
Submitted:
26 September 2023
Posted:
29 September 2023
You are already at the latest version
Abstract
CD74 is a type II cell surface receptor found to be highly expressed in several hematological and solid cancers, due to its ability to activate pathways associated with tumor cell survival and proliferation. Over the past 16 years, CD74 is emerging as a commonly detected fusion partner in multiple oncogenic fusion proteins. Studies have found CD74 fusion proteins in a range of cancers including lung adenocarcinoma, inflammatory breast cancer, and pediatric acute lymphoblastic leukemia. To date, there are five known CD74 fusion proteins, CD74-ROS1, CD74-NTRK1, CD74-NRG1, CD74-NRG2a, and CD74-PDGFRB, with a total of 16 different variants, each with unique genetic signatures. Importantly, the occurrence of CD74 in the formation of fusion proteins has not been well explored despite the fact that ROS1 and NRG1 families utilize CD74 as the primary partner for the formation of oncogenic fusions. Fusion proteins known to be oncogenic drivers, including those of CD74, are typically detected, and targeted after standard chemotherapeutic plans fail and the disease relapses. The analysis reported herein provides insights into early intervention of CD74 fusions and highlight the need for improved routine assessment methods so that targeted therapies can be applied while they are most effective.
Keywords:
Cluster of differentiation 74 (CD74)
; oncogenic fusion protein
; fusion gene
; CD74-ROS1
; CD74-NTRK1
; CD74-NRG1
; CD74-NRG2a
; CD74-PDGFRB
; cancer.
1. Introduction
A fusion protein is the outcome of a multistep process occurring at either the DNA or RNA level and involves the cleavage of two independent genes followed by their joining into a hybrid one. Upon translation, the newly formed fusion protein may or may not have a biological activity depending on multiple factors such as the protein’s localization, structural architecture, and functional domains [1]. Fusion proteins occurring in healthy individuals are of low significance as they are often inactive. However, in cancer the identification of such proteins, which are widely known as oncogenic fusion proteins, has a great diagnostic and therapeutic value [2]. Notably, oncogenic fusion proteins are cancer-specific, bioactive molecules that have been detected in both hematological and solid cancers [3]. Identification of fusion proteins is challenging and primarily performed by DNA or RNA next-generation sequencing (NGS), fluorescence in situ hybridization (FISH), reverse transcription-polymerase chain reaction (RT-PCR), and real-time polymerase chain reaction (RT-qPCR). Via these methods, which may be used alone or in combination, a large number of oncogenic fusions have been detected in either cancer patients or retrospective studies. Several oncogenic fusions protein families exist, with ROS proto-oncogene 1 (ROS1), anaplastic lymphoma kinase (ALK), neurotrophic tyrosine receptor kinase (NTRK), and neuregulin 1 (NRG1) being some of the most common partners that are primarily detected in lung cancers [4,5,6].
Cluster of Differentiation 74 (CD74) is a membrane bound protein expressed on the surface of human antigen presenting cells (APCs). Also known as the invariant chain (Ii), the soluble form of CD74, which is found in the endoplasmic reticulum (ER), associates with major histocompatibility class II (MHC II) molecules to ensure their proper folding. CD74 can act as a receptor for the tumorigenic and proinflammatory cytokines macrophage migration inhibitor factor (MIF) [7] and D-dopachrome tautomerase (D-DT, or MIF-2) [8]. After the formation of either the MIF-CD74 or D-DT-CD74 complex, intracellular signal transduction is achieved through the presence of the coreceptor proteins CD44 [9]. Other receptors capable of forming a complex with CD74 for MIF signaling including CXCR2 [10] and CXCR4 [11]. Consequently, the extracellular signal-regulated kinase (ERK)-1/2 mitogen-activated protein kinase (MAPK) [8, 12], nuclear factor kappa B (NF-κB) [13], phosphoinositide-3-kinase (PI3K)/Akt [14] and c-Jun N-terminal kinase (JNK) pathways are activated [15] with downstream effects in cell survival and proliferation [16].
Besides its expression on immune system cells, CD74 has also been expressed in a variety of cancer cells including thyroid carcinoma [17], bladder cancer [18], chronic lymphocytic leukemia (CLL) [19], multiple myeloma [20], breast cancer [21], gastrointestinal cancers [22], non-small cell lung cancer (NSCLC) [23], renal cell carcinoma (RCC) [24], prostate cancer [25], pancreatic cancer [26], and glioblastomas [27]. Elevated expression of CD74 can be a marker of tumor progression [28] as well as poor clinical prognosis [29]. Since the beginning of the 21st century, CD74 has also been reported to participate in the formation of oncogenic fusion proteins [30,31,32,33,34]. While the functional role of CD74 in these oncogenic fusion proteins is completely unknown, in certain protein families such as ROS1 and NRG1, its occurrence is very high.
Transcriptional splicing can produce four different human CD74 isoforms, p33, p35, p41 and p43, which are 216 [35], 232 [36], 280 [37], and 296 [38] amino acids long, respectively [37] (Figure 1 and Figure S1). Structurally, CD74 is encoded by 8-9 exons expressing an N-terminal intracellular cytoplasmic tail (exon 1), a single transmembrane segment (exon 2), and an extracellular C-terminal domain (exons 3-8) [39], which includes exon 6 that is critical for trimerization [40]. The ninth exon arises from alternative splicing that produces exon 6b, which encodes in both the longer CD74 isoforms, p41 and p43 [39]. Similarly, an alternative start codon gives rise to the 16 amino acid (aa) N-terminal extension, which is presented in CD74 isoforms p35 and p43 [41]. The biological assembly of CD74 is a homotrimer [42] and characterized by enhanced flexibility that negatively impacts all the efforts towards obtaining complete structural information. Instead, structural fragments of CD74 have been resolved by nuclear magnetic resonance (NMR) spectroscopy or protein crystallography and deposited in the protein data bank (PDB). These include the trimeric domain [43], class II associated invariant chain peptide (CLIP) [44-47], and p41 fragment [48,49] (Figure 1).
This review is focused on the CD74 oncogenic fusions (CD74-ROS1, CD74-NTRK1, CD74-NRG1, CD74-NRG2α, and CD74-PDGFRB) identified in various cancers. The structural features and expression patterns of all known CD74 fusion variants are described. Clinical patient characteristics, treatment plans, detection methods, as well as retrospective analyses of tumor samples are also included to provide insights into CD74 related oncogenic fusion proteins. While CD74 protein alone is an attractive molecular target in multiple cancers, the study of cancer-specific proteins, such as the ones described here, offer a great opportunity for refining the currently utilized cancer treatment plans and developing new methods for early detection.
2. CD74 Fusion Proteins in Cancer
To date, five in-frame CD74 fusion proteins have been reported in cancer. These include CD74-NRG2α, CD74-NTRK1, CD74-ROS1, CD74-PDGFRB, and CD74-NRG1 (Figure 2).
The discovery of CD74 fusion proteins began with the identification of CD74-ROS1 in an adult female lung adenocarcinoma patient in 2007 [30] (Figure 3). This fusion protein is characterized by the presence of two transmembrane domains, one obtained from CD74 and the other from ROS1. According to the structural architecture of this fusion, the kinase domain of ROS1 is localized in the cytoplasm (Figure 2). In 2013, CD74-NTRK1 was the next reported fusion, which was detected in an adult patient (unspecified gender) with lung adenocarcinoma (Figure 3) [31]. Similar to CD74-ROS1, this oncogenic fusion protein contains two transmembrane domains and an intracellular kinase domain [31] (Figure 2). An adult female invasive mucinous adenocarcinoma patient was found to harbor a CD74-NRG1 fusion in 2014 [32] (Figure 3). In contrast to the first two reported CD74 fusion proteins CD74-NRG1 has only one transmembrane domain, which is derived from CD74 [32]. Upon formation of CD74-NRG1 fusion, the epidermal growth factor (EGF)-like domain of NRG1 remains intact in the extracellular space, providing the ligand for ERBB2-ERBB3 receptor complexes. In 2016, a study focused on adult and pediatric patients with B-cell acute lymphoblastic leukemia (B-ALL) revealed 29 in-frame fusion proteins, one of which was the novel CD74-PDGFRB (Figure 3). This fusion was identified in a pediatric patient and according to its occurrence is significantly rarer in comparison to CD74-ROS1, CD74-NTRK1, and CD74-NRG1 [33]. From the structural point of view, CD74-PDGFRB contains two transmembrane domains, one from each protein partner and a kinase domain found in the cytoplasm (Figure 2). In 2020, CD74-NRG2α was identified in a female acinar adenocarcinoma patient from Japan [34] (Figure 3). This protein is the latest known CD74 fusion and similar to CD74-PDGFRB is very rare. CD74-NRG2α contains one transmembrane domain, from CD74, and an extracellular EGF motif encoded by exons 4 and 5 of NRG2α (Figure 2).
Noteworthy, the majority of CD74 fusion proteins found in patients were first reported in lung cancers and the same trend continues to date (Figure 3). Of the five fusions, only CD74-ROS1 was additionally identified in an inflammatory breast cancer patient (Figure S2), [52] besides its presentation in lung cancer. Interestingly, the rare CD74-PDGFRB oncogenic fusion remains to have cancer relevance in exclusively leukemia [53] (Figure S2).
CD74-ROS1 has seven variants reported in the literature, while CD74-NTRK1 and CD74-NRG1 have three and four variants each, respectively (Table 1). CD74-PDGFRB and CD74-NRG2α are very rare with only one known functional fusion (Table 1). Throughout most of the fusion variants, the first 6 exons of CD74, which include the intracellular, transmembrane, and extracellular trimeric domain are retained. Of the several isoforms of oncogenic fusion proteins identified, only the amino acid sequences of CD74-ROS1 (C6-R34 [30, 54], C6-R32 [54]) and CD74-NTRK1 (C8-N12 [31]) are published (Figure S3). The total amino acid residues reported for CD74-ROS1 variants C6-R34, C6-R32, and C6-R33 are 703 [30, 54], 806 [54], and 778 aa [55], respectively. Of the CD74-NTRK1 variants, the C8-N12 one has been documented as 627 aa long [31]; CD74-NRG1 C6-N6 was reported as 283 aa [32] and 282 aa [56] long in two separate publications. Of the remaining fusions, CD74-NRG1 C8-N6 is documented as 303 aa in length [56]. Of note, the amino acid length of CD74-PDGFRB and CD74-NRG2α have not been reported.
2.1. Analysis of CD74 Fusion Partners
In an expression study of human cancer cell lines, ROS1 was found highly expressed in many glioblastoma cell lines but was not seen in normal healthy brain tissue [65]. ROS1 has also been found to be overexpressed in oral carcinoma [66] and lung cancer tissue [67,68]. From the cell line SW-1088, ROS1 was further characterized to encode a 259 kDa (2347 aa) protein with a glycosylated extracellular domain, a single transmembrane domain, and an intracellular tyrosine kinase domain [69]. Despite the early characterization, ROS1 ligand is still unknown, making the protein an orphan receptor [70]. The ROS1 gene is encoded by 43 exons according to the NCBI Reference Sequence NM_002944.2. Exons 1-32 produce the extracellular domain of ROS1, while exon(s) 33 and 36-43 encode the transmembrane portion and C-terminal domain, respectively. The intracellular kinase domain or ROS1 is encoded by exons 36-41 [71]. Tumors positive for ROS1 are typically treated with Crizotinib, a tyrosine kinase receptor (TRK) inhibitor. This treatment plan was approved for clinical use in 2016 [72] based on a phase I clinical trial with ROS1 rearrangements in NSCLC that had a 72% objective response rate (ORR) to the drug [73]. In 2019, Entrectinib, a pan-TRK inhibitor, was approved for use against ROS1 NSCLC fusions based on its 77% ORR in clinical trials and ability to penetrate the central nervous system, making it suitable for ROS1 fusion NSCLC patients who experienced brain metastasis [74].
The NTRK1 gene encodes the tropomyosin kinase receptor A (TRKA) protein, which is involved in neural development [75]. This protein is produced in two isoforms due to alternative splicing [76], one is 790 aa long (trkAI) [76,77] and the more abundant form is 796 aa (trkAII) [76,78]. TRKA is composed of an extracellular domain for ligand binding, a transmembrane domain, and an intracellular tyrosine kinase domain [77]. Nerve growth factor (NGF) is the preferred ligand that binds with high affinity to the TRKA receptor [79, 80]. The main signaling pathways activated by TRKA include PI3K-AKT, Phospholipase C (PLC)-γ, ERK, and the MAPK pathways, which are all needed for neuronal survival and differentiation [81,82]. Human TRKAII, encoded by the NTRK1 gene that contains 17 exons. Exons 1-9 encode a unique extracellular domain, exons 10-11 yield the transmembrane domain, and exons 11-17 produce for the intracellular domain, which includes the tyrosine kinase domain [83]. In response to NGF, the tyrosine kinase domain becomes phosphorylated, which is imperative for intracellular signaling [83]. TRKA was identified as an oncogenic driver when it was found mutated in an acute myeloid leukemia (AML) patient [84]. The findings of the case study showed that deletion of a 75-amino acid segment from the extracellular domain led to constitutive tyrosine phosphorylation of the protein. Besides AML, TRKA has also been reported to participate in multiple fusions proteins [78]. Overexpression of TRKA is associated with many other cancers, including lung [85], thyroid [86], breast [87], and cervical cancer [88]. Oncogenic fusions typically incorporate the C-terminal portion of NTRK that contains the kinase domain with the N-terminal of a new protein partner. Consequently, the ligand-binding domain of NTRK is removed [72]. Larotrectinib, a TRK inhibitor, has been approved for use in patients harboring NTRK rearrangements based on its efficacy in clinical trials [89, 90] with a 79% ORR [91]. Entrectinib is also approved for use on NTRK fusions based on its anti-tumor activity and 57% ORR in clinical trials [92].
The NRG1 gene is known to produce many isoforms, which are expressed across various tissues through different promoters and alternative splicing [93,94]. Furthermore, expression of NRG1 is important to establish the cardiovascular and nervous system during embryonic development [95,96]. ERBB3 and ERBB4 are the EGF protein receptors for the NRG1 and NRG2 ligands [97]; This ligand binding induces receptor heterodimerization with ERBB2, causing activation of PI3K and MAPK pathways, which results in increased cell proliferation/ migration and resistance to apoptosis in cancer [97,98,99,100]. While the normal biological function of NRG2 remains to be fully understood [101], the structures of NRG1 and NRG2 both have an extracellular EGF-like domain, transmembrane domain, and cytosolic tail [94,102]. While there is currently no Food and Drug Administration (FDA) approved treatment for NRG1 fusions, Seribantumab, an anti-ERBB3 (HER3) monoclonal antibody (mAb) [103], is used in clinical trials for patients with NRG1 fusion-positive solid tumors with a 30% ORR [104,105]. Likewise, Zenocutuzumab, an ERBB2/ERBB3 (HER2/HER3) bispecific antibody, is under evaluation in clinical trials for patients with NRG1 positive fusions in cancer and has a 34% ORR [106].
The platelet-derived growth factor receptor β (PDGFRB) protein is encoded by the PDGFRB gene, and is known to express in mesenchymal [107] and smooth muscle cells [108]. This receptor tyrosine kinase (RTK) is made up of an extracellular ligand-binding domain, a transmembrane domain, and a split tyrosine kinase domain within its intracellular portion [109]. PDGFRB is 962 aa long, where exon 11 precedes the transmembrane domain, exon 12 encodes the juxta membrane domain, exon 14 encodes the first kinase domain, and exon 18 encodes for the second kinase domain [110]. Once bound to its ligand, PDGF-BB [111], the Ras-MAPK [112] signaling pathway is activated [109]. PDGFRB was found to be overexpressed in various cancers including lung [113], colon [114], melanoma [115], and breast carcinoma [116]. Studies found that Imatinib, a TKI, achieved durable response in patients with PDGFRB fusions implicated in chronic myeloproliferative disorders [117,118]. Besides Imatinib, Crenolanib is another TKI that is capable of inhibiting PDGFRB and has shown promising results in lung [119] cancer and colon [120] cancer cell lines.
2.2. CD74-ROS1 Expression
The first CD74 fusion protein, CD74-ROS1 was originally identified in NSCLC [30], and 14 years later was also reported in breast cancer [52]. This fusion appears in adult patients [30] and has a membrane bound cellular localization [30,121] with reports in the plasma membrane [121] and the endoplasmic reticulum [122]. Of the five known CD74 fusions, CD74-ROS1 is the most prevalent among ROS1 fusion proteins in NSCLC [71,123,124,125], and based on the data collected in this review, also appears to be the most prevalent partner among CD74-related fusions. CD74-ROS1 exists in seven different variants (Table 1) that are generated from rearrangements in both the CD74 and ROS1 genes. Of the CD74 isoforms, the p35 variant is involved in these fusions, and in patients has been found spliced at different exon breakpoints. Likewise, the ROS1 rearrangement occurs at different exons, resulting in the formation of unique CD74-ROS1 fusion proteins. CD74-ROS1 fusions typically have breakpoints located on ROS1 exon 32 or 34, retaining the ROS1 kinase domain [57] allowing for the use of tyrosine kinase inhibitors (TKIs).
In 2007, identification of CD74-ROS1 originally occurred in a 50-year-old female non-smoker with a stage IB lung adenocarcinoma [30]. The patient did not show signs of metastasis in the 2cm tumor resected, but DNA sequencing revealed the presence of a CD74-ROS1 fusion, exhibiting splicing of CD74 exon 6 to ROS1 exon 34. Interestingly, this fusion variant contained two transmembrane domains (Figure 2), the nucleotide sequence was deposited into GenBank (EU236945) [30]. The C6-R34 variant has been detected by RT-PCR in additional stages of lung adenocarcinoma, including IIA, IIIB, and IV [126].
The next identified CD74-ROS1 fusion breakpoint was a splicing between CD74 exon 6 and ROS1 exon 32, published in 2012 [54]. Through FISH analysis, and independent confirmation by RT-PCR, this C6-R32 fusion was detected in a 9mm diameter tumor removed from a 79-year-old female stage IA lung cancer patient [54]. Shortly thereafter, this breakpoint was also identified in a 44-year-old female never smoker with stage IV lung adenocarcinoma using RT-PCR [127]. A case report published in 2014 revealed a CD74-ROS1 variant of CD74 exon 7 fused to ROS1 exon 32 in a patient with three ROS1 fusion variants identified through RT-PCR [57]. Diagnosed with stage IV lung adenocarcinoma, this patient was a 61-year-old woman with no previous smoking history whose computerized tomography (CT) scan revealed a 3cm mass and magnetic resonance imaging (MRI) additionally showed evidence of brain metastases [57]. Of note, the DNA sequence for this C7-R32 fusion was not obtained [57]. The case of a CD74-ROS1 fusion in a 64-year-old inflammatory breast cancer patient (IBC) was found through NGS as a combination of CD74 exon 7 and ROS1 exon 34, which may have contributed to rapid disease progression as they died 18 months after tumor discovery [52].
In 2021, the most recently identified CD74-ROS1 variant was reported with a breakpoint fusing CD74 exon 3 to ROS1 exon 34 [58]. This novel fusion was identified through next generation sequencing in a 42-year-old female never-smoker with relapsed stage IVA lung adenocarcinoma [58]. While the patient underwent a partial pulmonary lobectomy after her original diagnosis four years prior, her lung cancer relapsed with lymph node, bone, and brain metastases [58]. Due to the presence of ROS1 tyrosine kinase domain, Crizotinib, was used effectively against the tumor. Additional brain metastases developed after Crizotinib, which was further treated with Entrectinib [58].
Expression of CD74-ROS1 has confirmed oncogenic transformation capacity towards fibroblast cells through activation of MAPK, SHP-2, and STAT-3 pathways [121]. Additionally, based on the oncoprotein’s expression in noninvasive NSCLC cell lines (H1755, H2009, and H1915), CD74-ROS1 induces an invasive mechanism via phosphorylation of E-Syt1; This mechanism was further validated in a xenograft model to be both invasive and metastatic [121]. Subcellular localization is critical for CD74-ROS1 variants, as those that localize in the ER have a compromised ability to activate the RAS/MAPK pathway [122].
Retrospective studies of tumor samples containing the CD74-ROS1 fusion were recorded to provide information about fusion variant, specimen source, detection method, age, gender, smoking status, and cancer stage (Table S1). Many specimens are either from formalin fixed embedded (FFPE) tumor tissue or frozen tumor samples. The oncogenic fusion was identified by means of FISH, using probes that detect the ROS1 portion of the fusion, followed by confirmation using RT-PCR. More recent studies verify fusions by NGS, and as recently as 2023, Nanostring has been able to detect fusions in plasma samples [5]. Many of the published cases of CD74-ROS1 are in female patients never-smokers, while the fusion has been detected from stages I-IV, highlighting the necessity of targeted therapy for early intervention. Of the seven different CD74-ROS1 variants, the C6-R34 one appears the most often (Table S1). The data also suggests that the occurrence of this fusion is equally likely in both smokers and nonsmokers (Table S1).
Clinical and case studies over the years outline additional details of CD74-ROS1 patients, such as the treatment sequence patients received, whether TKI resistance developed, and their overall survival (OS) (Table 2). Based on the cases reported in the literature, many CD74-ROS1 patients benefit from TKIs by having a greater OS and better response to treatment, compared to patients who receive traditional platinum-based chemotherapy, alone (Table 2). Mutations and metastasis can still arise after taking TKIs such as crizotinib, but can sometimes be overcome by introducing another TKI, such as entrectinib, dabrafenib, or cabozantinib (Table 2). A recently published clinical trial revealed that Unecritinib (TKI) achieved an 88.9% ORR for patients harboring CD74-ROS1 fusions, with a median progression free survival (PFS) of 21.2 months [128]. These results suggested a greater efficacy and lower toxicity compared to the previously used TKIs, crizotinib and entrectinib [128].
While the clinical and case studies (Table 2) show occurrence of CD74-ROS1 in many patients with relapsed, advanced stage cancer (III-IV), the retrospective studies done on patient tumor samples (Table S1) reveal that many CD74-ROS1 fusions can be detected at earlier cancer stages (I-II). The data suggests that even though fourth and even ninth line of treatment can have some success in patients, early detection, and intervention to target these fusions may yield improved patient outcomes without the necessity of going through traditional chemotherapies (Table 2).
2.3. CD74-NTRK1 Expression
CD74-NTRK1 was first detected by NGS in lung adenocarcinoma patient tumor samples and confirmed using RT-PCR, followed by FISH [31]. This fusion joined exon 8 of CD74 to exon 12 of NTRK1, thus retaining the NTRK1 kinase domain encoding for the TRKA receptor and was predicted to localize in the plasma membrane [31] (Table 1, Figure 2). The complete cDNA sequence of the fusion was further introduced into Ba/F3 cells, 293T cells, and NIH3T3 fibroblasts, proving that CD74-NTRK1 was oncogenic through the pERK pathway; the gene fusion was also found to induce tumorigenesis in nude mice [31]. Expression of the CD74-NTRK1 oncoprotein resulted in constitutive TRKA kinase activity and cell lines harboring the fusion were found to be sensitive to TRKA inhibitors ARRY-470, CEP-701, and crizotinib [31].
Much later, in 2020, next generation sequencing revealed a unique fusion of CD74 exon 7 with NTRK1 exon 8 (C7-N8) in a 41-year-old female stage IIIB adenocarcinoma patient [61] (Table 1). Additionally, NGS has identified a CD74-NTRK1 C6-N12 fusion mutation with a missense mutation in a lung cancer patient [62] without many further details (Table 1). These retrospective studies on tumor samples are detailed with the diagnosis, variant, method of detection, age, gender, and stage (Table S2). While not much detailed patient information is available on CD74-NTRK1, the three fusion variants are of interest for future studies as they may share similarities with other CD74 fusion proteins, and thus benefit from similar treatment plans.
2.4. CD74-NRG1 Expression
CD74-NRG1 was first identified in a 64-year-old female never-smoker stage IB invasive mucinous adenocarcinoma (IMA) lung cancer patient through transcriptome sequencing [32]. This fusion arose from a somatic genomic event resulting in a chromosomal rearrangement between CD74 exon 6 and NRG1 III-β3 exon 6 [32]. This novel CD74-NRG1 fusion protein was reported as 283 aa long, retained the transmembrane domain of CD74 and the EGF-like domain of NRG1, resulting in its membrane-bound cellular localization [32]. Another 2014 study published shortly thereafter, detected the C6-N6 fusion pairing in female never-smoker IMA patients aged 68 and 53, at cancer stages IIB and IA, respectively and reported the fusion as 282 aa long. [56].
H322 and H1568 lung cancer cell lines were virally transduced with the full CD74-NRG1 fusion, as well as with a truncated version of the fusion that lacked the EGF-like domain [32]. It was observed that CD74-NRG1 expressing cell lines had increased levels of p-AKT, p-ERBB2, and p-ERBB3 associated with PI3K/AKT pathway activation compared to cells that do not express the fusion [32]. Additional western blot analysis further supported that phosphorylated AKT and ERBB3 were dependent on the EGF-like domain being present in this fusion [32].
An alternative translocation produced a novel C8-N6 fusion pairing that incorporated the first 8 exons of CD74 and the EGF coding NRG1 exons, resulted in a 303 aa CD74-NRG1 fusion [56]. This fusion was detected in three IMA patients through RNA sequencing, and further validated through Sanger sequencing of RT-PCR products; Patient characteristics included a 55-year-old male 47 pack/year smoker at stage IA cancer, and two female never-smokers, one 78 years old at Stage IA IMA, and one 47-year-old at stage IB [56]. Additionally, in a 2023 published study, a fusion variant incorporating CD74 exon 6 joined to NRG1 exon 4 was identified in two female NSCLC patients in their 80s [63].
The same study treated EFM-19 reporter cells with media from H1299 lung cancer that expressed CD74-NRG1 and found that the fusion protein caused phosphorylation of ERBB2 and ERBB3, which suggested activation of HER2:HER3 autocrine receptor signaling; Phosphorylation of ERK and AKT was also observed [56]. Lapatinib and Afatinib (HER TKIs) were tested on the cells and as a result, phosphorylation events were suppressed [56]. CD74-NRG1 is capable of activating HER2 and HER3 autocrine signaling through the EGF-like domain of NRG1 in exons 6-7 interacting with HER3, which results in PI3K/AKT signaling and subsequent cell proliferation associated with oncogenic growth [63].
Multiple retrospective studies have been conducted on CD74-NRG1 patient tumor samples (Table S3). Many patients affected by these fusions were female non-smokers afflicted with either IMA or NSCLC. The clinical CD74-NRG1 patient characteristics of CD74-NRG1 are summarized in Table 3 with treatment courses and lifespan included. This fusion is detected in stages I-IV (Table S3, Table 3) and treated with platinum-based chemotherapy, monoclonal antibodies, and often responds well to afatinib (Table 3). Among NRG1 fusions in solid tumors, CD74 is found to be the most prevalent fusion partner [139]. Similar to CD74-ROS1, CD74-NRG1 fusions are capable of being identified as early as stage I cancer (Table S3). Considering the clinical patient characteristics, this fusion may also benefit from targeted therapy early on, rather than a platinum-based first line of treatment approach (Table 3).
2.5. CD74-PDGFRB Expression
RNA seq first identified a CD74-PDGFRB fusion in a 2.4-year-old male with B-cell acute lymphoblastic leukemia. The novel protein was found to be a fusion between CD74 exon 6 and PDGFRB exon 11 [33] (Table S4). Following two rounds of first-line treatment, the pediatric patient went into complete remission [33] (Table S4). Recently, another variant of CD74-PDGFRB,spliced between CD74 exon 1 and PDGFRB exon 11,was validated through RNA seq and polymerase chain reaction (PCR) in a 22-month-old female B-ALL patient, although it did not form a fusion protein [53]. Many fusion partners tend to fuse at PDGFRB exon 11 [110,141,142], thus retaining the PDGFRB transmembrane domain and split kinase domain.
Interestingly, in another case, Sanger sequencing showed a retained CD74 intron sequence that coded for a stop codon before the fusion breakpoint [53]. Referred to as CD74intr::PDGFRB, this fusion lacked an open reading frame to drive the translation of a functional fusion protein [53]. Additionally, a 2023 published case study identified a PDGFRB::CD74 fusion mRNA in a 19-month-old female patient diagnosed with an aggressive case of cutaneous non-Langerhans cell histiocytosis (NLCH) [143]. Although the biopsy specimen was benign, the mutations that led to the PDGFRB::CD74 fusion mRNA had not been previously associated with pediatric or adult NLCH [143].
While this fusion has been documented in the literature several times, its oncogenic variant remains to be further studied and characterized as the oncogenic signaling pathway that is activated is still unknown. But, it is likely that the presence of CD74-PDGFRB leads to activation of PDGFRB [143].
2.6. CD74-NRG2a Expression
The most recently identified CD74 fusion protein was CD74-NRG2α in a 70-year-old female, never smoker, stage IIIA acinar adenocarcinoma lung cancer patient [34] (Table S5). This fusion was identified through RNA-seq and confirmed through Sanger sequencing to fuse CD74 exon 6 to NRG2α exon 2, which retained the EGF-like domain of NRG2α and the transmembrane domain of CD74 [34] (Table S5, Figure 2). While the 43 mm tumor was surgically resected, the tumor returned and the patient was treated with Erlotinib, a tyrosine kinase inhibitor, but discontinued use a month later due to skin rash; The patient eventually died of disease 32 weeks later [34] (Table S5). Immunohistochemistry (IHC) analysis of tumor cells were moderately positive for p-HER4, and negative for p-HER2, p-HER3, and p-EGFR. As the single documented case of CD74-NRG2α, more research is needed to elucidate whether the fusion’s phosphorylation of HER4 alone promotes an oncogenic pathway, and if that is the case, how exactly it occurs [144].
3. Conclusions
Methods that are utilized to identify CD74 fusions includes DNA or RNA NGS, FISH, RT-PCR, and RT-qPCR. In clinical case studies, these methods typically identify the fusion after cancer recurrence. Both male and females are affected by CD74 fusions, with many female non-smokers reported as carriers.
CD74-ROS1 has the highest number of fusion variants (7) as well as patient data availability. Upon analysis of the published findings, it becomes clear that CD74-ROS1 is formed from very early to advanced stage cancers. This fusion is mostly detected in NSCLC and likely occurs equally in both smokers and non-smokers. CD74-NRG1 has the second greatest number of variants (4), followed by CD74-NTRK1 (3), while CD74-NR2α and CD74-PDGFRB have a single fusion protein variant each. CD74-NRG1 is detected often at stages I and II in retrospective studies of patient tumor tissue, while clinical studies report this fusion both in early and late-stage IMA and NSCLC. Based on patient data available, the CD74-NRG1 fusion occurs mostly in non-smokers with a few cases of smokers.
About half of the CD74 fusion proteins have been studied extensively and their signaling pathways elucidated, mostly in respect to their fusion partner. Likewise, inhibitors target the fusion partner rather than CD74. Throughout the five fusions expressed in cancer patients, all retain the transmembrane domain of CD74 that many times makes up for the transmembrane domain that biologically occurs in the fusion partner before splicing occurs. CD74 fusion proteins mainly manifest in lung cancer, apart from a single documented case in an aggressive case of inflammatory breast cancer and two cases of pediatric leukemia. Of note, the rare fusions CD74-PDGFRB and CD74-NRG2α require further studies to elucidate their exact mechanism of disease.
As many studies focus on the targetable fusion partners, the oncogenic role of CD74 in these CD74 fusion proteins remains to be fully understood. In ROS1 and NRG1 fusion families, CD74 is the most common partner, emphasizing the potential significance of CD74 occurrence in fusion proteins. The participation of CD74 as a partner in five different fusion proteins, with a total of 16 different variants, highlights its involvement in the formation of various types of fusions. The data on CD74 fusions offers insight for improved therapy plans for cancer patients and the development of additional methods for early detection to improve treatment success and patient survival.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1: Retrospective studies on tumor samples containing the CD74-ROS1 fusion, Table S2: Retrospective studies on tumor samples containing the CD74-NTRK1 fusion, Table S3: Retrospective studies on tumor samples containing the CD74-NRG1 fusion, Table S4: Retrospective study on a tumor sample containing the CD74-PDGFRB, Table S5: Retrospective study on a tumor sample containing the CD74-NRG2a fusion, Figure S1: Amino acid sequences of CD74 isoforms, Figure S2: Identified CD74 oncogenic fusions in the human body, Figure S3: Previously reported amino acid sequences of CD74 fusion proteins.
Author Contributions
Conceptualization, G.P. and J.V.; writing—original draft preparation, J.V.; writing—review and editing, G.P.; visualization, G.P and J.V.; supervision, G.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the Chemistry Department at the University of the Pacific for their support. All figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- de Mendíbil, I.O.; Vizmanos, J.L.; Novo, F.J. Signatures of Selection in Fusion Transcripts Resulting from Chromosomal Translocations in Human Cancer. PLOS ONE 2009, 4, e4805. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G.W. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2014, 34, 4845–4854. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.C.; Zhang, W. Fusion genes in solid tumors: an emerging target for cancer diagnosis and treatment. Chin. J. Cancer 2013, 32, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Farago, A.F.; et al. Clinicopathologic Features of Non-Small-Cell Lung Cancer Harboring an NTRK Gene Fusion. JCO Precis Oncol 2018. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Capitán, A.; et al. Detecting ALK, ROS1 and RET fusions and the METΔex14 splicing variant in liquid biopsies of non-small cell lung cancer patients using RNA-based techniques. Mol Oncol 2023. [Google Scholar] [CrossRef]
- Jonna, S.; Feldman, R.A.; Swensen, J.; Gatalica, Z.; Korn, W.M.; Borghaei, H.; Ma, P.C.; Nieva, J.J.; Spira, A.I.; Vanderwalde, A.M.; et al. Detection of NRG1 Gene Fusions in Solid Tumors. Clin. Cancer Res. 2019, 25, 4966–4972. [Google Scholar] [CrossRef]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF Signal Transduction Initiated by Binding to CD74. J. Exp. Med. 2003, 197, 1467–1476. [Google Scholar] [CrossRef]
- Merk, M.; Zierow, S.; Leng, L.; Das, R.; Du, X.; Schulte, W.; Fan, J.; Lue, H.; Chen, Y.; Xiong, H.; et al. The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc. Natl. Acad. Sci. USA 2011, 108, E577–E585. [Google Scholar] [CrossRef]
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 Is the Signaling Component of the Macrophage Migration Inhibitory Factor-CD74 Receptor Complex. Immunity 2006, 25, 595–606. [Google Scholar] [CrossRef]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef]
- Schwartz, V.; Lue, H.; Kraemer, S.; Korbiel, J.; Krohn, R.; Ohl, K.; Bucala, R.; Weber, C.; Bernhagen, J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009, 583, 2749–2757. [Google Scholar] [CrossRef] [PubMed]
- Lue, H.; et al. Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell Signal 2006, 18, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Gore, Y.; Starlets, D.; Maharshak, N.; Becker-Herman, S.; Kaneyuki, U.; Leng, L.; Bucala, R.; Shachar, I. Macrophage Migration Inhibitory Factor Induces B Cell Survival by Activation of a CD74-CD44 Receptor Complex. J. Biol. Chem. 2008, 283, 2784–2792. [Google Scholar] [CrossRef] [PubMed]
- Lue, H.; et al. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene 2007, 26, 5046–5059. [Google Scholar] [CrossRef]
- Lue, H.; Dewor, M.; Leng, L.; Bucala, R.; Bernhagen, J. Activation of the JNK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on CXCR4 and CD74. Cell. Signal. 2011, 23, 135–144. [Google Scholar] [CrossRef]
- Starlets, D.; Gore, Y.; Binsky, I.; Haran, M.; Harpaz, N.; Shvidel, L.; Becker-Herman, S.; Berrebi, A.; Shachar, I. Cell-surface CD74 initiates a signaling cascade leading to cell proliferation and survival. Blood 2006, 107, 4807–4816. [Google Scholar] [CrossRef]
- Cheng, S.-P.; Liu, C.-L.; Chen, M.-J.; Chien, M.-N.; Leung, C.-H.; Lin, C.-H.; Hsu, Y.-C.; Lee, J.-J. CD74 expression and its therapeutic potential in thyroid carcinoma. Endocrine-Related Cancer 2015, 22, 179–190. [Google Scholar] [CrossRef]
- Woolbright, B.L.; et al. Role of MIF1/MIF2/CD74 interactions in bladder cancer. J Pathol 2023, 259, 46–55. [Google Scholar] [CrossRef]
- Thavayogarajah, T.; Sinitski, D.; El Bounkari, O.; Torres-Garcia, L.; Lewinsky, H.; Harjung, A.; Chen, H.-R.; Panse, J.; Vankann, L.; Shachar, I.; et al. CXCR4 and CD74 together enhance cell survival in response to macrophage migration-inhibitory factor in chronic lymphocytic leukemia. Exp. Hematol. 2022, 115, 30–43. [Google Scholar] [CrossRef]
- Burton, J.D.; Ely, S.; Reddy, P.K.; Stein, R.; Gold, D.V.; Cardillo, T.M.; Goldenberg, D.M. CD74 Is Expressed by Multiple Myeloma and Is a Promising Target for Therapy. Clin. Cancer Res. 2004, 10, 6606–6611. [Google Scholar] [CrossRef]
- Greenwood, C.; Metodieva, G.; Al-Janabi, K.; Lausen, B.; Alldridge, L.; Leng, L.; Bucala, R.; Fernandez, N.; Metodiev, M.V. Stat1 and CD74 overexpression is co-dependent and linked to increased invasion and lymph node metastasis in triple-negative breast cancer. J. Proteom. 2011, 75, 3031–3040. [Google Scholar] [CrossRef] [PubMed]
- Gold, D.V.; et al. Enhanced expression of CD74 in gastrointestinal cancers and benign tissues. Int J Clin Exp Pathol 2010, 4, 1–12. [Google Scholar] [PubMed]
- McClelland, M.; Zhao, L.; Carskadon, S.; Arenberg, D. Expression of CD74, the Receptor for Macrophage Migration Inhibitory Factor, in Non-Small Cell Lung Cancer. Am. J. Pathol. 2009, 174, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Young, A.N.; Amin, M.B.; Moreno, C.S.; Lim, S.D.; Cohen, C.; Petros, J.A.; Marshall, F.F.; Neish, A.S. Expression Profiling of Renal Epithelial Neoplasms: A Method for Tumor Classification and Discovery of Diagnostic Molecular Markers. Am. J. Pathol. 2001, 158, 1639–1651. [Google Scholar] [CrossRef]
- Meyer-Siegler, K.L.; Iczkowski, K.A.; Leng, L.; Bucala, R.; Vera, P.L. Inhibition of Macrophage Migration Inhibitory Factor or Its Receptor (CD74) Attenuates Growth and Invasion of DU-145 Prostate Cancer Cells. J. Immunol. 2006, 177, 8730–8739. [Google Scholar] [CrossRef]
- Koide, N.; Yamada, T.; Shibata, R.; Mori, T.; Fukuma, M.; Yamazaki, K.; Aiura, K.; Shimazu, M.; Hirohashi, S.; Nimura, Y.; et al. Establishment of Perineural Invasion Models and Analysis of Gene Expression Revealed an Invariant Chain (CD74) as a Possible Molecule Involved in Perineural Invasion in Pancreatic Cancer. Clin. Cancer Res. 2006, 12, 2419–2426. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Decker, P.A.; Morlan, B.W.; Wu, W.; Ballman, K.V.; Giannini, C.; Sarkaria, J.N. Expression of CD74 in high grade gliomas: a potential role in temozolomide resistance. J. Neuro-Oncology 2010, 100, 177–186. [Google Scholar] [CrossRef]
- Hong, W.C.; Lee, D.E.; Kang, H.W.; Kim, M.J.; Kim, M.; Kim, J.H.; Fang, S.; Kim, H.J.; Park, J.S. CD74 Promotes a Pro-Inflammatory Tumor Microenvironment by Inducing S100A8 and S100A9 Secretion in Pancreatic Cancer. Int. J. Mol. Sci. 2023, 24, 12993. [Google Scholar] [CrossRef]
- Xu, S.; Li, X.; Tang, L.; Liu, Z.; Yang, K.; Cheng, Q. CD74 Correlated With Malignancies and Immune Microenvironment in Gliomas. Front. Mol. Biosci. 2021, 8, 706949. [Google Scholar] [CrossRef]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global Survey of Phosphotyrosine Signaling Identifies Oncogenic Kinases in Lung Cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cuesta, L.; et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov 2014, 4, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-F.; Wang, B.-Y.; Zhang, W.-N.; Huang, J.-Y.; Li, B.-S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.-J.; Dai, Y.-J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, S.; Hayashi, T.; Nagano, M.; Ueno, T.; Kojima, S.; Kawazu, M.; Shiraishi, Y.; Kishikawa, S.; Suehara, Y.; Takahashi, F.; et al. Identification of Novel CD74-NRG2α Fusion From Comprehensive Profiling of Lung Adenocarcinoma in Japanese Never or Light Smokers. J. Thorac. Oncol. 2020, 15, 948–961. [Google Scholar] [CrossRef]
- Claesson, L.; Larhammar, D.; Rask, L.; A Peterson, P. cDNA clone for the human invariant gamma chain of class II histocompatibility antigens and its implications for the protein structure. Proc. Natl. Acad. Sci. 1983, 80, 7395–7399. [Google Scholar] [CrossRef]
- Strubin, M.; Mach, B.; Long, E. The complete sequence of the mRNA for the HLA-DR-associated invariant chain reveals a polypeptide with an unusual transmembrane polarity. EMBO J. 1984, 3, 869–872. [Google Scholar] [CrossRef]
- Strubin, M.; Berte, C.; Mach, B. Alternative splicing and alternative initiation of translation explain the four forms of the Ia antigen-associated invariant chain. EMBO J. 1986, 5, 3483–3488. [Google Scholar] [CrossRef]
- O'Sullivan, D.M.; Larhammar, D.; Wilson, M.C.; A Peterson, P.; Quaranta, V. Structure of the human Ia-associated invariant (gamma)-chain gene: identification of 5' sequences shared with major histocompatibility complex class II genes. Proc Natl Acad Sci USA 1986, 83, 4484–4488. [Google Scholar] [CrossRef]
- Koch, N.; Lauer, W.; Habicht, J.; Dobberstein, B. Primary structure of the gene for the murine Ia antigen-associated invariant chains (Ii). An alternatively spliced exon encodes a cysteine-rich domain highly homologous to a repetitive sequence of thyroglobulin. EMBO J. 1987, 6, 1677–1683. [Google Scholar] [CrossRef]
- Gedde-Dahl, M.; Freisewinkel, I.; Staschewski, M.; Schenck, K.; Koch, N.; Bakke, O. Exon 6 Is Essential for Invariant Chain Trimerization and Induction of Large Endosomal Structures. J. Biol. Chem. 1997, 272, 8281–8287. [Google Scholar] [CrossRef]
- Kuwana, T.; Peterson, P.A.; Karlsson, L. Exit of major histocompatibility complex class II–invariant chain p35 complexes from the endoplasmic reticulum is modulated by phosphorylation. Proceedings of the National Academy of Sciences 1998, 95, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Marks, M.S.; Blum, J.S.; Cresswell, P. Invariant chain trimers are sequestered in the rough endoplasmic reticulum in the absence of association with HLA class II antigens. J. Cell Biol. 1990, 111, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Jasanoff, A.; Wagner, G.; Wiley, D.C. Structure of a trimeric domain of the MHC class II-associated chaperonin and targeting protein Ii. EMBO J. 1998, 17, 6812–6818. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; et al. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature 1995, 378, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Rudensky, A.Y.; Corper, A.L.; Teyton, L.; Wilson, I.A. Crystal Structure Of MHC Class II I-Ab in Complex with a Human CLIP Peptide: Prediction of an I-Ab Peptide-binding Motif. J. Mol. Biol. 2003, 326, 1157–1174. [Google Scholar] [CrossRef]
- Günther, S.; et al. Bidirectional binding of invariant chain peptides to an MHC class II molecule. Proc Natl Acad Sci USA 2010, 107, 22219–22224. [Google Scholar] [CrossRef]
- Nguyen, T.-B.; Jayaraman, P.; Bergseng, E.; Madhusudhan, M.S.; Kim, C.-Y.; Sollid, L.M. Unraveling the structural basis for the unusually rich association of human leukocyte antigen DQ2.5 with class-II-associated invariant chain peptides. J. Biol. Chem. 2017, 292, 9218–9228. [Google Scholar] [CrossRef]
- Gunčar, G.; Pungerčič, G.; Klemenčič, I.; Turk, V.; Turk, D. Crystal structure of MHC class II-associated p41 Ii fragment bound to cathepsin L reveals the structural basis for differentiation between cathepsins L and S. EMBO J. 1999, 18, 793–803. [Google Scholar] [CrossRef]
- Chiva, C.; et al. Synthesis and NMR structure of p41icf, a potent inhibitor of human cathepsin L. J Am Chem Soc 2003, 125, 1508–1517. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Ding, N.; Zhou, H.; Wang, S.; Tang, L.; Xiao, Z. A novel CD74-ROS1 gene fusion in a patient with inflammatory breast cancer: a case report. J. Med Case Rep. 2021, 15, 277. [Google Scholar] [CrossRef] [PubMed]
- Sadras, T.; Jalud, F.B.; Kosasih, H.J.; Horne, C.R.; Brown, L.M.; El-Kamand, S.; de Bock, C.E.; McAloney, L.; Ng, A.P.; Davidson, N.M.; et al. Unusual PDGFRB fusion reveals novel mechanism of kinase activation in Ph-like B-ALL. Leukemia 2023, 37, 905–909. [Google Scholar] [CrossRef]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Lan, S.; Li, H.; Liu, Y.; Xu, J.; Huang, Z.; Yan, S.; Zhang, Q.; Cheng, Y. A Novel ROS1-FBXL17 Fusion Co-Existing with CD74-ROS1 Fusion May Improve Sensitivity to Crizotinib and Prolong Progression-Free Survival of Patients with Lung Adenocarcinoma. OncoTargets Ther. 2020, 13, 11499–11504. [Google Scholar] [CrossRef] [PubMed]
- Nakaoku, T.; et al. Druggable oncogene fusions in invasive mucinous lung adenocarcinoma. Clin Cancer Res 2014, 20, 3087–3093. [Google Scholar] [CrossRef]
- Cai, W.; Li, W.; Ren, S.; Zheng, L.; Li, X.; Zhou, C. Coexistence of Three Variants Involving Two Different Fusion Partners of ROS1 Including a Novel Variant of ROS1 Fusions in Lung Adenocarcinoma: A Case Report. J. Thorac. Oncol. 2014, 9, e43–e46. [Google Scholar] [CrossRef]
- Hashiguchi, M.H.; Sato, T.; Watanabe, R.; Kagyo, J.; Matsuzaki, T.; Domoto, H.; Kato, T.; Nakahara, Y.; Yokose, T.; Hiroshima, Y.; et al. A case of lung adenocarcinoma with a novel CD74-ROS1 fusion variant identified by comprehensive genomic profiling that responded to crizotinib and entrectinib. Thorac. Cancer 2021, 12, 2504–2507. [Google Scholar] [CrossRef]
- Sehgal, K.; et al. Cases of ROS1 -rearranged lung cancer: when to use crizotinib, entrectinib, lorlatinib, and beyond? Precision Cancer Medicine 2020, 3. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, B.; Wang, C.; Liao, C.; Wang, S.; Cao, R.; Ma, T.; Wang, K. Case report: A novel reciprocal ROS1-CD74 fusion in a NSCLC patient partially benefited from sequential tyrosine kinase inhibitors treatment. Front. Oncol. 2022, 12, 1021342. [Google Scholar] [CrossRef]
- Xia, H.; Xue, X.; Ding, H.; Ou, Q.; Wu, X.; Nagasaka, M.; Shao, Y.W.; Hu, X.; Ou, S.-H.I. Evidence of NTRK1 Fusion as Resistance Mechanism to EGFR TKI in EGFR+ NSCLC: Results from a Large-Scale Survey of NTRK1 Fusions in Chinese Patients with Lung Cancer. Clin. Lung Cancer 2019, 21, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yan, S.; Liu, Y.; Ma, L.; Liu, X.; Liu, Y.; Cheng, Y. Analysis of NTRK mutation and clinicopathologic factors in lung cancer patients in northeast China. Int. J. Biol. Markers 2020, 35, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Severson, E.; Achyut, B.R.; Nesline, M.; Pabla, S.; Previs, R.A.; Kannan, G.; Chenn, A.; Zhang, S.; Klein, R.; Conroy, J.; et al. RNA Sequencing Identifies Novel NRG1 Fusions in Solid Tumors that Lack Co-Occurring Oncogenic Drivers. J. Mol. Diagn. 2023, 25, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Somwar, R.; Mangatt, B.P.; Edgren, H.; Desmeules, P.; Ruusulehto, A.; Smith, R.S.; Delasos, L.; Vojnic, M.; Plodkowski, A.J.; et al. Response to ERBB3-Directed Targeted Therapy in NRG1-Rearranged Cancers. Cancer Discov. 2018, 8, 686–695. [Google Scholar] [CrossRef]
- Birchmeier, C.; Sharma, S.; Wigler, M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc. Natl. Acad. Sci. 1987, 84, 9270–9274. [Google Scholar] [CrossRef]
- Shih, C.-H.; Chang, Y.-J.; Huang, W.-C.; Jang, T.-H.; Kung, H.-J.; Wang, W.-C.; Yang, M.-H.; Lin, M.-C.; Huang, S.-F.; Chou, S.-W.; et al. EZH2-mediated upregulation of ROS1 oncogene promotes oral cancer metastasis. Oncogene 2017, 36, 6542–6554. [Google Scholar] [CrossRef]
- Grenier, K.; Rivière, J.-B.; Bencheikh, B.O.A.; Corredor, A.L.G.; Shieh, B.C.; Wang, H.; Fiset, P.O.; Camilleri-Broët, S. Routine Clinically Detected Increased ROS1 Transcripts Are Related With ROS1 Expression by Immunohistochemistry and Associated With EGFR Mutations in Lung Adenocarcinoma. JTO Clin. Res. Rep. 2023, 4, 100530. [Google Scholar] [CrossRef]
- Lee, H.J.; Seol, H.S.; Kim, J.Y.; Chun, S.-M.; Suh, Y.-A.; Park, Y.-S.; Kim, S.-W.; Choi, C.-M.; Park, S.-I.; Kim, D.K.; et al. ROS1 Receptor Tyrosine Kinase, a Druggable Target, is Frequently Overexpressed in Non-Small Cell Lung Carcinomas Via Genetic and Epigenetic Mechanisms. Ann. Surg. Oncol. 2012, 20, 200–208. [Google Scholar] [CrossRef]
- Birchmeier, C.; O'Neill, K.; Riggs, M.; Wigler, M. Characterization of ROS1 cDNA from a human glioblastoma cell line. Proc Natl Acad Sci U S A 1990, 87, 4799–4803. [Google Scholar] [CrossRef]
- Acquaviva, J.; Wong, R.; Charest, A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim. et Biophys. Acta (BBA) - Rev. Cancer 2009, 1795, 37–52. [Google Scholar] [CrossRef]
- Drilon, A.; et al. ROS1-dependent cancers - biology, diagnostics and therapeutics. Nat Rev Clin Oncol 2021, 18, 35–55. [Google Scholar] [CrossRef]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; Román-Gil, M.S.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine Kinase Receptors in Oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef]
- Shaw, A.T.; et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014, 371, 1963–1971. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Dziadziuszko, R.; Barlesi, F.; Krebs, M.G.; Shaw, A.T.; de Braud, F.; Rolfo, C.; Ahn, M.-J.; Wolf, J.; et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2019, 21, 261–270. [Google Scholar] [CrossRef]
- Barbacid, M.; Lamballe, F.; Pulido, D.; Klein, R. The trk family of tyrosine protein kinase receptors. Biochim. et Biophys. Acta (BBA) - Rev. Cancer 1991, 1072, 115–127. [Google Scholar] [CrossRef]
- Barker, P.; Lomen-Hoerth, C.; Gensch, E.; Meakin, S.; Glass, D.; Shooter, E. Tissue-specific alternative splicing generates two isoforms of the trkA receptor. J. Biol. Chem. 1993, 268, 15150–15157. [Google Scholar] [CrossRef]
- Martin-Zanca, D.; et al. Molecular and biochemical characterization of the human trk proto-oncogene. Mol Cell Biol 1989, 9, 24–33. [Google Scholar]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Hempstead, B.L.; Martin-Zanca, D.; Chao, M.V.; Parada, L.F. The trk Proto-Oncogene Product: a Signal Transducing Receptor for Nerve Growth Factor. Science 1991, 252, 554–558. [Google Scholar] [CrossRef]
- Klein, R.; Jing, S.; Nanduri, V.; O'Rourke, E.; Barbacid, M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 1991, 65, 189–197. [Google Scholar] [CrossRef]
- Klesse, L.J.; Parada, L.F. Trks: signal transduction and intracellular pathways. Microsc Res Tech 1999, 45, 210–216. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed]
- Indo, Y.; Mardy, S.; Tsuruta, M.; Karim, M.A.; Matsuda, I. Structure and organization of the humanTRKA gene encoding a high affinity receptor for nerve growth factor. J. Hum. Genet. 1997, 42, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Reuther, G.W.; Lambert, Q.T.; Caligiuri, M.A.; Der, C.J. Identification and Characterization of an Activating TrkA Deletion Mutation in Acute Myeloid Leukemia. Mol. Cell. Biol. 2000, 20, 8655–8666. [Google Scholar] [CrossRef]
- Gao, F.; Griffin, N.; Faulkner, S.; Rowe, C.W.; Williams, L.; Roselli, S.; Thorne, R.F.; Ferdoushi, A.; Jobling, P.; Walker, M.M.; et al. The neurotrophic tyrosine kinase receptor TrkA and its ligand NGF are increased in squamous cell carcinomas of the lung. Sci. Rep. 2018, 8, 8135. [Google Scholar] [CrossRef]
- Faulkner, S.; Jobling, P.; Rowe, C.W.; Oliveira, S.R.; Roselli, S.; Thorne, R.F.; Oldmeadow, C.; Attia, J.; Jiang, C.C.; Zhang, X.D.; et al. Neurotrophin Receptors TrkA, p75NTR, and Sortilin Are Increased and Targetable in Thyroid Cancer. Am. J. Pathol. 2018, 188, 229–241. [Google Scholar] [CrossRef]
- Lagadec, C.; Meignan, S.; Adriaenssens, E.; Foveau, B.; Vanhecke, E.; Romon, R.; Toillon, R.-A.; Oxombre, B.; Hondermarck, H.; Le Bourhis, X. TrkA overexpression enhances growth and metastasis of breast cancer cells. Oncogene 2009, 28, 1960–1970. [Google Scholar] [CrossRef]
- Faulkner, S.; Griffin, N.; Rowe, C.W.; Jobling, P.; Lombard, J.M.; Oliveira, S.M.; Walker, M.M.; Hondermarck, H. Nerve growth factor and its receptor tyrosine kinase TrkA are overexpressed in cervical squamous cell carcinoma. FASEB BioAdvances 2020, 2, 398–408. [Google Scholar] [CrossRef]
- Hong, D.; Bauer, T.; Lee, J.; Dowlati, A.; Brose, M.; Farago, A.; Taylor, M.; Shaw, A.; Montez, S.; Meric-Bernstam, F.; et al. Larotrectinib in adult patients with solid tumours: a multi-centre, open-label, phase I dose-escalation study. Ann. Oncol. 2019, 30, 325–331. [Google Scholar] [CrossRef]
- Drilon, A.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Hong, D.S.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Steinthorsdottir, V.; Stefansson, H.; Ghosh, S.; Birgisdottir, B.; Bjornsdottir, S.; Fasquel, A.C.; Olafsson, O.; Stefansson, K.; Gulcher, J.R. Multiple novel transcription initiation sites for NRG1. Gene 2004, 342, 97–105. [Google Scholar] [CrossRef]
- Falls, D.L. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res 2003, 284, 14–30. [Google Scholar] [CrossRef]
- Meyer, D.; Birchmeier, C. Multiple essential functions of neuregulin in development. Nature 1995, 378, 386–390. [Google Scholar] [CrossRef]
- Kramer, R.; Bucay, N.; Kane, D.J.; E Martin, L.; E Tarpley, J.; E Theill, L. Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proceedings of the National Academy of Sciences 1996, 93, 4833–4838. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Holbro, T.; et al. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. NEW EMBO MEMBERS' REVIEW: The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef]
- Nagasaka, M.; Ou, S.-H.I. NRG1 and NRG2 fusion positive solid tumor malignancies: a paradigm of ligand-fusion oncogenesis. Trends Cancer 2022, 8, 242–258. [Google Scholar] [CrossRef]
- Busfield, S.J.; Michnick, D.A.; Chickering, T.W.; Revett, T.L.; Ma, J.; Woolf, E.A.; Comrack, C.A.; Dussault, B.J.; Woolf, J.; Goodearl, A.D.J.; et al. Characterization of a Neuregulin-Related Gene, Don-1, That Is Highly Expressed in Restricted Regions of the Cerebellum and Hippocampus. Mol. Cell. Biol. 1997, 17, 4007–4014. [Google Scholar] [CrossRef]
- Schoeberl, B.; et al. Systems biology driving drug development: From design to the clinical testing of the anti-ErbB3 antibody seribantumab (MM-121). NPJ Systems Biology and Applications 2017, 3. [Google Scholar] [CrossRef]
- Carrizosa, D.R.; Burkard, M.E.; Elamin, Y.Y.; Desai, J.; Gadgeel, S.M.; Lin, J.J.; Waqar, S.N.; Spigel, D.R.; Chae, Y.K.; Cheema, P.K.; et al. CRESTONE: Initial efficacy and safety of seribantumab in solid tumors harboring NRG1 fusions. J. Clin. Oncol. 2022, 40, 3006–3006. [Google Scholar] [CrossRef]
- Spigel, D.; Waqar, S.; Burkard, M.; Lin, J.; Chae, Y.; Socinski, M.; Gadgeel, S.; Reckamp, K.; Leland, S.; Plessinger, D.; et al. MO01.33 CRESTONE – Clinical Study of REsponse to Seribantumab in Tumors with NEuregulin-1 (NRG1) Fusions – A Phase 2 Study of the anti-HER3 mAb for Advanced or Metastatic Solid Tumors (NCT04383210). J. Thorac. Oncol. 2021, 16, S29–S30. [Google Scholar] [CrossRef]
- Schram, A.M.; Goto, K.; Kim, D.-W.; Martin-Romano, P.; Ou, S.-H.I.; O'Kane, G.M.; O'Reilly, E.M.; Umemoto, K.; Duruisseaux, M.; Neuzillet, C.; et al. Efficacy and safety of zenocutuzumab, a HER2 x HER3 bispecific antibody, across advanced NRG1 fusion (NRG1+) cancers. J. Clin. Oncol. 2022, 40, 105–105. [Google Scholar] [CrossRef]
- Arar, M.; et al. Platelet-derived growth factor receptor beta regulates migration and DNA synthesis in metanephric mesenchymal cells. J Biol Chem 2000, 275, 9527–9533. [Google Scholar] [CrossRef]
- Hellström, M.; Kalén, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar] [CrossRef]
- Hoch, R.V.; Soriano, P. Roles of PDGF in animal development. Development 2003, 130, 4769–4784. [Google Scholar] [CrossRef]
- Appiah-Kubi, K.; Lan, T.; Wang, Y.; Qian, H.; Wu, M.; Yao, X.; Wu, Y.; Chen, Y. Platelet-derived growth factor receptors (PDGFRs) fusion genes involvement in hematological malignancies. Crit. Rev. Oncol. 2017, 109, 20–34. [Google Scholar] [CrossRef]
- Heidaran, M.; Pierce, J.; Jensen, R.; Matsui, T.; Aaronson, S. Chimeric alpha- and beta-platelet-derived growth factor (PDGF) receptors define three immunoglobulin-like domains of the alpha-PDGF receptor that determine PDGF-AA binding specificity. J. Biol. Chem. 1990, 265, 18741–18744. [Google Scholar] [CrossRef]
- Lubinus, M.; Meier, K.; Smith, E.; Gause, K.; LeRoy, E.; Trojanowska, M. Independent effects of platelet-derived growth factor isoforms on mitogen-activated protein kinase activation and mitogenesis in human dermal fibroblasts. J. Biol. Chem. 1994, 269, 9822–9825. [Google Scholar] [CrossRef]
- Tsao, A.S.; Wei, W.; Kuhn, E.; Spencer, L.; Solis, L.M.; Suraokar, M.; Lee, J.J.; Hong, W.K.; Wistuba, I.I. Immunohistochemical Overexpression of Platelet-Derived Growth Factor Receptor–Beta (PDGFR-β) is Associated With PDGFRB Gene Copy Number Gain in Sarcomatoid Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2011, 12, 369–374. [Google Scholar] [CrossRef]
- Kim, M.S.; Choi, H.S.; Wu, M.; Myung, J.; Kim, E.J.; Kim, Y.S.; Ro, S.; Ha, S.E.; Bartlett, A.; Wei, L.; et al. Potential Role of PDGFRβ-Associated THBS4 in Colorectal Cancer Development. Cancers 2020, 12, 2533. [Google Scholar] [CrossRef]
- Nazarian, R.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Cristofanilli, M.; et al. Imatinib mesylate (Gleevec) in advanced breast cancer-expressing C-Kit or PDGFR-beta: clinical activity and biological correlations. Ann Oncol 2008, 19, 1713–1719. [Google Scholar] [CrossRef]
- David, M.; Cross, N.C.P.; Burgstaller, S.; Chase, A.; Curtis, C.; Dang, R.; Gardembas, M.; Goldman, J.M.; Grand, F.; Hughes, G.; et al. Durable responses to imatinib in patients with PDGFRB fusion gene–positive and BCR-ABL–negative chronic myeloproliferative disorders. Blood 2006, 109, 61–64. [Google Scholar] [CrossRef]
- Apperley, J.F.; Gardembas, M.; Melo, J.V.; Russell-Jones, R.; Bain, B.J.; Baxter, E.J.; Chase, A.; Chessells, J.M.; Colombat, M.; Dearden, C.E.; et al. Response to Imatinib Mesylate in Patients with Chronic Myeloproliferative Diseases with Rearrangements of the Platelet-Derived Growth Factor Receptor Beta. New Engl. J. Med. 2002, 347, 481–487. [Google Scholar] [CrossRef]
- Wang, P.; Song, L.; Ge, H.; Jin, P.; Jiang, Y.; Hu, W.; Geng, N. Crenolanib, a PDGFR inhibitor, suppresses lung cancer cell proliferation and inhibits tumor growth in vivo. OncoTargets Ther. 2014, 7, 1761–1768. [Google Scholar] [CrossRef]
- Fujino, S.; Miyoshi, N.; Ito, A.; Yasui, M.; Ohue, M.; Ogino, T.; Takahashi, H.; Uemura, M.; Matsuda, C.; Mizushima, T.; et al. Crenolanib Regulates ERK and AKT/mTOR Signaling Pathways in RAS/BRAF-Mutated Colorectal Cancer Cells and Organoids. Mol. Cancer Res. 2021, 19, 812–822. [Google Scholar] [CrossRef]
- Jun, H.J.; et al. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-Syt1 phosphorylation. Cancer Res 2012, 72, 3764–3774. [Google Scholar] [CrossRef]
- Neel, D.S.; Allegakoen, D.V.; Olivas, V.; Mayekar, M.K.; Hemmati, G.; Chatterjee, N.; Blakely, C.M.; McCoach, C.E.; Rotow, J.K.; Le, A.; et al. Differential Subcellular Localization Regulates Oncogenic Signaling by ROS1 Kinase Fusion Proteins. Cancer Res 2019, 79, 546–556. [Google Scholar] [CrossRef]
- Cui, M.; et al. Molecular and clinicopathological characteristics of ROS1-rearranged non-small-cell lung cancers identified by next-generation sequencing. Mol Oncol 2020, 14, 2787–2795. [Google Scholar] [CrossRef]
- Li, N.; Chen, Z.; Huang, M.; Zhang, D.; Hu, M.; Jiao, F.; Quan, M. Detection of ROS1 gene fusions using next-generation sequencing for patients with malignancy in China. Front. Cell Dev. Biol. 2022, 10, 1035033. [Google Scholar] [CrossRef]
- Muminovic, M.; et al. Importance of ROS1 gene fusions in non-small cell lung cancer. 2023.
- Bergethon, K.; Shaw, A.T.; Ou, S.-H.I.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 Rearrangements Define a Unique Molecular Class of Lung Cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef]
- Cai, W.; Li, X.; Su, C.; Fan, L.; Zheng, L.; Fei, K.; Zhou, C.; Manegold, C.; Schmid-Bindert, G. ROS1 fusions in Chinese patients with non-small-cell lung cancer. Ann. Oncol. 2013, 24, 1822–1827. [Google Scholar] [CrossRef]
- Lu, S.; Pan, H.; Wu, L.; Yao, Y.; He, J.; Wang, Y.; Wang, X.; Fang, Y.; Zhou, Z.; Wang, X.; et al. Efficacy, safety and pharmacokinetics of Unecritinib (TQ-B3101) for patients with ROS1 positive advanced non-small cell lung cancer: a Phase I/II Trial. Signal Transduct. Target. Ther. 2023, 8, 249. [Google Scholar] [CrossRef]
- Wang, V.; Bivona, T.; Ali, S.M.; Schrock, A.B.; Miller, V.A. CD74 - ROS1 Fusion in NSCLC Detected by Hybrid Capture–Based Tissue Genomic Profiling and ctDNA Assays. J. Thorac. Oncol. 2017, 12, e19–e20. [Google Scholar] [CrossRef]
- Meng, Z.-T.; Chen, P.; Zang, F.; Liu, Y.; Xu, X.; Su, Y.; Chen, J.; Lin, L.; Zhang, L.; Zhang, T. A patient with classic biphasic pulmonary blastoma harboring CD74–ROS1 fusion responds to crizotinib. OncoTargets Ther. 2017, 11, 157–161. [Google Scholar] [CrossRef]
- Mizuno, T.; Fujiwara, Y.; Yoshida, K.; Kohno, T.; Ohe, Y. Next-Generation Sequencer Analysis of Pulmonary Pleomorphic Carcinoma With a CD74-ROS1 Fusion Successfully Treated With Crizotinib. J. Thorac. Oncol. 2019, 14, e106–e108. [Google Scholar] [CrossRef]
- Wang, G.; Gao, J.; Lv, J.; Chen, X.; Wu, J.; Wang, R.; Jiang, J. Effective Treatment with Cabozantinib in an Advanced Non-Small-Cell Lung Cancer Patient Harboring a CD74-ROS1 Fusion: A Case Report. OncoTargets Ther. 2020, 13, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Huang, S.; Ye, X.; Feng, L.; Lu, Y.; Zhou, C.; Zhao, J.; He, T.; Wang, J.; Li, B. Crizotinib resistance conferred by BRAF V600E mutation in non–small cell lung cancer harboring an oncogenic ROS1 fusion. Cancer Treat. Res. Commun. 2021, 27, 100377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Z.; Han, X.; Li, J.; Guo, H.; Shi, J. Acquired MET D1228N Mutations Mediate Crizotinib Resistance in Lung Adenocarcinoma with ROS1 Fusion: A Case Report. Oncol. 2020, 26, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Yang, J.; Wang, D.; Yan, D. ROS1 fusion lung adenosquamous carcinoma patient with short-term clinical benefit after crizotinib treatment: a case report. Ann. Transl. Med. 2022, 10, 157. [Google Scholar] [CrossRef]
- Tyler, L.C.; Le, A.T.; Chen, N.; Nijmeh, H.; Bao, L.; Wilson, T.R.; Chen, D.; Simmons, B.; Turner, K.M.; Perusse, D.; et al. MET gene amplification is a mechanism of resistance to entrectinib in ROS1 + NSCLC. Thorac. Cancer 2022, 13, 3032–3041. [Google Scholar] [CrossRef]
- Tanaka, S.; et al. A case of CD74-ROS1-positive lung adenocarcinoma diagnosed by next-generation sequencing achieved long-term survival with pemetrexed regimens. Thorac Cancer 2023. [Google Scholar] [CrossRef]
- Pizzutilo, E.G.; Agostara, A.G.; Roazzi, L.; Romanò, R.; Motta, V.; Lauricella, C.; Marrapese, G.; Cerea, G.; Signorelli, D.; Veronese, S.M.; et al. Repotrectinib Overcomes F2004V Resistance Mutation in ROS1-Rearranged Non-Small Cell Lung Cancer: A Case Report. JTO Clin. Res. Rep. 2023, 100555. [Google Scholar] [CrossRef]
- Cha, Y.J.; Lee, C.; Joo, B.; A Kim, K.; Lee, C.-K.; Shim, H.S. Clinicopathological Characteristics of NRG1 Fusion-Positive Solid Tumors in Korean Patients. Cancer Res. Treat. 2023, 55, 1087–1095. [Google Scholar] [CrossRef]
- Chen, K.; Li, W.; Xi, X.; Zhong, J. A case of multiple primary lung adenocarcinoma with a CD74-NRG1 fusion protein and HER2 mutation benefit from combined target therapy. Thorac. Cancer 2022, 13, 3063–3067. [Google Scholar] [CrossRef]
- Abe, A.; Emi, N.; Tanimoto, M.; Terasaki, H.; Marunouchi, T.; Saito, H. Fusion of the platelet-derived growth factor receptor beta to a novel gene CEV14 in acute myelogenous leukemia after clonal evolution. Blood 1997, 90, 4271–4277. [Google Scholar] [CrossRef]
- Kim, H.-G.; Jang, J.-H.; Koh, E.-H. TRIP11-PDGFRB fusion in a patient with a therapy-related myeloid neoplasm with t(5;14)(q33;q32) after treatment for acute promyelocytic leukemia. Mol. Cytogenet. 2014, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Fayiga, F.F.; Reyes-Hadsall, S.C.; Moreno, B.A.; Oh, K.S.; Brathwaite, C.; Duarte, A.M. Novel ANKRD26 and PDGFRB gene mutations in pediatric case of non-Langerhans cell histiocytosis: Case report and literature review. J. Cutan. Pathol. 2023, 50, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, M.; Ou, S.I. Is NRG2α Fusion a "Doppelgänger" to NRG1α/β Fusions in Oncology? J Thorac Oncol 2020, 15, 878–880. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The CD74 isoforms P41 (280 aa – bottom left), P35 (232 aa – top left), P33 (216 aa – top right), and P43 (296 aa – bottom right) depicted on the cell surface of a healthy human cell. Experimentally resolved structural fragments of CD74 are illustrated in boxes along with their corresponding PDB numbers. The predicted structure of P43 produced by AlphaFold is also provided [50, 51]. IC: intracellular, EC: extracellular, CLIP: class II associated invariant chain peptide, TM: transmembrane domain, TD: trimerization domain, E6B: exon 6b, EXT: N-terminal extension.
Figure 1.
The CD74 isoforms P41 (280 aa – bottom left), P35 (232 aa – top left), P33 (216 aa – top right), and P43 (296 aa – bottom right) depicted on the cell surface of a healthy human cell. Experimentally resolved structural fragments of CD74 are illustrated in boxes along with their corresponding PDB numbers. The predicted structure of P43 produced by AlphaFold is also provided [50, 51]. IC: intracellular, EC: extracellular, CLIP: class II associated invariant chain peptide, TM: transmembrane domain, TD: trimerization domain, E6B: exon 6b, EXT: N-terminal extension.
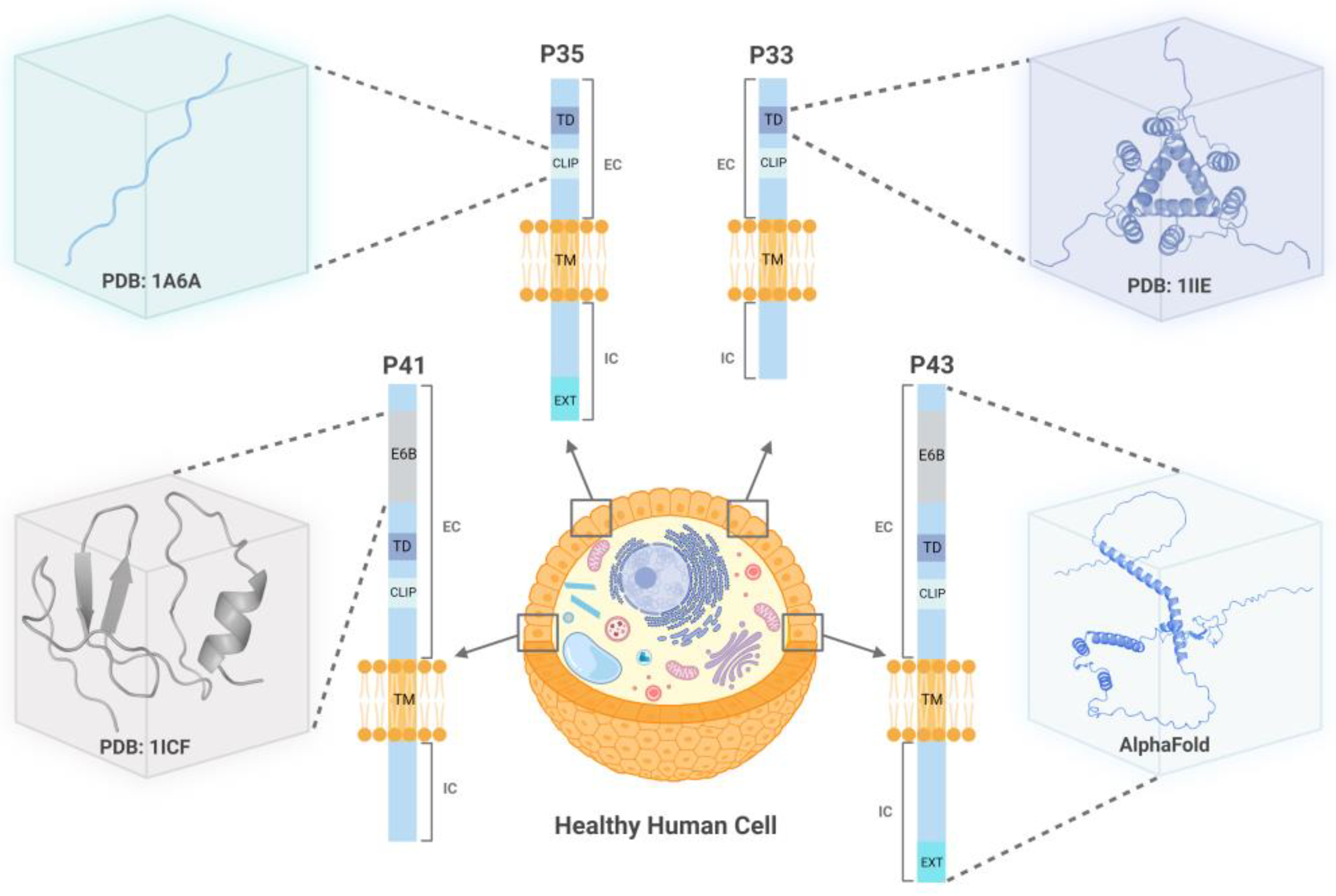
Figure 2.
CD74 oncogenic fusions. From left to right, CD74-NRG2α, CD74-NTRK1, CD74-ROS1, CD74-PDGFRB, and CD74-NRG1 are depicted on the membrane of a cancer cell. The CD74 segments participating in the formation of these fusions are enclosed within the dashed rectangles. For each fusion, the functional domain (EGF and KD) known to contribute to the protein’s oncogenic activity is illustrated. In all cases, this domain is derived from the protein partner of CD74. The size of each domain shown in the figure is drawn with no scale. IC: intracellular, TM: transmembrane domain, EC: extracellular, EGF: epidermal growth factor-like domain, KD: kinase domain.
Figure 2.
CD74 oncogenic fusions. From left to right, CD74-NRG2α, CD74-NTRK1, CD74-ROS1, CD74-PDGFRB, and CD74-NRG1 are depicted on the membrane of a cancer cell. The CD74 segments participating in the formation of these fusions are enclosed within the dashed rectangles. For each fusion, the functional domain (EGF and KD) known to contribute to the protein’s oncogenic activity is illustrated. In all cases, this domain is derived from the protein partner of CD74. The size of each domain shown in the figure is drawn with no scale. IC: intracellular, TM: transmembrane domain, EC: extracellular, EGF: epidermal growth factor-like domain, KD: kinase domain.
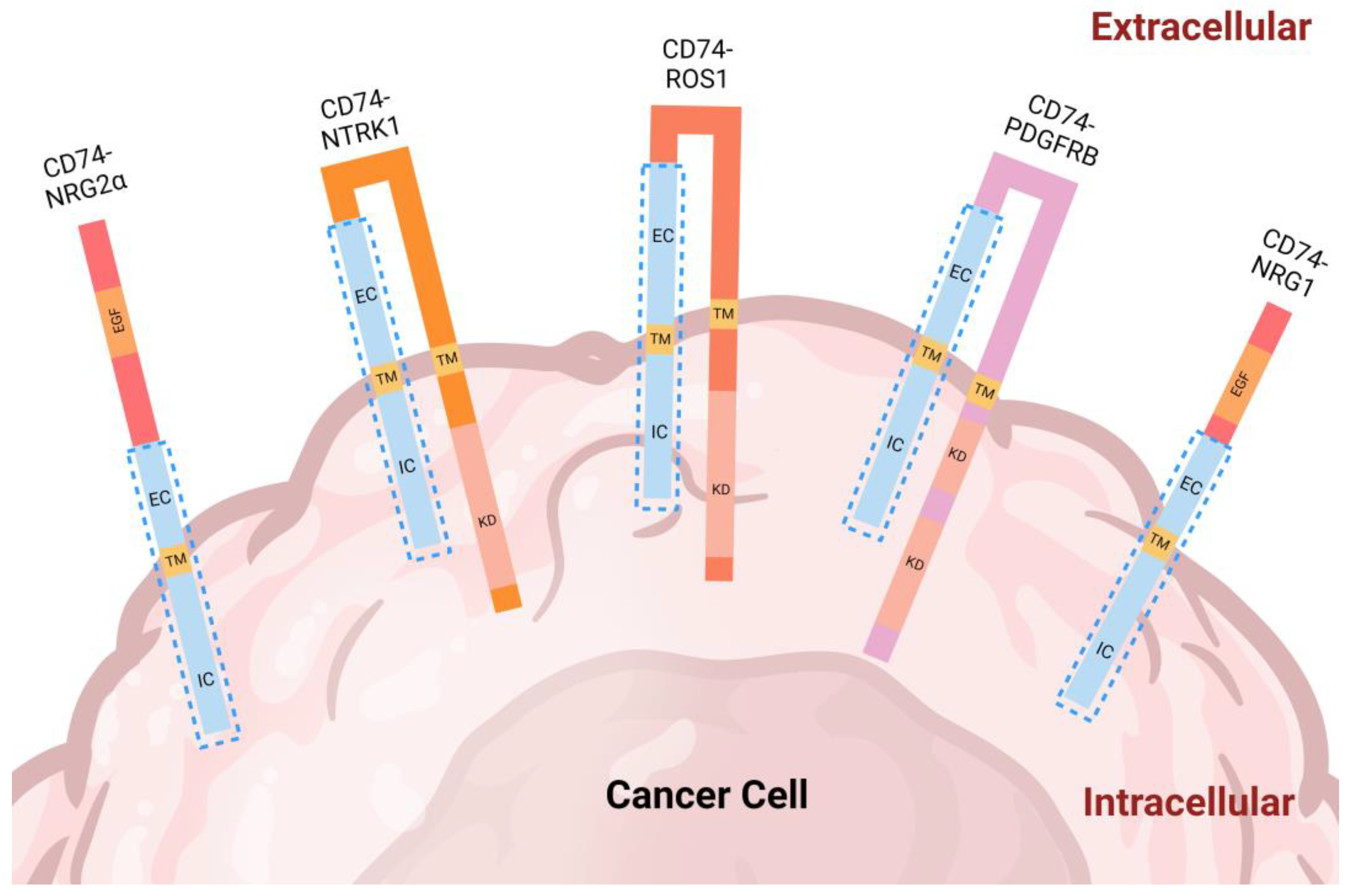
Figure 3.
Timeline for identification of CD74 fusions. The original study, gender of the patient, and type of cancer are also reported. Of note, the gender of the CD74-NTRK1 patient was not reported in the original study.
Figure 3.
Timeline for identification of CD74 fusions. The original study, gender of the patient, and type of cancer are also reported. Of note, the gender of the CD74-NTRK1 patient was not reported in the original study.
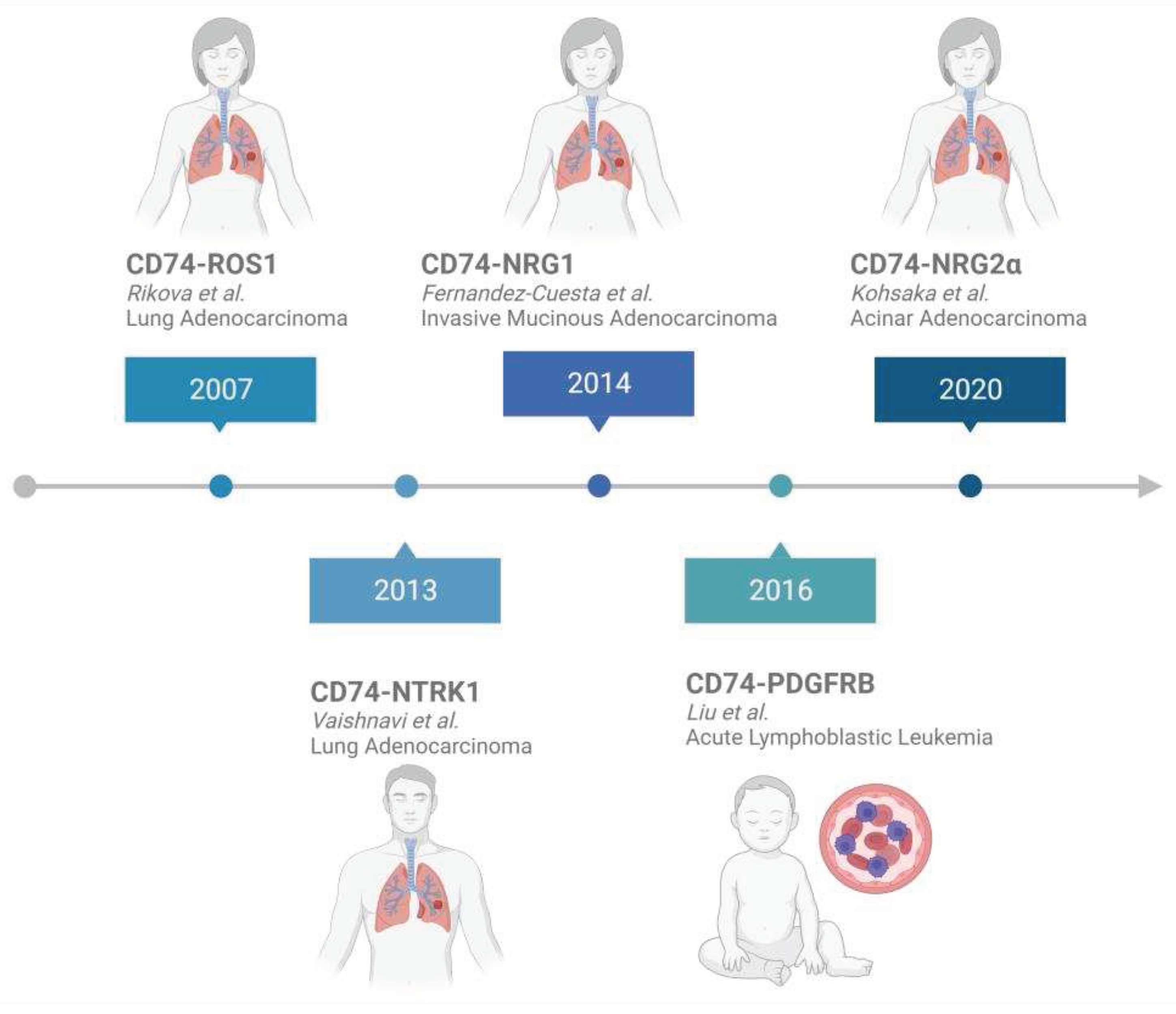

Table 1.
CD74 Fusion Protein Variants Detected in Cancer Patients.
| CD74 Fusion Protein | Exon Breakpoint | Cancer | Reference |
|---|---|---|---|
| CD74-ROS1 | C6-R34 C6-R32 C7-R32 C7-R34 |
NSCLC NSCLC NSCLC IBC |
[30] [54] [57] [52] |
| C3-R34 | NSCLC | [58] | |
| C6-R33 | NSCLC | [59] | |
| C6-R35 | NSCLC | [60] | |
| CD74-NTRK1 | C8-N12 C7-N8 C6-N12 |
NSCLC NSCLC NSCLC |
[31] [61] [62] |
| CD74-NRG1 | C6-N6 C8-N6 C6-N4 C7-N6 |
IMA IMA NSCLC IMA |
[32] [56] [63] [64] |
| CD74-PDGFRB | C6-P11 | B-ALL | [33] |
| CD74-NRG2a | C6-N2 | NSCLC | [34] |
Table 2.
CD74-ROS1 Clinical Patient Characteristics.
| Diagnosis | Variant | Detection Method |
Age | Gender | Smoker/ Pack Year (PY) |
Stage | Treatment | TKIResistance | LifespanOS | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| NSCLC | C7-R32 C6-R34 |
RT-PCR | 61 | F | 0 | IV (T4N3M1b) |
1st: pemetrexed + cisplatin*refused crizotinib for economic reason | - | - | [57] |
| IBC | C7-R34 | NGS | 64 | F | - | cT4N3M1 | 1st: paclitaxel2nd: capecitabine*refused crizotinib for economic reason | - | Died 4 mos. after dx |
[52] |
| NSCLC | C3-R34 | NGS | 38 | F | 0 | IVA (T1aN3M1A) |
1st: cisplatin, pemetrexed, + bevacizumab2nd: docetaxel + ramucirumab*3rd: crizotinib *4th: entrectinib |
Crizotinib resistance: brain metastasis | 1 mo. PFS | [58] |
| Metastatic NSCLC | - | CGP (NGS) ctDNA assay |
41 | F | 0 | - | 1st: carboplatin, pemetrexed + bevacizumab *2nd: crizotinib |
- | PFS 4 mos. after crizotinib | [129] |
| CBPB | - | NGS | 44 | F | 0 | IV (cT3N1M1b) |
*1st: crizotinib | Crizotinib resistance: ROS1 G2032R mutation |
PFS for 3 mos. PD after crizotinib resistance |
[130] |
| PPC | - | NGSRT-PCR | 56 | F | 0 | IIIA (T2aN2M0) |
1st: resection 2nd: paclitaxel, carboplatin + bevacizumab *3rd: crizotinib |
- | CR after 6 mos. | [131] |
| NSCLC | C6-R34 | NGS | 40 | F | 0 | - | 1st: gemcitabine + cisplatin 2nd: docetaxel 3rd: gefitinib *4th: cabozantinib *5th: crizotinib |
Cabozantinib resistance | SD after 2 mos. crizotinib | [132] |
| NSCLC | C6-R33 | NGSRT-qPCR | 51 | F | - | IVB (pT4N3M1c) |
1st: pemetrexed + cisplatin *2nd: crizotinib |
- | PFS 15.7 mos. after crizotinib | [55] |
| MetastaticNSCLC | C6-R33 | NGS | 60 | F | 0 | - | 1st: radiotherapy*2nd: entrectinib | - | 5 mos. SD | [59] |
| NSCLC | C6-R33 | NGS | 53 | F | - | IV | *1st: crizotinib *2nd: dabrefenib |
Crizotinib resistance: BRAF V600E mutation | 2 mos. PR after crizotinib. Dead 15 days after dabrefenib | [133] |
| Metastatic NSCLC | - | NGS | 30 | F | - | IIIC (T3N3M0) | 1st: cisplatin + pemetrexed 2nd: nedaplatin + pemetrexed *3rd: crizotinib *4th: cabozantinib |
Crizotinib resistance: MET D1228N mutation | Dead 21 months from dx | [134] |
| NSCLC- ASC | C6-R34 | NGS | 43 | F | 0 | IIIA (pT2aN2M0) | 1st: albumin-bound paclitaxel + camrelizumab *2nd: crizotinib 3rd: pemetrexed + carboplatin + bevacizumab4th: cisplatin + gemcitabine + bevacizumab + *crizotinib |
- | Dead, ~15 mos. After dx | [135] |
| NSCLC | C6-R32 C6-R35 |
DNA NGS RNA NGS FISH Sanger Seq |
44 | F | 0 | IVB | *1st: Crizotinib *2nd: Lorlatinib 3rd: chemotherapy |
Crizotinib resistance: bone metastasis Lorlatinib resistance: ROS1 G2032R mutation |
Dead, ~19 mos. After dx | [60] |
| NSCLC | - | Molecular testing- not specified | 54 | M | 0 | IV | *1st: crizotinib *2nd: entrectinib 3rd: Carboplatin + Pemetrexed 4th: pemetrexed + pembrolizumab *5th: Repotrectinib *6th: Cabozantinib |
Possible entrectinib resistance: MET amplification | Deceased ~50 months after dx | [136] |
| NSCLC | - | NGS | 54 | F | 30 | IVA (T2aN3M1a) |
1st: Cisplatin + pemetrexed 2nd: Carboplatin + paclitaxel 3rd: Pemetrexed 4th: Nivolumab 5th: Docetaxel 6th: Pemetrexed 7th: S-1 8th: Gemcitabine *9th: Entrectinib |
- | 1st: PR 2nd: SD 3rd: PR 4th: SD 5th: PR 6th: PD 7th: PD 8th: PD 9th: PR |
[137] |
| NSCLC | - | NGS | 49 | F | 0 | IV | *1st: Entrectinib *2nd: Crizotinib 3rd: Carboplatin *4th: Repotrectinib |
Possible Entrectinib resistance: ROS1 F2004V mutation | 1st: PR 2nd: SD 3rd: PD 4th: PR |
[138] |
PY: Pack-Years, CBPB: classic biphasic pulmonary blastoma, PPC: pulmonary pleomorphic carcinoma, PFS: progression free survival, CR: complete response, PD: progressive disease, WGS: whole-genome sequencing, SD: stable disease, PR: partial response, ASC: adenosquamous carcinoma, FFPE: formalin-fixed paraffin embedded, TMA: Tissue Microarray, TKI: tyrosine kinase inhibitor, BRAF: B-Raf proto-oncogene, serine/threonine kinase gene, dx: diagnosis, Seq: sequencing, CGP: comprehensive genomic profiling, ctDNA: circulating tumor DNA * asterisk indicates TKI prescribed after fusion identification. - dash indicates data not available. ~ denotes approximately equal to.
Table 3.
CD74- NRG1 Clinical Patient Characteristics.
| Diagnosis | Variant | Detection Method |
Age | Gender | Smoker/ Pack Year (PY) |
Stage | Treatment | Lifespan OS |
Reference |
|---|---|---|---|---|---|---|---|---|---|
| NSCLC | - | Genetic Testing | 62 | F | 0 | IV | 1st: pemetrexed + cisplatin 2nd: atezolizumab + albumin-bound paclitaxel + carboplatin *3rd: afatinib + pyrotinib |
PFS 5 mos. | [140] |
| IMA | C6-N6 | NGS | 86 | M | 0 | IIA (pT2bN0M0) |
1st: carboplatin + pemetrexed 2nd: paclitaxel + bevacizumab 3rd: nivolumab 4th: GSK2849330 (anti-ERBB3 mAb) *5th: afatinib |
PR 1 year and 7 mos. On GSK2849330 | [64] |
| IMA | C8-N6 | multiplex PCR | 81 | M | Former, smoked cigars for 1 year | IIB (pT3N0M0) |
*1st: afatinib | SD at 6 wks. PD at 13 wks. |
[64] |
| IMA | C7-N6 | NGS | 51 | M | <1 | IIIB(T4N2M0) | 1st: cisplatin + pemetrexed *2nd: afatinib |
PD 8 wks. After afatinib started | [64] |
PFS: progression free survival, PR: partial response, SD: stable disease, PD: progressive disease - dash indicates data not available.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



