
Submitted:
12 October 2023
Posted:
13 October 2023
You are already at the latest version
Abstract
Cardiomyopathies are major causes of heart failure. Chagas disease (CD) is caused by the parasite Trypanosoma cruzi, and it is endemic in Central, South America. Thirty percent of the cases evolve into chronic cardiomyopathy (CCC) with worse prognosis as compared with other cardiomyopathies. In vivo bioenergetic analysis and ex vivo proteomic analysis of myocardial tissues highlighted worse mitochondrial dysfunction in CCC, and previous studies identified nuclear-encoded mitochondrial gene variants segregating with CCC. Here, we assessed the role of the mitochondrial genome through mtDNA copy number variations and mtDNA haplotyping and sequencing from heart or blood tissues of severe, moderate CCC and asymptomatic/indeterminate Chagas disease as well as healthy controls as an attempt to help decipher mitochondrial-intrinsic genetic involvement in Chagas disease development. We have found that mtDNA copy number was significantly lower in CCC than in heart tissue from healthy individuals, while blood mtDNA content was similar among asymptomatic Chagas disease, moderate and severe CCC patients. MtDNA haplogrouping study has indicated that African haplogroups were over represented in the Chagas subject groups in comparison with Brazilian healthy individuals. The European lineage is associated to protection against cardiomyopathy and the macro haplogroup H is associated with increased risk towards CCC. By mitochondria DNA sequencing, 84 mtDNA-encoded protein sequence pathogenic variants were associated with CCC. Among them, two variants were associated to left ventricular non-compaction and two to hypertrophic cardiomyopathy. The finding that mitochondrial protein-coding SNPs and mitochondrial haplogroups associate with risk of evolving to CCC is consistent with a key role of mitochondrial DNA in the development of Chronic Chagas disease Cardiomyopathy.
Keywords:
chagas
; cardiomyopathy
; mitochondria
; haplgroups
; variants
; copy-number
1. Introduction
Cardiomyopathies are a heterogeneous group of myocardial diseases associated with mechanical or electrical dysfunction that usually exhibit inappropriate ventricular hypertrophy or dilation. Dilated cardiomyopathy is one of the most common causes of heart failure (1) and the most common indication for heart transplantation worldwide, with an estimated prevalence of 40 cases per 100 000 individuals and an annual incidence of 7 cases per 100 000 individuals. Dilated cardiomyopathy occurs more often among people in the third or fourth decade of life. Indeed, patients with dilated cardiomyopathy generally appear asymptomatic in the early stages, but symptoms gradually emerge with age as the heart function starts to decline due to progressive loss of function in the cardiomyocytes, impairing normal heart function (2, 3).
It is now also admitted that adult males seem to be more frequently affected than adult females (2, 3). Although most cases of dilated cardiomyopathy are considered idiopathic, approximately 20 to 35%, or potentially up to 50% of patient cases, could be attributed to the genetic basis, called familial dilated cardiomyopathy (4). More than 100 genes have been associated to dilated cardiomyopathies (5-8). Myocardial energy deprivation is a main feature of cardiomyopathies and heart failure, and multiple evidences indicate this is caused by mitochondrial dysfunction. Because the heart is an organ requiring a high energy demand, regulation of mitochondrial metabolism plays an important role in the pathogenesis of dilated cardiomyopathies. During the first stages of dilated cardiomyopathies, increased mitochondrial proliferation acts as a compensating mechanism for maintaining energy supply (9). However, the number of mitochondria declines during dilated cardiomyopathy progression leading to a reduction in ATP, decreased contractility and increased reactive oxidative species (ROS) all of which resulting in diastolic dysfunction and heart failure (10). In many cases mitochondrial cardiomyopathies have an underlying genetic component resulting in mitochondrial respiratory chain deficiencies (11-13), including fatty acid oxydation (11) or cardiolipin synthesis defects (14-16) and alterations of mitochondrial dynamics (11). Mitochondria are under dual genome control, where a small fraction of mitochondrial proteins are encoded by mitochondrial DNA (mtDNA), while more than 99% of them are encoded by nuclear DNA (nDNA). Mutations in over 250 mitochondria-related nDNA or mtDNA genes have been associated with mitochondrial disease, resulting in mitochondrial dysfunction leading to insufficient energy production required to meet the needs for organs with high energy requirements (17).
Cardiac involvement occurs in mitochondrial diseases, with cardiomyopathies being one of the most frequent cardiac manifestations found in these disorders occuring in 20–40% of patients. Mitochondrial cardiomyopathies can vary in severity from asymptomatic status to severe manifestations including heart failure, arrhythmias, and sudden cardiac death (11). MtDNA polymorphisms that have occurred over evolutionary time have permitted a distribution of mtDNA variants into mitochondrial haplogroups inherited from the maternal lineage, and these haplotypes may influence mitochondrial function (18).
Chagas disease (CD) is caused by the parasite Trypanosoma cruzi, and it is endemic in Central, South America, and Mexico. The clinical course of the disease comprises an acute phase, mostly asymptomatic, and a chronic phase, where 60% of the patients remain asymptomatic. Forty percent of the cases evolve into gastrointestinal disease and chronic cardiomyopathy with varying degrees of severity including refractory heart failure (19). Antiparasitic treatment with benznidazole or nifurtimox is most effective in the acute phase and prevents progression to chronic phase when given to children. However, these treatments had multiple adverse effects (20). Heart failure due to Chronic Chagas Cardiomyopathy (CCC) may have a worse prognosis with 50% shorter survival when compared to other cardiomyopathies of different etiologies such as ischemic cardiomyopathy and idiopathic dilated cardiomyopathy (21, 22). The major histopathological feature attending dilated cardiomyopathy in CCC is the presence of a diffuse myocarditis, with intense cardiomyocyte damage and hypertrophy, and significant fibrosis, in the presence of very scarce T. cruzi forms. The myocardial inflammatory infiltrate is thought to play a major role in disease development and progression (23, 24). The myocardial cytokine production profile suggests an IFNγ/TNFα Th1 type response, with interferon γ-induced chemokines (25-29). Moreover, our group has previously demonstrated that CCC myocardium presents a unique gene expression profile, distinct from other noninflammatory dilated cardiomyopathies, with a large number of interferon-gamma inducible genes among differentially expressed RNA (30, 31). The proteomic analysis of myocardial tissues revealed that CCC display a specific global protein expression profile. Pathway analysis of proteins differentially expressed in CCC highlighted mitochondrial dysfunction, cardiac hypertrophy, fibrosis, mitochondrial energy metabolism, fatty acid metabolism, the involvement of Creatine Kinase System and ATP Synthase Complex, variations of the mitochondrial membrane potential (mitochondrial ΔΨm) (32-35). IFN-γ and TNF-α treatment of the human cardiomyocyte cell line AC16 induces a dose-dependent reduction of mitochondrial ΔΨm and mtDNA copy number, suggesting a possible inflammatory origin for mitochondrial dysfunction in CCC (33). IFN-γ and TNF-α treatment of AC-16 cardiomyocytes’ mitochondria also induce an increased nitro-oxidative stress profile. The inhibition of STAT1/NF-κB/NOS2 axis and activation of AMPK, NRF2 and SIRT1 signaling pathways promoted protective effects in the IFN-γ/TNF-α-induced impairment of mitochondrial ΔΨm (36). This finding is especially crucial since the mitochondrial ΔΨm is the OXPHOS proton motive force that drives ATP production through ATP synthase is an essential mechanism for contraction and survival of cardiac cells (37). Gene and protein expression analyses in cytokine-treated AC-16 cardiomyocytes confirmed decreased levels of mitochondrial proteins involved in ATP generation, ion import and mitochondrial structural maintenance proteins and the overexpression of proteins involved in ATP catabolism and mitochondrial transition pore.
About 30% of Chagas disease patients develop CCC, suggesting the participation of modifier genes in differential disease progression. This was reinforced by the discovery of familial aggregation of cases of CCC in endemic area settings (38). Genetic susceptibility to CCC may result from functionally relevant genetic variants that lead to variations in the intensity of the innate or acquired immune response and in inflammatory cytokines and chemokines involved in the pathogenesis of the disease (39-41). Based on whole exome sequencing on multiple familial cases of Chagas disease, we have previously identified heterozygous, pathogenic variants linked to CCC in all tested families on 22 distinct nuclear-encoded genes, from which 20 were mitochondrial or inflammation-related - most of the latter involved in proinflammatory cytokine production (42). So, mitochondrial dysfunction and inflammation may be genetically determined in CCC, driven by rare genetic variants which increase mitochondrial susceptibility to IFN-γ-induced damage in the myocardium. Mutations in the mitochondrial genome accumulate over time in organs suffering mitochondrial dysfunction, resulting in decreased energy production and reactive oxygen species (ROS) overproduction. Defective copies of mtDNA might accumulate over time, raising the overall heteroplasmy level of deleterious mutations. Heteroplasmy is the concurrence of both normal (wt) and abnormal (mutant) mtDNA within one cell. In general, the level of mitochondrial dysfunction is commensurate with the mutational load of mtDNA within the tissue or cell.
In this paper, we assessed the role of the mitochondrial genome through mtDNA copy number variations and mtDNA haplotyping and sequencing from heart or blood tissues of severe, moderate CCC and asymptomatic/indeterminate Chagas disease as well as healthy controls as an attempt to help decipher mitochondrial-intrinsic genetic involvement in Chagas disease development.
2. Materials and Methods
2.1. Patients and Tissue Collection
Human left ventricular free wall heart tissue samples were obtained from patients with end-stage heart failure CCC at the time of heart transplantation (n = 34). These CCC patients underwent serological diagnosis of T. cruzi infection and standard electrocardiography and echocardiography, and tissues were subject to histopathological assessment (43). Biopsies from controls (n = 6) were obtained from healthy hearts of organ donors having no suitable recipient. The protocol was approved by the institutional review boards of the University of São Paulo School of Medicine and INSERM (French National Institute of Health and Medical Research). Written informed consent was obtained from all patients. All experimental methods comply with the Helsinki Declaration.
2.2. Heart tissue and blood DNA isolation
Heart tissue samples were crushed twice with lysis buffer. After proteinase K treatment, DNA was extracted with QIAamp DNA Mini Kit (Qiagen, Courtaboeuf, France) according to the manufacturer’s recommendations. For each subject, 5 to 15 ml of blood were collected into EDTA tube. Genomic DNA was isolated with FlexiGene DNA kit (Qiagen, Courtaboeuf, France) according to the manufacturer’s recommendations.
2.3. Mitochondrial DNA copy number quantification
The Quantitative PCR (qPCR) was conducted using SYBR Green/ROX qPCR Master Mix (2x) (Thermo Scientific), 1 ng of template DNA and primers for mitochondrial-encoded genes NADH: ubiquinone oxidoreductase subunit 1 (MT-ND1), cytochrome c oxidase subunit 1 (MT-COXI). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the endogenous control. The qPCR reactions were done in QuantStudio 12K (Applied Biosystem) with the following cycling program, 1 cycle of 50°C for 2 min, 95°C for 10 min, and 40 cycles of 50°C for 2 min, 95°C for 15s, and 60°C for 1 min. The ratio of mtDNA/nDNA was calculated using the formula 2x2ΔCT (44). The primer sequences were: MT-ND1forward: 5’ ATACCCATGGCCAACCTCCT 3’, MT-ND1 reverse: 5’ GGGCCTTTGCGTAGTTGTAT 3’; MT-COXI forward: 5’ TCCACTATGTCCTATCAATA 3’, MT-COXI reverse: 5’GGTGTAGCCTGAGAATAG 3’; GAPDH forward: 5’ CCCTGTCCAGTTAATTTC 3’, GAPDH reverse: 5’ CACCCTTTAGGGAGAAAA 3’.
2.4. Mitochondrial haplogroup determination
Mitochondrial haplogroups were determined according to MITOMASTER tool (https://www.mitomap.org/foswiki/bin/view/MITOMASTER/WebHome) which used HaploGrep2 with Phylotree 17 for haplogroup determination. Mitochondrial DNA genotyping was assessed using four different methods:
PCR amplicon sequencing: The genomic DNA was amplified by PCR. The primer sequences were: HmtL15997: 5’ CACCATTAGCACCCAAAGCT 3’, HmtH112: 5’ ACAGATACTGCGACATAGGG 3’. PCR amplifications were carried out with GoTaq polymerase (Promega, Charbonnières-les-Bains, France) and 1μM of each primer. 50μl reactions were carried out on Eppendorf thermocycler. The PCR products were loaded on agarose gel and purified with QIAEXII gel extraction kit (Qiagen) before Sanger sequencing. Sanger sequencing of the PCR amplicon was done with the same primers.
Direct sequencing: Hi-SNPseq, provided by CD Genomics, combines multiplex PCR and high-throughput sequencing (CD Genomics, Shirley, USA). Multiplex PCR amplification was done with 110 site-specific primers in two single tubes. These two reactions comprise the entire library covering the whole mtDNA sequence. Then, the amplicon pooled samples were purified to remove polymerase, primers, and short by- products. Different samples were distinguished by different barcoded primers. After mixing the samples, high-throughput sequencing was performed (pair ends 150). The sequences were aligned on the reference rCRS genome.
Whole Exome sequencing: Library preparation (Agilent SureSelect Human All Exon V6) and sequencing steps were commissioned to Genewiz (Leipzig, Germany). Raw BCL files generated by the sequencer were converted to fastq files for each sample using bcl2fastq v.2.19. Sequence reads were trimmed to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic v.0.38. The sequences were aligned on the reference rCRS genome.
Whole genome sequencing: For the Brazilian reference population (45), high-coverage WGS data were aligned on the human reference hg38 genome. For each sample, variant calling was done using the Genome Analysis Toolkit 4 (GATK4). Based on the mitochondria variants a mitochondria specific bam file.
2.5. Variant annotations
Sequencing data analysis was performed using a homemade nextflow pipeline that integrates all the classical NGS steps for molecular diagnosis. Mapping and quality control was done using BWA (46) and FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) respectively. Base recalibration, base quality score recalibration and duplicate marking processed with elPrep (47). Variant calling combines the results of GATK Mutect2 (48) in mitochondrial mode and Strelka2 (49). Variant annotation and prioritization were performed using an in-house tool KerMit that integrates various mitochondrial dedicated tools and databases like MITOMAP, HmtDB, ClinVar, MitoTip and HaploGrep for haplogroup prediction. Mitochondrial deletions were searched using eKLIPse (50).
2.6. Statistical analysis
All the statistical tests have been made using R 3.6.3. A first comparison on the presence/absence of genetic variants between two groups was carried out using the chi2 and fisher tests. A second analysis, based on allelic frequency, was done with a Kruskal Wallis test between all groups, and then with a Dunn test comparing phenotypes two by two using the rstatix R package. All the plots have been done with the ggplot2 R package.
Differences between groups are evaluated by statistical tests (Chi2 test with Yate’s correction, Fisher’s exact test, Kruskal Wallis test) using GraphPad Prism 9.3.0. Asterisk and the sharp sign (*) indicate p-value <0.05, (**p-value <0.001, (***) p-value <0.0001.
3. Results
3.1. MtDNA copy number is reduced in heart of Chagas patients
The quantification of mtDNA copy number was evaluated on heart tissue samples from end stage patient heart tissues (n=33) and on healthy hearts of organ donors having no suitable recipient (n=6) (Supplementary Table S1). MtDNA copy number using (MT-ND1) was significantly lower in CCC ((MT-ND1 39%; p= 0.0034) than control samples (Figure 1A) indicating a reduction of mtDNA copy number. Mitochondrial DNA (MT-COX1) gave a similar result (37%; p= 0.0019) (Figure 1B).
The mtDNA copy number was also evaluated in DNA extracted from blood of asymptomatic subjects (n=209), moderate CCC patients (n=235) and severe CCC patients (n=168) (Supplementary Table S2). The age of the individuals which ranges from 40 to 60 years old, was similar among the three groups (Figure 2A). As shown on Figure 2B and Figure 2C, the mtDNA content (and therefore mitochondrial mass) was similar between the three groups. So, the decrease of the mitochondrial mass detected on heart biopsies seems to be tissue specific.
This quantification was done on blood DNAs from patients between 40 and 60 years old. These patients were either asymptomatic for Chagas disease, or developed chronic cardiomyopathy stratified as moderate or severe based on left ventricular ejection fraction values [moderate CCC (0.40<EF<1) and severe CCC (0<EF<0.40)].
Human mtDNA haplogroups and their distributions have been extensively investigated across different nations and geographical regions, predominantly to define population origins and genetic structure. The rate at which mtDNA mutates is known as the mitochondrial molecular clock. The entire mtDNA was sequenced from 26 end stage heart tissue samples and 6 healthy hearts of organ donors. Table 1 describes the mitochondrial haplogroup lineage distribution between both groups.
This distribution suggests that African and European lineages may be enriched in end stage CCC patients compared to healthy controls. However, due to the limited size of our study group, the difference was not statistically significant.
In order to investigate this haplogroup distribution, a larger collection of blood DNA was then investigated. It included CCC patients (n=321), asymptomatic subjects (n=38) and we had access to a census-based sample of elderly individuals from São Paulo (Brazil) (Supplementary Table S3). This latest reference population includes 1171 healthy individuals (45). For each of them, the WGS was available and mitochondrial haplogrouping was assessed in a group of 289 individuals selected randomly from this cohort. The three main lineages were African, European and Native American/Asian (Figure 3). Chagas indivuduals (asymptomatic or with Chronic Chagas Cardiomyopathy) were compared to healthy controls, percentage of African lineage in healthy individuals reached 27% whereas this percentage was 55% in the Chagas patient groups. The Chi-square test with Yates' correction was significant (p<0.0001) with a relative risk of 2.004. This may imply that Chagas patients have a higher African mother lineage than healthy elderly individuals from the state of São Paulo, Brazil.
The percentage of European lineages in healthy individuals reached 44% whereas this percentage was 18% in the Chagas subject groups. The Chi-square test with Yates' correction gave a significant p value (p<0.0001) for a relative risk of 0.5292 (Figure 3).
We then stratified the Chagas subject group, in the asymptomatic/indeterminate subjects, the moderate CCC (0.40<EF<1) and severe CCC (0<EF<0.40) patients. The percentage of each lineage in each subgroup is shown on Figure 4. The African lineage was more represented in the asymptomatic sub-group than the CCC patient sub-group (71% vs 53%) (p=0.04). Similarly, the European lineage was overrepresented in the CCC patient subgroup (7.9% vs 18.7%) but this difference was not significant (p=0.12).
Focusing on African sub-haplogroups, the distribution was compared between ASY subjects, CCC patients, moderate CCC patients and severe CCC patients (Figure 5). This African sub-haplogroup distribution was not significantly different between the study groups (Supplementary Table S4). However, the percentage of L0 sub-haplogroup in ASY individuals reached 11% whereas this percentage was 4% in the CCC patient group. The percentage of L3 sub-haplogroup in ASY individuals reached 22% whereas this percentage was 33% in the CCC patient group. It may suggest that the L0 sub-haplogroup has a protective effect wheras the L3 sub-haplogroup may act as a disease mark.
In the same way, the European sub-haplogroup distribution was similar between the CCC study groups (Figure 6). However, when we compared the frequency of the European H sub-haplogroup, we detected some trends of association Table 2.
Whole mtDNA sequencing was performed on 112 samples (Supplementary Table S5). It included 38 asymptomatic subjects and 74 CCC patients. Among the CCC patients, 34 patients suffered from moderate cardiomyopathy whereas 36 patients had a severe cardiomyopathy.
Sequences obtained from on 112 samples were aligned according to the Cambridge Reference Sequence (rCRS). 12429 variants were detected. Three analyses were done. First of all, we compared the number of carriers of each variant between each group (ASY vs CCC; ASY vs Moderate CCC; ASY vs Severe CCC and Moderate CCC vs Severe CCC) (Supplementary Table S6). We also compared the heteroplasmy levels between the three groups (Kruskal Wallis between ASY, moderate CCC and severe CCC). Finally, we compared the heteroplasmy levels between the groups (two by two: ASY vs CCC; ASY vs Moderate CCC; ASY vs Severe CCC and Moderate CCC vs Severe CCC) (Supplementary Table S7). All these tests were corrected for multiple testing. 712 variants showed significant results in at least one test (adjusted p value < 0.05).
We annotated the associated variants, based on available information in VEP ClinVar, MitoMap and MitImpact databases (Supplementary Table S8 and S9). These variants are either located in intergenic region (n=220; 30.8%) or in coding region (n=493; 69.2%). Among these variants, only 84 variants change the protein sequence (Table 3). These protein sequence variants are enriched in a limited number of genes (MT-ND5 (n=21), MT-CYB (n=15), MT-ATP6 (n=11), MT-CO3 (n=8), MT-CO1 (n=7), MT-ND4 (n=6), MT-ATP8 (n=5), MT-ND6 (n=4) MT-ND4L (n=2), MT-ND1 (n=2), MT-CO2 (n=2), MT-ND2 (n=1)). The pathogenicity of each variant was defined with MitImpact which aggregate 24 databases (Clinvar, PolyPhen2, SIFT, FatHmmW, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, VEST, PANTHER, PhDSNP, SP, MutationTaster, SNPDryad, Condel, COVEC_WMV, MtoolBox, APOGEE, DEOGEN2, PolyPhen2 transf, SIFT transf, Mutatiossessor transf) (51). All the 84 protein modifier variants were classified as pathogenic in at least one database. 16 protein modifier variants were classified as pathogenic in more than 10 databases.
Among these 712 variants, 70 of them were associated to diseases (Supplementary Table S9). The 70 variants include 40 protein modifier variants. Among them, 2 were associated to Left Ventricular Non-Compaction disease and 2 to hypertrophic cardiomyopathy.
4. Discussion
We have found that mtDNA copy number in heart tissue was significantly lower in CCC than in healthy individuals, while blood mtDNA content was similar among asymptomatic Chagas disease, moderate and severe CCC patients. While mitochondrial haplogroup lineages of severe CCC patients and healthy heart donors were predominantly African, the percentage of African lineage was much higher among Chagas disease patients from the peripheral blood cohort than healthy individuals from the census-based São Paulo reference population. Conversely, the percentage of the European lineage was under-represented among the Chagas disease cohort than the reference population. The African lineage was over represented in the asymptomatic subgroup when compared with the CCC patient subgroup. Regarding European major haplogroups, H haplogroup was more prevalent among Chagas patients than in the reference population. Whole mtDNA sequencing disclosed that 84 CCC-specific variants were non-synonymous pathogenic changes, some of them previously associated with heart disease.
Here, we have shown that heart from end stage CCC patients display lower mitochondrial DNA copy number than heart tissues from organ donors having no suitable recipient. This result was also consistent with our previous findings that CCC showed even lower mtDNA copy number than other non-inflammatory cardiomyopathies (36) and heart proteomic analysis done on myocardial tissue (32-35). The finding that mtDNA copy number in blood is comparable among Chagas disease clinical groups indicates that the mtDNA copy number variation is tissue specific, and therefore mitochondrial dysfunction detected in CCC hearts is tissue-specific. Indeed, mtDNA copy number variation is depending on cell-type (52). mtDNA copy number variations have been associated to multiple diseases including cardiovascular diseases such as Myocardial Infraction (53), cardiomyopathy, heart failure, and arrhythmias (53). However, our data indicate that blood mtDNA copy number is not a useful clinical biomarker for CCC.
Cardiac mitochondria provide the most ATP in the heart. Therefore, mitochondrial dysfunction is pivotal in heart diseases because of energy supply shortage and increased production of reactive oxidative species (ROS). So, maintaining optimal mitochondrial homeostasis is essential for cardiomyocytes. The total mitochondrial content is mainly controlled by biogenesis and mitochondria-specific autophagy for degradation (54). Damaged mitochondria are normally removed by autophagy/lysosome machinery (55). Several studies described the importance of autophagy in parasite invasion (56). The most characterized mechanism of autophagy is the ATG5-, ATG7-, and LC3-mediated conventional autophagy pathway (57), recent studies have identified an Atg5/Atg7-independent pathway called alternative autophagy (58). In chronic Chagas patients, autophagy activity was not completely investigated. However, based on bulk RNA-seq we investigated the genetic deregulations present in the moderate and severe stages of CCC. Several genes, associated to mitophagy, are differentially expressed in CCC bulk tissues (such as ATG7, LC3, FIP200 and BECN1) (59). Severe CCC is associated to a myocardial inflammatory infiltrate and to an IFNγ/TNFα-rich Th1 type response (22-28). This exacerbated response will lead to ROS/NOS overproduction triggering mitochondrial damage and cardiomyocyte apoptosis. Defective mitophagy or autophagy may be associated to mtDNA release into the cytoplasm, outside of the cell, or into circulation. Circulating mtDNA enhances expression of type I interferons, and by NLRP3 inflammasome, which promotes the activation of pro-inflammatory cytokines Interleukin-1beta and Interleukin-18 (60). These trigger inflammatory responses in cardiomyocytes and may induce myocarditis, and dilated cardiomyopathy (61).
MtDNA haplogrouping study indicates that African haplogroups were over represented in the Chagas subject groups in compared to healthy individuals. Even if it is not statistically significant, the L0 African sub-haplogroup is more frequent in ASY individuals than in CCC patient groups. The L3 African sub-haplogroup is less frequent in ASY individuals than in the CCC patients. This sub-haplogroup distribution among the groups is consistent with the association of RNR1-921T/C which is a key marker of the L3 African sub-haplogroup. We cannot conclude that the African haplogroup is directly associated with an increased risk of developing severe chronic chagasic cardiomyopathy. It can be simply stated that individuals with African ancestry have a greater chance of lower socioeconomic status that exposes them to parasitic diseases such as Chagas disease. Therefore, the percentage of patients with CCC in the African ethnic group increases. Conversely, the reverse is observed for the European haplogroup. Our results suggest that European macro haplogroup H is associated with increased risk towards CCC. This result agrees with previous work on dilated and hypertrophic cardiomyopathy (62-64). Significantly, intrinsic mitochondrial function has been reported in skeletal muscle myocytes from individuals carrying haplogroup H as compared to haplogroup U (18). This may imply that different haplogroups display distinct mitochondrial function profiles. In the case of Haplogroup H, it is possible to hypothesize that increased OXPHOS is associated with increased ROS production, which may lead to mitochondrial dysfunction and heart damage in CCC patients.
The two main types of mitochondrial DNA mutation are large deletion and point mutations, mostly maternally transmitted, with some de novo mutations have also been reported. Here, no deletion was associated to Chagas disease. The finding of 84 mtDNA-encoded protein sequence pathogenic variants associated with CCC may indicate a role of mtDNA variants in CCC pathogenesis. At this level, the genetic contribution of each of them to the phenotype is undefined. It has been reported that mtDNA variants affect the expressivity of nDNA mutations, leading to experimental cardiomyopathy (65). The functional characterization is still an important bottleneck in mitochondrial diseases. Indeed, in the databases on mitochondrial variants, a very small proportion of these variants have been functionally characterized. Thus, for the vast majority of variants, indications of their functional implications remain putative. Moreover, as no clear variant is associated to disease here, may suggested an additive effet of the mt DNA variants. These studies are complicated by the fact that these variants have been shown to have incomplete penetrance (66). In addition, to complicate matters, mitochondrial disorders are clinically heterogeneous diseases associated with germline mutations in mitochondrial DNA (mtDNA) and/or nuclear DNA-encoded (nDNA) genes, with impaired mitochondrial structure and function. Therefore, mutations at the mtDNA or nDNA levels can have a variety of pathogenesis outcomes. We can thus postulate a synergic effect between these mtDNA variants and nuclear DNA mitochondrial variants previously associated to CCC (42). The description and the characterization of these variants is essential to define a susceptibility polygenic score analysis including all the associated variants.
When the proportion of harmful mtDNA variants reaches a critical level of heteroplasmy, the defects that can give rise to disease. The description of associations of pathogenic variants to multiple diseases, raise the concept of mitochondria genome editing technology. Gene editing technologies, commonly used for nuclear variants, are not so obvious to apply to mitochondria variants. The first approach leads to decrease the amount of variant mtDNA in mitochondria by specifically targeting and cleaving the mutant mtDNA molecules. Double-strand breaks in mutated mtDNA induce the rapid degradation of the linearized molecule. The residual mtDNA, mostly wild-type, replicates and the heteroplasmy level decreases. The critical point in this approach is the efficiency of the nucleases (mitoREs, mtZFNs, mitoTALENs) (67-69). The development of mitochondria targeted CRISPR/Cas9 systems has been hampered by the lack of efficiency to import guide RNA (gRNA) into the mitochondrial matrix (70, 71). The second approach does not try to decrease the heteroplasmy level but simply to correct the mutated base (72). This concept has been recently described by Kar et al. (73).
There is no cure available for mitochondrial diseases owing to the different genes and phenotypes associated with the cause of such disorders. Nevertheless, few symptomatic treatments have been proven by clinical trials as palliative therapies in the last decade. A mitochondrial cocktail, i.e. a combination of vitamins, cofactors, nutrients and antioxidants, may alleviate symptoms, limit disease progression, and overcome mitochondrial toxins. In the short term, specific dietary restrictions have been shown to ameliorate mitochondrial health in patients with mitochondrial disorders. Thus, high-carbohydrate diet increases oxidative stress, perhaps for individuals with impaired oxidative phosphorylation (74). The ketogenic diet (high-fat diet) has been shown to be beneficial for patients with pyruvate dehydrogenase deficiency, but not for pyruvate carboxylase deficiency and in the treatment of fatty acid oxidation disorders (75). Individuals with disorders of the respiratory chain could be treated with agents that enhance electron transport and substrate delivery and bypass of its components. For example, CoQ10 supplementation and have been shown to reduce high lactate levels after exercise and increase oxygen consumption (76, 77). A diet rich in vitamins and amino acids can be used as a source of redox agents and intracellular buffers for ATP (78). For stroke-like episodes, myopathy, diabetes and lactic acidosis a treatment using natural NO precursors (e.g. arginine and citrulline) was found to restore NO production (74, 79, 80).
Our results are consistent with our previous data based on proteomic, transcriptomic and genetic analyses (27, 33, 35, 36, 42, 59, 81-83). Mitochondrial dysfunction is clearly related to the compromise of mitochondria's ability to make appropriate levels of ATP and to an enhanced formation of reactive oxygen species (ROS). This dysfunction may be the result of mtDNA or nDNA variants, but may also occur as a response to aging and various disease and environmental stresses, leading to the development of cardiomyopathies (CMs), ROS accumulation and cell death. Mitochondrial dysfunction can have a direct deleterious effect on the contractile capacity of myocardial cells. Defects in mitochondrial regulation of calcium homeostasis can alter mechanical function and electrical conduction (84). Moreover, pathogenic inflammatory responses produced by damaged mitochondria can particularly trigger the atherosclerotic process. ROS generated in the mitochondria may lead to target-organ damage, dysfunction, hypertrophy, and inflammation. Hypertension and cardiac fibrosis are associated with depletion and inactivation of the key mitochondrial deacetylase, sirtuin 3, which is involved in the control of key metabolic steps (85-87).
The finding that pathogenic mitochondrial protein-coding SNPs and mitochondrial haplogroups associate with risk of evolving to CCC is consistent with a key role of mitochondrial DNA in the development of Chronic Chagas disease Cardiomyopathy and are in line with previous reports of nuclear DNA-encoded mitochondrial gene variants. These may interact and increase mitochondrial susceptibility to IFN-γ-induced damage in the CCC myocardium, leading to ventricular dysfunction.
5. Conclusions
This section is mandatory, with one or two paragraphs to end the main text.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Heart tissue samples used for mitochondria quantification. Table S2: Blood DNAs samples used for mitochondria quantification. Table S3: Blood samples used for haplogroup analysis. Table S4: Statistics on African sub-haplogroup distribution. Table S5: Clinical information on patients for whom the whole mitochondria genome has been sequenced (n=106). Table S6: Carrier analysis parformed on 12429 Variants. Table S7: Heteroplasmy level test between the three groups (ASY, moderate CCC and severe CCC) and heteroplasmy level analyses between the groups (two by two: ASY vs CCC; ASY vs Moderate CCC; ASY vs Severe CCC and Moderate CCC vs Severe CCC). Table S8: Variant annotation. Table S9: List of the variants associated to diseases.
Author Contributions
Experimental work: FG, JPSN, PA; Bioinformatic analysis: PB, DG; Biological sample collect: BMI, CM, AFF, RHBS, AK, SS, ANS, PP, AIF, EAB, CWP, BS, FCD, MFS, FAG, JADLP, FB, PB, RRA, HTLW, AS, MHH, EAD, ACP, VRJ, MM; Writing—original draft preparation: PB, PA, JPSN, ECN, CC; Writing—review and editing: MN, JK, VP, ECN, CC.
Funding
This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM); the Aix-Marseille University (grant number: AMIDEX “Internation-al_2018” MITOMUTCHAGAS); the French Agency for Research (Agence Nationale de la Recherche-ANR (grant numbers: “Br-Fr-Chagas”, “landscardio”). This work was founded by the Inserm Cross-Cutting Project GOLD. This project has received funding from the Ex-cellence Initiative of Aix-Marseille University - A*Midex a French “Investissements d’Avenir programme”- Institute MarMaRa AMX-19-IET-007. This work was supported by FAPESP (São Paulo State Research Funding Agency Brazil) (grant 2022/00758-0). Brazilian National Research Council (CNPq) provided a Productivity Award for ECN and JK. This work is supported by the CAPES-COFECUB program (Me987/23). CC is supported by the Inserm’s PRI/IRP 2022 program (Me987/23). The funders did not play any role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data Availability Statement
The data analyzed during the current study is available upon reasonable requests to the corresponding authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O'Connell, J.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996, 93, 841–2. [Google Scholar]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef]
- Mestroni, L.; Brun, F.; Spezzacatene, A.; Sinagra, G.; Taylor, M.R. Genetic Causes of Dilated Cardiomyopathy. Prog Pediatr Cardiol. 2014, 37, 13–8. [Google Scholar] [CrossRef]
- Bakalakos, A.; Ritsatos, K.; Anastasakis, A. Current perspectives on the diagnosis and management of dilated cardiomyopathy Beyond heart failure: a Cardiomyopathy Clinic Doctor's point of view. Hellenic J Cardiol 2018, 59, 254–61. [Google Scholar] [CrossRef]
- Akhtar, M.M.; Lorenzini, M.; Cicerchia, M.; Ochoa, J.P.; Hey, T.M.; Sabater Molina, M.; et al. Clinical Phenotypes and Prognosis of Dilated Cardiomyopathy Caused by Truncating Variants in the TTN Gene. Circ Heart Fail. 2020, 13, e006832. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, S.E.; Cho, M.C. Clinical Implication of Genetic Testing in Dilated Cardiomyopathy. Int J Heart Fail 2022, 4, 1–11. [Google Scholar] [CrossRef]
- Lu, J.T.; Muchir, A.; Nagy, P.L.; Worman, H.J. LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis Model Mech 2011, 4, 562–8. [Google Scholar] [CrossRef]
- Tabish, A.M.; Azzimato, V.; Alexiadis, A.; Buyandelger, B.; Knoll, R. Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys Rev 2017, 9, 207–23. [Google Scholar] [CrossRef]
- Zak, R.; Rabinowitz, M.; Rajamanickam, C.; Merten, S.; Kwiatkowska-Patzer, B. Mitochondrial proliferation in cardiac hypertrophy. Basic Res Cardiol 1980, 75, 171–8. [Google Scholar] [CrossRef]
- Goffart, S.; von Kleist-Retzow, J.C.; Wiesner, R.J. Regulation of mitochondrial proliferation in the heart: power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc Res 2004, 64, 198–207. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front Cardiovasc Med. 2016, 3, 25. [Google Scholar] [CrossRef]
- Potgieter, M.; Pretorius, E.; Pepper, M.S. Primary and secondary coenzyme Q10 deficiency: the role of therapeutic supplementation. Nutr Rev 2013, 71, 180–8. [Google Scholar] [CrossRef]
- Thorburn, D.R.; Sugiana, C.; Salemi, R.; Kirby, D.M.; Worgan, L.; Ohtake, A.; et al. Biochemical and molecular diagnosis of mitochondrial respiratory chain disorders. Biochim Biophys Acta. 2004, 1659, 121–8. [Google Scholar] [CrossRef]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol 2014, 171, 2080–90. [Google Scholar] [CrossRef]
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol 2014, 73, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; et al. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab 2014, 20, 158–71. [Google Scholar] [CrossRef]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef]
- Larsen, S.; Diez-Sanchez, C.; Rabol, R.; Ara, I.; Dela, F.; Helge, J.W. Increased intrinsic mitochondrial function in humans with mitochondrial haplogroup H. Biochim Biophys Acta 2014, 1837, 226–31. [Google Scholar] [CrossRef]
- Perez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Morillo, C.A.; Marin-Neto, J.A.; Avezum, A.; Sosa-Estani, S.; Rassi, A.; Rosas, F., Jr.; et al. Randomized Trial of Benznidazole for Chronic Chagas' Cardiomyopathy. N Engl J Med 2015, 373, 1295–306. [Google Scholar] [CrossRef]
- Bestetti, R.B.; Muccillo, G. Clinical course of Chagas' heart disease: a comparison with dilated cardiomyopathy. Int J Cardiol 1997, 60, 187–93. [Google Scholar] [CrossRef]
- Mady, C.; Cardoso, R.H.; Barretto, A.C.; da Luz, P.L.; Bellotti, G.; Pileggi, F. Survival and predictors of survival in patients with congestive heart failure due to Chagas' cardiomyopathy. Circulation 1994, 90, 3098–102. [Google Scholar] [CrossRef]
- Higuchi, M.L.; De Morais, C.F.; Pereira Barreto, A.C.; Lopes, E.A.; Stolf, N.; Bellotti, G.; et al. The role of active myocarditis in the development of heart failure in chronic Chagas' disease: a study based on endomyocardial biopsies. Clin Cardiol 1987, 10, 665–70. [Google Scholar] [CrossRef]
- Pereira Barretto, A.C.; Mady, C.; Arteaga-Fernandez, E.; Stolf, N.; Lopes, E.A.; Higuchi, M.L.; et al. Right ventricular endomyocardial biopsy in chronic Chagas' disease. Am Heart J 1986, 111, 307–12. [Google Scholar] [CrossRef]
- Abel, L.C.; Rizzo, L.V.; Ianni, B.; Albuquerque, F.; Bacal, F.; Carrara, D.; et al. Chronic Chagas' disease cardiomyopathy patients display an increased IFN-gamma response to Trypanosoma cruzi infection. J Autoimmun 2001, 17, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.C.; Ianni, B.M.; Abel, L.C.; Buck, P.; Mady, C.; Kalil, J.; et al. Increased plasma levels of tumor necrosis factor-alpha in asymptomatic/"indeterminate" and Chagas disease cardiomyopathy patients. Mem Inst Oswaldo Cruz 2003, 98, 407–11. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.G.; Santos, R.H.; Ianni, B.M.; Fiorelli, A.I.; Mairena, E.C.; Benvenuti, L.A.; et al. Myocardial chemokine expression and intensity of myocarditis in Chagas cardiomyopathy are controlled by polymorphisms in CXCL9 and CXCL10. PLoS Negl Trop Dis. 2012, 6, e1867. [Google Scholar] [CrossRef] [PubMed]
- Talvani, A.; Rocha, M.O.; Barcelos, L.S.; Gomes, Y.M.; Ribeiro, A.L.; Teixeira, M.M. Elevated concentrations of CCL2 and tumor necrosis factor-alpha in chagasic cardiomyopathy. Clin Infect Dis 2004, 38, 943–50. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.M.; Gazzinelli, R.T.; Silva, J.S. Chemokines, inflammation and Trypanosoma cruzi infection. Trends Parasitol 2002, 18, 262–5. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Neto, E.; Dzau, V.J.; Allen, P.D.; Stamatiou, D.; Benvenutti, L.; Higuchi, M.L.; et al. Cardiac gene expression profiling provides evidence for cytokinopathy as a molecular mechanism in Chagas' disease cardiomyopathy. Am J Pathol 2005, 167, 305–13. [Google Scholar] [CrossRef] [PubMed]
- Laugier, L.; Ferreira, L.R.P.; Ferreira, F.M.; Cabantous, S.; Frade, A.F.; Nunes, J.P.; et al. miRNAs may play a major role in the control of gene expression in key pathobiological processes in Chagas disease cardiomyopathy. PLoS Negl Trop Dis. 2020, 14, e0008889. [Google Scholar] [CrossRef]
- Cunha-Neto, E.; Teixeira, P.C.; Fonseca, S.G.; Bilate, A.M.; Kalil, J. Myocardial gene and protein expression profiles after autoimmune injury in Chagas' disease cardiomyopathy. Autoimmun Rev 2011, 10, 163–5. [Google Scholar] [CrossRef]
- Teixeira, P.C.; Ducret, A.; Langen, H.; Nogoceke, E.; Santos, R.H.B.; Silva Nunes, J.P.; et al. Impairment of Multiple Mitochondrial Energy Metabolism Pathways in the Heart of Chagas Disease Cardiomyopathy Patients. Front Immunol. 2021, 12, 755782. [Google Scholar] [CrossRef]
- Teixeira, P.C.; Iwai, L.K.; Kuramoto, A.C.; Honorato, R.; Fiorelli, A.; Stolf, N.; et al. Proteomic inventory of myocardial proteins from patients with chronic Chagas' cardiomyopathy. Braz J Med Biol Res 2006, 39, 1549–62. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.C.; Santos, R.H.; Fiorelli, A.I.; Bilate, A.M.; Benvenuti, L.A.; Stolf, N.A.; et al. Selective decrease of components of the creatine kinase system and ATP synthase complex in chronic Chagas disease cardiomyopathy. PLoS Negl Trop Dis. 2011, 5, e1205. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.P.S.; Andrieux, P.; Brochet, P.; Almeida, R.R.; Kitano, E.; Honda, A.K.; et al. Co-Exposure of Cardiomyocytes to IFN-gamma and TNF-alpha Induces Mitochondrial Dysfunction and Nitro-Oxidative Stress: Implications for the Pathogenesis of Chronic Chagas Disease Cardiomyopathy. Front Immunol. 2021, 12, 755862. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C.; Jr Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 2013, 113, 603–16. [Google Scholar] [CrossRef] [PubMed]
- Zicker, F.; Smith, P.G.; Netto, J.C.; Oliveira, R.M.; Zicker, E.M. Physical activity, opportunity for reinfection, and sibling history of heart disease as risk factors for Chagas' cardiopathy. Am J Trop Med Hyg 1990, 43, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Herrera, M.; Strauss, M.; Casares-Marfil, D.; Martin, J.; Chagas Genetics, C.N. Genomic medicine in Chagas disease. Acta Trop. 2019, 197, 105062. [Google Scholar] [CrossRef] [PubMed]
- Chevillard, C.; Nunes, J.P.S.; Frade, A.F.; Almeida, R.R.; Pandey, R.P.; Nascimento, M.S.; et al. Disease Tolerance and Pathogen Resistance Genes May Underlie Trypanosoma cruzi Persistence and Differential Progression to Chagas Disease Cardiomyopathy. Front Immunol. 2018, 9, 2791. [Google Scholar] [CrossRef]
- Ferreira, L.R.; Frade, A.F.; Baron, M.A.; Navarro, I.C.; Kalil, J.; Chevillard, C.; et al. Interferon-gamma and other inflammatory mediators in cardiomyocyte signaling during Chagas disease cardiomyopathy. World J Cardiol 2014, 6, 782–90. [Google Scholar] [CrossRef] [PubMed]
- Ouarhache, M.; Marquet, S.; Frade, A.F.; Ferreira, A.M.; Ianni, B.; Almeida, R.R.; et al. Rare Pathogenic Variants in Mitochondrial and Inflammation-Associated Genes May Lead to Inflammatory Cardiomyopathy in Chagas Disease. J Clin Immunol 2021, 41, 1048–63. [Google Scholar] [CrossRef] [PubMed]
- Frade, A.F.; Teixeira, P.C.; Ianni, B.M.; Pissetti, C.W.; Saba, B.; Wang, L.H.; et al. Polymorphism in the alpha cardiac muscle actin 1 gene is associated to susceptibility to chronic inflammatory cardiomyopathy. PLoS One 2013, 8, e83446. [Google Scholar] [CrossRef]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr Protoc Mouse Biol 2017, 7, 47–54. [Google Scholar] [CrossRef]
- Naslavsky, M.S.; Scliar, M.O.; Yamamoto, G.L.; Wang, J.Y.T.; Zverinova, S.; Karp, T.; et al. Whole-genome sequencing of 1,171 elderly admixed individuals from Sao Paulo, Brazil. Nat Commun. 2022, 13, 1004. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–95. [Google Scholar] [CrossRef] [PubMed]
- Herzeel, C.; Costanza, P.; Decap, D.; Fostier, J.; Verachtert, W. elPrep 4: A multithreaded framework for sequence analysis. PLoS One. 2019, 14, e0209523. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011, 43, 491–8. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Kallberg, M.; et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 2018, 15, 591–4. [Google Scholar] [CrossRef]
- Goudenege, D.; Bris, C.; Hoffmann, V.; Desquiret-Dumas, V.; Jardel, C.; Rucheton, B.; et al. eKLIPse: a sensitive tool for the detection and quantification of mitochondrial DNA deletions from next-generation sequencing data. Genet Med 2019, 21, 1407–16. [Google Scholar] [CrossRef]
- Castellana, S.; Ronai, J.; Mazza, T. MitImpact: an exhaustive collection of pre-computed pathogenicity predictions of human mitochondrial non-synonymous variants. Hum Mutat. 2015, 36, E2413–22. [Google Scholar] [CrossRef]
- Picard, M. Blood mitochondrial DNA copy number: What are we counting? Mitochondrion 2021, 60, 1–11. [Google Scholar] [CrossRef]
- Sundquist, K.; Sundquist, J.; Palmer, K.; Memon, A.A. Role of mitochondrial DNA copy number in incident cardiovascular diseases and the association between cardiovascular disease and type 2 diabetes: A follow-up study on middle-aged women. Atherosclerosis 2022, 341, 58–62. [Google Scholar] [CrossRef]
- Nah, J. The Role of Alternative Mitophagy in Heart Disease. Int J Mol Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C.; Marzetti, E. Contribution of impaired mitochondrial autophagy to cardiac aging: mechanisms and therapeutic opportunities. Circ Res 2012, 110, 1125–38. [Google Scholar] [CrossRef] [PubMed]
- Romano, P.S.; Cueto, J.A.; Casassa, A.F.; Vanrell, M.C.; Gottlieb, R.A.; Colombo, M.I. Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB Life 2012, 64, 387–96. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: machinery and regulation. Microb Cell 2016, 3, 588–96. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–8. [Google Scholar] [CrossRef] [PubMed]
- Brochet, P.; Ianni, B.M.; Laugier, L.; Frade, A.F.; Silva Nunes, J.P.; Teixeira, P.C.; et al. Epigenetic regulation of transcription factor binding motifs promotes Th1 response in Chagas disease cardiomyopathy. Front Immunol. 2022, 13, 958200. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; et al. Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–5. [Google Scholar] [CrossRef] [PubMed]
- Hagen, C.M.; Aidt, F.H.; Hedley, P.L.; Jensen, M.K.; Havndrup, O.; Kanters, J.K.; et al. Mitochondrial haplogroups modify the risk of developing hypertrophic cardiomyopathy in a Danish population. PLoS One 2013, 8, e71904. [Google Scholar] [CrossRef] [PubMed]
- Govindaraj, P.; Rani, B.; Sundaravadivel, P.; Vanniarajan, A.; Indumathi, K.P.; Khan, N.A.; et al. Mitochondrial genome variations in idiopathic dilated cardiomyopathy. Mitochondrion 2019, 48, 51–9. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Caggiano, M.; Barallobre-Barreiro, J.; Rego-Perez, I.; Crespo-Leiro, M.G.; Paniagua, M.J.; Grille, Z.; et al. Mitochondrial DNA haplogroup H as a risk factor for idiopathic dilated cardiomyopathy in Spanish population. Mitochondrion 2013, 13, 263–8. [Google Scholar] [CrossRef] [PubMed]
- McManus, M.J.; Picard, M.; Chen, H.W.; De Haas, H.J.; Potluri, P.; Leipzig, J.; et al. Mitochondrial DNA Variation Dictates Expressivity and Progression of Nuclear DNA Mutations Causing Cardiomyopathy. Cell Metab 2019, 29, 78–90 e5. [Google Scholar] [CrossRef]
- Garret, P.; Bris, C.; Procaccio, V.; Amati-Bonneau, P.; Vabres, P.; Houcinat, N.; et al. Deciphering exome sequencing data: Bringing mitochondrial DNA variants to light. Hum Mutat 2019, 40, 2430–43. [Google Scholar] [CrossRef] [PubMed]
- Yahata, N.; Matsumoto, Y.; Omi, M.; Yamamoto, N.; Hata, R. TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m. 13513G>A mutation. Sci Rep. 2017, 7, 15557. [Google Scholar]
- Gammage, P.A.; Minczuk, M. Enhanced Manipulation of Human Mitochondrial DNA Heteroplasmy In Vitro Using Tunable mtZFN Technology. Methods Mol Biol 2018, 1867, 43–56. [Google Scholar]
- Srivastava, S.; Moraes, C.T. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum Mol Genet 2001, 10, 3093–9. [Google Scholar] [CrossRef]
- Hussain, S.A.; Yalvac, M.E.; Khoo, B.; Eckardt, S.; McLaughlin, K.J. Adapting CRISPR/Cas9 System for Targeting Mitochondrial Genome. Front Genet. 2021, 12, 627050. [Google Scholar] [CrossRef]
- Bian, W.P.; Chen, Y.L.; Luo, J.J.; Wang, C.; Xie, S.L.; Pei, D.S. Knock-In Strategy for Editing Human and Zebrafish Mitochondrial DNA Using Mito-CRISPR/Cas9 System. ACS Synth Biol 2019, 8, 621–32. [Google Scholar] [CrossRef] [PubMed]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: advances and therapeutic opportunities. Nat Rev Drug Discov 2020, 19, 839–59. [Google Scholar] [CrossRef] [PubMed]
- Kar, B.; Castillo, S.R.; Sabharwal, A.; Clark, K.J.; Ekker, S.C. Mitochondrial Base Editing: Recent Advances towards Therapeutic Opportunities. Int J Mol Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Hsu, J.W.; Emrick, L.T.; Wong, L.J.; Craigen, W.J.; Jahoor, F.; et al. Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol Genet Metab 2012, 105, 607–14. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol 2006, 60, 223–35. [Google Scholar] [CrossRef] [PubMed]
- Rotig, A.; Appelkvist, E.L.; Geromel, V.; Chretien, D.; Kadhom, N.; Edery, P.; et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 2000, 356, 391–5. [Google Scholar] [CrossRef]
- Di Giovanni, S.; Mirabella, M.; Spinazzola, A.; Crociani, P.; Silvestri, G.; Broccolini, A.; et al. Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology 2001, 57, 515–8. [Google Scholar] [CrossRef]
- Parikh, S.; Saneto, R.; Falk, M.J.; Anselm, I.; Cohen, B.H.; Haas, R.; et al. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 2009, 11, 414–30. [Google Scholar] [CrossRef]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012, 2012, CD004426. [Google Scholar] [CrossRef] [PubMed]
- Almannai, M.; El-Hattab, A.W. Nitric Oxide Deficiency in Mitochondrial Disorders: The Utility of Arginine and Citrulline. Front Mol Neurosci. 2021, 14, 682780. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int J Mol Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Laugier, L.; Frade, A.F.; Ferreira, F.M.; Baron, M.A.; Teixeira, P.C.; Cabantous, S.; et al. Whole-Genome Cardiac DNA Methylation Fingerprint and Gene Expression Analysis Provide New Insights in the Pathogenesis of Chronic Chagas Disease Cardiomyopathy. Clin Infect Dis 2017, 65, 1103–11. [Google Scholar] [CrossRef]
- Nunes, J.P.S.; Moraes-Vieira, P.M.; Chevillard, C.; Cunha-Neto, E. Editorial: Mitochondria at the Crossroads of Immunity and Inflammatory Tissue Damage. Front Immunol. 2021, 12, 810787. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Crisosto, C.; Pennanen, C.; Vasquez-Trincado, C.; Morales, P.E.; Bravo-Sagua, R.; Quest, A.F.G.; et al. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat Rev Cardiol 2017, 14, 342–60. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, D.; Dong, X.; Zhang, X.; Liu, J.; Peng, L.; et al. LncDACH1 promotes mitochondrial oxidative stress of cardiomyocytes by interacting with sirtuin3 and aggravates diabetic cardiomyopathy. Sci China Life Sci 2022, 65, 1198–212. [Google Scholar] [CrossRef]
- Chen, W.J.; Cheng, Y.; Li, W.; Dong, X.K.; Wei, J.L.; Yang, C.H.; et al. Quercetin Attenuates Cardiac Hypertrophy by Inhibiting Mitochondrial Dysfunction Through SIRT3/PARP-1 Pathway. Front Pharmacol. 2021, 12, 739615. [Google Scholar] [CrossRef]
- Chen, J.; Chen, S.; Zhang, B.; Liu, J. SIRT3 as a potential therapeutic target for heart failure. Pharmacol Res. 2021, 165, 105432. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mitochondrial DNA quantification in heart tissues. We performed qPCR with primers for mitochondrial encoded genes NADH: ubiquinone oxidoreductase subunit 1 (MT-ND1), cytochrome c oxidase subunit 1 (MT-COXI) and endogenous nuclear-encoded control gene Glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The relative quantification of mitochondrial DNA was calculated by determining the ΔCt = Ct(endogenous nuclear-encoded control gene)-Ct(mitochondrial encoded genes) and then using the formula 2x2ΔCT.
Figure 1.
Mitochondrial DNA quantification in heart tissues. We performed qPCR with primers for mitochondrial encoded genes NADH: ubiquinone oxidoreductase subunit 1 (MT-ND1), cytochrome c oxidase subunit 1 (MT-COXI) and endogenous nuclear-encoded control gene Glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The relative quantification of mitochondrial DNA was calculated by determining the ΔCt = Ct(endogenous nuclear-encoded control gene)-Ct(mitochondrial encoded genes) and then using the formula 2x2ΔCT.
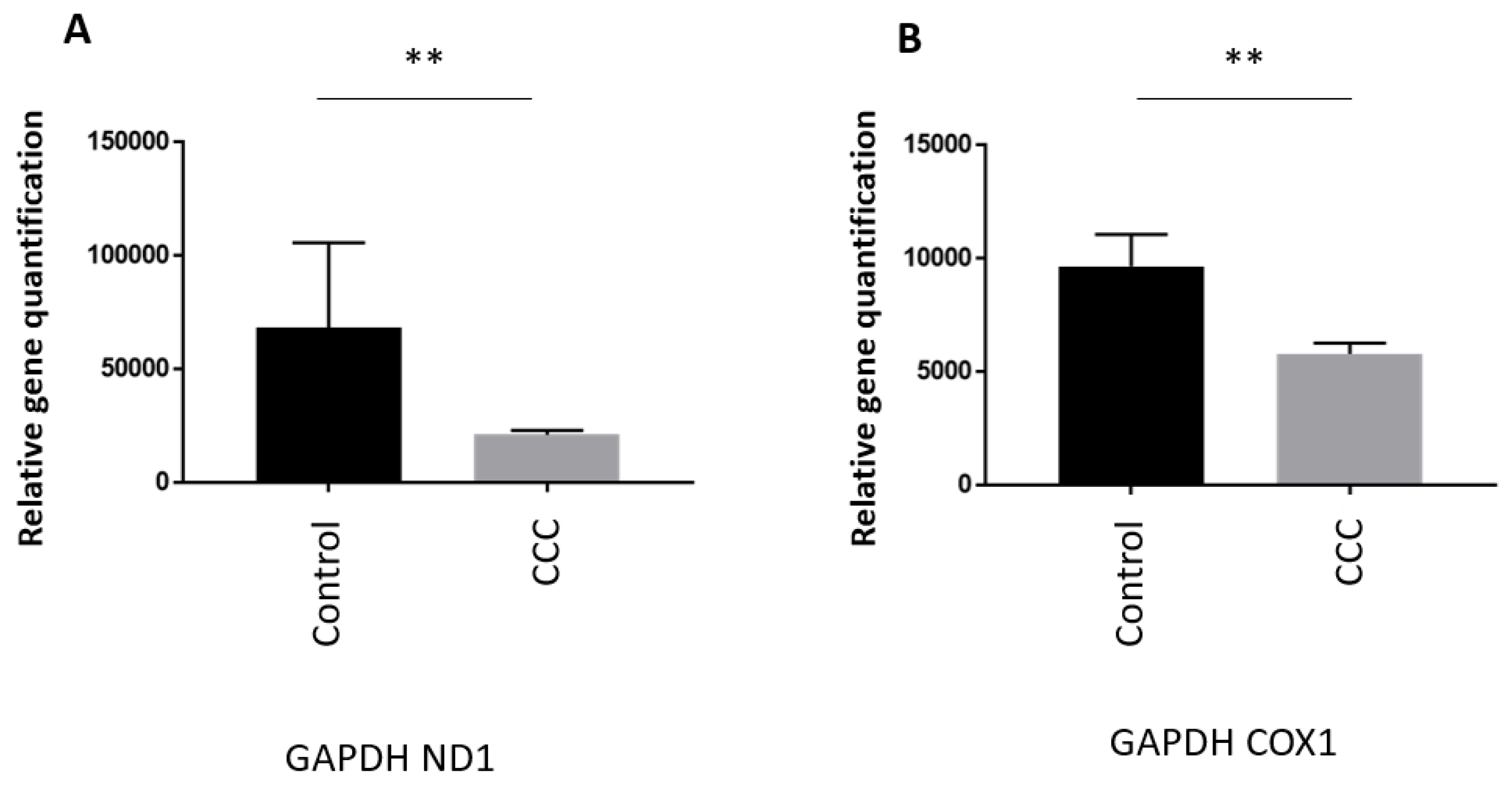
Figure 2.
Mitochondrial DNA quantification in blood DNA samples.
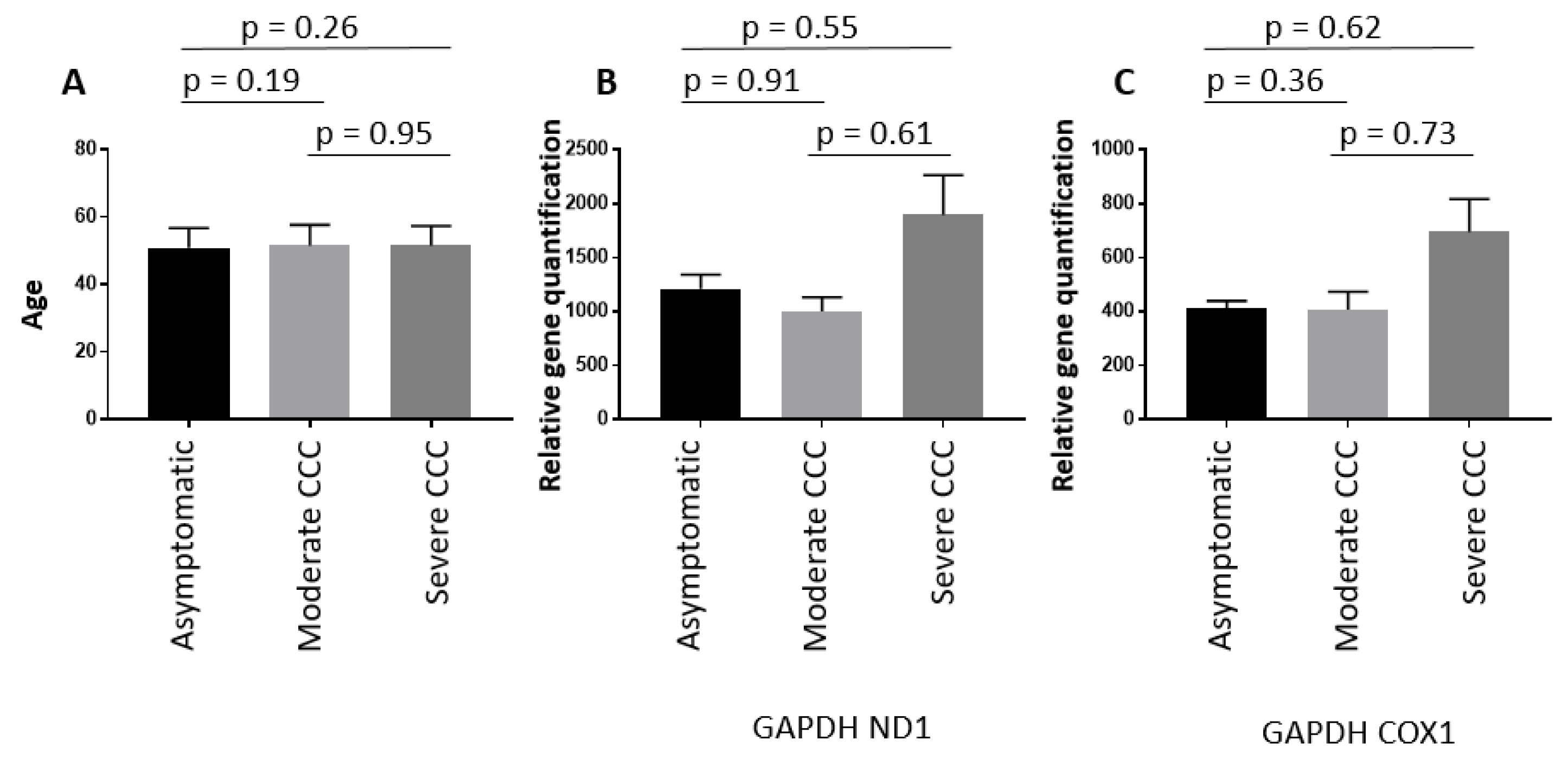
Figure 3.
Mitochondrial haplogroup distribution between the healthy Brazilian reference population and the Chagas cohort.
Figure 3.
Mitochondrial haplogroup distribution between the healthy Brazilian reference population and the Chagas cohort.
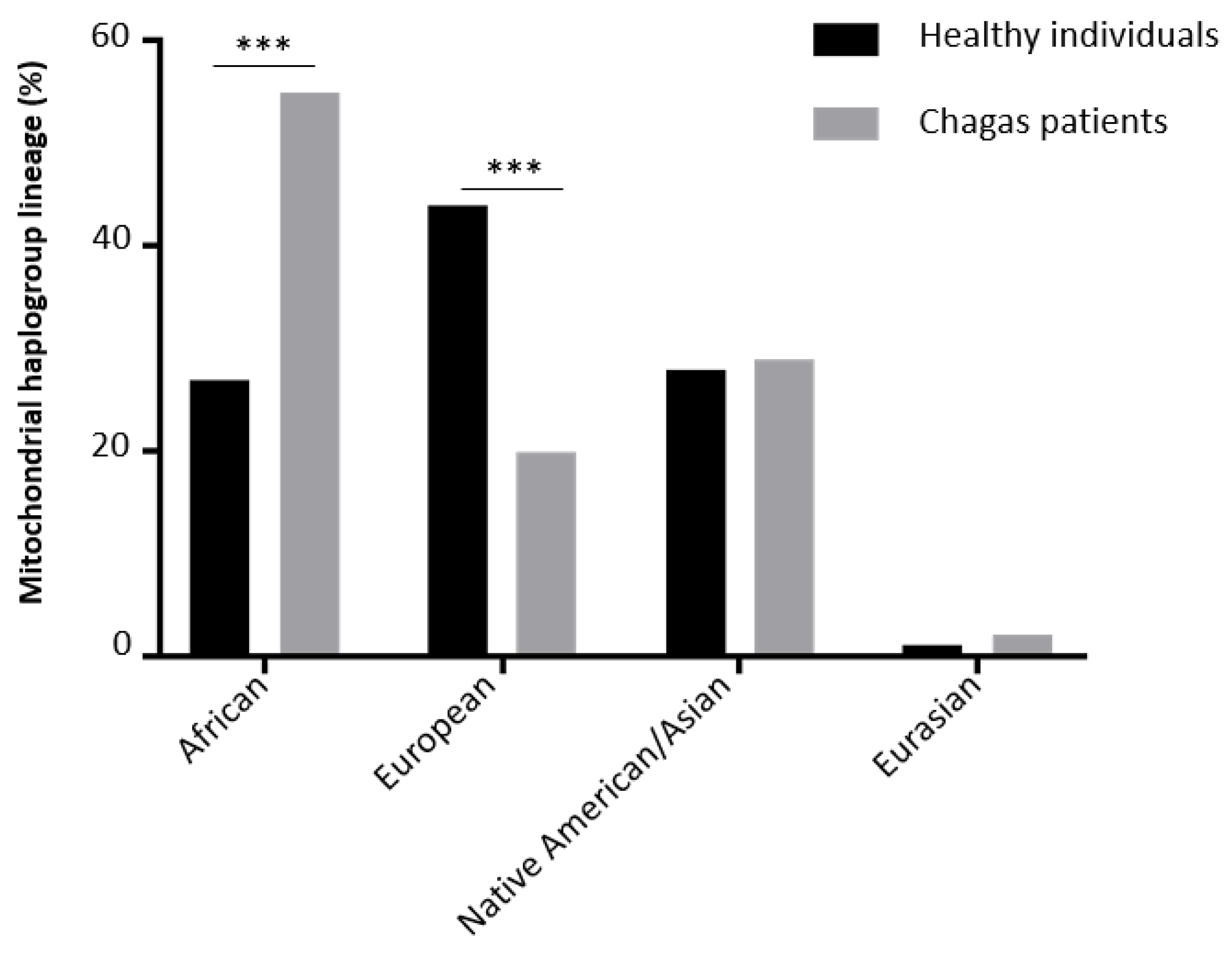
Figure 4.
Mitochondrial haplogroup distribution among the Chagas cohort.
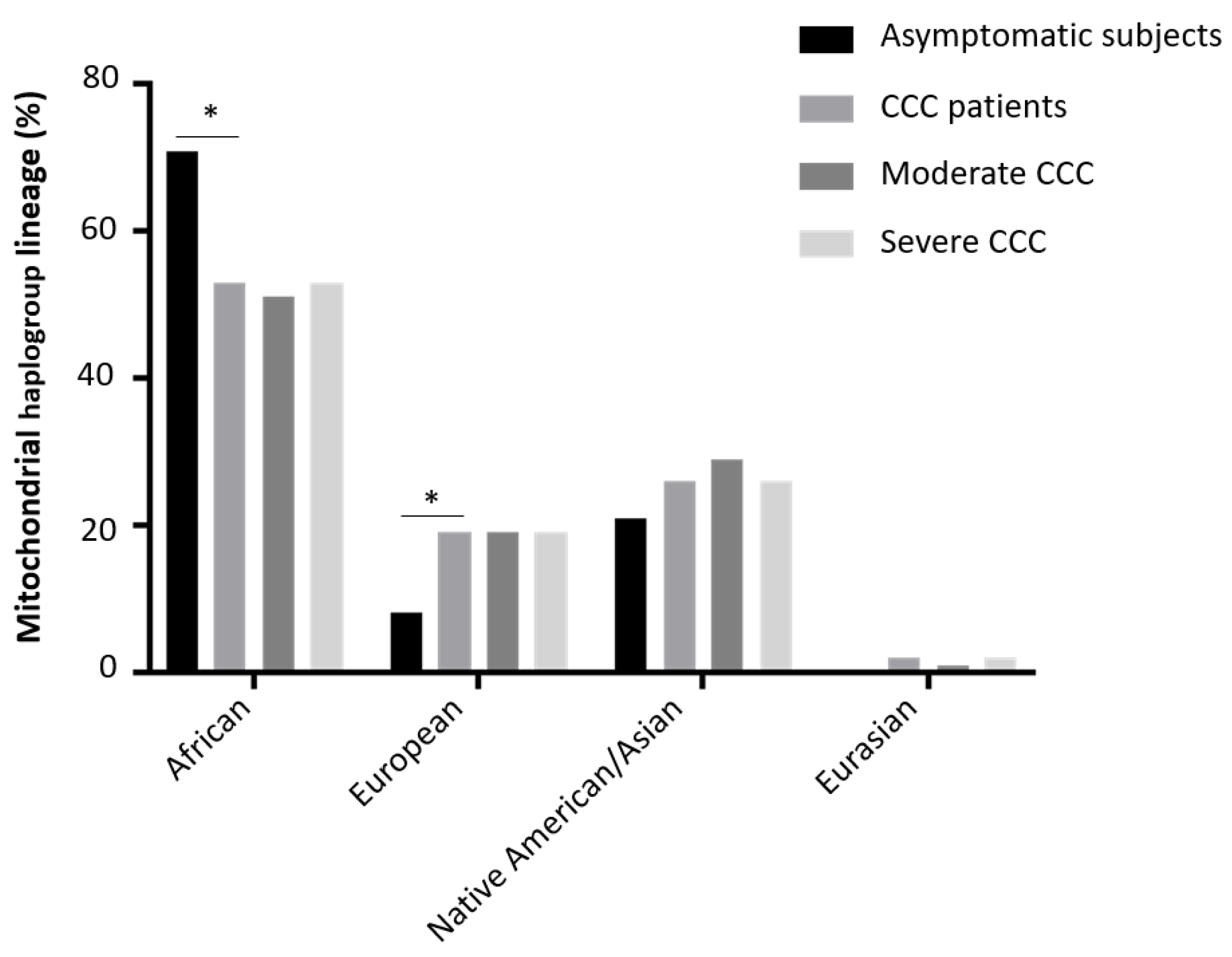
Figure 5.
African mitochondrial sub-haplogroup distribution in the Chagas cohort.
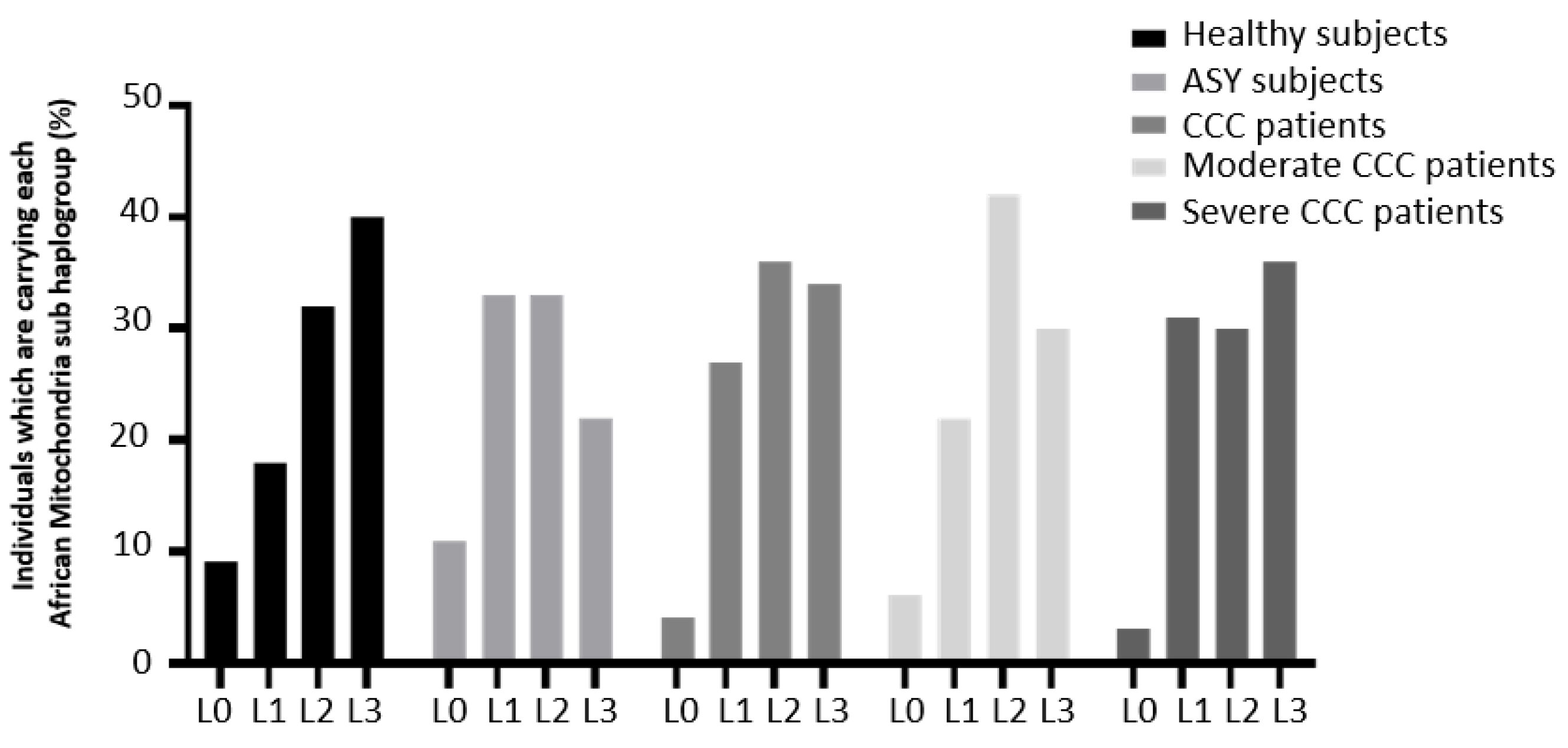
Figure 6.
European mitochondrial sub-haplogroup distribution in the Chagas cohort.
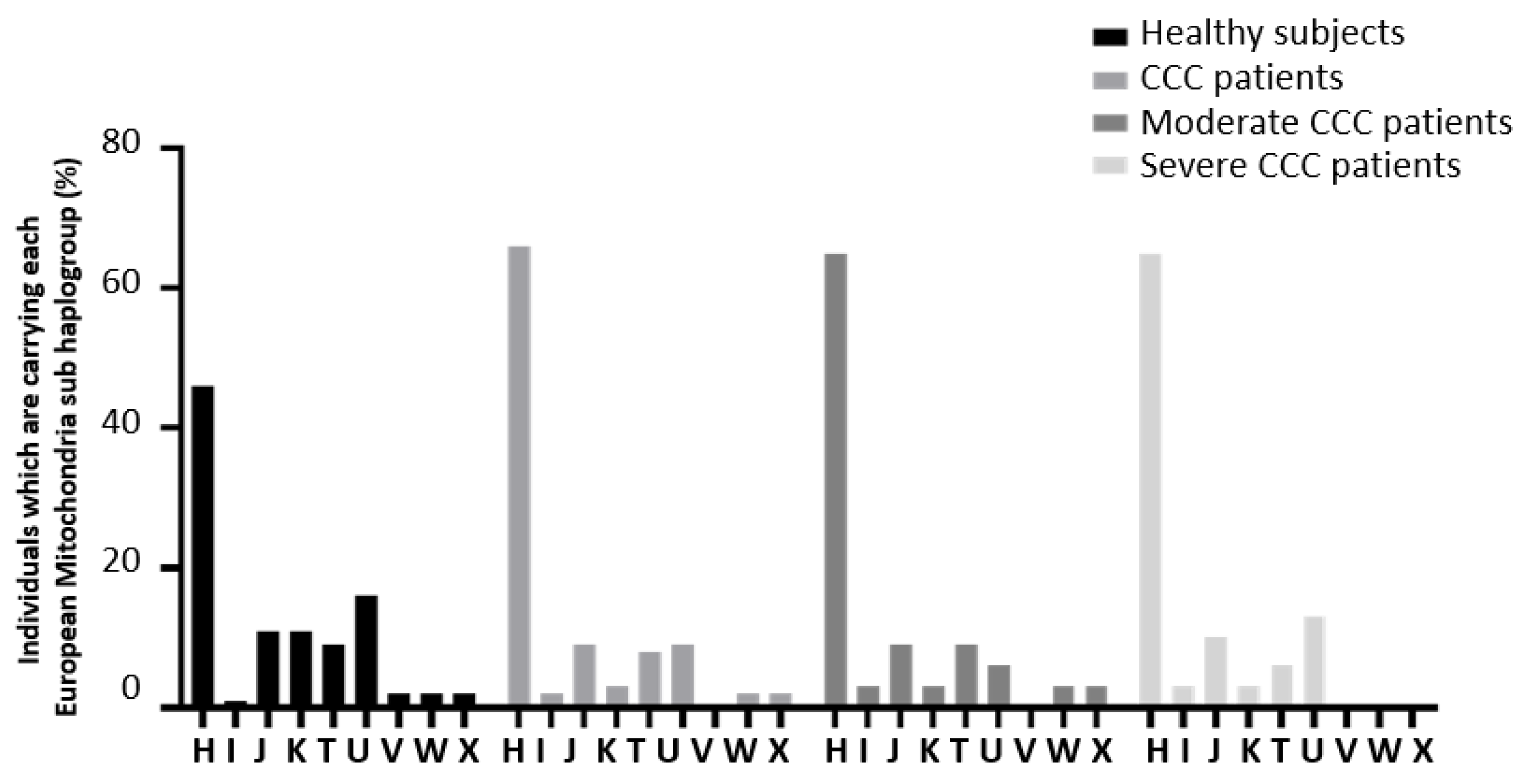

Table 1.
Haplogroup distribution in heart tissue samples.
| Haplogroup lineage | End stage heart tissue samples (CCC) | Healthy hearts of organ donors (control) |
|---|---|---|
| African | 19 (73%) | 4 (67%) |
| European | 4 (15%) | 0 (0%) |
| Native American/Asian | 2 (8%) | 1 (17%) |
| Eurasian | 1 (4%) | 1 (17%) |
Table 2.
European haplogroup H association study.
| Groups | p Value | Relative Risk |
|---|---|---|
| Healthy individuals versus chagas patients | 0.01 | 0.76 |
| Healthy individuals versus moderate chagas cardiomyopathy patients | 0.06 | 0.85 |
| Healthy individuals versus severe chagas cardiomyopathy patients | 0.07 | 0.86 |
Table 3.
Annotation of the 84 protein modifier variants.
| Pos | Ref | Alt | Csq | Gene symbol |
dbSNP ID |
Uniprot name |
Uniprot ID |
AA pos |
AA ref |
AA alt |
Described as pathogenic in the following databases. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3565 | A | C | RNA | MT-ND1 | NU1M | P03886 | 87 | T | P | PolyPhen2, SIFT, PROVEAN, CADD, PhDSNP, SP, COVEC_WMV, MtoolBox | |
| 3579 | A | C | RNA | MT-ND1 | NU1M | P03886 | 91 | M | I | CADD, PhDSNP, MutationTaster, SNPDryad, Condel | |
| 5262 | G | A | RNA | MT-ND2 | NU2M | P03891 | 265 | A | T | CADD, PANTHER, Condel, | |
| 6571 | C | T | RNA | MT-CO1 | COX1 | P00395 | 223 | A | V | PROVEAN, EFIN_SP, CADD, PhDSNP, MutationTaster, Condel, PolyPhen2 transf | |
| 6873 | C | T | RNA | MT-CO1 | COX1 | P00395 | 324 | L | F | PolyPhen2, SIFT, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, PANTHER, PhDSNP, MutationTaster, COVEC_WMV, MtoolBox, Mutatiossessor transf | |
| 6899 | G | C | RNA | MT-CO1 | rs1556423194 | COX1 | P00395 | 332 | M | I | Condel, PolyPhen2 transf, SIFT transf |
| 6909 | G | T | RNA | MT-CO1 | COX1 | P00395 | 336 | A | S | SIFT, CADD, PhDSNP | |
| 6915 | G | A | RNA | MT-CO1 | rs1603220687 | COX1 | P00395 | 338 | V | M | Condel, PolyPhen2 transf, SIFT transf |
| 7008 | G | T | RNA | MT-CO1 | COX1 | P00395 | 369 | D | Y | PolyPhen2, SIFT, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, PANTHER, PhDSNP, SP, MutationTaster, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 7072 | T | C | RNA | MT-CO1 | rs1603220760 | COX1 | P00395 | 390 | M | T | PolyPhen2, SIFT, EFIN_HD, CADD, PhDSNP, SP, MutationTaster, COVEC_WMV, MtoolBox |
| 7868 | C | T | RNA | MT-CO2 | rs1556423357 | COX2 | P00403 | 95 | L | F | PROVEAN, CADD, PANTHER, PhDSNP, Condel, PolyPhen2 transf |
| 8021 | A | G | RNA | MT-CO2 | rs1603221261 | COX2 | P00403 | 146 | I | V | Condel, PolyPhen2 transf |
| 8401 | A | C | INT | MT-ATP8 | ATP8 | P03928 | 12 | M | I | Condel, SIFT transf | |
| 8405 | A | G | INT | MT-ATP8 | rs1603221462 | ATP8 | P03928 | 14 | T | A | Condel, PolyPhen2 transf |
| 8527 | A | G | RNA | MT-ATP6 | rs878853003 | ATP6 | P00846 | 1 | M | V | PolyPhen2, MtoolBox |
| 8558 | C | G | RNA | MT-ATP8 | ATP8 | P03928 | 65 | P | A | PolyPhen2, FatHmm, PROVEAN, CADD, SP, COVEC_WMV, MtoolBox | |
| 8558 | C | G | RNA | MT-ATP8 | ATP6 | P00846 | 11 | A | G | CADD, PANTHER, PhDSNP | |
| 8566 | A | G | RNA | MT-ATP6 | rs3020563 | ATP6 | P00846 | 14 | I | V | SP, Condel |
| 8568 | C | T | RNA | MT-ATP8 | rs1603221589 | ATP8 | P03928 | 68 | S | F | PolyPhen2, FatHmm, EFIN_HD, CADD, SP, MtoolBox |
| 8618 | T | C | RNA | MT-ATP6 | rs28358885 | ATP6 | P00846 | 31 | I | T | Condel |
| 8632 | T | C | RNA | MT-ATP6 | rs1603221654 | ATP6 | P00846 | 36 | Y | H | PANTHER, Condel, PolyPhen2 transf |
| 8677 | A | C | RNA | MT-ATP6 | ATP6 | P00846 | 51 | K | Q | Condel, PolyPhen2 transf | |
| 8714 | C | T | RNA | MT-ATP6 | rs1603221724 | ATP6 | P00846 | 63 | T | I | FatHmm, PANTHER, Condel |
| 8795 | A | C | RNA | MT-ATP6 | ATP6 | P00846 | 90 | H | P | PolyPhen2, SIFT, PROVEAN, CADD, PANTHER, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox | |
| 8854 | G | A | RNA | MT-ATP6 | rs386829055 | ATP6 | P00846 | 110 | A | T | SIFT, CADD, PANTHER, PhDSNP, Condel |
| 8860 | A | G | RNA | MT-ATP6 | rs2001031 | ATP6 | P00846 | 112 | T | A | PROVEAN, PhDSNP, SP, Condel, PolyPhen2 transf |
| 8870 | T | C | RNA | MT-ATP6 | rs1556423560 | ATP6 | P00846 | 115 | M | T | Condel, PolyPhen2 transf |
| 8875 | T | C | RNA | MT-ATP6 | rs201123510 | ATP6 | P00846 | 117 | F | L | Condel, PolyPhen2 transf |
| 9261 | A | C | RNA | MT-CO3 | COX3 | P00414 | 19 | T | P | PolyPhen2, PROVEAN, EFIN_SP, CADD, PhDSNP, SP, SNPDryad, MtoolBox | |
| 9325 | T | C | RNA | MT-CO3 | rs879000531 | COX3 | P00414 | 40 | M | T | Condel, PolyPhen2 transf |
| 9717 | C | G | RNA | MT-CO3 | COX3 | P00414 | 171 | L | V | Condel, PolyPhen2 transf | |
| 9861 | T | C | RNA | MT-CO3 | rs878853060 | COX3 | P00414 | 219 | F | L | Condel, PolyPhen2 transf, SIFT transf |
| 9877 | T | A | RNA | MT-CO3 | COX3 | P00414 | 224 | M | K | MutationTaster, Condel | |
| 9880 | T | A | RNA | MT-CO3 | COX3 | P00414 | 225 | F | Y | Condel, PolyPhen2 transf | |
| 9896 | A | C | RNA | MT-CO3 | COX3 | P00414 | 230 | K | N | Condel, PolyPhen2 transf | |
| 9924 | T | G | RNA | MT-CO3 | COX3 | P00414 | 240 | W | G | PolyPhen2, PROVEAN, Mutatiossessor, EFIN_HD, CADD, VEST, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox | |
| 10677 | G | A | RNA | MT-ND4L | rs1603222944 | NU4LM | P03901 | 70 | E | K | FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, CADD, PANTHER, PhDSNP, SP, MutationTaster, SNPDryad, Condel, MtoolBox, Mutatiossessor transf |
| 10750 | A | G | RNA | MT-ND4L | rs372297272 | NU4LM | P03901 | 94 | N | S | PROVEAN, PhDSNP, MutationTaster, Condel |
| 10775 | G | A | RNA | MT-ND4 | rs879015842 | NU4M | P03905 | 6 | V | I | Condel, PolyPhen2 transf, SIFT transf |
| 10808 | C | T | RNA | MT-ND4 | rs2068723560 | NU4M | P03905 | 17 | L | F | CADD, Condel, MtoolBox |
| 10866 | T | C | RNA | MT-ND4 | rs1603222994 | NU4M | P03905 | 36 | I | T | Condel |
| 10972 | A | C | RNA | MT-ND4 | NU4M | P03905 | 71 | W | C | PolyPhen2, SIFT, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 11129 | A | G | RNA | MT-ND4 | rs1603223122 | NU4M | P03905 | 124 | T | A | Condel, SIFT transf |
| 11963 | G | A | RNA | MT-ND4 | rs201803948 | NU4M | P03905 | 402 | V | I | Condel, PolyPhen2 transf, SIFT transf |
| 12341 | C | A | RNA | MT-ND5 | NU5M | P03915 | 2 | T | N | Condel, PolyPhen2 transf | |
| 12346 | C | T | RNA | MT-ND5 | NU5M | P03915 | 4 | H | Y | Condel, PolyPhen2 transf, SIFT transf | |
| 12368 | C | T | RNA | MT-ND5 | NU5M | P03915 | 11 | T | I | FatHmm, PROVEAN, Condel, PolyPhen2 transf | |
| 12403 | C | T | RNA | MT-ND5 | NU5M | P03915 | 23 | L | F | Condel, PolyPhen2 transf | |
| 12802 | A | G | RNA | MT-ND5 | NU5M | P03915 | 156 | S | G | Condel | |
| 13034 | T | C | RNA | MT-ND5 | NU5M | P03915 | 233 | L | P | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, PANTHER, PhDSNP, SP, MutationTaster, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 13118 | T | A | RNA | MT-ND5 | NU5M | P03915 | 261 | I | N | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, CADD, PANTHER, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 13145 | G | A | RNA | MT-ND5 | NU5M | P03915 | 270 | S | N | Condel | |
| 13226 | A | C | RNA | MT-ND5 | NU5M | P03915 | 297 | D | A | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, CADD, PANTHER, PhDSNP, SP, MutationTaster, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 13303 | C | T | RNA | MT-ND5 | NU5M | P03915 | 323 | H | Y | PANTHER, PhDSNP, Condel, SIFT transf | |
| 13466 | G | A | RNA | MT-ND5 | NU5M | P03915 | 377 | S | N | FatHmm, PANTHER, Condel | |
| 13476 | A | T | RNA | MT-ND5 | NU5M | P03915 | 380 | L | F | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, PANTHER, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 13517 | A | T | RNA | MT-ND5 | NU5M | P03915 | 394 | H | L | PANTHER, Condel, PolyPhen2 transf | |
| 13600 | T | A | RNA | MT-ND5 | NU5M | P03915 | 422 | Y | N | PolyPhen2, FatHmmW, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, CADD, PANTHER, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, DEOGEN2, Mutatiossessor transf | |
| 13614 | A | T | RNA | MT-ND5 | NU5M | P03915 | 426 | M | I | Condel | |
| 13618 | C | T | RNA | MT-ND5 | NU5M | P03915 | 428 | L | F | Condel, MtoolBox | |
| 13651 | A | G | RNA | MT-ND5 | rs1569484594 | NU5M | P03915 | 439 | T | A | CADD, Condel, MtoolBox |
| 13763 | C | A | RNA | MT-ND5 | NU5M | P03915 | 476 | S | Y | PolyPhen2, PROVEAN, CADD, PANTHER, SP, Condel, MtoolBox, SIFT transf | |
| 13804 | G | A | RNA | MT-ND5 | rs1603224360 | NU5M | P03915 | 490 | A | T | PolyPhen2, PROVEAN, CADD, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox |
| 13816 | A | C | RNA | MT-ND5 | NU5M | P03915 | 494 | T | P | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, CADD, PANTHER, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox, Mutatiossessor transf | |
| 13886 | T | C | RNA | MT-ND5 | rs28359182 | NU5M | P03915 | 517 | L | P | PhDSNP, Condel |
| 14170 | A | T | RNA | MT-ND6 | NU6M | P03923 | 168 | I | M | PolyPhen2, MtoolBox | |
| 14172 | T | G | RNA | MT-ND6 | NU6M | P03923 | 168 | I | L | PolyPhen2, CADD, MtoolBox | |
| 14280 | A | G | RNA | MT-ND6 | NU6M | P03923 | 132 | S | P | FatHmm, PhDSNP, Condel | |
| 14562 | C | T | RNA | MT-ND6 | NU6M | P03923 | 38 | V | I | Condel | |
| 14757 | T | C | RNA | MT-CYB | rs1603224859 | CYB | P00156 | 4 | M | T | Condel, PolyPhen2 transf |
| 14793 | A | G | RNA | MT-CYB | rs2853504 | CYB | P00156 | 16 | H | R | PhDSNP, SP, Condel, PolyPhen2 transf |
| 14798 | T | C | RNA | MT-CYB | rs28357681 | CYB | P00156 | 18 | F | L | Condel, PolyPhen2 transf |
| 14871 | T | C | RNA | MT-CYB | rs28660155 | CYB | P00156 | 42 | I | T | PROVEAN, PhDSNP, Condel, PolyPhen2 transf |
| 14873 | C | A | RNA | MT-CYB | CYB | P00156 | 43 | L | I | Condel | |
| 14881 | C | G | RNA | MT-CYB | CYB | P00156 | 45 | I | M | PolyPhen2, FatHmm, PANTHER, PhDSNP, SP, MtoolBox | |
| 15077 | G | A | RNA | MT-CYB | rs201943501 | CYB | P00156 | 111 | E | K | PolyPhen2, PROVEAN, CADD, PhDSNP, SNPDryad, MtoolBox |
| 15596 | G | A | RNA | MT-CYB | rs1603225369 | CYB | P00156 | 284 | V | I | Condel |
| 15617 | G | A | RNA | MT-CYB | rs1556424625 | CYB | P00156 | 291 | V | I | PolyPhen2, CADD, PhDSNP, SP, COVEC_WMV, MtoolBox |
| 15664 | C | A | RNA | MT-CYB | rs1603225414 | CYB | P00156 | 306 | I | M | CADD, Condel |
| 15699 | G | A | RNA | MT-CYB | CYB | P00156 | 318 | R | H | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, EFIN_HD, CADD, PANTHER, PhDSNP, SNPDryad, COVEC_WMV, MtoolBox, Mutatiossessor transf | |
| 15725 | C | T | RNA | MT-CYB | rs1603225438 | CYB | P00156 | 327 | L | F | PANTHER, Condel |
| 15734 | G | A | RNA | MT-CYB | rs386829259 | CYB | P00156 | 330 | A | T | PhDSNP, Condel |
| 15777 | G | A | RNA | MT-CYB | rs879182710 | CYB | P00156 | 344 | S | N | CADD, PhDSNP, Condel |
| 15812 | G | A | RNA | MT-CYB | rs200336777 | CYB | P00156 | 356 | V | M | EFIN_SP, MutationTaster, Condel |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



