
Submitted:
02 October 2023
Posted:
03 October 2023
You are already at the latest version
Abstract
Fibrosis is the end result of persistent inflammatory responses induced by a variety of stimuli, including chronic infections, autoimmune reactions, and tissue injury. Fibrotic diseases affect all vital organs and are characterized by a high rate of morbidity and mortality in the developed world, although currently there are no approved antifibrotic therapies. In recent years, high levels of interleukin-17 (IL-17) have been associated with chronic inflammatory diseases with fibrotic complications, that culminate in organ failure. In this review, we provide an update on the role of IL-17 in fibrotic diseases, with particular attention to the most recent lines of research in the therapeutic field represented by the epigenetic mechanisms that control IL-17 levels in fibrosis. A better knowledge of the IL-17 signalling pathway implications in fibrosis could design new strategies for therapeutic benefits.
Keywords:
IL-17
; fibrosis
; autoimmune
; epigenetics
; biological drugs.
1. Introduction
Fibrosis is a process that develops slowly and leads to tissue degeneration, with severe consequences for organs such as the heart, lung, liver, kidney, and skin [1]. In the last few years, fibrotic disorders have significantly increased and negatively impact public health [2]. It is estimated that in the industrialized world, 45% of all deaths can be attributed to diseases where fibrosis plays a major etiological role [2,3]. Interestingly, in pathological disorders that are based on inflammatory processes, altered repair mechanisms can lead to the formation of fibrotic tissue upon wound healing [4] that may be responsible for aberrant tissue repair [1]. During the fibrotic process, an excessive accumulation of extracellular matrix (ECM) components occurs; collagen, fibronectin, and hyaluronic acid are released and synthesized to a greater level at the site of tissue injury, leading to organ failure and death [5].
In recent years, growing evidence has highlighted that aberrant fibrosis is also a major pathological feature of many chronic autoimmune diseases, including scleroderma, rheumatoid arthritis (RA), Crohn’s disease, systemic lupus erythematosus (SLE), and Sjӧgren’s syndrome (SS) [6,7]. These fibrotic diseases have, in common, a persistent inflammatory stimulus based on lymphocyte-monocyte interactions that produce growth factors and fibrogenic cytokines, inducing the deposition of connective tissue components that progressively destroy the healthy tissue structure [6,7]. These data confirmed that cytokines drive the acute and chronic inflammatory responses that culminate in fibrosis activation [8]. Recently, IL-17, a pro-inflammatory cytokine, has received growing attention derived from published results collected from the study of the correlation between inflammation and autoimmune diseases [9,10,11]; based on this evidence, herein we review recent discoveries on the role of the members of the IL-17 family in the crucial events of organ fibrosis.
2. The IL-17 cytokines family and its receptors
The IL-17 family is a recently identified system of secretory regulatory peptides that show homology in amino acid sequences, including an extremely preserved cysteine-knot fold structure [12,13]. IL-17A was first identified in 1993 [14] and named human cytotoxic T lymphocyte-associated antigen 8 (CTLA8). It was subsequently termed IL-17 in 1995 [15] and, more recently, IL-17A. The IL-17A gene is inserted on the 6p12 chromosome, and human IL-17A is a homodimeric protein of 35-kDa, that shares a different glycosylation [15,16]. Five members of the IL-17 family were known: IL-17B, IL-17C, IL-17D, IL-17E (also named IL-25), and IL-17F [16,17,18]. IL-17A and F are the closest members, with 50% homology, followed by IL-17B (29%), IL-17D (25%), and IL-17C (23%); IL-17E displays the lowest degree of sequence conservation (16%), implicating that IL-17E is the most dissimilar protein [19,20]. The functions of these five proteins moderately overlap with those of IL-17A, whose precise role in health and disease remains elusive. IL-17, through the binding to the IL-17 receptors (IL-17Rs), is involved in chronic and persistent inflammation, autoimmunity, and the maintenance of epithelial layer integrity [21]. These receptors share a unique protein-protein interaction domain in their cytoplasmic tail called the SEF/IL-17R (SEFIR) domain [22]. Among the IL-17R family members, IL-17RA, is the best-known receptor [23,24,25]. The IL-17R is a heterodimeric complex that is formed by the IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE, with the IL-17RA subunit in association with other subunits. IL-17A and IL-17F link to a dimeric IL17RA/RC system; IL-17B and IL-17E link to a dimeric 17RA/RB system; and IL-17C links to the IL17RA/RE complex [26]. In addition, IL-17D was recently reported to bind CD93 [27]. IL-17RA and IL-17RC, acting through the binding of the SEFIR domain with the ubiquitin ligase Act1 activate the TRAF6/TAK1/NF-κB pathway and the TRAF6/TAK1/MAPK/AP1 pathway [28]. Because IL-17RB, IL-17RD, and IL-17RE also contain a SEFIR domain, a similar mechanism of activation is probable. Moreover, a conserved intracellular subdomain homologous to Toll-IL-1R (TIR) domains was revealed, that could be essential for signalling downstream of the IL-1 receptor and Toll-like receptors (TLRs) [24,29].
3. Production and functions of the IL-17 family members
Functionally, IL-17 cytokines, critical for normal host immune responses, are potent drivers of inflammatory responses and cancer. Both in humans and in mice, IL-17 cytokines are produced by a broad spectrum of cell types that operate on multiple cellular targets [28,30,31], triggering the secretion of pro-inflammatory cytokines, chemokines, and prostaglandins [28,32]. In this context, IL-17A has been implicated in the pathogenesis of many disorders characterized by inflammatory complications, including cardiovascular and neurological diseases [33]. IL-17A is produced by CD4+ and CD8+ T cells, γδ T cells and it acts on endothelial cells, macrophages, fibroblasts, osteoblasts, and chondrocytes [33]. This stimulation by IL-17A enhances the production of pro-inflammatory proteins from monocytes, such as TNF-α, IL-6, IL-1β and IL-23. IL-17A also operates on mesenchymal cells derived from synovium and skin to stimulate the production of chemokines thus involving neutrophil (IL-8/CXCL8), lymphocyte (CCL20), and macrophage recruitment [25]. However, if dysregulated, IL-17A responses can promote the development and chronicity of inflammatory disorders in a number of autoimmune diseases [34,35]. IL-17B was demonstrated to be highly expressed during the intestinal inflammatory state and is able to induce neutrophil migration upon intraperitoneal administration, suggesting a pro-inflammatory key role [18,36,37]. Interestingly, enhanced IL-17B levels are linked with rare events in patients with various types of cancer, like breast, lung, and pancreatic [38]. IL-17B expression was also detected in synovial tissues from patients with RA, where it is mainly produced by neutrophils and chondrocytes [39]. Likewise, IL-17A, IL-17C, produced by epithelial cells and immune cells promote anti-microbial protective activity in the skin and in the intestine [28,40,41]. In addition, IL-17C is also secreted by keratinocytes and cutaneous neurons, but in a specific condition, represented by the reactivation of the herpes simplex virus [42]. IL-17D is the least understood of the IL-17 family of proteins. It is expressed in a broad variety of healthy tissues and has been found to be expressed at high levels in immunogenic cancer cells compared to poorly immunogenic tumour cells, leading to immune rejection mediated by NK cells. Therefore, it is well documented that IL-17D provokes exacerbated viral infections [43,44]. Some interesting studies have highlighted that IL-17D stimulating the endothelial cells promotes severe pro-inflammatory cytokine activity that leads to IL-6, IL-8, and GM-CSF secretion [43,45].IL-17E, also known as IL-25, is involved in the pathogenesis of fungal infections, allergies, and autoimmune disorders. IL-17E is diverse from other proteins of the IL-17 family; in fact, it was considered a “mucosal barrier” molecule that confers immunity against parasitic infections. Indeed, large levels of IL-17E are secreted following infection with the parasitic helminth Nippostrongylus or Aspergillus [28]. Therefore, IL-17E induces expression of IL-4, IL-5, and IL-13, all of which are associated with type 2 immunity [46], and is able to promote epithelial-cell hyperplasia by increasing mucus secretion and hyperreactivity of the airway epithelium [47].
4. Role of IL-17 in fibrotic evolution
All fibrotic tissues exhibit characteristics of chronic immunologically-mediated inflammatory status during the initial periods of their development. IL-17 is expressed in an altered manner, in several autoinflammatory diseases. Indeed, in inflammatory chronic conditions such as liver cirrhosis, idiopathic pulmonary fibrosis, and heart failure, IL-17 contributes to the severe fibrotic process through various mechanisms, including the induction of resident stromal cells and the progression of the inflammatory status. In liver fibrosis, IL-17, operating in synergy with IL-1β, IL-6, and IL-23, continues the inflammatory process by promoting transforming growth factor-β (TGFβ) expression in hepatic stellate cells [48,49]. IL-17 stimulates the hepatocytes, involved in fibroblast activation and collagen release, to secrete periostin [50]. Accordingly, inactivation of IL-17 signalling in hepatocytes diminished the fibrotic process in the liver of murine models affected by hepatitis-induced liver injury [51]. Therefore, IL-17 is markedly expressed in the bronchial mucosa of patients affected by severe asthma and drives the EMT process when used to induce human small airway epithelial cells in in vitro cultures [52]. Supporting the involvement of IL-17 in EMT-dependent fibrosis, Sisto et al. recently demonstrated that IL-17, through the support of IL-22, contributes to trigger the EMT-dependent fibrotic process in healthy human salivary gland epithelial cells, clarifying the role of IL-17 in the fibrotic evolution observed in SS [53]. It is interesting to note that, in patients with idiopathic pulmonary fibrosis (IPF), Th17 cells secrete TGFβ and IL-17A at high levels; in addition, when lung fibrosis was induced in vitro through bleomycin (BLM) treatment of a murine model or when Th17 cells were cultured simultaneously in the presence of human lung fibroblasts, an increase in collagen deposition and others ECM factors production was revealed [54]. Indeed, blocking IL-17 improves the fibrotic condition in the lung of murine models affected by pulmonary disease following a post-bone marrow transplant [54]. Finally, IL-17 secreted by γδ T cells and Th17 cells plays a key role in several conditions of heart fibrosis, probably involving various inflammatory events in these organs. In fact, IL-17 induces cardiac myofibroblast transformation in murine models, in which ischemia causes heart injury, as well as in experimental models of hypertension [55]. Blockade of the IL-17 signalling pathway reduces cardiac fibrosis and improves myocardial contractile function [56]. A schematic representation of the role of IL-17 in autoimmune-related fibrosis is reported in Figure 1.
5. Fibrosis mediated by several IL-17 family members
Dysregulated expression of IL-17 cytokines contributes to the triggering and exacerbation of fibrosis in a variable way, depending on the specific IL-17 family member. In this paragraph, we report the most recent knowledge regarding the role of IL-17 family members in the fibrotic evolution of inflammatory diseases.
5.1. IL-17A
Studies conducted using experimental animal models have demonstrated the role of IL-17A in regulating the complex interplay between lung inflammation and fibrosis. After BLM treatment to induce injury, IL-17A expression is upregulated, determining the release of pro-inflammatory cytokines and chemokines by endothelial cells and epithelial cells [54,57]. These factors recruit several types of inflammatory cells to the alveolar surface, and the resulting inflammation activates pulmonary fibrosis [58]. In addition, the same situations of inflammation, neutrophilia, and pulmonary fibrosis develop after IL-17A production following IL-1β treatment [54]. Confirming the pro-fibrotic role of IL-17A, blocking IL-17A through the intraperitoneal administration of an antibody against IL-17A reduces the acute inflammatory and fibrotic features in an experimental animal model [58]. Consequently, it is not surprising that the depletion of alveolar macrophages decreased the effects of IL-17 on the activation of lung fibrosis, supporting the hypothesis that these cells are involved first in the activation, producing pro-fibrotic factors such as of IL-1β and IL-23 [59]. The IL-17A receptor is also ubiquitously expressed on the membrane surface of epithelial cells and fibroblasts; these cells are involved in the EMT-mediated pulmonary process correlated with pulmonary fibrosis; furthermore, these cells regulate fibroblasts transformation into myofibroblasts, increasing extracellular matrix deposition [60]. Recently, it has been proposed that the IL-17A-mediated signalling and EMT of intrahepatic biliary epithelial cells are involved in the pathogenesis of primary biliary cirrhosis (PBC). The study demonstrated increased protein levels of the IL-17A receptor in intrahepatic biliary epithelial cells, and the IL-17A resulted accumulated around to these cells in the patients affected by PBC [48].
Additionally, IL-17A can act as a pro-fibrotic interleukin by suppressing autophagy in epithelial cells [59], although whether autophagy has a protective effect or not is yet to be determined. In addition, in some studies, after BLM treatment, an overexpression of IL-17R in fibroblasts was detected; furthermore, the addition of exogenous IL-17 can accelerate fibroblast proliferation, accompanied by an increased synthesis of specific proteins such as α-smooth muscle actin (α-SMA) and collagen [60]. The IL-17 stimulation of fibroblasts occurs, probably, via activation of NF-κB through the NF-κB activator 1 protein (Act1) [61], a critical mediator of IL-17 receptor family signalling, especially in autoimmune conditions [62].
5.2. IL-17B
Research on IL-17B’s role in fibrotic evolution has been limited, and in general, the function of IL-17B has not been thoroughly clarified. However, several studies support the possibility that the effects of IL-17A and IL-17B are very similar in the regulation of inflammation and fibrosis [63]. For example, IL-17B up-regulates the production of IL-6, IL-23, and IL-1α in the peritoneal neutrophils, macrophages and lymphocytes [61]; in addition, it determines TNF-α and IL-1β release by the human monocyte/macrophage cell line [64]. IL-17B promotes the recruitment of cells that express the chemokine receptors CXCR4 or CXCR5, and the experimental intraperitoneal administration of recombinant human IL-17B determines the chemoattraction of neutrophils, which release chemoattractants for other cells [65]. Additionally, IL-17B can synergize with IL-33 to regulate the T helper (Th)-mediated immune responses [66]. These pro-inflammatory functions suggest that IL-17B may influence the progression of fibrosis, which follows the early stages of inflammation. This hypothesis was confirmed by the research group of Yang, who recently reported that the expression of IL-17B was affected by dysbiosis; this situation induces lung fibrosis by interacting with TNF-α to stimulate the secretion of Th17-cell-promoting genes and neutrophil-recruiting genes [67].
5.3. IL-17C
IL-17C has been shown to be expressed in CD4+ T cells, dendritic cells (DCs), macrophages, and epithelial cells, which produce this interleukin during antimicrobial activity [68,69], determining an enhanced inflammatory response [68,69]. The activity of IL-17C occurs through binding to the IL-17 receptor complex, consisting of IL-17RA and IL-17RE subunits [70]. IL-17RE, in particular, was expressed mainly on epithelial cell surfaces and Th17 cells. Th17 cells react to stimulation with IL-17C, producing IL-17A and IL-17F, confirming that IL-17C might regulate the initial phase of the development of inflammation [68,69]. Once again, IL-17C production is dependent on NF-κB/Act1 activation, determined by IL-17C binding to the receptor complex IL-17RA/IL-17RE and, in turn, MAPK signalling molecules expression [70,71]. The function of the IL-17C isoform in the progression of fibrosis was mainly explored in IPF [72], using a lipopolysaccharide-induced lung injury as model of IPF. Data collected on epithelial cell damage, release of pro-inflammatory factors, and neutrophil recruitment mediated by the release of IL-17C confirmed the key role of IL-17C in lung inflammation. IL-17C’s role in Haemophilus influenzae and cigarette smoke-induced lung inflammation has been recently reported [73], mediating the expression of neutrophilic cytokines, the recruitment of neutrophils, and lung fibrotic damage [73].
5.4. IL-17D
IL-17D has a more limited expression and is detected in B lymphocytes and resting CD4+ T cells; IL-17D acts, in particular, on endothelial cells, regulating their secretion of pro-inflammatory factors [74]. However, knowledge of IL-17D’s role in the exacerbation of pulmonary fibrosis remains poorly investigated and needs clarifying studies.
5.5. IL-17E
IL-17E, recently correlated with pulmonary fibrosis, was secreted by Th2 cells, epithelial cells, endothelial cells, T cells, alveolar macrophages, DCs, eosinophils, and basophils [75]. Xu and collaborators [76] not only demonstrated an increase in IL-17E secretion, but also the activation of an EMT program in alveolar epithelial cells, determining EMT-dependent fibrosis activation in patients with IPF. These observations were confirmed by Hams [77] through the demonstration of increased levels of IL-17E in the lungs of IPF patients, which is correlated, interestingly, with IL-13 release that exacerbates collagen deposition during the IPF process. A schematic representation of the correlation of IL-17 subtypes A and E and EMT-dependent fibrosis was shown in Figure 2.
5.6. IL-17F
IL-17F shows a high homology of sequence and activities that mostly overlap with those of IL-17A [78]. The cells that produce IL-17F are the same of those releasing IL-17A. The IL-17F is able to trigger IL-6 and CXC chemokines release from tracheal epithelial cells, inflammatory cells, endothelial cells, and fibroblasts, supporting the hypothesis that IL-17F could modulate autoimmune and inflammatory diseases [79,80]. However, the field of investigation into the pro-fibrotic activity of IL-17F is still pioneering.
6. Epigenetic regulation of IL-17 in fibrotic diseases
Dysregulated Th17 cell responses contribute to the immunopathogenesis of multiple inflammatory and autoimmune diseases [81]. Following aberrant activation stimuli, T helpers appear to be involved in triggering autoimmune responses against many organs, such as joints, brain, skin, gut, pancreas, salivary and lachrymal glands, and the eye. This abnormal and persistent activation leads to the onset of multiple autoimmune diseases, including multiple sclerosis (MS), psoriasis, Crӧhn’s disease, type I diabetes, SS, uveitis, systemic sclerosis (SSc), and SLE [82]. It is now accepted that the fibrotic evolution of autoimmune diseases presents an excessive release of pro-fibrotic factors as a result of the activation of molecular cascades depending on chronic inflammation, and the recent challenge consists of identifying anti-fibrotic therapies that can also have value in autoimmune diseases. Previous research has significantly increased our understanding of genetic susceptibility to fibrotic diseases based on the identification of sequence variants, polymorphisms, and mutations in several genes [83,84]. However, the concordance rate for some fibrotic diseases in monozygotic twins is low, indicating that genetic predisposition is insufficient to explain disease development and suggesting a potential role of epigenetics as the missing link that connects environmental exposure to disease development [85]. The following paragraphs explore findings related to the epigenetic regulation of IL-17 in fibrosis.
6.1. Histone modification and IL-17 production
As an epigenetically modulated mechanism, differentiation of T helper cells was acknowledged to imply total changes in histone modifications such as H3K4me3 and H3K27me3 [86]. Recently, it was reported that the histone deacetylase (HDAC) inhibitor, which has potential effects on epigenetic alterations, had been shown to mitigate renal fibrotic conditions. Findings have observed the conversion of CD4+ forkhead box P3 (FOXP3)+ T regulatory (Treg) cells in T helper 17 cells (Th17), contributing to the progression of renal fibrosis. Worsening renal fibrosis was linked with the loss of CD4+FOXP3+IL-17+ T cells in splenic single-cell suspensions. FOXP3+IL-17+ T cells expressed TGF-β1 both in vitro and in vivo, and, indeed, the loss of TGF-β1 expression was confirmed using IL-17 siRNA. It is now well established that these cells play a critical role in converting Tregs into IL-17- and TGF-β1-secreting cells [87]. Therefore, targeting the epigenetic process that induces the pathogenic activation of CD4+ T helper cells may provide new therapeutic approaches to revolutionize the treatment of autoimmune conditions. Interestingly, removing acetyl groups by histone could occur through a series of HDACs that induce compact nucleosome structure and prevent active transcription; however, in some events, HDACs can directly activate transcription, although the exact processes by which they modulate transcription are currently poorly known [88]. In addition, HDACs seem to modulate the fibrotic process through fibroblast proliferation, senescence, and ECM production [89,90]. Based on this evidence, recent studies report that histone HDAC inhibitors can decrease the inflammatory status mediated by CD4+ T cells and subsequent fibrotic evolution [91,92] (Figure 3).
6.2. DNA demethylation in the control of IL-17 pro-fibrotic activity
DNA demethylation, generally linked with gene silencing, has recently been identified as a strategy in the regulation of IL-17-dependent fibrosis. Ichiyama et al. demonstrated DNA methylation at T helper-specific IFNγ, IL17A, and Foxp3 [93]. Therefore, it was demonstrated that the H3K27me3 demethylase JMJD3 [94] and DNA demethylases Ten-Eleven-Translocation (TET)2/TET35 are key activators of IL-17 expression, and recent data have evidenced that H3K9me3 may also modulate the expression of IL-17 [95]. It is now accepted that bromodomain antagonists [96] and histone H3K27me3 demethylase inhibitors [97] are able to decrease the inflammatory status linked to CD4+ T cell activation. Emerging evidence underscores the importance of bromodomain antagonists that interfere with epigenetic events on histones, related to transcriptional processes. The bromodomain and extraterminal domain (BET) family proteins, consisting of BRD2, BRD3, BRD4, and BRDT, are characterized by two bromodomains, that recognize and bind to lysine-acetylated histones and other acetylated proteins with different degrees of affinity. Small-molecule BET inhibitors interact on the acetyl moiety inserting into the bromodomain acetyl-lysine-binding pocket, which is specific to the BET family proteins, and this allows BET inhibitors to be perfect candidates for blocking the constitutively active regions that have active histone marks. Indeed, BET inhibition has been shown to reduce the differentiation of naive T cells into Th17 cells [98]. Interestingly, studies in vitro have demonstrated that BET inhibition potently suppressed Th17 cell responses in explanted lung tissue from cystic fibrosis’ patients with a history of chronic lung inflammation. Thus, these BET inhibitors are able to modulate T cell responses, specifically Th17-mediated inflammation, to inhibit IL-17-driven chemokine production in human bronchial epithelial cells through processes that include bromodomain-dependent inhibition of acetylated histones at the IL-17 gene locus [99]. In addition, in a murine model of acute pseudomonas aeruginosa lung infection, BET inhibition diminished inflammatory conditions without exacerbating infection, suggesting that BET inhibitors may be a potential therapeutic candidate in patients with cystic fibrosis [100].
Figure 3.
The main epigenetic changes associated with differentiation of T h17 cells and upregulation of IL-17 such as DNA methylation, histone post-translational modifications, and ncRNA (lncRNA and miRNAs). Bet proteins (bromodomain and extra-terminal domain); BRDs (bromodomains); DNMTs (DNA methyltransferases); ncRNA (non-coding RNA); lncRNA (long non-coding RNA); miRNA (microRNA); Dicer (Endoribonuclease Dicer C-terminal complex); TET (ten-eleven translocation); 2OG (2Oxoglutarate Oxygenase); IOX-1 (Histone demethylase inhibitor), Risc (RNA-induced silencing complex); T h17 cells (T helper 17 cells).
Figure 3.
The main epigenetic changes associated with differentiation of T h17 cells and upregulation of IL-17 such as DNA methylation, histone post-translational modifications, and ncRNA (lncRNA and miRNAs). Bet proteins (bromodomain and extra-terminal domain); BRDs (bromodomains); DNMTs (DNA methyltransferases); ncRNA (non-coding RNA); lncRNA (long non-coding RNA); miRNA (microRNA); Dicer (Endoribonuclease Dicer C-terminal complex); TET (ten-eleven translocation); 2OG (2Oxoglutarate Oxygenase); IOX-1 (Histone demethylase inhibitor), Risc (RNA-induced silencing complex); T h17 cells (T helper 17 cells).
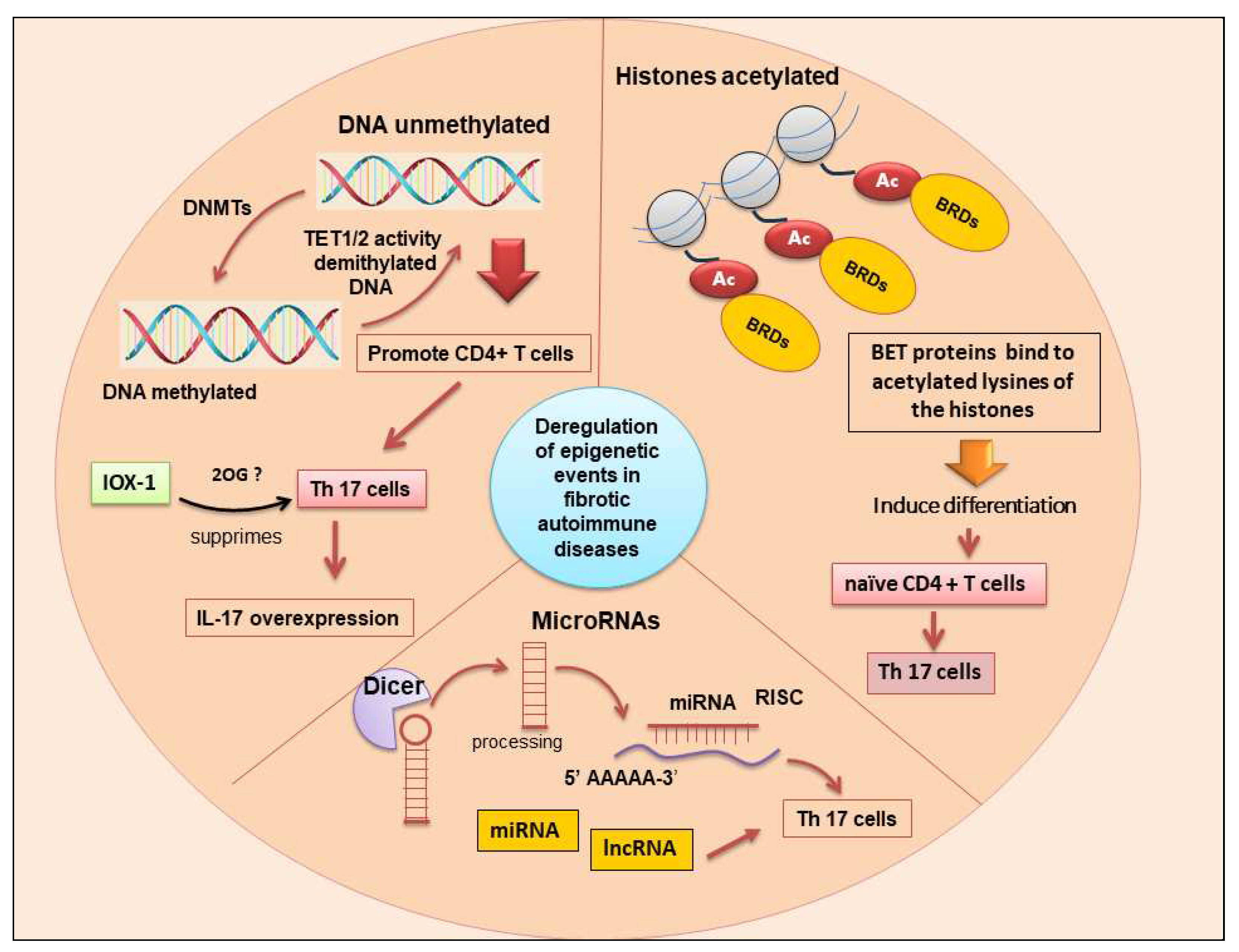
Based on this evidence, targeting Th17 cells as well as those molecules mediating the differentiation and inflammatory functions of these cells can become a plausible therapeutic approach for many autoimmune diseases. While classical biologic agents (monoclonal antibodies or recombinant proteins) targeting IL-17 and IL-23, as well as inhibitors of RORγt, have shown potent efficacy only in some fibrotic diseases such as psoriasis and RA, unfortunately, this potential has not been observed in other diseases, such as uveitis and Crӧhn’s disease [101]. This has led to the identification of new potential therapeutic targets, and recently, IOX1 (histone demethylase inhibitor) was identified as the potential candidate that suppresses Th17 function, targeting TET2 activity on the IL-17a promoter. The TET proteins TET1, TET2, and TET3 catalyze 5-methylcytosine (5 mC) conversion to 5-hydroxymethylcytosine (5 hmC) to regulate the DNA demethylation mechanism [102]. However, the potential therapeutic effect of the inhibitors of TET proteins has, until now, not been fully appreciated or developed. IOX1 is a general 2 Oxoglutarate Oxygenase (2OG) inhibitor that can also target other histone and DNA demethylases. Interestingly, IOX1 does not seem to have any direct interaction with demethylases could potentially activate IL-17A expression, but, probably, suppress Th17 cells through targeting other 2OG enzymes [103]. From early studies conducted in the laboratory, IOX1 appears to have the advantage of similar efficacy with less cellular toxicity when compared to previously known inhibitors of IL-17-mediated inflammation, such as Tofacitinib [103] (Figure 3).
6.3. Correlations of non-coding RNA expression and IL-17 levels in fibrosis
Different cascades and signalling pathways regulate fibrosis [6,104]. Recently, in addition to the large number of factors involved in fibrotic evolution, a number of non-coding RNAs (ncRNAs) have been found to affect fibrotic processes. The ncRNAs include a vast number of transcripts, within which long ncRNAs (lncRNAs) and microRNAs (miRNAs) have been extensively investigated in recent years, as they have demonstrated extensive regulatory activity on mRNA coding genes. miRNAs are single-stranded transcripts with sizes of about 22 nucleotides, produced from precursors of 60 -100 nucleotides by modifications done by an RNase III endonuclease, namely Dicer [105]. These small transcripts suppress the synthesis of proteins through base pairing to the 3′ untranslated region (3′UTR) of mRNA or, rarely, to the 5′UTR and coding regions [106]. When created, one or both strands of the miRNA duplex can be assimilated into the RNA-induced silencing complex regulating gene transcription [107]. On the other hand, lncRNAs have sizes greater than 200 nucleotides and represent a quantity higher than protein-coding genes [108]. With a total amount higher than that of protein coding genes, their variability is correlated with the complexity of the organism and with the type of molecular pathway that they regulate. In fact, lncRNAs influence fundamental biological processes such as imprinting, chromosomal configuration, and enzymatic activation [109]. Many studies have revealed that miRNAs and lncRNAs are key regulator of the development of fibrotic processes, often correlated with autoimmune conditions. Probably they act on the most common fibrotic pathway, mediated by TGF-β, phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), and Wnt/β-catenin. An example is represented by liver fibrosis, in which the activation and proliferation of hepatic stellate cells were regulated by miRNA or lncRNA that exerted their pro-fibrotic activity precisely by acting on the pathways just reported [110,111]. There is recent evidence that this pro- or anti-fibrotic activity may be mediated by a dysregulation of T helper cells with consequent variability in the release of IL-17. Positive correlations in miRNA expression and IL-17 levels have been observed in different studies related to fibrotic diseases or autoimmune diseases with a fibrotic evolution of the tissue or organs involved (Table 1). In experimental autoimmune uveoretinitis (EAU), an overexpression of miR-142-5p and miR-21 was detected to be correlated with increased IL-17 levels, but miR-182 was decreased [112]. In psoriasis, characterized by the fibrotic evolution of skin lesions, miR-1266 and miR-146, that are known to regulate IL-17A synthesis, were increased in the sera of these patients [113,114], also in association with RA [115,116]. A positive correlation was also observed in cardiac interstitial fibrosis, characterized by myocardial fibrosis, between IL-17 and lncRNA-AK081284 [117]. On the contrary, complicating the scenario, overexpression of ncRNA is accompanied by a reduction in IL-17 release in autoimmune conditions and other fibrotic diseases (Table 1), as observed, for example, in RA [116,118]. In an experimental model of autoimmune myasthenia gravis (EAMG), the administration of lentiviral miR-145 decreased EAMG disease, determining a concomitant decreased secretion of IL-17 [106]. In the prototypic fibrotic disease SSc, there is an increase in leucocytes in the skin, including primarily T cells. These T cells that are residing in the skin are in close proximity to the myofibroblasts, suggesting that they are governing their transdifferentiation [119] and may activate other immune cells in the inflammatory foci. It has been described in SSc fibroblasts that miRNA-129-5p is repressed compared with healthy control fibroblasts [120]. The authors also show that the T-cell cytokine IL-17 can increase miRNA-129-5p levels, and using siRNA to knock down IL-17 receptors in dermal fibroblasts reduced miRNA-129-5p levels. The actual target mRNA of miRNA-129-5p appears to be collagen alpha-1 [120]. This all suggests that the Th17 cells reduce collagen expression via the upregulation of the negative regulator miRNA-129-5p. Recently, Zhang and colleagues demonstrated that miR-125a-3p decreases levels of interlukin-17 and suppresses renal fibrosis via down-regulating TGF-β1 in Lupus nephritis (LN), an autoimmune disorder mediated by SLE. The condition of LN is accompanied by inflammation via a progressive suppression of kidney function, mediated by developing fibrosis [121]. In MS patients, the downregulation of miR-20b was revealed. In experimental autoimmune encephalomyelitis (EAE), primarily used as an animal model of human autoimmune inflammatory MS, miR-20b overexpression decreased disease severity by decreasing Th17 differentiation by targeting RORγt and STAT3 [122]. In the EAE model, miR-873 induced by IL-17 stimulation aggravated disease severity and increased inflammation by targeting Tumor Necrosis Factor Alpha-Induced Protein 3 or TNFAIP3 (A20)/NF-κ [123]. The same effect was obtained by the overexpression of miR-132 in the EAE [124]. Importantly, Du et al. reported that miR-326 expression correlated with MS disease severity in human patients, and in EAE mice, miR-326 regulates Th-17 cell differentiation through translational inhibition of Ets-1, a negative regulator of Th17 differentiation [125]. All these findings suggest that miRNA or lncRNA regulation and correlation with IL-17 are dependent on the fibrotic disease model (Table 1, Figure 3).
8. Conclusions
IL-17 is critical for host defence, but its role in the regulation of many chronic inflammatory, fibrotic and/or autoimmune diseases becomes more and more evident. Although the pivotal roles of IL-17 in chronic inflammatory conditions are increasingly enumerated, these new concepts are not enough to clarify the function of IL-17 in fibrosis, which often represents the evolution of autoimmune diseases. Overall, the thin differences in IL-17 cytokines and their receptors seem to influence their role in fibrotic evolution. A thriving field of research concerns the inflammatory mechanisms mediated by the activation of EMT, which much evidence links to fibrotic evolution. In addition, in recent years, various experimental data have demonstrated the key role of epigenetics in the genetic regulation of fibrosis [107]. Several epigenetic modifications are involved in this process, such as histone modifications, DNA demethylation, or miRNAs and lncRNAs. Currently, however, there is no known clinical research on fibrotic diseases that is based on epigenetics. This would be essential to identify not only new therapies but also predictive biomarkers for the diagnosis of fibrotic diseases, such as many autoimmune diseases. In an attempt to optimize the general application and effectiveness of therapies targeted against IL-17 subtypes and to clarify the multiple molecular pathways in which they appear to carry out their regulatory activity, it will be necessary to elucidate the immunological and genetic circumstances under which IL-17 becomes pro-fibrotic.
Author Contributions
all authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version for publication. M.S. and S.L. had full access to the data collected in the review, take responsibility for their integrity and performed a critical reading. All authors have read and agreed to the published version of the manuscript.
Funding
this research received no external funding.
Conflicts of Interest
the authors declare no conflict of interest.
References
- Distler, J.H.W.; Gyorfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Konigshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef] [PubMed]
- Mehal, W.Z.; Iredale, J.; Friedman, S.L. Scraping fibrosis: expressway to the core of fibrosis. Nat. Med. 2011, 17, 552–553. [Google Scholar] [CrossRef] [PubMed]
- Wick, G.; Grundtman, C.; Mayerl, C.; Wimpissinger, T.F.; Feichtinger,J. ; Zelger, B.; Sgonc, R.; Wolfram, D. The immunology of fibrosis. Annu. Rev. Immunol. 2013, 31, 107–135. [Google Scholar] [CrossRef]
- Enderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: from mechanisms to medicines. Nature. 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Miao, H.; Wu, X.Q.; Zhang, D.D.; Wang, Y.N.; Guo, Y.; Li, P.; Xiong, Q.; Zhao, Y.Y. Deciphering the cellular mechanisms underlying fibrosis-associated diseases and therapeutic avenues. Pharmacol. Res. 2021, 163, 105316. [Google Scholar] [CrossRef] [PubMed]
- Sisto,M. ; Ribatti, D.; Lisi, S. Organ Fibrosis and Autoimmunity: The Role of Inflammation in TGFβ-Dependent EMT. Biomolecules. 2021, 11, 310. [Google Scholar]
- Sisto, M.; Lisi, S. Towards a Unified Approach in Autoimmune Fibrotic Signalling Pathways. Int. J. Mol. Sci. 2023, 24, 9060. [Google Scholar] [CrossRef] [PubMed]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Kuwabara, T.; Ishikawa, F.; Kondo,M. ; Kakiuchi, T. The Role of IL-17 and Related Cytokines in Inflammatory Autoimmune Diseases. Mediators Inflamm. 2017, 2017, 3908061. [Google Scholar] [CrossRef]
- Ramani, K.; Biswas, P.S. Interleukin-17: Friend or foe in organ fibrosis. Cytokine. 2019, 120, 282–288. [Google Scholar] [CrossRef]
- Ruiz de Morales, J.M.G.; Puig, L.; Daudén, E.; Cañete, J.D.; Pablos, J.L.; Martín, A.O.; Juanatey, C.G.; Adán, A.; Montalbán, X.; Borruel, N.; Ortí, G.; Holgado-Martín, E.; García-Vidal, C.; Vizcaya-Morales, C.; Martín-Vázquez, V.; González-Gay, M.Á. Critical role of interleukin (IL)-17 in inflammatory and immune disorders: An updated review of the evidence focusing in controversies. Autoimmun. Rev. 2020, 19, 102429. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Adachi, M.; Oda, N.; Kokubu, F.; Huang, S.K. IL-17 cytokine family. J. Allergy Clin. Immunol. 2004, 114, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Rouvier, E.; Luciani,M. F.; Mattéi, M.G.; Denizot, F.; Golstein, P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J. Immunol. 1993, 150, 5445–5456. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Painter, S.L. Fanslow, W.C.; Ulrich, D.; Macduff, B.M.; Spriggs, M.K., Armitage, R.J. Human IL-17: a novel cytokine derived from T cells. J. Immunol. 1995, 155, 5483–5486. [Google Scholar] [CrossRef] [PubMed]
- Moseley, T.A.; Haudenschild, D.R.; Rose, L.; Reddi, A.H. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003, 14, 155–174. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; Filvaroff, E.H.; Yin, J.P.; Lee, J.; Cai, L.; Risser, P.; Maruoka, M.; Mao, W.; Foster, J.; Kelley, R.F.; Pan, G.; Gurney, A.L.; de Vos, A.M.; Starovasnik, M.A. IL-17s adopt a cystine knot fold: Structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001, 20, 5332–5341. [Google Scholar] [CrossRef]
- Lee, J.; Ho, W.H.; Maruoka, M.; Corpuz, R.T. , Baldwin, D.T., Foster, J.S., et al. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. J. Biol. Chem. 2001, 276, 1660–1664. [Google Scholar] [CrossRef]
- Bie, Q.; Jin, C.; Zhang, B.; Dong, H. IL-17B: A new area of study in the IL-17 family. Mol. Immunol. 2017, 90, 50–56. [Google Scholar] [CrossRef]
- Kolls, J.K.; Lindén, A. Interleukin-17 family members and inflammation. Immunity. 2004, 21, 467–476. [Google Scholar] [CrossRef]
- Huang, X.D.; Zhang, H.; He, M.X. Comparative and evolutionary analysis of the interleukin 17 gene family in invertebrates. PLos ONE. 2015, 10, e0132802. [Google Scholar] [CrossRef]
- Yang, X.O.; Chang, S.H.; Park, H.; Nurieva, R.; Shah, B.; Acero, L.; Wang, Y.H.; Schluns, K.S.; Broaddus, R.R.; Zhu, Z.; Dong, C. Regulation of inflammatory responses by IL-17F. J. Exp. Med. 2008, 205, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, A.; Lehmann, S.; Blank, J.; Kolbinger, F.; Rondeau, J.M. Structural Analysis Reveals that the Cytokine IL-17F Forms a Homodimeric Complex with Receptor IL-17RC to Drive IL-17RA-Independent Signaling. Immunity. 2020, 52, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Fanslow, W.C.; Seldin, M.F.; Rousseau, A.M.; Painter, S.L.; Comeau, M.R. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995, 3, 811–821. [Google Scholar] [CrossRef]
- Monin, L.; Gaffen, S.L. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb Perspect Biol. 2018, 10, a028522. [Google Scholar] [CrossRef]
- Beringer, A.; Noack, M.; Miossec, P. IL-17 in chronic inflammation: from discovery to targeting. Trends Mol. Med. 2016, 22, 230–241. [Google Scholar] [CrossRef]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lee, H.Y.; Zhao, X.; Han, J.; Su, Y.; Sun, Q.; Shao, J.; Ge, J.; Zhao, Y.; Bai, X.; He, Y.; Wang, X.; Wang, X.; Dong, C. Interleukin-17D regulates group 3 innate lymphoid cell function through its receptor CD93. Immunity. 2021, 54, 673–686. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 family of cytokines in health and disease. Immunity. 2019, 50, 892. [Google Scholar] [CrossRef]
- Pande, S.; Yang, X.; Friesel, R. Interleukin-17 receptor D (Sef) is a multi-functional regulator of cell signaling. Cell Commun. Signal. 2021, 19, 6. [Google Scholar] [CrossRef]
- Amatya, N.; Garg, A.V.; Gaffen, S. L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef]
- Nies, J. F, Panzer, U. IL-17C/IL-17RE: Emergence of a Unique Axis in TH17 Biology. Front. Immunol. 2020, 11, 341. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Kuchroo, V.K. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity, 2015, 43, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Lu, L.; Yu, X. Interleukin-17A Interweaves the Skeletal and Immune Systems. Front Immunol. 2021, 11, 625034. [Google Scholar] [CrossRef] [PubMed]
- Rosine, N.; Miceli-Richard, C. Innate Cells: The Alternative Source of IL-17 in Axial and Peripheral Spondyloarthritis? Front. Immunol. 2021, 11, 553742. [Google Scholar] [CrossRef]
- Mills, K.H.G. IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immunol. 2023, 23, 38–44. [Google Scholar] [CrossRef]
- Lee, Y.K.; Landuyt, A.E.; Lobionda, S.; Sittipo, P.; Zhao, Q.; Maynard, C.L. TCR-independent functions of Th17 cells mediated by the synergistic actions of cytokines of the IL-12 and IL-1 families. PLoS One. 2017, 12, e0186351. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Song, X.; Xiang, C.; He, C.; Xie, Y.; Zhou, Y.; Wang, N.; Guo, G.; Zhang, W.; Li, Y.; Liu, K.; Zou, Q.; Guo, H.; Shi, Y. Interleukin 17 B regulates colonic myeloid cell infiltration in a mouse model of DSS-induced colitis. Front Immunol. 2023, 14, 1055256. [Google Scholar] [CrossRef]
- Bie, Q.; Song, H.; Chen, X.; Yang, X.; Shi, S.; Zhang, L.; Zhao, R.; Wei, L.; Zhang, B.; Xiong, H.; Zhang, B. IL-17B/IL-17RB signaling cascade contributes to self-renewal and tumorigenesis of cancer stem cells by regulating Beclin-1 ubiquitination. Oncogene. 2021, 40, 220–2216. [Google Scholar] [CrossRef]
- Bastid, J. , Dejou, C.; Docquier, A; Bonnefoy, N. The Emerging Role of the IL-17B/IL-17RB Pathway in Cancer. Front. Immunol. 2020, 11, 718. [Google Scholar] [CrossRef]
- Chung, S.H.; Ye, X.Q.; Iwakura, Y. Interleukin-17 family members in health and disease. Int. Immunol. 2021, 33, 723–729. [Google Scholar] [CrossRef]
- Ramirez-Carrozzi, V.; Sambandam, A.; Luis, E.; Lin, Z.; Jeet, S.; Lesch, J.; Hackney, J.; Kim, J.; Zhou, M.; Lai, J.; Modrusan, Z.; Sai, T.; Lee, W.; Xu, M.; Caplazi, P.; Diehl, L.; de Voss, J.; Balazs, M.; Gonzalez, L. Jr.; Singh, H.; Ouyang, W.; Pappu, R. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 2011, 12, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Chanthaphavong, R.S.; Sun, S.; Trigilio, J.A.; Phasouk, K.; Jin, L.; Layton, E.D.; Li, A.Z.; Correnti, C.E.; De van der Schueren, W.; Vazquez, J.; O'Day, D.R.; Glass, I.A.; Knipe, D.M.; Wald, A.; Corey, L.; Zhu, J. Keratinocytes produce IL-17c to protect peripheral nervous systems during human HSV-2 reactivation. J. Exp. Med. 2017, 214, 2315–2329. [Google Scholar] [CrossRef] [PubMed]
- Saddawi-Konefka, R.; Seelige, R.; Gross, E.T.; Levy, E.; Searles, S.C.; Washington, A. Jr. Nrf2 Induces IL-17D to Mediate Tumor and Virus Surveillance. Cell Rep. 2016, 16, 2348–58. [Google Scholar] [CrossRef] [PubMed]
- Seelige, R.; Washington, A. Jr.; Bui, J.D. The ancient cytokine IL-17D is regulated by Nrf2 and mediates tumor and virus surveillance. Cytokine. 2017, 91, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Starnes, T.; Broxmeyer, H.E.; Robertson, M.J.; Hromas, R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J. Immunol. 2002, 169, 642–646. [Google Scholar] [CrossRef]
- Kleinschek, M.A. , Owyang, A.M., Joyce-Shaikh, B., Langrish, C.L., Chen, Y., Gorman, D.M.; Blumenschein, W.M.; McClanahan, T.; Brombacher, F.; Hurst, S.D.; Kastelein, R.A.;Cua, D.J.IL-25 regulates Th17 function in autoimmune inflammation. J. Exp. Med. 2007, 204, 161–170. [Google Scholar] [CrossRef]
- Owyang, A.M.; Zaph, C.; Wilson, E.H.; Guild, K.J.; McClanahan, T.; Miller, H.R. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J. Exp. Med. 2006, 203, 843–849. [Google Scholar] [CrossRef]
- Huang, Q.; Chu, S.; Yin, X.; Yu, X.; Kang, C.; Li, X.; Qiu, Y. Interleukin-17A-Induced Epithelial-Mesenchymal Transition of Human Intrahepatic Biliary Epithelial Cells: Implications for Primary Biliary Cirrhosis. Tohoku J. Exp. Med. 2016, 240, 269–275. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Amara, S.; Lopez, K.; Banan, B.; Brown, S.K.; Whalen, M.; Myles, E.; Ivy, M.T.; Johnson, T.; Schey, K.L.; Tiriveedhi, V. Synergistic effect of pro-inflammatory TNFα and IL-17 in periostin mediated collagen deposition: potential role in liver fibrosis. Mol. Immunol. 2015, 64, 26–35. [Google Scholar] [CrossRef]
- Tan, Z.; Qian, X.; Jiang, R.; Liu, Q.; Wang, Y.; Chen, C.; Wang, X.; Ryffel, B.; Sun, B. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J. Immunol. 2013, 191, 1835–1844. [Google Scholar] [CrossRef]
- Jiang, G.; Liu, C.T.; Zhang, W.D. IL-17A and GDF15 are able to induce epithelial-mesenchymal transition of lung epithelial cells in response to cigarette smoke. Exp. Ther. Med. 2018, 16, 12–20. [Google Scholar] [CrossRef]
- Sisto, M.; Lorusso, L.; Tamma, R.; Ingravallo, G.; Ribatti, D.; Lisi, S. Interleukin-17 and -22 synergy linking inflammation and EMT-dependent fibrosis in Sjögren's syndrome. Clin. Exp. Immunol. 2019, 198, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Madala, S.K.; Ramalingam, T.R.; Gochuico, B.R.; Rosas, I.O.; Cheever, A.W.; et al. Bleomycin and IL-1β-mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 2010, 207, 535–552. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, H.; Su, Z.; Sun, C.; Yin, J.; Yuan, H.; Sandoghchian, S.; Jiao, Z.; Wang, S.; Xu, H. IL-17 contributes to cardiac fibrosis following experimental autoimmune myocarditis by a PKCβ/Erk1/2/NF-κB-dependent signaling pathway. Int. Immunol. 2012, 24, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Yuan, J.; Liao, M.Y.; Xia, N.; Tang, T.T.; Li, J.J.; Jiao, J.; Dong, W.Y.; Nie, S.F.; Zhu, Z.F.; Zhang, W.C.; Lv, B.J.; Xiao, H.; Wang, Q.; Tu, X.; Liao, Y.H.; Shi, G.P.; Cheng, X. IL-17A promotes ventricular remodeling after myocardial infarction. J. Mol. Med. 2014, 92, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Gasse, P.; Riteau, N.; Vacher, R.; Michel, M.L.; Fautrel, A.; di Padova, F.; Fick, L.; Charron, S.; Lagente, V.; Eberl, G.; Le Bert, M.; Quesniaux, V.F.; Huaux, F.; Leite-de-Moraes, M.; Ryffel, B.; Couillin, I. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS One. 2011, 6, e23185. [Google Scholar] [CrossRef]
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair. 2012, 5, 11. [Google Scholar] [CrossRef]
- Gurczynski, S.J.; Moore, B.B. IL-17 in the lung: the good, the bad, and the ugly. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L6–L16. [Google Scholar] [CrossRef]
- Nie, Y.J.; Wu, S.H.; Xuan, Y.H.; Yan, G. Role of IL-17 family cytokines in the progression of IPF from inflammation to fibrosis. Mil. Med. Res. 2022, 9, 21. [Google Scholar] [CrossRef]
- Sønder, S.U.; Saret, S.; Tang, W.; Sturdevant, D.E.; Porcella, S.F.; Siebenlist, U. IL-17-induced NF-kappaB activation via CIKS/Act1: physiologic significance and signaling mechanisms. J. Biol. Chem. 2011, 286, 12881–12890. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zepp, J.; Li, X. Function of Act1 in IL-17 family signaling and autoimmunity. Adv. Exp. Med. Biol. 2012, 946, 223–235. [Google Scholar] [PubMed]
- Reynolds, J.M.; Lee, Y.H.; Shi, Y.; Wang, X.; Angkasekwinai, P.; Nallaparaju, K.C. ; Interleukin-17B Antagonizes Interleukin-25-Mediated Mucosal Inflammation. Immunity. 2015, 42, 692–603. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, J.; Huang, A.; Stinson, J.; Heldens, S.; Foster, J.; Dowd, P.; Gurney, A.L.; Wood, W.I. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc. Natl. Acad. Sci. U S A. 2000, 97, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ullrich, S.J.; Zhang, J.; Connolly, K.; Grzegorzewski, K.J.; Barber, M.C.; et al. A novel cytokine receptor-ligand pair. Identification, molecular characterization, and in vivo immunomodulatory activity. J. Biol. Chem. 2000, 275, 19167–19176. [Google Scholar] [CrossRef] [PubMed]
- Morrow, K.N.; Coopersmith, C.M.; Ford, M.L. IL-17, IL-27, and IL-33: A Novel Axis Linked to Immunological Dysfunction During Sepsis. Front. Immunol. 2019, 10, 1982. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, X.; Wang, J.; Lou, Q.; Lou, Y.; Li, L.; Wang, H.; Chen, J.; Wu, M.; Song, X.; Qian, Y. Dysregulated Lung Commensal Bacteria Drive Interleukin-17B Production to Promote Pulmonary Fibrosis through Their Outer Membrane Vesicles. Immunity. 2019, 50, 692–706. [Google Scholar] [CrossRef]
- Krohn, S.; Nies, J.F.; Kapffer, S.; Schmidt, T.; Riedel, J.H.; Kaffke, A.; Peters, A.; Borchers, A.; Steinmetz, O.M.; Krebs, C.F.; Turner, J.E.; Brix, S.R.; Paust, H.J.; Stahl, R.A.K.; Panzer, U. IL-17C/IL-17 Receptor E Signaling in CD4+ T Cells Promotes TH17 Cell-Driven Glomerular Inflammation. J. Am. Soc. Nephrol. 2018, 29, 1210–1222. [Google Scholar] [CrossRef]
- Vandeghinste, N.; Klattig, J.; Jagerschmidt, C.; Lavazais, S.; Marsais, F.; Haas, J.D.; Auberval, M.; Lauffer, F.; Moran, T.; Ongenaert, M.; Van Balen, M.; Dupont, S.; Lepescheux, L.; Garcia, T.; Härtle, S.; Eyerich, K.; Fallon, P.G.; Brys, R.; Steidl, S. Neutralization of IL-17C Reduces Skin Inflammation in Mouse Models of Psoriasis and Atopic Dermatitis. J. Invest. Dermatol. 2018, 138, 1555–1563. [Google Scholar] [CrossRef]
- Chang, S.H.; Reynolds, J.M.; Pappu, B.P.; Chen, G.; Martinez, G.J.; Dong, C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. 2011, 35, 611–621. [Google Scholar] [CrossRef]
- Conti, H.R.; Whibley, N.; Coleman, B.M.; Garg, A.V.; Jaycox, J.R.; Gaffen, S.L. Signaling through IL-17C/IL-17RE is dispensable for immunity to systemic, oral and cutaneous candidiasis. PLoS ONE. 2015, 10, e0122807. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, X.; Yang, C.; Wang, S.; Shen, H. Essential role of IL-17 in acute exacerbation of pulmonary fibrosis induced by non-typeable Haemophilus influenzae. Theranostics. 2022, 12, 5125–5137. [Google Scholar] [CrossRef]
- Vella, G.; Ritzmann, F.; Wolf, L.; Kamyschnikov, A.; Stodden, H.; Herr, C.; Slevogt, H.; Bals, R.; Beisswenger, C. IL-17C contributes to NTHi-induced inflammation and lung damage in experimental COPD and is present in sputum during acute exacerbations. PLoS One. 2021, 16, e0243484. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, S.; Liu, D. IL-17D: A Less Studied Cytokine of IL-17 Family. Int Arch Allergy Immunol. 2020, 181, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Dong, C. IL-25 in allergic inflammation. Immunol. Rev. 2017, 278, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Xu, X. .; Luo, S.; Li, B.; Dai, H.; Zhang, J. IL-25 contributes to lung fibrosis by directly acting on alveolar epithelial cells and fibroblasts. Exp. Biol. Med. 2019, 244, 770–780. [Google Scholar] [CrossRef]
- Hams, E.; Armstrong, M.E.; Barlow, J.L.; Saunders, S.P.; Schwartz, C.; Cooke, G.; Fahy, R.J.; Crotty, T.B.; Hirani, N.; Flynn, R.J.; Voehringer, D.; McKenzie, A.N.; Donnelly, S.C.; Fallon, P.G. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc. Natl. Acad. Sci U S A. 2014, 111, 367–372. [Google Scholar] [CrossRef]
- Yang, X.O.; Chang, S.H.; Park, H.; Nurieva, R. , Shah, B.; Acero, L, et al. Regulation of inflammatory responses by IL-17F. J. Exp. Med 2008, 205, 1063–1075. [Google Scholar] [CrossRef]
- Hot, A.; Miossec, P. Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Ann. Rheum. Dis. 2011, 70, 727–732. [Google Scholar] [CrossRef]
- Ritzmann, F.; Lunding, L.P.; Bals, R.; Wegmann, M.; Beisswenger, C. IL-17 Cytokines and Chronic Lung Diseases. Cells. 2022, 11, 2132. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector Th17 cells and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Joern, R.R.; Forsthuber, T.G. Memory CD4+ T Cells in Immunity and Autoimmune Diseases. Cells. 2020, 9, 531. [Google Scholar] [CrossRef]
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; Fingerlin, T.E.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.F.; Okamoto, T.; John, A.E.; et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir Med. 2017, 5, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Dagneaux, L.; Owen, A.R.; Bettencourt, J.W.; Barlow, J.D.; Amadio, P.C.; Kocher, J.P.; Morrey, M.E; Sanchez-Sotelo, J.; Berry, D.J.; van Wijnen, A.J.; Abdel, M.P. Human Fibrosis: Is There Evidence for a Genetic Predisposition in Musculoskeletal Tissues? J. Arthroplasty. 2020, 35, 3343–3352. [Google Scholar] [CrossRef]
- Liu, Y. , Wen, D., Ho, C. et al. Epigenetics as a versatile regulator of fibrosis. J. Transl. Med. 2023, 21, 164. [Google Scholar] [CrossRef]
- Wei, G.; Wei, L.; Zhu, J.; Zang, C.; Hu-Li, J.; Yao, Z.; Cui, K.; Kanno, Y.; Roh, T.Y.; Watford, W.T.; Schones, D.E.; Peng, W.; Sun, H.W.; Paul, W.E.; O'Shea, J.J.; Zhao, K. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009, 30, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.P.; Tsai, Y.G.; Lin, T.Y.; Wu, M.J.; Lin, C.Y. The attenuation of renal fibrosis by histone deacetylase inhibitors is associated with the plasticity of FOXP3+IL-17+ T cells. BMC Nephrol. 2017 18, 225. [CrossRef]
- Seto, E. , Yoshida, M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Felisbino, M.B.; McKinsey, T.A. Epigenetics in cardiac fibrosis. JACC Basic Transl. Sci. 2018, 3, 704–15. [Google Scholar] [CrossRef]
- Barcena-Varela, M.; Colyn, L.; Fernandez-Barrena, M.G. Epigenetic mechanisms in hepatic stellate cell activation during liver fibrosis and carcinogenesis. Int. J. Mol. Sci. 2019, 20, 2507. [Google Scholar] [CrossRef]
- Goschl, L.; Preglej, T.; Boucheron, N.; et al. Histone deacetylase 1 (HDAC1): a key player of T cell-mediated arthritis. J. Autoimmun. 2020, 108. [Google Scholar] [CrossRef]
- Regna, N.L.; Chafin, C.B.; Hammond, S.E.; Puthiyaveetil, A.G.; Caudell, D.L.; Reilly, C.M. Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo. Clin. Immunol. 2014, 151, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan X, Kim BS, Tanaka S, Ndiaye-Lobry D, Deng Y, Zou Y, Zheng P, Tian Q, Aifantis I, Wei L, Dong C. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity. 2015, 42, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zou, J.; Wang, M.; Ding, X.; Chepelev, I.; Zhou, X.; Zhao, W.; Wei, G.; Cui, J.; Zhao, K.; Wang, H.Y.; Wang, R.F. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nat. Commun. 2014, 5, 5780. [Google Scholar] [CrossRef] [PubMed]
- Takada, I. DGCR14 induces Il17a gene expression through the RORgamma/BAZ1B/RSKS2 complex. Mol. Cell Biol. 2015, 35, 344–355. [Google Scholar] [CrossRef]
- Bandukwala, H.S.; Gagnon, J.; Togher, S.; et al. Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc. Natl. Acad. Sci. U S A. 2012, 109, 14532–14537. [Google Scholar] [CrossRef]
- Cribbs, A.P.; Terlecki-Zaniewicz, S.; Philpott, M.; Baardman, J.; Ahern, D.; Lindow, M.; Obad, S.; Oerum, H.; Sampey, B.; Mander, P.K.; Penn, H.; Wordsworth, P.; Bowness, P.; de Winther, M.; Prinjha, R.K.; Feldmann, M.; Oppermann, U. Histone H3K27me3 demethylases regulate human Th17 cell development and effector functions by impacting on metabolism. Proc. Natl. Acad. Sci. U S A. 2020, 117, 6056–6066. [Google Scholar] [CrossRef]
- Mele, D.A.; Salmeron, A.; Ghosh, S.; Huang, H.R.; Bryant, B.M.; Lora, J.M. BET bromodomain inhibition suppresses TH17-mediated pathology. J. Exp. Med. 2013, 210, 2181–2190. [Google Scholar] [CrossRef]
- Klein, K. Bromodomain protein inhibition: a novel therapeutic strategy in rheumatic diseases. RMD Open. 2018, 4, e000744. [Google Scholar] [CrossRef]
- Chen, K.; Campfield, B.T.; Wenzel, S.E.; McAleer, J.P.; Kreindler, J.L.; Kurland, G.; Gopal. R.; Wang, T.; Chen, W., Eddens, T., Quinn, K.M.; Myerburg, M.M.; Horne, W.T.; Lora, J.M.; Albrecht, B.K.; Pilewski, J.M.; Kolls, J.K. Antiinflammatory effects of bromodomain and extraterminal domain inhibition in cystic fibrosis lung inflammation. JCI Insight. 2016, 1, e87168. [Google Scholar] [CrossRef]
- Fragoulis, G.E.; Siebert, S.; McInnes, I.B. Therapeutic targeting of IL-17 and IL-23 cytokines in immune-mediated diseases. Annu. Rev. Med. 2016, 67, 337–353. [Google Scholar] [CrossRef]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef]
- Hu, X.; Zou, Y.; Copland, D.A.; Schewitz-Bowers, L.P.; Li, Y.; Lait, P.J.P.; Stimpson, M.; Zhang, Z.; Guo, S.; Liang, J.; Chen, T.; Li, J.J.; Yuan, S.; Li, S.; Zhou, P.; Liu, Y.; Dick, A.D.; Wen, X.; Lee, R.W.J.; Wei, L. Epigenetic drug screen identified IOX1 as an inhibitor of Th17-mediated inflammation through targeting TET2. E Bio Medicine. 2022, 86, 104333. [Google Scholar] [CrossRef] [PubMed]
- Antar, S.A.; Ashour, N.A.; Marawan, M.E.; Al-Karmalawy, A.A. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. Int. J. Mol. Sci. 2023, 24, 4004. [Google Scholar] [CrossRef] [PubMed]
- Zapletal, D.; Kubicek, K.; Svoboda, P.; Stefl, R. Dicer structure and function: conserved and evolving features. EMBO Rep. 2023, 24, e57215. [Google Scholar] [CrossRef] [PubMed]
- Wang, B. Base Composition Characteristics of Mammalian miRNAs. J. Nucleic Acids. 2013, 2013, 951570. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S. .; Abak, A.; Talebi, S.F.; Shoorei, H.; Branicki, W.; Taheri, M.; Akbari Dilmaghani, N. Role of miRNA and lncRNAs in organ fibrosis and aging. Biomed Pharmacother. 2021, 143, 112132. [Google Scholar]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P. et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef]
- Li, Q.Y.; Gong, T.; Huang, Y.K.; Kang, L.; Warner, C.A.; Xie, H.; Chen, L.M.; Duan, X.Q. Role of noncoding RNAs in liver fibrosis. World J. Gastroenterol. 2023, 29, 1446–1459. [Google Scholar] [CrossRef]
- Yao, S.X.; Zhang, G.S.; Cao, H.X.; Song, G.; Li, Z.T.; Zhang, W.T. Correlation between microRNA-21 and expression of Th17 and Treg cells in microenvironment of rats with hepatocellular carcinoma. Asian Pac. J. Trop. Med. 2015 8, 762–765. [CrossRef]
- Ishida, W.; Fukuda, K.; Sakamoto, S.; Koyama, N.; Koyanagi,A. ; Yagita, H.; Fukushima, A. Regulation of experimental autoimmune uveoretinitis by anti-delta-like ligand 4 monoclonal antibody. Invest. Ophthalmol.Vis.Sci. 2011, 52, 8224–30. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, A.; Jinnin, M.; Oyama, R.; Yamane, K.; Fujisawa, A.; Sakai, K.; Masuguchi, S.; Fukushima, S.; Maruo, K.; Ihn, H. Increased serum levels of miR-1266 in patients with psoriasis vulgaris. Eur. J. Dermatol. 2012, 22, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Fang, X.; Zhang, Z.H.; Huang, Q.; Yan, K.X.; Kang, K.F.; Han, L.; Zheng, Z.Z. Dysregulation of miRNA146a versus IRAK1 induces IL-17 persistence in the psoriatic skin lesions. Immunol. Lett. 2012, 148, 151–162. [Google Scholar] [CrossRef]
- Niimoto, T.; Nakasa, T.; Ishikawa, M. , Okuhara, A.; Izumi, B.; Deie, M.; Suzuki, O.; Adachi, N.; Ochi, M. MicroRNA-146a expresses in interleukin-17 producing T cells in rheumatoid arthritis patients. BMC Musculoskelet. Disord. 2010, 11, 209. [Google Scholar] [CrossRef]
- Dong, L.; Wang, X.; Tan, J.; Li, H.; Qian, W.; Chen, J.; Chen, Q.; Wang, J.; Xu, W.; Tao, C.; Wang, S. Decreased expression of microRNA-21 correlates with the imbalance of Th17 and Treg cells in patients with rheumatoid arthritis. J. Cell Mol. Med. 2014, 18, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.Y.; Li, T.T.; Wang, J.; Jiang, Y.; Zhao, Y.; Jin, X.X.; Xue, G.L.; Yang, Y.; Zhang, X.F.; Sun, Y.Y.; Zhang, Z.R.; Gao, X.; Du, Z.M.; Lu, Y.J.; Yang, B.F.; Pan, Z.W. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J. Mol. Cell Cardiol. 2018, 115, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Castro-Villegas, C.; Pérez-Sánchez, C.; Escudero, A.; Filipescu, I.; Verdu, M.; Ruiz-Limón, P.; Aguirre, M.A.; Jiménez-Gomez, Y.; Font, P.; Rodriguez-Ariza, A.; Peinado, J.R.; Collantes-Estévez, E.; González-Conejero, R.; Martinez, C.; Barbarroja, N.; López-Pedrera, C. Circulating miRNAs as potential biomarkers of therapy effectiveness in rheumatoid arthritis patients treated with anti-TNFα. Arthritis Res. Ther. 2015, 17, 49. [Google Scholar] [CrossRef]
- O'Reilly, S.; Hügle, T.; van Laar, J.M. T cells in systemic sclerosis: a reappraisal. Rheumatology (Oxford). 2012, 51, 1540–1549. [Google Scholar] [CrossRef]
- Nakashima, T.; Jinnin, M.; Yamane, K.; Honda, N.; Kajihara, I.; Makino, T.; Masuguchi, S.; Fukushima, S.; Okamoto, Y.; Hasegawa, M.; Fujimoto, M.; Ihn, H. Impaired IL-17 signaling pathway contributes to the increased collagen expression in scleroderma fibroblasts. J. Immunol. 2012, 188, 3573–3583. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Deng, Y. miR-125a-3p decreases levels of interlukin-17 and suppresses renal fibrosis via down-regulating TGF-β1 in systemic lupus erythematosus mediated Lupus nephritic mice. Am. J. Transl. Res. 2019, 11, 1843–1853. [Google Scholar] [PubMed]
- Zhu, E.; Wang, X.; Zheng, B.; Wang, Q.; Hao, J.; Chen, S.; Zhao, Q.; Zhao, L.; Wu, Z.; Yin, Z. miR-20b suppresses Th17 differentiation and the pathogenesis of experimental autoimmune encephalomyelitis by targeting RORγt and STAT3. J. Immunol. 2014, 192, 5599–5609. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, F.; Pang, R.; Zhao, D.; Qiu, W.; Shan, K.; Zhang, J.; Lu, Y.; Li, Y.; Wang, Y. Interleukin-17 (IL-17)-induced microRNA 873 (miR-873) contributes to the pathogenesis of experimental autoimmune encephalomyelitis by targeting A20 ubiquitin-editing enzyme. J. Biol. Chem. 2014, 289, 28971–28986. [Google Scholar] [CrossRef]
- Hanieh, H.; Alzahrani, A. MicroRNA-132 suppresses autoimmune encephalomyelitis by inducing cholinergic anti-inflammation: a new Ahr-based exploration. Eur. J. Immunol. 2013, 43, 2771–2782. [Google Scholar] [PubMed]
- Du, C.; Liu, C.; Kang, J.; Zhao, G.; Ye, Z.; Huang, S.; Li, Z.; Wu, Z.; Pei, G. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat. Immunol. 2009, 10, 1252–1259. [Google Scholar] [CrossRef]
Figure 1.
Role of IL-17 cytokines in fibrotic autoimmune diseases. IL-17 is produced by epithelial cells, dendritic cells, macrophages, CD4 T helper 17 (Th17) and gamma/delta T cells (Tγδ17 cells). The IL-17 signalling promotes the production of pro-inflammatory factors as granulocyte-macrophage colony-stimulating (GM-CSF), tumour necrosis factor (TNFα), and the release of pro-inflammatory cytokines such as IL-6, IL-8 and IL-23. These pathogenic factors exacerbate chronic inflammation and, often through epithelial mesenchymal transition (EMT) process, cause the fibrotic evolution of autoimmune diseases.
Figure 1.
Role of IL-17 cytokines in fibrotic autoimmune diseases. IL-17 is produced by epithelial cells, dendritic cells, macrophages, CD4 T helper 17 (Th17) and gamma/delta T cells (Tγδ17 cells). The IL-17 signalling promotes the production of pro-inflammatory factors as granulocyte-macrophage colony-stimulating (GM-CSF), tumour necrosis factor (TNFα), and the release of pro-inflammatory cytokines such as IL-6, IL-8 and IL-23. These pathogenic factors exacerbate chronic inflammation and, often through epithelial mesenchymal transition (EMT) process, cause the fibrotic evolution of autoimmune diseases.
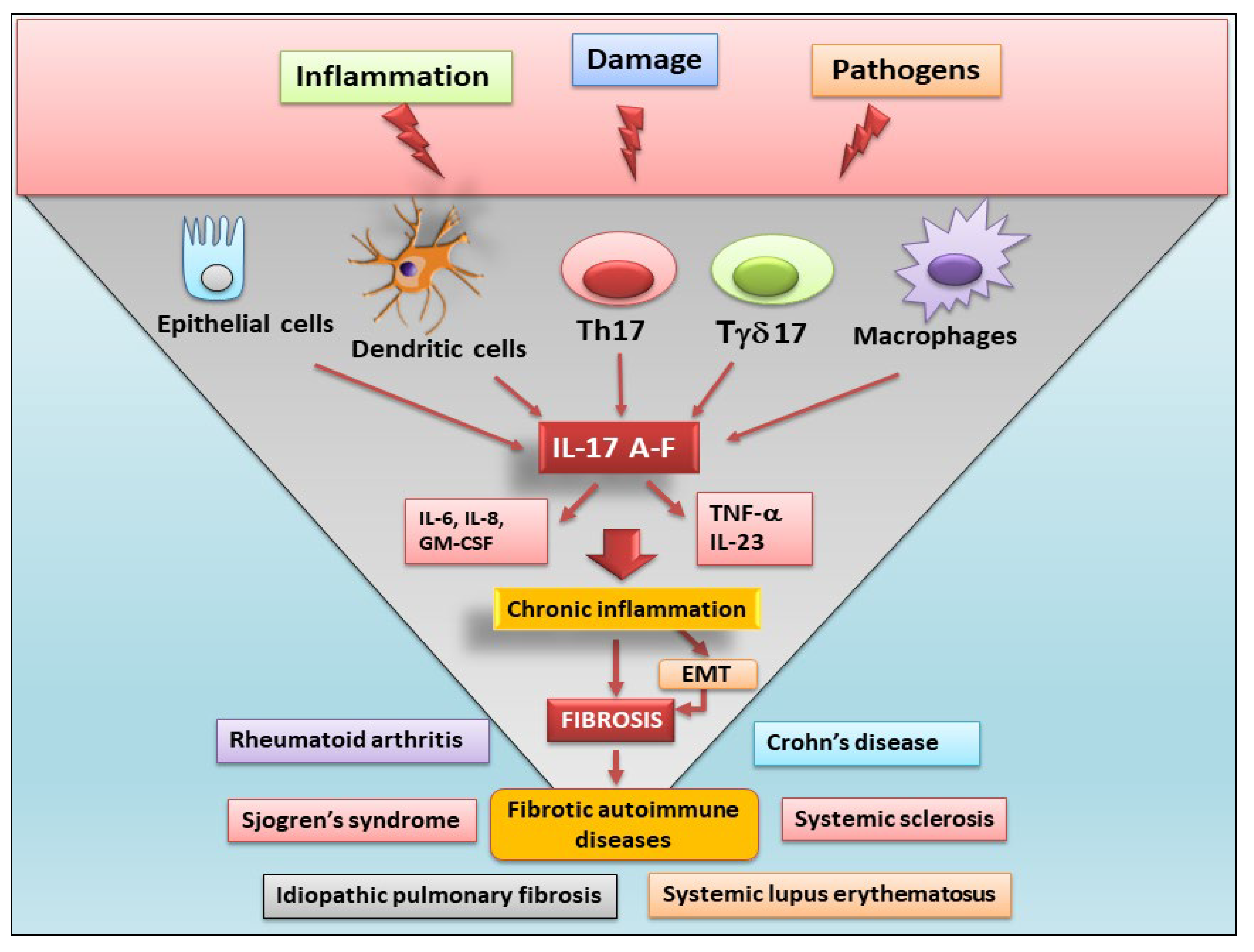
Figure 2.
Main cellular sources and targets of IL-17A and IL-17E in EMT-mediated fibrotic evolution of autoimmune diseases. IL-17A/E contribute to idiopathic pulmonary fibrosis, primary biliary cirrhosis and salivary gland fibrosis through the activation of EMT that leads to fibroblast proliferation and differentiation to myofibroblasts. EMT (epithelial mesenchymal transition); T h17 cells (T helper 17 cells).
Figure 2.
Main cellular sources and targets of IL-17A and IL-17E in EMT-mediated fibrotic evolution of autoimmune diseases. IL-17A/E contribute to idiopathic pulmonary fibrosis, primary biliary cirrhosis and salivary gland fibrosis through the activation of EMT that leads to fibroblast proliferation and differentiation to myofibroblasts. EMT (epithelial mesenchymal transition); T h17 cells (T helper 17 cells).
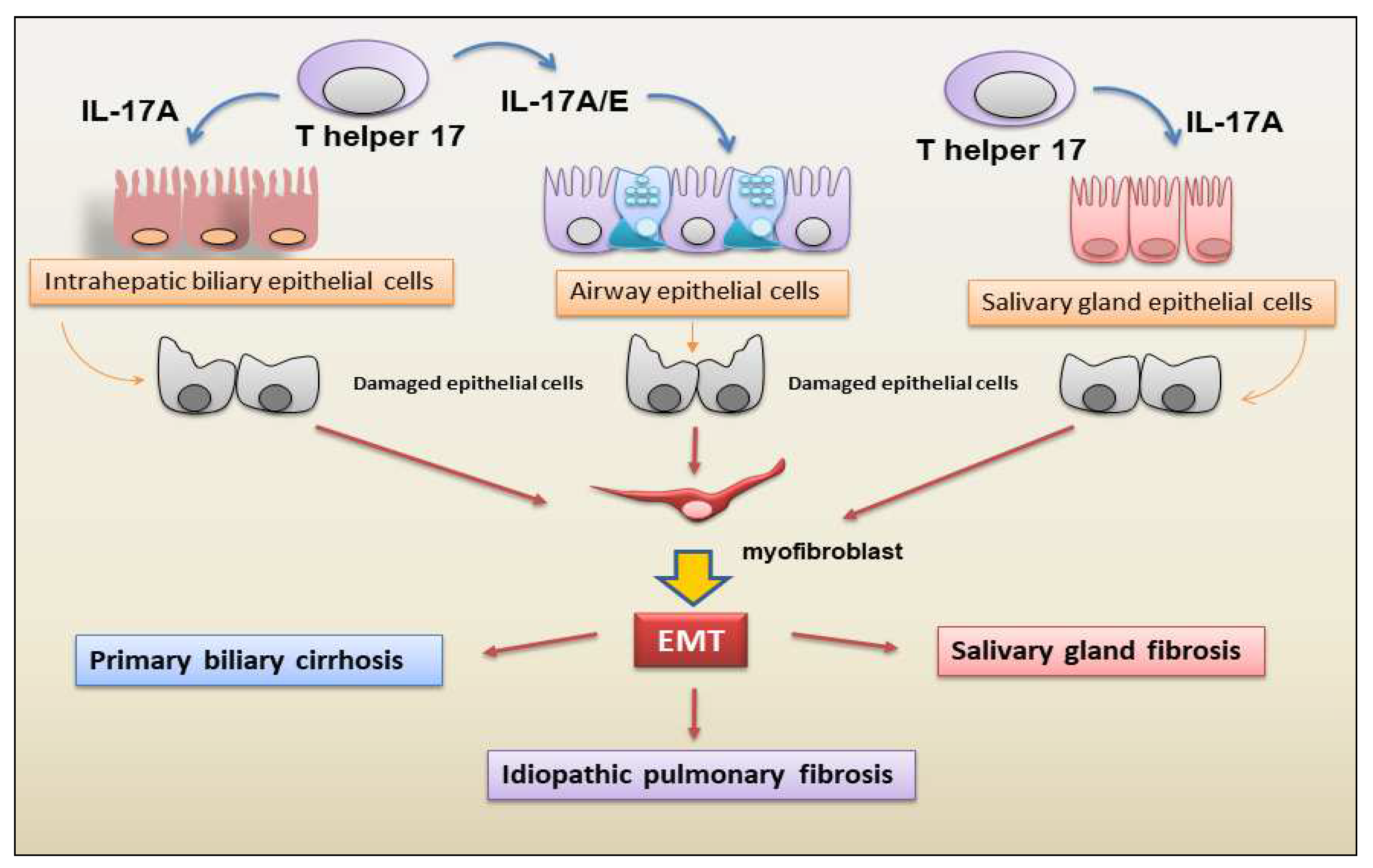

Table 1.
Positive or negative correlations in lncRNA/miRNA expression and IL-17 levels in fibrotic diseases or autoimmune fibrotic diseases.
Table 1.
Positive or negative correlations in lncRNA/miRNA expression and IL-17 levels in fibrotic diseases or autoimmune fibrotic diseases.
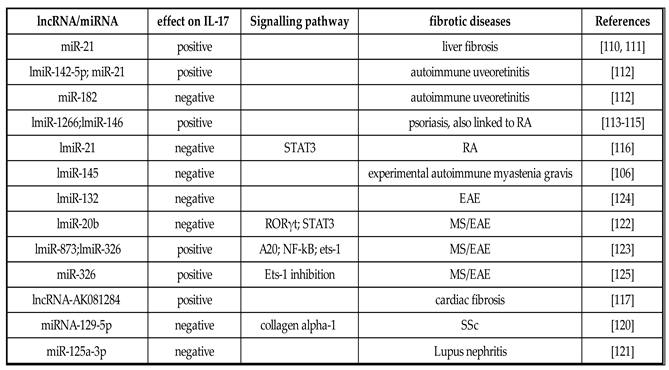
RA: rheumatoid arthritis; EAE: experimental autoimmune encephalomyelitis; MS: multiple sclerosis; SSc: systemic sclerosis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



