
Submitted:
29 September 2023
Posted:
03 October 2023
You are already at the latest version
Abstract
Amid global energy challenges, hydrogen evolution reaction (HER) is gaining traction for green hydrogen production. While catalyst research is ongoing, recognizing electrolyte effects remains crucial for sustainable hydrogen production via renewable-powered water electrolysis. This review delves into the intricate effects of electrolytes on the kinetics of HER. It examines key factors, including pH, cations, anions, impurities, and electrolyte concentration. It is discussed that electrolyte pH alters catalyst-electrolyte interactions and proton concentrations, thereby influencing factors such as hydrogen binding energy, water adsorption, and the overall reaction kinetics. Moreover, it is briefed that electrolyte cations, such as Li+, can impact HER positively or negatively, offering opportunities for improvement based on the metal substrate. Interestingly, there is a potential that HER can be tuned using Li+ ions to modify M-H bond energy, demonstrating flexibility beyond pH levels and counter ions. Varied adsorption energies of metal cations on metal electrodes are also found to influence HER kinetics. The effects of electrolyte anions and impurities are also discussed, emphasizing both positive and negative impacts on HER kinetics. Moreover, it is pointed out that the electrolyte engineering approach enhances the HER kinetics of both metal and carbon-based electrodes without permanent catalyst surface modifications. This review underscores the importance of electrolyte composition, highlighting both challenges and potential solutions in advancing HER research for sustainable energy production.
Keywords:
Hydrogen evolution reaction
; kinetics
; electrolyte
; electrolyte ions
; electrolyte impurities
; electrolyte engineering
1. Introduction
1.1. Background information on the hydrogen evolution reaction
In the 21st century, the pressing issues include the depletion of non-renewable energy sources, environmental harm from fossil fuels, and the escalating global energy demand. With fossil fuels responsible for 84% of energy consumption, their use leads to severe environmental issues. Researchers are investigating renewable energy sources such as solar, wind, and biomass to address the environmental problems and growing energy demands. However, these sources are intermittent, necessitating efficient energy transformation and storage technologies. Efficient energy storage methods are vital for reliable energy supply, especially with the rise of renewables, emphasizing electrochemical energy storage systems like batteries. However, these systems raise environmental issues due to material toxicity and energy-intensive manufacturing processes. Thus, hydrogen, renowned for its high energy density and emission-free combustion by-product, is a widely explored energy carrier, while water splitting utilizing surplus electricity from renewables holds promise for clean hydrogen production [1]. This method ensures high energy conversion efficiency [2,3,4], promoting its use in fuel cells or combustion for a greener future [5,6,7,8].
Water electrolysis involves two simultaneous electrochemical reactions, the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode, occurring in a typical cell with an anode, cathode, power source, and electrolyte [2,9]. The theoretical minimum energy for water splitting is 1.23 V at ambient temperature [9]. However, due to the polarization overpotential of the cathode and anode and the internal resistance of the electrolyte, additional energy is required, known as the cell overpotential [10,11]. Efficient electrocatalysts are crucial for both the HER and OER processes to improve energy conversion efficiency and minimize overpotential. In spite of the fact that the HER is vital for transforming electrical energy into chemical energy in the form of an H2 molecule, the OER also plays a significant part in the overall efficiency of an electrolyzer unit because of its intricate reaction mechanism [1]. Amid the global energy crisis, there is a rising interest in the HER to produce pure (green) hydrogen using renewable energy sources. Researchers are actively exploring new and cost-effective catalysts for HER to make this process economically viable. The HER volcano curves [12,13,14], based on Sabatier's principle, are valuable tools in understanding trends in HER activity, aiding in the search for optimal electrocatalysts by analyzing the reaction rate against the free energy of adsorption for intermediates [2].
Depending on the type of electrolyte employed, water electrolysis can take place in acidic, pH-neutral, or alkaline conditions. Equations (1) and (2) outline the corresponding HER reactions in acidic and alkaline electrolytes, respectively.
2H+ + 2e─ → H2
2H2O + 2e─ → H2 + 2OH─
The mechanism of HER, which is influenced by the pH of the solution [15], can be summarized in acidic environments as follows:
H+ + e─ + * → Hads
Hads + Hads → H2 + 2*
H+ + Hads + e─ → H2 + *
In an alkaline media, the HER mechanism can be presented in a similar way:
H2O + e─ + * → Hads + OH─
Hads + Hads → H2 + 2*
H2O + Hads + e− → H2 + OH- + *
Within the above-presented mechanisms, * signifies an unoccupied active site on the catalyst's surface. The reaction initiates with a Volmer step (Equations (3) and (6)), and the intermediate species (Hads) is subsequently eliminated from the surface via either the Tafel reaction (Equations (4) and (7)) or the Heyrovsky reaction (Equations (5) and (8)). HER on Pt and PGMs is rapid and reversible at low pH, but cost and scalability issues demand alternative catalysts. Cheaper metals like Ni hold potential as alternatives to Pt for the HER, but they exhibit lower performance and stability issues in acidic conditions. In alkaline media, the HER is notably slower, even on Pt, mainly due to the sluggish H2O dissociation step (Equation (6)). PGMs still have excellent HER performance in alkaline environments, but their activity declines by ~ 2 order of magnitude compared to acidic media [16], emphasizing the impact of the electrolyte in the HER.
Numerous water electrolysis techniques have been studied and can be categorized according to the electrolytes used. These methods encompass proton exchange membrane electrolysis (PEMEL) and alkaline electrolysis, which includes both traditional alkaline water electrolysis (AEL) and anion exchange membrane electrolysis (AEMEL) [2,7,9,17]. Although both AEL and PEMEL have attained high degrees of technological maturity [17], it is still challenging to produce hydrogen on a large scale using water electrolysis due to the high cost of PGM catalysts in acidic conditions and the low energy conversion efficiency of non-PGM catalysts in alkaline conditions. Moreover, the local acidic environment in PEMELs severely restricts the selection of electrocatalysts to a small subset of PGMs [18]. On the other hand, alkaline water splitting, employing a cost-effective KOH electrolyte, enables affordable non-PGMs such as Ni, offering advantages over PEMEL cells. In practical applications, large-scale water electrolysis is carried out in concentrated alkalis utilizing stable and inexpensive Ni-based catalysts, whose decreased activity relative to Pt is offset by their lower price [15].
The kinetics of the HER can be influenced by different factors, including the nature and composition of the electrolyte, the crystal shape and orientation of the electrode (single-crystal, polycrystalline, amorphous, etc.), and many other factors. This review will discuss the effect of the nature and concentration of electrolytes on the kinetics of HER.
1.2. Importance of studying the kinetics of HER
Based on the elementary processes (Equations (3) to (5)), it is conceivable to imagine two potential mechanisms in acidic conditions: Volmer-Tafel and Volmer-Heyrovsky [19], and the preferred mechanism depends on various factors, including the surface coverage of Hads and the overpotential applied. In situations where the surface coverage is high, there is a higher likelihood of the Volmer-Tafel mechanism occurring due to the increased probability of the surface recombination step. Conversely, when the surface coverage is low, the Volmer-Heyrovsky mechanism is favored [2]. The Volmer-Tafel pathway is more common at low overpotentials, while the Volmer-Heyrovsky becomes more prevalent as the overpotential increases [20]. The abovementioned mechanisms can result in three possible rate-determining steps (RDSs): Volmer, Heyrovsky, and Tafel. Both mechanisms exhibit an exponential increase in catalytic current with overpotential. Still, the rate of increase differs for different RDSs, allowing for the identification of the RDS and the surface mechanism of a catalyst [19]. Previous studies [21,22,23,24,25] have suggested that the Volmer step is the RDS for the kinetics of the HER/HOR on polycrystalline Pt surfaces. Conversely, some other studies [26,27] suggest that the Heyrovsky step controls the kinetics of the HER.
The Tafel slope provides information about the RDS and the plausible HER mechanism, specifically related to the electron-transfer kinetics in the catalytic reaction. A smaller Tafel slope indicates faster electrocatalytic kinetics, leading to higher current density (j) with lower overpotential. The exchange current density (j0) refers to the charge transfer rate under equilibrium conditions. A greater j0 signifies an accelerated charge transfer rate and a diminished reaction barrier [19]. Thus, a better electrocatalyst typically has a lower Tafel slope and a higher j0 [28]. In the HER potential region at 25°C, different Tafel slopes are theoretically predicted based on the RDS, i.e., −120 mV dec−1, −30 mV dec−1, −40 mV dec−1 for the Volmer, Tafel, and Heyrovsky limiting reactions, respectively. Typically, a Tafel slope of −120 mV dec−1 is observed on most materials at practical current densities. This finding is attributed to the slow discharge of protons or the sluggish electrochemical desorption of Hads atoms. However, in acidic environments where the Volmer step is exceptionally rapid, PGMs tend to exhibit a lower Tafel slope of −30 mV dec−1. While the literature commonly associates a Tafel slope of −120 mV dec−1 with the Volmer step for HER, Shinagawa et al. showed that this slope can also be obtained when the Heyrovsky step is the RDS at high Hads coverage (> 0.6) [27].
Moreover, there is a scenario where different steps proceed at a similar rate, making the Tafel slope analysis uncertain [16]. Watzele et al. discussed the challenges and uncertainties in understanding the HER. The authors used electrochemical impedance spectroscopy to study the relative contributions of two pathways (Volmer-Heyrovsky and Volmer-Tafel) to the HER at different electrode potentials and pH values. Their results showed that both pathways contribute similarly to the reaction, and neither dominates [29]. Electrocatalytic kinetics, unlike outer-sphere reactions, are intricate due to adsorbed intermediates. The Butler-Volmer equation must account for the variable surface coverage of these species influenced by electrode potential, posing challenges in Tafel slope interpretation, notably in HER and similar processes. Thus, not only the HER mechanism identification based on the Tafel slope is difficult, it can also be very misleading.
1.3. Definition and types of electrolytes used in HER studies
Electrolytes used for HER studies are various. They can differ by pH (acidic, neutral, or alkaline) or solvent (such as organic solvents or ionic liquids). The choice of electrolytes significantly impacts the efficiency of HER [11]. The majority of research on the HER mechanisms and activity of different materials has been conducted under kinetically preferred, extremely acidic [12,30,31,32,33], and alkaline aqueous conditions [33,34,35,36,37,38] while there has also been significant attention on studying the HER under near-neutral pH conditions [39,40,41,42,43]. Acidic electrolytes used for HER include H2SO4 [12,13,44,45], HCl [46,47] and HClO4 [33,46,48,49]; and the most commonly used alkaline electrolytes include NaOH [50,51] and KOH [14,33,36,46,48,52,53,54,55,56,57,58]. At the same time, the use of LiOH can also be found in the literature [46]. Besides the traditional liquid electrolytes, other types of electrolytes, such as solid electrolytes and ionic liquids (ILs), are also investigated for HER. ILs are utilized in electrochemical systems for efficient HER due to their exceptional properties, such as low vapor pressure, high electrical conductivity, and a wide variety of functional groups [11]. For example, Amaral et al. [59] studied the impact of adding an ionic liquid (IL) ([Emim][MeSO3]) to a Pt cathode in an 8 M KOH solution at temperatures from 25 to 85 ºC. The IL addition exhibited a catalytic effect, increasing j0 and reducing overall impedance up to 45 °C. The activation energy for HER in IL-added KOH was 10 kJ mol−1 than in the IL-free KOH solution.
1.4. Significance of electrolytes in HER kinetics
In general, HER is performed in highly acidic or highly alkaline electrolytes, and the nature of the electrolyte plays a significant role in the kinetic of HER. Due to the availability of higher concentrations of H3O+ ions, the HER on PGMs in acidic solutions is remarkably fast. However, its efficiency is limited by the diffusion of hydrogen ions from the bulk of the solution to the electrode-electrolyte interface is the concentration is not sufficiently high [33]. Furthermore, the acidic environment significantly limits catalyst options to only a few scarce and costly PGMs [18]. Conversely, the HER in alkaline electrolytes is relatively slow, but it allows us to use affordable non-PGMs. Besides strongly acidic/alkaline electrolytes, HER can also occur in neutral and near-neutral electrolytes. These electrolytes provide benefits like reduced corrosion and a wider range of electrocatalysts without the need for specialized membranes or acid/alkali-resistant catalysts [60,61]. Furthermore, neutral environments facilitate the utilization of seawater as an electrolyte and the desegregation of metal-based electrocatalysts with biocatalysts to produce biofuels [2]. Merrill et al. [62] found that protonated weak acids in microbial electrolysis cell (MEC) solutions affect HER through weak acid catalysis and reducing solution resistance. The study emphasized the importance of specific buffers in optimizing MEC efficiency across different pH ranges, with phosphate and acetate working better in acidic conditions and carbonate at higher pH due to increased conductivity.
Recent research has demonstrated that the interactions between the electrolyte and the electrocatalyst significantly influence the electrocatalytic properties. The electrolyte's impact on electrochemical reactions occurs through two mechanisms: (i) via chemisorption of adsorbents in the inner Helmholtz layer involving electron transfer, and (ii) through weak van der Waals interactions between the electrode and spectator (supporting) ions in the electrolyte at the outer Helmholtz layer [63,64]. The kinetics of HER in an aqueous medium are typically influenced by two adsorbates: Had in acidic conditions and OHad in alkaline conditions. For example, Strmcnik et al. found that OHad plays a crucial role in HER over Had for Pt in an alkaline medium [41]. However, conflicting reports have led to ongoing debates in this regard [65].
For a long time, the hydrogen binding energy (HBE) of the metal catalyst surface or the energy of the M-H bond (ΔEM-H) were outlined as a factor influencing HER rate, starting with the classical works of Trassati studying HER in acidic media. Sheng et al. experimentally demonstrated that the hydrogen desorption peaks of under potential deposited hydrogen (HUPD) on Pt(100) and Pt(110) are directly linked to metal-HBE and are pH-dependent [66]. Increasing pH levels led to increased HBE on Pt, resulting in a decrease in Pt's HER activity. Koper et al. demonstrated that spectator ions such as alkali metal cations viz., Li+, Na+, K+, and Cs+ have the ability to weaken the metal-OHad bond (M-OHad), resulting in an elevation of the interfacial pH and a subsequent positive shift in the HUPD desorption peak [67]. Later on, it was found that not only the pH values of the electrolyte but also the co-adsorption of alkali metals such as K are also responsible for the positive shift in the HUPD desorption peak of Pt by weakening the Pt-OHad bond. Recent research indicates that aqueous electrolytes containing alkali metal cations, specifically Li+, can effectively enhance various electrochemical reactions, including those in batteries, carbon dioxide reduction, and nitrogen reduction [64].
The use of Li+ containing aqueous electrolytes is not yet commercialized, making HER suppression necessary in certain processes [68]. Suo et al. discovered that highly concentrated lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) based aqueous electrolytes (> 5 M) can achieve a water-in-salt condition and enhance the water stability window to ~2.9 V [69]. However, at higher LiTFSI concentrations, TFSI− ions can split to form F─, which creates a LiF layer on the active electrode (stainless steel), selectively blocking the diffusion of H+ ions. Guha et al. discovered that the observed phenomenon in the presence of Li+ goes beyond the formation of LiF and is independent of the supporting anion (TFSI−), particularly when working with high Li+ concentrations rather than low concentrations as in previous reports [70]. They found that Pt electrodes suppress HER at all pH values, while for bulk gold (Au), HER activities are enhanced with Li+. The study suggested that the M/Li+ interaction plays a significant role in this phenomenon, and enhanced H2(g) production with Au was confirmed using gas chromatography-based quantifications.
2. Influence of Electrolyte Composition on HER Kinetics
2.1. Effect of electrolyte pH on HER kinetics
The influence of electrolyte pH on the rate of HER was overlooked due to experimental limitations in assessing rapid kinetics in acidic media. Nevertheless, it's now evident that pH strongly affects HER, particularly on highly active metals. For instance, when moving from pH = 0 to pH = 13, the HER activities of Pt, Ir, and Pd drop by factors of 210, 120, and 90, respectively [34,71]. On the other hand, non-PGM electrodes, such as Au, Ni, or Cu, experience a lesser impact, with a 10-fold reduction in HER activity [12,14,71]. Additional studies conducted in neutral solutions have shown that the decrease in activity directly correlates with the pH level [72]. Experimental findings show that the HER activity of various electrocatalysts progressively declines with the rise in pH [41]. A significant decrease in HER activity for Pt(111), Au(111), and polycrystalline Ir (Irpc) was observed with increasing pH (pH = 1 to 13) [73]. Au(111) displayed higher overpotentials than Pt(111) and Irpc at the same current density in acidic pH, attributed to the mass transport of reactive H3O+ species regulating the HER [40]. However, above pH = 5 and certain potentials, metal polarization curves became pH-independent, suggesting H2O to H2 transformation dominated HER currents. Thus, the main difference between the HER in alkaline and acidic media is that the HER in alkaline solutions is limited by a sluggish water dissociation step (Equation (6)) [41].
Examining the impact of surface-dependent kinetic rates is another way to investigate how pH of the electrolyte affects the HER. In particular, while the activity of Pt low-index single crystal surfaces varies only slightly in acidic media, it varies significantly in alkaline solutions [30,35,74]. According to Danilović et al. [75], the highly defected Pt(110) exhibits higher activity compared to the relatively “perfect” Pt(111). Moreover, the variation in activity is explained by the fact that the adsorption of hydroxyl and HUPD species is influenced by the structure of the crystal surface, which ultimately impacts the formation of the electroactive intermediate, HOPD [36]. For a more thorough understanding of the HER's structure-activity relationship, Marković et al. [73] have conducted a comparative study between the HER activity of Pt(111) and Pt(111) decorated by electrochemically deposited Pt islands (Pt-islands/Pt(111)). In alkaline media, the Pt-islands/Pt(111) surface exhibited 5 to 6 times higher HER activity compared to the pristine Pt(111), while in acidic media, the enhancement was only around 1.5 times. The pH effect shows that low-coordinated single-crystal Pt atoms play a crucial role in increasing the rate of the HER in alkaline solutions, promoting the dissociative adsorption of water [73].
In the current literature, several prevailing hypotheses explain why HER kinetics are slower in alkaline solutions than in acidic ones.
- (i)
- The HBE is pH-dependent [14,72,76]. This concept has helped to explain numerous experimental findings, even though some inconsistencies still exist [72,76,77]. For illustration, if the HBE were to increase, it would account for the positive potential shift of HUPD on PGM-electrodes when they change from an acidic to an alkaline electrolyte. Nevertheless, despite demonstrating considerably lower HER activity in alkaline electrolytes than in acidic ones, the Pt(111) surface remains largely unaffected by this shift caused by the HUPD [71,78,79]. Furthermore, if there were a universal increase in the HBE with pH, it would enhance the HER electrocatalytic activity of metals that weakly bind hydrogen (such as Au). However, this contradicts the experimental observations [71].
- (ii)
- The proton donor (H3O+ or H2O) is pH dependent [41]. In other words, the proton donor can switch from H3O+ in an acidic environment to H2O in an alkaline environment.
- (iii)
- At the electrode|electrolyte interface, there is a pH-dependent water reorganization energy. According to Koper et al. [80], the water-reorganization energy related to proton-electron transfer would be higher because interfacial fields are stronger in an alkaline environment. Rossmeisl et al. [81] initiated an attempt to address pH in density functional theory (DFT) calculation and applying the scheme to Pt(111)|electrolyte(water) interface as an example, they have observed that the adsorbate coverage and water orientation were affected by pH [81]. Recent studies by Rossmeisl et al. have associated the reduction in HER activity at high pH with changes in the configurational entropy of the proton as it crosses the outer Helmholtz plane [82]. Cheng et al. [83] carried out full solvent Quantum Mechanics Molecular Dynamics (QMMD) simulations to explicitly simulate the water/Pt(100) interface at applied voltage (U) from +0.29 V to -0.46 V, which is equivalent to pH from 0.2 to 12.8 at U = 0.3 V (RHE). The study deduced that the pH-dependent HBE on the noble metal is mostly caused by changes in water adsorption. They discovered that the electrode exhibited a tendency to repel water as the applied voltage was made more negative, which in turn boosted the hydrogen binding.
The HER mechanism in neutral solutions is proposed to be similar to that of the alkaline media, proceeding through the adsorption step (Equation (6)) followed by the desorption steps (Equation (7) or (8)). In near-neutral pH with typical supporting electrolytes like Na2SO4 or NaClO4, the HER relies on water as the primary reactant for significant hydrogen production. Due to reduced hydronium ion activity in these circumstances, reactant mass-transport flux becomes slower than the surface hydronium ion reduction rate. As a result, in unbuffered near-neutral pH electrolyte solutions, a significant amount of overpotential is needed to achieve greater current densities than in acidic or alkaline pH conditions [84]. In pH-neutral electrolytes, the HER process involves a two-step reduction process. In contrast, in strongly acidic or alkaline electrolytes, the reduction occurs in a single step with H3O+ ions or H2O molecules, respectively [85]. During the initial reduction phase of the HER, the main reactants are H3O+ ions, and this occurs at low cathodic overpotentials. As the overpotential increases, the HER process becomes diffusion-controlled, where constant current is observed [43]. The second reduction phase takes place at higher overpotentials, during which the principal reactants in HER change from H3O+ ions to H2O molecules, leading to a steady rise in reduction current [60]. For instance, the HER electrocatalytic performance of Pt in the pH range of 5 to 9 does not align with the predicted shift in thermodynamic potential (i.e., −59 mV per pH unit) [42]. The Mayrhofer group [39] discovered that in unbuffered or inadequately buffered electrolytes, the pH near electrode surfaces significantly deviates from the bulk electrolyte pH, particularly in the range of pH 4 to 10. These observations suggest that the kinetics of the HER in pH-neutral conditions behave uniquely.
Furthermore, Takanabe's research team uncovered that the HER processes in near pH-neutral solutions are influenced by the nature of the reactants, the state of the electrolytes (buffered or unbuffered), and their concentrations [42,43,86]. They investigated the relationship between HER and pH using various unbuffered 0.5 M Na2SO4 solutions. They found that HER activity is based on the activity of H3O+ ions rather than the nature of the supporting electrolyte. Depending on theoretical diffusion-limited current density, the authors categorized HER activity into three pH regions: acidic (1-5), neutral (5-9), and alkaline (9-13) [42]. The neutral region was found to have insufficient H3O+ ions, limiting HER activity. Yet, the supply of H3O+ ions can be enhanced by buffered electrolytes, which helps overcome the limited H3O+ ion availability near electrode surfaces [43,84,87]. Research has been focused on improved neutral pH performance using buffered solutions [39,40] such as phosphate, borate, and carbonate [62,88,89,90], yielding onset potentials similar to acidic and alkaline conditions [40,62]. The observed result stems from controlled pH near electrode surfaces due to ongoing reactions and buffering effects. On the other hand, some studies contend that the weak acid itself, e.g., phosphate species (H2PO4− and HPO42−), is likely the reactant in buffered conditions [62,86,91]. While it remains uncertain if weak acid ions directly interact on the surface, the HER current on a Pt catalyst is primarily determined by the mass-transport of a proton source (like phosphate ions) to the vicinity of the catalyst surface [84,86]. The HER activity in pH-neutral and alkaline conditions is significantly different, even though they are both thought to be influenced by the water-dissociation as the RDS. Some studies suggest that the HER activity is better in neutral electrolytes, while others claim the opposite.
Yan and colleagues proposed that HBE can serve as the sole factor to account for the gradual reduction in HER activity observed across PGM catalysts in different buffer electrolytes with a pH ranging from 0 to 13 [72,76]. According to them, HBE is higher in higher pH electrolytes, resulting in intermediate HER kinetics in neutral electrolytes. Additionally, they recommended OH− can tune the HBE and affect HER activity. Shao et al. [92] conducted a study using surface-enhanced infrared absorption spectroscopy and found that the HBE of Pt catalysts is influenced by the modified electric field, Hads coverage, Pt-H2O, as well as Hads-H2O interactions, leading to a weakened HBE with increased pH levels; this can cause slower reaction kinetics and lower HER activity in high pH environments. However, the HBE descriptor is insufficient to explain the HER catalytic behavior on well-defined Pt(111) surfaces [80]. According to Marković and Koper, the HER catalytic behavior on such surfaces is also determined by the presence of adsorbed hydroxyl molecules [38,41,80]. Marković et al. [73] suggested that in alkaline environments, HER/HOR require different types of sites for Hads and OHads, and the presence of OHads can affect the kinetics by rivaling for the same surface sites (blocking effect) or modifying adsorption energy (energetic effect) of the active intermediates [41,73]. They proposed that the activity of HER in alkaline solutions can be enhanced by carefully balancing the rate of H adsorption and OH desorption. Despite debates over the precise mechanisms involved, the combination of Ni(OH)2 (for the strongest OHads bond strength) with Pt (for optimal Hads adsorption free energy, ∆adsGH) has been found to enhance the activity of the HER in alkaline electrolytes significantly. This bi-functional tuning approach has also been favorably applied to pH-neutral solutions, suggesting that it is a viable method for speeding HER kinetics in pH-neutral solutions [60,93].
2.2. Impact of different cations and anions on HER kinetics
Researchers are working to improve catalyst activity by adjusting covalent adsorbate-surface interactions through surface electronic structure features. A current focus in electrocatalysis research is understanding the impact of spectator electrolyte species on electrode catalytic activity. In recent times, numerous instances of catalysts have been observed to exhibit catalytic activity that depends on cations [94] and pH [48,52,76]. Recent research reveals that apparently inactive components in electrolytes have a significant impact on catalytic performance. Specifically, alkali metal cations (Li+ to Cs+) in high-pH aqueous electrolytes can cause notable changes in reaction turnover frequency through noncovalent interactions with water molecules, spectator ions, surface adsorbates, and electrified interfaces [95]. Furthermore, the catalytic activity of Pt for HOR, ORR, and methanol oxidation, at 0.9 V versus RHE, is found to be cation-dependent, following the order of Li+ << Na+ < K+ < Cs+ [96]. Moreover, the kinetics of HER is found to be strongly affected by the nature of the cation, with Li+ having the most significant effect [63]. These observations suggest that the chemical composition of electrolytes presents promising opportunities to tune noncovalent interactions and solvation environments at the electrified interface, potentially leading to significant changes in catalytic activity and selectivity.
Diverse studies have explored the impact of different alkali metal cations, including Li+, Na+, K+, and Cs+, on the inherent HER performance of noble metals like Pt and Au. However, these reports often present conflicting findings, while investigations on non-noble metals remain scarce [97,98,99]. The presence of Li+ cations in the electrolyte was found to have a substantial impact on HER, especially in the presence of surface oxophilic groups, surpassing the benefits of surface decoration with M(OH)2 [37]. According to Subbaraman et al. [37], Li+ has the potential to boost the inherent HER performance of Pt-Ni(OH)2 composites, while the presence of Li+ had no effect on HER for Pt alone. The authors explained that the edges of Ni(OH)2 play a key role in water dissociation, which was further amplified by the introduction of Li+. However, the reason for (Li+)'s ineffectiveness in the presence of Pt alone remained unclear, and the HER enhancement phenomenon was restricted to an alkaline environment. Recently, Liu et al. [100] proposed that Li+ facilitates the elimination of adsorbed OHad from the double layer, thereby augmenting the intrinsic HER activity of the Pt-Ni(OH)2 system.
Xue et al. [101], studied the role of different alkali metal cation-containing electrolytes in the HER activity of the Pt(pc), Pt(111), Pt(221), Ir(111), Au(111), and Ag(pc) electrodes. The study showed the HER activity pattern of all the Pt-electrodes and that of Ir(111), regardless of their surface structure, was fairly linked to the hydration energy of the alkali metal cations in the electrolyte, following the sequence: Li+ > Na+ > K+ > Rb+ > Cs+. This pattern was reversed for the Au(111) and Ag(pc) electrodes. These findings prove that the presence of alkali metal cations indeed influences the HER performance of metal electrodes, with variations of ~4 times between electrolytes containing Li+ and Cs+. It was proposed that the observed influence may be attributed to non-covalent interactions between alkali metal cations near the catalytic centers and the adsorbed reaction intermediates at the electrode surface, or it could be due to co-adsorption of metal cations onto the electrode surface [63,101]. The presence of cations facilitates the removal of OHad from the OHad-(H2O)-AM+ adduct, resulting in higher HER activity with smaller and more acidic alkali cations [102]. Weber et al. [63] also found that an enhanced activity with LiOH is linked to a lower activation energy compared to the activity observed with NaOH and KOH electrolytes.
Huang et al. [103] used classical molecular dynamics (MD) simulation to investigate the effect of structure-making/breaking cations on the kinetics of the HER/HOR of Pt(111) in the pH range from 1 to 14. They observed that cations affected the kinetics, with j0 increasing in the order Cs+ < Rb+ < K+ < Na+ < Li+. Based on this study, electrolytes with larger (more structure-breaking) cations have more surface-bound cations than those with smaller (more structure-making) cations, leading to a cation-dependent interfacial hydrogen-bonding network. The resulting variations in interfacial water structure can influence the effective dielectric properties and fluctuations, affecting solvent reorganization energy (Figure 1). Figure 1a shows that larger cations (Cs+) lead to the removal of water molecules from the interface due to strong ion-surface interaction. Conversely, a stable interfacial water layer is formed with smaller cations (Li+) (Figure 1b). By applying the Born model of reorganization energy and reaction entropy, the interfacial static dielectric constant was estimated to be notably lower than in bulk electrolyte, with the order of increase being Li+ < Na+ < K+ < Rb+ < Cs+ on the negatively charged Pt RDE. This study suggests that as cations with a stronger structure-breaking tendency (e.g., Cs+) concentrate and partially desolvate at the electrified interface, it leads to higher static dielectric constants, increased reorganization energy, elevated entropic barrier for the formation of Had from H2O, and ultimately reduces the kinetics of HER/HOR.
Monteiro et al. [105] investigated the influence of cation type and concentration on HER kinetics on Pt and Au electrodes. They found that weakly hydrated cations (e.g., K+) promoted HER on gold only at low overpotentials, while strongly hydrated cations (e.g., Li+) facilitated HER at higher overpotentials (more alkaline pH). The same pattern was observed for Pt, but weakly hydrated cations inhibited HER early at lower alkalinity and cation concentrations. Weakly hydrated cations (K+) are proposed to stabilize the transition state of the water dissociation step since they are more concentrated towards the surface than strongly hydrated cations like Li+. However, when the pH and, hence, the near-surface cation concentrations are high, the buildup of these species at the outer Helmholtz plane inhibits HER. This is particularly evident on Pt, where a change in the RDS is shown around pH 13 when employing an electrolyte that contains Li+ or K+.
Guha et al. [106,107] conducted an extensive study on how supporting ions like Li+ can influence HER activities of metals. They have disclosed that highly concentrated electrolytes (sometimes called 'water-in-salt' type electrolytes) based catalysis significantly impacts the intrinsic catalytic activity of metals without causing permanent surface alterations. In one of their work, Guha et al. [107] investigated the impact of Li+ ion concentration on the HER of polycrystalline Pt and Au. They found that various lithium salts can modify the HER abilities of both materials. Specifically, increasing Li+ concentration suppresses Pt's HER activity while enhancing it in Au (Table 1). These effects were observed with various counter ions such as Li+, Na+, ClO4−, Cl−, and bis(trifluoromethanesulfonyl)-imide (TFSI−) ions and across different pH conditions (pH 2−13). The effects of the lithium salts, LiClO4, LiCl, and LiTFSI, on the HER process of Au were comparable. An increase in the concentration of LiClO4 from 0.01 to 5 M in 0.01 M HClO4 (pH = 2) and 0.1 M NaOH (pH = 13) has led to the shift in the HER onset potential in the positive direction, indicating the improvement in HER activity. The HER response shows a comparable effect of Li+ ions on the Au electrode with LiCl and lithium trifluoromethanesulfonate (LiOTf) electrolytes. Conversely, it is demonstrated that LiTFSI suppressed the HER on the Pt electrode, while LiClO4 or LiCl does not affect the HER of Pt. Moreover, the HER activities of Pt and Au were found to be unaffected by Na+ ions (originating from NaClO4). The authors deduced that the observed variations in the HER catalytic activity are caused by changes in the adsorption energies of various metal ions toward Au and Pt electrodes.
Additionally, Guha et al. [64] investigated the mechanism behind tunable HER on various metals at different pH levels using linear sweep voltammetry (LSV), electrochemical impedance spectroscopy (EIS), and Tafel analysis. The study investigated Pt, Ir, Pd, Au, Fe, and Ni catalysts, covering both sides of the Sabatier HER volcano plot, and assessed their HER efficiency under varying Li+ concentrations. The results revealed that Au, Fe, and Ni exhibited enhanced HER properties with higher Li+ concentration, while Pt, Pd, and Ir showed the opposite trend. Moreover, to study the role of anion and LiF formation, the authors have evaluated the effect of different Li+ concentrations using both LiCl and LiClO4. Similar to the results obtained with LiTFSI, an increase in Li+ concentration from 1 M to 5 M (in both LiClO4 and LiCl) has led to HER enhancement on Au, Fe, and Ni electrodes, while HER suppression was observed on Pt and Ir electrodes. These results offer evidence to support the authors’ claim that the suppression of HER in Pt and Ir is not solely caused by LiF. Figure 2a, displays the LSVs for Ir at two different LiTFSI and LiCl concentrations. The authors observed that the suppression of HER in Pt and Ir with LiTFSI was more significant compared to LiCl and LiClO4. Likewise, for the other metals (Au, Fe, and Ni), the HER enhancement in LiTFSI was lower than in LiCl and LiClO4 (see Figure 2b), indicating the potential formation of LiF, as reported by Suo et al. [69]. The authors confirmed the formation of LiF on Au, Pt, and Ir electrode surfaces through X-ray photoelectron spectroscopic (XPS) measurements and using attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) analyses after the electrolysis. The presence of LiF on Au electrodes led them to conclude that the LiF formation is not the sole reason for the HER suppression with LiTFSI-based aqueous electrolytes, as was previously reported [69]. To verify the effect of Li+ (using different LiTFSI concentrations) in shifting the HUPD desorption peak of Pt, the authors have also conducted various tests using CVs, LSVs, and EIS in acidic 0.5M H2SO4 (pH = 0) and alkaline 0.1M NaOH (pH = 13). The CVs of Pt confirm that the HBE of Pt decreased with the increase in Li+ concentration in both H2SO4 and NaOH electrolytes. The variation in metal HBE of the five metals with varied Li+ concentrations was also verified by the Authors theoretically using DFT and MD studies. Therefore, using theoretical studies, the authors deduced that there is a variation in metal HBE with changing Li+ concentration. At the same time, their experimental results demonstrated variations in Pt-H and Pd-H binding energies with Li+ concentration. Therefore, this study revealed that metals from both sides of the volcano plot can exhibit tunable HER properties, regardless of pH levels (0 and 13) and counter ions (TFSI−, Cl−, ClO4−, NO3−, and OH−), by modifying the M-H bond energy using Li+ ions.
Another contribution by Guha et al. [106] investigated the use of high-concentration Li+ ions-containing electrolytes for enhancing the electrocatalytic HER performance of different types of carbon nanotubes (CNTS), namely metallic multi-wall (MWCNTs) and semiconducting single wall (SWCNTs). The outcomes indicated that both the CNTs exhibited an enhancement in their HER performance with the increase in Li+ ions concentration. To validate the mechanism and establish the significance of Li+ in improving the HER of the CNTs, the researchers also explored several lithium salts with different counter ions, such as TFSI−, OTf−, ClO4−, Cl−, and OH−. Interestingly, they observed a similar enhancement in the HER characteristics of the CNTs. This study suggested that anions play a minor role in the observed phenomenon. Although Suo et al. [69] noted that LiF formation in LiTFSI and LiOTf-based electrolytes may present a kinetic barrier for proton reduction, higher concentrations of LiTFSI and LiOTf have actually improved the HER performance of the CNTs. Additionally, these electrodes exhibited excellent long-term stability in their HER performance. This study demonstrated that even the slow HER kinetics of CNTs in an alkaline solution can be improved by using an electrolyte engineering strategy without permanently modifying the surface of the catalyst.
In the context of electrolyte and cation effects on electrocatalysis, it is important to mention the study of Strmcnik et al. [98], who explored how Li+, Ba2+, and K+ impact Pt and Au during the ORR in an alkaline solution. They observed that in an alkaline environment, Li+ had a strong interaction with adsorbed OH− ions at the active sites of the Pt surface, leading to the blocking of these active sites for ORR on Pt. Therefore, the presence of Li+ results in a decrease in the ORR activity of the Pt surface. However, this effect was not observed on the Au surface due to the small coverage of OH− ions on the Au surface. A DFT study conducted by Matanović et al. [108] provides additional evidence, indicating that when H+ ions are present at low concentrations, alkali metal ions compete with them for adsorption on the Pt surface, blocking the active sites on the Pt surface and suppressing HER. As the Li+ concentration rises, the H+ concentration falls relative to the Li+ concentration, creating strong competition between Li+ and H+ for adsorption on the Pt surface [69]. The effect of alkali metal cations (originating from MClO4, where M refers to Li+, Na+, K+, Rb+, or Cs+) on the electric double layer (EDL) capacitance of Pt(111) and Au(111) electrodes was investigated by Garlyyev et al. [109]. The study revealed that the local effective concentrations of cations near the electrode for both Pt(111) and Au(111) electrodes can reach ~80 times higher than those in the bulk solution. The EDL capacitance increased linearly Li+ < Na+ < K+ < Rb+ < Cs+, indicating a significantly higher effective Li+ concentration within the double layer compared to the bulk solution. Another DFT-based theoretical work [110] revealed that Pt has a greater affinity to Li+ ions compared to Au. According to the DFT calculations, the adsorption potential of Li+ on the surfaces of Pt and Au is −1.30 V and −2.76 V vs. NHE, respectively. As a result, the strongly adsorbed Li+ ions block the active sites of Pt, hindering the HER. Additionally, Li+ has the capacity to destabilize water molecules [37], and the high Li+ concentration near the electrode surface may be favoring the breakdown of water molecules, which would then result in increased HER activity of Au where the surface is not obstructed by Li+ ions.
In our recent work [46], we have investigated the effect of the nature of the electrolyte (0.1 M HClO4, 0.1 M HCl, 0.5 M NaCl, 1 M KH2PO4, 0.1 M KOH, 1 M KOH and 0.1 M LiOH) on the HER activity of various monometallic polycrystalline electrodes (Pt, Ni, W, Co, Fe, Cr, Ag, Au and Zn) both for freshly polished and oxidatively treated electrodes. In order to compare the HER catalytic activities of the investigated metals in the various electrolytes, we have determined the overpotential values required to achieve a current density of −0.1 mA cm-2real (η0.1,real). The HER activity of the metals in the investigated electrolyte solutions, as determined by the η0.1,real, follows the following order: 0.1 M HClO4 > 0.1 M LiOH > 1 M KH2PO4 > 0.1 M HCl > 1 M KOH > 0.1M KOH > 0.5 M NaCl. The HER of the metals in LiOH was remarkably high, which is attributed to the effect of Li ions [104,111]. The higher HER activity of metals in a 1 M KH2PO4 solution can be attributed to the effect of K+ ions, the buffering properties of KH2PO4, and the involvement of weak acid components (H2PO4− and HPO42−) in the reduction process, as suggested by previous studies [62,86,91]. It is widely accepted that the accelerated formation of Hads intermediates from H3O+ significantly enhances the HER activity of metals in the acidic solutions (0.1 M HClO4 and 0.1 M HCl). While the higher activity of the metals in 0.1 M HClO4 can be ascribed to the non-adsorbing property of ClO4─ anion [66], conversely, the decreased activity of the metals in the HCl and NaCl solutions can be attributed to the negative effects of Cl─ ion poisoning. The higher HER activity of the metals in 1 M KOH compared to 0.1 M KOH is primarily due to the concentration effect of K+ ions [112].
There are conflicting views on the influence of electrolyte anions on the HER. Some studies suggest their effect is insignificant, as already exemplified, while others reveal that electrolytes can significantly influence the HER of metal catalysts. A comparable HER/HOR performance of Pt across three electrolytes (HClO4, HNO3, and H2SO4) is reported in Ref. [113]. Moreover, Ref. [114] shows that in contrast to the HOR, the HER current densities, which have been examined in low overpotential and underpotential sites, were found to be independent of the nature of the supporting electrolyte (HClO4, H2SO4, and HCl). Similar HOR/HER activities in the presence of H2SO4− and HClO4− ions were reported, which could be possibly because these counter anions may not adsorb on the catalyst surface at HOR/HER-relevant potentials in the vicinity of ≈ 0 VRHE [71]. Moreover, as already discussed, Guha et al. [106] revealed that the effect of the nature of anions (TFSI−, OTf−, ClO4−, Cl−, and OH−) on the HER of the CNTs was minor.
Several studies have explored how strong anion adsorption on PGM-surfaces influences the kinetics of pseudo-capacitive and Faradic processes. Anions can also adsorb on the electrode surface at potentials below the potential of zero charge, particularly within the HUPD region, impacting Pt-Had energetics and potentially causing alterations in the HER-kinetics [71]. In practical terms, anionic contamination from SO3− ions, which may be released during polymer electrolyte membrane degradation in electrolyzer/fuel cell operation, could adversely affect device performance and durability, especially with ultra-low PGM loadings. Furthermore, the presence of Br– and I– were reported to significantly reduce the overall rates of HER/HOR [71].
As already mentioned, our recent study [46] revealed the effect of both cations and anions on the HER of different metals. More specifically, the impact of Cl− ions was significant in both 0.1 M HCl and 0.5 M NaCl. This effect was evident on the volcano plots (a plot of HER overpotential values needed for the current density of 0.1 mA cm−2real, vs. the DFT calculated HBE) (Figure 3 and Figure 4). One can easily observe the impact of the electrolyte on the shape of the volcano curve, as evidenced by the broader overpotential range (form ~ 0 to > −0.6 V) required for HER in HCl (Figure 3b) compared to HClO4 (Figure 3a) which can be attributed to Cl− ion poisoning in 0.1 M HCl and the non-adsorbing property of ClO4─ anion in 0.1 M HClO4. The effect of electrolytes is also noticeable in neutral solutions, with NaCl (Figure 4a) exhibiting higher HER overpotentials than KH2PO4 (Figure 4b). Moreover, the volcano peak shifts by approximately −0.3 V, and unique characteristics such as flattened trends for W and Cr are observed in the NaCl solution. At the same time, Co exhibits unexpectedly low activities in both the pH-neutral solutions investigated. These findings align with the concepts proposed in Ref. [115], which discuss the activity of metals with highly exothermic hydrogen adsorption. The effect of SO42− and ClO4− anions on the HER activity of Pt(110) in 0.1 M KOH was examined by Sheng et al. [66]. ClO4─ was shown not to affect the HBE, while the addition of SO42− slightly altered the HBE of Pt (110).
2.3. Electrolyte Concentration and HER Kinetics
Arminio-Ravelo et al. [116] studied the effect of electrolyte composition and concentration on commercial Ir black nanoparticles using varying concentrations (0.05 M, 0.1 M, and 0.5 M) of H2SO4 and HClO4. It was found that H2SO4 hindered Ir oxidation and catalyst performance, while HClO4 showed minimal interference and better catalytic performance than H2SO4, regardless of its concentration. Varying concentrations of HClO4 showed no significant impact while increasing H2SO4 concentration led to decreased activity due to stronger adsorption of HSO4− and SO42− anions on the catalyst surface compared to ClO4− anions. HClO4 is suitable for catalytic performance as it has minimal impact, but concentrations higher than 0.1 M should be avoided to prevent potential anion interactions. The authors recommend using HClO4 as the electrolyte for benchmarking and reporting activity and stability trends in RDE measurements of Ir-based materials.
The effect of the nature of the electrode (polycrystalline and nanostructured), pH (12 to 14), and concentration of electrolyte (0.01 to 2 M KOH) on the HER activity of Ni-based catalysts was investigated by Faid et al. [117]. The findings showed that a pH and KOH concentration variation influenced the HER activity by affecting ECSA and Tafel slope. The nanostructured NiMo catalyst exhibited enhanced HER activity, and the Tafel slope was reduced from ~ −180 mV dec−1 to −60 mV dec−1 in the pH range of 12–14 and KOH concentrations of 0.01–1.0 M, indicating a promoting region for both ECSA and reaction order. Polycrystalline Ni showed different behaviors in different pH and KOH concentration regions, maintaining Tafel slopes around −120 mV dec−1. However, both catalysts' HER performances were inhibited under the conditions of decreased OHad transport kinetics. Higher HER activity of various metals in 1 M KOH than in 0.1 M KOH was also observed in our recent work [46]. Moreover, Li et al. [118] observed that the HER activity of Pt(pc) was improved with the increase in NaOH concentration from 0.01 M (pH 12) to 1.0 M (pH 14). Kuznetsov et al. [119] observed that the anodic charge and ECSA of NiCu/C catalysts were influenced by the NaOH concentration. They also found that the HER activity of partially oxidized Ni disc electrode improved with increasing pH/NaOH concentration when the potential was below −0.18 V vs. RHE. Moreover, current densities in cyclic voltammograms were found to vary with changes in electrolyte concentration, resulting in variations in the apparent ECSA of Ni nanostructures [120]. Goyal et al. [112] investigated the impact of electrolyte pH and alkali metal cation concentration on HER kinetics on Au electrodes. They found that increasing the cation concentration significantly enhances HER activity at moderately alkaline pH (pH = 11) by accelerating the sluggish Volmer step. It is highlighted that electrolyte pH and bulk cation concentration affect surface cation concentration and influence water dissociation kinetics, with interfacial cations stabilizing transition states. Yet, excess near-surface cations, particularly at elevated pH and cation levels, can impede HER activity by obstructing active sites.
Furthermore, studies indicate that optimizing the electrolyte composition can significantly enhance mass transport and improve the efficiency of HER, particularly when operating at near-neutral pH conditions [84,121]. Shinagawa et al. [86] studied, both experimentally and theoretically, the effect of electrolyte concentration on Pt electrode under neutral-buffered conditions employing sodium phosphate (0.2 − 4.2 M at pH 5) as electrolyte solution. Both experimental and theoretical research revealed a volcano-shaped relationship between solute concentration and HER activity. The HER performance increased with solute concentrations up to 2 M, but higher concentrations resulted in decreased performance. Their microkinetic model combined with a description of mass transport closely matched the experimental results, indicating the presence of notable concentration overpotentials during HER under buffered-neutral conditions. Their simulation model indicated that the concentration overpotential surpassed 46% even at 0.5 M, while the kinetic overpotential remains the lowest (∼10%) due to sluggish mass transport. The authors emphasized that mass transport is influenced by diffusion and activity coefficients, with the former determined by ion size and electrolyte viscosity. Thus, adjusting these parameters through effective electrolyte engineering can significantly enhance HER performance in neutral conditions, regardless of the electrocatalyst used.
During water splitting, oxygen is evolved at the anode (OER) and can migrate to the cathode, where catalysts for HER also facilitate oxygen reduction reaction (ORR). Oxygen crossover between anode and cathode in membraneless cells reduces the HER efficiency due to undesired competitive reactions, underscoring the need for oxygen-tolerant HER to improve efficiency. Shinagawa et al. [84] found that selecting the proper electrolyte enhances cell efficiency by controlling the mass transport of oxygen and the proton source (for example, weak acid). The study extensively analyzed the influence of different solutes on the HER and ORR under heavily-buffered conditions. They tested various electrolyte solutions, including KH2PO4, K2HPO4, K3PO4, LiH2PO4, NaH2PO4, NaHCO3, HClO4, KOH, and mixtures of them, in the concentration range from 0.01 to 3.0 M. Under specific conditions (1.5 M NaH2PO4 or 1.5 M 40% K2HPO4 + 60 % KH2PO4), the overpotential required to achieve −10 mA cm−2 was found to be less than −40 mV with 90% selectivity towards HER in oxygen-saturated electrocatalytic conditions. Using such highly concentrated buffers also reduced solution resistance, further enhancing the overall HER performance. The existing literature highlights that the concentration of electrolytes plays a crucial role in electrocatalytic HER, having a positive impact up to a certain threshold level, beyond which it adversely affects the HER.
2.4. Effect of electrolyte impurities
Electrolyte impurities can substantially impact the HER kinetics, causing reduced efficiency or complete inhibition of the reaction. Common impurities found in the electrolyte are metal ions, organic contaminants, and other foreign substances, which can modify the electrode's surface properties, affect reaction kinetics, and cause undesired side reactions during the HER. Impurities in electrolysis cells can originate from various sources. For instance, commercial KOH electrolytes may contain Zn as an impurity [6], while Pt- and Au-counter electrodes used in measurements can also introduce contaminants [71]. Moreover, impurities can arise from corrosion products of cell components due to the corrosive environment caused by highly alkaline electrolytes, high temperature, and the presence of molecular oxygen [122]. The HER activity of surfaces with low j0 values can be influenced by trace metal cations in the electrolyte, which can plate onto the surface due to the HER starting at low potentials [71]. Impurities can deposit as metallic species during H2 production through cathodic reduction or as salts/hydroxides through chemical precipitation, causing passivation of catalytically active sites [122]. Weber et al. [123] investigated the challenges in benchmarking HER/HOR activity of Pt-based catalysts in alkaline media viz., 0.1 M LiOH, NaOH, and KOH. They analyzed the electrochemical setup (such as cell material, hydrogen gas, and electrolyte solutions) to identify the source of impurities. They identified glass cells and hydrogen gas as non-significant sources of contamination. However, they noticed a significant reduction in the ECSA of Pt following HER/HOR measurements, especially in 0.1 M NaOH and LiOH solutions. Through long-term chronoamperometric experiments and X-ray photoelectron spectroscopy analysis, the authors discovered that trace metals (Cu, Zn, Pb, and Fe) from electrolyte salts were deposited on the Pt surface during the HER.
Studies show that the presence of trace metal cations can have both positive and negative effects on the HER activity of the substrate metal [71]. Li et al. [124] studied the influence of trace iron impurities and alkali metal cations (Na+ and Cs+) on the HER of polycrystalline Cu electrodes in alkaline conditions. The study found that during electrolysis in 0.1 M solutions of NaOH and CsOH, with the highest commercially available purity grades, small amounts of iron impurities were deposited on the Cu electrode. The presence of iron impurities significantly accelerated the HER rate in 0.1 M CsOH by up to five times over eleven CVs. The authors have pre-electrolyzed the electrolyte solution to remove iron impurities effectively, and after removing the iron impurities, the CVs stabilized with the cycle number. For purified electrolytes (0.1 M NaOH and CsOH), the HER current densities were found to be nearly identical, suggesting no significant cation effect on the HER rate on Cu. Klaus et al. [125] studied the influence of Fe incorporation on structure-activity relationships in Ni-(oxy)hydroxide by analyzing aged Ni(OH)2/NiOOH films in KOH using various characterization techniques. They discovered that aging in unpurified KOH led to >20% Fe incorporation after five weeks, resulting in higher OER activity, lower overpotential, and lower Tafel slope compared to samples aged in Fe-free KOH. Optimal catalyst activity was observed with 5-day aging in unpurified 1 M KOH. Similar findings are reported for other electrocatalytic reactions, like OER. Salmanion et al. [126] explored the OER of Co and Au in Ni and Fe-free KOH. The results demonstrated that cobalt oxide served as a relatively efficient catalyst for OER in a pure electrolyte, while gold does not exhibit good catalytic activity under the same conditions. Iron impurities in the form of FeO2─ present in commercial 1.0 M KOH electrolytes are actually known to react with Au electrode surfaces and create active sites for OER [127]. Moreover, Gong et al. [128] reported that Fe had a synergistic effect with Co, Ni, Cu, Ag, and Au (but not Ti) in enhancing the OER. Although the Co electrode showed good OER catalytic activity in a pure electrolyte, its Tafel slope decreased significantly in the presence of Ni-containing (Fe-free) KOH [126]. The study suggests that Ni ions precipitate on the electrode's surface, altering the redox-active sites, emphasizing the significance of trace electrolyte impurities and proposing the use of pure electrolytes for evaluating electrocatalysts' performance for OER. Extensive research has explored how arsenic compounds affect HER and HUPD on Pt, Ni, and steel electrodes. Even trace amounts of arsenic (~10-8 M) were consistently found to reduce j0 and increase overpotential for HER [129,130,131].
Moreover, both PEMEL and AEL cells can have two types of impurities: exogenous and endogenous. Electrolyzers usually need highly purified water, but the purified water may still contain low concentrations of ionic species and total organic carbon, considered as exogenous impurities [132]. Growing adoption of green hydrogen technology may raise pure water demand, thus creating public concerns in water-scarce areas. Re-using purified wastewater could address this, but it is energy-intensive and expensive. Opting out of proper purification for industrial processes risks contaminating electrocatalysts with impurities from untreated water [1]. Endogenous impurities in electrolyser systems, on the other hand, originate from internal sources. Throughout its operation, electrolyzers may undergo a gradual deterioration of their stack and balance of plant (BoP) parts, resulting in the production of impurities within the system. Additionally, impurities may arise from component leaching and contamination during the electrolyzer manufacturing, commissioning, and maintenance processes. Cations in PEMWEs present significant challenges as impurities, impacting the catalyst, ionomer, and membrane. Their presence can lead to performance degradation and reduced lifespan, as depicted in Figure 5. Anions are often associated with initiating side reactions, such as chlorine evolution, potentially affecting hydrogen quality and accelerating corrosion in metallic components [132]. In contrast to cations, anions cannot replace protons within the membrane and ionomer, leading to different operational mechanisms (Figure 5).
Therefore, to achieve the best HER performance, it is crucial to use high-purity electrolytes and meticulously control their quality to minimize the influence of impurities. Studies revealed that shielding the counter-electrode and pre-treating the electrolyte can help minimize the impact of metal cation impurities on HER. Moreover, an electrochemical purification step aimed at diminishing impurities originating from the electrolyte solution was devised by Weber et al. [123], which demonstrated that pre-electrolyzing the electrolyte solutions was found to be effective in removing iron impurities. The use of self-assembling and self-healing catalytically active films to overcome cathode deactivation triggered by electrolyte impurities is also mentioned in Ref. [133]. According to this study, introducing trace metal impurity (Zn) has raised the cell voltage. At the same time, adding an active material (NixB) formed a self-assembled catalyst film, restoring activity and lowering the voltage. To mitigate catalyst poisoning, using more resistant catalysts is an option. Metal oxide-based catalysts are less susceptible to poisoning compared to unmodified ones. Catalysts with carbonate, sulfate, and oxide compounds also display enhanced resilience against deactivation. For instance, the MoS2 electrocatalyst was found to be more tolerant to sulfur poisoning than Pt/C [1]. Carbon-based materials like nanotubes or graphene can also mitigate contaminant effects by serving as active HER electrodes. Despite lacking inherent strong catalytic properties, these materials can be modified with small catalyst amounts for highly efficient electrocatalysis. Doping graphene with heteroelements like nitrogen, sulfur, phosphorus, or boron enhances its electrocatalytic performance. Modified electrodes also display remarkable resistance to surface poisoning; intriguingly, impurities might improve their catalytic capabilities [134,135,136].
3. Summary and Outlook
This review explores the impact of electrolytes on hydrogen evolution reaction (HER) kinetics, emphasizing pH, cations, anions, and impurities. It briefly covers how electrolyte concentration affects HER kinetics, highlighting its beneficial impact up to a certain level, beyond which it hampers efficiency. Electrolyte pH significantly influences HER by altering catalyst-electrolyte chemistry and proton concentrations, affecting metal's hydrogen binding energy, proton donor, water adsorption, and interfacial water reorganization. These factors collectively determine the reaction kinetics, catalyst effectiveness, and overall HER efficiency. Although there are differing opinions, there is a general consensus that the HER activity tends to decrease with higher pH levels. Yet, the HER behavior in neutral solutions presents a unique phenomenon. Conversely, neutral solutions allow for applications such as seawater utilization and eliminate the need for specialized materials or acid/alkali-resistant catalysts. pH-neutral HER is affected by reactant properties, electrolyte state (buffered/unbuffered), and concentrations. The insufficient H3O+ ions restrict the HER activity in pH-neutral solutions at the electrode-electrolyte interface, but buffering enhances the H3O+ supply. Additionally, studies suggest weak acids like phosphate species (H2PO4− and HPO42−) likely act as reactants under buffered conditions. In water splitting, oxygen evolved at the anode (OER) can cross to the cathode in membraneless cells, affecting HER. For this purpose, heavily buffered electrolytes are found to exhibit better HER performance with very high selectivity towards HER in oxygen-saturated electrocatalytic conditions regulating oxygen transport and proton source (for example, weak acid).
Beyond electrolyte pH, electrolyte ions are also known to impact the kinetics of HER. Spectator ions can influence HER kinetics by co-adsorbing or interacting with adsorbed intermediates on catalyst surfaces. Recent studies show that aqueous electrolytes with Li+ can effectively enhance various electrochemical reactions, including HER. Notably, ~4 times HER performance difference is observed between Li+ and Cs+ containing electrolytes. Electrolytes with larger (more structure-breaking) cations exhibit more surface-bound cations than smaller (more structure-making) ones, influencing the interfacial hydrogen-bonding network and solvent reorganization energy. Larger cations like Cs+ remove water molecules at the interface through strong ion-surface interaction, while smaller cations like Li+ create a stable interfacial water layer. An optimal Li+ ion concentration improves HER on noble metals (such as Au) but affects the HER on platinum group metals (PGMs) (such as Pt, Ir, and Pd) less favorably. Pt has a greater affinity towards Li+ ions compared to Au, and thus, strongly adsorbed Li+ ions can block active sites of Pt surface. In contrast, high Li+ concentration near the electrode boosts the breakdown of water, increasing HER in unobstructed Au surfaces. These observations indicate that manipulating electrolyte composition offers the potential for modifying noncovalent interactions and solvation dynamics at the electrified interface, which could result in notable alterations in catalytic activity and selectivity.
The impact of electrolyte anions on HER is debated; some studies find them negligible, while others emphasize their substantial influence on the kinetics of HER. Anions can also adsorb on the electrode surface at potentials below the potential of zero charge, especially in the H-under potential (HUPD) region, affecting M-Had energetics and potentially reshaping HER kinetics. Sulphate (HSO4− and SO42−) and chloride (Cl−) ions are known to affect the HER negatively. Conversely, HClO4 is suggested as an ideal electrolyte for benchmarking and reporting activity and stability trends in rotating disk electrode (RDE) measurements due to the neutral or beneficial impact of ClO4− anions on HER kinetics. Anionic contamination from SO3− molecules during polymer electrolyte membrane degradation is also known to negatively impact device performance and durability of electrolyzers, particularly at low PGM-loadings.
Electrolyte impurities can also substantially impact the HER. Impurities can arise from multiple origins: from commercial electrolytes (e.g. Zn in KOH) or from Pt- and Au-counter electrodes. Additionally, impurities can result from corrosion products due to a corrosive electrochemical environment. Proton exchange membrane electrolysis and alkaline electrolysis cells can contain two impurity types: exogenous and endogenous. While electrolyzers require purified water, they may still contain slight levels of ionic species and organic carbon as exogenous impurities. In industrial-scale operations, compromising water quality and chemical selection can pollute electrocatalysts, posing a notable catalyst deactivation challenge. On the other hand, endogenous impurities stem from internal sources, often arising from gradual system deterioration and leaching in components. Although some studies highlight the positive impact of trace metal impurities on the HER activity of the substrate metal, electrolyte impurities are known to modify electrode surfaces, affect reaction kinetics, and trigger undesired side reactions during HER. Therefore, for optimal HER performance, utilizing high-purity electrolytes and rigorous quality control to minimize the impacts of impurities is essential. Moreover, employing pure electrolytes when assessing electrocatalyst performance is sensible, as impurities can either enhance or diminish the catalyst efficiency. Research suggests techniques such as electrolyte pre-treatment, electrochemical purification, shielding of counter-electrodes, and self-assembling and self-healing catalytically active films to mitigate the influence of impurities.
Clearly, depicting a comprehensive view of the electrolyte effects on HER kinetics is a difficult, if not impossible, task. However, it seems that a critical mass of fundamental knowledge has been acquired, and optimizing electrolytes looks like a tangible approach to boosting HER activity in the context of green hydrogen production. In fact, this could be the most straightforward approach to increase the efficiency of existing electrolyzer systems, as replacing electrolytes does not require a disruptive technology shift, while it can lead to significant improvements in the efficiency of the water electrolysis process. To reach this goal, the anode side must be investigated as well. That is, the electrolyte effects on OER should be addressed, too. However, OER is a much more complex reaction than HER. Here, we have mentioned several examples of electrolyte effects on oxygen electrode reactions (both ORR and OER). Due to its complexity, a comprehensive view of electrolyte effects on the oxygen electrode can remain elusive for a long time. However, this fact should not discourage future research in electrolyte engineering for improved water electrolysis efficiency.
Author Contributions
Conceptualization, I.A.P., and G.K.G; validation, I.A.P.; investigation, G.K.G., and A.Z.J; writing—original draft preparation, G.K.G., and A.Z.J; writing—review and editing, I.A.P.; visualization, G.K.G.; super-vision, I.A.P.; funding acquisition, I.A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Serbian Science Fund, grant RatioCAT (PROMIS program). A.Z.J and I.A.P. also acknowledge the financial support provided by the Serbian Ministry of Education, Science, and Technological Development (Contract number: 451-03-47/2023-01/200146).
Data Availability Statement
There are no additional data to be shared.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mikołajczyk, T. The impact of pollutants on catalyst performance during hydrogen evolution reaction: A brief review. Synth. Met. 2023, 296, 117379. [Google Scholar] [CrossRef]
- Angeles-Olvera, Z.; Crespo-Yapur, A.; Rodríguez, O.; Cholula-Díaz, J.L.; Martínez, L.M.; Videa, M. Nickel-Based Electrocatalysts for Water Electrolysis. Energies 2022, 15, 1609. [Google Scholar] [CrossRef]
- Khan, M.A.; Al-Attas, T.; Roy, S.; Rahman, M.M.; Ghaffour, N.; Thangadurai, V.; Larter, S.; Hu, J.; Ajayan, P.M.; Kibria, G. Seawater electrolysis for hydrogen production: a solution looking for a problem? Energy Environ. Sci. 2021, 14, 4831–4839. [Google Scholar] [CrossRef]
- Li, X.; Zhao, L.; Yu, J.; Liu, X.; Zhang, X.; Liu, H.; Zhou, W. Water Splitting: From Electrode to Green Energy System. Nano-Micro Lett. 2020, 12, 1–29. [Google Scholar] [CrossRef]
- Buriak, J.M.; Toro, C.; Choi, K.-S. Chemistry of Materials for Water Splitting Reactions. Chem. Mater. 2018, 30, 7325–7327. [Google Scholar] [CrossRef]
- Zeng, K.; Zhang, D. Recent progress in alkaline water electrolysis for hydrogen production and applications. Prog. Energy Combust. Sci. 2010, 36, 307–326. [Google Scholar] [CrossRef]
- Yan, Z.; Hitt, J.L.; Turner, J.A.; Mallouk, T.E. Renewable electricity storage using electrolysis. Proc. Natl. Acad. Sci. 2019, 117, 12558–12563. [Google Scholar] [CrossRef]
- Zhou, H.; Yu, F.; Zhu, Q.; Sun, J.; Qin, F.; Yu, L.; Bao, J.; Yu, Y.; Chen, S.; Ren, Z. Water splitting by electrolysis at high current densities under 1.6 volts. Energy Environ. Sci. 2018, 11, 2858–2864. [Google Scholar] [CrossRef]
- Cossar, E.; Murphy, F.; Baranova, E.A. Nickel-based anodes in anion exchange membrane water electrolysis: a review. J. Chem. Technol. Biotechnol. 2022, 97, 1611–1624. [Google Scholar] [CrossRef]
- Raveendran, A.; Chandran, M.; Dhanusuraman, R. A comprehensive review on the electrochemical parameters and recent material development of electrochemical water splitting electrocatalysts. RSC Adv. 2023, 13, 3843–3876. [Google Scholar] [CrossRef]
- Chen, K.; Xu, B.; Shen, L.; Shen, D.; Li, M.; Guo, L.-H. Functions and performance of ionic liquids in enhancing electrocatalytic hydrogen evolution reactions: a comprehensive review. RSC Adv. 2022, 12, 19452–19469. [Google Scholar] [CrossRef] [PubMed]
- Trasatti, S. Work function, electronegativity, and electrochemical behaviour of metals. J. Electroanal. Chem. Interfacial Electrochem. 1972, 39, 163–184. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef]
- Sheng, W.; Myint, M.; Chen, J.G.; Yan, Y. Correlating the hydrogen evolution reaction activity in alkaline electrolytes with the hydrogen binding energy on monometallic surfaces. Energy Environ. Sci. 2013, 6, 1509–1512. [Google Scholar] [CrossRef]
- Gutić, S.J.; Dobrota, A.S.; Fako, E.; Skorodumova, N.V.; López, N.; Pašti, I.A. Hydrogen Evolution Reaction-From Single Crystal to Single Atom Catalysts. Catalysts 2020, 10, 290. [Google Scholar] [CrossRef]
- Oshchepkov, “Investigation of the hydrogen electrode reactions on Ni electrocatalysts in alkaline medium,” 2017. [Online]. Available: https://tel.archives-ouvertes. 0200.
- Brauns, J.; Turek, T. Alkaline Water Electrolysis Powered by Renewable Energy: A Review. Processes 2020, 8, 248. [Google Scholar] [CrossRef]
- Li, Q.; Villarino, A.M.; Peltier, C.R.; Macbeth, A.J.; Yang, Y.; Kim, M.-J.; Shi, Z.; Krumov, M.R.; Lei, C.; Rodríguez-Calero, G.G.; et al. Anion Exchange Membrane Water Electrolysis: The Future of Green Hydrogen. J. Phys. Chem. C 2023, 127, 7901–7912. [Google Scholar] [CrossRef]
- Murthy, A.P.; Theerthagiri, J.; Madhavan, J. Insights on Tafel Constant in the Analysis of Hydrogen Evolution Reaction. J. Phys. Chem. C 2018, 122, 23943–23949. [Google Scholar] [CrossRef]
- Štrbac, S.; Smiljanić, M.; Wakelin, T.; Potočnik, J.; Rakočević, Z. Hydrogen evolution reaction on bimetallic Ir/Pt(poly) electrodes in alkaline solution. Electrochimica Acta 2019, 306, 18–27. [Google Scholar] [CrossRef]
- J. Durst et al., “(Invited) Hydrogen Oxidation and Evolution Reaction (HOR/HER) on Pt Electrodes in Acid vs. Alkaline Electrolytes: Mechanism, Activity and Particle Size Effects,” ECS Trans, vol. 64, no. 3, pp. 1069–1080, Aug. 2014. [CrossRef]
- Liu, L.; Liu, Y.; Liu, C. Enhancing the Understanding of Hydrogen Evolution and Oxidation Reactions on Pt(111) through Ab Initio Simulation of Electrode/Electrolyte Kinetics. J. Am. Chem. Soc. 2020, 142, 4985–4989. [Google Scholar] [CrossRef]
- Intikhab, S.; Snyder, J.D.; Tang, M.H. Adsorbed Hydroxide Does Not Participate in the Volmer Step of Alkaline Hydrogen Electrocatalysis. ACS Catal. 2017, 7, 8314–8319. [Google Scholar] [CrossRef]
- McCrum, I.T.; Koper, M.T.M. The role of adsorbed hydroxide in hydrogen evolution reaction kinetics on modified platinum. Nat. Energy 2020, 5, 891–899. [Google Scholar] [CrossRef]
- Rheinländer, P.J.; Herranz, J.; Durst, J.; Gasteiger, H.A. Kinetics of the Hydrogen Oxidation/Evolution Reaction on Polycrystalline Platinum in Alkaline Electrolyte Reaction Order with Respect to Hydrogen Pressure. J. Electrochem. Soc. 2014, 161, F1448–F1457. [Google Scholar] [CrossRef]
- Lamoureux, P.S.; Singh, A.R.; Chan, K. pH Effects on Hydrogen Evolution and Oxidation over Pt(111): Insights from First-Principles. ACS Catal. 2019, 9, 6194–6201. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 13801. [Google Scholar] [CrossRef]
- Murthy, A.P.; Madhavan, J.; Murugan, K. Recent advances in hydrogen evolution reaction catalysts on carbon/carbon-based supports in acid media. J. Power Sources 2018, 398, 9–26. [Google Scholar] [CrossRef]
- Watzele, S.; Fichtner, J.; Garlyyev, B.; Schwämmlein, J.N.; Bandarenka, A.S. On the Dominating Mechanism of the Hydrogen Evolution Reaction at Polycrystalline Pt Electrodes in Acidic Media. ACS Catal. 2018, 8, 9456–9462. [Google Scholar] [CrossRef]
- Marković, N.M.; Grgur, B.N.; Ross, P.N. Temperature-Dependent Hydrogen Electrochemistry on Platinum Low-Index Single-Crystal Surfaces in Acid Solutions. J. Phys. Chem. B 1997, 101, 5405–5413. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef]
- J. Durst, C. J. Durst, C. Simon, F. Hasché, and H. A. Gasteiger, “Hydrogen Oxidation and Evolution Reaction Kinetics on Carbon Supported Pt, Ir, Rh, and Pd Electrocatalysts in Acidic Media,” J Electrochem Soc, vol. 162, no. 1, pp. F190–F203, Dec. 2015. [CrossRef]
- Sheng, W.; Gasteiger, H.A.; Shao-Horn, Y. Hydrogen Oxidation and Evolution Reaction Kinetics on Platinum: Acid vs Alkaline Electrolytes. J. Electrochem. Soc. 2010, 157, B1529. [Google Scholar] [CrossRef]
- Durst, J.; Siebel, A.; Simon, C.; Hasché, F.; Herranz, J.; Gasteiger, H.A. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 2014, 7, 2255–2260. [Google Scholar] [CrossRef]
- Markovića, N.M.; Sarraf, S.T.; Gasteiger, H.A.; Ross, P.N. Hydrogen electrochemistry on platinum low-index single-crystal surfaces in alkaline solution. J. Chem. Soc. Faraday Trans. 1996, 92, 3719–3725. [Google Scholar] [CrossRef]
- Schmidt, T.; Ross, P.; Markovic, N. Temperature dependent surface electrochemistry on Pt single crystals in alkaline electrolytes: Part 2. The hydrogen evolution/oxidation reaction. J. Electroanal. Chem. 2002, 524-525, 252–260. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.-C.; Uchimura, M.; Paulikas, A.P.; Stamenkovic, V.; Markovic, N.M. Enhancing Hydrogen Evolution Activity in Water Splitting by Tailoring Li + -Ni(OH) 2 -Pt Interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef]
- Subbaraman, R.; Tripkovic, D.; Chang, K.-C.; Strmcnik, D.; Paulikas, A.P.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N.M. Trends in activity for the water electrolyser reactions on 3d M(Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts. Nat. Mater. 2012, 11, 550–557. [Google Scholar] [CrossRef]
- Katsounaros, I.; Meier, J.C.; Klemm, S.O.; Topalov, A.A.; Biedermann, P.U.; Auinger, M.; Mayrhofer, K.J. The effective surface pH during reactions at the solid–liquid interface. Electrochem. Commun. 2011, 13, 634–637. [Google Scholar] [CrossRef]
- Auinger, M.; Katsounaros, I.; Meier, J.C.; Klemm, S.O.; Biedermann, P.U.; Topalov, A.A.; Rohwerder, M.; Mayrhofer, K.J.J. Near-surface ion distribution and buffer effects during electrochemical reactions. Phys. Chem. Chem. Phys. 2011, 13, 16384–16394. [Google Scholar] [CrossRef]
- Strmcnik, D.; Uchimura, M.; Wang, C.; Subbaraman, R.; Danilovic, N.; van der Vliet, D.; Paulikas, A.P.; Stamenkovic, V.R.; Markovic, N.M. Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption. Nat. Chem. 2013, 5, 300–306. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Mechanistic Switching by Hydronium Ion Activity for Hydrogen Evolution and Oxidation over Polycrystalline Platinum Disk and Platinum/Carbon Electrodes. ChemElectroChem 2014, 1, 1497–1507. [Google Scholar] [CrossRef]
- Shinagawa, T.; Takanabe, K. Identification of intrinsic catalytic activity for electrochemical reduction of water molecules to generate hydrogen. Phys. Chem. Chem. Phys. 2015, 17, 15111–15114. [Google Scholar] [CrossRef]
- Fuentes-Aceituno, J.; Lapidus, G. A Kinetic-Mechanistic Study of The Hydrogen Evolution Reaction in Sulfuric Acid Solutions with Different Electrode Materials. J. New Mater. Electrochem. Syst. 2012, 15, 225–231. [Google Scholar] [CrossRef]
- Akbayrak, M.; Önal, A.M. High Durability and Electrocatalytic Activity Toward Hydrogen Evolution Reaction with Ultralow Rhodium Loading on Titania. J. Electrochem. Soc. 2020, 167, 156501. [Google Scholar] [CrossRef]
- Gebremariam, G.K.; Jovanović, A.Z.; Dobrota, A.S.; Skorodumova, N.V.; Pašti, I.A. Hydrogen Evolution Volcano(es)—From Acidic to Neutral and Alkaline Solutions. Catalysts 2022, 12, 1541. [Google Scholar] [CrossRef]
- Hall, D.M.; Beck, J.R.; Lvov, S.N. Electrochemical kinetics of the hydrogen reaction on platinum in concentrated HCl(aq). Electrochem. Commun. 2015, 57, 74–77. [Google Scholar] [CrossRef]
- Tang, Z.-Q.; Liao, L.-W.; Zheng, Y.-L.; Kang, J.; Chen, Y.-X. Temperature Effect on Hydrogen Evolution Reaction at Au Electrode. Chin. J. Chem. Phys. 2012, 25. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, Y.; Vasileff, A.; Qiao, S.-Z. The Hydrogen Evolution Reaction in Alkaline Solution: From Theory, Single Crystal Models, to Practical Electrocatalysts. Angew. Chem. Int. Ed. 2018, 57, 7568–7579. [Google Scholar] [CrossRef]
- Badawy, W.; Feky, H.; Helal, N.; Mohammed, H. Cathodic hydrogen evolution on molybdenum in NaOH solutions. Int. J. Hydrogen Energy 2013, 38, 9625–9632. [Google Scholar] [CrossRef]
- Meethal, R.P.; Saibi, R.; Srinivasan, R. Hydrogen evolution reaction on polycrystalline Au inverted rotating disc electrode in HClO4 and NaOH solutions. Int. J. Hydrogen Energy 2022, 47, 14304–14318. [Google Scholar] [CrossRef]
- Chanda, D.; Hnát, J.; Dobrota, A.S.; Pašti, I.A.; Paidar, M.; Bouzek, K. The effect of surface modification by reduced graphene oxide on the electrocatalytic activity of nickel towards the hydrogen evolution reaction. Phys. Chem. Chem. Phys. 2015, 17, 26864–26874. [Google Scholar] [CrossRef]
- Bao, F.; Kemppainen, E.; Dorbandt, I.; Bors, R.; Xi, F.; Schlatmann, R.; van de Krol, R.; Calnan, S. Understanding the Hydrogen Evolution Reaction Kinetics of Electrodeposited Nickel-Molybdenum in Acidic, Near-Neutral, and Alkaline Conditions. ChemElectroChem 2020, 8, 195–208. [Google Scholar] [CrossRef]
- Petrii, O.A.; Tsirlina, G.A. Electrocatalytic activity prediction for hydrogen electrode reaction: intuition, art, science. Electrochimica Acta 1994, 39, 1739–1747. [Google Scholar] [CrossRef]
- Miousse, D.; Lasia, A.; Borck, V. Hydrogen evolution reaction on Ni-Al-Mo and Ni-Al electrodes prepared by low pressure plasma spraying. J. Appl. Electrochem. 1995, 25, 592–602. [Google Scholar] [CrossRef]
- Tang, D.; Lu, J.; Zhuang, L.; Liu, P. Calculations of the exchange current density for hydrogen electrode reactions: A short review and a new equation. J. Electroanal. Chem. 2010, 644, 144–149. [Google Scholar] [CrossRef]
- Ernst, S.; Hamann, C. The pH-dependence of the hydrogen exchange current density at smooth platinum in alkaline solution (KOH). J. Electroanal. Chem. Interfacial Electrochem. 1975, 60, 97–100. [Google Scholar] [CrossRef]
- Santos, D.; Sequeira, C.; Macciò, D.; Saccone, A.; Figueiredo, J. Platinum–rare earth electrodes for hydrogen evolution in alkaline water electrolysis. Int. J. Hydrogen Energy 2013, 38, 3137–3145. [Google Scholar] [CrossRef]
- Amaral, L.; Cardoso, D.; Šljukić, B.; Santos, D.; Sequeira, C. Electrochemistry of hydrogen evolution in ionic liquids aqueous mixtures. 8th International Symposium on Materials (MATERIAIS) / 18th National Materials Conference of Sociedade-Portuguesa-de-Materiais. LOCATION OF CONFERENCE, COUNTRYDATE OF CONFERENCE; pp. 407–412.
- Zhou, Z.; Pei, Z.; Wei, L.; Zhao, S.; Jian, X.; Chen, Y. Electrocatalytic hydrogen evolution under neutral pH conditions: current understandings, recent advances, and future prospects. Energy Environ. Sci. 2020, 13, 3185–3206. [Google Scholar] [CrossRef]
- Xie, X.; Song, M.; Wang, L.; Engelhard, M.H.; Luo, L.; Miller, A.; Zhang, Y.; Du, L.; Pan, H.; Nie, Z.; et al. Electrocatalytic Hydrogen Evolution in Neutral pH Solutions: Dual-Phase Synergy. ACS Catal. 2019, 9, 8712–8718. [Google Scholar] [CrossRef]
- Merrill, M.D.; Logan, B.E. Electrolyte effects on hydrogen evolution and solution resistance in microbial electrolysis cells. J. Power Sources 2009, 191, 203–208. [Google Scholar] [CrossRef]
- Weber, D.J.; Janssen, M.; Oezaslan, M. Effect of Monovalent Cations on the HOR/HER Activity for Pt in Alkaline Environment. J. Electrochem. Soc. 2019, 166, F66–F73. [Google Scholar] [CrossRef]
- Guha, A.; Kaley, N.M.; Mondal, J.; Narayanan, T.N. Engineering the hydrogen evolution reaction of transition metals: effect of Li ions. J. Mater. Chem. A 2020, 8, 15795–15808. [Google Scholar] [CrossRef]
- Conway, B.; Tilak, B. Interfacial processes involving electrocatalytic evolution and oxidation of H2, and the role of chemisorbed H. Electrochimica Acta 2002, 47, 3571–3594. [Google Scholar] [CrossRef]
- Sheng, W. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat. Commun. 2015, 6, 5848. [Google Scholar] [CrossRef] [PubMed]
- McCrum, I.T.; Janik, M.J. pH and Alkali Cation Effects on the Pt Cyclic Voltammogram Explained Using Density Functional Theory. J. Phys. Chem. C 2015, 120, 457–471. [Google Scholar] [CrossRef]
- Kim, H.; Hong, J.; Park, K.-Y.; Kim, H.; Kim, S.-W.; Kang, K. Aqueous Rechargeable Li and Na Ion Batteries. Chem. Rev. 2014, 114, 11788–11827. [Google Scholar] [CrossRef]
- Suo, L.; Borodin, O.; Gao, T.; Olguin, M.; Ho, J.; Fan, X.; Luo, C.; Wang, C.; Xu, K. “Water-in-salt” electrolyte enables high-voltage aqueous lithium-ion chemistries. Science 2015, 350, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Laursen, A.B.; Varela, A.S.; Dionigi, F.; Fanchiu, H.; Miller, C.; Trinhammer, O.L.; Rossmeisl, J.; Dahl, S. Electrochemical Hydrogen Evolution: Sabatier’s Principle and the Volcano Plot. J. Chem. Educ. 2012, 89, 1595–1599. [Google Scholar] [CrossRef]
- Herranz, J.; Durst, J.; Fabbri, E.; Pătru, A.; Cheng, X.; Permyakova, A.A.; Schmidt, T.J. Interfacial effects on the catalysis of the hydrogen evolution, oxygen evolution and CO2-reduction reactions for (co-)electrolyzer development. Nano Energy 2016, 29, 4–28. [Google Scholar] [CrossRef]
- Sheng, W. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat. Commun. 2015, 6, 5848. [Google Scholar] [CrossRef]
- Strmcnik, D.; Lopes, P.P.; Genorio, B.; Stamenkovic, V.R.; Markovic, N.M. Design principles for hydrogen evolution reaction catalyst materials. Nano Energy 2016, 29, 29–36. [Google Scholar] [CrossRef]
- Marković, N.; Jr., P.R. Surface science studies of model fuel cell electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. [CrossRef]
- Danilovic, N.; Subbaraman, R.; Strmcnik, D.; Stamenkovic, V.; Markovic, N. Electrocatalysis of the HER in acid and alkaline media. J. Serbian Chem. Soc. 2013, 78, 2007–2015. [Google Scholar] [CrossRef]
- Zheng, J.; Sheng, W.; Zhuang, Z.; Xu, B.; Yan, Y. Universal dependence of hydrogen oxidation and evolution reaction activity of platinum-group metals on pH and hydrogen binding energy. Sci. Adv. 2016, 2, e1501602–1501602. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, G.; Li, G.; Huang, B.; Pan, J.; Liu, Q.; Han, J.; Xiao, L.; Lu, J.; Zhuang, L. Pt–Ru catalyzed hydrogen oxidation in alkaline media: oxophilic effect or electronic effect? Energy Environ. Sci. 2014, 8, 177–181. [Google Scholar] [CrossRef]
- van der Niet, M.J.; Garcia-Araez, N.; Hernández, J.; Feliu, J.M.; Koper, M.T. Water dissociation on well-defined platinum surfaces: The electrochemical perspective. Catal. Today 2013, 202, 105–113. [Google Scholar] [CrossRef]
- Marković, N.M.; Schmidt, T.J.; Grgur, B.N.; Gasteiger, H.A.; Behm, R.J.; Ross, P.N. Effect of Temperature on Surface Processes at the Pt(111)−Liquid Interface: Hydrogen Adsorption, Oxide Formation, and CO Oxidation. J. Phys. Chem. B 1999, 103, 8568–8577. [Google Scholar] [CrossRef]
- Ledezma-Yanez, I.; Wallace, W.D.Z.; Sebastián-Pascual, P.; Climent, V.; Feliu, J.M.; Koper, M.T.M. Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes. Nat. Energy 2017, 2. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Chan, K.; Ahmed, R.; Tripković, V.; Björketun, M.E. pH in atomic scale simulations of electrochemical interfaces. Phys. Chem. Chem. Phys. 2013, 15, 10321–10325. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Chan, K.; Skúlason, E.; Björketun, M.E.; Tripkovic, V. On the pH dependence of electrochemical proton transfer barriers. Catal. Today 2016, 262, 36–40. [Google Scholar] [CrossRef]
- Cheng, T.; Wang, L.; Merinov, B.V.; Goddard, W.A. Explanation of Dramatic pH-Dependence of Hydrogen Binding on Noble Metal Electrode: Greatly Weakened Water Adsorption at High pH. J. Am. Chem. Soc. 2018, 140, 7787–7790. [Google Scholar] [CrossRef]
- T. Shinagawa and K. Takanabe, “Electrolyte Engineering toward Efficient Hydrogen Production Elec-trocatalysis with Oxygen-Crossover Regulation under Densely Buffered Near-Neutral pH Conditions,” Journal of Physical Chemistry C, vol. 120, no. 3, pp. 1785–1794, Jan. 2016. [CrossRef]
- Murthy, A.P.; Govindarajan, D.; Theerthagiri, J.; Madhavan, J.; Parasuraman, K. Metal-doped molybdenum nitride films for enhanced hydrogen evolution in near-neutral strongly buffered aerobic media. Electrochimica Acta 2018, 283, 1525–1533. [Google Scholar] [CrossRef]
- Shinagawa, T.; Takanabe, K. Electrocatalytic Hydrogen Evolution under Densely Buffered Neutral pH Conditions. J. Phys. Chem. C 2015, 119, 20453–20458. [Google Scholar] [CrossRef]
- Shinagawa, T.; Takanabe, K. Towards Versatile and Sustainable Hydrogen Production through Electrocatalytic Water Splitting: Electrolyte Engineering. ChemSusChem 2017, 10, 1318–1336. [Google Scholar] [CrossRef] [PubMed]
- Kanan, M.W.; Nocera, D.G. In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co 2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Lutterman, D.A.; Surendranath, Y.; Nocera, D.G. A Self-Healing Oxygen-Evolving Catalyst. J. Am. Chem. Soc. 2009, 131, 3838–3839. [Google Scholar] [CrossRef] [PubMed]
- Kanan, M.W.; Yano, J.; Surendranath, Y.; Dincă, M.; Yachandra, V.K.; Nocera, D.G. Structure and Valency of a Cobalt−Phosphate Water Oxidation Catalyst Determined by in Situ X-ray Spectroscopy. J. Am. Chem. Soc. 2010, 132, 13692–13701. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, L.D.S.; Bergel, A.; Féron, D.; Basséguy, R. Hydrogen production by electrolysis of a phosphate solution on a stainless steel cathode. Int. J. Hydrogen Energy 2010, 35, 8561–8568. [Google Scholar] [CrossRef]
- S. Zhu, X. Qin, Y. Yao, and M. Shao, “PH-Dependent Hydrogen and Water Binding Energies on Platinum Surfaces as Directly Probed through Surface-Enhanced Infrared Absorption Spectroscopy,” J Am Chem Soc, vol. 142, no. 19, pp. 8748–8754, May 2020. [CrossRef]
- Y. Sun et al., “Electrodeposited cobalt-sulfide catalyst for electrochemical and photoelectrochemical hy-drogen generation from water,” J Am Chem Soc, vol. 135, no. 47, pp. 17699–17702, Nov. 2013. [CrossRef]
- Danilovic, N.; Subbaraman, R.; Strmcnik, D.; Stamenkovic, V.; Markovic, N. Electrocatalysis of the HER in acid and alkaline media. J. Serbian Chem. Soc. 2013, 78, 2007–2015. [Google Scholar] [CrossRef]
- Zhang, R.; Pearce, P.E.; Duan, Y.; Dubouis, N.; Marchandier, T.; Grimaud, A. Importance of Water Structure and Catalyst–Electrolyte Interface on the Design of Water Splitting Catalysts. Chem. Mater. 2019, 31, 8248–8259. [Google Scholar] [CrossRef]
- Strmcnik, D.; Kodama, K.; van der Vliet, D.; Greeley, J.; Stamenkovic, V.R.; Marković, N.M. The role of non-covalent interactions in electrocatalytic fuel-cell reactions on platinum. Nat. Chem. 2009, 1, 466–472. [Google Scholar] [CrossRef]
- Tymoczko, J.; Colic, V.; Ganassin, A.; Schuhmann, W.; Bandarenka, A.S. Influence of the alkali metal cations on the activity of Pt(111) towards model electrocatalytic reactions in acidic sulfuric media. Catal. Today 2015, 244, 96–102. [Google Scholar] [CrossRef]
- D. Strmcnik et al., “Effects of Li+, K+, and Ba2+ cations on the ORR at model and high surface area Pt and Au surfaces in alkaline solutions,” Journal of Physical Chemistry Letters, vol. 2, no. 21, pp. 2733–2736, Nov. 2011. [CrossRef]
- García, N.; Climent, V.; Orts, J.M.; Feliu, J.M.; Aldaz, A. Effect of pH and Alkaline Metal Cations on the Voltammetry of Pt(111) Single Crystal Electrodes in Sulfuric Acid Solution. Chemphyschem 2004, 5, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Li, J.; Jiao, L.; Doan, H.T.T.; Liu, Z.; Zhao, Z.; Huang, Y.; Abraham, K.M.; Mukerjee, S.; Jia, Q. Unifying the Hydrogen Evolution and Oxidation Reactions Kinetics in Base by Identifying the Catalytic Roles of Hydroxyl-Water-Cation Adducts. J. Am. Chem. Soc. 2019, 141, 3232–3239. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Garlyyev, B.; Watzele, S.; Liang, Y.; Fichtner, J.; Pohl, M.D.; Bandarenka, A.S. Influence of Alkali Metal Cations on the Hydrogen Evolution Reaction Activity of Pt, Ir, Au, and Ag Electrodes in Alkaline Electrolytes. ChemElectroChem 2018, 5, 2326–2329. [Google Scholar] [CrossRef]
- Dubouis, N.; Grimaud, A. The hydrogen evolution reaction: from material to interfacial descriptors. Chem. Sci. 2019, 10, 9165–9181. [Google Scholar] [CrossRef]
- Huang, B.; Rao, R.R.; You, S.; Myint, K.H.; Song, Y.; Wang, Y.; Ding, W.; Giordano, L.; Zhang, Y.; Wang, T.; et al. Cation- and pH-Dependent Hydrogen Evolution and Oxidation Reaction Kinetics. JACS Au 2021, 1, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Rao, R.R.; You, S.; Myint, K.H.; Song, Y.; Wang, Y.; Ding, W.; Giordano, L.; Zhang, Y.; Wang, T.; et al. Cation- and pH-Dependent Hydrogen Evolution and Oxidation Reaction Kinetics. JACS Au 2021, 1, 1674–1687. [Google Scholar] [CrossRef]
- Monteiro, M.C.O.; Goyal, A.; Moerland, P.; Koper, M.T.M. Understanding Cation Trends for Hydrogen Evolution on Platinum and Gold Electrodes in Alkaline Media. ACS Catal. 2021, 11, 14328–14335. [Google Scholar] [CrossRef]
- Guha, A.; Narayanan, T.N. Effect of ‘water-in-salt’ electrolytes in the electrochemical hydrogen evolution reaction of carbon nanotubes. J. Physics: Energy 2020, 2, 034001. [Google Scholar] [CrossRef]
- Guha, A.; Narayanaru, S.; Narayanan, T.N. Tuning the Hydrogen Evolution Reaction on Metals by Lithium Salt. ACS Appl. Energy Mater. 2018, 1, 7116–7122. [Google Scholar] [CrossRef]
- Matanovic, I.; Atanassov, P.; Garzon, F.; Henson, N.J. Density Functional Theory Study of the Alkali Metal Cation Adsorption on Pt(111), Pt(100), and Pt(110) Surfaces. ECS Trans. 2014, 61, 47–53. [Google Scholar] [CrossRef]
- Garlyyev, B.; Xue, S.; Watzele, S.; Scieszka, D.; Bandarenka, A.S. Influence of the Nature of the Alkali Metal Cations on the Electrical Double-Layer Capacitance of Model Pt(111) and Au(111) Electrodes. J. Phys. Chem. Lett. 2018, 9, 1927–1930. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.N.; McCrum, I.T.; Janik, M.J. Alkali cation specific adsorption onto fcc(111) transition metal electrodes. Phys. Chem. Chem. Phys. 2014, 16, 13699–13707. [Google Scholar] [CrossRef] [PubMed]
- Taji, Y.; Zagalskaya, A.; Evazzade, I.; Watzele, S.; Song, K.-T.; Xue, S.; Schott, C.; Garlyyev, B.; Alexandrov, V.; Gubanova, E.; et al. Alkali metal cations change the hydrogen evolution reaction mechanisms at Pt electrodes in alkaline media. Nano Mater. Sci. 2022. [Google Scholar] [CrossRef]
- Goyal, A.; Koper, M.T.M. The Interrelated Effect of Cations and Electrolyte pH on the Hydrogen Evolution Reaction on Gold Electrodes in Alkaline Media. Angew. Chem. Int. Ed. 2021, 60, 13452–13462. [Google Scholar] [CrossRef]
- Kamat, G.A.; Zeledón, J.A.Z.; Gunasooriya, G.T.K.K.; Dull, S.M.; Perryman, J.T.; Nørskov, J.K.; Stevens, M.B.; Jaramillo, T.F. Acid anion electrolyte effects on platinum for oxygen and hydrogen electrocatalysis. Commun. Chem. 2022, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- E. Lamy-Pitara, S. El Mouahid, and J. Barbier, “Effect of anions on catalytic and electrocatalytic hydro-genations and on the electrocatalytic oxidation and evolution of hydrogen on platinum,” 2000. [Online]. Available: www.elsevier.nl/locate/electacta.
- Quaino, P.; Juarez, F.; Santos, E.; Schmickler, W. Volcano plots in hydrogen electrocatalysis – uses and abuses. 2014, 5, 846–854. [CrossRef]
- Arminio-Ravelo, J.A.; Jensen, A.W.; Jensen, K.D.; Quinson, J.; Escudero-Escribano, M. Electrolyte Effects on the Electrocatalytic Performance of Iridium-Based Nanoparticles for Oxygen Evolution in Rotating Disc Electrodes. Chemphyschem 2019, 20, 2956–2963. [Google Scholar] [CrossRef] [PubMed]
- Faid, A.Y.; Foroughi, F.; Sunde, S.; Pollet, B. Unveiling hydrogen evolution dependence on KOH concentration for polycrystalline and nanostructured nickel-based catalysts. J. Appl. Electrochem. 2022, 52, 1819–1826. [Google Scholar] [CrossRef]
- Li, D.; Park, E.J.; Zhu, W.; Shi, Q.; Zhou, Y.; Tian, H.; Lin, Y.; Serov, A.; Zulevi, B.; Baca, E.D.; et al. Highly quaternized polystyrene ionomers for high performance anion exchange membrane water electrolysers. Nat. Energy 2020, 5, 378–385. [Google Scholar] [CrossRef]
- Kuznetsov, A.N.; Oshchepkov, A.G.; Cherstiouk, O.V.; Simonov, P.A.; Nazmutdinov, R.R.; Savinova, E.R.; Bonnefont, A. Influence of the NaOH Concentration on the Hydrogen Electrode Reaction Kinetics of Ni and NiCu Electrodes. ChemElectroChem 2020, 7, 1438–1447. [Google Scholar] [CrossRef]
- Cossar, E.; Houache, M.S.; Zhang, Z.; Baranova, E.A. Comparison of electrochemical active surface area methods for various nickel nanostructures. J. Electroanal. Chem. 2020, 870, 114246. [Google Scholar] [CrossRef]
- Shinagawa, T.; Takanabe, K. Impact of solute concentration on the electrocatalytic conversion of dissolved gases in buffered solutions. J. Power Sources 2015, 287, 465–471. [Google Scholar] [CrossRef]
- Barwe, S.; Mei, B.; Masa, J.; Schuhmann, W.; Ventosa, E. Overcoming cathode poisoning from electrolyte impurities in alkaline electrolysis by means of self-healing electrocatalyst films. Nano Energy 2018, 53, 763–768. [Google Scholar] [CrossRef]
- Weber, D.J.; Dosche, C.; Oezaslan, M. Fundamental Aspects of Contamination during the Hydrogen Evolution/Oxidation Reaction in Alkaline Media. J. Electrochem. Soc. 2020, 167, 024506. [Google Scholar] [CrossRef]
- Li, X.; Gunathunge, C.M.; Agrawal, N.; Montalvo-Castro, H.; Jin, J.; Janik, M.J.; Waegele, M.M. Impact of Alkali Metal Cations and Iron Impurities on the Evolution of Hydrogen on Cu Electrodes in Alkaline Electrolytes. J. Electrochem. Soc. 2020, 167, 106505. [Google Scholar] [CrossRef]
- Klaus, S.; Cai, Y.; Louie, M.W.; Trotochaud, L.; Bell, A.T. Effects of Fe Electrolyte Impurities on Ni(OH)2/NiOOH Structure and Oxygen Evolution Activity. J. Phys. Chem. C 2015, 119, 7243–7254. [Google Scholar] [CrossRef]
- Salmanion, M.; Najafpour, M.M. Oxygen-evolution reaction by gold and cobalt in iron and nickel free electrolyte. Int. J. Hydrogen Energy 2020, 46, 1509–1516. [Google Scholar] [CrossRef]
- Klaus, S.; Trotochaud, L.; Cheng, M.; Head-Gordon, M.; Bell, A.T. Experimental and Computational Evidence of Highly Active Fe Impurity Sites on the Surface of Oxidized Au for the Electrocatalytic Oxidation of Water in Basic Media. ChemElectroChem 2015, 3, 66–73. [Google Scholar] [CrossRef]
- Gong, L.; Koh, J.R.; Yeo, B.S. Mechanistic Study of the Synergy between Iron and Transition Metals for the Catalysis of the Oxygen Evolution Reaction. ChemSusChem 2018, 11, 3790–3795. [Google Scholar] [CrossRef]
- Bockris, J.O.; Conway, B.E. Studies in hydrogen overpotential. The effect of catalytic poisons at platinized platinum and nickel. Trans. Faraday Soc. 1949, 45, 989–999. [Google Scholar] [CrossRef]
- Gao, L.; Conway, B. Poisoning effects of arsenic species on H adsorption and kinetic behaviour of the H2 evolution reaction at Pt in KOH solution. J. Electroanal. Chem. 1995, 395, 261–271. [Google Scholar] [CrossRef]
- Gao, L.J.; Qian, S.Y.; Conway, B.E. Arsenic poisoning effects on cathodic polarization and hydrogen adsorption at platinum and steel electrodes in KF · 2HF melts. J. Appl. Electrochem. 1996, 26, 803–814. [Google Scholar] [CrossRef]
- Becker, H.; Murawski, J.; Shinde, D.V.; Stephens, I.E.L.; Hinds, G.; Smith, G. Impact of impurities on water electrolysis: a review. Sustain. Energy Fuels 2023, 7, 1565–1603. [Google Scholar] [CrossRef]
- Barwe, S.; Mei, B.; Masa, J.; Schuhmann, W.; Ventosa, E. Overcoming cathode poisoning from electrolyte impurities in alkaline electrolysis by means of self-healing electrocatalyst films. Nano Energy 2018, 53, 763–768. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Das, C.; Maji, T.K. MOF derived carbon based nanocomposite materials as efficient electrocatalysts for oxygen reduction and oxygen and hydrogen evolution reactions. RSC Adv. 2018, 8, 26728–26754. [Google Scholar] [CrossRef] [PubMed]
- Pumera, M. Materials Electrochemists’ Never-Ending Quest for Efficient Electrocatalysts: The Devil Is in the Impurities. ACS Catal. 2020, 10, 7087–7092. [Google Scholar] [CrossRef]
- Wang, L.; Sofer, Z.; Pumera, M. Will Any Crap We Put into Graphene Increase Its Electrocatalytic Effect? ACS Nano 2020, 14, 21–25. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The role of spectator ions, contained in the electrolyte, in modifying the outer Helmholtz layer (OHL) structure of the electrode/electrolyte interface. Schematic illustration (a) shows that larger cations (Cs+) lead to the removal of water molecules from the interface due to strong ion-surface interaction, while (b) demonstrates the formation of a stable interfacial water layer with smaller cations (Li+). According to Ref. [104].
Figure 1.
The role of spectator ions, contained in the electrolyte, in modifying the outer Helmholtz layer (OHL) structure of the electrode/electrolyte interface. Schematic illustration (a) shows that larger cations (Cs+) lead to the removal of water molecules from the interface due to strong ion-surface interaction, while (b) demonstrates the formation of a stable interfacial water layer with smaller cations (Li+). According to Ref. [104].
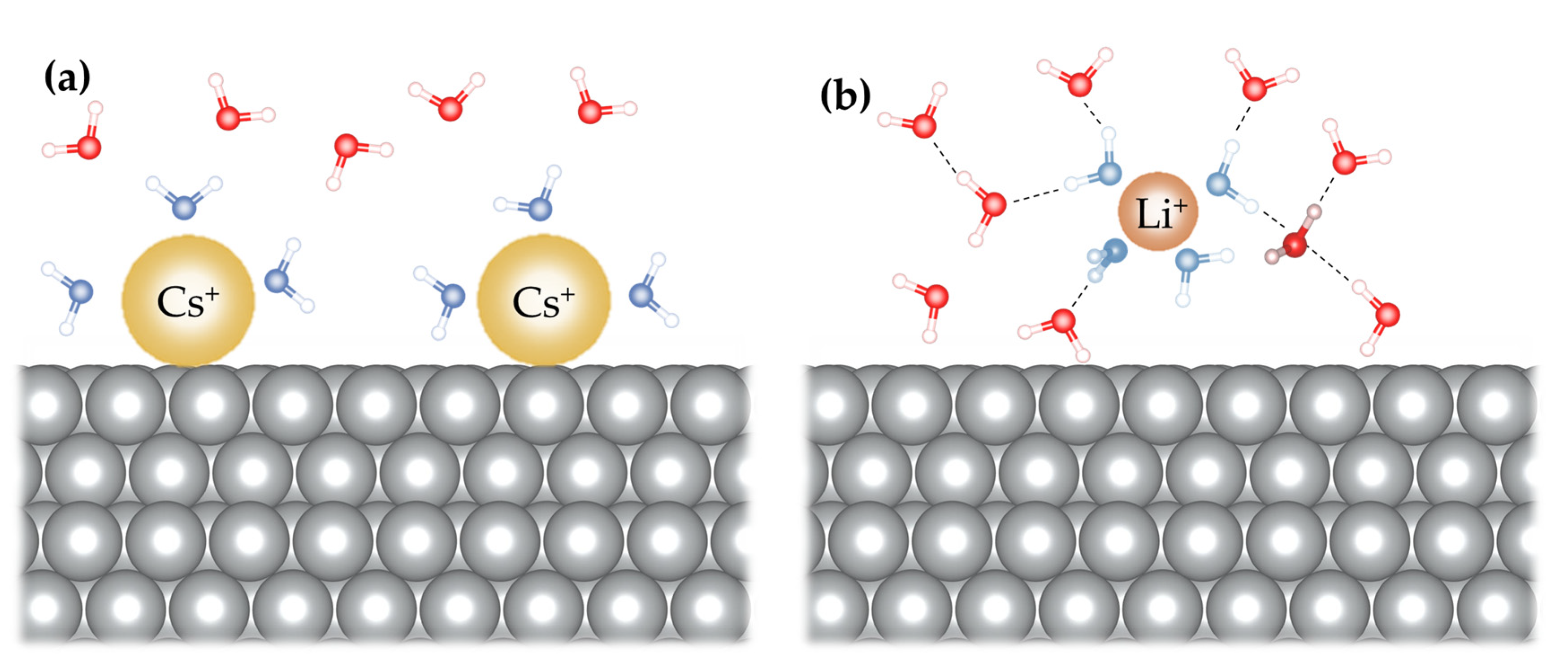
Figure 2.
Linear sweep voltammetries of a) Ir and b) Au in two different LiClO4 concentrations. Reproduced from Ref. [64] with permission from the Royal Society of Chemistry.
Figure 2.
Linear sweep voltammetries of a) Ir and b) Au in two different LiClO4 concentrations. Reproduced from Ref. [64] with permission from the Royal Society of Chemistry.
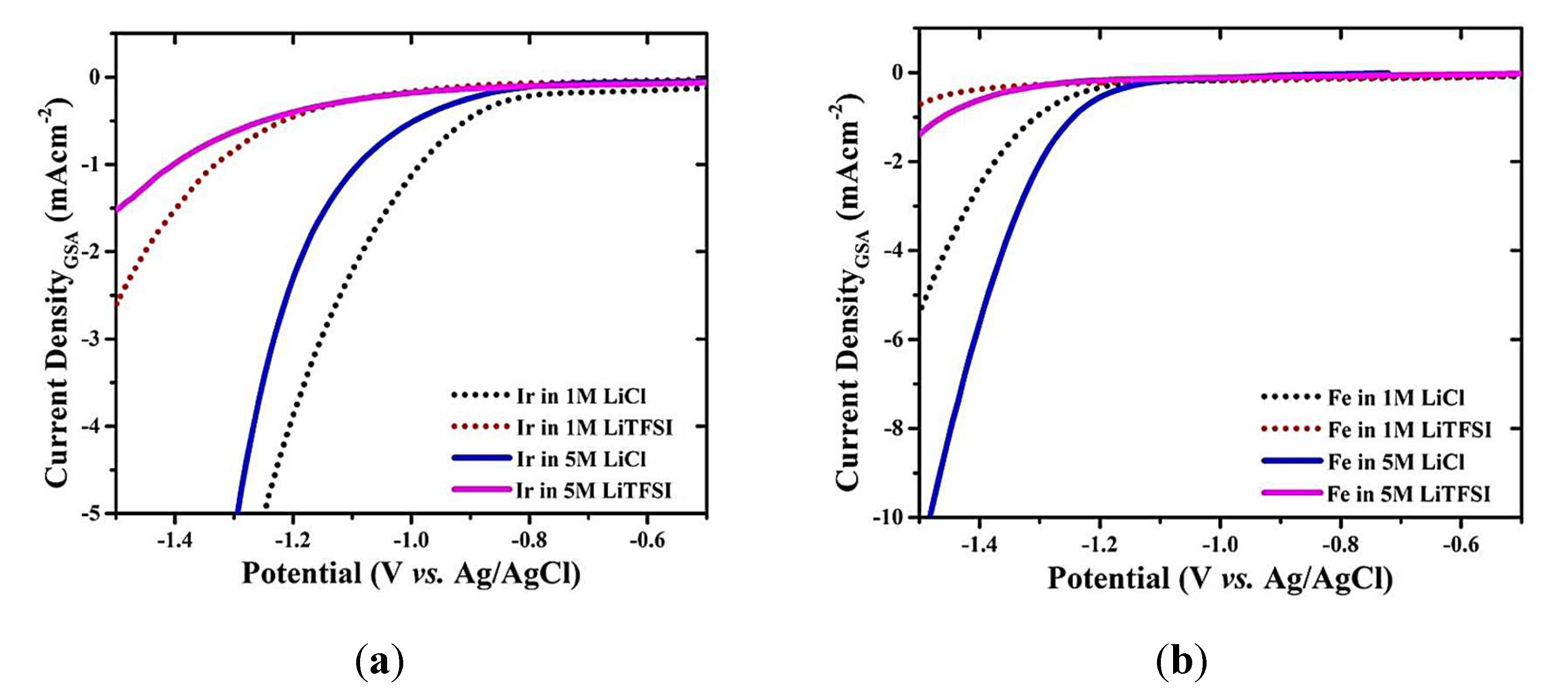
Figure 3.
The HER volcanoes in acidic solutions: (a) 0.1 M HClO4; (b) 0.1 M HCl. Freshly polished electrodes are represented by squares, and circles are used to represent electrodes after undergoing oxidative treatment. Reproduced from Ref [46]. This work open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY).
Figure 3.
The HER volcanoes in acidic solutions: (a) 0.1 M HClO4; (b) 0.1 M HCl. Freshly polished electrodes are represented by squares, and circles are used to represent electrodes after undergoing oxidative treatment. Reproduced from Ref [46]. This work open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY).
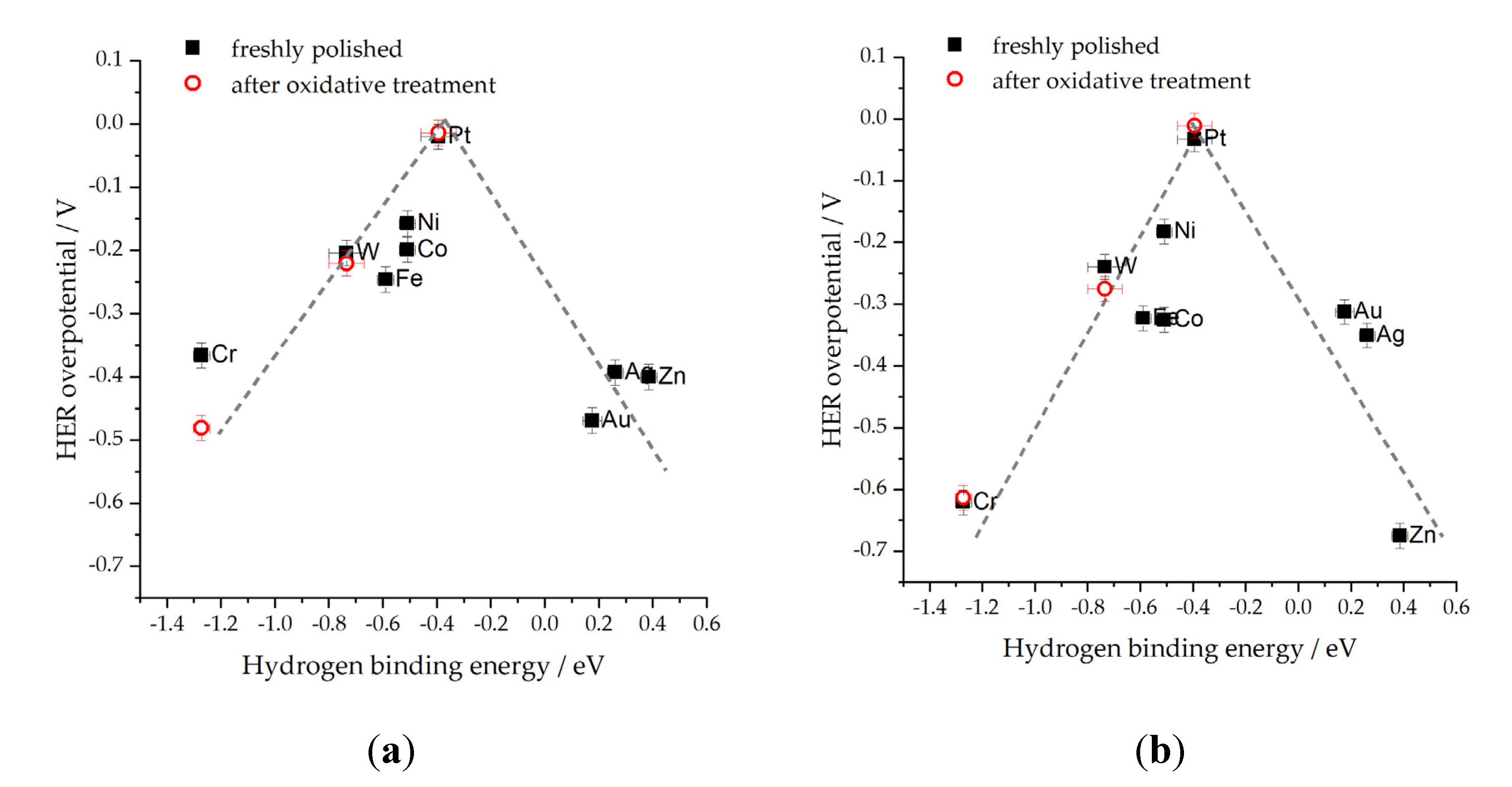
Figure 4.
The HER volcanoes in neutral solutions: (a) 0.5 M NaCl (simulated sea water); (b) 1 M KH2PO4 (pH was adjusted to 7.0 in both electrolytes.). Freshly polished electrodes are represented by squares, and circles are used to represent electrodes after undergoing oxidative treatment. Regarding the NaCl solution, significant changes in activity are indicated by arrows. Reproduced from [46]. This work open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY).
Figure 4.
The HER volcanoes in neutral solutions: (a) 0.5 M NaCl (simulated sea water); (b) 1 M KH2PO4 (pH was adjusted to 7.0 in both electrolytes.). Freshly polished electrodes are represented by squares, and circles are used to represent electrodes after undergoing oxidative treatment. Regarding the NaCl solution, significant changes in activity are indicated by arrows. Reproduced from [46]. This work open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY).
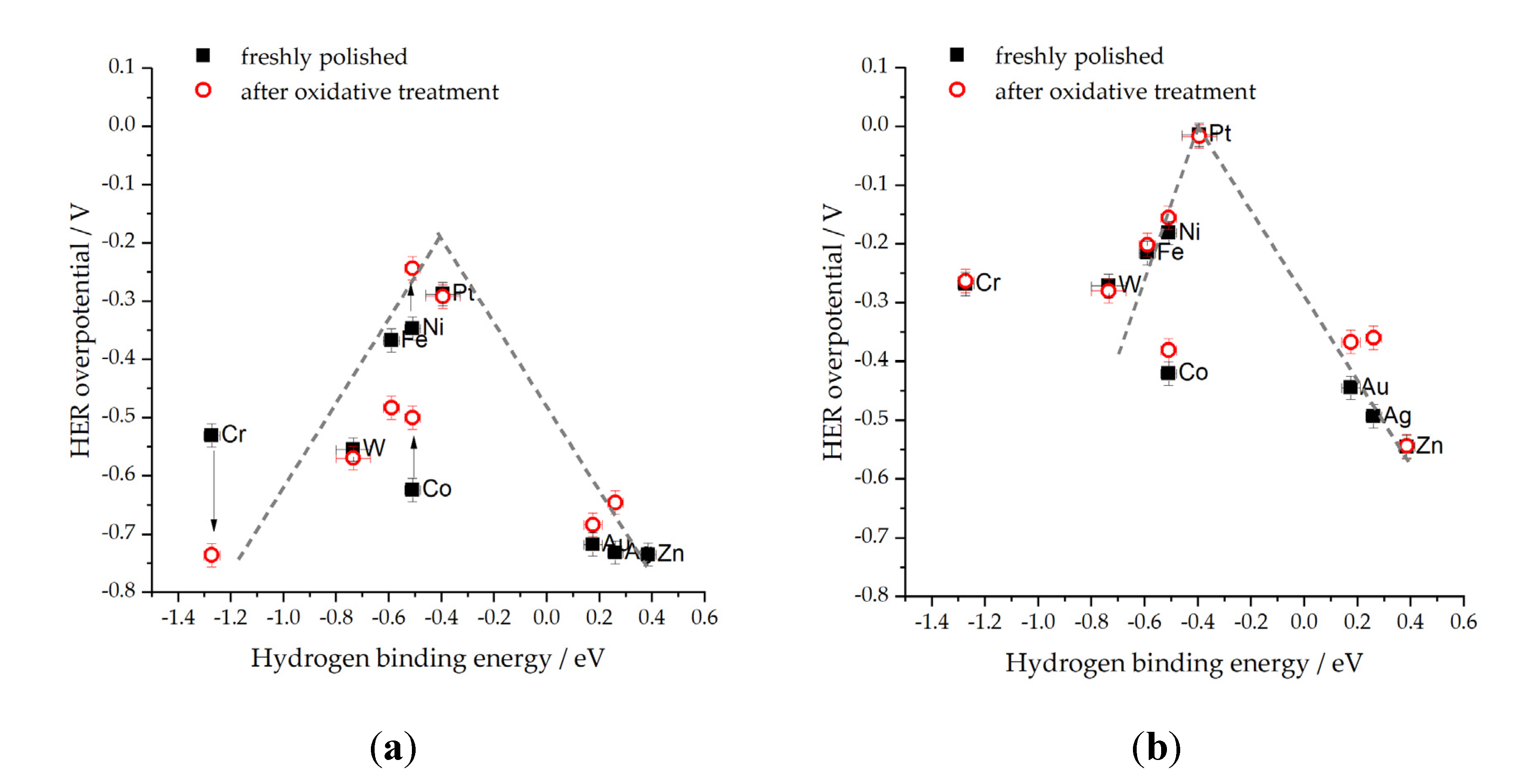
Figure 5.
Diagrammatic representation of the impact of different types of impurities on PEMWEs. Reproduced from Ref. [132]. with permission from the Royal Society of Chemistry.
Figure 5.
Diagrammatic representation of the impact of different types of impurities on PEMWEs. Reproduced from Ref. [132]. with permission from the Royal Society of Chemistry.
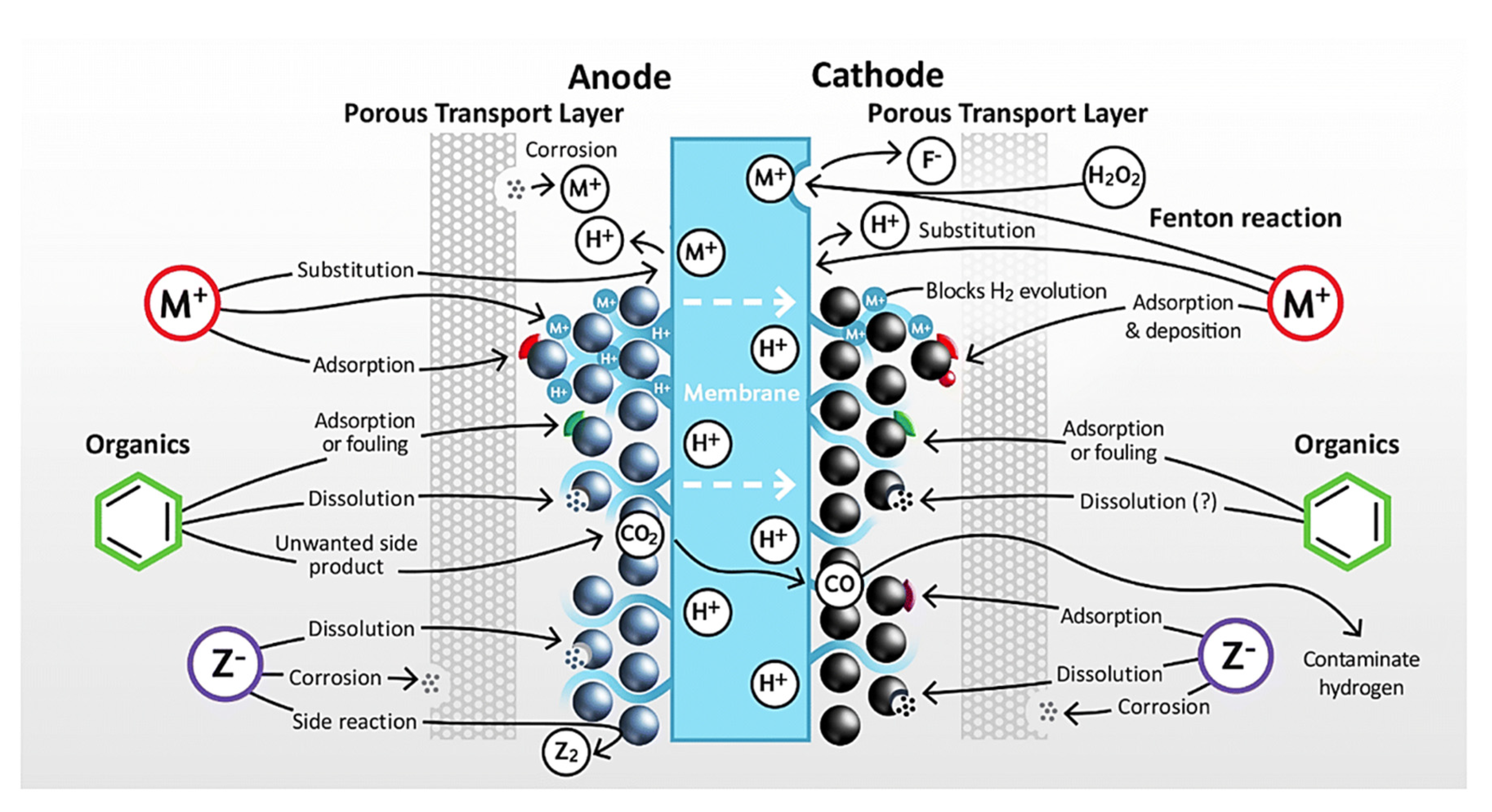

Table 1.
Hydrogen concentration and rate of hydrogen formation on Pt and Au electrodes in different electrolytes. Reprinted from Ref. [107]. Copyright 2018 American Chemical Society.
Table 1.
Hydrogen concentration and rate of hydrogen formation on Pt and Au electrodes in different electrolytes. Reprinted from Ref. [107]. Copyright 2018 American Chemical Society.
| Electrode | electrolyte | applied potential (V vs Ag/AgCl) |
duration (mins) | hydrogen concentrationn (ppm) |
rate of formation (×10−8 mol·s−1·cm−2) |
|---|---|---|---|---|---|
| Pt | 0.1 M LiTFSI | −1.1 | 5 | 16550 | 4056 |
| Pt | 20 M LiTFSI | −1.1 | 5 | 15799 | 3872 |
| Au | 0.1 M LiClO4 | −1.5 | 5 | 5755 | 2635 |
| Au | 5 M LiClO4 | −1.5 | 5 | 7773 | 3559 |
| Au | 0.1 M LiTFSI | −1.5 | 5 | 7950 | 3640 |
| Au | 20 M LiTFSI | −1.5 | 5 | 15577 | 7132 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



