
Submitted:
29 September 2023
Posted:
30 September 2023
You are already at the latest version
Abstract
The MMP-9-1562C/T polymorphism influence incidence and course of many diseases of the central nervous system. We found, using luciferase assays and Q-RT-PCR technique, allele-specific in-fluence of MMP-9-1562C/T polymorphism on the MMP-9 promoter activity and on the expression of MMP-9 mRNA in human neurons derived from SH-SY5Y cells. Then, we have elucidated the mechanism responsible for the allele-specific action of the MMP-9-1562C/T polymorphism on transcriptional regulation of the MMP-9 gene using pull-down assay combined with mass spec-trometry analysis, EMSA and EMSA supershift techniques, as well as DsiRNA-dependent gene silencing. We have found that MMP-9 promoter activity and MMP-9 mRNA expression are reg-ulated in human neurons in the MMP-9-1562C/T allele-specific manner with stronger upregulation conferred by the C allele. Moreover, we have revealed that the allele-specific action of the MMP-9-1562C/T polymorphism on the neuronal MMP-9 expression is related to HDAC1 and ZNF384 transcriptional regulators. We show that HDAC1 and ZNF384 bind differentially to the C and the T alleles forming in vitro different regulatory complexes. Moreover, our data demonstrate that HDAC1 and ZNF384 downregulate differentially the MMP-9 gene promoter activity and MMP-9 mRNA expression in vivo in human neurons acting mostly via the T allele.
Keywords:
MMP-9
; MMP-9-1562C/T polymorphism
; transcriptional regulation
; brain
; human neurons
1. Introduction
MMP-9-1562C/T is a single nucleotide polymorphism (SNP) localized at the -1562 position of the MMP-9 (Extracellular Matrix Metalloproteinase-9) gene promoter, which substitutes C for T [1] It is speculated that this polymorphism may exert a functional effect as it has been shown that reporter gene expression directed by the T allele is approximately 1.5-fold higher compared to reporter gene expression driven by the allele C in MALU cells [1]. Another hypothesis states that the presence of the C allele is responsible for a binding of nuclear repressor protein to the MMP-9 promoter resulting in its reduced transcriptional activity [1,2]. In contrast, lack of the protein binding to the T allele of the MMP-9 gene promoter was suggested to enhance its transcriptional activity [1,2].
It is known that MMP-9 has numerous functions in the development of the central nervous system (CNS) and may contribute to neurodevelopmental disorders [3,4,5,6,7,8]. Functional SNPs are found mainly in the regions of gene promoters, thus often affecting gene expression [9]. Several pieces of evidence have suggested the relationship between MMP-9-1562C/T polymorphism in the promoter of the MMP-9 gene and a risk of development of many diseases or a worsening of their course, which differs from population to population and between different studies in the same population. Such associations have been found in neurological disorders like multiple sclerosis (MS) [10,11,12,13], ischemic stroke (IS) [14,15], Guillain-Barré syndrome (GBS) [16], neurodegenerative diseases [17], schizophrenia (SZ) [18,19], or brain tumors [19].
The data show that the MMP-9 polymorphism, even if it is not the leading player responsible for the development of many diseases, certainly does matter [3]. Until now, no one has demonstrated regulation of MMP-9 expression by MMP-9-1562C/T polymorphism in brain cells.
2. Materials and Methods
SH-SY5Y cell culture and differentiation
Human SH-SY5Y neuroblastoma cells (ATCC® CRL-2266) were maintained in 1:1 EMEM/F12 (ATCC, Gibco), supplemented with 10% FBS and 1% penicillin/streptomycin 100X solution (Sigma Aldrich) at 37°C, 5% atmospheric CO2 in a humidified incubator. The ATCC CRL-2266 protocol was implemented for routine neuroblastoma subcultivation procedures. The cells were differentiated according to the protocol described by [20,21] with some modifications. Briefly, 2-3 X 104 cells/cm2 were seeded onto culture dishes and were allowed to be attached overnight in a standard medium with 10% FBS heat inactivated. The next day the FBS content of the culture medium was reduced to 5%, and the cells were exposed to 10µM all-trans retinoic acid (ATRA) (VWR). The cells were kept under these conditions for five days with a medium changes every second day. On the sixth day of differentiation, the cells were cultured in a standard medium without FBS, with addition of 10 µM ATRA and 50ng/ml brain-derived neurotropic factor (BDNF) (VWR) for an additional 3-4 days. Cells were ready to be used for further experiments.
Immunofluorescence microscopy
For immunofluorescence microscopy, the SH-SY5Y cultured cells were fixed with a fixing solution (4% paraformaldehyde, 4% sucrose in PBS, filtered) for 6-7 minutes at room temperature. After fixation, cells were washed three times in warmed PBS with 4% sucrose for 5 minutes at RT. Then SH-SY5Y cells were washed three times in PBS, permeabilized for 10 minutes in PBST (PBS with 0.3% Triton X-100; BioShop), and blocked for 2 h in 3% BSA (Sigma-Aldrich) in PBST at RT. The cells were then incubated with mouse monoclonal anti-MAP2 primary antibody (1:1000; M1406, Sigma-Aldrich) overnight at 4°C, washed three times in PBS, and thereafter incubated with the secondary polyclonal antibody conjugated with Alexa Fluor® 488 (goat anti-mouse, 1:500, Invitrogen) for 2h in RT. After washing, as indicated previously, coverslips with cells were mounted on glass slides with Fluoromount-G mounting medium, dried at room temperature for 5-6h and observed with ZEISS Spinning DISC confocal microscope with a 20x objective lens. Acquisition and analysis were performed with Java-based image processing program (ImageJ).
Preparation of nuclear extracts
Nuclear extracts from SH-SY5Y were prepared according to Bethyl Laboratories’ protocol with slight modifications. Briefly, cells were washed twice with cold PBS and resuspended in buffer A (1 M HEPES, pH 7.9, 1 M KCl, 0.5 M EDTA, 100% Nonindet-40 (Sigma), 1 M DTT (Sigma), and protease inhibitor cocktail (Sigma). Plate with cells was then incubated for 10 minutes at RT, scraped, and lysates were transferred to a microcentrifuge tube. Nuclei were pelleted at 4°C at 13 200 rpm for 3 minutes. Cytoplasmatic extracts were transferred to the new tube, and nuclei were resuspended in buffer B (1M HEPES, pH 7.9, 5M NaCl, O.5 M EDTA, glycerol, 1M DTT, and protease inhibitor cocktail). After pipetting, samples were vortexed every 15 minutes for 2 hours, centrifuged at 4°C at 13 200 rpm for 5 minutes, and supernatants were used as nuclear extracts. Protein concentration was estimated by The Pierce Detergent Compatible Bradford Assay Kit [Thermo Scientific #23246] using Synergy H4 (BioTek).
Transfection
Undifferentiated and differentiated SH-SY5Y cells were passaged at the same time and then cultured for five days. On the fifth day, cells were trypsinized, counted, and plated in 98-well plates at 4 x 104 cells per well so they were 70-90% confluent on the day of transfection. Undifferentiated SH-SY5Y cells were plated in a 100 µl standard medium containing serum with antibiotics. In contrast, differentiated SH-SY5Y cells were grown in a medium with 5% FBS, 10 µM ATRA and antibiotics (5th day of differentiation protocol – Figure 1a). The 6th-day medium was changed to antibiotics-free medium, and 10 µl of the mixture containing 0,3 µl Lipofectamine TM 3000 Reagent (Thermo Fischer Scientific), 200 ng appropriate construct (Addgene), and 2µl/µg DNA P3000TM Reagent diluted in 1:1 DMEM/F12 was added to each well. After 6 hours after transfection medium was changed to standard medium in case of undifferentiated SH-SY5Y cells, and to serum-free medium containing 10 µM ATRA, and 50ng/ml BDNF in case of differentiated SH-SY5Y cells.
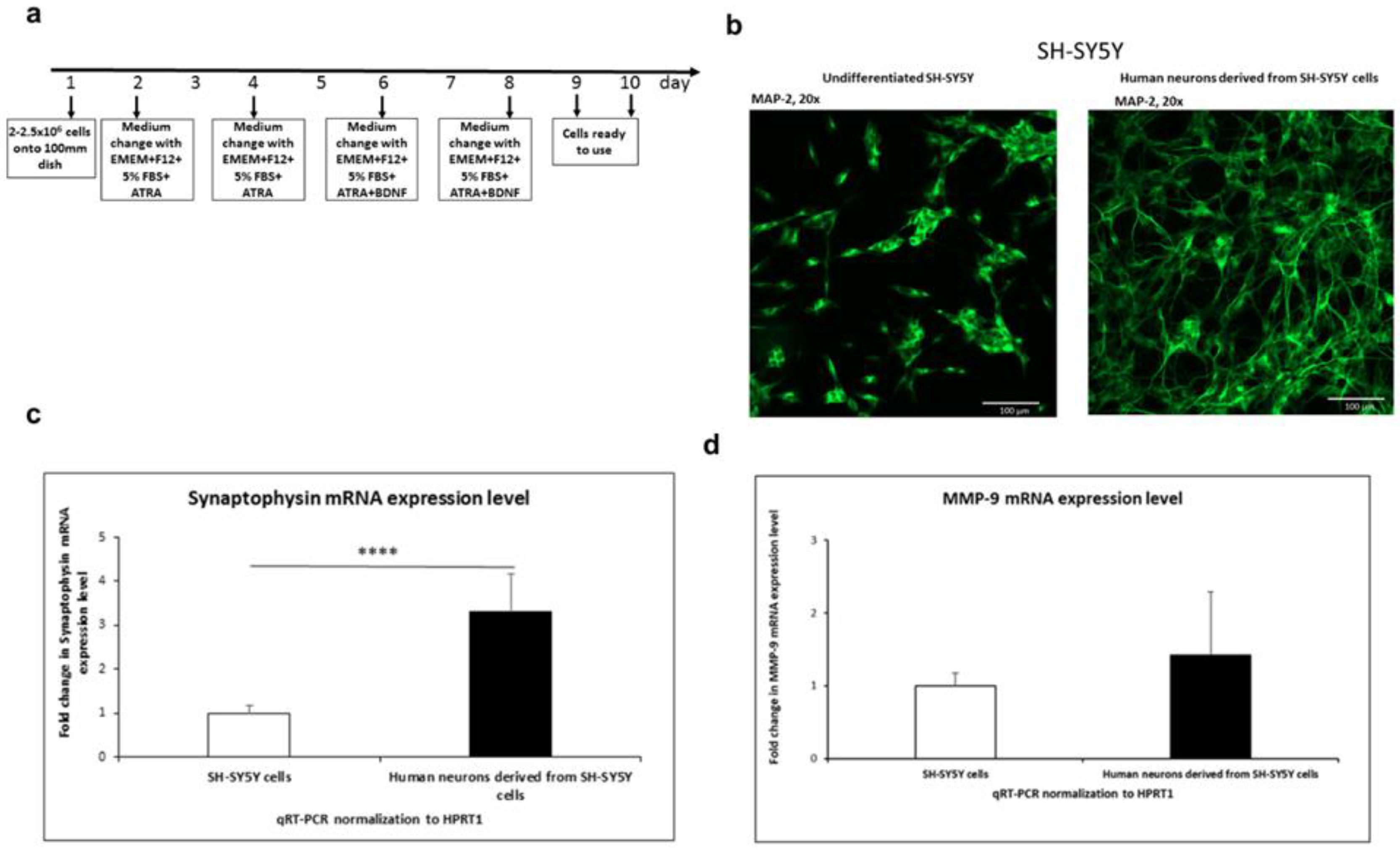
Figure 1.
SH-SY5Y differentiation into neurons. a. Timetable of SH-SY5Y human neuroblastoma cell line differentiation into neurons. b. The photographs of undifferentiated and differentiated SH-SY5Y neuroblastoma cells immunostained with the neuronal marker MAP-2 antibody. Alexa-488 was used to visualize MAP-2. Fluorescence images were obtained using a 20x objective. Scale bar: 100 µm. c. Expression of synaptophysin in SH-SY5Y cells and human neurons derived from SH-SY5Y cells analyzed by RT-qPCR. Data is presented as fold change in mRNA expression. The unpaired t-test test was followed by the D’Agostino and Pearson normality test. Values are given as mean ± SEM (****, p < 0.001; n=12). d. The overall expression level of the MMP-9 gene in SH-SY5Y cells and in human neurons. Data is presented as fold change in mRNA expression. The unpaired t-test test was followed by the D’Agostino and Pearson normality test. Values are given as mean ± SEM (n=9).
Figure 1.
SH-SY5Y differentiation into neurons. a. Timetable of SH-SY5Y human neuroblastoma cell line differentiation into neurons. b. The photographs of undifferentiated and differentiated SH-SY5Y neuroblastoma cells immunostained with the neuronal marker MAP-2 antibody. Alexa-488 was used to visualize MAP-2. Fluorescence images were obtained using a 20x objective. Scale bar: 100 µm. c. Expression of synaptophysin in SH-SY5Y cells and human neurons derived from SH-SY5Y cells analyzed by RT-qPCR. Data is presented as fold change in mRNA expression. The unpaired t-test test was followed by the D’Agostino and Pearson normality test. Values are given as mean ± SEM (****, p < 0.001; n=12). d. The overall expression level of the MMP-9 gene in SH-SY5Y cells and in human neurons. Data is presented as fold change in mRNA expression. The unpaired t-test test was followed by the D’Agostino and Pearson normality test. Values are given as mean ± SEM (n=9).
Luciferase reporter assay
DNA fragments corresponding to the region containing MMP-9-1562C/T SNP were amplified by PCR using heterozygous genomic DNA as a template and then cloned into KpnI and XhoI sites of the pGL3-promoter vector (Promega). DNA sequence analysis determined the insert's orientation and allele identity. The undifferentiated and differentiated SH-SY5Y cells were transfected with 200ng of appropriate construct using Lipofectamine TM 3000 Reagent. After 24h, the Dual-Glo® Luciferase Assay (Promega) was performed according to the manufacturer’s instructions.
Western blot
The nuclear or cytoplasmatic extracts (10 ug per well) were separated on 12.5% SDS-PAGE in Tris-glycine running buffer (25 mM Tris, 250 mM glycine (pH 8.3), 0.1% SDS) and electroblotted onto polyvinylidene difluoride membrane (Millipore) in transfer buffer (48 mM Tris, 39 mM glycine, 0.037% SDS, 20% methanol). Membranes were blocked in 5% nonfat milk/TBST (25 mm Tris-HCl (pH 8.0), 125 mm NaCl, 0.1% Tween 20) for 2 h at RT. Blots were then incubated with an appropriate primary antibody: TBP (TATA) Monoclonal Antibody (51841) 1: 1000 [Invitrogen #MA5-14739]; GAPDH 1:5000 [Millipore #MAB374] at 4 °C overnight. Peroxidase labeled Anti-mouse IgG secondary antibody [Vector Laboratories # PI-2000] was diluted 1:10000 in blocking buffer and incubated for over nihgt at 4°C. Proteins were detected by Western Bright Quantum HRP Substrate [Advansta #K-12042-D10] and chemiluminescence was measured using a UVITEC (Alliance) apparatus.
Gel shift assay
Gel shift assays were performed using LightShift® Chemiluminescent EMSA Kit (Thermo Scientific). Briefly, 5-10 µg of protein extracts prepared as described above were incubated with 20 fmol of 33-bp nucleotides labeled with biotin and 33-bp non-labeled competitor MMP-9-1562-C or –T (Supplementary Table1). Supershift assays were carried out by incubating 10 µg nuclear protein extracts with 1 µl of HDAC1 (D5C6U) XP® Rabbit mAb [Cell Signaling Technology #34589] or 0,5 µl of IgG normal Rabbit Ab [Santa Cruze sc-2027 X] (isotype control) for 30 min at RT. Then, DNA probes were added and incubated for 20 min at RT. Reactions with 5 µl ZNF384 Rabbit Ab [Invitrogen #PA-52044] or 0,5 µl of IgG normal Rabbit Ab (isotype control) were run at 4°C. The reactions with competitors (without Ab`s) were incubated for 20 min at RT before the electrophoretic run. DNA-protein and DNA protein-antibody complexes were resolved in 8% polyacrylamide gels in 0,5x TBE buffer (5xTBE: 450mM Tris, 450mM Boric Acid, 10 mM EDTA; pH = 8,3) for 1h with 100V at RT and transferred to Biodyne™ B Nylon Membrane (Thermo Scientific #77016) in 0,5x TBE buffer, on ice with 100V for 30 min. The complexes were visualized using UVITEC (Alliance).
Pull-down assay
60 pmol of 33 bp biotinylated nucleotides containing SNP of MMP-9-1562C/T (Supplementary Table 1.) were bound to 1mg of Dynabeads™ MyOne™ Streptavidin C1 (Thermo Fischer Scientific). 25 µl of SH-SY5Y nuclear extract was precleared in 1 ml of binding buffer (20 mM HEPES pH 7.6, 1mM EDTA, 10 mM (NH4)2SO4, 1mMDTT, 0,2% Tween-20, and 30mM KCl) in the presence of 1 mg of unbound beads and 288pmol of 33 bp non-labeled competitor MMP-9-1562-C or –T (Supplementary Table 1). Following magnetic separation, the supernatant was incubated with 1 mg of probe bound Dynabeads for 30 min at 4°C. The beads were then washed five times in 1 ml of binding buffer, frozen at -20°C and sent for mass spectrometry analysis.
Sample preparation for mass spectrometry analysis
Mass spectrometry experiments were performed in the Mass Spectrometry Laboratory at the Institute of Biochemistry and Biophysics PAS. Proteins were reduced by 1-hour incubation with 5 mM tris(2-carboxyethyl) phosphine (TCEP) at 60oC, followed by 10 min incubation at RT with 8 mM methyl methanethiosulfonate (MMTS). Proteins were digested on beads with 0.5 ugs of trypsin/Lys-C mix (Promega) overnight at 37 °C. Supernatants were transferred to a fresh tube and beads were washed with the extraction solution (2% acetonitrile, 0.1% formic acid) to assure maximum peptide recovery. Aliquots were dried on SpeedVac and reconstituted in 40 ul 10 mM HEPES buffer pH 8.0. Next steps were performed following SP3 protocol [Ultrasensitive proteome analysis using paramagnetic bead technology], with some modifications. Briefly, SP3 magnetic beads mix was prepared by combining equal parts of Sera-Mag Carboxyl hydrophilic and hydrophobic particles (09-981-121 and 09-981-123, GE Healthcare). The mix was washed three times with MS-grade water and resuspended in a working concentration of 10 ug/ul. Samples were mixed with 16 μl of the prepared bead mix and 1 ml of acetonitrile containing 0.1% formic acid to facilitate peptide binding. Beads were rinsed two times with 1 ml of isopropanol followed by acetonitrile. Peptides were eluted from the beads by subsequent incubation with MS-grade water and 2% DMSO with sonication during each step. Pulled aliquots were dried in SpeedVac and resuspended in 50 ul 2% acetonitrile and 0.1% formic acid. Peptide concentrations were measured using Pierce Quantitative Colorimetric Peptide Assay (Thermo Scientific).
Mass spectrometry and data analysis
1 ug of peptides from each sample was analyzed using nanoAcquity UPLC (Waters) directly coupled to a QExactive mass spectrometer (Thermo Scientific). Peptides were collected on a C18 trap column (180 µm x 20 mm, Waters) with 0.1% FA in water as a mobile phase and transferred to a nanocavity BEH C18 column (75 µm x 250 mm, 1.7 µm, Waters) using ACN gradient (0 – 35% ACN in 160 min) in the presence of 0.1% FA at a flow rate of 250 ml/min. Measurements were performed in the data-dependent mode with the top 12 precursors selected for MS2. Full MS scans covering the range of 300–1650 m/z were acquired at a resolution of 70,000 with a maximum injection time of 60 ms and an AGC target value of 1e6. MS2 scans were acquired at the resolution of 17,500 and the AGC target value of 5e5. Dynamic exclusion was set to 30 s. Obtained data were pre-processed with Mascot Distiller software (Matrixscience), and protein identification was performed using Mascot Server 2.5 (Matrixscience) against the Homo sapiens protein sequences (20496 sequences) deposited in the Swiss-Prot database (201904, 560118 sequences; 201292445 residues). The parameters settings were as follows: enzyme – Trypsin, missed cleavages – 2, fixed modifications – Methylthio (C), variable modifications –Oxidation(M), instrument – HCD. To reduce mass errors, peptide and fragment mass tolerance settings were established separately for each file after an off-line mass recalibration [22]. The confidence assessment was based on the target/decoy database search strategy [23], which provided q-value estimates for each peptide spectrum match. All queries with values> 0.01, subset proteins, and proteins identified with one peptide were discarded from further analysis. The mass recalibration, FDR computations, and data filtering were done with Mscan software and developed in-house (http://proteom.ibb.waw.pl/mscan/)
HDAC1 and ZNF384 silencing with DsiRNAs
Dicer-Substrate Short Interfering RNAs (DsiRNAs) and TriFECTa® Kits (Integrated DNA Technologies) were used for RNA silencing. For transfection, SH-SY5Y cells were differentiated as described above. On the fifth day, cells were transferred to 12-well dishes at a density of 450,000 cells/well in the presence of ATRA and antibiotics. The next day, medium was changed to a fresh one without antibiotics. Cells were transfected using the X-tremeGENE siRNA Transfection Reagent (Roche) according to manufacturer’s protocol. 10 nM DsiRNA against HDAC1, ZNF384, positive and negative controls, and 0,5ug of the pGL3 vector with the promoter of the MMP-9 gene containing the C or T allele, respectively, were added to the medium. Cells were incubated in the transfection mixture for about 6.5 h, followed by a change of medium with the addition of ATRA, BDNF, and an antibio tics. On the seventh day the medium was changed again to the same medium as above (1:1 EMEM/F12 +ATRA+BDNF + antibiotics) On the eighth day, ca. 48h after transfection, the cells were flooded with TRIZOL, and the dishes were placed at -80ºC until the subsequent RNA isolation.
RNA isolation and reverse transcription quantitative PCR
Total RNA was extracted from SH-SY5Y cells using TRIzol® Reagent [Life Technologies # 15596018] according to the manufacturer's protocol. 500 ng of RNA samples were subjected to RT reaction using TaqMan Reverse Transcription Kit [Applied Biosystem #N8080234]. Real-time PCR analysis was performed with 5× HOT FIREPol EvaGreen qPCR Mix Plus [Solis BioDyne # 08-25-00020] using the LightCycler480 System (Roche). All experiments were performed in triplicate. CT values were chosen in the linear range of amplification, and the comparative CT method was used to calculate differences in gene expression. Primers and conditions for RT-qPCR are listed in Supplementary Table 2.
Luciferase assay after HDAC1 and ZNF384 silencing
48h after transfection, luciferase activity was measured using the Dual-Glo® Luciferase Assay System (Promega) according to manufactured protocol. Each luciferase construct was assayed in triplicate, and each transfection experiment was repeated at least three times.
3. Results
3.1.
3.1.1. SH-SY5Y differentiation into neurons
To gain insight into the human neuronal -1562C/T-dependent regulation of MMP-9 expression we used human neuroblastoma SH-SY5Y cells. As it was already described, that SH-SY5Y cell line contains both neuroblast-like and epithelial-like cells [24]. Thus, we firstly differentiated SH-SY5Y cells into neurons. For this purpose, we used modified protocol based on the following ones: [25,26,27,28] (Figure 1A). It was previously shown that combination of serum starvation and the addition of retinoic acid (RA) led to removal of epithelial-like cells due to apoptosis [28]. To validate the differentiation procedure, SH-SY5Y cells were immunofluorescently stained for the neuronal marker MAP-2. As expected, the neuroblastoma cells differentiated into neurons according to our optimized procedure and exhibited significantly more MAP-2 than the undifferentiated SH-SY5Y cells (Figure 1B). Moreover, we found by RT-qPCR that expression of neuronal marker synaptophysin was significantly higher in differentiated neuronal cells compared to undifferentiated human neuroblastoma cells (Figure 1C). We further analyzed the overall expression level of the MMP-9 gene in SH-SY5Y cells and in neurons obtained by the differentiation of SH-SY5Y cells. We showed that the overall level of MMP-9 mRNA was slightly higher in mature neurons derived from SH-SY5Y cells than in the undifferentiated SH-SY5Y cells (Figure 1D).
3.1.2. Allele-specific effect of MMP-91562C/T polymorphism in luciferase and gel shift assay
To determine whether the -1562C/T polymorphism affects MMP-9 activity in neuronal cells derived from the SH-SY5Y cell line, the luciferase assay was performed. We showed that the C allele significantly increased the MMP-9 gene promoter activity as compared to the control (the empty pGl3 vector) (Figure 2A). Moreover, we observed that the C -1562C/T polymorphism allele conferred to the MMP-9 promoter higher activity than the T allele (Figure 2A). Next, we assessed the MMP-9 mRNA expression in human neurons derived from SH-SY5Y cells transfected with constructs containing the C or T allele of MMP-9-1562C/T polymorphism. RT-qPCR results revealed that the MMP-9 mRNA expression level was significantly higher in human neurons with the C allele as compared to control neurons (transfected with the empty pGL3 vector). On the other hand, the level of mRNA expression in human neurons transfected with the T allele was higher compared to the control, but lower compared to the neurons with the C allele, the differences were not statistically significant (Figure 2B). These findings indicate that the -1562C/T polymorphism affects the MMP-9 gene promoter activity in human neurons and, importantly, that the activation is allele-dependent.
Figure 2.
Allele-specific effect of MMP-91562C/T polymorphism in luciferase and gel shift assay. a. Luciferase activities recorded after transfection of the human neurons derived from SH-SY5Y cells with constructs containing either C or T allele of MMP-9 promoters controlling luciferase gene. The white bar indicates results obtained after transfection of MMP-9 promoter with the empty pGl3 vector. Outliers have been removed, Q=1%. Kruskal–Wallis test was followed by the D’Agostino and Pearson normality test. Values are mean ± SEM ( **, p < 0.05; n=30-35). b. RT-qPCR analysis of MMP-9 mRNA expression in human neurons transfected with constructs containing the C or T allele of MMP-9-1562C/T polymorphism. The white bar indicates results obtained after transfection of MMP-9 promoter with the empty pGl3 vector. Outliers have been removed, Q=1%. Kruskal–Wallis test was followed by the D’Agostino and Pearson normality test. Values are mean ± SEM (**, p < 0.05; n=10-12). c. Western blot validation of the purity of nuclear and cytoplasmatic extracts isolated from human neurons derived from SH-SY5Y cells. The 10 µg of nuclear and cytoplasmatic fraction protein was loaded on SDS-PAGE and processed by immunoblot analysis with anti-TATA and anti-GAPDH antibody, which are nuclear and cytoplasmic markers, respectively (Nakamura et al., 2017; Schläfli et al., 2015). d. Binding of nuclear proteins extracted from the SH-SY5Y cell line to the alleles of MM9-1562C/T analyzed by gel shift assay. The specifically shifted bands are indicated with the red arrowheads for the C allele and black arrowheads for the T allele. e. Binding of nuclear proteins extracted from human neurons derived from SH-SY5Y cells to the alleles of MM9-1562C/T analyzed by gel shift assay. The specifically shifted bands are indicated with the red arrowheads for the C allele and black arrowheads for the T allele.
Figure 2.
Allele-specific effect of MMP-91562C/T polymorphism in luciferase and gel shift assay. a. Luciferase activities recorded after transfection of the human neurons derived from SH-SY5Y cells with constructs containing either C or T allele of MMP-9 promoters controlling luciferase gene. The white bar indicates results obtained after transfection of MMP-9 promoter with the empty pGl3 vector. Outliers have been removed, Q=1%. Kruskal–Wallis test was followed by the D’Agostino and Pearson normality test. Values are mean ± SEM ( **, p < 0.05; n=30-35). b. RT-qPCR analysis of MMP-9 mRNA expression in human neurons transfected with constructs containing the C or T allele of MMP-9-1562C/T polymorphism. The white bar indicates results obtained after transfection of MMP-9 promoter with the empty pGl3 vector. Outliers have been removed, Q=1%. Kruskal–Wallis test was followed by the D’Agostino and Pearson normality test. Values are mean ± SEM (**, p < 0.05; n=10-12). c. Western blot validation of the purity of nuclear and cytoplasmatic extracts isolated from human neurons derived from SH-SY5Y cells. The 10 µg of nuclear and cytoplasmatic fraction protein was loaded on SDS-PAGE and processed by immunoblot analysis with anti-TATA and anti-GAPDH antibody, which are nuclear and cytoplasmic markers, respectively (Nakamura et al., 2017; Schläfli et al., 2015). d. Binding of nuclear proteins extracted from the SH-SY5Y cell line to the alleles of MM9-1562C/T analyzed by gel shift assay. The specifically shifted bands are indicated with the red arrowheads for the C allele and black arrowheads for the T allele. e. Binding of nuclear proteins extracted from human neurons derived from SH-SY5Y cells to the alleles of MM9-1562C/T analyzed by gel shift assay. The specifically shifted bands are indicated with the red arrowheads for the C allele and black arrowheads for the T allele.
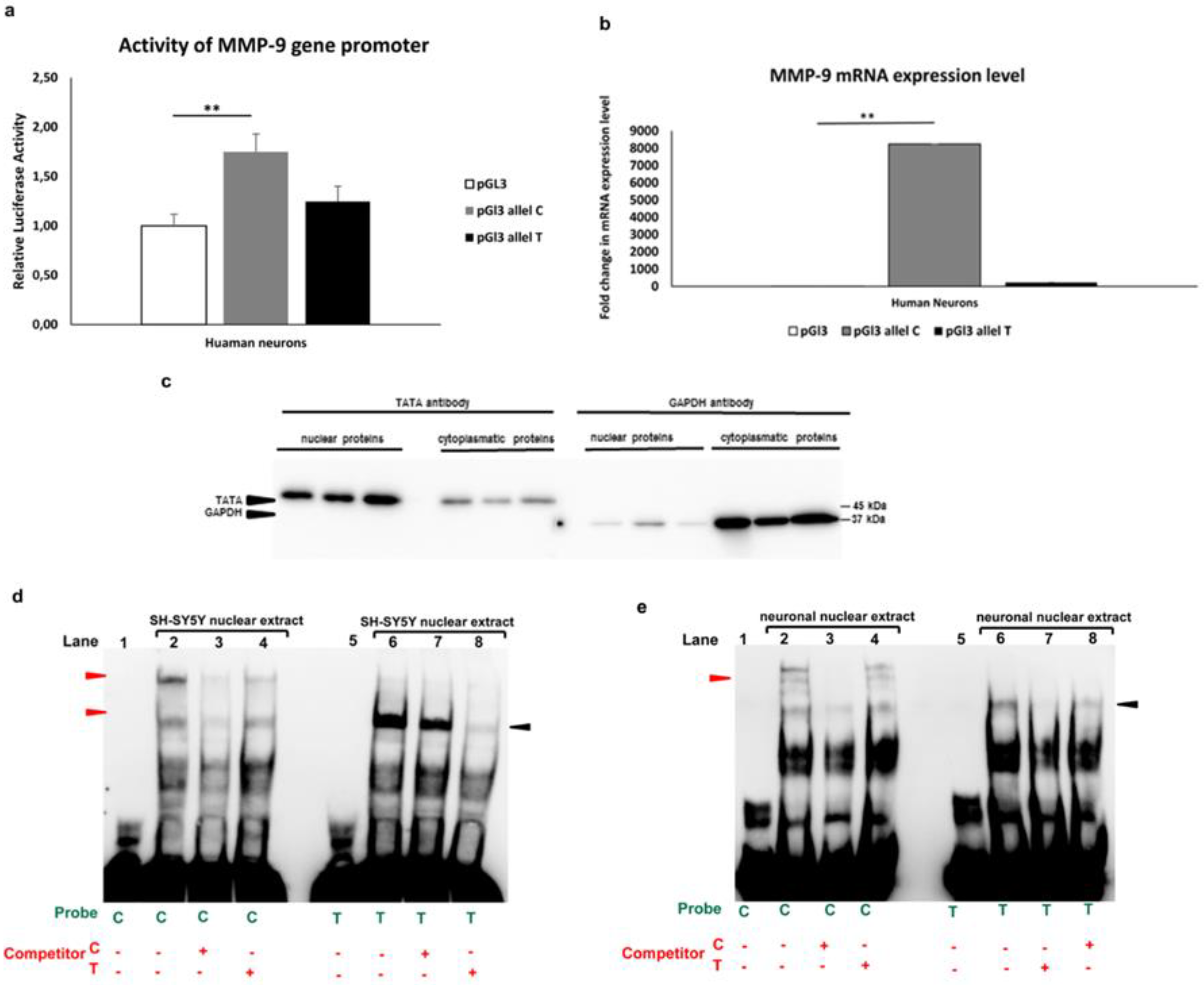
In the next step, we used the electrophoretic mobility shift assay (EMSA) to reveal proteins which can regulate MMP-9 gene expression via differential binding to the alleles of -1562C/T polymorphism [29]). To this end, we first verified the purity of nuclear extracts. The isolated protein fractions were examined for the presence of TATA and GAPDH, which are nuclear and cytoplasmic markers, respectively [30,31]. Western-blot analysis demonstrated a strong signal for TATA in nuclear extracts and a weak one in the cytoplasmic fractions (left panel Figure 2C). Whereas GAPDH showed a strong band in cytoplasmic extracts and almost undetectable in nuclear protein fraction (right panel Figure 2C). These results confirmed a high purity of isolated nuclear fractions which were further used in EMSA. The EMSA analysis revealed in the experiment with the C allele, two bands which were competed by the non-labeled C allele, but not by the T allele (red arrowheads). In comparison, the gel shift experiment performed with the T allele showed one shifted band, competed by non-labeled T allele but not by the C allele (black arrowheads) in SH-SY5Y nuclear extract (Figure 2D). Importantly, a similar conclusion was reached in the experiment with human neurons derived from SH-SY5Y cells showing three shifted bands with the C allele, which were competed by the non-labeled C allele and one band in the T allele, competed by non-labeled T allele but not by the C allele (Figure 2E). The results from the gel shift assays suggest that different sets of proteins regulate the MMP-9 expression in the MMP-9-1562C/T allele-specific manner (Figure 2D,E).
3.1.3. Identification of the MM9-1562C/T binding proteins in human neurons
In the next experiment, the pull-down assay followed by mass spectrometric analysis were used to identify MM9-1562C/T binding proteins in human neurons. We have found 554 proteins which were bound to the C allele of the MMP-9-1562C/T polymorphism, and 586 proteins bound to the T allele, of which 542 were shared between the groups (Figure 3A).
Figure 3.
Identification of the MM9-1562C/T binding proteins in human neurons. a. Venn diagram analysis showing the overlap between proteins bound to the C (green) and T (red) allele in the MMP-9-1562C/T polymorphism of the MMP-9 gene. b. The table depicting the proteins detected by pull-down assay combined with mass spectrometry and selected for EMSA supershift. c. Binding of human neuronal nuclear lysates to the alleles of MM9-1562C/T supershifted with HDAC1 antibody using EMSA method. Control reaction was conducted with an isotype antibody. The specifically shifted band is indicated with the red arrowhead for the C allele and black arrowhead for the T allele. d. Binding of human neuronal nuclear lysates to the alleles of MM9-1562C/T incubated with ZNF384 antibody and subjected to EMSA supershift. Control reaction was conducted with an isotype antibody. The specifically shifted band is indicated with the red arrowhead for the C allele and black arrowhead for the T allele.
Figure 3.
Identification of the MM9-1562C/T binding proteins in human neurons. a. Venn diagram analysis showing the overlap between proteins bound to the C (green) and T (red) allele in the MMP-9-1562C/T polymorphism of the MMP-9 gene. b. The table depicting the proteins detected by pull-down assay combined with mass spectrometry and selected for EMSA supershift. c. Binding of human neuronal nuclear lysates to the alleles of MM9-1562C/T supershifted with HDAC1 antibody using EMSA method. Control reaction was conducted with an isotype antibody. The specifically shifted band is indicated with the red arrowhead for the C allele and black arrowhead for the T allele. d. Binding of human neuronal nuclear lysates to the alleles of MM9-1562C/T incubated with ZNF384 antibody and subjected to EMSA supershift. Control reaction was conducted with an isotype antibody. The specifically shifted band is indicated with the red arrowhead for the C allele and black arrowhead for the T allele.
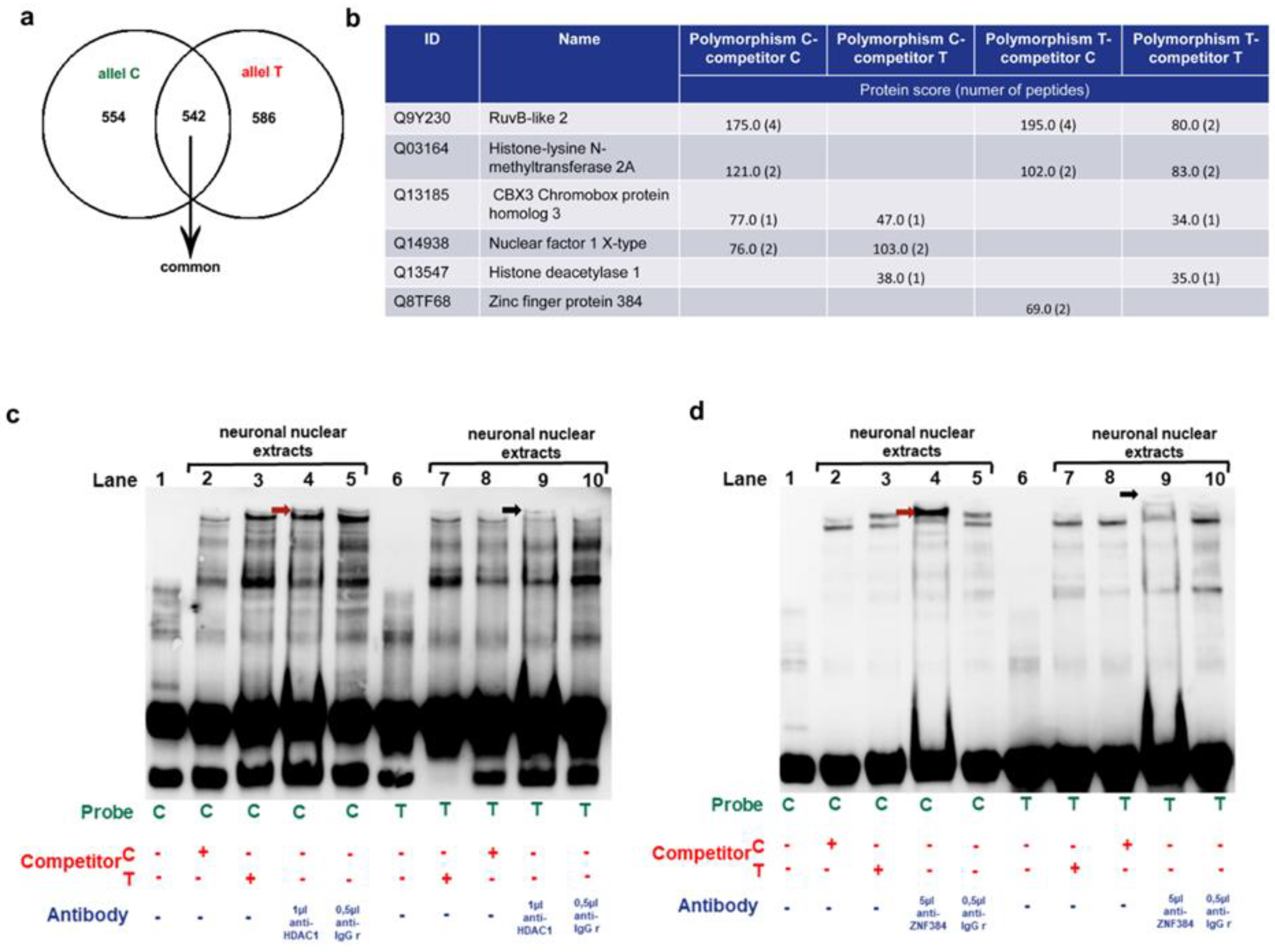
Next, we have selected from the data a few proteins which could be potentially interesting in the aspect of MMP-9 expression regulation (please see Figure 3B). It has been reported that YY1 (Ying Yang 1) is a critical repressor of MMP-9 transcription in brain neurons [32]. Thus, we checked YY1 protein association networks (String software). The analyses showed Histone deacetylase 1 (HDAC1) and RuvB-like 2 (RUVBL2) as YY1 partners and we selected these proteins for further studies. Moreover, Matinspector bioinformatics identification of transcription factors capable to bind to the MMP9-1562C/T polymorphism revealed Zinc finger protein 410 (ZNF410) as a transcription factor capable to bind to C or T allele. Interestingly, we identified ZNF-410-related DNA binding protein, Zinc finger protein 384 (ZNF384) using protein pull down assay combined with mass spectrometry analyses, so we also included this transcription factor in our further studies. Rest of chosen transcription factors selected for further analyses (KMT2A, NFX1, CBX3) are strongly involved in gene silencing or activation and therefore they could hypothetically influence MMP-9 expression. Moreover, the trabscription factors KMT2A and CBX3 are involved in chromatin remodeling, similarly to YY1(Figure 3B).
To check whether the selected proteins can bind to the MMP-9 gene promoter, we performed EMSA supershift analyses. We found that antibodies against four out of six chosen transcription factors (RUVBL2, KMT2A, CBX3, NFIX) did not generate gel shifts what meant that they did not bound to the promoter fragment of the MMP-9 gene in both the C and T allele (data not shown). However, we observed HDAC1-supershifted band in the C allele containing samples indicating for HDAC1/the C allele interaction (Figure 3C, lane 4). Regarding the T allele, the band was also visible, however it was localized higher and was weaker suggesting that HDAC1 binds to the T allele less intense and in the presence of other protein complexes than to the C allele (Figure 3C, lane 9). Similarly, the gel shift assay with the ZNF384 antibody showed a band shift with the C allele visible in lane 4 (Figure 3D, lane 4) and a weaker and higher supershifted band with the T allele (Figure 3D, lane 9). Therefore, we have found in vitro interaction of HDAC1 and ZNF384 DNA-binding regulatory proteins with the MMP9-1562C/T polymorphism, and we have also showed that it is the allele-dependent and stronger visualized with the C than the T allele in the experimental setting. Moreover, both HDAC1 as well as ZNF384 form in vitro different complexes with the C allele than with the T allele. Interestingly, the T allele complexes are larger than those containing the C allele.
3.1.4. Effects exerted by HDAC1 and ZNF384 on the MMP-9 promoter activity and its mRNA expression.
Next, we asked whether HDAC1 and ZNF384 can influence the MMP-9 promoter activity. Therefore, we knocked down HDAC1 and ZNF384 by DsiRNA in the human neurons and we measured the MMP-9 activity by luciferase assay. As a result, we observed higher activity of the MMP-9 gene promoter in a presence of the C allele, or the T allele as compared to control (human neurons transfected with the empty pGL3 vector) in HDAC1-silenced neurons (Figure 4A). Moreover, we found that, in ZNF384 silenced neurons, the activity of the C allele of the MMP-9 gene promoter was lower than in control (Figure 4a). On the contrary, our data showed that the T allele activity was higher as compared to control in ZNF384-silenced neurons (Figure 4A). Altogether, our results demonstrated that HDAC1 and ZNF384 proteins influence the MMP-9 promoter activity in vivo in the MMP9-1562C/T allele-dependent manner. The data suggests that HDAC-1 downregulates differentially both the C and the T allele activity, and ZNF384 upregulates the C allele activity and downregulates activity of the T allele in human neurons.
Figure 4.
Knockdown of HDAC1or ZNF384 by DsiRNA in human neurons derived from SH-SY5Y cells. a. MMP-9 activity measured by luciferase assay after silencing of HDAC1 or ZNF384 in human neurons with constructs containing either C or T allele of MMP-9 promoters controlling luciferase gene. Control reaction was conducted with constructs containing the empty pGl3 vector. Kruskal–Wallis test was followed by the D'Agostino and Pearson normality test. Values are mean ± SEM (n=20). b. MMP-9 mRNA expression measured by RT-qPCR after silencing of HDAC1 or ZNF384 in human neurons transfected with constructs containing either the C or T allele of the MMP9-1562C/T polymorphism. Control reaction was conducted with constructs containing the empty pGl3 vector. Outliers have been removed, Q=1%. One-way ANOVA was followed by Kolmogorov-Smirnov test. Values are mean ± SEM (****, p< 0.001; n = 6-8).
Figure 4.
Knockdown of HDAC1or ZNF384 by DsiRNA in human neurons derived from SH-SY5Y cells. a. MMP-9 activity measured by luciferase assay after silencing of HDAC1 or ZNF384 in human neurons with constructs containing either C or T allele of MMP-9 promoters controlling luciferase gene. Control reaction was conducted with constructs containing the empty pGl3 vector. Kruskal–Wallis test was followed by the D'Agostino and Pearson normality test. Values are mean ± SEM (n=20). b. MMP-9 mRNA expression measured by RT-qPCR after silencing of HDAC1 or ZNF384 in human neurons transfected with constructs containing either the C or T allele of the MMP9-1562C/T polymorphism. Control reaction was conducted with constructs containing the empty pGl3 vector. Outliers have been removed, Q=1%. One-way ANOVA was followed by Kolmogorov-Smirnov test. Values are mean ± SEM (****, p< 0.001; n = 6-8).
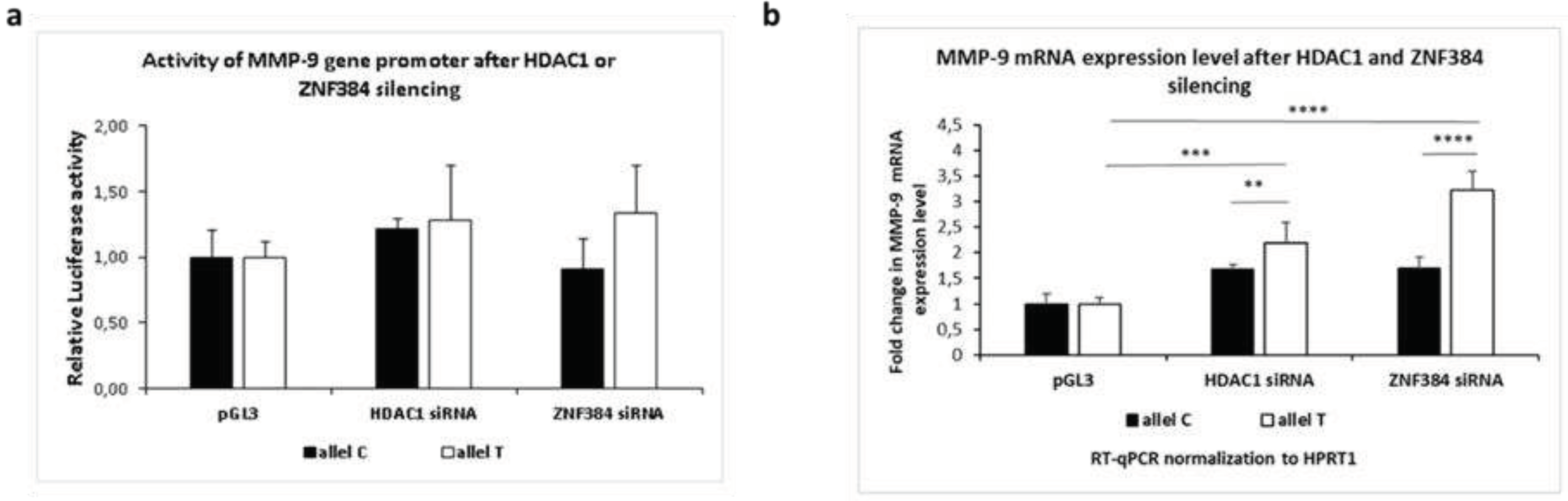
Then, we used RT-qPCR to examine whether HDAC1 and ZNF384 can influence the neuronal MMP-9 mRNA expression in vivo in the MMP9-1562C/T polymorphism-dependent manner. We knocked down HDAC1or ZNF384 expression by DsiRNA in human neurons derived from SH-SY5Y cells and we measured MMP-9 mRNA level. Interestingly, we observed an elevation of the MMP-9 mRNA expression for both HDAC1- or ZNF384-silenced neurons with both the C allele or the T allele as compared to control (non-silenced human neurons transfected with the pGL3 vector containing the T or C allele) (Figure 4B, white column). However, MMP-9 expression increase was statistically significant only in the T allele but not in the C allele transfected neurons (Figure 4B, black column). It suggests that HDAC1 and ZNF384 regulates gene expression in the MMP9-1562C/T-dependent manner and that this regulation is exerted in unstimulated human neurons mostly by their binding to the T allele. Moreover, the data imply that HDAC1 and ZNF384 are negative regulators of MMP-9 gene expression.
4. Discussion
Earlier studies have revealed the implication of MMP-9-1562C/T polymorphism in the pathophysiology of various diseases related to the central nervous system including brain [4]. Robert & Pelletier reported that the presence of SNPs in transcriptional regulatory elements influence the mRNA expression of this gene [33]. Therefore, the cytosine (C)>thymidine (T) single nucleotide polymorphism (SNP) at position -1562 in the MMP-9 promoter may affect the expression level of MMP-9 mRNA and consequently susceptibility to various diseases related to the incorrect expression of this protease.
This is the first study to examine the role of MMP-9-1562C/T gene promoter polymorphism in the expression of MMP-9 in brain cells and related transcriptional regulation. Here we showed that MMP-9 promoter activity was almost two-fold higher in the presence of the C allele as compared to control (human neurons transfected with empty pGl3 vector). In case of the T allele the MMP-9 promoter activity was 1.4-fold higher as compared to control although the differences were not statistically significant. Similarly, the mRNA expression of MMP-9 varied depending on MMP-9-1562C/T polymorphism. MMP-9 mRNA level was significantly higher when the C allele was present in human neurons as compared to control. The expression of MMP-9 mRNA increased also in human neurons transfected with a pGl3 vector containing the T allele as compared to control, even though the differences were not statistically significant. Experiments performed by Zhang et al., however, have produced conflicting results. They showed that in MALU cells, the reporter activity of the MMP-9 promoter was almost 1.5-times greater under the control of the T allele than the reporter activity controlled by the C allele [1]. Interestingly the allele-dependent effect of the MMP-9-1562C/T polymorphism was not shown in the studies conducted on primary amnion epithelial cell cultures, WISH, or THP-1 cells [34]. The discrepancy between their studies and ours in this respect is puzzling Thus, we speculate that, that mechanism responsible for the differences in the allele-dependent MMP-9 promoter activity is perhaps cell type-dependent.
Next, we investigated differences in transcriptional regulatory proteins binding to the MMP-9 gene promoter in relation to the C or T allele. Using the electrophoretic mobility shift assay, we showed two bands in case of allele C, which was competed by the non-labeled allele C but not by the allele T. In comparison, experiment with the allele T showed one shifted band, competed by non-labeled allele T but not by the allele C in purified SH-SY5Y nuclear extracts. Similar conclusion was reached from the experiment with neuronal nuclear extract showing three bands in case of allele C, which were competed by non-labeled allele C and one shifted band in allele T. The results suggest that different sets of proteins regulate the transcriptional mechanism in the promoter of the MMP-9 gene in an allele-specific manner. The results are similar to those obtained by Zhang et al. who showed that the bands present on the C allele were less prominent in the T allele [1].
We have extended our experiment further by identifying MM9-1562C/T binding proteins in human neurons using the pull-down assay followed by mass spectrometry technique. The analysis revealed 554 proteins which were bound to the C allele in the MMP-9-1562C/T polymorphism of the MMP-9 gene, and 586 proteins bound to the T allele, of which 542 were shared. After mass recalibration, FDR computations, and data filtering with Mscan software, we have selected proteins for further experiments. Additionally, we performed other bioinformatics analyzes that helped us find transcription factors that probably interact with the MMP-9 gene promoter, such as analysis in MatInpector and String software. For the C allele in the MMP-9 gene polymorphism, the Chromobox protein homolog 3 (CBX3), Nuclear factor 1 X-type (NFIX), and Histone deacetylase 1 (HDAC1) were chosen. These proteins as shown in Table 3b bound to the C allele in the presence of a competitor in the form of an MMP-9 promoter fragment with the T allele, so proteins that were not specific for the C allele were caught with the T allele. Reports have shown that CBX3 is involved in transcriptional silencing in heterochromatin-like complexes where recognizes and binds histone H3 tails methylated at 'Lys-9', leading to epigenetic repression [35]. It is also a repressor in mammalian cells when tethered to DNA [36]. In turn, NFIX has DNA-binding transcription factor activity [37,38] and regulates RNA polymerase II transcription [39], while HDAC1 is responsible for transcriptional repression and gene silencing [40,41]. On the other hand, for the T variant in the -1562C / T polymorphism of the MMP-9 gene, the following proteins were selected RuvB-like 2 (RUVBL2), Histone-lysine N-methyltransferase 2A (KMT2A) and Zinc finger protein 384 (ZNF384). It has been reported that RUVBL2 is a component of the NuA4 histone acetyltransferase complex, which is involved in transcriptional activation of selected genes mainly by acetylation of nucleosomal histones H4 and H2A [42]. Moreover, positively regulates transcription by RNA polymerase II [43]. Further, KMT2A in the MLL1/MLL complex mediates H3K4me, a specific tag for epigenetic transcriptional activation [44,45,46], positively regulates transcription [47]. The last one from the selected genes, ZNF 384 is not well studied, however it is described as a transcription factor that binds sequence-specific double-stranded DNA [48].
To investigate whether indeed selected transcription factors might specifically bind to the MMP-9 promoter, we performed EMSA supershift in the presence of RUVBL2, KMT2A, CBX3, NFIX, HDAC1 and ZNF384 specific antibodies. We have discovered that antibodies against RUVBL2, KMT2A, CBX3, NFIX do not bind to the promoter fragment of the MMP-9 gene in both the C and T allele polymorphism (data not shown). However, we observed complexes formed with antibodies directed to HDAC1 and ZNF384 in the C and T allele when human neurons were the source of nuclear proteins. Interestingly, in case of the T allele, the supershifts were much weaker as compared to the C allele suggesting that in the presence of the T allele, HDAC1 and ZNF384 may bind to the promoter of the MMP-9 to a lesser extent and/or in the presence of other transcription factors.
The results described above prompted us to evaluate the influence of HDAC1 and ZNF384 depletion onto MMP-9 promoter activity in human neurons. We depleted HDAC1 and ZNF384 expression by siRNA for HDAC1 and ZNF384 and measured the MMP-9 activity by luciferase assay. The MMP-9 promoter activity was higher under the control of the T allele after both HDAC1 and ZNF384 silencing as compared to control, i.e., cells transfected with empty pGL3 vector, although the increase in promoter activity was not statistically significant. As for the C allele, after HDAC1 knocking-down the MMP-9 activity was slightly higher compared to control. Opposite, after ZNF384 silencing, MMP-9 activity was lower compared to control. Previous data showed that overexpression of HDAC1 downregulated MMP-9 promoter activity in HT1080 fibrosarcoma cells in a dose-dependent manner [49]. Surprisingly, after silencing of both HDAC1 and ZNF 384, we have shown that the MMP-9 mRNA expression level significantly increased when the T allele was present in the promoter as compared to the control, i.e., cells transfected with the empty pGL3 vector. Although, MMP-9 expression was higher in the case of C allele compared to control, this increase was not statistically significant. The above results strongly suggests that HDAC1 and ZNF384 play an important role in the regulation of MMP-9 gene in the presence of the T allele. Previously performed experiment showed that knockdown of HDAC1 suppressed the expression and protein level of MMP-9 in human breast cancer MDA-MB-321, MCF-7 cells and in U251, T98G glioblastoma cell line [50,51]. Our data show that after ZNF384 and HDAC1 silencing, both promoter activity and MMP-9 gene expression levels increase significantly, but only in the presence of the T allele. In the case of the C allele, after HDAC1 and ZNF384 silencing, there are no major differences in promoter activity and gene expression levels of MMP-9. Data obtained by other researchers showed that when HDAC1 was silenced, promoter activity and MMP-9 gene expression decreased [49,50,51]. In the case of the T allele, as shown in the EMSA supershift image, HDAC1 and ZNF384 transcription factors bind to the MMP-9 gene promoter in smaller amounts, so differences in the activity of the MMP-9 gene promoter and its expression may depend strictly on a specific allele. Unfortunately, no other studies showing the effect of silencing ZNF384 on the activity of the MMP-9 promoter, or its expression have been published.
5. Conclusions
Together our present data clearly indicate that promoter activity and expression of MMP-9 are higher in human neurons under the control of the C allele. Moreover, it can be presumed that two transcription factors, HDAC1 and ZNF384, may play an essential role in regulating the MMP-9 transcriptional activity in human neurons only when the T allele is present in the promoter. It would be of great interest to further examine other transcription factors or mechanisms which may be important for the MMP-9-1562C/T dependent differential regulation of MMP-9 expression in human neurons.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Name and sequence of oligonucleotide probe used for EMSA, EMSA supershift, and pull-down with mass-spectrometry; Table S2: Primer sequences used for RT-qPCR as well as applied PCR conditions.
Author Contributions
Conceptualization, S.P-J. and M.R..; methodology, S.P-J., M.A-L, K.N; writing—original draft preparation, S.P-J.; writing—review and editing, A.B-P. and M.R.; visualization, S.P-J. and K.N.; supervision, M.R.; project administration, M. A-L..; funding acquisition, S.P-J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Science Centre, Grant number 2017/25/N/NZ3/02266
Data Availability Statement
The SH-SY5Y cell line was provided by LGC Standards, Ltd
Acknowledgments
We thank Laboratory of Mass Spectrometry, Institute of Biochemistry and Biophysics, Polish Academy of Science for mass spectrometry analysis.
References
- B. Zhang, A. Henney, P. Eriksson, A. Hamsten, H. Watkins, and S. Ye, “Genetic variation at the matrix metalloproteinase-9 locus on chromosome 20q12.2-13.1,” Hum Genet, vol. 105, no. 5, pp. 418–423, 1999. https://doi.org/10.1007/s004390051124. [CrossRef]
- S. Verma, K. Kesh, A. Gupta, and S. Swarnakar, “An overview of matrix metalloproteinase 9 polymorphism and gastric cancer risk,” Asian Pacific Journal of Cancer Prevention, vol. 16, no. 17, pp. 7393–7400, 2015. https://doi.org/10.7314/APJCP.2015.16.17.7393. [CrossRef]
- S. Pabian-Jewuła and M. Rylski, “Does the functional polymorphism-1562C/T of MMP-9 gene influence brain disorders?,” Frontiers in Cellular Neuroscience, vol. 17. Frontiers Media S.A., 2023. https://doi.org/10.3389/fncel.2023.1110967. [CrossRef]
- Beroun, S. Mitra, P. Michaluk, B. Pijet, M. Stefaniuk, and L. Kaczmarek, “MMPs in learning and memory and neuropsychiatric disorders,” Cellular and Molecular Life Sciences, vol. 76, no. 16, pp. 3207–3228, 2019. https://doi.org/10.1007/s00018-019-03180-8. [CrossRef]
- M. Go, B. S., Sirohi, S., & Walker, “The role of matrix metalloproteinase-9 in negative reinforcement learning and plasticity in alcohol dependence.,” Addiction biology, vol. 25, no. 3, p. 12715, 2020.
- S. V. Gore et al., “Role of matrix metalloproteinase-9 in neurodevelopmental deficits and experience-dependent plasticity in xenopus laevis,” Elife, vol. 10, pp. 1–24, 2021. https://doi.org/10.7554/eLife.62147. [CrossRef]
- S. M. Reinhard, K. Razak, and I. M. Ethell, “A delicate balance: Role of MMP-9 in brain development and pathophysiology of neurodevelopmental disorders,” Front Cell Neurosci, vol. 9, no. JULY, pp. 1–16, 2015. https://doi.org/10.3389/fncel.2015.00280. [CrossRef]
- Vafadari, A. Salamian, and L. Kaczmarek, “MMP-9 in translation: from molecule to brain physiology, pathology, and therapy,” J Neurochem, vol. 139, pp. 91–114, 2016. https://doi.org/10.1111/jnc.13415. [CrossRef]
- A. M. Langers, H. W. Verspaget, D. W. Hommes, and C. F. Sier, “Single-nucleotide polymorphisms of matrix metalloproteinases and their inhibitors in gastrointestinal cancer,” World J Gastrointest Oncol, vol. 3, no. 6, pp. 79–98, 2011. https://doi.org/10.4251/wjgo.v3.i6.79. [CrossRef]
- M. Živković, T. Djurić, E. Dinčić, R. Raičević, D. Alavantić, and A. Stanković, “Matrix metalloproteinase-9 -1562 C/T gene polymorphism in Serbian patients with multiple sclerosis,” J Neuroimmunol, vol. 189, no. 1–2, pp. 147–150, 2007. https://doi.org/10.1016/j.jneuroim.2007.06.022. [CrossRef]
- S. Sabbagh et al., “Association study between functional polymorphisms of MMP9 gene promoter and multiple sclerosis susceptibility in an Iranian population,” Iran J Public Health, vol. 48, no. 9, pp. 1697–1703, 2019. https://doi.org/10.18502/ijph.v48i9.3030. [CrossRef]
- K. S. da S. Fernandes et al., “Functional MMP-9 polymorphisms modulate plasma MMP-9 levels in multiple sclerosis patients,” J Neuroimmunol, vol. 249, no. 1–2, pp. 56–59, 2012. https://doi.org/10.1016/j.jneuroim.2012.04.001. [CrossRef]
- A. Valado et al., “Multiple sclerosis: Association of gelatinase B/matrix metalloproteinase-9 with risk and clinical course the disease,” Mult Scler Relat Disord, vol. 11, no. April 2016, pp. 71–76, 2017. https://doi.org/10.1016/j.msard.2016.12.003. [CrossRef]
- K. Buraczynska, J. Kurzepa, A. Ksiazek, M. Buraczynska, and K. Rejdak, “Matrix Metalloproteinase-9 (MMP-9) Gene Polymorphism in Stroke Patients,” Neuromolecular Med, vol. 17, no. 4, pp. 385–390, 2015. https://doi.org/10.1007/s12017-015-8367-5. [CrossRef]
- J. Montaner et al., “Safety Profile of Tissue Plasminogen Activator Treatment among Stroke Patients Carrying a Common Polymorphism (C-1562T) in the Promoter Region of the Matrix Metalloproteinase-9 Gene,” Stroke, vol. 34, no. 12, pp. 2851–2855, 2003. https://doi.org/10.1161/01.STR.0000098648.54429.1C. [CrossRef]
- S. Hayat, O. Ahmad, I. Mahmud, M. Z. H. Howlader, and Z. Islam, “Association of matrix metalloproteinase-9 polymorphism with severity of Guillain-Barré syndrome,” J Neurol Sci, vol. 415, no. May, p. 116908, 2020. https://doi.org/10.1016/j.jns.2020.116908. [CrossRef]
- A. Flex et al., “Effect of proinflammatory gene polymorphisms on the risk of Alzheimer’s disease,” Neurodegener Dis, vol. 13, no. 4, pp. 230–236, 2014. https://doi.org/10.1159/000353395. [CrossRef]
- H. Han et al., “The C(-1562)T polymorphism of matrix metalloproteinase-9 gene is associated with schizophrenia in China,” Psychiatry Res, vol. 190, no. 1, pp. 163–164, 2011. https://doi.org/10.1016/j.psychres.2011.04.026. [CrossRef]
- B. Glebauskiene et al., “Does MMP-9 Gene Polymorphism Play a Role in Pituitary Adenoma Development?,” Dis Markers, vol. 2017, 2017. https://doi.org/10.1155/2017/5839528. [CrossRef]
- A. Gimenez-Cassina, F. Lim, and J. Diaz-Nido, “Differentiation of a human neuroblastoma into neuron-like cells increases their susceptibility to transduction by herpesviral vectors,” J Neurosci Res, vol. 84, no. 4, pp. 755–767, Sep. 2006. https://doi.org/10.1002/jnr.20976. [CrossRef]
- M. M. Shipley, C. A. Mangold, and M. L. Szpara, “Differentiation of the SH-SY5Y human neuroblastoma cell line,” Journal of Visualized Experiments, vol. 2016, no. 108, Feb. 2016. https://doi.org/10.3791/53193. [CrossRef]
- A. Malinowska et al., “Diffprot - software for non-parametric statistical analysis of differential proteomics data,” J Proteomics, vol. 75, no. 13, pp. 4062–4073, Jul. 2012. https://doi.org/10.1016/j.jprot.2012.05.030. [CrossRef]
- J. E. Elias, W. Haas, B. K. Faherty, and S. P. Gygi, “Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations,” Nat Methods, vol. 2, no. 9, pp. 667–675, Sep. 2005. https://doi.org/10.1038/nmeth785. [CrossRef]
- J. L. Biedler, L. Helson, and B. A. Spengler, “Cancer Res Downloaded from,” 1973. [Online]. Available: http://cancerres.aacrjournals.org/content/33/11/2643http://cancerres.aacrjournals.org/content/33/11/2643#related-urls.
- M. Encinas et al., “Sequential Treatment of SH-SY5Y Cells with Retinoic Acid and Brain-Derived Neurotrophic Factor Gives Rise to Fully Differentiated, Neurotrophic Factor-Dependent, Human Neuron-Like Cells,” 2000.
- A. Gimenez-Cassina, F. Lim, and J. Diaz-Nido, “Differentiation of a human neuroblastoma into neuron-like cells increases their susceptibility to transduction by herpesviral vectors,” J Neurosci Res, vol. 84, no. 4, pp. 755–767, Sep. 2006. https://doi.org/10.1002/jnr.20976. [CrossRef]
- M. M. Shipley, C. A. Mangold, and M. L. Szpara, “Differentiation of the SH-SY5Y human neuroblastoma cell line,” Journal of Visualized Experiments, vol. 2016, no. 108, Feb. 2016. https://doi.org/10.3791/53193. [CrossRef]
- M. M. Shipley, C. A. Mangold, C. V Kuny, and M. L. Szpara, “Differentiated Human SH-SY5Y Cells Provide a Reductionist Model of Herpes Simplex Virus 1 Neurotropism,” 2017. https://doi.org/10.1128/JVI. [CrossRef]
- Y. Luo et al., “Rapid preparation of high-purity nuclear proteins from a small number of cultured cells for use in electrophoretic mobility shift assays,” BMC Immunol, vol. 15, no. 1, Dec. 2014. https://doi.org/10.1186/s12865-014-0062-z. [CrossRef]
- S. Nakamura, J. M. Hollander, T. Uchimura, H. C. Nielsen, and L. Zeng, “Pigment Epithelium-Derived Factor (PEDF) mediates cartilage matrix loss in an age-dependent manner under inflammatory conditions,” BMC Musculoskelet Disord, vol. 18, no. 1, Jan. 2017. https://doi.org/10.1186/s12891-017-1410-y. [CrossRef]
- A. M. Schläfli, S. Berezowska, O. Adams, R. Langer, and M. P. Tschan, “Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry,” European Journal of Histochemistry, vol. 59, no. 2, pp. 137–144, 2015. https://doi.org/10.4081/ejh.2015.2481. [CrossRef]
- M. Rylski et al., “Yin Yang 1 is a critical repressor of matrix metalloproteinase-9 expression in brain neurons,” Journal of Biological Chemistry, vol. 283, no. 50, pp. 35140–35153, 2008. https://doi.org/10.1074/jbc.M804540200. [CrossRef]
- Robert and J. Pelletier, “Exploring the Impact of Single-Nucleotide Polymorphisms on Translation,” Front Genet, vol. 9, no. October, pp. 1–11, 2018. https://doi.org/10.3389/fgene.2018.00507. [CrossRef]
- P. E. Ferrand et al., “A polymorphism in the matrix metalloproteinase-9 promoter is associated with increased risk of preterm premature rupture of membranes in African Americans,” Mol Hum Reprod, vol. 8, no. 5, pp. 494–501, 2002. https://doi.org/10.1093/molehr/8.5.494. [CrossRef]
- C. Bot, A. Pfeiffer, F. Giordano, D. Manjeera Edara, N. P. Dantuma, and L. Ström, “Independent Mechanisms Recruit the Cohesin Loader Protein NIPBL to Sites of DNA Damage,” 2017.
- N. Lehming, A. A. Le Saux, J. Schu¨llerschu¨ller, and M. Ptashne, “Chromatin components as part of a putative transcriptional repressing complex,” 1998. [Online]. Available: www.pnas.org.
- C. Seisenberger, E.-L. Winnacker, and H. Scherthan, “human .. genetics Localisation of the human nuclear factor I/X (NFI/X) gene to chromosome 19p13 and detection of five other related loci at lp21-22, lq42-43, 5q15, llp13 and 20q13 by FISH,” 1993.
- “qian1995”.
- A. K. Riffel, E. Schuenemann, and C. A. Vyhlidal, “Regulation of the CYP3A4 and CYP3A7 promoters by members of the nuclear factor I transcription factor family,” Mol Pharmacol, vol. 76, no. 5, pp. 1104–1114, Nov. 2009. https://doi.org/10.1124/mol.109.055699. [CrossRef]
- H. Yamaguchi et al., “Interferon-inducible protein IFIXα inhibits cell invasion by upregulating the metastasis suppressor maspin,” Mol Carcinog, vol. 47, no. 10, pp. 739–743, Oct. 2008. https://doi.org/10.1002/mc.20423. [CrossRef]
- H. Zhang, M. H. Muders, J. Li, F. Rinaldo, D. J. Tindall, and K. Datta, “Loss of NKX3.1 favors vascular endothelial growth factor-C expression in prostate cancer,” Cancer Res, vol. 68, no. 21, pp. 8770–8778, Nov. 2008. https://doi.org/10.1158/0008-5472.CAN-08-1912. [CrossRef]
- Y. Doyon, W. Selleck, W. S. Lane, S. Tan, and J. Côté, “Structural and Functional Conservation of the NuA4 Histone Acetyltransferase Complex from Yeast to Humans,” Mol Cell Biol, vol. 24, no. 5, pp. 1884–1896, Mar. 2004. https://doi.org/10.1128/mcb.24.5.1884-1896.2004. [CrossRef]
- M. Dalvai, L. Fleury, L. Bellucci, S. Kocanova, and K. Bystricky, “TIP48/Reptin and H2A.Z Requirement for Initiating Chromatin Remodeling in Estrogen-Activated Transcription,” PLoS Genet, vol. 9, no. 4, Apr. 2013. https://doi.org/10.1371/journal.pgen.1003387. [CrossRef]
- D. S. Nakamura, J. M. Hollander, T. Uchimura, H. C. Nielsen, and L. Zeng, “Pigment Epithelium-Derived Factor (PEDF) mediates cartilage matrix loss in an age-dependent manner under inflammatory conditions,” BMC Musculoskelet Disord, vol. 18, no. 1, Jan. 2017. https://doi.org/10.1186/s12891-017-1410-y. [CrossRef]
- S. Park, U. Osmers, G. Raman, R. H. Schwantes, M. O. Diaz, and J. H. Bushweller, “The PHD3 domain of MLL Acts as a CYP33-regulated switch between MLL-mediated activation and repression,” Biochemistry, vol. 49, no. 31, pp. 6576–6586, Aug. 2010. https://doi.org/10.1021/bi1009387. [CrossRef]
- S. Li et al., “Neuroprotective Effect of Osthole on Neuron Synapses in an Alzheimer???s Disease Cell Model via Upregulation of MicroRNA-9,” Journal of Molecular Neuroscience, vol. 60, no. 1, pp. 71–81, 2016. https://doi.org/10.1007/s12031-016-0793-9. [CrossRef]
- Y. Dou et al., “Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF,” Cell, vol. 121, no. 6, pp. 873–885, Jun. 2005. https://doi.org/10.1016/j.cell.2005.04.031. [CrossRef]
- Y. Yin et al., “Impact of cytosine methylation on DNA binding specificities of human transcription factors,” Science (1979), vol. 356, no. 6337, May 2017. https://doi.org/10.1126/science.aaj2239. [CrossRef]
- M. L. Mittelstadt and R. C. Patel, “AP-1 mediated transcriptional repression of matrix metalloproteinase-9 by recruitment of histone deacetylase 1 in response to interferon β,” PLoS One, vol. 7, no. 8, Aug. 2012. https://doi.org/10.1371/journal.pone.0042152. [CrossRef]
- S. Y. Park et al., “Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer,” Oncol Rep, vol. 25, no. 6, pp. 1677–1681, Jun. 2011. https://doi.org/10.3892/or.2011.1236. [CrossRef]
- X.-Q. Wang et al., “Knockdown of HDAC1 expression suppresses invasion and induces apoptosis in glioma cells,” 2017. [Online]. Available: www.impactjournals.com/oncotarget.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



