Submitted:
18 January 2024
Posted:
18 January 2024
You are already at the latest version
Abstract
Temporal lobe epilepsy (TLE) is associated with reorganization of hippocampal neuronal net-works, gliosis, neuroinflammation, loss of integrity of the blood-brain barrier (BBB) in humans and animal models. More than 30% of epilepsies remain intractable and characterization of the molecular mechanisms involved in BBB dysfunction is essential to the identification of new thera-peutic strategies. In this work, we induced status epilepticus in rats by injection of the proconvul-sant drug pilocarpine that leads to TLE. Using RT-qPCR, double immunohistochemistry and con-focal imaging, we studied at different time points of epileptogenesis (latent phase, 3, 7, 14 days; chronic phase, 1 and 3 months) the regulation of reactive glia and vascular markers. In the hippo-campus, increased expression of mRNA encoding the glial proteins GFAP and Iba1 confirmed neu-roinflammatory status. We report for the first time the concomitant induction in endothelial cells, pericytes and basement membrane of the BBB of specific proteins CD31, PDGFR and ColIV, that peaks at the same time points as inflammation. The altered expression of these proteins occurs ear-ly in TLE, during the latent phase, suggesting that they could be associated with early rupture and pathogenicity of the BBB that will contribute to the chronic phase of epilepsy.
Keywords:
Temporal lobe epilepsy
; Pilocarpine
; Hippocampus
; Epileptogenesis
; Gliosis
; Inflammation
; Blood-brain barrier
; Endothelial cells
; Pericytes
; Basement membrane
1. Introduction
Epilepsy is the third commonest neurological disorder, affecting more than 65 million people worldwide [1]. Despite progress in anti-epileptic treatments, up to 40% of epileptic patients remain resistant to all currently available therapies, and patients who respond to anti-epileptic treatments often complain of debilitating side effects [2]. TLE is the most prevalent form of epilepsy that affects particularly the hippocampus. It is characterized by the loss of principal neuronal cells and interneurons, structural reorganization such as sprouting, neo-spinogenesis and neurogenesis with cell dispersion. TLE is also associated with gliosis, neuroinflammation, and loss of the integrity of the BBB [3]. Each one of these characteristics has been observed in surgical or post-mortem samples obtained from patients with pharmaco-resistant TLE or from animal models of TLE [3,4,5]. The aforementioned histopathological alterations are thought to take place following an initial injury and contribute to epileptogenesis [5,6].
The BBB is a cellular barrier made up of endothelial cells that line brain capillaries and that are encased in a basement membrane (BM) –or basal lamina– in interaction with pericytes, that are closely linked to brain parenchyma cells such as astrocytes, microglia, and neurons [7]. Many proteins are involved in BBB function in physiopathological conditions which justifies our interest to study their regulation in epileptogenesis. Among those, we selected proteins involved in vascular and BBB maintenance and expressed in specific cell types of the BBB. Cluster of differentiation 31 (CD31), also known as platelet endothelial cell adhesion molecule-1 (PECAM-1) is expressed mainly by endothelial cells [8,9,10]. Platelet-derived growth factor receptor beta (PDGFRβ) is specific of pericytes, contractile cells that surround endothelial cells and that control blood-flow [11,12,13]. PDGFRβ is a biomarker of fibrotic modifications and stromal cell occupancy associated with scar formation [14,15]. We also followed the regulation of collagen IV (ColIV). It is the most abundant component of the BM, that is produced predominantly by endothelial cells, pericytes and astrocytes, other major constitutive cells of the BBB [16]. Astrocytes interact with, and surround, endothelial cells and pericytes, via specialized structures known as astrocytic end-feet.
The BBB protects the central nervous system (CNS) very efficiently. It is a selective filter between blood and brain parenchyma, it regulates the selective uptake of nutrients and proteins from the blood, the influx into the CNS of toxic xenobiotics and pathogens and the efflux of metabolic waste. BBB integrity is essential for CNS homeostasis, and many disorders of the CNS, including stroke, traumatic brain injuries, tumors, infectious diseases, neurodegenerative diseases and epilepsy entail BBB dysfunction [17]. BBB permeability or leakage is one of the earliest characteristics in these diseases [18,19]. BBB dysfunction may contribute to epileptogenesis via a cascade of events triggered by leakage of inflammatory mediators into the CNS, which causes neuroinflammation [20,21]. Conversely, seizures may further increase BBB breakdown [22,23,24]. Alterations of different neuronal and glial populations of the brain parenchyma and their interactions have been extensively studied in epilepsy [25,26]. However, considerably less attention has been given to the regulation of molecular components involved in the integrity and stabilization of the BBB, notably in TLE.
In the present work, we injected in rats with the proconvulsive drug pilocarpine to induce status epilepticus (PILO-SE) that leads to TLE. We investigated the modulation of reactive glia and vascular markers at several time points of epileptogenesis (latent phase, 3, 7, 14 days; chronic phase, 1 and 3 months, abbreviated PILO 3-14D; PILO 1M and 3M) using RT-qPCR, double immunohistochemistry, and confocal imaging. We report in the hippocampus of PILO-SE rats the increased expression of mRNA encoding the neuroinflammatory glial proteins GFAP and Iba1 and the concomitant induction in endothelial cells, pericytes and BM of the BBB, of specific proteins CD31, PDGFRβ and ColIV, respectively. These alterations could be associated with the rupture and pathogenicity of the BBB in TLE. Perivascular inflammation of reactive astrocytes and their end-feet in the PILO rat model was addressed previously [27], and was not addressed in the current study.
2. Results
All PILO rats that survived developed SE. From 10 min to 1 h following PILO injection, rats exhibited limbic motor seizures every few minutes. At 3 weeks, spontaneous recurrent seizures (SRSs) started to appear that could last up to 60 seconds, and developed into generalized seizures within the following days, that persisted for the lifetime of the animals, in agreement with previous reports [27,28,29,30,31]. We used 2 semi-quantitative approaches to follow the expression and regulation of BBB markers and/or cellular expression and distribution. First, we used RT-qPCR to study global changes of neuroinflammatory and BBB biomarkers at the level of the whole hippocampus. Second, we correlated these results with qualitative and semi-quantitative IHC regarding BBB biomarkers at the cellular level. We thus studied at different time points after PILO-induced SE, the expression of mRNAs encoding different neuroinflammatory and BBB markers using RT-qPCR (Figure 1) and specific primers listed in Table 1. In particular, we assessed the inflammatory and reactive glial markers GFAP and Iba1, and the vascular markers CD31, specific of endothelial cells, PDGFRβ, specific of pericytes, and ColIVa1 and ColIVa3, specific of the BBB basal lamina [32]. GFAP and Iba1 mRNA were increased at the latent phase (PILO 3 to 14D) and decreased at the chronic phase (PILO 1M and 3M). The mean GFAP and Iba1 mRNA levels were significantly increased in PILO rats when compared with control (CTL) rats (p<0.01; Dunnett’s test). Specifically, in PILO 3D, increases were ~11- and ~3-fold for GFAP and Iba1, respectively. In PILO 7D, GFAP and Iba1 increased ~9.7- and ~6-fold, respectively, whereas in PILO 14D, GFAP and Iba1 increased ~8- and ~5.7-fold, respectively. In PILO 1M, GFAP increased ~8-fold, whereas no difference was found for GFAP in PILO 3M and for Iba1 in PILO 1M and 3M (see histograms A and B, Figure 1). It is known that neuroinflammation may lead to alterations of the BBB [4,33]. Consistently, our data show that the upregulation of GFAP and Iba1 mRNA is associated with the significant increase of mRNAs encoding CD31 (PILO 3D and 7D, ~1.5-fold), PDGFRβ (PILO 7D, ~1.3-fold), and ColIVa1 (PILO 3D: ~2.2-fold; 7D: 1.8-fold). This upregulation follows the same temporal course as that of reactive glia markers although at different levels, suggesting that CD31, PDGFRβ, and ColIVa3 expression is associated with inflammatory processes at the BBB.
We next analyzed the regulation of the BBB protein markers, using qualitative and semi-quantitative immunohistochemistry with PDGFRβ, CD31, and Collagen IV (ColIV) antibodies to assess correlations with the RT-qPCR BBB markers and follow their distribution and the expression intensity at the cellular level of different areas of the hippocampus. Based on the RT-qPCR results showing a peak of the neuroinflammation markers GFAP and Iba1 during the latent period between PILO 3D and 7D, we conducted our immunohistochemistry analysis at PILO 3D. We first performed dual PDGFRβ and CD31 immunohisto-labeling in CTL and PILO 3D rats (Figure 2A-C, F-H). At low magnification, in all areas and layers of the hippocampus of CTL rats, weak to moderate PDGFRβ staining was observed in the nervous tissue parenchyma cells (Figure 2A,C). Note that numerous cells were also stained in the subgranular zone of the DG, both in CTL and PILO 3D rats. In addition, moderate PDGFRβ expression was also observed in blood vessels of all sizes and shapes (see insets in Figure 2A,C). PDGFRβ and CD31 staining were increased in all hippocampal areas and layers of PILO 3D rats, as shown by mosaic tile scans (Figure 2F,H). Indeed, increased PDGFRβ was observed in the oriens (O), pyramidal layer (P), radiatum (R), lacunosum-moleculare (LM), molecular layer (M), and in the dentate gyrus (DG). In CTL rats, immunohistochemical analysis of rat brain sections showed CD31 immunoreactivity in blood vessels of all sizes and shapes (Figure 2B,C), that increased in PILO 3D LM blood vessels, as well as in the CC (corpus callosum), O, P, and R (Figure 2G,H).
In the same line, we performed ColIV immunolabeling in CTL and PILO 3D rats (Figure 2D-E, I-J). In CTL rats, ColIV staining was observed only in the M of the DG lower blade (Figure 2D,E), whereas PILO 3D rats displayed strong labeling of the vasculature in the O, P, R, LM and M of the DG (Figure 2I,J). We refined our study by conducting semi-quantitative analysis in CTL and PILO 3D rats, to determine the extent of changes in the fluorescence intensity of the different vascular markers (Figure 2K). In PILO 3D animals (PDGFRβ, 27 vessels; CD31, 53 vessels; ColIV, 21 vessels), the expression of all vascular markers increased significantly in the blood vessels of the DG and the LM compared to CTL animals (PDGFRβ, 15 vessels; CD31, 29 vessels; ColIV, 15 vessels). Specifically, PDGFRβ, CD31 and ColIV increased 2.4-, ~2 and ~1.9-fold respectively (Figure 2K, p<0.001, Student’s t-test).
We used high magnification to analyze in detail the double immunolabeling of CD31 (red) and ColIV (green) in CTL animals compared with PILO 3D rats (Figure 3). The LM blood vessels displayed lower immunolabeling of CD31 (red) and ColIV (green) in CTL animals (A-F) when compared with PILO 3D rats (G-L). Increased CD31 and ColIV immunolabeling appeared within endothelial cells (red; arrows), and the basal lamina (green), respectively, as illustrated by higher magnification of the boxed-in area in L (M-R). Indeed, ColIV immunostaining was closely associated with CD31 staining (Figure 3M-R), with basal lamina surrounding endothelial cells as reported elsewhere [13,34]. In conclusion, in rat PILO 3D hippocampus, both CD31 and ColIV proteins were increased in rat blood vessel endothelial cells and basal membrane, respectively. Last, high magnification of CD31 (red) and PDGFRβ (green) double immunolabeling (Figure 4) show that the LM blood vessels displayed lower CD31 and PDGFRβ expression in CTL animals (A-F) when compared with PILO 3D rats (G-L).
It has been shown that PDGFRβ pericytes surround endothelial cells and are found between the abluminal surface of the endothelial tube and the BM. Also, CD31 endothelial cells stain at the inner lining of blood vessels, while they are located between the BM and the abluminal surface of the endothelial tube [13,34]. When analyzed at high magnification, the boxed-in region in L showed endothelial cells (red, arrows) encircled by a pericyte (green, arrow) (Figure 4M-R). To confirm that indeed CD31 and PDGFRβ were expressed in distinct cell types, kymographs were constructed and analyzed with ImageJ software from single-pixel width white lines taken from each channel of the confocal images (Figure 4K). Then, profiles of the signal intensities of CD31 (red line, K) and PDGFRβ (green line, K) were measured along the single-pixel width white lines drawn in (M) and (N), showing that immunolabeling of the two proteins is clearly distinct (Figure 4S). Thus, our results indicate that in PILO 3D, both CD31 and PDGFRβ increased specifically in endothelial cells and pericytes, respectively.
3. Discussion
In our present work, we investigated in the hippocampus the regulation of the neuroinflammatory GFAP and Iba1 markers and vascular proteins of the BBB such as CD31, PDGFRβ and ColIV, during the different phases of epilepsy. We used a well-characterized experimental model of TLE induced by PILO in adult rats and implemented RT-qPCR, qualitative and semi-quantitative immunohistochemistry, and confocal microscopy analysis. This PILO model was selected because it involves a dynamic reorganization of neuronal networks, gliosis, neuroinflammation, loss of integrity of the BBB and neurovascular rearrangements in the hippocampus. This reorganization begins after the initial period of SE following PILO injection, during the silent period/latent phase when animals display a normal behavior, and reaches a plateau at the chronic phase when the animals develop SRSs [3,4,5,35]. It is known that patients with epilepsy can experience respiratory alterations during seizures [36,37]. Indeed, seizures may cause apnoea that can reduce blood oxygen to a life-threatening level. Patients with epilepsy exhibit increased risk of myocardial infarction, stroke, and cardiovascular death [38]. In the PILO animal model, it is known that respiratory failure following convulsions is a common cause of acute death. Post-convulsive respiratory paralysis can be accompanied by hypothermia and by vascular congestion of the brain, heart, lungs, and kidneys [39]. More recently, it has been shown in rats with SE triggered by PILO, that 30–50% of epileptic rats exhibited a sharp decrease in oxygen consumption, low metabolic rate of oxygen, and slow regular ventilation [40]. These percentages are consistent with 55 to 60% animals surviving PILO administration as reported by us and others [41,42,43].
In the PILO treated rats, we used RT-qPCR to study the kinetics of mRNAs coding the GFAP and Iba1 reactive glial markers. We observed that mRNA levels of both markers were significantly increased in the hippocampus, at latent phase and decreased at chronic phase. These results are in agreement with previous studies including ours [27,44,45,46] and confirmed neuroinflammation in our PILO rats. The upregulation of glial markers in the hippocampus was accompanied by increased CD31, PDGFRβ and ColIVa1 mRNA, and concomitant protein expression at latent phase, when inflammation peaks, with increased GFAP and Iba1, between PILO 3D and 7D, and decreased at the chronic phase (PILO 1M and/or 3M), when glia reactivity subsides. CD31, PDGFRβ and ColIVa1 proteins were also increased in cells of the BBB. These findings demonstrate that in this rat model of TLE, all 3 markers exhibited a significant increase, which may be indicative of structural alteration, remodeling of the BBB or neuroprotection mechanisms. Our results question whether the 3 BBB markers whose expression is upregulated in our epilepsy model are beneficial or detrimental for vascular integrity.
CD31 is mainly expressed by endothelial cells. This protein is a cell-cell adhesion and signaling molecule involved in angiogenesis and transmigration [47]. In agreement with our results, CD31 was induced in cerebral blood vessels between 24 and 72 h after Kainate administration in a mouse model of TLE [48]. It has been reported that peripheral and central inflammation promote breakdown of the BBB due to the upregulation of inflammatory mediators [49]. Hence, the increased expression of the vascular proteins we observed may be involved in the breakdown of the BBB in PILO rats. However, BBB components may also be key players in the pathogenicity of TLE, independently of inflammation. For instance, the expression of CD31 by CNS endothelial cells is not required for inflammation initiation and clinical signs in an animal model of multiple sclerosis [50]. CD31 maintains vascular integrity during inflammation by engaging into pro-survival pathways and by inhibiting cytokine production and pro-inflammatory signaling [8,51]. Accordingly, CD31 induction in our TLE model may be associated with anti-inflammatory and neuroprotection processes.
PDGFRβ is a marker of pericytes that are spatially isolated contractile cells on capillaries that control cerebral blood low and BBB function in physiopathological conditions. Signaling through PDGFRβ regulates pericyte survival, proliferation, and migration. In TLE, we observed increased PDGFRβ expression in hippocampal blood vessels as expected, but also in nervous tissue parenchymal cells, suggestive of rearrangement in PDGFRβ distribution. The morphology and the distribution of these cells in all areas of the hippocampus including in the CC are reminiscent of glial cells. This observation is in agreement with Shen and colleagues [52] who detected PDGFRβ expression in cultured astrocytes isolated from neonatal mouse brain. Note that numerous cells were also stained in the subgranular zone of the DG both in CTL and PILO 3D rats, suggesting that PDGFRβ may be expressed in newly formed granule cells of the DG. Consistent with this observation, PDGFRβ is reported to be expressed in some neurons [52,53,54,55]. Outside the CNS, PDGFRβ protein expression has been proposed as a rescue response to cardiac vascular pathological insult [56]. In agreement with our results, Klement et al. [15] showed PDGFRβ mRNA increase in a mouse model of TLE induced by Kainate. In addition, within the regional scar that is a hallmark of epilepsy, a fibrotic-like PDGFRβ mesh was shown to develop around the capillaries, peaking at early stages post-SE, and regressing, but not resolving during the SRSs. Sakai and colleagues showed that the increased expression of PDGFRβ in the hippocampus after traumatic brain injury may lead to hypersensitivity to PILO in a relevant mouse model [57]. PDGFRβ was proposed as a possible pharmacological target in epilepsy and the PDGFRβ agonist PDGF-BB reduced mural cell loss, vascular pathology and epileptiform activity [15,57,58]. In brain specimens of patients with TLE and hippocampal sclerosis (TLE-HS), increased perivascular PDGFRβ positive pericytes and enlarged and tortuous vessels were observed compared to TLE-non HS. Similarly, brain specimens derived from epileptic subjects affected by intractable seizures associated with focal cortical dysplasia displayed high perivascular PDGFRβ immunoreactivity, typical of pericytes, and revealed ramified PDGFRβ positive cells proximal to microvessels [59]. The decreased expression of PDGFRβ from PILO 14D to PILO 3M may be due to the loss of mural pericytes known to occur in epilepsy [58]. Our results are also reminiscent of the expression of PDGFRβ in brain specimens of patients affected by drug resistant epilepsy where the increased expression and rearrangement of PDGFRβ labeling after SE suggest the involvement of pericytes in cerebrovascular modifications associated with epilepsy [11,60]. PDGFRβ labeling in animal experimental and human epileptic tissue is increased and appears to undergo distribution rearrangements in diseased tissue, associated with microvascular-pericyte-glia changes, scar formation, and inflammation. TNF-a, IL-6 and specially IL-1b, promote such pericyte-related modifications. Notably IL-1b, shown to be deeply involved in the pathogenesis of epilepsy [11] alters pericyte morphology and facilitates the formation of pericyte-microglia aggregates in ex vivo hippocampal slices [15,61]. In our PILO model, we have previously shown that IL-1b is one of the most expressed cytokines [27]. It is thus likely that the changes we observe in hippocampal pericytes are mediated by this cytokine. These changes may also result from pericyte-glia scaring where abnormal ColIII and ColIV accumulation or distribution have been associated with leaky capillaries during seizure progression [15].
ColIV is the most abundant component of the BM of endothelial and epithelial cells. In the brain, ColIV is produced predominantly by brain endothelial cells and pericytes and these cells are separated by a BM. Six ColIV alpha chains (ColIV4a1 to ColIV4a6) have been identified. We studied specifically ColIVa1 and ColIVa3 that are involved in brain vascular integrity [16]. Increased ColIV has been reported in brain blood vessels in mouse, rat and sheep animal models and human stroke tissue [62]. ColIV induction was shown inside fibrotic scars [63], and following spinal cord injury in rats, where it may participate in glial scar formation [64]. An association has also been shown between seizures and deposition of collagen in porcine brain with taenia-solium neurocysticercosis [63]. ColIV was increased in other models of pathology around the microvasculature in rat chronic hypertension [65] and ColIV expression has been shown in brains of TLE patients, albeit outside the vasculature and in meninges [66]. We thus find some discrepancies on vascular vs perivascular ColIV distribution in our results, considering that at the time points we studied, ColIV labeling was predominantly vascular in rat TLE. Interestingly, in epilepsy, collagen has been shown to have migrational properties, both in vitro and in vivo, on cells of the DG layer [67]. Since collagen is involved in scar formation typically associated with experimentally induced epilepsy [68], one can hypothesize that the increased ColIV that we observe in epileptic hippocampi is involved in TLE-associated scar formation, but also in the migration of cells, in particular dentate granule neurons that play a critical role in SRSs.
In conclusion, our data show that in brain diseases such as TLE with obvious reactive glial inflammation, glial scar formation, neuronal network reorganization and BBB dysfunction, several BBB proteins such as CD31, PDGFR and ColIV are increased. Although the RT-qPCR results were obtained in whole hippocampus which includes BBB but also brain parenchyma cells, we found good correlations in the expression levels using semi-quantitative IHC as an alternative approach. Besides protein expression levels, this approach yielded precious additional information at the level of the blood vessels and relative protein distribution in identified cell types, which other methods such as Western Blot or ELISA cannot provide. This study has some limitations. The proteins we studied have been evaluated in other pathological conditions, at different time points and in different species. It is thus difficult at this point to draw a consensus picture of the molecular and cellular processes at stake and to infer whether the observed changes at the molecular and cellular levels are detrimental or beneficial for BBB and brain parenchyma homeostasis. Many more components that control BBB pathophysiological properties need to be studied including components of the neurovascular unit such as extracellular matrix proteins, tight junction associated proteins etc. Some general principles are starting to emerge, raising the hope that a better understanding of BBB dysfunction in CNS diseases will help towards characterization of the mechanisms that control vascular integrity, the basis of BBB dysfunction during epilepsy, and the development of new therapeutic strategies.
4. Materials and Methods
4.1. Ethics Statement
All experimental procedures involving rats were approved by National and European regulations (EU directive N° 2010/63) and were in agreement with the authorization for animal experimentation granted to the laboratory by the Préfecture des Bouches du Rhône (permit number: D 13 055 08) and to the project (N° 00757.02) by the French Ministry of Research and Local Ethics Committee.
4.2. Rat pilocarpine TLE model
Adult male Wistar rats weighing (200-290 g, n= 40, Charles River, France) were housed in an animal facility at 22-25°C under a 12 h light/dark cycle, with free access to food and water. Rats were injected intraperitonally (i.p) with a low dose of the cholinergic antagonist scopolamine methyl nitrate (2 mg/kg; Sigma, Saint Louis, MO, USA) to minimize the peripheral effects of pilocarpine hydrochloride (PILO) (320 mg/kg; Sigma), a muscarinic cholinergic agonist diluted in 0.9% NaCl and administered i.p. 30 min after scopolamine. At 1 h after the onset of SE, animals received diazepam (10 mg/kg, i.p.), and thereafter were carefully monitored to ensure a high survival rate. Only animals that developed sustained SE after PILO injection were included in this study. Only seizures of grade 3 or greater on the Racine [69] scale were scored (i.e., forelimb clonus ± rearing ± falling). During the latent stage (PILO 3D - 14D), the animals recovered from SE and appeared normal regarding food and water intake, weight, and body temperature, while they did not develop seizures. During the chronic stage (PILO 1M and 3M), the animals developed SRSs. The onset was 2-3 weeks for PILO rats as previously described [70]. The frequency and intensity of seizures differed to some extent from one animal to another which is inherent to the model. In general, animals displayed an average of 3-4 seizures per day, similar to previous reports by our laboratory [41] and others [71]. Thirty rats were administered PILO, 8 animals died following cardiac and respiratory failure as previously described [39,40], 3 did not develop SE and 1 did not develop SRSs. Four animals did not reach the qualitative controls after fixation and brain observation (poor fixation or complete hippocampal sclerosis). Hence, a total of 18 PILO animals were included in the study, representing 55 to 60% as reported in other studies [42,43]. Six control rats received an injection of 0.9% NaCl. Each group of PILO-treated rats was compared to saline-treated animals used as CTL. PILO-treated animals were studied at several post-injection intervals during the latent and chronic stages using 2 semi-quantitative approaches to follow the regulation of expression of BBB markers and/or cellular expression and distribution. First, we used RT-qPCR and second we correlated with qualitative and semi-quantitative IHC. RT-qPCR yielded global results at the level of the whole hippocampus, while semi-quantitative IHC allowed us to follow the distribution and the expression intensity of BBB markers at the cellular level of different areas of the hippocampus, according to standard principles and approaches [72,73].
4.3. RNA extraction and RT-qPCR
Hippocampal tissue from CTL and PILO-treated rats were lysed using Qiazol lysis reagent (Qiagen France, Ulis, France) and total RNA was extracted using RNeasy Plus Universal Mini kit (Qiagen France) according to the manufacturer’s protocol. cDNA was synthesized from 500 ng of total RNA using the High-Capacity RNA-to-cDNAc kit (Applied Biosystems, CA, USA). We used 12.5 ng of cDNA for the RT-qPCR experiments that were performed as described in [27]. We used the 7500 Fast Real Time PCR System (Applied Biosystems) and TaqMan Fast Universal PCR Mix (Applied Biosystems) and TaqMan Assays probes (see Table 1) that were validated and purchased from Applied Biosystems. These probes were compliant with MIQE guidelines as they guarantee high specificity, sensibility, reproducibility, and high level of quantitative analysis.
The samples were run in duplicate on 96-well plates and analyzed with 7500 v2.0 software (Applied Biosystems). The conditions of the thermal cycle were as follows: initial denaturation at 95°C for 40 cycles, denaturation at 95°C, and hybridization and extension at 60°C. Relative expression levels were determined according to the ΔΔCt (Ct: cycle threshold) method where the expression level of the mRNA of interest is given by 2-ΔΔCT, where ΔΔCT = ΔCt target mRNA − ΔCt reference mRNA (RPL13, Ribosomal Protein L13, was used as internal mRNA reference) in the same sample.
For the Collagen RT-qPCR experiments, we selected primers that detect the ColIVa1 and ColIVa3 mRNAs encoding the vascular isoforms of ColIV. The results were presented as fold induction compared to control values, considered as being 1, and are presented as means ± SEM from three different rats (n= 6 hippocampi).
4.4. Tissue preparation for immunohistochemistry
Rats were deeply anesthetized with pentobarbital sodium injection (Nembutal, 120 mg/kg i.p., Ceva, France) and perfused through the heart with 0,9% NaCl followed with a fixative paraformaldehyde based solution (Antigenfix, Diapath, Italy). Following induction of SE, animals were subjected to a standard perfusion fixation protocol with NaCl, followed by 4% Antigenfix (paraformaldehyde-based; Diapath, Italy). The brains were removed 30 min later and were postfixed for 24 h at 4°C in the same fixative solution. Finally, brains were rinsed 3 times in Phosphate Buffer (0,12 M; PB, pH 7,2-7,4) and cryoprotected in 30% sucrose solution (Sigma) in phosphate buffer (PB) 0.12 M until fully dehydrated. Brains were frozen in isopentane solution (Sigma) at - 80°C, fixed in O.C.T. Tissue Tek (Sakura Finetek, Torrance, CA, USA) and sectioned coronally at 40 μm with a cryostat. The sections were collected sequentially in wells of culture plates containing an ethylene glycol-based cryoprotective solution and stored at -20°C until processing. Selected sections from each rat covering the dorsal hippocampus (from bregma -3,30 to -4,80 mm) according to the Rat Brain Atlas [74], were used for immunohistochemical staining. Free-floating sections from CTL and PILO rats were always processed in parallel.
4.5. Immunohistochemistry
Free floating sections were double-stained with anti-CD31 (PECAM-1; mouse, Abcam ab64543, 1:100, Cambridge, UK) and either PDGFRβ (rabbit, Abcam ab32570, 1:100) or Collagen IV (ColIV; goat, Southern Biotech 1340-01, 1:250, Birmingham, AL, USA). The ColIV antibody detects ColIVa1 to ColIVa6 (according to SouthernBiotech) and among these isoforms, ColIVa1 and ColIVa3 are expressed in brain vascular tissue (see https://brainrnaseq.org). Sections were permeabilized and saturated with PB 0.12 M containing 3% Bovine Serum Albumin (BSA) and 0.3% Triton X-100 in PB 0.12 M for 1 h at RT -0.3%) Triton X-100 for 1 h at RT. They were stained with the first primary antibody in PB 0.12 M, 3% BSA overnight at 4°C. The next day, sections were washed 3x in PB 0.12 M, stained with the appropriate second primary antibody, washed 3x in PB 0.12 M. The ColIV and CD31 labeling were visualized with a donkey anti-goat AlexaFluor A488 and donkey anti-mouse AlexaFluor A594 (1:800) respectively for 2 h at RT in dark. The PDGFRβ immunolabeling was visualized with a goat anti-rabbit biotin (1:200) for 2 h at RT followed by streptavidin AlexaFluor A488 (1/800). Tissue sections were washed 3x in PB 0.12 M, counterstained with with 5 mg/ml 4′,6-Diamidino-2-phenylindole (DAPI, Sigma) for 30 min at RT in dark. After 3 washes in PB 0.12 M, floating sections were mounted on Superfrost Plus glass slides using Fluoromount-G Mounting medium Thermo Fisher Scientific, Dardilly, France) and stored at -20°C until imaging and analysis. Immunohistochemical controls for double-labeling experiments included incubation of some sections in a mixture of a primary antibody and normal IgG (mouse/goat/rabbit normal IgG). In all cases, these sections exhibited the same pattern of immunolabeling as sections processed for single labeling. Methods are described in detail in [27].
4.6. Confocal microscopy and quantification
All cytochemical quantifications were performed blindly. Acquisitions were performed using a confocal laser-scanning LSM 700 Zeiss microscope with a x20 or x40 oil objective and analysis of immunostaining images was performed using ZEN software (Zeiss, Jena, Germany). The mosaic function was used to visualize the whole hippocampus. Z-stack function was also useful to determine precisely the co-expression of two markers in the same cells. Finally, ImageJ software (NIH) was used to quantify each immunolabeling. Pictures were binarized to 16-bit black and white images and a fixed intensity threshold was applied, defining each staining. Images were obtained with double averaging, frame size 1024x1024, and 1 AU pinhole for each channel. Quantification of blood vessel fluorescence intensity was obtained from at least 15 stained blood vessels of the DG and the LM from 2 sections per animal and on both hippocampi. This fluorescence intensity was measured by drawing and measuring around the inner and outer surface of various sized and shaped vessels and arterioles that were clearly identified morphologically. The mean fluorescence intensity in and around blood vessels was assessed using ImageJ software for the 3 markers as described in [23] from 3 CTL and 3 PILO rats at PILO 3D. Background fluorescence was set in the SR, in areas devoid of brain vessels in the same sections and was subtracted. An average value of 3 such areas was obtained from every image. Data are presented as the average fluorescence intensity of PDGFRβ, CD31, or ColIV, relative to CTL (percentage). Images were processed using Adobe Photoshop software. Using ImageJ, the fluorescence intensity profiles (arbitrary units, A.U.) as a function of distance (pixels) (as described in [75,76,77]), were obtained for PDGFRβ and CD31 to determine whether the different markers stain distinct cell types of the BBB.
4.7. Statistical analysis
Sample sizes and statistical power were determined according to Dell et al. [78], and Festing and Altman [79], using the Biosta TGV sofware (https://biostatgv.sentiweb.fr/?module=etude/sujets#). All experiments were performed on 3 rats per group. One-way ANOVA analysis followed by Dunnett’s post hoc test was used to compare the mean of each group with the mean of a CTL group (as used in [80]). Unpaired Student’s t-test was used to compare 2 independent groups (as used in [81]). All data are expressed as the mean ± SEM. Statistical significance was set at p<0,05. Histograms and statistical analyses were performed with PRISM GraphPad statistical software.
Author Contributions
GK: Conceptualization and design of the study, experimental methodology, acquisition and analysis of data, writing, review & editing. AB and AB: Experimental methodology. MK: Conceptualization and design of the study, writing - review & editing. LF: Conceptualization and design of the study, generation of figures, writing - review & editing.
Funding
This work was supported by a fellowship to GK from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No 642881, and AMIDEX (ICN PhD Program, ANR-11-IDEX-0001-02 grant) funded by the French Government «Investissements d’Avenir» program. Funding from the CNRS and Aix-Marseille Université (AMU) to the Institute of Neurophysiopathology (INP), UMR7051 is acknowledged.
Institutional Review Board Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Acknowledgments
We thank Dr. Mourad Mekaouche for advice and help with animal care.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
References
- Baulac, M.; de Boer, H.; Elger, C.; Glynn, M.; Kälviäinen, R.; Little, A.; Mifsud, J.; Perucca, E.; Pitkänen, A.; Ryvlin, P. Epilepsy Priorities in Europe: A Report of the ILAE-IBE Epilepsy Advocacy Europe Task Force. Epilepsia 2015, 56, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Ayalew, M.B.; Muche, E.A. Patient Reported Adverse Events among Epileptic Patients Taking Antiepileptic Drugs. SAGE Open Med. 2018, 6, 2050312118772471. [Google Scholar] [CrossRef] [PubMed]
- Ferhat, L. Potential Role of Drebrin a, an f-Actin Binding Protein, in Reactive Synaptic Plasticity after Pilocarpine-Induced Seizures: Functional Implications in Epilepsy. Int. J. Cell Biol. 2012, 2012, 474351. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Kuruba, R. Experimental Models of Status Epilepticus and Neuronal Injury for Evaluation of Therapeutic Interventions. Int. J. Mol. Sci. 2013, 14, 18284–18318. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Lucchi, C.; Vinet, J.; Gualtieri, F.; Marinelli, C.; Torsello, A.; Costantino, L.; Biagini, G. Pathophysiogenesis of Mesial Temporal Lobe Epilepsy: Is Prevention of Damage Antiepileptogenic? Curr. Med. Chem. 2014, 21, 663–688. [Google Scholar] [CrossRef] [PubMed]
- Weissberg, I.; Reichert, A.; Heinemann, U.; Friedman, A. Blood-Brain Barrier Dysfunction in Epileptogenesis of the Temporal Lobe. Epilepsy Res Treat. 2011, 143908. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and Function of the Blood-Brain Barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Privratsky, J.R.; Newman, D.K.; Newman, P.J. PECAM-1: Conflicts of Interest in Inflammation. Life Sci. 2010, 87, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lindenmayer, A.E.; Chaubal, A. Endothelial Cell Markers CD31, CD34, and BNH9 Antibody to H- and Y-Antigens--Evaluation of Their Specificity and Sensitivity in the Diagnosis of Vascular Tumors and Comparison with von Willebrand Factor. Mod. Pathol. 1994, 7, 82–90. [Google Scholar]
- Mbagwu, S.I.; Filgueira, L. Differential Expression of CD31 and Von Willebrand Factor on Endothelial Cells in Different Regions of the Human Brain: Potential Implications for Cerebral Malaria Pathogenesis. Brain Sci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Yamanaka, G.; Takata, F.; Kataoka, Y.; Kanou, K.; Morichi, S.; Dohgu, S.; Kawashima, H. The Neuroinflammatory Role of Pericytes in Epilepsy. Biomedicines 2021, 9, 759. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central Nervous System Pericytes in Health and Disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes Regulate the Blood-Brain Barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.O.; Kim, H.; Holl, D.; Werne Solnestam, B.; Lundeberg, J.; Carlén, M.; Göritz, C.; Frisén, J. Reducing Pericyte-Derived Scarring Promotes Recovery after Spinal Cord Injury. Cell 2018, 173, 153–165.e22. [Google Scholar] [CrossRef] [PubMed]
- Klement, W.; Blaquiere, M.; Zub, E.; deBock, F.; Boux, F.; Barbier, E.; Audinat, E.; Lerner-Natoli, M.; Marchi, N. A Pericyte-Glia Scarring Develops at the Leaky Capillaries in the Hippocampus during Seizure Activity. Epilepsia 2019, 60, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Liu, F.; Xu, W.; Liu, J.; Chen, S.; Wu, G. Transplanting GABAergic Neurons Differentiated from Neural Stem Cells into Hippocampus Inhibits Seizures and Epileptiform Discharges in Pilocarpine-Induced Temporal Lobe Epilepsy Model. World Neurosurg. 2019, 128, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.; Dingledine, R.; Aronica, E.; Bernard, C.; Blümcke, I.; Boison, D.; Brodie, M.J.; Brooks-Kayal, A.R.; Engel, J.; Forcelli, P.A.; et al. Commonalities in Epileptogenic Processes from Different Acute Brain Insults: Do They Translate? Epilepsia 2018, 59, 37–66. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.A.; Otte, W.M.; Gorter, J.A.; Dijkhuizen, R.M.; Wadman, W.J. Longitudinal Assessment of Blood-Brain Barrier Leakage during Epileptogenesis in Rats. A Quantitative MRI Study. Neurobiol. Dis. 2014, 63, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Gorter, J.A.; van Vliet, E.A.; Aronica, E. Status Epilepticus, Blood-Brain Barrier Disruption, Inflammation, and Epileptogenesis. Epilepsy Behav. 2015, 49, 13–16. [Google Scholar] [CrossRef]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like Receptor 4 and High-Mobility Group Box-1 Are Involved in Ictogenesis and Can Be Targeted to Reduce Seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional Morphology of the Blood-Brain Barrier in Health and Disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.A.; da Costa Araújo, S.; Redeker, S.; van Schaik, R.; Aronica, E.; Gorter, J.A. Blood-Brain Barrier Leakage May Lead to Progression of Temporal Lobe Epilepsy. Brain J. Neurol. 2007, 130, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, O.; Shelef, I.; Kaizerman, I.; Eliushin, A.; Afawi, Z.; Misk, A.; Gidon, M.; Cohen, A.; Zumsteg, D.; Friedman, A. Blood-Brain Barrier Disruption in Post-Traumatic Epilepsy. J. Neurol. Neurosurg. Psychiatry 2008, 79, 774–777. [Google Scholar] [CrossRef] [PubMed]
- Raabe, A.; Schmitz, A.K.; Pernhorst, K.; Grote, A.; von der Brelie, C.; Urbach, H.; Friedman, A.; Becker, A.J.; Elger, C.E.; Niehusmann, P. Cliniconeuropathologic Correlations Show Astroglial Albumin Storage as a Common Factor in Epileptogenic Vascular Lesions. Epilepsia 2012, 53, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron-Glia Interactions in the Pathophysiology of Epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef]
- Shen, W.; Pristov, J.B.; Nobili, P.; Nikolić, L. Can Glial Cells Save Neurons in Epilepsy? Neural Regen. Res. 2023, 18, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- Kyriatzis, G.; Bernard, A.; Bôle, A.; Pflieger, G.; Chalas, P.; Masse, M.; Lécorché, P.; Jacquot, G.; Ferhat, L.; Khrestchatisky, M. Neurotensin Receptor 2 Is Induced in Astrocytes and Brain Endothelial Cells in Relation to Neuroinflammation Following Pilocarpine-Induced Seizures in Rats. Glia 2021, 69, 2618–2643. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kastner, R.; Ingvar, M. Loss of Immunoreactivity for Glial Fibrillary Acidic Protein (GFAP) in Astrocytes as a Marker for Profound Tissue Damage in Substantia Nigra and Basal Cortical Areas after Status Epilepticus Induced by Pilocarpine in Rat. Glia 1994, 12, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Reams, R.Y.; Jordan, W.H.; Miller, M.A.; Thacker, H.L.; Snyder, P.W. Mesial Temporal Lobe Epilepsy: Pathogenesis, Induced Rodent Models and Lesions. Toxicol. Pathol. 2007, 35, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Knake, S.; Hamer, H.M.; Rosenow, F. Status Epilepticus: A Critical Review. Epilepsy Behav. 2009, 15, 10–14. [Google Scholar] [CrossRef]
- Papageorgiou, I.E.; Gabriel, S.; Fetani, A.F.; Kann, O.; Heinemann, U. Redistribution of Astrocytic Glutamine Synthetase in the Hippocampus of Chronic Epileptic Rats. Glia 2011, 59, 1706–1718. [Google Scholar] [CrossRef] [PubMed]
- Uzunalli, G.; Dieterly, A.M.; Kemet, C.M.; Weng, H.-Y.; Soepriatna, A.H.; Goergen, C.J.; Shinde, A.B.; Wendt, M.K.; Lyle, L.T. Dynamic Transition of the Blood-Brain Barrier in the Development of Non-Small Cell Lung Cancer Brain Metastases. Oncotarget 2019, 10, 6334–6348. [Google Scholar] [CrossRef] [PubMed]
- Mendes, N.F.; Pansani, A.P.; Carmanhães, E.R.F.; Tange, P.; Meireles, J.V.; Ochikubo, M.; Chagas, J.R.; da Silva, A.V.; Monteiro de Castro, G.; Le Sueur-Maluf, L. The Blood-Brain Barrier Breakdown During Acute Phase of the Pilocarpine Model of Epilepsy Is Dynamic and Time-Dependent. Front. Neurol. 2019, 10, 382. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhou, Y.; Chen, M.; Li, L.; Zhang, W. Pericytes for Therapeutic Approaches to Ischemic Stroke. Front. Neurosci. 2021, 15, 629297. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kastner, R.; Ingvar, M. Laminar Damage of Neurons and Astrocytes in Neocortex and Hippocampus of Rat after Long-Lasting Status Epilepticus Induced by Pilocarpine. Epilepsy Res. Suppl. 1996, 12, 309–316. [Google Scholar] [PubMed]
- Massey, C.A.; Sowers, L.P.; Dlouhy, B.J.; Richerson, G.B. Mechanisms of Sudden Unexpected Death in Epilepsy: The Pathway to Prevention. Nat. Rev. Neurol. 2014, 10, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Teran, F.A.; Bravo, E.; Richerson, G.B. Chapter 8 - Sudden Unexpected Death in Epilepsy: Respiratory Mechanisms. In Handbook of Clinical Neurology; Chen, R., Guyenet, P.G., Eds.; Respiratory Neurobiology: Physiology and Clinical Disorders, Part II; Elsevier, 2022; Vol. 189, pp. 153–176. [CrossRef]
- Olesen, J.B.; Abildstrøm, S.Z.; Erdal, J.; Gislason, G.H.; Weeke, P.; Andersson, C.; Torp-Pedersen, C.; Hansen, P.R. Effects of Epilepsy and Selected Antiepileptic Drugs on Risk of Myocardial Infarction, Stroke, and Death in Patients with or without Previous Stroke: A Nationwide Cohort Study. Pharmacoepidemiol. Drug Saf. 2011, 20, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.M.; Fulford, R.A. Pilocarpine-Induced Convulsions and Delayed Psychotic-like Reaction. Can. J. Biochem. Physiol. 1961, 39, 1287–1294. [Google Scholar] [CrossRef]
- Kouchi, H.; Ogier, M.; Dieuset, G.; Morales, A.; Georges, B.; Rouanet, J.-L.; Martin, B.; Ryvlin, P.; Rheims, S.; Bezin, L. Respiratory Dysfunction in Two Rodent Models of Chronic Epilepsy and Acute Seizures and Its Link with the Brainstem Serotonin System. Sci. Rep. 2022, 12, 10248. [Google Scholar] [CrossRef]
- Sbai, O.; Soussi, R.; Bole, A.; Khrestchatisky, M.; Esclapez, M.; Ferhat, L. The Actin Binding Protein α-Actinin-2 Expression Is Associated with Dendritic Spine Plasticity and Migrating Granule Cells in the Rat Dentate Gyrus Following Pilocarpine-Induced Seizures. Exp. Neurol. 2021, 335, 113512. [Google Scholar] [CrossRef]
- Esclapez, M.; Hirsch, J.C.; Ben-Ari, Y.; Bernard, C. Newly Formed Excitatory Pathways Provide a Substrate for Hyperexcitability in Experimental Temporal Lobe Epilepsy. J. Comp. Neurol. 1999, 408, 449–460. [Google Scholar] [CrossRef]
- Goffin, K.; Nissinen, J.; Van Laere, K.; Pitkänen, A. Cyclicity of Spontaneous Recurrent Seizures in Pilocarpine Model of Temporal Lobe Epilepsy in Rat. Exp. Neurol. 2007, 205, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Garzillo, C.; Mello, L. Characterization of Reactive Astrocytes in the Chronic Phase of the Pilocarpine Model of Epilepsy. Epilepsia 2002, 43, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Koh, S. Role of Brain Inflammation in Epileptogenesis. Yonsei Med. J. 2008, 49, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.A.; Wang, L.; Ribak, C.E. Rapid Astrocyte and Microglial Activation Following Pilocarpine-Induced Seizures in Rats. Epilepsia 2008, 49 Suppl 2, 33–41. [Google Scholar] [CrossRef]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial Functions of Platelet/Endothelial Cell Adhesion Molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.C.; Xu, P.; Gao, M.; Wang, J.; Jiang, D.; Zhu, X.; Won, M.-H.; Su, P.Q. Changes in the Blood-Brain Barrier Function Are Associated With Hippocampal Neuron Death in a Kainic Acid Mouse Model of Epilepsy. Front. Neurol. 2018, 9, 775. [Google Scholar] [CrossRef]
- Rana, A.; Musto, A.E. The Role of Inflammation in the Development of Epilepsy. J. Neuroinflammation 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.C.; Zhao, R.W.; Ueno, K.; Hickey, W.F. PECAM-1 (CD31) Expression in the Central Nervous System and Its Role in Experimental Allergic Encephalomyelitis in the Rat. J. Neurosci. Res. 1996, 45, 747–757. [Google Scholar] [CrossRef]
- Cheung, K.; Ma, L.; Wang, G.; Coe, D.; Ferro, R.; Falasca, M.; Buckley, C.D.; Mauro, C.; Marelli-Berg, F.M. CD31 Signals Confer Immune Privilege to the Vascular Endothelium. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E5815–5824. [Google Scholar] [CrossRef]
- Shen, J.; Ishii, Y.; Xu, G.; Dang, T.C.; Hamashima, T.; Matsushima, T.; Yamamoto, S.; Hattori, Y.; Takatsuru, Y.; Nabekura, J.; et al. PDGFR-β as a Positive Regulator of Tissue Repair in a Mouse Model of Focal Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2012, 32, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Iihara, K.; Sasahara, M.; Hashimoto, N.; Hazama, F. Induction of Platelet-Derived Growth Factor Beta-Receptor in Focal Ischemia of Rat Brain. J. Cereb. Blood Flow Metab. 1996, 16, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, J.; Issa, R.; Bujny, T.; Slevin, M.; Kumar, P.; Kumar, S.; Kaluza, J. A Putative Role for Platelet-Derived Growth Factor in Angiogenesis and Neuroprotection after Ischemic Stroke in Humans. Stroke 1997, 28, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Arimura, K.; Ago, T.; Kamouchi, M.; Nakamura, K.; Ishitsuka, K.; Kuroda, J.; Sugimori, H.; Ooboshi, H.; Sasaki, T.; Kitazono, T. PDGF Receptor β Signaling in Pericytes Following Ischemic Brain Injury. Curr. Neurovasc. Res. 2012, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chintalgattu, V.; Ai, D.; Langley, R.R.; Zhang, J.; Bankson, J.A.; Shih, T.L.; Reddy, A.K.; Coombes, K.R.; Daher, I.N.; Pati, S.; et al. Cardiomyocyte PDGFR-Beta Signaling Is an Essential Component of the Mouse Cardiac Response to Load-Induced Stress. J. Clin. Invest. 2010, 120, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Takata, F.; Yamanaka, G.; Yasunaga, M.; Hashiguchi, K.; Tominaga, K.; Itoh, K.; Kataoka, Y.; Yamauchi, A.; Dohgu, S. Reactive Pericytes in Early Phase Are Involved in Glial Activation and Late-Onset Hypersusceptibility to Pilocarpine-Induced Seizures in Traumatic Brain Injury Model Mice. J. Pharmacol. Sci. 2021, 145, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Arango-Lievano, M.; Boussadia, B.; De Terdonck, L.D.T.; Gault, C.; Fontanaud, P.; Lafont, C.; Mollard, P.; Marchi, N.; Jeanneteau, F. Topographic Reorganization of Cerebrovascular Mural Cells under Seizure Conditions. Cell Rep. 2018, 23, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Garbelli, R.; de Bock, F.; Medici, V.; Rousset, M.C.; Villani, F.; Boussadia, B.; Arango-Lievano, M.; Jeanneteau, F.; Daneman, R.; Bartolomei, F.; et al. PDGFRβ(+) Cells in Human and Experimental Neuro-Vascular Dysplasia and Seizures. Neuroscience 2015, 306, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Milesi, S.; Boussadia, B.; Plaud, C.; Catteau, M.; Rousset, M.-C.; De Bock, F.; Scheffer, M.; Lerner-Natoli, M.; Rigau, V.; Marchi, N. Redistribution of PDGFRβ Cells and NG2DsRed Pericytes at the Cerebrovasculature after Status Epilepticus. Neurobiol. Dis. 2014, 0, 151–158. [Google Scholar] [CrossRef]
- Klement, W.; Garbelli, R.; Zub, E.; Rossini, L.; Tassi, L.; Girard, B.; Blaquiere, M.; Bertaso, F.; Perroy, J.; de Bock, F.; et al. Seizure Progression and Inflammatory Mediators Promote Pericytosis and Pericyte-Microglia Clustering at the Cerebrovasculature. Neurobiol. Dis. 2018, 113, 70–81. [Google Scholar] [CrossRef]
- Michalski, D.; Spielvogel, E.; Puchta, J.; Reimann, W.; Barthel, H.; Nitzsche, B.; Mages, B.; Jäger, C.; Martens, H.; Horn, A.K.E.; et al. Increased Immunosignals of Collagen IV and Fibronectin Indicate Ischemic Consequences for the Neurovascular Matrix Adhesion Zone in Various Animal Models and Human Stroke Tissue. Front. Physiol. 2020, 11, 575598. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.M.; Trevisan, C.; Leifsson, P.S.; Johansen, M.V. The Association between Seizures and Deposition of Collagen in the Brain in Porcine Taenia Solium Neurocysticercosis. Vet. Parasitol. 2016, 228, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Liesi, P.; Kauppila, T. Induction of Type IV Collagen and Other Basement-Membrane-Associated Proteins after Spinal Cord Injury of the Adult Rat May Participate in Formation of the Glial Scar. Exp. Neurol. 2002, 173, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Özkan, E.; Çetin-Taş, Y.; Şekerdağ, E.; Kızılırmak, A.B.; Taş, A.; Yıldız, E.; Yapıcı-Eser, H.; Karahüseyinoğlu, S.; Zeybel, M.; Gürsoy-Özdemir, Y. Blood-Brain Barrier Leakage and Perivascular Collagen Accumulation Precede Microvessel Rarefaction and Memory Impairment in a Chronic Hypertension Animal Model. Metab. Brain Dis. 2021, 36, 2553–2566. [Google Scholar] [CrossRef] [PubMed]
- Veznedaroglu, E.; Van Bockstaele, E.J.; O’Connor, M.J. Extravascular Collagen in the Human Epileptic Brain: A Potential Substrate for Aberrant Cell Migration in Cases of Temporal Lobe Epilepsy. J. Neurosurg. 2002, 97, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, D.H.; Arsenault, L. Dentate Granule Cell Layer Collagen Explant Cultures: Spontaneous Axonal Growth and Induction by Brain-Derived Neurotrophic Factor or Basic Fibroblast Growth Factor. Neuroscience 1996, 74, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Hoeppner, T.J.; Morrell, F. Control of Scar Formation in Experimentally Induced Epilepsy. Exp. Neurol. 1986, 94, 519–536. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of Seizure Activity by Electrical Stimulation. II. Motor Seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Soussi, R.; Boulland, J.-L.; Bassot, E.; Bras, H.; Coulon, P.; Chaudhry, F.A.; Storm-Mathisen, J.; Ferhat, L.; Esclapez, M. Reorganization of Supramammillary-Hippocampal Pathways in the Rat Pilocarpine Model of Temporal Lobe Epilepsy: Evidence for Axon Terminal Sprouting. Brain Struct. Funct. 2015, 220, 2449–2468. [Google Scholar] [CrossRef]
- Bajorat, R.; Wilde, M.; Sellmann, T.; Kirschstein, T.; Köhling, R. Seizure Frequency in Pilocarpine-Treated Rats Is Independent of Circadian Rhythm. Epilepsia 2011, 52, e118–122. [Google Scholar] [CrossRef]
- Meyerholz, DK.; Beck, AP. Principles and approaches for reproducible scoring of tissue stains in research. Lab Invest. 2018 Jul;98(7):844-855. Epub 2018 May 30. [CrossRef] [PubMed]
- Kim, SW.; Roh, J.; Park, CS. Immunohistochemistry for Pathologists: Protocols, Pitfalls, and Tips. J Pathol Transl Med. 2016 Nov;50(6):411-418. Epub 2016 Oct 13. PMCID: PMC5122731. [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 4th Edition. Academic Press, Inc., San Diego, USA.; 1998, ISBN 0125476183.
- Sbai, O.; Ould-Yahoui, A.; Ferhat, L.; Gueye, Y.; Bernard, A.; Charrat, E.; Mehanna, A.; Risso, J.-J.; Chauvin, J.-P.; Fenouillet, E.; et al. Differential Vesicular Distribution and Trafficking of MMP-2, MMP-9, and Their Inhibitors in Astrocytes. Glia 2010, 58, 344–366. [Google Scholar] [CrossRef] [PubMed]
- Pagano, A.; Breuzard, G.; Parat, F.; Tchoghandjian, A.; Figarella-Branger, D.; De Bessa, T.C.; Garrouste, F.; Douence, A.; Barbier, P.; Kovacic, H. Tau Regulates Glioblastoma Progression, 3D Cell Organization, Growth and Migration via the PI3K-AKT Axis. Cancers 2021, 13, 5818. [Google Scholar] [CrossRef] [PubMed]
- Silvani, G.; Romanov, V.; Cox, C.D.; Martinac, B. Biomechanical Characterization of Endothelial Cells Exposed to Shear Stress Using Acoustic Force Spectroscopy. Front. Bioeng. Biotechnol. 2021, 9, 612151. [Google Scholar] [CrossRef] [PubMed]
- Dell, R.B.; Holleran, S.; Ramakrishnan, R. Sample Size Determination. ILAR J. 2002, 43, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Festing, M.F.W.; Altman, D.G. Guidelines for the Design and Statistical Analysis of Experiments Using Laboratory Animals. ILAR J. 2002, 43, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Sarodaya, N.; Karapurkar, J.K.; Suresh, B.; Jo, J.K.; Singh, V.; Bae, Y.S.; Kim, K.-S.; Ramakrishna, S. CYLD Destabilizes NoxO1 Protein by Promoting Ubiquitination and Regulates Prostate Cancer Progression. Cancer Lett. 2022, 525, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Sharvit, E.; Abramovitch, S.; Reif, S.; Bruck, R. Amplified Inhibition of Stellate Cell Activation Pathways by PPAR-γ, RAR and RXR Agonists. PloS One 2013, 8, e76541. [Google Scholar] [CrossRef]
Figure 1.
Histograms showing RT-qPCR quantification of the mean levels of mRNAs encoding the reactive glia markers GFAP (A), Iba1 (B) and the vascular markers CD31 (C), PDGFRβ (D), ColIVa1 (E) and ColIVa3 (F) at different time points after PILO-SE. GFAP and Iba1 mRNA were increased at early time points (latent phase, PILO 3D - 14D) and decreased at the chronic phase (PILO 1M and 3M). The expression of CD31, PDGFRβ and ColIVa1 mRNA follows the same trend as the glial markers. ColIVa3 mRNA levels were unchanged at early time points after PILO-SE but were decreased in the chronic phase at PILO 1M. Values are given as the mean ± SEM normalized to CTL. Asterisks indicate statistically significant differences: * p<0.05, ** p<0.01, *** p<0.001 (one-way Anova followed by Dunnett’s post hoc test); ns: not significant; in each histogram, we compared the means of controls to the means of each experimental condition; n=3 rats for each time point.
Figure 1.
Histograms showing RT-qPCR quantification of the mean levels of mRNAs encoding the reactive glia markers GFAP (A), Iba1 (B) and the vascular markers CD31 (C), PDGFRβ (D), ColIVa1 (E) and ColIVa3 (F) at different time points after PILO-SE. GFAP and Iba1 mRNA were increased at early time points (latent phase, PILO 3D - 14D) and decreased at the chronic phase (PILO 1M and 3M). The expression of CD31, PDGFRβ and ColIVa1 mRNA follows the same trend as the glial markers. ColIVa3 mRNA levels were unchanged at early time points after PILO-SE but were decreased in the chronic phase at PILO 1M. Values are given as the mean ± SEM normalized to CTL. Asterisks indicate statistically significant differences: * p<0.05, ** p<0.01, *** p<0.001 (one-way Anova followed by Dunnett’s post hoc test); ns: not significant; in each histogram, we compared the means of controls to the means of each experimental condition; n=3 rats for each time point.

Figure 2.
PDGFRβ, CD31 and ColIV protein expression increased in rat hippocampus following PILO-SE. A-J: PDGFRβ (green) and CD31 (red) double immunolabeling was performed in CTL (A-C) and PILO 3D (F-H). Similarly, ColIV (green) immunolabeling was performed in CTL (D,E) and PILO 3D rats (I-J). Cell nuclei were counterstained with DAPI (blue, C,E,H,J). CC, corpus callosum; O, stratum oriens; P, pyramidal neurons of CA1, CA2, and CA3; R, stratum radiatum; LM, stratum lacunosum-moleculare; M, molecular layer; DG, dentate gyrus; G, granule cell layer of the DG; H, hilus of the DG. PDGFRβ, CD31 and ColIV are markers of pericytes, endothelial cells and BM respectively. PDGFRβ staining was detected in the hippocampus of CTL rats (A,C) and increased in PILO 3D rats, particularly in and around blood vessels (F,H). CD31 staining was concentrated in blood vessels of the LM (B,C) in CTL animals and increased significantly in LM blood vessels as well as throughout O, P and R in PILO 3D rats (G,H). Weak ColIV staining was observed in the rat CTL hippocampus, in particular in blood vessels of the LM (D,E), whereas prominent labeling of the vasculature was noted in PILO 3D rats, in O, P, R, LM, M and DG (I,J). Scale bars: 250 μm. K: Quantification of increased PDGFRβ, CD31 and ColIV proteins in rat blood vessels following PILO-SE. Histograms showing average percentage of the fluorescence intensity of these 3 markers in DG and LM vessels in CTL and PILO 3D rats (K). Blood vessels of PILO 3D rats expressed significant PDGFRβ, CD31 and ColIV levels compared to CTL vessels. PDGFRβ, CD31 and CollV levels were increased compared to CTL. Values are given as the mean ± SEM as a percentage of CTL. Asterisks indicate statistically significant differences: *** p<0.001 (Student’s t-test).
Figure 2.
PDGFRβ, CD31 and ColIV protein expression increased in rat hippocampus following PILO-SE. A-J: PDGFRβ (green) and CD31 (red) double immunolabeling was performed in CTL (A-C) and PILO 3D (F-H). Similarly, ColIV (green) immunolabeling was performed in CTL (D,E) and PILO 3D rats (I-J). Cell nuclei were counterstained with DAPI (blue, C,E,H,J). CC, corpus callosum; O, stratum oriens; P, pyramidal neurons of CA1, CA2, and CA3; R, stratum radiatum; LM, stratum lacunosum-moleculare; M, molecular layer; DG, dentate gyrus; G, granule cell layer of the DG; H, hilus of the DG. PDGFRβ, CD31 and ColIV are markers of pericytes, endothelial cells and BM respectively. PDGFRβ staining was detected in the hippocampus of CTL rats (A,C) and increased in PILO 3D rats, particularly in and around blood vessels (F,H). CD31 staining was concentrated in blood vessels of the LM (B,C) in CTL animals and increased significantly in LM blood vessels as well as throughout O, P and R in PILO 3D rats (G,H). Weak ColIV staining was observed in the rat CTL hippocampus, in particular in blood vessels of the LM (D,E), whereas prominent labeling of the vasculature was noted in PILO 3D rats, in O, P, R, LM, M and DG (I,J). Scale bars: 250 μm. K: Quantification of increased PDGFRβ, CD31 and ColIV proteins in rat blood vessels following PILO-SE. Histograms showing average percentage of the fluorescence intensity of these 3 markers in DG and LM vessels in CTL and PILO 3D rats (K). Blood vessels of PILO 3D rats expressed significant PDGFRβ, CD31 and ColIV levels compared to CTL vessels. PDGFRβ, CD31 and CollV levels were increased compared to CTL. Values are given as the mean ± SEM as a percentage of CTL. Asterisks indicate statistically significant differences: *** p<0.001 (Student’s t-test).
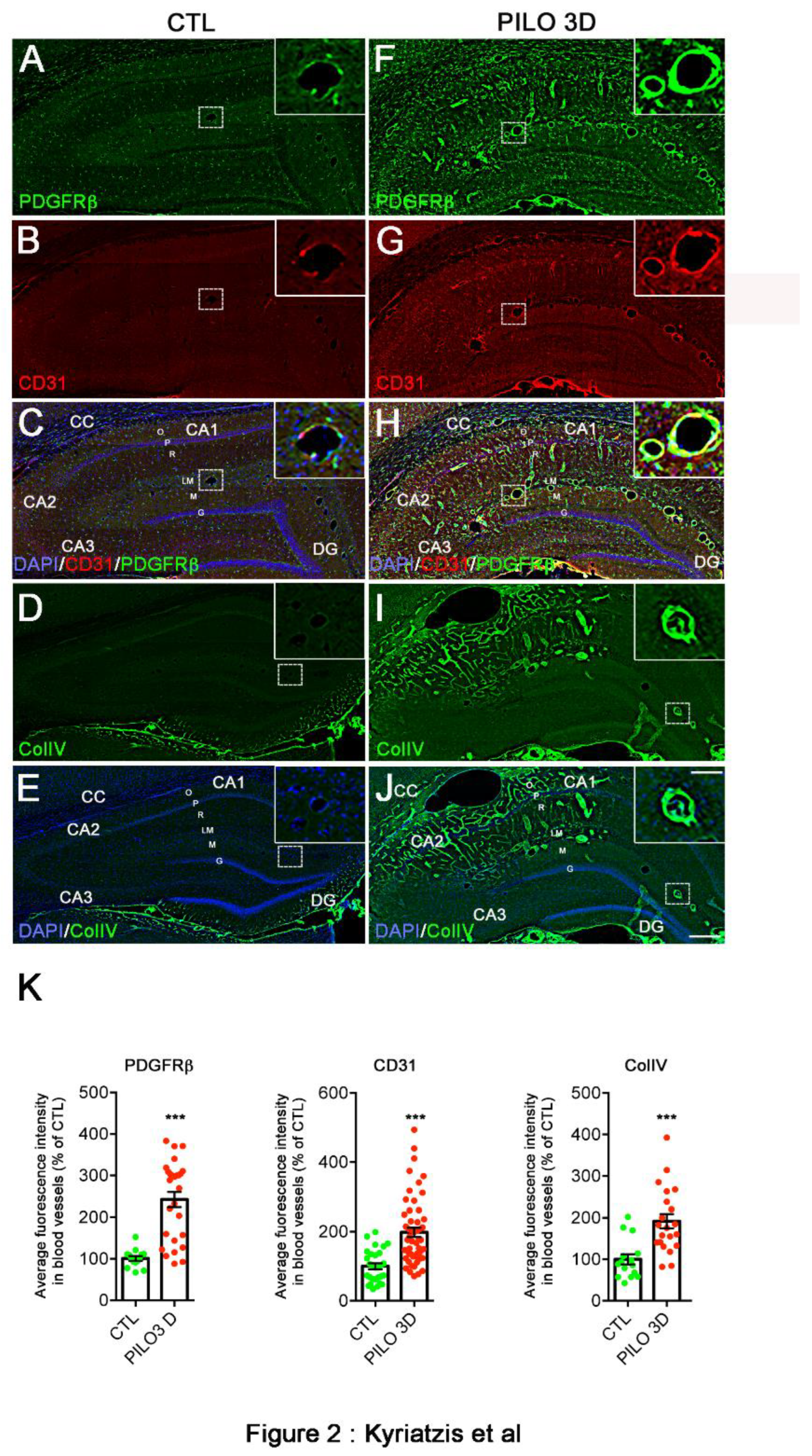
Figure 3.
Double immunostaining followed by high magnification of LM blood vessels showed higher immunolabeling of CD31 (red) and ColIV (green) in PILO 3D rats (G-L) compared to CTL animals (A-F). Increased CD31 and ColIV occurred essentially within endothelial cells (red, arrows), and the basal lamina (green) respectively, as illustrated by higher magnification of the boxed-in area in L (M-R). Cell nuclei were counterstained with DAPI (blue). Scale bars: 10 μm in A-L and 5 μm in M-R.
Figure 3.
Double immunostaining followed by high magnification of LM blood vessels showed higher immunolabeling of CD31 (red) and ColIV (green) in PILO 3D rats (G-L) compared to CTL animals (A-F). Increased CD31 and ColIV occurred essentially within endothelial cells (red, arrows), and the basal lamina (green) respectively, as illustrated by higher magnification of the boxed-in area in L (M-R). Cell nuclei were counterstained with DAPI (blue). Scale bars: 10 μm in A-L and 5 μm in M-R.
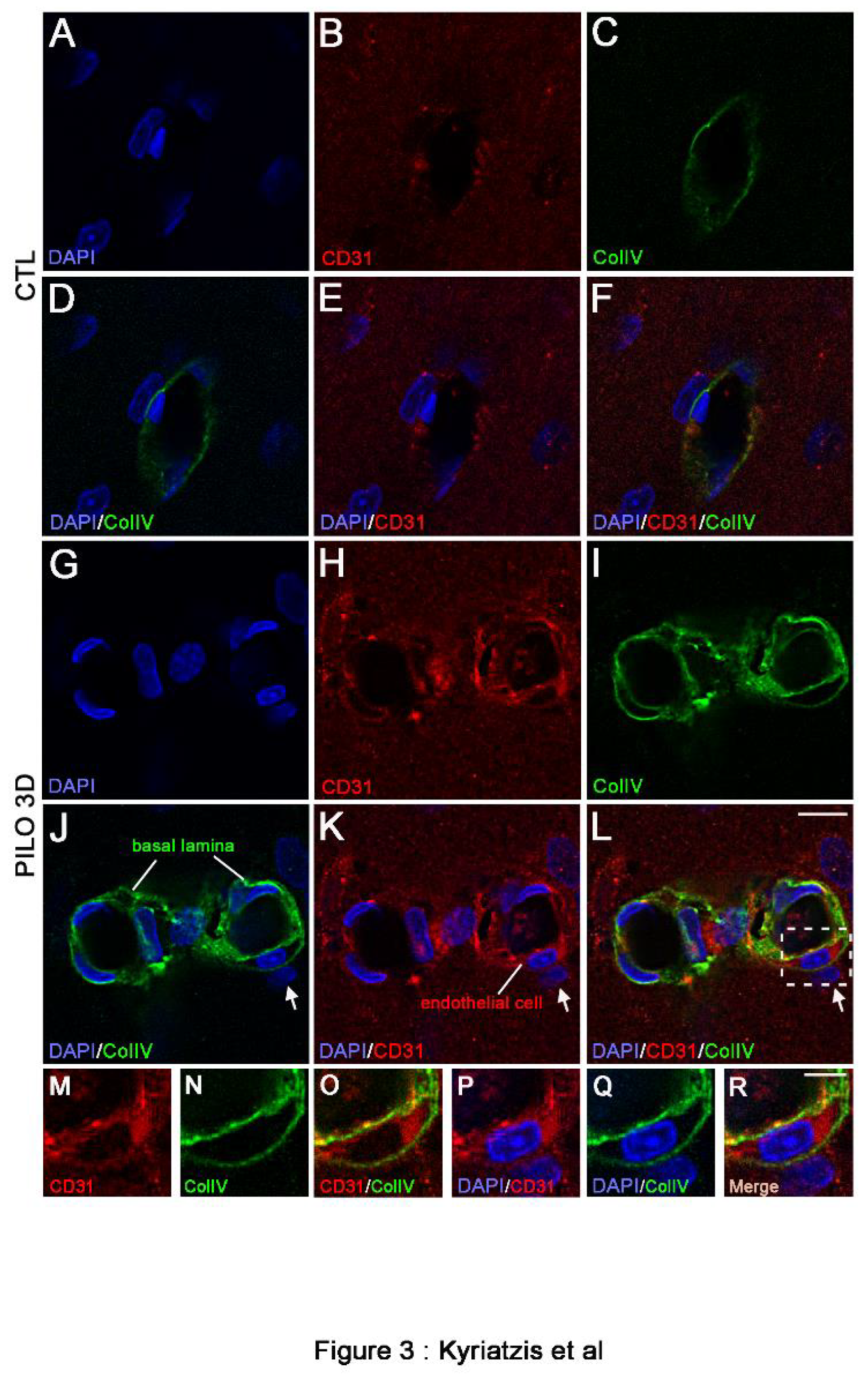
Figure 4.
Double immunostaining followed by high magnification of LM blood vessels showed higher immunolabeling of CD31 (red) and PDGFRβ (green) in PILO 3D rats (G-L) compared to CTL animals (A-F). Increased CD31 and PDGFRβ occurred essentially within endothelial cells (red), and pericytes (green) respectively, as illustrated by higher magnification of the boxed-in area in L (M-R). Cell nuclei were counterstained with DAPI (blue). Scale bars: 10 μm in A-L and 5 μm in M-R. White lines in M and R insets are representative scans across the cell-cell borders. Fluorescence intensity profiles (arbitrary units, A.U.) of CD31 and PDGFRβ, in relation to distance (pixels), were obtained to determine whether these markers indeed stained endothelial (red line) and pericytes (green line) respectively.
Figure 4.
Double immunostaining followed by high magnification of LM blood vessels showed higher immunolabeling of CD31 (red) and PDGFRβ (green) in PILO 3D rats (G-L) compared to CTL animals (A-F). Increased CD31 and PDGFRβ occurred essentially within endothelial cells (red), and pericytes (green) respectively, as illustrated by higher magnification of the boxed-in area in L (M-R). Cell nuclei were counterstained with DAPI (blue). Scale bars: 10 μm in A-L and 5 μm in M-R. White lines in M and R insets are representative scans across the cell-cell borders. Fluorescence intensity profiles (arbitrary units, A.U.) of CD31 and PDGFRβ, in relation to distance (pixels), were obtained to determine whether these markers indeed stained endothelial (red line) and pericytes (green line) respectively.
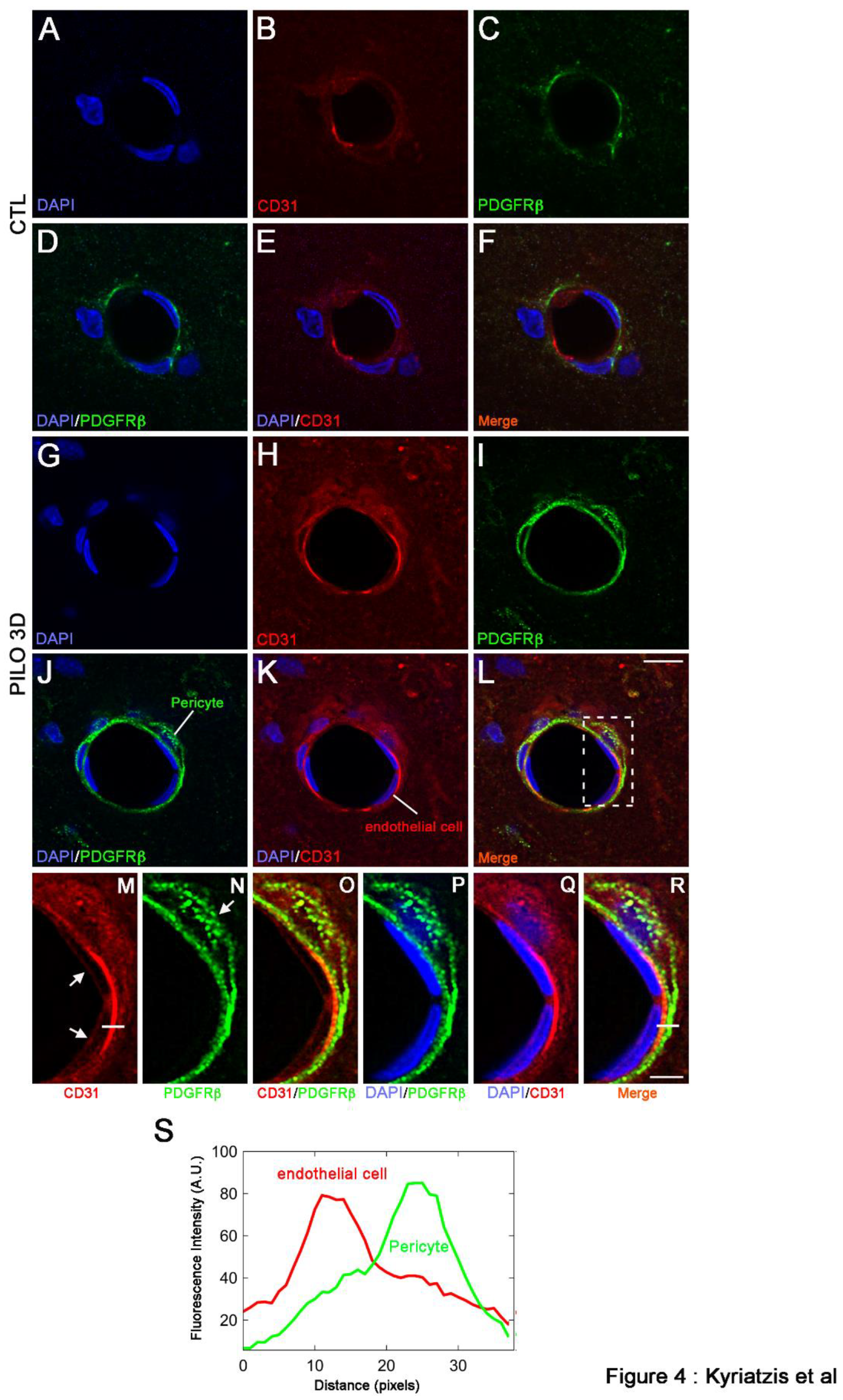

Table 1.
Rat TaqMan probes used for qPCR analysis.
| Gene name | Gene description | Probe ID |
| Gfap | Glial fibrillary acid protein | Rn01253033 |
| Iba1 | Ionized calcium binding adaptor molecule1 | Rn00574125 |
| CD31 or PECAM-1 | EndoCAM or Platelet endothelial cell adhesion molecule-1 | Rn01467262 |
| PDGFRβ | Platelet-derived growth factor beta | Rn01502596 |
| ColIV a1 | Collagen, type IV, a1 | Rn01482927 |
| ColIV a3 | Collagen, type IV, a3 | Rn01400991 |
| RPL13 | Ribosomal Protein L13 | Rn00821258 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



