
Submitted:
06 October 2023
Posted:
09 October 2023
You are already at the latest version
Abstract
Oral cancer ranks sixth among Taiwan's top 10 cancers, and most patients with poor prognosis acquire metastases. The essential fatty acid alpha-linolenic acid (ALA) has been found to diminish many cancer properties. However, the anti-cancer activity of ALA in oral cancer has yet to be determined. Migration and invasion assays confirmed OSCC cells' EMT capabilities, whereas flow cytometry and Western blotting identified the molecular pathways. ALA dramatically reduced cell growth in a concentration dependent manner, according to the findings. Low concentrations of ALA (100 or 200 μM) inhibit colony formation, expression of Twist and EMT-related proteins, expression of MMP2/-9 proteins and enzyme activity, as well as cell migration and invasion. Treatment with high concentrations of ALA (200 or 400 μM) greatly increases JNK phosphorylation and c-jun nuclear accumulation, then upregulates the FasL/caspase8/caspase3 and Bid/cytochrome c/caspase9/caspase3 pathways, leading to cell death. Low concentrations of ALA inhibit SAS and GNM cell migration and invasion by suppressing Twist and downregulating EMT-related proteins, or by decreasing the protein expression and enzyme activity of MMP-2/-9, whereas high concentrations of ALA promote apoptosis by activating the JNK/FasL/caspase 8/caspase 3-extrinsic pathway and the Bid/cytochrome c/caspase 9 pathway. ALA demonstrates potential as a treatment for OSCC patients.
Keywords:
α-linolenic acid
; epithelial-mesenchymal transition
; metastasis
; apoptosis
; oral cancer
; OSCC
1. Introduction
Oral cancer comprises cancers of the lips, tongue, and other oral structures, and it is the most prevalent form of head and neck cancer. According to the most recent report on cancer statistics worldwide, published in 2020 by the WHO's International Agency for Research on Cancer (IARC) [1], oral cancer ranks 16th among malignant tumors worldwide, with a global incidence rate of 2%, approximately 378,000 people; the mortality rate is 1.8%, around 178,000 people. Both the incidence and mortality rates are highest in the Asian region. Cancer originates from genetic mutations and is mainly associated with interactions between environmental, genetic, and metabolic factors[2,3,4,5,6].
The risk factors for oral cancer include heavy tobacco use [7] and excessive alcohol consumption [8,9,10]. Smoking and alcohol consumption have been proven to be the primary risk factors for oral squamous cell carcinoma, with a synergistic effect that increases the risk by 35 times [11]. Chewing betel nut, which contains betel nut extracts like ripe areca nut extracts (rANE) and tender areca nut extracts (tANE), increases the risk of oral cancer because it promotes the generation of reactive oxygen species (ROS) and the upregulation of the expression of inflammatory factors including cyclooxygenase-2 (COX-2), prostaglandin E2 (PGE2), and interleukin-1 (IL-1) [12,13].
Infection with the human papillomavirus (HPV) was linked to a rising rate of oral and oropharyngeal cancer in the past few decades, especially among young people [14,15,16]. HPV expresses viral oncoproteins E6 and E7, which inactivate the host's tumor suppressor genes (TSGs) p53 and pRb, leading to uncontrolled cell division and the formation of cancer [8,17,18]. Oral squamous cell carcinoma (OSCC) typically arises from premalignant lesions, resulting in significant histological alterations in the oral mucosa, and the vast majority of oral cancers are squamous cell carcinomas [19,20]. Most oral precancerous lesions are leukoplakia, erythroplakia, submucosal fibrosis, and a small percentage of patients may develop actinic keratosis [20,21]. If any of these lesions occur, surgical excision or further histological examination is necessary to prevent progression to tumors [15,21]. Surgical removal is the preferred treatment method for OSCC, and other therapies include surgery combined with radiotherapy (RT)/chemotherapy (CT), immunotherapy, and photodynamic therapy (PDT) [15,22,23].
The goal of the resection surgery is to completely remove local disease symptoms and neck lymph node metastasis, but the recurrence rate remains at 25% to 48% [20,24,25]. Among 20% to 30% of patients, it has also been observed that the recurrence at the primary site is more common than local and distant site recurrence. Local recurrence accounts for 10%-15% of deaths [20,26], while distant site recurrence accounts for 1% to 3.8% [26,27,28]. Surgical and radiation treatments can lead to tissue hypoxia and fibrosis, making it difficult to detect malignant cells in fibrotic tissue, which is the causes of local recurrence. Slootweg et al. examined 394 patients who underwent oral cancer tumor resection and found that the local recurrence rate was lower in patients with negative margins of adjacent tissues (3.9%) compared to patients with positive margins (21.9%) [20,29]. In addition to recurrent OSCC at the primary site, tongue cancer is most prone to neck lymph node metastasis and has the greatest tumor thickness. Neck lymph node metastasis and extracapsular spread (ECS) of lymph nodes are both major factors contributing to poor prognosis in oral cancer [22,30]. ECS can increase distant metastasis and mortality rates. Patients with neck metastases who are without ECS had a 52% chance of surviving for 5 years, while those who do have ECS have a 28% chance [20,31]. Lungs are the most frequent location of distant metastasis, and people who develop this condition typically only have a lifespan of 8–9 months [20,25]. Clinical research data indicates an 5-year survival rate of approximately 64% for all stages of OSCC patients [32]. However, the presence of neck lymph node metastasis decreases the 5-year survival rate by about 50% [20,33]. It also indicates that metastasis, or the spread of cancer cells from their original site to other organs, is a major contributor to cancer-related mortality [34]. ECS tumor EMT levels can predict the prognosis and survival of head and neck cancer, with shorter overall and disease-free survival, a greater recurrence rate, and wider spread shown in tumors with a higher EMT proportion [35].
The initial phase in metastasis is epithelial-mesenchymal transition (EMT), which can reduce cell-cell adhesion and increase tumor cell adherence to the matrix. To further their invasion and intravasation, tumor cells release matrix metalloproteinases (MMPs) to destroy the extracellular matrix (ECM). At this point, tumor cells can either expand inside blood arteries, bursting them open, or induce endothelial cell death, allowing tumor cells to extravasate via the spaces created by the dead endothelial cells and spread to other organs and tissues [36]. Finally, tumor cells form a favorable microenvironment for tumor colonization with immune cells and other stromal components, continuously proliferating to form a tumor[37,38]. TGF-, bone morphogenetic protein (BMP), NOTCH, Wnt/-catenin, STAT3, receptor tyrosine kinase (RTK), Hedgehog, extracellular matrix-mediated (ECM-mediated), Hypoxia, Hippo, ET-1, CAV-1, and Oxidative stress are just some of the signaling pathways that can regulate EMT [39,40,41]. These signaling pathways regulate the production of transcription factors like Zeb, Snail, and Twist, which then activate EMT by inducing stromal marker gene expression and inhibiting epithelial marker gene expression [40,42]. The TGF- signaling pathway, the zinc-finger E-box-binding homeobox ZEB1 and ZEB2, the zinc-finger binding transcription factors Snail and Slug, and the basic helix-loop-helix (bHLH) transcription factors Twist all play critical roles in upregulating EMT and can synergize with one another to promote EMT expression. Essential steps in the initiation of EMT include the binding of these proteins to the promoter regions of cell adhesion-related genes and the subsequent regulation of the expression of EMT-related proteins [40,42,43]. Twist1 can activate EMT by upregulating Vimentin [40,44]. Twist is mainly activated by TGF-β, Wnt/β-catenin, STAT3, and hypoxia signaling pathways, initiating EMT [45]. Studies have shown that Twist can downregulate E-cadherin and promote the expression of N-cadherin, fibronectin, and Vimentin [46]. In human pulmonary arterial endothelial cells (HPAE), activation of the TGF-β signaling pathway can induce Twist1 expression [40]. Twist is also associated with promoting cancer cell metastasis, angiogenesis, invasion, extravasation, and chromosomal instability. Moreover, high expression of Twist can protect cancer cells from apoptosis [47]. Research has shown that increased expression of Twist1/2 in cancer cells can inhibit the expression of p53 and Rb, leading to anti-cellular senescence and death [45,47]. In BALB/c mice implanted with mouse breast cancer cells such as 67NR, 168FARN, 4TO7, and 4T1, Twist1 was found to bind to the promoter of Snail2, upregulating Snail2 protein expression to induce EMT and promote breast tumor metastasis [43,48]. Relevant studies have shown that the transcription factors SNAIL and ZEB2, associated with EMT, can upregulate MMP expression, further promoting cancer cell invasion [42]. It has been demonstrated through studies [49] that tumors with high MMP expression have a poor prognosis and a higher chance of recurrence. Head and neck squamous cell carcinoma (HNSCC) also has a role for MMP-9 in tumor invasion and metastasis [49,50]. Several studies have shown that MMP-2, MMP-3, and MMP-9 are expressed in OSCC cells and tissues [51]. In addition, SAS human tongue squamous cell carcinoma cells and GNM human gingival cancer with neck lymph node metastases have been demonstrated to exhibit MMP-2 and MMP-9, which is linked to migration and invasion [52,53].
Cell apoptosis is an irreversible process and is considered a unique and important programmed cell death mode that maintains cellular homeostasis within tissues. The pathways involved can be broadly classified as either extrinsic (involving death receptors) or intrinsic (involving mitochondria). Cell apoptosis is crucial in various developmental processes and requires strict regulation. Developmental defects, autoimmune diseases, neurological disorders, and cancer are all possible outcomes of apoptotic dysregulation [54]. Transmembrane death receptors, mostly derived from the tumor necrosis factor receptor gene superfamily, mediate the extrinsic pathway. The death receptors of the TNF receptor family contain a death domain rich in cysteine residues in their extracellular structure, which binds to procaspase-8 and self-catalyzes into cleaved caspase-8. This further cleaves and activates caspase-3 and promotes the cleavage of Poly(ADP-ribose) Polymerases (PARP), leading to cell apoptosis. Multiple non-receptor stimuli, such as growth factors, hormones, radiation, toxins, hypoxia, viral infection, and free radicals, are involved in the intrinsic pathway. These conditions trigger the release of cytochrome c into the cytoplasm by increasing the permeability of the mitochondrial membrane, leading to a loss of the mitochondrial membrane potential. Apoptosomes are formed when cytochrome c binds to Apaf-1 and procaspase-9. [55,56]. After being cleaved, procaspase-9 then activates caspase-3, which in turn induces cell death. Cytoplasmic adapter proteins with death domains are recruited by ligands and death receptors such as FasL/FasR, TNF-/TNFR1, Apo3L/DR3, Apo2L/DR4, and Apo2L/DR5. Through death domain dimerization, Fasligand (FasL) binds to procaspase-8 and establishes a connection with the Fas-associated death domain (FADD). As a result, procaspase-8 is cleaved and activated by the death-inducing signaling complex (DISC), which in turn activates caspase-3 and promotes cell apoptosis [54]. NF-kB, Sp1, IFN, c-Myc, FKHRL1, and c-jun are only some of the transcription factors that can influence FasL production [57,58]. These transcription factors can increase the expression of FasL and potentially further activate downstream caspases to promote cell apoptosis. Recent study has revealed an important regulatory role for the JNK signaling pathway in Fas-mediated cell death. Researchers Suhara et al. reported that knocking down FasL expression in VSMCs using adenoviral transfection of a dominant negative c-jun mutant (Adeno-TAM67) was successful. Since c-jun is primarily regulated by JNK, when JNK is inhibited, c-jun is unable to enter the nucleus for transcriptional activity, leading to decreased FasL expression. Based on these results, it has been demonstrated that JNK can upregulate the transcription of the FasL gene [57]. Current research also indicates that the JNK pathway can induce cell apoptosis in OSCC [59]. Therefore, this pathway deserves further investigation.
Substances found in nature have been shown to have anti-tumor properties against several distinct cancers. They also improve immunological function, increase survival, and increase chemotherapy's effectiveness [60,61,62,63,64]. For example, Juniperus communis extract can promote apoptosis in OSCC cells through the exogenous Fas/FasL pathway [65]. The important fatty acid ALA is a polyunsaturated fatty acid (PUFA). Because the human body cannot produce it on its own, ALA must be taken in through food or other means [66]. Walnuts, flaxseeds, hemp seeds, and their oils all contain ALA, an important polyunsaturated fatty acid obtained from plants. Green leafy vegetables, canola oil, soybean oil, and it all contain trace amounts [67]. Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are two of the most essential long-chain fatty acids produced from ALA through a sequence of desaturation and elongation processes [68]. Membrane fluidity, protein and cellular function, eicosanoid metabolism, gene expression, and cell signaling are all crucially regulated by EPA and DHA, which are derived from fish oil or ALA-rich plant lipids. EPA and DHA integrate into a cascade that operates in parallel with the inflammation cascade governed by the metabolism of arachidonic acid (AA) [69,70]. Previous studies into the EPA cascade's anti-inflammatory effects found that it mitigated the AA cascade's inflammatory effects, suggesting that the EPA cascade had this property [71]. Numerous studies have indicated the anticancer effects of ALA, which can inhibit the growth of human breast cancer cells and suppress the proliferation of human renal cancer cells [72,73,74,75,76]. ALA upregulates the expression of caspase-3 to induce apoptosis in human colon cancer cells (HT-29) [77]. ALA also induces apoptosis in MCF-7 human breast cancer cells by increasing Bax expression and decreasing Bcl-2 expression, which results in the release of cytochrome c, which in turn activates caspase-3 and promotes PARP cleavage [78]. Studies have also shown that ALA inhibits EMT in MDA-MB-231 and Hs578T human breast cancer cells by upregulating E-cadherin expression and downregulating Twist1, Snail2, N-cadherin, Vimentin, and fibronectin expression [79]. The capacity of SiHa and HeLa human cervical cancer cells to migrate is inhibited by ALA because it reduces the production of MMP-2 and MMP-9 [80]. The growth, adhesion, and invasion of HT29, HCT116, and MCA38 human and mouse colon cancer cells, respectively, are all suppressed by this compound [81]. Animal studies have also shown that a diet high in ALA from fish oil (1 g/day, containing 54.5% ALA) slows the progression of breast cancer in rats [82], and that a diet high in ALA from mice (content ranging from 17.2% to 43.12%) slows the progression of prostate cancer in mice. [83]. Although ALA has been demonstrated to combat various types of cancer, there is limited research on its relevance to oral cancer, and further investigation is warranted.
2. Materials and Methods
2.1. Cell Culture
SAS and GNM human oral squamous cell carcinoma cell lines were provided by Professor Chen-Chia Yu of Chung Shan Medical University for this study. The cells were grown in DMEM with 10% FBS and 1% penicillin added. Cells were incubated in a humidified incubator at 37°C. Additionally, the medium was changed every one to two days.
2.2. Cell Viability Analysis
Cell metabolic activity was measured using the MTT assay. SAS (2.5 × 105) and GNM (4 × 105) cell lines were treated with serial concentrations of ALA (0–800 μM) for 24 hours. Then, the cells were assessed for viability using the MTT reagent. To ascertain the results, the absorbance was detected at 570 nm using a microplate reader.
2.3. Total Protein Extraction
SAS (6 × 105 in 6 cm dish) and GNM (1 × 106 in 6 cm dish) cell lines were treated with different concentrations of ALA (0–200 μM) for different time durations. After treatment, the cells were scraped and lysed using an ultrasonic cell disruptor (Vibra-Cell™, Sonics & Materials, Inc., CT, USA). After cell lysis, the samples were centrifuged at 4°C and 15,000 rpm (20,600g) for 30 minutes using a refrigerated microcentrifuge (3,500 Centrifuge, KUBOTA, OSA, Japan). The resulting supernatant can be used for protein quantification, sample preparation, or stored at -20°C.
2.4. Nuclear and Cytosolic Protein Extraction
SAS and GNM cell lines were seeded at density of 3 × 106 and 3.6 × 106 cells in 10 cm culture dishes, treated with 200 and 400 μM ALA for 0, 15, 30, and 60 minutes, and then centrifuged at 4°C and 8,500 rpm (6,600g) for 5 minutes using a refrigerated microcentrifuge. Then the sample was subjected to 30 minutes of shaking at 4°C using an orbital shaker (Intelli-Mixer RM-2, Daigger Scientific, NJ, USA). Cytosolic protein was extracted from the supernatant, and nuclear protein was extracted from the pellet. Both protein extracts could be used for protein quantification, sample preparation or stored at -20°C.
2.5. Mitochondrial Protein Extraction
SAS and GNM cells were seeded at a density of 1.8 × 106 and 2.1 × 106, respectively, in 10 cm culture dishes and treated with various concentrations of ALA (0, 50, 100, and 200 μM) for 16 hours. The cells were then harvested and cytosol was extracted using a buffer mix. The supernatant was collected and saved. The supernatant was further centrifuged at 4°C and 10,400 rpm (10,000g) for 30 minutes, and the resulting supernatant was collected as the cytoplasmic fraction. Next, 80 μL of mitochondria extraction buffer mix was added to the Eppendorf tube containing the pellet. The tube was vortexed (Vortex-Genie 2, Scientific Industries, Inc., NYC, USA) for 10 seconds to separate the mitochondria, which could be directly used for sample preparation or stored at -20°C in a freezer.
2.6. Western Blotting
Semidry transfer to a polyvinylidene fluoride (PVDF) membrane was used to transfer the proteins that had been separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The transferred PVDF membranes were blocked with 5% non-fat dry milk in PBS. Primary antibodies were selected based on the research objectives and incubated with the membranes overnight at 4°C. The membranes were then washed three times with PBST at room temperature. Secondary antibodies were selected based on the research objectives and incubated with the membranes for 1 hour at room temperature. After three washes with PBST, the antibodies bound to the protein blots were detected using ImageQuant (LAS-4000, FUJIFILM, TYO, Japan). The following primary antibodies and reagents were used in this study: caspase-8 antibody (9746) (1:500), Anti-phospho-JNK (Thr183/Tyr185, Thr221/Tyr223) antibody (07-175) (1:1000), β-actin antibody (MAB1501) (1:4000), Fas antibody (GTX13550) (1:1000), Fas Ligand antibody (GTX66619) (1:1000), Bid antibody (GTX110568) (1:1000), E-cadherin antibody (GTX100443) (1:1000), Vimentin antibody (GTX100619) (1:5000), Twist 1/2 antibody (GTX127310) (1:1000), MMP-2 antibody (GTX104577) (1:1000), c-jun antibody (SC-1694) (1:1000), GAPDH antibody (SC-32233) (1:500), Anti-JNK 1/2 antibody (A1HO1362) (1:500), Anti-caspase 9 antibody (ab32539) (1:500), Anti-cleaved-caspase 3 antibody (ab2302) (1:500), Anti-cytochrome c antibody (ab13575) (1:1000), Anti-Poly-(ADP-Ribose)-Polymerase antibody (11835238001) (1:1000), COXIV Polyclonal antibody (11242-1-AP) (1:1000), MMP-9 antibody (NBP2-13173) (1:1000). The following secondary antibodies were used: Mouse IgG antibody (GTX213111-01) (1:6000), Rabbit IgG H&L antibody (ab97051) (1:6000).
2.7. Wound Healing Assay
Culture-Inserts were attached to 3 cm culture dishes, and 70 μL of SAS (1.5 × 104) and GNM (3 × 104) cells were seeded into the grids on both sides of the Culture-Insert. After incubation for 24 hours, 2 mL of DMEM medium was added to each plate, and the Culture-Insert was removed vertically. The images were taken at a 100X magnification level using a microscope. Subsequently, serial concentrations of 0–100 μM or 0–200 μM ALA were added to the dishes, and they were incubated in a cell culture incubator for 24 or 48 hours. The cells were observed and photographed under a microscope at 100x magnification, and the images were quantified using Image J.
2.8. Cell Invasion Assay
An 8.0 μm pore invasion chamber coated with Matrigel was set up then SAS (1.5 × 104) and GNM (3 × 104) cells were seeded into it. The cells were incubated for 24 hours and then treated with serial concentrations of ALA (0–100 μM or 0–200 μM). After a further day of incubation, the cells were fixed and stained for microscopic examination. Image J was then used to perform the analysis on the images.
2.9. Analysis of Cell Apoptosis
SAS and GNM cell lines were separately seeded in 6 cm culture dishes at a density of 6 × 105 and 1 × 106 cells, respectively. Serial concentrations of ALA (0–200 μM or 0–400 μM) were added to the dishes and the cells were treated for 24 hours or 0, 12, or 24 hours with 200, 400 μM ALA. The morphology of cells was examined and photographed at 100x magnification. After treatment, the cells were detached, collected, and detected by flow cytometry. Flow cytometry was used for the detection and quantification of cell apoptosis. The scatter plot displayed annexin V fluorescence-labeled cells on the x-axis and propidium iodide fluorescence-labeled cells on the y-axis. By combining annexin V and propidium iodide staining the results could be distinguished using flow cytometry. Finally, the data were quantified using Flow JO analysis software.
2.10. Cell Colony Formation Assay
SAS and GNM cell lines were separately seeded in a 6-well plate at a density of 2 × 102 and 2 × 103 cells, respectively. Serial concentrations of ALA (0–100 μM, or 0–200 μM) were added to the wells and cells were cultured for 9 days. And the absorbance was measured at 645 nm using a microplate reader. It is reasonable to calculate the colony-forming fraction by comparing the absorbance readings of the control and treatment groups. After treatment, colonies were quantified using Image J software. The percentage of colonies was determined by comparing the absorbance values of the control group and the treatment group.
2.11. Gelatinase Zymography Analysis
SAS and GNM cell lines were separately seeded in a 6-cm culture dish at a density of 6 × 105 and 1×106 cells, respectively. Then treated with serial concentrations of ALA (0–200 μM, or 0–400 μM), and incubated for 24 hours. Electrophoresis was performed on the collected conditioned medium after it had been combined with a loading buffer. The gel was washed twice, incubated in reaction buffer (containing 1% NaN3, 2 M Tris base pH 8.0, and 1 M CaCl2), stained (containing Coomassie Brilliant Blue R-250, naphthol blue black methanol, and acetic acid), and destained (containing methanol and acetic acid). The enzyme activity was quantified using Alphaeasefc Software.
2.12. Statistical Analysis
The experimental data for each group were presented as mean ± SD. SPSS was used for statistical analysis on the experimental data. One-way ANOVA followed by Tukey's test was used for analysis. Significant differences between groups were denoted by different letters when the p-value was <0.05. The statistical data in all Figures were analyzed using the t-test (Student's two-tailed t-test), and significance was indicated by asterisks (*P<0.05, **P<0.01, ***P<0.001) compared to control. Graphs were generated using GraphPad Prism software, and the pathway diagram for the conclusion was created using BioRender (http://biorender.com).
3. Results
3.1. Effects of ALA on cell morphology, cell viability, and cell colony formation in SAS and GNM cells
In this study, the impact of different doses of ALA on the cell morphology and viability of SAS and GNM was observed using microscopy and MTT assay. The results showed (Figure 1A,B) that treatment with 0, 25, 50, 100, 200, 400, 600, and 800 μM ALA for 24 hours caused noticeable shrinkage and rounding of SAS and GNM cells, as well as a relative decrease in cell number, particularly at doses above 200 μM ALA. ALA significantly reduced the cell viability of SAS (Figure 1C), with a survival rate of 67.9% at a dose of 200 μM ALA, while at 600 μM ALA, the survival rate was only about 3.7%. Similarly, ALA treatment significantly decreased the cell viability of GNM (Figure 1D), with a survival rate of 79.1% at a dose of 400 μM ALA, and a survival rate of approximately 5% at 800 μM ALA, accompanied by cell shrinkage and even suspension. Therefore, for further research, SAS and GNM cells were treated with 0, 50, 100, and 200 μM ALA, and 0, 100, 200, and 400 μM ALA, respectively. After 9 days of treatment, changes in cell growth and proliferation were observed. Colony formation assay results (Figure 1E–J) revealed that as the dose of ALA increased, the number of cell colonies decreased, with the highest dose of ALA exhibiting the most significant inhibitory effect. These results indicate that ALA significantly inhibits cell proliferation and colony formation of both SAS (Figure 1E–G) and GNM (Figure 1H–J) cells.
3.2. Effects of ALA on cell migration, invasion, and related protein expression in SAS and GNM cells
The results of the wound healing assay and the Boyden chamber assay were analyzed to determine if ALA affected cell migration and invasion. The results showed that ALA significantly inhibited the migration of SAS and GNM cells (Figure 2A–F). Compared to the control group (100%), treatment with 50 and 100 μM ALA reduced cell migration to 72.9% and 49.3% for SAS (Figure 2C,E), and 100 and 200 μM ALA reduced GNM cell migration to 89.1% and 78.6% (Figure 2D,F). The Boyden chamber assay results (Figure 2G,H) revealed that ALA effectively suppressed the invasion ability of SAS and GNM cells. Compared to the control group (100%), treatment with 50 and 100 μM ALA reduced cell invasion to 61.7% and 35.6%, while 100 and 200 μM ALA reduced GNM cell invasion to 62.1% and 22.1% (Figure 2I,J). These results demonstrate that ALA can effectively inhibit the migration and invasion abilities of SAS and GNM cells.
Poor tumor differentiation and high invasiveness have both been linked to the expression of EMT-related proteins in individuals with oral cancer, according to previous research [84]. Protein expression for E-cadherin and Vimentin was measured after treating SAS and GNM cells with 0, 50, 100, and 200 μM ALA and 0, 100, 200, and 400 μM ALA, respectively. The results showed that E-cadherin expression and Vimentin expression reduced in SAS cells when ALA concentration increased (Figure 3A–C). Although ALA had no effect on E-cadherin expression in GNM cells, it did have an inhibitory effect on Vimentin protein expression (Figure 3D–F). As a result, EMT in SAS and GNM cells can be successfully blocked by ALA. The expression of the protein Twist has been shown to substantially correlate with clinical stage and lymph node metastatic aggressiveness in oesophageal squamous cell carcinoma [85]. Cells were treated with 0, 50, 100, and 200 μM ALA and 0, 100, 200, and 400 μM ALA in SAS and GNM cells, respectively (Figure 3G–J), and the expression of Twist protein was monitored. Figure 3G–J reveal that SAS and GNM cell Twist protein expression drastically decreased as ALA dose increased. It has been hypothesized that ALA can suppress EMT in SAS and GNM cells via influencing Twist transcriptionally. Multiple studies have linked the elevated expression and enzymatic activity of MMP-2 and MMP-9 to oral cancer cell motility and invasion [53]. Cells were treated with 0, 50, 100, and 200 μM ALA and 0, 100, 200, and 400 μM ALA, respectively, to determine if ALA influences the expression and enzymatic activity of MMP-2 and MMP-9 proteins. The results demonstrated that the expression of MMP-2 and pro-MMP-9 proteins, as well as their enzymatic activity, considerably decreased as the dose of ALA increased (Figure 3K–P,Q–V). ALA may prevent SAS and GNM cell migration and invasion by decreasing MMP-2 and MMP-9 expression and activity.
3.3. Effects of ALA on cell apoptosis and related protein expression in SAS and GNM cells
Figure 1 shows that excessive ALA concentrations induce cell death. Cells were treated with 0, 50, 100, and 200 μM ALA and 0, 100, 200, and 400 μM ALA, respectively, and the expression of apoptosis-related proteins was detected to elucidate the potential pathways by which ALA promotes apoptosis in SAS and GNM cells. In addition, apoptosis induction by ALA was verified by flow cytometry when cells were treated with 0, 50, 100, 200, 400 μM and 0, 100, 200, 400, 600 μM ALA for 24 hours. Figure 4A–D demonstrated that the percentage of apoptotic cells increased with increasing ALA concentration, with statistically significant differences being seen between the 200 μM and 400 μM ALA-treated SAS cells (Figure 4C) and the 400 μM and 600 μM ALA-treated GNM cells (Figure 4D). To further investigate the effects of ALA on cell shape and apoptosis, SAS and GNM cells were treated with 200 μM and 400 μM ALA for 0, 12, and 24 hours. After 24 hours of ALA treatment, both SAS and GNM cells shrank noticeably and showed an increase in the number of floating cells (Figure S1A–D), with an accompanying rise in the number of apoptotic cells (Figure S1C,D). This demonstrates that within 24 hours, ALA promotes apoptosis in SAS and GNM cells. Cleaved-caspase 8 (Figure 4F,I), cleaved-caspase 9 (Figure 4G,J), and cleaved-caspase 3 (Figure 4K,M) protein expression was shown to increase with increasing ALA dose (Figure 4E–N). Cleaved-caspase PARP was most strongly induced in SAS and GNM cells by 200 μM and 400 μM ALA, respectively (Figure 4L,N). According to these findings, ALA has the ability to trigger apoptosis in SAS and GNM cells via endogenous mechanisms. Cells were treated with 0, 50, 100, and 200 μM ALA and 0, 100, 200, and 400 μM ALA, respectively, and the expression of mitochondrial and cytoplasmic cytochrome c proteins was detected to assess if ALA regulates the endogenous apoptotic pathway to start apoptosis in SAS and GNM cells. The results indicated (Figure 4O–T) that the amount of cytochrome c in the mitochondria greatly decreased and the amount of cytochrome c in the cytoplasm significantly increased as the dose of ALA was raised. It is believed that ALA promotes cell death by inducing mitochondrial permeability, which in turn causes the release of cytochrome c from the mitochondria into the cytoplasm.
3.4. ALA's ability to regulate the expression of Fas, FasL, Bid, and apoptosis-related proteins in SAS and GNM cells
Previous studies had shown that extrinsic apoptosis in oral cancer cells can be induced by upregulating Fas/FasL/Bid expression [65,86]. Fas, FasL, and Bid protein expression was measured after cells were treated with varying concentrations of ALA for 24 hours. The results revealed that both SAS cells (Figure 5A–D) and GNM cells (Figure 6A–D) expressed less Fas protein and more FasL protein when the dose of ALA increased. The expression of the protein Bid was also boosted by ALA. Initiating extrinsic apoptosis in SAS and GNM cells may be possible due to ALA's potential to activate caspase 8 via upregulation of Fas/FasL. ALA may also induce intrinsic apoptosis in SAS and GNM cells by elevating Bid protein expression, followed by further Bid activation via caspase 8. Figure 4E–N, 5 and 6 show that ALA can cause apoptosis in SAS and GNM cells via both extrinsic and intrinsic apoptotic mechanisms, respectively. To further investigate the influence of ALA treatment period on the expression of apoptosis-related proteins, SAS and GNM cells were treated with 200 μM and 400 μM ALA for 0, 12, and 24 hours. Figure 5E–L and 6E-L show that as time went on, ALA inhibited Fas expression and induced FasL protein expression, and that it also induced the expression of cleaved-caspase 8 (Figure 5H) and Bid (Figure 5I), cleaved-caspase 9 (Figure 5J), cleaved-caspase 3 (Figure 5K), and PARP (Figure 5L). The 24-hour ALA treatment group showed the most dramatic improvements. Within 24 hours, ALA induced Fas/FasL-mediated apoptosis in both SAS and GNM cells.
3.5. The effects of different treatment times of ALA on the expression of phosphorylated JNK protein, nuclear c-jun protein accumulation, and their association with FasL in SAS and GNM cells
Previous research has linked c-jun overexpression and FasL upregulation to JNK signaling activity [57]. A significant association was found between JNK and FasL when analyzing samples from clinical head and neck squamous cell carcinoma patients using the cBioPortal gene database, which shed light on the nature of the interaction between JNK and FasL [87]. Additionally, JNK expression in oral cancer was analyzed using GENT2 (Gene Expression database of Normal and Tumor tissues 2). In contrast to normal tissues, tumor tissues were shown to express JNK at much lower levels [88]. SAS and GNM cells were treated with 200 μM and 400 μM ALA for 0 (control), 15, 30, and 60 minutes, respectively, and their expression of p-JNK and nuclear c-jun proteins was analyzed by Western blotting (Figure 7A–H). Figure 7A–D shows that p-JNK protein expression was enhanced after 15 minutes of ALA treatment compared to the 0-minute ALA treatment group, and that c-jun protein accumulation in the nucleus was considerably raised between 15 and 30 minutes of ALA therapy (Figure 7E–H). These findings provide further support for the hypothesis that FasL protein expression can be upregulated by activation of the JNK signaling pathway. In this investigation, we used the JNK inhibitor SP600125 to learn more about the connection between JNK and FasL. The data demonstrated that p-JNK expression was higher in the ALA-treated group compared to the control group. Both p-JNK (Figure 7I–L) and FasL expression (Figure 7M–P) were dramatically suppressed by pretreatment with SP600125 in response to ALA. In conclusion, the data presented above demonstrate that ALA stimulates cell apoptosis via regulation of FasL via JNK activation.
4. Discussion
The basic structure of the cell membrane is lipids, with PUFAs, especially n-6 PUFA arachidonic acid (AA), being one of the most essential components [89]. AA can be converted into eicosanoids by phospholipase A2 (PLA2), which are linked to tumor growth, progression, and metastasis [90,91]. Recent studies on the anticancer properties of n-3 PUFAs has focused mostly on EPA and DHA present in fish oil, and these fatty acids have been demonstrated to have an effect against many different types of cancer [92,93,94]. It has also been demonstrated that they improve the efficacy and tolerability of chemotherapy[95]. Researchers have found that n-3 PUFAs influence immunological responses, inflammation, cell proliferation, apoptosis, metastasis, and angiogenesis in cancer cells via blocking the production of prostaglandins [96]. n-3 PUFAs have been shown to modulate membrane fluidity or modify the structure and composition of lipid rafts, thereby inhibiting the growth of breast cancer cells [97,98]. The maximum concentration of alpha-linolenic acid (ALA) is found in flaxseed oil, which is an excellent source of ALA supplementation [72,99]. According to research, ALA is advantageous to the human body and has anticancer properties. A case-control study suggested that ALA consumption may protect against the development of bladder cancer [100]. ALA can inhibit liver cancer cell COX-2 expression and promote apoptosis [101]. In ACI-T mice implanted with subcutaneous hepatoma 3924A cells, a diet high in 10% ALA for 28 days significantly suppressed the expression of fatty acid synthase and promoted death in tumor tissues [101]. In BALB/c mice with MCF-7 human breast cancer xenografts and thymectomy, feeding them ALA-rich flaxseed oil (40 g/kg) for 8 weeks inhibited tumor growth and induced apoptosis [102]. A recent study found that ALA inhibits the growth of human breast cancer cells (MCF-7 and MDA-MB-231), as well as cervical cancer cells (SiHa and HeLa), by lowering the generation of nitric oxide (NO) and triggering lipid peroxidation [103]. ALA can also stabilize HIF-1 expression in MCF-7 breast cancer cells and downregulate fatty acid synthase to initiate mitochondrial apoptosis [68]. Although numerous studies have verified ALA's anticancer properties, its effects on oral cancer are still unknown.
In this study, SAS and GNM cells were used as experimental models to investigate the anticancer effects and mechanisms of ALA. 24 hours were spent treating cells with variable concentrations of ALA (0, 25, 50, 100, 200, 400, 600, and 800 μM). It was discovered that 100 and 200 μM ALA did not induce cytotoxicity in SAS and GNM cells, whereas 200 and 400 μM ALA significantly reduced cell viability. In the 800 μM ALA treatment group, only about 5% of the cells were viable (Figure 1). Consequently, non-lethal concentrations of ALA (50, 100, and 100, 200 μM) were chosen for further study of their effects on cell migration. As shown in Figure 2, treatment of SAS and GNM cells with 100 and 200 μM ALA for 24 hours inhibited migration and invasion. These results suggest that GNM cells are more malignant than SAS cells because they require larger amounts of ALA to prevent migration and invasion. Human osteosarcoma (MG63, 143B, and U2OS) cells' proliferation and invasion have been demonstrated to be suppressed by 80 μM ALA in previous research [104]. Proliferation of breast cancer cells (MCF-7, BT-474, MDA-MB 231, and MDA-MB 468) can be suppressed by ALA at concentrations ranging from 20 to 200 μM by downregulating the expression of proteins such cyclin-D1, progesterone receptor, and caveolin-1 and upregulating the expression of estrogen receptor [73,74]. SiHa and HeLa human cervical cancer cell migration can be blocked by ALA (10–80 μM) by downregulation of VEGF, MMP-2, and MMP-9 expression. [80]. Human esophageal cancer cell lines OE19 and OE33 had their proliferation, colony size, adhesion, and migration suppressed by 0.5–5 mM ALA because to its ability to activate the AMPK signaling pathway and increase expression of tumor suppressor genes p53, p21, and p27 [105]. Different cell lines have varying ALA tolerances, and ALA inhibits cancer cell proliferation and metastasis by regulating diverse signaling pathways. Downregulation of E-cadherin and upregulation of N-cadherin, fibronectin, and vimentin expression has been linked to Twist1, which is one of the primary transcription factors that induce EMT. It plays an important part in cancer development and spread [46,47]. Studies show that E-cadherin is downregulated and Twist is overexpressed in OSCC tumor tissues relative to normal tissues[106]. Nuclear expression of Twist is significantly correlated with clinical stage and lymph node metastasis in OSCC cells compared to normal oral mucosal cells [85], suggesting that Twist plays a crucial role in the development and lymph node metastasis of OSCC. Nuclear expression of Twist is significantly correlated with clinical stage and lymph node metastasis in OSCC cells compared to normal oral mucosal cells[85], indicating that Twist plays a crucial role in the development and lymph node metastasis of OSCC. Recent work in our group found that ALA suppresses EMT, migration, and invasion of MDA-MB-231 human breast cancer cells [79]. However, it is unclear if ALA controls EMT in human oral squamous cell carcinoma cells. Figure 3 shows that a dose-dependent inhibition of Twist expression was seen when ALA (100 μM and 200 μM) was applied to SAS and GNM human oral squamous cell carcinoma cells. The expression of Vimentin was downregulated while that of E-cadherin was upregulated as a result of ALA therapy. Oral cancer cell migration and invasion can be encouraged by elevated MMP-2 and MMP-9 expression and activity [53]. The expression and enzymatic activity of MMP-2 and MMP-9 in SAS and GNM cells are suppressed by ALA, as shown in Figure 5. These results suggest that ALA can reduce the expression and activity of MMP-2 and MMP-9, promote the expression of E-cadherin, and so prevent the migration and invasion of SAS and GNM cells. DHA has been proven in studies to reduce MMP-9 production and activity in MCF-7 human breast cancer cells via blocking DNA-binding activity of NF-B and AP. More research is needed to confirm whether or not ALA can suppress MMP expression in SAS and GNM cells by modulating NF-B and AP-1 [107].
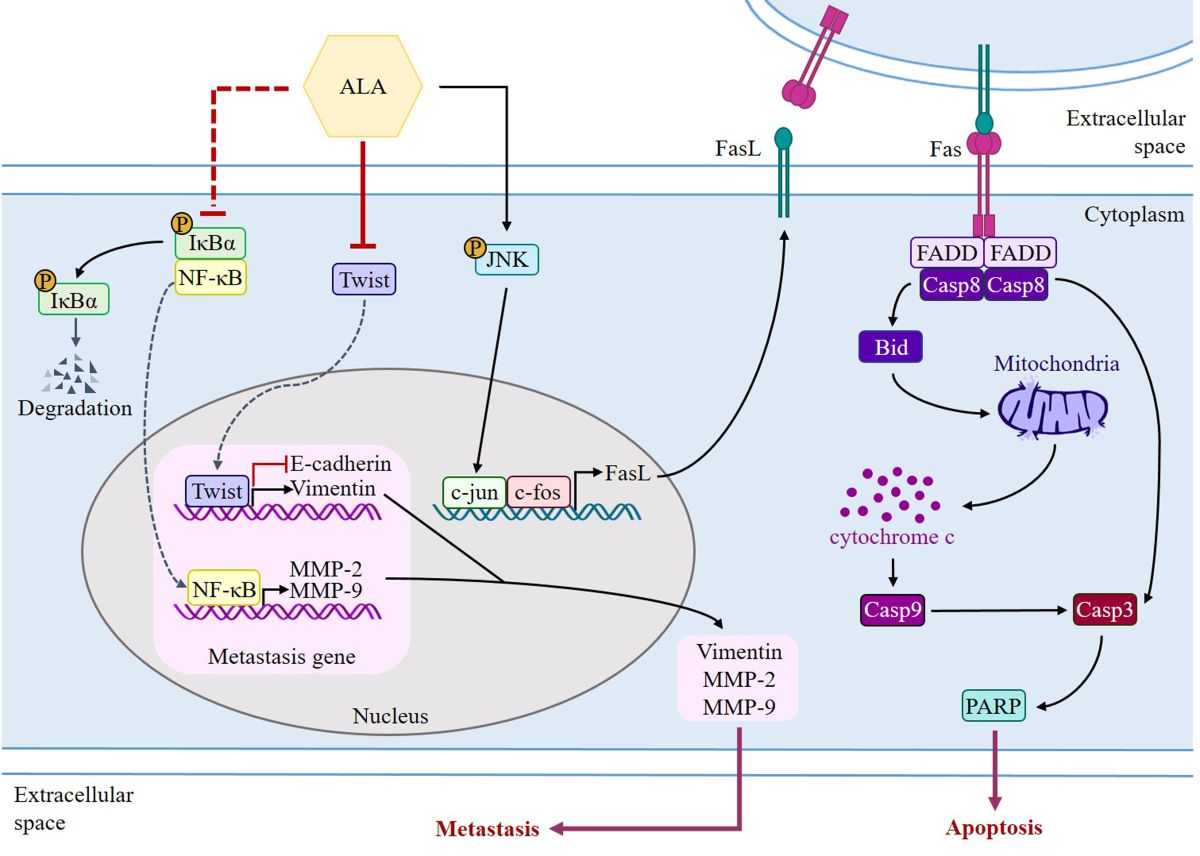
Auto-activation of procaspase-8, caspase-3, and PARP activation are all known to follow FasL interaction to the Fas receptor and begin extrinsic apoptosis [54]. However, the release of cytochrome c and the activation of the intrinsic apoptosis pathway are both triggered by the cleavage of Bid, which occurs upon caspase-8 activation, to generate tBid, which then inserts into the mitochondrial membrane. As a result, Bid has been singled out as an integral connector between extrinsic and intrinsic pathways [108]. Previous studies have shown that the caspase-8/Bid pathway can trigger apoptosis in oral cancer cells [86]. Increased cell proliferation and invasion capabilities, as well as a poor prognosis, have all been linked to a low FasL/Fas ratio in patients with OSCC [109]. ALA was administered at various concentrations to SAS and GNM human oral squamous cell carcinoma cells. High concentrations of ALA substantially decreased Fas expression and increased FasL protein expression, resulting in the activation of caspase-8 and extrinsic apoptosis (Figure 5 and Figure 6). Additionally, ALA treatment further activates Bid via caspase-8, resulting in intrinsic apoptosis. These results are consistent with Lee et al.'s findings [65]. FasL is known to be expressed by T cells during immune responses; it can bond to Fas on the membranes of tumor cells and induce apoptosis. There are two forms of FasL: membrane-bound FasL (mFasL) and soluble FasL (sFasL). mFasL is cleaved by MMP to produce the latter [58]. Increased sFasL can compete with mFasL on T cells to bind to Fas receptors on the membranes of cancer cells, thereby inhibiting the signaling that induces cell apoptosis and suppressing the immune system's attack on malignancies [109]. The findings of this study demonstrated that ALA effectively inhibited the expression of MMPs, suggesting that ALA may also inhibit the production of sFasL to reduce competitive binding with membrane-bound mFasL. Through the interaction between mFasL and Fas receptors on cancer cells, ALA may induce apoptosis. Future experiments can investigate this hypothesis. Previous research has demonstrated that JNK can upregulate the expression of FasL and induce apoptosis in OSCC cells [57,59] Pretreatment with SP600125 substantially inhibited JNK phosphorylation and decreased the expression of FasL induced by ALA, according to the findings of this study. ALA induces JNK phosphorylation, upregulates FasL expression, and promotes cell apoptosis. In a 4-week human investigation, participants consumed 6 g of flaxseed oil cake (containing approximately 5.74 g of ALA) daily; at the conclusion of the study, their plasma ALA concentration increased significantly from 85.13 μM to 249.97 μM [110]. On the basis of these findings, it can be estimated that to increase the plasma ALA concentration by 100, 200, or 400 μM, an additional daily intake of 3.75, 7.5, or 15 g of flaxseed oil (each containing 3.6, 7.2, or 14.4 g of ALA) for 4 weeks would be necessary to achieve the target values, which could improve the prognosis of patients with oral squamous cell carcinoma. In previous human trials, 30 mL of flaxseed oil was administered for 4 weeks, and in another study, flaxseed pastries with a high flaxseed content (containing 59% ALA, approximately 23.6 g) were consumed for 30 days [99,111]. It has been reported that 50 grams of flaxseed consumed daily has no negative effects on the human organism [112]. Multiple studies have found that ALA can prevent the migration and invasion of SAS and GNM oral squamous cell carcinoma cells, as well as downregulate the expression of Twist and EMT-related proteins. Apoptosis in SAS and GNM oral squamous cell carcinoma cells can be induced by ALA through elevation of FasL expression and activation of the FasL/caspase signaling pathway. The use of ALA as a targeted therapeutic in clinical settings is feasible. Additional study can be conducted to discover whether ALA can influence the development of oral squamous cell carcinoma via other signaling pathways, and more in-depth animal tests can be performed, all with the hope that ALA can serve as an auxiliary therapy for OSCC patients.
5. Conclusions
By decreasing expression and activity of MMP-2 and MMP-9 as well as Twist and EMT-related proteins, ALA prevents SAS and GNM cells from migrating and invading. ALA can also induce apoptosis in SAS and GNM cells by upregulating FasL through the FasL/caspase 8/caspase 3-extrinsic apoptotic pathway or the Bid/cytochrome c/caspase 9/caspase 3-intrinsic apoptotic pathway.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org
Author Contributions
Li C-C (Principal Investigator) and Yeh E-L designed the research; Su C-C, Shih Y-W, Liu K-L, Chen H-W, Wu C-C and Yang Y-C performed the experiment; Su C-C, Shih Y-W, Yu C-C, Yeh E-L and Li C-C analyzed the data; Yu C-C and Li C-C wrote the paper; Yeh E-L and Li C-C revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants CSMU-TSMH-107-02 from Antai Tian-Sheng Memorial Hospital, Pingtung, Taiwan. This study was also supported by grant CSMU-INT-111-010 from Chung Shan Medical University and by grant CMU102-ASIA-11 from China Medical University and Asia University, Taichung, Taiwan.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
Flow cytometry and chemiluminescence/fluorescence imaging analyzer were performed in the Instrument Center of Chung Shan Medical University, which is supported by National Science Council, Ministry of Education and Chung Shan Medical University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Nowell, P.C. Diagnostic and prognostic value of chromosome studies in cancer. Ann. Clin. Lab. Sci. 1974, 4, 234–240. [Google Scholar]
- Bajaj, J.; Diaz, E. Stem cells in cancer initiation and progression. J. Cell Biol. 2019, 219, e201911053. [Google Scholar] [CrossRef]
- Tzanakakis, G.; Neagu, M. Proteoglycans and immunobiology of cancer—therapeutic implications. Front. Immunol. 2019, 10, 875. [Google Scholar] [CrossRef]
- Tzanakakis, G.; Giatagana, E.-M. Proteoglycans in the pathogenesis of hormone-dependent cancers: mediators and effectors. Cancers (Basel) 2020, 12, 2401. [Google Scholar] [CrossRef]
- Giatagana, E.-M.; Berdiaki, A. Lumican in carcinogenesis—revisited. Biomolecules 2021, 11, 1319. [Google Scholar] [CrossRef]
- Lin, W.-J.; Jiang, R.-S. Smoking, alcohol, and betel quid and oral cancer: a prospective cohort study. J. Oncol. 2011, 2011. [Google Scholar] [CrossRef]
- Dhanuthai, K.; Rojanawatsirivej, S. Oral cancer: A multicenter study. Med. Oral Patol. Oral Cir. Bucal. 2018, 23, e23. [Google Scholar] [CrossRef]
- Grewal, P.; Viswanathen, V.A. Liver cancer and alcohol. Clin. Liver Dis. 2012, 16, 839–850. [Google Scholar] [CrossRef]
- Stornetta, A.; Guidolin, V. Alcohol-derived acetaldehyde exposure in the oral cavity. Cancers (Basel) 2018, 10, 20. [Google Scholar] [CrossRef]
- Curado, M.P.; Hashibe, M. Recent changes in the epidemiology of head and neck cancer. Curr. Opin. Oncol. 2009, 21, 194–200. [Google Scholar] [CrossRef]
- Verma, P.; Kumar, A. Assessment of relationship of ABO blood groups in oral cancer patients-a retrospective study. Annals of Maxillofacial Surgery 2021, 11, 80. [Google Scholar] [CrossRef]
- Zhong, X.; Lu, Q. Oral microbiota alteration associated with oral cancer and areca chewing. Oral Dis. 2021, 27, 226–239. [Google Scholar] [CrossRef]
- Joseph, B.K. Oral cancer: prevention and detection. Med. Princ. Pract. 2002, 11, 32–35. [Google Scholar] [CrossRef]
- Wong, T.; Wiesenfeld, D. Oral cancer. Aust. Dent. J. 2018, 63, S91–S99. [Google Scholar] [CrossRef]
- Chow, L.Q. Head and neck cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef]
- Hübbers, C.U.; Akgül, B. HPV and cancer of the oral cavity. Virulence 2015, 6, 244–248. [Google Scholar] [CrossRef]
- Sarode, G.; Maniyar, N. Epidemiologic aspects of oral cancer. Dis. Mon. 2020, 66, 100988. [Google Scholar] [CrossRef]
- Jones, K.R.; Lodge-Rigal, R.D. Prognostic factors in the recurrence of stage I and II squamous cell cancer of the oral cavity. Archives of Otolaryngology–Head & Neck Surgery 1992, 118, 483–485. [Google Scholar]
- Kademani, D. Oral cancer. In Proceedings of the Mayo Clin. Proc. 2007; pp. 878–887. [Google Scholar]
- D’souza, S.; Addepalli, V. Preventive measures in oral cancer: An overview. Biomed. Pharmacother. 2018, 107, 72–80. [Google Scholar] [CrossRef]
- Montero, P.H.; Patel, S.G. Cancer of the oral cavity. Surgical Oncology Clinics 2015, 24, 491–508. [Google Scholar] [CrossRef]
- Chamoli, A.; Gosavi, A.S. Overview of oral cavity squamous cell carcinoma: Risk factors, mechanisms, and diagnostics. Oral Oncol. 2021, 121, 105451. [Google Scholar] [CrossRef]
- Pearlman, N.W. Treatment outcome in recurrent head and neck cancer. Arch. Surg. 1979, 114, 39–42. [Google Scholar] [CrossRef]
- Younes, R.N.; Gross, J.L. Surgical treatment of lung metastases of head and neck tumors. The American journal of surgery 1997, 174, 499–502. [Google Scholar] [CrossRef]
- Leitner, C.; Rogers, S. Death certification in patients whose primary treatment for oral andoropharyngeal carcinoma was operation: 1992–1997. Br. J. Oral Maxillofac. Surg. 2001, 39, 204–209. [Google Scholar] [CrossRef]
- León, X.; Quer, M. Distant metastases in head and neck cancer patients who achieved loco-regional control. Head & Neck: Journal for the Sciences and Specialties of the Head and Neck 2000, 22, 680–686. [Google Scholar]
- Kowalski, L.P.; Carvalho, A.L. Predictive factors for distant metastasis from oral and oropharyngeal squamous cell carcinoma. Oral Oncol. 2005, 41, 534–541. [Google Scholar] [CrossRef]
- Slootweg, P.J.; Hordijk, G.J. Treatment failure and margin status in head and neck cancer. A critical view on the potential value of molecular pathology. Oral Oncol. 2002, 38, 500–503. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Beckett, M.A. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981, 41, 1657–1663. [Google Scholar]
- Johnson, J.T.; Barnes, E.L. The extracapsular spread of tumors in cervical node metastasis. Arch. Otolaryngol. 1981, 107, 725–729. [Google Scholar] [CrossRef]
- Tan, M.T.; Wu, J.G. A PIK3CA transgenic mouse model with chemical carcinogen exposure mimics human oral tongue tumorigenesis. Int. J. Exp. Pathol. 2020, 101, 45–54. [Google Scholar] [CrossRef]
- Bugshan, A.; Farooq, I. Oral squamous cell carcinoma: metastasis, potentially associated malignant disorders, etiology and recent advancements in diagnosis. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Suhail, Y.; Cain, M.P. Systems biology of cancer metastasis. Cell systems 2019, 9, 109–127. [Google Scholar] [CrossRef]
- Lee, W.-Y.; Shin, D.-Y. Prognostic significance of epithelial-mesenchymal transition of extracapsular spread tumors in lymph node metastases of head and neck cancer. Ann. Surg. Oncol. 2014, 21, 1904–1911. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Tavazoie, S.F. Endothelial-cell killing promotes metastasis. Nature 2016, 536, 154–155. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016. [Google Scholar]
- Fares, J.; Fares, M.Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal transduction and targeted therapy 2020, 5, 28. [Google Scholar] [CrossRef]
- Diepenbruck, M.; Christofori, G. Epithelial–mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Babaei, G.; Aziz, S.G.-G. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed. Pharmacother. 2021, 133, 110909. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nature reviews Molecular cell biology 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Science signaling 2014, 7, re8–re8. [Google Scholar] [CrossRef]
- Usman, S.; Waseem, N.H. Vimentin is at the heart of epithelial mesenchymal transition (EMT) mediated metastasis. Cancers (Basel) 2021, 13, 4985. [Google Scholar] [CrossRef]
- Merindol, N.; Riquet, A. The emerging role of Twist proteins in hematopoietic cells and hematological malignancies. Blood cancer journal 2014, 4, e206–e206. [Google Scholar] [CrossRef]
- Yang, F.; Sun, L. SET8 promotes epithelial–mesenchymal transition and confers TWIST dual transcriptional activities. The EMBO journal 2012, 31, 110–123. [Google Scholar] [CrossRef]
- Khan, M.A.; Chen, H.-c. Twist: a molecular target in cancer therapeutics. Tumor Biol. 2013, 34, 2497–2506. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Paolillo, M.; Schinelli, S. Extracellular matrix alterations in metastatic processes. International journal of molecular sciences 2019, 20, 4947. [Google Scholar] [CrossRef]
- Van Tubergen, E.A.; Banerjee, R. Inactivation or loss of TTP promotes invasion in head and neck cancer via transcript stabilization and secretion of MMP9, MMP2, and IL-6. Clin. Cancer Res. 2013, 19, 1169–1179. [Google Scholar] [CrossRef]
- Ogbureke, K.U.; Nikitakis, N.G. Up-regulation of SIBLING proteins and correlation with cognate MMP expression in oral cancer. Oral Oncol. 2007, 43, 920–932. [Google Scholar] [CrossRef]
- Liu, G.; Li, J. The effects of MMPs and TIMPs on the metastasis of oral squamous cell carcinoma to neck lymph nodes. Hua xi kou Qiang yi xue za zhi= Huaxi Kouqiang Yixue Zazhi= West China Journal of Stomatology 2001, 19, 216–218, 224. [Google Scholar]
- Lai, W.-W.; Hsu, S.-C. Quercetin inhibits migration and invasion of SAS human oral cancer cells through inhibition of NF-κB and matrix metalloproteinase-2/-9 signaling pathways. Anticancer Res. 2013, 33, 1941–1950. [Google Scholar]
- Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M. The apoptosome: heart and soul of the cell death machine. Neoplasia 1999, 1, 5–15. [Google Scholar] [CrossRef]
- Hill, M.M.; Adrain, C. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. The EMBO journal 2004, 23, 2134–2145. [Google Scholar] [CrossRef]
- Suhara, T.; Kim, H.-S. Suppression of Akt signaling induces Fas ligand expression: involvement of caspase and Jun kinase activation in Akt-mediated Fas ligand regulation. Mol. Cell. Biol. 2002, 22, 680–691. [Google Scholar] [CrossRef]
- Kavurma, M.; Khachigian, L. Signaling and transcriptional control of Fas ligand gene expression. Cell Death Differ. 2003, 10, 36–44. [Google Scholar] [CrossRef]
- Ho, H.-Y.; Lin, C.-C. Apoptotic effects of dehydrocrenatidine via JNK and ERK pathway regulation in oral squamous cell carcinoma. Biomed. Pharmacother. 2021, 137, 111362. [Google Scholar] [CrossRef]
- Wang, X.; Xie, J. Melittin inhibits tumor growth and decreases resistance to gemcitabine by downregulating cholesterol pathway gene CLU in pancreatic ductal adenocarcinoma. Cancer Lett. 2017, 399, 1–9. [Google Scholar] [CrossRef]
- Gao, S.; Li, X. Cepharanthine induces autophagy, apoptosis and cell cycle arrest in breast cancer cells. Cell. Physiol. Biochem. 2017, 41, 1633–1648. [Google Scholar] [CrossRef]
- Fan, J.; Bao, Y. Mechanism of modulation through PI3K-AKT pathway about Nepeta cataria L.’s extract in non-small cell lung cancer. Oncotarget 2017, 8, 31395. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Yang, Y.-H. Anti-cancer effect of danshen and dihydroisotanshinone I on prostate cancer: targeting the crosstalk between macrophages and cancer cells via inhibition of the STAT3/CCL2 signaling pathway. Oncotarget 2017, 8, 40246. [Google Scholar] [CrossRef]
- Mi, C.; Ma, J. Imperatorin suppresses proliferation and angiogenesis of human colon cancer cell by targeting HIF-1α via the mTOR/p70S6K/4E-BP1 and MAPK pathways. J. Ethnopharmacol. 2017, 203, 27–38. [Google Scholar] [CrossRef]
- Lee, C.-C.; Hsiao, C.-Y. Suppression of oral cancer by induction of cell cycle arrest and apoptosis using Juniperus communis extract. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Swanson, D.; Block, R. Omega-3 fatty acids EPA and DHA: health benefits throughout life. Advances in nutrition 2012, 3, 1–7. [Google Scholar] [CrossRef]
- Welch, A.A.; Shakya-Shrestha, S. Dietary intake and status of n–3 polyunsaturated fatty acids in a population of fish-eating and non-fish-eating meat-eaters, vegetarians, and vegans and the precursor-product ratio of α-linolenic acid to long-chain n–3 polyunsaturated fatty acids: results from the EPIC-Norfolk cohort. The American journal of clinical nutrition 2010, 92, 1040–1051. [Google Scholar]
- Roy, S.; Rawat, A.K. Alpha-linolenic acid stabilizes HIF-1 α and downregulates FASN to promote mitochondrial apoptosis for mammary gland chemoprevention. Oncotarget 2017, 8, 70049. [Google Scholar] [CrossRef]
- Calder, P.C. The role of marine omega-3 (n-3) fatty acids in inflammatory processes, atherosclerosis and plaque stability. Mol. Nutr. Food Res. 2012, 56, 1073–1080. [Google Scholar] [CrossRef]
- Wall, R.; Ross, R.P. Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr. Rev. 2010, 68, 280–289. [Google Scholar] [CrossRef]
- Yadav, S.; Tiwari, V. Comparative efficacy of alpha-linolenic acid and gamma-linolenic acid to attenuate valproic acid-induced autism-like features. J. Physiol. Biochem. 2017, 73, 187–198. [Google Scholar] [CrossRef]
- Yuan, Q.; Xie, F. The review of alpha-linolenic acid: Sources, metabolism, and pharmacology. Phytother. Res. 2022, 36, 164–188. [Google Scholar] [CrossRef]
- Mason-Ennis, J.K.; LeMay-Nedjelski, L.P. Exploration of mechanisms of α-linolenic acid in reducing the growth of oestrogen receptor positive breast cancer cells (MCF-7). Journal of functional foods 2016, 24, 513–519. [Google Scholar] [CrossRef]
- Wiggins, A.K.; Kharotia, S. α-Linolenic acid reduces growth of both triple negative and luminal breast cancer cells in high and low estrogen environments. Nutr. Cancer 2015, 67, 1001–1009. [Google Scholar] [CrossRef]
- Wiggins, A.K.; Mason, J.K. Growth and gene expression differ over time in alpha-linolenic acid treated breast cancer cells. Exp. Cell Res. 2015, 333, 147–154. [Google Scholar] [CrossRef]
- Yang, L.; Yuan, J. α-linolenic acid inhibits human renal cell carcinoma cell proliferation through PPAR-γ activation and COX-2 inhibition. Oncol. Lett. 2013, 6, 197–202. [Google Scholar] [CrossRef]
- González-Fernández, M.J.; Ortea, I. α-Linolenic and γ-linolenic acids exercise differential antitumor effects on HT-29 human colorectal cancer cells. Toxicology Research 2020, 9, 474–483. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, H.D. Growth-Inhibitory and Proapoptotic Effects of Alpha-Linolenic Acid on Estrogen-Positive Breast Cancer Cells: Second Look at n-3 Fatty Acid. Ann. N. Y. Acad. Sci. 2009, 1171, 190–195. [Google Scholar] [CrossRef]
- Wang, S.-C.; Sun, H.-L. α-Linolenic acid inhibits the migration of human triple-negative breast cancer cells by attenuating Twist1 expression and suppressing Twist1-mediated epithelial-mesenchymal transition. Biochem. Pharmacol. 2020, 180, 114152. [Google Scholar] [CrossRef]
- Deshpande, R.; Mansara, P. Alpha-linolenic acid regulates Cox2/VEGF/MAP kinase pathway and decreases the expression of HPV oncoproteins E6/E7 through restoration of p53 and Rb expression in human cervical cancer cell lines. Tumor Biol. 2016, 37, 3295–3305. [Google Scholar] [CrossRef]
- Chamberland, J.P.; Moon, H.-S. Down-regulation of malignant potential by alpha linolenic acid in human and mouse colon cancer cells. Fam. Cancer 2015, 14, 25–30. [Google Scholar] [CrossRef]
- Schiessel, D.L.; Yamazaki, R.K. α-Linolenic fatty acid supplementation decreases tumor growth and cachexia parameters in Walker 256 tumor-bearing rats. Nutr. Cancer 2015, 67, 839–846. [Google Scholar] [CrossRef]
- Li, J.; Gu, Z. Dietary supplementation of α-linolenic acid induced conversion of n-3 LCPUFAs and reduced prostate cancer growth in a mouse model. Lipids Health Dis. 2017, 16, 1–9. [Google Scholar] [CrossRef]
- Liu, P.-F.; Kang, B.-H. Vimentin is a potential prognostic factor for tongue squamous cell carcinoma among five epithelial–mesenchymal transition-related proteins. PLoS One 2017, 12, e0178581. [Google Scholar] [CrossRef]
- Seyedmajidi, M.; Seifi, S. Immunohistochemical expression of TWIST in oral squamous cell carcinoma and its correlation with clinicopathologic factors. J. Cancer Res. Ther. 2018, 14, 964–969. [Google Scholar] [CrossRef]
- Won, D.-H.; Kim, L.-H. In vitro and in vivo anti-cancer activity of silymarin on oral cancer. Tumor Biol. 2018, 40, 1010428318776170. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 2013, 6, pl1–pl1. [Google Scholar] [CrossRef]
- Park, S.-J.; Yoon, B.-H. GENT2: an updated gene expression database for normal and tumor tissues. BMC Med. Genomics 2019, 12, 1–8. [Google Scholar] [CrossRef]
- Das, U.N. “Cell membrane theory of senescence” and the role of bioactive lipids in aging, and aging associated diseases and their therapeutic implications. Biomolecules 2021, 11, 241. [Google Scholar] [CrossRef]
- Panigrahy, D.; Greene, E.R. EET signaling in cancer. Cancer Metastasis Rev. 2011, 30, 525–540. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal transduction and targeted therapy 2021, 6, 94. [Google Scholar] [CrossRef]
- Kato, I.; Akhmedkhanov, A. Prospective study of diet and female colorectal cancer: the New York University Women's Health Study. 1997.
- Takezaki, T.; Inoue, M. Diet and lung cancer risk from a 14-year population-based prospective study in Japan: with special reference to fish consumption. Nutr. Cancer 2003, 45, 160–167. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Omega-3 polyunsaturated fatty acids and their health benefits. Annual review of food science and technology 2018, 9, 345–381. [Google Scholar] [CrossRef]
- Mocellin, M.C.; de Quadros Camargo, C. Fish oil effects on quality of life, body weight and free fat mass change in gastrointestinal cancer patients undergoing chemotherapy: a triple blind, randomized clinical trial. Journal of functional foods 2017, 31, 113–122. [Google Scholar] [CrossRef]
- Larsson, S.C.; Kumlin, M. Dietary long-chain n− 3 fatty acids for the prevention of cancer: a review of potential mechanisms. The American journal of clinical nutrition 2004, 79, 935–945. [Google Scholar] [CrossRef]
- Gutiérrez, S.; Svahn, S.L. Effects of omega-3 fatty acids on immune cells. International journal of molecular sciences 2019, 20, 5028. [Google Scholar] [CrossRef]
- Corsetto, P.A.; Cremona, A. Chemical–physical changes in cell membrane microdomains of breast cancer cells after omega-3 PUFA incorporation. Cell Biochem. Biophys. 2012, 64, 45–59. [Google Scholar] [CrossRef]
- Edel, A.L.; Patenaude, A.F. The effect of flaxseed dose on circulating concentrations of alpha-linolenic acid and secoisolariciresinol diglucoside derived enterolignans in young, healthy adults. Eur. J. Nutr. 2016, 55, 651–663. [Google Scholar] [CrossRef]
- Brinkman, M.T.; Karagas, M.R. Intake of α-linolenic acid and other fatty acids in relation to the risk of bladder cancer: results from the New Hampshire case–control study. Br. J. Nutr. 2011, 106, 1070–1077. [Google Scholar] [CrossRef]
- Vecchini, A.; Ceccarelli, V. Dietary α-linolenic acid reduces COX-2 expression and induces apoptosis of hepatoma cells. J. Lipid Res. 2004, 45, 308–316. [Google Scholar] [CrossRef]
- Truan, J.S.; Chen, J.M. Flaxseed oil reduces the growth of human breast tumors (MCF-7) at high levels of circulating estrogen. Mol. Nutr. Food Res. 2010, 54, 1414–1421. [Google Scholar] [CrossRef]
- Deshpande, R.; Mansara, P. Alpha-linolenic acid regulates the growth of breast and cervical cancer cell lines through regulation of NO release and induction of lipid peroxidation. Journal of Molecular Biochemistry 2013, 2. [Google Scholar]
- Fan, H.; Huang, W. α-Linolenic Acid Suppresses Proliferation and Invasion in Osteosarcoma Cells via Inhibiting Fatty Acid Synthase. Molecules 2022, 27, 2741. [Google Scholar] [CrossRef]
- Moon, H.-S.; Batirel, S. Alpha linolenic acid and oleic acid additively down-regulate malignant potential and positively cross-regulate AMPK/S6 axis in OE19 and OE33 esophageal cancer cells. Metabolism 2014, 63, 1447–1454. [Google Scholar] [CrossRef]
- Fan, C.-C.; Wang, T.-Y. Expression of E-cadherin, Twist, and p53 and their prognostic value in patients with oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 1735–1744. [Google Scholar] [CrossRef]
- Chen, H.-W.; Chao, C.-Y. Inhibition of matrix metalloproteinase-9 expression by docosahexaenoic acid mediated by heme oxygenase 1 in 12-O-tetradecanoylphorbol-13-acetate-induced MCF-7 human breast cancer cells. Arch. Toxicol. 2013, 87, 857–869. [Google Scholar] [CrossRef]
- Reed, J.C. Mechanisms of apoptosis. The American journal of pathology 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Chien, M.-H.; Chang, W.-M. A Fas ligand (FasL)-fused humanized antibody against tumor-associated glycoprotein 72 selectively exhibits the cytotoxic effect against oral cancer cells with a low FasL/Fas ratio. Mol. Cancer Ther. 2017, 16, 1102–1113. [Google Scholar] [CrossRef]
- Patenaude, A.; Rodriguez-Leyva, D. Bioavailability of α-linolenic acid from flaxseed diets as a function of the age of the subject. Eur. J. Clin. Nutr. 2009, 63, 1123–1129. [Google Scholar] [CrossRef]
- Schwab, U.S.; C. Callaway, J. Effects of hempseed and flaxseed oils on the profile of serum lipids, serum total and lipoprotein lipid concentrations and haemostatic factors. Eur. J. Nutr. 2006, 45, 470–477. [Google Scholar] [CrossRef]
- Martinchik, A.; Baturin, A. Nutritional value and functional properties of flaxseed. Vopr. Pitan. 2012, 81, 4–10. [Google Scholar]
Figure 1.
Effect of ALA on morphology and cell viability in SAS cells and GNM cells. (A) The morphology of SAS cells (B) The morphology of GNM cells. (C) The cell viability was determined using the MTT assay of SAS. (D) The cell viability was determined using the MTT assay of GMN. (E-G) The SAS cells growth and proliferation were determined using the colony formation assay. (H-J) The GNM cells growth and proliferation was determined using the colony formation assay. Values are expressed as mean ± standard deviation. Significance of difference in weeks of different tests was evaluated by Tukey’s multiple range test statistical analysis. Different superscript letters a, b, c, d indicate that the statistically different from each other (P<0.05).
Figure 1.
Effect of ALA on morphology and cell viability in SAS cells and GNM cells. (A) The morphology of SAS cells (B) The morphology of GNM cells. (C) The cell viability was determined using the MTT assay of SAS. (D) The cell viability was determined using the MTT assay of GMN. (E-G) The SAS cells growth and proliferation were determined using the colony formation assay. (H-J) The GNM cells growth and proliferation was determined using the colony formation assay. Values are expressed as mean ± standard deviation. Significance of difference in weeks of different tests was evaluated by Tukey’s multiple range test statistical analysis. Different superscript letters a, b, c, d indicate that the statistically different from each other (P<0.05).
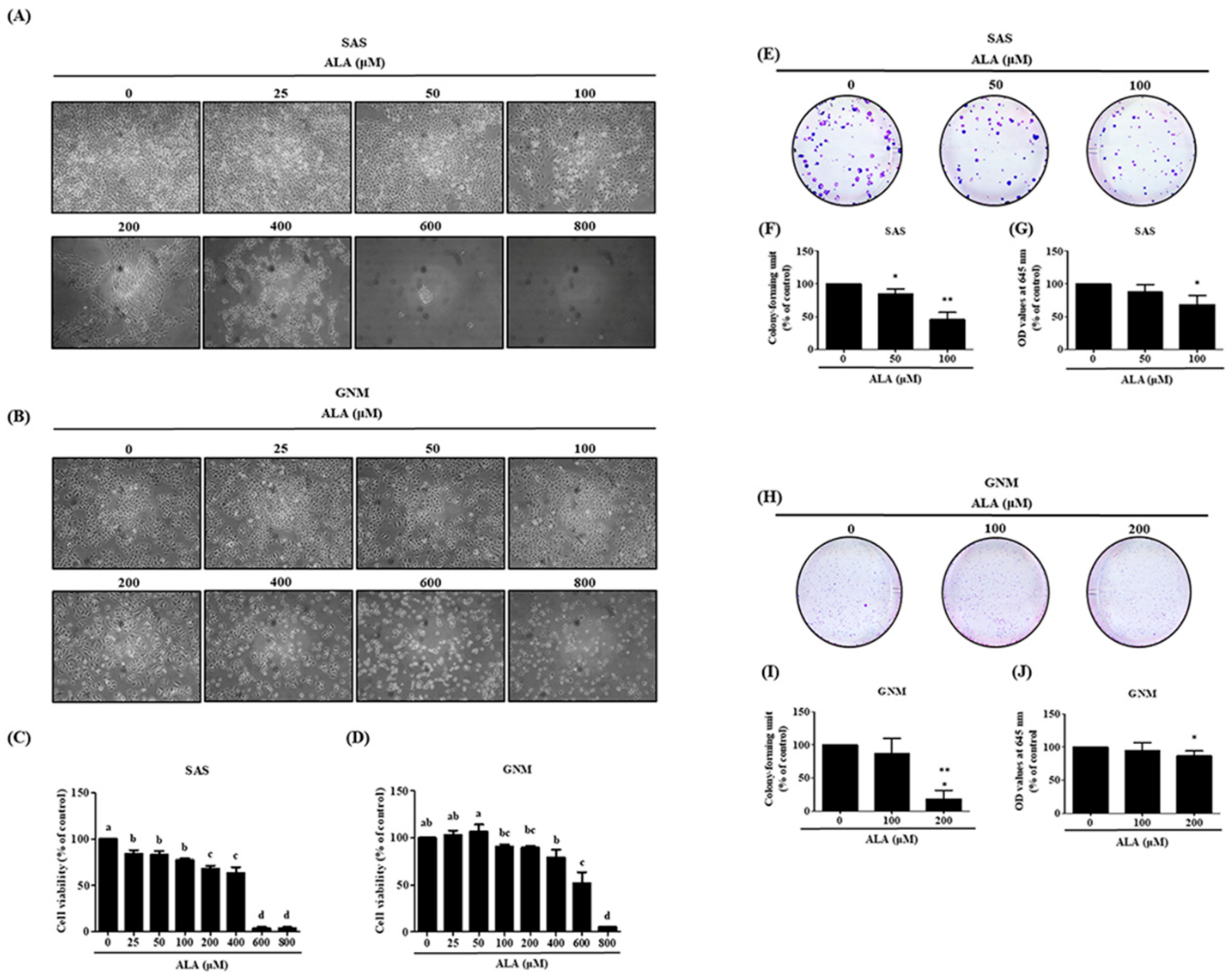
Figure 2.
Depicts the effect of ALA on cell migration and invasion in SAS and GNM cells. SAS cells (A) and GNM cells (B) were treated for 24 or 48 hours with 0, 50, 100, or 0, 100, 200 μM ALA. (C-F) The wound healing assay was used to measure cell migration. (G) SAS cells and (H) GNM cells were treated for 24 hours with 0, 50, 100, or 0, 100, 200 μM ALA. The cell invasion was measured using the Boyden chamber assay (I, J). The mean and standard deviation are used to express the values. Tukey's multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b, and c show that the are statistically different (P<0.05).
Figure 2.
Depicts the effect of ALA on cell migration and invasion in SAS and GNM cells. SAS cells (A) and GNM cells (B) were treated for 24 or 48 hours with 0, 50, 100, or 0, 100, 200 μM ALA. (C-F) The wound healing assay was used to measure cell migration. (G) SAS cells and (H) GNM cells were treated for 24 hours with 0, 50, 100, or 0, 100, 200 μM ALA. The cell invasion was measured using the Boyden chamber assay (I, J). The mean and standard deviation are used to express the values. Tukey's multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b, and c show that the are statistically different (P<0.05).
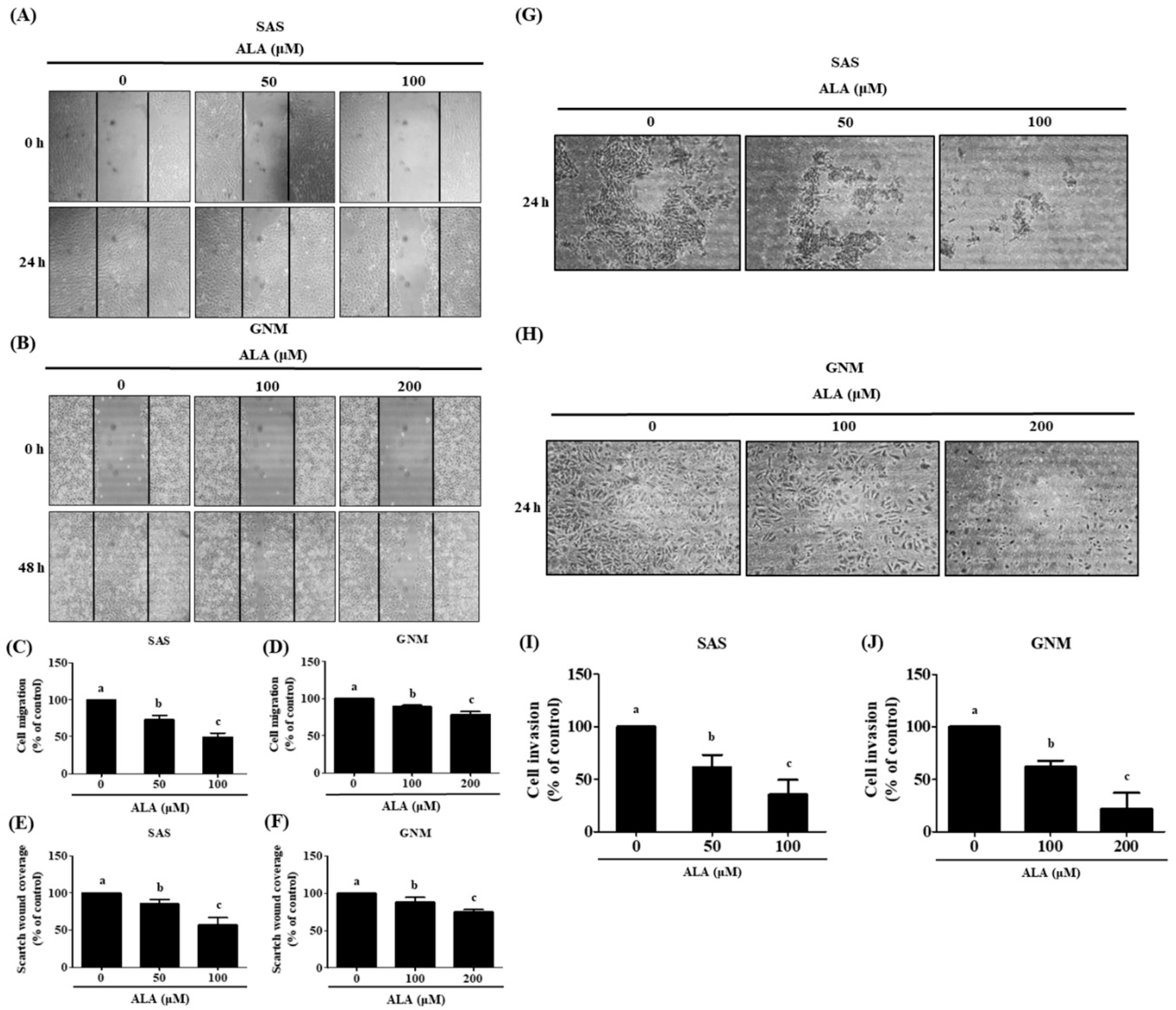
Figure 3.
The effect of ALA on the expression of EMT-related proteins in SAS and GNM cells. For 24 hours, cells were treated with 0, 50, 100, 200 μM or 0, 100, 200, 400 μM ALA. (A) Expression of E-cadherin and vimentin in SAS cells. (B) SAS cell E-Cadherin expression quantification. (C) SAS vimentin expression quantification. (D) Expression of E-cadherin and vimentin in GNM cells. (E) Quantification of E-Cadherin expression in GNM cells. (F) Quantification of vimentin expression in GNM cells. (G) Twist expression in SAS cells. (H) SAS cell twist expression quantification. (I) Twist expression in GNM cells. (J) Quantification of twist expression in GNM cells. (K) SAS cell MMP-2 and MMP-9 expression. (L) SAS cell pro-MMP9 expression quantification. (M) SAS cell MMP-2 expression measurement. (N) Expression of MMP-2 and MMP-9 in GNM cells. (O) Quantification of pro-MMP9 expression in GNM cells. (P) Quantification of MMP-2 expression in GNM cells. (Q) Protein expression and enzyme activity of MMP-2 and MMP-9 in SAS cells. (R) MMP-2 enzyme activity in SAS cells. (S) MMP-9 enzyme activity in SAS cells. (T) Protein expression and enzyme activity of MMP-2 and MMP-9 in GNM cells. (U) MMP-2 enzyme activity in GNM cells. (V) MMP-9 enzyme activity in GNM cells. Western blotting was used to evaluate the protein expression of various proteins. Gelatin zymography was used to assess enzyme activity. The mean and standard deviation are used to express the values. Tukey‘s multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b, c show that the are statistically different (P<0.05).
Figure 3.
The effect of ALA on the expression of EMT-related proteins in SAS and GNM cells. For 24 hours, cells were treated with 0, 50, 100, 200 μM or 0, 100, 200, 400 μM ALA. (A) Expression of E-cadherin and vimentin in SAS cells. (B) SAS cell E-Cadherin expression quantification. (C) SAS vimentin expression quantification. (D) Expression of E-cadherin and vimentin in GNM cells. (E) Quantification of E-Cadherin expression in GNM cells. (F) Quantification of vimentin expression in GNM cells. (G) Twist expression in SAS cells. (H) SAS cell twist expression quantification. (I) Twist expression in GNM cells. (J) Quantification of twist expression in GNM cells. (K) SAS cell MMP-2 and MMP-9 expression. (L) SAS cell pro-MMP9 expression quantification. (M) SAS cell MMP-2 expression measurement. (N) Expression of MMP-2 and MMP-9 in GNM cells. (O) Quantification of pro-MMP9 expression in GNM cells. (P) Quantification of MMP-2 expression in GNM cells. (Q) Protein expression and enzyme activity of MMP-2 and MMP-9 in SAS cells. (R) MMP-2 enzyme activity in SAS cells. (S) MMP-9 enzyme activity in SAS cells. (T) Protein expression and enzyme activity of MMP-2 and MMP-9 in GNM cells. (U) MMP-2 enzyme activity in GNM cells. (V) MMP-9 enzyme activity in GNM cells. Western blotting was used to evaluate the protein expression of various proteins. Gelatin zymography was used to assess enzyme activity. The mean and standard deviation are used to express the values. Tukey‘s multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b, c show that the are statistically different (P<0.05).
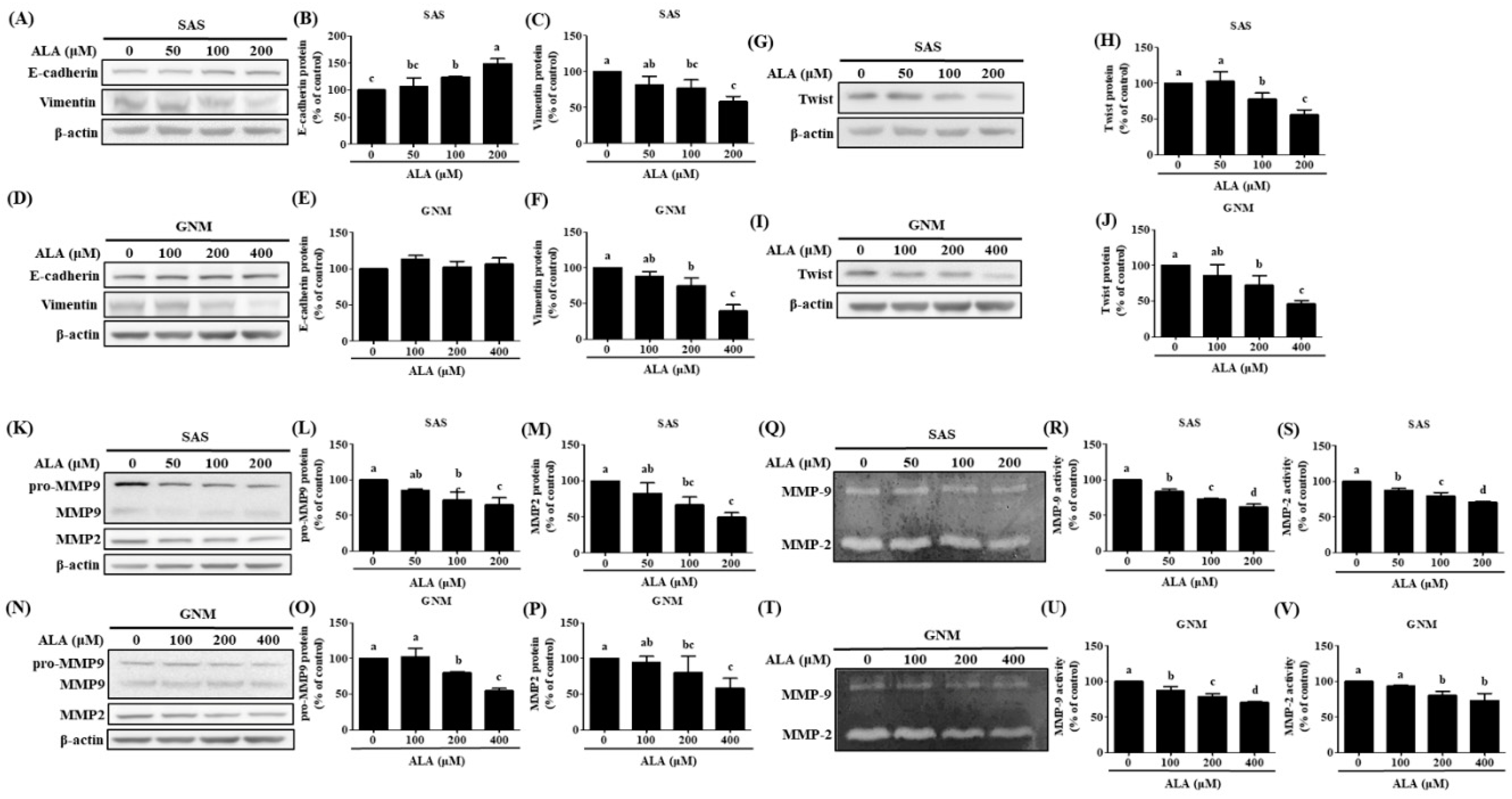
Figure 4.
Effect of ALA on the degree of apoptosis in SAS cells and GNM cells. SAS and GNM cells were treated for 24 hours with 0, 50, 100, 200, 400, or 600 μM ALA. (A) The level of apoptosis in SAS cells. (B) Apoptosis levels in GNM cells. (C) SAS cell apoptosis %. (D) GNM cell apoptosis %. (E) Apoptosis-related protein expression in SAS cells. (F) SAS cell cleaved-caspase 8 expression quantification. (G) SAS cell cleaved-caspase 9 expression was quantified. (H) Apoptosis-related protein expression in GNM cells. (I) Quantification of cleaved-caspase 8 expression in GNM cells. (J) Quantification of cleaved caspase 9 expression in GNM cells. (K) SAS cell cleaved-caspase 3 expression quantification. (L) SAS cell cleaved-caspase PARP expression was quantified. (M) GNM cell cleaved-caspase 3 expression was quantified. (N) GNM cell cleaved-caspase PARP expression was quantified. (O) Cytochrome c protein expression in mitochondria in SAS cells. (P) Cytochrome c cytosol protein expression in SAS cells. (Q) SAS cell mitochondria and cytoplasm protein expression of cytochrome c quantitation. (R) Cytochrome c protein expression in mitochondria in GNM cells. (S) Cytochrome c cytosol protein expression in GNM cells. (T) Quantification of mitochondrial and cytosol protein expression of cytochrome c in GNM cells. The mean and standard deviation are used to express the values. Tukey's multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b, and c show that the are statistically different (P<0.05).
Figure 4.
Effect of ALA on the degree of apoptosis in SAS cells and GNM cells. SAS and GNM cells were treated for 24 hours with 0, 50, 100, 200, 400, or 600 μM ALA. (A) The level of apoptosis in SAS cells. (B) Apoptosis levels in GNM cells. (C) SAS cell apoptosis %. (D) GNM cell apoptosis %. (E) Apoptosis-related protein expression in SAS cells. (F) SAS cell cleaved-caspase 8 expression quantification. (G) SAS cell cleaved-caspase 9 expression was quantified. (H) Apoptosis-related protein expression in GNM cells. (I) Quantification of cleaved-caspase 8 expression in GNM cells. (J) Quantification of cleaved caspase 9 expression in GNM cells. (K) SAS cell cleaved-caspase 3 expression quantification. (L) SAS cell cleaved-caspase PARP expression was quantified. (M) GNM cell cleaved-caspase 3 expression was quantified. (N) GNM cell cleaved-caspase PARP expression was quantified. (O) Cytochrome c protein expression in mitochondria in SAS cells. (P) Cytochrome c cytosol protein expression in SAS cells. (Q) SAS cell mitochondria and cytoplasm protein expression of cytochrome c quantitation. (R) Cytochrome c protein expression in mitochondria in GNM cells. (S) Cytochrome c cytosol protein expression in GNM cells. (T) Quantification of mitochondrial and cytosol protein expression of cytochrome c in GNM cells. The mean and standard deviation are used to express the values. Tukey's multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b, and c show that the are statistically different (P<0.05).
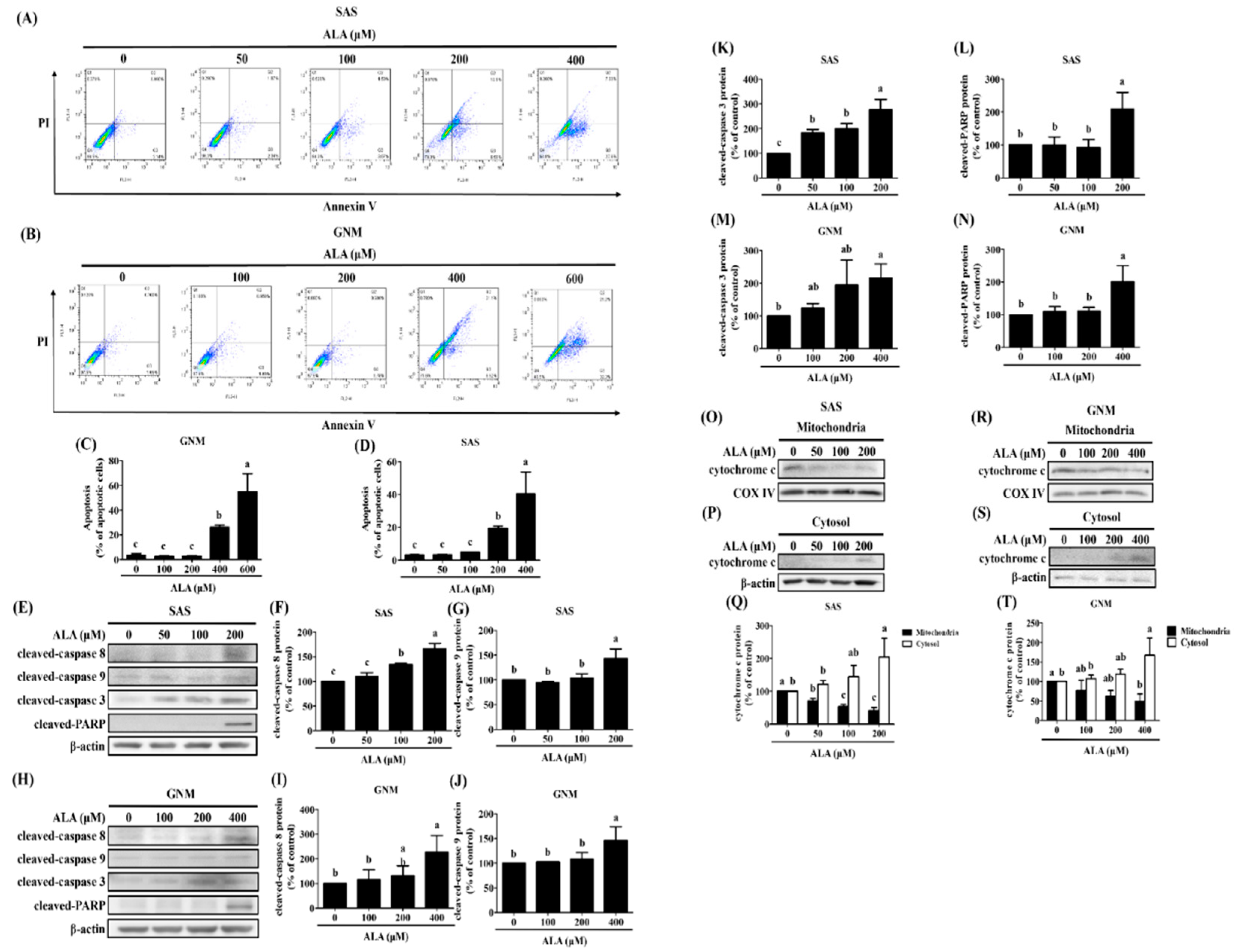
Figure 5.
The effect of ALA on Fas, FasL, Bid, and apoptosis-related protein expression in SAS cells at various time points and ALA concentrations. (A) Fas, FasL, and Bid expression in SAS cells at various ALA concentrations. (B) Quantification of Fas expression in SAS cells at various ALA doses. (C) SAS cell FasL expression measurement under various ALA concentrations. (D) Bid expression measurement of SAS cells at various ALA concentrations. (E) Fas, FasL, Bid, and apoptosis-related protein expression in SAS cells at various time points after ALA treatment. (F) Quantification of Fas expression in SAS cells at various time points after ALA treatment. SAS cell (G) FasL expression measurement at various timepoints after ALA treatment. (H) cleaved-caspase 8 expression in SAS cells at various time points after ALA treatment. (I) Bid expression measurement of SAS cells at various timepoints after ALA treatment. (J) Quantification of cleaved-caspase 9 expression in SAS cells at various time periods after ALA treatment. (K) SAS cells cleaved-caspase 3 expression measurement at various timepoints after ALA treatment. (L) Quantification of cleaved-PARP expression in SAS cells at various time periods after ALA treatment. The mean and standard deviation are used to express the values. Tukey's multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b, and c show that the are statistically different (P<0.05).
Figure 5.
The effect of ALA on Fas, FasL, Bid, and apoptosis-related protein expression in SAS cells at various time points and ALA concentrations. (A) Fas, FasL, and Bid expression in SAS cells at various ALA concentrations. (B) Quantification of Fas expression in SAS cells at various ALA doses. (C) SAS cell FasL expression measurement under various ALA concentrations. (D) Bid expression measurement of SAS cells at various ALA concentrations. (E) Fas, FasL, Bid, and apoptosis-related protein expression in SAS cells at various time points after ALA treatment. (F) Quantification of Fas expression in SAS cells at various time points after ALA treatment. SAS cell (G) FasL expression measurement at various timepoints after ALA treatment. (H) cleaved-caspase 8 expression in SAS cells at various time points after ALA treatment. (I) Bid expression measurement of SAS cells at various timepoints after ALA treatment. (J) Quantification of cleaved-caspase 9 expression in SAS cells at various time periods after ALA treatment. (K) SAS cells cleaved-caspase 3 expression measurement at various timepoints after ALA treatment. (L) Quantification of cleaved-PARP expression in SAS cells at various time periods after ALA treatment. The mean and standard deviation are used to express the values. Tukey's multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b, and c show that the are statistically different (P<0.05).
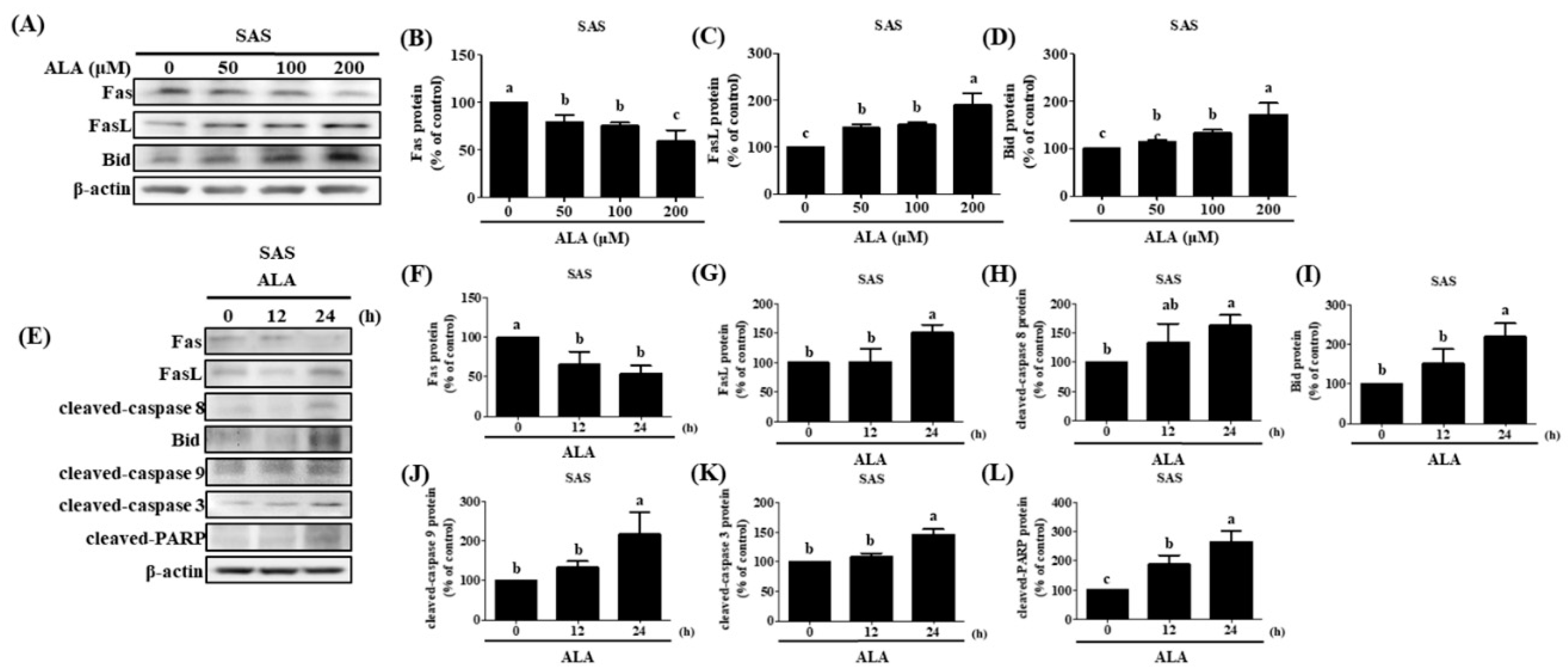
Figure 6.
The effect of ALA on Fas, FasL, Bid, and apoptosis-related protein expression in GNM cells at various time points and ALA concentrations. (A) Fas, FasL, and Bid expression in GNM cells at various ALA concentrations. (B) Quantification of Fas expression in GNM cells at various ALA doses. (C) GNM cell FasL expression measurement under various ALA concentrations. (D) Bid expression measurement of GNM cells at various ALA concentrations. (E) Fas, FasL, Bid, and apoptosis-related protein expression in GNM cells at various time points after ALA treatment. (F) Quantification of Fas expression in GNM cells at various time points after ALA treatment. GNM cell (G) FasL expression measurement at various timepoints after ALA treatment. (H) cleaved-caspase 8 expression in GNM cells at various time points after ALA treatment. (I) Bid expression measurement of GNM cells at various timepoints after ALA treatment. (J) Quantification of cleaved-caspase 9 expression in GNM cells at various time periods after ALA treatment. (K) GNM cells cleaved-caspase 3 expression measurement at various timepoints after ALA treatment. (L) Quantification of cleaved-PARP expression in GNM cells at various time periods after ALA treatment. The mean and standard deviation are used to express the values. Tukey's multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b, and c show that the are statistically different (P<0.05).
Figure 6.
The effect of ALA on Fas, FasL, Bid, and apoptosis-related protein expression in GNM cells at various time points and ALA concentrations. (A) Fas, FasL, and Bid expression in GNM cells at various ALA concentrations. (B) Quantification of Fas expression in GNM cells at various ALA doses. (C) GNM cell FasL expression measurement under various ALA concentrations. (D) Bid expression measurement of GNM cells at various ALA concentrations. (E) Fas, FasL, Bid, and apoptosis-related protein expression in GNM cells at various time points after ALA treatment. (F) Quantification of Fas expression in GNM cells at various time points after ALA treatment. GNM cell (G) FasL expression measurement at various timepoints after ALA treatment. (H) cleaved-caspase 8 expression in GNM cells at various time points after ALA treatment. (I) Bid expression measurement of GNM cells at various timepoints after ALA treatment. (J) Quantification of cleaved-caspase 9 expression in GNM cells at various time periods after ALA treatment. (K) GNM cells cleaved-caspase 3 expression measurement at various timepoints after ALA treatment. (L) Quantification of cleaved-PARP expression in GNM cells at various time periods after ALA treatment. The mean and standard deviation are used to express the values. Tukey's multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b, and c show that the are statistically different (P<0.05).
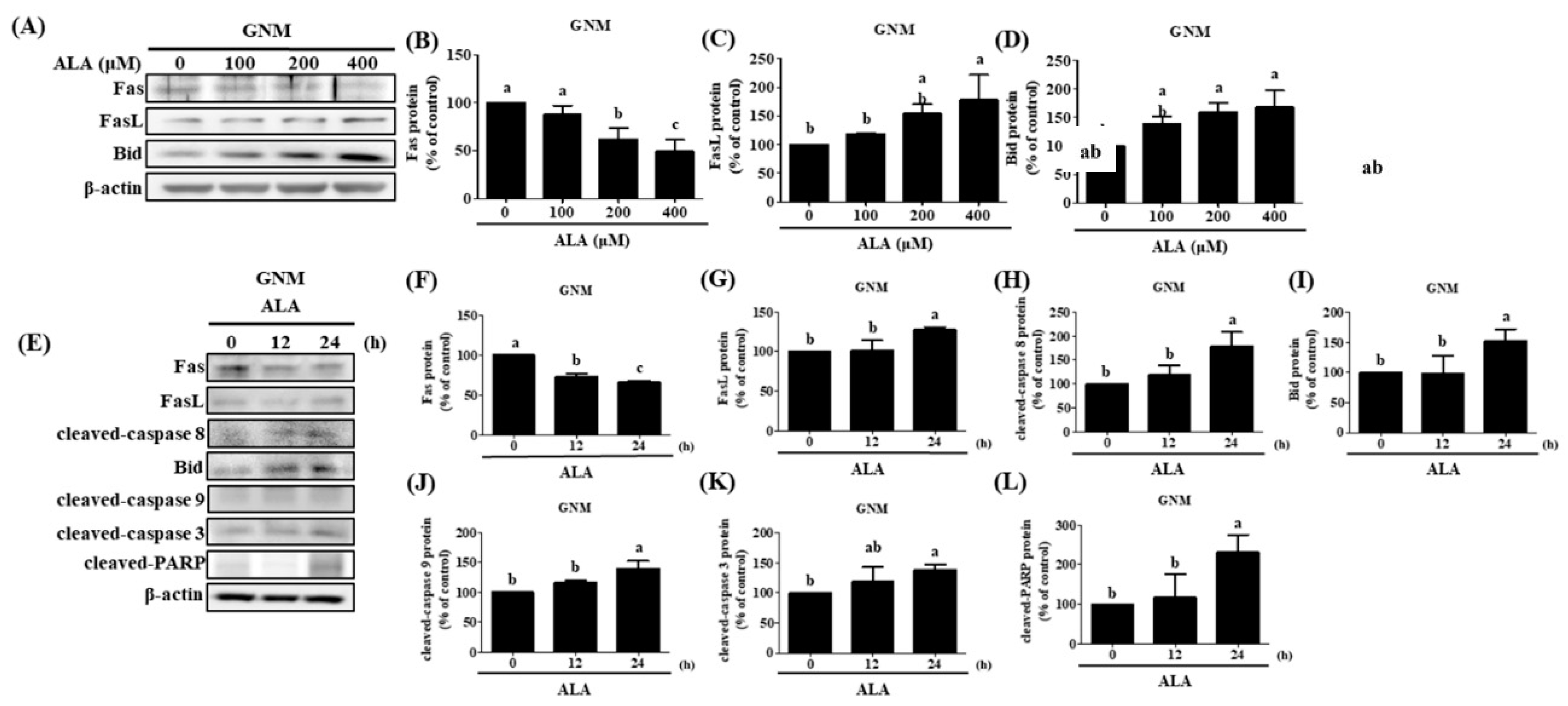
Figure 7.
Effect of ALA on p-JNK protein expression and c-jun protein accumulation of nucleus and JNK inhibitor on p-JNK and FasL protein expression in SAS and GNM cells at different time points. SAS and GNM cells were exposed to 200 or 400 μM ALA for 0, 15, 30, and 60 minutes. (A) p-JNK, JNK1, and JNK2 protein expression in SAS cells at various time periods after ALA treatment. (B) Quantification of p-JNK protein expression in SAS cells at various time periods after ALA treatment. (C) p-JNK, JNK1, and JNK2 protein expression in GNM cells at various time periods after ALA treatment. (D) Quantification of p-JNK protein expression in GNM cells at various time points after ALA treatment. (E) c-jun and PARP protein expression in SAS cells at various time periods after ALA treatment. (F) Quantification of c-jun protein expression in SAS cells at various time periods after ALA treatment. (G) c-jun and PARP expression in GNM cells at various time periods after ALA treatment. (H) Quantification of c-jun and protein expression in GNM cells at various time periods after ALA treatment. (I) Protein expression of p-JNK, JNK1, and JNK2 in SAS cells after treatment with a JNK inhibitor. (J) Quantification of p-JNK protein expression in SAS after treatment with a JNK inhibitor. (K) p-JNK, JNK1, and JNK2 protein expression in GNM cells after JNK inhibitor treatment. (L) Quantification of p-JNK protein expression in GNM cells after treatment with a JNK inhibitor. (M) FasL protein expression in SAS cells after JNK inhibitor treatment (N) FasL protein expression quantification in SAS cells after JNK inhibitor treatment. (O) FasL protein expression in GNM cells after JNK inhibitor treatment (P) FasL protein expression quantification in GNM cells after JNK inhibitor treatment. Western blotting was used to determine protein expression. The mean and standard deviation are used to express the values. Tukey's multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b imply that the are statistically different (P<0.05).
Figure 7.
Effect of ALA on p-JNK protein expression and c-jun protein accumulation of nucleus and JNK inhibitor on p-JNK and FasL protein expression in SAS and GNM cells at different time points. SAS and GNM cells were exposed to 200 or 400 μM ALA for 0, 15, 30, and 60 minutes. (A) p-JNK, JNK1, and JNK2 protein expression in SAS cells at various time periods after ALA treatment. (B) Quantification of p-JNK protein expression in SAS cells at various time periods after ALA treatment. (C) p-JNK, JNK1, and JNK2 protein expression in GNM cells at various time periods after ALA treatment. (D) Quantification of p-JNK protein expression in GNM cells at various time points after ALA treatment. (E) c-jun and PARP protein expression in SAS cells at various time periods after ALA treatment. (F) Quantification of c-jun protein expression in SAS cells at various time periods after ALA treatment. (G) c-jun and PARP expression in GNM cells at various time periods after ALA treatment. (H) Quantification of c-jun and protein expression in GNM cells at various time periods after ALA treatment. (I) Protein expression of p-JNK, JNK1, and JNK2 in SAS cells after treatment with a JNK inhibitor. (J) Quantification of p-JNK protein expression in SAS after treatment with a JNK inhibitor. (K) p-JNK, JNK1, and JNK2 protein expression in GNM cells after JNK inhibitor treatment. (L) Quantification of p-JNK protein expression in GNM cells after treatment with a JNK inhibitor. (M) FasL protein expression in SAS cells after JNK inhibitor treatment (N) FasL protein expression quantification in SAS cells after JNK inhibitor treatment. (O) FasL protein expression in GNM cells after JNK inhibitor treatment (P) FasL protein expression quantification in GNM cells after JNK inhibitor treatment. Western blotting was used to determine protein expression. The mean and standard deviation are used to express the values. Tukey's multiple range test statistical analysis was used to assess the significance of differences in weeks across different tests. Different superscript letters a, b imply that the are statistically different (P<0.05).
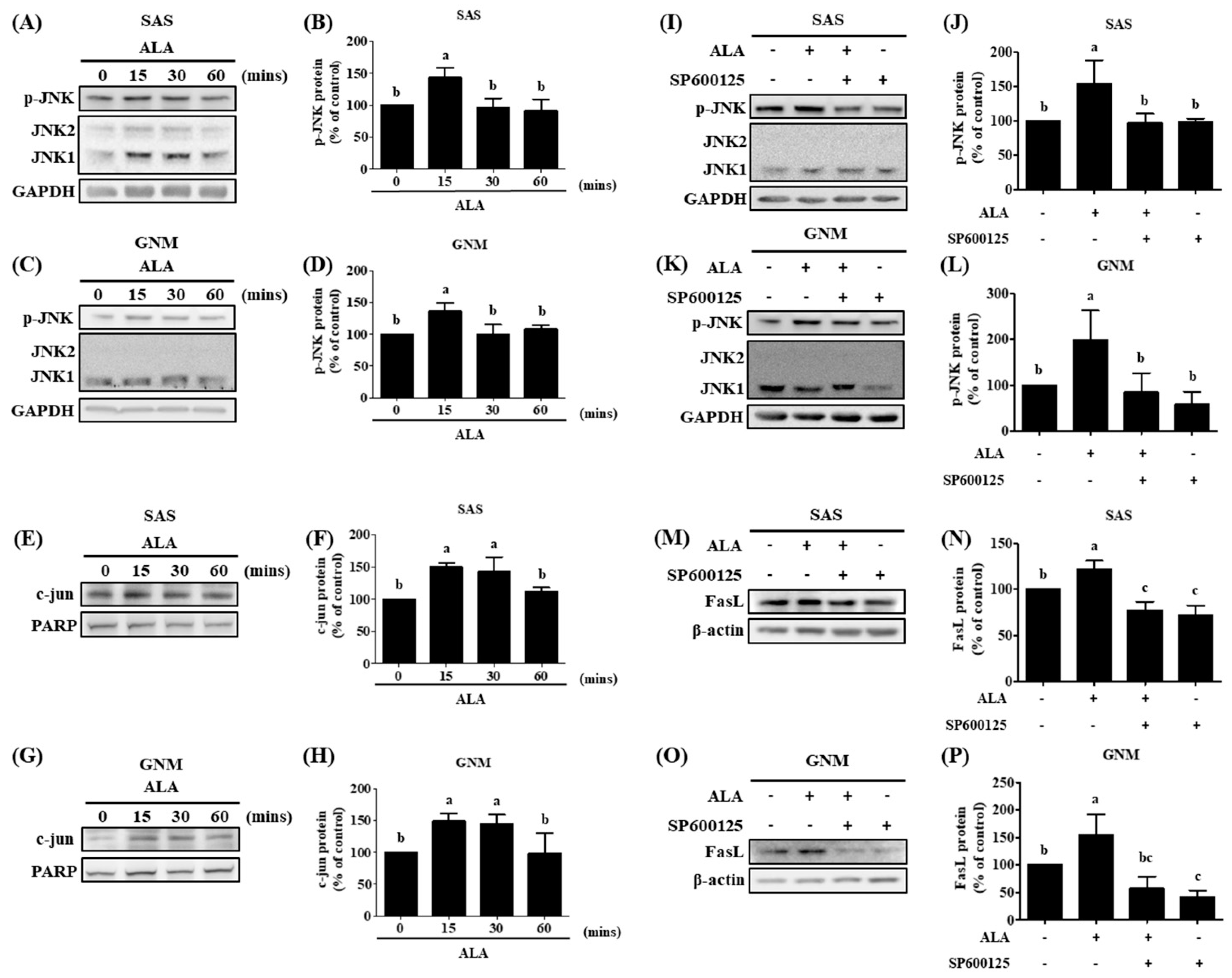
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



