
Submitted:
12 October 2023
Posted:
13 October 2023
You are already at the latest version
Abstract
Gabapentin (GBP), a GABA analogue, is primarily used as an anticonvulsant to treat partial seizures and neuropathic pain. While a majority of the side effects are associated with the nervous system, emerging evidence suggests a high risk for heart diseases in the patients taking GBP. In the present study, we used a preclinical model of rats to investigate (1) the acute cardiovascular responses to GBP (bolus i.v. injection, 50 mg/kg) and (2) the effects of chronic GBP treatment (i.p. 100 mg/kg/day x 7 days) on cardiovascular function and the myocardial proteome. Under isoflurane-anesthesia, rat blood pressure (BP), heart rate (HR), and left ventricular (LV) hemodynamics were measured using Millar pressure transducers. The LV myocardium and brain cortex were analyzed by proteomics, bioinformatics, and western blot to explore the molecular mechanisms underlying GBP-induced cardiac dysfunction. In experiment (1), we found that i.v. GBP significantly decreased BP, HR, maximal LV pressure, and maximal and minimal dP/dt, whereas it increased IRP-AdP/dt, Tau, systolic, diastolic, and cycle durations (*p < 0.05 and **p < 0.01 vs baseline; n = 4/group). In experiment (2), we found that chronic GBP treatment resulted in hypotension, bradycardia, and LV systolic dysfunction, with no change in plasma norepinephrine. In the myocardium, we identified 109 differentially expressed proteins involved in calcium pathways, cholesterol metabolism, and galactose metabolism. Particularly, we found that calmodulin, a key protein of intracellular calcium signaling, was significantly upregulated by GBP in the heart but not in the brain. In summary, we found that acute and chronic GBP treatments suppressed cardiovascular function in rats, which is attributed to abnormal calcium signaling in cardiomyocytes. These data reveal a novel side effect of GBP independent of the nervous system, providing important translational evidence to suggest that GBP can evoke adverse cardiovascular events by depression of myocardial function.
Keywords:
Gabapentin
; Arterial Blood Pressure
; Heart Rate
; Left Ventricular Function
; Proteomics
; Bioinformatics
; Calmodulin
Introduction
Gabapentin (GBP) is a 3,3-disubstituted derivative of gamma-aminobutyric acid (GABA), recommended as a first-line treatment for chronic neuropathic pain, particularly in diabetic neuropathy and post-herpetic neuralgia1, and as an anticonvulsant to help control partial seizures in the treatment of epilepsy2. GBP is also used to treat an array of other central nervous associated disorders, including insomnia, drug and alcohol addiction, anxiety, bipolar disorder, borderline personality disorder, menopausal conditions, vertigo, pruritic disorders, and migraines3. On the other hand, increasing evidence indicates that GBP is being misused and abused, with various side effects such as central hypoventilation, deficits in visual field, suicidal behavior, somnolence, dizziness, mood changes, and tiredness4.
Although most GBP therapeutic and side effects, are evoked by alterations in neural function, increasing clinical and animal studies suggest non-neural effects by directly acting on peripheral tissues and organs, particularly the cardiovascular system. Designed as an analog of GABA, GBP binds with high affinity to the α2δ subunits of voltage-gated Ca2+ channels (VGCCs)5 and reduces the high-threshold Ca2+ currents of presynaptic membranes. This leads to an inhibition of the release of the excitatory amino acids glutamate and aspartate1. Since the α2δ subunits are expressed in all subtypes of VGCCs6, which are present not only in neurons but also in all other excitable cells, including skeletal muscle, vascular smooth muscle, and ventricular myocytes7, an influence of GBP on the function of non-nervous systems and relevant side effects should not be ignored.
Indeed, a retrospective cohort study revealed a significantly increased risk of adverse cardiovascular events, including heart failure, myocardial infarction, peripheral vascular disease, stroke, deep venous thrombosis, and pulmonary embolism in the patients with diabetic neuropathy following long-term use of GBP8. Furthermore, several clinical cases of GBP-induced atrial fibrillation9, acute heart failure10, 11, cardiomyopathy12, and intraoperative hypotension13 have been recently reported. Thus, we postulate that these events may be attributed, in part, to the effects of GBP on Ca2+ currents in cardiomyocytes or vascular smooth muscle.
Evidence from animal studies suggest that GBP can impact cardiovascular function by both central and peripheral mechanisms. Chen et al.14 found that microinjection of GBP into the nucleus tractus solitarii (NTS) of the brainstem induced prominent dose-related depressor and bradycardic responses in spontaneously hypertensive rats (SHR) via nitric oxide (NO)-dependent mechanisms. Behuliak et al.15 demonstrated that acute intravenous injection of GBP lowered blood pressure and heart rate by a sympatho-inhibitory mechanism in both SHR and normotensive rats (WKY), with a greater effect observed in the former. Interestingly, by utilizing pressure arteriography and whole-cell voltage-clamp techniques, Largeau et al.16 found that GBP dramatically decreased the myogenic tone of isolated mesenteric arteries and evoked weak vasodilation in endothelium-denuded vessels, but had no significant effect on Cav1.2 currents in ventricular cardiomyocytes. These studies suggest that GBP-induced peripheral edema and acute heart failure may be due to the effects of GBP on heart and blood vessels. However, to the best of our knowledge, no studies investigate GBP’s influence on left ventricular (LV) function in intact animals. In the present study, we evaluated the effects of acute and chronic administration of GBP on hemodynamics in isoflurane-anesthetized rats to address the hypothesis that GBP can suppress cardiac function. In addition, we performed label-free mass spectrometry-based proteomics of the LV myocardium followed by bioinformatic analysis to explore the molecular mechanisms and intracellular pathways underlying the GBP effects on cardiomyocytes.
Materials and Methods
All animal procedures were conducted in accordance with the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and conformed to ARRIVE Guidelines (https://www.nc3rs.org.uk/arrive-guidelines, accessed on May 9, 2023), as approved by the Animal Care and Use Committee of the University of Nebraska Medical Centre (UNMC-IACUC Protocol #17-080-09).
Eighteen Fisher 344 rats (3 - 4 months old), which included both sexes, were assigned to two groups. Group 1 (8 rats) was used to determine the acute cardiovascular responses to a bolus intravenous injection (i.v.) of GBP (50 mg/kg). Group 2 (10 rats) was used to determine the effects of 7-day intraperitoneal injection (i.p.) of GBP (100 mg/kg) on cardiovascular function and to determine the proteomic profile of the myocardium.
- Arterial blood pressure (AP), heart rate (HR), and left ventricular (LV) hemodynamics measurements.
Rats were anesthetized with 3% isoflurane delivered in 100% oxygen, lying on an electric heating pad to maintain body temperature at 37 °C. A PE10 catheter was inserted into the superior vena cava through the right jugular for vein saline supplementation and i.v. injection of GBP. A catheter-tipped transducer (SPR-407, Millar Instruments, Houston, TX) was advanced via the right femoral artery into the abdominal aorta to measure BP and HR, while another catheter-tipped transducer (SPR-320NR, Millar Instruments, Houston, TX) was advanced via right carotid artery into the LV chamber to measure cardiac function. The transducer signals were input into a PowerLab® data-acquisition system with sampling rate at 2 k/s, from which the data were monitored, recorded, and saved in a computer by the LabChart® 8 software and analyzed using the Blood Pressure Module software v1 (AD Instruments, Colorado Springs, CO) with an average of 50 – 100 cycles.
- 2.
- Plasma norepinephrine assay
300 μL plasma was used to measure norepinephrine (NE) concentration employing a Norepinephrine Enzyme Immunoassay kit (Labor Diagnostika Nord KG, Nordhorn, Germany), based on the manufacturer’s instructions and performed in duplicates for each sample.
- 3.
- Proteomics analysis
- (1)
- Sample preparation for mass spectrometry
The protein concentration was estimated in each sample using BCA Protein Assay Kit (Pierce). 100 μg protein from each sample was diluted to 100 μL volume with 100 mM ammonium bicarbonate (ambic). Proteins were reduced with 5 μL of 200 mM tris(2-carboxyethyl) phosphine (TCEP) (1 h incubation, 55 ⁰C) and alkylated with 5 μL of 375 mM iodoacetamide (IAA) (30 min incubation in the dark, room temperature). The reduced and alkylated proteins were purified with acetone precipitation at -20 ⁰C overnight. The next day, protein precipitates were collected by centrifugation at 8000 × g for 10 minutes at 4 ⁰C and pellets were briefly air-dried and resuspended in 100 μL of 50 mM ambic. The protein digestion was carried out using 2.5 μg of trypsin per sample (16 h incubation, 37 ⁰C). The samples were dried out using a speed vacuum and then desalted with C18 spin columns (Pierce). Clean peptides were dried out again with a speed vacuum and resuspended in 100 mM TEAB. The TMT label reagents were equilibrated at room temperature for 10-15 min with the lid sealed, following which to each 0.8 mg TMT label reagent 42 μL anhydrous acetonitrile was added, vortexed briefly to make sure the reagents were fully dissolved. The peptides were labeled by adding 42 μL TMT label reagent to every 100 μL resuspended peptides and incubated for 1 h at room temperature. Quenching of the reaction was then performed by adding 8 μL quench buffer to the reaction and incubating for 15 min at room temperature. Each labeled peptide sample from the two treatment groups were combined to be compared in each experimental set containing different tags. The pooled peptide sample was then desalted using high pH reverse-phase HPLC, and eluted peptides were collected and dried using a speed vac. The dried peptides were finally dissolved in 40 μl 0.1% (v/v) aqueous FA and analyzed using a high-resolution mass spectrometry nano-LC-MS/MS Orbitrap Exploris 480™ coupled with UltiMate 3000 HPLC system (Thermo Scientific).
- (2)
- LC-MS/MS
1.5 μg of each sample was loaded onto trap column Acclaim PepMap 100 (75μm×2 cm C18 LC Columns, Thermo Fisher Scientific) at a flow rate of 4 μl/min, and next separated with a Thermo RSLC Ultimate 3000 (Thermo Fisher Scientific) on a Thermo Easy-Spray PepMap RSLC C18 column (75μm×50 cm C-18 2 μm, Thermo Fisher Scientific) at a flow rate 0.3 μl/min and 50 °C, with a step gradient of 9%–25% solvent B (0.1% FA in 80% acetonitrile) from 10–20 min and 25%–40% solvent B from 15–120 min, with a 155 min total run time. The MS scan was done using a detector: Orbitrap resolution 120000; scan range 350-1800 m/z; RF lens 30%; AGC target 4.0 e5; maximum injection time 100 ms. The most intense ions with charge state 2-6 isolated in 3 s cycles were selected in the MS scan for further fragmentation. MS2 scan parameters set: activation HCD with 35% normalized collision energy, detected at a mass resolution of 30000 The AGC target for MS/MS was set at 5.0 e4, and ion filling time was set to 60 ms.
- (3)
- Data Analysis
Protein identification was performed by searching MS/MS data against the UniProt database (Rat) in Proteome Discoverer (Thermo Fisher Sci, vs 3.0.), assuming the digestion enzyme trypsin and the 10plex TMT reporter ion MS2 mode as the searching mode. The parameters for Sequest HT were set as follows: Enzyme: trypsin, Max missed cleavage: 2, Precursor mass tolerance: 10 ppm, Peptide tolerance: ± 0.02 Da, Fixed modifications: carbamidomethyl (C); Dynamic modifications: oxidation (M), acetyl (N-term) and TMT (K) – 229.163Da. The parameters for the Precursor ions quantifier were set as follows: peptides to use unique + razor, precursor abundance based on intensity; normalization mode: total peptide amount; scaling mode: on all average.
- 4.
- Bioinformatics analysis
Proteins were recognized as differentially expressed if the p-value of the t-test was ≤ 0.05 and the absolute fold change was ≥1.5. A gene ontology (GO) analysis of differentially expressed proteins (DEPs) was conducted using the Cytoscape plug-in ClueGO17. Biological processes and molecular functions were included for the GO enrichment analysis. A canonical pathway analysis was performed using Ingenuity Pathway Analysis (IPA) software (Ingenuity® Systems, Redwood City, CA, USA, www.ingenuity.com, accessed on August 8, 2023) by comparing the DEPs against known canonical pathways (signaling and metabolic) within the IPA knowledgebase. For further analysis, enriched pathways with a Benjamini–Hochberg false discovery rate (FDR) p-value ≤ 0.05 were considered.
- 5.
- Western blotting analysis
Protein isolation from the LV myocardium and brain cortex of saline- and GBP- treated animals was isolated using RIPA buffer (50 mM TrisHCl, 195 mM NaCl, 2 mM EDTA, 1% NP-40, 0.1% SDS) with 1% protease inhibitor cocktail (Abcam, ab65621) and then centrifuged at 20,000 g × 20 min at 4 °C. Protein concentration was estimated using the Pierce bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA, USA). 10 μg and 5 μg of proteins separately from the LV myocardium and brain cortex of each animal were loaded onto a 10% Bis-Tris wells (Invitrogen, Waltham, MA, USA) under reducing conditions, followed by transfer to a nitrocellulose membrane using iBlot2 (Invitrogen). Post transfer, the membranes were stained with Ponceau S stain (Thermo Fisher Scientific) to assess for equal protein loading detection and quantification. The membranes were treated with 5% fat free milk to block nonspecific antibody binding for 1 h at room temperature on a rocker. After blocking, membranes were incubated overnight at 4 °C with 1:1000 calmodulin (CaM) antibody (A4885; ABclonal). The next day, membranes were washed and treated with 1:2500 HRP conjugated anti-rabbit IgG for 1 h followed by additional washes. Blots were developed with 1:1 solution of Radiance Chemiluminescent Substrate and Luminol/Enhancer (Azure Biosystems, Dublin, CA, USA). A c300 imaging system (Azure Biosystems) was used to visualize the blots, and images acquired were quantified using the Image-J software version 1.52a.
- 6.
- Statistical Analyses
All data are expressed as mean ± SD. Student’s t test was used to compare the difference between 2 groups, with the aid of Prism 8 software. A P value of < 0.05 was taken as indicative of statistical significance.
Results
- Effect of acute bolus intravenous injection of GBP on cardiovascular function.
We first determined acute responses of BP, HR, and LV hemodynamics to an intravenous injection of GBP (50 mg/kg; 100 mg/ml x 0.1 ml in 3 min). The results are shown in Figure 1 where the left panel is a representative original recording, and the right panel shows combined summary data (table 1).
The recording shows that GBP administration evoked a transient and slight suppression of most hemodynamic parameters, followed by a prolonged and significant inhibition, showing a biphasic effect. Since the i.v. catheter tip was placed into the superior vena cava, we believe the inhibitory effect of the 1st phase was induced directly by the outflow of GBP solution from the catheter tip going through the heart chamber and vessel system. Accordingly, when the injection was completed (indicated by green arrow), this effect disappears. At approximately 7 min post injection (indicated by blue arrow) when the GBP solution was completely mixed with circulating blood and GBP concentration in blood reached a steady state, we observed the inhibitory effect of the 2nd phase, which lasted at least 60 min with a maximal effect appearing at 30 min post injection (yellow arrow). Since the half-life of GBP is between 5 - 7 hours, we speculate that i.v. GBP-induced cardiovascular inhibition could potentially continue to last longer than one hour.
Table 1 shows that BP, HR, and most LV hemodynamic parameters are significantly reduced after GBP treatment (the values were calculated at 30 min post injection indicated by the yellow arrow) as compared with the baseline (before GBP treatment). The decreased systolic pressure (SP) and pulse pressure (PP) represent impaired cardiac function and reduced cardiac output, while the decreased diastolic pressure suggests vasodilation. The lower maximal and minimal dP/dt values suggest depressed myocardial contractility and impaired LV relaxation, which are further confirmed by a significantly decreased IRP-AdP/dt, the slope of a straight line fit to the pressure over the isovolumic relaxation period, as well as a significantly increased Tau, the exponential time constant of relaxation. However, there was no significant difference in LVEDP between pre- and post-GBP, suggesting that acute GBP-induced depression of cardiac function did not develop into heart failure. It should be noted that i.v. injection of 0.1 ml saline (vehicle) did not evoke significant changes in BP, HR, and LV hemodynamics over the same time period as rats that underwent GBP injection.
- 2.
- Effect of chronic i.p. treatment of GBP on cardiovascular function.
We determined chronic effects of GBP administration (i.p. 100 mg/kg/day for 7-consecutive days) on cardiovascular function compared with saline as the vehicle control. The measurements were carried out 6 hours after last GBP administration on day 7. The results are shown in Figure 2 where the left panel is representative of original recordings and the right panel shows the combined data (table 2). GBP-treated rats exhibited significantly lower BP, HR, and LV hemodynamics as compared with the rats receiving i.p. saline, suggesting that the inhibitory effects of chronic GBP treatment on cardiovascular function was similar to that which was induced by acute GBP-treatment. However, some parameters, which were significantly reduced in acute GBP-treated rats (table 1), did not display a significant change in chronic GBP-treated rats (table 2), such as PP, dP/dtmin, IRP-Average dP/dt, Tau, DD, and CD, suggesting a different cardiovascular response to acute- and chronic- GBP administration. This difference, however, could also be attributed to different time points between the two groups that were selected to calculate the parameters since we evaluated the acute response after 30 min post GBP treatment (table 1) but evaluated the chronic response at 6 hr after GBP administration (table 2).
- 3.
- Plasma NE concentration
To determine if sympathetic system is involved in GBP-induced cardiovascular dysfunction, we next measured the plasma NE concentration in the rats receiving chronic GPB treatment. No significant differences in plasma NE concentration were observed between the two groups (Figure 3), suggesting that 7-day i.p. treatment of GBP in a dose of 100 mg/kg did not change circulating catecholamine and sympathetic tone.
- 4.
- Proteomics and bioinformatics analyses
To explore the molecular mechanisms underlying chronic GBP treatment-induced cardiac dysfunction, we employed Tandem Mass Tag (TMT) multiplexing coupled to liquid chromatography-mass spectrometry based quantitative proteomics to analyze LV myocardium. A total of 2262 proteins with a minimum of 2+ unique peptides were identified, of which 109 proteins were found to be differentially expressed between groups (Supplemental Table 1). Panel A of Figure 4 shows principal component analysis (PCA), which represents an overall separation and reproducibility among the biological replicates occurred between GBP- and saline-treated rats with 65.2% of variation. Panel B of Figure 4 is a volcano plot indicating significantly upregulated (red dots, n = 45) or downregulated (blue dots, n = 64) proteins in GBP rats as compared with saline rats. Panel C of Figure 4 is heatmap analysis of the top 121 proteins, revealing a good consistency of protein profiles between groups. Next, using ClueGO, a user friendly cytoscape plug in, we analyzed both the molecular and biological processes enriched within the differentially expressed proteins. Amongst the molecular functions, we found enrichment of ribonucleoprotein complex (11.8%), heat shock binding (10.75%) and RNA helicase activity (10.75%). Similarly, in the biological functions, we found enrichment in the translation regulator activity (31.34%), followed by nucleic acid transport (14.93%) and negative regulation of chromatin silencing (11.94%). These data are presented in Figure 5.
- 5.
- Western blotting analysis
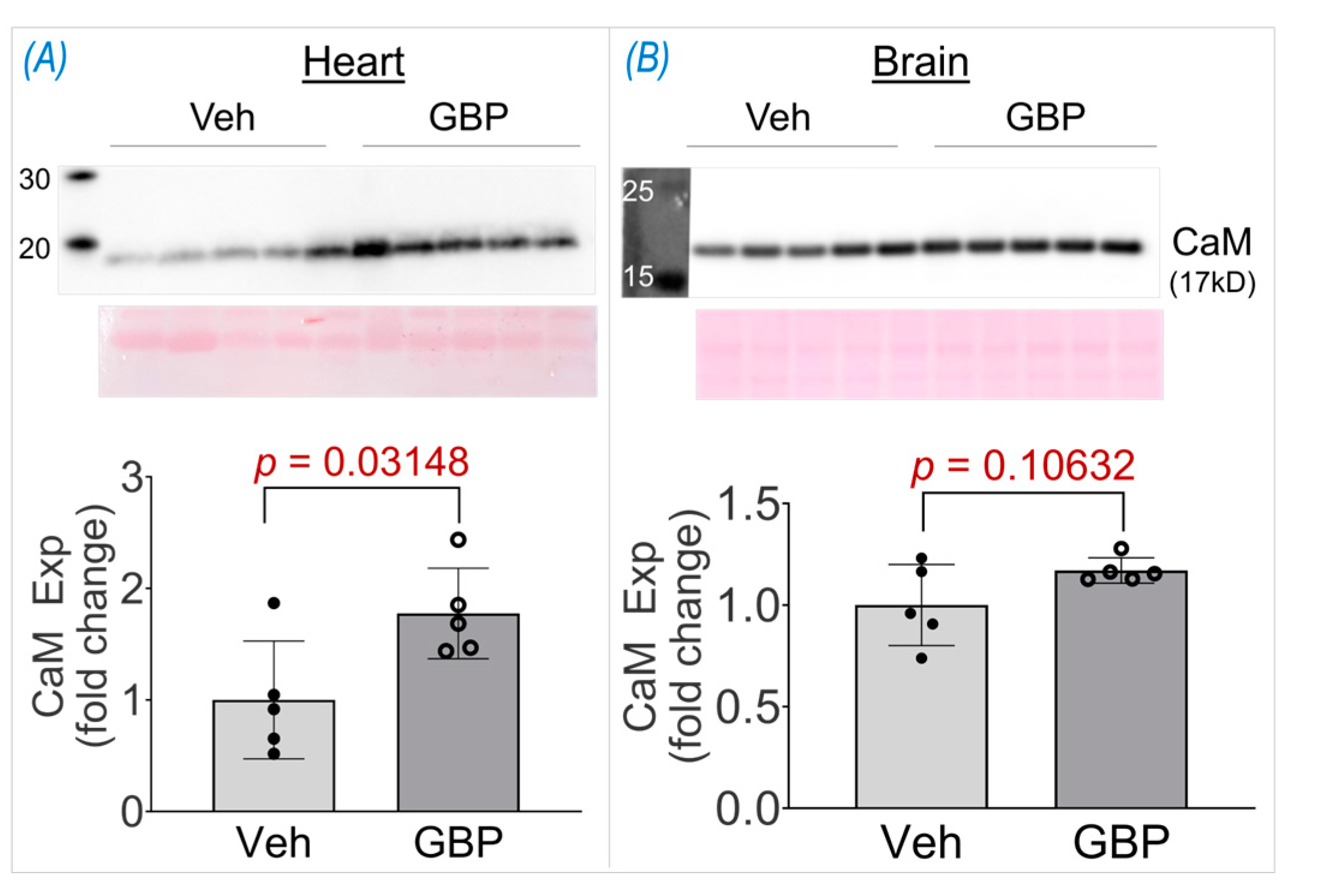
High throughput studies generate vast data and the initial potential hits identified thus need to be further validated. One such protein hit we identified from the proteomics screen was calmodulin (CaM), which was +1.52 fold upregulated in the GBP group. To further validate its upregulation, we performed a western blot on the heart lysates from the two groups. As seen in Figure 6, CaM expression was significantly elevated (+1.77 fold) in the heart lysates of the GBP-treated animals thus validating our proteomics screen data. To further ascertain if the upregulation of CaM is heart specific, we also examined its expression in the brain cortical lysates of the two groups. Interestingly, there was no change in CaM expression in the brain lysatesof the GBP animals, suggesting that 7-day i.p. treatment of GBP with a dose of 100 mg/kg/day significantly upregulated CaM specifically in the myocardium that may contribute to the subsequent cardiac dysfunction.
Discussion
Although GBP has long been used in a neurosetting, its cardiovascular side effects have recently attracted attention in human and veterinary medicine, and basic medical science areas8, 18. However, the exact actions and underlying mechanisms of GBP on the heart, blood vessels, and autonomic regulation remain to be completely elucidated. The novel findings of the present study are that both acute and chronic systemic administration of GBP in rats evoked a significant inhibition of cardiac function. This included bradycardia, depressed myocardial contractility, and LV systolic/diastolic dysfunction, which were accompanied by a decrease in BP without a change in plasma norepinephrine concentration. These data imply a direct effect of GBP on the heart. In addition, using mass spectrometry-based proteomics, we identified 109 differentially expressed proteins in heart samples of rats receiving chronic GBP treatment. Bioinformatics analysis further suggests that these proteins are involved in calcium signaling pathways, cholesterol metabolism, galactose metabolism, and other biological processes associated with cardiac function. One of the most important pathways that we identified using proteomics and validated by western blot in this study was CaM, a highly conserved protein that senses intracellular Ca2+ and relays signals to various Ca2+-sensitive enzymes, ion channels, and other proteins19. We found that CaM was markedly upregulated in the myocardium, but not in the brain cortices of the rats receiving chronic GBP treatment. This finding strongly suggests an association of the observed cardiac dysfunction with impaired regulation of cardiomyocyte Ca2+ signaling. To the best of our knowledge, this is the first study to provide physiological and molecular evidence suggesting a direct effect of GBP on cardiac function and protein profiles by combining whole animal experiments with omics techniques and molecular validation. Consistent with our findings in normal rats, a recent study by Allen et al.20 demonstrated that a single dose of oral GBP in healthy cats produced a modest decrease in LV systolic function, displaying a significant decrease in two-dimensional fractional shortening and a significant increase in both LV internal diameter in systole and left atrial volume.
The effects of GBP on cardiovascular function in normotensive and hypertensive rats has been investigated recently by several studies. In conscious spontaneously hypertensive rats (SHR) and Wistar-Kyoto (WKY) rats, Behuliak et al.15 found that acute intravenous injection of GBP significantly reduced BP and HR, with greater effects in SHR than WKY. They further demonstrated that the hypotension and bradycardia were mediated by attenuating sympathetic nerve transmission and modulating the arterial baroreceptor-HR reflex. To explore the central mechanisms of GBP-evoked cardiovascular depression, Chen et al14 found that microinjection of GBP into the nucleus tractus solitarii (NTS) of SHR rats induced a dose-dependent decrease in BP and HR, which was completely abolished by pretreatment with L-NAME, a NOS inhibitor. These data suggest a critical role of GBP in central autonomic regulation via a NO-dependent pathway. On the other hand, to explore the peripheral mechanisms of GBP-induced edema and acute heart failure, Largeau et al16 utilized pressure arteriography and whole-cell patch clamp to study the direct effects of GBP on vasomotor capacity of third-order mesenteric arteries and L-type calcium current in LV cardiomyocytes isolated from adult male Wistar rats. They found that GBP evoked a marked decrease in myogenic tone and a weak vasodilation but had no effects on cellular Ca2+ current. These data provide the first evidence demonstrating that GBP can modulate cardiovascular function through a direct effect on non-nervous tissues. In the present study by simultaneously measuring BP, HR, and LV hemodynamics in anesthetized rats, we found an immediate hypotension, bradycardia, and impaired LV contractility/relaxation in response to intravenous injection of GBP, further confirming the direct effects of GBP on the heart and blood vessels.
While there are no clinical studies that have investigated the side effects or toxicity of GBP on cardiovascular function, several case reports describe adverse cardiovascular events induced by GBP or its structurally similar compound, pregabalin. These include development of new onset congestive heart failure10, 11, decompensation of pre-existing CHF21-23, cardiac conduction disturbances24, 25, atrial fibrillation9, and hypotension13, 26. Analysis of large-scale nation-wide database of de-identified patients (TriNetZ EHRs), Pan et al.8 recently found a significant increase in risk for cardiovascular diseases, including heart failure, myocardial infarction, peripheral vascular disease, stroke, deep venous thrombosis, and pulmonary embolism in patients with diabetic neuropathy following long-term use of GBP and pregabalin. They further found that there were significant associations between short-term (3 month) GBP use and heart failure, myocardial infarction, peripheral vascular disease, deep venous thrombosis, and pulmonary embolism. The data in the present study provide animal experimental evidence to support the cardiovascular dysfunction described by these clinical case reports and high risk of cardiovascular diseases found by the retrospective cohort study.
Although autonomic dysfunction has been proposed to explain the negative influence of GBP on the cardiovascular system14, 15, the exact mechanisms remain to be explored. A decreased peripheral sympathetic nerve transmission15 and suppressed central sympathetic nerve outflow14 can explain GBP-induced hypotension and bradycardia, but is likely not the cause of GBP-evoked new onset congestive heart failure10, 11, decompensation of pre-existing heart failure21-23, or increase in the risk of heart failure8, since sympatho-inhibition is generally believed to ameliorate, instead of exacerbate, the cardiac dysfunction in the heart failure state. Based on our data showing acute cardiac dysfunction by GBP and marked upregulation of CaM in the myocardium of rats receiving chronic GBP, we hypothesize that GBP-induced cardiovascular pathological consequences may be attributed to a direct effect of GBP on myocardial contractility induced by abnormal Ca2+ signaling in cardiomyocytes.
Despite its structural similarity to GABA, GBP cannot bind to GABA receptors or affect the neuronal uptake or degradation of GABA, suggesting that the effects of GBP are not mediated by interacting directly with GABA receptors or with high-affinity Na+-dependent GABA transporters27. Instead, GBP directly inhibits the voltage-gated calcium channels (VGCCs) of neurons, leading to reduction in presynaptic Ca2+ influx, a decrease in the release of excitatory neurotransmitters, and a relief of neuropathic pain28. VGCCs are composed of a complex, consisting of an α1 and associated β and α2δ subunits, where the α1 subunit constitutes the channel pore and allows calcium influx from the extracellular space into the cells. The β and α2δ subunits support channel trafficking and tune the kinetic properties of Ca2+ currents29, 30. By binding to the α2δ subunits of the VGCC complex, GBP blocks neuronal Ca2+ influx, exerting its analgesic efficacy28, 31. In addition to the nervous system, VGCCs are also found in other excitable tissues, such as cardiac muscle, skeletal muscle, smooth muscle, and endocrine cells, playing a critical role in regulation of muscle contraction and hormone secretion30. In the present study, we propose that the cardiac dysfunction observed in rats and in other studies in cats20 is related to blockade by GBP on cardiomyocytes L-type Ca2+ current by binding to the α2δ subunit of the CaV1.2 channel, a cardiac subtype of VGCCs. Indeed, by employing a [3H]GBP binding assay and α2δ-null mice, Fuller-Bicer et al.32 demonstrated a binding capacity of GBP with VGCCs in cardiomyocytes. They further found that α2δ-deficient hearts display a significantly lower basal contractility and relaxation (+dP/dt and -dP/dt) as compared with the wild type32. Actually, VGCCs participate in and are critical for almost all aspects of cardiac physiology, pathology, and pharmacology, through regulating extracellular Ca2+ influx and subsequent intracellular signaling transduction33, 34.
Cardiomyocyte VGCCs, the CaV1.2 channel subunit, mediates excitation-contraction coupling (ECC), a highly coordinated process whereby membrane depolarization leads to contraction35. During the period of action potential generation, small amounts of extracellular Ca2+ ions enter the cytosol via the CaV1.2 and trigger the release of massive amounts of intracellular Ca2+ from the sarcoplasmic reticulum (SR) into the cytosol via the ryanodine receptor (RyR, a Ca2+ release channels resided in SR), known as Ca2+-induced Ca2+ release (CICR)36. These Ca2+ ions released from SR bind to troponin-C and enable actin-myosin cross-bridging and sliding of the myofilaments, resulting in sarcomere shortening and myocardial contraction37. CICR is a complex process critical for ECC, not only involving in CaV1.2 and RyR but also CaM. CaM is a Ca2+ binding protein, which crosstalks with both CaV1.2 and RyR, forming a triangular interaction to regulate Ca2+ behavioral and kinetics 38. On one hand, the functional communications between the RyR and CaV1.2 are believed to be reciprocal. CaV1.2 opens RyR, defined as orthograde signaling, and RyR can prevent CaV1.2 inactivation, defined as retrograde signaling39, 40. On the other hand, CaM in both Ca2+-bound and Ca2+-free states can bind and regulate both RyR and CaV1.2. RyR activity is increased by CaM at low Ca2+ concentrations (nM; Ca2+-free CaM) but inhibited by CaM at high Ca2+ concentrations (μM; Ca2+-bound CaM), while CaV1.2 can be inactivated and facilitated by CaM via its binding to channel C-terminal domain as a Ca2+ sensor38. One of the important findings from the present study is a marked upregulation of CaM in the hearts of rats receiving 7-day GBP treatment, suggesting an impact of GBP on cardiomyocyte Ca2+ signaling which we believe underlies the subsequent cardiac dysfunction. Indeed, it has been found that hyper-activated Ca2+/CaM signaling contributed to pathology in several cardiac diseases, including heart failure, cardiac hypertrophy, and arrhythmia41, 42. Although the exact mechanisms by which GBP upregulates CaM and the resulting functional consequences by the upregulated CaM remain unclear, our data does suggest an involvement of intracellular Ca2+ signaling dysfunction in GBP-induced cardiac side effects and will open novel avenues for treating in the future.
Supplementary Materials
Table S1: Supplementary Table 1.
Author Contributions
L.G. and V.V.P. conceived the project and co-wrote the article; L.G. and S.P. carried out cardiovascular experiments and generated Figure 1 and Figure 2; V.S. made the preclinical rat model. K.S. performed proteomic analysis and generated Figure 4 under the guidance of V.K; S.J. performed bioinformatics analysis and generated Figure 5 under the guidance of C.G.; Western blot analyses were performed by V.V.P. and V.S. Plasma norepinephrine concentration was measured by L.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by National Institutes of Health (NIH) grant R01HL160820 awarded to L.G. The Bioinformatics and Systems Biology Core is partially supported by NIH awards to C.G. (5P20GM103427, 5P30CA036727, 5U54GM115458).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of University of Nebraska Medical Center (protocol code 17-080-09-FC approved on 09/01/2023).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained within the article and supplementary materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Baillie JK and Power IJCoiid. The mechanism of action of gabapentin in neuropathic pain. 2006;7:33-39.
- Ziganshina LE, Abakumova T, Hoyle CHJIJoR and Medicine Si. Gabapentin monotherapy for epilepsy: A review. 2023:1-44. [CrossRef]
- Smith RV, Havens JR and Walsh SLJA. Gabapentin misuse, abuse and diversion: a systematic review. 2016;111:1160-1174. [CrossRef]
- Quintero GCJJoep. Review about gabapentin misuse, interactions, contraindications and side effects. 2017:13-21. [CrossRef]
- Calandre EP, Rico-Villademoros F and Slim MJEron. Alpha2delta ligands, gabapentin, pregabalin and mirogabalin: a review of their clinical pharmacology and therapeutic use. 2016;16:1263-1277. [CrossRef]
- Isom LL, De Jongh KS and Catterall WAJN. Auxiliary subunits of voltage-gated ion channels. 1994;12:1183-1194. [CrossRef]
- Catterall WAJAroc and biology d. Structure and regulation of voltage-gated Ca2+ channels. 2000;16:521-555. [CrossRef]
- Pan Y, Davis PB, Kaebler DC, Blankfield RP and Xu RJCD. Cardiovascular risk of gabapentin and pregabalin in patients with diabetic neuropathy. 2022;21:170. [CrossRef]
- Park SH, Hunter K, Berry H and Martins YCJJoMCR. Atrial fibrillation induced by gabapentin: a case report. 2023;17:236. [CrossRef]
- Al-Bast B, Deshpande R, Hu YY and Siddique MJJotACoC. GABAPENTIN INDUCED HEART FAILURE. 2022;79:2231-2231.
- Barold S, Barold D, Hon RJCRCCJ, J. CRCC and org wc. Gabapentin-Induced Congestive Heart Failure. 2.
- Tellor KB, Ngo-Lam R, Badran D, Armbruster AL, Schwarze MWJJocp and therapeutics. A rare case of a gabapentin-induced cardiomyopathy. 2019;44:644-646. [CrossRef]
- Patrick J, Tobias JJPA and Journal CC. Gabapentin and intraoperative hypotension. 2021;9.
- Chen H-H, Li Y-D, Cheng P-W, Fang Y-C, Lai C-C, Tseng C-J, Pan J-Y and Yeh T-CJACS. Gabapentin reduces blood pressure and heart rate through the nucleus tractus solitarii. 2019;35:627. [CrossRef]
- Behuliak M, Bencze M, Polgárová K, Kuneš J, Vaněčková I and Zicha JJH. Hemodynamic Response to Gabapentin in Conscious Spontaneously Hypertensive Rats: The Role of Sympathetic Nervous System. 2018;72:676-685. [CrossRef]
- Largeau B, Bordy R, Pasqualin C, Bredeloux P, Cracowski J-L, Lengelle C, Gras-Champel V, Auffret M, Maupoil V, Jonville-Bera A-PJB and Pharmacotherapy. Gabapentinoid-induced peripheral edema and acute heart failure: a translational study combining pharmacovigilance data and in vitro animal experiments. 2022;149:112807. [CrossRef]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z and Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091-3. [CrossRef]
- Veronezi TM, Lopes DJ, Zardo IL, Ferronatto JV, Trojan MM, Franck KR, de Azevedo AF, Spiering AG, Nunes LN, Fadel LJJoFM and Surgery. Evaluation of the effects of gabapentin on the physiologic and echocardiographic variables of healthy cats: a prospective, randomized and blinded study. 2022;24:e498-e504. [CrossRef]
- Vetter SW and Leclerc EJEJoB. Novel aspects of CaM target recognition and activation. 2003;270:404-414. [CrossRef]
- Allen ME, LeBlanc NL and Scollan KFJJotAAHA. Hemodynamic, echocardiographic, and sedative effects of oral gabapentin in healthy cats. 2021;57:278-284. [CrossRef]
- Robert Lee Page I, Cantu M, Lindenfeld J, Hergott LJ and Lowes BDJJoCM. Possible heart failure exacerbation associated with pregabalin: case discussion and literature review. 2008;9:922-925. [CrossRef]
- De Smedt RH, Jaarsma T, Van Den Broek SA and Haaijer-Ruskamp FMJBjocp. Decompensation of chronic heart failure associated with pregabalin in a 73-year-old patient with postherpetic neuralgia: a case report. 2008;66:327. [CrossRef]
- Murphy N, Mockler M, Ryder M, Ledwidge M and McDonald KJJocf. Decompensation of chronic heart failure associated with pregabalin in patients with neuropathic pain. 2007;13:227-229. [CrossRef]
- Rasimas JJ and Burkhart KKJTAjoem. Cardiac conduction disturbances after an overdose of nefazodone and gabapentin. 2006;24:886-888. [CrossRef]
- Aksakal E, Bakirci EM, Emet M and Uzkeser MJTAJoEM. Complete atrioventricular block due to overdose of pregabalin. 2012;30:2101. e1-4. [CrossRef]
- Klein-Schwartz W, Shepherd JG, Gorman S and Dahl BJJoTCT. Characterization of gabapentin overdose using a poison center case series. 2003;41:11-15. [CrossRef]
- Taylor CP, Gee NS, Su T-Z, Kocsis JD, Welty DF, Brown JP, Dooley DJ, Boden P and Singh LJEr. A summary of mechanistic hypotheses of gabapentin pharmacology. 1998;29:233-249. [CrossRef]
- Cui W, Wu H, Yu X, Song T, Xu X and Xu FJFiCN. The calcium channel α2δ1 subunit: interactional targets in primary sensory neurons and role in neuropathic pain. 2021;15:699731. [CrossRef]
- Lanzetti S and Di Biase VJM. Small molecules as modulators of voltage-gated calcium channels in neurological disorders: state of the art and perspectives. 2022;27:1312.
- Catterall WAJCSHpib. Voltage-gated calcium channels. 2011;3:a003947.
- Chincholkar M. Analgesic mechanisms of gabapentinoids and effects in experimental pain models: a narrative review. Br J Anaesth. 2018;120:1315-1334. [CrossRef]
- Fuller-Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N, Muth JN, Mikala G, Petrashevskaya NN, Jordan MA, Zhang SP, Qin N, Flores CM, Isaacsohn I, Varadi M, Mori Y, Jones WK and Schwartz A. Targeted disruption of the voltage-dependent calcium channel alpha2/delta-1-subunit. Am J Physiol Heart Circ Physiol. 2009;297:H117-24.
- Roden DM, Balser JR, George AL, Jr. and Anderson ME. Cardiac ion channels. Annu Rev Physiol. 2002;64:431-75.
- Eisner D. Calcium in the heart: from physiology to disease. Exp Physiol. 2014;99:1273-82. [CrossRef]
- Maier LS and Bers DM. Role of Ca2+/CaM-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res. 2007;73:631-40.
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1-14.
- Otsuka M, Ebashi F and Imai S. Cardiac Myosin B and Calcium Ions. J Biochem. 1964;55:192-4. [CrossRef]
- Tang W, Sencer S and Hamilton SLJFiB-L. CaM modulation of proteins involved in excitation-contraction coupling. 2002;7:1583-1589.
- Nakai J, Sekiguchi N, Rando TA, Allen PD and Beam KG. Two regions of the ryanodine receptor involved in coupling with L-type Ca2+ channels. J Biol Chem. 1998;273:13403-6.
- Nakai J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG and Allen PD. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;380:72-5. [CrossRef]
- Zhang T and Brown JH. Role of Ca2+/CaM-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res. 2004;63:476-86.
- Schulman H and Anderson ME. Ca/CaM-dependent Protein Kinase II in Heart Failure. Drug Discov Today Dis Mech. 2010;7:e117-e122.
Figure 1.
Effects of a bolus i.v. injection of GBP on cardiovascular activity. Left panel is a representative of an original recording. Red and green arrows indicate start and end of GBP injection respectively. Yellow arrow indicates the maximal effect of GBP. Table 1 shows combined data presented as Mean ± SD; *p < 0.05, and **p < 0.01, compared with before GBP. n = 4/group. ns: no statistical significance. CI: Contractility Index; PTI: Pressure Time Index; IRP-AdP/dt: Isovolumetric relaxation average dP/dt. SD, DD, and CD: systolic, diastolic, and cycle duration.
Figure 1.
Effects of a bolus i.v. injection of GBP on cardiovascular activity. Left panel is a representative of an original recording. Red and green arrows indicate start and end of GBP injection respectively. Yellow arrow indicates the maximal effect of GBP. Table 1 shows combined data presented as Mean ± SD; *p < 0.05, and **p < 0.01, compared with before GBP. n = 4/group. ns: no statistical significance. CI: Contractility Index; PTI: Pressure Time Index; IRP-AdP/dt: Isovolumetric relaxation average dP/dt. SD, DD, and CD: systolic, diastolic, and cycle duration.
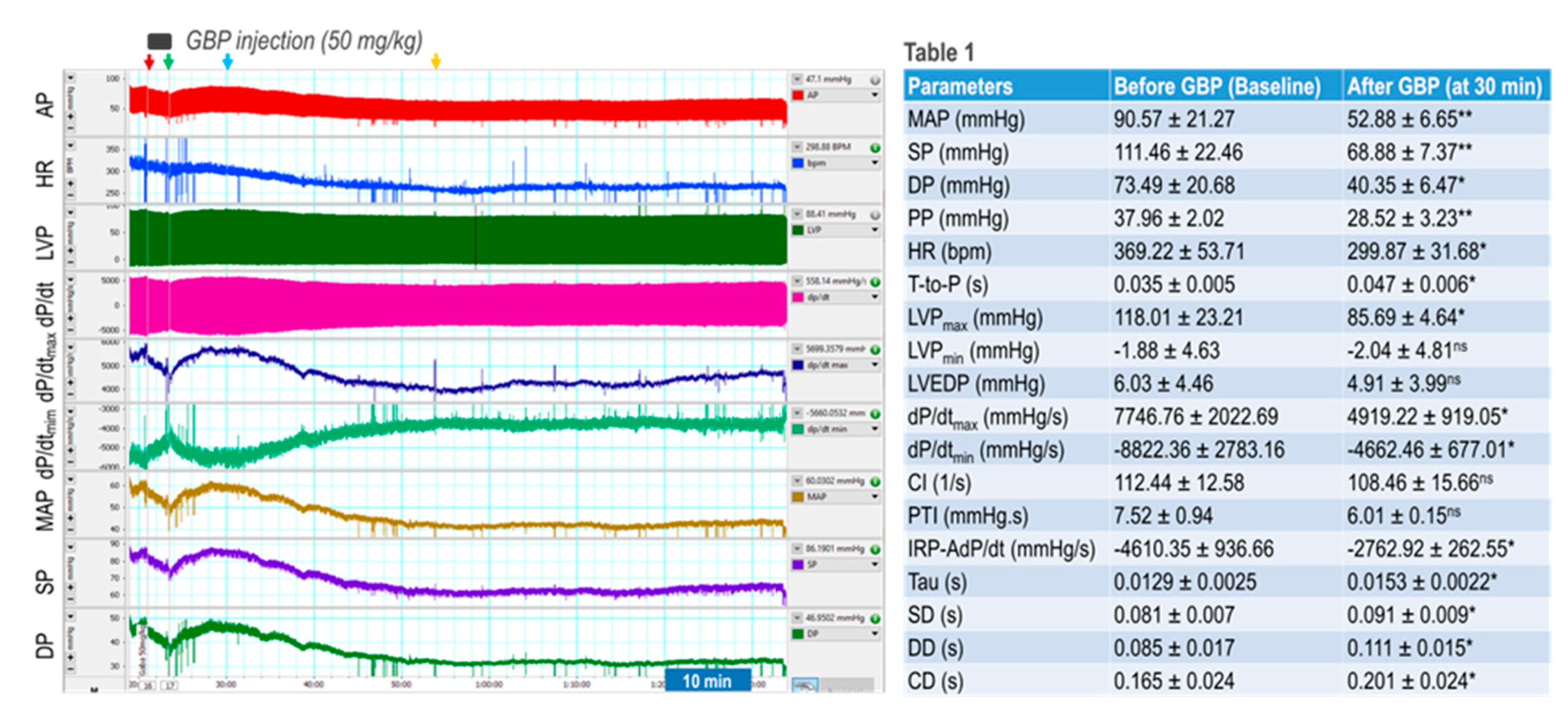
Figure 2.
Effects of 7-day i.p. injection of GBP on cardiovascular function. Left panel is a representative original recording. Table 2 shows combined data presented as Mean ± SD; *p < 0.05, **p < 0.01, and ***p < 0.001, compared with saline-treated group. n = 5/group. ns: no statistical significance.
Figure 2.
Effects of 7-day i.p. injection of GBP on cardiovascular function. Left panel is a representative original recording. Table 2 shows combined data presented as Mean ± SD; *p < 0.05, **p < 0.01, and ***p < 0.001, compared with saline-treated group. n = 5/group. ns: no statistical significance.
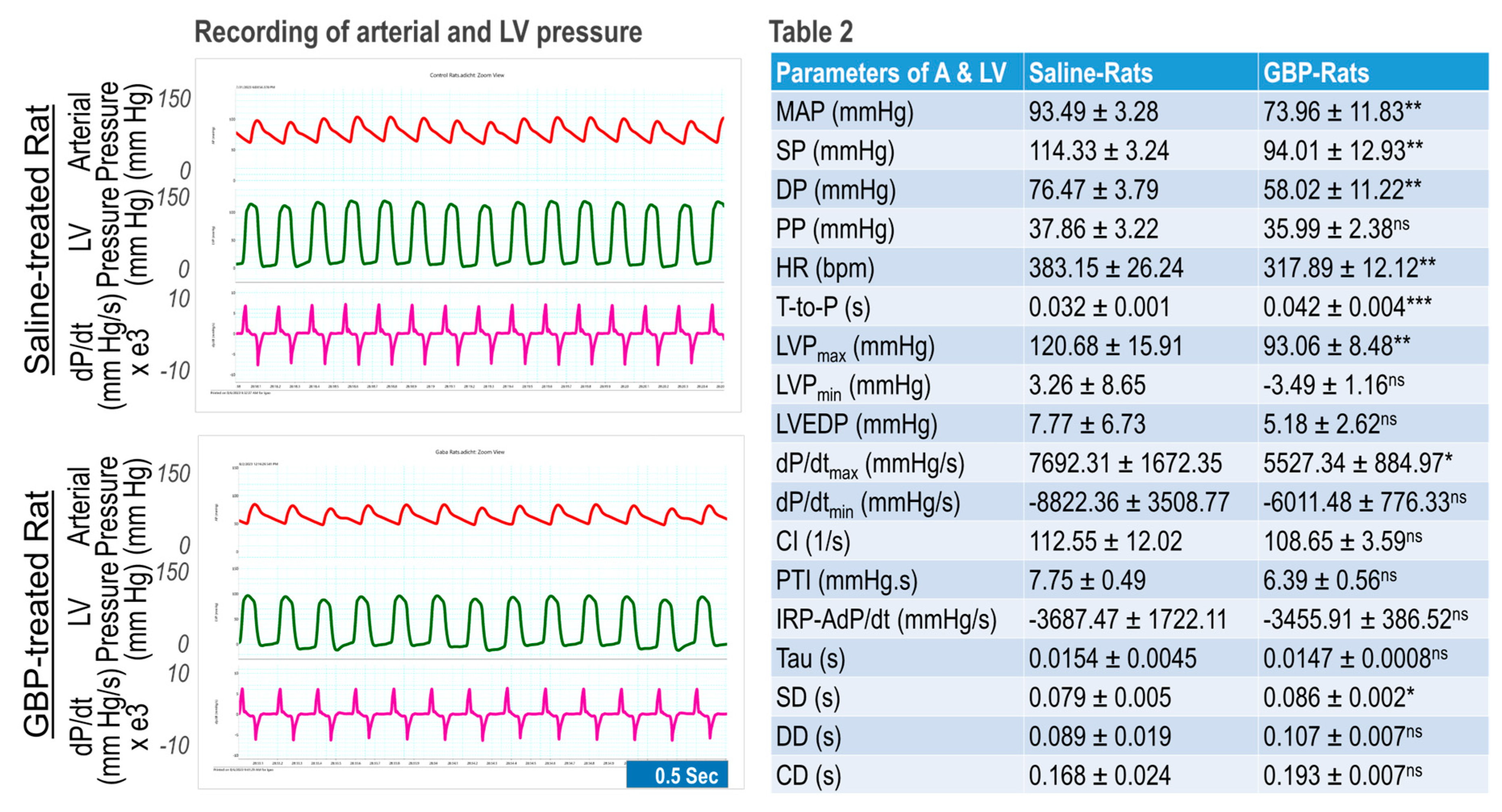
Figure 3.
Plasma norepinephrine concentration of the rats receiving 7-day saline or GBP treatment. n = 5/group.
Figure 3.
Plasma norepinephrine concentration of the rats receiving 7-day saline or GBP treatment. n = 5/group.
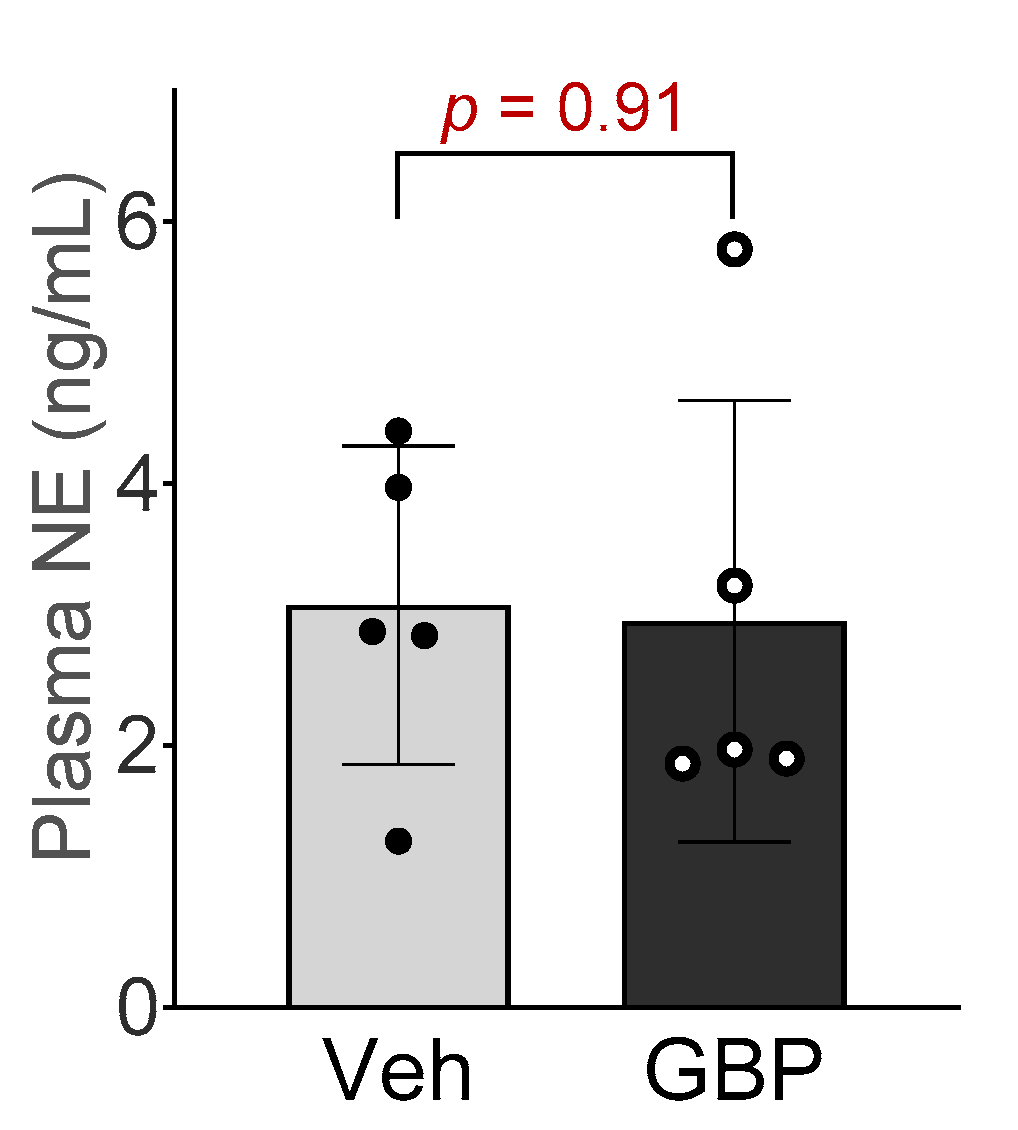
Figure 4.
Differentially expressed proteins in LV myocardium of rats receiving GBP or saline treatment for 7 days. (A) Principal component analysis (PCA) showing a separation occurred between groups. (B) Volcano plot showing the log2 fold change plotted against the -log10 p value; (C) Heatmap showing the log2 of the top 121 differentially expressed proteins with 55 up- and 66 down- regulation. n = 5/group.
Figure 4.
Differentially expressed proteins in LV myocardium of rats receiving GBP or saline treatment for 7 days. (A) Principal component analysis (PCA) showing a separation occurred between groups. (B) Volcano plot showing the log2 fold change plotted against the -log10 p value; (C) Heatmap showing the log2 of the top 121 differentially expressed proteins with 55 up- and 66 down- regulation. n = 5/group.
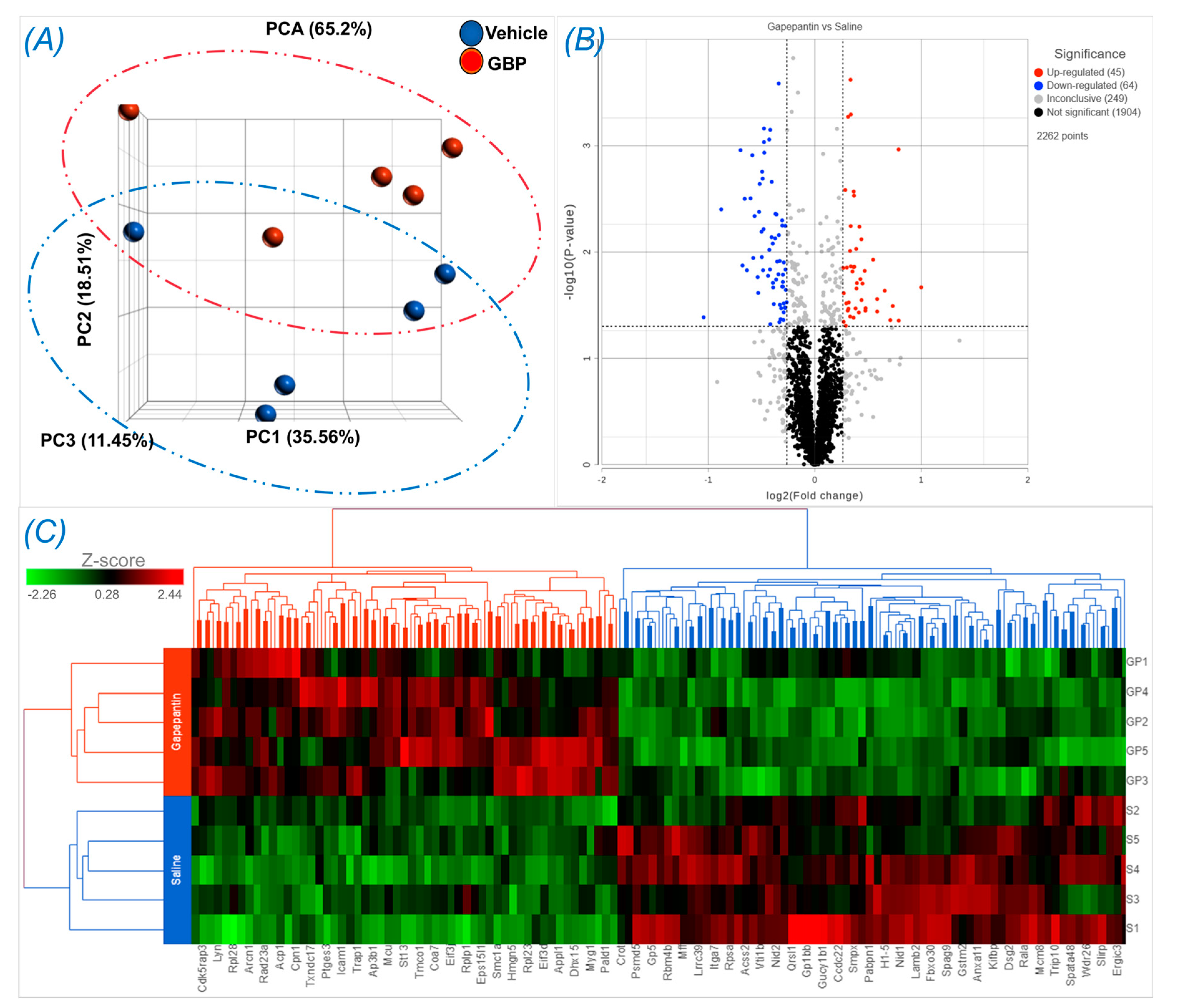
Figure 5.
Pie chart representing the distribution of differentially expressed proteins identified in the LV myocardium of GBP-rats as compared with saline-rats, according to these proteins’ molecular functions (A) and biological processes (B) by using ClueGO analysis.
Figure 5.
Pie chart representing the distribution of differentially expressed proteins identified in the LV myocardium of GBP-rats as compared with saline-rats, according to these proteins’ molecular functions (A) and biological processes (B) by using ClueGO analysis.
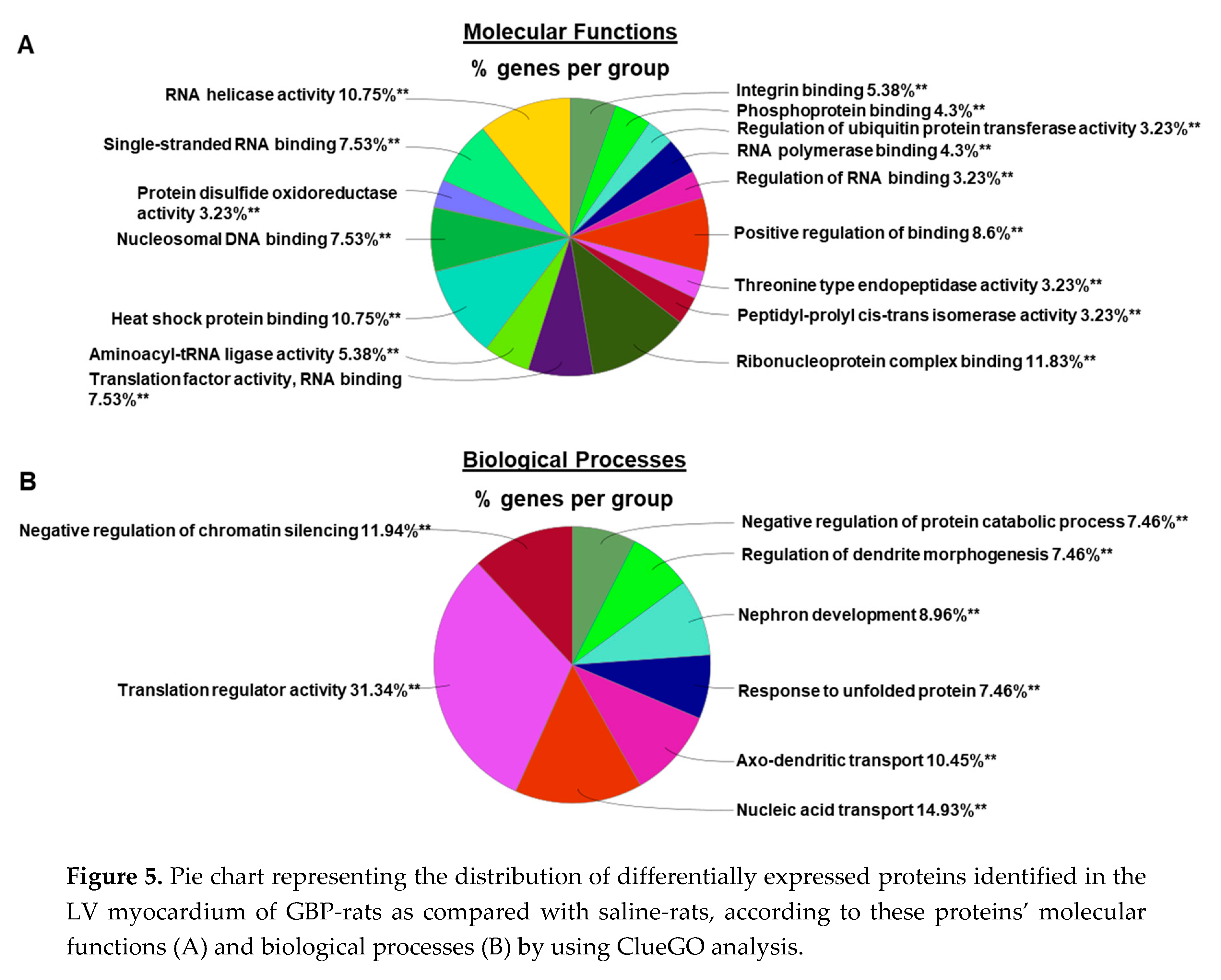
Figure 6.
Western blot data showing CaM expression in LV myocardium (A) and brain cortex (B) of rats receiving 7-day GBP or saline (Veh) treatment. n = 5/group.
Figure 6.
Western blot data showing CaM expression in LV myocardium (A) and brain cortex (B) of rats receiving 7-day GBP or saline (Veh) treatment. n = 5/group.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



