
Submitted:
05 October 2023
Posted:
05 October 2023
You are already at the latest version
Abstract
Norovirus (NoV) is recognised as a major cause of epidemic and sporadic acute gastroenteritis (AGE) in all age groups. Information on the genetic diversity of the NoVs circulating in the 1980s and 1990s, before the development and adoption of dedicated molecular assays, is limited compared to the last decades. In the period 1986-2020, uninterrupted viral surveillance was conducted in symptomatic children hospitalized for AGE in Palermo, Italy, providing a unique time capsule for exploring the epidemiological and evolutionary dynamics of enteric viruses. In this 35-year long time span NoVs of genogroup II (GII) were detected in 15.6% of AGE requiring hospitalization, whilst GI NoVs in 1.4%. Overall, the predominant NoV capsid (Cap) genotype was GII.4 (60.8%), with temporal replacement of the GII.4 Cap variants and associated polymerase (Pol) types. The chronology of emergence and circulation of the different GII.4 variants were consistent with the data available in literature. Also, for GII.3 and GII.2 NoVs the circulation of different lineages/strains, differing in either the Cap or Pol genes or in both, was observed. This long-term study revealed the ability of NoVs to modify continuously and rapidly their genomic makeup and highlights the importance of surveillance activities for vaccine design.
Keywords:
norovirus
; evolution
; genotypes
; GII.4
; GII.3
; GII.2
; Italy
1. Introduction
Noroviruses (NoVs) are a major cause of acute gastroenteritis (AGE) worldwide. NoVs belong to the family Caliciviridae and are non-enveloped viruses with a positive-sense single-stranded RNA genome (7.3 to 7.5 kb), organized into three open reading frames (ORFs). ORF1 encodes a large non-structural polyprotein that includes the viral RNA-dependent RNA polymerase (RdRp), whilst ORF2 encodes the major capsid protein (VP1) and ORF3 encodes the minor capsid protein (VP2) [1]. NoVs were first identified upon electron microscopy observation of stools of patients involved in an outbreak of AGE at an elementary school in Norwalk, Ohio, USA in 1968 and the causative agent was therefore named Norwalk virus [2]. With the development of specific molecular assays in the 1990s the role of NoV as causative agent of AGE has been clarified [3]. NoVs are responsible for more than 90% of non-bacterial AGE epidemics worldwide and are considered the first or second most important cause of diarrhoea in children, along with rotaviruses [4,5]. NoVs have been estimated to cause around 1.1 million hospitalizations and up to 200,000 deaths per year, mostly in children less than 5 years of age in developing countries [4,5]. The genetic diversity of NoVs is a challenge for diagnostics, for classification, and for the development of vaccines. Based on the complete amino acid (aa) sequence of VP1, NoVs are classified into ten genogroups (GI-GX) and 49 capsid (Cap) genotypes. Based on partial nucleotide (nt) sequences of the polymerase region, 60 confirmed polymerase (Pol) types have also been described [6]. The majority of NoV strains associated with disease in humans belong to genogroups GI and GII, and are further classified into more than 40 human genotypes [7]. Multiple NoV genotypes co-circulate in human populations but GII genotype 4 (GII.4) has been associated with most (>80%) outbreaks and sporadic cases of gastroenteritis in both developed and developing countries [8,9,10]. Since the mid-1990s, six global epidemics of NoV GII.4 have been documented and each has been associated with periodic emergence of novel GII.4 variants at intervals of 3-4 years. The pandemic GII.4 variants include US95_96, which emerged in the late 1990s [11,12], followed by Farmington Hills 2002 in 2002 [13,14], Hunter 2004 in 2004 [15], Den Haag 2006b in 2007 [16,17], New Orleans 2009 in 2009 [18] and finally by Sydney 2012 in 2011-12 [19,20]. It has been proposed that new pandemic GII.4 NoV variants generally evolve through the acquisition of residue substitutions in the capsid protein VP1 that alter the antigenicity enabling evasion of host population immunity [19,21,22,23,24,25,26] and/or modify affinity to histo-blood group antigens (HBGA) receptor [27]. In addition to the pandemic GII.4 variants of global relevance, minor GII.4 variants have been described in epidemics restricted to specific geographical regions, namely the variant Asia 2003 [28], Yerseke_2006a [16], Osaka 2007 [29] and Apeldoorn 2008 [30]. NoV genotyping has been complicated by the emergence of recombinant strains that have polymerase and capsid regions derived from separate ancestral strains [20]. The global molecular epidemiology of emerging GII.4 strains is largely based on data from outbreak surveillance programmes, that have been enacted worldwide in the 2000s and 2010s. Improvements in diagnostics, with the development and large adoption of molecular assays for NoVs, have provided valuable information on NoV epidemiology in the last two decades, but information on the diversity of NoVs in the 1980s and 1990s is more limited. Uninterrupted surveillance for AGE in hospitalized children has been carried out in Palermo, Italy, since the mid-1980s, providing a unique collection spanning more than 35 consecutive years that can be used as a time machine to investigate retrospectively the genetic evolution of enteric viruses. In this study, we generated epidemiological and sequence data on the NoV strains circulating in the local paediatric population of Palermo since the end of the last century.
2. Material and Methods
During 35 consecutive years, from 1986 to 2020, uninterrupted NoV surveillance was conducted in Palermo, South of Italy. A total of 8433 stool samples were collected from paediatric patients (<5 years old) hospitalized with AGE at the ‘‘G. Di Cristina’’ Children’s Hospital. AGE was defined by at least 3 watery stools with or without bouts of vomiting in 24 h and of less than 7 days of duration, with no identifiable symptoms other than those related to infective gastroenteritis. Stool samples were collected within 12 h after admission to the hospital to avoid inclusion of nosocomial cases and stored at -20 or -80°C until processing. Viral RNA from stool samples collected from 1986 to 2000, was extracted using ELITE InGenius automated extraction platform (ELITechGroup, Inc., Bothell, WA). For samples collected from 2001 to 2020, viral RNA was extracted from 140μl of a 10% stool suspension using a QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. Random hexamers were used for reverse transcription reaction to obtain complementary DNA (cDNA), by MMLV reverse transcriptase (Invitrogen, Carlsbad, CA). A quantitative reverse transcription (RT)-PCR assay (qRT-PCR), able to differentiate between GI and GII NoV-positive samples, was used to detect NoV RNA [19]. NoV-positive specimens were genotyped with a multi-target strategy, generating sequence data on the diagnostic region A (spanning the ORF1 region coding for the polymerase) and region C (encompassing the initial part of the ORF2 and coding for the capsid), using primers JV12/JV13 and COG2F/G2SKR, respectively [31,32,33,34,35]. A selection of 40 samples representative of different NoV GII.4 variants, observed during the study period, were also tested in the hypervariable capsid P2 domain using primer EVP2F and EVP2R, as previously described [36].
Sequence alignment was performed with CLUSTAL W [37]. Phylogenetic analysis was carried out using the MEGAX software [38], using the Kimura 2-parameter model as a method of substitution and the phylogenetic trees of partial sequences of Pol and Cap were constructed using the Maximum-likelihood method with 1,000 bootstrap replicates. Genotype assignment was performed using the Noronet automated genotyping tool (https://www.rivm.nl/en/noronet/databases) and the CDC calicivirus typing tool (https://norovirus.ng.philab.cdc.gov/).
3. Results
3.1. Prevalence and typing of NoVs
Out of 8433 stools of symptomatic children hospitalized with AGE, GII NoV was detected in 15.6% (1317/8433) of patients, whilst GI NoV was detected in 1.4% (117/8433). The temporal distribution of NoV GI and GII infections is shown in Figure 1. Genotyping of the Cap and Pol regions of NoV GII positive samples was obtained in 64.3% (847/1317) and 61.6% (812/1317) of the strains, respectively. The prevalent Cap genotype was GII.4 (60.8%), followed by GII.3 (13.3%), GII.2 (12.4%), GII.6 (4.7%), GII.17 (2%) and GII.1 (1.9%). Among the 812 Pol sequences analysed, GII.P4 represented the prevalent type (34.7%), followed by GII.P16 (28.2%), GII.P31 (14.6%), GII.P2 (4.9%), GII.P21 (3.7%), GII.P7 (2.7%) and GII.P33 (2.5%). Overall, 49.5% (652/1317) of the NoV GII strains were fully typed, obtaining the Cap/Pol combination. GII.4[P4] accounted for 28.3% of the fully typed strains, followed by GII.4[P16] (19.9%), GII.4[P31] (15.3%), GII.2[P16] (8.2%), GII.2[P2] (5,3%), GII.3[P21] (3,5%) and GII.6[P7] (3%). The temporal distribution of Cap and Pol genotypes and of fully Cap/Pol typed strains is shown in Figure 2a–c, respectively.
3.2. Focus on the early stages of Norovirus circulation
NoV RNA was not detected from the 191 available stool samples collected from 1986 to 1988. NoVs were first detected in Palermo in 1989 stool samples and circulated at low prevalence (up to 5.4%) until 1994, showing a high genotype diversity in the ORF2 (Figure 1). In particular, GII.4 and GII.6 Cap genotypes were detected in 1989, but were replaced by GII.2 in 1990 and by GII.6 in 1993. In 1994, the most prevalent genotype was GII.3 (47.4%) followed by GII.2 and GII.8 (15.8%), GII.5 (10.5%), GII.4 and GII.13 (5.3%). From 1995 the GII.4 genotype became predominant, with the exception of the years 2003, 2004 and 2016 (Figure 2a). Based on sequence analysis of region A (ORF1), the GII.P4 Pol type was predominant from 1989 to 2011, with the only exception of 1994, when GII.P3 (64.3%), GII.P5 (21.4%), and GII.P8 (14.3%) co-circulated (Figure. 2b).
3.3. Genetic evolution and diversification of NoV genotypes
Phylogenetic analyses based on sequences generated from the diagnostic region C (ORF2 gene) were performed in order to decipher the genetic diversification over time of the three predominant NoV Cap types, i.e. GII.4, GII.3, and GII.2.
3.3.1. Analyses of GII.3 and GII.2 NoVs
Upon phylogenetic analysis, the Cap gene sequences of the Italian GII.3 NoVs segregated into three different clusters (II-IV) previously defined by Boon et al. [39]. The GII.3 strains circulating from 1994 to 1997 segregated within Cap cluster II together with GII.3[P3] strains emerged in the 1980s and 1990s in Japan, USA, and Mexico, and were all characterized by a P3 Pol gene. The GII.3 strains detected from 2003 onward segregated within Cap clusters III and IV, and showed different Cap-Pol combinations. In particular, all the Italian GII.3 strains circulating from 2004 to 2006 and from 2014 to 2016 showed a GII.P21 Pol gene and segregated in cluster III, whilst the GII.3 NoVs detected in 2012 and 2019 segregated within cluster IV in association with different Pol types, as follows: in 2012 they were associated with P4_2006b, P12, P16 and P21 Pol types, whilst in 2019 with P12, P16 and P30 (Figure 3b).
The Italian GII.2 strains detected over the whole study period segregated in five different Cap lineages (a-e) in the GII.2 phylogenetic tree. GII.2 lineages generally included strains isolated in consecutive years and showing the same Pol type except for lineage a and c where two different Pol types were included, with P2 being replaced by P16 and P34, respectively (Figure 3c).
3.3.2. Analysis of the GII.4 NoV variants
The first GII.4 strain was detected in Palermo in a 1989 faecal sample and was characterised as a Lordsdale variant, displaying 99.8% nt identity to the reference strain (X86557), detected in UK four years after, in 1993. A significant heterogeneity (97.4-100% nt identity) was observed among the 39 GII.4 NoVs detected in Palermo from 1994 to 1997, segregating into different branches of the GII.4 US95/96 variant sub-tree, together with contemporary NoVs circulating in Europe and USA in the same time spam (Figure 3a). In 2002, the GII.4 variant Farmington Hills 2002 became predominant, whilst the variant GII.4 Hunter 2004 was predominant from 2004 to 2006. In 2006, the variant Den Haag 2006b emerged and co-circulated with the variant Yerseke 2006a in 2007 and with the GII.4 variant Apeldoorn 2007 in 2008. From 2009 onwards, the GII.4 variant New Orleans 2009 became predominant for a couple of years, being replaced by the GII.4 variant Sydney 2012, which emerged in 2011 in Palermo [40]. GII.4 Sydney 2012 variant stably circulated until 2020 (Figure 3a). However, while initially the Cap variant Sydney 2012 was associated to a P31 (Pe) Pol-type, from 2013 onwards recombinant strains emerged with a Pol gene New Orleans 2009, followed, from 2017 onwards, by recombinant strains with a GII.P16 Pol gene (Figure 3a).
3.4. Antigenic variation in the hypervariable P2 domain of GII.4 variants
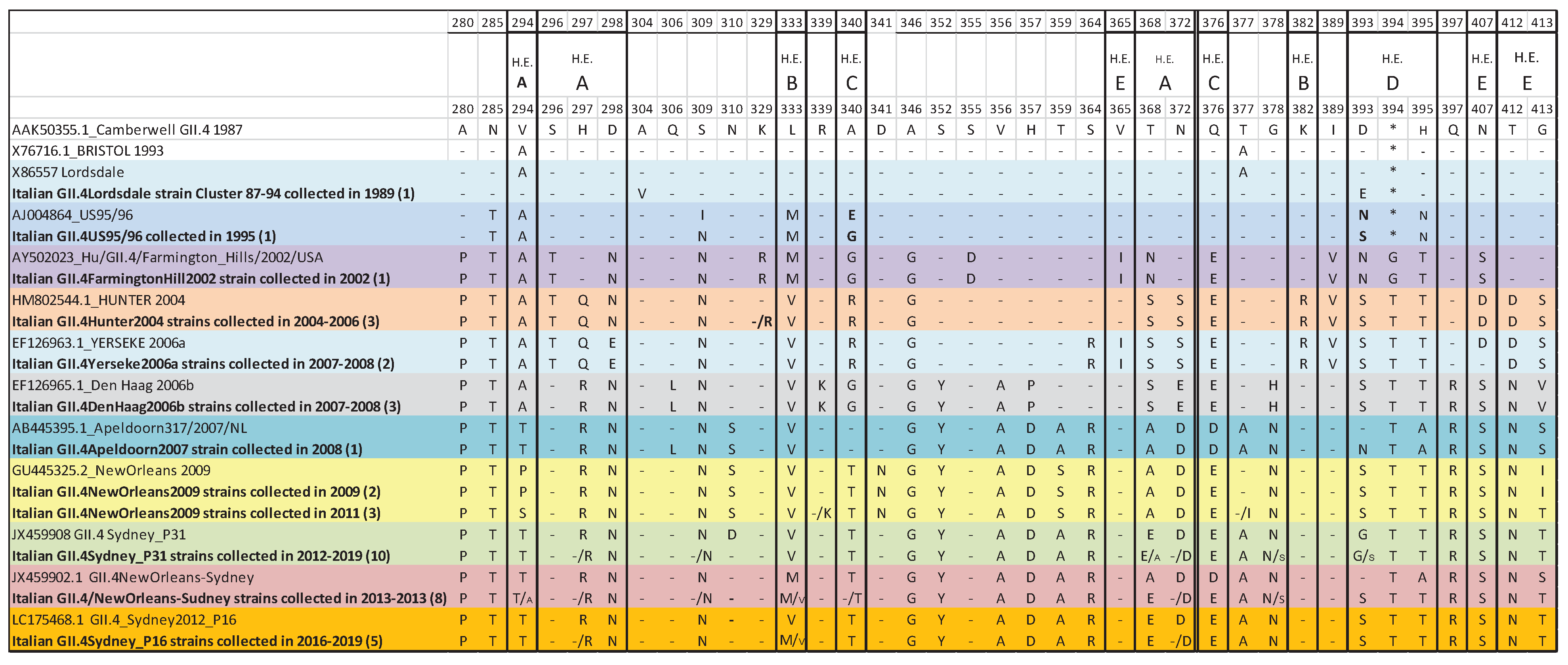
Analysis of the aa sequences of the hypervariable P2 domain of the reference strains for GII.4 NoV variants showed a high aa identity between the ancestral GII.4 NoV strain Camberwell and the older GII.4 variants (Lordsdale and US95/96) (94.9-98.1% id), whilst several aa substitutions accumulated since 2002, with the emergence of later GII.4 NoV variants (Figure 4). In particular, an insertion at position 394, located in the D hypervariable domain, appeared in 2002 Farmington Hills variant, together with several conserved aa substitutions (i.e, D298N/E, L333M/V, Q376E/D and N407S/D) in hypervariable domains. Additional aa mutations were observed in the Den Haag and Apeldoorn GII.4 NoV variants, emerging in 2006 and 2007, in the A epitope of the P2 domain (i.e. T368A, N372D). When the aa sequences of the Italian GII.4 strains collected over the study period were compared to the reference sequences, the Italian Lordsdale strain differed in two residues located in the A (N372D) and E (D393E) hypervariable domains from the prototype (X86557) whilst the Italian GII.4 US95/96 strain differed in three residues (S309N, E340G and N393S) from the prototype AJ004864. The sequences of the Italian GII.4 Farmington Hills 2002, Yerseke 2006a, Den Haag 2006b and New Orleans 2009 strains were strictly conserved with respect to the prototype sequences (AY502023, EF126963, EF126965 and GU445325, respectively), whilst several polymorphisms were observed in the P2 epitopes of Italian GII.P31-GII.4 Sydney 2012 and GII.P4New Orleans-GII.4 Sydney 2012 strains, that circulated over a longer time frame (Figure 4).
Aminoacid substitutions in small characters have been found in a minority of the sequences analysed.
4. Discussion
After the first identification by electron microscopy of NoVs as cause of AGE in symptomatic children in 1970 [2], NoVs have long been underestimated, until the development and adoption of specific molecular assays for routine diagnostics in the 1990s. In parallel, the literature on NoV has grown significantly after the year 2000, with an average of 30 manuscripts per year, versus less than 4 manuscripts per year in the second half of the 1990s (https://pubmed.ncbi.nlm.nih.gov/?term=norovirus, searched on 01/01/2023). However, information on the epidemiology of the NoVs circulating before the 2000s is limited and fragmentary [39,41,42].
In this archival retrospective study, NoV molecular epidemiology was investigated over 35 consecutive years, from 1986 to 2020. This archive of stool specimens and/or genetic material extracted from faecal samples derives from one of the longest enteric virus surveillances conducted in the European continent, providing an essential tool to investigate the evolution of NoVs.
Over the study period, NoV infection was detected in 17% of paediatric patients (<5 years old) hospitalized in Palermo, Italy. GII NoV, first detected in Palermo in 1989, represented the prevalent genogroup, accounting for 15.6% of paediatric gastroenteritis and reaching the highest rate (30%) in 2006 (Figure 1). GI NoVs were first detected in Palermo in 1994 and they were found occasionally and scattered over the remaining study period, but in 2002-2004 and 2011 when they were responsible for 3.2-7% of AGEs. The absence of NoV in the very first years of the surveillance activity in Sicily, from 1986 to 1988, could be ascribed to the low number of samples tested in 1986 and 1987, but in 1988 the considerable number of 182 faecal samples were tested negative for NoV. Although climate variations may affect NoV seasonal circulation and long-term storage of samples can affect the stability of nucleic acids, our results could simply reflect the local viral epidemiology of that period, suggesting the introduction of NoVs in Palermo only at the very end of the 1980s and their limited circulation until the mid-1990s. Alternatively, mutations in the primers/probe binding sites could have hindered the detection of earliest NoV strains with the molecular assays used in this study.
In order to investigate the genetic variability of GII NoVs over time, Cap (ORF2) sequence analysis was performed, unveiling a high genotype diversity until 1994, followed by the predominance of GII.4 genotype from 1995 to 2020, with sporadic peaks of activity of GII.3 and GII.2 genotypes in 2003-2004 and 2016, respectively (Figure 2a). The persisting epidemiological relevance of GII.4 genotype in Palermo was characterized by a fast rate of evolution, due to accumulation of punctate mutations within the protruding (P) domain of the capsid (10-3 nt substitutions/site/year), coupled with intra- and inter-genotype recombination at the ORF1–ORF2 overlap in more recent years, starting from 2012 with the Sydney strain. These mechanisms have been proposed for the effective selection of strains with improved fitness and with the ability to evade the immune response [20,43]. As previously observed worldwide, in Palermo nine pandemic variants of GII.4 NoVs (Lordsdale, US95/96, Farmington Hills 2002, Hunter 2004, Yerseke 2006a, Den Haag 2006b, Apeldoorn 2007, New Orleans 2009 and Sydney 2012) emerged consecutively [5,8,22,44,45], completely replacing each other over the study period every two-three years (Figure 3A). The first NoV detected in this study (in 1989) was a GII.4 with a Cap gene genetically related to Lordsdale genotype (99.8% nt identity), identified in the UK in 1993 (X86557) [46]. Lindesmith et al. hypothesized that pre-1995 Camberwell-like strains typically produced low-level endemic diseases in human populations, whereas since the mid-1990s the accumulation of point mutations has promoted the spread of post-1996 Lordsdale/Grimsby strains [23]. However, the limited availability of NoV sequences from the 1980s, makes it difficult to date back the emergence of such an ancient genotype [47]. Recombination events were rarely detected in the older Italian strains, which usually carried their canonical GII.P4 polymerase, with the exception of a single strain GII.4_US95/96[GII.P2] detected in 1994. By converse, in the last decade sequential recombination events repeatedly affected the variant GII.4 Sydney 2012. As already reported, this variant actually emerged in Italy in 2011 as a pre-epidemic strain with the original GII.P31 polymerase, anticipating the Australian and global circulation [19,40].
Thereafter, the local circulation of the Sydney variant was sustained by the acquisition of a GII.P4 New Orleans 2009 polymerase in 2013 and a GII.P16 polymerase in 2017 [35,48,49,50]. The sequential acquisition of such Pol genes may have been the key to the success of the Sydney variant and boosted its global emergence and spread.
The protruding P2 domain of the Cap protein possesses the epitopes involved in binding to the host cell, responsible for virus antigenicity [21,51]. The P2 domain was sequenced to better understand the evolution of GII.4 NoV strains over time. The aa alignment of 22 GII.4 Italian NoV strains selected over the study period showed punctate mutations accumulating over the time and associated with the sequential emergence of GII.4 variants every two-three years. In particular, a conserved aa insertion at position 394, in Epitope D (amino acids 393-395), which is mostly a threonine residue, was observed in all GII.4 strains since the emergence of the Farmington Hills GII.4 variant in 2002 [52]. A change at residue 395 has been shown to alter GII.4 NoV antigenic profile [23]. Crystal structure of the putative Epitope D has shown its strategic position on the surface of the capsid, since this epitope interacts with the histo-blood group antigen (HBGA) binding site, suggesting the role of such mutations in both receptor switching and escape from herd immunity [53,54]. It was previously described that the older GII.4 variants (i.e., Camberwell, Bristol, Lordsdale and US95/96) bound strongly only to antigen H of HBGA while the new GII.4 variants extended their capability to bind also A and B antigens [54]. With respect to the ancestral strain GII.4 Camberwell 1987, no aa changes were observed in the epitopes A and E among the older Italian GII.4 strains, while, since the detection of GII.4 US95/96 variant, several aa changes in epitopes B, C and D were observed. Interestingly, the majority of aa substitutions were accumulated since 2002 with the emerging strain Farmington Hills and the mutation H395T represented the key shift in the antigenic milieu of the GII.4 NoVs. Since 2006, additional amino acid mutations were observed also in Epitope A, located on the surface ridge of the capsid and probably involved in the evolution and adaptation of the novel GII.4 variants [55]. The direct role of the escape phenotype of epitope A was further demonstrated by the DenHaag 2006b variant which carried amino acid changes at positions 294, 296-298, 368 and 372 [9].
Over the 35 years of surveillance, GII.3 NoVs represented the second most relevant genotype detected in Palermo, as also reported in other epidemiological studies elsewhere [56,57]. By phylogenetic analyses of Cap gene, four different clusters (I-IV) have been described among GII.3 strains [39]. Clusters I and II contained the oldest GII.3 strains, detected in the 1970s, 1980s and 1990s, while clusters III and IV included the strains circulating since the 2000s. In Palermo GII.3 strains belonging to the four clusters described in literature were detected over the study period, with clusters III and IV temporally overlapping from 2012 to 2016 and an exclusive circulation of cluster IV thereafter. Italian GII.3 strains circulating from 1994 to 1997 in Palermo exhibited a P3 Pol gene. GII.3[P3] strains emerged globally in the 1980s and 1990s [58]. After 5 years of apparent absence of circulation, from 1998 to 2002, since 2003 a succession of recombination events affecting GII.3 strains were detected in Palermo, with the acquisition of P21, P12, and P16 Pol genes. The GII.3[P21] Cap/Pol combination represented one of the most successful GII.3 variants, being associated with symptomatic infections in children worldwide from 2000 to 2009 [59,60,61]. The increased mutation rate observed in the recombinant GII.3[P21] strains probably improved viral fitness [62]. As observed in Palermo, the progressive substitution of strains belonging to different Cap clusters and the acquisition of Pol genes being the issue of recombination events possibly allowed the persistent detection of GII.3 strains. Recombinant GII.3[P21] strains were detected from 2003 to 2006 and then from 2013 to 2016 and in 2018, while GII.3[P12] strains, circulated in 2012 and 2019, and GII.3[P16] strains in 2011-2013. The latter strains were closely related to NoVs detected in Parma (Italy) and Bangladesh in the same period [63,64].
GII.2 NoV represented the third most relevant genotype detected over the study period, showing different Cap lineages and Cap/Pol combinations (Figure 3c). In particular, the strain GII.2[P2] circulated from 1990 to 1994 and again in 2011, whilst recombinant strains with polymerase GII.P34, GII.P4_2006b, and GII.P16 appeared in 1996, 2009 and 2016, respectively. GII.2 NoVs usually account for <1-1.5% of infections globally, with sporadic peaks of circulation [65,66,67,68,69]. On analysis of the Cap gene, the GII.2[P2] Italian strains collected in 2011-2016 were closely related to the GII.2[P16] Nashville strain (KY865307), supposed to be the donor of the polymerase for recombinant GII.4[P16] viruses [70,71]. Starting from 2011 the circulation of GII.2 genotype in Palermo was sustained by a variety of strains combining two different Cap lineages and Cap/Pol combinations.
5. Conclusions
In conclusion, a unique 35-year collection of specimens was used to explore long-term trends in NoV genetic diversity and evolution. Despite the large number of NoV genotypes co-circulating in human populations, specific genotypes GII.4 and GII.3 have predominated over time [9,39]. As already shown in previous studies, our findings confirm the predominant role of GII.4 Cap type starting from 1995, but GII.3 and GII.2 retained a relevant epidemiological role over long time periods, emerging and re-emerging over time with different Cap and Pol determinants. NoVs continuously and rapidly change, likely in order to escape the immunity elicited in a settled population, yet in an intricate balance with host genetic resistance factors [27]. Although GII.4 genotype is possibly the most successful NoV strain for its evolution ability, in this study also GII.3 and GII.2 NoVs demonstrated to be able to persist in a settled population over time through genetic evolution using the accumulation of point mutation and Cap/Pol recombination. Studying the evolutionary dynamics of NoVs not only can help to predict the emergence of new epidemic strains but it is also pivotal to conceiving effective norovirus vaccines.
Author Contributions
Conceptualization, Methodology, Data Curation, Writing - Original Draft, Writing - Review & Editing: Floriana Bonura and Simona De Grazia
Data: Curation:Writing - Review & Editing: Simona De Grazia, Giovanni M. Giammanco, and Vito Martella. Methodology and Technical approaches: Chiara Filizzolo, Mariangela Pizzo, Giuseppa L. Sanfilippo, Federica Cacioppo, Emilia Palazzotto, Francesca Di Bernardo, Antonina Collura.All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Ethical approval was granted by University Hospital Ethical Committee Palermo 1 (No. 02/2017).
Informed Consent Statement
Although the collection of faecal samples was part of the general process of diagnosis of the acute gastroenteritis affecting the minors/children, we obtained verbal informed consent from the family or caretakers.
Data Availability Statement
The data are available under reasonable request to the corresponding authors.
Conflicts of Interest
The authors declare no conflict of interest
References
- Green, K.Y. Caliciviridae: The Noroviruses. In Fields virology; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott Williams and Wilkins: Philadelphia, 2013; pp. 582–608. [Google Scholar]
- Kapikian AZ; Wyatt RG; Dolin R; Thornhill TS; Kalica AR; Chanock RM Visualization by Immune Electron Microscopy of a 27-Nm Particle Associated with Acute Infectious Nonbacterial Gastroenteritis. J Virol 1972, 10, 1075–1781. [CrossRef] [PubMed]
- Estes, M.K.; Prasad, B.V.; Atmar, R.L. Noroviruses Everywhere: Has Something Changed? Curr Opin Infect Dis 2006, 19, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Widdowson, M.A.; Glass, R.I.; Akazawa, K.; Vinje, J.; Parashar, U.D. Systematic Literature Review of Role of Noroviruses in Sporadic Gastroenteritis. Emerg Infect Dis 2008, 14, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity. PLoS Pathog 2017, 13, e1006136. [Google Scholar] [CrossRef]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.-W.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated Classification of Norovirus Genogroups and Genotypes. J Gen Virol 2019, 100, 1393–1406. [Google Scholar] [CrossRef]
- de Graaf, M.; van Beek, J.; Koopmans, M.P.G. Human Norovirus Transmission and Evolution in a Changing World. Nat Rev Microbiol 2016, 14, 421–433. [Google Scholar] [CrossRef]
- Siebenga, J.J.; Vennema, H.; Zheng, D.-P.; Vinje, J.; Lee, B.E.; Pang, X.-L.; Ho, E.C.M.; Lim, W.; Choudekar, A.; Broor, S.; et al. Norovirus Illness Is a Global Problem: Emergence and Spread of Norovirus GII.4 Variants, 2001-2007. J Infect Dis 2009, 200, 802–812. [Google Scholar] [CrossRef]
- Bok, K.; Abente, E.J.; Realpe-Quintero, M.; Mitra, T.; Sosnovtsev, S.V.; Kapikian, A.Z.; Green, K.Y. Evolutionary Dynamics of GII.4 Noroviruses over a 34-Year Period. J. Virol. 2009, 83, 11890–11901. [Google Scholar] [CrossRef]
- Farahmand, M.; Moghoofei, M.; Dorost, A.; Shoja, Z.; Ghorbani, S.; Kiani, S.J.; Khales, P.; Esteghamati, A.; Sayyahfar, S.; Jafarzadeh, M.; et al. Global Prevalence and Genotype Distribution of Norovirus Infection in Children with Gastroenteritis: A Meta-Analysis on 6 Years of Research from 2015 to 2020. Rev Med Virol 2022, 32, e2237. [Google Scholar] [CrossRef]
- Noel, J.S.; Lee, T.W.; Kurtz, J.B.; Glass, R.I.; Monroe, S.S. Typing of Human Astroviruses from Clinical Isolates by Enzyme Immunoassay and Nucleotide Sequencing. J Clin Microbiol 1995, 33, 797–801. [Google Scholar] [CrossRef]
- White, P.A.; Hansman, G.S.; Li, A.; Dable, J.; Isaacs, M.; Ferson, M.; McIver, C.J.; Rawlinson, W.D. Norwalk-like Virus 95/96-US Strain Is a Major Cause of Gastroenteritis Outbreaks in Australia. J Med Virol 2002, 68, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Lopman, B.; Vennema, H.; Kohli, E.; Pothier, P.; Sanchez, A.; Negredo, A.; Buesa, J.; Schreier, E.; Reacher, M.; Brown, D.; et al. Increase in Viral Gastroenteritis Outbreaks in Europe and Epidemic Spread of New Norovirus Variant. Lancet 2004, 363, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Widdowson, M.-A.; Cramer, E.H.; Hadley, L.; Bresee, J.S.; Beard, R.S.; Bulens, S.N.; Charles, M.; Chege, W.; Isakbaeva, E.; Wright, J.G.; et al. Outbreaks of Acute Gastroenteritis on Cruise Ships and on Land: Identification of a Predominant Circulating Strain of Norovirus--United States, 2002. J Infect Dis 2004, 190, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Tu, E.T.; McIver, C.J.; Rawlinson, W.D.; White, P.A. Emergence of a New Norovirus Genotype II.4 Variant Associated with Global Outbreaks of Gastroenteritis. Journal Clinical Microbiology 2006, 44, 327–333. [Google Scholar] [CrossRef]
- Tu, E.T.-V.; Bull, R.A.; Greening, G.E.; Hewitt, J.; Lyon, M.J.; Marshall, J.A.; McIver, C.J.; Rawlinson, W.D.; White, P.A. Epidemics of Gastroenteritis during 2006 Were Associated with the Spread of Norovirus GII.4 Variants 2006a and 2006b. Clin Infect Dis 2008, 46, 413–420. [Google Scholar] [CrossRef]
- Eden, J.S.; Bull, R.A.; Tu, E.; McIver, C.J.; Lyon, M.J.; Marshall, J.A.; Smith, D.W.; Musto, J.; Rawlinson, W.D.; White, P.A. Norovirus GII.4 Variant 2006b Caused Epidemics of Acute Gastroenteritis in Australia during 2007 and 2008. J Clin Virol 2010, 49, 265–271. [Google Scholar] [CrossRef]
- Yen, C.; Wikswo, M.E.; Lopman, B.A.; Vinje, J.; Parashar, U.D.; Hall, A.J. Impact of an Emergent Norovirus Variant in 2009 on Norovirus Outbreak Activity in the United States. Clin Infect Dis 2011, 53, 568–571. [Google Scholar] [CrossRef]
- Eden, J.S.; Hewitt, J.; Lim, K.L.; Boni, M.F.; Merif, J.; Greening, G.; Ratcliff, R.M.; Holmes, E.C.; Tanaka, M.M.; Rawlinson, W.D.; et al. The Emergence and Evolution of the Novel Epidemic Norovirus GII.4 Variant Sydney 2012. Virology 2014, 450–451, 106–113. [Google Scholar] [CrossRef]
- Eden, J.S.; Tanaka, M.M.; Boni, M.F.; Rawlinson, W.D.; White, P.A. Recombination within the Pandemic Norovirus GII.4 Lineage. J Virol 2013, 87, 6270–6282. [Google Scholar] [CrossRef]
- Siebenga, J.J.; Vennema, H.; Renckens, B.; de Bruin, E.; van der Veer, B.; Siezen, R.J.; Koopmans, M. Epochal Evolution of GGII.4 Norovirus Capsid Proteins from 1995 to 2006. J Virol 2007, 81, 9932–9941. [Google Scholar] [CrossRef]
- White, P.A. Evolution of Norovirus. Clin Microbiol Infect 2014, 20, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Donaldson, E.F.; Lobue, A.D.; Cannon, J.L.; Zheng, D.P.; Vinje, J.; Baric, R.S. Mechanisms of GII.4 Norovirus Persistence in Human Populations. PLoS Med 2008, 5, e31. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Beltramello, M.; Donaldson, E.F.; Corti, D.; Swanstrom, J.; Debbink, K.; Lanzavecchia, A.; Baric, R.S. Immunogenetic Mechanisms Driving Norovirus GII.4 Antigenic Variation. PLoS Pathog 2012, 8, e1002705. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Costantini, V.; Swanstrom, J.; Debbink, K.; Donaldson, E.F.; Vinje, J.; Baric, R.S. Emergence of a Norovirus GII.4 Strain Correlates with Changes in Evolving Blockade Epitopes. J Virol 2013, 87, 2803–2813. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Lindesmith, L.C.; Donaldson, E.F.; Costantini, V.; Beltramello, M.; Corti, D.; Swanstrom, J.; Lanzavecchia, A.; Vinje, J.; Baric, R.S. Emergence of New Pandemic GII.4 Sydney Norovirus Strain Correlates With Escape From Herd Immunity. J Infect Dis 2013, 208, 1877–1887. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, W.B.; Zhang, J.; Hou, J.W.; Tang, F.; Zhang, X.F.; Du, L.F.; Su, J.G.; Li, Q.M. Evolution of the Interactions between GII.4 Noroviruses and Histo-Blood Group Antigens: Insights from Experimental and Computational Studies. PLoS Pathog 2021, 17, e1009745. [Google Scholar] [CrossRef]
- Seto, Y.; Iritani, N.; Kubo, H.; Kaida, A.; Murakami, T.; Haruki, K.; Nishio, O.; Ayata, M.; Ogura, H. Genotyping of Norovirus Strains Detected in Outbreaks between April 2002 and March 2003 in Osaka City, Japan. Microbiol Immunol 2005, 49, 275–283. [Google Scholar] [CrossRef]
- Motomura, K.; Yokoyama, M.; Ode, H.; Nakamura, H.; Mori, H.; Kanda, T.; Oka, T.; Katayama, K.; Noda, M.; Tanaka, T.; et al. Divergent Evolution of Norovirus GII/4 by Genome Recombination from May 2006 to February 2009 in Japan. J Virol 2010, 84, 8085–8097. [Google Scholar] [CrossRef]
- Belliot, G.; Kamel, A.H.; Estienney, M.; Ambert-Balay, K.; Pothier, P. Evidence of Emergence of New GGII.4 Norovirus Variants from Gastroenteritis Outbreak Survey in France during the 2007-to-2008 and 2008-to-2009 Winter Seasons. J Clin Microbiol 2010, 48, 994–998. [Google Scholar] [CrossRef]
- Vinje, J.; Koopmans, M.P. Molecular Detection and Epidemiology of Small Round-Structured Viruses in Outbreaks of Gastroenteritis in the Netherlands. Journal Infection Disease 1996, 174, 610–615. [Google Scholar] [CrossRef]
- Kageyama, T.; Kojima, S.; Shinohara, M.; Uchida, K.; Fukushi, S.; Hoshino, F.B.; Takeda, N.; Katayama, K. Broadly Reactive and Highly Sensitive Assay for Norwalk-like Viruses Based on Real-Time Quantitative Reverse Transcription-PCR. Journal Clinical Microbiology 2003, 41, 1548–1557. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Kageyama, T.; Fukushi, S.; Hoshino, F.B.; Shinohara, M.; Uchida, K.; Natori, K.; Takeda, N.; Katayama, K. Genogroup-Specific PCR Primers for Detection of Norwalk-like Viruses. Journal Virological Methods 2002, 100, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Vennema, H.; de Bruin, E.; Koopmans, M. Rational Optimization of Generic Primers Used for Norwalk-like Virus Detection by Reverse Transcriptase Polymerase Chain Reaction. J Clin Virol 2002, 25, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Bonura, F.; Urone, N.; Bonura, C.; Mangiaracina, L.; Filizzolo, C.; Sciortino, G.; Sanfilippo, G.L.; Martella, V.; Giammanco, G.M.; De Grazia, S. Recombinant GII.P16 Genotype Challenges RT-PCR-Based Typing in Region A of Norovirus Genome. J Infect 2021, 83, 69–75. [Google Scholar] [CrossRef]
- Vega, E.; Barclay, L.; Gregoricus, N.; Williams, K.; Lee, D.; Vinje, J. Novel Surveillance Network for Norovirus Gastroenteritis Outbreaks, United States. Emerg Infect Dis 17, 1389–1395. [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Boon, D.; Mahar, J.E.; Abente, E.J.; Kirkwood, C.D.; Purcell, R.H.; Kapikian, A.Z.; Green, K.Y.; Bok, K. Comparative Evolution of GII.3 and GII.4 Norovirus over a 31-Year Period. J Virol 2011, 85, 8656–8666. [Google Scholar] [CrossRef]
- Giammanco, G.M.; De Grazia, S.; Tummolo, F.; Bonura, F.; Calderaro, A.; Buonavoglia, A.; Martella, V.; Medici, M.C. Norovirus GII.4/Sydney/2012 in Italy, Winter 2012-2013. Emerg Infect Dis 2013, 19, 1348–1349. [Google Scholar] [CrossRef]
- Mori, K.; Nagano, M.; Kimoto, K.; Somura, Y.; Akiba, T.; Hayashi, Y.; Sadamasu, K.; Kai, A. Detection of Enteric Viruses in Fecal Specimens from Nonbacterial Foodborne Gastroenteritis Outbreaks in Tokyo, Japan between 1966 and 1983. Jpn J Infect Dis 2017, 70, 143–151. [Google Scholar] [CrossRef]
- Siqueira, J.A.M.; Bandeira, R. da S.; Oliveira, D. de S.; Dos Santos, L.F.P.; Gabbay, Y.B. Genotype Diversity and Molecular Evolution of Noroviruses: A 30-Year (1982-2011) Comprehensive Study with Children from Northern Brazil. PLoS One 2017, 12, e0178909. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; White, P.A. Mechanisms of GII.4 Norovirus Evolution. Trends Microbiol 2011, 19, 233–240. [Google Scholar] [CrossRef]
- Kroneman, A.; Verhoef, L.; Harris, J.; Vennema, H.; Duizer, E.; van Duynhoven, Y.; Gray, J.; Iturriza, M.; Böttiger, B.; Falkenhorst, G.; et al. Analysis of Integrated Virological and Epidemiological Reports of Norovirus Outbreaks Collected within the Foodborne Viruses in Europe Network from 1 July 2001 to 30 June 2006. J Clin Microbiol 2008, 46, 2959–2965. [Google Scholar] [CrossRef] [PubMed]
- Motoya, T.; Nagasawa, K.; Matsushima, Y.; Nagata, N.; Ryo, A.; Sekizuka, T.; Yamashita, A.; Kuroda, M.; Morita, Y.; Suzuki, Y.; et al. Molecular Evolution of the VP1 Gene in Human Norovirus GII.4 Variants in 1974-2015. Front Microbiol 2017, 8, 2399. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Clarke, I.N.; Caul, E.O.; Lambden, P.R. Human Enteric Caliciviruses Have a Unique Genome Structure and Are Distinct from the Norwalk-like Viruses. Arch Virol 1995, 140, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, E.F.; Lindesmith, L.C.; Lobue, A.D.; Baric, R.S. Norovirus Pathogenesis: Mechanisms of Persistence and Immune Evasion in Human Populations. Immunol Rev 2008, 225, 190–211. [Google Scholar] [CrossRef] [PubMed]
- Martella, V.; Medici, M.C.; De Grazia, S.; Tummolo, F.; Calderaro, A.; Bonura, F.; Saporito, L.; Terio, V.; Catella, C.; Lanave, G.; et al. Evidence for Recombination between the Pandemic GII.4 Norovirus Strains New Orleans 2009 and Sydney 2012. J Clin Microbiol 2013, 51, 3855–3857. [Google Scholar] [CrossRef] [PubMed]
- De Grazia, S.; Lanave, G.; Giammanco, G.M.; Medici, M.C.; De Conto, F.; Tummolo, F.; Calderaro, A.; Bonura, F.; Urone, N.; Morea, A.; et al. Sentinel Hospital-Based Surveillance for Norovirus Infection in Children with Gastroenteritis between 2015 and 2016 in Italy. PLoS One 2018, 13, e0208184. [Google Scholar] [CrossRef]
- Medici, M.C.; Tummolo, F.; De Grazia, S.; Calderaro, A.; De Conto, F.; Terio, V.; Chironna, M.; Bonura, F.; Pucci, M.; Banyai, K.; et al. Epidemiological Dynamics of Norovirus GII.4 Variant New Orleans 2009. J Gen Virol 2015, 96, 2919–2927. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, X. The p Domain of Norovirus Capsid Protein Forms a Subviral Particle That Binds to Histo-Blood Group Antigen Receptors. J Virol 2005, 79, 14017–14030. [Google Scholar] [CrossRef]
- Dingle, K.E. Mutation in a Lordsdale Norovirus Epidemic Strain as a Potential Indicator of Transmission Routes. J Clin Microbiol 2004, 42, 3950–3957. [Google Scholar] [CrossRef] [PubMed]
- Vinje, J.; Green, J.; Lewis, D.C.; Gallimore, C.I.; Brown, D.W.; Koopmans, M.P. Genetic Polymorphism across Regions of the Three Open Reading Frames of “Norwalk-like Viruses. ” Arch Virol 2000, 145, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Donaldson, E.F.; Lindesmith, L.C.; Baric, R.S. Genetic Mapping of a Highly Variable Norovirus GII.4 Blockade Epitope: Potential Role in Escape from Human Herd Immunity. J Virol 2012, 86, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Donaldson, E.F.; Baric, R.S. Norovirus GII.4 Strain Antigenic Variation. J Virol 85, 231–242. [CrossRef]
- Zhou, H.-L.; Zhen, S.-S.; Wang, J.-X.; Zhang, C.-J.; Qiu, C.; Wang, S.-M.; Jiang, X.; Wang, X.-Y. Burden of Acute Gastroenteritis Caused by Norovirus in China: A Systematic Review. J Infect 2017, 75, 216–224. [Google Scholar] [CrossRef]
- Wangchuk, S.; Matsumoto, T.; Iha, H.; Ahmed, K. Surveillance of Norovirus among Children with Diarrhea in Four Major Hospitals in Bhutan: Replacement of GII.21 by GII.3 as a Dominant Genotype. PLoS One 2017, 12, e0184826. [Google Scholar] [CrossRef]
- Mahar, J.E.; Bok, K.; Green, K.Y.; Kirkwood, C.D. The Importance of Intergenic Recombination in Norovirus GII.3 Evolution. J Virol 2013, 87, 3687–3698. [Google Scholar] [CrossRef]
- Gallimore, C.I.; Cubitt, D.; du Plessis, N.; Gray, J.J. Asymptomatic and Symptomatic Excretion of Noroviruses during a Hospital Outbreak of Gastroenteritis. J Clin Microbiol 2004, 42, 2271–2274. [Google Scholar] [CrossRef]
- Ambert-Balay, K.; Bon, F.; Le Guyader, F.; Pothier, P.; Kohli, E. Characterization of New Recombinant Noroviruses. Journal Clinical Microbiology 2005, 43, 5179–5186. [Google Scholar] [CrossRef]
- Gallimore, C.I.; Cheesbrough, J.S.; Lamden, K.; Bingham, C.; Gray, J.J. Multiple Norovirus Genotypes Characterised from an Oyster-Associated Outbreak of Gastroenteritis. Int J Food Microbiol 2005, 103, 323–330. [Google Scholar] [CrossRef]
- Mahar, J.E.; Kirkwood, C.D. Characterization of Norovirus Strains in Australian Children from 2006 to 2008: Prevalence of Recombinant Strains. J Med Virol 2011, 83, 2213–2219. [Google Scholar] [CrossRef] [PubMed]
- Medici, M.C.; Tummolo, F.; Martella, V.; Giammanco, G.M.; De Grazia, S.; Arcangeletti, M.C.; De Conto, F.; Chezzi, C.; Calderaro, A. Novel Recombinant GII.P16_GII.13 and GII.P16_GII.3 Norovirus Strains in Italy. Virus Res 2014, 188, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Nahar, S.; Afrad, M.H.; Begum, N.; Al-Mamun, F.; Sarker, A.K.; Das, S.K.; Faruque, A.S.G.; Pourkarim, M.R.; Choudhuri, M.S.K.; Azim, T.; et al. High Prevalence of Noroviruses among Hospitalized Diarrheal Patients in Bangladesh, 2011. J Infect Dev Ctries 2013, 7, 892–896. [Google Scholar] [CrossRef]
- Qin, S.-W.; Chan, T.-C.; Cai, J.; Zhao, N.; Miao, Z.-P.; Chen, Y.-J.; Liu, S.-L. Genotypic and Epidemiological Trends of Acute Gastroenteritis Associated with Noroviruses in China from 2006 to 2016. Int J Environ Res Public Health 2017, 14. [Google Scholar] [CrossRef]
- Hoa Tran, T.N.; Trainor, E.; Nakagomi, T.; Cunliffe, N.A.; Nakagomi, O. Molecular Epidemiology of Noroviruses Associated with Acute Sporadic Gastroenteritis in Children: Global Distribution of Genogroups, Genotypes and GII.4 Variants. J Clin Virol 2013, 56, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.-G.; Shi, C.; Xu, C.; Lin, Q.; Zhang, J.; Yi, Q.-H.; Zhang, J.; Bao, C.-J.; Huo, X.; Zhu, Y.-F.; et al. Outbreaks of Acute Gastroenteritis Associated with a Re-Emerging GII.P16-GII.2 Norovirus in the Spring of 2017 in Jiangsu, China. PLoS One 2017, 12, e0186090. [Google Scholar] [CrossRef] [PubMed]
- Hata, M.; Nakamura, N.; Kobayashi, S.; Onouchi, A.; Saito, T.; Hirose, E.; Adachi, H.; Saito, N.; Ito, M.; Yasui, Y.; et al. Emergence of New Recombinant Noroviruses GII.P16-GII.2 and GII.P16-GII.4 in Aichi, Japan, during the 2016/17 Season. Jpn J Infect Dis 2018, 71, 319–322. [Google Scholar] [CrossRef]
- Medici, M.C.; Tummolo, F.; Martella, V.; De Conto, F.; Arcangeletti, M.C.; Pinardi, F.; Ferraglia, F.; Chezzi, C.; Calderaro, A. Emergence of Novel Recombinant GII.P16_GII.2 and GII. P16_GII.4 Sydney 2012 Norovirus Strains in Italy, Winter 2016/2017. New Microbiol 2018, 41, 71–72. [Google Scholar]
- Giammanco, G.M.; Bonura, F.; Urone, N.; Purpari, G.; Cuccia, M.; Pepe, A.; Li Muli, S.; Cappa, V.; Saglimbene, C.; Mandolfo, G.; et al. Waterborne Norovirus Outbreak at a Seaside Resort Likely Originating from Municipal Water Distribution System Failure. Epidemiol Infect 2018, 146, 879–887. [Google Scholar] [CrossRef]
- Iritani, N.; Kaida, A.; Abe, N.; Sekiguchi, J.-I.; Kubo, H.; Takakura, K.-I.; Goto, K.; Ogura, H.; Seto, Y. Increase of GII.2 Norovirus Infections during the 2009-2010 Season in Osaka City, Japan. J Med Virol 2012, 84, 517–525. [Google Scholar] [CrossRef]
Figure 1.
Prevalence of NoV GI and GII infections in children hospitalized for AGE over the study period in Palermo, Italy.
Figure 1.
Prevalence of NoV GI and GII infections in children hospitalized for AGE over the study period in Palermo, Italy.
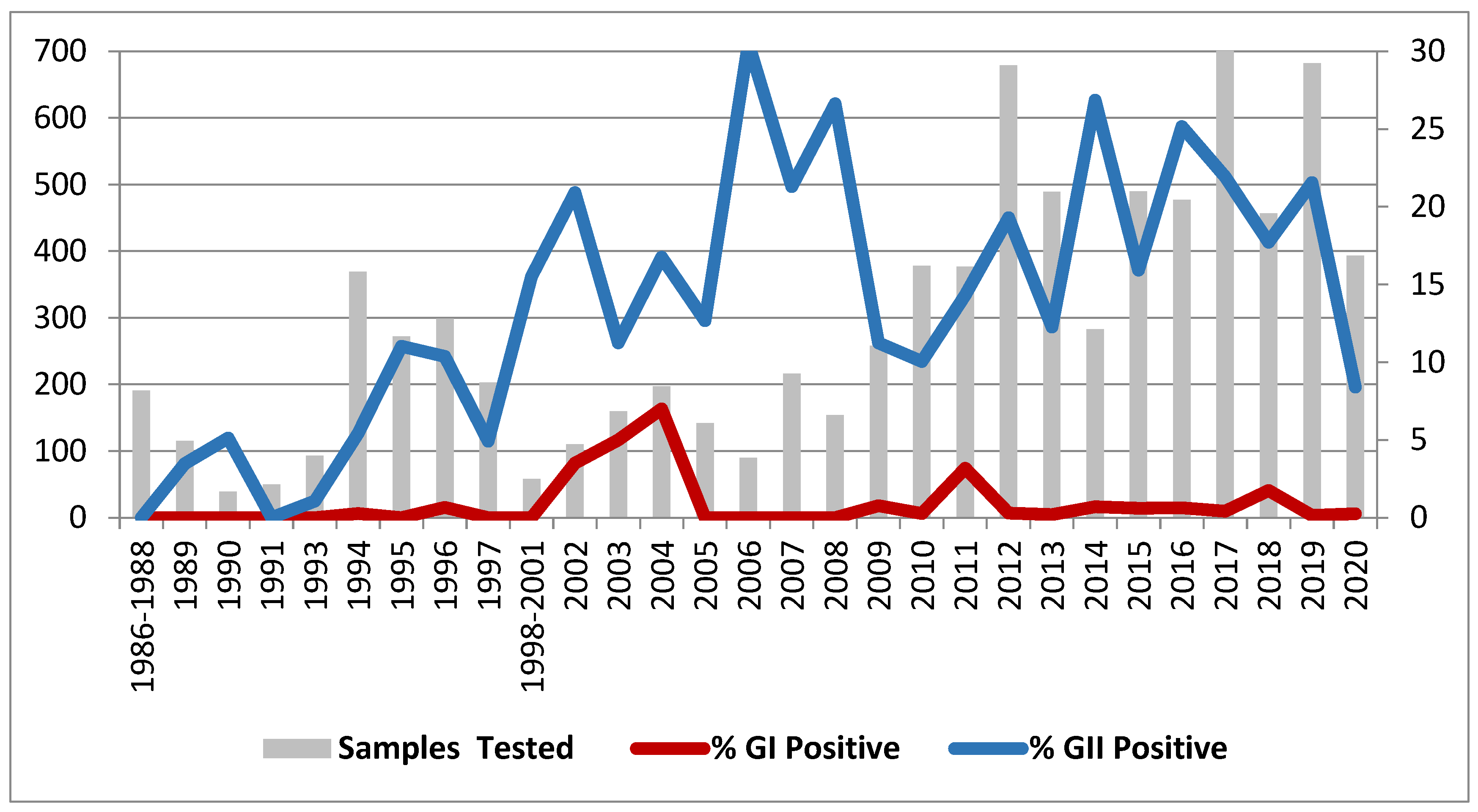
Figure 2.
Temporal distribution of Cap (a) and Pol (b) NoV genotypes and Cap/Pol combinations (c) over the study period.
Figure 2.
Temporal distribution of Cap (a) and Pol (b) NoV genotypes and Cap/Pol combinations (c) over the study period.
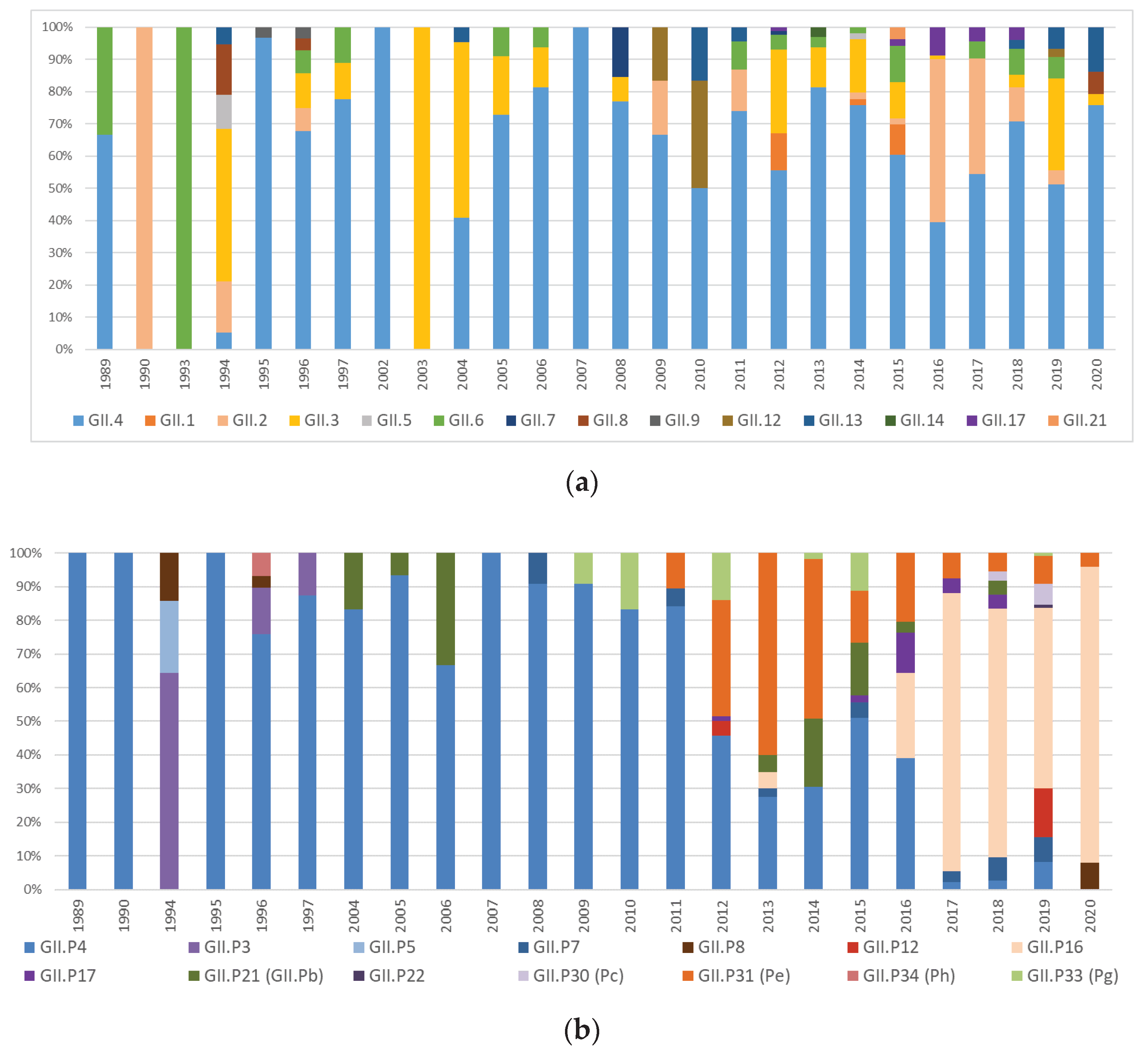
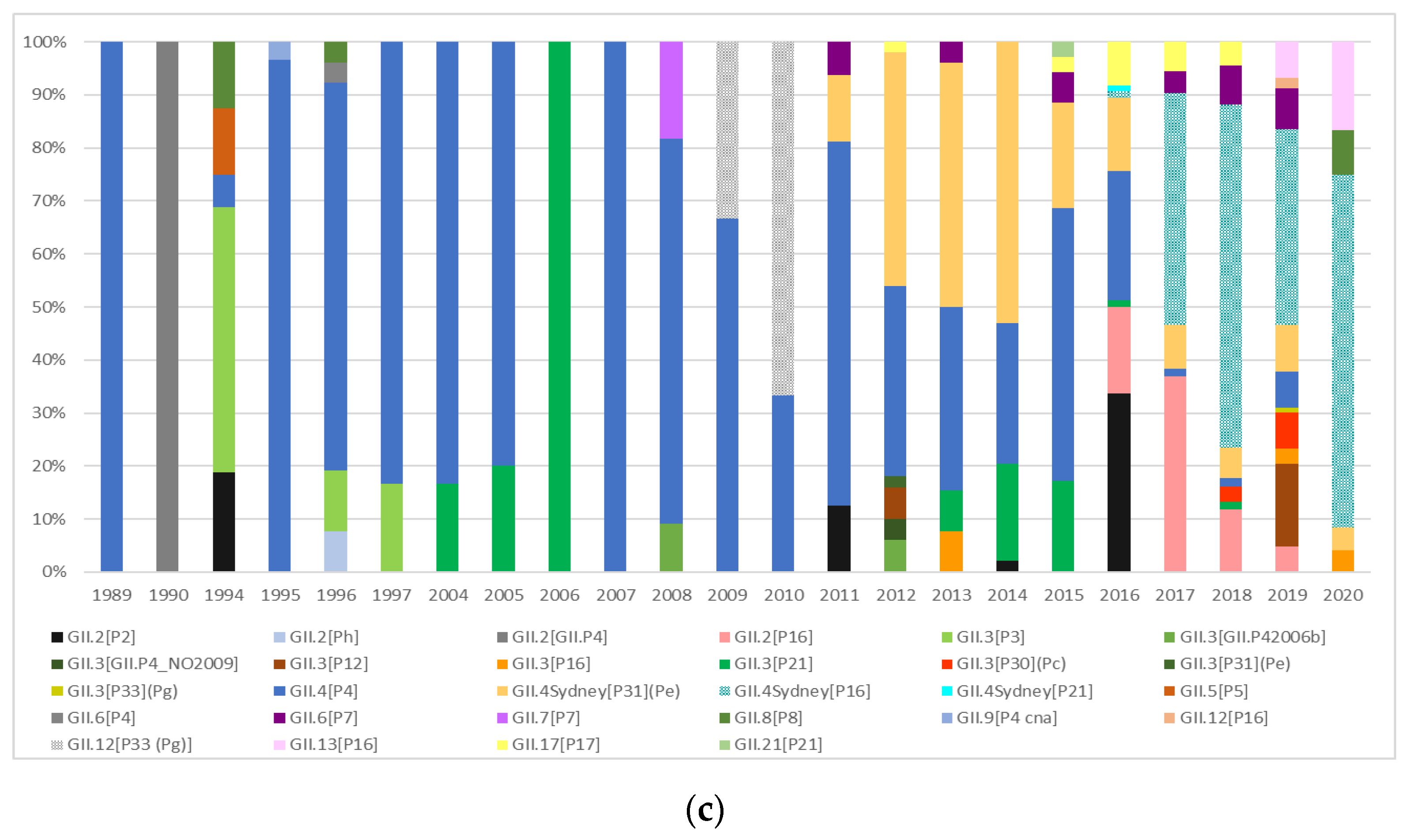
Figure 3.
Phylogenetic analysis of partial ORF2 region in NoV Italian GII.4 (a), GII.3 (b) and GII.2 (c) strains.
Figure 3.
Phylogenetic analysis of partial ORF2 region in NoV Italian GII.4 (a), GII.3 (b) and GII.2 (c) strains.
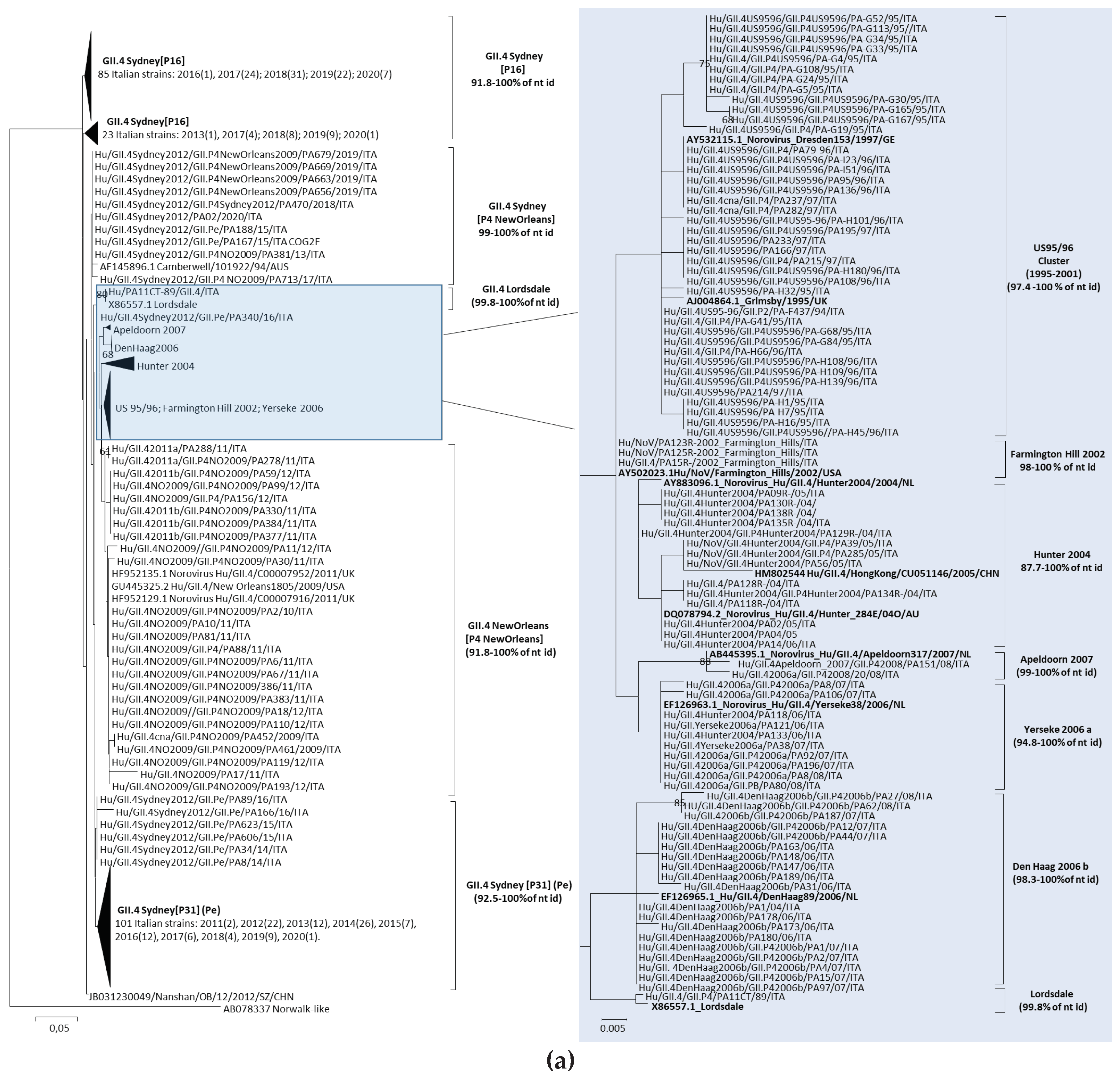

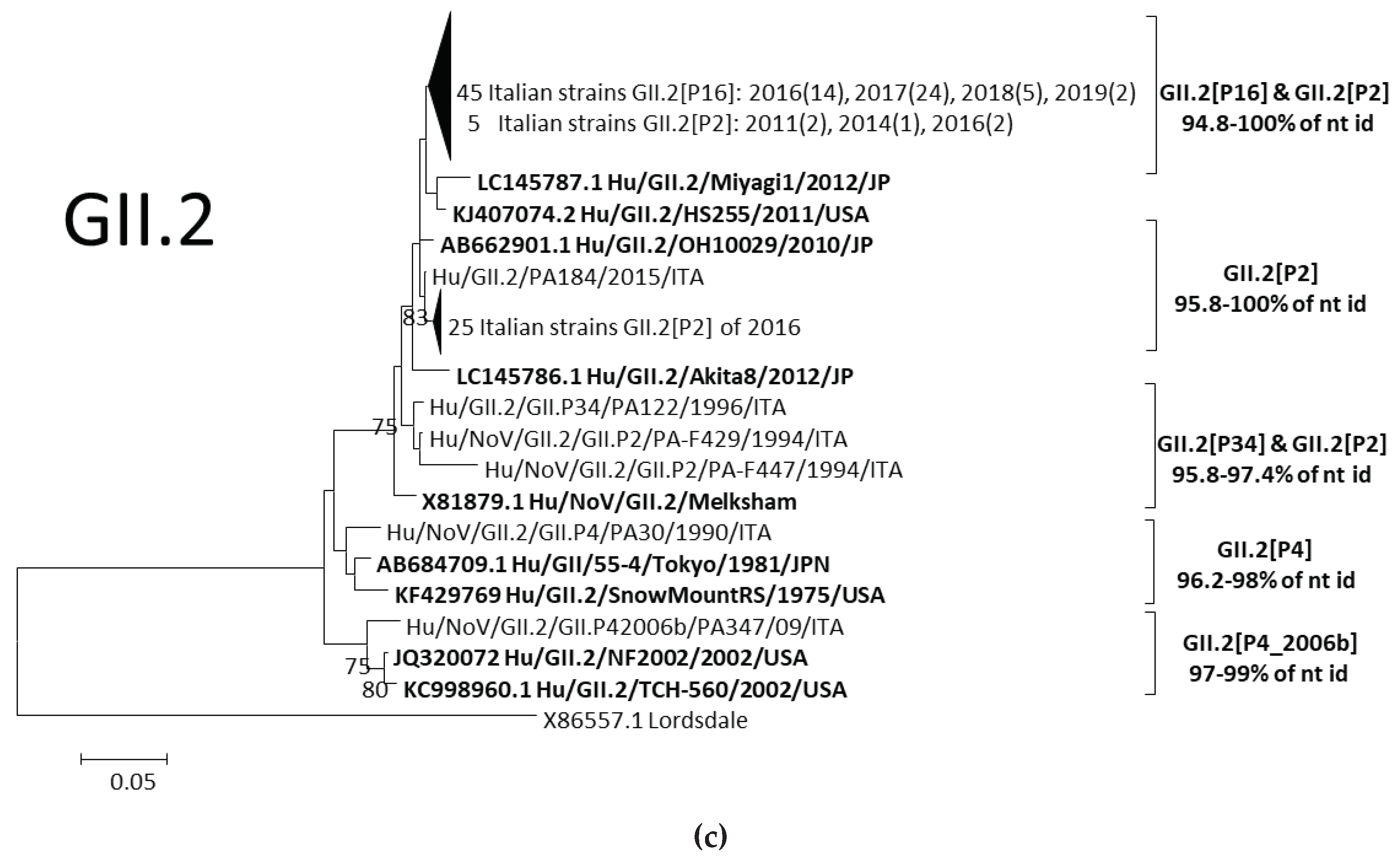
Figure 4.
Evolutionary amino acid analysis of representative GII.4 strains collected from 1987 to 2020.
Figure 4.
Evolutionary amino acid analysis of representative GII.4 strains collected from 1987 to 2020.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



