative PERG grand-average waveforms recorded in WT eyes at 7d after either spOHT (red) or stOHT (yellow) challenges. C. Dynamics of PERG amplitude at 7, 14 and 21 dpi in C57Bl6/J eyes after spiking vs steady OHT challenges. Mean ± SE, *P <0.05 D. SpOHT-induced RGC loss at 1-3 weeks after spike or steady OHT. Mean ± SE, *P <0.05.
Figure 2.
PERG changes after single spOHT challenge are stable over time. Electro-physiological recordings of changes in PERG amplitude in the retinas exposed to either spOHT stress or stOHT (green bars) elevation for 7, 14 and 21 dpi. *P<0.05 . A. Representative PERG grand-average waveforms recorded in WT eyes prior to (baseline, blue) or 7d (red) after the challenge. B. Representative PERG grand-average waveforms recorded in WT eyes at 7d after either spOHT (red) or stOHT (yellow) challenges. C. Dynamics of PERG amplitude at 7, 14 and 21 dpi in C57Bl6/J eyes after spiking vs steady OHT challenges. Mean ± SE, *P <0.05 D. SpOHT-induced RGC loss at 1-3 weeks after spike or steady OHT. Mean ± SE, *P <0.05.
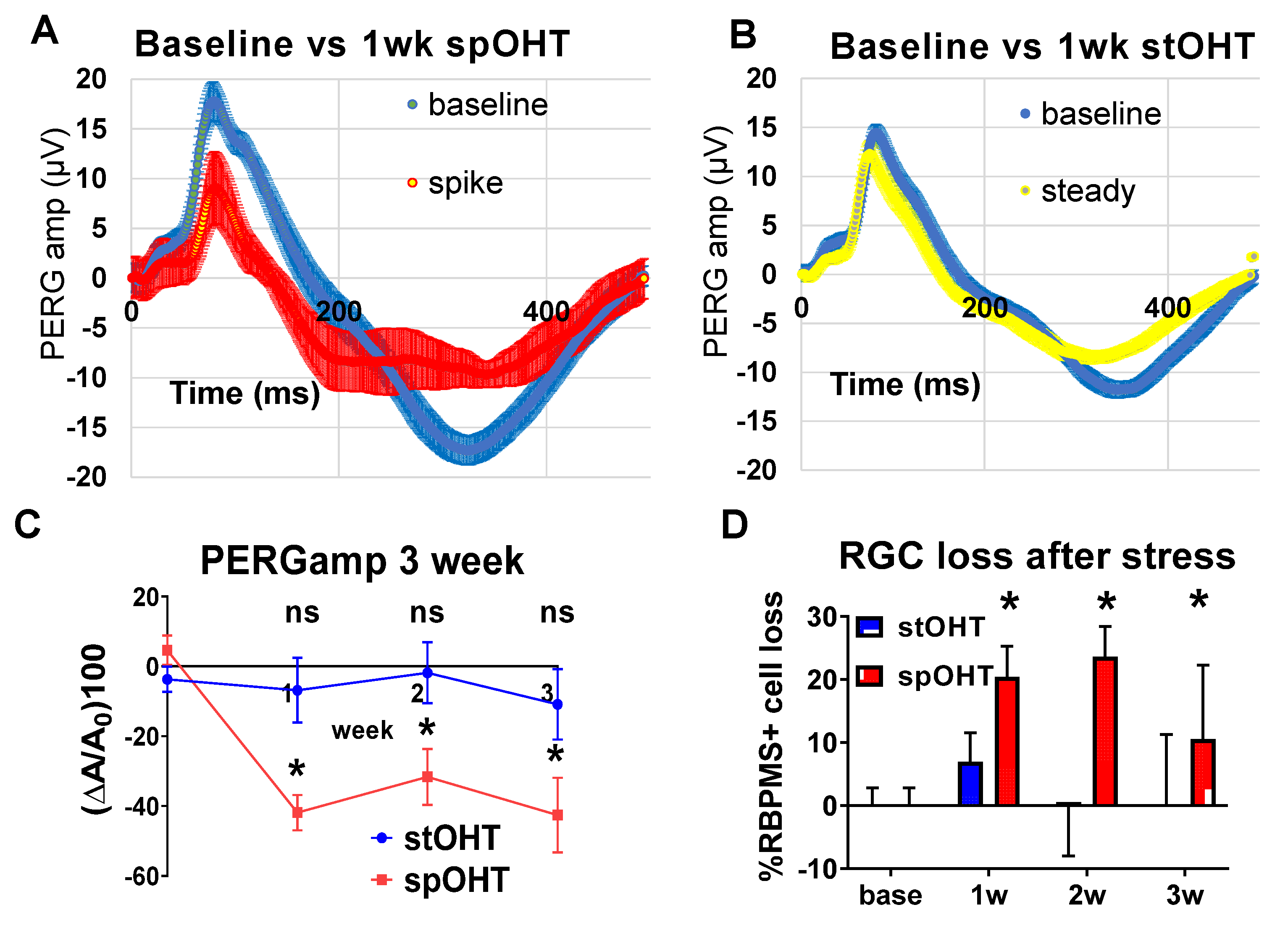
Figure 3.
Cytokine release into the vitreous strongly correlate with inflammasome activity. Cytokine levels in the vitreous of WT, NLRP1-/- , Casp1-/-, GsdmD-/- and Panx1-/- mice were measured simultaneously by multiplex ELISA at 24h after spOHT. Mean intravitreal concentration was measured as pg/ml (± SE) in a given transgenic mouse line and normalized by internal standards for every plate to allow between plate comparison. P<0.05, n=3-5.
Figure 3.
Cytokine release into the vitreous strongly correlate with inflammasome activity. Cytokine levels in the vitreous of WT, NLRP1-/- , Casp1-/-, GsdmD-/- and Panx1-/- mice were measured simultaneously by multiplex ELISA at 24h after spOHT. Mean intravitreal concentration was measured as pg/ml (± SE) in a given transgenic mouse line and normalized by internal standards for every plate to allow between plate comparison. P<0.05, n=3-5.
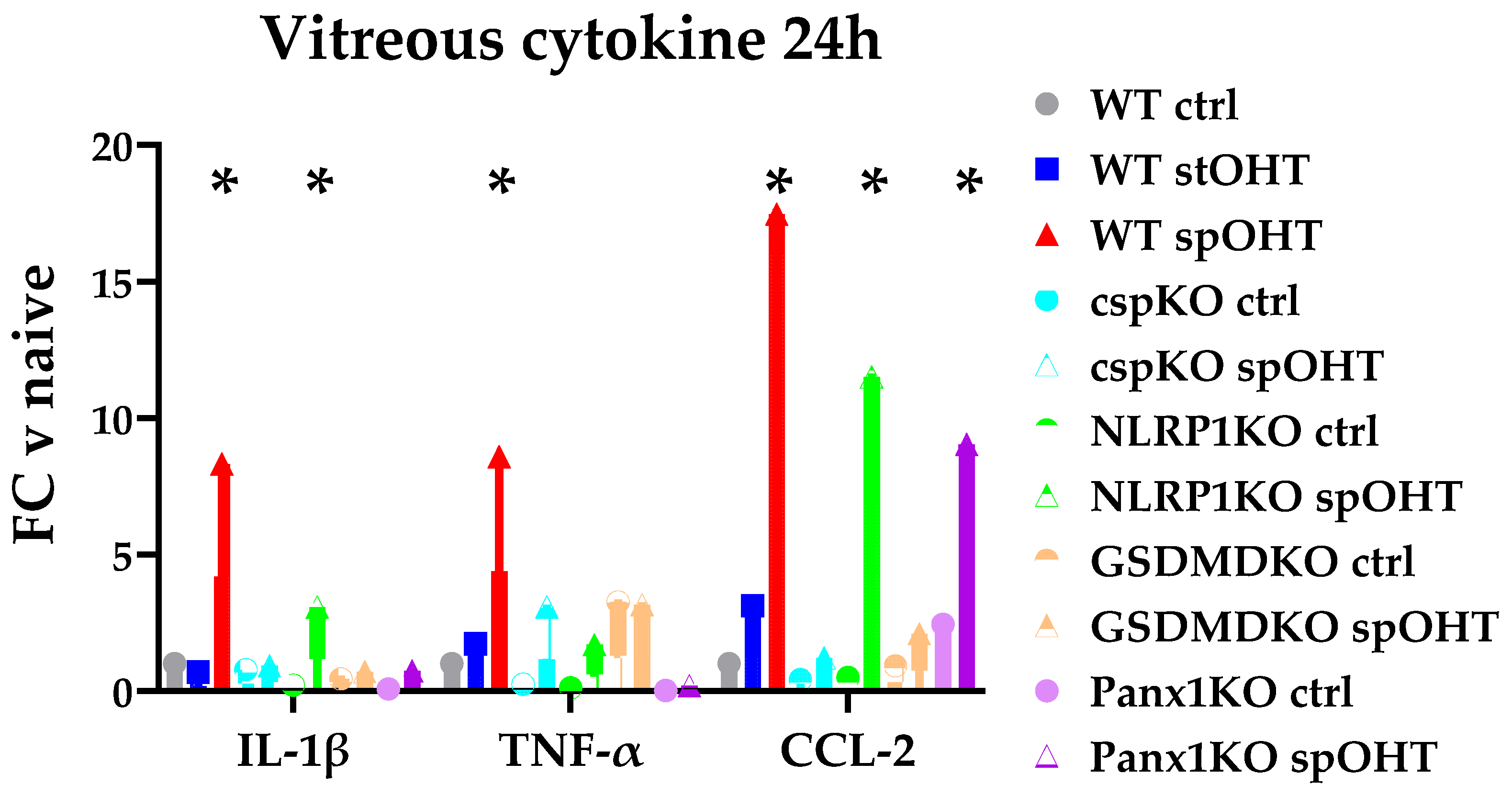
Figure 4.
Panx1 is essential for transcriptional activation of cytokines and inflammasome pathways in response to spOHT. Gene expression analysis was performed by RT-PCR in retinal samples collected 24 hrs after spOHT. Significant increases in il11b, tnfα, ccl2, casp1, nlrp3 and gsdmD transcripts found in WT mice (A, red) were blocked by PANX1 channel inactivation in Panx1-/- retinas (A, purple). Panx1 inactivation did not affect the upregulation of the gsdmD gene (B). *p<0.05, n=3-5.
Figure 4.
Panx1 is essential for transcriptional activation of cytokines and inflammasome pathways in response to spOHT. Gene expression analysis was performed by RT-PCR in retinal samples collected 24 hrs after spOHT. Significant increases in il11b, tnfα, ccl2, casp1, nlrp3 and gsdmD transcripts found in WT mice (A, red) were blocked by PANX1 channel inactivation in Panx1-/- retinas (A, purple). Panx1 inactivation did not affect the upregulation of the gsdmD gene (B). *p<0.05, n=3-5.
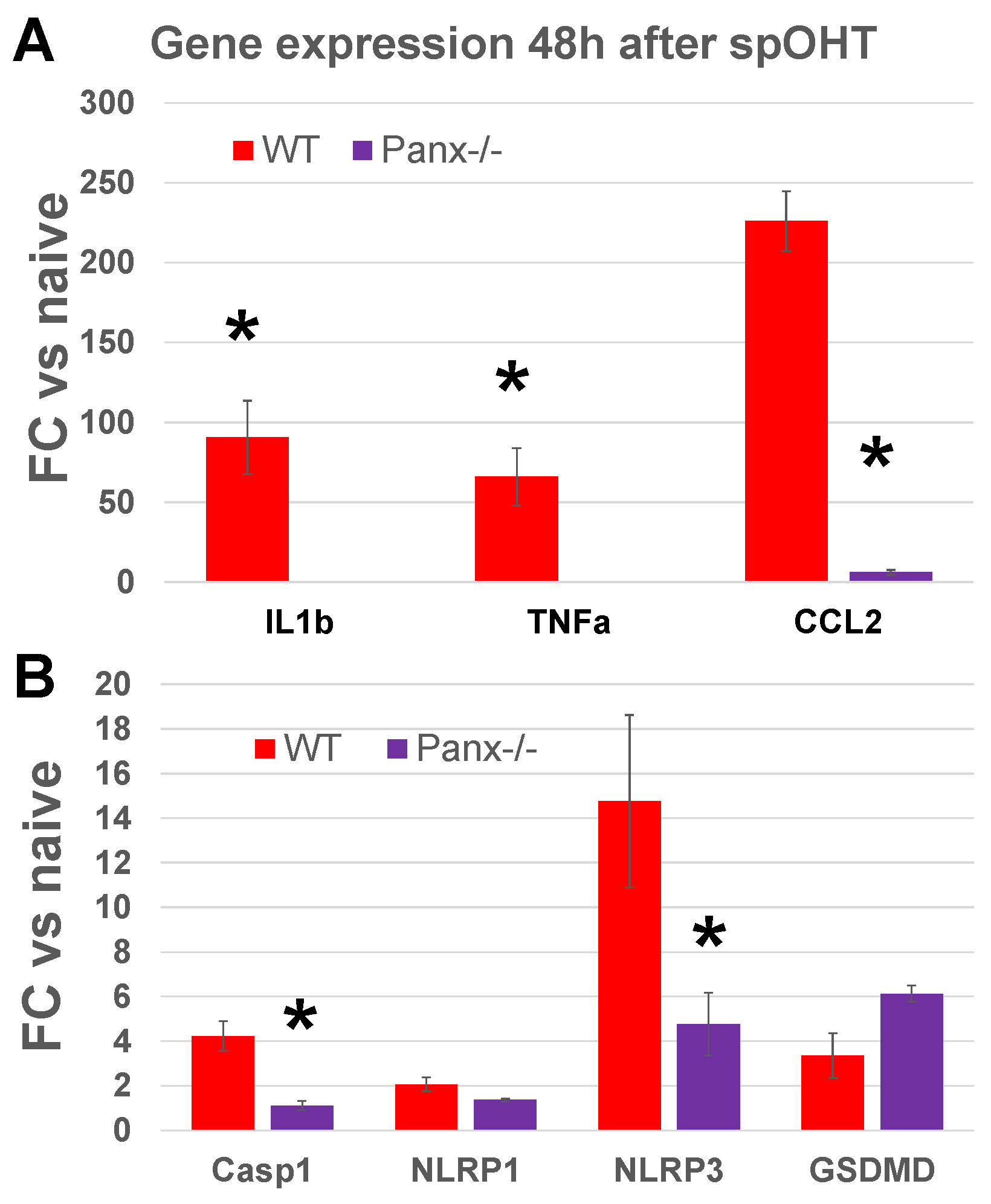
Figure 5.
PERG and RGC after spOHT injury with inflammasome knockout. A. significant PERGamp deficit was detected in WT and GsdmD-/- eyes at 1w after spOHT. No significant change in PERGamp was detected in spOHT-challenged eyes of the inflammasome-deficient strains NLRP1-/-, Casp1-/-, and Panx1-/- , in WT eyes pre-treated by Casp3 inhibitor DEVD and in control WT eye with stOHT challenge. B. RGC loss is significantly suppressed by inactivation of all key inflammasome proteins, the IL-1 release pore protein GsdmD, or caspase 3. The density of RBPMS+ RGCs was assayed by direct counting in wholemounts 7d after OHT challenge.
Figure 5.
PERG and RGC after spOHT injury with inflammasome knockout. A. significant PERGamp deficit was detected in WT and GsdmD-/- eyes at 1w after spOHT. No significant change in PERGamp was detected in spOHT-challenged eyes of the inflammasome-deficient strains NLRP1-/-, Casp1-/-, and Panx1-/- , in WT eyes pre-treated by Casp3 inhibitor DEVD and in control WT eye with stOHT challenge. B. RGC loss is significantly suppressed by inactivation of all key inflammasome proteins, the IL-1 release pore protein GsdmD, or caspase 3. The density of RBPMS+ RGCs was assayed by direct counting in wholemounts 7d after OHT challenge.
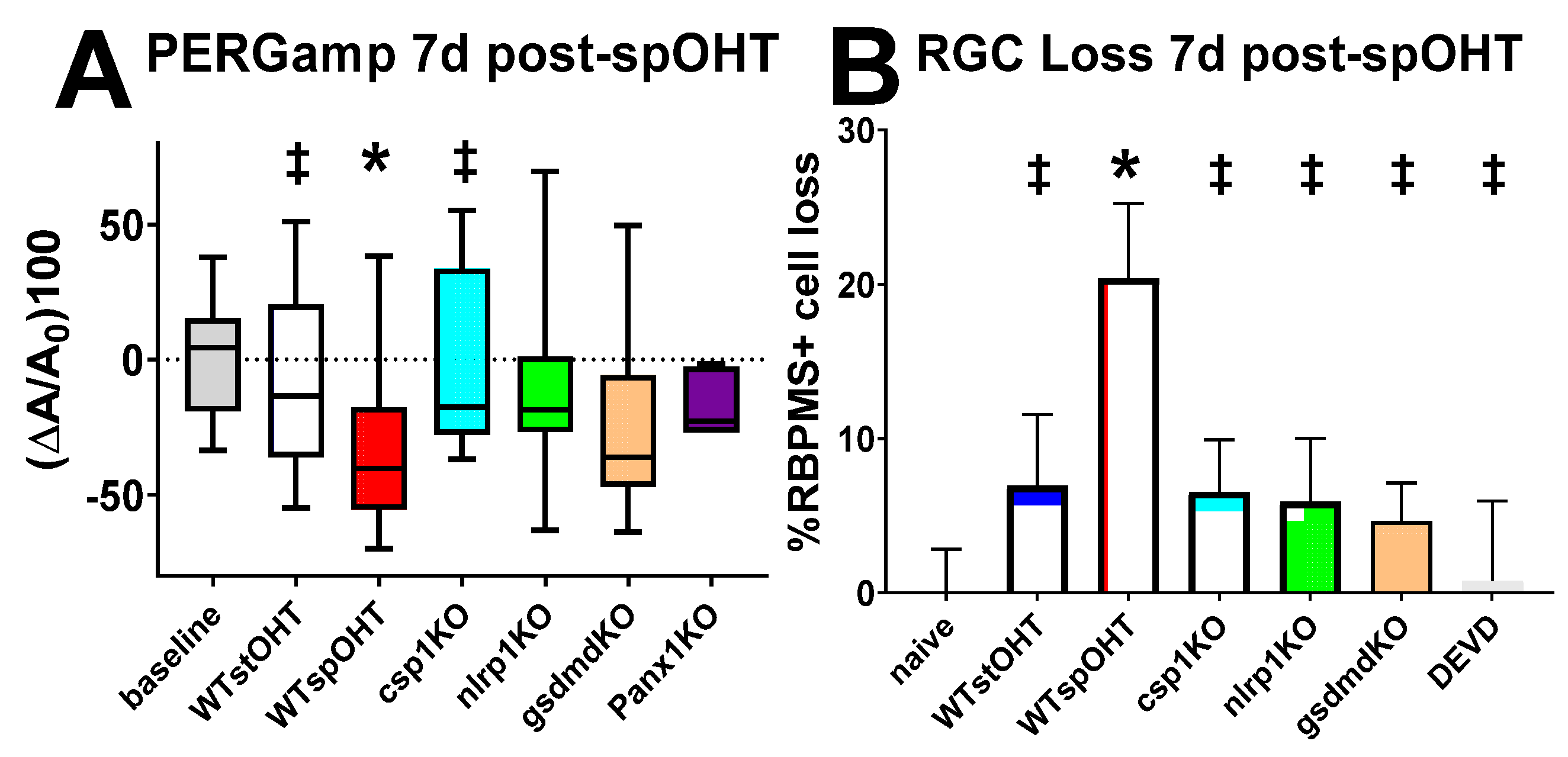
Figure 6.
Acute caspase activation in the GCL of spOHT-challenged retinas. A. Caspase-1 (red) and caspase-3/7 (green) activity in wholemount retinas was detected by an in vivo injection of FLICA substrate. Retinas were co-stained with GFAP (light grey, top panels) and RBPMS (light grey, bottom panels) white/merge to identify astrocytes and RGCs, respectively. White arrows indicate cells that co-activate caspases 1 and 3/7; yellow arrows-cells point at Casp3/7 activity only. D. Pre-treatment with VX765 casp1 blocked both Casp1 and Casp3/7 activity after spOHT. Size bar, 20 µm.
Figure 6.
Acute caspase activation in the GCL of spOHT-challenged retinas. A. Caspase-1 (red) and caspase-3/7 (green) activity in wholemount retinas was detected by an in vivo injection of FLICA substrate. Retinas were co-stained with GFAP (light grey, top panels) and RBPMS (light grey, bottom panels) white/merge to identify astrocytes and RGCs, respectively. White arrows indicate cells that co-activate caspases 1 and 3/7; yellow arrows-cells point at Casp3/7 activity only. D. Pre-treatment with VX765 casp1 blocked both Casp1 and Casp3/7 activity after spOHT. Size bar, 20 µm.
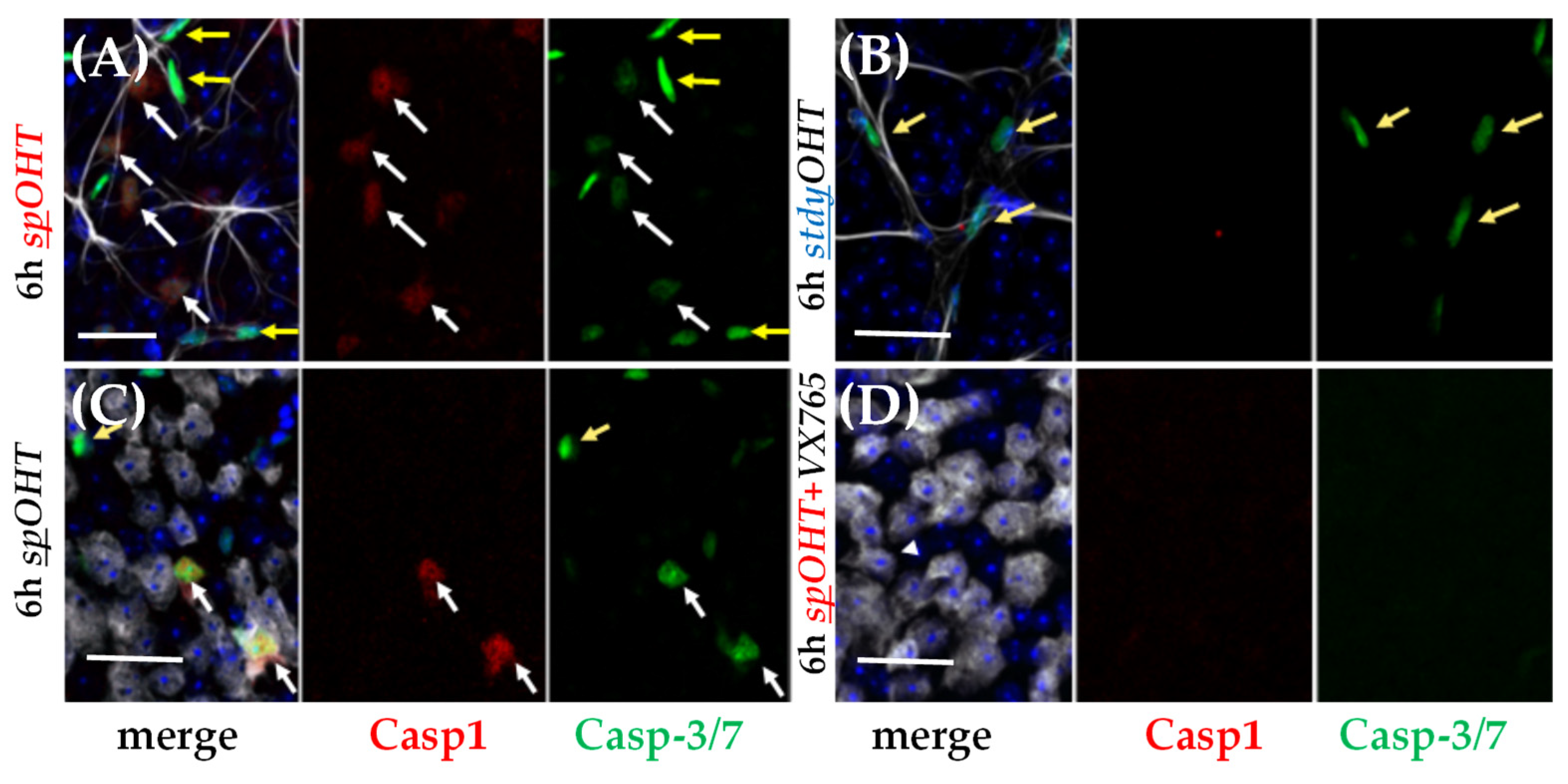
Figure 7.
Pro-inflammatory cytokines in the spOHT-challenged retina. Immunolabeling for IL-1β (blue) co-localized with neurons in the GCL and INL; TNFα-specific labelling (green) confined to GFAP+ astroglia (arrows), infiltrating monocytes (arrowheads) in the retina 48h after the rspOHT challenge. ASC-citrine specks (magenta) co-localized with the GFAP+ TNFα+ astrocytes and Muller glia (yellow arrows) in the inner retina, and with astrocytes in the optic nerve. In control stOHT-treated retinas (bottom panels) both TNFα and IL1β -specific labeling were reduced, no infiltrating monocytes were detected. Size bars in µm.
Figure 7.
Pro-inflammatory cytokines in the spOHT-challenged retina. Immunolabeling for IL-1β (blue) co-localized with neurons in the GCL and INL; TNFα-specific labelling (green) confined to GFAP+ astroglia (arrows), infiltrating monocytes (arrowheads) in the retina 48h after the rspOHT challenge. ASC-citrine specks (magenta) co-localized with the GFAP+ TNFα+ astrocytes and Muller glia (yellow arrows) in the inner retina, and with astrocytes in the optic nerve. In control stOHT-treated retinas (bottom panels) both TNFα and IL1β -specific labeling were reduced, no infiltrating monocytes were detected. Size bars in µm.
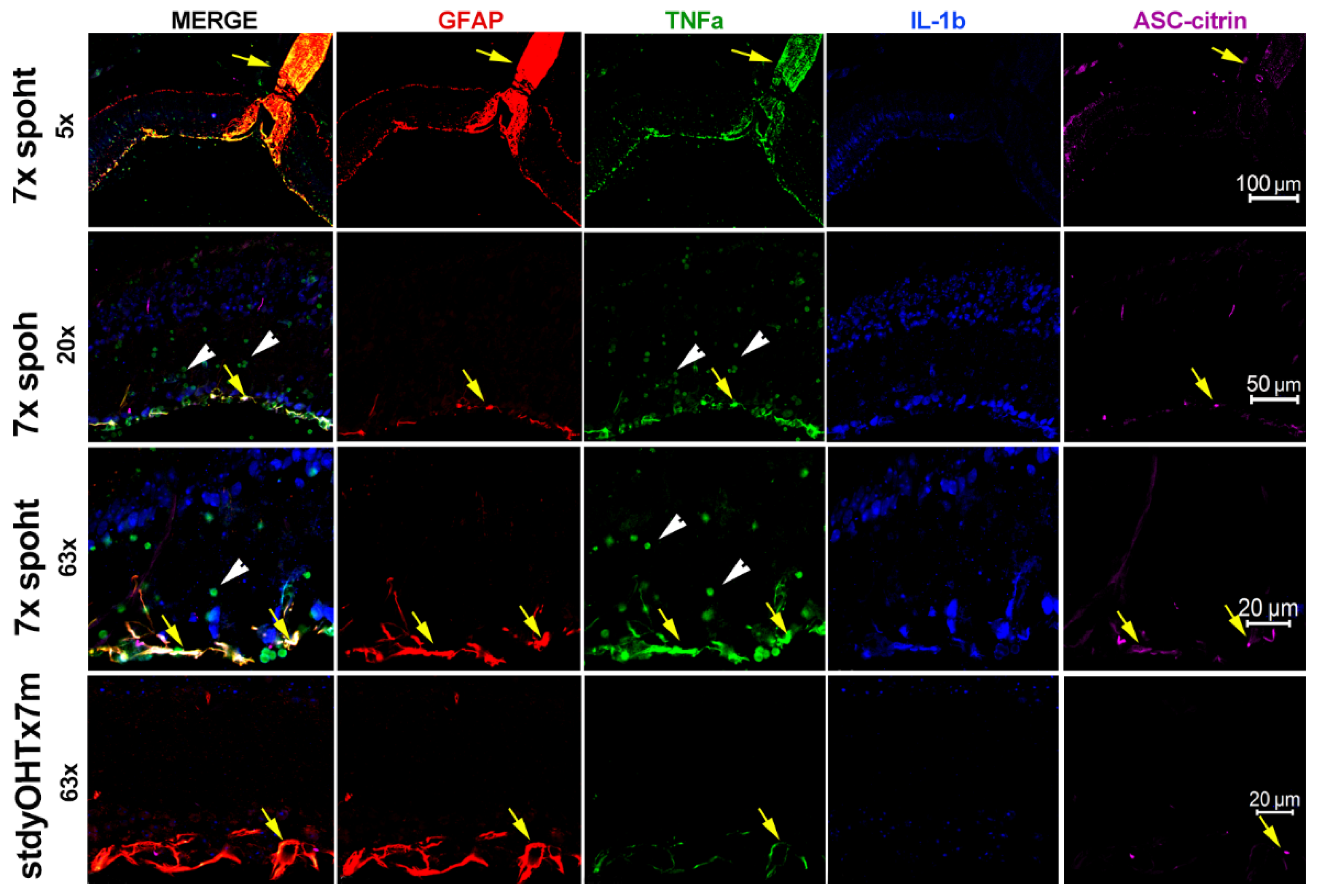
Figure 8.
Inflammasome sensor proteins in the spOHT-challenged inner retina. A. Immunolabeling specific for NLRP1 co-localized with TUJ1+ neurons, their axons, GFAP+ astrocytes (yellow arrowheads) in the GCL of both spOHT and sham-treated control retinas; in spOHT samples NLRP1 was also detected in infiltrating monocytes (yellow arrowheads). B. NLRP3 labeling co-localized with TUJ1+ neurons and infiltrating monocytes (yellow arrowheads) in the GCL of spOHT-challenged retinas; in sham controls it also labeled glial cells (white arrows). C. AIM2 sensor co-localized with GlutSyn+ Muller glia (white arrows) in both sham-treated controls and spOHT retinas, and showed an increased accumulation in the GCL region after spOHT challenge. Size bars, 20µm.
Figure 8.
Inflammasome sensor proteins in the spOHT-challenged inner retina. A. Immunolabeling specific for NLRP1 co-localized with TUJ1+ neurons, their axons, GFAP+ astrocytes (yellow arrowheads) in the GCL of both spOHT and sham-treated control retinas; in spOHT samples NLRP1 was also detected in infiltrating monocytes (yellow arrowheads). B. NLRP3 labeling co-localized with TUJ1+ neurons and infiltrating monocytes (yellow arrowheads) in the GCL of spOHT-challenged retinas; in sham controls it also labeled glial cells (white arrows). C. AIM2 sensor co-localized with GlutSyn+ Muller glia (white arrows) in both sham-treated controls and spOHT retinas, and showed an increased accumulation in the GCL region after spOHT challenge. Size bars, 20µm.
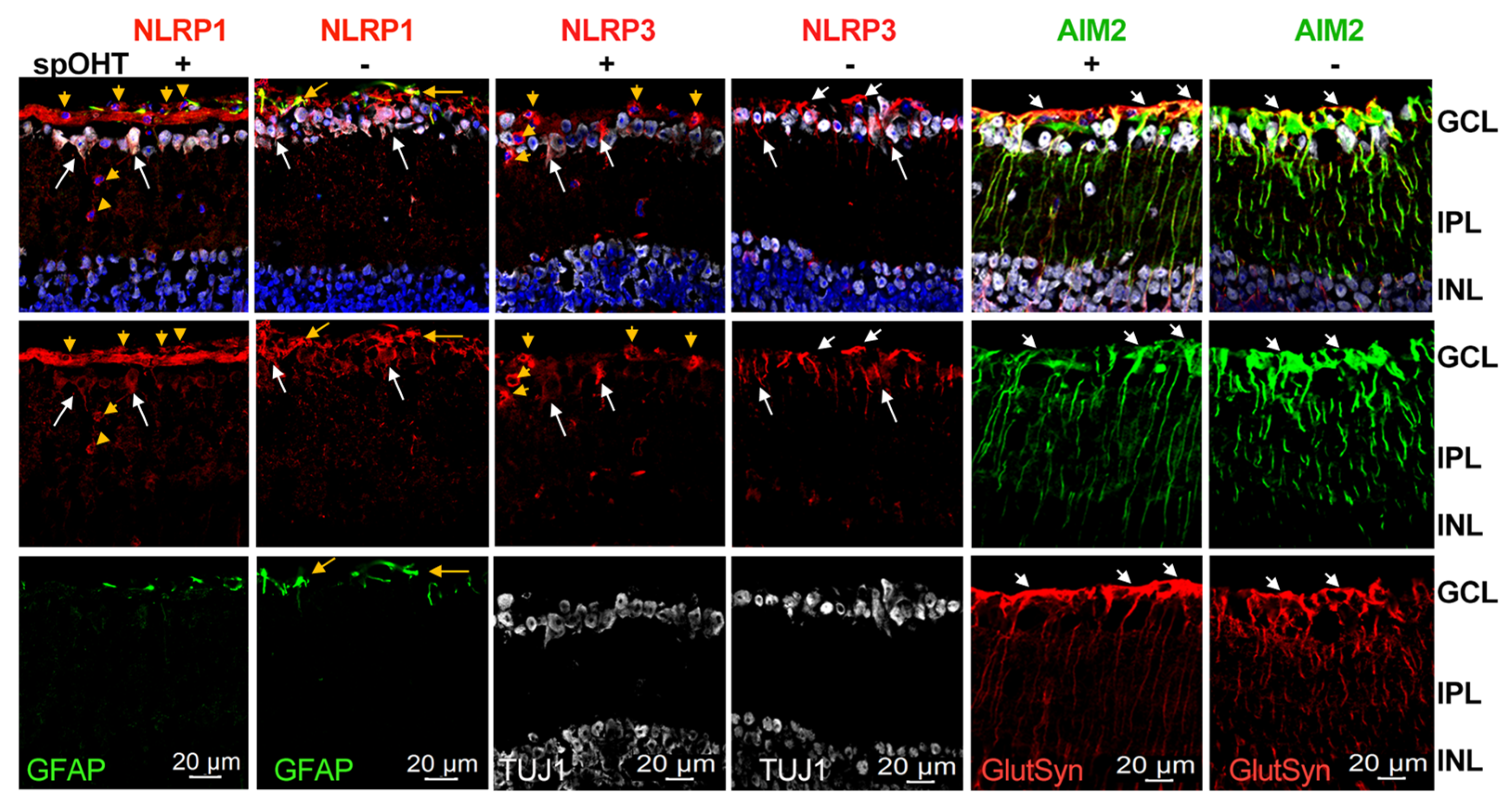
Figure 9.
Monocyte/macrophages infiltration into the inner retina and optic nerves after spOHT challenge. A,B. Representative micrographs for cells expressing CD45 (red) and CD11b (green) markers in rounded monocyte/macrophage cells that were abundant at the inner retinal surface, in the GCL, INL and in the optic nerves of the spOHT-challenged eyes. Co-staining with GFAP (light grey, top panels) identified astrocytes. C,D. Control immunostaining in stdyOHT-challenged optic nerves showed no CD45+ monocytes in both retina and optic nerves. Small punctate CD45+CD11b+ cells are ramified microglia. Bright green puncta represent citrin-labelled ASC-specks of mature inflammasomes that are abundant in the optic nerve astrocytes and in infiltrating cells (yellow arrows).
Figure 9.
Monocyte/macrophages infiltration into the inner retina and optic nerves after spOHT challenge. A,B. Representative micrographs for cells expressing CD45 (red) and CD11b (green) markers in rounded monocyte/macrophage cells that were abundant at the inner retinal surface, in the GCL, INL and in the optic nerves of the spOHT-challenged eyes. Co-staining with GFAP (light grey, top panels) identified astrocytes. C,D. Control immunostaining in stdyOHT-challenged optic nerves showed no CD45+ monocytes in both retina and optic nerves. Small punctate CD45+CD11b+ cells are ramified microglia. Bright green puncta represent citrin-labelled ASC-specks of mature inflammasomes that are abundant in the optic nerve astrocytes and in infiltrating cells (yellow arrows).
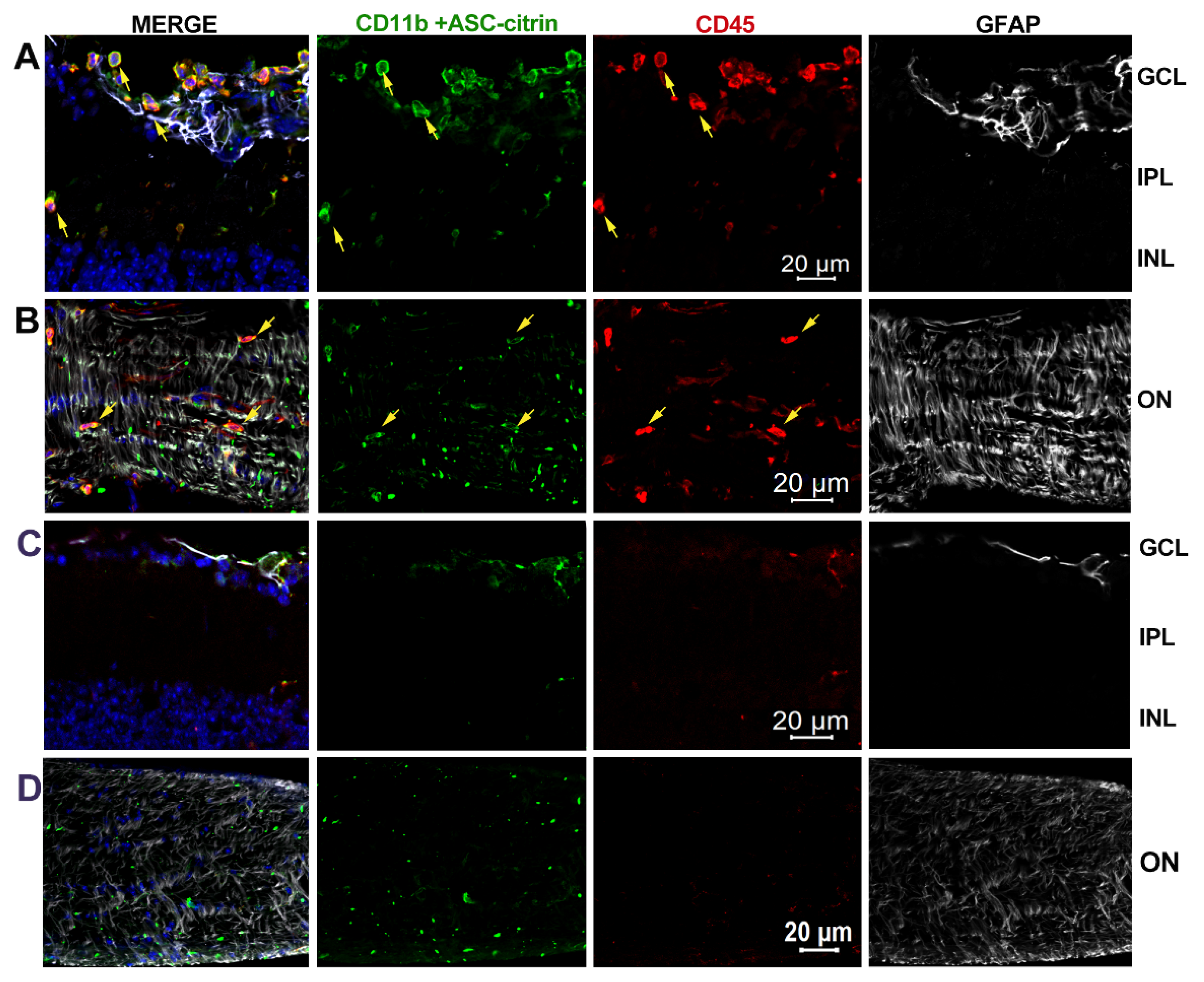
Figure 10.
Inflammasome activation in the Ad5-MYOC-indiced chronic OHT/glaucoma model. Dynamics of initial IOP changes (A) was similar in WT, Casp1- and NLRP1- strains with relatively higher levels of IOP elevation in knockouts at 2-4 wks. An increase in IOP in experimental eyes temporally correlated with RGC dysfunction detected as with a ~50% decline in PERGamp (B)in WT but not in Casp1- and NLRP1- mice at 2 wks post-induction. C. IL-1β release was detectable 3-8 wk post-OHT induction, peaking at 5wks at ~60% of that in positive control eyes with injection of ATP or ischemia-reperfusion (IR) injury. D. RGC density changes in experimental eyes vs eyes from naïve controls showed 32% and 19.1% loss in WT center (/C) and periphery (/P). The loss was significantly less in Casp1- and Nlrp1- retinas at 7dpi after spOHT challenge, *P<0.05 (E-F). ASC-speck labeling (green specks/filaments) revealed mature inflammasome complexes co-localizing with RBPMS+(red) RGCs (yellow arrows), GFAP+ (grey) astrocytes (white arrows) and blood vessels (unstained dark grey structures, blue “BV”) G. Quantification of ASC-citrin+ cell types in retinal wholemounts at 6 and 8wks post-induction indicated predominant localization to RGCs and astrocytes, *P<0.05.
Figure 10.
Inflammasome activation in the Ad5-MYOC-indiced chronic OHT/glaucoma model. Dynamics of initial IOP changes (A) was similar in WT, Casp1- and NLRP1- strains with relatively higher levels of IOP elevation in knockouts at 2-4 wks. An increase in IOP in experimental eyes temporally correlated with RGC dysfunction detected as with a ~50% decline in PERGamp (B)in WT but not in Casp1- and NLRP1- mice at 2 wks post-induction. C. IL-1β release was detectable 3-8 wk post-OHT induction, peaking at 5wks at ~60% of that in positive control eyes with injection of ATP or ischemia-reperfusion (IR) injury. D. RGC density changes in experimental eyes vs eyes from naïve controls showed 32% and 19.1% loss in WT center (/C) and periphery (/P). The loss was significantly less in Casp1- and Nlrp1- retinas at 7dpi after spOHT challenge, *P<0.05 (E-F). ASC-speck labeling (green specks/filaments) revealed mature inflammasome complexes co-localizing with RBPMS+(red) RGCs (yellow arrows), GFAP+ (grey) astrocytes (white arrows) and blood vessels (unstained dark grey structures, blue “BV”) G. Quantification of ASC-citrin+ cell types in retinal wholemounts at 6 and 8wks post-induction indicated predominant localization to RGCs and astrocytes, *P<0.05.
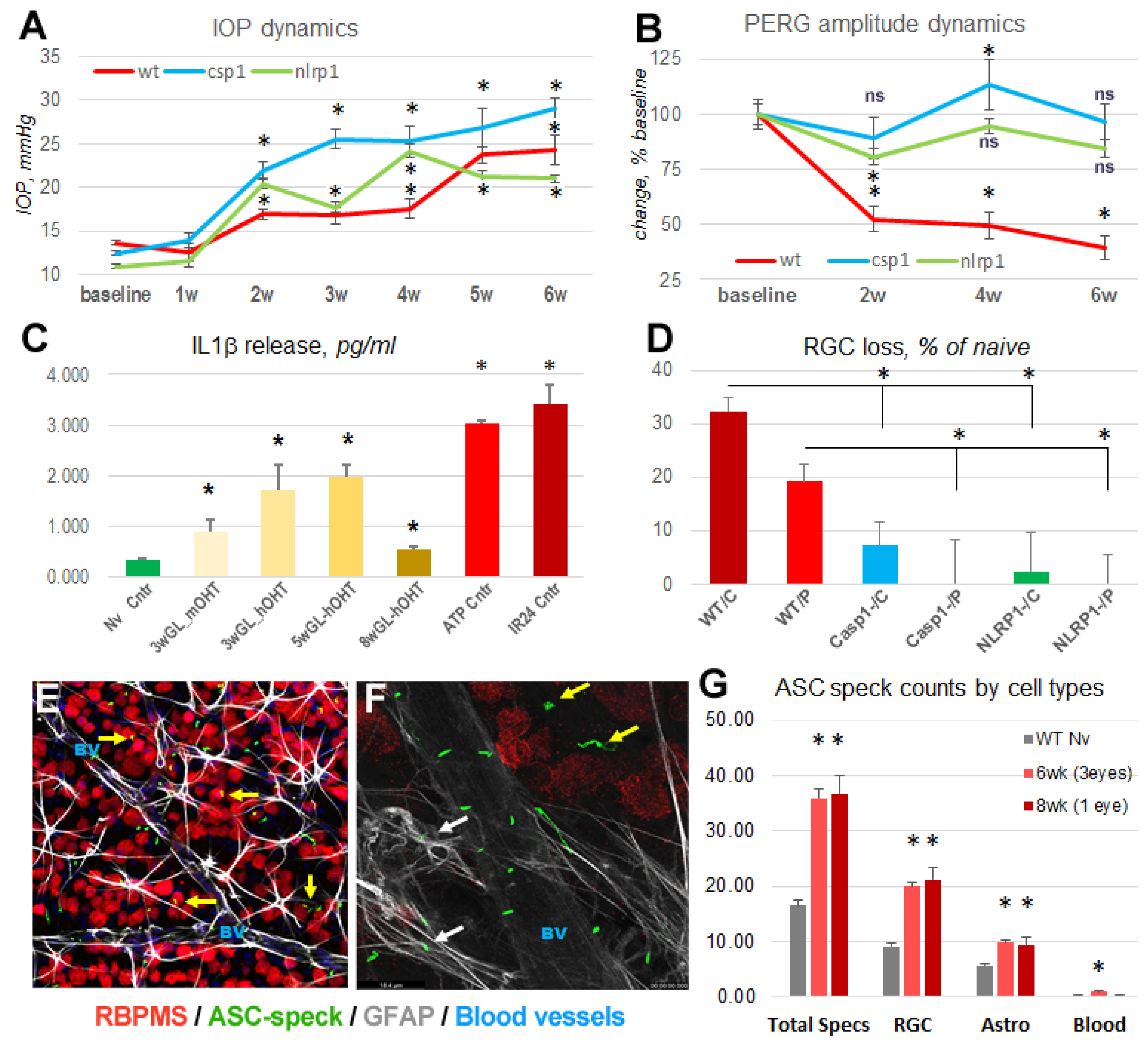
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



