
Submitted:
02 October 2023
Posted:
02 October 2023
You are already at the latest version
Abstract
: Early and accurate detection of infectious diseases is a key step for surveillance, epidemiology, and control, notably timely disease diagnosis, patient management and follow-up. In this study, we aimed to develop hand-held ultra-fast duplex PCR assays coupled to amplicon detection by lateral flow (LF) immunoassay to deliver a rapid, and simple molecular diagnostic test for con-comitant detection and identification of the main Leishmania parasites encountered in Tunisia and the Old World. We selected two DNA targets to amplify L. major/L. tropica and L. infantum/L. tropica group of species DNAs, respectively. We optimized the experimental conditions of a du-plex ultra-fast PCR. The amplification is performed by a portable Palm convection PCR machine within 18 min and the products are detected by a LF cassette within 10 minutes. The test allows the identification of the infecting species according to the position and number of test lines re-vealed. Tested on a selection of DNAs of representative Leishmania strains of the three studied species (N=37), the ultra-fast duplex PCR-LF showed consistent, stable, and reproducible results. The analytical limit of detection of the test was 0.4pg for L. major, 4pg for L. infantum and 40pg for L. tropica.
Keywords:
Cutaneous leishmaniases
; Molecular diagnosis
; Point of care
; Leishmania
; Molecular target
; Palm PCR
; Duplex PCR
; Lateral Flow immunoassay
1. Introduction
Early and accurate detection of infectious diseases is a key step for surveillance, epidemiology and control but notably for timely disease diagnosis, patient management and follow-up. According to the World Health Organization Special Programme for Research and Training in Tropical Diseases (WHO/TDR) an ideal diagnostic tool should be used at point of care (POC) and fulfill the ASSURED criteria: Affordability, Sensitivity, Specificity, User friendliness, Rapidity and Robustness, Equipment free and Deliverable to end-users. During the last decade, several diagnostic tests satisfying these criteria were developed to identify major human pathogens such as HIV, TB and malaria [1] as these diseases received much attention compared to neglected tropical diseases such as leishmaniases. Cutaneous leishmaniases (CL) are a group of vector- borne parasitic diseases. It is a major but worldwide neglected public health problem. In the Old World (OW) more than 1 million CL cases are annually reported and 80% occur in MENA. Challenges in clinical CL patient management are essentially due to diverse clinical manifestations, multiple causing agents and their co-endemicity. They are complicated by the continuous change in Leishmania epidemiology in Tunisia [2] and Worldwide [3,4,5] making the surveillance, epidemiology and disease control challenging. In addition, the primary drugs employed for CL treatment are toxic and their efficiency may depend on parasite species/strains [6,7,8], which emphasizes even more on the relevance of CL etiology. However, CL Diagnosis is routinely done by microscopy direct examination on Giemsa stained smears, a time consuming technique that needs trained personnel and cannot identify the parasites. Species identification and taxonomical differentiation can only be done by molecular tests. Conventional polymerase chain reaction (PCR) is the molecular gold standard technique used to detect the parasites but should be complemented in a second step by other lengthy and laborious tests (RFLP, sequencing...)for species identification [9,10]. At present, there is only one commercially available CL diagnosis tool (CL Detect™ Rapid Test, InBios) meeting POC criteria for generic Leishmania detection. It is a lateral flow based immunoassay that detect amastigotes antigens present in skin lesions of individuals infected with Leishmania parasites. However, this test was not recommended for use by some studies [11]; or it was shown that it should be complemented by additional methods because of its low sensitivity [12]. Other POC format tools based on isothermal amplification were also developed [13,14,15,16,17,18]. Nevertheless all these tools are generic and detect Leishmania parasites without identifying them. Consequently, a simple, reliable and rapid DNA test that detects and identifies the species while minimizing time to result does not yet exist for CL.
Despite PCR is a laboratory-based technique, advances in technologies adapted its use for POC testing. In addition, recent pandemic and outbreaks has given us a clear reminder that, there is an increasing need for portable PCR solutions for remote testing for surveillance and diseases control. Palm PCR is a battery powered and pocket sized convective PCR machine able to perform DNA amplification in ultra-fast speed (10-18min). Using a ready to use mix for amplification and lateral flow immunoassays for Leishmania parasites detection, make the palm PCR a very promising option for On-site testing [19,20,21].
Therefore, in this study we aim to deliver novel CL molecular diagnosis assays that satisfy POC criteria for timely patient management and disease control. Indeed, this study describes handheld ultra-fast duplex PCR assays coupled to amplicon detection by lateral flow (LF) chromatography on a generic cassette (PCRD). We demonstrated their potential as rapid and simple molecular diagnostic tests for the concomitant detection and identification of the main Leishmania parasites encountered in Tunisia and the Old World including L. major, L. tropica, and L. infantum/donovani. Our test intend to equip areas with low resources and poor laboratories infrastructure with an equitable access to high quality patient diagnosis and management.
2. Materials and Methods
2.1. Ethical statement
The study is approved by the Ethic Committee of: Institut Pasteur de Tunis (Ref:2016/24/I/LRIPT04).
2.2. Parasite strains
We used a selection of 37 well-characterized Leishmania DNAs belonging to L. major (N=12), L. infantum (N=11), L. tropica (N=10), L. donovani (N=1), L. aethiopica (N=1), L. arabica (N=1) and L. turanica (N=1) species for test development (Table 1). Analyzed DNAs were extracted from Leishmania strains obtained from reference centers in Monpellier, clinical isolates from health centers in Tunisia and strains isolated from reservoirs in the frame of field study in Tunisia [22] . Species assignment was undertaken by isoenzyme and/or PCR-and RFLP typing [9].
2.3. Assays design
We aimed to design assays based on ultra-fast PCRs coupled to PCRD lateral flow for the concomitant detection and identification of the 3 studied Leishmania species (L. major, L. tropica and L. infantum). As the PCRD (and most of the commercially available lateral flow test has only two test lines), we sought to identify two groups of DNA targets. The first group would correspond to DNA targets showing higher level of similarity in their DNA sequences between L. major and L. tropica as compared to L. infantum to identify an L. major/L. tropica specific target. The second group would show higher level of similarity in their DNA sequences between L. infantum and L. tropica as compared to L. major to identify an L. infantum/L. tropica specific target. The two specific targets will be first used to set up group of species specific simplex assays that will be than combined for the set up of a ultra fast/LF duplex assays. Lateral flow detection is a sandwich immunochromatografic based assay that relies on the apropriately labelled primers (FAM/Biotin and DIG/Biotin) used for the PCR assays. The labels we used are recommanded by the PCRD manufacturer so that the labelled amplicons are captured by the antibodies immobilized at the test lines (anti-Biotin) to form a couloured complex and therefore a visible line. Principles underlying the ultra-fast PCR and LF assays for the simultaneous detection and identification of Leishmania parasites and the expected results are shown in Figure 1.
2.4. Target selection and primers design
DNA targets were identified based on bibliography search and were selected essentially from specific genes published by [23]and listed among the species specific ones [23] or genes used in other published molecular methods and were described as showing inter-species sequence polymorphisms [24] (Table 2). Sequences of these targets were available in our local database in addition to those retrieved from TritrypDB public database (Release 59, 30 Aug 2022). We used Geneious 3.6.2 computer program for sequence alignment and SNPs identification.
Primers were designed manually to specifically amplify DNA targets in L. major/L. tropica or L. infantum/L. tropica group of species. The design took in consideration sequence polymorphisms across multiple sequence alignments of strains and species. Priming sites were selected to be conserved within a group of species (eg. L. major/L. tropica) and presenting polymorphisms in the DNA sequences of the remaining studied species (eg. L. infantum). Moreover, primers design was undertaken considering other criteria including expected amplicon size (should be different for the two DNA targets involved in the duplex PCR), melting temperature (should be the same for the two DNA targets involved in the duplex PCR) and the absence of secondary structures and internal hybridization for optimal PCR efficiency.
2.5. Ultra-fast simplex PCR assays for target screening
To screen for Leishmania group of species simplex specific amplification assays, we used non- labeled primers (RAN Biolinks, Tunisia) (Table 3) and agarose gels visualization; the retained primers were then labeled to be able to detect the amplicons by lateral flow assay.
The 20µl PCR mixture contained a ready to use 1X PalmTaq Express Master Mix (Ahram Biosystems, Inc. Korea) that includes 0.8U hot start Palm Taq high speed DNA polymerase, 2.5mM MgCl2 and 0.2mM dNTPs to which we add 0.5µM each primer and 2µl of 20ng/µl parasite’s DNA. PCR was run in a battery-operated Palm PCR G3 Ultra-fast mobile PCR system (Ahram Biosystems, Inc. Korea). In the Palm PCR system, each run is controlled by a protocol, as defined by a set of control parameters. A PCR protocol is defined by four control parameters including PCR speed or Turbo (T1, T2 or T3), annealing temperature (52-60°C), cycle’s number (max 100), and a preheat step. After selecting a protocol on the Palm PCR device, the PCR time is automatically set by the operating software linked to the speed and cycles respectively. The duration of the PCR is 10-18 minutes depending on the Turbo (T) and the cycle's number selected. We used Turbo 3 (T3) and 45 cycles to run our simplex PCRs. Annealing temperature was set depending on the primer pairs used. The reaction time was 13min 30s. Then, PCR products were analyzed by 2% agarose gel electrophoresis (1h, 80V). For each primer pair, reactivity was tested on a selection of 3 strains’ DNA per each species and the selection was based on their ability to react according to taxonomic specificity.
2.6. Ultra-Fast duplex PCR assays screening and optimization
To set up duplex assays, most relevant simplex assays were engaged in ultra-fast duplex PCR where 2 primer pairs were added in the same reaction mix. Ultra-fast duplex PCR highly depends on the target sequence, amplicon size and primers properties. Therefore, different primers combinations (Table 4), equimolar concentrations (0.5µM, 0.4µM, 0.3µM, 0.25µM and 0.2µM) and ratios (0.35:0.15 and 0.3:0.2) were tested in order to select primers combination/concentration having the best analytical sensitivity and specificity. The reactions were set up at room temperature and run using the battery-powered, Palm portable PCR machine (Ahram Biosystems Inc., Seoul, Korea). We used Turbo 1 (T1), 45 cycles and an annealing temperature of 60°C to run our PCRs. PCRs run time using T1 speed and 45 cycles parameters is 18min. Visualization of the PCR products was undertaken by agarose gel electrophoresis (1h, 80V).
2.7. Ultra- fast duplex PCR and LF reading
The 20µl PCR mixture contained a ready to use 1X PalmTaq Express Master Mix (Ahram Biosystems, Inc. Korea) to which we add 0.3µM of primers amplifying mt30 marker, 0.2µM of primers amplifying it20 marker and 2µl of 20ng/µl parasite’s DNA. The ultra-fast duplex PCR was run in the Palm PCR device employing the previously described control parameters (T1, 45 cycles, Ta=60°C). Then 6µl of the PCR products were diluted in 84µl dilution buffer (Abingdon Health, UK), 75µl of which were transferred in the sample pad of the PCRD cassette as recommended by the PCRD manufacturer. Amplicons were then captured on a two- test lines neutravidin- coated carbon nanoparticle- based LF chromatography system (PCRD, Abingdon Health). The result was read with the naked eye after 10 minutes flow migration and a picture was immediately taken for our records.
2.8. Analytical specificity and sensitivity
The retained ultra-fast PCR protocol using the retained primer pairs was validated for its taxonomic specificity on panels of representative well-described Leishmania strains belonging to different species from diverse geographical origins and hosts (Table 1). Analytical sensitivity was also tested on 1/10 serial dilutions starting from 20ng/µl to 2x10-5ng/µl of input DNA of Leishmania parasites belonging to the 3 species: L. major, L. tropica and L. infantum.
To mimic a real situation of the detection of parasites' DNA from CL samples and test the impact of human DNA on our ultra-fast PCR assays, 50µl of human blood was spiked with 4x108 of L. major cultured promastigotes. Then total DNA (human and parasite's DNAs) was extracted using QIAmp DNA mini kit (Qiagen) as recommended by the manufacturer. The extracted DNA was 10-fold serially diluted and used to investigate the analytical sensitivity of our developed ultra-fast duplex PCR/LF test.
3. Results
3.1. Target selection and primers design
Based on multiple alignments of Leishmania species DNA sequences, we identified in total four promising DNA targets with relevance to develop our expected specific ultra-fast PCR assays. They correspond to coding and intergenic regions with high level of conservation (>90%) between L. major and L. tropica DNA sequences (mt30, mt7SL and mt22) and others with high level of conservation (>90%) between L. tropica and L. infantum DNA sequences (it20). Our local database from an ongoing project contained sequence information covering different strains of the species of interest. Within these targets, we searched for sequence polymorphisms to design primer pairs specific to L. major/L. tropica group of species and others specific to L. infantum/L. tropica group of species. We designed in total 6 primer pairs, they include 6 primer pairs targeting L. major and L. tropica group of species (mt30FR and mt22F1R1, mt22F2R2, mt22F1R2, mt22F2R1 and mt7SLFR) and 2 targeting L. infantum and L. tropica group of species (it20F1R1 and it20F2R2) (Table 3).
3.2. Specific simplex ultra fast PCRs screening assays
For cost effectiveness, simplex PCR assays were first set up using non-labeled primers (RAN Biolinks, Tunisia) in order to test the taxonomic specificity of the designed primers. For each primer pair, taxonomic specificity was tested on a selection of 3 strains for each species.
Simplex PCR assays were set up to target L. major and L. tropica group of species and L. infantum and L. tropica group of species. They were run using PalmTaq Express master mix and non-labeled primers in the Palm PCR device. Amplicons were visualized on agarose gels. Results showed that primer pair mt7SLFR is reacting with the 3 tested Leishmania species and primer pair mt22F1R2 is showing non-specific amplifications. So these primer pairs were rejected. Six primer pairs gave specific profiles as expected. Indeed, primer pairs mt22F1R1, mt22F2R2, mt22F1R2 and mt30F/R reacted with L. major and L. tropica DNAs without reacting with L. infantum DNA; primers pairs it20F1/R1 and it20F2/R2 reacted with L. infantum and L. tropica DNAs without reacting with L. major DNA (Figure 2). These primers were therefore selected for the ultra-fast duplex PCR assays development.
3.3. Ultra-fast duplex PCR and LF detection assays set up
To set up the ultra-fast duplex PCR, we tested 8 different combinations of PCRs primers including mt22F1R1/it20F1R1 (A), mt22F2R2/it20F2/R2 (B) mt22F1R1/it20F2R2 (C), mt22F2R2/it20F1R1 (D), mt22-F2R1/it20-F1R1 (E), mt22-F2/R1/it20-F2R2 (F), mt30-FR/it20-F1R1 (G) and mt30FR/it20F2R2 (H) (Table 4).
Most of the combinations in the duplex PCRs did not give the expected results especially with L. tropica species. Some of the combinations (C, F and H) gave non-specific amplifications (Figure 3.a). In other cases, (B, D and E), we observed only one band for L. tropica where 2 bands are expected (Figure 3.b) In one case (A), we observed a significant imbalance in band intensities (Figure 3.c).
The best results obtained in term of species identification and differentiation was observed with the ultra-fast duplex PCR combining mt30FR and it20F1R1 (G) primer pairs. The selected ultra-fast duplex PCR was optimized by varying primers ratios to balance band intensities in agarose gel (Figure 4). The retained protocol is as follow: primers final concentrations mt30FR: 0.3µM, it20F1R1: 0.2µM (Figure 4), T1 turbo, Ta=60°C, 45 cycles. Then, the selected primers were differently labeled (mt30F-Dig/R-Biotin and it20F1-Fam/R1-Biotin) (RAN Biolinks, Tunisia) to allow detection by LF chromatography on the generic two test-line PCRD cassette used in this study (Abington Health, UK). The amplicons were first visualized on agarose gels then on PCRD lateral flow
Th selected protocol was tested on a selection of 33 well characterized Leishmania DNAs belonging to L. major (N=12), L. infantum (N=11) and L. tropica (N=10). They showed stable and reproducible results within each species (Figure 5).
Other Leishmania species were also tested including L. donovani (N=1), L. aethiopica (N=1), L. arabica (N=1) and L. turanica (N=1). Results showed that L. aethiopica has the same amplification profile as L. tropica while L. donovani, L. arabica and L. turanica share the same profile as L. infantum (Figure 6).
The analytical sensitivity was tested on the three species L. major, L. tropica and L. infantum and was 0.4pg for L. major, 4pg for L. infantum and 40pg for L. tropica (Figure 7). It corresponds to 0,5 parasites, 5 parasites and 50 parasites for L. infantum, L. major and L. tropica respectively if we assume that an average diploid genome mass of 80fg as stated by [25]. We notice that the test is less sensitive with L. tropica as two targets are amplified in the DNA sequences of this species.
3.4 Impact of human DNA on the ultra-fast duplex PCR and LF assays In order to test the impact of human DNA on our ultra-fast PCR assays and mimic a CL sample, cultured L. major promastigotes were spiked with human blood. Total DNA (human and parasite's DNAs) was extracted from this reconstitution, serially diluted and used to test the analytical sensitivity of our developed ultra-fast duplex PCR/LF assay. It showed a detection limit of 8 parasites (Figure 8) which is comparable to the limit of detection observed when we tested L. major DNA alone (5 parasites). No cross-reactivity was observed when we tested our assay with human DNA (Figure 8).
4. Discussion
In this study, we aimed at developing a simple, rapid, sensitive and specific DNA based test to detect and identify the most frequent Leishmania parasites causing cutaneous leishmaniasis (CL) in the OW; L. major, L. tropica and L. infantum. Current advances in enzymes technology and in equipment miniaturization have made PCR rapid to perform and feasible at the POC to guide decisions on treatment and clinical management of infectious diseases [20]. Accurate detection and identification of Leishmania parasites in endemic areas that lack appropriate resources is a global public health problem. In Tunisia and many other countries of the OW, direct examination using microscopy is the most commonly used technique. It is specific but lacks sensitivity in addition to the fact that it does not allow species identification. Molecular methods are needed to fill this gap, but their use is limited to few reference laboratories due to the cost of the required equipment to perform such experiments. Therefore, there is an urgent need for simple, rapid and accurate tests with high sensitivity and specificity that can be used at the point of care, without requiring special expertise neither sophisticated equipment, given the conditions prevailing in many disease- endemic areas.
Our study focused on the development of an alternative to the conventional PCR, based on duplex convective PCR using the Palm PCR device. The reaction operates with battery power at room temperature and uses a very simple system with ready-to-use mixes. It also has the advantage of being fast since the reaction takes place in 18 minutes (equivalent to 45 cycles of amplification) hence it is considered an ultra-fast method. The amplicons visualization is done using PCRD lateral flow immunoassay in 10 minutes. To our knowledge, this is the first study that associates convective PCR technology with lateral flow detection for Leishmania parasites detection and identification. The unique study describing an ultra-fast PCR for CL diagnosis used E-gel reader electrophoresis system for the detection of Leishmania Viania and some mimickers such as fungal or mycobacterial infections [26]. Moreover, it is the fastest PCR based technology method so far that could be used for the concomitant detection and identification of Leishmania parasites. Indeed, a conventional PCR reaction and other PCR- based techniques used for the same purpose require on average between 1.5 and 2 hours for parasites detection and additional steps and time for species identification [10,27]. Moreover, in our case, DNA target amplification and PCRD visualization take less than 30 minutes for Leishmania parasites detection and species identification. In the same context, a study aiming at delivering a CL diagnostic test in POC format, developed a method based on recombinase polymerase isothermal amplification coupled to lateral flow detection. The test took 50 minutes including 40 minutes for the amplification and 10 minutes for lateral flow detection of amplicons [15]. The described test is able to detect Leishmania parasites, but does not identify the species. Thus, our tool offers a time saving even compared to other POC format methods.
In addition, LF testing accessibility and feasibility have been demonstrated especially during the COVID19 pandemic. It showed to be an easy to use, affordable and accurate system that should be used for the next generation tests [28]. It offers a simple method for decentralized diagnosis of infectious diseases and control strategies [29,30,31]. Nevertheless, coupling ultra-fast duplex PCR to LF is challenging and the most arduous step in the development process is primers selection that avoid the formation of the non-specific band in the LF. The production of artefacts is mainly due to inter and intra-molecular interactions of primers [32]. It is very important to carefully design and check primers properties in silico in order to maximize the probability of duplex PCR experiment success [33].
Duplex PCR success depends mainly on the target sequence, amplicon size and primers properties [32]. The selected duplex combining the primer pairs mt30-F/R and it20-F1/R1 allowed to have 3 different amplification profiles and thus to distinguish the 3 species studied. The difference is based on the number of amplicons and their specificities. Indeed, by using the pair of primers mt30F/R and it20F1/R1, we obtained on agarose gel specific profiles of species including a band of 350 bp for L. major, two bands of 350 bp and 209 bp for L. tropica and one band of 209 bp for L. infantum. In addition, detection of amplicons by the PCRD LF assay yielded results in agreement with those obtained on agarose gel. The LF test allows the identification of the infecting species according to the number and position of test lines revealed: L. infantum (line1), L. major (line 2) and L. tropica (lines 1 and 2) which makes the result read out very simple and easy to interpret. As L. infantum and L. donovani group of species and L. tropica and L. aethipica group of species have a high level of sequence conservation [34,35] we had PCR tests reacting similarly with the two pairs of species. This should be useful if their application would be extended to other MENA and African regions where L. aethiopica and L. donovani are predominant [36].
One of our primer selection criteria was that primers specificities were shared by 2 species like L. major and L. tropica, or by L. tropica and L. infantum. We took advantages of sequences similarity between pairs of Leishmania species in order to identify our DNA targets and design the group of species-specific primers. Therefore, by duplexing the two types of assays, we were able to detect and identify the three Leishmania species encountered in Tunisia (and in Africa and Middle East) in one single reaction. On the other hand, some combinations of primers in a duplex PCR reaction did not give the expected profile, especially for L. tropica where 2 amplicons are expected. In the majority of combinations, we observed the appearance of a single amplification band instead of two. This result could be explained by the fact that, generally, there is a certain competitiveness between primers as the two of them are competing for the same pool of reagents. There is also a preferential amplification of certain specific targets due to their GC content leading to preferential denaturation [33]; or a differential accessibility of targets within genomes due to secondary structures [33]. Therefore, a single intense band was obtained following a duplex amplification by ultra-fast convective PCR. This results in an unbalanced amplifications leading to a single band or different intensities of the obtained bands [33,37].Another hypothesis is that there is a deficiency in the spontaneous circulation of molecules using the convection principle on which Palm-PCR is based. These molecules will therefore not be able to reach the appropriate temperature zone for the hybridization of the primer to the target. This hypothesis is supported by the fact that some of the primer combinations that performed poorly by convective PCR performed very well by conventional PCR (data not shown).
Our data suggest that the ultra-fast PCR method is sensitive and that the sensitivity varies according to the species. The limit of detection of our assay is 0.4pg for L. major, 4pg for L. infantum and 40pg for L. tropica; the equivalent of 5, 50 and 500 parasites respectively. The test showed to be less sensitive with L. tropica. For this species, we had two targets that are competing with the same reagent mixture that reach their depletion more quickly compared to L. major and L. infantum where a unique target is amplified. When tested on DNA extracted from human blood spiked with cultured Leishmania parasites, our assay detected 8 parasites. We noticed that human DNA did not affect the sensitivity of our test as we had a limit of detection of 5 parasites when we tested our assay with L. major DNA. Furthermore, no cross-reactivity was observed with human DNA. Other comparable results in terms of analytical sensitivity have been obtained by other studies using different sophisticated methods. For example, a study describing a probe-based allele-specific real-time PCR for Leishmania species identification was able to detect 12 parasites per reaction [38]. Another study describing a multiplex PCR targeting Leishmania sp. kDNA and a conserved region of the mammalian gapdh gene, detected 0.1ng of Leishmania DNA diluted in 100 ng of mammalian DNA [39]. Nevertheless, other studies based on isothermal amplifications showed a higher analytical sensitivity by detecting as low as 0.1 parasites per reaction [40]. It is known that the sensitivity of a multiplex PCR assay is reduced with increased numbers of target genes in the reaction [41]. In a recent study describing a Palm PCR assay coupled to agarose gel electrophoresis for Leishmania spp. detection in cutaneous ulcers achieved a specificity and a sensitivity of 90% and 91.7% respectively when tested in lab conditions [26]. In the field the same assay showed a sensitivity of 100% and a specificity of 25% [26]. The false positivity rate noted was assigned to contamination during DNA extraction in the field [26]. Our assay is combining the accuracy of a PCR test and the rapidity of an isothermal method. In addition to the time saving, Palm PCR System is a portable device that reduces the cost required for diagnostics. The average price of a conventional PCR or qPCR devices is typically between $5000 and $15000 with additional $ 0.6-2.5 USD for reagents per reaction, while that of Palm PCR is around $3000-3500 and reagents cost $0.6 per reaction [26]. Energy saving is also a strong point of Palm-PCR. A conventional thermocycler consumes on average up to 700W of energy, whereas the Palm PCR device consumes about 5W on mains power with the possibility of operating on rechargeable battery.
The CL diagnostic test based on ultra-fast duplex PCR through the Palm PCR System and lateral flow detection by PCRD appears prone for operation in low resource areas. This potential tool complies with the WHO "ASSURED" criteria to control and manage these infectious skin diseases beyond the framework of specialized laboratories. However, test evaluation and performance description are yet to be performed by validating our method using an adequate panel of cutaneous samples including from CL lesions. This will define the usefulness of the method and define its accuracy.
5. Conclusions
Through this study, we bring a proof of concept demonstration that ultra-fast duplex PCR method coupled to lateral flow chromatography read out would be a valuable test that provide fast on-site sample DNA analysis, with small sample consumption, short analysis time, and high sensitivity. It will potentially enable CL diagnosis at community health centers reducing the need of referring patients to a regional hospital for disease confirmation. Ultra-fast duplex PCR assays using a handheld PCR device coupled to LF read out hold promises as valuable POC test for CL diagnosis and species-adapted therapy guidance.
Author Contributions
Conceptualization: IBA, IG and YS; Methodology: IBA and YS; Validation: IBA and YS; Investigation: IBA, ZH and OR; Resources: IG and ASC; Visualization: IBA; Supervision: IBA; Project administration: IBA; Funding acquisition: IBA and IG; Writing—original draft preparation: IBA; writing—review and editing, IBA, YS and IG. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by USAID-PEER Women in Sciences Seed Grant, the NAS-USAID-PEER (AID-OAA-A-11-00012-PEER 518) and the Ministry of Higher Education and Research, Tunisia
Institutional Review Board Statement
The study is approved by the Biomedical Ethic Committee of Institut Pasteur de Tunis (Ref: 2016/24/I/LRIPT04).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Land, K.J.; Boeras, D.I.; Chen, X.-S.; Ramsay, A.R.; Peeling, R.W. REASSURED Diagnostics to Inform Disease Control Strategies, Strengthen Health Systems and Improve Patient Outcomes. Nat Microbiol 2019, 4, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Fathallah-Mili, A.; Saghrouni, F.; BenSaid, Z.; BenAoun, Y.S.-; Guizani, I.; BenSaid, M.; Mili, A.F.; Saghrouni, F.; BenSaid, Z.; BenAoun, Y.S.-; et al. Retrospective Analysis of Leishmaniasis in Central Tunisia: An Update on Emerging Epidemiological Trends; IntechOpen, 2012; ISBN 978-953-51-0274-8.
- Garni, R.; Tran, A.; Guis, H.; Baldet, T.; Benallal, K.; Boubidi, S.; Harrat, Z. Remote Sensing, Land Cover Changes, and Vector-Borne Diseases: Use of High Spatial Resolution Satellite Imagery to Map the Risk of Occurrence of Cutaneous Leishmaniasis in Ghardaïa, Algeria. Infect Genet Evol 2014, 28, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Haddad, N.; Saliba, H.; Altawil, A.; Villinsky, J.; Al-Nahhas, S. Cutaneous Leishmaniasis in the Central Provinces of Hama and Edlib in Syria: Vector Identification and Parasite Typing. Parasit Vectors 2015, 8, 524. [Google Scholar] [CrossRef] [PubMed]
- Rhajaoui, M.; Nasereddin, A.; Fellah, H.; Azmi, K.; Amarir, F.; Al-Jawabreh, A.; Ereqat, S.; Planer, J.; Abdeen, Z. New Clinico-Epidemiologic Profile of Cutaneous Leishmaniasis, Morocco. Emerg Infect Dis 2007, 13, 1358–1360. [Google Scholar] [CrossRef] [PubMed]
- Koff, A.; Rosen, T. Treatment of Cutaneous Leishmaniasis. Journal of the American Academy of Dermatology 1994, 31. [Google Scholar] [CrossRef]
- Arevalo, J.; Ramirez, L.; Adaui, V.; Zimic, M.; Tulliano, G.; Miranda-Verástegui, C.; Lazo, M.; Loayza-Muro, R.; De Doncker, S.; Maurer, A.; et al. Influence of Leishmania (Viannia) Species on the Response to Antimonial Treatment in Patients with American Tegumentary Leishmaniasis. J Infect Dis 2007, 195, 1846–1851. [Google Scholar] [CrossRef]
- Mosimann, V.; Neumayr, A.; Hatz, C.; Blum, J.A. Cutaneous Leishmaniasis in Switzerland: First Experience with Species-Specific Treatment. Infection 2013, 41, 1177–1182. [Google Scholar] [CrossRef]
- Schönian, G.; Nasereddin, A.; Dinse, N.; Schweynoch, C.; Schallig, H.D.F.H.; Presber, W.; Jaffe, C.L. PCR Diagnosis and Characterization of Leishmania in Local and Imported Clinical Samples. Diagn Microbiol Infect Dis 2003, 47, 349–358. [Google Scholar] [CrossRef]
- Bel Hadj Ali, I.; H, C.; Y, S.B.A.; E, H.-S.; H, S.; A, Y.; O, E.D.; Z, H.; Mm, M.; M, B.S.; et al. Dipeptidyl Peptidase III as a DNA Marker to Investigate Epidemiology and Taxonomy of Old World Leishmania Species. PLoS neglected tropical diseases 2021, 15. [Google Scholar] [CrossRef]
- van Henten, S.; Fikre, H.; Melkamu, R.; Dessie, D.; Mekonnen, T.; Kassa, M.; Bogale, T.; Mohammed, R.; Cnops, L.; Vogt, F.; et al. Evaluation of the CL Detect Rapid Test in Ethiopian Patients Suspected for Cutaneous Leishmaniasis. PLoS Negl Trop Dis 2022, 16, e0010143. [Google Scholar] [CrossRef]
- Bennis, I.; Verdonck, K.; El Khalfaoui, N.; Riyad, M.; Fellah, H.; Dujardin, J.-C.; Sahibi, H.; Bouhout, S.; Van der Auwera, G.; Boelaert, M. Accuracy of a Rapid Diagnostic Test Based on Antigen Detection for the Diagnosis of Cutaneous Leishmaniasis in Patients with Suggestive Skin Lesions in Morocco. Am J Trop Med Hyg 2018, 99, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Gonzalez, A.; Saldarriaga, O.A.; Tartaglino, L.; Gacek, R.; Temple, E.; Sparks, H.; Melby, P.C.; Travi, B.L. A Novel Molecular Test to Diagnose Canine Visceral Leishmaniasis at the Point of Care. Am J Trop Med Hyg 2015, 93, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Ghosh, P.; Khan, M.A.A.; Hossain, F.; Böhlken-Fascher, S.; Matlashewski, G.; Kroeger, A.; Olliaro, P.; Abd El Wahed, A. Mobile Suitcase Laboratory for Rapid Detection of Leishmania Donovani Using Recombinase Polymerase Amplification Assay. Parasit Vectors 2016, 9, 281. [Google Scholar] [CrossRef]
- Cossio, A.; Jojoa, J.; Castro, M.d.M.; Castillo, R.M.; Osorio, L.; Shelite, T.R.; Saravia, N.G.; Melby, P.C.; Travi, B.L. Diagnostic Performance of a Recombinant Polymerase Amplification Test—Lateral Flow (RPA-LF) for Cutaneous Leishmaniasis in an Endemic Setting of Colombia. PLOS Neglected Tropical Diseases 2021, 15, e0009291. [Google Scholar] [CrossRef]
- Khan, M.A.A.; Faisal, K.; Chowdhury, R.; Nath, R.; Ghosh, P.; Ghosh, D.; Hossain, F.; Abd El Wahed, A.; Mondal, D. Evaluation of Molecular Assays to Detect Leishmania Donovani in Phlebotomus Argentipes Fed on Post-Kala-Azar Dermal Leishmaniasis Patients. Parasit Vectors 2021, 14, 465. [Google Scholar] [CrossRef]
- Mesa, L.E.; Manrique, R.; Robledo, S.M.; Tabares, J.; Pineda, T.; Muskus, C. The Performance of the Recombinase Polymerase Amplification Test for Detecting Leishmania Deoxyribonucleic Acid from Skin Lesions of Patients with Clinical or Epidemiological Suspicion of Cutaneous Leishmaniasis. Trans R Soc Trop Med Hyg 2021, 115, 1427–1433. [Google Scholar] [CrossRef]
- Erber, A.C.; Sandler, P.J.; de Avelar, D.M.; Swoboda, I.; Cota, G.; Walochnik, J. Diagnosis of Visceral and Cutaneous Leishmaniasis Using Loop-Mediated Isothermal Amplification (LAMP) Protocols: A Systematic Review and Meta-Analysis. Parasites Vectors 2022, 15, 34. [Google Scholar] [CrossRef]
- Kim, T.-H.; Hwang, H.J.; Kim, J.H.; Department of Life and Nanopharmaceutical Sciences, Graduate School, Kyung Hee University, Seoul 02447, Republic of Korea. Optimization of Ultra-Fast Convection Polymerase Chain Reaction Conditions for Pathogen Detection with Nucleic Acid Lateral Flow Immunoassay. Intern. J. Oral Biol. 2019, 44, 8–13. [Google Scholar] [CrossRef]
- Lim, S.; Nan, H.; Lee, M.-J.; Kang, S.H. Fast On-Site Diagnosis of Influenza A Virus by Palm PCR and Portable Capillary Electrophoresis. J Chromatogr B Analyt Technol Biomed Life Sci 2014, 963, 134–139. [Google Scholar] [CrossRef]
- Mahale, P.; Warke, R.; Ramaiya, M.; Balasubramanian, D.; Shetty, S.; Mankeshwar, R.; Chowdhary, A. Assessment of Efficacy of Palm Polymerase Chain Reaction with Microscopy, Rapid Diagnostic Test and Conventional Polymerase Chain Reaction for Diagnosis of Malaria. Indian Journal of Medical Microbiology 2019, 37, 192–197. [Google Scholar] [CrossRef]
- Ben-Ismail, R.; Ben Rachid, M.S.; Gradoni, L.; Gramiccia, M.; Helal, H. Leishmaniose cutanée zoonotique en Tunisie : Étude du réservoir dans le foyer de Douara. 1987.
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and Gene Copy Number Variation Allow Major Structural Change between Species and Strains of Leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef]
- Zelazny, A.M.; Fedorko, D.P.; Li, L.; Neva, F.A.; Steven; Fischer, H. Evaluation of 7sl Rna Gene Sequences for the Identification of Leishmania Spp. 2005.
- Tupperwar, N.; Vineeth, V.; Rath, S.; Vaidya, T. Development of a Real-Time Polymerase Chain Reaction Assay for the Quantification of Leishmania Species and the Monitoring of Systemic Distribution of the Pathogen. Diagn Microbiol Infect Dis 2008, 61, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Kariyawasam, R.; Valencia, B.M.; Lau, R.; Shao, E.; Thompson, C.A.; Stevens, M.; Kincaid, L.; Del Castillo, A.L.Q.; Cruz-Arzapalo, L.O.; Llanos-Cuentas, A.; et al. Evaluation of a Point-of-Care Molecular Detection Device for Leishmania Spp. and Intercurrent Fungal and Mycobacterial Organisms in Peruvian Patients with Cutaneous Ulcers. Infection 2021, 49, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Schönian, G.; Nasereddin, A.; Dinse, N.; Schweynoch, C.; Schallig, H.D.F.H.; Presber, W.; Jaffe, C.L. PCR Diagnosis and Characterization of Leishmania in Local and Imported Clinical Samples11Part of This Work Has Been Presented at the Second World Congress on Leishmaniosis in Crete, Greece, May 2001. Diagnostic Microbiology and Infectious Disease 2003, 47, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Budd, J.; Miller, B.S.; Weckman, N.E.; Cherkaoui, D.; Huang, D.; Decruz, A.T.; Fongwen, N.; Han, G.-R.; Broto, M.; Estcourt, C.S.; et al. Lateral Flow Test Engineering and Lessons Learned from COVID-19. Nat Rev Bioeng 2023, 1, 13–31. [Google Scholar] [CrossRef]
- Ahmed, M.; Pollak, N.M.; Hugo, L.E.; van den Hurk, A.F.; Hobson-Peters, J.; Macdonald, J. Rapid Molecular Assays for the Detection of the Four Dengue Viruses in Infected Mosquitoes. Gates Open Res 2022, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, N.J.; Menting, S.; Wentink-Bonnema, E.M.S.; Broekhuizen-van Haaften, P.E.; Withycombe, E.; Schallig, H.D.F.H.; Mens, P.F. Laboratory Evaluation of the Miniature Direct-on-Blood PCR Nucleic Acid Lateral Flow Immunoassay (Mini-DbPCR-NALFIA), a Simplified Molecular Diagnostic Test for Plasmodium. Malar J 2023, 22, 98. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, Q.; Yuan, W.; Shi, Y.; Dong, S.; Luo, X. Visual and Rapid Identification of Chlamydia Trachomatis and Neisseria Gonorrhoeae Using Multiplex Loop-Mediated Isothermal Amplification and a Gold Nanoparticle-Based Lateral Flow Biosensor. Front Cell Infect Microbiol 2023, 13, 1067554. [Google Scholar] [CrossRef]
- Sint, D.; Raso, L.; Traugott, M. Advances in Multiplex PCR: Balancing Primer Efficiencies and Improving Detection Success. Methods Ecol Evol 2012, 3, 898–905. [Google Scholar] [CrossRef]
- Elnifro, E.M.; Ashshi, A.M.; Cooper, R.J.; Klapper, P.E. Multiplex PCR: Optimization and Application in Diagnostic Virology. Clin Microbiol Rev 2000, 13, 559–570. [Google Scholar] [CrossRef]
- Fernández-Arévalo, A.; El Baidouri, F.; Ravel, C.; Ballart, C.; Abras, A.; Lachaud, L.; Tebar, S.; Lami, P.; Pratlong, F.; Gállego, M.; et al. The Leishmania Donovani Species Complex: A New Insight into Taxonomy☆. International Journal for Parasitology 2020, 50, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Krayter, L.; Schnur, L.F.; Schönian, G. The Genetic Relationship between Leishmania Aethiopica and Leishmania Tropica Revealed by Comparing Microsatellite Profiles. PLoS ONE 2015, 10, e0131227. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.M.; Welburn, S.C. Leishmaniasis Beyond East Africa. Frontiers in Veterinary Science 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, Z.; Kim, T.-H.; Wang, X.; Bu, Z.; Li, S.; Hwang, H.J.; Kim, J.H. Ultra-Fast Detection and Differentiation of Brucella Genus Bacteria, B. Abortus, B. Melitensis, and B. Suis, Using Multiplex Convection Polymerase Chain Reaction 2019.
- Wu, Y.; Jiang, M.; Li, S.; Waterfield, N.R.; Yang, G. Establish an Allele-Specific Real-Time PCR for Leishmania Species Identification. Infectious Diseases of Poverty 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Cássia-Pires, R. de; Melo, M. de F.A.D. de; Barbosa, R. da H.; Roque, A.L.R. Multiplex PCR as a Tool for the Diagnosis of Leishmania Spp. KDNA and the Gapdh Housekeeping Gene of Mammal Hosts. PLoS ONE 2017, 12, e0173922. [Google Scholar] [CrossRef]
- Saldarriaga, O.A.; Castellanos-Gonzalez, A.; Porrozzi, R.; Baldeviano, G.C.; Lescano, A.G.; Santos, M.B. de L.; Fernandez, O.L.; Saravia, N.G.; Costa, E.; Melby, P.C.; et al. An Innovative Field-Applicable Molecular Test to Diagnose Cutaneous Leishmania Viannia Spp. Infections. PLOS Neglected Tropical Diseases 2016, 10, e0004638. [Google Scholar] [CrossRef]
- Phuektes, P.; Mansell, P.D.; Browning, G.F. Multiplex Polymerase Chain Reaction Assay for Simultaneous Detection of Staphylococcus Aureus and Streptococcal Causes of Bovine Mastitis. Journal of Dairy Science 2001, 84, 1140–1148. [Google Scholar] [CrossRef]
Figure 1.
Principles underlying simplex and duplex ultra-fast PCR and lateral low PCRD assays for the simultaneous detection and identification of Leishmania parasites and expected results. 1: L. major (L.m), 2: L. infantum (L.i), 3: L. tropica (L.t), N: No template control, M: Molecular weight, PCRD test line 1 detects L. major/L. tropica (L.m/L.t) using DIG/Biotin labeled primers, PCRD test line 2 detects L. infantum/L. tropica (L.i/L.t) using FAM/Biotin labeled primers, C: Control line.
Figure 1.
Principles underlying simplex and duplex ultra-fast PCR and lateral low PCRD assays for the simultaneous detection and identification of Leishmania parasites and expected results. 1: L. major (L.m), 2: L. infantum (L.i), 3: L. tropica (L.t), N: No template control, M: Molecular weight, PCRD test line 1 detects L. major/L. tropica (L.m/L.t) using DIG/Biotin labeled primers, PCRD test line 2 detects L. infantum/L. tropica (L.i/L.t) using FAM/Biotin labeled primers, C: Control line.
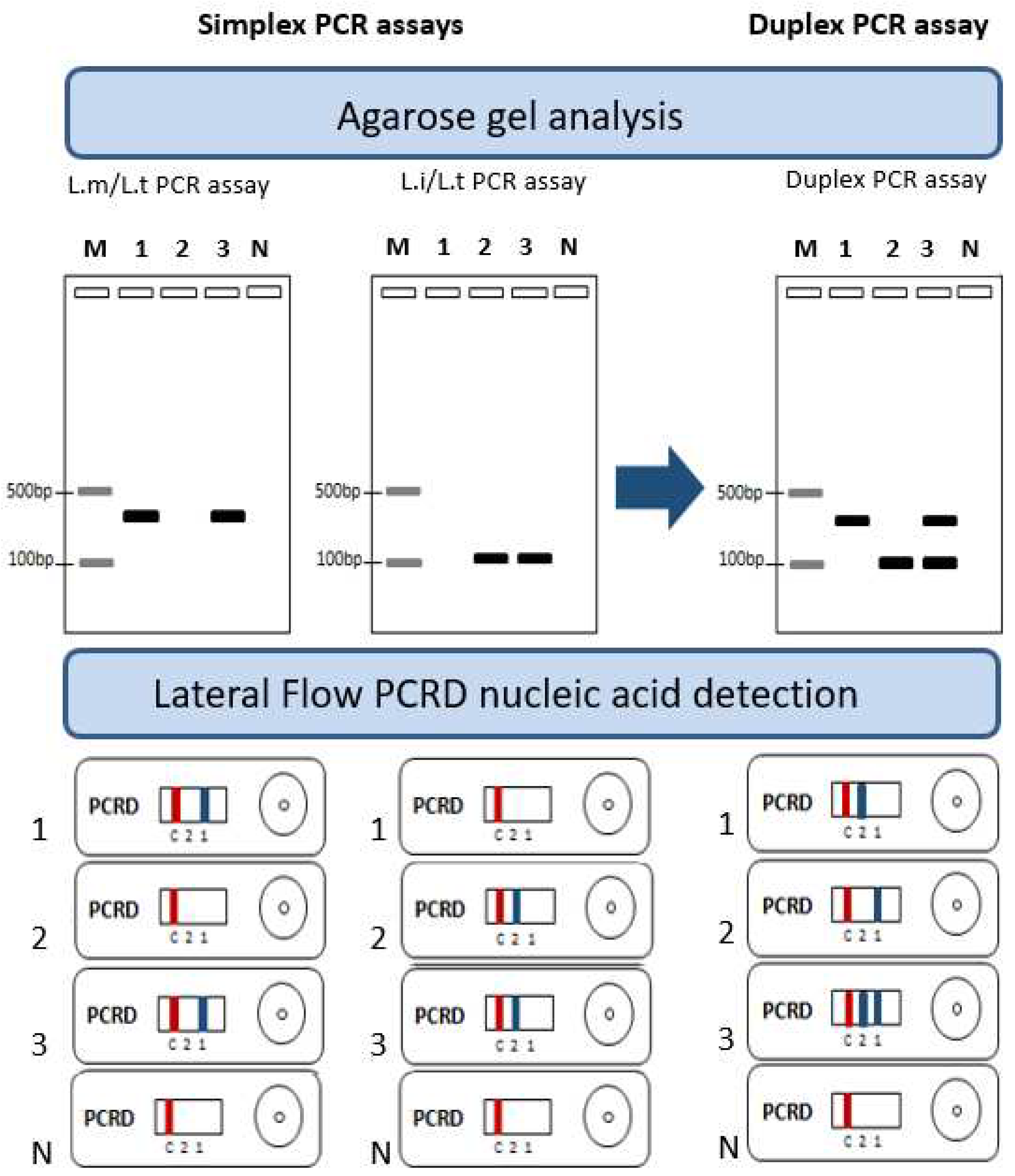
Figure 2.
Amplification results of the selected primer pairs used for the ultra-fast simplex PCR assays set in this study visualized on a 2% agarose gel electrophoresis. (a) mt22-F1/R1, (b) mt22-F2/R2, (c) mt30-F/R, (d): mt22F2R1 (e) it20-F2/R2, (f) it20-F1/R1,. L. major [1 : P-strain, 2 : IL53, 3 : R115], L. tropica [4 : DBKM, 5 : A Sinai III, 6 : Adhanis], L. infantum [7 : LV08, 8 : LV49, 9 : LV10], M : 100bp Molecular weight, N : No template control.
Figure 2.
Amplification results of the selected primer pairs used for the ultra-fast simplex PCR assays set in this study visualized on a 2% agarose gel electrophoresis. (a) mt22-F1/R1, (b) mt22-F2/R2, (c) mt30-F/R, (d): mt22F2R1 (e) it20-F2/R2, (f) it20-F1/R1,. L. major [1 : P-strain, 2 : IL53, 3 : R115], L. tropica [4 : DBKM, 5 : A Sinai III, 6 : Adhanis], L. infantum [7 : LV08, 8 : LV49, 9 : LV10], M : 100bp Molecular weight, N : No template control.
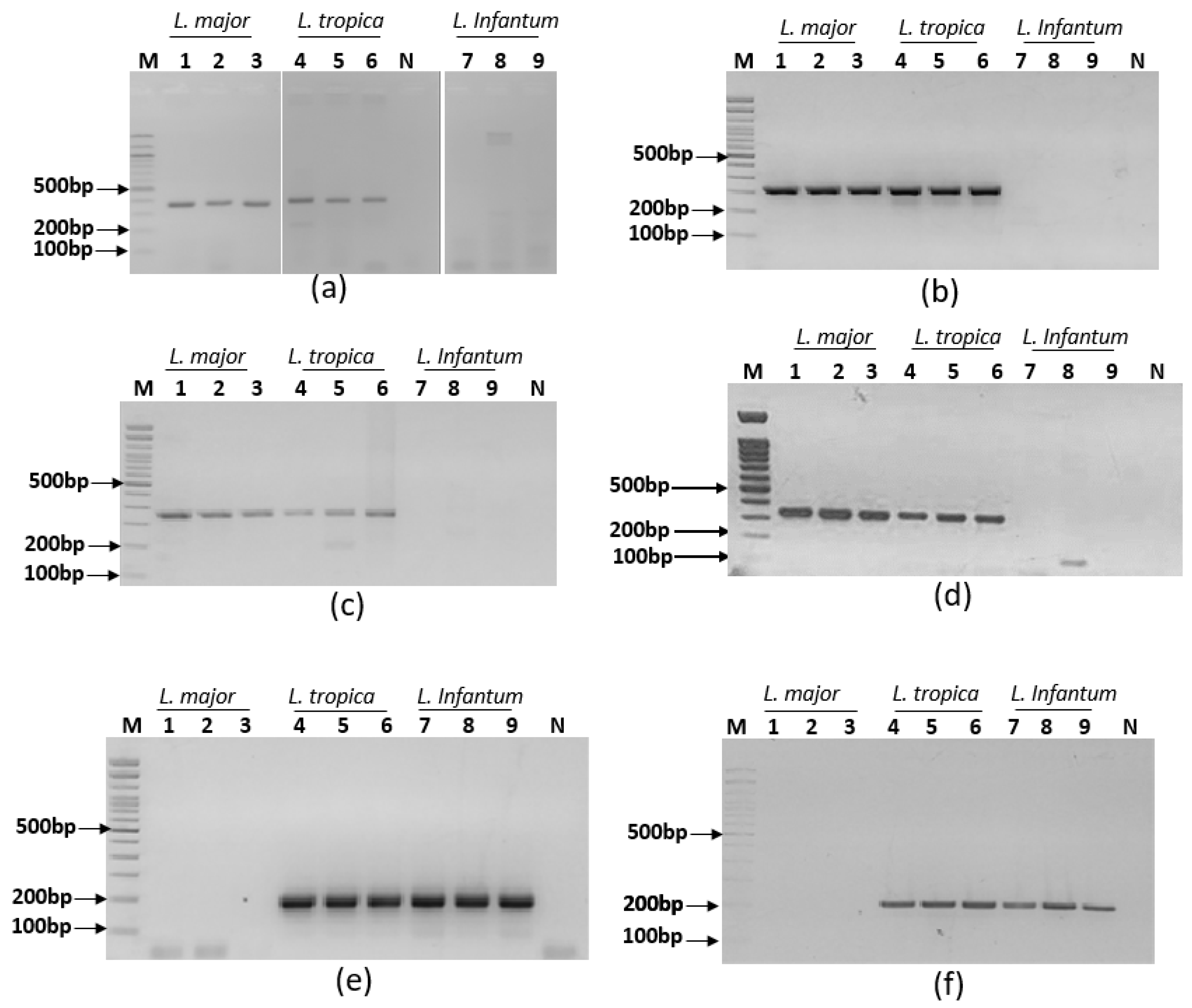
Figure 3.
Different scenarios of ultra fast duplex PCR with L. tropica species DNA using different combinations of primers. (a): Duplex PCR C showing non-specific amplification. (b) Duplex PCR D showing one band with L. tropica. (c): Duplex PCR A showing significant unbalanced amplification. 1: Bum30, 2: DBKM, M: 100bp molecular weight, N: No template control.
Figure 3.
Different scenarios of ultra fast duplex PCR with L. tropica species DNA using different combinations of primers. (a): Duplex PCR C showing non-specific amplification. (b) Duplex PCR D showing one band with L. tropica. (c): Duplex PCR A showing significant unbalanced amplification. 1: Bum30, 2: DBKM, M: 100bp molecular weight, N: No template control.
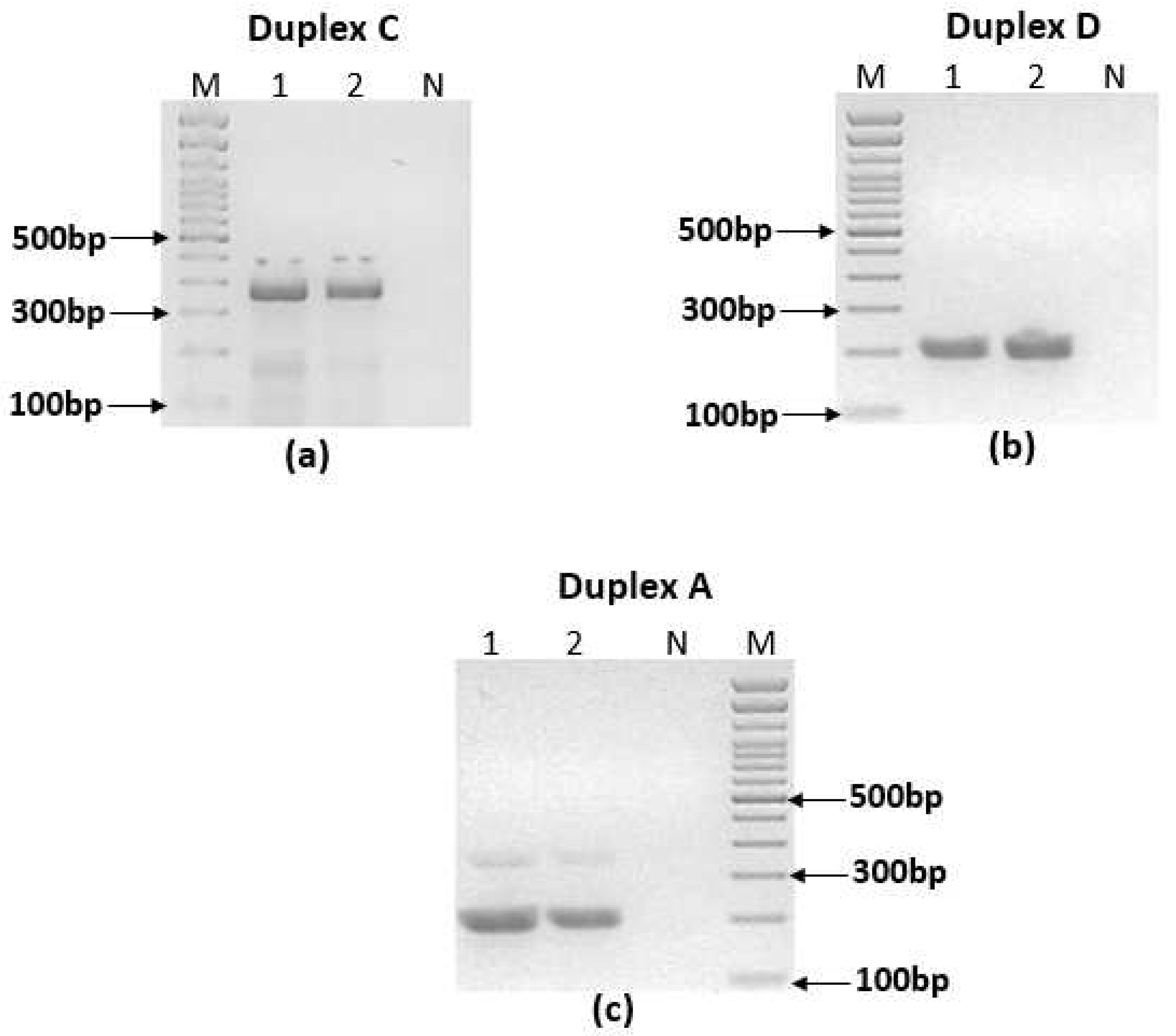
Figure 4.
Agarose gel profiles of the mt30FR/it20F1R1 ultra-fast duplex PCR using different primer’s concentrations. (a) Using equimolar concentrations. (b) Using 2 different ratios. N: No template control; M: 100bp Molecular weight.
Figure 4.
Agarose gel profiles of the mt30FR/it20F1R1 ultra-fast duplex PCR using different primer’s concentrations. (a) Using equimolar concentrations. (b) Using 2 different ratios. N: No template control; M: 100bp Molecular weight.
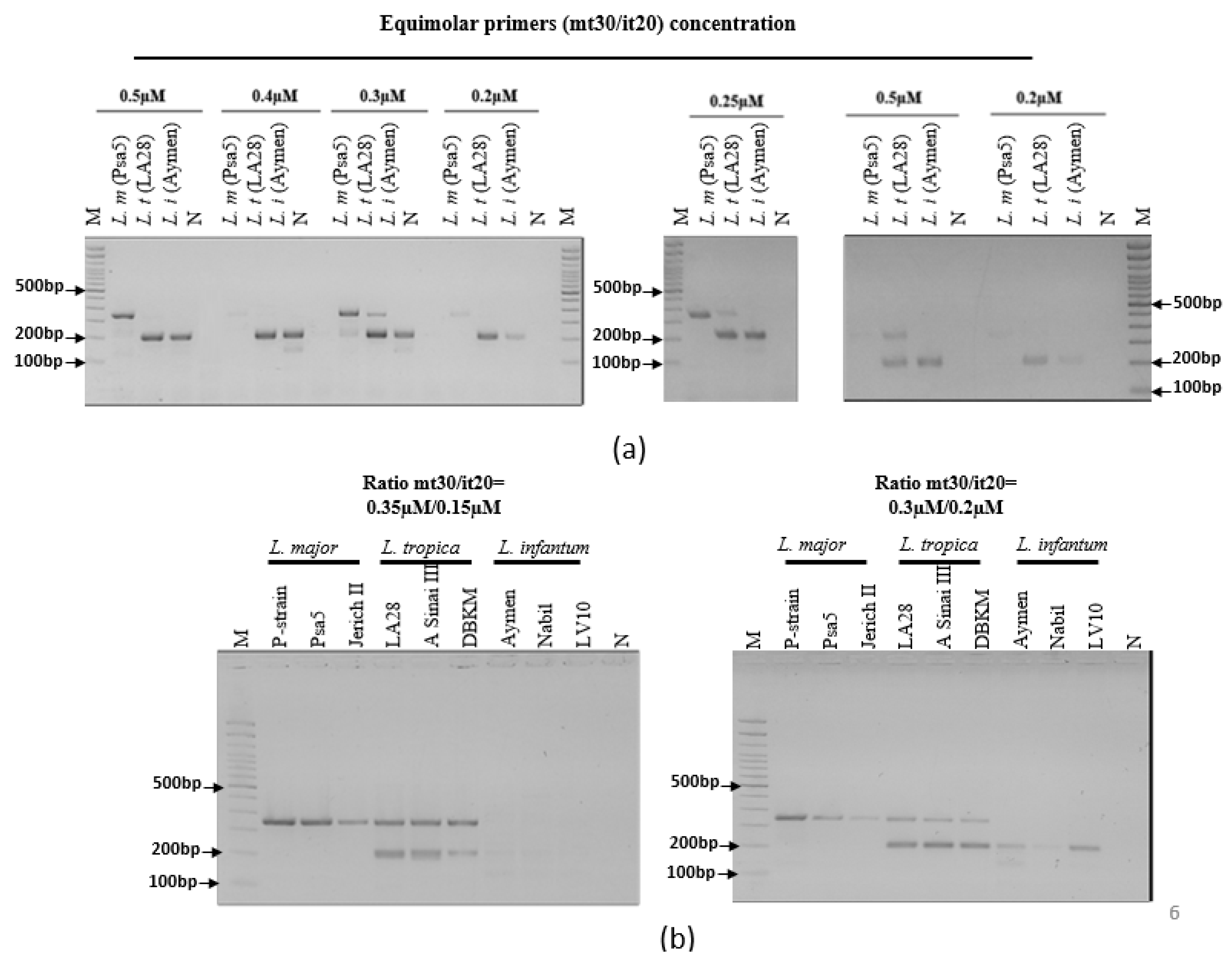
Figure 5.
Ultra-Fast duplex PCR mt30FR/it20F1R1: Test of analytical specificity. (a) 2% agarose gel electrophoresis. (b) PCRD detection. M: 100bp Molecular weight, N: No template control. 1: R44, 2: R99, 3: R155, 4: Bag17, 5:Bag9, 6: DBKM7: LV49, 8: D13, 9: D16, arrow: positive test.
Figure 5.
Ultra-Fast duplex PCR mt30FR/it20F1R1: Test of analytical specificity. (a) 2% agarose gel electrophoresis. (b) PCRD detection. M: 100bp Molecular weight, N: No template control. 1: R44, 2: R99, 3: R155, 4: Bag17, 5:Bag9, 6: DBKM7: LV49, 8: D13, 9: D16, arrow: positive test.

Figure 6.
Amplification profile of other Leishmaia species tested with the ultra-fast duplex PCR mt30FR/it20F1R1. (a) 2% agarose gel electrophoresis. (b) PCRD detection. 1 : New World L. infantum (PP75), 2 : L. donovani (L1005), 3 : L. aethiopica (L100), 4 : L. arabica (J238), 5 : L. turanica (95A), 6 : L. major (R44), 7 : L. tropica (Bag17), 8 : L. infantum (LV49), M : 100bp Molecular weight, N: No template control, arrow: positive test.
Figure 6.
Amplification profile of other Leishmaia species tested with the ultra-fast duplex PCR mt30FR/it20F1R1. (a) 2% agarose gel electrophoresis. (b) PCRD detection. 1 : New World L. infantum (PP75), 2 : L. donovani (L1005), 3 : L. aethiopica (L100), 4 : L. arabica (J238), 5 : L. turanica (95A), 6 : L. major (R44), 7 : L. tropica (Bag17), 8 : L. infantum (LV49), M : 100bp Molecular weight, N: No template control, arrow: positive test.

Figure 7.
Analytical sensitivity of the ultra-fast duplex PCR mt30FR/it20F1R1 using a 10-fold serial dilution of DNA of: (a) L. major, (b) L. infantum and (c) L. tropica. M: 100bp Molecular weight, 1: 40ng, 2: 4ng, 3: 0.4ng, 4: 4x10-2ng, 5: 4x10-3ng, 6: 4x10-4ng, 7: 4x10-5ng, N: No template control.
Figure 7.
Analytical sensitivity of the ultra-fast duplex PCR mt30FR/it20F1R1 using a 10-fold serial dilution of DNA of: (a) L. major, (b) L. infantum and (c) L. tropica. M: 100bp Molecular weight, 1: 40ng, 2: 4ng, 3: 0.4ng, 4: 4x10-2ng, 5: 4x10-3ng, 6: 4x10-4ng, 7: 4x10-5ng, N: No template control.
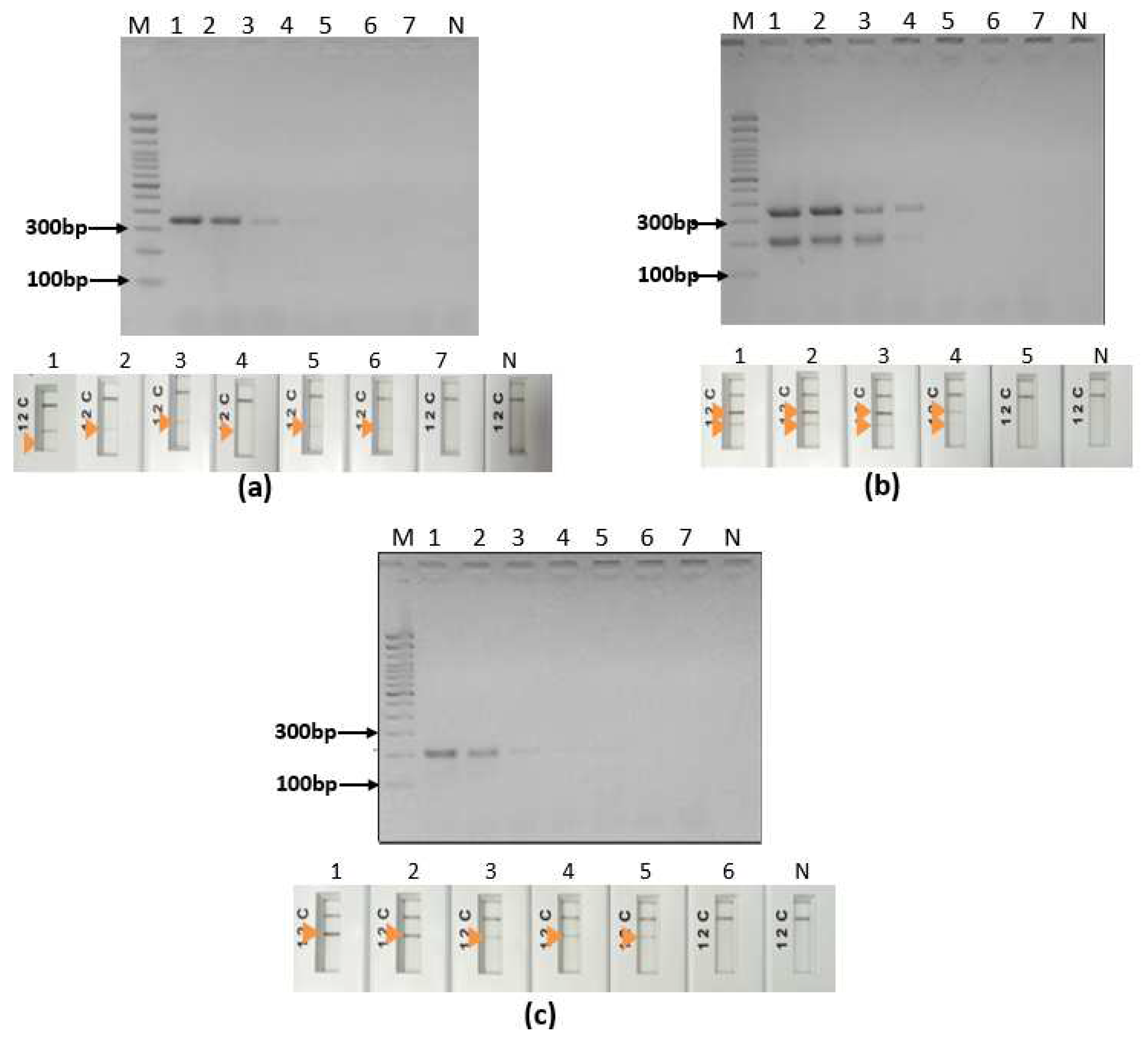
Figure 8.
Limit of detection of the ultra-fast duplex PCR mt30F/R-it20F1R1 using a serial dilution of DNA extracted from a mixture of 4x108 parasites and 50µl of human blood, 2µl of the total extracted DNA is added in the reaction. (a) 2% Agarose gel detection. (b) Lateral Flow detection (PCRD) MW: Molecular weight, p: parasites, N: No template control.
Figure 8.
Limit of detection of the ultra-fast duplex PCR mt30F/R-it20F1R1 using a serial dilution of DNA extracted from a mixture of 4x108 parasites and 50µl of human blood, 2µl of the total extracted DNA is added in the reaction. (a) 2% Agarose gel detection. (b) Lateral Flow detection (PCRD) MW: Molecular weight, p: parasites, N: No template control.
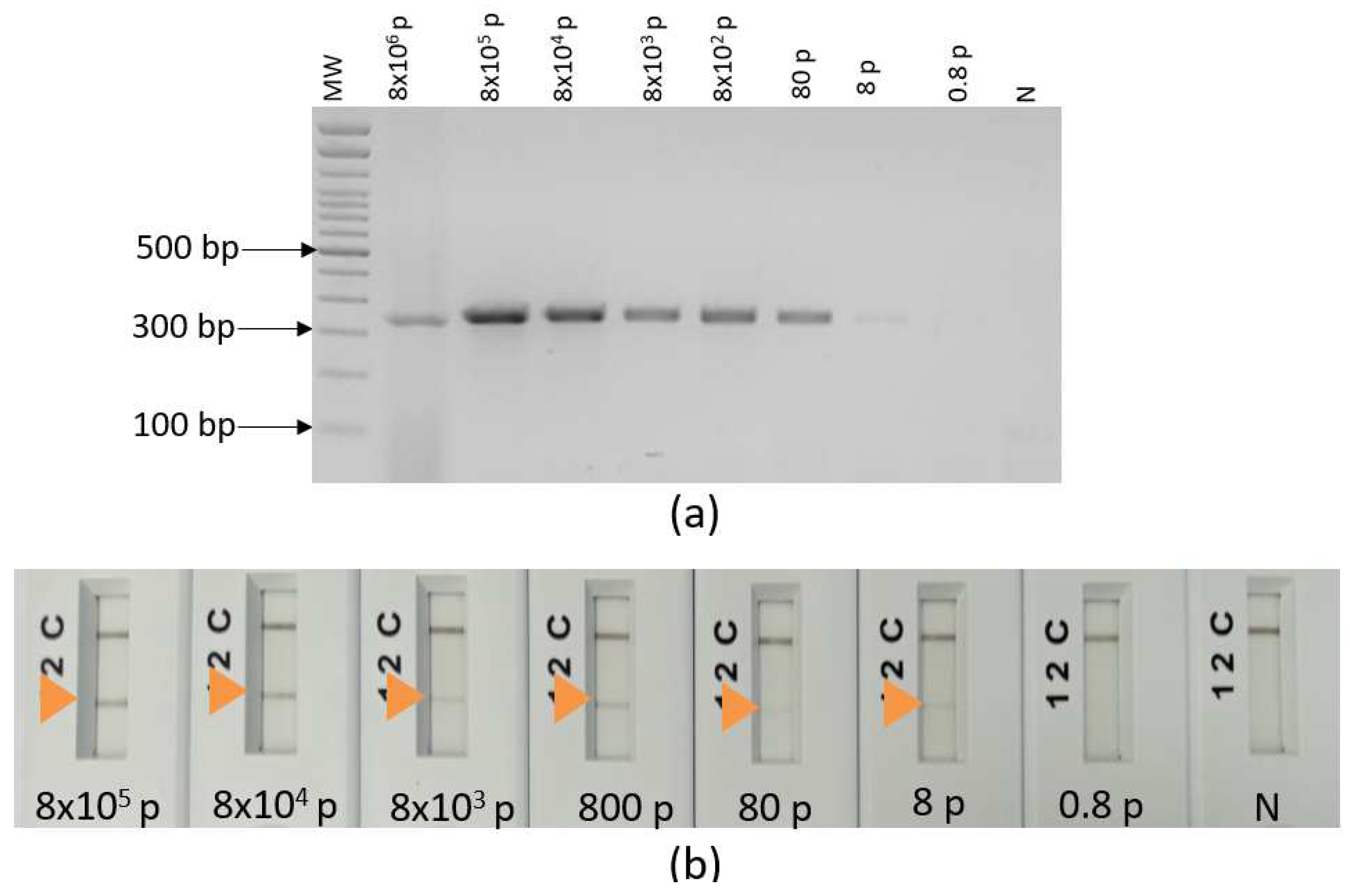

Table 1.
Strain’s DNAs used for the tool development process.
| WHO Code | Lab Code | Species | Zymodem | Clinical manifestation |
| MMER/TN/87/Ron114 | R114 | L. major | MON-25 | NA |
| MPSA/TN/87/Ron99 | R99 | L. major | MON-25 | NA |
| MPSA/TN/87/Ron44 | R44 | L. major | MON-25 | NA |
| MPSA/TN/87/Ron155 | R155 | L. major | MON-25 | NA |
| MPSA/TN/87/Ron 102 | R102 | L. major | MON-25 | NA |
| MRHO/SU/59/P-Strain | P-strain | L. major | MON-4 | NA |
| MHOM/IL/83/IL24 | IL24 | L. major | MON-66 | CL |
| MHOM/IL/83/IL53 | IL53 | L. major | MON-67 | CL |
| MHOM/IL/67/Jericho II | Jerichll | L. major | MON-26 | CL |
| MPSA/TN/89/Psa1 | Psa1 | L. major | NT | NA |
| MPSA/TN/89/Psa5 | Psa5 | L. major | NT | NA |
| MHOM/TN/11/EMPA12 | EMPA12 | L. major | NT | CL |
| MHOM/TN/80/IPT1 | IPT1 | L. infantum | MON-1 | VL |
| MHOM/TN/88/Aymen | Aymen | L. infantum | MON-1 | VL |
| MHOM/TN/88/Nabil | Nabil | L. infantum | MON-1 | VL |
| MHOM/TN/92/LV08 | LV08 | L. infantum | NT | VL |
| MHOM/TN/92/LV10 | LV10 | L. infantum | MON-80 | VL |
| MHOM/TN/94/LV49 | LV49 | L. infantum | MON-24 | VL |
| MHOM/TN/94/LV50 | LV50 | L. infantum | MON-1 | VL |
| MHOM/TN/97/Drep 13 | D13 | L. infantum | MON-24 | CL |
| MHOM/TN/98/Drep16 | D16 | L. infantum | MON-24 | CL |
| MHOM/TN87/KA412 | KA412 | L. infantum | MON-1 | VL |
| MHOM/BR/74/PP75 | PP75 | L. infantum | MON-1 | VL |
| MHOM/IQ/76/BAG17 | Bag17 | L. tropica | LON-24 | CL |
| MRAT/IQ/73/Adhanis I | Adhanis | L. tropica | MON-5 | NA |
| MCAN/IN/71/DBKM | DBKM | L. tropica | MON-62 | NA |
| MHOM/IL/00/Gabaï159 | Gabai 159 | L. tropica | LON-9 | CL |
| MHOM/GR/00/LA28 | LA28 | L. tropica | LON-16 | CL |
| MHOM/IQ/73/A Sinaï III | A Sinai III | L. tropica | LON-11 | CL |
| MHOM/IQ/76/BAG9 | Bag 9 | L. tropica | MON-53 | CL |
| MHOM/SU/74/SAF K27 | K27 | L. tropica | MON-60 | CL |
| MHOM/IQ//73/Bumm30 | Bumm30 | L. tropica | LON-17 | VL |
| MHOM/IL/78/Rachnan | Rachnan | L. tropica | MON-60 | CL |
| MHOM/ET/72/GEBRE1 | L1005 | L. donovani | MON-82 | VL |
| MPSA/SA/84/Jisha 238 | J238 | L. arabica | LON-64 | NA |
| MHOM/ET/72/L100 | L100 | L. aethiopica | MON-14 | CL |
| MRHO/SU/74/95-A | 95A | L. turanica | MON-64 | NA |
NA: Not Applicable, NT: Not typed, CL: Cutaneous leishmaniasis, VL: Visceral leishmaniasis.
Table 2.
Identified targets for the set-up of the specific PCR assays.
| Target | Gene | Protein | ||
| L. major | L. infantum | L. tropica | ||
| mt30 | Intergenic region between LmjF30.0190 & LmjF30.0200 | Intergenic region between LINF_300006850 & LINF_300006900 |
Intergenic region between LTRL590_300007200 & LTRL590_300007300 |
None |
| mt22 | Non coding sequence | LinJ.22.0300 | LTRL590_220009300 | Hypothetical protein in L. infantum and L. tropica Non coding sequence in L. major |
| it20 | Absent | LinJ.20.0040 | LTRL590_200005300 | Phosphate-Repressible Phosphate Permease-like protein Absent in L. major |
| mt7SL | LmjF.05.SRP.RNA | LINJ_05_snRNA1 | 7SL gene Partial sequence | 7SL RNA |
Table 3.
Designed primer pairs and their specificity.
| Primer pairs | Sequences (5'-3') | Size (bp) | Expected specificity | ||
|---|---|---|---|---|---|
| L. major | L. tropica | L. infantum | |||
| mt22F1 | ACCGAACCCAACGCTGAAG | 366 | + | + | - |
| mt22R1 | AGTGCATGAGGCGTGTATGG | ||||
| mt22F2 | CACTCATGCGTGTCCATTCT | 319 | + | + | - |
| mt22R2 | GTATGGGAAGGTGGGGGT | ||||
| mt22F1 | 352 | + | + | - | |
| mt22R2 | |||||
| mt22F2 | 333 | + | + | - | |
| mt22R1 | |||||
| mt30F | GGTGCAATGTGCGCATG | 350 | + | + | - |
| mt30R | GCTTGGCGCTCTCGAAAAG | ||||
| mt7sLF | TTGGTGGTGGTGGGATGGAC | 191 | + | + | - |
| mt7sLR | CACCACGTCAACGCAGCAAA | ||||
| it20F1 | TCTGGATTGCAGTCGTCGG | 209 | - | + | + |
| it20R1 | CTTGGCGATACCTCCTGAT | ||||
| it20F2 | AGCCTTGGTGGTGTCTTTTG | 195 | - | + | + |
| it20R2 | CAAAGAAGACGGCAGACACA | ||||
+: Positive PCR, -: Negative PCR.
Table 4.
Primers combinations tested for the set-up of the ultra-fast duplex PCR and expected amplicons size.
Table 4.
Primers combinations tested for the set-up of the ultra-fast duplex PCR and expected amplicons size.
| Duplex PCRs | Primer pairs | Size (bp) | Expected specificity | ||
|---|---|---|---|---|---|
| L. major | L. tropica | L. infantum | |||
| A | mt22F1 | 366 | + | + | - |
| mt22R1 | |||||
| it20F1 | 209 | - | + | + | |
| it20R1 | |||||
| B | mt22F2 | 319 | + | + | - |
| mt22R2 | |||||
| it20F2 | 195 | - | + | + | |
| it20R2 | |||||
| C | mt22F1 | 366 | + | + | - |
| mt22R1 | |||||
| it20F2 | 195 | - | + | + | |
| it20R2 | |||||
| D | mt22F2 | 319 | + | + | - |
| mt22R2 | |||||
| it20F1 | 209 | - | + | + | |
| it20R1 | |||||
| E | mt22F2 | 333 | + | + | - |
| mt22R1 | |||||
| it20F1 | 209 | - | + | + | |
| it20R1 | |||||
| F | mt22F2 | 333 | + | + | - |
| mt22R1 | |||||
| it20F2 | 195 | - | + | + | |
| it20R2 | |||||
| G | mt30F | 350 | + | + | - |
| mt30R | |||||
| it20F1 | 209 | - | + | + | |
| it20R1 | |||||
| H | mt30F | 350 | + | + | - |
| mt30R | |||||
| it20F2 | 195 | - | + | + | |
| it20R2 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



