
Submitted:
06 October 2023
Posted:
09 October 2023
You are already at the latest version
Abstract
The embryonic epicardium originates from the proepicardium, an extracardiac primordium constituted by a cluster of mesothelial cells. In early embryos, the embryonic epicardium is characterized by a squamous cell epithelium resting on the myocardium surface. Subsequently, it invades the subepicardial space and thereafter the embryonic myocardium by means of epithelial-mesenchymal transition. Within the myocardium, epicardial-derived cells present multilineage potential, later differentiating into smooth muscle cells and contributing both to coronary vasculature and cardiac fibroblasts in the mature heart. Over the last decades, we have progressively increased our understanding of those cellular and molecular mechanisms driving proepicardial/embryonic epicardium formation. This study provides a state-of-the-art review of the transcriptional and emerging post-transcriptional mechanisms involved in the formation and differentiation of the embryonic epicardium.
Keywords:
proepicardium
; epicardium
; transcriptional
; post-transcriptional
Origin of the embryonic epicardium
During cardiac development, the epicardium originates from an extracardiac primordium, the proepicardium (PE), which is constituted by a cluster of mesothelial cells located on the cephalic and ventral surfaces of the liver-sinus venosus limit in avian embryos [1,2,3,4,5,6,7,8], and the pericardial side of the septum transversum in mammalian embryos [9]. In early embryos, the epicardium acquires the form of a squamous cell epithelium that either rests directly on the surface of the myocardium or covers a subepicardial space that appears to be densely populated by mesenchymal cells [10]. It is also assumed that the epicardium is not a simple mesothelium but is, in fact, made up of discrete clusters of heterogeneous cell types, which include haematopoietic contribution from distinct origins, encased by an extracellular matrix (ECM) that anatomically resembles a stem or progenitor cell ‘niche’ [11].
It has been reported that PE originates in the periphery of the heart forming fields in the lateral plate mesoderm (LPM), as part of an early cardiac progenitor lineage [12]. A single PE bud is formed during zebrafish cardiogenesis [13], while in other fish -such as the sturgeons- bilateral primordia are formed, which subsequently converge into a single PE structure in the embryonic midline [14]. Noticeably, in mice, bilateral PE anlage is also established and it further develops similarly to sturgeons, while in chick embryos only the right-side anlage develops [15]. Interestingly, chicken PE arises both from the splanchnic layer of the LPM and the somatic mesoderm, which also contributes to the mesothelial portion of the PE that forms the typical villous protrusions [16,17,18]. The above observations suggest that the embryonic left-right signal might play a significant role during PE development. Furthermore, while all proepicardial cells in a given species (e.g., in mouse, zebrafish and chick) are morphologically similar, they exhibit a distinct differentiation potential due to various marker expression [19,20,21]. Therefore, detailed composition of the embryonic epicardium is not well known.
After PE formation, cells translocate to the myocardial surface of the looping heart, where they adhere, migrate and proliferate to form a squamous epithelial layer: the embryonic epicardium [22]. It has been described that PE translocation to the myocardium takes place through distinct mechanisms among species, including direct contact and/or the release of free floating cell clusters (or cysts) into the pericardial cavity, or even PE cells migration from the sinus venosus towards the heart along the surface of the inflow tract [23,24,25,26]. After attachment to the myocardial surface, the cells start to migrate laterally, until the complete heart is enveloped by the epicardium [5]. It has been described that the epicardium surrounding the arterial pole does not originate from the PE, but from the coelomic/pericardial mesothelium at the area where the aortic sac leaves the pericardial cavity [27,28]. This cell population will contribute to the outer mesenchymal layer of the arterial pole within the pericardial cavity, contributing to the arterial epicardium formation, whereas the PE-derived epicardium will cover the myocardial outflow tract (Figure 1A).
Derivates of the embryonic epicardium
Once the epicardium is established, epicardial cells will be directly involved in the formation of the myocardium. A group of epithelial cells will undergo an epithelial-mesenchymal transition (EMT), giving rise to the epicardium derived cells (EPDCs), and then migrate into the matrix in the subepicardial layer to form the subepicardium [22]. The subepicardium thickness will eventually vary according to the underlying heart structure to be covered, and may vary among species. In particular, in chick embryos, the subepicardium is relatively thin in the atrial and ventricular myocardium. However, in the atrioventricular sulcus the subepicardium is thicker in order to provide those EPDCs needed for coronary formation [29,30].
From the subepicardium, mesenchymal EPDCs will form migratory processes and invade the myocardium, in a spatio-temporary regulated fashion, where several factors expressed in the underlying myocardium will define the permissiveness for EPDCs. These migrate into the underlying myocardium in a tangential pattern, and most of them are retained directly underneath the local area of the subepicardium [5,30,31].
Within the myocardium, EPDCs present multi-lineage potential, differentiating into smooth muscle cells (SMCs) and contributing to the coronary vasculature and cardiac fibroblasts (CF) of the mature heart [22,26]. Most EPDCs reach their final positions: i) around the coronary arteries as smooth muscle cells (SMCs) and adventitial fibroblasts [27,29,32,33,34]; ii) in the atrioventricular cushions [27,31,35]; iii) in the subendocardium of the ventricular trabeculae and atria [27,30,31]; and iv) in the ventricular myocardium as interstitial fibroblasts [31]. Other contributions of EPDCs to cardiac endothelial cells (ECs) [36,37,38] and cardiomyocytes (CMs) [39,40] have also been described, although this issue needs further research [41,42]. Therefore, both the sinus venosus and ventricular endocardium are considered major contributors to ECs [43], while EPDCs have a low contribution, if any [44]. With respect to epicardial-derived cardiomyocytes, lineage tracing studies -by using Scleraxis, WT1 and TBX18- have indicated possible epicardial-derived cardiomyocyte labeling, although their contribution is still controversial [39,41,42].
Additionally, after the embryonic epicardium has covered the developing heart, the epicardial cells will produce cytokines and growth factors in order to induce the myocardial development. In this sense, impaired embryonic epicardium development and/or cytokines and growth factor delivery results in deficient ventricular chamber maturation [45,46,47,48,49,50].
In contrast to the embryonic epicardium, the postnatal mammalian epicardium seems to be a dormant single-cell layer, since most genes involved in epicardial activation, such as WT1, Tbx18 and Raldh2 are rapidly downregulated postnatally, being scarcely detectable only during the first 3 months, in mice [51].
Transcriptional regulation of the embryonic epicardium
The development of the embryonic epicardium and its cellular derivatives is a highly complex and regulated process. In the former section it has been shown the crucial importance of the epicardial derivatives for both, the constitution of the fibrous skeleton of the heart and its vascularization. Furthermore, the epicardium and EPDCs are the origin of molecular signals towards the developing myocardium. The correct growth and compaction of the cardiac wall depend from these pathways. Thus, the precise regulation of these cellular and molecular mechanisms requires a precise orchestration of a set of transcription factors in order to activate or inhibit the genes involved in all these processes.
Transcription factors act at different levels during the development of the epicardium and its derivatives [52]. We can distinguish the development of the proepicardium and the migration of epicardial cells over the myocardium, the epithelial-mesenchymal transition (EMT) giving rise to EPDCs, the differentiation of these EPDCs mainly into fibroblasts and vascular SMCs, and the invasion of the epicardial derivatives into the myocardial wall. We will describe below the main transcription factors involved in the control of all these processes.
Proepicardial development
As we have described above, the proepicardium develops in the ventral side of the venous pole of the heart. Little is known about the molecular mechanisms governing location and development of this cluster of cells that expresses most of the characteristic epicardial genes. Liver-derived signals induce ectopic expression of proepicardial markers in chick embryos [53], and this can explain the proximity of the proepicardial bud to the developing liver. BMP4 signaling has also been proposed as a proepicardial inductor [15,54]. In Xenopus, the LIM homeodomain protein Lhx9 is essential for the correct position of the proepicardial organ on the septum transversum [55]. Its deficiency leads to proepicardial malposition and loss of attachment to the heart surface.
The hypothesis of an evolutionary origin of the proepicardium from an ancestral external glomerulus should also be considered in order to explain the mechanisms of its development [56,57]. For example, Wilms’ tumor 1 interacting protein (WTIP), an interacting partner of Wilms’ tumor protein (WT1) essential for the development of podocytes is also necessary for PE specification in the zebrafish heart. Overexpression of WTIP mRNA, induces ectopic expression of PE markers in the cardiac and pharyngeal arch regions [58].
Epicardial migration and epicardial EMT
The function of the Wilms tumor suppressor gene Wt1 was originally related with the development of kidneys and gonads [59]. However, its importance for epicardial development was soon evidenced. WT1 acting as a transcriptional activator or repressor is involved in a number of genetic mechanisms leading to the transformation of the epicardium into EPDCs (reviewed in [60]). WT1 promotes the expression of integrin 4, a receptor for myocardial VCAM. This regulates adhesion of the epicardial cells to the myocardial wall and their migration. Other targets of WT1 are nestin (an intermediate filament) and coronin-1B (an actin-binding protein), related with cell motility and migration [61,62]. WT1 activates expression of TrkB, the receptor of BDNF, involved in coronary vascularization [63]. But the main processes activated by WT1 in the epicardium are directly related to the core mechanisms of the epithelial-mesenchymal transition. These are the Snail/E-cadherin and the Wnt/retinoic acid pathways.
WT1 is a direct transcriptional activator of Snail1 and a repressor of E-cadherin in mice [64]. Snail1 plays a pivotal role in the regulation of EMT in mammals, through repression of epithelial genes and activation of genes related with mesenchymal phenotype and cell motility. Repression of E-cadherin also leads to loss of the epithelial phenotype. However, it has been shown that Snail1 silencing in the epicardium does not impairs the epicardial EMT [65], suggesting that this process can be activated by different ways. In fact, Snail2 have also been suggested as involved in the EMT of the murine epicardial cells, although this transcription factor would be activated by Tbx18 and inhibited by WT1 [66]. On the other hand, there is evidence of the importance of Snail1 in avian epicardial EMT [67]. Thus, the roles of Snail1 and Snail2 as direct executors of the epicardial EMT, repressing the epithelial phenotype and promoting a mesenchymal phenotype, need further investigation.
Retinoic acid and the canonical Wnt pathway have also been involved in the epicardial EMT. WT1 has a significant role in the retinoic acid signaling pathway since it promotes the epicardial expression of RALDH2, a key retinoic acid-synthesizing enzyme highly expressed in the epicardium [68]. Retinoic acid promotes a cytoskeletal rearrangement in epicardial cells necessary for EMT in a RhoA-dependent fashion [69]. Systemic loss of the nuclear receptor RXR leads to malformation of the epicardium and deficient EMT [70]. Conditional deletion of this receptor in the epicardium causes a similar phenotype [47]. These authors identified a retinoid-dependent Wnt signaling pathway cooperating in the epicardial EMT. In fact, -catenin is essential for this process [71]. Thus, in parallel with the role played by WT1 in the control of Snail1 and E-cadherin expression, WT1 regulates epicardial EMT through canonical Wnt, non-canonical Wnt, and retinoic acid signaling pathways [72].
The retinoic acid signaling pathway must be carefully regulated both positive and negatively for a correct epicardial and coronary vessel development [69]. The above mentioned factor WTIP, a WT1 partner expressed in the proepicardium and epicardium, can be relevant in the inhibition of this pathway. WTIP blocks ASXL2, a chromatin factor highly expressed in the heart that promotes the retinoic acid signaling [73].
Invading the myocardium
A key event in the epicardial contribution to heart development is the invasion of the myocardial wall by the EPDCs. This is necessary in order to establish the fibrous skeleton of the heart and to organize the complex vascularization of the myocardium. The transcription factor NFATC1 becomes activated and moves to the nucleus when it is dephosphorylated by calcineurin. NFATC1 is required for myocardial invasion of EPDCs, by induction of cathepsin K expression, an enzyme that degrades extracellular matrix. Loss of NFATc in murine EPDCs causes loss of cathepsin K expression in the myocardial interstitium and embryonic death. The mutant embryos show reduced coronary vessel and fibrous matrix penetration into myocardium [74]. Conditional depletion of calcineurin b1 (a NFAT activator) in the epicardium also provokes defects in the coronary smooth muscle [75]. This study also showed a direct role for NFATc in the transcription of Smad2, another transcription factor necessary for transduction of TGFβ-Alk5 signaling.
The hypoxia inducible transcription factor-1α (HIF-1α) seems to be a negative regulator of the myocardial invasion by EPDCs. Expression of constitutively active HIF-1α into the embryonic avian epicardium reduced EPDCs migration into the myocardium probably through upregulation of the VEGFR1, a decoy receptor that sequesters VEGFR [76]. A balance between signals promoting and inhibiting myocardial invasion by EPDCs may be necessary for a correct patterning of the coronary vasculature.
The protein NFB plays also a role in the acquisition of invasive ability of epicardial cells. Epicardial cells incubated with an inhibitor of NF-kB signaling cannot invade a collagen gel in response to TGFβ2 or BMP2. TGFR3-null mice fail to activate the NFB pathway in the epicardium and show reduction of EPDCs and coronary vascularization [77].
Finally, the myocardin-related transcription factors MRTF-A and B have been related with the control of EPDCs motility and the activation of the migratory and invasive program. Conditional ablation of MRTF in the epicardium leads to a decreased migration of EPDCs and subepicardial haemorrhage due to depletion of pericytes, a cell type derived from the EPDCs in an MRTF-dependent process [78].
Differentiation of EPDCs
The EPDCs mainly contribute to fibroblasts and coronary smooth muscle. It is uncertain if different lineages of EPDCs are already established in the proepicardium or if the EPDCs are multipotent and differentiate in response to local cues [79,80]. A number of transcription factors, mainly belonging to the bHLH family, are critically involved in the control of their differentiation.
The bHLH protein TCF21 (aka epicardin, POD1, capsulin) is required for normal epicardial development and it regulates EPDCs differentiation (reviewed in [81]). TCF21 loss of function leads to premature differentiation of EPDCs. TCF21 heterodimerizes with E12, another bHLH factor, for repression of transcription. In the cardiac interstitium downregulation of TCF21 leads to differentiation of EPDCs into smooth muscle, while persistence of its expression promotes fibroblast identity. Although targets of TCF21 have not been identified, in mesenchymal cells the smooth muscle markers SM22a, calponin andSMA are targets repressed by TCF21, suggesting a similar role in EPDCs. Interestingly, retinoic acid activates expression of TCF21, thus keeping the EPDCs in an undifferentiated state [82]. This repressor role of TCF21 on the EPDCs has also been demonstrated in Xenopus [83]. Loss of TCF21 function in zebrafish also reduces FGF and VEGF signaling in the heart, reducing myocardial growth [84].
The epicardial expression of TCF21 is negatively regulated by basonuclin-1, a zinc-finger transcription factor, and this can be related with the balance in the differentiation of fibroblasts and smooth muscle cells. Loss of basonuclin-1 in epicardium derived from human pluripotent stem cells leads to a predominance of TCF21+ cells, and a reduction of smooth muscle progenitors [85].
The T-Box transcription factor Tbx18 has also a role in maintaining the progenitor status of EPDCs, similar to that described above for TCF21. The role of Tbx18 in murine epicardial EMT is controversial, it may induce the process by activation of Snail2 [66], but other studies consider Tbx18 dispensable for epicardial EMT [86,87]. However, Tbx18 has a critical role to avoid the premature differentiation of smooth muscle cells. Tbx18-null mice show defects in the remodeling of the coronary vascular plexus and alteration in the expression of genes related with vascularisation [86,87]. Hypoxia provokes upregulation of Tbx18 in the epicardium, inducing the expression of Snail1 and enhancing EMT and motility [88]. Another T-Box factor, Tbx5, probably plays also a role in epicardial development, since its deletion in the proepicardium leads to low production of EPDCs, reduced migration into the myocardial wall, and low density of the coronary vessels [89].
Other bHLH transcription factors are involved in the origin and differentiation of EPDCs. For example, Twist1 is expressed in EPDCs of avian embryos and it promotes mesenchymal cell proliferation and migration [90]. Scleraxis is expressed in a proepicardial subdomain and during the early stages of the epicardium. The loss of function in mice leads to persistent expression of EMT markers, suggesting a role in the differentiation of EPDCs, mainly towards fibroblasts [91]. In fact, Scleraxis induces the expression of Col1a2 in adult cardiac fibroblasts [92]. Finally, Hand2 is necessary for a normal development of the epicardium, where it activates PDGFR [93]. This receptor of PDGF, is required for epicardial EMT and differentiation of EPDCs [94].
Other transcription factors involved in epicardial development
A number of transcription factors have been recently identified as participating in different phases of epicardial development. For example, the SRY-box protein Sox9 is expressed in EPDCs and it must play some role in epicardial EMT since its overexpression rescues the defective EMT caused by PDGF receptor ablation in the epicardium [94]. This role appears to be dispensable, since Sox9 loss of function has no consequences in this process.
The epicardial deletion of GATA4 and GATA6 leads to a drastic reduction of the number of coronary endothelial cells [95]. The replacement of GATA4 by GATA6 in the systemic GATA4-null mice does not rescue the epicardial phenotype suggesting that they are not playing redundant roles in this tissue [96].
CDX1, a caudal-related family member, has recently been involved in epicardial EMT. Its expression promotes EMT, but a low-dose CDX1 is required for enhanced migration and differentiation of EPDCs into vascular smooth muscle. Both, continued high-level expression of CDX1 or CDX1 deficiency reduce the ability of EPDCs to migrate and to differentiate [97].
The kinases LATS1 and LATS2 are important regulators of cell fate. Epicardial deletion of LATS1/2 in mice embryos is lethal due to defective coronary artery remodelling. These kinases inhibit the transcriptional function of the factor YAP, a Hippo pathway effector that prevents EPDCs differentiation into fibroblasts [98]. In fact, YAP inhibition reduces proliferation in EPDCs [99]. These studies reveal the involvement of the Hippo signaling pathway in the regulation of the EPDCs differentiation.
Finally, TFEB, a member of the microphthalmia-associated transcription factor family has been involved in the negative regulation of the epicardial EMT by activation of the TGIF1 promoter. TGIF1 (thymine-guanine-interacting factor 1) is a repressor of the TGF/SMAD signaling pathway [100]. Thus, epicardial overexpression of TFEB is lethal due to defective EMT.
Post-transcriptional control of epicardial development
Transcriptional regulation is main molecular mechanism driving cell specification and determination during embryonic development. The identification of novel players, i.e. non-coding RNAs, that modulate post-transcriptional gene regulation has added an additional layer of complexity to the understanding of the molecular mechanisms driving these morphogenetic processes. Non coding RNAs are currently classified according to their length into two subclasses, a) small non-coding RNAs (<200nt) including herein piwiRNAs, microRNAs, snoRNAs among other and long non-coding RNAs (>200nt), including herein lncRNAs and circRNAs [101,102]. microRNAs represent the most abundant and well-studied class of small non coding RNAs. microRNAs are nuclearly encoded and transcribed, exported to the cytoplasm when maturation occurs. Mature microRNAs exert their function by base-pair complementary with target transcripts leading to RNA instability and/or translation blockage [103]. On the other hand, lncRNAs are also nuclearly transcribed but they can exert their function both within the nucleus as well as in the cytoplasm [104,105]. Finally, circular RNAs (circRNAs) are normally generated by exon-exon back-splicing and they have been found in a wide range of eukaryotic species, exerting a variety of biological functions, upon which is particularly relevant their function as microRNAs sponges [106].
Multiple evidences have demonstrated that non-coding RNAs, including particularly herein both microRNAs and lncRNAs display differential expression profiles in homeostasis and pathological conditions [107,108,109]. Within the cardiovascular field, multiple evidences have demonstrated tissue-specific expression in both normal and pathological conditions [110,111,112], as well as, during cardiac development and regeneration [113,114,115,116]. Similarly, ample evidence is available about the functional role of distinct microRNAs an lncRNAs during both cardiogenesis as well as in distinct pathophysiological conditions, such as cardiac structural and arrhythmogenic diseases [117,118,119,120,121]. However, in the context of PE and epicardial development, limited information is yet available.
First evidence on the functional role of microRNAs was reported by the seminal work of Singh et al. [122] demonstrating that conditional deletion of Dicer, a key microRNA processing exonuclease in the developing epicardium was essential for correct development of the coronary vessels in mice. Subsequently, several studies have provided additional evidences on the role of microRNAs in key epicardial-derived biological processes such as epithelial-to-mesenchymal transition (EMT) [123,124,125], cardiac tissue repair [126,127,128] and cardiomyocyte proliferation [129,130].
EMT is required for the colonization of the embryonic subepicardial space emanating from the nascent embryonic epicardial layer, leading to the formation of EPDCs. Bronnum et al. [131] identified miR-21 as a key microRNA regulating Pdcd4 and Spry1 and thus controlling fibrogenic EMT while more recently Pontemezzo et al. [132] reported that Tgf-1 induced EMT resulted in miR-200c inhibition that, in turn, modulated Fstl1 impacting thus on mouse epicardial cell transition.
Epicardial deployment is essential for myocardial wall growth, as indicated in previous sections. Absence or impaired development of the epicardial layer results in thin myocardium and reduced compact myocardium development [133]. Jang et al. [134] recently reported that HDAC3 regulation of miR-322 and miR-503 in the epicardial layer is essential for the modulation of two distinct growth factors, i.e. Igf2 and Fgf9, that if impaired restrict myocardial growth.
Activation of the epicardial layer is required for cardiac repair in different species. Curiously, proepicardial cells, if cultured in isolation, can spontaneously generate beating cardiomyocytes, a process that is promoted by Bmp and halted by Fgf signaling [135]. Furthermore, adult epicardial cells, if primed with thymosin 4, can differentiate into mature and fully functional integrated cardiomyocytes in adverse conditions, i.e. in myocardial infarction, yet in a very limited yield [136]. All these evidences nonetheless support the notion of a key role of the epicardial cells in cardiac regeneration. Dueñas et al. [137] demonstrated that Bmp and Fgf regulated miR-195 expression and furthermore miR-195 administration could promote PE to enhance cardiomyogenesis, a process modulated by Smurf1 and Smad3 in chicken explants. More recently, Garcia-Padilla et al. [138] reported that while Bmp and Fgf signaling could similarly modulate miR-195 expression in mouse PE explants, they failed in augment cardiomyogenesis, reporting thus species-specific differences in this microRNA modulated pathway.
Cardiomyocyte proliferation represents a key biological cornerstone to heal the broken heart. Diverse experiments in different species have reported an essential role of the epicardium promoting cardiomyocyte proliferation in several cardiac injury models [139,140,141,142]. Del Campo et al. [143] reported that epicardial-derived extracellular vesicles (EVs) loaded with cardiomyogenic reparative microRNAs, i.e. miR-30a, miR-30e, miR-27a and miR-100, were capable of inducing cardiomyocyte cell cycle reentry in a mouse model of myocardial infarction. Similarly and more recently, Zhu et al. [144] reported that lncRNA TARID could modulate Tcf21 transcription faction function in EPDCs leading to improve cardiac function in a mouse model of myocardial infarction.
In sum these studies reported the emerging role of non-coding RNAs modulating key biological processes orchestrated by or with contribution of the epicardium. In coming years, we will witness increasing evidence of the functional role of these types as well as of other types of non-coding RNAs, including therein lncRNAs and circRNAs in epicardial development in homeostasis and disease.
Conclusions and perspectives
For a long time, it was believed that the epicardium derived from the outer layer of the embryonic heart, and so labelled as 'epimyocardium'. However, more recent studies have revealed an extracardiac origin of the epicardium - derived from a proepicardium located at the posterior end of the cardiac tube- together with evidence of an epicardial EMT process, opening a new highly productive research field. We now understand that a major portion of cardiac cells, primarily coronary smooth muscle and fibroblasts, originate from the embryonic epicardium. There is also evidence suggesting the existence of a minor EPDCs differentiation into other cell types, such as coronary endothelium. In addition to this large-scale cellular contribution, the epicardium and its mesenchymal derivatives play a crucial signaling role in myocardial growth and maturation. Therefore, the role of epicardium is fundamental for cardiac morphogenesis, implying complex molecular mechanisms of control and regulation. As described in this review, numerous signaling and regulatory pathways -both at transcriptional and post-transcriptional levels- have been uncovered in the last three decades.
Despite the present knowledge of the epicardial contribution to cardiac development, many aspects still remain unclear. In particular, it is not well known whether different derivatives from the embryonic epicardium belong to distinct cell lineages established in the proepicardium, or that, alternatively, EPDCs are in fact pluripotential [see for a recent review 145]. The signaling mechanisms between the epicardium/EPDCS and the embryonic myocardium are not fully understood as yet. Noticeably, the importance of small and long non-coding RNAs in epicardium and EPDCs development is just beginning to be fruitful. As an interesting approach, is the adult epicardium capable of recovering the potential to acquire embryonic features? What is more, would it be able to transdifferentiate or produce signals for cardiac repair?
These and other questions related to the epicardium will continue to drive research on the embryonic epicardium and its crucial roles in cardiac morphogenesis and regeneration.
References
- Männer, J. The development of pericardial villi in the chick embryo. Anat Embryol (Berl) 1992, 186, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Männer, J. Experimental study on the formation of the epicardium in chick embryos. Anat Embryol (Berl) 1993, 187, 281–289. [Google Scholar] [CrossRef]
- Männer, J. Perez-Pomares, J.M., Macias, D., and Munoz-Chapuli, R. The origin, formation and developmental significance of the epicardium: a review. Cells Tissues Organs 2001, 169, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Männer J, Schlueter J, Brand T. Experimental analyses of the function of the proepicardium using a new microsurgical procedure to induce loss-of-proepicardial-function in chick embryos. Dev Dyn. 2005, 233, 1454–63. [CrossRef] [PubMed]
- Lie-Venema H, van den Akker NM, Bax NA, Winter EM, Maas S, Kekarainen T, Hoeben RC, deRuiter MC, Poelmann RE, Gittenberger-de Groot AC. Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. Scientific World Journal. 2007, 7, 1777–98. [CrossRef]
- Bax NA, Lie-Venema H, Vicente-Steijn R, Bleyl SB, Van Den Akker NM, Maas S, Poelmann RE, Gittenberger-de Groot AC. Platelet-derived growth factor is involved in the differentiation of second heart field-derived cardiac structures in chicken embryos. Dev Dyn. 2009, 238, 2658–69. [CrossRef]
- Carmona R, Guadix JA, Cano E, Ruiz-Villalba A, Portillo-Sánchez V, Pérez-Pomares JM, Muñoz-Chápuli R. The embryonic epicardium: an essential element of cardiac development. J Cell Mol Med. 2010, 14, 2066–72. [CrossRef]
- Niderla-Bielińska J, Jankowska-Steifer E, Flaht-Zabost A, Gula G, Czarnowska E, Ratajska A. Proepicardium: Current Understanding of its Structure, Induction, and Fate. Anat Rec (Hoboken). 2019, 302, 893–903. [CrossRef]
- Komiyama M, Ito K, Shimada Y. Origin and development of the epicardium in the mouse embryo. Anat Embryol (Berl) 1987, 176, 183–189. [CrossRef]
- Muñoz-Chápuli R, Macías D, González-Iriarte M, Carmona R, Atencia G, Pérez-Pomares JM. The epicardium and epicardial-derived cells: multiple functions in cardiac development. Rev Esp Cardiol. 2002, 55, 1070–82. [CrossRef]
- Balmer GM, Bollini S, Dubé KN, Martinez-Barbera JP, Williams O, Riley PR. Dynamic haematopoietic cell contribution to the developing and adult epicardium. Nat Commun. 2014, 5, 4054. [CrossRef] [PubMed]
- Mommersteeg MT, Domínguez JN, Wiese C, Norden J, de Gier-de Vries C, Burch JB, Kispert A, Brown NA, Moorman AF, Christoffels VM. The sinus venosus progenitors separate and diversify from the first and second heart fields early in development. Cardiovasc Res. 2010, 87, 92–101. [CrossRef] [PubMed]
- Serluca, F. C. Development of the proepicardial organ in the zebrafish. Dev. Biol. 2008, 315, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Icardo, J. M., Guerrero, A, Durán, A. C., Colvee, E., Domezain, A., and Sans-Coma, V. The development of the epicardiumin the sturgeon Acipenser naccarii. Anat. Rec. (Hoboken). 2009, 292, 1593–1601. [CrossRef]
- Schulte, I., Schlueter, J, Abu-Issa, R., Brand, T., and Männer, J. Morphological and molecular left-right asymmetries in the development of the proepicardium: a comparative analysis on mouse and chick embryos. Dev. Dyn. 2007, 236, 684–695. [CrossRef]
- vanWijk B, van den Berg G, Abu-Issa R, Barnett P, van der Velden S, Schmidt M, Ruijter JM, Kirby ML, Moorman AF, van den Hoff MJ. Epicardium and myocardium separate from a common precursor pool by crosstalk between bone morphogenetic protein- and fibroblast growth factor-signaling pathways. Circ Res 2009, 105, 431–441. [CrossRef]
- Maya-Ramos L, Cleland J, Bressan M, Mikawa T. Induction of the Proepicardium. J Dev Biol. 2013, 1, 82–91. [CrossRef]
- Schlueter J, Brand T. Subpopulation of proepicardial cells is derived from the somatic mesoderm in the chick embryo. Circ Res. 2013, 113, 1128–37. [CrossRef]
- Torlopp A, Schlueter J, Brand T. 2010. Role of fibroblast growth factor signaling during proepicardium formation in the chick embryo. Dev Dyn 239: 2393–2403. [CrossRef]
- Liu J, Stainier DY. Tbx5 and Bmp signaling are essential for proepicardium specification in zebrafish. Circ Res. 2010, 106, 1818–28. [CrossRef]
- Katz TC, Singh MK, Degenhardt K, Rivera-Feliciano J, Johnson RL, Epstein JA, Tabin CJ. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Developmental Cell. 2012, 22, 639–650. [CrossRef]
- Smits AM, Dronkers E, Goumans MJ. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol Res. 2018, 127, 129–140. [CrossRef]
- Plavicki JS, Hofsteen P, Yue MS, Lanham KA, Peterson RE, Heideman W. Multiple modes of proepicardial cell migration require heartbeat. BMC Dev Biol. 2014 ;14:18. 15 May. [CrossRef]
- Li J, Miao L, Zhao C, Shaikh Qureshi WM, Shieh D, Guo H, Lu Y, Hu S, Huang A, Zhang L, et al. CDC42 is required for epicardial and pro-epicardial development by mediating FGF receptor trafficking to the plasma membrane. Development 2017, 144, 1635–1647. [CrossRef]
- Cao Y, Duca S, Cao J. Epicardium in Heart Development. Cold Spring Harb Perspect Biol. 2020, 12, a037192. [CrossRef]
- Sanchez-Fernandez C, Rodriguez-Outeiriño L, Matias-Valiente L, Ramirez de Acuña F, Hernandez-Torres F, Lozano-Velasco E, Dominguez JN, Franco D, Aranega AE. Regulation of Epicardial Cell Fate during Cardiac Development and Disease: An Overview. Int J Mol Sci. 2022, 23, 3220. [CrossRef]
- Männer, J. Does the subepicardial mesenchyme contribute myocardioblasts to the myocardium of the chick embryo heart?A quail-chick chimera study tracing the fate op the epicardial primordium. Anat. Rec. 1999, 255, 212–226. [Google Scholar] [CrossRef]
- Perez-Pomares, J.M., Phelps, A., Sedmerova, M., and Wessels, A. Epicardial-like cells on the distal arterial end of the cardiac outflow tract do not derive from the proepicardium but are derivatives of the cephalic pericardium. Dev. Dyn. 2003, 227, 56–68. [CrossRef] [PubMed]
- Vrancken Peeters, M.-P.F.M., Gittenberger-de Groot, A.C., Mentink, M.M.T., and Poelmann, R.E. Smooth muscle cells and fibroblasts of the coronary arteries derive from epithelial-mesenchymal transformation of the epicardium. Anat. Embryol. 1999, 199, 367–378. [CrossRef]
- Lie-Venema, H., Eralp, I., Maas, S., Gittenberger-de Groot, A.C., Poelmann, R.E., and DeRuiter, M.C. Myocardial heterogeneity in permissiveness for epicardium-derived cells and endothelial precursor cells along the developing heart tube at the onset of coronary vascularization. Anat. Rec. 2005; 282A, 120–129.
- Gittenberger-de Groot, A.C. , Vrancken Peeters, M. -P.F.M., Mentink, M.M.T., Gourdie, R.G., and Poelmann, R.E. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar]
- Poelmann, R.E. , Gittenberger-de Groot, A. C., Mentink, M.M.T., Bökenkamp, R., and Hogers, B. Development of the cardiac coronary vascular endothelium, studied with antiendothelial antibodies, in chicken-quail chimeras. Circ. Res. 1993, 73, 559–568. [Google Scholar]
- Poelmann, R.E. , Lie-Venema, H. , and Gittenberger-de Groot, A.C. The role of the epicardium and neural crest as extracardiac contributors to coronary vascular development. Tex. Heart Inst. J. 2002, 29, 255–261. [Google Scholar]
- Mikawa T, Gourdie RG. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev Biol. 1996, 174, 221–32. [CrossRef] [PubMed]
- Wessels A, van den Hoff MJ, Adamo RF, Phelps AL, Lockhart MM, Sauls K, Briggs LE, Norris RA, van Wijk B, Perez-Pomares JM, Dettman RW, Burch JB. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev Biol. 2012, 366, 111–24. [CrossRef]
- Pérez-Pomares JM, Carmona R, González-Iriarte M, Atencia G, Wessels A, Muñoz-Chápuli R. Origin of coronary endothelial cells from epicardial mesothelium in avian embryos. Int J Dev Biol. 2002, 46, 1005–13.
- Carmona R, Barrena S, López Gambero AJ, Rojas A, Muñoz-Chápuli R. Epicardial cell lineages and the origin of the coronary endothelium. FASEB J. 2020, 34, 5223–5239. [CrossRef]
- Cano E, Carmona R, Ruiz-Villalba A, Rojas A, Chau YY, Wagner KD, Wagner N, Hastie ND, Muñoz-Chápuli R, Pérez-Pomares JM. Extracardiac septum transversum/proepicardial endothelial cells pattern embryonic coronary arterio- venous connections. Proc Natl Acad Sci U S A. 2016, 113, 656–61. [CrossRef]
- Cai CL, Martin JC, Sun Y, Cui L, Wang L, Ouyang K, Yang L, Bu L, Liang X, Zhang X, Stallcup WB, Denton CP, McCulloch A, Chen J, Evans SM. A myocardial lineage derives from Tbx18 epicardial cells. Nature. 2008, 454, 104–8. [CrossRef]
- Villa Del Campo C, Lioux G, Carmona R, Sierra R, Muñoz-Chápuli R, Clavería C, Torres M. Myc overexpression enhances of epicardial contribution to the developing heart and promotes extensive expansion of the cardiomyocyte population. Sci Rep. 2016 Oct 18;6:35366. doi: 10.1038/srep35366. Erratum in: Sci Rep. 2016 Dec 09;6:37880. PMID: 27752085; PMCID: PMC5082763.
- Christoffels VM, Grieskamp T, Norden J, Mommersteeg MT, Rudat C, Kispert A. Tbx18 and the fate of epicardial progenitors. Nature. 2009, 458, E8-E9; discussion E9-E10. [CrossRef]
- Rudat C, Kispert A. Wt1 and epicardial fate mapping. Circ Res. 2012, 111, 165–9. [CrossRef]
- Red-Horse K, Ueno H, Weissman IL, Krasnow MA. Coronary arteries form by Developmental reprogramming of venous cells. Nature 2010, 464, 549–553. [CrossRef]
- Chen HI, Sharma B, Akerberg BN, Numi HJ, Kivelä R, Saharinen P, Aghajanian H, McKay AS, Bogard PE, Chang AH, Jacobs AH, Epstein JA, Stankunas K, Alitalo K, Red-Horse K. The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Development. 2014, 141, 4500–12. [CrossRef]
- Pennisi DJ, Ballard VL, Mikawa T. Epicardium is required for the full rate of myocyte proliferation and levels of expression of myocyte mitogenic factors FGF2 and its receptor, FGFR-1, but not for transmural myocardial patterning in the embryonic chick heart. Dev Dyn. 2003, 228, 161–72. [CrossRef]
- Stuckmann I, Evans S, Lassar AB. Erythropoietin and retinoic acid, secreted from the epicardium, are required for cardiac myocyte proliferation. Dev Biol. 2003, 255, 334–49. [CrossRef] [PubMed]
- Merki E, Zamora M, Raya A, Kawakami Y, Wang J, Zhang X, Burch J, Kubalak SW, Kaliman P, Izpisua Belmonte JC, Chien KR, Ruiz-Lozano P. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc Natl Acad Sci U S A. 2005, 102, 18455–60. [CrossRef]
- Lavine KJ, Ornitz DM. Fibroblast growth factors and Hedgehogs: at the heart of the epicardial signaling center. Trends Genet. 2008, 24, 33–40. [CrossRef]
- Pennisi DJ, Mikawa T. FGFR-1 is required by epicardium-derived cells for myocardial invasion and correct coronary vascular lineage differentiation. Dev Biol. 2009, 328, 148–59. [CrossRef] [PubMed]
- Cavallero S, Shen H, Yi C, Lien CL, Kumar SR, Sucov HM. CXCL12 Signaling Is Essential for Maturation of the Ventricular Coronary Endothelial Plexus and Establishment of Functional Coronary Circulation. Dev Cell. 2015, 33, 469–77. [CrossRef] [PubMed]
- Smart, N. Bollini, S, K.N. Dubé, J.M. Vieira, B. Zhou, S. Davidson, D. Yellon, J.Riegler, A.N. Price, M.F. Lythgoe, W.T. Pu, P.R. Riley, De novo cardiomyocytes from within the activated adult heart after injury, Nature 474 (2011)640–644. [CrossRef]
- Braitsch, C.M.; Yutzey, K.E. Transcriptional control of cell lineage development in epicardium-derived Cells. J Dev Biol 2013, 1, 92–111. [Google Scholar] [CrossRef]
- Ishii, Y.; Langberg, J.D.; Hurtado, R.; Lee, S.; Mikawa, T. Induction of proepicardial marker gene expression by the liver bud. Development 2007, 134, 3627–37. [Google Scholar] [CrossRef]
- Schlueter, J.; Männer, J.; Brand, T. BMP is an important regulator of proepicardial identity in the chick embryo. Dev Biol 2006, 295, 546–58. [Google Scholar] [CrossRef]
- Tandon, P.; Wilczewski, C.M.; Williams, C.E.; Conlon, F.L. The Lhx9-integrin pathway is essential for positioning of the proepicardial organ. Development 2016, 143, 831–40. [Google Scholar] [CrossRef]
- Pombal MA, Carmona R, Megías M, Ruiz A, Pérez-Pomares JM, Muñoz-Chápuli R. Epicardial development in lamprey supports an evolutionary origin of the vertebrate epicardium from an ancestral pronephric external glomerulus. Evol Dev. 2008, 10, 210–6. [CrossRef] [PubMed]
- Cano, E.; Carmona, R.; Velecela, V.; Martínez-Estrada, O.; Muñoz-Chápuli, R. The proepicardium keeps a potential for glomerular marker expression which supports its evolutionary origin from the pronephros. Evol Dev 2015, 17, 224–30. [Google Scholar] [CrossRef] [PubMed]
- Powell, R.; Bubenshchikova, E.; Fukuyo, Y.; Hsu, C.; Lakiza, O.; Nomura, H.; Renfrew, E.; Garrity, D.; Obara, T. Wtip is required for proepicardial organ specification and cardiac left/right asymmetry in zebrafish. Mol Med Rep 2016, 14, 2665–78. [Google Scholar] [CrossRef] [PubMed]
- Kreidberg, J.A.; Sariola, H.; Loring, J.M.; Maeda, M.; Pelletier, J.; Housman, D.; Jaenisch, R. WT-1 is required for early kidney development. Cell 1993, 74, 679–91. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.D. Every Beat You Take-The Wilms' Tumor Suppressor WT1 and the Heart. Int J Mol Sci 2021, 22, 7675. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Wagner, K.D.; Scholz, H.; Kirschner, K.M.; Schedl, A. Intermediate filament protein nestin is expressed in developing kidney and heart and might be regulated by the Wilms' tumor suppressor Wt1. Am J Physiol Regul Integr Comp Physiol 2006, 291, R779–87. [Google Scholar] [CrossRef]
- Hsu, W.H.; Yu, Y.R.; Hsu, S.H.; Yu, W.C.; Chu, Y.H.; Chen, Y.J.; Chen, C.M.; You, L.R. The Wilms' tumor suppressor Wt1 regulates Coronin 1B expression in the epicardium. Exp Cell Res 2013, 319, 1365–81. [Google Scholar] [CrossRef]
- Wagner N, Wagner KD, Theres H, Englert C, Schedl A, Scholz H. Coronary vessel development requires activation of the TrkB neurotrophin receptor by the Wilms' tumor transcription factor Wt1. Genes Dev. 2005, 19, 2631–42. [CrossRef]
- Martínez-Estrada, O.M.; Lettice, L.A.; Essafi, A.; Guadix, J.A.; Slight, J.; Velecela, V.; Hall, E.; Reichmann, J.; Devenney, P.S.; Hohenstein, P.; Hosen, N.; Hill, R.E.; Muñoz-Chapuli, R.; Hastie, N.D. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat Genet 2010, 42, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Casanova, J.C.; Travisano, S.; de la Pompa, J.L. Epithelial-to-mesenchymal transition in epicardium is independent of Snail1. Genesis 2013, 51, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, M.; Nimura, K.; Mori, M.; Nakagami, H.; Kaneda, Y. The transcription factors Tbx18 and Wt1 control the epicardial epithelial-mesenchymal transition through bi-directional regulation of Slug in murine primary epicardial cells. PLoS One 2013, 8, e57829. [Google Scholar] [CrossRef] [PubMed]
- Tao, G.; Miller, L.J.; Lincoln, J. Snai1 is important for avian epicardial cell transformation and motility. Dev Dyn 2013, 242, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Guadix, J.A.; Ruiz-Villalba, A.; Lettice, L.; Velecela, V.; Muñoz-Chápuli, R.; Hastie, N.D.; Pérez-Pomares, J.M.; Martínez-Estrada, O.M. Wt1 controls retinoic acid signaling in embryonic epicardium through transcriptional activation of Raldh2. Development 2011, 138, 1093–7. [Google Scholar] [CrossRef]
- Wang, S.; Yu, J.; Jones, J.W.; Pierzchalski, K.; Kane, M.A.; Trainor, P.A.; Xavier-Neto, J.; Moise, A.R. Retinoic acid signaling promotes the cytoskeletal rearrangement of embryonic epicardial cells. FASEB J 2018, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.J.; Hutson, D.R.; Kubalak, S.W. Analysis of the proepicardium-epicardium transition during the malformation of the RXRalpha-/- epicardium. Dev Dyn 2005, 233, 1091–101. [Google Scholar] [CrossRef]
- Zamora, M.; Männer, J.; Ruiz-Lozano, P. Epicardium-derived progenitor cells require beta-catenin for coronary artery formation. Proc Natl Acad Sci U S A 2007, 104, 18109–14. [Google Scholar] [CrossRef]
- Von Gise, A.; Zhou, B.; Honor, L.B.; Ma, Q.; Petryk, A.; Pu, W.T. WT1 regulates epicardial epithelial to mesenchymal transition through β-catenin and retinoic acid signaling pathways. Dev Biol 2011, 356, 421–31. [Google Scholar] [CrossRef]
- Khan, F.F.; Li. Y.; Balyan, A.; Wang, Q.T. WTIP interacts with ASXL2 and blocks ASXL2-mediated activation of retinoic acid signaling. Biochem Biophys Res Commun 2014, 451, 101–6. [Google Scholar] [CrossRef]
- Combs, M.D.; Braitsch, C.M.; Lange, A.W.; James, J.F.; Yutzey, K.E. NFATC1 promotes epicardium-derived cell invasion into myocardium. Development 2011, 138, 1747–57. [Google Scholar] [CrossRef]
- Yang, J.; Zeini, M.; Lin, C.Y.; Lin, C.J.; Xiong, Y.; Shang, C.; Han, P.; Li, W.; Quertermous, T.; Zhou, B.; Chang, C.P. Epicardial calcineurin-NFAT signals through Smad2 to direct coronary smooth muscle cell and arterial wall development. Cardiovasc Res 2014, 101, 120–9. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Doughman, Y.; Yang, K.; Ramirez-Bergeron, D.; Watanabe, M. Epicardial HIF signaling regulates vascular precursor cell invasion into the myocardium. Dev Biol 2013, 376, 136–49. [Google Scholar] [CrossRef] [PubMed]
- DeLaughter, D.M.; Clark, C.R.; Christodoulou, D.C.; Seidman, C.E.; Baldwin, H.S.; Seidman, J.G.; Barnett, J.V. Transcriptional Profiling of Cultured, Embryonic Epicardial Cells Identifies Novel Genes and Signaling Pathways Regulated by TGFβR3 In Vitro. PLoS One 2016, 11, e0159710. [Google Scholar] [CrossRef] [PubMed]
- Trembley, M.A.; Velasquez, L.S.; de Mesy Bentley, K.L.; Small, E.M. Myocardin-related transcription factors control the motility of epicardium-derived cells and the maturation of coronary vessels. Development 2015, 142, 21–30. [Google Scholar] [CrossRef]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev Cell 2012, 22, 639–50. [Google Scholar] [CrossRef] [PubMed]
- Lupu, I.E.; Redpath, A.N.; Smart, N. Spatiotemporal analysis reveals overlap of key proepicardial markers in the developing murine heart. Stem Cell Reports 2020, 14, 770–787. [Google Scholar] [CrossRef]
- Hu, H.; Lin, S.; Wang, S.; Chen, X. The role of transcription factor 21 in epicardial cell differentiation and the development of coronary heart disease. Front Cell Dev Biol 2020, 8, 457. [Google Scholar] [CrossRef]
- Braitsch, C.M.; Combs, M.D.; Quaggin, S.E.; Yutzey, K.E. Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev Biol 2012, 368, 345–57. [Google Scholar] [CrossRef]
- Tandon, P.; Miteva, Y.V.; Kuchenbrod, L.M.; Cristea, I.M.; Conlon, F.L. Tcf21 regulates the specification and maturation of proepicardial cells. Development 2013, 140, 2409–21. [Google Scholar] [CrossRef]
- Boezio, G.L.M.; Zhao, S.; Gollin, J.; Priya, R.; Mansingh, S.; Guenther, S.; Fukuda, N.; Gunawan, F.; Stainier, D.Y.R. The developing epicardium regulates cardiac chamber morphogenesis by promoting cardiomyocyte growth. Dis Model Mech 2023, 16, dmm049571. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, L.; McManus, S.A.; Moignard, V.; Sebukhan, D.; Delaune, A.; Andrews, S.; Bernard, W.G.; Morrison, M.A.; Riley, P.R.; Göttgens, B.; Gambardella Le Novère, N.; Sinha, S. BNC1 regulates cell heterogeneity in human pluripotent stem cell-derived epicardium. Development 2019, 146, dev174441. [Google Scholar] [CrossRef] [PubMed]
- Greulich, F.; Farin, H.F.; Schuster-Gossler, K.; Kispert, A. Tbx18 function in epicardial development. Cardiovasc Res 2012, 96, 476–83. [Google Scholar] [CrossRef] [PubMed]
- Greulich, F.; Rudat, C.; Farin, H.F.; Christoffels, V.M.; Kispert, A. Lack of genetic Interaction between Tbx18 and Tbx2/Tbx20 in mouse epicardial development. PLoS One 2016, 11, e0156787. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Gao, Y.; Xiao, S.; Qin, Q.; Wei, X.; Yan, Y.; Wu, L.; Deng, S.; Du, J.; Liu, Y.; She, Q. Hypoxia induced the differentiation of Tbx18-positive epicardial cells to CoSMCs. Sci Rep 2016, 6, 30468. [Google Scholar] [CrossRef] [PubMed]
- Diman, N.Y.; Brooks, G.; Kruithof, B.P.; Elemento, O.; Seidman, J.G.; Seidman, C.E.; Basson, C.T.; Hatcher, C.J. Tbx5 is required for avian and mammalian epicardial formation and coronary vasculogenesis. Circ Res 2014, 115, 834–44. [Google Scholar] [CrossRef] [PubMed]
- Shelton, E.L.; Yutzey, K.E. Twist1 function in endocardial cushion cell proliferation, migration, and differentiation during heart valve development. Dev Biol 2008, 317, 282–95. [Google Scholar] [CrossRef]
- Levay, A.K.; Peacock, J.D.; Lu, Y.; Koch, M.; Hinton, R.B. Jr; Kadler, K.E.; Lincoln, J. Scleraxis is required for cell lineage differentiation and extracellular matrix remodeling during murine heart valve formation in vivo. Circ Res 2008, 103, 948–56. [Google Scholar] [CrossRef]
- Espira, L.; Lamoureux, L.; Jones, S.C.; Gerard, R.D.; Dixon, I.M.; Czubryt, M.P. The basic helix-loop-helix transcription factor scleraxis regulates fibroblast collagen synthesis. J Mol Cell Cardiol 2009, 47, 188–95. [Google Scholar] [CrossRef]
- Barnes, R.M.; Firulli, B.A.; VanDusen, N.J.; Morikawa, Y.; Conway, S.J.; Cserjesi, P.; Vincentz, J.W.; Firulli, A.B. Hand2 loss-of-function in Hand1-expressing cells reveals distinct roles in epicardial and coronary vessel development. Circ Res 2011, 108, 940–9. [Google Scholar] [CrossRef]
- Smith, C.L.; Baek, S.T.; Sung, C.Y.; Tallquist, M.D. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ Res 2011, 108, e15–26. [Google Scholar] [CrossRef] [PubMed]
- Kolander, K.D.; Holtz, M.L.; Cossette, S.M.; Duncan, S.A.; Misra, R.P. Epicardial GATA factors regulate early coronary vascular plexus formation. Dev Biol 2014, 386, 204–15. [Google Scholar] [CrossRef] [PubMed]
- Borok, M.J.; Papaioannou, V.E.; Sussel, L. Unique functions of Gata4 in mouse liver induction and heart development. Dev Biol 2016, 410, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Wang, L.; Wang, H.; Shen, T.; Yang, Y.; Sun, Y.; Tang, N.; Ni, T.; Zhu, J.; Mailman, R.B.; Wang, Y. A novel role of CDX1 in embryonic epicardial development. PLoS One 2014, 9, e103271. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Hill, M.C.; Zhang, M.; Martin, T.J.; Morikawa, Y.; Wang, S.; Moise, A.R.; Wythe, J.D.; Martin, J.F. Hippo signaling plays an essential role in cell state transitions during cardiac fibroblast development. Dev Cell 2018, 45, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Liu, B.; Deng, S.; Du, J.; She, Q. Agrin Yes-associated Protein Promotes the Proliferation of Epicardial Cells. J Cardiovasc Pharmacol 2021, 77, 94–99. [Google Scholar] [CrossRef]
- Astanina, E.; Doronzo, G.; Corà, D.; Neri, F.; Oliviero, S.; Genova, T.; Mussano, F.; Middonti, E.; Vallariello, E.; Cencioni, C.; Valdembri, D.; Serini, G.; Limana, F.; Foglio, E.; Ballabio, A.; Bussolino, F. The TFEB-TGIF1 axis regulates EMT in mouse epicardial cells. Nat Commun 2022, 13, 5191. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, 17–29. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Mattick JS, Amaral PP, Carninci P, Carpenter S, Chang HY, Chen LL, Chen R, Dean C, Dinger ME, Fitzgerald KA, et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol. 2023, 24, 430–447. [CrossRef]
- Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021, 22, 96–118. [CrossRef]
- Liu CX, Chen LL. Circular RNAs: Characterization, cellular roles, and applications. Cell. 2022, 185, 2016–2034. [CrossRef] [PubMed]
- Lee YS, Dutta A. MicroRNAs in cancer. 2009;4:199-227. Annu Rev Pathol. 2009, 4, 199–227. [CrossRef]
- Xu J, Wu KJ, Jia QJ, Ding XF. Roles of miRNA and lncRNA in triple-negative breast cancer. J Zhejiang Univ Sci B. 2020, 21, 673–689. [CrossRef] [PubMed]
- Wu KL, Tsai YM, Lien CT, Kuo PL, Hung AJ. The Roles of MicroRNA in Lung Cancer. Int J Mol Sci. 2019, 20, 1611. [CrossRef]
- Wojciechowska A, Braniewska A, Kozar-Kamińska K. MicroRNA in cardiovascular biology and disease. Adv Clin Exp Med. 2017, 26, 865–874. [CrossRef] [PubMed]
- Huang, Y. The novel regulatory role of lncRNA-miRNA-mRNA axis in cardiovascular diseases. J Cell Mol Med. 2018, 22, 5768–5775. [Google Scholar] [CrossRef]
- Gomes CPC, Schroen B, Kuster GM, Robinson EL, Ford K, Squire IB, Heymans S, Martelli F, Emanueli C, Devaux Y; EU-CardioRNA COST Action (CA17129). Regulatory RNAs in Heart Failure. Circulation. 2020, 141, 313–328. [CrossRef]
- Chinchilla A, Lozano E, Daimi H, Esteban FJ, Crist C, Aranega AE, Franco D. MicroRNA profiling during mouse ventricular maturation: a role for miR-27 modulating Mef2c expression. Cardiovasc Res. 2011, 89, 98–108. [CrossRef] [PubMed]
- Bonet F, Hernandez-Torres F, Esteban FJ, Aranega A, Franco D. Comparative Analyses of MicroRNA Microarrays during Cardiogenesis: Functional Perspectives. Microarrays (Basel). 2013, 2, 81–96. [CrossRef]
- Lozano-Velasco E, Garcia-Padilla C, Del Mar Muñoz-Gallardo M, Martinez-Amaro FJ, Caño-Carrillo S, Castillo-Casas JM, Sanchez-Fernandez C, Aranega AE, Franco D. Post-Transcriptional Regulation of Molecular Determinants during Cardiogenesis. Int J Mol Sci. 2022, 23, 2839. [CrossRef]
- Caño-Carrillo S, Lozano-Velasco E, Castillo-Casas JM, Sánchez-Fernández C, Franco D. The Role of ncRNAs in Cardiac Infarction and Regeneration. J Cardiovasc Dev Dis. 2023, 10, 123. [CrossRef]
- Fang Y, Xu Y, Wang R, Hu L, Guo D, Xue F, Guo W, Zhang D, Hu J, Li Y, Zhang W, Zhang M. Recent advances on the roles of LncRNAs in cardiovascular disease. J Cell Mol Med. 2020, 24, 12246–12257. [CrossRef]
- Franco D, Aranega A, Dominguez JN. Non-coding RNAs and Atrial Fibrillation. Adv Exp Med Biol. 2020;1229:311-325. [CrossRef] [PubMed]
- Garcia-Padilla C, Lozano-Velasco E, Garcia-Lopez V, Aranega A, Franco D, Garcia-Martinez V, Lopez-Sanchez C. Comparative Analysis of Non-Coding RNA Transcriptomics in Heart Failure. Biomedicines. 2022, 10, 3076. [CrossRef]
- Zeng Y, Wu N, Zhang Z, Zhong L, Li G, Li Y. Non-coding RNA and arrhythmias: expression, function, and molecular mechanism. Europace. 2023, 25, 1296–1308. [CrossRef]
- Dueñas A, Expósito A, Aranega A, Franco D. The Role of Non-Coding RNA in Congenital Heart Diseases. J Cardiovasc Dev Dis. 2019, 6, 15. [CrossRef]
- Singh MK, Lu MM, Massera D, Epstein JA. MicroRNA-processing enzyme Dicer is required in epicardium for coronary vasculature development. J Biol Chem. 2011, 286, 41036–45. [CrossRef]
- Expósito-Villén A, E Aránega A, Franco D. Functional Role of Non-Coding RNAs during Epithelial-To-Mesenchymal Transition. Noncoding RNA. 2018, 4, 14. [CrossRef]
- Zou XZ, Liu T, Gong ZC, Hu CP, Zhang Z. MicroRNAs-mediated epithelial- mesenchymal transition in fibrotic diseases. Eur J Pharmacol. 2017 Feb 5;796:190-206. [CrossRef] [PubMed]
- Vettori S, Gay S, Distler O. Role of MicroRNAs in Fibrosis. Open Rheumatol J. 2012;6:130-9. [CrossRef]
- Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, Ali H, Braga L, Gorgodze N, Bernini F, Burchielli S, Collesi C, Zandonà L, Sinagra G, Piacenti M, Zacchigna S, Bussani R, Recchia FA, Giacca M. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature. 2019, 569, 418–422. [CrossRef]
- Gao L, Qiu F, Cao H, Li H, Dai G, Ma T, Gong Y, Luo W, Zhu D, Qiu Z, Zhu P, Chu S, Yang H, Liu Z. Therapeutic delivery of microRNA-125a-5p oligonucleotides improves recovery from myocardial ischemia/reperfusion injury in mice and swine. Theranostics. 2023, 13, 685–703. [CrossRef] [PubMed]
- Wadley GD, Lamon S, Alexander SE, McMullen JR, Bernardo BC. Noncoding RNAs regulating cardiac muscle mass. J Appl Physiol (1985). 2019, 127, 633–644. [CrossRef] [PubMed]
- Qu S, Zeng C, Wang WE. Noncoding RNA and Cardiomyocyte Proliferation. Stem Cells Int. 2017;2017:6825427. [CrossRef]
- Abbas N, Perbellini F, Thum T. Non-coding RNAs: emerging players in cardiomyocyte proliferation and cardiac regeneration. Basic Res Cardiol. 2020, 115, 52. [CrossRef]
- Brønnum H, Andersen DC, Schneider M, Sandberg MB, Eskildsen T, Nielsen SB, Kalluri R, Sheikh SP. miR-21 promotes fibrogenic epithelial-to-mesenchymal transition of epicardial mesothelial cells involving Programmed Cell Death 4 and Sprouty-1. PLoS One. 2013, 8, e56280. [CrossRef]
- Pontemezzo E, Foglio E, Vernucci E, Magenta A, D'Agostino M, Sileno S, Astanina E, Bussolino F, Pellegrini L, Germani A, Russo MA, Limana . miR-200c-3p Regulates Epitelial-to-Mesenchymal Transition in Epicardial Mesothelial Cells by Targeting Epicardial Follistatin-Related Protein 1. Int J Mol Sci. 2021, 22, 4971. [CrossRef]
- Takahashi M, Yamagishi T, Narematsu M, Kamimura T, Kai M, Nakajima Y. Epicardium is required for sarcomeric maturation and cardiomyocyte growth in the ventricular compact layer mediated by transforming growth factor β and fibroblast growth factor before the onset of coronary circulation. Congenit Anom (Kyoto). 2014, 54, 162–71. [CrossRef] [PubMed]
- Jang J, Song G, Pettit SM, Li Q, Song X, Cai CL, Kaushal S, Li D. Epicardial HDAC3 Promotes Myocardial Growth Through a Novel MicroRNA Pathway. Circ Res. 2022, 131, 151–164. [CrossRef]
- Kruithof BP, van Wijk B, Somi S, Kruithof-de Julio M, Pérez Pomares JM, Weesie F, Wessels A, Moorman AF, van den Hoff MJ. BMP and FGF regulate the differentiation of multipotential pericardial mesoderm into the myocardial or epicardial lineage. Dev Biol. 2006, 295, 507–22. [CrossRef] [PubMed]
- Smart N, Bollini S, Dubé KN, Vieira JM, Zhou B, Davidson S, Yellon D, Riegler J, Price AN, Lythgoe MF, Pu WT, Riley PR. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 2011, 474, 640–4. [CrossRef]
- Dueñas A, Expósito A, Muñoz MDM, de Manuel MJ, Cámara-Morales A, Serrano-Osorio F, García-Padilla C, Hernández-Torres F, Domínguez JN, Aránega A, Franco D. MiR-195 enhances cardiomyogenic differentiation of the proepicardium/septum transversum by Smurf1 and Foxp1 modulation. Sci Rep. 2020, 10, 9334. [CrossRef]
- Garcia-Padilla C, Hernandez-Torres F, Lozano-Velasco E, Dueñas A, Muñoz-Gallardo MDM, Garcia-Valencia IS, Palencia-Vincent L, Aranega A, Franco D. The Role of Bmp- and Fgf Signaling Modulating Mouse Proepicardium Cell Fate. Front Cell Dev Biol. 2022 Jan 4;9:757781. [CrossRef]
- Boezio GLM, Zhao S, Gollin J, Priya R, Mansingh S, Guenther S, Fukuda N, Gunawan F, Stainier DYR. The developing epicardium regulates cardiac chamber morphogenesis by promoting cardiomyocyte growth. Dis Model Mech. 2023, 16, dmm049571. [CrossRef] [PubMed]
- Sun J, Peterson EA, Wang AZ, Ou J, Smith KE, Poss KD, Wang J. <i>hapln1</i> Defines an Epicardial Cell Subpopulation Required for Cardiomyocyte Expansion During Heart Morphogenesis and Regeneration. Circulation. 2022, 146, 48–63. [CrossRef]
- Huang Y, Harrison MR, Osorio A, Kim J, Baugh A, Duan C, Sucov HM, Lien CL. Igf Signaling is Required for Cardiomyocyte Proliferation during Zebrafish Heart Development and Regeneration. PLoS One. 2013, 8, e67266. [CrossRef]
- Itou J, Akiyama R, Pehoski S, Yu X, Kawakami H, Kawakami Y. Regenerative responses after mild heart injuries for cardiomyocyte proliferation in zebrafish. Dev Dyn. 2014, 243, 1477–86. [CrossRef]
- Del Campo CV, Liaw NY, Gunadasa-Rohling M, Matthaei M, Braga L, Kennedy T, Salinas G, Voigt N, Giacca M, Zimmermann WH, Riley PR. Regenerative potential of epicardium-derived extracellular vesicles mediated by conserved miRNA transfer. Cardiovasc Res. 2022, 118, 597–611. [CrossRef]
- Zhu D, Liu S, Huang K, Li J, Mei X, Li Z, Cheng K. Intrapericardial long non-coding RNA-Tcf21 antisense RNA inducing demethylation administration promotes cardiac repair. Eur Heart J. 2023, 44, 1748–1760. [CrossRef] [PubMed]
- Sanchez-Fernandez, C. Rodriguez-Outeiriño, L. Matias-Valiente, L. de Acuña, F.R., Franco, D. Aránega, A.E. Understanding Epicardial Cell Heterogeneity during Cardiogenesis and Heart Regeneration. J. Cardiovasc. Dev. Dis. 2023, 10, 376. [CrossRef]
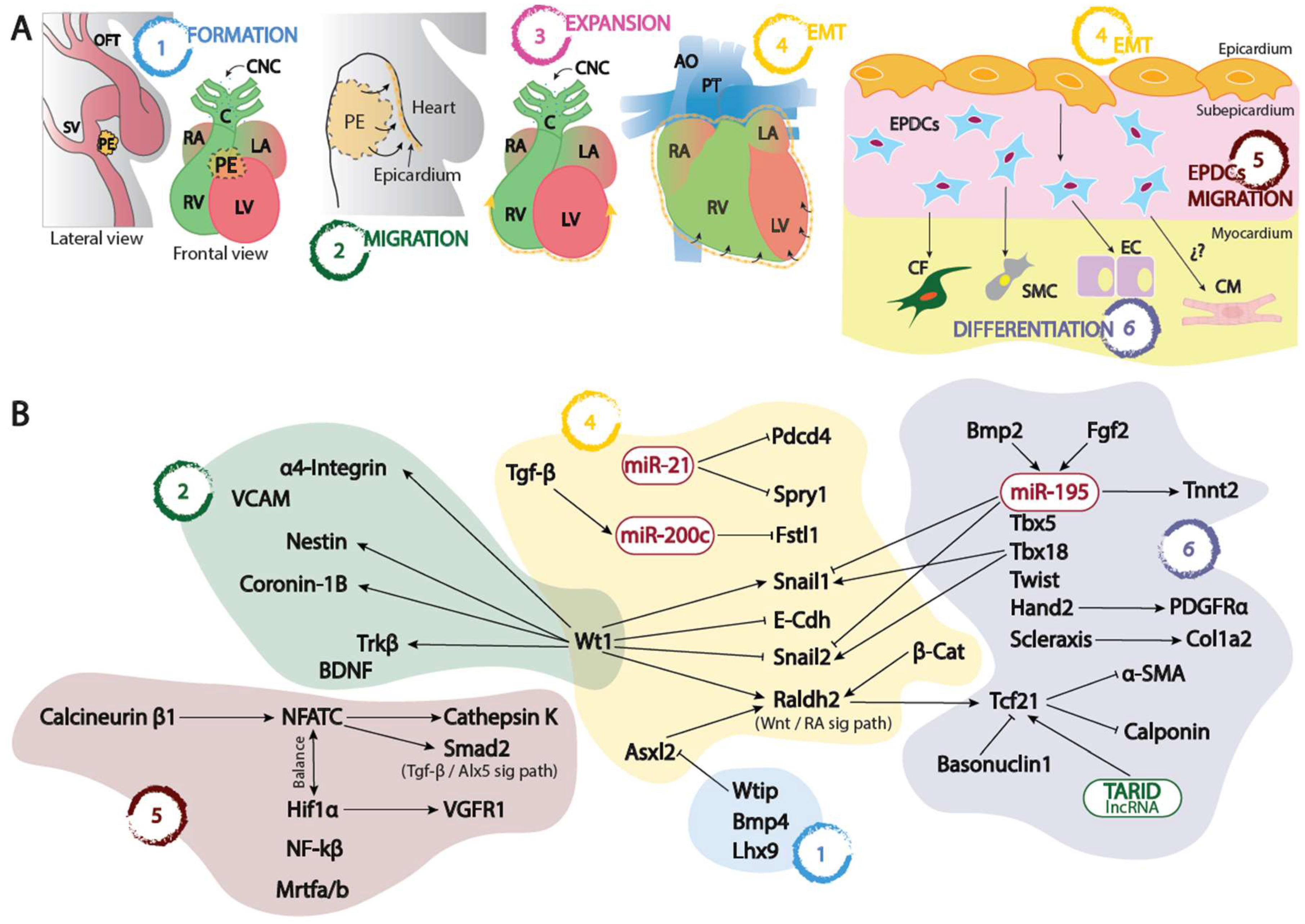
Figure 1.
Panel A. Schematic representation of the proepicardium (PE) and embryonic epicardium formation since its origin at the sinus venosus-septum transversum (1), its migration (2) and expansion (3) into the naked myocardium, its epithelial to mesenchymal transition leading to the formation of the EPCDs (4) and finally the migration and invasion (5) of the embryonic myocardium differentiating into distinct cell types (6). Panel B. Schematic representation of the distinct transcriptional and post-transcriptional regulatory mechanisms involved in each of the distinct processes depicted in panel A. OFT, outflow tract; SV, sinus venosus; PE, proepicardium; RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; CNC, cardiac neural crest, C, conus; AO, aorta; PT, pulmonary trunk; EPDCs, epicardial derived cells; CF, cardiac fibroblasts; SMC, smooth muscle cells; EC, endothelial cells; CM, cardiomyocytes; EMT, epithelial to mesenchymal transition.
Figure 1.
Panel A. Schematic representation of the proepicardium (PE) and embryonic epicardium formation since its origin at the sinus venosus-septum transversum (1), its migration (2) and expansion (3) into the naked myocardium, its epithelial to mesenchymal transition leading to the formation of the EPCDs (4) and finally the migration and invasion (5) of the embryonic myocardium differentiating into distinct cell types (6). Panel B. Schematic representation of the distinct transcriptional and post-transcriptional regulatory mechanisms involved in each of the distinct processes depicted in panel A. OFT, outflow tract; SV, sinus venosus; PE, proepicardium; RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; CNC, cardiac neural crest, C, conus; AO, aorta; PT, pulmonary trunk; EPDCs, epicardial derived cells; CF, cardiac fibroblasts; SMC, smooth muscle cells; EC, endothelial cells; CM, cardiomyocytes; EMT, epithelial to mesenchymal transition.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



