
Submitted:
09 October 2023
Posted:
10 October 2023
You are already at the latest version
Abstract
Background: Aging rate is affected by genetic changes, epigenetic modifications, oxidative stress, inflammation, lifestyle and environmental factors. In accelerated aging (AA), biological age exceeds the chronological one. Objective: To undertake a critical reappraisal of the AA concept and to reveal its weaknesses and limitations. Methods: We reviewed over 300 recent articles dealing with physiology of brain aging and pathophysiology of neurodegeneration. Results: (1) Organ systems age at different rates, which interferes with application of the AA concept to individual organs. (2) Aging rate can be decelerated due to individual structure–functional reserves built with cognitive, physical training or pharmacological interventions. (3) The AA concept lacks standardised terminology and methodology. (4) Specific molecular biomarkers (MBM) reflect aging. To consolidate the AA theory, researchers should validate numerous MBM candidates. (5) Aging factors, mechanisms and the exact nature of their biological outcomes are not well understood. Conclusion: Although considered as a perspective theory, the AA concept has serious limitations and it requires an update.
Keywords:
aging
; accelerated aging
; brain aging
; neurodegeneration
; epigenetics
; biological clocks
; molecular biomarkers
; rejuvenation
; structural reserves
; functional reserves
1. Introduction
Aging is associated with structural and functional changes that increase risks of dis- eases and death [1,2,3]. Multiple factors contribute to aging. Accumulation of damage and dysfunction may happen due to genetic mutations, epigenetic modifications, oxida- tive stress and inflammation [4,5]. Physiological or biological age (BA) is defined as the current state of the individual as a biological system. A combination of detectable lifetime- dependent biological parameters characterizes the system state. These parameters include the current profile of genomic DNA methylation, age-associated structural changes in the brain, brain functional reserve, etc.
In normal aging, BA is equal to chronological age. If the process of getting older is accelerated, BA exceeds the chronological age. In decelerated aging, BA becomes lower than the official age [6,7,8]. Accelerated aging (AA) shares common features with the normal one, but protein aggregation and excitotoxicity are specific to AA [9,10,11]. Understanding mechanisms of aging opens opportunities for targeted treatment of the diseases that occur late in life [9].
AA is the area of research with unresolved issues such as non-standardized terminol- ogy [12] and understudied mechanisms [13]. Researchers have not reached an agreement on whether neurodegeneration (ND) is a type of AA [14,15] or its outcome [14,16,17,18]. The latter view argues that certain biomarkers (BMs) are ND-specific and they do not detect AA [17].
Different theories were proposed to explain AA pathogenesis [19,20,21]. The genetic theory assumes that accumulation of DNA mutations and/or gene dysregulation are the major causes of AA [19,22]. The theory considers random DNA changes but ignores chro- mosomal, multifactorial and monogenic alterations [13,23,24]. The multi-proteinopathies theory describes aggregation of misfolded proteins as a leading cause of cell dysfunction in age-related diseases [20,25]. The free radical theory postulates that the primary accelerator of aging is oxidative damage of DNA and proteins [15,26,27,28]. But the concept fails to explain difference between normal and abnormal levels of reactive oxygen species [21,29]. In practice, no diagnostic BMs can identify and prognosticate AA reliably [20,30].
2. Biomolecular Aspects of Aging
Neurocentric and neurovascular hypotheses describe aetiology of ND at sub-, cellular, and supra-cellular levels. Initially, research efforts were focused mainly on neurons. Then, investigators recognized the importance of non-neural cells in higher brain functions. Neu- rovascular (NV) view refers to a neurovascular unit (NVU) which is a dynamic multicellular structure mediating functional interactions between brain tissues per se and blood vessels. The NVU includes astrocytes, microglia, oligodendrocytes, precursor cells, endothelial cells, pericytes, excitatory and inhibitory neurons [31]. The NV hypothesis proposes that neural cells in the NVU and circulating immune cells secrete proinflammatory mediators contributing to age-related neuroinflammation [32], cell degeneration [33,34] and endothe- lial impairment [34,35]. These changes disrupt molecular networks, induce damage to the blood–brain barrier [36,37] and lead to NVU dysfunction which is a major cause of ND [38]. But the exact role of an NVU in ND remains unclear [39]. A research for ND-associated BMs is difficult due to high complexity and molecular heterogeneity of the NVU network. It requires whole genome studies, e.g., global transcriptome analysis followed by hierarchical data clustering [40] or single-cell/single-nucleus transcriptomics [41,42].
Molecular biomarkers (MBMs) are biomolecules, their components, fragments or modifications with the associated measurable parameters that serve as a tool to diagnose pathologies and monitor biological processes. MBMs can be used to evaluate aging, partic- ularly to estimate the rate of its progression [43]. Aging MBMs include mRNA transcripts, proteins [44], telomere length, serum markers of DNA damage [45], DNA methylation profiles [46,47], histone modifications [48,49,50,51,52,53,54,55,56,57,58,59], differentially expressed genes [42,60], non- coding RNAs [61,62,63,64] and other biomolecules (Figure 1).
Despite a large number of suggested MBMs of ND, only few of them are validated. In most studies, sample sizes were too small to justify the accuracy and reproducibility of MBM data. For example, a recent study aimed at determining whether age affects different cell types in the NVU. The study resulted in a model discriminating patients with Alzheimer’s disease (AD) from healthy controls (HCs). The model revealed 15 genes related to AA (AAG): IGF1R, MXI1, RB1, PPARA, NFE2L2, STAT5B, FOS, PRKCD, YWHAZ, HTT, MAPK9,
HSPA9, SDHC, PRKDC and PDPK1 [42]. Differential expression of IGF1R, MXI1, PPARA, YWHAZ and MAPK9 correlated with ND progression. Therefore, they may function as facilitators or inhibitors of AD. But the research neither demonstrated cell-specific roles of the discovered AAGs nor identified their contribution to AD pathogenesis and interactions in the NVU. Moreover, the study failed to justify the AAGs as MBMs since it had an insufficient sample size of only 11 AD patients and 7 HCs [42]. ND results from multiple structural changes at different genetic loci over a period of time [65,66]. AD accounts for 90% of ND cases. High risk of developing AD is associated with alterations in 15 genes that predispose to ND (NDG): GBA1, APP, PSEN1, MAPT, GRN, SETX, SPAST, CSF1R, C9orf72 [67], TET2 [68], TBK1 [69], TOMM40, APOC1 [70], APOE [70,71] and TREM2 [72,73,74,75,76,77,78]. Surprisingly, the gene sets do not overlap across the studies on AAGs and NDGs. Researchers found that the APOE e4 allele and mutation spectrum for TREM2 gene are risk factors for developing dementia with Lewy bodies, multi-cognitive decline and corticobasal degeneration [78,79,80,81,82,83,84,85,86,87]. DNA methylation level reflects rate of aging. Approximately 1.5% of genomic DNA contains 5-methylcytosine (5-mC), and the level decreases during ontogenesis [88]. The level of 5-mC is the highest in embryos, and then it reduces gradually across life [89,90]. In aging, global genomic DNA hypomethylation proceeds along with hypermethylation of CpG islands. These changes in DNA methylation patterns are called “epigenetic drift” [91,92]. In the mammalian genome, 60% of the CpG islands are associated with gene promoters and regulate gene transcription [91].
In normal aging, age-predictive models demonstrate gradual linear changes in the DNA methylation profile, but environmental or genetic risk factors can accelerate the process of getting older [93]. In monozygotic twins, the divergence of the methylome increases at different rates [94]. Change of the DNA methylation profile was proposed as a mechanism of an epigenetic clock [95,96,97] by analogy with a biological clock [98,99]. Monitoring deviation between biological and chronological age helps to study development and aging across the lifespan [100]. Horvath [101], Hannum [93] and PhenoAge [102] epigenetic clocks serve as markers of ND [102,103,104,105,106], with the first of these showing the strongest correlation between epigenetic and chronological age [107].
Histone modifications can serve as potential MBMs of aging, however, heterogeneity of animal models used to develop the biomarkers limits their applicability. For example, a drop in highly abundant transcription activation mark H3K4me3 [48] correlated with an extended lifespan in Caenorhabditis elegans [49]. Contrarily, an increase in the H3K4me3 level was linked with AA in Drosophila melanogaster [50]. The level of heterochromatin- associated histone transcription repression mark H3K9me3 gradually decreases during aging in haematopoietic stem cells of humans and mice [51]. In C. elegans and other models of senescance, the most significant loss of H3K9me3 occurs in repressive regions [52,53]. H3K27me3 is associated with transcriptional silencing in aging [54]. The role of H3K27me3 is controversial, as studies showed its bidirectional lifelong changes [55,56,57,58,59].
Increased levels of H4K20me3 and H3K4me3 and decreased levels of H3K9me1 and H3K27me3 are common age-associated epigenetic marks [108,109,110]. Research showed an increase in H3K4me3 promoter methylation in a CK-p25 tauopathy mouse model and hippocampus of AD patients [111,112]. The following histone methylation marks can also be found in an Alzheimer brain: H4K20me2, H3K4me2, H3K27me3, H3K79me1, H3K79me2, H3K36me2, H4K20me3, H3K27me1 and H3K56me1 [113,114]. Besides, histone acetylation marks H3K9ac, H3K14ac and H4K16ac are associated with normal and accelerated aging [110,111,113,114,115]. Histone phosphorylation marks H4S47p and H3S10p and histone ubiquitination mark H2BK120ub are observed in AD [114,116,117]. Further systematic research should elucidate regulatory mechanisms of histone modifications, their interaction, and interplay between histone marks and other factors.
Long non-coding RNAs (lncRNAs) are presented with the growth-arrest-specific transcript 5 (GAS5) which plays a significant role in cell proliferation and apoptosis [122,123,124]. Its down-regulation leads to phosphorylation of the tau protein in ND [125,126]. Long intergenic brain cytoplasmic RNA 1 (BCYRN1) expressed in the dendritic domains of neurons is down-regulated in aging [127].
MicroRNAs (miRNAs) impact neuronal plasticity, influence tau protein metabolism and mediate brain aging through regulation of gene expression [128,129,130,131,132,133,134,135,136,137]. Regulation of miR-145a and miR-375 depends on age in mouse brains [138,139,140]. The MIR29 family, MIR339-5p, MIR195 and MIR107 modulate expression of beta-secretase 1 involved in cleaving the amyloid precursor protein [141,142,143,144,145,146]. Interestingly, miR-34 plays a protective role in Drosophila [147] and MIR144/MIR451 regulates ADAM metallopeptidase domain 10 in AD [148]. Hypothalamic stem cells secret over 20 miRNAs into the cerebrospinal fluid. These miRNAs control aging rate in mice [149], which should also be relevant to human brain [150]. Future studies should verify miRNA MBMs in humans [151].
Circular RNAs (circRNAs) are abundant in the brain, and their expression changes with age in skeletal muscles [152,153]. CircRNAs contribute to ND through interaction with miRNAs. For example, ciRS-7 potentially functions as a sponge for MIR7-1 [154] and its level is reduced dramatically in an AD brain [155]. Cerebral circRNAs are linked with neurotransmitter function, synaptic activities and neuronal maturation. They target the expression and availability of specific age-related mRNAs in the brain. At least four circRNAs are involved in postoperative neurocognitive disorders [156]. Another study revealed nearly 1200 cerebral circRNAs in a rat model of aging [157]. Various biomarker candidates including circRNAs await validation in the clinical arena.
3. Aging of Organs and Systems beyond Neurodegeneration
Aging affects organs and systems with different rates of change; therefore, the AA concept needs to be adjusted when applied to individual organs. For example, ovarian aging implies a loss of follicle numbers and decreased oocyte viability. Typically, an accelerated decline in fertility begins around the age of 38 years and continues until the climacteric [158]; however, a non-uniform decrease in follicle numbers results in a large variation in menopause onset. BA of the male reproduction system can also be assessed by fertility, but the arrest of reproductive capacity is reversible in older men, with lifestyle and disease factors prevailing over other determinants of aging [159]. In mice, oxidative stress, inflammation, DNA damage and de novo mutations accelerate testicular aging [160,161,162,163], while growth differentiation factor 11 enhances antioxidant enzyme activity and protects the testes [164]. A progressive age-related drop in Leydig and Sertoli cell function [165], testicular size [166] or testosterone level was demonstrated in older men [167]; but no decrease in testicle size or the levels of testosterone was observed in a cohort of older men with healthy lifestyle and affordable healthcare service [168,169].
The cumulative effect of a disease rather than age may account for changes in male fertility throughout life. Chronological age inaccurately reflects reproductive BA. Therefore, the AA concept cannot be adopted to the reproductive system. This illustrates a challenge in assessing BA at the organ and system levels.
Sex hormones that affect fertility are a part of the endocrine system. Susceptibility to aging differ among endocrine glands. In men, hypothalamic–pituitary–testicular axis does not undergo dramatic chronobiological changes: only 35–50% of men over 80 have reduced testosterone levels [170,171]. Diabetes mellitus and obesity predispose to accelerated adipose tissue dysfunction affecting telomere length [172,173]. Adrenal and thyroid functions undergo less prominent age-related transformations than their hypothalamic regulation [174]; therefore, BA assessment from hormonal findings is challenging. With aging, hormone activity decreases and endocrine alterations are established [175]. BA is affected by the level of glycosylated haemoglobin, glucose, triglycerides, low-density lipoproteins and total cholesterol [176,177,178]. The modulation of these parameters, lifestyle and environmental factors can prevent or contribute to AA [179]. Effectiveness of hormone replacement therapy for aging reversal is questionable though [180].
Environmental and endocrinological factors affect BA of connective tissue. Status of the skeletal system reflects an individual endocrine profile and micronutrient balance [181,182,183] as well as environmental and occupational attainments [184]. For example, bone resorption in astronauts prevails over its formation due to microgravity; however, bone density normalises after the flight [185,186]. Skin elasticity serves as a marker of aging which rate can be modified due to estrogen deficiency, metabolic alterations and exogenous factors (burns) [187,188,189,190,191]. Fibroblasts constitute a natural cell stock that allows skin rejuvenation, repair and decelerated aging [192]. In connective tissue, a combinatory effect of internal and external factors determines BA more accurately than the chronological one [193,194]. Therefore, an inability to account for the decreased aging rate reveals a weakness of the AA concept.
Studies in other systems have also reported reversibility of age-related changes in them. For example, physical training can rejuvenate the respiratory system by expanding the alveolar space. However, studies on these issues did not comprehensively evaluate BA of the lung since the impact of muscle atrophy on results in the spirometry test was not considered [195,196,197]. Lifestyle changes (e.g., calorie restriction and physical activity) could also reverse aging in patients with early stages of chronic kidney disease [198]. Another example is shown by the discovered potential to rejuvenate kidneys with up-regulation of the Klotho gene [199]. These evidences speak for a limited generalisation of the AA concept. AA affects various systems and cross-organ communication. The interaction between systems can impede the atrophy of an organ through compensatory mechanisms in other organs. Several studies have demonstrated the role of the central nervous system in reversing the aging of other systems and organs [200,201,202]. Endocrine and cardiovascular diseases promote renal aging [203]. Conversely, kidney transplantation can revive other parts of the body [204,205]. The characterisation of organ- and system-specific aging processes is challenging and it will require combinatory approaches that are largely missing in the AA concept.
4. Limitations of the AA Concept
The AA concept should be considered within the context of individual capacities and personalised structure–functional reserve mechanisms (Figure 2). A generalized term “structure-functional reserves” is introduced to denote an observed variability in multiple structural and functional parameters at different levels in a population: expression of genetically and epigenetically regulated genes, number, viability and functionality of cells, amount of synapses and intercellular contacts, secretion of cytokines, potency of physiological responses, etc. The term definition could be further developed and linked to statistical distributions of measurable biological parameters in the population and to a norm of reaction.
Physiological reserves reflect the remaining capacity of an organ to perform its function. Aging and diseases lead to atrophy due to a reduction in the number of cells and supracellular structures [206,207]. In the context of brain aging, physiological cognitive reserve reflects the level of education, occupational and environmental attainments and performance in cognitive tests [206]. Reversible forms of mild cognitive impairment (MCI) and dementia represent clinical examples of restoring individual reserve potential. The examples are not aligned with AA theory [208,209]. Neural compensation in the elderly leads to formation of secondary brain networks [210], which decelerate the aging of the brain [206,211]. In elderly patients, reversion of MCI results from specific lifestyle activities and cognitive stimulation throughout life [212,213].
Age assessment requires an accurate estimation of individual reserves that account for biological and chronological age differences. In neuroscience, machine learning models predicted total years lived in good health from brain-imaging data with error of 2.1–4.9 years [214,215]. Individual brain age can also be calculated as a difference between chronological age and the predicted BA [216]. In obstetrics, the evaluation of gynecological status takes into account both reproductive health and potential fertility. Overall BA depends on reserve capacities of individual systems and organs [217,218].
Variance in reserve capacities complicates precise BA assessment. Criteria of AA of the brain are unclear since indicators of normal aging are still missing [219]. Methods for BA assessment are not standardized, they do not take into account individual reserve potential, and reference curves for brain changes have not yet been created. Methodological discrepancies lead to contradictory findings in different studies. For example, AD adds 1.5 years to brain age, MCI adds 1 year, multiple sclerosis—0.41 years, Parkinson’s disease (PD)—3 years and schizophrenia—5.5 years. The last two pathologies impact cognition in a milder and slower way than AD [220,221,222]. Another research on AD revealed the added brain age between 6 and 9 years [223].
Several methodologic limitations demonstrate the need for caution when assessing research findings. For example, studies on age-related brain atrophy commonly have a cross-sectional design that is less accurate compared to the longitudinal one [224]. Many studies are based on small non-representative cohorts [225,226,227]; therefore, applicability of the designed mathematical models is low. Certain studies of brain aging focus on the middle-aged and elderly population and they fail to report individual prenatal pathologies and childhood trauma affecting brain health and BA of study participants [228]. Application of the concept of AA to localised degeneration presents a challenge since different brain parts become older unevenly [229]. For example, in localised ND, BA assessment reflects the level of damage to the most vulnerable brain parts (e.g., substancia nigra and ruber nuclei in PD) [230,231,232]; however, one should also consider the brain resources that can minimise the atrophy effects [233]. In systemic ND, the brain ages faster than in localised ND [234,235], and the difference in the speed of atrophic changes is apparent [236].
ND has polyetiological nature, and contemporary neuroscience lacks a clear explanation of cooperation between different causal factors. It is still unclear whether chronic diseases lead to or result from ND [237,238] since the genetic, environmental and lifestyle factors interact in an undefined way [18,239,240]. Several articles have revealed a misalignment between dementia risk, cognitive performance and MBM levels [241,242]. Another disadvantage of AA studies is an inability to report an impact of medications on study results [243]. Last but not least, AA represents a diagnostic but not pathognomonic signature in ND and in psychiatric diseases such as schizophrenia, bipolar disorder and major depressive disorder [222,244,245]. The entire range of symptoms observed in these patients cannot be explained by brain aging only [222,246,247].
Drug therapy could extend reserves. For instance, antidepressants can help to re- verse MCI [248]. Certain cognitive disorders demonstrated a reversible pattern in cognitive performance upon treatment [248,249]. Sex hormone replacement in ND reduces risk of cognitive impairment, delays symptom onset and slows the progression [250]. Antioxidant-based therapy also alleviates severity of the disease [251,252]. Recent ND studies described a number of novel therapeutic options targeting mitophagy, protein aggregation and cellular senescence. These options include specific antibodies, inducers of cell proliferation and NAD+ supplementations [9,18,253]. The observed treatment effects contradict the idea of irreversibility of changes claimed by the AA concept.
5. Recommendations for Further Development and Improvement of AA Concept
5.1. Statistical Models
Building highly accurate machine learning models is the most common solution for distinguishing AA from normal aging. Recent articles suggested ways to improve the models. These are identification of potential gaps or inconsistencies in study methodology, search for additional data sources, rigorous statistical analysis. Following the suggestions, scientists will reveal the trends or correlations that have been overlooked. Below, we list a set of model parameters that should be controlled:
- o
- Sample size. The number of individuals in the study can affect the statistical power of the analysis, and larger sample sizes generally provide more robust results.
- o
- Biomarker types. The choice of biomarkers can impact the diagnostic model used, as different types of biomarkers may require different statistical analyses.
- o
- Age range. The age range of the study population can influence the types of biomarkers identified, as some biomarkers may be more prevalent in certain age groups.
- o
- Data normalization. Normalization of the data is critical to ensure that data collection or processing differences do not affect the analysis.
- o
- Statistical methods. The choice of statistical methods can impact accuracy of analysis, and different methods may be more appropriate for different data types.
Careful consideration of these parameters will advance diagnostic models based on specific MBMs of AA. Recommendations also include functional characterisation of all types of molecular clocks: organ-tissue-specific, single-cell-specific and disease-specific. Studies can benefit from integration of epigenetics, exploration of additional epigenomic markers of aging, and generation of data in robust non-human aging models.
5.2. Molecular Clocks
Molecular clocks could help to study aging in specific organs and tissues. Organ- and tissue-specific clocks will unravel the complexity of aging in multicellular biological system. Animal studies reported a number of useful techniques that use the mutation rate of biomolecules to deduce the time [254]. These include organ-specific clocks for liver [255,256,257], lungs [255,256], blood [256,258], heart and cortex [255], adipose, kidney, muscle tissues [256], and multiple tissue [259].
5.3. Single-Cell Epigenomics
Analysis of gene expression at a single cell level provides a deep insight into aging [260,261]. For instance, lifetime-dependent cell-to-cell variability in methylation, or so called “epigenomic noise”, occurs in human immune cells in blood and in mouse muscle stem cells [262,263]. Epigenomic noise results in increased transcriptional heterogeneity, especially in stem cell niche genes [262]. A recent trend is construction of epigenetic clocks at a single-cell level by applying novel methods [264,265] and deep-learning computer algorithms [266,267,268].
5.4. New Epigenetic Biomarkers
Search for new epigenetic marks of aging represents another challenge and opens exciting opportunities. Connections between aging and DNA modifications other than methylation are puzzling. Evidently, such connections exist. In the mouse, senescence of hippocampus cells deregulated histone H4 acetylation (H4K12) [269] and accumulated histone variant H2A.Z [270]. In the brain of AD patients, researchers found acetylated histones H3 (H3K9ac) and H4 (H4K16ac) [113,271]. Longevity in mammals is linked to histone acetylation by SIRT6 HDAC, and this discovery unlocks potential for development of pharmacological agents targeting AA [272,273,274].
5.5. Consideration of Ageotypes
Recently, longitudinal, deep multiomic profiling enabled identification of distinct aging phenotypes, termed ‘ageotypes’. These personalised physiological subsets of aging reflect impact of various individual factors on aging rate which depends on genetics, epigenetic changes, lifestyle habits and environmental exposure. Models reflecting age will improve diagnostic accuracy as new information is added [44]. Potential biomarkers of aging and health metrics can be incorporated into the model with ageotypes, and this approach will allow us to monitor the intervention effectiveness in each subset [44,275,276].
5.6. Genetic Predisposition to AA
Human mutations in prototypical progeroid syndromes deepen our knowledge on mechanisms underlying aging. Some autosomal recessive mutations are associated with in- effective genome maintenance systems, deficient DNA helicase activity or aberrant nuclear architecture. Genome instability disorders caused by these mutations are classified into groups three of which include sunlight hypersensitivity disorders: (i) Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy; (ii) ionizing radiation hypersensitivity disorders such as Ataxia telangiectasia, Nijmegen breakage syndrome; (iii) progeroid disorders such as Werner syndrome, Hutchinson–Gilford progeria syndrome, Bloom syndrome, Rothmund–Thompson syndrome and Fanconi anemia [277,278,279]. Studies on the aforelisted disorders can also lead to the discovery of anti-aging treatment.
5.7. Application of Animal Models
Future studies will benefit from emergence of new animal models exhibiting age- related features – accelerated senescence, damage of nuclear envelope, increased accumulation of genomic lesions [280]. Interventions and modulators are commonly tested with well-developed mouse aging models [254]. Mouse models demonstrated epigenetic clock acceleration by a high-fat diet, effects of rapamycin and caloric restriction [255,257]. Killifish(Nothobranchius furzeri) is a vertebrate with the shortest captive lifespan, which makes the species suitable for modeling senescence [281,282,283,284,285,286,287]. The following animals can also mimic aspects of human aging in longevity models: naked mole rats (Heterocephalus glaber, Fukomys mechowii) [288,289,290], Brandt’s bat (Myotis brandtii) [291,292,293], olm (Proteus anguinus) [294,295,296], bivalve (Arctica islandica) [297,298], Hydra (Hydra vulgaris/Hydra mag- nipapillata) [299,300,301,302] and Planaria (Schmidtea mediterranea) [303,304,305]. These animal models can provide robust data on aging.
6. Conclusions
- i.
- The review revealed certain weaknesses and limitations of AA concept. In particular, no unified methodology and terminology has been established in the field. The studies that justify the AA concept have too low sample sizes. Under certain conditions, some changes reverse with age.
- ii.
- Age-related diseases and exhaustion of individual reserves can indicate accelerated senescence. Specific molecular biomarkers reflect aging in individual organs, especially in the brain. Still, validation of a biomarker candidate remains a challenge. Scientists struggle to provide clinical interpretation and apply biomarkers to dis- ease subtyping. Mechanisms of getting older and their exact nature are not well understood.
- iii.
- Activation of regenerative mechanisms is a potential way to decelerate brain aging. Another opportunity for rejuvenation is to restore metabolic homeostasis and energy reserves at a molecular level with novel therapeutic options. For example, neurodegeneration can be delayed with sex hormone replacement, antioxidants, targeted therapy, lifestyle improvement and safe environment. Future longitudinal studies could pro- vide clinics and society with novel therapeutic options on preventing accelerated aging and slowing aging rate.
7. Afterword: Aging Science History and Theories
Several theories have been postulated to explain a possible biological meaning or evolutionary role of aging: evolutionary advantage of species (1890s, Weisman), accumulated mutations (1952, Medawar), antagonistic pleiotropy (1957, Williams), replicative senescence (1965, Hayflick), and the disposable soma theory (1972, Kirkwood) [306]. Theories about what causes aging commonly fall into either of two categories: genetic or stochastic. The genetic, or programmed, group refers to endocrine, immunological and programmed longevity theories. They suggest that aging is predetermined through genetics and that organisms have a built-in clock which dictates life expectancy. Stochastic, or damage, theories propose that random errors and damage accumulate over time and limit longevity. This group includes wear-and-tear, rate of living, cross-linking, free radical and somatic DNA damage hypotheses [307].
Theories of aging can also be classified by biological level. Gene regulation, codon restriction, error catastrophe, somatic mutation and dysdifferentiation theories describe molecular-level processes. Cellular senescence–telomere, free radical, wear-and-tear and apoptosis theories focus on the cellular level. Neuroendocrine, immunologic and rate of living theories conceptualise changes at the system level [308].
In 1920s, Laboratories of the Rockefeller Institute for Medical Research conducted experiments on AA and published findings. The author applied the terms “normal aging” and “aging rate” to effects of light on Drosophila inbred in the dark [309,310]. Since then, the numbers of references on “aging”, “aging rate” and “AA” has reached 614,132; 56,088 and 21,401, respectively [311].
In 1928, a Professor of Neurology of the Columbia University, Frederick Tilney, published the work “The aging of the human brain”, where an AD patient brain was compared to the normally aged one in the diagnostic context of the number of plaques. In this work, he also claimed an abundance of senile plaques in all human brains after the age of 90 years, influence of unfavourable factors and diseases on the brain. Recorded a century ago, his words are worth repeating today: “It is amazing how little general or particular interest man has shown in the most important organ of his body and life. Up to the present time he has devoted relatively little attention and much less capital to the understanding of that part of his machinery which is the secret of his success and the only hope for his future progress, if not his actual salvation. . . The ridiculous stupidity of annually consecrating appalling sums of money to the savage purposes of destruction should in time shock human intelligence out of patronizing such futilities and into wiser realizations. Certainly, one liberally supported and effective brain institute would prove an incomparably more profitable investment for civilization than the most powerful battle fleet that ever sailed the seas.” (Tilney, 1928 [312]).
Author Contributions
Y.S., S.A.A., B.S.E. and M.L. contributed to the conceptual idea of the paper. Y.S., K.N.-V.G. and R.H. formulated the objectives. Y.S., N.V.K., D.M. and K.L. wrote the manuscript. N.V.K., D.M. and K.L. prepared the figures for data presentation. N.V.K., G.L.S., D.S., A.K., S.M. and F.I. contributed to the literature review. All authors contributed to the writing of the original draft article. N.V.K. and B.S.E. performed the review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by ASPIRE, the technology program management pillar of Abu Dhabi’s Advanced Technology Research Council (ATRC), via the ASPIRE Precision Medicine Research Institute Abu Dhabi (ASPIREPMRIAD) award grant number VRI-20-10.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 5-mC | 5-methylcytosine |
| AA | accelerated aging |
| AAG | gene related to accelerated aging |
| AD | Alzheimer’s disease |
| BA | biological age |
| BM | biomarker |
| circRNA | circular RNA |
| HC | healthy control |
| lncRNA | long non-coding RNA |
| MBM | molecular biomarkers |
| MCI | mild cognitive impairment |
| miRNA | microRNA |
| ncRNA | non-coding RNA |
| ND | neurodegeneration |
| NDG | gene that predispose to neurodegeneration |
| NV | neurovascular |
| NVU | neurovascular unit |
| PD | Parkinson’s disease |
References
- Isaev, N.K.; Stelmashook, E.V.; Genrikhs, E.E. Neurogenesis and brain aging. Rev. Neurosci. 2019, 30, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Brivio, P.; Paladini, M.S.; Racagni, G.; Riva, M.A.; Calabrese, F.; Molteni, R. From healthy aging to frailty: In search of the underlying mechanisms. Curr. Med. Chem. 2019, 26, 3685–3701. [Google Scholar] [CrossRef]
- Feltes, B.C.; de Faria Poloni, J.; Bonatto, D. Development and aging: Two opposite but complementary phenomena. Aging Health-A Syst. Biol. Perspect. 2015, 40, 74–84. [Google Scholar]
- Bogeska, R.; Mikecin, A.M.; Kaschutnig, P.; Fawaz, M.; Büchler-Schäff, M.; Le, D.; Ganuza, M.; Vollmer, A.; Paffenholz, S.V.; Asada, N.; et al. Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell 2022, 29, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Adelman, E.R.; Figueroa, M.E. Human hematopoiesis: Aging and leukemogenic risk. Curr. Opin. Hematol. 2021, 28, 57. [Google Scholar] [CrossRef]
- Hooten, N.N.; Pacheco, N.L.; Smith, J.T.; Evans, M.K. The accelerated aging phenotype: The role of race and social determinants of health on aging. Ageing Res. Rev. 2022, 73, 101536. [Google Scholar] [CrossRef]
- Forrester, S.N.; Zmora, R.; Schreiner, P.J.; Jacobs, D.R., Jr.; Roger, V.L.; Thorpe, R.J., Jr.; Kiefe, C.I. Accelerated aging: A marker for social factors resulting in cardiovascular events? SSM-Popul. Health 2021, 13, 100733. [Google Scholar] [CrossRef] [PubMed]
- Hamczyk, M.R.; Nevado, R.M.; Barettino, A.; Fuster, V.; Andres, V. Biological versus chronological aging: Jacc focus seminar. J. Am. Coll. Cardiol. 2020, 75, 919–930. [Google Scholar] [CrossRef]
- Vaquer-Alicea, J.; Diamond, M.I. Propagation of protein aggregation in neurodegenerative diseases. Annu. Rev. Biochem. 2019, 88, 785–810. [Google Scholar] [CrossRef]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the extra (synaptic) mile: Excitotoxicity as the road toward neurodegenerative diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef]
- Margolick, J.B.; Ferrucci, L. Accelerating aging research: How can we measure the rate of biologic aging? Exp. Gerontol. 2015, 64, 78–80. [Google Scholar] [CrossRef]
- Melzer, D.; Pilling, L.C.; Ferrucci, L. The genetics of human ageing. Nat. Rev. Genet. 2020, 21, 88–101. [Google Scholar] [CrossRef]
- Miller, M.W.; Sadeh, N. Traumatic stress, oxidative stress and post-traumatic stress disorder: Neurodegeneration and the accelerated-aging hypothesis. Mol. Psychiatry 2014, 19, 1156–1162. [Google Scholar] [CrossRef]
- Ghosh, C.; De, A. Basics of aging theories and disease related aging-an overview. PharmaTutor 2017, 5, 16–23. [Google Scholar]
- Wadhwa, R.; Gupta, R.; Maurya, P.K. Oxidative stress and accelerated aging in neurodegenerative and neuropsychiatric disorder. Curr. Pharm. Des. 2018, 24, 4711–4725. [Google Scholar] [CrossRef]
- Bersani, F.S.; Mellon, S.H.; Reus, V.I.; Wolkowitz, O.M. Accelerated aging in serious mental disorders. Curr. Opin. Psychiatry 2019, 32, 381. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Wang, X.; Ma, Z.; Cheng, J.; Lv, Z. A genetic program theory of aging using an RNA population model. Ageing Res. Rev. 2014, 13, 46–54. [Google Scholar] [CrossRef]
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. Handb. Clin. Neurol. 2018, 145, 301–307. [Google Scholar]
- Sanz, A.; Stefanatos, R.K. The mitochondrial free radical theory of aging: A critical view. Curr. Aging Sci. 2008, 1, 10–21. [Google Scholar] [CrossRef]
- Libertini, G.; Shubernetskaya, O.; Corbi, G.; Ferrara, N. Is evidence supporting the subtelomere–telomere theory of aging? Biochemistry 2021, 86, 1526–1539. [Google Scholar] [CrossRef]
- Xie, L.; Wu, S.; He, R.; Li, S.; Lai, X.; Wang, Z. Identification of epigenetic dysregulation gene markers and immune landscape in kidney renal clear cell carcinoma by comprehensive genomic analysis. Front. Immunol. 2022, 13, 901662. [Google Scholar] [CrossRef]
- Ru˚ žicˇka, M.; Kulhánek, P.; Radová, L.; Cˇ echová, A.; Špacˇková, N.; Fajkusová, L.; Réblová, K. Dna mutation motifs in the genes associated with inherited diseases. PLoS ONE 2017, 12, e0182377. [Google Scholar]
- Korb, M.K.; Kimonis, V.E.; Mozaffar, T. Multisystem proteinopathy: Where myopathy and motor neuron disease converge. Muscle Nerve 2021, 63, 442–454. [Google Scholar] [CrossRef]
- Barja, G. The mitochondrial free radical theory of aging. Prog. Mol. Biol. Transl. Sci. 2014, 127, 1–27. [Google Scholar]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef]
- Esmaeili, Y.; Yarjanli, Z.; Pakniya, F.; Bidram, E.; Łos, M.J.; Eshraghi, M.; Klionsky, D.J.; Ghavami, S.; Zarrabi, A. Targeting autophagy, oxidative stress, and er stress for neurodegenerative diseases treatment. J. Control. Release 2022, 345, 147–175. [Google Scholar] [CrossRef]
- Pomatto, L.C.; Davies, K.J. Adaptive homeostasis and the free radical theory of ageing. Free Radic. Biol. Med. 2018, 124, 420–430. [Google Scholar] [CrossRef]
- Simpson, D.J.; Chandra, T. Epigenetic age prediction. Aging Cell 2021, 20, e13452. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef]
- Campisi, J. Cancer, aging and cellular senescence. In Vivo 2000, 14, 183–188. [Google Scholar] [PubMed]
- Zlokovic, B.V. New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurotherapeutics 2008, 5, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; De Silva, T.M.; Chen, J.; Faraci, F.M. Cerebral vascular disease and neurovascular injury in ischemic stroke. Circ. Res. 2017, 120, 449–471. [Google Scholar] [CrossRef]
- Lähteenvuo, J.; Rosenzweig, A. Effects of aging on angiogenesis. Circ. Res. 2012, 110, 1252–1264. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 887–900. [Google Scholar] [CrossRef]
- Wilhelm, I.; Nyúl-Tóth, Á.; Kozma, M.; Farkas, A.E.; Krizbai, I.A. Role of pattern recognition receptors of the neurovascular unit in inflamm-aging. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1000–H1012. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Wang, D.Q.; Tan, E.K. The role of neurovascular unit in neurodegeneration. Front. Cell. Neurosci. 2022, 16, 870631. [Google Scholar] [CrossRef]
- Spitzer, D.; Guérit, S.; Puetz, T.; Khel, M.I.; Armbrust, M.; Dunst, M.; Macas, J.; Zinke, J.; Devraj, G.; Jia, X.; et al. Profiling the neurovascular unit unveils detrimental effects of osteopontin on the blood–brain barrier in acute ischemic stroke. Acta Neuropathol. 2022, 144, 305–337. [Google Scholar] [CrossRef]
- Jeong, H.W.; Diéguez-Hurtado, R.; Arf, H.; Song, J.; Park, H.; Kruse, K.; Sorokin, L.; Adams, R.H. Single-cell transcriptomics reveals functionally specialized vascular endothelium in brain. eLife 2022, 11, e57520. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, Y.Z.; Liu, Y.S. Accelerated aging-related transcriptome alterations in neurovascular unit cells in the brain of Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 949074. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Chen, W.; McDermott, J.; Han, J.D.J. Molecular and phenotypic biomarkers of aging. F1000Research 2017, 6, 860. [Google Scholar] [CrossRef] [PubMed]
- Ahadi, S.; Zhou, W.; Schüssler-Fiorenza Rose, S.M.; Sailani, M.R.; Contrepois, K.; Avina, M.; Ashland, M.; Brunet, A.; Snyder, M. Personal aging markers and ageotypes revealed by deep longitudinal profiling. Nat. Med. 2020, 26, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; von Figura, G.; Liu, Y.; Kraus, J.M.; Torrice, C.; Dillon, P.; Rudolph-Watabe, M.; Ju, Z.; Kestler, H.A.; Sanoff, H.; et al. Lifestyle impacts on the aging-associated expression of biomarkers of dna damage and telomere dysfunction in human blood. Aging Cell 2010, 9, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on dna methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, 1–18. [Google Scholar] [CrossRef]
- Day, K.; Waite, L.L.; Thalacker-Mercer, A.; West, A.; Bamman, M.M.; Brooks, J.D.; Myers, R.M.; Absher, D. Differential dna methylation with age displays both common and dynamic features across human tissues that are influenced by cpg landscape. Genome Biol. 2013, 14, 1–19. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Greer, E.L.; Maures, T.J.; Hauswirth, A.G.; Green, E.M.; Leeman, D.S.; Maro, G.S.; Han, S.; Banko, M.R.; Gozani, O.; Brunet, A. Members of the h3k4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 2010, 466, 383–387. [Google Scholar] [CrossRef]
- Li, L.; Greer, C.; Eisenman, R.N.; Secombe, J. Essential functions of the histone demethylase lid. PLoS Genet. 2010, 6, e1001221. [Google Scholar] [CrossRef]
- Djeghloul, D.; Kuranda, K.; Kuzniak, I.; Barbieri, D.; Naguibneva, I.; Choisy, C.; Bories, J.C.; Dosquet, C.; Pla, M.; Vanneaux, V.; et al. Age-associated decrease of the histone methyltransferase suv39h1 in hsc perturbs heterochromatin and b lymphoid differentiation. Stem Cell Rep. 2016, 6, 970–984. [Google Scholar] [CrossRef]
- Li, C.L.; Pu, M.; Wang, W.; Chaturbedi, A.; Emerson, F.J.; Lee, S.S. Region-specific h3k9me3 gain in aged somatic tissues in caenorhabditis elegans. PLoS Genet. 2021, 17, e1009432. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, E.W.; Croteau, D.L.; Bohr, V.A. Heterochromatin: An epigenetic point of view in aging. Exp. Mol. Med. 2020, 52, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone h3 lysine 27 methylation in polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Siebold, A.P.; Banerjee, R.; Tie, F.; Kiss, D.L.; Moskowitz, J.; Harte, P.J. Polycomb repressive complex 2 and trithorax modulate Drosophila longevity and stress resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 169–174. [Google Scholar] [CrossRef]
- Ni, Z.; Ebata, A.; Alipanahiramandi, E.; Lee, S.S. Two set domain containing genes link epigenetic changes and aging in caenorhabditis elegans. Aging Cell 2012, 11, 315–325. [Google Scholar] [CrossRef]
- Maures, T.J.; Greer, E.L.; Hauswirth, A.G.; Brunet, A. The h3k27 demethylase utx-1 regulates C. elegans lifespan in a germline- independent, insulin-dependent manner. Aging Cell 2011, 10, 980–990. [Google Scholar] [CrossRef]
- Liu, L.; Cheung, T.H.; Charville, G.W.; Hurgo, B.M.; Leavitt, T.; Shih, J.; Brunet, A.; Rando, T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013, 4, 189–204. [Google Scholar] [CrossRef]
- Baumgart, M.; Groth, M.; Priebe, S.; Savino, A.; Testa, G.; Dix, A.; Ripa, R.; Spallotta, F.; Gaetano, C.; Ori, M.; et al. Rna-seq of the aging brain in the short-lived fish N. furzeri–conserved pathways and novel genes associated with neurogenesis. Aging Cell 2014, 13, 965–974. [Google Scholar] [CrossRef]
- Peters, M.J.; Joehanes, R.; Pilling, L.C.; Schurmann, C.; Conneely, K.N.; Powell, J.; Reinmaa, E.; Sutphin, G.L.; Zhernakova, A.; Schramm, K.; et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Li, X.; Khanna, A.; Li, N.; Wang, E. Circulatory mir-34a as an RNA-based, noninvasive biomarker for brain aging. Aging 2011, 3, 985. [Google Scholar] [CrossRef]
- Dhahbi, J.M. Circulating small noncoding rnas as biomarkers of aging. Ageing Res. Rev. 2014, 17, 86–98. [Google Scholar] [CrossRef]
- Grammatikakis, I.; Panda, A.C.; Abdelmohsen, K.; Gorospe, M. Long noncoding rnas (lncrnas) and the molecular hallmarks of aging. Aging 2014, 6, 992. [Google Scholar] [CrossRef]
- Kour, S.; Rath, P.C. Long noncoding rnas in aging and age-related diseases. Ageing Res. Rev. 2016, 26, 1–21. [Google Scholar] [CrossRef]
- Finkel, D.; Pedersen, N.L.; Plomin, R.; McClearn, G.E. Longitudinal and cross-sectional twin data on cognitive abilities in adulthood: The swedish adoption/twin study of aging. Dev. Psychol. 1998, 34, 1400. [Google Scholar] [CrossRef]
- Reynolds, C.A.; Finkel, D. A meta-analysis of heritability of cognitive aging: Minding the “missing heritability” gap. Neuropsychol. Rev. 2015, 25, 97–112. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Pletnikova, O.; Geiger, J.T.; Murphy, N.A.; Abramzon, Y.; Rudow, G.; Mamais, A.; Sabir, M.S.; Crain, B.; Ahmed, S.; et al. Genetic analysis of neurodegenerative diseases in a pathology cohort. Neurobiol. Aging 2019, 76, 214–e1. [Google Scholar] [CrossRef]
- Cochran, J.N.; Geier, E.G.; Bonham, L.W.; Newberry, J.S.; Amaral, M.D.; Thompson, M.L.; Lasseigne, B.N.; Karydas, A.M.; Roberson, E.D.; Cooper, G.M.; et al. Non-coding and loss-of-function coding variants in tet2 are associated with multiple neurodegenerative diseases. Am. J. Hum. Genet. 2020, 106, 632–645. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.J.; Kim, M.J.; Kim, J.; Kim, Y.J.; You, S.; Koh, J.; Kim, S.Y.; Lee, J.H. Exome array study did not identify novel variants in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1958.e13–1958.e14. [Google Scholar] [CrossRef] [PubMed]
- Nikolac Perkovic, M.; Pivac, N. Genetic Markers of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1192, 27–52. [Google Scholar]
- Song, W.; Hooli, B.; Mullin, K.; Jin, S.C.; Cella, M.; Ulland, T.K.; Wang, Y.; Tanzi, R.E.; Colonna, M. Alzheimer’s disease-associated trem2 variants exhibit either decreased or in-creased ligand-dependent activation. Alzheimer’s Dement. 2017, 13, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Dols-Icardo, O.; Bullido, M.J.; Pastor, P.; Rodríguez-Rodríguez, E.; López de Munain, A.; de Pancorbo, M.M.; Pérez-Tur, J.; Alvarez, V.; Antonell, A.; et al. Assessing the role of the trem2 p. r47h variant as a risk factor for Alzheimer’s disease and frontotemporal dementia. Neurobiol. Aging 2014, 35, 444.e1–444.e4. [Google Scholar] [CrossRef] [PubMed]
- Mehrjoo, Z.; Najmabadi, A.; Abedini, S.S.; Mohseni, M.; Kamali, K.; Najmabadi, H.; Khorram Khorshid, H.R. Association study of the trem2 gene and identification of a novel variant in exon 2 in iranian patients with late-onset Alzheimer’s disease. Med. Princ. Pract. 2015, 24, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. Trem2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of trem2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tan, L.; Chen, Q.; Tan, M.S.; Zhou, J.S.; Zhu, X.C.; Lu, H.; Wang, H.F.; Zhang, Y.D.; Yu, J.T. A rare coding variant in trem2 increases risk for Alzheimer’s disease in han chinese. Neurobiol. Aging 2016, 42, 217–e1. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Carrasquillo, M.M.; Benitez, B.A.; Skorupa, T.; Carrell, D.; Patel, D.; Lincoln, S.; Krishnan, S.; Kachadoorian, M.; Reitz, C.; et al. Trem2 is associated with increased risk for Alzheimer’s disease in african amer-icans. Mol. Neurodegener. 2015, 10, 1–7. [Google Scholar] [CrossRef]
- Berge, G.; Sando, S.B.; Rongve, A.; Aarsland, D.; White, L.R. Onset of dementia with lewy bodies is delayed for carriers of the apolipoprotein e ε2 genotype in a norwegian cohort. Mov. Disord. 2014, 29, S220. [Google Scholar]
- Calvo, A.; Chiò, A. Sclerosi laterale amiotrofica come modello di gestione interdisciplinare. SALUTE E SOCIETÀ 2015, 3, 173–184. [Google Scholar] [CrossRef]
- Borroni, B.; Ferrari, F.; Galimberti, D.; Nacmias, B.; Barone, C.; Bagnoli, S.; Fenoglio, C.; Piaceri, I.; Archetti, S.; Bonvicini, C.; et al. Heterozygous trem2 mutations in frontotemporal dementia. Neurobiol. Aging 2014, 35, 934.e7–934.e10. [Google Scholar] [CrossRef]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. Trem2 in neurodegeneration: Evidence for association of the p. r47h variant with frontotemporal dementia and parkinson’s disease. Mol. Neurodegener. 2013, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Cady, J.; Koval, E.D.; Benitez, B.A.; Zaidman, C.; Jockel-Balsarotti, J.; Allred, P.; Baloh, R.H.; Ravits, J.; Simpson, E.; Appel, S.H.; et al. Trem2 variant p. r47h as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014, 71, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Slattery, C.F.; Beck, J.A.; Harper, L.; Adamson, G.; Abdi, Z.; Uphill, J.; Campbell, T.; Druyeh, R.; Mahoney, C.J.; Rohrer, J.D.; et al. R47h trem2 variant increases risk of typical early-onset Alzheimer’s disease but not of prion or frontotemporal dementia. Alzheimer’s Dement. 2014, 10, 602–608. [Google Scholar] [CrossRef]
- Gonzalez Murcia, J.D.; Schmutz, C.; Munger, C.; Perkes, A.; Gustin, A.; Peterson, M.; Ebbert, M.T.; Norton, M.C.; Tschanz, J.T.; Munger, R.G.; et al. Assessment of trem2 rs75932628 association with Alzheimer’s disease in a population-based sample: The cache county study. Neurobiol. Aging 2013, 34, 2889. [Google Scholar] [CrossRef] [PubMed]
- Walton, R.L.; Soto-Ortolaza, A.I.; Murray, M.E.; Lorenzo-Betancor, O.; Ogaki, K.; Heckman, M.G.; Rayaprolu, S.; Rademakers, R.; Ertekin-Taner, N.; Uitti, R.J.; et al. Trem2 p. r47h substitution is not associated with dementia with lewy bodies. Neurol. Genet. 2016, 2, e85. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhu, Z.; Chen, K.; Wei, D.; Li, X.; Li, H.; Zhang, J.; Chen, X.; Chen, Y.; Zhang, Z. Apoe ε4 allele accelerates age-related multi-cognitive decline and white matter damage in non-demented elderly. Aging 2020, 12, 12019. [Google Scholar] [CrossRef]
- Goel, N.; Karir, P.; Garg, V.K. Role of DNA methylation in human age prediction. Mech. Ageing Dev. 2017, 166, 33–41. [Google Scholar] [CrossRef]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 1–8. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Zampieri, M.; Ciccarone, F.; Calabrese, R.; Franceschi, C.; Bürkle, A.; Caiafa, P. Reconfiguration of DNA methylation in aging. Mech. Ageing Dev. 2015, 151, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Ryan, C.P. “Epigenetic clocks”: Theory and applications in human biology. Am. J. Hum. Biol. 2021, 33, e23488. [Google Scholar] [CrossRef] [PubMed]
- Martino, D.; Loke, Y.J.; Gordon, L.; Ollikainen, M.; Cruickshank, M.N.; Saffery, R.; Craig, J.M. Longitudinal, genome-scale analysis of dna methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013, 14, R42. [Google Scholar] [CrossRef]
- Bjornsson, H.T.; Sigurdsson, M.I.; Fallin, M.D.; Irizarry, R.A.; Aspelund, T.; Cui, H.; Yu, W.; Rongione, M.A.; Ekström, T.J.; Harris, T.B.; et al. Intra-individual change over time in dna methylation with familial clustering. JAMA 2008, 299, 2877–2883. [Google Scholar] [CrossRef] [PubMed]
- Wynford-Thomas, D. Telomeres, p53 and cellular senescence. Oncol Res. 1996, 8, 387–398. [Google Scholar]
- von Zglinicki, T. Telomeres: Influencing the rate of aging. Ann. N. Y. Acad. Sci. 1998, 854, 318–327. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; West, J.; Beck, S. Age-associated epigenetic drift: Implications, and a case of epigenetic thrift? Hum. Mol. Genet. 2013, 22, R7–R15. [Google Scholar] [CrossRef]
- Horvath, S. Dna methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Ritchie, S.J.; Muniz-Terrera, G.; Harris, S.E.; Gibson, J.; Redmond, P.; Cox, S.R.; Pattie, A.; et al. The epigenetic clock is correlated with physical and cognitive fitness in the lothian birth cohort 1936. Int. J. Epidemiol. 2015, 44, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Bennett, D.A.; Horvath, S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging 2015, 7, 1198. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Langfelder, P.; Kwak, S.; Aaronson, J.; Rosinski, J.; Vogt, T.F.; Eszes, M.; Faull, R.L.; Curtis, M.A.; Waldvogel, H.J.; et al. Huntington’s disease accelerates epigenetic aging of human brain and disrupts dna methylation levels. Aging 2016, 8, 1485. [Google Scholar] [CrossRef]
- Grodstein, F.; Lemos, B.; Yu, L.; Klein, H.U.; Iatrou, A.; Buchman, A.S.; Shireby, G.L.; Mill, J.; Schneider, J.A.; De Jager, P.L.; et al. The association of epigenetic clocks in brain tissue with brain pathologies and common aging phenotypes. Neurobiol. Dis. 2021, 157, 105428. [Google Scholar] [CrossRef]
- Grodstein, F.; Lemos, B.; Yu, L.; Iatrou, A.; De Jager, P.L.; Bennett, D.A. Characteristics of epigenetic clocks across blood and brain tissue in older women and men. Front. Neurosci. 2021, 14, 555307. [Google Scholar] [CrossRef]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet. 2007, 23, 413–418. [Google Scholar] [CrossRef]
- Han, S.; Brunet, A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012, 22, 42–49. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Gjoneska, E.; Pfenning, A.R.; Mathys, H.; Quon, G.; Kundaje, A.; Tsai, L.H.; Kellis, M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 2015, 518, 365–369. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, W.; Williams, J.B.; Yang, F.; Wang, Z.J.; Yan, Z. Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer’s disease. Sci. Adv. 2020, 6, eabc8096. [Google Scholar] [CrossRef] [PubMed]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Santana, D.A.; Smith MD, A.C.; Chen, E.S. Histone modifications in Alzheimer’s disease. Genes 2023, 14, 347. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Dean, B.; Thomas, E. Disease-and age-related changes in histone acetylation at gene promoters in psychiatric disorders. Transl. Psychiatry 2011, 1, e64. [Google Scholar] [CrossRef]
- Chaput, D.; Kirouac, L.; Stevens, S.M., Jr.; Padmanabhan, J. Potential role of PCTAIRE-2, PCTAIRE-3 and P-Histone H4 in amyloid precursor protein-dependent Alzheimer pathology. Oncotarget 2016, 7, 8481. [Google Scholar] [CrossRef]
- Ogawa, O.; Zhu, X.; Lee, H.G.; Raina, A.; Obrenovich, M.E.; Bowser, R.; Smith, M.A. Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: A mitotic catastrophe? Acta Neuropathol. 2003, 105, 524–528. [Google Scholar] [CrossRef]
- D’haene, E.; Vergult, S. Interpreting the impact of noncoding structural variation in neurodevelopmental disorders. Genet. Med. 2021, 23, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Sherazi, S.A.M.; Abbasi, A.; Jamil, A.; Uzair, M.; Ikram, A.; Qamar, S.; Olamide, A.A.; Arshad, M.; Fried, P.J.; Ljubisavljevic, M.; et al. Molecular hallmarks of long non-coding RNAs in aging and its significant effect on aging-associated diseases. Neural Regen. Res. 2023, 18, 959–968. [Google Scholar]
- Wang, D.Q.; Fu, P.; Yao, C.; Zhu, L.S.; Hou, T.Y.; Chen, J.G.; Lu, Y.; Liu, D.; Zhu, L.Q. Long non-coding RNAs, novel culprits, or bodyguards in neurodegenerative diseas-es. Mol. Ther. Nucleic Acids 2018, 10, 269–276. [Google Scholar] [CrossRef]
- Mishra, P.; Kumar, S. Association of lncRNA with regulatory molecular factors in brain and their role in the pathophysiology of schizophrenia. Metab. Brain Dis. 2021, 36, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.M.; Cicala, C.; Charlesworth, A.; Ciccarelli, C.; Rossi, G.B.; Philipson, L.; Sorrentino, V. Regulation and expression of a growth arrest-specific gene (gas5) during growth, differentiation, and development. Mol. Cell. Biol. 1992, 12, 3514–3521. [Google Scholar] [PubMed]
- Pickard, M.; Mourtada-Maarabouni, M.; Williams, G. Long non-coding RNA gas5 regulates apoptosis in prostate cancer cell lines. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Mourtada-Maarabouni, M.; Pickard, M.; Hedge, V.; Farzaneh, F.; Williams, G. Gas5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 2009, 28, 195–208. [Google Scholar] [CrossRef]
- Tang, S.; Buchman, A.S.; De Jager, P.L.; Bennett, D.A.; Epstein, M.P.; Yang, J. Novel variance-component TWAS method for studying complex human diseases with applications to Alzheimer’s dementia. PLoS Genet. 2021, 17, e1009482. [Google Scholar] [CrossRef]
- Liang, W.S.; Dunckley, T.; Beach, T.G.; Grover, A.; Mastroeni, D.; Walker, D.G.; Caselli, R.J.; Kukull, W.A.; McKeel, D.; Morris, J.C.; et al. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol. Genom. 2007, 28, 311–322. [Google Scholar] [CrossRef]
- Mus, E.; Hof, P.R.; Tiedge, H. Dendritic bc200 RNA in aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 10679–10684. [Google Scholar] [CrossRef]
- Maoz, R.; Garfinkel, B.P.; Soreq, H. Alzheimer’s disease and ncRNAs. Neuroepigenomics Aging Dis. 2017, 978, 337–361. [Google Scholar]
- Fiore, R.; Khudayberdiev, S.; Saba, R.; Schratt, G. Micro-RNA function in the nervous system. Prog. Mol. Biol. Transl. Sci. 2011, 102, 47–100. [Google Scholar]
- Goodall, E.F.; Heath, P.R.; Bandmann, O.; Kirby, J.; Shaw, P.J. Neuronal dark matter: The emerging role of microRNAs in neurodegeneration. Front. Cell. Neurosci. 2013, 7, 178. [Google Scholar] [CrossRef]
- Dickson, J.R.; Kruse, C.; Montagna, D.R.; Finsen, B.; Wolfe, M.S. Alternative polyadenylation and mir-34 family members regulate tau expression. J. Neurochem. 2013, 127, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; Dorval, V.; Parsi, S.; Morin, F.; Planel, E.; et al. Mir-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 2015, 24, 6721–6735. [Google Scholar] [CrossRef]
- Santa-Maria, I.; Alaniz, M.E.; Renwick, N.; Cela, C.; Fulga, T.A.; Van Vactor, D.; Tuschl, T.; Clark, L.N.; Shelanski, M.L.; McCabe, B.D.; et al. Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J. Clin. Investig. 2015, 125, 681–686. [Google Scholar] [CrossRef]
- Hébert, S.S.; Papadopoulou, A.S.; Smith, P.; Galas, M.C.; Planel, E.; Silahtaroglu, A.N.; Sergeant, N.; Buée, L.; De Strooper, B. Genetic ablation of dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 2010, 19, 3959–3969. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, Y.; Zhao, B. Roles of glycogen synthase kinase 3 in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 864–879. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, J.S.; Lopez, M.A.; Boriek, A.M. Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3β. J. Biol. Chem. 2010, 285, 29336–29347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.B.; Wu, L.; Xiong, R.; Wang, L.L.; Zhang, B.; Wang, C.; Li, H.; Liang, L.; Chen, S.D. MicroRNA-922 promotes tau phos- phorylation by downregulating ubiquitin carboxy-terminal hydrolase l1 (uchl1) expression in the pathogenesis of Alzheimer’s disease. Neuroscience 2014, 275, 232–237. [Google Scholar] [CrossRef]
- Law, P.T.; Ching, A.K.; Chan, A.W.; Wong, Q.W.; Wong, C.K.; To, K.F.; Wong, N. Mir-145 modulates multiple components of the insulin-like growth factor pathway in hepatocellular carcinoma. Carcinogenesis 2012, 33, 1134–1141. [Google Scholar] [CrossRef]
- El Ouaamari, A.; Baroukh, N.; Martens, G.A.; Lebrun, P.; Pipeleers, D.; Van Obberghen, E. mir-375 targets 3-phosphoinositide– dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic β-cells. Diabetes 2008, 57, 2708–2717. [Google Scholar] [CrossRef]
- Inukai, S.; de Lencastre, A.; Turner, M.; Slack, F. Novel microRNAs differentially expressed during aging in the mouse brain. PLoS ONE 2012, 7, e40028. [Google Scholar] [CrossRef]
- Yang, G.; Song, Y.; Zhou, X.; Deng, Y.; Liu, T.; Weng, G.; Yu, D.; Pan, S. MicroRNA-29c targets β-site amyloid precursor protein-cleaving enzyme 1 and has a neuroprotective role in vitro and in vivo. Mol. Med. Rep. 2015, 12, 3081–3088. [Google Scholar] [CrossRef]
- Lei, X.; Lei, L.; Zhang, Z.; Zhang, Z.; Cheng, Y. Downregulated mir-29c correlates with increased bace1 expression in sporadic Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 1565. [Google Scholar] [PubMed]
- Zong, Y.; Wang, H.; Dong, W.; Quan, X.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Qin, C. mir-29c regulates bace1 protein expression. Brain Res. 2011, 1395, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; De Strooper, B. MicroRNA regulation of Alzheimer’s amyloid precursor protein expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting bace1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA mir-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of β-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Kennerdell, J.R.; Liu, N.; Bonini, N.M. Mir-34 inhibits polycomb repressive complex 2 to modulate chaperone expression and promote healthy brain aging. Nat. Commun. 2018, 9, 4188. [Google Scholar] [CrossRef]
- Cheng, C.; Li, W.; Zhang, Z.; Yoshimura, S.; Hao, Q.; Zhang, C.; Wang, Z. MicroRNA-144 is regulated by activator protein-1 (ap-1) and decreases expression of Alzheimer disease-related a disintegrin and metalloprotease 10 (adam10). J. Biol. Chem. 2013, 288, 13748–13761. [Google Scholar] [CrossRef]
- Zhang, Y.; Kim, M.S.; Jia, B.; Yan, J.; Zuniga-Hertz, J.P.; Han, C.; Cai, D. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 2017, 548, 52–57. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic programming of systemic ageing involving ikk-β, nf-κb and gnrh. Nature 2013, 497, 211–216. [Google Scholar] [CrossRef]
- Mohammed, C.P.D.; Park, J.S.; Nam, H.G.; Kim, K. MicroRNAs in brain aging. Mech. Ageing Dev. 2017, 168, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Panda, A.C.; De, S.; Grammatikakis, I.; Kim, J.; Ding, J.; Noh, J.H.; Kim, K.M.; Mattison, J.A.; de Cabo, R.; et al. Circular RNAs in monkey muscle: Age-dependent changes. Aging 2015, 7, 903. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynami-cally expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Lukiw, W.J. Circular RNA (circRNA) in Alzheimer’s disease (ad). Front. Genet. 2013, 4, 307. [Google Scholar] [CrossRef]
- Bao, N.; Liu, J.; Peng, Z.; Zhang, R.; Ni, R.; Li, R.; Wu, J.; Liu, Z.; Pan, B. Identification of circRNA-miRNA-mRNA networks to explore the molecular mechanism and immune regulation of postoperative neurocognitive disorder. Aging 2022, 14, 8374. [Google Scholar] [CrossRef]
- Mahmoudi, E.; Fitzsimmons, C.; Geaghan, M.P.; Shannon Weickert, C.; Atkins, J.R.; Wang, X.; Cairns, M.J. Circular RNA biogenesis is decreased in postmortem cortical gray matter in schizophrenia and may alter the bioavailability of associated miRNA. Neuropsychopharmacology 2019, 44, 1043–1054. [Google Scholar] [CrossRef]
- Broekmans, F.; Soules, M.; Fauser, B. Ovarian aging: Mechanisms and clinical consequences. Endocr. Rev. 2009, 30, 465–493. [Google Scholar] [CrossRef]
- Frungieri, M.B.; Calandra, R.S.; Bartke, A.; Matzkin, M.E. Male and female gonadal ageing: Its impact on health span and life span. Mech. Ageing Dev. 2021, 197, 111519. [Google Scholar] [CrossRef]
- De la Rochebrochard, E.; Thonneau, P. Paternal age 40 years: An important risk factor for infertility. Am. J. Obstet. Gynecol. 2003, 189, 901–905. [Google Scholar] [CrossRef]
- Brahem, S.; Mehdi, M.; Elghezal, H.; Saad, A. The effects of male aging on semen quality, sperm dna fragmentation and chromosomal abnormalities in an infertile population. J. Assist. Reprod. Genet. 2011, 28, 425–432. [Google Scholar] [CrossRef]
- Paul, C.; Robaire, B. Ageing of the male germ line. Nat. Rev. Urol. 2013, 10, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Frungieri, M.B.; Calandra, R.S.; Bartke, A.; Matzkin, M.E. Ageing and inflammation in the male reproductive tract. Andrologia 2018, 50, e13034. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ni, S.; Li, C.; Song, L.; Zhang, S. Gonadal rejuvenation of mice by growth differentiation factor 11. J. Gerontol. Ser. A 2022, 77, 892–901. [Google Scholar] [CrossRef]
- Mularoni, V.; Esposito, V.; Di Persio, S.; Vicini, E.; Spadetta, G.; Berloco, P.; Fanelli, F.; Mezzullo, M.; Pagotto, U.; Pelusi, C.; et al. Age-related changes in human Leydig cell status. Hum. Reprod. 2020, 35, 2663–2676. [Google Scholar] [CrossRef]
- Mahmoud, A.; Goemaere, S.; El-Garem, Y.; Van Pottelbergh, I.; Comhaire, F.; Kaufman, J. Testicular volume in relation to hormonal indices of gonadal function in community-dwelling elderly men. J. Clin. Endocrinol. Metab. 2003, 88, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Golan, R.; Scovell, J.M.; Ramasamy, R. Age-related testosterone decline is due to waning of both testicular and hypothalamic- pituitary function. Aging Male 2015, 18, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, D.J.; Staraj, S. Testicular size: The effects of aging, malnutrition, and illness. J. Androl. 1985, 6, 144–151. [Google Scholar] [CrossRef]
- Ilacqua, A.; Izzo, G.; Emerenziani, G.P.; Baldari, C.; Aversa, A. Lifestyle and fertility: The influence of stress and quality of life on male fertility. Reprod. Biol. Endocrinol. 2018, 16, 1–11. [Google Scholar] [CrossRef]
- Kaufman, J. Ageing of the hypothalamo-pituitary-testicular axis in men. Horm. Res. Paediatr. 1995, 43, 25–28. [Google Scholar]
- Harman, S.M.; Metter, E.J.; Tobin, J.D.; Pearson, J.; Blackman, M.R. Longitudinal effects of aging on serum total and free testosterone levels in healthy men. J. Clin. Endocrinol. Metab. 2001, 86, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, R.; Parrillo, L.; Longo, M.; Florese, P.; Desiderio, A.; Zatterale, F.; Miele, C.; Raciti, G.A.; Beguinot, F. Molecular basis of ageing in chronic metabolic diseases. J. Endocrinol. Investig. 2020, 43, 1373–1389. [Google Scholar] [CrossRef] [PubMed]
- Emami, M.; Agbaedeng, T.A.; Thomas, G.; Middeldorp, M.E.; Thiyagarajah, A.; Wong, C.X.; Elliott, A.D.; Gallagher, C.; Hendriks, J.M.L.; Lau, D.H.; et al. Accelerated biological aging secondary to cardiometabolic risk factors is a predictor of cardiovascular mortality: A systematic review and meta-analysis. Can. J. Cardiol. 2022, 38, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Kahn, C.R. Endocrine regulation of ageing. Nat. Rev. Mol. Cell Biol. 2007, 8, 681–691. [Google Scholar] [CrossRef]
- Van den Beld, A.W.; Kaufman, J.M.; Zillikens, M.C.; Lamberts, S.W.; Egan, J.M.; van der Lely, A.J. The physiology of endocrine systems with ageing. Lancet Diabetes Endocrinol. 2018, 6, 647–658. [Google Scholar] [CrossRef]
- Park, J.; Cho, B.; Kwon, H.; Lee, C. Developing a biological age assessment equation using principal component analysis and clinical biomarkers of aging in korean men. Arch. Gerontol. Geriatr. 2009, 49, 7–12. [Google Scholar] [CrossRef]
- Nakamura, E.; Moritani, T.; Kanetaka, A. Effects of habitual physical exercise on physiological age in men aged 20–85 years as estimated using principal component analysis. Eur. J. Appl. Physiol. Occup. Physiol. 1996, 73, 410–418. [Google Scholar] [CrossRef]
- Nakamura, E.; Moritani, T.; Kanetaka, A. Biological age versus physical fitness age in women. Eur. J. Appl. Physiol. Occup. Physiol. 1990, 61, 202–208. [Google Scholar] [CrossRef]
- Nunn, A.V.; Bell, J.D.; Guy, G.W. Lifestyle-induced metabolic inflexibility and accelerated ageing syndrome: Insulin resistance, friend or foe? Nutr. Metab. 2009, 6, 1–26. [Google Scholar] [CrossRef]
- Chahal, H.; Drake, W. The endocrine system and ageing. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2007, 211, 173–180. [Google Scholar] [CrossRef]
- Chandra, A.; Rajawat, J. Skeletal aging and osteoporosis: Mechanisms and therapeutics. Int. J. Mol. Sci. 2021, 22, 3553. [Google Scholar] [CrossRef] [PubMed]
- Gaffney-Stomberg, E.; Hughes, J.M.; Guerriere, K.I.; Staab, J.S.; Cable, S.J.; Bouxsein, M.L.; McClung, J.P. Once daily calcium (1000 mg) and vitamin D (1000 iu) supplementation during military training prevents increases in biochemical markers of bone resorption but does not affect tibial microarchitecture in Army recruits. Bone 2022, 155, 116269. [Google Scholar] [CrossRef]
- Wang, S.; Luo, Z.; Luo, H.; Li, Z.; Yuan, Z.; Tang, J.; Lin, L.; Du, Z.; Zhou, J.R. Effects of a calcium/vitamin D/zinc combination on anti-osteoporosis in ovariectomized rats. J. Trace Elem. Med. Biol. 2023, 77, 127138. [Google Scholar] [CrossRef]
- Sfeir, J.G.; Drake, M.T.; Khosla, S.; Farr, J.N. Skeletal aging. Mayo Clin. Proc. 2022, 97, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.I.; Hinton, P.S. Physical activity and bone health. MO Med. 2014, 111, 59. [Google Scholar]
- Orwoll, E.S.; Adler, R.A.; Amin, S.; Binkley, N.; Lewiecki, E.M.; Petak, S.M.; Shapses, S.A.; Sinaki, M.; Watts, N.B.; Sibonga, J.D. Skeletal health in long-duration astronauts: Nature, assessment, and management recommendations from the NASA bone summit. J. Bone Miner. Res. 2013, 28, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Boismal, F.; Serror, K.; Dobos, G.; Zuelgaray, E.; Bensussan, A.; Michel, L. Skin aging: Pathophysiology and innovative therapies. Med. Sci. M/S 2020, 36, 1163–1172. [Google Scholar]
- Baumann, L. Skin ageing and its treatment. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2007, 211, 241–251. [Google Scholar] [CrossRef]
- Kohl, E.; Steinbauer, J.; Landthaler, M.; Szeimies, R.M. Skin ageing. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 873–884. [Google Scholar] [CrossRef]
- Brincat, M.; Muscat Baron, Y.; Galea, R. Estrogens and the skin. Climacteric 2005, 8, 110–123. [Google Scholar] [CrossRef]
- Park, H.Y.; Kim, J.H.; Jung, M.; Chung, C.H.; Hasham, R.; Park, C.S.; Choi, E.H. A long-standing hyperglycaemic condition impairs skin barrier by accelerating skin ageing process. Exp. Dermatol. 2011, 20, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Bonté, F.; Girard, D.; Archambault, J.C.; Desmoulière, A. Skin changes during ageing. Biochem. Cell Biol. Ageing Pt II Clin. Sci. 2019, 91, 249–280. [Google Scholar]
- Shpichka, A.; Butnaru, D.; Bezrukov, E.A.; Sukhanov, R.B.; Atala, A.; Burdukovskii, V.; Zhang, Y.; Timashev, P. Skin tissue regeneration for burn injury. Stem Cell Res. Ther. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bae, C.Y.; Kang, Y.G.; Piao, M.H.; Cho, B.; Cho, K.H.; Park, Y.K.; Yu, B.Y.; Lee, S.W.; Kim, M.J.; Lee, S.H.; et al. Models for estimating the biological age of five organs using clinical biomarkers that are commonly measured in clinical practice settings. Maturitas 2013, 75, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Naskalova, S.; Shatilo, V.; Pisaruk, A.; Antoniuk-Shcheglova, I.; Bondarenko, O.; Bodretska, L.; Shapovalenko, I. Estimating the functional age of the respiratory system. Ageing Longev. 2022, 3, 71–76. [Google Scholar] [CrossRef]
- Sprung, J.; Gajic, O.; Warner, D.O. Age related alterations in respiratory function–anesthetic considerations. Can. J. Anesth. 2006, 53, 1244. [Google Scholar] [CrossRef]
- Charansonney, O.L. Physical activity and aging: A life-long story. Discov. Med. 2011, 12, 177–185. [Google Scholar] [PubMed]
- Negri, A.L. The klotho gene: A gene predominantly expressed in the kidney is a fundamental regulator of aging and cal- cium/phosphorus metabolism. J. Nephrol. 2005, 18, 654–658. [Google Scholar]
- Wei, S.Y.; Pan, S.Y.; Li, B.; Chen, Y.M.; Lin, S.L. Rejuvenation: Turning back the clock of aging kidney. J. Formos. Med. Assoc. 2020, 119, 898–906. [Google Scholar] [CrossRef]
- Alcedo, J.; Flatt, T.; Pasyukova, E.G. The role of the nervous system in aging and longevity. Front. Genet. 2013, 4, 124. [Google Scholar] [CrossRef]
- Bouchard, J.; Villeda, S.A. Aging and brain rejuvenation as systemic events. J. Neurochem. 2015, 132, 5–19. [Google Scholar] [CrossRef]
- Kimura, K.; Ieda, M.; Kanazawa, H.; Yagi, T.; Tsunoda, M.; Ninomiya, S.; Kurosawa, H.; Yoshimi, K.; Mochizuki, H.; Yamazaki, K.; et al. Cardiac sympathetic rejuvenation: A link between nerve function and cardiac hypertrophy. Circ. Res. 2007, 100, 1755–1764. [Google Scholar] [CrossRef]
- Martin, J.; Sheaff, M. Renal ageing. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2007, 211, 198–205. [Google Scholar] [CrossRef]
- Harciarek, M.; Biedunkiewicz, B.; Lichodziejewska-Niemierko, M.; De˛bska-S´lizien´, A.; Rutkowski, B. Continuous cognitive improvement 1 year following successful kidney transplant. Kidney Int. 2011, 79, 1353–1360. [Google Scholar] [CrossRef]
- Griva, K.; Thompson, D.; Jayasena, D.; Davenport, A.; Harrison, M.; Newman, S.P. Cognitive functioning pre-to post-kidney transplantation—A prospective study. Nephrol. Dial. Transplant. 2006, 21, 3275–3282. [Google Scholar] [CrossRef] [PubMed]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef]
- Amanvermez, R.; Tosun, M. An update on ovarian aging and ovarian reserve tests. Int. J. Fertil. Steril. 2016, 9, 411. [Google Scholar] [PubMed]
- Chilovi, B.V.; Caratozzolo, S.; Mombelli, G.; Zanetti, M.; Rozzini, L.; Padovani, A. Does reversible mci exist? Alzheimer’s Dement. 2011, 7, 5. [Google Scholar] [CrossRef]
- Vermunt, L.; van Paasen, A.J.L.; Teunissen, C.E.; Scheltens, P.; Visser, P.J.; Tijms, B.M.; Alzheimer’s Disease Neuroimaging Initiative. Alzheimer disease biomarkers may aid in the prognosis of mci cases initially reverted to normal. Neurology 2019, 92, e2699–e2705. [Google Scholar] [CrossRef] [PubMed]
- Zarahn, E.; Rakitin, B.; Abela, D.; Flynn, J.; Stern, Y. Age-related changes in brain activation during a delayed item recognition task. Neurobiol. Aging 2007, 28, 784–798. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, E.J.; Slade, K.; May, P.J.; Nuttall, H.E. Towards an understanding of healthy cognitive ageing: The importance of lifestyle in cognitive reserve and the scaffolding theory of aging and cognition. J. Gerontol. Ser. B 2022, 78, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Doi, T.; Lee, S.; Makizako, H. Reversible predictors of reversion from mild cognitive impairment to normal cognition: A 4-year longitudinal study. Alzheimer’s Res. Ther. 2019, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, M.J.; Sachdev, P.; Wen, W.; Chen, X.; Brodaty, H. Lifespan mental activity predicts diminished rate of hippocampal atrophy. PLoS ONE 2008, 3, e2598. [Google Scholar] [CrossRef] [PubMed]
- Aycheh, H.M.; Seong, J.K.; Shin, J.H.; Na, D.L.; Kang, B.; Seo, S.W.; Sohn, K.A. Biological brain age prediction using cortical thickness data: A large scale cohort study. Front. Aging Neurosci. 2018, 10, 252. [Google Scholar] [CrossRef]
- Franke, K.; Gaser, C. Ten years of brainage as a neuroimaging biomarker of brain aging: What insights have we gained? Front. Neurol. 2019, 10, 789. [Google Scholar] [CrossRef]
- Elliott, M.L.; Belsky, D.W.; Knodt, A.R.; Ireland, D.; Melzer, T.R.; Poulton, R.; Ramrakha, S.; Caspi, A.; Moffitt, T.E.; Hariri, A.R. Brain-age in midlife is associated with accelerated biological aging and cognitive decline in a longitudinal birth cohort. Mol. Psychiatry 2021, 26, 3829–3838. [Google Scholar] [CrossRef] [PubMed]
- Saxon, S.V.; Etten, M.J.; Perkins, E.A. Physical Change and Aging: A Guide for Helping Professions; Springer Publishing Company: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Berezina, T.N.; Rybtsov, S.A. Use of personal resources may influence the rate of biological aging depending on individual typology. Eur. J. Investig. Health Psychol. Educ. 2022, 12, 1793–1811. [Google Scholar] [CrossRef]
- Bethlehem, R.A.I.; Seidlitz, J.; White, S.R.; Vogel, J.W.; Anderson, K.M.; Adamson, C.; Adler, S.; Alexopoulos, G.S.; Anagnostou, E.; Areces-Gonzalez, A. Brain charts for the human lifespan. Nature 2022, 604, 525–533. [Google Scholar] [CrossRef]
- Gaser, C.; Franke, K.; Klöppel, S.; Koutsouleris, N.; Sauer, H.; Initiative, A.D.N. Brainage in mild cognitive impaired patients: Predicting the conversion to Alzheimer’s disease. PLoS ONE 2013, 8, e67346. [Google Scholar] [CrossRef]
- Eickhoff, C.R.; Hoffstaedter, F.; Caspers, J.; Reetz, K.; Mathys, C.; Dogan, I.; Amunts, K.; Schnitzler, A.; Eickhoff, S.B. Advanced brain ageing in Parkinson’s disease is related to disease duration and individual impairment. Brain Commun. 2021, 3, fcab191. [Google Scholar] [CrossRef]
- Koutsouleris, N.; Davatzikos, C.; Borgwardt, S.; Gaser, C.; Bottlender, R.; Frodl, T.; Falkai, P.; Riecher-Rössler, A.; Möller, H.J.; Reiser, M.; et al. Accelerated brain aging in schizophrenia and beyond: A neuroanatomical marker of psychiatric disorders. Schizophr. Bull. 2014, 40, 1140–1153. [Google Scholar] [CrossRef]
- Franke, K.; Gaser, C. Longitudinal changes in individual brainage in healthy aging, mild cognitive impairment, and Alzheimer’s disease. GeroPsych 2012, 25, 235–245. [Google Scholar] [CrossRef]
- Scahill, R.I.; Frost, C.; Jenkins, R.; Whitwell, J.L.; Rossor, M.N.; Fox, N.C. A longitudinal study of brain volume changes in normal aging using serial registered magnetic resonance imaging. Arch. Neurol. 2003, 60, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Coffey, C.E.; Wilkinson, W.E.; Parashos, I.A.; Soady, S.A.; Sullivan, R.J.; Patterson, L.J.; Figiel, G.S.; Webb, M.C.; Spritzer, C.E.; Djang, W.T. Quantitative cerebral anatomy of the aging human brain: A cross-sectional study using magnetic resonance imaging. Neurology 1992, 42, 527. [Google Scholar] [CrossRef]
- Good, C.D.; Johnsrude, I.S.; Ashburner, J.; Henson, R.N.; Friston, K.J.; Frackowiak, R.S. A voxel-based morphometric study of ageing in 465 normal adult human brains. Neuroimage 2001, 14, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Foxman, E.F.; Molony, R.D. Early local immune defences in the respiratory tract. Nat. Rev. Immunol. 2017, 17, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.; Patro, N.; Patro, I.K. Cumulative multiple early life hits-a potent threat leading to neurological disorders. Brain Res. Bull. 2019, 147, 58–68. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. A timeline for parkinson’s disease. Park. Relat. Disord. 2010, 16, 79–84. [Google Scholar] [CrossRef]
- Turknett, J.; Wood, T.R. Demand coupling drives neurodegeneration: A model of age-related cognitive decline and dementia. Cells 2022, 11, 2789. [Google Scholar] [CrossRef]
- Schneider, C.B.; Donix, M.; Linse, K.; Werner, A.; Fauser, M.; Klingelhoefer, L.; Löhle, M.; von Kummer, R.; Reichmann, H.; Storch, A.; et al. Accelerated age-dependent hippocampal volume loss in parkinson disease with mild cognitive impairment. Am. J. Alzheimer’s Dis. Other Dement. 2017, 32, 313–319. [Google Scholar] [CrossRef]
- Smith, S.M.; Elliott, L.T.; Alfaro-Almagro, F.; McCarthy, P.; Nichols, T.E.; Douaud, G.; Miller, K.L. Brain aging comprises many modes of structural and functional change with distinct genetic and biophysical associations. eLife 2020, 9, e52677. [Google Scholar] [CrossRef] [PubMed]
- Bayati, A.; Berman, T. Localized vs. systematic neurodegeneration: A paradigm shift in understanding neurodegenerative diseases. Front. Syst. Neurosci. 2017, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, I.; Mishra, S.; Sone, D.; Khanna, P.; Matsuda, H. T1-weighted mri-driven brain age estimation in Alzheimer’s disease and parkinson’s disease. Aging Dis. 2020, 11, 618. [Google Scholar] [CrossRef] [PubMed]
- McCartney, D.L.; Stevenson, A.J.; Walker, R.M.; Gibson, J.; Morris, S.W.; Campbell, A.; Murray, A.D.; Whalley, H.C.; Porteous, D.J.; McIntosh, A.M.; et al. Investigating the relationship between dna methylation age acceleration and risk factors for Alzheimer’s disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2018, 10, 429–437. [Google Scholar] [CrossRef]
- Norrara, B.; Doerl, J.G.; Guzen, F.P.; Cavalcanti, J.R.L.P.; Freire, M.A.M. Commentary: Localized vs. systematic neurodegeneration: A paradigm shift in understanding neurodegenerative diseases. Front. Syst. Neurosci. 2017, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Jung, Y.S.; Ock, M.; Yoon, S.J. Daly estimation approaches: Understanding and using the incidence-based approach and the prevalence-based approach. J. Prev. Med. Public Health 2022, 55, 10. [Google Scholar] [CrossRef]
- Gao, T.; Wang, X.C.; Chen, R.; Ngo, H.H.; Guo, W. Disability adjusted life year (daly): A useful tool for quantitative assessment of environmental pollution. Sci. Total Environ. 2015, 511, 268–287. [Google Scholar] [CrossRef]
- Jonsson, B.A.; Bjornsdottir, G.; Thorgeirsson, T.E.; Ellingsen, L.M.; Walters, G.B.; Gudbjartsson, D.F.; Stefansson, H.; Stefansson, K.; Ulfarsson, M.O. Brain age prediction using deep learning uncovers associated sequence variants. Nat. Commun. 2019, 10, 5409. [Google Scholar] [CrossRef]
- Hung, C.W.; Chen, Y.C.; Hsieh, W.L.; Chiou, S.H.; Kao, C.L. Ageing and neurodegenerative diseases. Ageing Res. Rev. 2010, 9, S36–S46. [Google Scholar] [CrossRef]
- Sibbett, R.A.; Altschul, D.M.; Marioni, R.E.; Deary, I.J.; Starr, J.M.; Russ, T.C. Dna methylation-based measures of accelerated biological ageing and the risk of dementia in the oldest-old: A study of the lothian birth cohort 1921. BMC Psychiatry 2020, 20, 1–15. [Google Scholar] [CrossRef]
- Soldan, A.; Pettigrew, C.; Li, S.; Wang, M.C.; Moghekar, A.; Selnes, O.A.; Albert, M.; O’Brien, R. Relationship of cognitive reserve and cerebrospinal fluid biomarkers to the emergence of clinical symptoms in preclinical Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2827–2834. [Google Scholar] [CrossRef]
- Fries, G.R.; Bauer, I.E.; Scaini, G.; Valvassori, S.S.; Walss-Bass, C.; Soares, J.C.; Quevedo, J. Accelerated hippocampal biological aging in bipolar disorder. Bipolar Disord. 2020, 22, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Tønnesen, S.; Kaufmann, T.; de Lange, A.G.; Richard, G.; Doan, N.T.; Alnæs, D.; van der Meer, D.; Rokicki, J.; Moberget, T.; Maximov, I.I.; et al. Brain age prediction reveals aberrant brain white matter in schizophrenia and bipolar disorder: A multisample diffusion tensor imaging study. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2020, 5, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Whalley, H.C.; Gibson, J.; Marioni, R.; Walker, R.M.; Clarke, T.K; Howard, D.M.; Adams, M.J.; Hall, L.; Morris, S.; Deary, I.J.; et al. Accelerated epigenetic ageing in major depressive disorder. bioRxiv 2017, 210666. [Google Scholar]
- Dudley, J.A.; Lee, J.H.; Durling, M.; Strakowski, S.M.; Eliassen, J.C. Age-dependent decreases of high energy phosphates in cerebral gray matter of patients with bipolar i disorder: A preliminary phosphorus-31 magnetic resonance spectroscopic imaging study. J. Affect. Disord. 2015, 175, 251–255. [Google Scholar] [CrossRef]
- Masuda, Y.; Okada, G.; Takamura, M.; Shibasaki, C.; Yoshino, A.; Yokoyama, S.; Ichikawa, N.; Okuhata, S.; Kobayashi, T.; Yamawaki, S.; et al. Age-related white matter changes revealed by a whole-brain fiber-tracking method in bipolar disorder compared to major depressive disorder and healthy controls. Psychiatry Clin. Neurosci. 2021, 75, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Muangpaisan, W.; Petcharat, C.; Srinonprasert, V. Prevalence of potentially reversible conditions in dementia and mild cognitive impairment in a geriatric clinic. Geriatr. Gerontol. Int. 2012, 12, 59–64. [Google Scholar] [CrossRef]
- Gauthier, S.; Touchon, J. Mild cognitive impairment is not a clinical entity and should not be treated. Arch. Neurol. 2005, 62, 1164–1166. [Google Scholar] [CrossRef]
- Bates, K.; Harvey, A.R.; Carruthers, M.; Martins, R. Androgens, andropause and neurodegeneration: Exploring the link between steroidogenesis, androgens and Alzheimer’s disease. Cell Mol. Life Sci. 2005, 62, 281–292. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the interdependence between oxidative stress and inflammation explain the antioxidant paradox? Oxidative Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, M.; Scarian, E.; Rey, F.; Gagliardi, S.; Carelli, S.; Pansarasa, O.; Cereda, C. Biomaterials in neurodegenerative disorders: A promising therapeutic approach. Int. J. Mol. Sci. 2020, 21, 3243. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, T.M.; Bonder, M.J.; Stark, A.K.; Krueger, F.; von Meyenn, F.; Stegle, O.; Reik, W. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 2017, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Chwiałkowska, K.; Rubbi, L.; Lusis, A.J.; Davis, R.C.; Srivastava, A.; Korstanje, R.; Churchill, G.A.; Horvath, S.; Pellegrini, M. A multi-tissue full lifespan epigenetic clock for mice. Aging 2018, 10, 2832. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tsui, B.; Kreisberg, J.F.; Robertson, N.A.; Gross, A.M.; Yu, M.K.; Carter, H.; Brown-Borg, H.M.; Adams, P.D.; Ideker, T. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 2017, 18, 57. [Google Scholar] [CrossRef]
- Petkovich, D.A.; Podolskiy, D.I.; Lobanov, A.V.; Lee, S.G.; Miller, R.A.; Gladyshev, V.N. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab. 2017, 25, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Meer, M.V.; Podolskiy, D.I.; Tyshkovskiy, A.; Gladyshev, V.N. A whole lifespan mouse multi-tissue DNA methylation clock. eLife 2018, 7, e40675. [Google Scholar] [CrossRef]
- Bahar, R.; Hartmann, C.H.; Rodriguez, K.A.; Denny, A.D.; Busuttil, R.A.; Dollé, M.E.; Calder, R.B.; Chisholm, G.B.; Pollock, B.H.; Klein, C.A.; et al. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 2006, 441, 1011–1014. [Google Scholar] [CrossRef]
- Rimmelé, P.; Bigarella, C.L.; Liang, R.; Izac, B.; Dieguez-Gonzalez, R.; Barbet, G.; Donovan, M.; Brugnara, C.; Blander, J.M.; Sinclair, D.A.; et al. Aging-like phenotype and defective lineage specification in sirt1-deleted hematopoietic stem and progenitor cells. Stem Cell Rep. 2014, 3, 44–59. [Google Scholar] [CrossRef]
- Cheung, P.; Vallania, F.; Warsinske, H.C.; Donato, M.; Schaffert, S.; Chang, S.E.; Dvorak, M.; Dekker, C.L.; Davis, M.M.; Utz, P.J.; et al. Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell 2018, 173, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Hernando-Herraez, I.; Evano, B.; Stubbs, T.; Commere, P.H.; Jan Bonder, M.; Clark, S.; Andrews, S.; Tajbakhsh, S.; Reik, W. Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat. Commun. 2019, 10, 4361. [Google Scholar] [CrossRef] [PubMed]
- Trapp, A.; Kerepesi, C.; Gladyshev, V.N. Profiling epigenetic age in single cells. Nat. Aging 2021, 1, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; An, Q.; Guo, Y.; Zhong, J.; Fan, S.; Rao, P.; Liu, X.; Liu, Y.; Fan, G. Simultaneous profiling of mRNA transcriptome and DNA methylome from a single cell. Single Cell Methods Seq. Proteom. 2019, 1979, 363–377. [Google Scholar]
- Angermueller, C.; Lee, H.J.; Reik, W.; Stegle, O. Deepcpg: Accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017, 18, 1–13. [Google Scholar]
- Hamidouche, Z.; Rother, K.; Przybilla, J.; Krinner, A.; Clay, D.; Hopp, L.; Fabian, C.; Stolzing, A.; Binder, H.; Charbord, P.; et al. Bistable epigenetic states explain age-dependent decline in mesenchymal stem cell heterogeneity. Stem Cells 2017, 35, 694–704. [Google Scholar] [CrossRef] [PubMed]
- de Lima Camillo, L.P.; Lapierre, L.R.; Singh, R. A pan-tissue DNA-methylation epigenetic clock based on deep learning. npj Aging 2022, 8, 4. [Google Scholar] [CrossRef]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef]
- Stefanelli, G.; Azam, A.B.; Walters, B.J.; Brimble, M.A.; Gettens, C.P.; Bouchard-Cannon, P.; Cheng, H.M.; Davidoff, A.M.; Narkaj, K.; Day, J.J.; et al. Learning and age-related changes in genome-wide h2a.z binding in the mouse hippocampus. Cell Rep. 2018, 22, 1124–1131. [Google Scholar] [CrossRef]
- Klein, H.U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers tau pathology-driven changes of chromatin organization in the aging human brain. Biorxiv 2018, 22, 273789. [Google Scholar]
- Roichman, A.; Elhanati, S.; Aon, M.A.; Abramovich, I.; Di Francesco, A.; Shahar, Y.; Avivi, M.Y.; Shurgi, M.; Rubinstein, A.; Wiesner, Y.; et al. Restoration of energy homeostasis by sirt6 extends healthy lifespan. Nat. Commun. 2021, 12, 3208. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.; Finigan, A.; Figg, N.L.; Uryga, A.K.; Bennett, M.R. Sirt6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ. Res. 2021, 128, 474–491. [Google Scholar] [CrossRef] [PubMed]
- Soto-Palma, C.; Niedernhofer, L.J.; Faulk, C.D.; Dong, X. Epigenetics, DNA damage, and aging. J. Clin. Investig. 2022, 132, e158446. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Hartmann, C.; Secci, R.; Hermann, A.; Fuellen, G.; Walter, M. Ranking biomarkers of aging by citation profiling and effort scoring. Front. Genet. 2021, 12, 686320. [Google Scholar] [CrossRef]
- Bürkle, A.; Moreno-Villanueva, M.; Bernhard, J.; Blasco, M.; Zondag, G.; Hoeijmakers, J.H.; Toussaint, O.; Grubeck-Loebenstein, B.; Mocchegiani, E.; Collino, S.; et al. MARK-AGE biomarkers of ageing. Mech. Ageing Dev. 2015, 151, 2–12. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Human Genetic Disorders Associated with Genome Instability, Premature Aging and Cancer Predispo- sition. Open Cancer J. 2008, 2, 42–52. [Google Scholar] [CrossRef]
- Tiwari, V.; Wilson, D.M. DNA damage and associated DNA repair defects in disease and premature aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef]
- Rizza, E.R.; DiGiovanna, J.J.; Khan, S.G.; Tamura, D.; Jeskey, J.D.; Kraemer, K.H. Xeroderma pigmentosum: A model for human premature aging. J. Investig. Dermatol. 2021, 141, 976–984. [Google Scholar] [CrossRef]
- Han, Z.Z.; Fleet, A.; Larrieu, D. Can accelerated ageing models inform us on age-related tauopathies? Aging Cell 2023, 22, e13830. [Google Scholar] [CrossRef]
- Platzer, M.; Englert, C. Nothobranchius furzeri: A model for aging research and more. Trends Genet. 2016, 32, 543–552. [Google Scholar] [CrossRef]
- Reichwald, K.; Petzold, A.; Koch, P.; Downie, B.R.; Hartmann, N.; Pietsch, S.; Baumgart, M.; Chalopin, D.; Felder, M.; Bens, M.; et al. Insights into sex chromosome evolution and aging from the genome of a short-lived fish. Cell 2015, 163, 1527–1538. [Google Scholar] [CrossRef]
- Valenzano, D.R.; Benayoun, B.A.; Singh, P.P.; Zhang, E.; Etter, P.D.; Hu, C.K.; Clément-Ziza, M.; Willemsen, D.; Cui, R.; Harel, I.; et al. The African turquoise killifish genome provides insights into evolution and genetic architecture of lifespan. Cell 2015, 163, 1539–1554. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Reichwald, K.; Groth, M.; Taudien, S.; Hartmann, N.; Priebe, S.; Shagin, D.; Englert, C.; Platzer, M. The transcript catalogue of the short-lived fish Nothobranchius furzeri provides insights into age-dependent changes of mRNA levels. BMC Genom. 2013, 14, 185. [Google Scholar] [CrossRef] [PubMed]
- Genade, T.; Benedetti, M.; Terzibasi, E.; Roncaglia, P.; Valenzano, D.R.; Cattaneo, A.; Cellerino, A. Annual fishes of the genus Nothobranchius as a model system for aging research. Aging Cell 2005, 4, 223–233. [Google Scholar] [CrossRef]
- Di Cicco, E.; Tozzini, E.T.; Rossi, G.; Cellerino, A. The shortlived annual fish Nothobranchius furzeri shows a typical teleost aging process reinforced by high incidence of age-dependent neoplasias. Exp. Gerontol. 2011, 46, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Dodzian, J.; Kean, S.; Seidel, J.; Valenzano, D.R. A protocol for laboratory housing of Turquoise Killifish (Nothobranchius furzeri). J. Vis. Exp. 2018, 134, e57073. [Google Scholar]
- Tan, L.; Ke, Z.; Tombline, G.; Macoretta, N.; Hayes, K.; Tian, X.; Lv, R.; Ablaeva, J.; Gilbert, M.; Bhanu, N.V.; et al. Naked mole rat cells have a stable epigenome that resists iPSC reprogramming. Stem Cell Rep. 2017, 9, 1721–1734. [Google Scholar] [CrossRef]
- Dammann, P.; Šumbera, R.; Massmann, C.; Scherag, A.; Burda, H. Extended longevity of reproductives appears to be common in Fukomys molerats (Rodentia. Bathyergidae). PLoS ONE 2011, 6, e18757. [Google Scholar]
- Ruby, J.G.; Smith, M.; Buffenstein, R. Naked mole-rat mortality rates defy gompertzian laws by not increasing with age. eLife 2018, 7, e31157. [Google Scholar] [CrossRef]
- Munshi-South, J.; Wilkinson, G.S. Bats and birds: Exceptional longevity despite high metabolic rates. Ageing Res. Rev. 2010, 9, 12–19. [Google Scholar] [CrossRef]
- Seim, I.; Fang, X.; Xiong, Z.; Lobanov, A.V.; Huang, Z.; Ma, S.; Feng, Y.; Turanov, A.A.; Zhu, Y.; Lenz, T.L.; et al. Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nat. Commun. 2013, 4, 2212. [Google Scholar] [CrossRef]
- Podlutsky, A.J.; Khritankov, A.M.; Ovodov, N.D.; Austad, S.N. A new field record for bat longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 1366–1368. [Google Scholar] [CrossRef] [PubMed]
- Holtze, S.; Lukacˇ, M.; Cizelj, I.; Mutschmann, F.; Szentiks, C.A.; Jelic´, D.; Hermes, R.; Göritz, F.; Braude, S.; Hildebrandt, T.B. Monitoring health and reproductive status of olms (Proteus anguinus) by ultrasound. PLoS ONE 2017, 12, e0182209. [Google Scholar] [CrossRef] [PubMed]
- Mulec, J. Welcome to the -omics era of the 21st century: Will Proteus anguinus finally reveal all its mysteries? Acta Carsol. 2020, 1, 49. [Google Scholar] [CrossRef]
- Voituron, Y.; de Fraipont, M.; Issartel, J.; Guillaume, O.; Clobert, J. Extreme lifespan of the human fish (Proteus anguinus): A challenge for ageing mechanisms. Biol. Lett. 2011, 7, 105–107. [Google Scholar] [CrossRef]
- Philipp, E.E.; Wessels, W.; Gruber, H.; Strahl, J.; Wagner, A.E.; Ernst, I.M.; Rimbach, G.; Kraemer, L.; Schreiber, S.; Abele, D.; et al. Gene expression and physiological changes of different populations of the long-lived bivalve Arctica islandica under low oxygen conditions. PLoS ONE 2012, 7, e44621. [Google Scholar] [CrossRef] [PubMed]
- Lutz, R.A.; Goodsell, J.G.; Mann, R.; Castagna, M. Experimental culture of the ocean quahog, Arctica islandica. J. World Maricult. Soc. 1981, 12, 196–205. [Google Scholar] [CrossRef]
- Chapman, J.A.; Kirkness, E.F.; Simakov, O.; Hampson, S.E.; Mitros, T.; Weinmaier, T.; Rattei, T.; Balasubramanian, P.G.; Borman, J.; Busam, D.; et al. The dynamic genome of Hydra. Nature 2010, 464, 592–596. [Google Scholar] [CrossRef]
- Schaible, R.; Sussman, M.; Kramer, B.H. Aging and potential for self-renewal: Hydra living in the age of aging—A mini-review. Gerontology 2014, 60, 548–556. [Google Scholar] [CrossRef]
- Bellantuono, A.J.; Bridge, D.; Martínez, D.E. Hydra as a tractable, long-lived model system for senescence. Invert. Reprod. Dev. 2015, 59, 39–44. [Google Scholar] [CrossRef]
- Klimovich, A.; Rehm, A.; Wittlieb, J.; Herbst, E.-M.; Benavente, R.; Bosch, T.C.G. Non-senescent Hydra tolerates severe distur- bances in the nuclear lamina. Aging 2018, 10, 951–972. [Google Scholar] [CrossRef] [PubMed]
- Grohme, M.A.; Schloissnig, S.; Rozanski, A.; Pippel, M.; Young, G.R.; Winkler, S.; Brandl, H.; Henry, I.; Dahl, A.; Powell, S.; et al. The genome of Schmidtea mediterranea and the evolution of core cellular mechanisms. Nature 2018, 554, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Fincher, C.T.; Wurtzel, O.; de Hoog, T.; Kravarik, K.M.; Reddien, P.W. Cell type transcriptome atlas for the planarian Schmidtea mediterranea. Science 2018, 360, eaaq1736. [Google Scholar] [CrossRef] [PubMed]
- Merryman, M.S.; Alvarado, A.S.; Jenkin, J.C. Culturing Planarians in the Laboratory. Methods Mol. Biol. 2018, 1774, 241–258. [Google Scholar]
- Lipsky, M.S.; King, M. Biological theories of aging. Dis.-A-Mon. 2015, 61, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Jin, K. Modern biological theories of aging. Aging Dis. 2010, 1, 72. [Google Scholar]
- Weinert, B.T.; Timiras, P.S. Invited review: Theories of aging. J. Appl. Physiol. 2003, 95, 1706–1716. [Google Scholar] [CrossRef]
- Northrop, J.H. The influence of the intensity of light on the rate of growth and duration of life of Drosophila. J. Gen. Physiol. 1925, 9, 81. [Google Scholar] [CrossRef]
- Northrop, J.H. Duration of life of an aseptic Drosophila culture inbred in the dark for 230 generations. J. Gen. Physiol. 1926, 9, 763–765. [Google Scholar] [CrossRef]
- NCBI PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/ (accessed on 31 August 2023).
- Tilney, F. The Aging of the Human Brain. Bull. N. Y. Acad. Med. 1928, 4, 1125–1143. [Google Scholar]
Figure 1.
Examples of epigenetic changes and factors affected by accelerated aging at different levels that could serve as potential biomarkers of aging. (I) DNA nucleotide modification level: cytosine. (C) can be converted to 5-methylcytosine (5-mC) and oxidised further to 5-hydroxymethylcytosine (5-hmC). (II) Histone modification level: Three examples of histone 3 (H3) lysine methylation marks are indicated. Lysine 4 (K4) H3K4me3 mark is associated with active genes, lysine 9 (K9) H3K9me3 is an established mark of transcriptionally repressed heterochromatin and lysine 27 (K27) H3K27me3 mark is linked to both transcriptional activation and repression. (III) Regulatory RNAs level: several types of ncRNAs, long non-coding RNA (lncRNA), microRNA (miRNA), small interfering RNA (siRNA), circular RNA (circRNA), shown schematically.
Figure 1.
Examples of epigenetic changes and factors affected by accelerated aging at different levels that could serve as potential biomarkers of aging. (I) DNA nucleotide modification level: cytosine. (C) can be converted to 5-methylcytosine (5-mC) and oxidised further to 5-hydroxymethylcytosine (5-hmC). (II) Histone modification level: Three examples of histone 3 (H3) lysine methylation marks are indicated. Lysine 4 (K4) H3K4me3 mark is associated with active genes, lysine 9 (K9) H3K9me3 is an established mark of transcriptionally repressed heterochromatin and lysine 27 (K27) H3K27me3 mark is linked to both transcriptional activation and repression. (III) Regulatory RNAs level: several types of ncRNAs, long non-coding RNA (lncRNA), microRNA (miRNA), small interfering RNA (siRNA), circular RNA (circRNA), shown schematically.
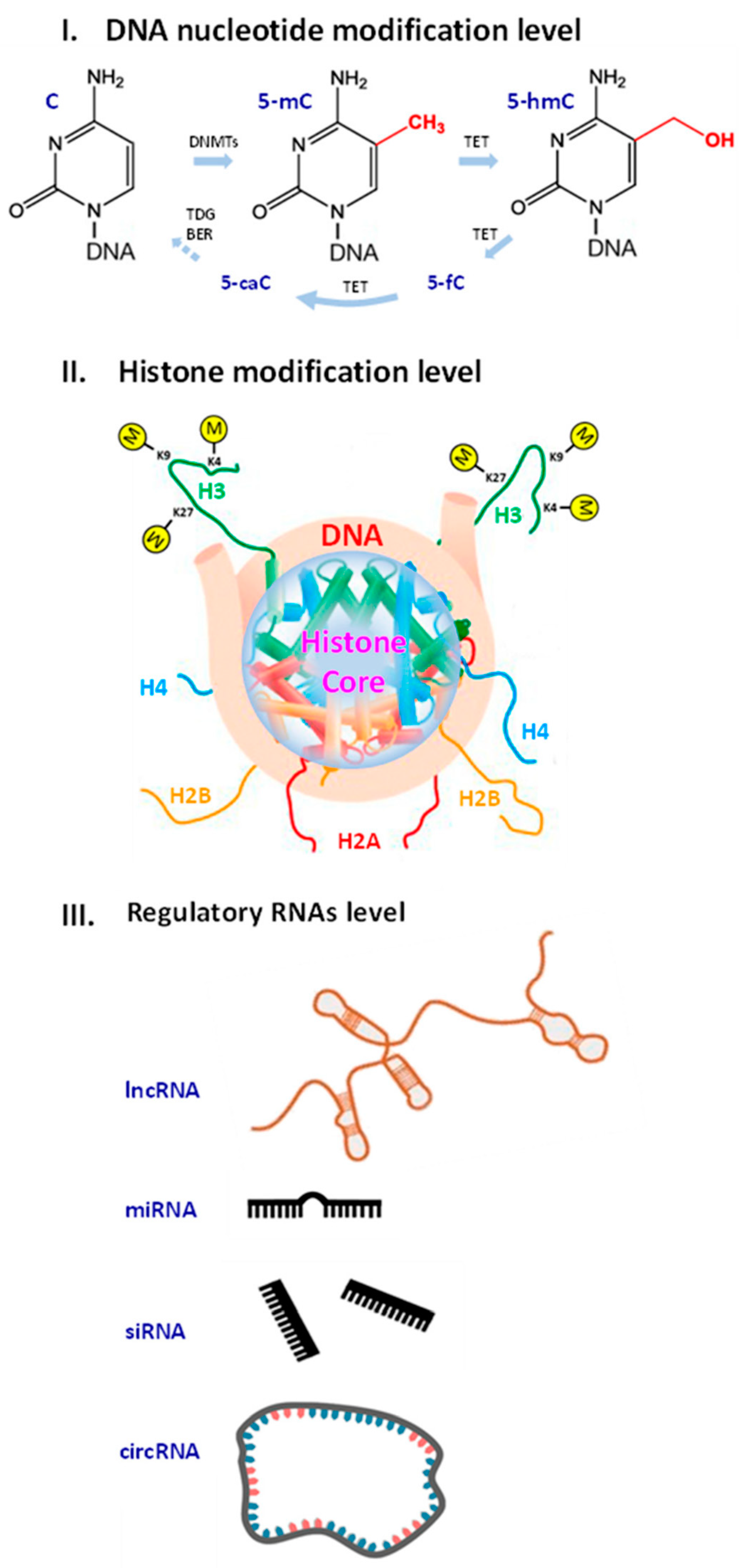
Figure 2.
Factors influencing the rate of aging. Factors accelerating aging include: genetic mutations, epigenetic changes, protein aggregation, oxidative stress, inflammation and excitotoxity. Factors decelerating aging include: a healthy lifestyle, favourable environment, hygiene and immunisation, stem cell regenerative capacities, internal resources of cell stocks and drug therapy.
Figure 2.
Factors influencing the rate of aging. Factors accelerating aging include: genetic mutations, epigenetic changes, protein aggregation, oxidative stress, inflammation and excitotoxity. Factors decelerating aging include: a healthy lifestyle, favourable environment, hygiene and immunisation, stem cell regenerative capacities, internal resources of cell stocks and drug therapy.
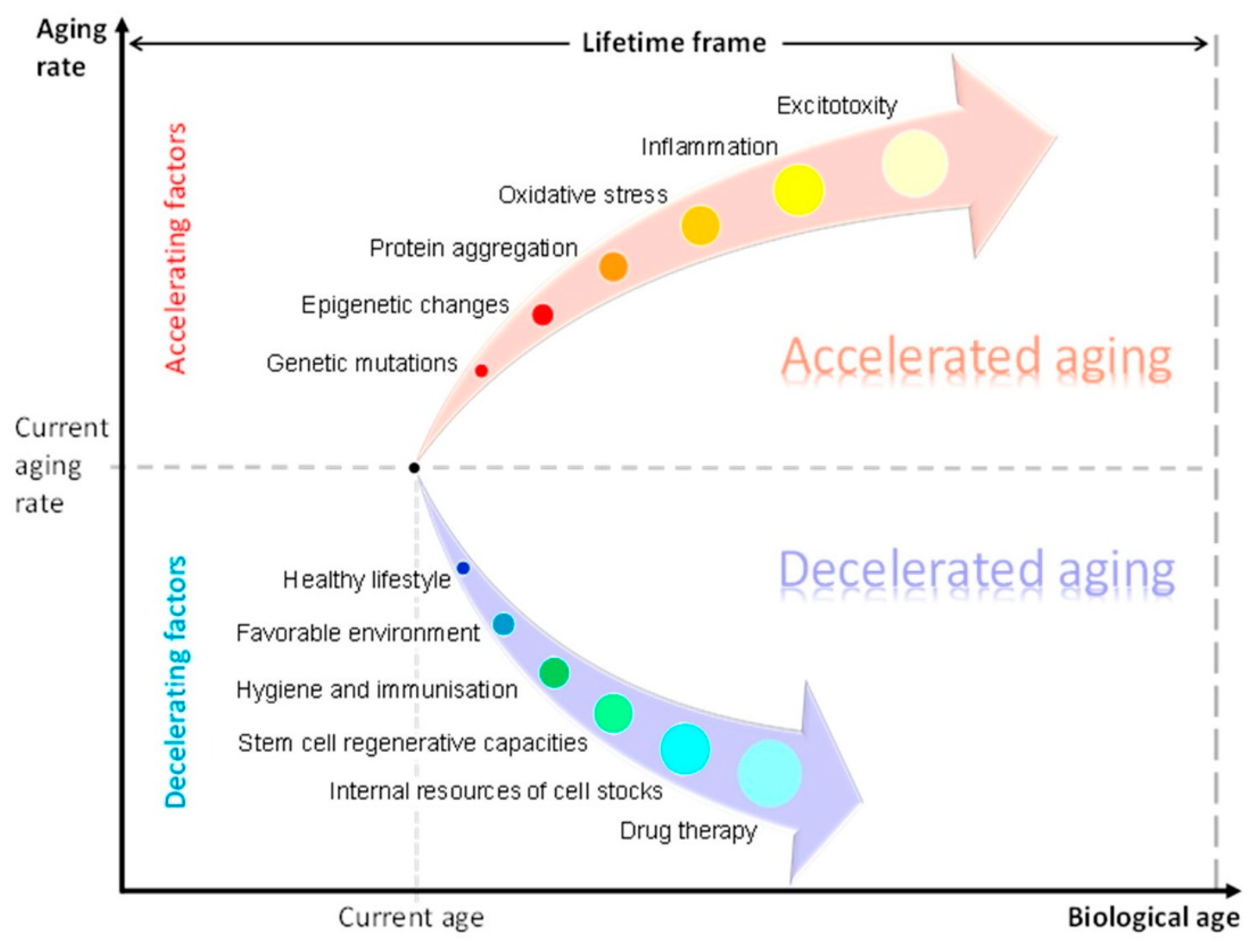
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



