
Submitted:
07 October 2023
Posted:
08 October 2023
You are already at the latest version
Abstract
Background: Chronic Hepatitis B Virus (HBV) infection is a global health concern, associated with severe liver diseases, necessitating ongoing research on novel drug candidates. This study aims to identify potential drug candidates targeting HBV core protein (HBcAg) and disrupting capsid assembly, a critical step in the virus's life cycle. Methods: HBcAg in complex with HBV inhibitors were obtained from the Protein Data Bank (PDB). CavityPlus server was used for analysis of druggable cavity. Structure-based pharmacophores were extracted from identified cavities, and potential allosteric ligand binding sites were assessed using CavPharmer, CorrSite, and CovCys. LigandScout was employed for ligand-based pharmacophore screening against an FDA-approved library. The ZINC database was screened with features extracted from CavPharmer. Molecular docking studies were conducted using Autodock Vina. Lead compounds were selected based on docking scores, binding modes, and interactions within the druggable cavity. Results: Strong druggable pockets were found for Ciclopirox, while Compound 24, NVR10-001E2, and others showed medium to weak pockets. Ligand-based pharmacophores varied in size and complexity. Screening revealed potential hits matching these pharmacophores, including Ciclopirox olamine, Voriconazole, Enasidenib, and Statins. A large compound database search yielded additional hits like ZINC86859997 and ZINC63280172. Docking analyses confirmed these hits' potential, highlighting their interactions with critical HBc protein residues, offering promising leads for hepatitis B drug development. Conclusions: Voriconazole, Enasidenib, and Lovastatin have shown promises. These hits displayed favorable interactions with crucial HBc protein residues, indicating their potential as lead compounds The mechanism of action of statins with anti-HBV activities also highlighted. This comprehensive approach offers valuable insights into targeting HBc protein for antiviral drug discovery.
Keywords:
hepatitis B
; antiviral
; drug discovery
; HBc protein
; pharmacophore-based screening
; drug repurposing
1. Introduction
Chronic Hepatitis B Virus (CHB) infection as a significant health issue affected ~300 million of people worldwide, posing significant risks for liver diseases, including fibrosis, cirrhosis, and hepatocellular carcinoma [1]. In this regard, the primary goal of HBV treatment is to prevent the already mentioned serious HBV-associated liver diseases.
The therapies for HBV infection are aimed for a functional and complete cure, by inducing sustained HBV surface antigen (HBsAg) loss or seroconversion with no formation or detection of new covalently closed circular DNA (cccDNA) molecules [2,3,4]. While vaccines have reduced HBV prevalence, challenges like limited access to vaccination and vaccine non-response still exist [5]. Current treatments for chronic HBV infection include pegylated interferons (PEG-IFNs) and nucleoside/nucleotide analogs (NAs) such as lamivudine (3TC), adefovir (ADF), entecavir (ETV), tenofovir disoproxil (TDF), and tenofovir alafenamide (TAF). Curative outcome of these interventions has limited success due to the development of drug-resistant mutants, potential side effects, long-term therapy, and presence of cccDNA. Interferons are effective but often poorly tolerated, especially at high doses [6].
Therefore, there is still needs for more anti-HBV research, introducing novel therapeutic options. The HBV genome encodes four overlapping open reading frames (ORFs), including P (polymerase), C (core), S (surface), and X (HBxAg). Core antigen (HBcAg) assembles into capsids with T = 4 or T = 3 symmetry, composed of HBc dimers [7,8]. The icosahedral HBcAg comprises three domains, consisting of a 149-amino acid N-terminal assembly domain (Cp149), a linker region, and a 34-residue disordered nucleic acid-binding C-terminal domain essential for viral genome replication and stability. HBcAg is essential for genome packaging and reverse transcription [9], and the capsid assembly process in HBV life cycle is a critical target for the treatment of CHB.
Capsid assembly modulators (CAMs) have shown potential for curing HBV infection. CAMs are compounds designed to disrupt nucleocapsid assembly by affecting the kinetics and interactions between core dimers [10,11,12]. CAMs are categorized into two families (I and II) based on their mechanisms of action. CAM I molecules (e.g., Bay 41-4109 and GLS4) induce misassembly of core dimers, leading to aberrant capsid structures, while CAM II molecules (e.g., PPA and SBA) stabilize core dimer interactions, promoting immature hasty nucleocapsid assembly [13]. Collectively, CAMs bind to a hydrophobic pocket at the core dimerization interface, causing structural changes that block capsid assembly and potentially inhibit HBV replication. The advent of CAMs as a potential treatment strategy for chronic HBV infection has garnered significant attention in recent research. Some CAMs have advanced to different clinical trials phases (Yuen et al., 2022; Zoulim et al., n.d.). Among these CAMs, GST-HG141, a novel compound undergoing phase I clinical evaluation, has demonstrated remarkable promise. GST-HG141 promotes the formation of "empty" capsids that lack genetic material by acceleration of HBV capsid assembly [19]. This unique mode of action, coupled with its favorable pharmacokinetic properties, has further bolstered its potential as an HBV treatment. In vivo studies in an AAV-HBV mouse model have showcased its dose-dependent reduction in serum and liver HBV DNA levels, albeit with a rebound effect following treatment cessation. Additionally, GST-HG141 has exhibited promising synergistic effects when used in combination with other HBV inhibitors and has displayed specificity for HBV, sparing other DNA and RNA viruses in vitro [19]. A further study highlighted HEC72702, with a unique chirality, as a potent anti-HBV with improved pharmacokinetics, and reduced side effects. Additionally, it demonstrated higher stability, induced fewer structural changes, and had a stronger binding affinity to HBcAg, making it a promising candidate for HBV inhibition [20]. Further computational study by Tomaya et al demonstrated a novel CAM I pyrimidotriazine derivatives, Compound 2b with significant anti-HBV activity in cell cultures. The compound inhibited capsid formation, reduced capsid-associated HBV RNA and DNA levels, and showed effectiveness against an ETV/3TC-resistant HBV mutant [21].
Further inhibitors of HBcAg have also issued with promising results both in vivo and in vitro. These are included Yhhu6669 [22], pyrazolyl-thiazole (PT), glyoxamide-pyrrolamide (GPA), and dibenzo-thiazepin-2-one (DBT) [23], JNJ-64530440 [24], Compound A and B [25], Ciclopirox [26], NVR 3-778 [27], HEC72702 [28], JNJ-827 and JNJ-890 [29], Cetylpyridinium chloride (CPC) [30], Bay 41-4109 [18,31], isothiafludine (NZ-4) [5], and other derivatives [3,32]. These interests in developing direct HBcAg-binders show the novelty of such approaches, and repurposing drugs with same or enhanced direct-action on HBV core protein still holds much promises.
The present study contributes significantly in the field of anti-HBV research by introducing novel potential lead compounds. By utilizing computational-aided approaches like structure-based and ligand-based pharmacophore drug discovery, this study helps to accelerate the drug development process by screening a vast library of compounds to identify those with the potential to disrupt HBV capsid assembly, a crucial step in the virus's life cycle. Such approaches enable the rapid identification of promising drug candidates, reducing the time and resources required for experimental drug discovery. Ultimately, this study paves the way of the repurposing FDA-approved drugs for HBV, addressing a critical gap in the field and offering new hope for individuals affected by this persistent and challenging viral infection.
2. Materials and Methods
Preparation of Capsid protein structures
Crystallographic structures of HBcAg were retrieved from protein database (PDB; https://www.rcsb.org/). Accordingly, crystallographic structures of the protein in complex with the new HBV inhibitors, including Ciclopirox (PDB_ID: 6J10), NVR10-001E2 (PDB_ID: 5E0I), sulfamoylbenzamide (PDB_ID: 5T2P), heteroaryldihydropyrimidine (PDB_ID: 5WRE), N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide (PDB_ID: 7K5M), and Compound 24 or (6S,8R)-N-(3-bromo-4-fluorophenyl)-8-fluoro-10-methyl-11-oxo-1,3,4,7,8,9,10,11-octahydro-2H-pyrido[4',3':3,4]pyrazolo[1,5-a][1,4]diazepine-2-carboxamide (PDB_ID: 8GIH) were collected with the resolutions of 2.30 Å, 1.95 Å, 1.69 Å, 1.95 Å, 2.65 Å, and 2.65 Å, respectively [26,41,42,43,44,45].
The protein chain in complex with the respective inhibitor were cleaned from the other non-standard residues, HOH molecules, and other amino acid chains as previously reported [46]. The structures were energy minimized with Swiss-Pdb viewer v4.1.0 (https://spdbv.unil.ch/) to make sure there are no clashes within the cleaned structures. A monomer of HBcAg in complex with the ligands were used for analyzing the pharmaceutical druggable cavities in the further study.
Prediction of strong druggable cavities
We employed the CavityPlus server (accessible at http://www.pkumdl.cn:8000/cavityplus/#/computation) to conduct a more accurate and robust analysis of protein druggable cavities and assess the functionality of the energy-minimized HBcAg structures when they were complexed with the ligands mentioned earlier [47,48]. To identify the druggable cavities of the ligands’ binding sites on the S protein, CavPharmer was employed to extract pharmacophore features from the selected cavities, while CorrSite identified possible allosteric ligand binding sites by analyzing motion correlations. CovCys was employed to automatically detect druggable cysteine residues, offering a valuable strategy for discovering new binding sites suitable for covalent allosteric ligand development. It's worth noting that throughout our study, we maintained the server settings at their default values, ensuring consistency with prior research findings [49,50].
ZINC database screening
To increase the likelihood of identifying promising compounds within specific pharmacophores, the extracted features from CavPharmer were adopted in screening with ZINCPharmer, as previously outlined in [49]. In this context, the ZINC Purchasable database (last updated on December 20, 2014), comprising a substantial collection of 206,433,075 conformations belonging to 21,777,093 compounds was utilized for the screening process. In cases where the number of hits was substantial, the RMSD threshold reduced (> 0.5Å), and conversely, increased it when there were fewer hits. Molecular docking analysis was conducted on the ZINC-derived hits with the most-fitted compounds and RMSD values.
Stablishing chemical libraries and virtual screening
For identifying pharmacophore of the HBcAg-complexed ligands within the crystallographic structures, LigandScout 4.4.7 [21] software was used. Furthermore, the software used for preparation of a large chemical library of small molecules and ligand-based pharmacophore screening. Briefly, the predicted pharmacophores were imported into the software screening utility to perform high-throughput screening (HTS). An FDA-approved library filtered by Lipinski’s role of five (Drugbank with 1751 compounds) was prepared based on the LigandScout’s iCon best conformer generation type like it was reported before [51]. Duplicate compounds, if there were any, identified and removed during library generation.
The scoring function of hit identification was set on Pharmacophore-Fit score with the Screening mode of Match all query features. Additionally, best matching conformation of hits was retrieved, for which maximum number of omitted pharmacophore feature was rationally modified for obtaining hits preserving maximum number of pharmacophore features. Further molecular docking analysis was conducted, focusing on hits with the highest pharmacophore-fit score.
Molecular docking
The hits in their energy-minimized, 3D confirmation, and explicit orders were used for molecular docking as ligands to HBsAg monomer as the receptor. Molecular docking studies using Autodock Vina v1.2.0 [52,53] was conducted to investigate the binding interactions between the compounds identified through LigandScout or ZINCPharmer screenings. For high-throughput molecular docking, the Galaxy v1.5.7 server, accessible at https://usegalaxy.eu was utilized. Additionally, vina scoring function was employed, generating five docking poses for each ligand within the Galaxy server environment. We meticulously analyzed the docking results, considering scoring functions, visual inspection, and factors such as binding affinity, inter/intra molecular scores, and interaction patterns [40].
To encompass the druggable cavity, a grid box parameter was determined using MGLTools v1.5.7 [54,55]. A cut-off was measured by a same process of docking, determining the affinity of the ligands within the crystallographic structures to HBc monomer. The lead compounds prioritized based on favorable docking scores, optimal binding modes within the relevant pharmacophore features, and potential critical interactions with residues in the cavity.
3. Results
Search of druggable pockets
Six energy-minimized complex structures of HBc that were fixed for their side chains, and cleaned from non-standard/structural atoms and HOH molecules were measured for the pharmacophore features of the binding pockets of the ligands within the viral protein. Two different strategies were employed for screening two separate chemical databases. Firstly, the cavities in which the ligands were bonded with HBcAg were scanned for putative druggable cavities, and virtual screening of ZINC database. In this regards, druggable cavities were predicted and their respective pharmacophore features were extracted. The druggable pocket of the Ciclopirox (chain ID: B4O) in complex with HBc was strong (DrugScore of 1233.00). The highlighted pharmacophore was consisted of only seven features, including one H-Bond donor (HBD) center, one H-Bond acceptor (HBA) center, and five Hydrophobic regions. No allosteric site was detected (Table 1).
Same process was repeated for NVR10-001E2 (chain ID: 5J6) in complex with HBc. A medium druggable (DrugScore of 568.00) pocket was detected where the ligand was in contact with the receptor. The surface area of the druggable site was a little larger from that of Ciclopirox, resulted in six pharmacophore features consisted of three HBAs and three Hydrophobic centers. No allosteric site was also detected. The druggability of the cavity pocket in which Sulfamoylbenzamide (chain ID: K89) was bonded with HBcAg was also measured for its strength. The results demonstrated a weak druggable pocket (DrugScore of -430.00). The number and composition of the pharmacophore features was similar to that of NVR10-001E2, three HBAs and three Hydrophobic centers. More results on heteroaryldihydropyrimidine (Chain ID: 7TL) showed a medium druggable cavity (DrugScore of 30.00) consisted with six features in the pharmacophore, including four HBAs and two Hydrophobic centers.
Further investigation for more pharmacophore was obtained by measuring the strength of N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide (Chain ID: VXJ) in complex with HBV core protein. The cavity results showed a relatively medium-strength druggable cavity (DrugScore of 111.00) consisted of six, two HBAs and four Hydrophobic centers. No allosteric site was recognized. Last retrieved structure of HBcAg was in complex with Compound 24 (Chain ID: ZMH). Additional medium druggable pocket was recognized with a DrugScore of 287.00 at the binding site of the ligand within HBcAg. The pharmacophore was comprised of four features, including two HBAs and two Hydrophobic centers.
Ligand-based Pharmacophores
Second phase of drug discovery was consisted of extracting ligand-based pharmacophores from the compounds within the HBcAg binding sites from the crystallographic structures. In this regard, one pharmacophore was extracted for each ligand reflecting the inhibitors’ pharmacophore features.
The findings (Figure 2) showed variety of pharmacophore seizes for each ligand. In this regard, Ciclopirox-derived pharmacophore was consisted of thirteen features, including four hydrophobic centers, two HBA, one HBD, and eight exclusion volumes. Furthermore, fifteen features were predicted for Compound 24 that was comprised of four hydrophobic centers, two HBAs, and eight exclusion volumes. For heteroaryldihydropyrimidine, the number of features was sixteen, and comprised of four hydrophobic regions, two HBAs, and ten exclusion volumes. Fourteen pharmacophore features were also highlighted for N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide. These were four hydrophobic centers, two HBAs, and eight exclusion volumes. Two further ligands, NVR10-001E2 and sulfamoylbenzamide both with seventeen features had the largest pharmacophores. The pharmacophores were constructed by four hydrophobic centers, three HBAs, and ten exclusion centers for NVR10-001E2, and five hydrophobic centers, two HBAs, and ten exclusion volumes for sulfamoylbenzamide.
Screened hits
FDA-approved Drugbank library was screening for putative inhibitors through ligand-based extracted pharmacophore matching all the features. In this regard, HTS of Ciclopirox-extracted pharmacophore resulted in 10 hits. Ciclopirox olamine was the top of the list with the highest pharmacophore-fit score of 58.73, indicating efficiency and accuracy of the extracted pharmacophore, reliability of the generated library, and performance of screening process. Further hits are listed in the Table 2. Besides, the result of the screening for drug compounds matching with the pharmacophore features of NVR10-001E2 showed two compounds Voriconazole and 2-methyl-1-({4-[6-(trifluoromethyl)pyridin-2-yl]-6-{[2-(trifluoromethyl)pyridin-4-yl]amino}-1,3,5-triazin-2-yl}amino)propan-2-ol (Enasidenib) with pharmacophore-fit scores of 76.33 and 76.18, respectively.
For the pharmacophore features obtained from sulfamoylbenzamide no hit was identified in the first attempt. For enhancing the chance of hit identification the exclusion volumes were ignored. This, resulted in two hits namely Enasidenib and Dasatinib both with a fit score of 75.44. Heteroaryldihydropyrimidine was the other ligand that the predicted pharmacophore of which was screening for proposing further anti-HBV compounds. The screening demonstrated four FDA-approved drugs with completely matched all pharmacophore features. These were Lesinurad, Belzutifan, Simvastatin, and Lovastatin with fit scores of more than 65 (Figure 3). Extra screening of N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide-based pharmacophore led to identifying two more hits, Elvitegravir and Sotagliflozin, completely match with the ligand’s binding cavity features. Lastly, the pharmacophore retrieved from Compound 24 complexed with HBcAg was screened for repurposing drugs targeting same pocket within the viral protein. The result showed only one hit, Dolutegravir, matching seamlessly to the pharmacophore features of Compound 24 binding site.
Screening large ZINC database
Due to the narrow numbers of the hit identified through screening of the pharmacophores with LigandScout, the ZINC database was searched through ZINCPharmer. The resulted screening is reported in the Table 3.
Screening of Ciclopirox-complex HBcAg-obtained pharmacophore led to identification of one hit matching all features. Also, not hit was identified by screening the NVR10-001E2-drived pharmacophore. However, by reducing one distant hydrophobic region, two hits were identified at RMSD ≤ 0.5Å. Additionally, one hits was also identified by screening the pharmacophore of sulfamoylbenzamide, matching all the features. No hits were identified by screening the heteroaryldihydropyrimidine-derived pharmacophore, not either by reducing number of features. By discarding one distant hydrophobic feature in screening the N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide’s pharmacophore, 34 hits were identified with RMSD ≤ 0.5Å.
Docking with Vina
In the present study, two sets of ligand-based and cavity-extracted pharmacophores were obtained from each of the crystallographic structures of HBcAg complexes with six different experimentally approved inhibitors. The screening of FDA-approved drugs consisted with Lipinski’s role of five in the Ciclopirox-based pharmacophore (Table 2), showed ten drugs with a variety of affinity scores, ranging from -4.663 Kcal.mol-1 (Hydroxycitronellal) to -7.268 Kcal.mol-1 (Isavuconazole). Masoprocol and Isavuconazole were two most active compounds with the highest numbers of 11 and 7 torsions, indicating the high flexibilities and potential as the lead compounds. Additionally, Isavuconazole exhibited the most suitable inter+intra molecular interaction energy (-11.374 Kcal.mol-1).
The druggable cavity of Ciclopirox within HBcAg was extracted with CavPharmer for screening a massive small molecule database of ZINC. The identified hit, ZINC86859997, showed relatively low binding energy (-4.696 Kcal.mol-1), with nine active torsions.
The docking results of the Compound 24-based pharmacophore involved the evaluation of compounds, including Compound 24, Dolutegravir, and 38 compounds from the ZINK database. The findings demonstrated that Dolutegravir was the top candidate due to its slightly higher docking score (-7.736 Kcal.mol-1) compared to Compound 24 (-7.480 Kcal.mol-1). Dolutegravir also exhibits strong intermolecular and intramolecular interactions (INTER + INTRA: -9.998 Kcal.mol-1), suggesting a stable binding configuration. The presence of five active torsions indicates potential moderate flexibility to optimize binding. On the other hand, Compound 24 as a potential HBcAg inhibitor shows a decent docking score (-7.480 Kcal.mol-1), but its overall interaction energy (INTER + INTRA: -8.048 Kcal.mol-1) was slightly weaker than Dolutegravir. Additionally, it had fewer active torsions (2), which might indicate a more rigid structure. Besides, most compounds from the ZINC database had weaker binding affinities, with varying numbers of active torsions (Table 3). However, there was some promising hits in the screening, including ZINC63280172 (-7.629 Kcal.mol-1) with relatively desirable intermolecular energy (-10.338) and 7 active torsions. Moreover, two further compounds, ZINC39774429 (-7.629 Kcal.mol-1) and ZINC63270292 (-7.527 Kcal.mol-1) had slightly higher affinity to HBcAg that that of Compound 24.
Four FDA-approved drugs were matched the heteroaryldihydropyrimidine-based pharmacophore, and proceeded to docking tests. Accordingly, the docking results for the main compound, heteroaryldihydropyrimidine, and four other screened compounds (Lesinurad, Belzutifan, Simvastatin, and Lovastatin) are presented in the Table 2.
As a result, Heteroaryldihydropyrimidine had the most favorable affinity of -7.473 Kcal.mol-1, indicating strong binding to the receptor. It also had a relatively low INTER + INTRA score, suggesting that a significant portion of its binding energy comes from intermolecular interactions (-10.192 Kcal.mol-1). Still, this compound with 8 active torsions represents a good flexibility during binding. Furthermore, Belzutifan had a similar affinity like that observed in Heteroaryldihydropyrimidine and a moderate INTER + INTRA score with 6 active torsions, indicating lower flexibility during binding. Simvastatin and Lovastatin both had affinities close to Belzutifan (-6.587 Kcal.mol-1 and -7.519 Kcal.mol-1, respectively). Also, their INTER + INTRA scores were substantially high (-12.046 Kcal.mol-1 and -12.464 Kcal.mol-1), indicative of a better contribution of intramolecular interactions, possibly suggesting less favorable binding. Both compounds also had a significant number of active torsions (13 and 12, respectively), indicating substantial structural flexibility during binding.
Further docking results of pharmacophores derived from the HBcAg-complexed with N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide is provided in Table 2 and Table 3. Despite having a favorable Vina score, the compound had a positive INTER + INTRA score, indicating a greater contribution from intramolecular interactions (12.915 Kcal.mol-1), which was not desirable for a binding compound. Additionally, it has fewer active torsions, suggesting more structural stability during binding. Additionally, Elvitegravir and Sotagliflozin both had a similar binding affinity, but their INTER + INTRA scores were more negative, suggesting that a significant portion of their binding energy comes from intermolecular interactions between the ligands and the receptor. As an indication of flexibility, they had a high number of active torsions. Among the hits from ZINC database, ZINC39996725 (-7.223 Kcal.mol-1) and ZINC13321334 (-7.092 Kcal.mol-1), with grater intermolecular interaction energies along with their high numbers of active torsions were highlighted as two potential HBcAg inhibitors.
The docking results of NVR10-001E2, as well as other hits with the same pharmacophore, including Voriconazole, Enasidenib, ZINC59676742, and ZINC59676742 is presented in Table 2 and Table 3. Enasidenib exhibits the highest negative vina score (-7.867 Kcal.mol-1), indicating strong binding affinity. NVR10-001E2 and Voriconazole also show good affinities. Enasidenib shows the highest negative INTER + INTRA score (-12.006), suggesting a favorable binding energy. This is followed by ZINC59676742. Both had strong binding energies but with higher INTRA contributions. Enasidenib had high active torsions (9).
Docking findings of the cavity-derived pharmacophore of the Sulfamoylbenzamide exhibited the most negative binding affinity (-7.404 Kcal.mol-1) of Dasatinib. It is followed closely by Enasidenib and Sulfamoylbenzamide, with slightly lower but still favorable binding affinities. Also, Dasatinib exhibits the most negative INTER + INTRA score (-10.459 Kcal.mol-1), reflecting a strong overall binding energy. Sulfamoylbenzamide and Enasidenib also had favorable binding energies, with Sulfamoylbenzamide having a lower degree of structural flexibility (INTRA) compared to the other compounds. Furthermore, Dasatinib has the most active torsions (8), followed by Enasidenib (7), and Sulfamoylbenzamide (5). ZINC63844353 has six active torsions.
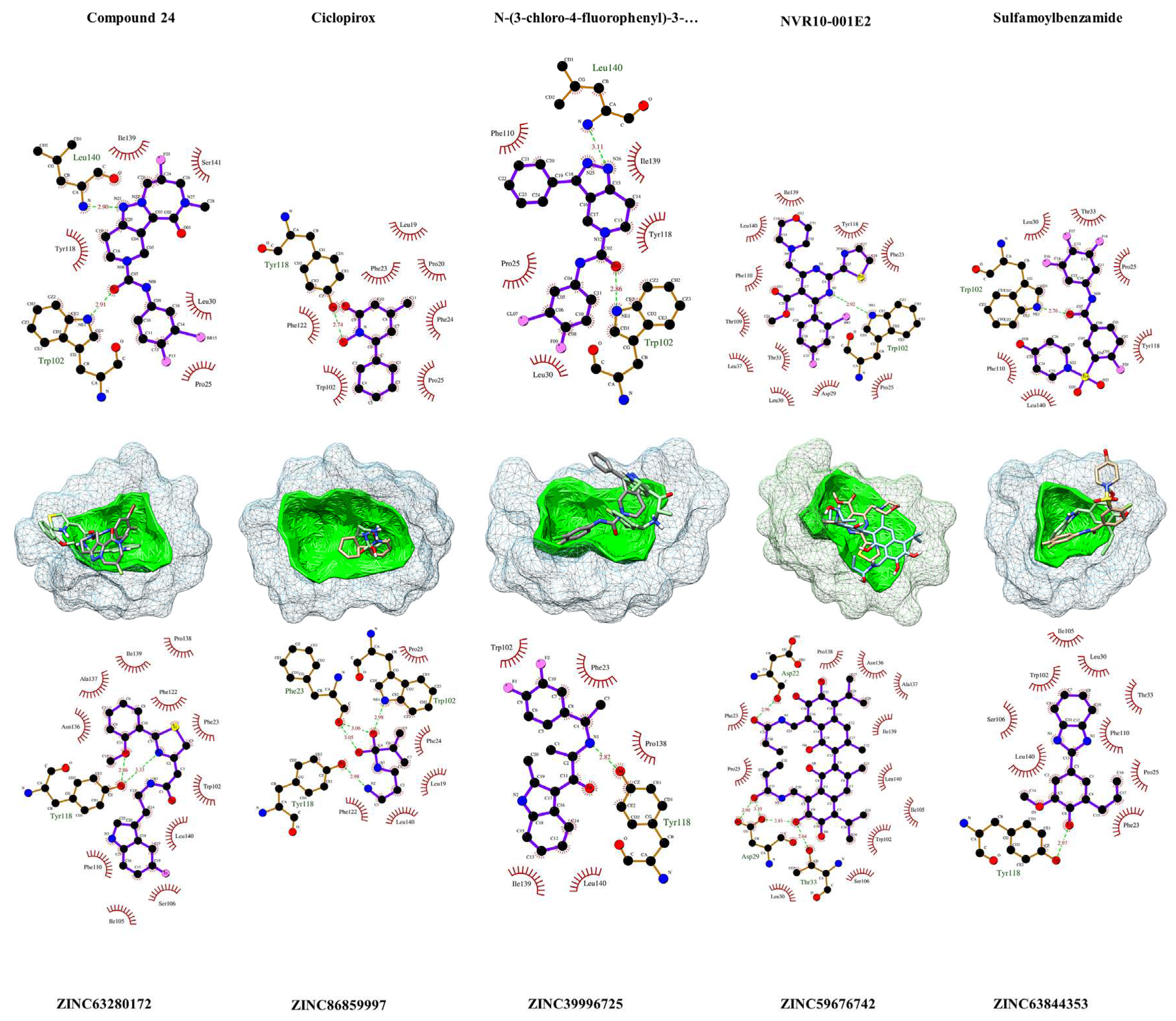
The ligand and hits interaction analysis
As depicted in Figure 4, Ciclopirox demonstrated robust binding to HBcAg, engaging in hydrogen-bonding interactions with critical residues Phe23, Trp102, and Tyr118, while fostering hydrophobic contacts with Pro25, Phe25, Leu19, Phe122, and Leu140. Compound 24 exhibited affinity by forming hydrogen bonds with Leu140 and Trp102, complemented by hydrophobic interactions involving Ile139, Ser141, Tyr118, Pro25, and Leu30. In contrast, NVR10-001E2 primarily established a hydrogen bond with Trp102, with Figure 4 depicting numerous hydrophobic interactions with neighboring residues. Sulfamoylbenzamide displayed a pivotal hydrogen bond with Trp102, and hydrophobic contacts with Pro25, Leu30, Thr33, Phe110, Tyr118, and Leu140. Similarly, N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide engaged in hydrogen bonding with Trp102 and Leu140, while fostering hydrophobic interactions with Pro25, Leu30, Phe110, Tyr118, and Ile139. These findings highlight the intricate binding profiles of the ligands within the HBcAg binding pocket.
In the pursuit of potent antiviral agents, we evaluated five lead compounds derived from distinct pharmacophore screenings. ZINC86859997, originating from the Ciclopirox-derived pharmacophore, exhibited binding prowess by engaging in hydrogen bonds with pivotal HBcAg residues, Phe23, Trp102, and Tyr118, accompanied by hydrophobic interactions with Phe24, Pro25, Leu19, Fhe122, and Leu140. Meanwhile, ZINC63280172, the Compound 24-based lead, established two hydrogen bonds with Tyr118 and intriguing proximal interactions. ZINC59676742, guided by the NVR10-001E2-derived pharmacophore, showcased five hydrogen bonds with Asp22, Asp29, and Thr33, along with eleven hydrophobic contacts. Within the sulfamoylbenzamide-derived pharmacophore, ZINC63844353 engaged in a hydrogen bond with Tyr118 and neighboring hydrophobic interactions. Lastly, ZINC39996725, originating from the N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide-based pharmacophore, exhibited a hydrogen bond with Tyr118 and five hydrophobic interactions involving Phe23, Trp102, Pro138, Ile139, and Leu140. These results elucidate the multifaceted binding profiles of these compounds with HBcAg, offering promising insights for antiviral drug development.
Figure 1.
Crystallographic representation of six different HBc structures retrieved for ligand-based pharmacophore extraction. The ligands (denoted in A to F) are shown in their respective binding cavities. The druggable cavities are shown in green and the surface of the cavities are in red. The ligands are colored by elements. Pharmacophore features are in golden spheres around the cavities. Also, the features are labeled in magenta. In this regard, N stands for HBA centers, O for HBA centers, C for hydrophobic centers, F represent root of h-bond roots, P shows positive electrostatic centers, S represent negative electrostatic centers, and B denote excluded Volume centers.
Figure 1.
Crystallographic representation of six different HBc structures retrieved for ligand-based pharmacophore extraction. The ligands (denoted in A to F) are shown in their respective binding cavities. The druggable cavities are shown in green and the surface of the cavities are in red. The ligands are colored by elements. Pharmacophore features are in golden spheres around the cavities. Also, the features are labeled in magenta. In this regard, N stands for HBA centers, O for HBA centers, C for hydrophobic centers, F represent root of h-bond roots, P shows positive electrostatic centers, S represent negative electrostatic centers, and B denote excluded Volume centers.
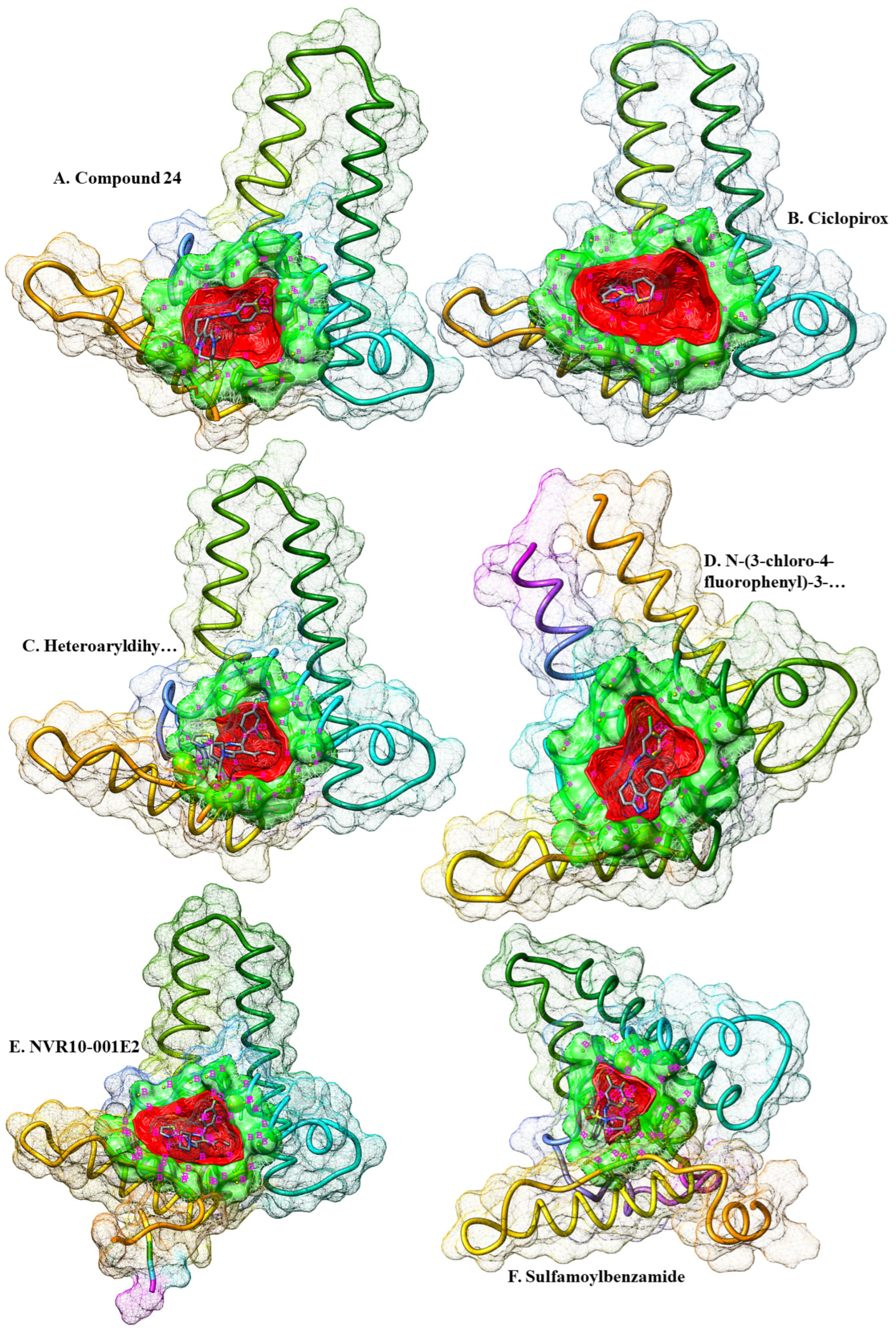
Figure 2.
Ligand-based pharmacophores of HBcAg-complexed with six ligands. The close residues near the ligands’ features are also depicted in rectangles. Hydrophobic centers are in yellow spheres, the acceptors of h-bonds are highlighted with red arrows, the acceptors of h-bonds are demonstrated with green arrows, ionizable regions are in Blue, and exclusion volumes are presented with grey spheres.
Figure 2.
Ligand-based pharmacophores of HBcAg-complexed with six ligands. The close residues near the ligands’ features are also depicted in rectangles. Hydrophobic centers are in yellow spheres, the acceptors of h-bonds are highlighted with red arrows, the acceptors of h-bonds are demonstrated with green arrows, ionizable regions are in Blue, and exclusion volumes are presented with grey spheres.
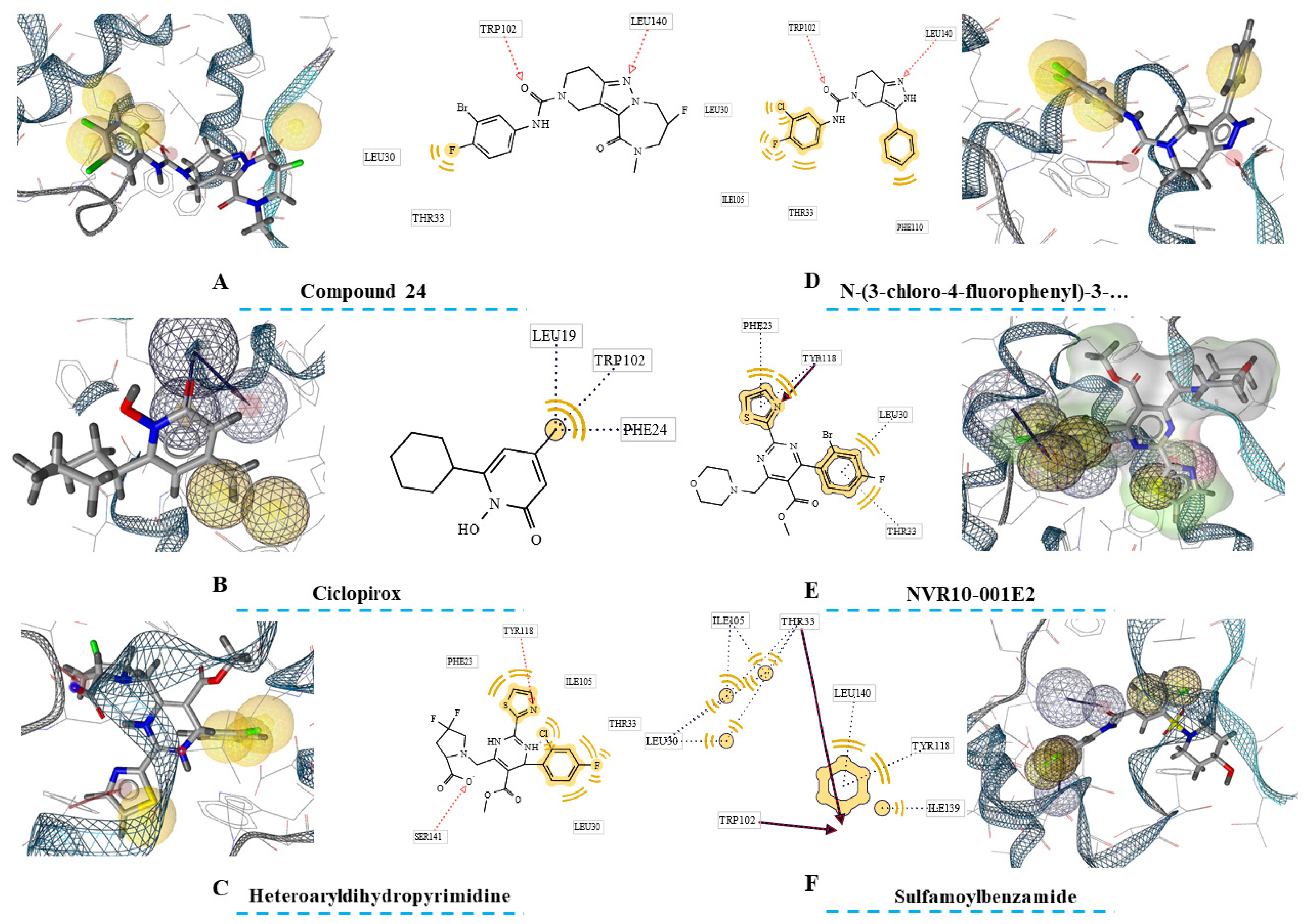
Figure 3.
The screening results of the ligand-based pharmacophores. The largest number of identified hits was from Ciclopirox-based pharmacophore, while the pharmacophore from Compound 24 with one hit had the lowest. In the middle of the circles are aligned hits with the extracted pharmacophores. Matching features of the hits are depicted within the rectangles in 2D forms around the circles.
Figure 3.
The screening results of the ligand-based pharmacophores. The largest number of identified hits was from Ciclopirox-based pharmacophore, while the pharmacophore from Compound 24 with one hit had the lowest. In the middle of the circles are aligned hits with the extracted pharmacophores. Matching features of the hits are depicted within the rectangles in 2D forms around the circles.
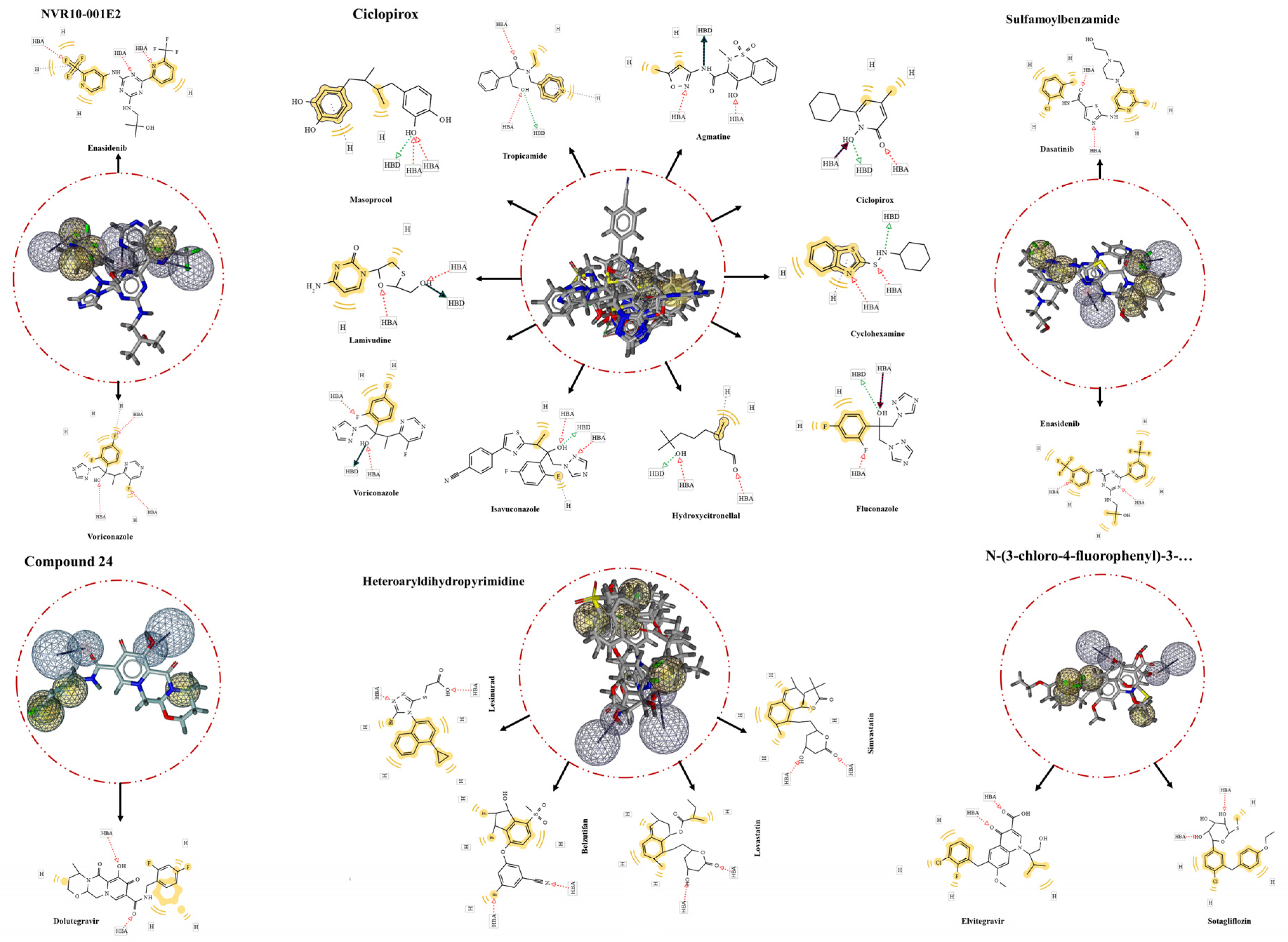
Figure 4.
Visualization of the HBcAg residues in contact or in close proximity with the ligands and leads. In above, the results of LigPlot+ v2.2.8 [56] analysis of ligands with HBcAg is provided, and the interaction of identified ZINC-screened compounds is shown below. The compounds are within their predicted binding pockets (mesh structures) highlighted with the respective cavity surfaces (green). The plots show the interaction of the compounds with the protein. The proxy residues with hydrophobic interactions are highlighted in brick-red colors. Hydrogen-bonds are depicted with light-green dashes, along with the resolved h-bonds’ distance (Å) in-between.
Figure 4.
Visualization of the HBcAg residues in contact or in close proximity with the ligands and leads. In above, the results of LigPlot+ v2.2.8 [56] analysis of ligands with HBcAg is provided, and the interaction of identified ZINC-screened compounds is shown below. The compounds are within their predicted binding pockets (mesh structures) highlighted with the respective cavity surfaces (green). The plots show the interaction of the compounds with the protein. The proxy residues with hydrophobic interactions are highlighted in brick-red colors. Hydrogen-bonds are depicted with light-green dashes, along with the resolved h-bonds’ distance (Å) in-between.
4. Discussion
In this study, a comprehensive exploration of druggable pockets within HBcAg using six structures was conducted. These structures were prepared to identify binding pockets where potential ligands could interact with HBcAg. Two different strategies were employed, each targeting distinct chemical databases. The initial structure-based pharmacophore strategy involved scanning the binding pockets where the ligands were already observed to interact with HBcAg. This method helped to identify all possible pharmacophore features in proximity of where the ligands are bonded with HBcAg, and expand screening to larger chemical library and discovery of novel lead compounds. The results highlighted several druggable pockets with varying strengths. Notably, the Ciclopirox-binding pocket exhibited a robust druggable score, followed by medium-strength pockets for NVR10-001E2, heteroaryldihydropyrimidine, and N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide. However, the sulfamoylbenzamide pocket demonstrated weaker druggability.
The second approach, ligand-based pharmacophore was employed to screen for compounds that matched all the requisite pharmacophore features. This strategy helped to highlight the FDA-approved drugs, which can be repurposed for anti-HBV researches by having same pharmacological properties shared with the primary ligands in each structure. Accordingly, the Ciclopirox-based pharmacophore yielded 10 hits, with Ciclopirox olamine exhibiting the highest pharmacophore-fit score, affirming the accuracy of the extracted pharmacophore. The NVR10-001E2 pharmacophore identified two promising compounds, Voriconazole and Enasidenib. Also, Enasidenib and Dasatinib were identified for the sulfamoylbenzamide pharmacophore. Heteroaryldihydropyrimidine's pharmacophore matched four FDA-approved drugs, namely Lesinurad, Belzutifan, Simvastatin, and Lovastatin. Finally, the pharmacophore derived from N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide led to the identification of two hits, Elvitegravir and Sotagliflozin, and the pharmacophore extracted from Compound 24 revealed Dolutegravir as the sole hit.
While this study primarily focused on the molecular interactions of potential inhibitors with HBcAg, it is important to acknowledge the broader landscape of drug discovery and target identification. The integration of bioinformatic approaches has opened up new avenues for drug repurposing in various diseases. For instance, in the context of CHB, recent research has shown the potential of utilizing genomic variants to drive drug repurposing efforts for CHB treatment. In this regard, Irham et al., identified eighteen clinical drugs targeting six different druggable targets for treatment of CHB [33]. Here, it was found that Lamivudine and Simvastatin match the Ciclopirox-derived and Heteroaryldihydropyrimidine-derived pharmacophores, respectively, and both are reported by the mentioned study to can be repurposed for HBV treatment. This suggest dual potential anti-CHB effects of these drugs by either direct acting on HBcAg or introduced druggable genes. Studies on drug repurposing anti-HBV agents are handful, especially on those targeting HBcAg. Ciclopirox was one of the first FDA-approved drugs screened from hundreds of other drugs to be repurposed in HBV infection [26]. As a hydroxypyridone, Ciclopirox olamine is using for treating skin fungal infections [34]. Here, three further antifungals, including Fluconazole, Voriconazole, and Isavuconazole were found to match the ligand-derived pharmacophores. Voriconazole was identified in two Ciclopirox-based and NVR10-001E2-based screening, and it had a comparable high affinity to HBcAg compare to the main ligands. This suggests the potential of antifungals with common pharmacological features to have anti-HBV activities. The antiviral effects of some antifungals are shown before [26,35], indicating the potential ancestry druggable site shared in viruses and fungi that opens a vast field in the future researches.
Among other FDA-approved lead compounds that may have the potential to suppress HBV capsid assembly, the fitting results of statins within the pharmacophores with high affinities were interesting. A systematic-review and meta-analysis has suggested that the risk of HCC in HBV patients who had been received statins was > 50% lower than non-users [36]. Simvastatin, especially, has a strong synergy toward enhancing anti-HBV activity of nucleos(t)ide analogues [37]. The findings of the present study suggest that Lovastatin is more potentiate to have anti-HBV activity. Both Simvastatin and Lovastatin are pharmacologically match the features of Heteroaryldihydropyrimidine-derived pharmacophore, and Lovastatin had higher affinity to HBV core protein in compare to that of either Heteroaryldihydropyrimidine or Simvastatin. This also highlight the probable anti-HBV mechanism of action of statins through inhibiting viral capsid assembly. More approved drugs were including antiviral hits like Dolutegravir and Elvitegravir with considerable affinities and feature-fitting that necessitates further research. Moreover, it is worth noting that the anti-HIV properties of HBcAg inhibitors, such as Compound 24 and N-(3-chloro-4-fluorophenyl)-3-phenyl-, which their pharmacophore served as a source for screening of Dolutegravir and Elvitegravir, present intriguing avenues for future research.
Given the limited number of hits obtained in the initial screening and to introduce novel HBcAg inhibitors, a broader search was conducted in the ZINC database using ZINCPharmer. The cavity-derived pharmacophores were comprised of more features, extending the screening to more potentially hits with higher scores. Results varied across different pharmacophores, with some identifying hits matching all features and others requiring adjustments to improve hit identification. These hits demonstrated varying binding affinities and structural flexibility. Molecular docking was performed to evaluate the binding affinity and interactions of hits with HBcAg. Results revealed the binding preferences and potential lead compounds for each pharmacophore. For example, Ciclopirox cavity-based pharmacophore screening identified ZINC86859997 as a potential inhibitor with a comparable binding energy. N-(3-chloro-4-fluorophenyl)-3-phenyl-2,4,6,7-tetrahydro-5H-pyrazolo[4,3-c]pyridine-5-carboxamide cavity-based pharmacophore suggested ZINC39996725 and ZINC13321334 as potential HBcAg inhibitors. These lead compounds may have anti-HBV activities, and development of novel candidates’ small molecules with these compounds as scaffolds could also broad the antiviral field of research.
The binding sites of the identified hits within HBcAg needed a focused investigation. Zhou et al,. examined the mechanisms of action of two reference compounds, (heteroaryldihydropyrimidines) HAP_R01 and (sulfamoylbenzamides) SBA_R01, in the context of HBV capsid assembly inhibition. HAP_R01 demonstrates potent efficacy by reducing capsid-associated DNA levels and causing degradation of core proteins [38]. Amino acid substitutions are predicted to affect susceptibility to CAMs, with experimental data confirming these predictions. The clinical relevance of these substitutions in HBV-infected patients is evaluated, with some changes found at low levels in treatment-naïve patients. The affected domains, "concave" and "cap" pockets, formed by specific amino acid residues in the capsid protein, including F23, P25, D29, L30, T33, L37, W102, I105, S106, T109, F110, Y118, F122, I139, L140, and S141 in one chain and V124, W125, R127, T128, P129, and Y132A in another chain [38]. Further study by Pen et al., in discovery of novel CAMs, it was showed that the compounds tent to engage in hydrophobic and aromatic interactions with hydrophobic pockets within the HBV capsid protein. Such interactions play a pivotal role in disrupting the assembly of HBV capsid proteins. Furthermore, these compounds establish crucial hydrogen bonds with specific amino acid residues, particularly Ser121, which significantly contribute to their inhibitory activity against HBV capsid assembly [39]. Furthermore, it was showed that Ciclopirox in its crystallographic structure, and its derivatives from a fragment-based drug discovery approach tent to interact at the specific capsid protein residues, including F23, F24, Y118, F122, P25, W102, and L19 [26,40]. The findings in the present study highlights the importance of large number hydrophobic interactions in proximity of the ligands/hits binding sites within HBcAg. Of such residues, Leu140, Trp102, Y118, Thr33, Asp29, Phe23, Asp22 found to have important roles in, prominently, hydrogen bonds between ligands and the receptor. Above all, Trp102 was involved in most of the ligand/hit hydrogen bonding interactions. These indicate that mutations in this site might affect the anti-viral activities of the ligands and hits.
5. Conclusions
The findings of the present study contribute to the search for novel anti-HBV agents and highlight the potential for repurposing existing drugs to target HBcAg. However, it is essential to validate these computational results through experimental assays to confirm their efficacy and safety as HBV inhibitors. Further optimization and in-depth studies are needed to develop these compounds into potential therapeutics for HBV infection. Additionally, the results emphasize the importance of exploring multiple strategies, including structure-based and ligand-based approaches, in drug discovery efforts.
Author Contributions
Conceptualization, A.Mo..; methodology, A.Mo.; software, A.Mo, M.N, K.S, F.B, M.M, A.G, F.S.A, and A.M.; validation, A.Mo; formal analysis, A.Mo, and M.N.; investigation, A.Mo.; resources, A.Mo, M.N, K.S, F.B, M.M, A.G, F.S.A, and A.M.; data curation, A.M and S.H.M.; writing—original draft preparation, A.Mo, M.N, K.S, F.B, M.M, A.G, F.S.A, and A.M.; writing—review and editing, A.Mo and S.H.M.; visualization, A.Mo, M.N, K.S, F.B, M.M, A.G, F.S.A, and A.M.; supervision, S.H.M.; project administration, S.H.M.; funding acquisition, A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Iran University of Medical Sciences, Tehran, Iran, grant number 24817.
Institutional Review Board Statement
“The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Ethics Committee of Iran University of Medical Sciences, Tehran, Iran with approval code of IR.IUMS.REC.1401.812.
Informed Consent Statement
Not applicable
Data Availability Statement
Data are represented within the manuscript. However, any additional data will be provided if required by the authors.
Acknowledgments
Not Applicable.
Conflicts of Interest
Authors A.M and F.S.A are affiliated with Vista Aria Rena Gene Inc., Iran. However, the remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Jeng, W.J.; Papatheodoridis, G. V.; Lok, A.S.F. Hepatitis B. The Lancet 2023, 401, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Mohebbi, A.; Lorestani, N.; Tahamtan, A.; Kargar, N.L.; Tabarraei, A. An Overview of Hepatitis B Virus Surface Antigen Secretion Inhibitors. Front Microbiol 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Lu, D.; Xu, Y. Bin; Xing, W.Q.; Tong, X.K.; Wang, G.F.; Feng, C.L.; He, P.L.; Yang, L.; Tang, W.; et al. A Novel Pyridazinone Derivative Inhibits Hepatitis B Virus Replication by Inducing Genome-Free Capsid Formation. Antimicrob Agents Chemother 2015, 59, 7061–7072. [Google Scholar] [CrossRef] [PubMed]
- Mohebbi, A.; Azadi, F.; Hashemi, M.M.; Askari, F.S.; Razzaghi, N. Havachoobe (Onosma Dichroanthum Boiss) Root Extract Decreases the Hepatitis B Virus Surface Antigen Secretion in the PLC/PRF/5 Cell Line. Intervirology 2021, 64, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Y.; Chen, H.; Shi, L.; research, X.T.-A. ; 2016, undefined Effect of a Hepatitis B Virus Inhibitor, NZ-4, on Capsid Formation. Elsevier.
- Ye, J.; Chen, J. Interferon and Hepatitis B: Current and Future Perspectives. Front Immunol 2021, 12, 733364. [Google Scholar] [CrossRef]
- Jiang, B.; Hildt, E. Intracellular Trafficking of HBV Particles. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jin, L.; Jih, J.; Shih, C.; Hong Zhou, Z. 3. 5Å CryoEM Structure of Hepatitis B Virus Core Assembled from Full-Length Core Protein. PLoS One 2013, 8, e69729. [Google Scholar] [CrossRef]
- Qazi, S.; Schlicksup, C.J.; Rittichier, J.; Vannieuwenhze, M.S.; Zlotnick, A. An Assembly-Activating Site in the Hepatitis B Virus Capsid Protein Can Also Trigger Disassembly. ACS Chem Biol 2018, 13, 2114–2120. [Google Scholar] [CrossRef]
- Lecoq, L.; Brigandat, L.; Huber, R.; Fogeron, M.L.; Wang, S.; Dujardin, M.; Briday, M.; Wiegand, T.; Callon, M.; Malär, A.; et al. Molecular Elucidation of Drug-Induced Abnormal Assemblies of the Hepatitis B Virus Capsid Protein by Solid-State NMR. Nature Communications 2023 14:1 2023, 14, 1–14. [Google Scholar] [CrossRef]
- Taverniti, V.; Ligat, G.; Debing, Y.; Kum, D.B.; Baumert, T.F.; Verrier, E.R. Capsid Assembly Modulators as Antiviral Agents against HBV: Molecular Mechanisms and Clinical Perspectives. J Clin Med 2022, 11. [Google Scholar] [CrossRef]
- Berke, J.M.; Dehertogh, P.; Vergauwen, K.; Van Damme, E.; Mostmans, W.; Vandyck, K.; Pauwels, F. Capsid Assembly Modulators Have a Dual Mechanism of Action in Primary Human Hepatocytes Infected with Hepatitis B Virus. Antimicrob Agents Chemother 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Taverniti, V.; Ligat, G.; Debing, Y.; …, D.K.-J. of clinical; 2022, undefined Capsid Assembly Modulators as Antiviral Agents against HBV: Molecular Mechanisms and Clinical Perspectives. mdpi.comV Taverniti, G Ligat, Y Debing, DB Kum, TF Baumert, ER VerrierJournal of clinical medicine, 2022•mdpi.com.
- Zoulim, F.; Lenz, O.; Vandenbossche, J.; Gastroenterology, W.T.-. ; 2020, undefined JNJ-56136379, an HBV Capsid Assembly Modulator, Is Well-Tolerated and Has Antiviral Activity in a Phase 1 Study of Patients with Chronic Infection. Elsevier.
- Huang, Q.; Cai, D.; Yan, R.; Li, L.; Zong, Y.; Guo, L.; Mercier, A.; Zhou, Y.; Tang, A.; Henne, K.; et al. Preclinical Profile and Characterization of the Hepatitis B Virus Core Protein Inhibitor ABI-H0731. Antimicrob Agents Chemother 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Berke, J.M.; Dehertogh, P.; Vergauwen, K.; Mostmans, W.; Vandyck, K.; Raboisson, P.; Pauwels, F. Antiviral Properties and Mechanism of Action Studies of the Hepatitis B Virus Capsid Assembly Modulator JNJ-56136379. Antimicrob Agents Chemother 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Locarnini, S.; Lim, T.H.; Strasser, S.I.; Sievert, W.; Cheng, W.; Thompson, A.J.; Given, B.D.; Schluep, T.; Hamilton, J.; et al. Combination Treatments Including the Small-Interfering RNA JNJ-3989 Induce Rapid and Sometimes Prolonged Viral Responses in Patients with CHB. J Hepatol 2022, 77, 1287–1298. [Google Scholar] [CrossRef]
- Stray, S.J.; Zlotnick, A. BAY 41-4109 Has Multiple Effects on Hepatitis B Virus Capsid Assembly. Journal of Molecular Recognition 2006, 19, 542–548. [Google Scholar] [CrossRef]
- Li, C.; Wu, M.; Zhang, H.; Mai, J.; Yang, L.; Ding, Y.; Niu, J.; Mao, J.; Wu, W.; Zhang, D.; et al. Safety, Tolerability, and Pharmacokinetics of the Novel Hepatitis B Virus Capsid Assembly Modulator GST-HG141 in Healthy Chinese Subjects: A First-in-Human Single- and Multiple-Dose Escalation Trial. Antimicrob Agents Chemother 2021, 65. [Google Scholar] [CrossRef]
- Farrokhzadeh, A.; Badichi Akher, F.; Olotu, F.A.; Van Heerden, F.R. Impact of HEC72702 Chirality on the Selective Inhibition of Hepatitis B Virus Capsid Dimer: A Dynamics–Structure–Energetics Perspective. Chem Biol Drug Des 2021, 97, 167–183. [Google Scholar] [CrossRef]
- Toyama, M.; Sakakibara, N.; Takeda, M.; research, M.O.-V. ; 2019, undefined Pyrimidotriazine Derivatives as Selective Inhibitors of HBV Capsid Assembly. Elsevier.
- Chen, W.; Gong, Y.; Long, G.; Wang, X.; Yang, Y.; Liu, J.; Li, H.; Tong, X.; Zhao, Q.; Yang, L.; et al. A Prodrug of the Capsid Assembly Modulator Improved Druggability and Lowing HBsAg and HBeAg for the Treatment of Chronic Hepatitis B. Eur J Med Chem 2023, 257. [Google Scholar] [CrossRef]
- Corcuera, A.; Stolle, K.; Hillmer, S.; Seitz, S.; research, J.L.-A. ; 2018, undefined Novel Non-Heteroarylpyrimidine (HAP) Capsid Assembly Modifiers Have a Different Mode of Action from HAPs in Vitro. Elsevier.
- Gane, E.; Schwabe, C.; …, E.B.-J. of; 2022, undefined Safety, Antiviral Activity and Pharmacokinetics of JNJ-64530440, a Novel Capsid Assembly Modulator, as 4 Week Monotherapy in Treatment-Naive Patients with Chronic. academic.oup.comEJ Gane, C Schwabe, E Berliba, P Tangkijvanich, A Jucov, N Ghicavii, T Verbinnen, O LenzJournal of Antimicrobial Chemotherapy, 2022•academic.oup.com.
- Vermes, T.; Kielpinski, M.; Henkel, T.; …, M.P.-A. ; 2022, undefined An Automated Microfluidic Platform for the Screening and Characterization of Novel Hepatitis B Virus Capsid Assembly Modulators. pubs.rsc.orgT Vermes, M Kielpinski, T Henkel, MA Pericàs, E Alza, A Corcuera, H Buschmann, T GoldnerAnalytical Methods, 2022•pubs.rsc.org.
- Kang, J.A.; Kim, S.; Park, M.; Park, H.J.; Kim, J.H.; Park, S.; Hwang, J.R.; Kim, Y.C.; Jun Kim, Y.; Cho, Y.; et al. Ciclopirox Inhibits Hepatitis B Virus Secretion by Blocking Capsid Assembly. Nature Communications 2019 10:1 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Lam, A.M.; Espiritu, C.; Vogel, R.; Ren, S.; Lau, V.; Kelly, M.; Kuduk, S.D.; Hartman, G.D.; Flores, O.A.; Klumpp, K. Preclinical Characterization of NVR 3-778, a First-in-Class Capsid Assembly Modulator against Hepatitis B Virus. Antimicrob Agents Chemother 2019, 63. [Google Scholar] [CrossRef]
- Ren, Q.; Liu, X.; Yan, G.; Nie, B.; Zou, Z.; Li, J.; Chen, Y.; Wei, Y.; Huang, J.; Luo, Z.; et al. 3-((R)-4-(((R)-6-(2-Bromo-4-Fluorophenyl)-5-(Ethoxycarbonyl)-2-(Thiazol-2-Yl)-3,6-Dihydropyrimidin-4-Yl)Methyl)Morpholin-2-Yl)Propanoic Acid (HEC72702), a Novel Hepatitis B Virus Capsid Inhibitor Based on Clinical Candidate GLS4. J Med Chem 2018, 61, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Lahlali, T.; Berke, J.M.; Vergauwen, K.; Foca, A.; Vandyck, K.; Pauwels, F.; Zoulim, F.; Durantel, D. Novel Potent Capsid Assembly Modulators Regulate Multiple Steps of the Hepatitis b Virus Life Cycle. Antimicrob Agents Chemother 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Seo, J.; Cho, Y.; Ko, E.; Kim, Y.; research, G.J.-V. ; 2019, undefined Cetylpyridinium Chloride Interaction with the Hepatitis B Virus Core Protein Inhibits Capsid Assembly. Elsevier.
- Wu, G.Y.; Zheng, X.J.; Yin, C.C.; Jiang, D.; Zhu, L.; Liu, Y.; Wei, L.; Wang, Y.; Chen, H.S. Inhibition of Hepatitis B Virus Replication by Bay 41-4109 and Its Association with Nucleocapsid Disassembly. Journal of Chemotherapy 2008, 20, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Liu, X.; Luo, Z.; Li, J.; Wang, C.; …, S.G.-B. & M.; 2017, undefined Discovery of Hepatitis B Virus Capsid Assembly Inhibitors Leading to a Heteroaryldihydropyrimidine Based Clinical Candidate (GLS4). Elsevier.
- Irham, L.; Adikusuma, W.; Reports, D.P.-. … and B.; 2022, undefined The Use of Genomic Variants to Drive Drug Repurposing for Chronic Hepatitis B. Elsevier.
- Abrams, B.; Hänel, H.; dermatology, T.H.-C. in; 1991, undefined Ciclopirox Olamine: A Hydroxypyridone Antifungal Agent. Elsevier.
- Shen, S.; Zhang, Y.; Yin, Z.; Zhu, Q.; Zhang, J.; Wang, T.; Fang, Y.; Wu, X.; Bai, Y.; Dai, S.; et al. Antiviral Activity and Mechanism of the Antifungal Drug, Anidulafungin, Suggesting Its Potential to Promote Treatment of Viral Diseases. BMC Med 2022, 20. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Y.; Li, X.; Zhang, L.; Zhao, N.; Du, H.; Zhou, B.; Ye, Y. Statins in Hepatitis B or C Patients Is Associated With Reduced Hepatocellular Carcinoma Risk: A Systematic Review and Meta-Analysis. The Turkish Journal of Gastroenterology 2022, 33, 136. [Google Scholar] [CrossRef]
- Bader, T.; Korba, B. Simvastatin Potentiates the Anti-Hepatitis B Virus Activity of FDA-Approved Nucleoside Analogue Inhibitors in Vitro. Antiviral Res 2010, 86, 241. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hu, T.; Zhou, X.; Wildum, S.; Garcia-Alcalde, F.; Xu, Z.; Wu, D.; Mao, Y.; Tian, X.; Zhou, Y.; et al. Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms. Scientific Reports 2017 7:1 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Pan, T.; Ding, Y.; Wu, L.; Liang, L.; He, X.; Li, Q.; …, C.B.-E.J. of; 2019, undefined Design and Synthesis of Aminothiazole Based Hepatitis B Virus (HBV) Capsid Inhibitors. Elsevier.
- Mohebbi, A.; Ghorbanzadeh, T.; Naderifar, S.; Khalaj, F.; Askari, F.S.; Salehnia Sammak, A. A Fragment-Based Drug Discovery Developed on Ciclopirox for Inhibition of Hepatitis B Virus Core Protein: An in Silico Study. PLoS One 2023, 18, e0285941. [Google Scholar] [CrossRef]
- Kuduk, S.D.; DeRatt, L.G.; Stoops, B.; Shaffer, P.; Lam, A.M.; Espiritu, C.; Vogel, R.; Lau, V.; Flores, O.A.; Hartman, G.D. Diazepinone HBV Capsid Assembly Modulators. Bioorg Med Chem Lett 2022, 72, 128823. [Google Scholar] [CrossRef]
- Kuduk, S.D.; Stoops, B.; Alexander, R.; Lam, A.M.; Espiritu, C.; Vogel, R.; Lau, V.; Klumpp, K.; Flores, O.A.; Hartman, G.D. Identification of a New Class of HBV Capsid Assembly Modulator. Bioorg Med Chem Lett 2021, 39, 127848. [Google Scholar] [CrossRef]
- Zhou, Z.; Hu, T.; Zhou, X.; Wildum, S.; Garcia-Alcalde, F.; Xu, Z.; Wu, D.; Mao, Y.; Tian, X.; Zhou, Y.; et al. Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms. Scientific Reports 2017 7:1 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Zhou, Z.; Hu, T.; Zhou, X.; Wildum, S.; Garcia-Alcalde, F.; Xu, Z.; Wu, D.; Mao, Y.; Tian, X.; Zhou, Y.; et al. Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms. Scientific Reports 2017 7:1 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, K.; Lam, A.M.; Lukacs, C.; Vogel, R.; Ren, S.; Espiritu, C.; Baydo, R.; Atkins, K.; Abendroth, J.; Liao, G.; et al. High-Resolution Crystal Structure of a Hepatitis B Virus Replication Inhibitor Bound to the Viral Core Protein. Proc Natl Acad Sci U S A 2015, 112, 15196–15201. [Google Scholar] [CrossRef] [PubMed]
- Mohebbi, A.; Mohammadi, S.; Memarian, A. Prediction of HBF-0259 Interactions with Hepatitis B Virus Receptors and Surface Antigen Secretory Factors. Virusdisease 2016, 27, 234–241. [Google Scholar] [CrossRef]
- Wang, S.; Xie, J.; Pei, J.; Lai, L. CavityPlus 2022 Update: An Integrated Platform for Comprehensive Protein Cavity Detection and Property Analyses with User-Friendly Tools and Cavity Databases. J Mol Biol 2023, 435, 168141. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A Web Server for Protein Cavity Detection with Pharmacophore Modelling, Allosteric Site Identification and Covalent Ligand Binding Ability Prediction. Nucleic Acids Res 2018, 46, W374–W379. [Google Scholar] [CrossRef]
- Askari, F.S.; Ebrahimi, M.; Parhiz, J.; Hassanpour, M.; Mohebbi, A.; Mirshafiey, A. Digging for the Discovery of SARS-CoV-2 Nsp12 Inhibitors: A Pharmacophore-Based and Molecular Dynamics Simulation Study. Future Virol 2022. [CrossRef]
- Mohebbi, A.; Askari, F.S.; Sammak, A.S.; Ebrahimi, M.; Najafimemar, Z. Druggability of Cavity Pockets within SARS-CoV-2 Spike Glycoprotein and Pharmacophore-Based Drug Discovery. Future Virol 2021, 16, 389–397. [Google Scholar] [CrossRef]
- Mohebbi, A. Ligand-Based 3D Pharmacophore Modeling, Virtual Screening, and Molecular Dynamic Simulation of Potential Smoothened Inhibitors. J Mol Model 2023, 29. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1. 2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J Chem Inf Model 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J Comput Chem 2009, 31, NA–NA. [Google Scholar] [CrossRef] [PubMed]
- Mohebbi, A.; Mirarab, A.; Shaddel, R.; Shafaei Fallah, M.; Memarian, A. Molecular Dynamic Simulation and Docking of Cyclophilin A Mutants with Its Potential Inhibitors. Journal of Clinical and Basic Research 2021, 5, 26–41. [Google Scholar] [CrossRef]
- Mohebbi, A.; Ebrahimi, M.; Askari, F.S.; Shaddel, R.; Mirarab, A.; Oladnabi, M. QSAR Modeling of a Ligand-Based Pharmacophore Derived from Hepatitis B Virus Surface Antigen Inhibitors; 2022; Vol. 38;
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J Chem Inf Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]

Table 1.
The cavity results of the predicted druggable cavity pockets for the known inhibitors of HBcAg.
Table 1.
The cavity results of the predicted druggable cavity pockets for the known inhibitors of HBcAg.
| PDB_ID | Pred.Max.pKd | Pred.Ave.pKd | DrugScore | Druggability | Surface Area (Å2) | Volume (Å3) |
|---|---|---|---|---|---|---|
| 6j10 | 10.69 | 6.95 | 1233.00 | Strong | 374.25 | 529.25 |
| Res. | A34,L101,Y118,L30,W102,L19,T33,S106,S141,L140,F24,L37,I126,F110,I139,D29,P138,F122,Y38,L16,P20,F23,S21,C107,F103,W125,P25,L119,L31,S26,D22,A137,I105,T109 | |||||
| 5E0I | 11.67 | 6.86 | 568.00 | Medium | 282.50 | 406.75 |
| Res. | T33,VAL115,L140,C107,L108,L37,I105,F24,F110,D22,F122,D29,W102,S106,I126,L30,P25,A34,F23,F103,L31,L101,Y38,Y118,S141,A137,S26,T109,P138,H104,I139 | |||||
| 5T2P | 8.22 | 5.44 | 430.00 | Weak | 148.00 | 155.00 |
| Res. | A34,L101,Y118,L30,W102,T33,L140,F24,L37,F110,D29,Y38,F23,C107,F103,P25,H104,S26,S106,I105,T109 | |||||
| 5WRE | 9.98 | 6.04 | 30.00 | Medium | 163.75 | 210.00 |
| Res. | T33,L140,C107,L37,I105,F24,F110,D29,W102,S106,L30,P25,A34,F23,F103,L31,L101,Y38,Y118,S141,S26,T109,H104 | |||||
| 7K5M | 9.03 | 5.71 | 111.00 | Medium | 235.25 | 245.75 |
| Res. | T33,L140,C107,L37,I105,F24,F110,D22,F122,D29,W102,S106,L19,L30,P25,A34,F23,F103,L31,L101,Y38,Y118,S141,A137,S26,T109,P138,I139 | |||||
| 8GIH | 10.67 | 6.27 | 287.00 | Medium | 223.50 | 264.75 |
| Res. | T33,L140,C107,L37,D32,I105,F24,F110,L119,F122,D29,W102,S106,L30,P25,A34,F23,F103,L31,L101,Y38,Y118,S141,S26,T109,P138,H104,I139 | |||||
Table 2.
The docking results of the hits from the ligand-based pharmacophore screening of Drugbank dataset with HBcAg.
Table 2.
The docking results of the hits from the ligand-based pharmacophore screening of Drugbank dataset with HBcAg.
| Compound | Vina scores (Kcal.mol-1) | INTER+INTRA€ (Kcal.mol-1) | INTER¥ (Kcal.mol-1) | INTRA£ (Kcal.mol-1) | Active Torsions ŧ |
|---|---|---|---|---|---|
| Ciclopirox-derived pharmacophore | |||||
| Ciclopirox | -5.808 | -6.633 | -6.316 | -0.318 | 3 |
| Fluconazole | -5.878 | -8.963 | -7.956 | -1.007 | 6 |
| Voriconazole | -6.630 | -9.561 | -8.500 | -1.061 | 7 |
| Thiohexam | -6.677 | -8.027 | -7.818 | -0.209 | 3 |
| Lamivudine | -5.134 | -6.160 | -6.135 | -0.025 | 3 |
| Isavuconazole | -7.268 | -11.374 | -9.694 | -1.679 | 9 |
| Tropicamide | -5.723 | -9.028 | -8.062 | -0.966 | 7 |
| Isoxicam | -7.058 | -8.011 | -7.469 | -0.542 | 3 |
| Hydroxycitronellal | -4.663 | -6.675 | -5.954 | -0.721 | 7 |
| Masoprocol | -6.194 | -9.921 | -8.165 | -1.756 | 11 |
| Compound 24-derived pharmacophore | |||||
| Compound 24 | -7.480 | -8.048 | -7.940 | -0.108 | 2 |
| Dolutegravir | -7.736 | -9.998 | -9.316 | -0.682 | 5 |
| Heteroaryldihydropyrimidine-derived pharmacophore | |||||
| Heteroaryldihydropyrimidine | -7.473 | -11.848 | -10.192 | -1.655 | 8 |
| Lesinurad | -6.256 | -9.382 | -8.277 | -1.105 | 5 |
| Belzutifan | -7.519 | -10.573 | -9.529 | -1.044 | 6 |
| Simvastatin | -6.587 | -12.046 | -10.065 | -1.981 | 13 |
| Lovastatin | -7.519 | -12.464 | -11.044 | -1.420 | 12 |
| N-(3-chloro-4-fluorophenyl)-3-phenyl-...-derived pharmacophore | |||||
| N-(3-chloro-4-fluorophenyl)-3- ... | -6.820 | 5.337 | -7.578 | 12.915 | 2 |
| Elvitegravir | -6.685 | -10.846 | -9.776 | -1.070 | 11 |
| Sotagliflozin | -6.691 | -10.823 | -9.563 | -1.259 | 11 |
| NVR10-001E2-derived pharmacophore | |||||
| NVR10-001E2 | -6.470 | -10.278 | -8.941 | -1.337 | 7 |
| Voriconazole | -6.387 | -9.840 | -8.611 | -1.229 | 7 |
| Enasidenib | -7.867 | -12.006 | -10.858 | -1.149 | 9 |
| Sulfamoylbenzamide-derived pharmacophore | |||||
| Sulfamoylbenzamide | -7.194 | -10.096 | -9.223 | -0.873 | 5 |
| Enasidenib | -7.236 | -11.182 | -10.028 | -1.154 | 7 |
| Dasatinib | -7.404 | -10.459 | -9.740 | -0.719 | 8 |
|
€ The INTER + INTRA score shows the combined energy contribution from intermolecular and intramolecular interactions. ¥ INTER represents intermolecular interactions, which are typically the binding interactions between the compound and the target. £ INTRA represents intramolecular interactions within the compound itself. ŧ Active torsions represent the number of flexible bonds within each compound that can rotate or flex during binding. | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



