
Submitted:
16 October 2023
Posted:
16 October 2023
You are already at the latest version
Abstract
: With the increase in demand for non-dairy starter cultures and probiotic bacteria as carriers, the use of legumes (also called pulses) as an alternative has gained momentum. In this study, we investigated the diversity of bacterial communities in samples of pigeon pea (Cajanus cajan L. Millsp.) soaked in water for 12 h and 24 h. We soaked 500 g of pigeon pea in sterile distilled water at room temperature (± 25 °C) for 12 h and 24 h; 10 mL of the soaking water was then collected to measure the bacterial diversity using a metataxonomic analysis. The V1–V9 regions on the 16S ribosomal RNA gene were amplified using 27F and 1492R primers under specific polymerase chain reaction conditions for the bacterial identification. Genomic DNA (130 ng) was sequenced on a R9.4 flow cell by Oxford Nanopore Technologies using a GridION sequencer. Library preparations were initiated using a Native Barcoding Kit 24 V14 (SQK-NBD114.24). Primary data were acquired using MinKNOW version 22.05.7. A total of 13 bacterial families and 89 genera were identified in the pigeon pea sample soaked for 12 h; 26 families and 90 genera were identified in the pigeon pea sample soaked for 24 h. Among the bacterial families identified, the five predominant families in both samples were Enterobacteriaceae, Erwiniaceae, Yersiniaceae, Pectobacteriaceae, and Lactobacillaceae. According to the relative abundance of the identified bacterial genera, the following nine genera were predominant in both samples: Enterobacter, Klebsiella, Citrobacter, Pantoea, Kosakonia, Pseudoenterobacter, Pluralibacter, Leclercia, and Kluyvera. At a genus level, a slight increase in the abundance of Klebsiella, Kosakonia, and Pluralibacter and a slight decrease in the abundance of Citrobacter were observed after prolonged incubation from 12 h to 24 h. The values of five diversity indices revealed that the sample soaked in water for 24 h had a richer bacterial abundance and diversity than the 12 h sample. Shannon and Simpson values revealed a higher bacterial diversity in the sample collected at 24 h than the sample collected at 12 h. Species observations and abundance-based coverage estimator (ACE) values demonstrated that the sample collected at 24 h harbored a higher bacterial richness than the sample collected at 12 h. These findings indicated that the bacterial diversity in the pigeon pea samples increased with the soaking time. The bacterial communities during the soaking of the pigeon pea samples were dominated by the Enterobacteriaceae family and Enterobacter genus. The presence of bacterial genera such as Lacticaseibacillus, Lentilactobacillus, and Secundilactobacillus was notable because of their importance as starter cultures for fermented plant-based milk products, including pigeon pea beverages for lactose-intolerant individuals or individuals with malnutrition.
Keywords:
pigeon pea
; bacterial diversity
; metataxonomic analysis
1. Introduction
Functional foods have positive effects on human health. Their physiological properties are determined by their bioactive components. These include dietary fiber, antioxidants, phytochemicals, polyunsaturated fatty acids, prebiotics, probiotics, postbiotics, and synbiotics. In Indonesia, the utilization of local food sources for functional foods is essential to meet the protein needs of rural communities. One example of functional foods produced using local food sources is tempeh, a traditional fermented soybean dish prepared using Rhizopus that is consumed in various countries around the world. [1] reported the health benefits of tempeh and other fermented soy foods due to their antioxidative, antihypertensive, anti-inflammatory, anticancer, and neuroprotective properties. Certain microbes degrade proteins to amino acids during tempeh fermentation; this determines the flavor and aroma of tempeh. The native microbes in soybeans affect the chemical composition and aroma of the final product [2].
Pigeon pea (Cajanus cajan L. Millsp.; local name: kacang gude) is another legume that is widely used as a raw ingredient for tempeh fermentation. It belongs to the Fabaceae family [3]. Indonesian pigeon pea has several advantages over other legume crops, including being tolerant of drought and infertile soils [4] due to its deep roots [5]. Pigeon pea crops can be cultivated in dry areas where soybean plants do not grow well. Its deep rooting does not interfere with the nutrition absorption of other plants; thus, pigeon pea can be intercropped [6].
Pigeon pea is cultivated in several regions in Indonesia due to its edible seeds. The fruit of pigeon pea is a pod that is 4–10 cm long, hairy, flat, and green in color. Pigeon pea seeds are round and small; the number of seeds per pod ranges from four to nine [3]. The pods are straight or crescent in shape and the seed-coat color can be grayish white, cream, yellow, purplish brown, or black. The seed coats are smooth and shiny; the seed weight varies between 4 and 26 g per 100 grains [7]. Pigeon pea seeds comprise a seed coat (14%), an embryo (1%), and cotyledons (85%). The nutritional content of pigeon pea seeds per 100 g is 21.7 g protein, 1.5 g fat, 62.8 g carbohydrates, 12.7 g water content, and 336 kcal total energy [8]. Pigeon pea has traditionally been used as a medicinal plant [9]; however, it contains cyanide and antinutritional compounds. Processing is required to reduce these compounds. Pigeon pea fermentation increases the availability of nutrients as well as the health benefits of pigeon pea as a functional food [10].
Soybean and many other pulses are excellent food sources due to their high amount of dietary fibers, proteins, or micronutrients and phytochemicals. The consumption of pulse products has been somewhat limited due to intestinal disturbances such as flatulence. This is caused by the presence of oligosaccharides such as raffinose and stachyose, which are non-digestible and not assimilated in the small intestine by human GI enzymes; rather, they are fermented by the microbiota and are thus responsible for flatulence [11].
Fermentation is a food biotransformation process that provides positive nutritional and sensory properties to products depending on the microbes being used as starter cultures [12,13]. Certain microbes can synthesize vitamins such as folic acid, riboflavin, niacin, thiamin [14,15], and B12 [16] from the fermentation of grain legumes as a substrate. Other microbes can metabolize n-hexanal and pentanal, which assign a beany flavor to the products [17]. Fermentation can reduce the level of oligosaccharides that cause postprandial flatulence from legumes [18,19] and can decrease antinutritional components such as tannins, phytic acid [20], and trypsin inhibitors [21]. Different strains of the same species can have completely distinct metabolic patterns; consequently, this affects the taste and texture of the product [22].
The process of producing tempeh from pigeon pea is performed in two stages. The first stage is to soak the pigeon pea for approximately 24 h. Soaking results in pigeon pea acidification, which occurs through bacterial activity. At this stage, the pH drops from 7 to 4, which is important for the growth of Rhizopus oryzae. This species is inoculated with pigeon pea in the second stage, which lasts approximately 48 h. Information regarding the types of bacteria that play a role in the acidification of pigeon pea tempeh is limited. Our research intention was to determine the bacteria responsible for pigeon pea acidification and select those that could be used as starter cultures for the preparation of pigeon-pea-based functional food products. In this study, we performed a metataxonomic analysis by sequencing 16S ribosomal RNA (16S rRNA) genes that have been widely used to monitor microbial populations. Our aims were to detect and identify all the bacteria that played a role in the acidification of pigeon pea samples soaked for 12 and 24 h and to measure their abundance in water-immersed pigeon pea samples.
2. Materials and Methods
2.1. Pigeon Pea Collection and Sample Preparation
Pigeon pea samples were collected from Nusa Tenggara Timur Province in Indonesia. These were sorted to obtain peas with good or intact pods. In brief, 500 g of pigeon pea was soaked in sterile distilled water at room temperature (± 25 °C) for 12 and 24 h. Afterward, 10 mL of the soaking water was collected to measure the bacterial diversity using a metagenomic analysis.
2.2. Genomic DNA Extraction
In brief, 10 mL of each soaking-water sample was centrifuged at 2500 × g for 10 min. The obtained pellets were first washed with saline and then with sterile distilled water. These were then used for genomic DNA extraction with a ZymoBIOMICS DNA Miniprep Kit D4300 (Zymo Research, Cambridge, UK). The DNA concentration was determined using NanoDrop spectrophotometers and a Qubit fluorometer (Thermo Fisher, Waltham, MA, USA). The DNA quality was assessed via agarose gel electrophoresis followed by visualization using a Gel-Doc EZ imager (Bio-Rad, CA, USA).
2.2.1. Amplification of the 16s rRNA V1–V9 Regions
The V1–V9 regions of the 16S rRNA gene for the identification of bacterial species were amplified using 27F and 1492R primers under the following PCR conditions: preliminary denaturation at 95 °C for 3 min followed by 5 cycles at 95 °C for 15 s, 55 °C for 15 s, and 72 °C for 30 s; 30 cycles at 95 °C for 15 s, 62 °C for 15 s, and 72 °C for 30 s; and a final extension at 72 °C for 1 min.
2.2.2. Library Preparation and Sequencing
A library was prepared using a Native Barcoding Kit 24 V14 (SQK-NBD114.24) (Oxford Nanopore Technologies, Oxford, UK). The genomic DNA (130 ng) was sequenced using Oxford Nanopore Technology (ONT, Oxford, UK), which provided the long-read sequencing that covered the full-length sequence of the 16S rRNA gene. Sequencing was conducted on an R9.4 flow cell by ONT using a GridION sequencer (ONT, Oxford, UK). Primary data were acquired using MinKNOW version 22.05.7; this is the software used with nanopore sequencing devices.
2.2.3. Bioinformatics Analysis
Base calling was performed using Guppy version 6.1.5 with a high-accuracy model [23]. The quality of FASTQ files was visualized using NanoPlot [24]. The reads were classified using a centrifuge classifier [25]. An index for bacteria and archaea was built using the NCBI 16S RefSeq database (https://ftp.ncbi.nlm.nih.gov/refseq/TargetedLoci/). The downstream analysis and visualizations were performed using Pavian (https://github.com/fbreitwieser/pavian), Krona Tools (https://github.com/marbl/Krona), and RStudio (R version 4.2.0) (https://www.R-project.org/).
3. Results and Discussion
3.1. Metataxonomic Data
The bacterial diversity in the soaking water of the pigeon pea samples soaked for different lengths of time (12 and 24 h) was examined by sequencing the hypervariable regions (V1–V9) of the 16S rRNA gene. The 16S rRNA gene is well-preserved and uniquely found in all bacteria and archaea, suggesting that it could specifically target and identify the bacteria and archaea present in the samples. As all the informative sites of the 16S rRNA gene were considered, the full-length 16S rRNA sequences provided a high level of taxonomic and phylogenetic resolution for our bacterial identification [26].
Shannon and Simpson diversity indices were applied to measure the bacterial diversity in the samples. Both consider the number of species living in a habitat and their relative abundance [27]. The observed species, Chao1, and abundance-based coverage estimator (ACE) indices reflected the sample richness. The values of these five diversity indices are shown in Figure 1. The sample collected at 24 h had a richer genus abundance and diversity than the sample collected at 12 h. The Shannon and Simpson values revealed that the sample collected at 24 h harbored a higher bacterial diversity than the sample collected at 12 h. The species observation and ACE values revealed that the sample collected at 24 h harbored a higher sample richness than the sample collected at 12 h. These findings indicated that the bacterial diversity in the pigeon pea samples increased with the soaking time.
After being soaked in water for 12 and 24 h, the dominant phylum of the pigeon pea samples was Proteobacteria (Figure 2a,b). Within Proteobacteria, Enterobacteriaceae was the predominant family and Enterobacter and Klebsiella were the most abundant genera in the samples (Figure 2a,b). At the species level, Enterobacter ludwigii (15%), Enterobacter asburiae (11%), and Enterobacter cloacae (8%) were the most abundant in the sample soaked in water for 12 h. These three Enterobacter species remained the most dominant species after the pigeon pea sample was soaked in water for 24 h. Other phyla were detected, including Firmicutes and Actinobacter; however, their relative abundance remained below 1.00%. Nur et al. [28] reported that Firmicutes and Actinobacteria were consistently observed in tempeh fermentation; Firmicutes was the most dominant bacteria.
3.2. Bacterial Diversity
The bacterial 16S rRNA gene sequences were classified at the family and genus levels to discover the contents of the bacterial communities observed in pigeon pea samples soaked in water. A total of 13 families (Table 1) and 89 genera (Table 2) were identified in the pigeon pea sample soaked in water for 12 h; 26 bacterial families (Table 3) and 90 genera were identified in the pigeon pea sample soaked in water for 24 h (Table 4).
The relative abundances of the different bacterial communities in all the samples at the family and genus levels are shown in Figure 3. Among the bacterial families observed and identified from the pigeon pea samples soaked for 12 and 24 h, five were predominant. These were Enterobacteriaceae, Erwiniaceae, Yersiniaceae, Pectobacteriaceae, and Lactobacillaceae (Figure 3a). According to the relative abundance of the observed and identified bacterial genera, nine were predominant (Figure 3b). These were Enterobacter, Klebsiella, Citrobacter, Pantoea, Kosakonia, Pseudoenterobacter, Pluralibacter, Leclercia, and Kluyvera (Figure 3b). At the genus level, a slight increase in the abundance of Klebsiella, Kosakonia, and Pluralibacter and a slight decrease in the abundance of Citrobacter were observed after prolonged incubation from 12 h to 24 h (Figure 3b).
The bacterial communities in the soaking water of the pigeon pea samples were dominated by the bacterial genus Enterobacter. The Enterobacter species reported in this study included E. ludwigii, E. cloaceae, E. asburiae, E. chuandaensis, E. sichuanensis, E. quasiroggenkampii, E. oligotrophicus, E. wuhouensis, E. bugandensis, E. cancerogenus, E. mori, and E. kobei. Other genera of the Enterobacteriaceae family were also identified in the pigeon pea samples, including Salmonella and Shigella; these are considered to be pathogenic microbes that cause foodborne diseases (Table 2 and Table 4). The Enterobacteriaceae family is the major causative agent of foodborne diseases and it is widely dispersed in nature. The high abundance of this family in the pigeon pea samples was not surprising because they are present and have been detected in natural ecosystems, including in the gastrointestinal tract of vertebrates as well as in vegetation and aquatic habitats [29].
Small differences in the bacterial communities were observed in the pigeon pea samples with different soaking times. Figure 4 reveals that 231 and 296 bacteria were identified in the pigeon pea samples soaked in water for 12 and 24 h, respectively. From the total number of observed and identified bacteria, those detected in the pigeon pea sample soaked in water for 12 h accounted for 55% and those detected in the sample soaked for 24 h accounted for 70.4%. This difference increased with the soaking time and was consistent with the results of the diversity indices.
The presence of a few members of the Lactobacillaceae family in the soaking water of the pigeon pea samples was notable because certain bacterial species of this family are industrially important as starter cultures for dairy fermentation (Figure 5). The bacterial species belonging to the Lactobacillaceae family observed and identified in this study were Lacticaseibacillus paracasei (32%), Lentilactobacillus parabuchneri (14%), Lentilactobacillus hilgardii (7%), Liquorilactobacillus vini (9%), Liquorilactobacillus nagelii (7%), and Liquorilactobacillus sicerae (2%) (Figure 5). The occurrence of a few lactic acid bacteria species during soybean fermentation has also been reported [30,31]. Lactic acid bacteria belong to the Firmicutes phylum. This group is the dominant bacterial group in tempeh and has a role in the acidification of pigeon pea during soaking [2]. The lactic acid bacteria genera Lacticaseibacillus, Lentilactobacillus, and Liquoriactobacillus observed in this study might have important roles in pigeon pea acidification. Radita et al. [32] reported that during fermentation, the low pH of pigeon pea inhibited the growth of spoilage microorganisms.
The Actinobacteria phylum is another subdominant group in tempeh, but it is present in lesser amounts compared with Firmicutes and Proteobacteria. All three bacterial phyla comprise the lipolytic bacteria in tempeh that play an important role in flavor production [28]. Several Proteobacteria species, including Klebsiella pneumoniae and Citrobacter freundii, play a major role in the production of vitamin B12 in tempeh. Different food products exhibit certain differences in their bacterial communities. Li et al. [33] reported that Firmicutes and Proteobacteria were the predominant phyla in a Chinese traditional fermented broad bean (Vicia faba L.) paste. Peng et al. [34] reported a unique microbial diversity of fermented vegetables obtained from different regions of Hainan, China; Lactobacillus was the most dominant genus. Within this genus, Lactobacillus plantarum was the most abundant species, followed by L. fermentum and L. pentosaceus. Similarly, an analysis of the microbial composition of kimchi revealed that the main bacteria were lactic acid bacteria, including Lactobacillus, Leuconostoc, and Weissella [35].
Previous studies on the microflora of pigeon pea grains have frequently detected Lentilactobacillus and Lactococcus [36,37]. Demarinis et al. [36] reported the isolation and identification of lactic acid bacteria and acetic acid bacteria from pigeon pea grains. Among the identified lactic acid bacteria species, Lentilactobacillus casei, Leuconostoc mesenteroides, and Gluconobacter hansenii were the most abundant. Balogun et al. [37] reported the presence of Lactococcus lactis and Enterococcus faecalis in fermented pigeon pea grains. The Lentilactobacillus genus accounted for approximately 30% of the microbes observed in fermented pigeon pea samples examined in North America. One Lentilactobacillus species, Lentilactobacillus hilgardii, is known to produce exopolysaccharide; this affects the physicochemical quality of products during fermentation. Marsh et al. [38] reported that Leuconostoc species are rarely observed in pigeon pea grains. Other bacterial genera observed at low levels in pigeon pea grains included Acetobacter and Gluconoacetobacter [39]. Gluconoacetobacter was initially present in the grain but disappeared after fermentation. Acetobacteria was detected at low levels in pigeon pea grains but its role remained unclear [39].
Our metataxonomic analysis results revealed the presence of diverse bacterial families and genera in pigeon pea samples soaked in water. During fermentation, certain microbes with a metabolic activity are able to synthesize vitamins, increase the nutritional content, and produce metabolites that affect the taste and texture of the products. Several lactic acid bacteria were observed in our study, suggesting their importance in pigeon pea fermentation. These species should be isolated to assess their capability as starter cultures to develop pigeon-pea-based functional foods or drinks.
4. Conclusions
The metataxonomic analysis of pigeon pea samples soaked in water for 12 and 24 h revealed a diversity of bacterial families and genera, mainly from the Proteobacteria phylum. The main bacterial families observed and detected were Enterobacteriaceae, Erwiniaceae, Yersiniaceae, Pectobacteriaceae, and Lactobacillaceae. The pigeon pea sample collected after 24 h of soaking revealed a higher bacterial diversity and richness than the sample collected after 12 h. Overall, the bacterial communities in the soaking water of the pigeon pea samples were dominated by the Enterobacteriaceae family and the Enterobacter genus. The presence of Lacticaseibacillus, Lentilactobacillus, and Secundilactobacillus was notable because of their importance as starter cultures for the development of fermented pigeon pea beverages for lactose-intolerant individuals. This may prevent malnutrition (e.g., low lysine in pulses) and reduce antinutritional and flatulence factors.
Author Contributions
Conceptualization, W.W., D.W., Y.A.P., and Y.S.; methodology, Y.S., W.W., D.W., Y.A.P., and B.H.L.; validation, Y.S., W.W., D.W., Y.A.P., and B.H.L.; formal analysis, Y.S., W.W., D.W., and Y.A.P.; investigation, Y.S., W.W., D.W., and Y.A.P.; resources, Y.S., and W.W.; data curation, Y.S., W.W., Y.A.P., D.W., and B.H.L.; writing—original draft preparation, Y.S., D.W., Y.A.P., D.W., H.R.P., and B.H.L.; supervision, W.W., D.W., and Y.A.P.; funding acquisition, Y.S., and W.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
All data generated or analyzed during this study have been included in the paper. The raw data are available from the corresponding author on reasonable request.
Acknowledgments
Yuni Sine is a recipient of the LPDP scholarship provided by the Indonesian Government that enables her to pursue a doctoral degree at the Graduate School of Biotechnology, Universitas Gadjah Mada, in Yogyakarta, Indonesia.
Conflicts of Interest
The authors declare no competing interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Qiao, Y.; Zhang, K.; Zhang, Z.; Zhang, C.; Sun, Y.; Feng, Z. Fermented soybean foods: A review of their functional components, mechanism of action and factors influencing their health benefits. Food Res. Int. 2022, 158, 111575. [Google Scholar] [CrossRef]
- Nurdini, A.L.; Nuraida, L.; Suwanto, A.; Suliantari. Microbial growth dynamics during tempe fermentation in two different home industries. Int. Food Res. J. 2015, 22, 1668–1674, https://api.semanticscholar.org/CorpusID:27006181. [Google Scholar]
- Valenzuela, H.; Smith, J. Cowpea: Cooperative Extension Service; College of Tropical Agriculture and Human Resources, University of Hawai: Honolulu, HI, USA, 2002; pp. 1–4. [Google Scholar]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.A.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeon pea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83–89. [Google Scholar] [CrossRef]
- Odeny, D.A. The potential of pigeon pea (Cajanus cajan (L.) Millsp.) in Africa. Nat. Resour. Forum. 2007, 31, 297–305. [Google Scholar] [CrossRef]
- Sheahan, C.M. Plant Guide for Pigeon pea (Cajanus cajan). USDA-Natural Resources Conservation Service; Cape May Plant Materials Center: Cape May, NJ, USA, 2012; p. 08210. [Google Scholar]
- Van der Maesen, L.J.G. Taxonomy of Cajanus. In Proceedings of the International Workshop on Pigeonpeas, Patancheru, India, 15–19 December 1980; Volume 2, pp. 9–14. [Google Scholar]
- Abebe, B.K. The dietary use of pigeon pea for human and animal diets. Sci. World J. 2022, 4873008. [Google Scholar] [CrossRef]
- Akande, K.E.; Abubakar, M.M.; Adegbola, T.A.; Bogoro, S.E.; Doma, U.D. Chemical evaluation of the nutritive quality of pigeon pea (Cajanus cajan (L). Millsp.). Int. Poult. Sci. 2010, 9, 63–65, https://api.semanticscholar.org/CorpusID:86930724. [Google Scholar] [CrossRef]
- Nwosu, J.N.; Ojukwu, M.; Ogueke, C.C.; Ahaotu, I.; Owuamanam, C.I. The antinutritional properties and ease of dehulling on the proximate composition of pigeon pea (Cajanus cajan) as affected by malting. Int. J. Life Sci. 2013, 2, 60–67, http://www.crdeepjournal.org/wp-content/uploads/2013/05/Vol-2-2-1-IJLS.pdf. [Google Scholar]
- Liu, L.; Chen, X.; Hao, L.; Zhang, G.; Jin, Z.; Li, C.; Yang, Y.; Rao, J.; Chen, B. Traditional fermented soybean products: Processing, flavor formation, nutritional and biological activities. Crit. Rev. Food Sci. Nutr. 2022, 62, 1971–1989. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.Y.; Li, H.B.; Gunaratne, A.; Sui, Z.Q.; Corke, H. Effects of fermented edible seeds and their products on human health: Bioactive components and bioactivities. Compreh. Rev. Food Sci. Food Saf. 2017, 16, 489–531. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, B.K.; Gowen, A.; McKenna, B. Pulse Foods: Processing, Quality and Nutraceutical Applications; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Hou, J.W.; Yu, R.C.; Chou, C.C. Changes in some components of soymilk during fermentation with bifidobacteria. Food Res. Int. 2000, 33, 393–397. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, L.; Ziao, G.; Feng, J.; Zhou, H.; Huang, F. Changes in some nutritional components of soymilk during fermentation by the culinary and medicinal mushroom Grifola frondosa. LWT 2015, 62, 468–473. [Google Scholar] [CrossRef]
- Gu, Q.; Zhang, C.; Song, D.; Li, P.; Zhu, X. Enhancing vitamin B12 content in soy-yogurt by Lactobacillus reuteri. Int. J. Food Microbiol. 2015, 206, 56–59. [Google Scholar] [CrossRef]
- Desai, A.; Small, D.M.; MCGill, A.E.J.; Shah, N.P. Metabolism of Raffinose and Stachyose in Reconstituted Skim Milk and of n-Hexanal and Pentanal in Soymilk by Bifidobacteria. Biosci. Microflora 2002, 21, 245–250. [Google Scholar] [CrossRef]
- Sandberg, A.S. Developing functional ingredients: A case study of pea protein. In Functional Foods; Woodhead Publishing: Sawston, UK, 2011; pp. 358–382. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, R.; Yang, H.; Chou, C.C. Sugar and acid contents in soymilk fermented with lactic acid bacteria alone or simultaneously with bifidobacteria. Food Microbiol. 2002, 20, 333–338. [Google Scholar] [CrossRef]
- Fredrikson, M.; Andlid, T.; Haikara, A.; Sandberg, A.S. Phytate degradation by micro-organisms in synthetic media and pea flour. J. Appl. Microbiol. 2002, 93, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumari, S.; Wongputtisin, P.; Nout, M.J.; Sarkar, P.K. Optimization of soybean processing into kinema, a Bacillus-fermented alkaline food, with respect to a minimum level of antinutrients. J. Appl. Microbiol. 2015, 119, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Hickisch, A.; Beer, R.; Vogel, R.F.; Toelstede, S. Influence of lupin milk heat treatment and exopolysaccharide-producing lactic acid bacteria on the physical characteristics of lupin-based yogurt alternatives. Food Res. Int. 2016, 84, 180–188. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network base calling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef]
- de Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef]
- Bahram, M.; Anslan, S.; Hildebrand, F.; Bork, P.; Tedersoo, L. Newly designed 16S rRNA metabarcoding primers amplify divers and novel archaeal taxa from the environment. Environ. Microbiol. Rep. 2018, 11, 487–494. [Google Scholar] [CrossRef]
- Fang, R.S.; Chen, Y.C.; Chen, Q.H. Bacterial diversity analysis during the fermentation of traditional Chinese yellow rice wine revealed by 16S rDNA 454 pyrosequencing. J. Food Sci. 2015, 80, M2265–M227. [Google Scholar] [CrossRef]
- Nur, N.; Meryandini, A.; Suhartono, M.T.; Suwanto, A. Lipolytic bacteria and the dynamics of flavor production in Indonesian tempeh. Biodiversitas J. Biol. Divers. 2020, 21, 3818–3825. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. The changing face of the family Enterobacteriaceae (Order: “Enterobacterales”): New members, taxonomical issues, geographic expansion, and new diseases and disease syndromes. Clin. Microbiol. Rev. 2021, 34, 2. [Google Scholar] [CrossRef]
- Sirilun, S.; Sivamaruthi, B.S.; Kesika, P.; Peerajan, S.; Chaiyasut, C. Lactic acid bacteria mediated fermented soybean as a potent nutraceutical candidate. Asian Pac. J. Trop. Biomed. 2017, 7, 930–936. [Google Scholar] [CrossRef]
- Ma, H.; Wang, L.; Yu, H.; Wang, W.; Wu, G.; Qin, G.; Tan, Z.W.Y.; Pang, H. Protease-producing lactic acid bacteria with antibacterial properties and their potential use in soybean meal fermentation. Chem. Biol. Technol. Agric. 2022, 9, 1–17. [Google Scholar] [CrossRef]
- Radita, R.; Suwanto, A.; Wahyudi, A.T.; Rusmana, I. Firmicutes is the predominant bacteria in tempeh. Int. Food Res. J. 2018, 25, 2313–2320, https://api.semanticscholar.org/CorpusID:202544934. [Google Scholar]
- Li, Z.; Rui, J.; Li, X.; Li, J.; Dong, L.; Huang, Q.; Huang, C.; Wang, Z.; Li, L.; Xuan, P.; et al. Bacterial community succession and metabolite changes during doubanjiang-meju fermentation, a Chinese traditional fermented broad bean (Viciafaba L. ) paste. Food Chem. 2017, 218, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Jiang, S.; Chen, J.; Ma, C.; Huo, D.; Shao, Y.; Zhiang, J. Unique microbial diversity and metabolic pathway features of fermented vegetables from Hainan, China. Front. Microbiol. 2018, 9, 309. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Chun, J.B. Bacterial community structure in Kimchi, a Korean fermented vegetable food, as revealed by 16S rRNA gene analysis. Int. J. Food Microbiol. 2005, 103, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Demarinis, C.; Verni, M.; Pinto, L.; Rizzello, C.G.; Baruzzi, F. Use of Selected Lactic Acid Bacteria for the Fermentation of Legume-Based Water Extracts. Foods. 2022, 11, 21–3346. [Google Scholar] [CrossRef] [PubMed]
- Balogun, M.A.; Ahmed, R.N.; Akintayo, O.A.; Aruna, T.E.; Omovbude, M.O.; Shittu, T. Microbial, chemical and sensory evaluation of pigeon pea condiment from wild and controlled fermentation. Ceylon J. Sci. 2021, 50, 269–277. [Google Scholar] [CrossRef]
- Marsh, A.J.; O’Sullivan, O.; Hill, C.; Ross, R.P.; Cotter, P.D. Sequencing-based analysis of the bacterial and fungal composition of kefir grains and milks from multiple sources. PLoS ONE 2013, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gulitz, A.; Stadie, J.; Wenning, M.; Ehrmann, M.A.; Vogel, R.F. The microbial diversity of water kefir. Int. J. Food Microbiol. 2011, 151, 284–288. [Google Scholar] [CrossRef]
Figure 1.
Diversity index box figure of bacteria in pigeon pea samples at different soaking times: (a) observed species index; (b) Chao1 index; (c) ACE index; (d) Shannon index; (e) Simpson index.
Figure 1.
Diversity index box figure of bacteria in pigeon pea samples at different soaking times: (a) observed species index; (b) Chao1 index; (c) ACE index; (d) Shannon index; (e) Simpson index.
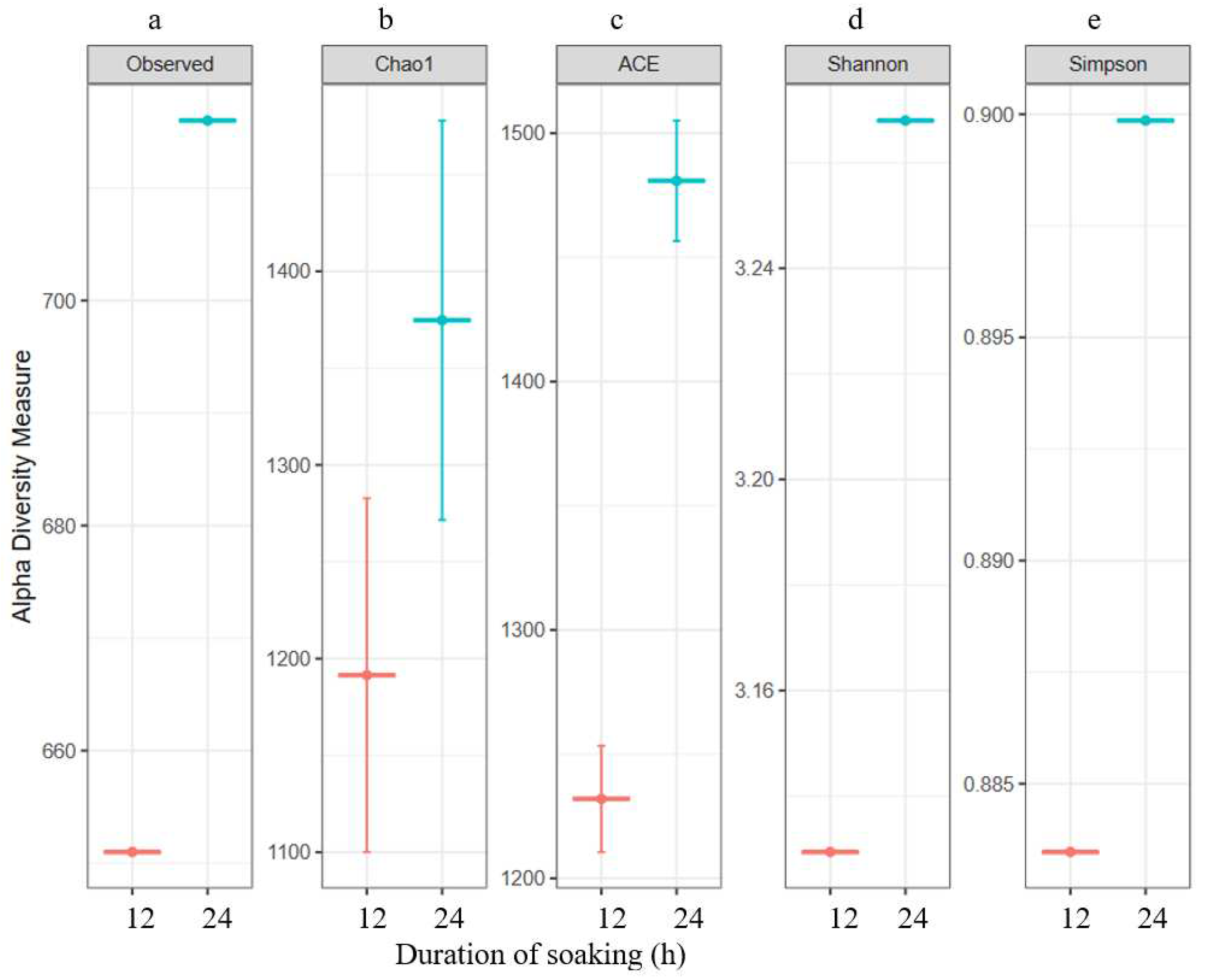
Figure 2.
Bacterial composition of pigeon pea samples after being soaked in water: (a) 12 h; (b) 24 h.
Figure 2.
Bacterial composition of pigeon pea samples after being soaked in water: (a) 12 h; (b) 24 h.
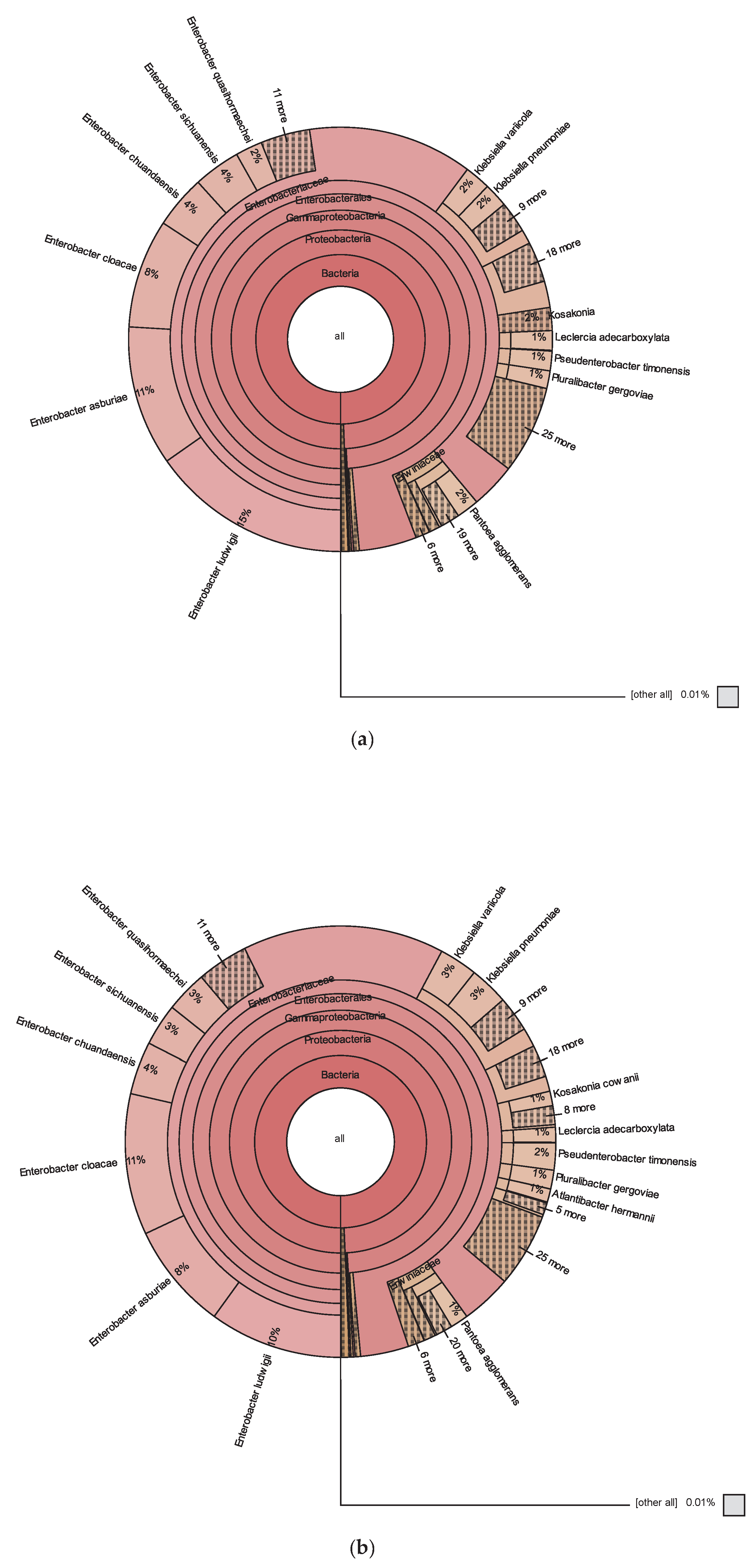
Figure 3.
Relative abundance of bacterial families and genera in pigeon pea samples soaked: (a) 12 h; (b) 24 h.
Figure 3.
Relative abundance of bacterial families and genera in pigeon pea samples soaked: (a) 12 h; (b) 24 h.
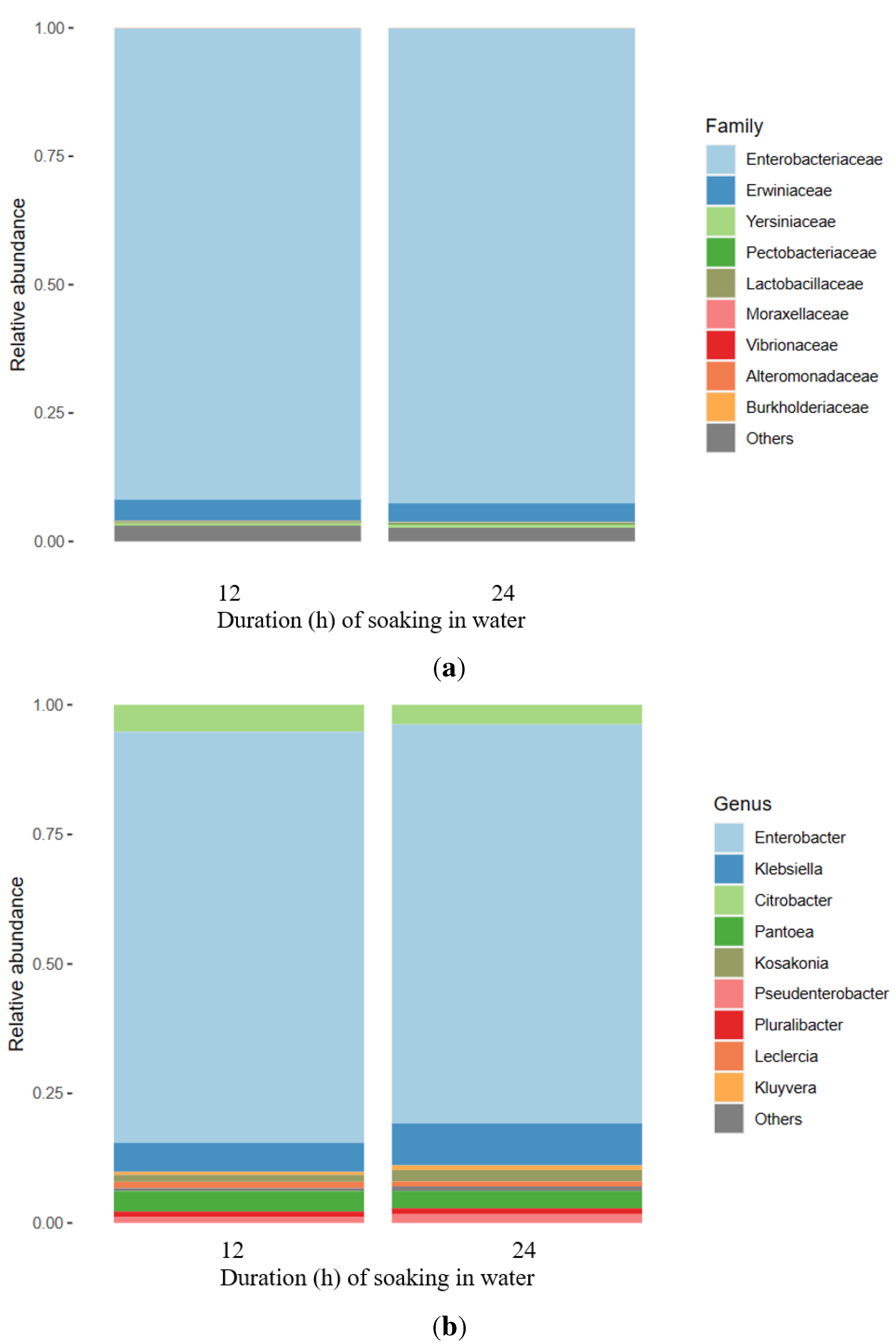
Figure 4.
Venn diagram of pigeon pea samples soaked in water for different times.
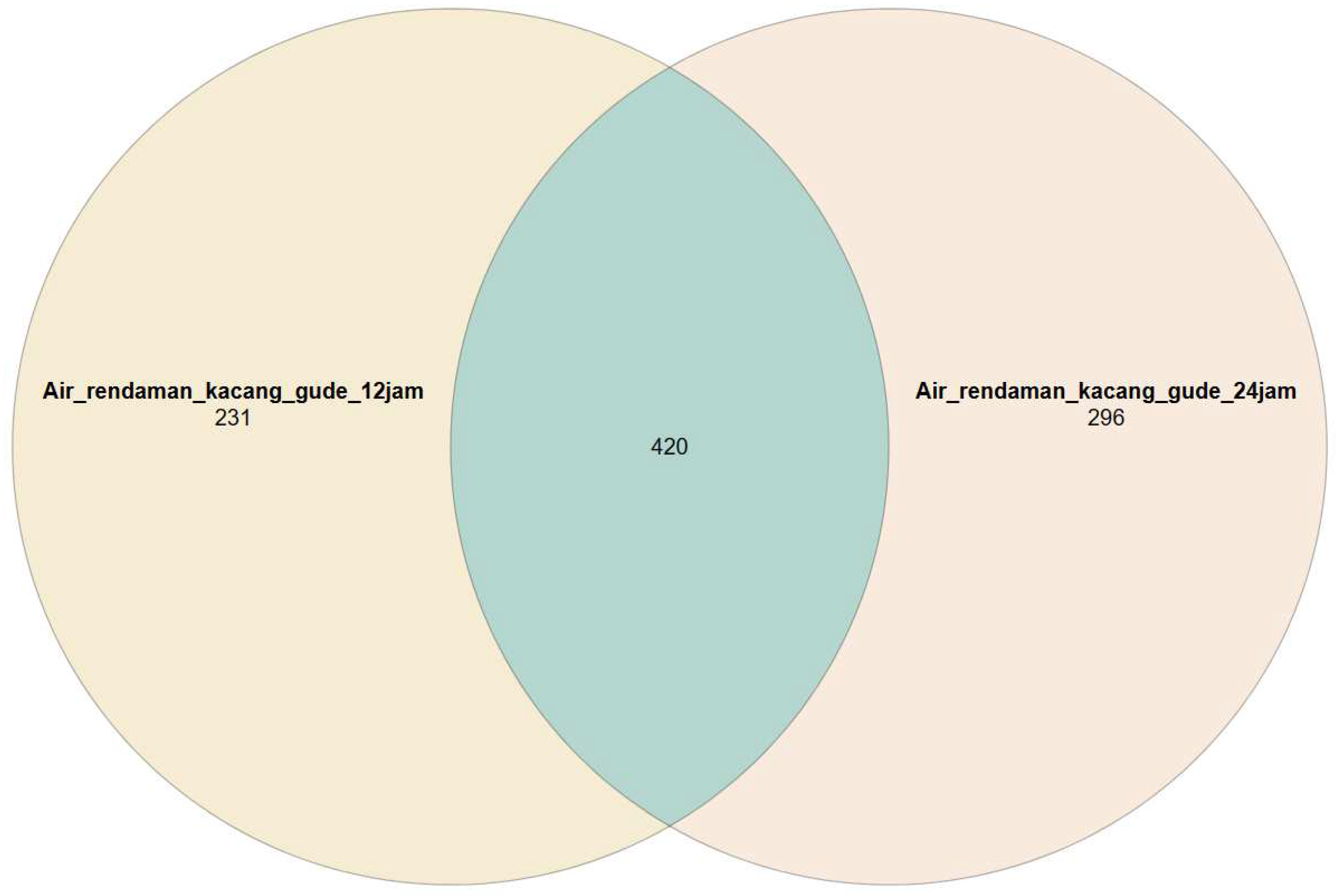
Figure 5.
Bacterial composition of Lactobacillaceae family in pigeon pea sample soaked for 24 h.
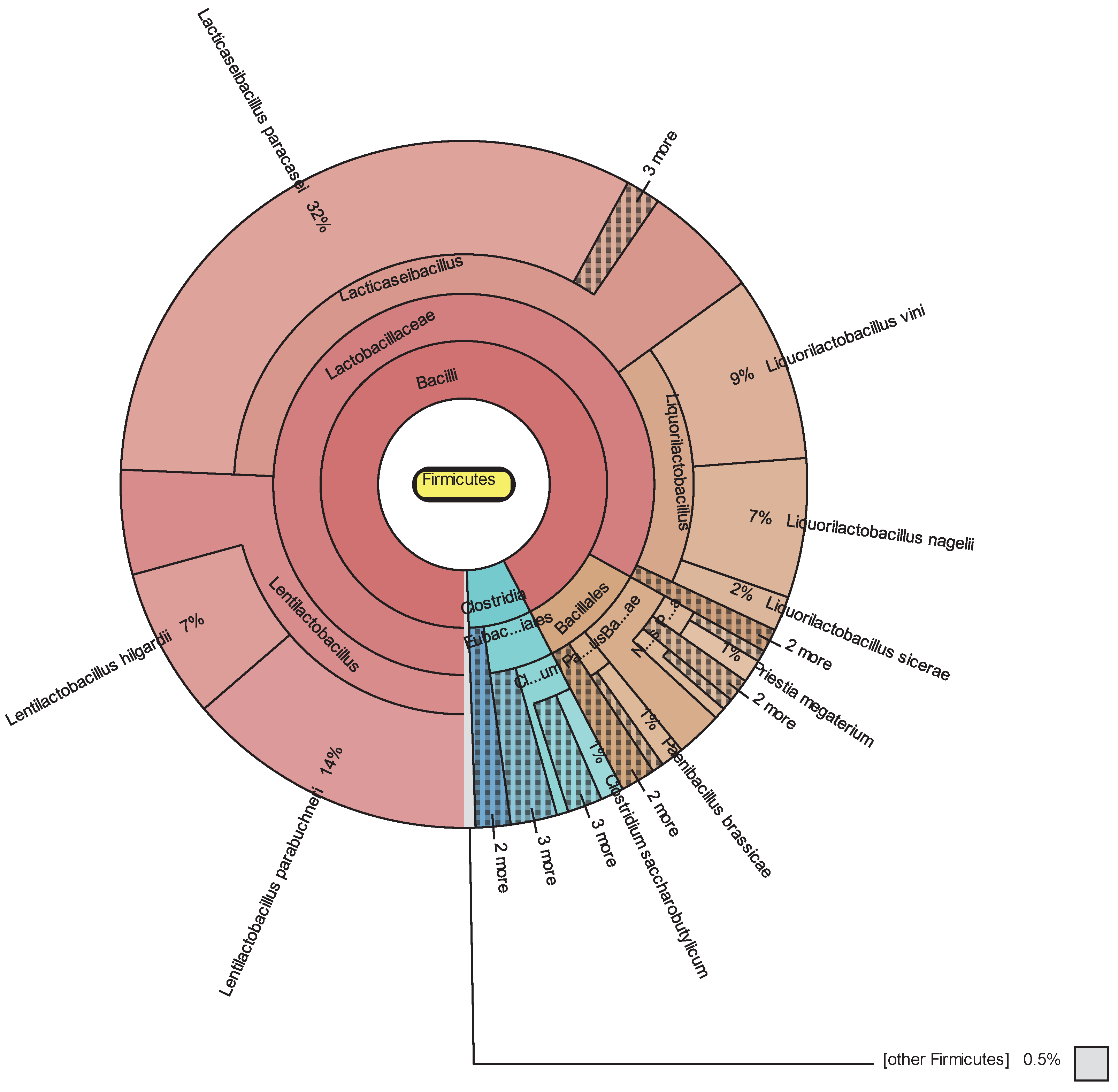

Table 1.
Classification of the bacterial families of the pigeon pea sample soaked in water for 12 h using the Centrifuge nt database.
Table 1.
Classification of the bacterial families of the pigeon pea sample soaked in water for 12 h using the Centrifuge nt database.
| Family Name | taxID | taxRank | Number of Uniquely Classified Reads |
|---|---|---|---|
| Enterobacteriaceae | 543 | Family | 2607 |
| Erwiniaceae | 1903409 | Family | 22 |
| Pectobacteriaceae | 1903410 | Family | 10 |
| Calotrichaceae | 2661849 | Family | 4 |
| Halomonadaceae | 28256 | Family | 2 |
| Morganellaceae | 1903414 | Family | 2 |
| Vibrionaceae | 641 | Family | 2 |
| Alteromonadaceae | 72275 | Family | 1 |
| Balneolaceae | 1813606 | Family | 1 |
| Flavobacteriaceae | 49546 | Family | 1 |
| Intrasporangiaceae | 85021 | Family | 1 |
| Kofleriaceae | 224464 | Family | 1 |
| Micrococcaceae | 1268 | Family | 1 |
Table 2.
Classification of the bacterial genera of the pigeon pea sample soaked in water for 12 h using the Centrifuge nt database.
Table 2.
Classification of the bacterial genera of the pigeon pea sample soaked in water for 12 h using the Centrifuge nt database.
| Genus Name | taxID | taxRank | Number of Uniquely Classified Reads |
|---|---|---|---|
| Enterobacter | 547 | Genus | 9484 |
| Citrobacter | 544 | Genus | 1516 |
| Klebsiella | 570 | Genus | 819 |
| Pantoea | 53335 | Genus | 138 |
| Kluyvera | 579 | Genus | 100 |
| Kosakonia | 1330547 | Genus | 98 |
| Serratia | 613 | Genus | 85 |
| Tatumella | 82986 | Genus | 54 |
| Erwinia | 551 | Genus | 46 |
| Cronobacter | 413496 | Genus | 30 |
| Vibrio | 662 | Genus | 23 |
| Pectobacterium | 122277 | Genus | 21 |
| Bacillus | 1386 | Genus | 18 |
| Dickeya | 204037 | Genus | 14 |
| Rahnella | 34037 | Genus | 14 |
| Buttiauxella | 82976 | Genus | 13 |
| Mangrovibacter | 451512 | Genus | 12 |
| Planctopirus | 1649480 | Genus | 12 |
| Pseudocitrobacter | 1504576 | Genus | 11 |
| Cedecea | 158483 | Genus | 10 |
| Lentilactobacillus | 2767893 | Genus | 10 |
| Mixta | 2100764 | Genus | 9 |
| Acinetobacter | 469 | Genus | 9 |
| Franconibacter | 1649295 | Genus | 8 |
| Chimaeribacter | 2716544 | Genus | 7 |
| Lelliottia | 1330545 | Genus | 7 |
| Edwardsiella | 635 | Genus | 6 |
| Actinobacillus | 713 | Genus | 5 |
| Escherichia | 561 | Genus | 5 |
| Lacticaseibacillus | 2759736 | Genus | 5 |
| Pseudomonas | 286 | Genus | 4 |
| Yersinia | 629 | Genus | 4 |
| Izhakiella | 1780190 | Genus | 3 |
| Leclercia | 83654 | Genus | 3 |
| Shigella | 620 | Genus | 3 |
| Acetomicrobium | 49894 | Genus | 2 |
| Bradyrhizobium | 374 | Genus | 2 |
| Brenneria | 71655 | Genus | 2 |
| Brevitalea | 2048911 | Genus | 2 |
| Gibbsiella | 929812 | Genus | 2 |
| Haemophilus | 724 | Genus | 2 |
| Lonsdalea | 1082702 | Genus | 2 |
| Paraburkholderia | 1822464 | Genus | 2 |
| Psychromonas | 67572 | Genus | 2 |
| Raoultella | 160674 | Genus | 2 |
| Trabulsiella | 158851 | Genus | 2 |
| Xenorhabdus | 626 | Genus | 2 |
| Actinoplanes | 1865 | Genus | 1 |
| Aeribacillus | 1055323 | Genus | 1 |
| Aeromonas | 642 | Genus | 1 |
| Atlantibacter | 1903434 | Genus | 1 |
| Brucella | 234 | Genus | 1 |
| Burkholderia | 32008 | Genus | 1 |
| Caldimonas | 196013 | Genus | 1 |
| Catenulispora | 414878 | Genus | 1 |
| Crinalium | 241421 | Genus | 1 |
| Croceicoccus | 1295327 | Genus | 1 |
| Flavisolibacter | 398041 | Genus | 1 |
| Gloeobacter | 33071 | Genus | 1 |
| Halomonas | 2745 | Genus | 1 |
| Humidesulfovibrio | 356 | Genus | 1 |
| Hyphomicrobium | 81 | Genus | 1 |
| Iningainema | 1932705 | Genus | 1 |
| Ktedonobacter | 363276 | Genus | 1 |
| Leucothrix | 45247 | Genus | 1 |
| Marichromatium | 85076 | Genus | 1 |
| Massilia | 149698 | Genus | 1 |
| Mathylobacillus | 404 | Genus | 1 |
| Micromonospora | 1873 | Genus | 1 |
| Microvirga | 186650 | Genus | 1 |
| Morganella | 581 | Genus | 1 |
| Motilimonas | 1914248 | Genus | 1 |
| Niastella | 354354 | Genus | 1 |
| Novosphingobium | 165696 | Genus | 1 |
| Oceanisphaera | 225143 | Genus | 1 |
| Paraglaciecola | 1621534 | Genus | 1 |
| Pedomicrobium | 47494 | Genus | 1 |
| Permianibacter | 1649479 | Genus | 1 |
| Phenylobacterium | 20 | Genus | 1 |
| Providencia | 586 | Genus | 1 |
| Pseudoxanthomonas | 83618 | Genus | 1 |
| Rhabdothermincola | 2820403 | Genus | 1 |
| Rhodoplanes | 29407 | Genus | 1 |
| Salmonella | 590 | Genus | 1 |
| Sphingomonas | 13687 | Genus | 1 |
| Thermithiobacillus | 119979 | Genus | 1 |
| Thermosynthropha | 54293 | Genus | 1 |
| Virgisporangium | 65504 | Genus | 1 |
Table 3.
Classification of the bacterial families of the pigeon pea sample soaked in water for 24 h using the Centrifuge nt database.
Table 3.
Classification of the bacterial families of the pigeon pea sample soaked in water for 24 h using the Centrifuge nt database.
| Family Name | taxID | taxRank | Number of Uniquely Classified Reads |
|---|---|---|---|
| Enterobacteriaceae | 543 | Family | 2836 |
| Yersiniaceae | 1903411 | Family | 34 |
| Erwiniaceae | 1903409 | Family | 23 |
| Vibrionaceae | 641 | Family | 6 |
| Calotrichaceae | 2661849 | Family | 5 |
| Pectobacteriaceae | 1903410 | Family | 5 |
| Bacillaceae | 186817 | Family | 4 |
| Burkholderiaceae | 119060 | Family | 2 |
| Methylococcaceae | 403 | Family | 2 |
| Acidobacteriaceae | 204434 | Family | 1 |
| Aeromonadaceae | 84642 | Family | 1 |
| Anaerolineaceae | 292628 | Family | 1 |
| Bruguierivoracaceae | 2812006 | Family | 1 |
| Flavobacteriaceae | 49546 | Family | 1 |
| Gomontiellaceae | 1892255 | Family | 1 |
| Halanaerobiaceae | 972 | Family | 1 |
| Halomonadaceae | 28256 | Family | 1 |
| Heliobacteriaceae | 31984 | Family | 1 |
| Hyphomicrobiaceae | 45401 | Family | 1 |
| Intrasporangiaceae | 85021 | Family | 1 |
| Microbacteriaceae | 85023 | Family | 1 |
| Morganellaceae | 1903414 | Family | 1 |
| Pasteurellaceae | 712 | Family | 1 |
| Pseudomonadaceae | 135621 | Family | 1 |
| Rhodospirillaceae | 41295 | Family | 1 |
| Succinivibrionaceae | 83763 | Family | 1 |
Table 4.
Classification of the bacterial genera of the pigeon pea sample soaked in water for 24 h using the Centrifuge nt database.
Table 4.
Classification of the bacterial genera of the pigeon pea sample soaked in water for 24 h using the Centrifuge nt database.
| Genus Name | taxID | taxRank | Number of Uniquely Classified Reads |
|---|---|---|---|
| Enterobacter | 547 | Genus | 11233 |
| Klebsiella | 570 | Genus | 1061 |
| Citrobacter | 544 | Genus | 842 |
| Kluyvera | 579 | Genus | 150 |
| Kosakonia | 1330547 | Genus | 144 |
| Pantoea | 53335 | Genus | 98 |
| Serratia | 613 | Genus | 68 |
| Tatumella | 82986 | Genus | 58 |
| Erwinia | 551 | Genus | 45 |
| Buttiauxella | 82976 | Genus | 33 |
| Cronobacter | 413496 | Genus | 31 |
| Dickeya | 204037 | Genus | 23 |
| Mangrovibacter | 451512 | Genus | 21 |
| Pectobacterium | 122277 | Genus | 18 |
| Vibrio | 662 | Genus | 17 |
| Planctopirus | 1649480 | Genus | 16 |
| Edwardsiella | 635 | Genus | 13 |
| Rahnella | 34037 | Genus | 12 |
| Shigella | 620 | Genus | 12 |
| Pseudocitrobacter | 1504576 | Genus | 11 |
| Lacticaseibacillus | 2759736 | Genus | 10 |
| Lentilactobacillus | 2767893 | Genus | 9 |
| Acetomicrobium | 49894 | Genus | 8 |
| Lelliottia | 1330545 | Genus | 8 |
| Escherichia | 561 | Genus | 7 |
| Trabulsiella | 158851 | Genus | 6 |
| Chimaeribacter | 2716544 | Genus | 5 |
| Brevitalea | 2048911 | Genus | 4 |
| Rosenbergiella | 1356488 | Genus | 4 |
| Yersinia | 629 | Genus | 4 |
| Gibbsiella | 929812 | Genus | 3 |
| Raoultella | 160674 | Genus | 3 |
| Salmonella | 590 | Genus | 3 |
| Xenorhabdus | 626 | Genus | 3 |
| Bradyrhizobium | 374 | Genus | 2 |
| Burkholderia | 32008 | Genus | 2 |
| Crinalium | 241421 | Genus | 2 |
| Franconibacter | 1649295 | Genus | 2 |
| Gloeobacter | 33071 | Genus | 2 |
| Legionella | 445 | Genus | 2 |
| Microbacterium | 33882 | Genus | 2 |
| Miltoncostaea | 2843200 | Genus | 2 |
| Providencia | 586 | Genus | 2 |
| Pseudaeromonas | 1929090 | Genus | 2 |
| Reyranella | 445219 | Genus | 2 |
| Salinicola | 404432 | Genus | 2 |
| Shewanella | 22 | Genus | 2 |
| Thalassotalea | 1518149 | Genus | 2 |
| Achromobacter | 222 | Genus | 1 |
| Aeromonas | 642 | Genus | 1 |
| Alkalimonas | 265980 | Genus | 1 |
| Ammonifex | 42837 | Genus | 1 |
| Bosea | 85413 | Genus | 1 |
| Caballeronia | 1827195 | Genus | 1 |
| Cephalothrix | 1844514 | Genus | 1 |
| Chloroflexus | 1107 | Genus | 1 |
| Clostridium | 1485 | Genus | 1 |
| Conexibacter | 191494 | Genus | 1 |
| Defluviitalea | 1185408 | Genus | 1 |
| Dongia | 1146845 | Genus | 1 |
| Euryhalinema | 2661529 | Genus | 1 |
| Halomonas | 2745 | Genus | 1 |
| Iningainema | 1932705 | Genus | 1 |
| Laceyella | 292635 | Genus | 1 |
| Leclercia | 83654 | Genus | 1 |
| Lonsdalea | 1082702 | Genus | 1 |
| Lysobacter | 68 | Genus | 1 |
| Marinicella | 863253 | Genus | 1 |
| Massilia | 149698 | Genus | 1 |
| Motilibacter | 1434021 | Genus | 1 |
| Motilimonas | 1914248 | Genus | 1 |
| Neobacillus | 2675232 | Genus | 1 |
| Nitrospira | 1234 | Genus | 1 |
| Oceanisphaera | 225143 | Genus | 1 |
| Paraburkholderia | 1822464 | Genus | 1 |
| Pelagicoccus | 455433 | Genus | 1 |
| Phenylobacterium | 20 | Genus | 1 |
| Photobacterium | 657 | Genus | 1 |
| Pseudoalteromonas | 53246 | Genus | 1 |
| Pseudomonas | 286 | Genus | 1 |
| Psychromonas | 67572 | Genus | 1 |
| Rhizobium | 379 | Genus | 1 |
| Secundilactobacillus | 2767892 | Genus | 1 |
| Shimwellia | 1335483 | Genus | 1 |
| Solirubrobacter | 207599 | Genus | 1 |
| Tepidimonas | 114248 | Genus | 1 |
| Thermoanaerobacter | 1754 | Genus | 1 |
| Thermodesulfovibrio | 28261 | Genus | 1 |
| Trinickia | 2571160 | Genus | 1 |
| Tumebacillus | 432330 | Genus | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



