
Submitted:
30 September 2023
Posted:
02 October 2023
You are already at the latest version
Abstract
JC polyomavirus (JCPyV) is a human-specific polyomavirus that establishes a silent lifelong infection in multiple peripheral organs, predominantly those of the urinary tract, of immunocompetent individuals. In immunocompromised settings, however, JCPyV can infiltrate the central nervous system (CNS), where it causes several encephalopathies of high morbidity and mortality. JCPyV-induced progressive multifocal leukoencephalopathy (PML), a devastating demyelinating brain disease, was an AIDS-defining disease pre-antiretroviral therapy that has “reemerged” as a complication of immunomodulating and chemotherapeutic agents. No effective anti-polyomavirus therapeutics are currently available. How depressed immune status sets the stage for JCPyV resurgence in the urinary tract, how the virus evades pre-existing antiviral antibodies to become viremic, and where/how it enters the CNS are incompletely understood. Addressing these questions requires a tractable animal model of JCPyV CNS infection. Although no animal model can replicate all aspects of any human disease, mouse polyomavirus (MuPyV) in mice and JCPyV in humans share key features of peripheral and CNS infection and antiviral immunity. In this review, we will discuss evidence suggesting how JCPyV migrates from the periphery to the CNS, innate and adaptive immune responses to polyomavirus infection, and how the MuPyV-mouse model is providing insights into the pathogenesis of JCPyV CNS disease.
Keywords:
JCPyV
; MuPyV
; polyomavirus
; progressive multifocal leukoencephalopathy (PML)
; CNS
; antiviral immunity
; neurotropic virus
1. Introduction
JCPyV is a member of the family Polyomaviridae, a group of non-enveloped double-stranded DNA viruses that are highly host-specific and are found in a wide range of species [1]. One of fourteen human-specific polyomaviruses, JCPyV is notable due to its pathogenic neurotropism. The virus can cause a multitude of diseases in the CNS, such as JCPyV meningitis, JCPyV granule cell neuronopathy, and, most prominently, PML, a demyelinating disease involving lytic infection of glial cells [2,3]. These conditions are relatively rare, despite the ubiquitous presence of JCPyV in humans worldwide. Indeed, seroprevalence for JCPyV ranges from 40 to 80 percent and increases with age, with initial exposure likely occurring in childhood [4,5,6,7]. Transmission of JCPyV presumably occurs through the fecal-oral route, as viral particles have been found in the urine and stool [8,9]. Moreover, JCPyV is a frequent contaminant of urban sewage [10]. Transmission via the respiratory route is also possible as JCPyV has been found in the tonsillar tissue of various populations [11,12].
In an immunocompetent individual, JCPyV infection is typically asymptomatic. The virus can establish several sites of infection in tissues such as the kidney, bone marrow, and lymphoid tissue [3]. Viral infiltration of the CNS occurs in an immunocompromised setting, which allows the virus to leave peripheral sites of infection and travel to the CNS. Where and how JCPyV infiltrates the brain are unknown and is challenging to explore due to the historical lack of an easily experimentally manipulatable small animal model. Such a model would provide important insight into aspects such as early viral pathogenesis— prior to manifestation of neurological symptoms— and immune mechanisms associated with control of JCPyV.
Mouse polyomavirus (MuPyV), the inaugural polyomavirus, has been established as a model for studying CNS-associated polyomavirus infection [13]. MuPyV is prevalent in wild mouse populations [14] and, like JCPyV, causes an asymptomatic and persistent infection in its immunocompetent host [15,16]. This review explores the outstanding question of how JCPyV may infiltrate the CNS from the periphery as implicated by findings employing the MuPyV model. We will cover peripheral polyomavirus persistence, how the virus resurges in peripheral sites, escapes antiviral immunity, and invades the CNS, which CNS-resident and infiltrating cells the virus infects, and immunological control of polyomaviruses in the CNS.
2. Structure and Lifecycle of Polyomaviruses: JCPyV and MuPyV
Both JCPyV and MuPyV MuPyV have nonenveloped icosahedral T=7 capsids encasing a covalently closed circular genome packaged into a supercoiled minichromosome. Both genomes are similar in size, with JCPyV being 5130 base pairs long and MuPyV being 5300 base pairs long [17,18]. Diagrammed in Figure 1, the genomes of both viruses are divided into an early region and a late region based on the temporal expression of proteins during their replication cycle. For JCPyV, the early region encodes two nonstructural proteins, large T antigen (LTAg) and small T antigen (STAg), and three T antigen splice variants, T'135, T'136, and T'165 [4,19]. The late region comprises three structural capsid proteins — VP1, VP2, and VP3— as well as an agnoprotein and two microRNAs (miRNAs) that are processed from a pre-miRNA designated pre-miRJ-1 [17,20]). Because the miRNAs are perfectly complimentary to LTAg, they regulate the stability of LTAg transcripts [20]. MuPyV also encodes for a precursor miRNA that is processed into two mature miRNAs, which coordinate the cleavage of early mRNA transcripts [20]. The capsid is organized into 72 pentamers of VP1, and each pentamer is associated with either VP2 or VP3 [21]. The early and late regions are separated by a non-coding control region (NCCR), which contains the origin of replication and binding sites for host proteins that determine species and cell tropism [22]. While the common nonpathogenic strain, referred to as the archetype or wild-type (WT) strain, contains a canonically organized NCCR, pathogenic variations of JCPyV found in the brain display extensively rearranged NCCRs, indicating that such rearrangements facilitate neurotropism [23,24,25,26].
Likewise, the genome of MuPyV is divided into an early and late region separated by an NCCR, with the early region encompassing STAg and LTAg and the late region comprising the structural proteins VP1, VP2, and VP3 [18]. Unlike JCPyV, MuPyV lacks an agnoprotein and encodes for a middle T antigen (MTAg), a membrane-localized oncoprotein that behaves as a constitutively active growth factor receptor [27,28]. Figure 1 depicts the similarities and differences between the MuPyV genome and JCPyV genomes.
Attachment and entry of both MuPyV and JCPyV are possible through receptors containing sialic acid residues. In the case of JCPyV, attachment involves binding to the ⍺2,6-linked sialic acid-containing lactoseries tetrasaccharide c (LSTc) receptor motif, though certain isolates of JCPyV have been found to bind sialic acid-containing ganglioside receptors [29]. Attachment to this receptor motif occurs through VP1, the secondary structure of which is composed of antiparallel β strands connected by loops that contain residues critical for receptor binding [30]. Viral entry is then assisted by interaction with 5-hydroxytryptamine 2 (5-HT2) receptors, which bind serotonin [31]. Similarly, MuPyV binds ganglioside receptors containing terminal sialic acid residues. Two such receptors have been identified as GD1a and GT1b, and viral entry is facilitated by the α4β1 integrin [32,33]. Interestingly, immunohistochemistry of C57BL/6 mouse brains has shown that
Figure 1.
Comparison of JCPyV and MuPyV genomes and how each virus enters its host cell. Both viruses initially attach to a receptor containing a sialic acid. Entrance is then facilitated by a secondary receptor. Both viruses are then internalized through different means. While JCPyV is internalized via clathrin-mediated endocytosis, MuPyV is internalized through caveolin-mediated endocytosis. The viruses then enter the early endosomes and transition into late endosomes. Both MuPyV and JCPyV are transmitted through similar routes and establish infections in similar tissues. Made in BioRender.
Figure 1.
Comparison of JCPyV and MuPyV genomes and how each virus enters its host cell. Both viruses initially attach to a receptor containing a sialic acid. Entrance is then facilitated by a secondary receptor. Both viruses are then internalized through different means. While JCPyV is internalized via clathrin-mediated endocytosis, MuPyV is internalized through caveolin-mediated endocytosis. The viruses then enter the early endosomes and transition into late endosomes. Both MuPyV and JCPyV are transmitted through similar routes and establish infections in similar tissues. Made in BioRender.
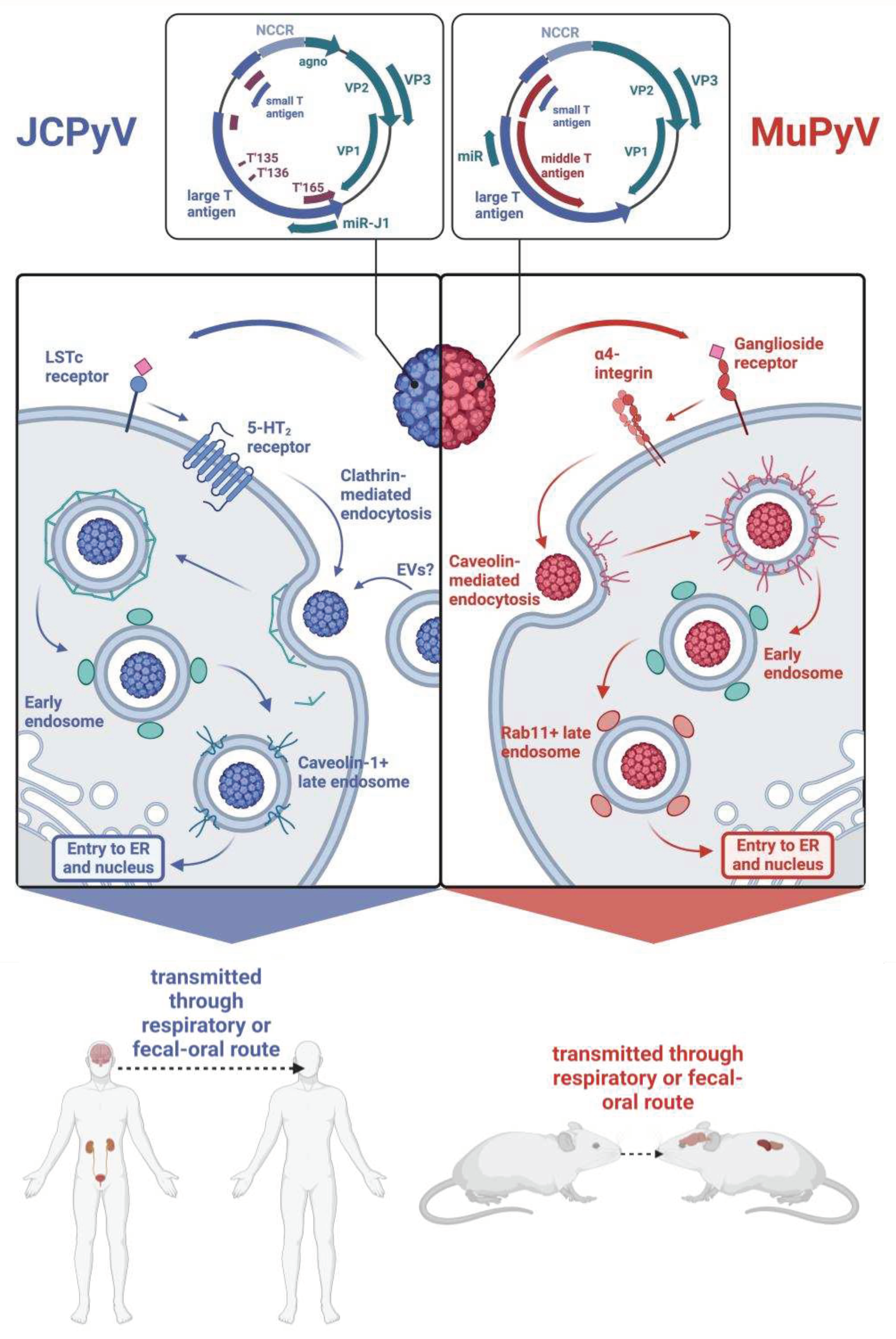
GT1b and GD1a are both expressed highly in neuronal tissue, with the former being expressed in more specific parts of the brain, such as the cerebral cortex and hippocampus, and the latter being more broadly expressed in both white and gray matter [34]. The distribution of these gangliosides supports the capability of MuPyV to infect brain tissue of its host.
JCPyV and MuPyV differ in internalization processes (Figure 1). While MuPyV is internalized through caveolin-mediated endocytosis, JCPyV is internalized through clathrin-mediated endocytosis [35]. Recent work suggests that JCPyV may also be internalized through extracellular vesicles (EVs), which are cell membrane-derived vesicles that provide a means of intercellular communication and whose structure and contents are heterogeneous [36,37].
After internalization, both JCPyV and MuPyV transition to Rab5+ early endosomes, but whereas MuPyV moves to Rab11+ endosomes, a transition that appears to be dependent on the acidic endosomal environment, JCPyV enters a caveolin-1+ late endosome [38,39]. Both viruses localize to the ER and utilize the ER-associated degradation pathway (ERAD) to enter the cytosol. Throughout this process, partial degradation of the viral capsid occurs. Much of what is known regarding uncoating of polyomaviruses within the ER is from studies of MuPyV and simian virus 40 (SV40), a well-studied polyomavirus that is naturally found in rhesus monkeys but can be transmitted to humans [40,41]. The architecture of MuPyV and SV40 is partly stabilized by disulfide bonds as well as calcium ions [40]. Both viruses interact with members of the protein disulfide isomerase family, which disrupt the aforementioned disulfide bonds [40,42]. Uncoating also involves dissociation of calcium ions from the capsid, which, in the case of SV40, may be expedited by lower calcium concentration in the cytosol than the ER [43,44]. Uncoating of JCPyV may also be dependent on calcium, as thapsigargin, which blocks calcium transport into the ER, reduces in vitro infection of a JCPyV-based pseudovirus [45]. Both JCPyV and MuPyV ultimately end up in the nucleus, where the viral genome is released, and transcription of the early region occurs, resulting in expression of the T antigens specific to each virus [46,47].
The functions of both LTAg and STAg in JCPyV can be ascertained from studies of SV40. In SV40, LTAg inhibits the cell cycle through interactions with the tumor suppressor proteins retinoblastoma-associated protein (RB) and p53. SV40 LTAg also exhibits helicase activity, unwinding viral DNA and subsequently facilitating DNA replication [48]. The STAg in JCPyV also contributes to cell cycle arrest by binding to Rb proteins as well as protein phosphatase 2A, a significant regulator of the cell cycle [49]. Replication is supported by the three T antigen splice variants, without which JCPyV replication efficiency is reduced [50]. As the virus appropriates the host cell replication machinery to replicate its genome, the late region is transcribed, producing new viral capsid proteins that eventually enclose newly produced viral genomes. Late region transcription of JCPyV also produces agnoprotein. While its function is not fully known, agnoprotein contains a mitochondrial targeting sequence, localizing to mitochondria and altering metabolic functions as a result [51].
3. How Is JCPyV Transmitted?
JCPyV can be transmitted through the respiratory route. The respiratory tract expresses an abundance of ⍺2,6-linked sialic acid binding receptors [52]. JCPyV DNA has also been found in tonsillar tissue of individuals [11]. The tropism of JCPyV for respiratory tract tissue can be examined through the lens of cancer. While many studies have found various polyomaviruses in multiple oropharyngeal and lung cancers, only one study has detected JCPyV, albeit infrequently, in adenoid cystic carcinoma, an aggressive cancer that commonly affects the salivary glands [53,54,55,56,57,58,59]. Moreover, JCPyV virus-like particles (VLPs) carrying miRNA therapies were successfully used to treat lung adenocarcinoma growth and metastasis in vitro and in mice, demonstrating the possibility of transferring genetic material via JCPyV VLPs to the lungs [60]. Similarly, MuPyV is able to cause systemic infection via respiratory inoculation [15,61]. MuPyV VLPs were also used as a vaccine vector against group A Streptococcus, which was delivered intranasally [62]. More work is needed in vivo to elucidate the relationship between JCPyV and respiratory transmission mechanisms [54,63].
JCPyV may also be transmitted via the fecal-oral route. JCPyV infection rates within populations have been monitored through community sewage surveillance, as the virus is readily found in urine and stool [8,64,65,66,67]. During the coronavirus disease 2019 (COVID-19) pandemic, social and contact isolation protocols have decreased the transmission of respiratory borne illnesses, including rhinovirus, influenza, and respiratory syncytial virus [68]. JCPyV seropositivity rates did not follow this trend and remained consistent instead, regardless of isolation measures [69]. On the other hand, another study, which examined JCPyV seropositivity rates in multiple sclerosis (MS) patients during COVID-19, discovered that conversion rates declined during lock down [70]. Other studies have found an increasing prevalence of JCPyV LTAg in colorectal cancers [71,72,73,74,75]. Our laboratory has demonstrated that MuPyV can cause systemic infection after oral gavage inoculation (AE Lukacher, unpublished observations). MuPyV is suggested to be transmitted through the fecal-oral route as well, evidenced by its shedding in the urine and its availability in drinking water [76,77]. Regardless of the route of infection, JCPyV and MuPyV can both reside in the kidney and other peripheral tissues. MuPyV can be used as a model for JCPyV. Utilizing MuPyV in mice would be key to understanding how polyomaviruses are spread and are able to cause systemic infection.
3.1. Polyomaviruses in Peripheral Tissues
The kidney is a major reservoir for JCPyV infection. Outside the kidney, JCPyV persists in various other tissues, including the spleen, lymph node, lung, and bone marrow [78,79,80]. Since the majority of studies utilize kidney tubules, we will mainly discuss peripheral polyomavirus infection in the kidney. Various studies have diagnosed persistent JCPyV infection via viruria in about 19-45% of healthy seropositive adults [81,82,83,84]. Moreover, a study that examined JCPyV VLP migration in mice found that mice injected with JCPyV VLPs through the tail vein demonstrated accumulation of VLPs largely in the kidney and the liver [85]. Because viruria is common in asymptomatic carriers and JCPyV is known to attach to and infect kidney epithelium, we can conclude that the kidney is a significant reservoir of JCPyV persistence. Little is known about JCPyV’s life cycle in the kidney. Not only is the kidney a major site of persistent JCPyV infection, but it is also a source of graft failure in kidney transplant patients. Case reports have demonstrated that, while rare, transplant patients can experience JCPyV-associated nephropathy (JC-PVAN) [86,87,88,89,90,91]. The mechanism behind how JCPyV infiltrates the kidney and how JC-PVAN may develop is unknown.
BK polyomavirus (BKPyV), a non-neurotropic human polyomavirus, is a major culprit of BKPyV-associated nephropathy (BK-VAN) in renal transplant patients [92,93]. BK-VAN results in 1 to 10% of graft losses within 12 months [94,95,96,97]. Because this disease is a major cause of infectious graft loss, many studies focusing on BK-VAN have been conducted and could shed light on JCPyV affinity and persistence in the kidney. BKPyV shares about 70% sequence homology to JCPyV [98,99]. Like JCPyV, BKPyV has a seroprevalence of 80-90% in adults and is similarly transmitted via respiratory or fecal-oral routes [100,101,102,103]. The mechanism behind BKPyV infection and latency in the kidney has been proposed and discussed [104,105]. The kidney tubules were found to be the most permissive to BKPyV replication. The tubules facilitate polyomavirus replication via increased glycolysis and constitutive expression of transcription factors that steer cells into S phase [106]. Kidney tubules are also considered to be immune-privileged, allowing the virus to replicate freely undetected by the innate or adaptive immune system 9/30/2023 11:32:00 PM. Evading surveillance would allow polyomaviruses to replicate in the kidney tubule and be shed in the urine, which is seen for both JCPyV and BKPyV. The mechanism that results in asymptomatic shedding or in the development of end stage renal disease (ESRD) is unknown and is limited due to the lack of a proper animal model.
Similarly, MuPyV resides persistently in the kidneys of infected mice [21,107,108,109,110]. Intracranial (IC) infection of MuPyV results in persistent viral DNA in the kidney [111]. How the virus enters the glomerulus, infects the tubules, and establishes persistent infection has yet to be unraveled. Pinpointing the mechanism would be key to develop new ways to diagnose and treat transplant patients at risk of graft-loss due to PVAN. MuPyV, however, has been used in a murine kidney transplant model [107]. Major conclusions from this study reveal that the dominant mechanism of transplant injury in PVAN is due to both host-immunity and virus-induced inflammation, as each alone was insufficient in resulting in permanent graft loss. More studies in vivo need to be done to understand the pathophysiology of PVAN.
4. How Do Polyomaviruses Maintain Long-Term Persistence?
In an immunocompetent host, transmission of JCPyV ultimately results in a persistent infection that is likely kept in check by the immune system, given that JCPyV does not exhibit true latency. JCPyV and MuPyV undergo active lytic infection in vitro, while in vivo we do not see signs of viremia unless the hosts are immunocompromised. Interestingly, the literature demonstrates high rates of JCPyV-specific antibodies in the blood [5,112,113,114]. How the virus elicits this strong humoral response is unknown. To elicit a lifelong response, JCPyV and MuPyV may establish a series of lytic infections that continuously prime the immune system; MuPyV viruria appears persistently and episodically [77]. One study demonstrates that MuPyV utilizes its miRNA to promote viruria during acute and persistent infection [77]. Virally-encoded miRNAs have been cited for polyomaviruses as a means to both establish persistence and evade host-immunity. MuPyV persistence is also known to be partly driven by enhancers in kidneys. Alterations in these enhancers are shown to diminish both acute and persistent MuPyV infection [115]. Alternatively, the immune system maintains a subclinical smoldering infection. In an immunocompetent host, B cells and T cells control lytic infection because deficiencies in either B cells or T cells allow MuPyV to cause disease [13,21,110,116,117,118,119,120].
Whether persistent infection is elicited by many episodes of lysis, a controlled smoldering infection, or some combination has yet to be answered. Answering these questions are unanswerable in vitro and are key to uncovering the role of lifelong polyomavirus infection and to the development of diagnostics and potential therapeutics to patients at risk of developing PML.
5. How Does Polyomavirus Travel from the Periphery to the Brain?
Although JCPyV infects the kidney, the brain is typically the site of pathology, so the virus must travel between the organs. In patients with PML, the JCPyV in the brain is genotypically different from the virus in the kidney [17,23,121,122,123,124,125,126]. Polyomaviruses residing in the kidney typically exhibit the archetype strain. When patients are immunosuppressed, viral replication is amplified and results in mutations in the NCCR or VP1 [127,128,129]. Viral shedding increases and results in lytic infection. Patient studies have shown that the NCCR undergoes rearrangements in two HIV-positive patients with PML [123,130,131]. One study found that HIV-negative MS patients with PML did not have complex rearrangements, suggesting that either T helper cell depletion or HIV itself may play a role in NCCR rearrangement [25].
Specific mutations in the capsid protein VP1 are also associated with shifts in tropism and can promote neurovirulence and brain infection. JCPyV variants found in the cerebrospinal fluid (CSF) of PML patients exhibit VP1 mutations [132,133]. In mice, VP1 mutations in MuPyV emerge after serially passaging MuPyV in vitro and in the presence of a VP1-specific neutralizing monoclonal antibody (mAB) [21]. Building on this study, our recent work demonstrates that in an immunocompromised setting, MuPyV infected mice developed MuPyV variants with VP1 mutations, with varying effects on tissue tropism. Mice intracranially infected with a double-mutant VP1+ virus displayed more infected cells in the brain. Double mutant virus-infected mice also developed hydrocephalus to a higher degree, indicative of increased neurovirulence of the VP1 double-mutant virus [110]. Taken together, the virus is likely only able to travel to the brain after immunosuppression, which promotes conditions in which the NCCR and VP1 are mutated. This requirement is substantiated with previous studies that show that increasing immunosuppression is directly correlated with increased JCPyV DNA in the brain in individuals without PML [134,135,136,137].
Contrary evidence demonstrates that during autopsy of healthy individuals and in sampling of pediatric brain cancers that JCPyV resides in the brain with no evidence of brain pathology [122,138,139,140,141,142,143,144,145,146,147,148,149,150]. In mice, peripheral infection with MuPyV does not lead to detectable brain infections. During immunosuppression, MuPyV follows similar trends to JCPyV where viral replication, serum presence, and viruria are all increased [21,77,110]. Further studies need to examine how MuPyV is able to travel from the kidney to the brain to shed light on JCPyV pathogenesis.
6. How Does Polyomavirus Invade the Brain?
To enter and infect the brain, JCPyV must cross one of its multiple barrier sites. The blood-brain barrier (BBB) is the most studied barrier site consisting of vascular endothelial cells, pericytes and astrocytic end feet [151]. The blood-CSF-brain barrier (BCSFB) exists in the ventricles as well as on the surface of the brain with separate tissues in each site. Within the ventricles, endothelial and epithelial cell layers of the choroid plexus (ChP) separate the blood from the CSF, while ciliated epithelial cells of the ependyma divide the CSF from the parenchyma. On the outer surface of the brain, the meninges are divided into three layers: the dura layer closest to the skull with blood vessels and resident immune cells; the arachnoid layer with permeating CSF; and the pial layer closest to the brain. All three barriers are depicted in Figure 2. The barrier JCPyV uses to establish brain infection is unknown; multiple theories have been proposed, detailed below.
6.1. Via the Blood-Brain Barrier?
One potential entry site for polyomaviruses into the CNS is the BBB. The BBB exists throughout the brain, dividing the blood vessels from the surrounding parenchyma. The vascular endothelial cells are interconnected by tight junctions and adherens junctions, restricting pathogens from crossing [151,152]. Pericytes, astrocytes, and neurons complement the endothelial cells, sensing and regulating blood flow, forming the overall neurovascular unit. Under homeostasis, few immune cells are permitted to cross this barrier; however, during neuroinflammation additional immune cells are able to enter.
One proposed mechanism of initial CNS infection by JCPyV is the direct infection of the neuroendothelial cells of the BBB. Cell-free JCPyV has been detected in plasma [153,154]. In culture, JCPyV productively infects primary human brain microvascular endothelial cells [155]. JCPyV has also been shown to bind to sialic acid receptors on the surface of endothelial cells in human brain tissue sections [156]. These same cells lack expression of the 5-HT2 receptors necessary for viral entry. Several studies have described neuroendothelial cells expressing JCPyV transcripts in patient postmortem tissue, as detected by in situ hybridization [157,158]. It is unclear whether this infection occurred from the luminal or parenchymal side of the cells. Given the infrequency of infected neuroendothelial cells and the lack of suitable receptors for binding and internalization, direct infection of the BBB remains an unlikely mechanism for viral entry to the CNS.
Extravasion of infected immune cells via the BBB into the brain has been proposed as a “Trojan horse” model of initial CNS infection. Primary B lymphocytes and CD34+ hematopoietic progenitor cells in culture are susceptible to JCPyV infection [159,160] while CD34+ progenitors and B cells from PML patient spleens [78], bone marrow, blood [153,159,161,162,163,164,165], and brains [166] were positive for JCPyV DNA. However, the virus does not appear to replicate well within B cells or CD34+ cells in vitro; virus bound to B cells mostly remains at the cell surface [160] and JCPyV in primary B cells or B cell lines results in an infection rate under 5% [159,160,167,168]. This low infection rate also occurs in vivo, as few HIV+ or PML patients have viral mRNA in blood even when JCPyV DNA is detectable [161]. The limited infection rate does not necessarily preclude CD34+ progenitor cell or B cell-mediated entry, as virus attached to or within B cells is sufficient to infect glial cells in culture [168]. The more recent development of PML in patients receiving monoclonal antibody therapy also complicates JCPyV entry via B lymphocytes. Rituximab is a chimeric mouse-human CD20 antibody that binds and depletes B cells [169] and is used to treat non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, and pemphigus vulgaris [170]. Since its approval by the FDA in 1997, other CD20-targeting antibodies have been introduced to the market, including murine, human, and humanized forms [171]. Because these antibodies deplete B cells, and therefore would limit lymphocyte entry into the CNS, it is unclear whether a sufficient number of cells persist during treatment to allow for establishment of brain infection by JCPyV. However, following the cessation of anti-CD20 therapy, CD34+ progenitors, immature and naïve B cell populations expanded between 6-10 months post-treatment while memory B cells remained reduced [172,173]. The expansion of B cells from JCPyV infected bone marrow may then lead to the dissemination of virus to the brain after CD20 depletion.
6.2. Via the ChP-CSF-Ependyma Barriers?
Another proposed entry site of polyomaviruses is the BCSFB within the ventricles. The ChP contains fenestrated vascular endothelium unlike the BBB, and a stromal layer between the endothelium and the epithelium. The epithelial cells form a barrier with tight and adherens junctions preventing pathogens from entering the CSF. The ependymal cells lining the ventricles are less tightly connected than the ChP epithelial cells, joined by adherens junctions and gap junctions without any tight junctions.
As with the BBB, one potential mechanism of brain entry is the direct infection of the ChP, either of the blood vessels or of the epithelial cells via the fenestrated endothelium. The ChP epithelium and blood vessels express the receptors necessary for JCPyV to bind [174] and JCPyV can productively infect cultured ChP epithelial cells in vitro [37,150] in vivo [175,176]. A particularly interesting feature of ChP infection is the production of JCPyV packaged within EVs [37], which eliminates the need for specific receptors for viral entry. As mentioned earlier, recent studies found that EVs are involved in transmitting the virus to cells independent of 5-HT2 receptors [177]. JCPyV-infected ChP cells were able to release EVs containing virus, which were capable of infecting glial cells [37]. EVs with JCPyV DNA have also been found in human plasma samples [126]. JCPyV-containing EVs have been shown to carry JCPyV-associated DNA, miRNA, and whole viral particles [178]. These data suggest a route of infection between permissive and nonpermissive cell types.
Once the ChP is infected, virus can be secreted into the CSF. JCPyV DNA has been detected in the CSF of PML patients [179,180] and patients with JCV-associated meningitis [175,181] and is included in the diagnostic criteria for PML [179] although the predictive value of positive or negative PCR results is debated [182,183,184]. Productive MuPyV infection occurs in the ependyma lining the ventricles following intracerebroventricular infection [21,110,185]. In addition to direct infection of the BCSFB, this route of infection may also occur via infected leukocytes, particularly CD34+ hematopoietic progenitor cells [186]. As in the blood, anti-CD20 therapy depletes B cells in the CSF, and cessation of antibody treatment leads to an expansion of CD34+, pre- and immature B cells [187].
6.3. Via the Meninges?
A third potential entry site for JCPyV is through the meninges on the surface of the brain. The dura mater contains fenestrated vasculature similar to the ChP and also contains lymphatic vessels that drain to the cervical lymph nodes. Below the dura mater, the arachnoid mater is interconnected by tight junctions, restricting pathogens to the dura. CSF flows in the subarachnoid space between the arachnoid and pial layers, or collectively the leptomeninges, and therefore, pathogens already in the CSF can access these layers. In contrast with the fenestrated dural vessels, subpial vessels are similar to the BBB with tight and adherens junctions preventing pathogens from entering the meninges. The pia mater is more permeable to CSF and allows for the exchange of interstitial fluid in the brain and CSF.
Polyomaviruses may enter first at the dura. Recent literature has found that the dural meninges are a site of infection for Borrelia burgdorferi in Lyme disease [188]. The presence of polyomaviruses in the dura has never been studied. Interestingly, B cells within the dura are replenished by the bone marrow within the calvaria of the skull [189,190]; if this bone marrow also is a site of persistent JCPyV, then infected B cells may cross directly from the calvaria to the dura. Polyomaviruses within the CSF can access the leptomeningeal cells within the arachnoid and pial layers. Leptomeninges are susceptible to JCPyV [150,156,175,176] and MuPyV [21,185] infection in vivo and in vitro. The virus may also enter via direct infection of the subpial vessels; JCPyV has been shown to bind sialic receptors on the surface of endothelial cells within the meninges in tissue sections [174].
7. What Brain Cells Are Infected by Neurotrophic Polyomaviruses, and What Pathologies Does This Infection Cause?
The pathology of brain polyomavirus infection is driven in part by the specific cells infected. JCPyV and MuPyV virions bind to sialylated gangliosides and serotonergic 5-HT2 receptors to be internalized via endocytosis. Polyomaviruses can bypass this requirement, however, by releasing the virus within EVs [37]. These vesicles can then fuse with cells, allowing the virus to enter. Although JCPyV brain infection is associated with PML, the virus can also cause meningitis [175,181,185], granule cell neuronopathy [191,192,193,194], and encephalopathy [195,196]. The presentation of the illness is largely determined by the type of cells infected, which is driven in part by the aforementioned NCCR and VP1 mutations.
7.1. Oligodendrocytes
Oligodendrocytes are the myelinating cell of the CNS, providing insulation for the saltatory conduction of axons and trophic support for the underlying neurons. The death of these cells results in loss of neuronal conductance, eventually leading to the development of white matter lesions. In PML, oligodendrocytes are absent from the center of lesions, and at the periphery infected cells display enlarged nuclei dense with replicating JCPyV [197,198]. The lytic infection of oligodendrocytes causes demyelination of the axons and ultimately many of the symptoms of PML. Mature human oligodendrocytes as well as their precursors do not express the sialic acid LSTc but do express 5-HT2 receptors; in tissue sections, JCPyV is not able to bind these cells [174]. This contradicts abundant examples of polyomavirus-infected oligodendrocytes in patient tissue [199,200]. Similarly, MuPyV does not infect cultured oligodendrocytes but is seen within infected cells from mice [116,201]. These conflicting results can be explained by infection via EVs, likely released from the infected ChP [37]. This phenomenon is further exemplified in nude mouse experiments where the route of infection altered the pathology. Human tumor-implanted nude mice showed demyelination and paralysis as a result of MuPyV infection [202,203] while bypassing the meninges and ChP via stereotaxic infections in the striatum resulted in reduced inflammation and demyelination [204]. Human glia-mouse chimeras [205,206] infected with JCPyV in the corpus callosum developed focal demyelination, although fewer oligodendrocytes were LTAg+ or VP1+ than other cell types [207]. While this may be due to astrocytic cell death or forced cell cycle entry by the infected oligodendrocytes, an additional explanation is the direct injection of the virus into the corpus callosum limited oligodendrocyte infection by EVs.
7.2. Astrocytes
Astrocytes support the functions of many of the other cells in the brain, including sequestering excess neurotransmitter at synapses, taking up glucose from blood vessels and processing it to lactate for neuronal use, and regulating myelination by oligodendrocytes via glutamate signaling [208,209]. These cells also interact with many immune cells, particularly with microglia, the CNS resident myeloid cells, and peripheral immune cells after infection. Such interactions can induce astrocyte reactivity [210,211]. Loss of astrocytes via viral infection disrupts many other cells and can cause neuronal cell death via loss of trophic support. Like oligodendrocytes, astrocytes lack sialic acid receptors and cannot bind JCPyV in tissue sections [212] but express viral proteins in post-mortem tissue [197,199]. These cells are also likely infected by EVs [37]. Lesions within PML brains contain “bizarre astrocytes,” cells with enlarged nuclei and stellate processes which are often multinucleated with apparent mitotic spindles [198]. Although oligodendrocytes often undergo lytic cell death after JCPyV infection with virions present in their nuclei, astrocytes typically express LTAg in their cytoplasm with little viral protein in their nuclei [198,199]. In JCPyV infection of astrocytes in vitro, normal human astrocytes showed delayed production of VP1 compared to SV40-transformed SVG-A cells [213], suggesting a cell-intrinsic delay in viral replication. In particular, the bizarre astrocytes present in lesions produce little virus but instead have been transformed by viral infection [197,214]. The conserved LTAg in polyomaviruses has been shown to transform and immortalize cells through its interactions with RB [215,216] and p53 [217,218], preventing checkpoint inhibition to allow for continuous divisions. It is not known why viral infection leads to the formation of these bizarre astrocytes rather than immortalization and tumor formation as may be suggested by in vitro experiments, and how these bizarre astrocytes function relative to healthy cells.
7.3. Glial Progenitor Cells
Neural progenitor cells (NPCs) are multipotential stem cells that can differentiate into astrocytes, oligodendrocytes, or neurons, and are important to replace damaged cells after an injury. JCPyV can non-productively infect NPCs in vitro [219], although direct infection of these cells or a bipotential glial precursor line CG-4 results in a reduced ability for the precursors to differentiate [219,220]. Under homeostatic conditions, oligodendrocyte precursor cells can migrate and differentiate to replace damaged oligodendrocytes to maintain myelin. Mice continue to undergo myelination after the first 6 weeks [221] in humanized glia mice, the murine oligodendrocytes are replaced by human cells over time [206]. Humanized glia-mice 12 weeks post-infection with JCPyV showed hypomyelination in the corpus callosum and internal capsule compared to uninfected chimeras [207], suggesting a defect in the ability of precursors to differentiate and myelinate. In an inflammatory MS model, oligodendrocyte precursor cells exposed to interferon-γ (IFN-γ) failed to differentiate to mature, myelin-producing cells and instead presented peptide on major histocompatibility complex (MHC) class I and were eliminated by CD8+ T cells [222]. It is possible the inflammatory conditions from JCPyV infection also limit the differentiation of precursor cells, thereby limiting remyelination.
7.4. ChP Epithelium and Ependyma
The ChP epithelium is the primary secreter of CSF as well as driving CSF flow throughout the brain via its ciliated surface. As mentioned previously, these cells provide a barrier from the fenestrated blood vessels to the CSF. Either direct infection or inflammation of ChP epithelial cells can cause hypersecretion of CSF leading to the development of hydrocephalus [223,224,225]. ChP epithelia express both LSTc and 5-HT2 receptors [174] and are permissive for JCPyV infection in vitro [37,150] and in vivo [175,176]. Infection of the ChP leads to the development of hydrocephalus in JCPyV meningitis rather than typical symptoms associated with PML. The ChP epithelium expresses multiple pattern recognition receptors (PRRs), including toll-like receptors (TLRs) [223,226,227,228,229] and C-type lectin receptors [230] but the immune sensing of ChP cells during polyomavirus infection has not been explored. Immunodeficient mice intracerebroventricularly infected with MuPyV do not show direct infection of ChP epithelium but do develop severe hydrocephalus [185], suggesting the inflammatory signaling by MuPyV alone is sufficient to lead to CSF overproduction. In addition to altering CSF secretion, infection of the ChP can lead to the downregulation of tight junctions, increasing the permeability of the BCSFB. Specifically, inflammation in a stroke mouse model [231], infection of cultured human ChP epithelial cells with Borrelia burgdorferi [232] or mice modeling cerebral toxoplasmosis [233] lead to reduced expression of tight junction genes and proteins. The effect of polyomavirus infection on ChP epithelial permeability has not been determined.
The ependyma is a ciliated cell layer that lines the ventricles and promotes the flow of CSF. This layer also regulates the exchange of CSF and interstitial fluid within the brain parenchyma, and likely produces CSF. A defective ependyma barrier therefore also contributes to hydrocephalus and ventriculomegaly [234,235,236,237]. While direct infection of the ependyma by JCPyV has not been seen, MuPyV infects the ependymal lining causing denudation in immunodeficient mice [185]. Inflammation of the ependyma via stimulation of its PRRs also leads to hydrocephalus and ventriculomegaly [226,238]. As with the ChP, inflammatory signaling at the ependyma may contribute to the pathology seen with JCPyV meningitis, but this has not been investigated.
7.5. Leptomeningeal Cells
The leptomeninges include the arachnoid and pial meninges layers surrounding the exterior surface of the brain. The arachnoid mater regulates CSF exchange to the glymphatic system to be drained, while the pia mater regulates interstitial fluid-CSF exchange. Inflammation and infection of the leptomeninges can disrupt both flow and clearance of the CSF [239,240] and can lead to hydrocephalus in addition to the development of meningitis [241,242]. Leptomeninges are productively infected by JCPyV in vitro [174,176,243] and in vivo [175,176,243], leading to JCPyV meningitis without the symptoms of PML. Mouse meningeal infection with MuPyV in vivo and in vitro replicates this finding [21,185]. Considering the importance of the meninges to the clearance of the CSF, determining the potential disruption caused by polyomavirus infection could help with understanding pathology in JCPyV meningitis and PML.
7.6. Neurons
The neurons are electrochemically coupled cells that carry messages along their axons to neighboring neurons. Different regions of the brain correspond to various functions, and therefore the location of infection in or around neurons will determine its impact. One subset of neurons, the granule cells of the cerebellum, receive sensory inputs from the body and project its axons to the molecular layer [244]. These cells in particular are susceptible to JCPyV infection both with PML [245,246] alone, causing JCPyV granule cell neuronopathy [191,192,193,194]. With the neuronal cell death, granule cell neuronopathy causes cerebellar dysfunction with symptoms like gait ataxia, loss of coordination, and dysarthria. Specific mutations in the C-terminus of VP1 are often associated with granule cell infection that reduce the ability of the virus to infect glia [247,248]. JCPyV can also infect cortical pyramidal neurons, leading to the development of JCPyV encephalopathy [195,196]. Patients with infected cortices develop cognitive decline and aphasia.
8. What Is the Innate Immune Response to Polyomavirus?
All viruses are obligate intracellular pathogens. The antiviral immune response thus involves a series of evolved mechanisms that not only dampen viral replication but also signal the infection to neighboring cells and recruit the innate and adaptive immune system. While some of the signaling mechanisms for the innate immune response have been determined, many remain unstudied.
8.1. Sensing, Reporting, and Alarming Innate Immunity
Antiviral immunity can be divided into three systems: sensing, reporting, and alarming. For polyomaviruses, the sensing system is an active field of research with many questions unanswered. Sensing is performed by PRRs, a diverse group of receptors which sense either extracellular or intracellular molecules associated with pathogens. On the extracellular front, TLRs, especially TLR4 and TLR9, are important in sensing neuropathic polyomaviruses like JCPyV and MuPyV [249,250,251,252]. One JCPyV vaccine study has looked into the interaction of TLR4 with JCPyV VP1 capsid protein as a mechanism of priming immunity [251]. Single nucleotide polymorphisms in TLR3 and 4 contribute to BKPyV viremia in transplant patients [253,254]. Although TLR sensing of MuPyV has not been extensively studied, one study demonstrated a direct interaction between MuPyV VP1 and murine TLR4 in primary and untransformed mouse embryonic fibroblasts and showed that TLR4-mediated recognition of the virus led to cancer-like phenotypes [249].
Intracellularly, PRRs sense both DNA and RNA viruses in the cytoplasm. In a healthy host cell, DNA should be localized within either the nucleus or the mitochondria. The PRR cyclic GMP–AMP synthase (cGAS), a double-stranded DNA sensor, appears to be a key sensor of polyomavirus [255,256]. cGAS drives interferon-β (IFN-β) production during MuPyV infection in vitro when genotoxic stress is achieved in murine fibroblasts [255]. Early in its replication cycle, however, MuPyV fails to induce IFN-β due to lack of interaction with cGAS until the virus undergoes replication in the nucleus.
Another study suggests that JCPyV T antigens do not interact with the cGAS sensing pathway [256]. Retinoic-acid inducible gene I (RIG-I), an RNA sensing PRR, has been implicated in the role of JCPyV infection of SVG-A human cell lines [256]. JCPyV STAg interacts with RIG-I directly to suppress its signaling. Outside of these two studies, not much is known about how neurotropic polyomaviruses interact with antiviral sensors and downstream effectors. One study has found that endothelial cells are the first line of defense against BKPyV infection and can release low levels of IFNs [257]. An additional study found that double-stranded RNA sensors, like TLR3, RIG-I and melanoma differentiation-associated protein 5 (MDA5), all play a role in BKPyV infection [258]. Studies have not been conducted for JCPyV and MuPyV and present a gap in the field that needs to be addressed. Understanding the antiviral and innate immune system responses is essential to understanding how to resolve persistent infections at the cellular level and develop novel therapeutics to utilize these systems against polyomavirus diseases.
Sensing mechanisms target signaling mechanisms to release respective alarms, also known as IFNs. There are three types of IFNs, conveniently named type I, type II, and type III. All cells express type I IFNs via PRRs and reporter pathways. With potentially hundreds of type I IFNs, we will focus on the most understood: IFN-α and IFN-β. Type II IFNs include IFN-γ, which are derived from natural killer (NK) cells and cytotoxic T cells. Type III IFNs are a recently discovered type that cover all the IFN-λ subtypes that are uniquely expressed by epithelial cells. All of the aforementioned DNA and RNA sensing molecules stimulate the expression of type I IFNs. Interferon regulatory factor 3 (IRF 3) is essential for the expression of IFN-β [255,259]. DNA sensing through cGAS stimulates production of cyclic GMP-AMP (cGAMP), which binds to its adapter, stimulator of interferon genes (STING), thus leading to activation of IRF3 to initiate transcription of IFN-β. RNA sensing through RIG-I and MDA5, another PRR, initiates binding with their adapter, mitochondrial antiviral-signaling protein (MAVS), which also leads to IRF3 activation. Additionally, TLR4 is also known to activate IRF3, while TLR9 stimulates the phosphorylation of IRF7, helping initiate transcription of IFN-α [260,261]. Downstream adapters have been implicated but not adequately studied in polyomaviruses. As previously discussed, MuPyV interacts with cGAS and p204 during infection of murine fibroblasts, also activating IRF3 and stimulating IFN-β production [255]. Another study found that IFN-α and IFN-β are both negative transcriptional regulators of JCPyV infection in human glial cells via induction of CAAT/enhancer binding protein beta (C/EBPβ) and liver inhibitory protein (LIP) [262]. Downregulation of C/EBPβ-LIP via shRNA treatment caused reactivation of JCPyV. The field of polyomaviruses and antiviral immunity is steadily growing, and studies have not articulated which sensing-adaptor pathways prevail and what their function might be within a more complex host-model. More work needs to be done especially in the context of neurovirulence, as this pathway is crucial in intracellular reporting of viral activity, which could play unique targets in treatment and therapeutics.
Janus kinase (JAK) proteins and signal transducer and activator of transcription (STAT) transduce signals from all IFN receptors. Gain-of-function mutations in STAT1 have been associated with the development of PML [263]. One case report has linked JAK-STAT inhibitors with the incidence of fatal encephalopathy due to JCPyV [195], so understanding the effects of JAK-STATs in the context of neurovirulent polyomavirus infection is imperative to understanding the development of PML. In patient samples, JCPyV infectivity increased in renal tubules during type I IFN blockade [264]. Activated STAT1 and IRF9 were found to initiate transcription of interferon-stimulated genes (ISGs) which were thought to dampen JCPyV replication in primary renal proximal tubular epithelial cells and establish latency. Without a mouse model, conducting these studies in vivo presents a challenge.
MuPyV has provided an avenue to study the effects of types I and II IFNs against MuPyV-induced encephalitis. Similar to what has been shown regarding IFN regulation of JCPyV, MuPyV has been found to be regulated by type I IFN in vitro and played a role in the recruitment of cytotoxic CD8+ T cells in vivo. Transgenic mice lacking the interferon-α/β receptor (IFNAR) demonstrated increased viral titers in vivo but also demonstrated increased proliferation, functionality, and memory differentiation of CD8+ cells [117]. Additional studies with MuPyV show that type II IFNs increase viral load and the presence of MuPyV-induced tumors in vivo [265]. These pathways both point to the importance of STAT1 within the infected cell. During MuPyV intracerebroventricular infection, STAT1 and CD8+ T cells work in concert to dampen neurovirulent polyomavirus load and ameliorate neurodegeneration [185]. STAT1 null mice infected with MuPyV demonstrated increased brain viral loads compared with type I and type II IFN receptor- deficient and WT mice infected with MuPyV. Additionally, MuPyV-infected STAT1-null mice developed severe ventriculomegaly and elevated CD8+ T cell infiltrates. STAT1 null mice depleted of CD8+ T cells die within two weeks of infection. Because the ependyma is both directly infected with MuPyV [185] and can be inflamed indirectly via activation of TLRs [226,229], loss of STAT1 specifically at this barrier leads to increased neuroinflammation after MuPyV infection [SA Spencer and AE Lukacher, unpublished observations].
8.2. The Innate Immune Enzyme Family APOBEC3: A Driver of Polyomavirus VP1 Mutations?
Apolipoprotein B mRNA-editing catalytic polypeptide-like 3 (APOBEC3) genes are antiviral ISGs that code for a family of cytidine deaminases [266,267]. APOBEC3s act on single-stranded DNA, inducing mutagenic damage by converting cytosine to uracil [268]. They are well-studied in the context of retrovirus restriction, with one member having initially been identified as an inhibitor of HIV-1 infection [269]. Due to their influence on viral genomes, APOBEC3s are thought to play a role in the evolution of multiple viruses, including polyomaviruses. For instance, the genomes of JCPyV and other human polyomaviruses, such as BKPyV, KI polyomavirus, and WU polyomavirus all show footprints of APOBEC3 activity [270]. APOBEC3s have particularly been implicated in causing BKPyV mutations that may bestow a selective advantage to the virus. In settings of immunosuppression, BKPyV can abundantly replicate and cause nephropathy, a scenario that occurs in 1 to 10% of kidney transplant patients [102]. Observed VP1 mutations in BKPyV variants isolated from nephropathy patients were consistent with APOBEC3-associated damage [271]. Moreover, BKPyV infection of human urothelium results in increased APOBEC3A and APOBEC3B expression [272].
The involvement of APOBEC3s in influencing JCPyV intra-host evolution in the context of pathogenicity has not been studied. As discussed previously, changes in VP1 can alter viral tropism, receptor binding, and recognition by the host’s neutralizing antibody response [133,273]. Similar to what was observed in BKPyV, APOBEC3s may not actually control JCPyV, instead presenting a driver for VP1 mutations that inadvertently confer an advantage to the virus, generating variants that can escape the host’s neutralizing antibody response.
9. What Is the Adaptive Immune Response to Polyomavirus Infection?
Prolonged immunosuppression is a critical risk factor in the development of PML. Historically, PML was associated with acquired immunodeficiency syndrome (AIDS), with 2 to 5 % of patients developing PML after AIDS onset [1]. While incidence of HIV-associated PML has decreased since the implementation of highly active antiretroviral therapy, which manages AIDS through suppression of HIV at different viral life cycle stages, PML has become a possible complication of relatively recently developed immunomodulatory drugs. For instance, natalizumab, an immunomodulator used to treat MS, inhibits lymphocyte trafficking into the brain with the intent of reducing inflammation [274]. When combined with risk factors such as high JCV seropositivity and prolonged and/or previous use of immunosuppressants, inhibition of lymphocyte trafficking can lead to the development of PML [274]. The adaptive immune response is carried out by cytotoxic CD8+ T cells, CD4+ T helper cells, and antibody-producing B cells; we will discuss the contribution of each cell type to control of polyomavirus infection.
9.1. CD8+ T Cells
The significance of CD8+ T cells in polyomavirus control is emphasized by studies demonstrating a strong association between an early JCPyV-specific CD8+ T cell response and a favorable clinical outcome in PML patients [275,276]. In clinical studies exploring pembrolizumab, an inhibitor of the immunosuppressive protein programmed cell death protein 1 (PD-1), as a treatment for PML, an increased JCPyV-specific CD8+ T cell response was linked with lower viral loads and, ultimately, clinical improvement [3]. CD8+ T cells have been shown to colocalize with JCPyV-infected glial cells, further validating the importance of CD8+ T cells in controlling JCPyV [277,278]. The necessity of CD8+ T cells to viral control in an immunocompromised setting is demonstrated in vivo in STAT1 null mice, which are immunocompromised and exhibit high viral loads in the brain when infected with MuPyV. When depleted of CD8+ T cells, these mice display severe weight loss and increased mortality by two weeks post-infection compared to WT animals [185].
CD8+ resident memory T (TRM) cells constitute a subset of CD8+ T cells and are critical responders to viral reinfection in non-lymphoid tissues, such as the brain. TRM cells are semi-permanent fixtures of non-lymphoid tissues and are responsible for rapidly countering repeated antigen encounters [279,280]. The expression of chemokine receptor CXCR6 is associated with the transition of CD8+ T cells from circulating effectors to resident memory [281,282]. Previous studies have shown CXCR6 and its chemokine ligand CXCL16 to be important for TRM formation in the lung [283], skin [284,285] and liver [286]. In the brain, TRM cells require CXCL16-CXCR6 signaling after resolution of West Nile virus infection for maintenance, as loss of CXCR6 leads to reduced numbers of CD8+ cells [287]. This chemokine signaling was also required for TRM formation in an Alzheimer’s disease mouse model, and CXCR6 null mice had reduced CD8+ T cells with increased cognitive decline [288]. The dominant source of CXCL16 in the brain is myeloid cells including microglia [287,288]. With MuPyV infection, we have observed CXCR6+ CD8+ T cells in the kidney and brains of infected mice, as well as expression of CXCL16 by brain microglia (M Lauver, S Spencer and A Lukacher, unpublished observations). In CXCR6 deficient mice, CD8+ T cells show reduced expression of TRM marker CD69 [289] after infection, suggesting a requirement of CXCR6 for the establishment of TRM cells. Mice without CXCR6 signaling infected with MuPyV showed reduced expression of PD-1 on CD8+ T cells in the brain (S Spencer and A Lukacher, unpublished observations).
PD-1, a classic inhibitory immune receptor, is an intrinsic attribute of virus-specific CD8+ brain-resident TRM (bTRM) cells established during MuPyV infection [111]. Notably, PD-1 expression is upregulated on JCPyV-specific CD8+ cells in PML patients [289]. Increased PD-1 and PD-L1 expression have also been observed in autopsied PML lesions [3]. MuPyV-specific CD8+ bTRM cells highly express PD-1, a characteristic not shared by MuPyV-specific memory CD8+ cells in the spleen, supporting PD-1 as a brain-intrinsic trait of CD8+ TRM cells [111]. Disrupting PD-1 signaling influences bTRM cell differentiation. MuPyV-infected mice lacking programmed cell death ligand 1 (PD-L1), one of the two canonical ligands for PD-1, present a higher frequency of virus-specific CD103+ CD8+ cells compared to wild-type mice [111]. CD103 is an alpha integrin considered to be a broad marker of TRM cells [290]. Wild-type mice treated with a blocking PD-L1 antibody also exhibit this increase in CD103+ cell frequency, an observation recapitulated by PD-1fl/fl Rosa-CreERT2 mice, in which PD-1 can be conditionally deleted with tamoxifen administration. Upon PD-1 deletion after MuPyV infection, these mice demonstrate an increase in CD103+ cell frequency compared to vehicle-treated animals [291]; A Butic and AE Lukacher, unpublished observations]. PD-1 has also been shown to constrain the effector functions of MuPyV-specific CD8+ bTRM cells ex vivo. Bone marrow-derived dendritic cells (BMDCs) from wild-type and PD-L1-/- mice were used to stimulate brain CD8+ cells isolated from MuPyV-infected mice. PD-L1 null BMDCs incited a lower frequency of cells that were positive for IFN-γ, a classically inflammatory and antiviral cytokine, compared to wild-type BMDCs [291]. Inflammatory gene expression in wild-type and PD-L1 deficient mice indicate that inflammatory genes were upregulated in PD-L1-/- mice during acute infection [291]. Similarly, brain sections from acutely infected, tamoxifen-treated PD-1fl/fl Rosa-CreERT2 mice exhibit increased glial fibrillary acidic protein (GFAP) and ionized calcium binding adaptor molecule 1 (Iba1) immunofluorescent staining compared to vehicle-treated mice [A Butic and AE Lukacher, unpublished observations]. GFAP and Iba1 are markers of reactive gliosis, an aspect of neuroinflammation. More specifically, GFAP is upregulated in activated astrocytes, while Iba1 is increased in microglia [292,293]. Unexpectedly, inflammatory gene expression is reduced in PD-L1 null mice during persistent infection [291]. Collectively, these findings portray PD-1 as a dynamic regulator of MuPyV-induced neuroinflammation and a potentially important factor in MuPyV-specific bTRM cell differentiation. Exploring the factors that contribute to bTRM cell differentiation and also function and maintenance is critical to designing therapies for PML as well as improving patient stratification for therapies such as pembrolizumab. Although pembrolizumab has shown clinical effectiveness in some PML patients, its effects are still inconsistent, with some patients showing no response [3,294].
9.2. CD4+ T Cells
Given that PML is associated with HIV, which classically infects CD4+ T cells and thus depletes this crucial cell type over time, the requirement of CD4+ T cells in JCPyV control is increasingly appreciated [295]. PML patients who survived more than a year after onset appeared 4.8 times more likely to exhibit a discernable CD4+ T cell response compared to PML patients who passed away within a year of onset—an unsurprising observation considering the interplay between CD4+ and CD8+ T cells [275]. Furthermore, CD4+ T cell numbers were found to be inversely correlated with JCPyV DNA levels in the brain, implicating CD4+ T cells in promotion of viral infiltration of the CNS [137].
CD4+ T cells support several aspects of a robust CD8+ T cell response, from initially facilitating antigen presentation by dendritic cells to aiding expansion of memory CD8+ T cells [296]. Mice depleted of CD4+ T cells have increased virus levels in the kidney along with higher rates of viremia compared to mice depleted only of CD8+ T cells [110]. Viral control in the kidneys may thus be dependent on the help provided by CD4+ T cells. During chronic viral infection as modeled by lymphocytic choriomeningitis virus, CD4+ T cells are a critical source of interleukin (IL) -21, without which CD8+ T cell responses are severely impaired [297,298]. In MuPyV infection of the CNS, a subset of virus-specific CD4+ T cells secrete IL-21 and disrupted IL-21 signaling results in fewer CD8+ bTRM cells [299]. As CD4+ T cells drive B cell differentiation, they are important for the development of a broad antiviral neutralizing antibody repertoire.
9.3. B Cells
Despite being potential disseminators of JCPyV, B cells may also be involved in control of polyomavirus, a role supported by rituximab-associated PML [300]. B cells constitute an important part of the antiviral response, as they are responsible for generating antigen-specific antibodies and are part of immunological memory, which is critical for responding to previously encountered pathogens. In a study following four PML patients, three died within a few months after onset of PML; the survivor exhibited an increase in neutralizing titers over time in both plasma and CSF, suggesting that the humoral response may be predictive of PML outcome [301]. Moreover, MS patients with PML exhibited a lack of expansion of B and T cells [302].
MuPyV infection of B cell-deficient mice leads to chronic viremia and lack of a neutralizing antibody response [303]. B cell deficiency with T cell impairment leads to the emergence of antibody-resistant and possibly neurotropic MuPyV mutants [110]. MuPyV-infected B cell-deficient mice were immunized with a VP1 monoclonal antibody to mimic a restricted antibody response, often seen in PML patients that are unable to neutralize JCPyV variants with VP1 mutations [110,304,305]. When these mice were additionally depleted of both CD4+ and CD8+ T cells, antibody-resistant MuPyV variants with VP1 mutations arose. Overall, a restricted antibody response in concert with T cell deficiency provides the ideal setting for resurgent MuPyV replication and emergence of mutations in VP1 antibody epitopes.
How B cells control polyomavirus infection in the CNS remains to be seen. A reservoir of developmentally immature B cells exists in the calvaria bone marrow, presenting a non-systemic source of surveilling B cells [189,190]. Using MuPyV as a model, it would be interesting to explore how this immunological niche may control polyomavirus infection.
10. Conclusions
The route that JCPyV follows from the periphery to the CNS remains unanswered. JCPyV enters the periphery likely through respiratory or fecal-oral route, establishing a persistent infection in the urinary tract and other peripheral sites. The mechanisms behind JCPyV persistence are not fully understood, but it is possible that the virus establishes a low-level infection through undefined means. In an immunocompromised setting, unchecked viral replication can lead to mutations in NCCR and VP1 that shift tissue tropism, allowing mutants to evade humoral immunity and become viremic. Deficiencies in both T and B cells can promote a narrowed neutralizing antibody response, reducing chances of viral neutralization and giving virus access to the CNS. It is not clear how the virus enters the CNS, but theories have been proposed for its ability to penetrate each of the three CNS barriers: the BBB, the BCSFB, and the meninges. Infection may occur directly, with the virus infecting endothelial cells or indirectly, with the virus potentially infecting B cells and CD34+ hematopoietic progenitor cells to transit these barriers. As prolonged immunosuppression is a significant factor in JCPyV infection of the CNS, PML is associated with some but not all agents that cause depressed or modulated immunity; thus, it is critical to understand the immune response to polyomavirus infection. Both the innate and adaptive response offer various control mechanisms, from sensing the virus and inducing IFN-I expression to trigger antiviral effector ISGs, to a broad neutralizing antibody repertoire and the formation of bTRM cells. Exploring polyomavirus CNS pathogenesis and immunity is being advanced by the MuPyV model. Studying a neurotropic polyomavirus in its natural host has yielded valuable insight into early CNS pathogenesis and may be the key to improving therapeutic strategies for PML.
Author Contributions
Writing—original draft preparation, A.B.B., S.A.S., S.K.S.; writing—review and editing, A.B.B., S.A.S., S.K.S., A.E.L.; visualization— A.B.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institute of Neurologic Disorders and Stroke grant R35 NS127217.
Acknowledgments
We thank members of the Lukacher laboratory (Matthew Lauver, Katelyn Ayers, Zoe Katz, and Kalynn Alexander) for helpful feedback.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 5-HT2 | 5-hydroxytryptamine 2 |
| AIDS | Acquired imunodeficiency syndrome |
| APOBEC3 | Apolipoprotein B mRNA-editing catalytic polypeptide-like 3 |
| BBB | Blood-brain barrier |
| BCSFB | Blood-CSF-brain barrier |
| BK-VAN | BKPyV-associated nephropathy |
| BKPyV | BK polyomavirus |
| BMDC | Bone marrow-derived dendritic cells |
| bTrm | brain-resident memory |
| C/EBPβ | CAAT/enhancer binding protein beta |
| cGAMP | Cyclic GMP-AMP |
| cGAS | Cyclic GMP-AMP synthase |
| ChP | Choroid plexus epithelium |
| CNS | Central nervous system |
| COVID-19 | Coronavirus disease 2019 |
| CSF | Cerebrospinal fluid |
| ERAD | ER-associated degradation pathway |
| ESRD | End stage renal disease |
| EV | Extracellular vesicle |
| GFAP | Glial fibrillary acidic protein |
| Iba1 | Ionized calcium binding adaptor molecule 1 |
| IFN | Interferon |
| IFN-α | Interferon-α |
| IFN-β | Interferon-β |
| IFN-γ | Interferon-γ |
| IFNAR | Interferon-α/β receptor |
| IL | Interleukin |
| IRF3 | Interferon regulatory factor 3 |
| ISG | Interferon-stimulated gene |
| JAK | Janus kinase |
| JC-PVAN | JCPyV-associated nephropathy |
| JCPyV | JC polyomavirus |
| LIP | Liver inhibitory protein |
| LSTc | Lactoseries tetrasaccharide c |
| LTAg | Large T antigen |
| mAB | Monoclonal antibody |
| MDA5 | Melanoma differentiation-associated protein 5 |
| MHC | Major histocompatibility complex |
| miRNA | microRNA |
| MS | Multiple sclerosis |
| MTAg | Middle T antigen |
| MuPyV | Mouse polyomavirus |
| NCCR | Non-coding control region |
| NK | Natural killer |
| NPC | Neural progenitor cells |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death ligand 1 |
| PML | Progressive multifocal leukoencephalopathy |
| PRR | Pattern recognition receptor |
| RB | Retinoblastoma-associated protein |
| RIG-I | Retinoic-acid inducible gene I |
| STAg | Small T antigen |
| STAT | Signal transducers and activators of transcription |
| STING | Stimulatory of interferon genes |
| SV40 | Simian virus 40 |
| TLR | Toll-like receptor |
| Trm | Resident memory |
| VLP | Virus-like particle |
| WT | Wild-type |
References
- Atkinson, A.L.; Atwood, W.J. Fifty Years of JC Polyomavirus: A Brief Overview and Remaining Questions. Viruses 2020, 12, 969. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Koralnik, I.J. Progressive Multifocal Leukoencephalopathy and Other Disorders Caused by JC Virus: Clinical Features and Pathogenesis. Lancet Neurol 2010, 9, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Cortese, I.; Muranski, P.; Enose-Akahata, Y.; Ha, S.-K.; Smith, B.; Monaco, M.; Ryschkewitsch, C.; Major, E.O.; Ohayon, J.; Schindler, M.K.; et al. Pembrolizumab Treatment for Progressive Multifocal Leukoencephalopathy. N Engl J Med 2019, 380, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Kardas, P.; Kranz, D.; Leboeuf, C. The Human JC Polyomavirus (JCPyV): Virological Background and Clinical Implications. APMIS 2013, 121, 685–727. [Google Scholar] [CrossRef] [PubMed]
- Laine, H.K.; Waterboer, T.; Syrjänen, K.; Grenman, S.; Louvanto, K.; Syrjänen, S. Seroprevalence of Polyomaviruses BK and JC in Finnish Women and Their Spouses Followed-up for Three Years. Sci Rep 2023, 13, 879. [Google Scholar] [CrossRef] [PubMed]
- Kamminga, S.; van der Meijden, E.; Feltkamp, M.C.W.; Zaaijer, H.L. Seroprevalence of Fourteen Human Polyomaviruses Determined in Blood Donors. PLoS One 2018, 13, e0206273. [Google Scholar] [CrossRef]
- Zheng, H.-Y.; Kitamura, T.; Takasaka, T.; Chen, Q.; Yogo, Y. Unambiguous Identification of JC Polyomavirus Strains Transmitted from Parents to Children. Arch Virol 2004, 149, 261–273. [Google Scholar] [CrossRef]
- Ijaz, R.; Shahzad, N.; Farhan Ul Haque, M. Detection of BK and JC Polyomaviruses in Sewage Water of the Urban Areas of Lahore, Pakistan. Biologia (Bratisl) 2023, 1–8. [Google Scholar] [CrossRef]
- Prezioso, C.; Ciotti, M.; Obregon, F.; Ambroselli, D.; Rodio, D.M.; Cudillo, L.; Gaziev, J.; Mele, A.; Nardi, A.; Favalli, C.; et al. Polyomaviruses Shedding in Stool of Patients with Hematological Disorders: Detection Analysis and Study of the Non-Coding Control Region’s Genetic Variability. Med Microbiol Immunol 2019, 208, 845–854. [Google Scholar] [CrossRef]
- Bofill-Mas, S.; Pina, S.; Girones, R. Documenting the Epidemiologic Patterns of Polyomaviruses in Human Populations by Studying Their Presence in Urban Sewage. Appl Environ Microbiol 2000, 66, 238–245. [Google Scholar] [CrossRef]
- Kato, A.; Kitamura, T.; Takasaka, T.; Tominaga, T.; Ishikawa, A.; Zheng, H.-Y.; Yogo, Y. Detection of the Archetypal Regulatory Region of JC Virus from the Tonsil Tissue of Patients with Tonsillitis and Tonsilar Hypertrophy. J Neurovirol 2004, 10, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, F.; Carbone, D.; Mugavero, R.; Palmieri, A.; Lauritano, D.; Baggi, L.; Nardone, M.; Martinelli, M.; Carinci, F. Human Polyomavirus in Tonsillar Microbiota of an Afghan Population Group. J Biol Regul Homeost Agents 2018, 32, 185–190. [Google Scholar] [PubMed]
- Ayers, K.N.; Carey, S.N.; Lukacher, A.E. Understanding Polyomavirus CNS Disease - a Perspective from Mouse Models. FEBS J 2022, 289, 5744–5761. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Dey, D.; Kreisman, L.; Velupillai, P.; Dahl, J.; Telford, S.; Bronson, R.; Benjamin, T. Receptor-Binding and Oncogenic Properties of Polyoma Viruses Isolated from Feral Mice. PLoS Pathog 2007, 3, e179. [Google Scholar] [CrossRef] [PubMed]
- Swanson, P.A.; Lukacher, A.E.; Szomolanyi-Tsuda, E. Immunity to Polyomavirus Infection: The Polyomavirus-Mouse Model. Semin Cancer Biol 2009, 19, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Drake, D.R.; Lukacher, A.E. Beta 2-Microglobulin Knockout Mice Are Highly Susceptible to Polyoma Virus Tumorigenesis. Virology 1998, 252, 275–284. [Google Scholar] [CrossRef]
- Frisque, R.J.; Bream, G.L.; Cannella, M.T. Human Polyomavirus JC Virus Genome. J Virol 1984, 51, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, G.G. Gene Regulation and Quality Control in Murine Polyomavirus Infection. Viruses 2016, 8, 284. [Google Scholar] [CrossRef]
- Trowbridge, P.W.; Frisque, R.J. Identification of Three New JC Virus Proteins Generated by Alternative Splicing of the Early Viral mRNA. J Neurovirol 1995, 1, 195–206. [Google Scholar] [CrossRef]
- Seo, G.J.; Fink, L.H.L.; O’Hara, B.; Atwood, W.J.; Sullivan, C.S. Evolutionarily Conserved Function of a Viral microRNA. J Virol 2008, 82, 9823–9828. [Google Scholar] [CrossRef]
- Lauver, M.D.; Lukacher, A.E. JCPyV VP1 Mutations in Progressive MultifocalLeukoencephalopathy: Altering Tropismor Mediating Immune Evasion? Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.J.; Moore, L.D.; Mirsky, M.M.; Major, E.O. JC Virus Promoter/Enhancers Contain TATA Box-Associated Spi-B-Binding Sites That Support Early Viral Gene Expression in Primary Astrocytes. J Gen Virol 2012, 93, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Yogo, Y.; Kitamura, T.; Sugimoto, C.; Ueki, T.; Aso, Y.; Hara, K.; Taguchi, F. Isolation of a Possible Archetypal JC Virus DNA Sequence from Nonimmunocompromised Individuals. J Virol 1990, 64, 3139–3143. [Google Scholar] [CrossRef] [PubMed]
- Daniel, A.M.; Swenson, J.J.; Mayreddy, R.P.; Khalili, K.; Frisque, R.J. Sequences within the Early and Late Promoters of Archetype JC Virus Restrict Viral DNA Replication and Infectivity. Virology 1996, 216, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Ciardi, M.R.; Zingaropoli, M.A.; Iannetta, M.; Prezioso, C.; Perri, V.; Pasculli, P.; Lichtner, M.; d’Ettorre, G.; Altieri, M.; Conte, A.; et al. JCPyV NCCR Analysis in PML Patients with Different Risk Factors: Exploring Common Rearrangements as Essential Changes for Neuropathogenesis. Virol J 2020, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Kardas, P.; Major, E.O.; Hirsch, H.H. Rearranged JC Virus Noncoding Control Regions Found in Progressive Multifocal Leukoencephalopathy Patient Samples Increase Virus Early Gene Expression and Replication Rate. J Virol 2010, 84, 10448–10456. [Google Scholar] [CrossRef]
- Treisman, R.; Novak, U.; Favaloro, J.; Kamen, R. Transformation of Rat Cells by an Altered Polyoma Virus Genome Expressing Only the Middle-T Protein. Nature 1981, 292, 595–600. [Google Scholar] [CrossRef]
- Dilworth, S.M. Polyoma Virus Middle T Antigen and Its Role in Identifying Cancer-Related Molecules. Nat Rev Cancer 2002, 2, 951–956. [Google Scholar] [CrossRef]
- Ströh, L.J.; Maginnis, M.S.; Blaum, B.S.; Nelson, C.D.S.; Neu, U.; Gee, G.V.; O’Hara, B.A.; Motamedi, N.; DiMaio, D.; Atwood, W.J.; et al. The Greater Affinity of JC Polyomavirus Capsid for A2,6-Linked Lactoseries Tetrasaccharide c than for Other Sialylated Glycans Is a Major Determinant of Infectivity. J Virol 2015, 89, 6364–6375. [Google Scholar] [CrossRef]
- Neu, U.; Maginnis, M.S.; Palma, A.S.; Ströh, L.J.; Nelson, C.D.S.; Feizi, T.; Atwood, W.J.; Stehle, T. Structure-Function Analysis of the Human JC Polyomavirus Establishes the LSTc Pentasaccharide as a Functional Receptor Motif. Cell Host Microbe 2010, 8, 309–319. [Google Scholar] [CrossRef]
- Maginnis, M.S.; Haley, S.A.; Gee, G.V.; Atwood, W.J. Role of N-Linked Glycosylation of the 5-HT2A Receptor in JC Virus Infection. J Virol 2010, 84, 9677–9684. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Gilbert, J.M.; Stehle, T.; Lencer, W.; Benjamin, T.L.; Rapoport, T.A. Gangliosides Are Receptors for Murine Polyoma Virus and SV40. EMBO J 2003, 22, 4346–4355. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Belloni, L.; Sthandier, O.; Amati, P.; Garcia, M.-I. A4β1 Integrin Acts as a Cell Receptor for Murine Polyomavirus at the Postattachment Level. J Virol 2003, 77, 3913–3921. [Google Scholar] [CrossRef] [PubMed]
- Vajn, K.; Viljetić, B.; Degmečić, I.V.; Schnaar, R.L.; Heffer, M. Differential Distribution of Major Brain Gangliosides in the Adult Mouse Central Nervous System. PLoS One 2013, 8, e75720. [Google Scholar] [CrossRef] [PubMed]
- Pho, M.T.; Ashok, A.; Atwood, W.J. JC Virus Enters Human Glial Cells by Clathrin-Dependent Receptor-Mediated Endocytosis. J Virol 2000, 74, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat Rev Mol Cell Biol 2018, 19, 213–228. [Google Scholar] [CrossRef]
- O’Hara, B.A.; Morris-Love, J.; Gee, G.V.; Haley, S.A.; Atwood, W.J. JC Virus Infected Choroid Plexus Epithelial Cells Produce Extracellular Vesicles That Infect Glial Cells Independently of the Virus Attachment Receptor. PLoS Pathog 2020, 16, e1008371. [Google Scholar] [CrossRef]
- Liebl, D.; Difato, F.; Horníková, L.; Mannová, P.; Stokrová, J.; Forstová, J. Mouse Polyomavirus Enters Early Endosomes, Requires Their Acidic pH for Productive Infection, and Meets Transferrin Cargo in Rab11-Positive Endosomes. J Virol 2006, 80, 4610–4622. [Google Scholar] [CrossRef]
- Querbes, W.; O’Hara, B.A.; Williams, G.; Atwood, W.J. Invasion of Host Cells by JC Virus Identifies a Novel Role for Caveolae in Endosomal Sorting of Noncaveolar Ligands. J Virol 2006, 80, 9402–9413. [Google Scholar] [CrossRef]
- Kilcher, S.; Mercer, J. DNA Virus Uncoating. Virology 2015, 479–480, 578–590. [Google Scholar] [CrossRef]
- Vilchez, R.A.; Butel, J.S. Emergent Human Pathogen Simian Virus 40 and Its Role in Cancer. Clin Microbiol Rev 2004, 17, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Walczak, C.P.; Tsai, B. A PDI Family Network Acts Distinctly and Coordinately with ERp29 to Facilitate Polyomavirus Infection. J Virol 2011, 85, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Kuksin, D.; Norkin, L.C. Disassembly of Simian Virus 40 during Passage through the Endoplasmic Reticulum and in the Cytoplasm. J Virol 2012, 86, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tsai, B. A Large and Intact Viral Particle Penetrates the Endoplasmic Reticulum Membrane to Reach the Cytosol. PLoS Pathog 2011, 7, e1002037. [Google Scholar] [CrossRef]
- Gee, G.V.; O’Hara, B.A.; Derdowski, A.; Atwood, W.J. Pseudovirus Mimics Cell Entry and Trafficking of the Human Polyomavirus JCPyV. Virus Res 2013, 178, 10–1016. [Google Scholar] [CrossRef]
- Dupzyk, A.; Tsai, B. How Polyomaviruses Exploit the ERAD Machinery to Cause Infection. Viruses 2016, 8, 242. [Google Scholar] [CrossRef]
- Chen, Z.; Ji, Z.; Ngiow, S.F.; Manne, S.; Cai, Z.; Huang, A.C.; Johnson, J.; Staupe, R.P.; Bengsch, B.; Xu, C.; et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 2019, 51, 840–855. [Google Scholar] [CrossRef]
- Ramsperger, U.; Stahl, H. Unwinding of Chromatin by the SV40 Large T Antigen DNA Helicase. EMBO J 1995, 14, 3215–3225. [Google Scholar] [CrossRef]
- Bollag, B.; Hofstetter, C.A.; Reviriego-Mendoza, M.M.; Frisque, R.J. JC Virus Small t Antigen Binds Phosphatase PP2A and Rb Family Proteins and Is Required for Efficient Viral DNA Replication Activity. PLoS One 2010, 5, e10606. [Google Scholar] [CrossRef]
- Prins, C.; Frisque, R.J. JC Virus T’ Proteins Encoded by Alternatively Spliced Early mRNAs Enhance T Antigen-Mediated Viral DNA Replication in Human Cells. J Neurovirol 2001, 7, 250–264. [Google Scholar] [CrossRef]
- Saxena, R.; Saribas, S.; Jadiya, P.; Tomar, D.; Kaminski, R.; Elrod, J.W.; Safak, M. Human Neurotropic Polyomavirus, JC Virus, Agnoprotein Targets Mitochondrion and Modulates Its Functions. Virology 2021, 553, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Eash, S.; Tavares, R.; Stopa, E.G.; Robbins, S.H.; Brossay, L.; Atwood, W.J. Differential Distribution of the JC Virus Receptor-Type Sialic Acid in Normal Human Tissues. Am J Pathol 2004, 164, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Hametoja, H.; Hagstrom, J.; Haglund, C.; Back, L.; Makitie, A.; Syrjanen, S. Polyomavirus JCPyV Infrequently Detectable in Adenoid Cystic Carcinoma of the Oral Cavity and the Airways. Virchows Arch 2019, 475, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Abdel Aziz, H.O.; Nakanishi, Y.; Masuda, S.; Saito, H.; Tsuneyama, K.; Takano, Y. Oncogenic Role of JC Virus in Lung Cancer. J Pathol 2007, 212, 306–315. [Google Scholar] [CrossRef]
- Csoma, E.; Bidiga, L.; Mehes, G.; Katona, M.; Gergely, L. Survey of KI, WU, MW, and STL Polyomavirus in Cancerous and Non-Cancerous Lung Tissues. Pathobiology 2018, 85, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Joh, J.; Jenson, A.B.; Moore, G.D.; Rezazedeh, A.; Slone, S.P.; Ghim, S.J.; Kloecker, G.H. Human Papillomavirus (HPV) and Merkel Cell Polyomavirus (MCPyV) in Non Small Cell Lung Cancer. Exp Mol Pathol 2010, 89, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.; Waterboer, T.; Pawlita, M.; Michel, A.; Cai, Q.; Zheng, W.; Gao, Y.T.; Lan, Q.; Rothman, N.; Langseth, H.; et al. Serum Biomarkers of Polyomavirus Infection and Risk of Lung Cancer in Never Smokers. Br J Cancer 2016, 115, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Lasithiotaki, I.; Antoniou, K.M.; Derdas, S.P.; Sarchianaki, E.; Symvoulakis, E.K.; Psaraki, A.; Spandidos, D.A.; Stathopoulos, E.N.; Siafakas, N.M.; Sourvinos, G. The Presence of Merkel Cell Polyomavirus Is Associated with Deregulated Expression of BRAF and Bcl-2 Genes in Non-Small Cell Lung Cancer. Int J Cancer 2013, 133, 604–611. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.S.; de Campos, G.M.; Giovanetti, M.; Zucherato, V.S.; Lima, A.R.J.; Santos, E.V.; Haddad, R.; Ciccozzi, M.; Carlos Junior Alcantara, L.; Elias, M.C.; et al. Viral Metagenomics Unveils MW (Malawi) Polyomavirus Infection in Brazilian Pediatric Patients with Acute Respiratory Disease. J Med Virol 2023, 95, e28688. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-C.; Wang, M.; Chou, M.-C.; Chao, C.-N.; Fang, C.-Y.; Chen, P.-L.; Chang, D.; Shen, C.-H. Gene Therapy for Castration-Resistant Prostate Cancer Cells Using JC Polyomavirus-like Particles Packaged with a PSA Promoter Driven-Suicide Gene. Cancer Gene Ther 2019, 26, 208–215. [Google Scholar] [CrossRef]
- Gottlieb, K.; Villarreal, L.P. The Distribution and Kinetics of Polyomavirus in Lungs of Intranasally Infected Newborn Mice. Virology 2000, 266, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Hernandez, T.; Carnathan, D.G.; Jones, S.; Cork, A.J.; Davies, M.R.; Moyle, P.M.; Toth, I.; Batzloff, M.R.; McCarthy, J.; Nizet, V.; et al. An Experimental Group A Streptococcus Vaccine That Reduces Pharyngitis and Tonsillitis in a Nonhuman Primate Model. mBio 2019, 10, e00693–19. [Google Scholar] [CrossRef] [PubMed]
- Sinagra, E.; Raimondo, D.; Gallo, E.; Stella, M.; Cottone, M.; Rossi, F.; Messina, M.; Spada, M.; Tomasello, G.; Ferrara, G.; et al. JC Virus and Lung Adenocarcinoma: Fact or Myth? Anticancer Res 2017, 37, 3311–3314. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C.; Xue, H.; Zhang, C.Y. The Oncogenic Roles of JC Polyomavirus in Cancer. Front Oncol 2022, 12, 976577. [Google Scholar] [CrossRef] [PubMed]
- Levican, J.; Levican, A.; Ampuero, M.; Gaggero, A. JC Polyomavirus Circulation in One-Year Surveillance in Wastewater in Santiago, Chile. Infect Genet Evol 2019, 71, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.M.; Correia, A.; Pereira, M.S.; Almeida, C.R.; Alves, I.; Pinto, V.; Catarino, T.A.; Mendes, N.; Leander, M.; Oliva-Teles, M.T.; et al. Metabolic Control of T Cell Immune Response through Glycans in Inflammatory Bowel Disease. Proc Natl Acad Sci U S A 2018, 115, E4651–E4660. [Google Scholar] [CrossRef]
- Hewitt, J.; Greening, G.E.; Leonard, M.; Lewis, G.D. Evaluation of Human Adenovirus and Human Polyomavirus as Indicators of Human Sewage Contamination in the Aquatic Environment. Water Res 2013, 47, 6750–6761. [Google Scholar] [CrossRef]
- Chiu, N.C.; Chi, H.; Tai, Y.L.; Peng, C.C.; Tseng, C.Y.; Chen, C.C.; Tan, B.F.; Lin, C.Y. Impact of Wearing Masks, Hand Hygiene, and Social Distancing on Influenza, Enterovirus, and All-Cause Pneumonia During the Coronavirus Pandemic: Retrospective National Epidemiological Surveillance Study. J Med Internet Res 2020, 22, e21257. [Google Scholar] [CrossRef]
- Vigiser, I.; Piura, Y.; Kolb, H.; Shiner, T.; Komarov, I.; Karni, A.; Regev, K. JCV Seroconversion Rate during the SARS COVID-19 Pandemic. Mult Scler Relat Disord 2022, 68, 104244. [Google Scholar] [CrossRef]
- Dwyer, C.; Sharmin, S.; Kalincik, T. Rates of John Cunningham Virus Seroconversion Greatly Reduced in Natalizumab-Treated Patients during COVID-19-Related Lockdowns. Eur J Neurol 2023. [Google Scholar] [CrossRef]
- Esmailzadeh, N.; Ranaee, M.; Alizadeh, A.; Khademian, A.; Saber Amoli, S.; Sadeghi, F. Presence of JC Polyomavirus in Nonneoplastic Inflamed Colon Mucosa and Primary and Metastatic Colorectal Cancer. Gastrointest Tumors 2020, 7, 30–40. [Google Scholar] [CrossRef]
- Uleri, E.; Piu, C.; Caocci, M.; Ibba, G.; Sanges, F.; Pira, G.; Murgia, L.; Barmina, M.; Giannecchini, S.; Porcu, A.; et al. Multiple Signatures of the JC Polyomavirus in Paired Normal and Altered Colorectal Mucosa Indicate a Link with Human Colorectal Cancer, but Not with Cancer Progression. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.Y.; Chen, S.Y.; Hsiao, B.X.; Huang, H.Y.; Chen, Y.J.; Tung, C.L.; Fang, C.Y. Unusually High Incidence of Polyomavirus JC Infection in the Higher Grade of Colorectal Cancer Tissues in Taiwan. Eur J Med Res 2022, 27, 127. [Google Scholar] [CrossRef] [PubMed]
- Ksiaa, F.; Allous, A.; Ziadi, S.; Mokni, M.; Trimeche, M. Assessment and Biological Significance of JC Polyomavirus in Colorectal Cancer in Tunisia. J BUON 2015, 20, 762–769. [Google Scholar] [PubMed]
- Muhsen, M.A.Z.; Ghaith Sachit, H.; Jaafar Hussein, M.; Mohammed Ali, S.H.; Mohammed Al-Alwany, S.H.; Khalil Hussein, A. Molecular Interplay of John Cunningham Virus with Interleukin 1 Beta in Colorectal Carcinomatous Tissues from a Group of Iraqi Patients. Arch Razi Inst 2022, 77, 2299–2306. [Google Scholar] [CrossRef]
- Rowe, W.P.; Hartley, J.W.; Brodsky, I.; Huebner, R.J.; Law, L.W. Observations on the Spread of Mouse Polyoma Virus Infection. Nature 1958, 182, 1617. [Google Scholar] [CrossRef]
- Burke, J.M.; Bass, C.R.; Kincaid, R.P.; Ulug, E.T.; Sullivan, C.S. The Murine Polyomavirus MicroRNA Locus Is Required To Promote Viruria during the Acute Phase of Infection. J Virol 2018, 92, e02131–17. [Google Scholar] [CrossRef]
- Houff, S.A.; Major, E.O.; Katz, D.A.; Kufta, C.V.; Sever, J.L.; Pittaluga, S.; Roberts, J.R.; Gitt, J.; Saini, N.; Lux, W. Involvement of JC Virus-Infected Mononuclear Cells from the Bone Marrow and Spleen in the Pathogenesis of Progressive Multifocal Leukoencephalopathy. N Engl J Med 1988, 318, 301–305. [Google Scholar] [CrossRef]
- Grinnell, B.W.; Padgett, B.L.; Walker, D.L. Distribution of Nonintegrated DNA from JC Papovavirus in Organs of Patients with Progressive Multifocal Leukoencephalopathy. J Infect Dis 1983, 147, 669–675. [Google Scholar] [CrossRef]
- Tan, C.S.; Dezube, B.J.; Bhargava, P.; Autissier, P.; Wuthrich, C.; Miller, J.; Koralnik, I.J. Detection of JC Virus DNA and Proteins in Bone Marrow of HIV-Positive and HIV-Negative Patients. J Infect Dis 2009, 199, 881–888. [Google Scholar] [CrossRef]
- Prezioso, C.; Ciotti, M.; Brazzini, G.; Piacentini, F.; Passerini, S.; Grimaldi, A.; Landi, D.; Nicoletti, C.G.; Zingaropoli, M.A.; Iannetta, M.; et al. Diagnostic Value of JC Polyomavirus Viruria, Viremia, Serostatus and microRNA Expression in Multiple Sclerosis Patients Undergoing Immunosuppressive Treatment. J Clin Med 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Dolci, M.; Favero, C.; Bollati, V.; Campo, L.; Cattaneo, A.; Bonzini, M.; Villani, S.; Ticozzi, R.; Ferrante, P.; Delbue, S. Particulate Matter Exposure Increases JC Polyomavirus Replication in the Human Host. Environ Pollut 2018, 241, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Delbue, S.; Franciotta, D.; Giannella, S.; Dolci, M.; Signorini, L.; Ticozzi, R.; D’Alessandro, S.; Campisciano, G.; Comar, M.; Ferrante, P.; et al. Human Polyomaviruses in the Cerebrospinal Fluid of Neurological Patients. Microorganisms 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Boukoum, H.; Nahdi, I.; Sahtout, W.; Skiri, H.; Segondy, M.; Aouni, M. BK and JC Virus Infections in Healthy Patients Compared to Kidney Transplant Recipients in Tunisia. Microb Pathog 2016, 97, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Zimmermann, T.; Demina, V.; Sotnikov, S.; Ried, C.L.; Rahn, H.; Stapf, M.; Untucht, C.; Rohe, M.; Terstappen, G.C.; et al. Trafficking of JC Virus-like Particles across the Blood-Brain Barrier. Nanoscale Adv 2021, 3, 2488–2500. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Abdulkhalek, S. Kidney Allograft Dysfunction Due to John Cunningham (JC) Virus Nephropathy. Cureus 2022, 14, e32021. [Google Scholar] [CrossRef]
- Hocker, B.; Tabatabai, J.; Schneble, L.; Oh, J.; Thiel, F.; Pape, L.; Rusai, K.; Topaloglu, R.; Kranz, B.; Klaus, G.; et al. JC Polyomavirus Replication and Associated Disease in Pediatric Renal Transplantation: An International CERTAIN Registry Study. Pediatr Nephrol 2018, 33, 2343–2352. [Google Scholar] [CrossRef]
- Seppala, H.M.; Helantera, I.T.; Laine, P.K.S.; Lautenschlager, I.T.; Paulin, L.G.; Jahnukainen, T.J.; Auvinen, P.O.V.; Auvinen, E. Archetype JC Polyomavirus (JCPyV) Prevails in a Rare Case of JCPyV Nephropathy and in Stable Renal Transplant Recipients With JCPyV Viruria. J Infect Dis 2017, 216, 981–989. [Google Scholar] [CrossRef]
- Janphram, C.; Worawichawong, S.; Disthabanchong, S.; Sumethkul, V.; Rotjanapan, P. Absence of JC Polyomavirus (JCPyV) Viremia in Early Post-Transplant JCPyV Nephropathy: A Case Report. Transpl Infect Dis 2017, 19. [Google Scholar] [CrossRef]
- Lautenschlager, I.; Jahnukainen, T.; Kardas, P.; Lohi, J.; Auvinen, E.; Mannonen, L.; Dumoulin, A.; Hirsch, H.H.; Jalanko, H. A Case of Primary JC Polyomavirus Infection-Associated Nephropathy. Am J Transplant 2014, 14, 2887–2892. [Google Scholar] [CrossRef]
- Munker, D.; Veit, T.; Schonermarck, U.; Arnold, P.; Leuschner, G.; Barton, J.; Mummler, C.; Briegel, I.; Mumm, J.N.; Zoller, M.; et al. Polyomavirus Exerts Detrimental Effects on Renal Function in Patients after Lung Transplantation. J Clin Virol 2021, 145, 105029. [Google Scholar] [CrossRef] [PubMed]
- Purighalla, R.; Shapiro, R.; McCauley, J.; Randhawa, P. BK Virus Infection in a Kidney Allograft Diagnosed by Needle Biopsy. Am J Kidney Dis 1995, 26, 671–673. [Google Scholar] [CrossRef] [PubMed]
- Honsova, E.; Lodererova, A.; Havrdova, T.; Voska, L.; Boucek, P. [Recurrent Polyomavirus Infections in Kidney Transplants (BK Virus Nephropathy)]. Cesk Patol 2004, 40, 25–28. [Google Scholar] [PubMed]
- Hirsch, H.H.; Knowles, W.; Dickenmann, M.; Passweg, J.; Klimkait, T.; Mihatsch, M.J.; Steiger, J. Prospective Study of Polyomavirus Type BK Replication and Nephropathy in Renal-Transplant Recipients. N Engl J Med 2002, 347, 488–496. [Google Scholar] [CrossRef]
- Ramos, E.; Drachenberg, C.B.; Wali, R.; Hirsch, H.H. The Decade of Polyomavirus BK-Associated Nephropathy: State of Affairs. Transplantation 2009, 87, 621–630. [Google Scholar] [CrossRef]
- Shen, C.L.; Wu, B.S.; Lien, T.J.; Yang, A.H.; Yang, C.Y. BK Polyomavirus Nephropathy in Kidney Transplantation: Balancing Rejection and Infection. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Gras, J.; Le Flecher, A.; Dupont, A.; Verine, J.; Amara, A.; Delaugerre, C.; Molina, J.M.; Peraldi, M.N. Characteristics, Risk Factors and Outcome of BKV Nephropathy in Kidney Transplant Recipients: A Case-Control Study. BMC Infect Dis 2023, 23, 74. [Google Scholar] [CrossRef]
- Barth, H.; Solis, M.; Lepiller, Q.; Sueur, C.; Soulier, E.; Caillard, S.; Stoll-Keller, F.; Fafi-Kremer, S. 45 Years after the Discovery of Human Polyomaviruses BK and JC: Time to Speed up the Understanding of Associated Diseases and Treatment Approaches. Crit Rev Microbiol 2017, 43, 178–195. [Google Scholar] [CrossRef]
- Broekema, N.M.; Imperiale, M.J. Efficient Propagation of Archetype BK and JC Polyomaviruses. Virology 2012, 422, 235–241. [Google Scholar] [CrossRef]
- Knowles, W.A.; Pipkin, P.; Andrews, N.; Vyse, A.; Minor, P.; Brown, D.W.; Miller, E. Population-Based Study of Antibody to the Human Polyomaviruses BKV and JCV and the Simian Polyomavirus SV40. J Med Virol 2003, 71, 115–123. [Google Scholar] [CrossRef]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of Polyomavirus BK and JC Infection and Replication in 400 Healthy Blood Donors. J Infect Dis 2009, 199, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H. BK Virus: Opportunity Makes a Pathogen. Clin Infect Dis 2005, 41, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Siguier, M.; Sellier, P.; Bergmann, J.F. BK-Virus Infections: A Literature Review. Med Mal Infect 2012, 42, 181–187. [Google Scholar] [CrossRef]
- Alcendor, D.J. BK Polyomavirus Virus Glomerular Tropism: Implications for Virus Reactivation from Latency and Amplification during Immunosuppression. J Clin Med 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, C.; Orio, J.; Collette, S.; Senecal, L.; Hebert, M.J.; Renoult, E.; Tibbles, L.A.; Delisle, J.S. BK Polyomavirus and the Transplanted Kidney: Immunopathology and Therapeutic Approaches. Transplantation 2016, 100, 2276–2287. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, X.; Zhang, H.; Zhao, G.D.; Yang, H.; Qiu, J.; Meng, S.; Wu, P.; Tao, L.; Wang, Q.; et al. Single-Cell Transcriptome Identifies the Renal Cell Type Tropism of Human BK Polyomavirus. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Wang, J.; Dong, Y.; Mathews, D.V.; Albrecht, J.A.; Breeden, C.P.; Farris, A.B.; Lukacher, A.E.; Ford, M.L.; Newell, K.A.; et al. Alloimmunity But Not Viral Immunity Promotes Allograft Loss in a Mouse Model of Polyomavirus-Associated Allograft Injury. Transplant Direct 2017, 3, e161. [Google Scholar] [CrossRef] [PubMed]
- Maru, S.; Jin, G.; Desai, D.; Amin, S.; Shwetank, null; Lauver, M. D.; Lukacher, A.E. Inhibition of Retrograde Transport Limits Polyomavirus Infection In Vivo. mSphere 2017, 2, e00494–17. [Google Scholar] [CrossRef] [PubMed]
- Frost, E.L.; Kersh, A.E.; Evavold, B.D.; Lukacher, A.E. Cutting Edge: Resident Memory CD8 T Cells Express High-Affinity TCRs. J Immunol 2015, 195, 3520–3524. [Google Scholar] [CrossRef]
- Lauver, M.D.; Jin, G.; Ayers, K.N.; Carey, S.N.; Specht, C.S.; Abendroth, C.S.; Lukacher, A.E. T Cell Deficiency Precipitates Antibody Evasion and Emergence of Neurovirulent Polyomavirus. Elife 2022, 11. [Google Scholar] [CrossRef]
- Shwetank, N.; Abdelsamed, H.A.; Frost, E.L.; Schmitz, H.M.; Mockus, T.E.; Youngblood, B.A.; Lukacher, A.E. Maintenance of PD-1 on Brain-Resident Memory CD8 T Cells Is Antigen Independent. Immunol Cell Biol 2017, 95, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Bonek, R.; Guenter, W.; Jalowinski, R.; Karbicka, A.; Litwin, A.; Maciejowski, M.; Zajdel, R.; Zajdel, K.; Petit, V.; Rejdak, K. JC Virus Seroprevalence and JCVAb Index in Polish Multiple Sclerosis Patients Treated with Immunomodulating or Immunosuppressive Therapies. J Clin Med 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Bonek, R.; Guenter, W.; Jalowinski, R.; Karbicka, A.; Litwin, A.; Maciejowski, M.; Zajdel, R.; Petit, V.; Rejdak, K. JC Virus Seroprevalence and JCVAb Index in Polish Multiple Sclerosis Treatment-Naive Patients. J Clin Med 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Hanaei, S.; Sahraian, M.A.; Mohammadifar, M.; Ramagopalan, S.V.; Ghajarzadeh, M.; Ghajarzadeh, M. Prevalence of Anti-JC Virus Antibody Seropositivity in Patients with Multiple Sclerosis: A Systematic Review and Meta-Analysis. Intervirology 2019, 62, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Rochford, R.; Moreno, J.P.; Peake, M.L.; Villarreal, L.P. Enhancer Dependence of Polyomavirus Persistence in Mouse Kidneys. J Virol 1992, 66, 3287–3297. [Google Scholar] [CrossRef]
- Shwetank, N.; Frost, E.L.; Mockus, T.E.; Ren, H.M.; Toprak, M.; Lauver, M.D.; Netherby-Winslow, C.S.; Jin, G.; Cosby, J.M.; Evavold, B.D.; et al. PD-1 Dynamically Regulates Inflammation and Development of Brain-Resident Memory CD8 T Cells During Persistent Viral Encephalitis. Front Immunol 2019, 10, 783. [Google Scholar] [CrossRef]
- Qin, Q.; Shwetank, *!!! REPLACE !!!*; Frost, E.L.; Maru, S.; Lukacher, A.E. Type I Interferons Regulate the Magnitude and Functionality of Mouse Polyomavirus-Specific CD8 T Cells in a Virus Strain-Dependent Manner. J Virol 2016, 90, 5187–5199. [Google Scholar] [CrossRef]
- Qin, Q.; Lauver, M.; Maru, S.; Lin, E.; Lukacher, A.E. Reducing Persistent Polyomavirus Infection Increases Functionality of Virus-Specific Memory CD8 T Cells. Virology 2017, 502, 198–205. [Google Scholar] [CrossRef]
- Mockus, T.E.; Shwetank, N.; Lauver, M.D.; Ren, H.M.; Netherby, C.S.; Salameh, T.; Kawasawa, Y.I.; Yue, F.; Broach, J.R.; Lukacher, A.E. CD4 T Cells Control Development and Maintenance of Brain-Resident CD8 T Cells during Polyomavirus Infection. PLoS Pathog 2018, 14, e1007365. [Google Scholar] [CrossRef]
- Mockus, T.E.; Netherby-Winslow, C.S.; Atkins, H.M.; Lauver, M.D.; Jin, G.; Ren, H.M.; Lukacher, A.E. CD8 T Cells and STAT1 Signaling Are Essential Codeterminants in Protection from Polyomavirus Encephalopathy. J Virol 2020, 94, e02038–19. [Google Scholar] [CrossRef]
- Boldorini, R.; Caldarelli-Stefano, R.; Monga, G.; Zocchi, M.; Mediati, M.; Tosoni, A.; Ferrante, P. PCR Detection of JC Virus DNA in the Brain Tissue of a 9-Year-Old Child with Pleomorphic Xanthoastrocytoma. J Neurovirol 1998, 4, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Passerini, S.; Prezioso, C.; Prota, A.; Babini, G.; Bargiacchi, L.; Bartolini, D.; Moens, U.; Antonelli, M.; Pietropaolo, V. Detection of Human Neurotropic JCPyV DNA Sequence in Pediatric Anaplastic Xanthoastrocytoma. J Neurovirol 2023, 29, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Passerini, S.; Prezioso, C.; Prota, A.; Babini, G.; Coppola, L.; Lodi, A.; Epifani, A.C.; Sarmati, L.; Andreoni, M.; Moens, U.; et al. Detection Analysis and Study of Genomic Region Variability of JCPyV, BKPyV, MCPyV, HPyV6, HPyV7 and QPyV in the Urine and Plasma of HIV-1-Infected Patients. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, L.; Khalili, K. Induction of Brain Tumors by the Archetype Strain of Human Neurotropic JCPyV in a Transgenic Mouse Model. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Signorini, L.; Dolci, M.; Favi, E.; Colico, C.; Ferraresso, M.; Ticozzi, R.; Basile, G.; Ferrante, P.; Delbue, S. Viral Genomic Characterization and Replication Pattern of Human Polyomaviruses in Kidney Transplant Recipients. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Scribano, S.; Guerrini, M.; Arvia, R.; Guasti, D.; Nardini, P.; Romagnoli, P.; Giannecchini, S. Archetype JC Polyomavirus DNA Associated with Extracellular Vesicles Circulates in Human Plasma Samples. J Clin Virol 2020, 128, 104435. [Google Scholar] [CrossRef]
- Moens, U.; Prezioso, C.; Pietropaolo, V. Genetic Diversity of the Noncoding Control Region of the Novel Human Polyomaviruses. Viruses 2020, 12. [Google Scholar] [CrossRef]
- Prezioso, C.; Bianchi, M.; Obregon, F.; Ciotti, M.; Sarmati, L.; Andreoni, M.; Palamara, A.T.; Pascarella, S.; Moens, U.; Pietropaolo, V. Structural Analysis of Merkel Cell Polyomavirus (MCPyV) Viral Capsid Protein 1 (VP1) in HIV-1 Infected Individuals. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Maginnis, M.S.; Stroh, L.J.; Gee, G.V.; O’Hara, B.A.; Derdowski, A.; Stehle, T.; Atwood, W.J. Progressive Multifocal Leukoencephalopathy-Associated Mutations in the JC Polyomavirus Capsid Disrupt Lactoseries Tetrasaccharide c Binding. mBio 2013, 4, e00247–13. [Google Scholar] [CrossRef]
- Hu, C.; Huang, Y.; Su, J.; Wang, M.; Zhou, Q.; Zhu, B. Detection and Analysis of Variants of JC Polyomavirus in Urine Samples from HIV-1-Infected Patients in China’s Zhejiang Province. J Int Med Res 2018, 46, 1024–1032. [Google Scholar] [CrossRef]
- Karalic, D.; Lazarevic, I.; Banko, A.; Cupic, M.; Jevtovic, D.; Jovanovic, T. Analysis of Variability of Urinary Excreted JC Virus Strains in Patients Infected with HIV and Healthy Donors. J Neurovirol 2018, 24, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.E.; Li, H.; Sur, G.; Carmillo, P.; Bushnell, S.; Tizard, R.; McAuliffe, M.; Tonkin, C.; Simon, K.; Goelz, S.; et al. Sequencing and Analysis of JC Virus DNA from Natalizumab-Treated PML Patients. J Infect Dis 2011, 204, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Reid, C.; Testa, M.; Brickelmaier, M.; Bossolasco, S.; Pazzi, A.; Bestetti, A.; Carmillo, P.; Wilson, E.; McAuliffe, M.; et al. Progressive Multifocal Leukoencephalopathy (PML) Development Is Associated with Mutations in JC Virus Capsid Protein VP1 That Change Its Receptor Specificity. J Infect Dis 2011, 204, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, J.; Karasoulos, T.; Bowden, S.; Glogowski, I.; McLean, C.A. Immunosuppression Increases Latent Infection of Brain by JC Polyomavirus. Pathology 2011, 43, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, J.; Harrison, E.; McLean, C.A. Progressive Multifocal Leukoencephalopathy Development Is Associated with Mutations in JC Virus Capsid Protein VP1 That Change the Receptor Specificity of the Virus. J Infect Dis 2011, 204, 1643–1644. [Google Scholar] [CrossRef]
- Bayliss, J.; Karasoulos, T.; McLean, C.A. Frequency and Large T (LT) Sequence of JC Polyomavirus DNA in Oligodendrocytes, Astrocytes and Granular Cells in Non-PML Brain. Brain Pathol 2012, 22, 329–336. [Google Scholar] [CrossRef]
- Bayliss, J.; Karasoulos, T.; McLean, C.A. Immunosuppression Increases JC Polyomavirus Large T Antigen DNA Load in the Brains of Patients without Progressive Multifocal Leukoencephalopathy. J Infect Dis 2013, 207, 133–136. [Google Scholar] [CrossRef]
- Brassesco, M.S.; Darrigo, L.G., Jr.; Valera, E.T.; Oliveira, R.S.; Yamamoto, Y.A.; de Castro Barros, M.V.; Tone, L.G. Giant-Cell Glioblastoma of Childhood Associated with HIV-1 and JC Virus Coinfection. Childs Nerv Syst 2013, 29, 1387–1390. [Google Scholar] [CrossRef]
- Okamoto, H.; Mineta, T.; Ueda, S.; Nakahara, Y.; Shiraishi, T.; Tamiya, T.; Tabuchi, K. Detection of JC Virus DNA Sequences in Brain Tumors in Pediatric Patients. J Neurosurg 2005, 102, 294–298. [Google Scholar] [CrossRef]
- Rollison, D.E.; Utaipat, U.; Ryschkewitsch, C.; Hou, J.; Goldthwaite, P.; Daniel, R.; Helzlsouer, K.J.; Burger, P.C.; Shah, K.V.; Major, E.O. Investigation of Human Brain Tumors for the Presence of Polyomavirus Genome Sequences by Two Independent Laboratories. Int J Cancer 2005, 113, 769–774. [Google Scholar] [CrossRef]
- Del Valle, L.; Enam, S.; Lara, C.; Ortiz-Hidalgo, C.; Katsetos, C.D.; Khalili, K. Detection of JC Polyomavirus DNA Sequences and Cellular Localization of T-Antigen and Agnoprotein in Oligodendrogliomas. Clin Cancer Res 2002, 8, 3332–3340. [Google Scholar] [PubMed]
- Del Valle, L.; Delbue, S.; Gordon, J.; Enam, S.; Croul, S.; Ferrante, P.; Khalili, K. Expression of JC Virus T-Antigen in a Patient with MS and Glioblastoma Multiforme. Neurology 2002, 58, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, L.; Gordon, J.; Enam, S.; Delbue, S.; Croul, S.; Abraham, S.; Radhakrishnan, S.; Assimakopoulou, M.; Katsetos, C.D.; Khalili, K. Expression of Human Neurotropic Polyomavirus JCV Late Gene Product Agnoprotein in Human Medulloblastoma. J Natl Cancer Inst 2002, 94, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Krynska, B.; Otte, J.; Franks, R.; Khalili, K.; Croul, S. Human Ubiquitous JCV(CY) T-Antigen Gene Induces Brain Tumors in Experimental Animals. Oncogene 1999, 18, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, D.J.; Finegold, M.J.; Butel, J.S.; Kupsky, W.J.; Garcea, R.L. DNA Sequences Similar to Those of Simian Virus 40 in Ependymomas and Choroid Plexus Tumors of Childhood. N Engl J Med 1992, 326, 988–993. [Google Scholar] [CrossRef]
- White, F.A., 3rd; Ishaq, M.; Stoner, G.L.; Frisque, R.J. JC Virus DNA Is Present in Many Human Brain Samples from Patients without Progressive Multifocal Leukoencephalopathy. J Virol 1992, 66, 5726–5734. [Google Scholar] [CrossRef]
- Perez-Liz, G.; Del Valle, L.; Gentilella, A.; Croul, S.; Khalili, K. Detection of JC Virus DNA Fragments but Not Proteins in Normal Brain Tissue. Ann Neurol 2008, 64, 379–387. [Google Scholar] [CrossRef]
- White, M.K.; Khalili, K. Pathogenesis of Progressive Multifocal Leukoencephalopathy--Revisited. J Infect Dis 2011, 203, 578–586. [Google Scholar] [CrossRef]
- Pietropaolo, V.; Prezioso, C.; Bagnato, F.; Antonelli, G. John Cunningham Virus: An Overview on Biology and Disease of the Etiological Agent of the Progressive Multifocal Leukoencephalopathy. New Microbiol 2018, 41, 179–186. [Google Scholar]
- O’Hara, B.A.; Gee, G.V.; Atwood, W.J.; Haley, S.A. Susceptibility of Primary Human Choroid Plexus Epithelial Cells and Meningeal Cells to Infection by JC Virus. J Virol 2018, 92, e00105–18. [Google Scholar] [CrossRef]
- Ampie, L.; McGavern, D.B. Immunological Defense of CNS Barriers against Infections. Immunity 2022, 55, 781–799. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.W.; McGavern, D.B. Immune Dynamics in the CNS and Its Barriers during Homeostasis and Disease. Immunol Rev 2022, 306, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Koralnik, I.J.; Boden, D.; Mai, V.X.; Lord, C.I.; Letvin, N.L. JC Virus DNA Load in Patients with and without Progressive Multifocal Leukoencephalopathy. Neurology 1999, 52, 253–260. [Google Scholar] [CrossRef]
- Dubois, V.; Dutronc, H.; Lafon, M.E.; Poinsot, V.; Pellegrin, J.L.; Ragnaud, J.M.; Ferrer, A.M.; Fleury, H.J. Latency and Reactivation of JC Virus in Peripheral Blood of Human Immunodeficiency Virus Type 1-Infected Patients. J Clin Microbiol 1997, 35, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Chapagain, M.L.; Verma, S.; Mercier, F.; Yanagihara, R.; Nerurkar, V.R. Polyomavirus JC Infects Human Brain Microvascular Endothelial Cells Independent of Serotonin Receptor 2A. Virology 2007, 364, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Haley, S.A.; O’Hara, B.A.; Nelson, C.D.; Brittingham, F.L.; Henriksen, K.J.; Stopa, E.G.; Atwood, W.J. Human Polyomavirus Receptor Distribution in Brain Parenchyma Contrasts with Receptor Distribution in Kidney and Choroid Plexus. Am J Pathol 2015, 185, 2246–2258. [Google Scholar] [CrossRef]
- Dorries, K.; Johnson, R.T.; Ter Meulen, V. Detection of Polyoma Virus DNA in PML-Brain Tissue by (in Situ) Hybridization. Journal of General Virology 1979, 42, 49–57. [Google Scholar] [CrossRef]
- RW, von E.; IW, S.; M, P.; B, Z.; M, D.; HV, V. New JC Virus Infection Patterns by in Situ Polymerase Chain Reaction in Brains of Acquired Immunodeficiency Syndrome Patients with Progressive Multifocal Leukoencephalopathy. Journal of NeuroVirology 2004, 10, 1–11. [CrossRef]
- Monaco, M.C.; Atwood, W.J.; Gravell, M.; Tornatore, C.S.; Major, E.O. JC Virus Infection of Hematopoietic Progenitor Cells, Primary B Lymphocytes, and Tonsillar Stromal Cells: Implications for Viral Latency. J Virol 1996, 70, 7004–7012. [Google Scholar] [CrossRef]
- Wei, G.; Liu, C.K.; Atwood, W.J. JC Virus Binds to Primary Human Glial Cells, Tonsillar Stromal Cells, and B-Lymphocytes, but Not to T Lymphocytes. J Neurovirol 2000, 6, 127–136. [Google Scholar] [CrossRef]
- Dubois, V.; Dutronc, H.; Lafon, M.E.; Poinsot, V.; Pellegrin, J.L.; Ragnaud, J.M.; Ferrer, A.M.; Fleury, H.J. Latency and Reactivation of JC Virus in Peripheral Blood of Human Immunodeficiency Virus Type 1-Infected Patients. J Clin Microbiol 1997, 35, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Lafon, M.E.; Ragnaud, J.M.; Pellegrin, J.L.; Damasio, F.; Baudouin, C.; Michaud, V.; Fleury, H.J. Detection of JC Virus DNA in the Peripheral Blood Leukocytes of HIV-Infected Patients. AIDS 1996, 10, 353–358. [Google Scholar] [CrossRef]
- Dubois, V.; Moret, H.; Lafon, M.E.; Janvresse, C.B.; Dussaix, E.; Icart, J.; Karaterki, A.; Ruffault, A.; Taoufik, Y.; Vignoli, C.; et al. Prevalence of JC Virus Viraemia in HIV-Infected Patients with or without Neurological Disorders: A Prospective Study. J Neurovirol 1998, 4, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Frohman, E.M.; Monaco, M.C.; Remington, G.; Ryschkewitsch, C.; Jensen, P.N.; Johnson, K.; Perkins, M.; Liebner, J.; Greenberg, B.; Monson, N.; et al. JC Virus in CD34+ and CD19+ Cells in Patients with Multiple Sclerosis Treated with Natalizumab. JAMA Neurol 2014, 71, 596–602. [Google Scholar] [CrossRef]
- Tornatore, C.; Berger, J.R.; Houff, S.A.; Curfman, B.; Meyers, K.; Winfield, D.; Major, E.O. Detection of JC Virus DNA in Peripheral Lymphocytes from Patients with and without Progressive Multifocal Leukoencephalopathy. Ann Neurol 1992, 31, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Major, E.O.; Amemiya, K.; Elder, G.; Houff, S.A. Glial Cells of the Human Developing Brain and B Cells of the Immune System Share a Common DNA Binding Factor for Recognition of the Regulatory Sequences of the Human Polyomavirus, JCV. J Neurosci Res 1990, 27, 461–471. [Google Scholar] [CrossRef]
- Atwood, W.J.; Amemiya, K.; Traub, R.; Harms, J.; Major, E.O. Interaction of the Human Polyomavirus, JCV, with Human B-Lymphocytes. Virology 1992, 190, 716–723. [Google Scholar] [CrossRef]
- Chapagain, M.L.; Nerurkar, V.R. Human Polyomavirus JC (JCV) Infection of Human B Lymphocytes: A Possible Mechanism for JCV Transmigration across the Blood-Brain Barrier. J Infect Dis 2010, 202, 184–191. [Google Scholar] [CrossRef]
- Reff, M.E.; Carner, K.; Chambers, K.S.; Chinn, P.C.; Leonard, J.E.; Raab, R.; Newman, R.A.; Hanna, N.; Anderson, D.R. Depletion of B Cells in Vivo by a Chimeric Mouse Human Monoclonal Antibody to CD20. Blood 1994, 83, 435–445. [Google Scholar] [CrossRef]
- Hanif, N.; Anwer, F. StatPearls. In; Treasure Island: StatPearls Publishing: Florida, 2023.
- Administration, U.S.F. and D. List of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations (Purple Book); FDA, 2020.
- Anolik, J.H.; Friedberg, J.W.; Zheng, B.; Barnard, J.; Owen, T.; Cushing, E.; Kelly, J.; Milner, E.C.; Fisher, R.I.; Sanz, I. B Cell Reconstitution after Rituximab Treatment of Lymphoma Recapitulates B Cell Ontogeny. Clin Immunol 2007, 122, 139–145. [Google Scholar] [CrossRef]
- Roll, P.; Palanichamy, A.; Kneitz, C.; Dorner, T.; Tony, H.P. Regeneration of B Cell Subsets after Transient B Cell Depletion Using Anti-CD20 Antibodies in Rheumatoid Arthritis. Arthritis Rheum 2006, 54, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Haley, S.A.; O’Hara, B.A.; Nelson, C.D.S.; Brittingham, F.L.P.; Henriksen, K.J.; Stopa, E.G.; Atwood, W.J. Human Polyomavirus Receptor Distribution in Brain Parenchyma Contrasts with Receptor Distribution in Kidney and Choroid Plexus. Am J Pathol 2015, 185, 2246–2258. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.P.; Wuthrich, C.; Dang, X.; Nauen, D.; Karimi, R.; Viscidi, R.; Bord, E.; Batson, S.; Troncoso, J.; Koralnik, I.J. A Fatal Case of JC Virus Meningitis Presenting with Hydrocephalus in a Human Immunodeficiency Virus-Seronegative Patient. Ann Neurol 2014, 76, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Corbridge, S.M.; Rice, R.C.; Bean, L.A.; Wüthrich, C.; Dang, X.; Nicholson, D.A.; Koralnik, I.J. JC Virus Infection of Meningeal and Choroid Plexus Cells in Patients with Progressive Multifocal Leukoencephalopathy. J Neurovirol 2019, 25, 520–524. [Google Scholar] [CrossRef]
- Morris-Love, J.; Gee, G.V.; O’Hara, B.A.; Assetta, B.; Atkinson, A.L.; Dugan, A.S.; Haley, S.A.; Atwood, W.J. JC Polyomavirus Uses Extracellular Vesicles To Infect Target Cells. mBio 2019, 10. [Google Scholar] [CrossRef]
- Giannecchini, S. Evidence of the Mechanism by Which Polyomaviruses Exploit the Extracellular Vesicle Delivery System during Infection. Viruses 2020, 12. [Google Scholar] [CrossRef]
- Berger, J.R.; Aksamit, A.J.; Clifford, D.B.; Davis, L.; Koralnik, I.J.; Sejvar, J.J.; Bartt, R.; Major, E.O.; Nath, A. PML Diagnostic Criteria: Consensus Statement from the AAN Neuroinfectious Disease Section. Neurology 2013, 80, 1430–1438. [Google Scholar] [CrossRef]
- Tornatore, C.; Berger, J.R.; Houff, S.A.; Curfman, B.; Meyers, K.; Winfield, D.; Major, E.O. Detection of JC Virus DNA in Peripheral Lymphocytes from Patients with and without Progressive Multifocal Leukoencephalopathy. Ann Neurol 1992, 31, 454–462. [Google Scholar] [CrossRef]
- Viallard, J.F.; Ellie, E.; Lazaro, E.; Lafon, M.E.; Pellegrin, J.L. JC Virus Meningitis in a Patient with Systemic Lupus Erythematosus. Lupus 2005, 14, 964–966. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Amenta, E.; Chavez, V.; Fukuta, Y.; Hasbun, R. Undetectable JC Virus CSF PCR in Patients with JC Virus-Induced Progressive Multifocal Leukoencephalopathy. J Neurovirol 2023, 29, 94–99. [Google Scholar] [CrossRef]
- Mornese Pinna, S.; Trunfio, M.; Imperiale, D.; Calcagno, A. JC Virus DNA in Cerebrospinal Fluid: Insight into Clinical Significance. Diagn Microbiol Infect Dis 2020, 97, 115017. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Saegeman, V.; Beuselinck, K.; Wouters, A.; Cypers, G.; Meyfroidt, G.; Schrooten, M. Predictive Value of JC Virus PCR in Cerebrospinal Fluid in the Diagnosis of PML. Diagn Microbiol Infect Dis 2019, 95, 114859. [Google Scholar] [CrossRef] [PubMed]
- Mockus, T.E.; Netherby-Winslow, C.S.; Atkins, H.M.; Lauver, M.D.; Jin, G.; Ren, H.M.; Lukacher, A.E. CD8 T Cells and STAT1 Signaling Are Essential Codeterminants in Protection from Polyomavirus Encephalopathy. J Virol 2020, 94, e02038–19. [Google Scholar] [CrossRef] [PubMed]
- Chalkias, S.; Dang, X.; Bord, E.; Stein, M.C.; Kinkel, R.P.; Sloane, J.A.; Donnelly, M.; Ionete, C.; Houtchens, M.K.; Buckle, G.J.; et al. JC Virus Reactivation during Prolonged Natalizumab Monotherapy for Multiple Sclerosis. Ann Neurol 2014, 75, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.H.; Stark, J.L.; Lauber, J.; Ramsbottom, M.J.; Lyons, J.A. Rituximab Reduces B Cells and T Cells in Cerebrospinal Fluid of Multiple Sclerosis Patients. J Neuroimmunol 2006, 180, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Casselli, T.; Divan, A.; Vomhof-DeKrey, E.E.; Tourand, Y.; Pecoraro, H.L.; Brissette, C.A. A Murine Model of Lyme Disease Demonstrates That Borrelia Burgdorferi Colonizes the Dura Mater and Induces Inflammation in the Central Nervous System. PLoS Pathog 2021, 17, e1009256. [Google Scholar] [CrossRef] [PubMed]
- Cugurra, A.; Mamuladze, T.; Rustenhoven, J.; Dykstra, T.; Beroshvili, G.; Greenberg, Z.J.; Baker, W.; Papadopoulos, Z.; Drieu, A.; Blackburn, S.; et al. Skull and Vertebral Bone Marrow Are Myeloid Cell Reservoirs for the Meninges and CNS Parenchyma. Science 2021, 373. [Google Scholar] [CrossRef]
- Brioschi, S.; Wang, W.L.; Peng, V.; Wang, M.; Shchukina, I.; Greenberg, Z.J.; Bando, J.K.; Jaeger, N.; Czepielewski, R.S.; Swain, A.; et al. Heterogeneity of Meningeal B Cells Reveals a Lymphopoietic Niche at the CNS Borders. Science 2021, 373. [Google Scholar] [CrossRef]
- Dang, L.; Dang, X.; Koralnik, I.J.; Todd, P.K. JC Polyomavirus Granule Cell Neuronopathy in a Patient Treated with Rituximab. JAMA Neurol 2014, 71, 487–489. [Google Scholar] [CrossRef]
- Granot, R.; Lawrence, R.; Barnett, M.; Masters, L.; Rodriguez, M.; Theocharous, C.; Pamphlett, R.; Hersch, M. What Lies beneath the Tent? JC-Virus Cerebellar Granule Cell Neuronopathy Complicating Sarcoidosis. J Clin Neurosci 2009, 16, 1091–1092. [Google Scholar] [CrossRef]
- Hecht, J.H.; Glenn, O.A.; Wara, D.W.; Wu, Y.W. JC Virus Granule Cell Neuronopathy in a Child with CD40 Ligand Deficiency. Pediatr Neurol 2007, 36, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Koralnik, I.J.; Wüthrich, C.; Dang, X.; Rottnek, M.; Gurtman, A.; Simpson, D.; Morgello, S. JC Virus Granule Cell Neuronopathy: A Novel Clinical Syndrome Distinct from Progressive Multifocal Leukoencephalopathy. Ann Neurol 2005, 57, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Reoma, L.B.; Trindade, C.J.; Monaco, M.C.; Solis, J.; Montojo, M.G.; Vu, P.; Johnson, K.; Beck, E.; Nair, G.; Khan, O.I.; et al. Fatal Encephalopathy with Wild-Type JC Virus and Ruxolitinib Therapy. Ann Neurol 2019, 86, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, C.; Dang, X.; Westmoreland, S.; McKay, J.; Maheshwari, A.; Anderson, M.P.; Ropper, A.H.; Viscidi, R.P.; Koralnik, I.J. Fulminant JC Virus Encephalopathy with Productive Infection of Cortical Pyramidal Neurons. Ann Neurol 2009, 65, 742–748. [Google Scholar] [CrossRef]
- ZURHEIN, G.; CHOU, S.M. PARTICLES RESEMBLING PAPOVA VIRUSES IN HUMAN CEREBRAL DEMYELINATING DISEASE. Science 1965, 148, 1477–1479. [Google Scholar] [CrossRef]
- ÅSTRÖM, K.-E.; MANCALL, E.L.; RICHARDSON, E.P., JR. PROGRESSIVE MULTIFOCAL LEUKO-ENCEPHALOPATHY: A HITHERTO UNRECOGNIZED COMPLICATION O CHRONIC LYMPHATIC LEUKÆMIA AND HODGKIN’S DISEASE1. Brain 1958, 81, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Dörries, K.; Johnson, R.T.; ter Meulen, V. Detection of Polyoma Virus DNA in PML-Brain Tissue by (in Situ) Hybridization. J Gen Virol 1979, 42, 49–57. [Google Scholar] [CrossRef]
- Ironside, J.W.; Lewis, F.A.; Blythe, D.; Wakefield, E.A. The Identification of Cells Containing JC Papovavirus DNA in Progressive Multifocal Leukoencephalopathy by Combined in Situ Hybridization and Immunocytochemistry. J Pathol 1989, 157, 291–297. [Google Scholar] [CrossRef]
- McCance, D.J. The Types of Mouse Brain Cells Susceptible to Polyoma Virus Infection in Vitro. J Gen Virol 1984, 65 ( Pt 1) Pt 1, 221–226. [Google Scholar] [CrossRef]
- McCance, D.J.; Sebesteny, A.; Griffin, B.E.; Balkwill, F.; Tilly, R.; Gregson, N.A. A Paralytic Disease in Nude Mice Associated with Polyoma Virus Infection. J Gen Virol 1983, 64 (Pt 1) Pt 1, 57–67. [Google Scholar] [CrossRef]
- Sebesteny, A.; Tilly, R.; Balkwill, F.; Trevan, D. Demyelination and Wasting Associated with Polyomavirus Infection in Nude (Nu/Nu) Mice. Lab Anim 1980, 14, 337–345. [Google Scholar] [CrossRef]
- Nakamichi, K.; Takayama-Ito, M.; Nukuzuma, S.; Kurane, I.; Saijo, M. Long-Term Infection of Adult Mice with Murine Polyomavirus Following Stereotaxic Inoculation into the Brain. Microbiol Immunol 2010, 54, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.J.; McClain, C.R.; Schanz, S.J.; Protack, T.L.; Windrem, M.S.; Goldman, S.A. CD140a Identifies a Population of Highly Myelinogenic, Migration-Competent and Efficiently Engrafting Human Oligodendrocyte Progenitor Cells. Nat Biotechnol 2011, 29, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Windrem, M.S.; Schanz, S.J.; Guo, M.; Tian, G.F.; Washco, V.; Stanwood, N.; Rasband, M.; Roy, N.S.; Nedergaard, M.; Havton, L.A.; et al. Neonatal Chimerization with Human Glial Progenitor Cells Can Both Remyelinate and Rescue the Otherwise Lethally Hypomyelinated Shiverer Mouse. Cell Stem Cell 2008, 2, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Windrem, M.S.; Zou, L.; Chandler-Militello, D.; Schanz, S.J.; Auvergne, R.M.; Betstadt, S.J.; Harrington, A.R.; Johnson, M.; Kazarov, A.; et al. Human Glial Chimeric Mice Reveal Astrocytic Dependence of JC Virus Infection. J Clin Invest 2014, 124, 5323–5336. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb Perspect Biol 2015, 7, a020370. [Google Scholar] [CrossRef]
- Molina-Gonzalez, I.; Miron, V.E. Astrocytes in Myelination and Remyelination. Neurosci Lett 2019, 713, 134532. [Google Scholar] [CrossRef]
- Han, R.T.; Kim, R.D.; Molofsky, A.V.; Liddelow, S.A. Astrocyte-Immune Cell Interactions in Physiology and Pathology. Immunity 2021, 54, 211–224. [Google Scholar] [CrossRef]
- Lawrence, J.M.; Schardien, K.; Wigdahl, B.; Nonnemacher, M.R. Roles of Neuropathology-Associated Reactive Astrocytes: A Systematic Review. Acta Neuropathol Commun 2023, 11, 42. [Google Scholar] [CrossRef]
- Haley, S.A.; O’Hara, B.A.; Nelson, C.D.; Brittingham, F.L.; Henriksen, K.J.; Stopa, E.G.; Atwood, W.J. Human Polyomavirus Receptor Distribution in Brain Parenchyma Contrasts with Receptor Distribution in Kidney and Choroid Plexus. Am J Pathol 2015, 185, 2246–2258. [Google Scholar] [CrossRef]
- Wilczek, M.P.; DuShane, J.K.; Armstrong, F.J.; Maginnis, M.S. JC Polyomavirus Infection Reveals Delayed Progression of the Infectious Cycle in Normal Human Astrocytes. J Virol 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Mázló, M.; Tariska, I. Are Astrocytes Infected in Progressive Multifocal Leukoencephalopathy (PML)? Acta Neuropathol 1982, 56, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Bernards, R.; Friend, S.H.; Gooding, L.R.; Hassell, J.A.; Major, E.O.; Pipas, J.M.; Vandyke, T.; Harlow, E. Large T Antigens of Many Polyomaviruses Are Able to Form Complexes with the Retinoblastoma Protein. J Virol 1990, 64, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.; Huang, J.; Wu, S.Q.; Hauser, P.; Reznikoff, C.A. Role of SV40 T Antigen Binding to pRB and P53 in Multistep Transformation in Vitro of Human Uroepithelial Cells. Carcinogenesis 1993, 14, 2297–2302. [Google Scholar] [CrossRef]
- Reich, N.C.; Levine, A.J. Specific Interaction of the SV40 T Antigen-Cellular P53 Protein Complex with SV40 DNA. Virology 1982, 117, 286–290. [Google Scholar] [CrossRef]
- Tan, T.H.; Wallis, J.; Levine, A.J. Identification of the P53 Protein Domain Involved in Formation of the Simian Virus 40 Large T-Antigen-P53 Protein Complex. J Virol 1986, 59, 574–583. [Google Scholar] [CrossRef]
- Ferenczy, M.W.; Johnson, K.R.; Marshall, L.J.; Monaco, M.C.; Major, E.O. Differentiation of Human Fetal Multipotential Neural Progenitor Cells to Astrocytes Reveals Susceptibility Factors for JC Virus. J Virol 2013, 87, 6221–6231. [Google Scholar] [CrossRef]
- Tretiakova, A.; Krynska, B.; Gordon, J.; Khalili, K. Human Neurotropic JC Virus Early Protein Deregulates Glial Cell Cycle Pathway and Impairs Cell Differentiation. J Neurosci Res 1999, 55, 588–599. [Google Scholar] [CrossRef]
- Sturrock, R.R. Myelination of the Mouse Corpus Callosum. Neuropathol Appl Neurobiol 1980, 6, 415–420. [Google Scholar] [CrossRef]
- Kirby, L.; Jin, J.; Cardona, J.G.; Smith, M.D.; Martin, K.A.; Wang, J.; Strasburger, H.; Herbst, L.; Alexis, M.; Karnell, J.; et al. Oligodendrocyte Precursor Cells Present Antigen and Are Cytotoxic Targets in Inflammatory Demyelination. Nat Commun 2019, 10, 3887. [Google Scholar] [CrossRef]
- Karimy, J.K.; Zhang, J.; Kurland, D.B.; Theriault, B.C.; Duran, D.; Stokum, J.A.; Furey, C.G.; Zhou, X.; Mansuri, M.S.; Montejo, J.; et al. Inflammation-Dependent Cerebrospinal Fluid Hypersecretion by the Choroid Plexus Epithelium in Posthemorrhagic Hydrocephalus. Nat Med 2017, 23, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.M.; Reeves, B.C.; Kiziltug, E.; Duy, P.Q.; Karimy, J.K.; Mansuri, M.S.; Marlier, A.; Allington, G.; Greenberg, A.B.W.; DeSpenza, T.; et al. The Choroid Plexus Links Innate Immunity to CSF Dysregulation in Hydrocephalus. Cell 2023, 186, 764–785. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Brissette, C.A.; Watt, J.A. The Choroid Plexus and Its Role in the Pathogenesis of Neurological Infections. Fluids Barriers CNS 2022, 19, 75. [Google Scholar] [CrossRef]
- Chakravarty, S.; Herkenham, M. Toll-like Receptor 4 on Nonhematopoietic Cells Sustains CNS Inflammation during Endotoxemia, Independent of Systemic Cytokines. J Neurosci 2005, 25, 1788–1796. [Google Scholar] [CrossRef]
- Laflamme, N.; Rivest, S. Toll-like Receptor 4: The Missing Link of the Cerebral Innate Immune Response Triggered by Circulating Gram-Negative Bacterial Cell Wall Components. FASEB J 2001, 15, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Rivest, S. Molecular Insights on the Cerebral Innate Immune System. Brain Behav Immun 2003, 17, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yamagata, K.; Krukoff, T.L. Differential Expression of the CD14/TLR4 Complex and Inflammatory Signaling Molecules Following i.c.v. Administration of LPS. Brain Res 2006, 1095, 85–95. [Google Scholar] [CrossRef]
- Quintana, E.; Fernández, A.; Velasco, P.; de Andrés, B.; Liste, I.; Sancho, D.; Gaspar, M.L.; Cano, E. DNGR-1(+) Dendritic Cells Are Located in Meningeal Membrane and Choroid Plexus of the Noninjured Brain. Glia 2015, 63, 2231–2248. [Google Scholar] [CrossRef]
- Wang, A.Z.; Bowman-Kirigin, J.A.; Desai, R.; Kang, L.I.; Patel, P.R.; Patel, B.; Khan, S.M.; Bender, D.; Marlin, M.C.; Liu, J.; et al. Single-Cell Profiling of Human Dura and Meningioma Reveals Cellular Meningeal Landscape and Insights into Meningioma Immune Response. Genome Med 2022, 14, 49. [Google Scholar] [CrossRef]
- Thompson, D.; Sorenson, J.; Greenmyer, J.; Brissette, C.A.; Watt, J.A. The Lyme Disease Bacterium, Borrelia Burgdorferi, Stimulates an Inflammatory Response in Human Choroid Plexus Epithelial Cells. PLoS One 2020, 15, e0234993. [Google Scholar] [CrossRef]
- Figueiredo, C.A.; Steffen, J.; Morton, L.; Arumugam, S.; Liesenfeld, O.; Deli, M.A.; Kröger, A.; Schüler, T.; Dunay, I.R. Immune Response and Pathogen Invasion at the Choroid Plexus in the Onset of Cerebral Toxoplasmosis. J Neuroinflammation 2022, 19, 17. [Google Scholar] [CrossRef] [PubMed]
- Banizs, B.; Pike, M.M.; Millican, C.L.; Ferguson, W.B.; Komlosi, P.; Sheetz, J.; Bell, P.D.; Schwiebert, E.M.; Yoder, B.K. Dysfunctional Cilia Lead to Altered Ependyma and Choroid Plexus Function, and Result in the Formation of Hydrocephalus. Development 2005, 132, 5329–5339. [Google Scholar] [CrossRef] [PubMed]
- Medina-Bolívar, C.; González-Arnay, E.; Talos, F.; González-Gómez, M.; Moll, U.M.; Meyer, G. Cortical Hypoplasia and Ventriculomegaly of P73-Deficient Mice: Developmental and Adult Analysis. J Comp Neurol 2014, 522, 2663–2679. [Google Scholar] [CrossRef] [PubMed]
- Tissir, F.; Qu, Y.; Montcouquiol, M.; Zhou, L.; Komatsu, K.; Shi, D.; Fujimori, T.; Labeau, J.; Tyteca, D.; Courtoy, P.; et al. Lack of Cadherins Celsr2 and Celsr3 Impairs Ependymal Ciliogenesis, Leading to Fatal Hydrocephalus. Nat Neurosci 2010, 13, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Wodarczyk, C.; Rowe, I.; Chiaravalli, M.; Pema, M.; Qian, F.; Boletta, A. A Novel Mouse Model Reveals That Polycystin-1 Deficiency in Ependyma and Choroid Plexus Results in Dysfunctional Cilia and Hydrocephalus. PLoS One 2009, 4, e7137. [Google Scholar] [CrossRef]
- Xia, Y.; Yamagata, K.; Krukoff, T.L. Differential Expression of the CD14/TLR4 Complex and Inflammatory Signaling Molecules Following i.c.v. Administration of LPS. Brain Res 2006, 1095, 85–95. [Google Scholar] [CrossRef]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The Glymphatic Pathway in Neurological Disorders. Lancet Neurol 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.J.; Hua, J.; Cao, D.; Ho, M.L. Neurofluids and the Glymphatic System: Anatomy, Physiology, and Imaging. Br J Radiol 2023, 20230016. [Google Scholar] [CrossRef] [PubMed]
- Bystritsky, R.J.; Chow, F.C. Infectious Meningitis and Encephalitis. Neurol Clin 2022, 40, 77–91. [Google Scholar] [CrossRef]
- Gundamraj, V.; Hasbun, R. Viral Meningitis and Encephalitis: An Update. Curr Opin Infect Dis 2023, 36, 177–185. [Google Scholar] [CrossRef]
- Ballesta, B.; González, H.; Martín, V.; Ballesta, J.J. Fatal Ruxolitinib-Related JC Virus Meningitis. J Neurovirol 2017, 23, 783–785. [Google Scholar] [CrossRef] [PubMed]
- Consalez, G.G.; Goldowitz, D.; Casoni, F.; Hawkes, R. Origins, Development, and Compartmentation of the Granule Cells of the Cerebellum. Front Neural Circuits 2020, 14, 611841. [Google Scholar] [CrossRef] [PubMed]
- Du Pasquier, R.A.; Corey, S.; Margolin, D.H.; Williams, K.; Pfister, L.A.; De Girolami, U.; Mac Key, J.J.; Wüthrich, C.; Joseph, J.T.; Koralnik, I.J. Productive Infection of Cerebellar Granule Cell Neurons by JC Virus in an HIV+ Individual. Neurology 2003, 61, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Tagliati, M.; Simpson, D.; Morgello, S.; Clifford, D.; Schwartz, R.L.; Berger, J.R. Cerebellar Degeneration Associated with Human Immunodeficiency Virus Infection. Neurology 1998, 50, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Koralnik, I.J. A Granule Cell Neuron-Associated JC Virus Variant Has a Unique Deletion in the VP1 Gene. J Gen Virol 2006, 87, 2533–2537. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Vidal, J.E.; Penalva de Oliveira, A.C.; Simpson, D.M.; Morgello, S.; Hecht, J.H.; Ngo, L.H.; Koralnik, I.J. JC Virus Granule Cell Neuronopathy Is Associated with VP1 C Terminus Mutants. J Gen Virol 2012, 93, 175–183. [Google Scholar] [CrossRef]
- Janovec, V.; Ryabchenko, B.; Skarkova, A.; Pokorna, K.; Rosel, D.; Brabek, J.; Weber, J.; Forstova, J.; Hirsch, I.; Huerfano, S. TLR4-Mediated Recognition of Mouse Polyomavirus Promotes Cancer-Associated Fibroblast-Like Phenotype and Cell Invasiveness. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Velupillai, P.; Sung, C.K.; Andrews, E.; Moran, J.; Beier, D.; Kagan, J.; Benjamin, T. Polymorphisms in Toll-like Receptor 4 Underlie Susceptibility to Tumor Induction by the Mouse Polyomavirus. J Virol 2012, 86, 11541–11547. [Google Scholar] [CrossRef]
- Kanse, S.; Khandelwal, M.; Pandey, R.K.; Khokhar, M.; Desai, N.; Kumbhar, B.V. Designing a Multi-Epitope Subunit Vaccine against VP1 Major Coat Protein of JC Polyomavirus. Vaccines (Basel) 2023, 11. [Google Scholar] [CrossRef]
- Shahzad, N.; Shuda, M.; Gheit, T.; Kwun, H.J.; Cornet, I.; Saidj, D.; Zannetti, C.; Hasan, U.; Chang, Y.; Moore, P.S.; et al. The T Antigen Locus of Merkel Cell Polyomavirus Downregulates Human Toll-like Receptor 9 Expression. J Virol 2013, 87, 13009–13019. [Google Scholar] [CrossRef]
- Yaghobi, R.; Khodavandi, A.; Alizadeh, F. Association of TLR4 Polymorphisms and Polyomavirus BK Infection in Liver Transplant Patients. Trop Biomed 2017, 34, 886–894. [Google Scholar] [PubMed]
- Redondo, N.; Rodriguez-Goncer, I.; Parra, P.; Lopez-Medrano, F.; Gonzalez, E.; Hernandez, A.; Trujillo, H.; Ruiz-Merlo, T.; San Juan, R.; Folgueira, M.D.; et al. Genetic Polymorphisms in TLR3, IL10 and CD209 Influence the Risk of BK Polyomavirus Infection after Kidney Transplantation. Sci Rep 2022, 12, 11338. [Google Scholar] [CrossRef] [PubMed]
- Ryabchenko, B.; Soldatova, I.; Sroller, V.; Forstova, J.; Huerfano, S. Immune Sensing of Mouse Polyomavirus DNA by P204 and cGAS DNA Sensors. FEBS J 2021, 288, 5964–5985. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Dvorkin, S.; Chiang, J.J.; Potter, R.B.; Gack, M.U. The Small t Antigen of JC Virus Antagonizes RIG-I-Mediated Innate Immunity by Inhibiting TRIM25’s RNA Binding Ability. mBio 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Saenz Robles, M.T.; Duray, A.M.; Cantalupo, P.G.; Pipas, J.M. Human Polyomavirus BKV Infection of Endothelial Cells Results in Interferon Pathway Induction and Persistence. PLoS Pathog 2019, 15, e1007505. [Google Scholar] [CrossRef] [PubMed]
- Heutinck, K.M.; Rowshani, A.T.; Kassies, J.; Claessen, N.; van Donselaar-van der Pant, K.A.; Bemelman, F.J.; Eldering, E.; van Lier, R.A.; Florquin, S.; Ten Berge, I.J.; et al. Viral Double-Stranded RNA Sensors Induce Antiviral, pro-Inflammatory, and pro-Apoptotic Responses in Human Renal Tubular Epithelial Cells. Kidney Int 2012, 82, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Huerfano, S.; Ryabchenko, B.; Forstova, J. Nucleofection of Expression Vectors Induces a Robust Interferon Response and Inhibition of Cell Proliferation. DNA Cell Biol 2013, 32, 467–479. [Google Scholar] [CrossRef]
- Jiang, Z.; Mak, T.W.; Sen, G.; Li, X. Toll-like Receptor 3-Mediated Activation of NF-kappaB and IRF3 Diverges at Toll-IL-1 Receptor Domain-Containing Adapter Inducing IFN-Beta. Proc Natl Acad Sci U S A 2004, 101, 3533–3538. [Google Scholar] [CrossRef]
- Chen, H.C.; Zhan, X.; Tran, K.K.; Shen, H. Selectively Targeting the Toll-like Receptor 9 (TLR9)--IRF 7 Signaling Pathway by Polymer Blend Particles. Biomaterials 2013, 34, 6464–6472. [Google Scholar] [CrossRef]
- May, D.; Bellizzi, A.; Kassa, W.; Cipriaso, J.M.; Caocci, M.; Wollebo, H.S. IFNalpha and Beta Mediated JCPyV Suppression through C/EBPbeta-LIP Isoform. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Zerbe, C.S.; Marciano, B.E.; Katial, R.K.; Santos, C.B.; Adamo, N.; Hsu, A.P.; Hanks, M.E.; Darnell, D.N.; Quezado, M.M.; Frein, C.; et al. Progressive Multifocal Leukoencephalopathy in Primary Immune Deficiencies: Stat1 Gain of Function and Review of the Literature. Clin Infect Dis 2016, 62, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Assetta, B.; De Cecco, M.; O’Hara, B.; Atwood, W.J. JC Polyomavirus Infection of Primary Human Renal Epithelial Cells Is Controlled by a Type I IFN-Induced Response. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.J.; Lin, E.; Pack, C.D.; Frost, E.L.; Hadley, A.; Swimm, A.I.; Wang, J.; Dong, Y.; Breeden, C.P.; Kalman, D.; et al. Gamma Interferon Controls Mouse Polyomavirus Infection in Vivo. J Virol 2011, 85, 10126–10134. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Lei, K.J.; Jin, W.; Greenwell-Wild, T.; Wahl, S.M. Induction of APOBEC3 Family Proteins, a Defensive Maneuver Underlying Interferon-Induced Anti–HIV-1 Activity. J Exp Med 2006, 203, 41–46. [Google Scholar] [CrossRef]
- Okeoma, C.M.; Low, A.; Bailis, W.; Fan, H.Y.; Peterlin, B.M.; Ross, S.R. Induction of APOBEC3 in Vivo Causes Increased Restriction of Retrovirus Infection. J Virol 2009, 83, 3486–3495. [Google Scholar] [CrossRef]
- Sadeghpour, S.; Khodaee, S.; Rahnama, M.; Rahimi, H.; Ebrahimi, D. Human APOBEC3 Variations and Viral Infection. Viruses 2021, 13, 1366. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a Human Gene That Inhibits HIV-1 Infection and Is Suppressed by the Viral Vif Protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Poulain, F.; Lejeune, N.; Willemart, K.; Gillet, N.A. Footprint of the Host Restriction Factors APOBEC3 on the Genome of Human Viruses. PLoS Pathog 2020, 16, e1008718. [Google Scholar] [CrossRef]
- Peretti, A.; Geoghegan, E.M.; Pastrana, D.V.; Smola, S.; Feld, P.; Sauter, M.; Lohse, S.; Ramesh, M.; Lim, E.S.; Wang, D.; et al. Characterization of BK Polyomaviruses from Kidney Transplant Recipients Suggests a Role for APOBEC3 in Driving In-Host Virus Evolution. Cell Host Microbe 2018, 23, 628–635. [Google Scholar] [CrossRef]
- Baker, S.C.; Mason, A.S.; Slip, R.G.; Skinner, K.T.; Macdonald, A.; Masood, O.; Harris, R.S.; Fenton, T.R.; Periyasamy, M.; Ali, S.; et al. Induction of APOBEC3-Mediated Genomic Damage in Urothelium Implicates BK Polyomavirus (BKPyV) as a Hit-and-Run Driver for Bladder Cancer. Oncogene 2022, 41, 2139–2151. [Google Scholar] [CrossRef]
- Jelcic, I.; Jelcic, I.; Kempf, C.; Largey, F.; Planas, R.; Schippling, S.; Budka, H.; Sospedra, M.; Martin, R. Mechanisms of Immune Escape in Central Nervous System Infection with Neurotropic JC Virus Variant. Ann Neurol 2016, 79, 404–418. [Google Scholar] [CrossRef] [PubMed]
- Dunham, S.R.; Schmidt, R.; Clifford, D.B. Treatment of Progressive Multifocal Leukoencephalopathy Using Immune Restoration. Neurotherapeutics 2020, 17, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Gheuens, S.; Bord, E.; Kesari, S.; Simpson, D.M.; Gandhi, R.T.; Clifford, D.B.; Berger, J.R.; Ngo, L.; Koralnik, I.J. Role of CD4+ and CD8+ T-Cell Responses against JC Virus in the Outcome of Patients with Progressive Multifocal Leukoencephalopathy (PML) and PML with Immune Reconstitution Inflammatory Syndrome ▿. J Virol 2011, 85, 7256–7263. [Google Scholar] [CrossRef] [PubMed]
- Du Pasquier, R.A.; Kuroda, M.J.; Zheng, Y.; Jean-Jacques, J.; Letvin, N.L.; Koralnik, I.J. A Prospective Study Demonstrates an Association between JC Virus-Specific Cytotoxic T Lymphocytes and the Early Control of Progressive Multifocal Leukoencephalopathy. Brain 2004, 127, 1970–1978. [Google Scholar] [CrossRef]
- Wüthrich, C.; Kesari, S.; Kim, W.-K.; Williams, K.; Gelman, R.; Elmeric, D.; De Girolami, U.; Joseph, J.T.; Hedley-Whyte, T.; Koralnik, I.J. Characterization of Lymphocytic Infiltrates in Progressive Multifocal Leukoencephalopathy: Co-Localization of CD8(+) T Cells with JCV-Infected Glial Cells. J Neurovirol 2006, 12, 116–128. [Google Scholar] [CrossRef]
- Martin-Blondel, G.; Bauer, J.; Cuvinciuc, V.; Uro-Coste, E.; Debard, A.; Massip, P.; Delisle, M.-B.; Lassmann, H.; Marchou, B.; Mars, L.T.; et al. In Situ Evidence of JC Virus Control by CD8+ T Cells in PML-IRIS during HIV Infection. Neurology 2013, 81, 964–970. [Google Scholar] [CrossRef]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.-Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. T Cell Memory. Skin-Resident Memory CD8+ T Cells Trigger a State of Tissue-Wide Pathogen Alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Masopust, D. Tissue-Resident Memory T Cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef]
- Abdelbary, M.; Hobbs, S.J.; Gibbs, J.S.; Yewdell, J.W.; Nolz, J.C. T Cell Receptor Signaling Strength Establishes the Chemotactic Properties of Effector CD8. Nat Commun 2023, 14, 3928. [Google Scholar] [CrossRef]
- Evrard, M.; Becht, E.; Fonseca, R.; Obers, A.; Park, S.L.; Ghabdan-Zanluqui, N.; Schroeder, J.; Christo, S.N.; Schienstock, D.; Lai, J.; et al. Single-Cell Protein Expression Profiling Resolves Circulating and Resident Memory T Cell Diversity across Tissues and Infection Contexts. Immunity 2023, 56, 1664–1680. [Google Scholar] [CrossRef]
- Wein, A.N.; McMaster, S.R.; Takamura, S.; Dunbar, P.R.; Cartwright, E.K.; Hayward, S.L.; McManus, D.T.; Shimaoka, T.; Ueha, S.; Tsukui, T.; et al. CXCR6 Regulates Localization of Tissue-Resident Memory CD8 T Cells to the Airways. J Exp Med 2019, 216, 2748–2762. [Google Scholar] [CrossRef] [PubMed]
- Heim, T.A.; Lin, Z.; Steele, M.M.; Mudianto, T.; Lund, A.W. CXCR6 Promotes Dermal CD8. bioRxiv 2023. [Google Scholar] [CrossRef]
- Zaid, A.; Hor, J.L.; Christo, S.N.; Groom, J.R.; Heath, W.R.; Mackay, L.K.; Mueller, S.N. Chemokine Receptor-Dependent Control of Skin Tissue-Resident Memory T Cell Formation. J Immunol 2017, 199, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Tse, S.W.; Radtke, A.J.; Espinosa, D.A.; Cockburn, I.A.; Zavala, F. The Chemokine Receptor CXCR6 Is Required for the Maintenance of Liver Memory CD8+ T Cells Specific for Infectious Pathogens. J Infect Dis 2014, 210, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Rosen, S.F.; Soung, A.L.; Yang, W.; Ai, S.; Kanmogne, M.; Davé, V.A.; Artyomov, M.; Magee, J.A.; Klein, R.S. Single-Cell RNA Transcriptome Analysis of CNS Immune Cells Reveals CXCL16/CXCR6 as Maintenance Factors for Tissue-Resident T Cells That Drive Synapse Elimination. Genome Med 2022, 14, 108. [Google Scholar] [CrossRef]
- Su, W.; Saravia, J.; Risch, I.; Rankin, S.; Guy, C.; Chapman, N.M.; Shi, H.; Sun, Y.; Kc, A.; Li, W.; et al. CXCR6 Orchestrates Brain CD8+ T Cell Residency and Limits Mouse Alzheimer’s Disease Pathology. Nat Immunol 2023, 24, 1735–1747. [Google Scholar] [CrossRef]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From Activation Marker to Metabolic Gatekeeper. Eur J Immunol 2017, 47, 946–953. [Google Scholar] [CrossRef]
- Szabo, P.A.; Miron, M.; Farber, D.L. Location, Location, Location: Tissue Resident Memory T Cells in Mice and Humans. Sci. Immunol. 2019, 4, eaas9673. [Google Scholar] [CrossRef]
- Shwetank, *!!! REPLACE !!!*; Frost, E.L.; Mockus, T.E.; Ren, H.M.; Toprak, M.; Lauver, M.D.; Netherby-Winslow, C.S.; Jin, G.; Cosby, J.M.; Evavold, B.D.; et al. PD-1 Dynamically Regulates Inflammation and Development of Brain-Resident Memory CD8 T Cells During Persistent Viral Encephalitis. Front Immunol 2019, 10, 783. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, K.K.W. Glial Fibrillary Acidic Protein: From Intermediate Filament Assembly and Gliosis to Neurobiomarker. Trends Neurosci 2015, 38, 364–374. [Google Scholar] [CrossRef]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-Specific Localisation of a Novel Calcium Binding Protein, Iba1. Molecular Brain Research 1998, 57, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pawlitzki, M.; Schneider-Hohendorf, T.; Rolfes, L.; Meuth, S.G.; Wiendl, H.; Schwab, N.; Grauer, O.M. Ineffective Treatment of PML with Pembrolizumab: Exhausted Memory T-Cell Subsets as a Clue? Neurol Neuroimmunol Neuroinflamm 2019, 6, e627. [Google Scholar] [CrossRef] [PubMed]
- Vidya Vijayan, K.K.; Karthigeyan, K.P.; Tripathi, S.P.; Hanna, L.E. Pathophysiology of CD4+ T-Cell Depletion in HIV-1 and HIV-2 Infections. Front. Immunol. 2017, 8, 580. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The Multifaceted Role of CD4+ T Cells in CD8+ T Cell Memory. Nat Rev Immunol 2016, 16, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Elsaesser, H.; Sauer, K.; Brooks, D.G. IL-21 Is Required to Control Chronic Viral Infection. Science 2009, 324, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, A.; Kisielow, J.; Schmitz, I.; Freigang, S.; Shamshiev, A.T.; Weber, J.; Marsland, B.J.; Oxenius, A.; Kopf, M. IL-21R on T Cells Is Critical for Sustained Functionality and Control of Chronic Viral Infection. Science 2009, 324, 1576–1580. [Google Scholar] [CrossRef]
- Ren, H.M.; Kolawole, E.M.; Ren, M.; Jin, G.; Netherby-Winslow, C.S.; Wade, Q.; Shwetank, *!!! REPLACE !!!*; Rahman, Z.S.M.; Evavold, B.D.; Lukacher, A.E.C.-T.; et al. IL-21 from High-Affinity CD4 T Cells Drives Differentiation of Brain-Resident CD8 T Cells during Persistent Viral Infection. Sci Immunol 2020, 5. [Google Scholar] [CrossRef]
- Bennett, C.L.; Focosi, D.; Socal, M.P.; Bian, J.C.; Nabhan, C.; Hrushesky, W.J.; Bennett, A.C.; Schoen, M.W.; Berger, J.R.; Armitage, J.O. Progressive Multifocal Leukoencephalopathy in Patients Treated with Rituximab: A 20-Year Review from the Southern Network on Adverse Reactions. The Lancet Haematology 2021, 8, e593–e604. [Google Scholar] [CrossRef]
- Solis, M.; Guffroy, A.; Lersy, F.; Soulier, E.; Gallais, F.; Renaud, M.; Douiri, N.; Argemi, X.; Hansmann, Y.; De Sèze, J.; et al. Inadequate Immune Humoral Response against JC Virus in Progressive Multifocal Leukoencephalopathy Non-Survivors. Viruses 2020, 12, 1380. [Google Scholar] [CrossRef]
- Bertoli, D.; Sottini, A.; Capra, R.; Scarpazza, C.; Bresciani, R.; Notarangelo, L.D.; Imberti, L. Lack of Specific T- and B-Cell Clonal Expansions in Multiple Sclerosis Patients with Progressive Multifocal Leukoencephalopathy. Sci Rep 2019, 9, 16605. [Google Scholar] [CrossRef]
- Lauver, M.D.; Goetschius, D.J.; Netherby-Winslow, C.S.; Ayers, K.N.; Jin, G.; Haas, D.G.; Frost, E.L.; Cho, S.H.; Bator, C.M.; Bywaters, S.M.; et al. Antibody Escape by Polyomavirus Capsid Mutation Facilitates Neurovirulence. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Jelcic, I.; Combaluzier, B.; Jelcic, I.; Faigle, W.; Senn, L.; Reinhart, B.J.; Ströh, L.; Nitsch, R.M.; Stehle, T.; Sospedra, M.; et al. Broadly Neutralizing Human Monoclonal JC Polyomavirus VP1–Specific Antibodies as Candidate Therapeutics for Progressive Multifocal Leukoencephalopathy. Sci Transl Med 2015, 7, 306ra150. [Google Scholar] [CrossRef] [PubMed]
- Ray, U.; Cinque, P.; Gerevini, S.; Longo, V.; Lazzarin, A.; Schippling, S.; Martin, R.; Buck, C.B.; Pastrana, D.V. JC Polyomavirus Mutants Escape Antibody-Mediated Neutralization. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Hypothesized infection routes through the three barriers of the CNS: the BBB, the ChP-CSF-ependyma barrier, and the meninges. Generally, for all three barriers, infection may occur directly via free virions or through indirect means like EVs or infected leukocytes. Made in BioRender.
Figure 2.
Hypothesized infection routes through the three barriers of the CNS: the BBB, the ChP-CSF-ependyma barrier, and the meninges. Generally, for all three barriers, infection may occur directly via free virions or through indirect means like EVs or infected leukocytes. Made in BioRender.
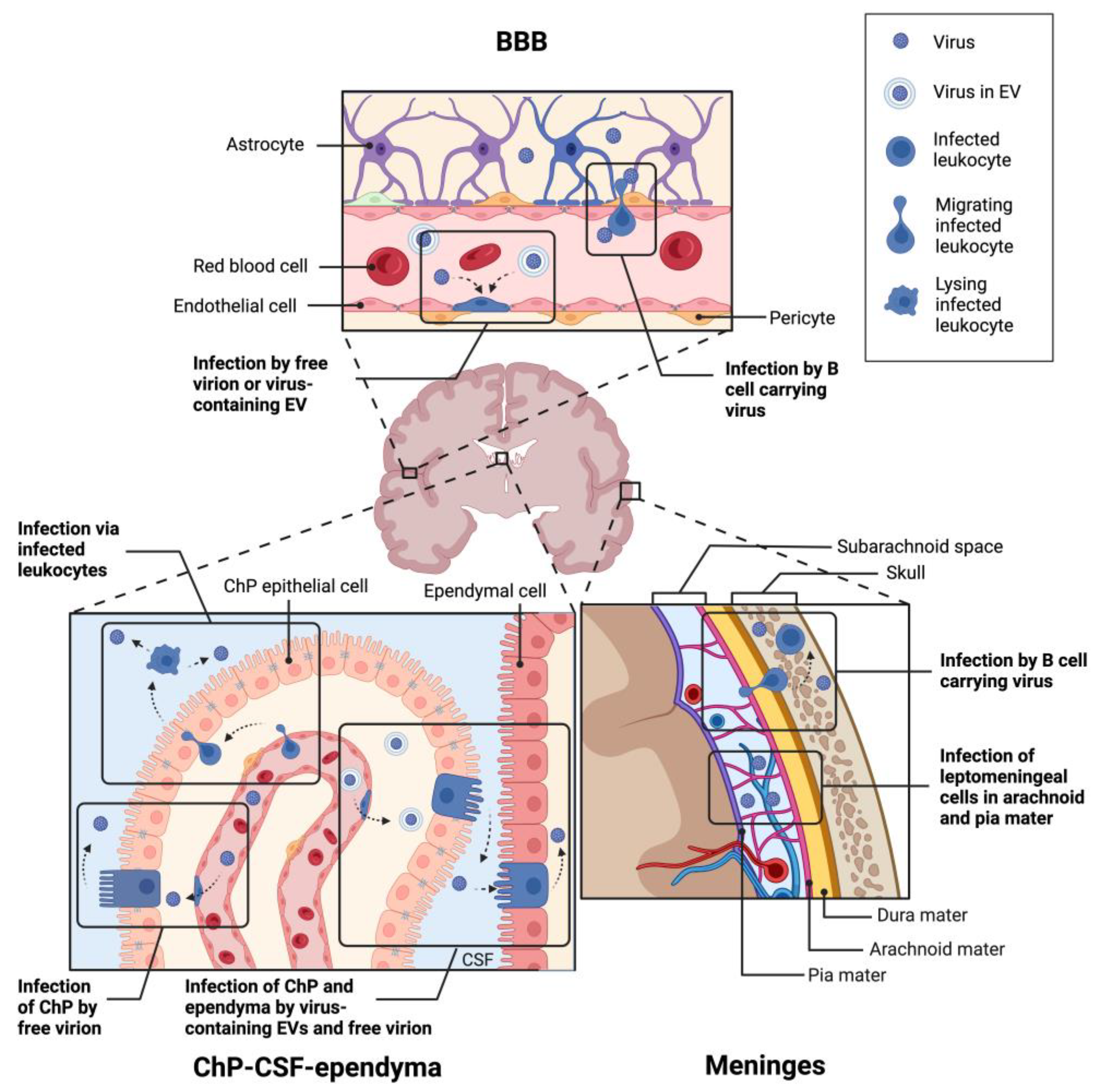
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



