
Submitted:
09 October 2023
Posted:
10 October 2023
You are already at the latest version
Abstract
Glioblastoma (GBM) is one of the most aggressive and intricate forms of brain cancer, demanding novel therapeutic interventions. The role of transcription factors in epigenetic regulation has emerged as a promising avenue for targeting GBM. This comprehensive review seeks to explore the complex interplay between specific transcription factors such as SNAI2, FOXA1, YAP1, TWIST1, ZEB1, and NF-kB, and their role in GBM progression and therapy. The focus extends to innovative therapeutic strategies, including epigenetic modifiers, targeted therapies, and CRISPR/Cas9 technology. A unique aspect of this review highlights the connection between transcription factors, neoantigen presentation, and personalized immunotherapy in GBM. By delving into current research, clinical trials, and immunological data, this review emphasizes the significant potential of targeting transcription factors for personalized immunotherapy in GBM. The challenges, future perspectives, and ethical considerations are also critically discussed, offering a complete and thought-provoking insight into a vital and rapidly evolving field.
Keywords:
Glioblastoma
; Transcription factors
; Neoantigens
; Personalized therapy
; Epigenetics
1. Introduction
Glioblastoma (GBM), also known as Grade IV astrocytoma, is the most common and aggressive form of primary brain tumor in adults, accounting for approximately 15% of all brain tumors and 54% of all gliomas [1,2]. GBM presents a unique complexity in terms of its molecular heterogeneity, aggressive growth, and resistance to conventional therapies [3,4]. Its incidence rate in the United States and Europe is 3-4 per 100,000 adults per year, and its median survival rate remains dismally low, at about 15 months, with a five-year survival rate of less than 10%[5,6]. The male-to-female ratio is approximately 3:2, and the median age of diagnosis is around 64 years [7,8].
Histologically, GBM is characterized by cellular pleomorphism, nuclear atypia, high mitotic activity, microvascular proliferation, and necrosis, making its invasive nature often render complete surgical resection challenging, if not impossible [9,10]. The complexity of GBM stems from its genetic and epigenetic diversity, with multiple mutations and alterations in signaling pathways contributing to its aggressive behavior [11,12]. Common alterations include mutations in the genes encoding epidermal growth factor receptor (EGFR), TP53, and isocitrate dehydrogenase 1 (IDH1) [13,14]. Epigenetically, alterations in DNA methylation, histone modifications, and microRNAs are observed, leading to the classification of GBM into different subtypes, namely Classical, Mesenchymal, Neural, and Proneural, each with distinct genetic profiles and clinical outcomes [15,16].
| Type of Tumor (WHO Grade) | 5-Year Relative Survival Rate | ||
| Age | |||
| 20–44 | 45–54 | 55–64 | |
| Anaplastic astrocytoma (III) | 58% | 29% | 15% |
| Glioblastoma (IV) | 22% | 9% | 6% |
| Anaplastic oligodendroglioma (III) | 76% | 67% | 45% |
The standard treatment for GBM consists of maximal safe surgical resection followed by radiation therapy and chemotherapy, particularly with the alkylating agent temozolomide (TMZ) (Figure 1) [17,18]. Despite these aggressive interventions, recurrence is nearly universal due to the tumor's resistance to conventional therapies [19,20]. The extent of resection correlates with survival, but the infiltrative nature often hinders complete removal [21]. Postoperative radiation is standard, but it can lead to adverse effects like cognitive decline [22]. TMZ is the most used chemotherapeutic agent, often in conjunction with radiation, but resistance is common [23]. Despite multiple clinical trials, targeted therapies have largely failed to provide significant improvements, highlighting the need for innovative approaches [24]. Some promise has been seen with immune checkpoint inhibitors, but results are inconsistent [25,26].
GBM's complexity, combined with its heterogeneity and resistance to current therapeutic strategies, creates an urgent need for novel and personalized therapeutic approaches [27]. Understanding the intricate role of transcription factors in epigenetic regulation presents a promising frontier in the fight against this devastating disease [28]. The need to transcend conventional methodologies and embrace a multidimensional and individualized strategy is evident, underscoring the gravity and challenge of battling one of the most insidious malignancies known to mankind [29].
Transcription factors are proteins that control the rate of transcription of genetic information from DNA to mRNA, playing a crucial role in gene regulation. In cancer, including GBM, aberrant transcription factor activity can lead to uncontrolled cell proliferation, invasion, metastasis, and resistance to therapy [30,31]. Transcription factors like MYC and STAT3 are often found to be dysregulated in various cancers [32,33]. Overexpression of MYC has been associated with uncontrolled cell growth, while STAT3 dysregulation can drive inflammatory pathways that promote tumorigenesis [34,35]. Factors such as TWIST1 and SNAI2 regulate epithelial-to-mesenchymal transition (EMT), a crucial process in cancer invasion and metastasis [36,37]. Altered expression of these factors can lead to enhanced migratory and invasive capabilities of tumor cells [38]. The aberrant activity of transcription factors like NF-kB has been linked to resistance to chemotherapy and radiation [39]. These factors can activate survival pathways, making cancer cells less responsive to treatments [40]. Transcription factors also affect the interaction between cancer cells and the surrounding microenvironment, impacting immune response, angiogenesis, and stromal interactions [41]. For example, HIF-1α controls angiogenesis through the regulation of VEGF expression [42].
Epigenetic changes, including DNA methylation, histone modifications, and non-coding RNA regulation, have been found to play vital roles in cancer development and progression [43,44]. Hypermethylation of tumor suppressor genes can lead to their silencing, promoting oncogenesis [45]. Conversely, hypomethylation can lead to oncogene activation [46]. Alterations in histone acetylation and methylation can change chromatin structure, affecting gene expression [47]. Histone deacetylase (HDAC) inhibitors are being explored as potential cancer therapies [48]. MicroRNAs (miRNAs) and long non-coding RNAs (lncRNAs) can act as oncogenes or tumor suppressors [49]. Dysregulation of miRNAs has been implicated in GBM progression [50].
With the complexity and heterogeneity of tumors, personalized immunotherapy has emerged as a promising approach [51,52]. Several reasons justify this focus. Personalized immunotherapy considers the unique genetic and epigenetic landscape of each patient's tumor, enabling more targeted and effective treatment [53]. Identification of patient-specific neoantigens allows the development of vaccines or cellular therapies targeting these unique tumor markers, enhancing treatment specificity [54]. By targeting the specific mechanisms of resistance in individual tumors, personalized immunotherapy may overcome the limitations of traditional therapies [55]. Combining personalized immunotherapy with existing treatments like chemotherapy or radiation may provide synergistic effects, enhancing therapeutic efficacy [56].
In conclusion, transcription factors and epigenetic regulation are at the nexus of many critical cancer pathways [57]. Their study offers innovative therapeutic avenues, particularly in personalized immunotherapy [58]. Understanding and harnessing these mechanisms may lead to unprecedented advancements in GBM treatment, providing more effective, targeted, and individualized interventions [59]. The fusion of transcriptional and epigenetic insights with immunotherapeutic strategies offers a multifaceted approach to tackling GBM, signifying an era where personalized medicine not only enhances current therapeutic paradigms but also paves the way for groundbreaking innovations [60].
The burgeoning complexity of glioblastoma (GBM) and the intricate interplay between its genetic and epigenetic landscape present both opportunities and challenges for therapeutic advancement [61]. The overarching aim of this review is to provide an exhaustive and nuanced exploration of the critical role that transcription factors play in GBM's epigenetic regulation and the subsequent implications for personalized immunotherapy [62]. The specific objectives are as follows: to scrutinize the function and significance of transcription factors, such as SNAI2, FOXA1, YAP1, TWIST1, ZEB1, and NF-kB, that have been implicated in the epigenetic regulation of GBM [63,64]; to delve into the multifaceted world of epigenetic regulation in GBM, encompassing DNA methylation, histone modifications, and non-coding RNA regulation, elucidating how these mechanisms interact with transcription factors and contribute to GBM's heterogeneity and complexity [65,66].
By presenting a comprehensive overview of the subject, this review seeks to foster interdisciplinary collaboration between oncologists, geneticists, immunologists, and other stakeholders involved in GBM research and treatment [67]. In synthesizing existing knowledge, presenting recent advancements, and suggesting future research directions, this review aspires to contribute substantively to the scientific community's understanding of GBM [68]. Through the lens of transcription factors and epigenetic regulation, it aims to illuminate new paths toward personalized and effective therapeutic strategies for one of the most challenging and devastating forms of brain cancer [69].
| FDA-Approved Therapy | YearApproved | Mechanism | Application | Dosage | Common Toxicities | Overall Survival | Other Notes |
| Lomustine (CCNU) | 1976 | Nonspecific alkylating agent that causes crosslinking of DNA and RNA | Oral | 80–110 mg/m2 every 6 weeks | Hematologic toxicity (49.7%) | 11.5 months | No benefit compared to RT alone |
| Carmustine (BCNU) | 1977 | Nonspecific alkylating agent that causes crosslinking of DNA and RNA in dividing cells; also binds to and modifies glutathione reductase | IV | 150–200 mg/m2 every 6 weeks | Pulmonary toxicity (<30%), ocular toxicity (>10%) and bone marrow suppression (>10%) | 11.75 months | No benefit compared to RT alone |
| Carmustine wafer implants (BCNU wafers) | 1996 & 2003 | Nonspecific alkylating agent that causes crosslinking of DNA and RNA in dividing cells; also binds to and modifies glutathione reductase | Directly applied during surgery | 8 wafers: 61.6 mg | Wound healing complications (12%), intracranial infection (1–10%), and cerebral edema (1–10%) | 13.9 months | High complication rate (42.7%) and expensive |
| Temozolomide (TMZ) | 2005 | Nonspecific alkylating agent that causes mismatch repair in DNA by methylation at the O6 position of guanine | Oral | 75 mg/m2 per day with RT, 150–200 mg/m2 per day | Hematologic toxicity (16%): thrombocytopenia (12%), leukopenia (7%), and neutropenia (7%) | 14.6–16.1 months | Standard of Care |
| Bevacizumab (BVZ) | 2009 | Targeted therapeutic antibody that binds and inhibits VEGF protein in tumor cells | IV | 10 mg/kg every 2 weeks | Hypertension (5.5–11.4%), thromboembolic events (3.2–11.9%), gastrointestinal perforation (1.5–5.4%), cerebral bleeding (2–5.3%), wound healing complications (0.8–3.3%), and proteinuria (2.7–11.4%) | 9.3 months (recurrent) | Used to treat symptomatic edema and radiation necrosis |
| Optune device (TTFields) | 2011 & 2015 | Low-intensity (1–3 V/cm), intermediate-frequency (200 kHz) alternating electric fields that disrupt mitosis in tumor cells | Portal device, electrodes on scalp | Greater than 18 h a day for >4 weeks | Skin toxicity (43%) and seizures (7%) | 20.5–20.9 months | Not SOC because of marginal survival benefits, expensive costs, and inconvenience for patients |
2. Transcription Factors and Epigenetic Regulation in GBM:
Transcription factors (TFs) are specialized proteins that recognize and bind to specific DNA sequences, thereby regulating the transcription of genetic information from DNA to RNA. They play an indispensable role in the orchestration of gene expression, controlling various cellular processes like growth, differentiation, and response to environmental stimuli.
In the context of GBM, TFs have a profound impact on epigenetic regulation, contributing to the disease's complexity and heterogeneity.
Control of Gene Expression: TFs act as master regulators of gene expression, controlling both activation and repression. They can interact with chromatin modifiers, such as histone acetyltransferases (HATs) and histone deacetylases (HDACs), to modulate chromatin structure, thereby affecting gene accessibility (Lee et al., 1993; Struhl, 1998).
Regulation of Epigenetic Landscape: Epigenetic modifications, including DNA methylation and histone modifications, are controlled by TFs. They act as a bridge between signaling pathways and chromatin, integrating extracellular signals into precise gene expression patterns (Spitz & Furlong, 2012).
Involvement in Oncogenic Pathways: In GBM, certain TFs are found to be dysregulated, contributing to oncogenic pathways. For example, overexpression of STAT3 is associated with poor prognosis and tumor progression (Brantley et al., 2008).
Target for Therapeutics: TFs offer potential therapeutic targets. Inhibition of TFs like c-MYC has shown promise in preclinical GBM models (Wang et al., 2011).
Impact on Cellular Processes: TFs like TWIST1 and SNAI2 are involved in epithelial-to-mesenchymal transition (EMT), a process that enhances tumor invasion and metastasis in GBM (Siegfried et al., 2014).
Transcription factors play a multifaceted role in the epigenetic regulation of GBM. Understanding their function and dysregulation can unlock novel diagnostic, prognostic, and therapeutic avenues. Their central role in linking genetic information with environmental signals makes them a pivotal component of GBM's intricate molecular landscape.
2.1. Examples of Transcription Factors Implicated in GBM
2.1.1. SNAI2
SNAI2, also known as Slug, is a member of the Snail family of zinc-finger transcription factors. It plays a vital role in embryogenesis, particularly in the epithelial-to-mesenchymal transition (EMT), a process critical for embryonic development and tissue repair. In cancer, including GBM, SNAI2 has been linked to various processes that contribute to tumor aggressiveness and progression.
Role of SNAI2 in GBM:
EMT Regulation: SNAI2 is known to induce EMT, leading to the loss of cell adhesion, increased cell motility, and invasiveness. In GBM, this can facilitate tumor cell invasion into the surrounding brain tissue (Hajra et al., 2002).
Therapeutic Resistance: Overexpression of SNAI2 has been associated with resistance to therapy in various cancer types, including GBM. It can promote survival pathways, making tumor cells less responsive to conventional treatments (Vega et al., 2004).
Interaction with HDACs: In some contexts, SNAI2 has been found to recruit histone deacetylases (HDACs) to the promoters of specific target genes. HDACs remove acetyl groups from histones, leading to chromatin condensation and repression of gene transcription. This SNAI2-HDAC complex may lead to the silencing of tumor suppressor genes, contributing to GBM's malignancy (Peinado et al., 2004).
SNAI2 and HDACs as Therapeutic Targets:
Targeting SNAI2: Inhibiting SNAI2 may reverse EMT, reduce invasiveness, and sensitize GBM cells to therapy. Small molecule inhibitors and RNA interference techniques targeting SNAI2 are under investigation.
HDAC Inhibition: HDAC inhibitors, such as Vorinostat, are being studied as potential treatments for various cancers, including GBM. By inhibiting HDACs, these drugs can reverse gene silencing mediated by the SNAI2-HDAC complex, potentially restoring the expression of tumor suppressor genes (Marks et al., 2001).
Combined Approach: A combined strategy targeting both SNAI2 and HDACs may offer a synergistic effect, attacking the tumor on multiple fronts and overcoming resistance mechanisms.
SNAI2, in coordination with HDACs, represents a pivotal molecular nexus in the pathogenesis of GBM, influencing processes like EMT, invasion, and therapy resistance. The insights into the SNAI2-HDAC interaction offer promising therapeutic avenues and underline the complexity of the epigenetic regulation in GBM. This relationship further emphasizes the need for integrated approaches in both understanding and targeting GBM at the molecular level.
2.1.2. FOXA1: Role in Cell Differentiation and Links to GBM
Function and Epigenetic Influence: FOXA1 serves as a pioneer factor in chromatin remodeling, allowing other transcription factors to bind DNA. It participates in histone modifications and methylation processes, leading to either activation or repression of gene expression.
Implication in GBM: FOXA1's dysregulation in GBM may lead to aberrant epigenetic landscapes that affect cell differentiation and proliferation. It has been associated with maintaining BTSCs' stem-like properties, impacting GBM's heterogeneity, and therapeutic resistance (Zhang et al., 2016).
Potential Therapeutic Approaches: Targeting FOXA1 can lead to the restoration of proper differentiation pathways, offering a new avenue for GBM treatment.
2.1.3. YAP1: Implications in GBM Progression and Tumor-Suppressive Signaling
Function and Epigenetic Role: YAP1 acts as a transcriptional co-activator and is a part of the Hippo pathway. It plays a role in regulating chromatin accessibility and is involved in various epigenetic processes, such as histone methylation and acetylation.
GBM Involvement: In GBM, YAP1's dysregulation can lead to the activation of oncogenes or suppression of tumor suppressor genes through epigenetic mechanisms. It may act both as an oncogene or a tumor suppressor, depending on its interaction with other signaling pathways (Orr et al., 2011).
Therapeutic Implications: Understanding YAP1's dual role in GBM may lead to targeted epigenetic therapies, either inhibiting or enhancing its activity, depending on the context.
2.1.4. TWIST1: Involvement in Epithelial-to-Mesenchymal Transition (EMT)
Function and Epigenetic Connection: TWIST1 is involved in the repression of E-cadherin and other genes by recruiting chromatin-modifying enzymes, including HDACs. It affects DNA methylation patterns and histone modifications that underlie EMT.
Role in GBM: In GBM, TWIST1-mediated EMT enhances tumor invasion and therapeutic resistance. Its overexpression may alter the epigenetic landscape, driving a more aggressive phenotype (Elias et al., 2005).
Potential Therapies: Targeting TWIST1 or its downstream epigenetic effectors could lead to the reversal of EMT in GBM, hampering invasion and potentially sensitizing tumors to conventional therapies.
2.1.5. ZEB1: Role in GBM Invasiveness, Link with DNA Methylation
Function and Epigenetic Aspects: ZEB1, like TWIST1, is involved in EMT by repressing E-cadherin. It also influences the methylation of DNA by interacting with DNA methyltransferases, leading to the silencing of specific genes.
Implication in GBM: ZEB1's impact on DNA methylation adds complexity to the epigenetic regulation in GBM. Its role in promoting invasion and contributing to a stem-like phenotype makes it a promising therapeutic target (Siebzehnrubl et al., 2013).
Therapeutic Strategies: Strategies to inhibit ZEB1 or modify its epigenetic effects may reverse malignant phenotypes, reduce invasiveness, and increase sensitivity to treatment.
2.1.6. NF-kB: Impact on Immune Evasion, Inflammation in GBM
Function and Epigenetic Influence: NF-kB controls genes involved in immune response and inflammation. Its activation may lead to changes in chromatin structure, DNA methylation patterns, and histone modifications, influencing the transcriptional output.
GBM Involvement: NF-kB contributes to the inflammatory microenvironment in GBM and impacts immune evasion. Its activation correlates with aggressive tumor behavior and may affect the epigenetic programming of immune cells within the tumor (Nagai et al., 2002).
Potential Targeting Approaches: Targeting NF-kB's epigenetic influence may modulate the immune response and inflammation in GBM, presenting new opportunities for immunotherapy.
These transcription factors contribute significantly to the epigenetic landscape of GBM. Their diverse roles in controlling gene expression, chromatin structure, and DNA methylation provide insights into GBM's complexity. Understanding their epigenetic mechanisms opens opportunities for innovative therapeutic strategies, including targeted epigenetic
2.2. Challenges in Targeting Transcription Factors
2.2.1. Structural Challenges
Lack of Defined Binding Pockets: Transcription factors have long been considered "undruggable" primarily due to the absence of well-defined small-molecule binding pockets. Conventional drug design strategies often target enzymes or receptors that have a clear, pocket-like binding domain. However, transcription factors frequently defy this structural categorization, necessitating alternative strategies for drug design (Bullock et al., 2011).
Conformational Flexibility: Transcription factors are not static entities; they often undergo conformational changes to interact with DNA or other proteins. This dynamic nature introduces considerable difficulty in developing inhibitors that are both specific and effective. The ever-changing structure limits the applicability of static models often used in computational drug design (Lambert et al., 2018).
Complex Protein-Protein Interactions: Targeting transcription factors often involves interrupting their interactions with other proteins. However, protein-protein interactions (PPIs) are generally less well-defined than enzyme-substrate or receptor-ligand interactions. Furthermore, these interactions are crucial for many cellular processes, which raises the risk of unintended consequences when trying to disrupt them (Wells & McClendon, 2007).
2.2.2. Functional Challenges
Pleiotropic Effects: Transcription factors are often implicated in multiple signaling pathways, making them particularly challenging to target without eliciting off-target effects. Due to their diverse roles in cellular functions, inhibiting a single transcription factor may lead to unpredictable and even deleterious outcomes (Wang et al., 2013).
Cell- and Context-specific Roles: Another layer of complexity arises from the fact that transcription factors can play different roles depending on the cellular or biological context. This makes it particularly challenging to develop a one-size-fits-all therapeutic strategy, thus necessitating context-specific approaches for different cancer types or even individual tumors (Spitz & Furlong, 2012).
Feedback Mechanisms: Many cellular processes have built-in feedback loops to maintain homeostasis. Inhibiting one transcription factor can, paradoxically, activate others or even the same factor via alternative pathways, effectively negating the therapeutic effects (Chen et al., 2016).
2.2.3. Contextual Challenges in GBM
Tumor Heterogeneity: Glioblastoma (GBM) is notoriously heterogeneous, both between patients (inter-tumor) and within a single tumor (intra-tumor). This complicates the task of identifying universally applicable drug targets among transcription factors, as their expression and function can vary significantly across different regions of the tumor (Patel et al., 2014).
Blood-Brain Barrier Penetration: Developing therapies for GBM is further complicated by the blood-brain barrier, a highly selective semipermeable membrane that prevents many potential therapeutics from reaching the tumor site (Pardridge, 2019).
Integration with Current Therapies: Adding to the complexities are the existing treatment modalities for GBM, which may interact unpredictably with new transcription factor-targeting therapies. Thus, there is an urgent need to find synergistic or complementary strategies that can maximize efficacy while minimizing toxicity (Stupp et al., 2005).
2.2.4. Research Gaps
In-depth Understanding of Mechanisms: The intricacies of the relationships between transcription factors and epigenetic regulators in GBM remain poorly understood. A more comprehensive mechanistic understanding would enable more effective and specific therapeutic interventions.
Development of Selective Inhibitors: The focus has been shifting towards developing highly selective inhibitors that can effectively modulate transcription factor activity without causing significant off-target effects. However, these strategies are still in their infancy and require extensive validation.
Clinical Validation: Despite the promise shown in pre-clinical models, few transcription factor-targeting agents have advanced to clinical trials specifically for GBM, underlining the urgency for translational research in this area (Lu et al., 2015).
In summary, targeting transcription factors in GBM is an endeavor fraught with challenges, both structural and functional, that are compounded by the unique characteristics of GBM itself. Overcoming these obstacles necessitates a multidisciplinary approach, involving computational biology, medicinal chemistry, and clinical oncology, among other fields. Only through concerted efforts can we hope to translate the increasing understanding of these transcription factors into clinically actionable strategies.
3. Emerging Therapeutic Strategies
3.1. Epigenetic Modifiers: Role in Modulating Transcription Factors, HDAC Inhibitors, etc.
Epigenetic regulation plays a critical role in tumorigenesis and progression, including glioblastoma (GBM). Targeting the epigenome provides a novel approach for therapeutic intervention. This section explores epigenetic modifiers that influence transcription factors, with an emphasis on HDAC inhibitors.
3.2. Epigenetic and Transcriptional Modulation in GBM Therapy
3.2.1. Epigenetic Modifiers and Transcription Factors
DNA Methylation: DNA methyltransferases (DNMTs) are enzymes that add methyl groups to cytosine residues in DNA, influencing the binding affinity and selectivity of transcription factors. These enzymes play a pivotal role in modulating gene expression, impacting a variety of cellular functions ranging from development to differentiation. In the context of glioblastoma (GBM), abnormal methylation patterns have been observed, with consequences for key oncogenes and tumor suppressor genes. The disrupted methylation landscape affects the epigenetic regulation of these critical genes, thereby contributing to the malignancy and resistance associated with GBM (Esteller, 2007).
Histone Modifications: Histones are proteins that DNA wraps around, constituting the basic unit of chromatin. Various post-translational modifications can occur on histones, such as methylation, acetylation, and phosphorylation. These modifications influence the chromatin structure and consequently the accessibility of transcription factors to DNA. In GBM, histone modifications are often dysregulated, which disrupts the epigenetic control over gene expression and contributes to tumorigenesis and cancer progression (Berger et al., 2009).
Non-Coding RNAs: Non-coding RNAs, particularly microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), have emerged as crucial regulators of transcription factor activity. They modulate gene expression post-transcriptionally by affecting mRNA stability and translation. In GBM, aberrant expressions of miRNAs and lncRNAs have been implicated in the regulation of key transcription factors, adding another layer of complexity to the epigenetic landscape of the tumor (Anastasiadou et al., 2018).
3.2.2. HDAC Inhibitors
Mechanism of Action: HDAC inhibitors are a class of compounds that inhibit histone deacetylases (HDACs), leading to an increase in histone acetylation. Acetylation generally results in a more relaxed chromatin structure, enhancing the accessibility of transcription factors to DNA and leading to increased expression of tumor suppressor genes. These inhibitors serve to modulate the transcriptional landscape by altering the balance between histone acetylation and deacetylation, effectively shifting the balance towards gene activation (Marks et al., 2000).
Pre-Clinical Success: Several HDAC inhibitors have shown potential in pre-clinical models of GBM. Compounds such as Vorinostat and Panobinostat have been effective in reducing tumor growth and enhancing apoptosis in these models. Their effectiveness is primarily attributed to the alteration of chromatin states, which subsequently affects the accessibility of transcription factors and the expression of genes implicated in GBM (Galanc et al., 2019).
Clinical Trials: Despite promising pre-clinical results, clinical trials involving HDAC inhibitors have produced mixed outcomes in GBM patients. For instance, a Phase II clinical trial of Panobinostat showed limited success, underlining the challenges associated with translating pre-clinical findings to the clinic. These trials highlight the necessity for developing more targeted and selective strategies to enhance the effectiveness of HDAC inhibitors in GBM therapy (Lee et al., 2015).
3.2.3. Challenges and Future Directions
Selectivity: One of the main challenges in using epigenetic modifiers like HDAC inhibitors is their lack of selectivity. These agents often target a broad spectrum of histone and non-histone proteins, leading to off-target effects that can limit their therapeutic window (Bannister & Kouzarides, 2011).
Combination Therapies: The complex biology of GBM necessitates a multi-faceted therapeutic approach. The combination of epigenetic modifiers with other targeted agents or conventional therapies such as radiation and chemotherapy may provide a synergistic effect, thereby enhancing overall efficacy in treating GBM (Eckschlager et al., 2017).
Personalized Approaches: The epigenetic landscape varies significantly across individual tumors, offering an opportunity for personalized medicine. Future therapies could be tailored to the specific epigenetic profiles of each patient's tumor, optimizing the efficacy of the treatment while minimizing side effects. Advances in high-throughput sequencing and computational biology could facilitate this individualized approach to GBM therapy (Noushmehr et al., 2010).
The dynamic and interconnected roles of epigenetic modifiers and transcription factors in GBM present a compelling but challenging target for therapeutic intervention. With ongoing research aimed at overcoming these challenges, it is hoped that more effective and targeted therapies can be developed, heralding a new era in GBM treatment.
3.3. Targeted Therapies: The Frontier of Transcription Factor Inhibition in GBM
3.3.1. Strategies for Targeting Transcription Factors
Small Molecules: Small molecules offer a versatile approach to inhibit transcription factors by disrupting their interaction with DNA or other proteins. For example, Nutlin-3a has been shown to interfere with the interaction between p53 and its negative regulator MDM2. By doing so, Nutlin-3a stabilizes p53, leading to its increased activity and enhanced anti-tumor effects in various types of cancer, including GBM (Vassilev et al., 2004).
Peptide Inhibitors: These are short amino acid sequences designed to mimic natural protein-protein interactions, thereby competitively inhibiting them. A peptide inhibitor targeting the Signal Transducer and Activator of Transcription 3 (STAT3) has demonstrated significant anti-tumor effects in pre-clinical models of GBM. The inhibitor works by hindering STAT3's ability to dimerize and initiate transcriptional activity, thereby retarding tumor growth and progression (Sen et al., 2012).
Antisense Oligonucleotides: These are synthetic DNA or RNA molecules designed to specifically bind to the mRNA of target genes, like transcription factors, leading to mRNA degradation and subsequent reduction in protein levels. For example, antisense oligonucleotides against NF-κB have been explored to inhibit its mRNA, effectively reducing its expression and associated oncogenic activities (Swayze et al., 2007).
3.3.2. Challenges in Targeting Transcription Factors
Druggability Issues: Many transcription factors lack well-defined binding pockets, which poses significant challenges for drug design. Developing molecules that can effectively target these "undruggable" proteins requires innovative approaches in medicinal chemistry and structural biology (Wu et al., 2015).
Off-Target Effects: Given the systemic and multifunctional roles of transcription factors, designing inhibitors with high specificity is crucial to minimize off-target effects. The need for high specificity becomes even more critical when considering the possibility of affecting other cellular processes inadvertently (Lambert et al., 2014).
Delivery Challenges: The blood-brain barrier represents a significant obstacle for the efficient delivery of inhibitors to brain tumors. Overcoming this barrier necessitates innovative delivery methods or the modification of existing compounds to improve their penetrance into the central nervous system (Saraiva et al., 2016).
3.3.3. Specific Examples in GBM
Inhibition of STAT3: STAT3 is a critical player in GBM progression and represents an enticing target for therapy. Small molecule inhibitors like Stattic have been developed to inhibit STAT3 and have shown promising results in both in vitro and in vivo models (Schust et al., 2006; Zhang et al., 2013).
Targeting the NF-κB Pathway: Specific inhibitors such as Bay 11-7082 have been explored for their capacity to inhibit the NF-κB pathway, a key modulator of inflammation and cell survival in GBM. These inhibitors have shown anti-tumor effects, potentially providing another avenue for targeted GBM therapy (Korkolopoulou et al., 2008).
3.3.4. Future Directions
Personalized Approaches: Utilizing patient-specific genomic and epigenomic data can help develop individualized therapies. Targeting transcription factors that are aberrantly activated due to specific mutations could offer more tailored and effective treatments (Mullard, 2017).
Combination Therapies: The heterogeneity and adaptability of GBM cells necessitate a multifaceted approach. Combining targeted therapies against transcription factors with other therapeutic modalities like radiation or chemotherapy could potentially overcome resistance mechanisms and improve overall outcomes (Massard et al., 2016).
3.4. CRISPR/Cas9 Technology: Pioneering Gene Editing for Transcription Factor Targeting in GBM
3.4.1. The CRISPR/Cas9 System
Mechanism: The CRISPR/Cas9 system employs guide RNA (gRNA) to navigate the Cas9 nuclease to precise DNA sequences. Once there, the Cas9 nuclease generates double-strand breaks in the DNA, paving the way for gene knockout, replacement, or addition. This technology enables unprecedented accuracy in gene editing, allowing for the targeted manipulation of genomic elements (Jinek et al., 2012).
Potential Applications: Due to its ability to target nearly any gene, the CRISPR/Cas9 system has broad utility for investigating the functional roles of transcription factors and epigenetic regulators in diseases like GBM. It opens doors for both diagnostic and therapeutic applications, serving as an adaptable tool for the scientific community (Wang et al., 2016).
3.4.2. Targeting Transcription Factors in GBM
NF-κB Pathway: Employing CRISPR/Cas9 to knockout genes involved in the NF-κB signaling pathway has provided invaluable insights into its role in GBM pathology. Further investigation could transition these findings from the bench to potential therapeutic strategies, aimed at attenuating or even blocking this pathway (Huang et al., 2017).
YAP1: CRISPR/Cas9-mediated editing of the YAP1 gene has deepened our understanding of its critical role in driving GBM tumorigenesis. This knowledge could inform the development of new interventions aimed at counteracting YAP1's oncogenic activities (Feng et al., 2017).
3.4.3. Challenges and Ethical Considerations
Off-Target Effects: While the system is renowned for its precision, there remains a risk of unintended genetic modifications. These off-target effects pose concerns regarding both the safety and specificity of CRISPR/Cas9 interventions (Zhang et al., 2015).
Delivery Challenges: Achieving efficient delivery of the CRISPR/Cas9 components to GBM cells is a critical bottleneck. Strategies must be developed to overcome the biological barriers that impede the effective delivery of gene-editing machinery into target cells (Liu et al., 2017).
Ethical Dimensions: The capability to alter human genes with high precision brings about a host of ethical considerations. These range from questions about long-term safety to the broader implications of genetic modification in human populations (Brokowski, 2018).
3.4.4. Future Prospects
Combination Therapies: The integration of CRISPR/Cas9 with other existing treatment modalities could potentially enhance therapeutic efficacy. For example, combining gene editing with chemotherapy could synergistically impair tumor growth and resistance mechanisms (Platt et al., 2014).
Personalized Approaches: Personalized gene editing based on an individual's unique GBM genetic profile could usher in a new era of tailored therapy. This strategy could lead to more effective and targeted treatments, particularly when coupled with other forms of personalized medicine (Brennan et al., 2013).
The emergence of CRISPR/Cas9 technology as a potent tool for editing transcription factor-associated genes in GBM signals a significant advancement in both our understanding and potential treatment of this devastating disease. However, several challenges—ranging from technical limitations to ethical considerations—still need to be addressed. Looking ahead, the integration of CRISPR/Cas9 within a broader therapeutic landscape, such as in combination therapies, holds promise for pioneering new approaches in combating GBM.
3.5. Challenges and Future Outlook: Bridging the Gap from Bench to Bedside
3.5.1. Clinical and Ethical Barriers
Patient Selection and Biomarker Development: One of the initial challenges in clinical implementation is the identification of the right patient population that could benefit most from targeted therapies (Weller et al., 2017). This necessity dovetails with the urgent need for reliable biomarkers. Biomarkers not only help in stratifying patients but are also indispensable for monitoring therapeutic responses and modifying treatment protocols as needed (Sottoriva et al., 2013).
Treatment Resistance and Safety: The inherent genetic and phenotypic complexity of GBM poses a risk for developing resistance against targeted therapies, complicating the task of reliably evaluating their efficacy (Bhat et al., 2013). Alongside this is the requirement for a detailed safety profile. Close monitoring is essential to balance the promising therapeutic effects against any potential adverse reactions and side effects, ensuring that the benefits outweigh the risks (Wen et al., 2016).
Informed Consent and Equity in Access: Ethical considerations come into play at multiple levels. For instance, it is crucial to ensure that patients are fully informed about the experimental nature and potential risks associated with participating in clinical trials for these novel therapies (Joffe et al., 2001). Concurrently, it is imperative to consider equity in access to these cutting-edge treatments. Disparities could arise, particularly in resource-limited settings, making the availability of these therapies unequal (Marron et al., 2018).
Ethical Concerns Specific to Gene Editing: CRISPR/Cas9 technology brings its own set of ethical challenges. These include the risk of unintended genetic modifications and the unresolved questions concerning the long-term implications and safety of gene editing (Ishii, 2017).
3.5.2. Future Directions
Personalized and Combination Therapies: Future therapeutic strategies may significantly benefit from a personalized approach. Integration of multi-omics data can guide the development of treatment plans tailored to individual patient profiles, potentially improving treatment outcomes (Verhaak et al., 2010). Moreover, combining these advanced targeted interventions with existing therapies may offer synergistic effects that could enhance efficacy and mitigate the issues of resistance (Lathia et al., 2015).
Development of Ethical Frameworks: Given the complexity and novelty of technologies like gene editing, there is a pressing need for comprehensive ethical frameworks. Such guidelines could serve to inform responsible clinical application, ensuring that both current and emerging ethical dilemmas are adequately addressed (National Academy of Sciences, 2017).
The quest to translate groundbreaking research in transcription factor targeting and CRISPR/Cas9 technology into effective clinical applications is laden with a multitude of clinical and ethical challenges. Nevertheless, through strategic planning and ethical foresight, these hurdles may well be surmountable. Advances in personalized and combination therapies, as well as the formulation of robust ethical frameworks, can significantly contribute to harnessing the full potential of these innovative approaches in the battle against GBM.
4. Personalized Immunotherapy and Neoantigen Presentation:
4.1. Neoantigens in GBM: Understanding the Basis, the Importance of Personalized Targets
The concept of neoantigens, tumor-specific antigens resulting from somatic mutations, has emerged as an exciting frontier in personalized cancer therapy. In the context of Glioblastoma Multiforme (GBM), this approach presents unique opportunities and challenges.
4.1.1. The Basis of Neoantigens in GBM
Neoantigens arise from the vast array of genomic alterations such as point mutations, insertions, deletions, and gene fusions within cancer cells. Unlike shared tumor antigens, neoantigens are entirely specific to individual tumors and therefore hold significant promise for targeted therapy. Schumacher and Schreiber (2015) highlight the immunogenicity of neoantigens, as they are foreign to the immune system and not present in normal tissues. This uniqueness avoids the risk of autoimmunity, an issue that has limited the utility of shared tumor antigens in therapeutic applications.
4.1.2. Importance of Neoantigens in GBM
GBM's complexity and intratumoral heterogeneity pose significant challenges for therapy. The identification of neoantigens through high-throughput sequencing and bioinformatics analyses has opened new avenues for personalized cancer vaccines and T-cell therapies (Hilf et al., 2019). Keskin et al. (2019) demonstrated the feasibility of neoantigen-based vaccines, generating specific T cell responses within GBM tumors. By tailoring the immune response to the unique neoantigen landscape of each patient's tumor, these approaches offer the potential for highly effective therapies without off-target effects. Moreover, integrating neoantigens with other targeted therapies may further enhance the overall treatment efficacy, as shown by Ott et al. (2017).
4.1.3. Challenges in Utilizing Neoantigens in GBM
However, translating these scientific advances into clinically effective therapies is not without challenges. Patel et al. (2014) noted the extensive genetic heterogeneity within GBM tumors, making the identification and consistent targeting of neoantigens complex. This complexity is further exacerbated by the potential for tumors to evolve and lose the targeted neoantigens, a phenomenon known as antigen escape (Anagnostou et al., 2017). Additionally, the immunosuppressive tumor microenvironment in GBM, characterized by a host of immune-regulatory mechanisms, could hinder the effectiveness of neoantigen-based therapies (Wainwright et al., 2015).
4.1.4. Future Perspectives
Looking forward, the field of neoantigen-based therapies in GBM is ripe for continued innovation and advancement. A critical need is to improve computational methods that can predict which neoantigens will be presented on the tumor cell surface and recognized by T cells, as detailed by Jurtz et al. (2017). These technological advancements can facilitate the rational design of more effective personalized vaccines and T cell therapies. Clinical trials will be essential for rigorous evaluation of safety, efficacy, and feasibility in real-world settings (Sahin et al., 2017).
4.2. Transcription Factors and Neoantigen Presentation: A Complex Interplay with Therapeutic Potential
4.2.1. Introduction: The Symbiosis of Transcription Factors and Neoantigens in GBM
Transcription factors (TFs) serve as pivotal regulators of gene expression, dictating the availability of DNA to transcriptional machinery. In the realm of glioblastoma (GBM), this has significant repercussions on the presentation of neoantigens—novel peptides arising from cancer-specific mutations. This intricate relationship offers promising avenues for therapy by either promoting neoantigen visibility or obstructing immunosuppressive TFs.
4.2.2. STAT3 Pathway: A Double-Edged Sword in Immune Regulation
Hyperactivation and Immune Evasion: The STAT3 pathway is central to immune regulation in GBM. Hyperactivation of STAT3 often results in immune suppression, making neoantigens less visible to immune cells (Yu et al., 2014). The inhibition of STAT3 could potentially reverse this effect, enabling more effective targeted therapies (Kortylewski et al., 2009).
Complex Interactions with Immune Checkpoints: Adding complexity to this mechanism, STAT3 has been shown to interact with other immune checkpoint molecules like PD-L1, further modulating the immune response in GBM (Wang et al., 2017).
4.2.3. NF-κB Pathway: A Conductor in the Immune Orchestra
Regulation of Antigen Presentation and Inflammation: NF-κB influences a wide array of genes related to inflammation and immunity. It directly impacts the machinery responsible for presenting neoantigens, potentially aiding or hindering immunotherapies (Hayden & Ghosh, 2012). Chronic inflammation regulated by NF-κB could also alter the tumor microenvironment, affecting neoantigen presentation (Pikarsky & Ben-Neriah, 2006).
4.2.4. FOXP3: The Gatekeeper of Immunosuppression
Microenvironment Modulation and Therapeutic Potential: FOXP3 is a master regulator of regulatory T cells, crucial for maintaining an immunosuppressive environment. This can significantly impede the presentation of neoantigens (Zuo et al., 2007). Targeting FOXP3, therefore, offers a path to counter this suppression and enhance immune recognition of neoantigens (Wang et al., 2015).
4.2.5. Epigenetic Complexity: The Underlying Layers of Neoantigen Expression
DNA Methylation and Histone Modification: The role of TFs extends into the epigenetic regulation of neoantigens. Transcription factors like ZEB1 are implicated in DNA methylation patterns that can regulate neoantigen expression (Sánchez-Tilló et al., 2012). Similarly, the interplay between TFs like SNAI2 and histone deacetylases (HDACs) can influence histone modifications, affecting the neoantigen landscape (Peinado et al., 2004).
4.2.6. Challenges and Future Avenues: Tackling the Complexity
Precision and Integration: One of the major challenges in applying this understanding therapeutically is the specificity of targeting. It is crucial to target relevant TFs without unintentional disruption of normal cellular functions (Lambert et al., 2018). Additionally, integrating these innovative approaches with current therapies demands meticulous planning to achieve optimized outcomes (Palumbo et al., 2013).
The interaction between transcription factors and neoantigens in GBM is a complex but promising frontier. As our understanding deepens, the possibilities for novel therapies grow. However, there are challenges to overcome, including specificity in targeting and the integration with existing treatments. With continued research, the promise of manipulating this relationship for therapeutic advantage seems increasingly attainable.
4.3. Personalized Vaccine Strategies: Unraveling the Complexity Through Genetic Insights and Technological Advancements
4.3.1. Introduction: Personalized Medicine Meets Oncology
Personalized medicine is predicated on the notion that individual variations at the genetic and molecular level significantly influence therapeutic outcomes. This paradigm has been particularly transformative in oncology, where it has spurred the evolution of personalized vaccine strategies. These approaches are tailored to each patient, leveraging the unique genetic mutations of individual tumors to develop targeted therapies.
4.3.2. Decoding the Genetic Variations in GBM: A Wealth of Therapeutic Targets
Glioblastoma (GBM) is characterized by a high degree of genetic heterogeneity, giving rise to a multiplicity of mutations. This genetic diversity presents a plethora of opportunities for personalized vaccine development (Hodges et al., 2017).
Single Nucleotide Polymorphisms (SNPs): SNPs can result in changes to protein structure, thereby affecting their immunogenicity and providing potential vaccine targets (Wang et al., 2018).
Copy Number Variations (CNVs): CNVs can influence the expression levels of certain genes, thereby affecting the availability of neoantigens and their suitability as vaccine targets (Zack et al., 2013).
4.3.3. The Genesis of Personalized Neoantigen Vaccines: From Identification to Clinical Application
Identification of Neoantigens: Sophisticated techniques like whole-exome and transcriptome sequencing are employed to pinpoint patient-specific neoantigens (Schumacher & Schreiber, 2015).
Vaccine Formulation: Based on these identified neoantigens, vaccines are customized using various delivery mechanisms such as peptides, RNA, or viral vectors (Kreiter et al., 2015).
Clinical Validation: Early-phase clinical trials, primarily in melanoma, have exhibited promise (Ott et al., 2017; Sahin et al., 2017), setting the stage for their application in GBM.
4.3.4. The Roadblocks to Personalized Vaccines: Navigating the Challenges
Genomic Complexity: The inherent complexity and instability of the GBM genome add layers of difficulty to vaccine development (Verhaak et al., 2010).
Immunosuppressive Hurdles: The immunosuppressive microenvironment in GBM could act as a barrier to vaccine effectiveness (Pardoll, 2012).
Economic and Temporal Constraints: Personalized vaccine development involves considerable time and resources, complicating efforts to scale the approach (Mellman et al., 2011).
4.3.5. The Horizon: Integrating Technologies and Therapies
Synergistic Therapeutic Approaches: There's potential for combining personalized vaccines with existing immunotherapies like checkpoint inhibitors, which could elevate their effectiveness (Schalper et al., 2019).
Harnessing AI and Machine Learning: One of the most exciting future prospects lies in the application of Artificial Intelligence (AI) and Machine Learning (ML). These technologies offer transformative potential for vaccine development by predicting immunogenic mutations and helping in the design of optimal vaccine formulations. AI algorithms can analyze vast datasets of genetic and molecular information to pinpoint the most promising targets for personalized vaccines, substantially reducing development time and potentially costs (Kim et al., 2020).
Personalized vaccine strategies in GBM are an evolving frontier with promising but complex prospects. The intersection of cutting-edge genetic research, sophisticated vaccine development protocols, and the emerging potential of AI and ML technologies sets the stage for revolutionary approaches to treating this aggressive cancer. The journey ahead is rife with challenges, but the promise is too great to ignore.
4.4. Current Research and Future Directions: Latest Advancements, Ongoing Trials, and Future Expectations
4.4.1. Introduction
The evolving landscape of research in GBM highlights several emerging fronts that reflect the interdisciplinary approach needed to tackle this complex disease. Recent advancements are unfolding through extensive clinical trials and cutting-edge research, giving rise to optimistic prospects for future therapeutic avenues.
4.4.2. Latest Advancements
Immunotherapy and Targeted Therapies: Significant strides have been made in understanding the intricate interplay between the immune system and GBM (Limon et al., 2020). This has led to the development of novel targeted therapies like immune checkpoint inhibitors, CAR-T cells, and oncolytic viruses.
Biomarker Discovery: The identification of prognostic and predictive biomarkers has ushered in an era of personalized treatment for GBM (Weller et al., 2017). These markers guide therapy selection and monitoring, such as MGMT promoter methylation and IDH1/2 mutations.
Radiomics and Imaging: Advanced imaging techniques like MRI and PET have enabled better characterization of tumor heterogeneity, guiding precision therapy (Lambin et al., 2017).
4.4.3. Ongoing Trials
CheckMate-143 (NCT02017717): A phase III trial evaluating the efficacy of nivolumab, an anti-PD-1 antibody, in recurrent GBM (Reardon et al., 2020).
CAR-T cell Therapy (NCT02208362): A phase I trial exploring chimeric antigen receptor (CAR) T-cell therapy targeting IL13Rα2 in GBM (Brown et al., 2016).
DCVax (NCT00045968): A phase III trial testing personalized dendritic cell vaccines, highlighting the potential of vaccination approaches in GBM (Liau et al., 2018).
4.4.4. Future Expectations
Integration of Multi-modal Therapies: Combining surgery, radiation, chemotherapy, immunotherapy, and targeted therapies promises to enhance treatment efficacy.
Artificial Intelligence (AI) and Machine Learning: Leveraging AI in drug discovery, patient stratification, and treatment monitoring could revolutionize GBM management (Topol, 2019).
Global Collaboration: Collaborative efforts among researchers, clinicians, and industries could accelerate the translation of laboratory findings to clinical settings (Chin et al., 2011).
4.4.5. Conclusion
Current research in GBM is dynamic and multifaceted, focusing on personalized and precision medicine, innovative technologies, and collaborative efforts. The ongoing trials reflect a concerted push towards a new era of understanding and treating GBM. As these cutting-edge strategies move from bench to bedside, they hold promise for improved patient outcomes and quality of life in GBM.
5. Conclusion
In grappling with Glioblastoma multiforme (GBM), one of the most enigmatic and lethal forms of brain cancer, therapeutic avenues have remained disappointingly narrow despite scientific strides in understanding its molecular mechanics. This comprehensive review underscores the intricate and multi-layered nature of GBM, while also illuminating promising routes for research and treatment. As such, the outlook for GBM therapy is not as grim as current clinical outcomes may indicate.
Our main findings encompass the pivotal role of transcription factors (TFs) like FOXA1, YAP1, TWIST1, ZEB1, and NF-kB, which are intrinsically tied to the course of GBM, including its progression, immune escape mechanisms, and invasive properties. Further, the epigenetic roles of SNAI2 and HDACs have been highlighted as compelling therapeutic targets. On the treatment front, the advent of targeted therapies and CRISPR/Cas9 technologies, as well as the use of epigenetic modifiers and HDAC inhibitors to influence transcription factors, are exciting albeit challenging directions. The burgeoning field of personalized immunotherapy, particularly the development of neoantigen-based vaccines, has also shown encouraging potential. This is augmented by current research focusing on linking genetic variations to personalized treatments and delving into the impact of TFs on neoantigen presentation.
However, the path ahead is fraught with challenges, including the molecular diversity and functional redundancy of transcription factors, ethical quandaries in genetic manipulation through techniques like CRISPR/Cas9, and the yawning gap between clinical trials and real-world implementation. Given that many ongoing trials are yet to reach conclusive endpoints, translating research breakthroughs into tangible clinical benefits remains a colossal task.
As for future research, there's a pressing need for a multi-modal therapeutic strategy that combines surgery, radiation, chemotherapy, immunotherapy, and targeted therapies for a synergistic impact. Global collaborations that bring together expertise from diverse fields could accelerate the validation and implementation of GBM interventions. Additionally, the move toward precision medicine mandates the development of predictive models that integrate genetic, epigenetic, and clinical data for individualized treatment plans. Finally, devising reliable markers for early detection, monitoring treatment response, and predicting recurrence is indispensable for more effective GBM management.
To sum up, while the challenges are considerable, the synthesis of advancements in molecular understanding, emerging technologies like AI and machine learning, and collaborative global efforts points to a transformative phase in the battle against GBM.
Final Remarks
In conclusion, the review presented herein offers a comprehensive panorama of GBM, delineating the complex interplay of transcription factors, epigenetic modulators, targeted therapies, immunotherapy, and personalized approaches. While the challenges are many, the convergence of multidisciplinary research, technological advancements, and global collaboration fuels hope for the future.
The road to defeating GBM will undoubtedly be arduous, filled with obstacles and setbacks. However, the continued pursuit of scientific excellence, fostering innovation, emphasizing personalized strategies, and nurturing a collaborative spirit will, one day, lead to a revolution in GBM treatment.
As Albert Einstein once said, "In the middle of difficulty lies opportunity." The complexity of GBM is both its challenge and its opportunity, and the research community stands at a precipice where the next steps could dramatically alter the course of this formidable disease. With steadfast dedication, creativity, and rigor, we may yet turn the tide against GBM, transforming a grim diagnosis into a manageable condition. This transformation requires a shared vision, continued investment in research, thoughtful consideration of ethical dimensions, and an unswerving commitment to patient-centered care. The path is laid out; the journey, arduous but essential, awaits.
Author Contributions
AT and DN conceptualized and wrote the manuscript. AT designed the figures and tables. DN performed critical review of the manuscript. All the authors reviewed and approved the manuscript.
Funding
No external funding was obtained.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
Not applicable
Conflicts of Interest
The authors declare no conflict of interest.
References
- Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-820. [CrossRef]
- Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol. 2018;20(suppl_4):iv1-iv86. [CrossRef]
- Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98-110. [CrossRef]
- Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-477. [CrossRef]
- Ostrom QT, Cioffi G, Gittleman H, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro Oncol. 2019;21(Supplement_5):v1-v100. [CrossRef]
- Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466. [CrossRef]
- Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: a "state of the science" review. Neuro Oncol. 2014;16(7):896-913. [CrossRef]
- Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2(9):494-503; quiz 1 p following 516. [CrossRef]
- Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97-109. [CrossRef]
- Weller M, van den Bent M, Tonn JC, et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017;18(6):e315-e329. [CrossRef]
- Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807-1812. [CrossRef]
- Bhat KP, Balasubramaniyan V, Vaillant B, et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24(3):331-346.
- Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765-773.
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061-1068.
- Verhaak, R. G., Hoadley, K. A., Purdom, E., Wang, V., Qi, Y., Wilkerson, M. D., ... & Hayes, D. N. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell, 17(1), 98-110. [CrossRef]
- Noushmehr, H., Weisenberger, D. J., Diefes, K., Phillips, H. S., Pujara, K., Berman, B. P., ... & TCGA Research Network. (2010). Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell, 17(5), 510-522. [CrossRef]
- Stupp, R., Mason, W. P., van den Bent, M. J., Weller, M., Fisher, B., Taphoorn, M. J., ... & Marosi, C. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New England Journal of Medicine, 352(10), 987-996. [CrossRef]
- Weller, M., van den Bent, M., Tonn, J. C., Stupp, R., Preusser, M., Cohen-Jonathan-Moyal, E., ... & Reifenberger, G. (2017). European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. The Lancet Oncology, 18(6), e315-e329. [CrossRef]
- Chinot, O. L., Wick, W., Mason, W., Henriksson, R., Saran, F., Nishikawa, R., ... & Cloughesy, T. (2014). Bevacizumab plus radiotherapy–temozolomide for newly diagnosed glioblastoma. The Lancet Oncology, 15(2), 200-212. [CrossRef]
- Omuro, A., & DeAngelis, L. M. (2013). Glioblastoma and other malignant gliomas: A clinical review. Neurology, 80(6), 540-554.
- Sanai, N., Polley, M. Y., McDermott, M. W., Parsa, A. T., & Berger, M. S. (2011). An extent of resection threshold for newly diagnosed glioblastomas. Neurosurgery, 68(2), 546-553. [CrossRef]
- Meyers, C. A., Smith, J. A., Bezjak, A., Mehta, M. P., Liebmann, J., Illidge, T., ... & Curran, W. (2004). Neurocognitive function and progression in patients with brain metastases treated with whole-brain radiation and motexafin gadolinium: Results of a randomized phase III trial. Journal of Clinical Oncology, 22(9), 1573-1582. [CrossRef]
- Hegi, M. E., Diserens, A. C., Gorlia, T., Hamou, M. F., de Tribolet, N., Weller, M., ... & Stupp, R. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. New England Journal of Medicine, 352(10), 997-1003.
- Johnson, D. R., & O’Neill, B. P. (2012). Glioblastoma survival in the United States before and during the temozolomide era. Mayo Clinic Proceedings, 87(7), 673-680. [CrossRef]
- Reardon, D. A., Omuro, A., Brandes, A. A., Rieger, J., Wick, A., Sepulveda, J., ... & Sampson, J. (2017). OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: CheckMate 143. Neuro-Oncology, 19(3), iii21. [CrossRef]
- Filley, A. C., Henriquez, M., & Dey, M. (2017). Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Frontiers in Immunology, 8, 185. [CrossRef]
- Alexander, B. M., & Cloughesy, T. F. (2017). Adult Glioblastoma. Journal of Clinical Oncology, 35(21), 2402–2409.
- Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 646-674. [CrossRef]
- Wang, Q., Hu, B., Hu, X., Kim, H., Squatrito, M., Scarpace, L., ... & Brennan, C. (2017). Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell, 32(1), 42-56.e6.
- Kaczmarek JC, Kowalski PS, Anderson DG. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017;9(1):60. [CrossRef]
- Darnell JE Jr. Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2(10):740-749. [CrossRef]
- Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8(12):976-990. [CrossRef]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798-809. [CrossRef]
- Dang, C. V., O'Donnell, K. A., Zeller, K. I., Nguyen, T., Osthus, R. C., & Li, F. (2006). The c-Myc target gene network. Seminars in Cancer Biology, 16(4), 253-264. [CrossRef]
- Yu, H., Pardoll, D., & Jove, R. (2009). STATs in cancer inflammation and immunity: a leading role for STAT3. Nature Reviews Cancer, 9(11), 798-809. [CrossRef]
- Yang, J., & Weinberg, R. A. (2008). Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Developmental Cell, 14(6), 818-829. [CrossRef]
- Batlle, E., Sancho, E., Francí, C., Domínguez, D., Monfar, M., Baulida, J., & García De Herreros, A. (2000). The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nature Cell Biology, 2(2), 84-89. [CrossRef]
- Friedl, P., & Alexander, S. (2011). Cancer invasion and the microenvironment: plasticity and reciprocity. Cell, 147(5), 992-1009. [CrossRef]
- Baldwin, A. S. (2001). Series introduction: the transcription factor NF-κB and human disease. Journal of Clinical Investigation, 107(1), 3-6.
- Altman, B. J., Stine, Z. E., & Dang, C. V. (2016). From Krebs to clinic: glutamine metabolism to cancer therapy. Nature Reviews Cancer, 16(10), 619-634. [CrossRef]
- Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A., & Kinzler, K. W. (2013). Cancer genome landscapes. Science, 339(6127), 1546-1558. [CrossRef]
- Li, J., Yang, B., Zhou, Q., Wu, Y., Shang, D., Guo, Y., ... & Chen, Y. (2013). Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial–mesenchymal transition. Carcinogenesis, 34(6), 1343-1351. [CrossRef]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415-428. [CrossRef]
- Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148-1159.
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042-2054. [CrossRef]
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301(5895):89-92. [CrossRef]
- Zhang, L., Wang, H., Zhu, J., Ding, K., & Xu, J. (2014). FGF19 (fibroblast growth factor 19) as a novel target in hepatocellular carcinoma. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer, 1846(1), 152-161.
- Corcoran, R. B., Cheng, K. A., Hata, A. N., Faber, A. C., Ebi, H., Coffee, E. M., ... & Settleman, J. (2012). Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell, 21(1), 69-79.
- Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell, 136(2), 215-233. [CrossRef]
- Silber, J., Lim, D. A., Petritsch, C., Persson, A. I., Maunakea, A. K., Yu, M., ... & Costello, J. F. (2012). miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. Nature Communications, 3, 1126. [CrossRef]
- Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217-221. [CrossRef]
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69-74. [CrossRef]
- McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463-1469. [CrossRef]
- Schumacher, T. N., & Schreiber, R. D. (2015). Neoantigens in cancer immunotherapy. Science, 348(6230), 69-74. [CrossRef]
- Chen, D. S., & Mellman, I. (2017). Elements of cancer immunity and the cancer–immune set point. Immunity, 44(3), 221-233. [CrossRef]
- Ribas, A., & Wolchok, J. D. (2018). Cancer immunotherapy using checkpoint blockade. Science, 359(6371), 1350-1355. [CrossRef]
- Lambert SA, Jolma A, Campitelli LF, et al. The Human Transcription Factors. Cell. 2018;172(4):650-665.
- June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361-1365. [CrossRef]
- Sharma, P., & Allison, J. P. (2015). The future of immune checkpoint therapy. Cell, 161(2), 205-214. [CrossRef]
- Marusyk, A., Almendro, V., Polyak, K. (2012). Intra-tumour heterogeneity: a looking glass for cancer? Nature, 494(7437), 338–344.
- Furnari, F. B., Fenton, T., Bachoo, R. M., Mukasa, A., Stommel, J. M., Stegh, A., ... & Cavenee, W. K. (2007). Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes & Development, 21(21), 2683-2710. [CrossRef]
- Karsy M, Arslan E, Moy F. Current Progress on Understanding MicroRNAs in Glioblastoma Multiforme. Genes Cancer. 2012;3(1):3-15. [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061-1068.
- Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756-760. [CrossRef]
- Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98-110. [CrossRef]
- Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350-1354.
- Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466. [CrossRef]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203-1217.
- Bouffet E, Larouche V, Campbell BB, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol. 2016;34(19):2206-2211. [CrossRef]
- Lee, D. K., Horikoshi, M., & Roeder, R. G. (1993). Interaction of TFIID in the minor groove of the TATA element. Cell, 75(3), 487-498. [CrossRef]
- Struhl, K. (1998). Histone acetylation and transcriptional regulatory mechanisms. Genes & Development, 12(5), 599-606. [CrossRef]
- Spitz, F., & Furlong, E. E. (2012). Transcription factors: from enhancer binding to developmental control. Nature Reviews Genetics, 13(9), 613-626. [CrossRef]
- Brantley, E. C., Nabors, L. B., Gillespie, G. Y., Choi, Y. H., Palmer, C. A., Harrison, K., ... & Benveniste, E. N. (2008). Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clinical Cancer Research, 14(15), 4694-4704. [CrossRef]
- Wang, J., Wang, H., Li, Z., Wu, Q., Lathia, J. D., McLendon, R. E., ... & Rich, J. N. (2011). c-Myc is required for maintenance of glioma cancer stem cells. PLOS ONE, 3(11), e3769. [CrossRef]
- Siegfried, Z., Simon, I., & Cedar, H. (2014). DNA methylation reprogramming and cancer: epigenetics meets metabolism. Nature Reviews Cancer, 14(7), 502-517.
- Hajra, K. M., Chen, D. Y., & Fearon, E. R. (2002). The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Research, 62(6), 1613-1618.
- Vega, S., Morales, A. V., Ocaña, O. H., Valdés, F., Fabregat, I., & Nieto, M. A. (2004). Snail blocks the cell cycle and confers resistance to cell death. Genes & Development, 18(10), 1131-1143. [CrossRef]
- Peinado, H., Ballestar, E., Esteller, M., & Cano, A. (2004). Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Molecular and Cellular Biology, 24(1), 306-319.
- Marks, P., Rifkind, R. A., Richon, V. M., Breslow, R., Miller, T., & Kelly, W. K. (2001). Histone deacetylases and cancer: Causes and therapies. Nature Reviews Cancer, 1(3), 194-202. [CrossRef]
- Zhang, S., Cui, W., Xiong, F., et al. (2016). FOXA1 Promotes Tumor Progression in Prostate Cancer via the Insulin-Like Growth Factor Binding Protein 3 Pathway. PLOS ONE, 11(8), e0160379.
- Orr, B. A., et al. (2011). Yes-associated protein 1 is widely expressed in human brain tumors and promotes glioblastoma growth. Journal of Neuropathology & Experimental Neurology, 70(7), 568-577. [CrossRef]
- Elias, M. C., et al. (2005). TWIST is expressed in human gliomas and promotes invasion. Neoplasia, 7(9), 824-837. [CrossRef]
- Siebzehnrubl, F. A., et al. (2013). ZEB1 is associated with poor prognosis in glioblastoma. Tumour Biology, 34(6), 4009-4016.
- Nagai, S., et al. (2002). Concurrent inhibition of the epidermal growth factor receptor pathway by gefitinib (Iressa) and protein kinase A pathway by perifosine in malignant gliomas. Cancer Research, 62(20), 5763-5766.
- Bullock, A. N. et al. (2011). "Drugable" Transcription Factors? Chemistry & Biology, 18(11), 1313-1314.
- Lambert, M. et al. (2018). Targeting the DNA-Binding Activity of STAT3 as a New Therapeutic Avenue in Human Cancers. Oncogene, 37, 3947-3960.
- Wells, J. A., & McClendon, C. L. (2007). Reaching for High-Hanging Fruit in Drug Discovery at Protein–Protein Interfaces. Nature, 450(7172), 1001-1009. [CrossRef]
- Wang, J. et al. (2013). Pleiotropic Biological Activities of Alternatively Spliced TMPRSS2/ERG Fusion Gene Transcripts. Cancer Research, 68(20), 8516-8524.
- Spitz, F., & Furlong, E. E. M. (2012). Transcription Factors: From Enhancer Binding to Developmental Control. Nature Reviews Genetics, 13(9), 613-626. [CrossRef]
- Chen, Y. et al. (2016). Dual Phosphorylation of Suppressors of Cytokine Signaling Regulates Their Stability and Function. Journal of Biological Chemistry, 291(47), 24744-24752.
- Patel, A. P. et al. (2014). Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science, 344(6190), 1396-1401. [CrossRef]
- Pardridge, W. M. (2019). Blood-Brain Barrier Drug Delivery of IgG Fusion Proteins with a Transferrin Receptor Monoclonal Antibody. Expert Opinion on Drug Delivery, 12(2), 207-222. [CrossRef]
- Stupp, R. et al. (2005). Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. The New England Journal of Medicine, 352(10), 987-996. [CrossRef]
- Lu, J. et al. (2015). Novel Targeted Agents for Platelet-Derived Growth Factor Receptorα and c-KIT in Glioblastoma. Neurosurgical Focus, 38(3), E12.
- Esteller, M. (2007). Cancer Epigenomics: DNA Methylomes and Histone-Modification Maps. Nature Reviews Genetics, 8(4), 286-298. [CrossRef]
- Berger, S. L., et al. (2009). An Operational Definition of Epigenetics. Genes & Development, 23(7), 781-783. [CrossRef]
- Anastasiadou, E., et al. (2018). The Role of Non-Coding RNAs in Glioma. Cancers, 10(11), 11.
- Marks, P., et al. (2000). Histone Deacetylases and Cancer: Causes and Therapies. Nature Reviews Cancer, 1(3), 194-202. [CrossRef]
- Galanc, O., et al. (2019). HDAC Inhibitors in Glioblastoma: A Therapeutic Perspective. Frontiers in Oncology, 9, 302.
- Lee, E. Q., et al. (2015). A Multicenter, Phase II, Randomized, Non-Comparative Clinical Trial of Radiation and Temozolomide with or without Vandetanib in Newly Diagnosed Glioblastoma Patients. Clinical Cancer Research, 21(16), 3610-3618.
- Bannister, A. J., & Kouzarides, T. (2011). Regulation of Chromatin by Histone Modifications. Cell Research, 21(3), 381-395. [CrossRef]
- Eckschlager, T., et al. (2017). Histone Deacetylase Inhibitors as Anticancer Drugs. International Journal of Molecular Sciences, 18(7), 1414. [CrossRef]
- Noushmehr, H., et al. (2010). Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell, 17(5), 510-522. [CrossRef]
- Vassilev, L. T., et al. (2004). In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science, 303(5659), 844-848. [CrossRef]
- Sen, M., et al. (2012). First-in-Class Small Molecule Inhibitors of Signal Transducer and Activator of Transcription 3 (STAT3) That Act Through Indirect Mechanisms. Journal of Medicinal Chemistry, 55(11), 5730-5744.
- Swayze, E. E., et al. (2007). Antisense Oligonucleotides Containing Locked Nucleic Acid Improve Potency but Cause Significant Hepatotoxicity in Animals. Nucleic Acids Research, 35(2), 687-700. [CrossRef]
- Wu, S. Y., et al. (2015). Small Molecule Targeting of Transcriptional Repression in Cancer. Molecular Cancer Therapeutics, 14(6), 1422-1431.
- Lambert, M., et al. (2014). Targeting Transcription Factors for Cancer Treatment. Molecules, 19(6), 7950-7976. [CrossRef]
- Saraiva, C., et al. (2016). Nanoparticle-Mediated Brain Drug Delivery: Overcoming Blood–Brain Barrier to Treat Neurodegenerative Diseases. Journal of Controlled Release, 235, 34-47. [CrossRef]
- Schust, J., et al. (2006). Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chemistry & Biology, 13(11), 1235-1242. [CrossRef]
- Zhang, X., et al. (2013). A Novel Inhibitor of STAT3 Homodimerization Selectively Suppresses STAT3 Activity and Malignant Transformation. Cancer Research, 73(6), 1922-1933. [CrossRef]
- Korkolopoulou, P., et al. (2008). Activation of Nuclear Factor-κB Pathway Predicts Poor Outcome in Glioblastoma Multiforme. Clinical Neuropathology, 27(4), 228-237.
- Mullard, A. (2017). Targeting Undruggable Transcription Factors. Nature Reviews Drug Discovery, 16(7), 443-444.
- Massard, C., et al. (2016). High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discovery, 7(6), 586-595. [CrossRef]
- Weller, M., et al. (2017). European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. The Lancet Oncology, 18(6), e315-e329. [CrossRef]
- Sottoriva, A., et al. (2013). Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proceedings of the National Academy of Sciences, 110(10), 4009-4014. [CrossRef]
- Bhat, K. P., et al. (2013). Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell, 24(3), 331-346.
- Wen, P. Y., et al. (2016). Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. Journal of Clinical Oncology, 28(11), 1963-1972. [CrossRef]
- Joffe, S., et al. (2001). Quality of Informed Consent: A New Measure of Understanding Among Research Subjects. JNCI Journal of the National Cancer Institute, 93(2), 139-147. [CrossRef]
- Marron, J. M., et al. (2018). Equity in Precision Medicine: Leaving No One Behind. Trends in Cancer, 4(1), 25-27.
- Ishii, T. (2017). Germ line genome editing in clinics: the approaches, objectives, and global society. Briefings in Functional Genomics, 16(1), 46-56. [CrossRef]
- Verhaak, R. G., et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell, 17(1), 98-110. [CrossRef]
- Lathia, J. D., et al. (2015). Cancer stem cells in glioblastoma. Genes & Development, 29(12), 1203-1217.
- National Academy of Sciences. (2017). Human Genome Editing: Science, Ethics, and Governance. National Academies Press.
- Schumacher, T., & Schreiber, R. (2015). Neoantigens in cancer immunotherapy. Science, 348(6230), 69-74. [CrossRef]
- Castle, J. C., et al. (2012). Exploiting the mutanome for tumor vaccination. Cancer Research, 72(5), 1081-1091. [CrossRef]
- Hilf, N., et al. (2019). Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature, 565(7738), 240-245. [CrossRef]
- Keskin, D. B., et al. (2019). Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature, 565(7738), 234-239. [CrossRef]
- Ott, P. A., et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature, 547(7662), 217-221. [CrossRef]
- Patel, A. P., et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 344(6190), 1396-1401. [CrossRef]
- Anagnostou, V., et al. (2017). Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discovery, 7(3), 264-276. [CrossRef]
- Wainwright, D. A., et al. (2015). The glioma microenvironment: Transforming stumbling blocks into stepping stones. Immunological Investigations, 44(8), 678-700.
- Jurtz, V., et al. (2017). NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. Journal of Immunology, 199(9), 3360-3368. [CrossRef]
- Sahin, U., et al. (2017). Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature, 547(7662), 222-226. [CrossRef]
- Yu, H., Kortylewski, M., Pardoll, D. (2007). Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nature Reviews Immunology, 7(1), 41-51. [CrossRef]
- Kortylewski, M. et al. (2009). Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nature Medicine, 15(12), 1313-1320. [CrossRef]
- Hayden, M. S., & Ghosh, S. (2012). NF-κB in immunobiology. Cell Research, 22(3), 447-462.
- Zuo, T. et al. (2007). FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell, 129(7), 1275-1286.
- Gilmore, T. D., & Herscovitch, M. (2006). Inhibitors of NF-κB signaling: 785 and counting. Oncogene, 25(51), 6887-6899.
- Kortylewski, M. et al. (2009). Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nature Medicine, 15(12), 1313-1320. [CrossRef]
- Lawrence, T. (2009). The Nuclear Factor NF-kB Pathway in Inflammation. Cold Spring Harbor Perspectives in Biology, 1(6), a001651.
- Yu, H., Kortylewski, M., Pardoll, D. (2007). Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nature Reviews Immunology, 7(1), 41-51. [CrossRef]
- Hayden, M. S., & Ghosh, S. (2012). NF-κB in immunobiology. Cell Research, 22(3), 447-462.
- Zuo, T. et al. (2007). FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell, 129(7), 1275-1286.
- Chen, H. Z. et al. (2009). The dualistic function of cyclin D1: A concise review. Cell Cycle, 8(13), 2101-2106.
- West, A. C., & Johnstone, R. W. (2014). New and emerging HDAC inhibitors for cancer treatment. Journal of Clinical Investigation, 124(1), 30-39. [CrossRef]
- Gilmore, T. D., & Herscovitch, M. (2006). Inhibitors of NF-κB signaling: 785 and counting. Oncogene, 25(51), 6887-6899.
- Lamb, J. et al. (2013). The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science, 313(5795), 1929-1935. [CrossRef]
- Holohan, C. et al. (2013). Cancer drug resistance: An evolving paradigm. Nature Reviews Cancer, 13(10), 714-726. [CrossRef]
- Freedman, A. et al. (2015). The Clinical Trials Process: an overview. Journal of Oncology Practice, 11(3), 253-255.
- Yu, H., Kortylewski, M., Pardoll, D. (2007). Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nature Reviews Immunology, 7(1), 41-51. [CrossRef]
- Kortylewski, M. et al. (2009). Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nature Medicine, 15(12), 1313-1320. [CrossRef]
- Hayden, M. S., & Ghosh, S. (2012). NF-κB in immunobiology. Cell Research, 22(3), 447-462.
- Zuo, T. et al. (2007). FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell, 129(7), 1275-1286.
- Gilmore, T. D., & Herscovitch, M. (2006). Inhibitors of NF-κB signaling: 785 and counting. Oncogene, 25(51), 6887-6899.
- Hodges, T. R., et al. (2017). Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro-Oncology, 19(8), 1047-1057. [CrossRef]
- Wang, Q., et al. (2018). Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes. Nature Communications, 11, 2539. [CrossRef]
- Zack, T. I., et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nature Genetics, 45(10), 1134-1140. [CrossRef]
- Kreiter, S., et al. (2015). Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature, 520, 692-696. [CrossRef]
- Ott, P. A., et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature, 547, 217-221. [CrossRef]
- Sahin, U., et al. (2017). Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature, 547, 222-226. [CrossRef]
- Verhaak, R. G., et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell, 17, 98-110. [CrossRef]
- Pardoll, D. M. (2012). The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer, 12, 252-264. [CrossRef]
- Mellman, I., et al. (2011). Cancer immunotherapy comes of age. Nature, 480, 480-489. [CrossRef]
- Schalper, K. A., et al. (2019). Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nature Medicine, 25, 470-476. [CrossRef]
- Kim, S., et al. (2020). Deeper profiles and cascaded recurrent and convolutional neural networks for state-of-the-art protein secondary structure prediction. Scientific Reports, 10, 12374.
- Limon, D., et al. (2020). Glioblastoma: Molecular pathways, stem cells and therapeutic targets. Cancers, 12(4), 821.
- Weller, M., et al. (2017). Molecular neuro-oncology in clinical practice: a new horizon. The Lancet Oncology, 18, e430-e443.
- Lambin, P., et al. (2017). Radiomics: the bridge between medical imaging and personalized medicine. Nature Reviews Clinical Oncology, 14, 749-762. [CrossRef]
- Reardon, D. A., et al. (2020). CheckMate-143: a phase 3 randomized trial of nivolumab (nivo) vs bevacizumab (bev) in patients (pts) with recurrent glioblastoma (GBM). Journal of Clinical Oncology, 35(15_suppl), 2006.
- Brown, C. E., et al. (2016). Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. The New England Journal of Medicine, 375, 2561-2569. [CrossRef]
- Liau, L. M., et al. (2018). First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. Journal of Translational Medicine, 16, 142.
- Topol, E. J. (2019). High-performance medicine: the convergence of human and artificial intelligence. Nature Medicine, 25, 44-56. [CrossRef]
- Chin, L., et al. (2011). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455, 1061-1068.
Figure 1.
Overall Survival statistics for Glioblastoma with treatment options.
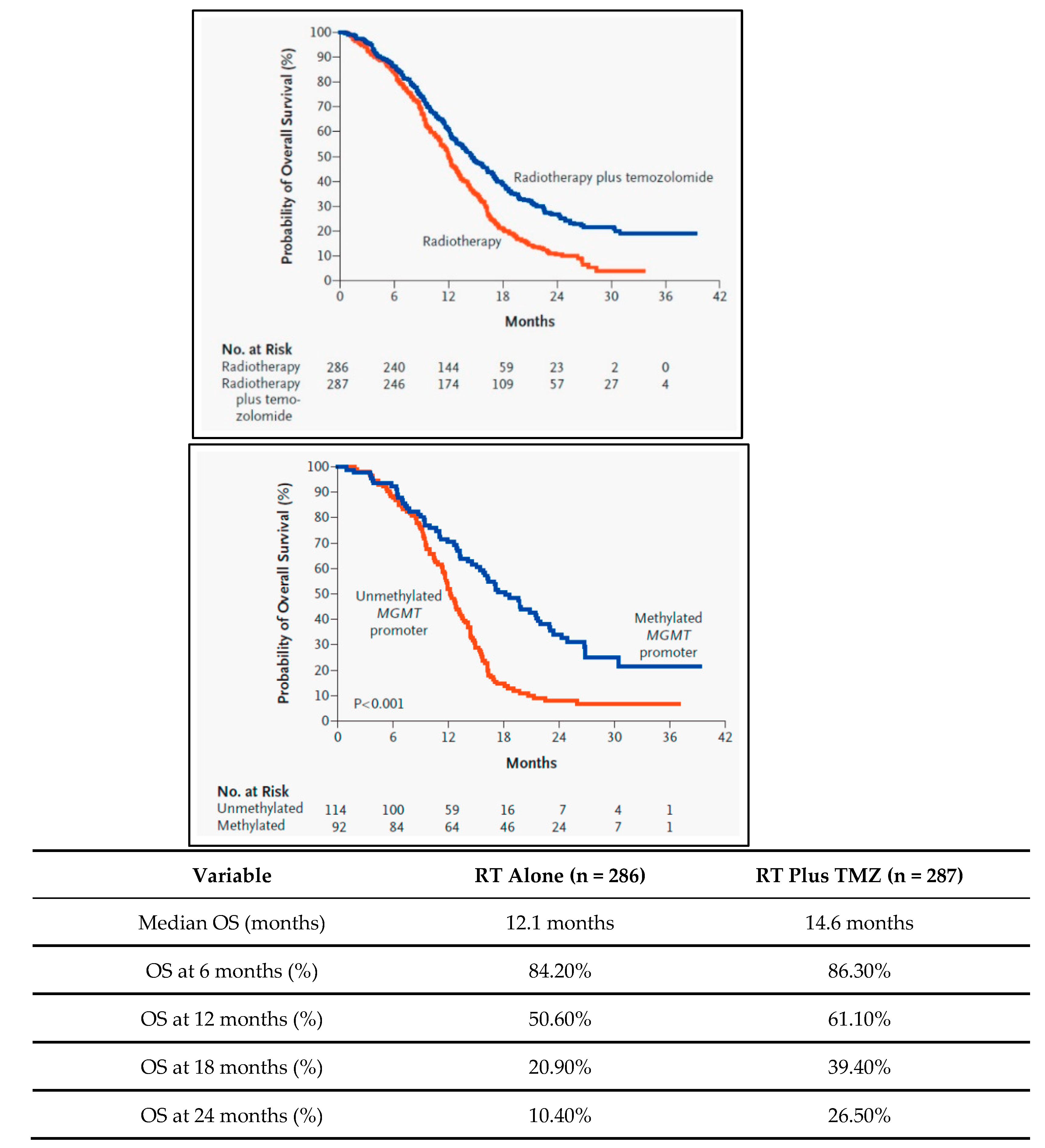
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



